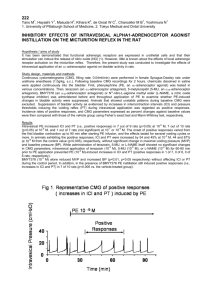
European Journal of Pharmacology 546 (2006) 120 – 126 www.elsevier.com/locate/ejphar EDHF-mediated rapid restoration of hypotensive response to acetylcholine after chronic, but not acute, nitric oxide synthase inhibition in rats Kaushik M. Desai a,⁎, Venkat Gopalakrishnan a , Linda M. Hiebert b , J. Robert McNeill a , Thomas W. Wilson c a Department of Pharmacology, University of Saskatchewan, Saskatoon, Canada Department of Veterinary Biomedical Sciences, University of Saskatchewan, Saskatoon, Canada Department of Medicine, Royal University Hospital, University of Saskatchewan, Saskatoon, Canada b c Received 24 March 2006; received in revised form 22 June 2006; accepted 27 June 2006 Available online 5 July 2006 Abstract Several in vitro studies have shown that endothelium-dependent vasodilatation is maintained by endothelium-derived hyperpolarizing factor (EDHF) or prostacyclin in vessels isolated from endothelial nitric oxide synthase knockout mice. Since this has not been addressed by in vivo studies, we sought to define the magnitude and the onset time of this compensation by recording blood pressure responses to endotheliumdependent vasodilators in rats treated acutely or chronically with the NOS inhibitor, Nω-nitro-L-arginine methyl ester (L-NAME). Groups of male Sprague–Dawley rats were given plain water (control) or L-NAME (0.7 mg/ml) in drinking water for 1 day, 5 days, 3 wks or 6 wks. Dosedependent hypotensive responses to acetylcholine, bradykinin and sodium nitroprusside were determined in anesthetized rats before and after acute intravenous infusion of either L-NAME or a combination of apamin plus charybdotoxin that would selectively inhibit EDHF. Acute LNAME treatment increased the mean arterial pressure and inhibited acetylcholine- and bradykinin-induced fall in blood pressure in control but not in chronic L-NAME treated rats. The endothelium-dependent hypotensive responses to acetylcholine and bradykinin were restored in rats treated with L-NAME after a time period of 24 h along with increased sensitivity to sodium nitroprusside and reduced plasma nitrate + nitrite levels. While apamin + charybdotoxin pretreatment inhibited the responses to acetylcholine and bradykinin in both acute and chronic L-NAME treated groups, it was more pronounced in the latter group. In conclusion, chronic inhibition of nitric oxide synthase results in the development of a compensatory hypotensive response to acetylcholine within 24 h and this is mediated by EDHF. © 2006 Elsevier B.V. All rights reserved. Keywords: Nitric oxide; EDHF; Endothelium; Vasodilation; Nitric oxide synthase; In vivo 1. Introduction Acetylcholine induces an endothelium-dependent vasodilatation and a transient hypotensive response in vivo (Furchgott and Zawadzki, 1980; Rees et al., 1990). Endothelial nitric oxide synthase (eNOS) mediates this in vivo response to acetylcholine through nitric oxide (NO) release causing stimulation of soluble guanylate cyclase and formation of cyclic guanosine monophosphate (cGMP) (Murad, 1994; Rees et al., 1990; Wang et al., ⁎ Corresponding author. A120 Health Sciences Building, Department of Pharmacology, University of Saskatchewan, 107 Wiggins Road, Saskatoon, SK, Canada S7N 5E5. Tel.: +1 306 966 2723; fax: +1 306 966 1440. E-mail address: k.desai@usask.ca (K.M. Desai). 0014-2999/$ - see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.ejphar.2006.06.072 1993). In vitro experiments have shown that in large conduit vessels such as the aorta, acetylcholine-induced vasodilatation is predominantly mediated by NO (Freitas et al., 2003; Nagao et al., 1992; Shimokawa et al., 1996). However, in small resistance type vessels such as the mesenteric, hindlimb, coronary and brain pial vessels, besides NO, other mediators such as the endotheliumderived hyperpolarizing factor (EDHF) or prostacyclin, contribute to the endothelium-dependent vasodilator response to agonists (Brandes et al., 2000; Gödecke et al., 1998; Lamping et al., 2000; Meng et al., 1996). EDHF has been proposed to mediate its vasodilator action through the initial activation of small conductance and intermediate conductance calcium-activated potassium channels (KCa) that are present on the endothelium and are sensitive to inhibition by a combination of optimal concentrations K.M. Desai et al. / European Journal of Pharmacology 546 (2006) 120–126 of apamin and charybdotoxin (Busse et al., 2002; Garland and Plane, 1996). A compensatory increase in EDHF and or prostacyclin-mediated vasodilatation to acetylcholine has been demonstrated in blood vessels of eNOS knockout mice (Busse et al., 2002; Gödecke et al., 1998; Iwakiri et al., 2002; Koller et al., 1994; Lamping et al., 2000; Meng et al., 1996; Sun et al., 1999) and high salt treated rats (Katusic, 2002; Sofola et al., 2002). In hypertensive patients, impaired NO release is compensated by an endothelium-derived hyperpolarizing vasodilator mediator (Taddei et al., 1999). In animal models of hypertension, atherosclerosis, hyperlipidemia and diabetes as well as in clinical settings with patients with these disease conditions, a large number of studies have shown evidence of endothelial dysfunction, measured as a reduced level of NO-mediated endothelium-dependent vasodilatation, while others have shown unimpaired endothelium-dependent vasodilation (Boulanger, 1999; Brunner et al., 2005; Chan et al., 2000). This controversy remains unresolved. The relative roles of NOindependent endothelial mediators such as EDHF and/or prostacyclin contributing to endothelium-dependent hypotensive response under such in vivo situations have not been adequately explored. The time-course of development of compensatory hypotensive response, especially after in vivo inhibition of NOS, has not been reported. Here, we attempted to outline the time when the compensatory mechanisms begin and/or when endothelial dysfunction, measured as reduced hypotensive responses to endothelium-dependent agonists, develops after acute and chronic inhibition of NOS over different time periods in Sprague–Dawley rats. In this study, we have used Nω-nitro-L-arginine methyl ester (L-NAME) to inhibit NO formation in adult rats, a situation that more closely, though not ideally, reflects the gradual impairment of NO production that can occur in disease states, as opposed to eNOS knockout animals. 2. Methods and materials The experimental protocols used here were approved and carried out under the guidelines of the Animal Care Committee of the University of Saskatchewan and conform with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). 2.1. In vivo experiments Male Sprague–Dawley rats weighing 300–350 g were used. Different groups of rats (n = 4–6 each group) were given plain drinking water (control) or Nω-nitro-L-arginine methyl ester (LNAME, 0.7 mg/ml, corresponding to a daily intake of approximately 65 mg/kg) (Ribeiro et al., 1992) in drinking water for 1 day, 3 days, 5 days, 3 weeks or 6 weeks. They were anaesthetized with intraperitoneal (i.p.) thiopental sodium, 100 mg/kg (dissolved in saline at 25 mg/ml) (Laight et al., 2000). The rat was placed on a heated pad to maintain the temperature at 37 °C measured by a rectal probe. The trachea was cannulated and the rat was allowed to breathe spontaneously. The right carotid artery and the left jugular vein were cannulated with polythene cannulas 121 (Portex Ltd., Hythe, England). The carotid cannula (id 0.4, od 0.8 mm) was filled with heparinised saline (50 U/ml) and connected to a pressure transducer to record mean arterial pressure using the Powerlab data acquisition system (AD Instruments Pvt. Ltd., Sydney, Australia). Hypotensive responses to endotheliumdependent agonists, acetylcholine and bradykinin, in vivo were recorded as a transient fall in mean arterial pressure (Laight et al., 2000; Rees et al., 1990). The jugular vein cannula (id 0.5, od 0.63 mm) was used to administer drugs as intravenous (i.v.) bolus injections. After a 30 min stabilization period, responses were obtained to various hypotensive agents. Enough time was allowed between responses for the mean arterial pressure to recover to the resting level. 2.2. Responses to drugs Drug doses were calculated as μg/kg body weight. Drugs were injected i.v. in a volume of 0.4 ml/kg body weight and flushed with 0.1 ml saline. Dose-related responses were obtained to acetylcholine (0.02 to 2 μg/kg), bradykinin (0.1 to 30 μg/kg), and to NO-releasing endothelium-independent vasodilators nitroglycerine (0.1 to 50 μg/kg) and sodium nitroprusside (0.2 to 20 μg/kg). A dose that represented the mid-part of the dose–response curve was selected for some agents, for single dose responses in some cases. Thus, responses to acetylcholine (0.02 to 2 μg/kg, whole dose–response curve), bradykinin (3 μg/kg), nitroglycerine (5 μg/ kg) and sodium nitroprusside (2 μg/kg) were obtained before (control) and after L-NAME (100 mg/kg i.v.) (Rees et al., 1990) or apamin (25 μg/kg i.v.) + charybdotoxin (25 μg/kg i.v.) administration in different groups of rats. The minimum effective doses for apamin plus charybdotoxin were determined in preliminary experiments based on previously used doses (Shinde et al., 2005). Apamin plus charybdotoxin were used at only one time-point of chronic L-NAME treatment in one group of rats since their cost was very high for in vivo type studies. 2.3. Measurement of plasma nitrate plus nitrite Levels of plasma nitrate plus nitrite were obtained by the Griess method using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI). 0.5 ml jugular vein blood samples were collected into 10% sodium citrate. The tubes were centrifuged at 4 °C and 3000 rpm for 5 min, and the recovered plasma was stored at − 70 °C. Subsequently, the samples were thawed and transferred to Nanosep® 10K Omega centrifuge filter tubes (Pall Corp., Ann Arbor, Michigan) which employ 10 kDa molecular weight cut-off filters. Tubes were centrifuged at 4 °C and 15,000 g for 20 min to remove the hemoglobin. The filtrate was then transferred in 40 μl aliquots to a 96 well plate, with each sample performed in duplicate. To each well, 10 μl of Enzyme Cofactor mixture and 10 μl of nitrate reductase mixture were added, and the plate was covered and incubated at room temperature for 3 h. After the incubation was complete, 50 μl each of 2% sulphanilamide in 5% phosphoric acid and 0.2% naphthylethylenediamine dihydrochloride was added to each well. After allowing 10 min for optimal color development, we read the absorbance at 540 nm using the Anthos II Plate Reader. 122 K.M. Desai et al. / European Journal of Pharmacology 546 (2006) 120–126 Table 1 Basal values of mean arterial pressure (mean ± S.E.M.) in different groups of rats Treatment group Mean arterial pressure (mmHg) Control Acute L-NAME Chronic L-NAME — 6 weeks Chronic L-NAME — 3 weeks Chronic L-NAME — 5 days Chronic L-NAME — 1 day Apa + ChTx L-NAME + Apa + ChTx 107 ± 5 (n = 10) 149 ± 3 (n = 6)a 135 ± 5 (n = 5)b 133 ± 5 (n = 6)b 133 ± 2 (n = 7)b 138 ± 3 (n = 4)b 111 ± 8 (n = 4) 144 ± 2 (n = 4)a,c Chronic L-NAME was given in drinking water (0.7 mg/ml) for the time indicated. Acute L-NAME (100 mg/kg), apamin (25 μg/kg) and charybdotoxin (25 μg/kg) were administered intravenously. ω L-NAME — N -nitro-L-arginine methyl ester, Apa — apamin, ChTx — charybdotoxin, n = number of rats, aP < 0.001, bP < 0.01 vs. control group, c P < 0.01 vs. Apa + ChTx group. 2.4. Materials Acetylcholine, Nω-nitro-L-arginine methyl ester (L-NAME), apamin, indomethacin, sodium nitroprusside dihydrate and bradykinin acetate were purchased from Sigma-Aldrich Canada Ltd. (Oakville, Ontario, Canada). Charybdotoxin was purchased from Ana Spec (San Jose, CA). Nitroglycerine was purchased from SABEX (Boucherville, Québec, Canada). Thiopental sodium was from Abbott Laboratories Ltd. (SaintLaurent, Québec, Canada). in a group of rats that received acute L-NAME infusion (i.v.) subsequent to receiving chronic L-NAME treatment (Fig. 1A and B). The mean arterial pressures were significantly higher after acute L-NAME, compared to their respective control groups, in both chronic L-NAME treatment group (155± 3 vs. 135± 5 mmHg, P < 0.05, n = 5 each) and acute L-NAME treatment group (153± 3 vs. 103 ± 9 mmHg, P < 0.001, n = 5 each). In the chronic L-NAME treated rats, a single dose of sodium nitroprusside, 2 μg/kg, induced a greater hypotensive response (40± 3% of the baseline, n = 5, P < 0.01) compared to the responses seen in the control group (29 ± 2% of the baseline, n = 5). Plasma nitrate + nitrite levels were significantly lower (P < 0.01) in chronic L-NAME treated group (1.54± 0.31 μM, n = 6) compared to the control group (4.55 ± 0.73 μM, n = 6). In preliminary experiments we have ascertained that pretreatment with indomethacin (10 mg/kg i.p.) did not affect either the basal mean arterial pressure values or the responses to acetylcholine in either the control group or in the chronic LNAME treated rats (data not shown). 3.2. Chronic L-NAME for 3 weeks Acute L-NAME administration led to a significant rightward shift in acetylcholine-evoked fall in mean arterial pressure 2.5. Data analysis Hypotensive responses were calculated as % fall in mean arterial pressure with respect to the baseline mean arterial pressure before each response. We, and others, have shown that the dose-dependent percent fall in mean arterial pressure after acetylcholine, bradykinin, sodium nitroprusside and nitroglycerine is unrelated to the baseline mean arterial pressure (Laight et al., 2000; Weldon et al., 1995). This is also supported by the fact that the mean arterial pressure values were comparable in the rats given acute L-NAME alone and in the rats that were given L-NAME chronically (Table 1). The former group showed inhibition of hypotensive responses to acetylcholine, the later did not, as discussed in results. The values are expressed as mean ± SEM with the number of experiments (n) shown in brackets. The data was analyzed for statistical significance using one-way analysis of variance (ANOVA) with Dunnett's post-test, or Student's unpaired two-tailed t-test, as appropriate. A P value less than 0.05 was deemed significant. 3. Results 3.1. Chronic L-NAME for 6 weeks The hypotensive responses to acetylcholine and bradykinin were inhibited with a significant rightward shift in their dose– response curves in rats that received acute L-NAME treatment. In contrast, the dose–response curves to acetylcholine and bradykinin remained unaffected in chronic L-NAME treatment group or Fig. 1. Dose–response curves for acetylcholine (ACh, A) and bradykinin (BK, B) evoked hypotension in Sprague–Dawley rats after 6 wks of chronic nitric oxide synthase (NOS) inhibition. Two groups of rats (n = 6 each) were given either plain drinking water (Control) or N ω-nitro-L-arginine methyl ester (L-NAME, 0.7 mg/ml water) for 6 wks (Chronic L-NAME). After determining the responses to ACh and BK, both groups were given acute L-NAME (100 mg/kg, i.v.) and the responses were repeated. ⁎P < 0.05, ⁎⁎P < 0.01, ⁎⁎⁎P < 0.001 vs. paired response in the same rat before acute L-NAME. K.M. Desai et al. / European Journal of Pharmacology 546 (2006) 120–126 123 inhibited the response to a single dose of bradykinin, 3 μg/kg, selected from the mid-part of its dose–response curve, in the control group of rats. In the chronic L-NAME group the response to bradykinin was comparable to the control group and was not inhibited by acute L-NAME treatment (Fig. 2B). The responses to single doses of nitroglycerine (5 μg/kg) and sodium nitroprusside (2 μg/kg), selected from the mid-part of their dose–response curves, were significantly greater in chronic L-NAME treated group compared to the control group; acute LNAME treatment did not modify these responses (Fig. 2C). In the control group, after acute L-NAME treatment the hypotensive responses to nitroglycerine and sodium nitroprusside were greater. These observations are consistent with the data reported earlier (Moncada et al., 1991). 3.3. Chronic L-NAME for 5 days When L-NAME was given for 5 days, the fall in mean arterial pressure evoked by acetylcholine was comparable or even greater than that observed in the control group (Fig. 3A). Administration of acute L-NAME to control rats increased the mean arterial pressure significantly whereas in the chronic L- Fig. 2. Dose–response curves for acetylcholine (ACh)-induced hypotension (A) and hypotensive responses to single doses (selected from the mid-part of their dose–response curves) of bradykinin (BK, B), nitroglycerine (NTG, C) and sodium nitroprusside (SNP, C) in Sprague–Dawley rats after 3 wks of chronic NOS inhibition. Two groups of rats (n = 6 each) were given either plain drinking water (Control) or N ω-nitro-L-arginine methyl ester (L-NAME, 0.7 mg/ml water) for 3 wks (Chronic L-NAME). After taking responses to all the agonists, both groups were given acute (ac.) L-NAME (100 mg/kg, i.v.) and the responses were repeated. ⁎P < 0.05, ⁎⁎P < 0.01, ⁎⁎⁎P < 0.001 vs. paired response in the same rat before acute L-NAME (A and B) or chronic L-NAME vs. paired plain water control group (C). (Fig. 2A). However, 3 weeks of chronic L-NAME treatment failed to alter the dose–response curve to acetylcholine from its respective control group. Acute L-NAME treatment given to chronic L-NAME treated rats for 3 weeks did not inhibit the fall in mean arterial pressure to acetylcholine (Fig. 2A). Acute L-NAME administration led to a significant increase in the mean arterial pressure only in the control group (146 ± 2 vs. 100 ± 3 mmHg, P < 0.001, n = 4) but not in the chronic L-NAME group (143 ± 7 vs. 133 ± 5 mmHg, n = 6). Acute L-NAME also Fig. 3. Dose–response curves for acetylcholine (ACh)-induced hypotension (A) and mean arterial pressure (MAP, B) in Sprague–Dawley rats. Two groups of rats (n = 5 each) were given plain drinking water (Control) for 1 day or 5 days (not shown) and two groups (n = 5–6 each) were given Nω-nitro-L-arginine methyl ester (L-NAME, 0.7 mg/ml water) for 1 day or 5 days. After taking responses to ACh, both groups were given acute L-NAME (100 mg/kg, i.v.) and the responses were repeated. Acute L-NAME did not inhibit the responses to ACh in the 1 day or the 5 day chronic L-NAME groups (not shown). ⁎P < 0.05, ⁎⁎P < 0.01, ⁎⁎⁎P < 0.001 vs. paired response in the same rat before acute L-NAME (A) or vs. control group (B). 124 K.M. Desai et al. / European Journal of Pharmacology 546 (2006) 120–126 NAME treated rats there was no further increase in the mean arterial pressure (Fig. 3B). 3.4. L-NAME given acutely or for 1 day The dose–response curves to acetylcholine-evoked hypotension were similar between L-NAME treated groups (L-NAME given for 1 day or 5 days) and the control group (Fig. 3A). On the other hand, acute L-NAME administration to the control group of rats showed a significant level of inhibition with a rightward shift in the dose–response curve to acetylcholine (Fig. 3A). 3.5. Apamin plus charybdotoxin inhibit the responses to acetylcholine The hypotensive response curves to acetylcholine were quite similar between chronic L-NAME treatment (given for 3 days) and the control group (Fig. 4). Preinfusion of apamin plus charybdotoxin combination either in the control group or chronic L-NAME treated group led to significant rightward shifts in the dose–response curves to acetylcholine (Fig. 4). Interestingly, the shift in the dose–response curve after apamin plus charybdotoxin infusion was more pronounced in the chronic L-NAME treated group compared to the shift attained in the control group. Administration of apamin plus charybdotoxin in acute L-NAME treated rats was invariably lethal. Therefore, the responses to acetylcholine could not be determined after the inclusion of apamin plus charybdotoxin combination in this group. There were no significant differences in the fall in mean arterial pressure evoked by a fixed concentration of sodium nitroprusside before and after the infusion of apamin plus charybdotoxin in the control (35 ± 2% vs. 28 ± 5%, n = 4 each) as well as chronic L-NAME treated groups (40 ± 2% vs. 34 ± 9%, n = 3 each). Fig. 4. Dose–response curves to acetylcholine (ACh)-induced hypotension after chronic NOS inhibition for 3 days in Sprague–Dawley rats. Two groups of rats (n = 4–6 each) were given plain drinking water (Control) for 3 days (pooled data shown for control graph) while one group (n = 4) was given N ω-nitro-L-arginine methyl ester chronically (Ch. L-NAME, 0.7 mg/ml water) for 3 days. After taking responses to acetylcholine, one control group was given acute L-NAME (Ac. L-NAME, 100 mg/kg, i.v., n = 6), while the other control group (n = 4) and the chronic L-NAME group (n = 3) were given apamin (Apa, 25 μg/kg i.v.) plus charybdotoxin (ChTx, 25 μg/kg i.v.) and the responses were repeated. ⁎P<0.05, ⁎⁎P < 0.01, ⁎⁎⁎P<0.001 vs paired response in the same rat before acute l-NAME or apamin plus charybdotoxin. 4. Discussion We report the rapid development of a compensatory endothelium-dependent agonist-induced hypotension within one day after chronic NOS inhibition. This compensatory hypotension is mediated via activation of endothelial calcium-activated potassium channels, probably through the release of EDHF. Endothelial dysfunction, in terms of reduced agonist-induced endotheliumdependent hypotension was not observed even after 6 weeks of chronic NOS inhibition. To our knowledge this time-frame of compensatory changes in endothelial function in vivo is being reported for the first time in the literature. The intravenous injection of a small dose of acetylcholine produces a transient fall in blood pressure owing to generalized vasodilatation. A considerably larger dose is required to elicit bradycardia or block of AV nodal conduction from a direct action of acetylcholine on the heart (Brown and Taylor, 2006). We used doses of acetylcholine up to 2 μg/kg body wt. (Laight et al., 1998), less than previously reported use (Rees et al., 1989). Moreover, for the entire dose range of acetylcholine that we have used, the heart rate did not change at the peak of the hypotensive response as compared to the baseline value (unpublished data) (Rees et al., 1989). Thus, bradycardia or tachycardia does not seem to be modulating the response. Also, the hypotensive response is endothelium-dependent since acetylcholine produces contraction of vascular smooth muscle in the absence of the endothelium (Furchgott and Zawadzki, 1980). The effectiveness of oral administration of L-NAME in the drinking water (Ribeiro et al., 1992; Shinde et al., 2005) and the inhibition of NOS are supported by several observations. In chronic L-NAME treated rats there was a significant increase in the mean arterial pressure, a reduction in plasma nitrate + nitrite levels and no further increases in the mean arterial pressure after acute L-NAME administration in chronic L-NAME treated rats, especially after 5 days of L-NAME treatment. Moreover, after chronic NOS inhibition, the responses to NO-releasing but endothelium-independent vasodilators such as sodium nitroprusside and nitroglycerine were significantly enhanced indicating increased sensitivity of soluble guanylate cyclase after chronic NOS inhibition (Moncada et al., 1991). Thus, in the presence of NOS inhibition, the normal response to acetylcholine, as compared to control rats, can be safely assumed to be mediated by non-NO mediators such as EDHF and/or prostacyclin. In support of this observation, acute L-NAME inhibited the responses to acetylcholine and bradykinin in the control rats but not in chronic LNAME treated rats (Figs. 1A,B and 2A,B). These kind of compensated normal responses have been reported in vitro in vessels such as the coronary (Gödecke et al., 1998; Lamping et al., 2000), brain pial (Meng et al., 1996), skeletal muscle (Koller et al., 1994), the mesenteric (Sofola et al., 2002) and other vessels (Iwakiri et al., 2002) in eNOS knockout mice. The role of prostacyclin in the in vivo response to acetylcholine seems to be none or negligible since administration of diclofenac (Brandes et al., 2000) or indomethacin (Rees et al., 1990) to inhibit cyclooxygenase-mediated prostanoid production did not affect the mean arterial pressure or the hypotensive responses to acetylcholine. The compensatory response was also K.M. Desai et al. / European Journal of Pharmacology 546 (2006) 120–126 observed when bradykinin, another endothelium-dependent agonist, was used. This compensatory response was observed as early as 1 day after NOS inhibition with L-NAME. However, acute administration of L-NAME in control (given plain water) rats significantly inhibited the responses to acetylcholine and bradykinin, indicating involvement of NO in the hypotensive response to both agonists under normal circumstances. It should be pointed out that acetylcholine- and bradykinin-induced hypotensive responses taken even 3 h after acute L-NAME were significantly inhibited. Normally, the responses to acetylcholine and bradykinin were determined 1 h after acute i.v. administration of a single dose of L-NAME. The response after 1 day of L-NAME was close to normal and was not significantly inhibited (Fig 3A). This confirms that that the compensatory response by EDHF begins within 1 day of NOS inhibition. It is noteworthy that the increase in mean arterial pressure is not completely normalized even 6 weeks after chronic NOS inhibition (Table 1). When mean arterial pressure is measured 3 or 6 weeks after NOS inhibition, it is lower than the values for mean arterial pressure determined after acute NOS inhibition (Table 1). This suggests differential regulation of the basal tone in resistance vessels, which determines the mean arterial pressure, and the agonist-induced endothelium-dependent vasodilatation which determines acute changes in blood flow to organs in response to neurohumoral regulation. Whether this also means a more fundamental role of NO in basal tone and steady-state blood pressure as compared to EDHF and a more pronounced role of EDHF in acute blood flow changes remains to be established. It is worth noting that most studies on EDHF relate to its role in endothelium-dependent vasodilatation. This kind of differential compensatory changes cannot be revealed by in vitro experiments. The compensated response to acetylcholine after chronic NOS inhibition was almost completely inhibited by blockade of endothelial KCa channels with the combination of apamin plus charybdotoxin. The inhibition was greater in magnitude than that in control rats by apamin plus charybdotoxin. This strongly suggests a predominant involvement of KCa channels contributing to the compensated response during NOS inhibition. This also agrees with several in vitro studies on eNOS knockout mice which report a compensatory increase in the release of EDHF in different vascular beds (Brandes et al., 2000; Huang et al., 2001; Woodman et al., 2000) and in high salt treated Sprague–Dawley rats (Katusic, 2002; Sofola et al., 2002). In the control rats the response seems to be equally mediated by NO and KCa channels, probably activated by EDHF. Interestingly, even after prolonged NOS inhibition by chronic L-NAME treatment for 6 weeks, we did not see endothelial dysfunction as indicated by an apparently normal endotheliumdependent hypotensive response. We did not look at other indicators of endothelial dysfunction besides reduced endothelium-dependent responses in view of our specific objective. Thus, endothelial dysfunction might develop only if the compensatory vasodilation mediated by KCa channels or prostacyclin fails. There are several animal models and human studies involving different pathologies that show normal or impaired endothelium-dependent relaxation (Boulanger, 1999; Brunner et al., 2005; Chan et al., 2000). These differences might reflect 125 different stages of the development of endothelial dysfunction. Thus, it may be worthwhile to investigate the roles of EDHF as well as prostacyclin signaling pathways besides NO signaling in models of endothelial dysfunction. This may help better planning of treatment approaches in cardiovascular disease states. In conclusion, the hypotensive response to endothelium-dependent agonists, such as acetylcholine and bradykinin, is rapidly compensated within 1 day after chronic inhibition of nitric oxide synthase. The compensatory relaxation is mediated by activation of KCa channels. Endothelial dysfunction, measured as reduced endothelium-dependent hypotensive response, does not develop after inhibition of NOS activity for at least up to 6 weeks in Sprague–Dawley rats. Acknowledgements This work is supported by a group grant from the Heart and Stroke Foundation of Saskatchewan and a CIHR grant to Dr Gopalakrishnan. References Boulanger, C.M., 1999. Secondary endothelial dysfunction: hypertension and heart failure. J. Mol. Cell. Cardiol. 31, 39–49. Brandes, R.P., Schmitz-Winnenthal, F.H., Feletou, M., Gödecke, A., Huang, P.L., Vanhoutte, P.M., Fleming, I., Busse, R., 2000. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endotheliumdependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc. Natl. Acad. Sci. U. S. A. 97, 9747–9752. Brown, J.H., Taylor, P., 2006. Muscarinic receptor agonists and antagonists, In: Brunton, L.L., Lazo, J.S., Parker, K.L. (Eds.), Goodman And Gilman's The Pharmacological Basis of Therapeutics, 11th Edition. McGraw Hill, USA, p. 184. Brunner, H., Cockroft, J.R., Deanfield, J., Donald, A., Ferrannini, E., Halcox, J., Kiowski, W., Lüscher, T.F., Mancia, G., Natali, A., Oliver, J.J., Pessina, A.C., Rizzoni, D., Rossi, G.P., Salvetti, A., Spieker, L.E., Taddei, S., Webb, D.J., 2005. Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the working group on endothelins and endothelial factors of the European Society of hypertension. J. Hypertens. 23, 233–246. Busse, R., Edwards, G., Feletou, M., Fleming, I., Vanhoutte, P.M., Weston, A.H., 2002. EDHF: bringing the concepts together. Trends Pharmacol. Sci. 23, 374–380. Chan, N.N., Vallance, P., Colhoun, H.M., 2000. Nitric oxide and vascular responses in Type I diabetes. Diabetologia 43, 137–147. Freitas, M.R., Schott, C., Corriu, C., Sassard, J., Stoclet, J.C., Andriantsitohaina, R., 2003. Heterogeneity of endothelium-dependent vasorelaxation in conductance and resistance arteries from Lyon normotensive and hypertensive rats. J. Hypertens. 21, 1505–1512. Furchgott, R.F., Zawadzki, J.V., 1980. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376. Garland, C.J., Plane, F., 1996. Relative importance of endothelium-derived hyperpolarizing factor for the relaxation of vascular smooth muscle in different arterial beds. In: Vanhoutte, P.M. (Ed.), Endothelium-Derived Hyperpolarizing Factor 1. Harwood Academic Publishers, pp. 173–179. Gödecke, A., Decking, U.K.M., Ding, Z., Hirchenhain, J., Bidmon, H.J., Gödecke, S., Schrader, J., 1998. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ. Res. 82, 186–194. Huang, A., Sun, D., Carroll, M.A., Jiang, H., Smith, C.J., Connetta, J.A., Falck, J.R., Shesely, E.G., Koller, A., Kaley, G., 2001. EDHF mediates flowinduced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am. J. Physiol. 280, H2462–H2469. 126 K.M. Desai et al. / European Journal of Pharmacology 546 (2006) 120–126 Iwakiri, Y., Cadelina, G., Sessa, W.C., Groszmann, R.J., 2002. Mice with targeted deletion of eNOS develop hyperdynamic circulation associated with portal hypertension. Am. J. Physiol. 283, G1074–G1081. Katusic, Z.S., 2002. Back to the salt mines — endothelial dysfunction in hypertension and compensatory role of endothelium-derived hyperpolarizing factor (EDHF). J. Physiol. 543 (Pt 1), 1. Koller, A., Sun, D., Huang, A., Kaley, G., 1994. Corelease of nitric oxide and prostaglandins mediates flow-dependent dilation of rat gracilis muscle arterioles. Am. J. Physiol. 266, H326–H332. Laight, D.W., Kengatharan, K.M., Gopaul, N.K., Anggärd, E.E., Carrier, M.J., 1998. Investigation of oxidant stress and vasodepression to glyceryl trinitrate in the obese Zucker rat in vivo. Br. J. Pharmacol. 125, 895–901. Laight, D.W., Desai, K.M., Anggärd, E.E., Carrier, M.J., 2000. Endothelial dysfunction accompanies a pro-oxidant, pro-diabetic challenge in the insulin resistant, obese Zucker rat in vivo. Eur. J. Pharmacol. 402, 95–99. Lamping, K., Nuno, D., Shesely, E.G., Maeda, N., Faraci, F., 2000. Vasodilator mechanisms in the coronary circulation of endothelial nitric oxide synthasedeficient mice. Am. J. Physiol. 279, H1906–H1912. Meng, W., Ma, J., Ayata, C., Hara, H., Huang, P.L., Fishman, M.C., Moskowitz, M.A., 1996. ACh dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am. J. Physiol. 271, H1145–H1150. Moncada, S., Rees, D.D., Schulz, R., Palmer, R.M.J., 1991. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc. Natl. Acad. Sci. U. S. A. 88, 2166–2170. Murad, F., 1994. Regulation of cytosolic guanylyl cyclase by nitric oxide: the NO-cyclic GMP signal transduction system. Adv. Pharmacol. 26, 19–33. Nagao, T., Illiano, S., Vanhoutte, P.M., 1992. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am. J. Physiol. 263, H1090–H1094. Rees, D.D., Palmer, R.M., Moncada, S., 1989. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. U. S. A. 86, 3375–3378. Rees, D.D., Palmer, R.M., Schulz, R., Hodson, H.F., Moncada, S., 1990. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 101, 746–752. Ribeiro, M.O., Antunes, E., de Nucci, G., Lovisolo, S.M., Zatz, R., 1992. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension 20, 298–303. Shimokawa, H., Yasutake, H., Fujii, K., Owada, M.K., Nakaike, R., Fukumoto, Y., Takayanagi, T., Nagao, T., Egashira, K., Fujishima, M., Takeshita, A., 1996. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 28, 703–711. Shinde, U.A., Desai, K.M., Yu, C., Gopalakrishnan, V., 2005. Nitric oxide synthase inhibition exaggerates the hypotensive response to ghrelin: role of calcium-activated potassium channels. J. Hypertens. 23, 779–784. Sofola, O.A., Knill, A., Hainsworth, R., Drinkhill, M., 2002. Change in endothelial function in mesenteric arteries of Sprague–Dawley rats fed a high salt diet. J. Physiol. 543, 255–260. Sun, D., Huang, A., Smith, C.J., Stackpole, C.J., Connetta, J.A., Shesely, E.G., Koller, A., Kaley, G., 1999. Enhanced release of prostaglandins contributes to flow-induced arteriolar dilation in eNOS knockout mice. Circ. Res. 85, 288–293. Taddei, S., Ghiadoni, L., Virdis, A., Buralli, S., Salvetti, A., 1999. Vasodilation to bradykinin is mediated by an ouabain-sensitive pathway as a compensatory mechanism for impaired nitric oxide availability in essential hypertensive patients. Circulation 100, 1400–1405. Wang, Y.-X., Poon, C.I., Pang, C.C.Y., 1993. Vascular pharmacodynamics of NG-nitro-L-arginine methyl ester in vitro and in vivo. J. Pharmacol. Exp. Ther. 267, 1091–1099. Weldon, S.M., Winquist, R.J., Madwed, J.B., 1995. Differential effects of LNAME on blood pressure and heart rate responses to acetylcholine and bradykinin in cynomolgus primates. J. Pharmacol. Exp. Ther. 272, 126–133. Woodman, O.L., Wongsawatkul, O., Sobey, C.G., 2000. Contribution of nitric oxide, cyclic GMP and K+ channels to acetylcholine-induced dilatation of rat conduit and resistance arteries. Clin. Exp. Pharmacol. Physiol. 27, 34–40.