PCTH 325
Pharmacokinetics 2 and
Drug Dosing
Dr. Shabbits jennifer.shabbits@ubc.ca
September 11, 2014
Learning objectives
1. Describe the mechanisms of drug metabolism, the significance of enzyme induction & inhibition, and how the first-pass effect can influence dosing
2. Describe how drugs are excreted from the body and how pH can be manipulated to influence this process
3. Define and describe MTC, MEC and the therapeutic range
4. Describe, interpret and apply the C vs t equation and graph
5. Define and calculate t
½ fraction eliminated
, C max
, t max
, fraction remaining and
6. Describe the significance of steady state and how it can be achieved
7. Calculate loading and maintenance doses and describe how and when they are used
Metabolism
the irreversible biotransformation of drug
• makes it more polar to ↑ renal (urinary) excretion
Occurs primarily in the liver via 2 (usually sequential) enzyme-catalyzed processes:
• Phase I oxidation/reduction/hydrolysis
• Phase II conjugation
Phase I: cytochrome P450 enzymes
A superfamily of related enzymes that add on or uncover small polar groups (–OH, –NH
2
, –COOH) water solubility to
Phase 1
Metabolite Parent Drug
P450 Enzymes
P450 enzyme induction and inhibition
• some P450 enzymes can be induced or inhibited by other drugs, foods, pregnancy or disease
Induction: metabolic activity of enzymes
[Drug] (ex: alcohol)
Inhibition: metabolic activity of enzymes
[Drug] (ex: grapefruit juice)
Primary cause of drug interactions
Requires drug dosing to be increased or decreased
Phase II: conjugative enzymes
Mediated by various non-P450 liver enzymes
• covalently add large, polar, endogenous molecules to
Phase I metabolite
• ensures that metabolite is ready for excretion phenytoin
(glucuronide, glutathione, sulfate, acetate, amino acids etc)
Phase II glucuronyl transferase
Phase I
P450 p-OH-phenytoin phenytoin-ether-glucuronide
Drug metabolism
• usually inactivates the drug
• is required to activate prodrugs
Metabolite + Receptor
≠ MR complex
CYP2D6
Drug metabolism
• may be harmful if the metabolite(s) are toxic
Tylenol ® =
Acetaminophen
CYP2E1
Induced by alcohol
Toxic semiquinone
First pass metabolism
Most drugs absorbed from the GI tract are delivered to the liver before reaching the systemic circulation
IV Drug delivered to target receptors
oral doses > iv doses to account for loss due to metabolism
Some drug is metabolised
(lost)
Oral
Drug
Drug metabolism in the gut
Some drugs undergo significant metabolism by bacterial enzymes in the gut (ex: digoxin)
What effect might a course of antibiotic therapy have in a person taking digoxin?
Excretion – kidney (renal excretion)
The irreversible loss of drug from the body
Blood Proximal tubule
1. Passive Glomerular Filtration diffusion of small drugs <20kDa
Afferent arteriole
2. Active Tubular Secretion transport systems for large drugs Drug in blood
3. Passive Tubular Reabsorption concentration gradient may drive uncharged drug back into blood
*urine pH is key
Efferent arteriole
Urine
Weak acid:
Absorbed or Excreted?
HA
⇋
H + + A -
Weak base: B + H +
⇋
BH +
Changing urine pH to treat an overdose
increasing urine pH shifts equilibrium to promote excretion of weak acid drugs
(ex: aspirin)
• iv sodium bicarbonate raises pH from 6-8
HA H + +
A Ionized form is water soluble excreted in urine
*Remember: pH = [H + ]
Changing urine pH to treat an overdose
How would you modify the urine pH to treat an overdose of a weak base drug?
B + H + BH +
Changing urine pH to treat an overdose
urine pH shifts equilibrium to promote excretion of weak base drugs
(ex: amphetamine)
•
Summary
Administration of agonist or antagonist
Enteral, parenteral, inhaled, topical, etc.
Steady state
Phase I, Phase II, prodrugs
Ionized drug - pH
Unionized drug - pH
Drug binds receptor(s)
Response desired? side effect?
TD
50
, ED
50
,
Part 4: Drug Dosing
Designing drug dosing regimens
How much drug?
• Magnitude of therapeutic (and toxic) effects depends on drug dose
How often?
• Magnitude of effect declines over time as drug levels decrease
For how long?
• Continuous drug use has a cost (economic, side effects, toxicity)
0
Concentration – time relationships
• follow and predict drug concentration in the body
• blood is the reference → delivers drug to receptors
? iv
? non-iv
Administer drug
Take blood samples at various times
Measure [drug]
Plot data
Time
∞
Concentration – time relationships
• allow us to ‘visualize’ the ADME processes
0
Time
∞
Concentration – time relationships
• show us the magnitude & duration of the effect
C max
MTC (minimum toxic conc.)
Therapeutic Range
MEC (minimum effective conc.)
0 t max Time
∞
The C vs t equation – iv administration
C t
= C
0 e -kt
C
0
e = base of the natural logarithm
C t
= drug conc at time ‘t’
C
0
= drug conc at time ‘0’ k = first order elimination rate constant
→ the fraction of drug eliminated per unit time
C t
0
0 t
Time
Linearizing the C vs t equation
Exponential: C t
= C
0 e -kt
Log e
(or ln): lnC t
= -kt + lnC
0
Y = mX + b lnC
0 use these equations to predict drug concentration at various times
Slope = -k
Time (t)
Application of k: half-life ( t
½
)
Elimination half-life (t
½
): the time required for drug concentration to decrease by half t
½
= ln2 = 0.693
k k
Sample Calculation:
The first order elimination rate constant for acetaminophen (Tylenol®) is 0.23 hr -1 .
What is its half-life?
Note units k: time -1 t
½
: time
Application of t
½
:
drug washout
# Elimination half-lives
0
1
2
3
4
5
% Drug
Remaining
100
50
25
12.5
6.25
3.13
% Drug
Eliminated
0
50
75
87.5
93.75
96.87
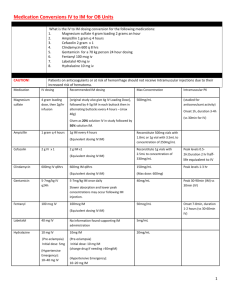
Used to estimate “drug washout” prior to surgery, following a drug overdose etc. ~5 half-lives for drug to be ‘completely’ eliminated
Question…
A person presents to the ER following an overdose of Tylenol ( t
½
= 3 hrs). How long will it take for the drug to get out of his system without any medical intervention?
Application of C vs t eqn: predicting [drug]
If the initial plasma concentration of Tylenol is 20 μ g/ml, what will the plasma concentration be after 8 hours?
Use C t
= C
0 e -kt or lnC t
= -k t + lnC
0
Step 1: lnC
8h
= -kt + lnC
0
Step 2:
Step 3:
Step 4:
Drugs can be taken in single doses
Single drug dosing often puts drug concentration in the therapeutic range for too short a time, if at all
Not even reached
Too brief
Time Time
Drugs can be taken in multiple doses
Peak (max) & trough (min) concentrations fluctuate around a prolonged steady state mean (C ss
)
Peaks
Troughs
Steady state concentration
Steady state occurs when the rate of drug administration = the rate of drug elimination, which takes ~ 5 half-lives
Time
Css
Drug input
Drug output
Administration = Elimination
(rate in = rate out)
Steady state concentration
Steady state can occur at ANY concentration. The goal is to have C ss fall within the therapeutic range
The concentration is a function of:
• drug dose
• dosing interval
Css
Time
Getting to steady state
A proper dosing regimen will put C ss in the therapeutic range. There are 2 approaches:
5
6
3
4
0
1
2
1. Exponential approach: give small, repeat doses at intervals ≈ the drug’s half-life (ex: 200 mg every 4 hr)
# t
½
12
16
20
24
4
8
Time (hr) Amount previous dose left (mg)
0 0
100
150
175
187.5
193.75
196.88
200
200
200
200
New Dose
(mg)
200
200
200
Total amount in body (mg)
200
300
350
375
387.5
393.8
396.9
2. Give a loading dose followed by maintenance doses
Loading dose (LD)
• A large dose of drug used to raise the plasma concentration to a therapeutic (target) level faster than with smaller repeat doses
LD = C target x V d where C target
= desired C ss
Intermittent, non-iv administration
Maintenance dose (MD)
• Smaller repeat doses are then used to maintain the desired plasma concentration
• The MD replaces the drug that is eliminated by the body during the dosing interval ‘t’ (ie the time between doses)
MD = (fraction eliminated) x LD = (1-e -kt ) x LD
Rearranging C t
= C
0 e -kt gives us the:
Fraction remaining (C t
/C
0
) = e -kt
Fraction eliminated = 1-e -kt
Practice
problem
The target concentration of a drug to be given 3 times a day is 35 mg/L. The Vd = 25 L and t
1/2
= 12 h.
What loading and maintenance doses would be appropriate?
Practice
problem – cont’d
MD = (1-e -kt ) x LD
What is k?
What is t?
the technical dosing regimen would be:
the practical dosing regimen would be:
Therapeutic drug monitoring
• Provides individualized (patient-specific) dosing information
Give an initial dose based on expected, published averages
Measure the patient’s actual plasma concentration
Revise subsequent dosing
• Useful for drugs with narrow therapeutic range & special populations
(ex: geriatrics, pregnancy, pediatrics)
Derivation of the half-life equation ~ FYI
lnC t
= lnC
0
-kt ln(C t
/C
0
) = -kt multiply through by -1 ln(C
0
/C t
) = kt when t = t
½
C t
= ½C
0 or C
0
= 2C t ln(2C t
/C t
) = kt
½ ln2 = kt
½ t
½
= ln2/k = 0.693/k