The alignment of mass spectra and identification of biomarkers of
advertisement

The alignment of mass spectra and identification of biomarkers of pancreatic cancer M.S. Plan B Project Report July 5th, 2011 Junmin Shi M.S. Applied and Computational Mathematics Candidate Dr. Ronald Regal Advisor University of Minnesota Duluth Department of Mathematics and Statistics Page | 1 ACKNOWLEDGEMENTS ................................................................................................ 3 ABSTRACT .................................................................................................................... 4 1 INTRODUCTION ..................................................................................................... 5 2 BACKGROUND ....................................................................................................... 6 2.1 2.2 3 PANCREATIC CANCER ............................................................................................................ 6 MALDI-­‐TOF ...................................................................................................................... 7 EXPLORATORY DATA ANALYSIS ............................................................................. 8 3.1 3.2 3.3 SCATTER AND GRAY-­‐SCALE PLOTS ............................................................................................ 8 CONTOUR PLOT ................................................................................................................. 10 PCA SCORE PLOT ............................................................................................................... 11 4 BASELINE CORRECTION, NORMALIZATION AND SMOOTHING ............................. 13 5 CLUSTERING ........................................................................................................ 17 6 ALIGNMENT ........................................................................................................ 21 6.1 6.2 6.3 6.4 7 FFT CROSS-­‐CORRELATION .................................................................................................... 21 PARAMETRIC TIME WARPING (PTW) .................................................................................... 24 CORRELATION OPTIMIZED WARPING ..................................................................................... 26 RESULTS AND COMPARISONS ............................................................................................... 28 DISCRIMINATION ANALYSIS ................................................................................ 31 7.1 7.2 FISHER’S DISCRIMINANT ANALYSIS ......................................................................................... 31 STEPWISE DISCRIMINANT ANALYSIS ....................................................................................... 34 8 CONCLUSION ....................................................................................................... 38 9 REFERENCES ........................................................................................................ 39 10 10.1 10.2 10.3 10.4 APPENDICES .................................................................................................... 40 R CODES ........................................................................................................................... 40 SAS CODES ....................................................................................................................... 42 MATLAB CODES ................................................................................................................. 48 PROOF ............................................................................................................................. 54 Page | 2 Acknowledgements First and foremost I offer my sincerest gratitude to my advisor, Dr. Ronald Regal, who has supported me throughout my project with his patience, passion and expertise whilst allowing me the room to work in my own way. I attribute the level of my Masters degree to his encouragement and effort and without him this project, too, would not have been completed or written. I also give thanks to Michael Madden, who is from the Medical School at the University of Minnesota, provided me with the MS spectra dataset for my project. He also gave me lots of helpful information by which my work has always been guided in a proper direction. I deeply appreciate Dr. Richard Green and Dr. Guihua Fei for their willingness to be on my committee. I also would like to give thanks to Dr. Steven Trogdon for his help on testing C++ codes and fixing Matlab on the Linux machine while I have been working on my project. The Department of Mathematics and Statistics has provided the support and equipment I have needed to produce and complete my project. I have learned a lot in mathematics and statistics through taking courses from the faculty members at the department: Dr. Barry James, Dr. Kang James, Dr. Guihua Fei, Dr. Richard Green, Dr. Marshall Hampton, and Dr. Ronald Regal. Finally, I thank my peer graduate students for helping, working with, and sharing with me with their warm hearts. Page | 3 Abstract A joint research team of the Medical School at University of Minnesota Duluth and St. Mary’s Hospital has generated mass spectra (MS) datasets consisting of 96 samples taken from pancreatic cancer patients. A series of exploratory data analysis methods has been employed to help “clean” and prepare the data for further statistical analysis: gray-­‐scale plot, Pearson’s correlation coefficient, PCA score plot, and moving average smoothing. Furthermore, asymmetric least-­‐squares (ALS) baseline estimation was used for MS baseline removal. Then the set of 96 samples was classified into six subgroups by clustering analysis with Euclidean distance and Ward’s linkage method. However, variability in the mass-­‐to-­‐charge ratio (m/z) in mass spectra poses the largest challenge and impedes further exploring of the usefulness of MS data. Therefore, four distinct software packages of alignment were used to correct the misalignment in m/z values in order that the same informational peaks can be cross-­‐referenced in different samples: SpecAlign, Parametric Time Warping (PTW), Correlation Optimized Warping (COW), and Icoshift. Then, by comparing the squared ratios of Pearson’s correlation coefficient before and after alignment relative to the reference spectrum, we conclude that COW performed the “best” but most computation-­‐expensive alignment. The COW-­‐aligned MS dataset was imported to SpecAlign for picking major peaks and a matrix of 34 peaks with their m/z values and intensities included was exported. Then we performed Fisher’s discriminant analysis and stepwise discriminant analysis. By Fisher’s method, we found a combination of peaks or proteins that are important in separating the pre-­‐assigned groups the most and the peaks with larger discriminant coefficients might be good candidates for medical researchers who seek for the biomarkers of pancreatic cancer. By the stepwise method, we found a series of peaks that are important on discriminating among the clinical impression groups. Page | 4 1 Introduction Proteomics, which is characterized as a multifaceted, rapidly evolving and open-­‐ended field, in general aims in the large-­‐scale determination of gene and cellular function and structure directly at the protein level (Aebersold, et al, 2003; Feng, et al, 2009). Proteomic profiling technologies, such as matrix-­‐assisted laser desorption ionization time-­‐of-­‐flight (MALDI-­‐TOF) and surface-­‐enhanced laser desorption ionization time-­‐of-­‐
flight (SELDI-­‐TOF), have been proven to be indispensible by tremendous recent success in their ability to identify and precisely quantify thousands of proteins from complex samples (Wong, et al, 2005). In particular, mass spectrometry-­‐based proteomics has become a unique tool for discovering proteomic patterns or features which may be relevant for the identification of biomarkers of disease and diagnostic purposes. Here in consideration of my project, I present a mass-­‐spectrometry (MS) dataset consisting of 96 samples, which were collected and measured on a MALDI spectrometer by a joint research team of St. Mary’s Hospital and the Medical School, University of Minnesota, Duluth. Each of the 96 MS spectra consists of two columns, one the mass-­‐to-­‐
charge (m/z) ratio, the other intensity. Basically, the first column indexed by an approximate of 20,000 m/z values ranging from 999 to 20,094 identifies distinct protein ions or peptides which are measured in the second column with respect to their relative abundance in individual samples. For instance, the m/z ratio (45.5) of the simple positively-­‐charged ion, (C2H7)2+, can be calculated by dividing its mass number (91) by its charge number (2). The 96 samples were collected from different parts of pancreas of patients who were diagnosed with pancreatic cancer. In general, the goals of my project can be formulated as below. The first step is to properly correct the misalignment of m/z values by means of multiple algorithms of alignment such that the same informative peaks or signals can be cross-­‐referenced in the MS spectra under different conditions and hence to determine the optimal method(s) of alignment. The second goal of my project is to use multivariate statistical models to identify the biomarkers of pancreatic cancer. Page | 5 2 Background 2.1 Pancreatic cancer The pancreas is a small gland organ, approximately six inches long, located in the upper abdomen and adjacent to the small intestine. It functions in the digestive and endocrine system of human beings. The pancreas is an endocrine gland secreting several important hormones that regulate sugar levels in the blood, as well as an exocrine gland producing pancreatic enzymes combined with juices from the intestines completing the function of breaking down carbohydrates, proteins, and lipids in the chyme, and chemicals that neutralize stomach acids that pass from the stomach into the small intestine. Cancer that starts in the pancreas is called pancreatic cancer, which is a malignant neoplasm of the pancreas. Pancreatic cancer is sometimes called a “silent killer” because early pancreatic cancer often does not cause symptoms, and the later symptoms are usually nonspecific and varied. Therefore, pancreatic cancer is often not diagnosed until it is advanced. Common symptoms include weight loss, back pain, and jaundice. Risk factors for pancreatic cancer include age, sex, smoking, diet, as well as obesity. The 96 samples were collected by two distinct sampling technologies, surgical and endoscopic, from different parts of the pancreas of individual patients. Among them 46 were sampled by means of endoscopy, 49 were collected by means of surgery, but with one sample collected with an unknown method. Some samples (coded as D) are characterized as fluid from pancreatic ducts through which enzymes secreted by the pancreas are conducted to the intestines; some are coded as M standing for mucinous cyctadenoma, others S standing for serous cycstadenoma, most of them were sampled from pancreas cysts; samples with clinical impression IPMN (intraductal papillary mucinous neoplasm) are subdivided into three categories: I (IPMN), Ib (IPMN benign), and Im (IPMN malignant); the last category of samples is called P standing for pseudocyst. Furthermore, the accuracy of clinical descriptions and classification is dependent upon the sampling technologies: we are in general more confident in the Page | 6 information of samples obtained by surgery than those obtained by endoscopy. For a complete description of sample attributes, please refer to Table 2.1. Table 2.1 Summary of clinical impressions Category Percent Category Percent Category Percent Malignant 13.54 Serous 9.38 Duct 21.88 Benign 13.54 Mucinous_
Cystadenoma 11.46 Fluid 50.00 IPMN 20.83 Serous_Cystadenoma 8.33 Cyst 50.00 IPMN_Benign 11.46 Body 7.29 Pseudocyst 7.29 IPMN_Malignant 9.38 Tail 13.54 Serum 8.33 Mucinous 13.54 Head 16.67 2.2 MALDI-­‐TOF Next it would be helpful for us to introduce some background information about the working mechanisms of MALDI-­‐TOF (matrix-­‐assisted laser desorption/ionization time-­‐of-­‐
flight). First of all, the proteins to be analyzed are isolated from cell lysate or tissues by biochemical fractionation or affinity selection and then degraded enzymatically to peptides. Then MALDI sublimates and ionizes the peptides out of a dry, crystalline matrix via laser pulses. The charged ions are accelerated through the magnetic field, deflected and hence separated on the basis of m/z ratio, and eventually counted by a detector. A generic MS spectrum consists of the x-­‐axis indexed by the m/z ratio and the y-­‐axis representing the relative abundance of proteins. Page | 7 3 Exploratory Data Analysis The sort and amount of information which can be extracted from MS data depend upon the way in which they are investigated. Most commonly, MS spectra are compared with respect to major peaks appearing at different m/z values. However, to accomplish this can be tedious and inefficient for gaining insightful knowledge on how they are similar or dissimilar to each other. A more compact way of comparing MS spectra would be a plot of pair-­‐wise correlation coefficients for all samples , such that samples that are lowly or highly correlated to the others will appear to be some patterns striking to viewers due to contrast in colors. Nevertheless, its weakness lies in the heavy loss of information due to the fact that correlation coefficients solely tells us the linear dependence among different variables and reduce to one scalar the multi-­‐dimensional features of MS spectra data. An alternative would be PCA (Principal Component Analysis) score plot by which the dimensions of MS data can be decreased substantially without loss of significant information, patterns and structures among the samples can be visualized in 2 or 3-­‐dimension by projecting original data points on the axis of principal components and thereafter outliers can be readily inspected with greater confidence. 3.1 Scatter and gray-­‐scale plots Figure 3.1 shows the scatter plot of sample B5 and its gray-­‐scale plot with brightness proportional to intensity and therefore corresponding to peaks in the scatter plot. It is apparent from the scatter plot that structures are more dynamic in m/z values ranging from 1000 to 8000, that major peaks are superimposed upon some level of background, and that there exists an bulge in the beginning of the spectrum. Caution should be addressed to the initial “peak” which could be an artifact of measurements (personal communications with Dr. Higgins). Therefore, subsequent computations will be implemented on data with the “problematic” segment truncated for all the 96 samples. Since it is tremendously tedious and blurring to plot all the spectra in scatter form, we employ a gray-­‐scale plot of the spectra (Figure 3.2) illustrating similar features stated above. It is apparent from Figure 3.2 that all the major peaks occur at m/z values smaller than 8,000. Page | 8 Figure 3.1 Scatter plot (top) and gray scale plot (bottom) of sample B5 Figure 3.2 Gray scale plot of all the 96 MS spectra together Page | 9 3.2 Contour plot One of the methods calculating correlation coefficient, Pearson product-­‐moment correlation coefficient, is employed as the measure of the linear dependence between two samples. The formula for calculating Pearson’s r is written as: n
r=
∑ (x
i
− x )( y i − y )
i =1
n
∑ (x
i =1
i
− x)
2
n
∑(y
i
− y)
2
i =1
A contour plot of the correlations is shown below (Figure 3.3). Judging from the blue and yellow, we can conclude that several samples are lowly correlated with the others: A6 (blue), A14 (blue), A24 (yellow), B13 (blue), B17 (blue), C3 (yellow), C17 (yellow), D7 (yellow), and D23 (yellow). The off-­‐diagonal dark-­‐red indicates a correlation coefficient of 1 because they are correlated with themselves. Figure 3.3 Contour plot of Pearson’s correlation among the 96 MS spectra Page | 10 3.3 PCA score plot The goal of principle component analysis (PCA) is to reveal the internal structure of data generated from complex systems by means of re-­‐expressing them on the most meaningful basis and meanwhile filtering out noisy and even redundant information. Intuitively, the goal of PCA is to determine which vector basis, dimension, or dynamics can describe the complex systems in the most representative way. Algebraically, the goal is achieved by linearly combining the original random variables in a way which has the maximum variance and geometrically forms a new coordinate system representing the directions with maximum variability. Figure 3.4 PCA score plot of the raw MS data (numbers indicating groups by clustering analysis in Section 5) In consideration of our 96 MS spectra, each of them is depicted in a set of roughly 20,000 variables in terms of intensities indexed by m/z values. By applying PCA to the dataset, we intend to discern their inherent structure in three or fewer dimensions without significant loss of information filtered out from the “garbled” confounds. Figure 3.4 shows some graphic results of PCA: several data points are well-­‐dispersed around Page | 11 the main cloud implying that the correspondent samples are more dissimilar to the majority. Furthermore, the PCA score plot is more or less consistent with the Pearson’s correlation plot in terms of the samples identified as “outliers”, but delivers extra information about how those samples are distributed on the coordinate system of the first two principle components. Page | 12 4 Baseline correction, normalization and smoothing While factors giving rise to spectral background are poorly constrained (potential causes include chemical noises in the matrix or ion overloading), the existence of high-­‐level background noise is necessarily undesirable. For example, the issues may include but are not limited to artificially high values of pair-­‐wise correlation coefficient and poor performance of profile-­‐based algorithms of alignment (it will be discussed in subsequent sections) due to some cumulative effects. Since all our 96 MS spectra have background levels that complicate comparison of spectra, we will perform baseline subtraction to each of them. In addition, as it was mentioned in Chapter 3 that the existence of initial bulge in each spectrum could be some measuring artifacts, baseline estimation will be carried out on data after the initial segments (approximately corresponding to 999-­‐1100 in m/z values) truncated. Furthermore, experience has shown that measurements of MS intensity have errors which can reach up to 50%. So we take the logarithms of the intensity to suppress the variance due to measuring errors on abundance. There are a variety of algorithms available for baseline removal, some of which may include quantile regression (QR) baseline correction and asymmetric least-­‐squares (ALS) baseline estimation (Eilers, 2004). Each of them has its own advantages and disadvantages, but I intend to employ the ALS approach. The estimation of baseline through ALS approach is achieved by fitting a smooth trend, the so-­‐called Whittaker smoother, to the discrete series of MS spectra, which in turn can be completed by minimizing the function Q = ∑ vi ( yi − f i ) 2 + λ ∑ (Δ2 f i ) 2 i
i
where y i refers to the data, f i the Whittaker smoother, vi prior weights, λ the controller of the degree of smoothing, and Δ2 f i second-­‐order difference in the Whittaker smoother. The rule for choosing the asymmetric weights, vi , is the following ⎧ p
vi = ⎨
⎩1 − p
yi > f i
yi ≤ f i
where the asymmetry parameter p lies in the range between 0 and 1, inclusively; and p refers to weight for data points above the Whittaker smoother f i , 1 − p the weight Page | 13 for data points below f i . The solution to finding both vi and f i is achieved by iterating between them until a convergence, i.e., the minimization of Q , is reached because the function Q is convex in f (Eilers, 2004). Here, the initial value for v is commonly chosen to be 1. It is obvious for a baseline estimate that a choice of near-­‐zero p and rather large λ will make f follow the valleys of y . Figure 4.1 Plots of Sample B23 and B24 before (top) and after (bottom) baseline removal The ALS algorithm included in Package ‘PTW’ (Bloemberg, et al, 2010) was run on R 2.13.0 for my dataset with λ = 1x107 and p = 0.001. Plots of Samples B23 and B24 are shown in Figure 4.1 for before and after baseline subtraction. The top panel shows that B23 appears to be distinct from B24 due to the existence of uneven backgrounds; it is however obvious from the bottom panel that B23 almost mimics every major and minor peak in B24 after baseline correction likely suggesting that they have much in common in terms of chemical compositions at the molecular level. Therefore, the visual impression after baseline removal is consistent with the fact that B23 and B24 are replicate samples of the same type from the same patient. Furthermore, it is highly desirable for MS spectra to be smoothed because sharp peaks or spikes may cause failure of convergence of the integral functions of profile-­‐based algorithm of alignment (Eilers, 2004). The smoother I used is 11-­‐point moving average Page | 14 which should produce strong smoothing for our MS dataset. Figure 4.2 shows the result of smoothing for Sample B23. The visual impression that the graph gives us in terms of smoothing effects is that the peaks are broadened and vertical shifts are flattened out. Figure 4.2 Plot of Sample B23 with its smoothed counterpart superimposed It would be beneficial to inspect the contour plot of Pearson correlation coefficient after a series of steps of data “cleaning”. Figure 4.3 shows pair-­‐wise Pearson’s correlation among the 96 MS spectra, which has much in common with its counterpart before data cleaning in terms of the general pattern, that is, the ones dissimilar to the others. The difference lies in the substantial amount of reduction in correlation coefficient indicated by the all-­‐around dark blue, which is caused largely by baseline removal. Page | 15 Figure 4.3 Pair-­‐wise Pearson’s correlation among the 96 MS spectra after data cleaning Page | 16 5 Clustering We have learned from previous sections that Pearson’s correlation coefficient is one way of inspecting how our MS spectra are similar or dissimilar to each other. However, we gain no information about how one spectrum is more similar to some than to others, how many and what groups they can be classified into. Moreover, alignment also requires clustering because it is only necessary to align the same informational peaks which appear at distinct m/z values due to errors of measurements across different spectra and hence clustering will maximize the possibility of identical peaks staying in one cluster to be aligned. Figure 5.1 A dendrogram for the 96 MS spectra by Ward’s hierarchical clustering method The goal of clustering analysis is to group a set of data objects into clusters such that those objects are similar to one another staying within the same cluster and dissimilar to the objects in other clusters. Since we have no prior information about classes for our MS dataset, discovering intrinsic structures of MS data will be unsupervised learning. Experience has shown that there has to be some degree of subjectivity in deciding which measure of distance and linkage method to use for any particular dataset. Here for our Page | 17 MS dataset we choose the combination of Euclidean distance and Ward’s linkage method. The Euclidean distance is computed as
d ( x , y ) = ( x1 − y1 ) 2 + ( x 2 − y 2 ) 2 + + ( x p − y p ) 2 = ( x − y )' ( x − y )
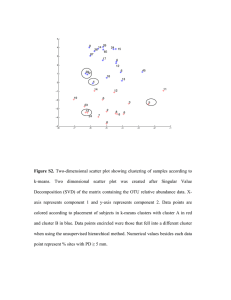
where x and y are two objects with p dimensional observations. The ordinary Euclidean distance is sometimes called straight-­‐line distance in the sense that each coordinate contributes equally to the calculation of distance. It is often preferable for clustering analysis in the case when we do not have prior knowledge of the distinct objects. The Ward’s linkage method is superior to others such as single, complete and average linkage methods in that it produces a dendrogram in which objects within the same cluster are more similar to each other and different clusters are less similar to one another. It is the case because the criterion of error sum of squares (ESS) is implemented for Ward’s method. The merge of any pair of clusters occurs only when the combination results in the smallest increase in ESS. Figure 5.1 shows the dendrogram with vertical axis labeled as ESS and horizontal axis as MS numbers. The grouping is indicated by different colors which are distinguished at a threshold of 40 in ESS. The colors suggest that it is appropriate to subdivide the 96 objects into 6 subgroups. Table 5.1 lists sample IDs in the 6 subgroups. The results of the hierarchical clustering analysis are consistent with those of correlation and PCA score plot in the sense that a few outliers, including B13 and B17, stay in Group 1 which is small and well-­‐
separated from Group 6 in which some very similar spectra are clustered together. Table 5.1 Samples clustered in the 6 subgroups. Group1 A1, B13, B17 Group2 A6, A14, A15, A24, C6 Group3 A2, B1, B3, B4, B5, B9, B15, B18, B22, C2, C3, D6, D7, D23, D24 Group4 A8, A19, A20, A21, B6, B8, B12, B21, C9, C10, C13, C18, C20, C21, D14, D22 Group5 A10, A23, B2, B10, B14, B20, C5, C8, C19, C23, C24, D1, D3, D8, D9, D10, D19, D21 Group6 A3, A4, A5, A7, A9, A11, A12, A13, A16, A17, A18, A22, B7, B11, B16, B19, B23, B24, C1, C4, C7, C11, C12, C14, C15, C16, C17, C22, D2, D4, D5, D11, D12, D13, D15, D16, D17, D18, D20 Page | 18 Gray-­‐scale plots of MS spectra in the same cluster will produce some visual impression of how well the clustering analysis has done. Those plots of the Group 2 spectra are shown as the following. It is apparent from the plots that some major peaks indicated by the brightness appear at the same m/z values across all the MS spectra in the same clusters, indicating that those spectra are chemically similar to one another. Figure 5.2 Gray-­‐scale plot of 5 MS spectra in Group 2 The effect of clustering analysis is more obvious in the contour plot of Pearson’s correlation coefficient among the 96 MS samples. It is in general true from the plot that samples are highly correlated in the same groups and weakly correlated between different clusters, which can be visualized by the six rectangles of color contrast. However, group 4 as a whole seems to have higher correlation with group 2 than any other groups. Page | 19 Figure 5.2 Contour plot of Pearson’s correlation coefficient among the 96 MS samples in cluster order Page | 20 6 Alignment One of the challenging issues arising from MS spectra data is the variability or misalignment in m/z values such that the same protein will have different m/z values in different samples. To analyze the data, we need a rectangular array where rows are spectra and the values for one protein are in a single column. Experience has shown that the variance of m/z values may reach up to ± 0.1% − 0.2% at any point (Feng, et al., 2009; Wong, et al., 2005). Factors causing the shifts are unavoidable errors of measurements, differences in timing of measurements, variations of operations from one instrument to another, and aging of the instruments. There have been a variety of algorithms in the literature for alignment of spectra. These algorithms fall into two broad categories: the peak-­‐based alignment approach and the profile-­‐based alignment approach (Kong, et al., 2009). Peak-­‐based alignment is characterized as aligning only “significant” peaks that have to be chosen in advance. Profile-­‐based alignment attempts to achieve the goal of alignment by minimizing some target functions either globally or on a cumulative local scale. Some algorithms of profile-­‐based alignment include FFT cross correlation (Wong, et al., 2005; Wong, et al., 2005; Savorani, et al., 2010), Parametric Time Warping (PTW, Eilers, et al., 2004; Bloemberg, et al., 2010), and Correlation Optimized Warping (COW, Nederkassel, et al., 2006). I will use my dataset to test the performance of each package and compare the results. The criteria for judging the performance of alignment will be the ratios of correlation coefficient before and after alignment relative to the mean spectrum and PCA score plot. Below follow some brief introductions to the theories behind those algorithms mentioned above and graphical results of alignment. 6.1 FFT cross-­‐correlation The cross-­‐correlation function between any two functions, r (x) and s(x) , at any shift position, u , is defined by ∞
Corr (r , s) u = ∫ r ( x) s( x + u )dx −∞
Page | 21 Furthermore, the forward and reverse Fourier transforms for any function h(x ) are given by ∞
H ( χ ) = ∫ h ( x )e
2πiχx
−∞
∞
h( x) = ∫ H ( χ )e
dx −2πiχx
−∞
dx h( x) ⇔ H ( χ ) Alternatively, the cross-­‐correlation function between r (x) and s(x) at any shift position can be obtained by firstly applying forward Fourier transforms to r (x) and s(x) , then multiplying the transformed functions R ( χ ) and S * ( χ ) , and then finally performing reverse Fourier transform of this product. The whole process is typically denoted as Corr (r , s ) u ⇔ R( χ ) S * ( χ ) where S * ( χ ) refers to the complex conjugate. The optimal shift position, u op , between r (x) and s(x) can be found by uop = max u (Corr (r , s )u ) In practice, the algorithm is applied recursively to spectra segments of both the reference spectrum and the sample spectrum to be aligned. The software, SpecAlign, is employed to run our clustered spectra dataset; and the mean spectrum of all spectra in the same cluster, calculated by the equation 1
θi (t ) =
ni
ni
∑X
ij
(t ) for cluster i with ni spectra within it j =1
is taken as the reference spectrum for that cluster. Page | 22 Figure 6.1 Comparison of alignment for group4 between the whole spectra raw (upper left), SpecAlign aligned (lower left), zoom-­‐in raw (upper right), and zoom-­‐in aligned (lower right). The alignment was obtained with a maximally-­‐allowed shift of 50 points (One sampling interval is approximately equal to one unit on m/z scale). Figure 6.1 shows the comparison before and after alignment for group4. The efficiency of alignment is more obvious in the zoom-­‐in plots illustrating that peaks occurring at around 15,000 merged together into the right major peak after alignment. The maximally-­‐allowed shift is determined by empirical result. It is self-­‐evident that a larger maximum shift would be more flexible for more peaks to be aligned. The theory of cross-­‐correlation by FFT is also essential for the Icoshift software package which has greater flexibility where users are able to align separate, non-­‐overlapping intervals independently, or align the entire spectrum based on only a reference interval instead of the whole reference. The latter option is not trivial because in some cases a certain segment of the spectrum is more shifted than another, so correcting only based on the problematic section and leaving others untouched will give rise to a superior result of alignment. The Icoshift package is run on Matlab 2008a for testing out MS spectra dataset. Page | 23 Figure 6.2 shows the comparison before and after alignment for group4. It is safe to conclude by visual inspection that there is not a great deal of misalignment that has been corrected for the peaks centering at around 15,000, as compared to those aligned by SpecAlign. The reason is due to the choice of small maximally-­‐allowed shift. However, it is not intended to choose a larger maximum shift on the basis of experience of measurement errors. Figure 6.2 Comparison of alignment for group4 between the whole spectra raw (upper left), icoshift aligned (lower left), zoom-­‐in raw (upper right), and zoom-­‐in aligned (lower right). The alignment was obtained with a maximally-­‐allowed shift of 50 points. 6.2 Parametric Time Warping (PTW) The PTW algorithm views signals as stretched or squeezed relative to a reference signal along the m/z value. Therefore, a warping function is applied to the signals such that they can be corrected as similar as possible to the reference signal. Experience has shown that a warping function, w(ti ) , of a polynomial with degree 2 in t is sufficient in many applications (Eilers, et al., 2004). The warping function is typically denoted as below: Page | 24 K
w(ti ) = ∑ ak tik = a0 + a1ti + a2ti2 k =0
where ak refers to the warping coefficients and ti represents the vector with sampling points. Now suppose that we have a sample spectrum, xi = x(ti ) and a reference spectrum, yi = y(ti ) . We attempt to align the sample spectrum to the reference by means of the warping function w(ti ) and obtain the desired interpolated or aligned spectrum x( w(ti )) . Then the problem reduces to finding the warping coefficients, which in turn is achieved by minimizing the objective function S = ∑[ yi − x(w(ti ))]2 i∈H
where H refers to the set of indices i at which the spectra are sampled. Rewriting x( w(ti )) by use of Taylor expansion gives x( w(ti )) = x( w%(t ) + Δw(t )) ≈ x( w%(t )) + x ' ( w%(t ))Δw(t )
K
= x( w%(t )) + ∑ Δak x ' ( w%(t ))t k
k =0
Substituting the interpolated spectrum in the target function S with the approximation above yields K
S = ∑ [ yi − x( w%(ti )) − ∑ Δak x ' ( w%(ti ))ti k ]2 i∈H
k =0
By minimizing the least square function above, corrections to the warping coefficients can be obtained through recursive regression until convergence. Finally, the aligned spectrum can be obtained by linear interpolation with respect to the corresponding reference interval. Our MS spectra dataset is run by use of the PTW package loaded onto R 2.13.0 (PTW, Eilers, et al., 2004 & Bloemberg, et al., 2010). Figure 6.3 shows the comparison before and after alignment for group4. In comparison with alignment by other methods, the improvement after alignment is not so obvious by visual impression in both the whole and zoom-­‐in plots. Page | 25 Figure 6.3 Comparison of alignment for group4 between the whole spectra raw (upper left), PTW aligned (lower left), zoom-­‐in raw (upper right), and zoom-­‐in aligned (lower right). The alignment was obtained with an equivalent of maximally-­‐allowed shift of 50 points. 6.3 Correlation Optimized Warping Similar to other profile-­‐based algorithms, COW aims to correct misalignments along the m/z value in MS spectra without requiring peak detection. The COW algorithm was originally developed by Nielsen et al. (1998). The goal of alignment is achieved by dynamic programming (DP) which requires breaking down the global problem into a segment-­‐wise correlation optimization. Suppose that we have a sample MS spectrum, si = s(ti ) , which needs to be aligned relative to a reference MS spectrum, ri = r (ti ) . The DP algorithm requires both signals to be divided into a user-­‐
specified number of sections, which do not have to be equal in length and will be after alignment due to linear interpolations. Each section in the sample signal is stretched or squeezed by shifting the position of its border point a limited length, which in turn is controlled by a so-­‐called slack parameter. Then the shifted sections of the sample spectrum are linearly interpolated to the length of the corresponding section in the reference spectrum and the Pearson’s correlation coefficients among them are calculated. The same process can be applied progressively to other sections with the Page | 26 exception that the initial and final boundaries are fixed so that the first and last points in the sample and reference signals are forced to match. Finally, a global solution of alignment can be obtained by tracing backwards from the last section by determining the maximum values of the cumulative sum of Pearson’s correlation coefficients calculated forwards and their corresponding border points. It is self-­‐evident in COW that the slack parameter controls the flexibility of alignment and that the degree of flexibility decreases for segments toward the terminals of the signal compared to the middles. The COW algorithm is run on Matlab R2008a for testing our MS spectra dataset. Figure 6.4 shows the comparison before and after alignment for group4. The efficiency of alignment is more obvious in the zoom-­‐in plots illustrating that peaks occurring at around 15,000 merged together into the right major peak after alignment. Figure 6.4 Comparison of alignment for group4 between the whole spectra raw (upper left), COW aligned (lower left), zoom-­‐in raw (upper right), and zoom-­‐in aligned (lower right). The alignment was obtained with an equivalent of maximally-­‐allowed shift of 100 points. Page | 27 6.4 Results and comparisons It would be difficult to determine whether one algorithm does better alignment than another only by visual inspection. Alternatively, one can calculate the square of Pearson’s correlation between each sample and the corresponding group mean spectrum, and then take the ratio of squared correlation before and after alignment (Kong et al, 2009). Intuitively, good alignment will produce the ratio of greater than 1. Figure 6.5 shows the plot of the ratios calculated for each sample tested on each of the four algorithms. In general, the performance of PTW is inferior to that of the others, which is indicated by the ratios lightly less than 1. The reason could be that the quadratic functions used in PTW are not flexible enough to capture non-­‐quadratic m/z distortions in our complex MS spectra. Of the three more superior algorithms, there are a number of ratios calculated from Icoshift slightly less than 1; and Icoshift seems to produce more stable alignments than the others do. Several ratios with values greater than 1.5, indicated by the spikes in the plot, could result from the fact that a very low correlation before alignment is observed and taking square exaggerates the effect. One noteworthy thing about the comparison is that all the algorithms except PTW are consistent in terms of producing better alignment on the same number of samples. Conclusively, COW and SpecAlign did better work than the others, and COW is more favorable than SpecAlign in the sense that SpecAlign may sometimes over-­‐aligned some intervals of some spectra. However, COW will be the last algorithm to be considered for our dataset because it is the most computation-­‐expensive one (in about 10-­‐20 minutes for each group). Page | 28 Figure 6.5 A comparison of the performance of alignment by four distinct algorithms. The sample numbers on the x-­‐axis are ordered with respect to those shown in the clusters. Nederkassel et al. (2006) employed PCA score plots to investigate the effect of alignment and concluded by comparing the PCA score plots before and after alignment that alignment produces a better discrimination of distinct groups of samples. The same effect is also observed for out MS samples on PCA score plot. Figure 6.6 shows the PCA score plot after alignment by COW. Comparison of the plots before (See Figure 3.4) and after alignment reveals that the COW algorithm results in a more separated pattern of the 96 MS samples on the first two PCs, which is potentially superior to discriminating different groups of samples. Page | 29 Figure 6.6 PCA score plot of the 96 MS samples after alignment by COW Page | 30 7 Discrimination analysis 7.1 Fisher’s discriminant analysis The ultimate goal is to find the biomarkers for pancreatic cancer. Discrimination analysis can fulfill the objective of detecting biomarkers of certain disease. Algebraically, the objective can be described as finding the linear combinations of the variables to achieve maximum separation of the group means. In our case, we have six groups which consist of different number of samples with similar attributes in the same group and dissimilar among groups; by discrimination analysis we attempt to find a combination of distinct proteins indexed by m/z values, which will contribute to separating the groups the most; the significance of certain proteins in separating the groups is proportional to the absolute values of the coefficients of the discriminants. Therefore, by evaluating the linear discriminants, we can determine which proteins are important in detecting the biomarker. Figure 7.1 Scatter plot of Sample D16 with its selected peaks superimposed Page | 31 Fisher’s discriminant analysis reduces the dimension of the data from a very large number of variables to a relatively fewer linear combinations by representing the populations properly without, however, losing significant information. An appropriate choice of fewer linear combinations of the observations follows finding the coefficients aˆ ' Baˆ
, is maximized (please refer to the appendix for the ˆ' ˆ
aWa
derivation of Fisher’s population discriminants). The number of Fisher’s discriminants required for separating populations as much as possible is determined by whichever is the minimum: g-­‐1 (the number of populations minus one) and p (the number of variables). Fisher’s discriminant function has advantages over many other methods in that it does not require the populations be normally distributed, although it does implicitly assume equal population covariance since a pooled estimate of the common covariance matrix is used (Johnson, et al., 2002). a’s such that the ratio,
The MS dataset after alignment by COW was imported to SpecAlign in order to pick major peaks. A series of 34 peaks was exported with their intensities and positions at m/z value included. This newly generated matrix was used for Fisher’s discriminant analysis. Figure 7.1 shows one example in terms of how well the peaks selected by SpecAlign match those by visual impression. It is obvious from the plot that a few peaks in D16 are not picked up, which could result from the fact that those unselected peaks are not important in other samples, and on the contrary that those selected but minor peaks in D16 are major peaks in other samples. In any case, there is a balance that has to be reached as one attempts to pick peaks from multiple MS samples with distinct features. The algorithm of Fisher’s linear discriminant analysis was coded in Matlab and the matrix consisting of those peaks was run in Matlab2008a. Figure 7.2 shows the plot of groups of samples on the reduced two-­‐discriminant space. It is very clear from the plot that the separation of the six groups of individual observations is fully exhibited in the two-­‐dimensional discriminant space, with the exception of one sample from group3 which is well-­‐separated from the main cloud of group3 and attached to the margin of group 6. It reveals that the 34 variables (peaks) used as input for discriminant analysis are significant in separating those groups. The relative significance of those peaks is revealed by the coefficients of the first two discriminants (Table 7.1). For example, the Page | 32 peaks centered at 6669, 7941, 15141, and 15933 are more important than the rest in terms of contributing to the separation of the groups. Thus, they may serve as a clue to the medical researchers that the proteins represented by those peaks are potential candidates of biomarkers of the pancreatic cancer. Figure 7.1 The 96 MS samples plotted on the reduced two-­‐discriminant space Table 7.1 List of peaks indexed by m/z values and their correspondent discriminant coefficients m/z 2157 2437 2829 3157 3485 3685 st
1 -­‐
0.0085 0.1301 -­‐
0.0362 -­‐
discriminant 0.0296 0.1351 0.1332 2nd 0.1539 0.2754 0.1966 0.3115 0.0970 -­‐
discriminant 0.0201 m/z 5125 5349 5573 5925 6261 6669 1st 0.0088 -­‐
-­‐
0.3960 -­‐
0.2721 discriminant 0.1073 0.2770 0.1492 2nd -­‐
-­‐
0.0863 0.2131 0.1299 -­‐
discriminant 0.0755 0.8609 1.0961 m/z 8349 8581 9061 9685 10341 11093 4165 4421 4877 -­‐
-­‐
-­‐
0.2860 0.4335 0.0021 -­‐
-­‐
-­‐
0.1582 0.0317 0.1287 7301 7741 7941 0.3961 0.7813 -­‐
0.3292 -­‐
0.1348 -­‐
0.1216 1.1897 11845 12517 12789 Page | 33 1 0.0749 -­‐
discriminant 0.3607 nd
2 -­‐
0.0532 discriminant 0.2167 m/z 13461 14237 st
1 -­‐
0.4203 discriminant 1.4782 2nd 0.2927 -­‐
discriminant 0.5808 st
0.2640 -­‐
0.0080 0.1924 -­‐
0.1753 14653 15141 0.6337 -­‐
1.6657 0.1227 0.7100 0.3290 0.1447 -­‐
-­‐
0.1563 0.3684 1.0912 -­‐
0.0609 0.6651 0.0573 0.1876 0.2826 15933 19590 19990 1.3799 -­‐
0.4889 0.9663 -­‐
0.7430 -­‐
0.4838 0.8208 7.2 Stepwise discriminant analysis Although Fisher’s discriminant analysis is very useful with respect to identifying a set of peaks or proteins that may separate the pre-­‐assigned groups the most, it is weak in providing us with any statistical inference. In addition, we were unable to use any information about clinical impressions through Fisher’s discriminant analysis. However, the stepwise discriminant method performs a stepwise selection of “significant” peaks or proteins for each clinical impression. The clinicians may benefit from stepwise analysis in terms of predicting which proteins are important in discriminating among the groups of clinical impressions. The SAS procedure, STEPDISC, was run for the matrix of peaks selected from SpecAlign and clinical impressions. For example, the SAS output gives three peaks important in discriminating for “Duct”: Peak_6669, Peak_4877, and Peak_5125. Table 7.2 gives a complete list of peaks important in discriminating for each clinical impression. Page | 34 Table 7.2 A summary of the results of stepwise discriminant analysis Clinical impression Duct Peaks 6669, 4877, 5125 Clinical impression Malignant Peaks Clinical impression Cyst Peaks 15933, 7301 9061, 3485, 19990 Clinical impression Benign Peaks 11845, 12789 Clinical impression Serous Clinical Peaks Clinical impression impression Mucinous_cysta 5349, 6669 IPMN_malignant Clinical impression Tail Peaks 2829, 3685, 8349, 2157 Clinical impression Mucinous Peaks 11093, 15141 Peaks 10341, 3485 Peaks 5349, 4421, 3485, 2829, 6261 Clinical impression Peaks Clinical impression Peaks Clinical impression Peaks Fluid 6669, 9061, 5925, 3485, 19990, 12789, 11093 Head 9061, 7301, 3685, 4421, 2157, 12517 Serum 2157, 9685, 4877, 4165, 13461, 15141, 3485, 12789 Clinical impression Peaks Clinical impression Peaks Clinical impression Peaks IPMN_benign 8581, 11845, 3485, 12517, 14237 Serous_cysta 15933, 11093, 19990, 14653, 6261 Pseudocyst 2437, 8581, 6669, 4165, 3157, 5349 To assess how well these proteins discriminate between the groups of clinical impressions, SAS proc DISCRIM is used to perform cross-­‐validation error rates. Each case is “held out” from the data being used to generate the decision rule. Using all other cases, we develop the classification rule and then apply the rule to predict the classification of the held-­‐out case. Cross-­‐validation is used to avoid having very complicated rules fitting the data well but it is not useful for predicting new cases. How well the proteins are able to classify cases correctly can be summarized in many ways. Two of these are false positive and false negative rates. The false positives are the cases Page | 35 we classify as positive for the trait, for example classified as malignant, when they are really negative for the trait, really not malignant, X10 in the table given below. The false positive rate is then the fraction of negative cases in the cases we classify as positive. Classified from data Truth Not Malignant Malignant Not Malignant X00 X10 Total Malignant X01 X11 X0+ X1+ Similarly, the false negative rate is the fraction of cases we classify as negative that are actually positive for the trait. For example, the false positive and negative rates for “Duct” are 0.3810 and 0.1333, respectively. Table 7.3 gives a complete list of those rates for each clinical impression. Table 7.3 A summary of false positive and negative rates for each clinical impression Clinical impression Duct Clinical impression Fluid Clinical impression Cyst Clinical impression Serum Clinical False False positive negative rate rate 0.1333 0.3810 False False positive negative rate rate 0.2292 0.2708 False False positive negative rate rate 0.2917 0.4167 False False positive negative rate rate 0.0568 0.1250 False False positive Clinical impression False negative rate False positive rate Benign Clinical impression 0.2289 0.3077 False negative rate False positive rate 0.2169 0.3846 False negative rate False positive rate 0.1500 0.4375 False negative rate False positive rate 0.2771 0.0000 False negative False Tail Clinical impression Head Clinical impression Mucinous Clinical Page | 36 impression negative rate rate impression rate 0.2222 0.2000 IPMN_malignant 0.2414 IPMN_benign Clinical False False positive Clinical False negative impression negative rate impression rate rate Serous_cysta Clinical impression Mucinous_cysta Clinical impression Pseudocyst 0.1023 0.3750 False False positive negative rate rate 0.4824 0.0000 False False positive negative rate rate 0.0899 0.2857 positive rate 0.1818 False positive rate Malignant Clinical impression 0.3012 0.3077 False negative rate False positive rate Serous 0.1379 0.2222 Page | 37 8 Conclusion Before any more advanced statistical analysis is performed, those methods of data cleaning are necessary and have been proved efficient: truncation, normalization, smoothing, resampling, and baseline removal. The asymmetric least-­‐squares (ALS) baseline estimation properly divides MS signals from background noises. Ward’s hierarchical clustering method appears to be more favorable than other linkage methods because it produced a “neat” result of clustering. The contour plot of Pearson’s correlation coefficient among the 96 MS samples reveals that the clustering is appropriate in the sense that samples are highly correlated within the same group and lowly correlated between different groups. Among the four different software packages of alignment, COW has been proved to be the most powerful method in terms of correcting the misalignment in m/z values, as indicated by higher squared ratios of Pearson’s correlation coefficient before and after alignment relative to the reference spectrum. However, it is also the most time-­‐consuming method. The PCA score plot is an appropriate way of investigating the effect of alignment: alignment produces a better discrimination of distinct groups of samples. The goal of identifying biomarkers of the pancreatic cancer is achieved through discrimination analysis. By Fisher’s method, we found a combination of peaks or proteins that are important in separating the pre-­‐
assigned groups the most and the peaks with larger discriminant coefficients might be good candidates for medical researchers who seek for the biomarkers of pancreatic cancer. By the stepwise method, we found a series of peaks that are important in discriminating among the clinical impression groups. Page | 38 9 References [1] Aebersold R. and Matthias M. (2003) Mass spectrometry-­‐based proteomics, Nature, 422, 198-­‐206. [2] Bloemberg, T.G., et al. (2010) Improved parametric time warping for proteomics, Chemometrics and Intelligent Laboratory Systems, 104, 65-­‐74. [3] Eilers, P.H. (2004) Parametric time warping, Anal. Chem., 76, 404-­‐411. [4] Feng, Y., et al., (2009) Alignment of protein mass spectrometry data by integrated Markov chain shifting method, Statistics and Its Interface, 2, 329-­‐340. [5] Johnson, R.A. and Wichern D.W. (2002) Applied and multivariate statistical analysis (Fifth edition), Prentice-­‐Hall, 426-­‐745. [6] Kong, X. and Reilly, C. (2009) A Baysian approach to the alignment of mass spectra, Bioinformatics, 25, 3213-­‐3220. [7] Savorani, F., et al. (2010) icoshift: A versatile tool for the rapid alignment of 1D NMR spectra, Journal of Magnetic Resonance, 202, 190-­‐202. [8] Tomasi, G., et al. (2004) Correlation optimized warping and dynamic time warping as preprocessing methods for chromatographic data, Journal of Chemometrics, 18, 231-­‐241. [9] van Nederkassel, A.M., et al. (2006) Chemometric treatment of vanillin fingerprint chromatograms effect of different signal alignments on principal component analysis plots, Journal of Chromatography A, 1120, 291-­‐298. [10] van Nederkassel, A.M., et al. (2006) A comparison of three algorithms for chromatograms alignment, Journal of Chromatography A, 1118, 199-­‐210. [11] Vest Nielsen, N., et al. (1998) Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimized warping, Journal of Chromatography A, 805, 17-­‐35. [12] Wong, W.H., et al. (2005) SpecAlign-­‐processing and alignment of mass spectra datasets, Bioinformatics, 21, 2088-­‐2090. [13] Wong, W.H., et al. (2005) Application of fast Fourier transform cross-­‐correlation for the alignment of large chromatographic and spectral datasets, Anal. Chem., 77, 5655-­‐5661. Page | 39 10 Appendices 10.1 R codes Baseline subtraction #Baseline correction for Samples B23 and B24--------------------------library(ptw)
y <- read.table("All_trunc.csv", header=TRUE, sep=",")
y <- t(y)
y_cal <- log(y[2:97,])
z1 <- asysm(y_cal[47,], lambda = 1e7, p=0.001)
plot(y[1,],y_cal[47,], type = "l")
lines(y[1,],z1, col = 2)
z2 <- asysm(y_cal[48,], lambda = 1e7, p=0.001)
plot(y[1,],y_cal[48,], type = "l")
lines(y[1,],z2, col = 2)
library(xlsReadWrite)
write.xls(z1, "B23.xls")
write.xls(z2, "B24.xls")
#---------------------------------------------------------------------x1 <- baseline.corr(y_cal[47,], lambda = 1e7, p=0.001)
x2 <- baseline.corr(y_cal[48,], lambda = 1e7, p=0.001)
library(xlsReadWrite)
write.xls(x1, "B23.corr.xls")
write.xls(x2, "B24.corr.xls")
#---------------------------------------------------------------------#Baseline subtraction for all 96 MS spectra
x <- baseline.corr(y_cal, lambda = 1e7, p = 0.001)
x <- t(x)
write.xls(x, "all_bline_corr.xls")
#---------------------------------------------------------------------z11 <- y_cal[47,]
z21 <- y_cal[48,]
write.xls(z11, "B23o.xls")
write.xls(z21, "B24o.xls")
Parametric time warping # Group 1-------------------------------------------------------------library(ptw)
g1 <- read.table("group1.csv", header=TRUE, sep=",")
g1 <- t(g1)
ref <- g1[5,]
samp <- g1[2:4,]
g1.ptw <- ptw(ref, samp, warp.type = "global", optim.crit = "WCC",
try = FALSE, verbose = TRUE,
trwdth = 50, init.coef = c(0, 1, 0, 0, 0 ,0))
summary(g1.ptw)
# Plot the results
plot(g1.ptw, what = "signal", type = "individual", ask = TRUE)
plot(g1.ptw, what = "function")
Page | 40 # Export warped signals
library(xlsReadWrite)
g1.warped <- g1.ptw$warped.sample
g1.warped <- t(g1.warped)
write.xls(g1.warped, "g1w.xls")
#Group 2--------------------------------------------------------------rm(list = ls())
objects()
g2 <- read.table("group2.csv", header=TRUE, sep=",")
g2 <- t(g2)
ref <- g2[7,]
samp <- g2[2:6,]
g2.ptw <- ptw(ref, samp, warp.type = "global", optim.crit = "WCC",
try = FALSE, verbose = TRUE,
trwdth = 50, init.coef = c(0, 1, 0, 0, 0 ,0))
summary(g2.ptw)
# Plot the results
plot(g2.ptw, what = "signal", type = "individual", ask = TRUE)
plot(g2.ptw, what = "function")
# Export warped signals
library(xlsReadWrite)
g2.warped <- g2.ptw$warped.sample
g2.warped <- t(g2.warped)
write.xls(g2.warped, "g2w.xls")
#Group 3--------------------------------------------------------------rm(list = ls())
ls()
g3 <- read.table("group3.csv", header=TRUE, sep=",")
g3 <- t(g3)
ref <- g3[17,]
samp <- g3[2:16,]
g3.ptw <- ptw(ref, samp, warp.type = "global", optim.crit = "WCC",
try = FALSE, verbose = TRUE,
trwdth = 50, init.coef = c(0, 1, 0, 0, 0 ,0))
summary(g3.ptw)
# Plot the results
plot(g3.ptw, what = "signal", type = "individual", ask = TRUE)
plot(g3.ptw, what = "function")
# Export warped signals
library(xlsReadWrite)
g3.warped <- g3.ptw$warped.sample
g3.warped <- t(g3.warped)
write.xls(g3.warped, "g3w.xls")
#Group 4--------------------------------------------------------------rm(list = ls())
objects()
g4 <- read.table("group4.csv", header=TRUE, sep=",")
g4 <- t(g4)
ref <- g4[18,]
samp <- g4[2:17,]
g4.ptw <- ptw(ref, samp, warp.type = "global", optim.crit = "WCC",
try = FALSE, verbose = TRUE,
trwdth = 50, init.coef = c(0, 1, 0, 0, 0 ,0))
summary(g4.ptw)
# Plot the results
plot(g4.ptw, what = "signal", type = "individual", ask = TRUE)
plot(g4.ptw, what = "function")
Page | 41 # Export warped signals
library(xlsReadWrite)
g4.warped <- g4.ptw$warped.sample
g4.warped <- t(g4.warped)
write.xls(g4.warped, "g4w.xls")
#Group 5--------------------------------------------------------------rm(list = ls())
objects()
g5 <- read.table("group5.csv", header=TRUE, sep=",")
g5 <- t(g5)
ref <- g5[20,]
samp <- g5[2:19,]
g5.ptw <- ptw(ref, samp, warp.type = "global", optim.crit = "WCC",
try = FALSE, verbose = TRUE,
trwdth = 50, init.coef = c(0, 1, 0, 0, 0 ,0))
summary(g5.ptw)
# Plot the results
plot(g5.ptw, what = "signal", type = "individual", ask = TRUE)
plot(g5.ptw, what = "function")
# Export warped signals
library(xlsReadWrite)
g5.warped <- g5.ptw$warped.sample
g5.warped <- t(g5.warped)
write.xls(g5.warped, "g5w.xls")
#Group 6--------------------------------------------------------------rm(list = ls())
objects()
g6 <- read.table("group6.csv", header=TRUE, sep=",")
g6 <- t(g6)
ref <- g6[41,]
samp <- g6[2:40,]
g6.ptw <- ptw(ref, samp, warp.type = "global", optim.crit = "WCC",
try = FALSE, verbose = TRUE,
trwdth = 50, init.coef = c(0, 1, 0, 0, 0 ,0))
summary(g6.ptw)
# Plot the results
plot(g6.ptw, what = "signal", type = "individual", ask = TRUE)
plot(g6.ptw, what = "function")
# Export warped signals
library(xlsReadWrite)
g6.warped <- g6.ptw$warped.sample
g6.warped <- t(g6.warped)
write.xls(g6.warped, "g6w.xls")
10.2 SAS codes Stepwise discriminant analysis (by Dr. Ronald Regal) libname specs 'C:\Documents and
Settings\mathuser\Desktop\Regal\Junmin\Spec\March
11\June\specsclustered';
run;
/* Clear Graphics Window
proc greplay igout=work.gseg nofs;
delete _all_;
Page | 42 run;
*/
proc import replace
datafile="C:\Documents and
Settings\mathuser\Desktop\Regal\Junmin\Spec\March
11\June\specsclustered\pancreatic_info.xls"
out=specs.spec_classify;
run;
proc print data= specs.spec_classify;
run;
proc import replace
datafile="C:\Documents and
Settings\mathuser\Desktop\Regal\Junmin\Spec\March
11\June\specsclustered\clusters.xls"
out=specs.clusters;
run;
proc print data= specs.clusters;
run;
data specs.classify;
set specs.spec_classify;
specs = tranwRD(spec,"A ","A");
specs = tranwRD(specs,"B ","B");
specs = tranwRD(specs,"C ","C");
specs = tranwRD(specs,"D ","D");
clin_low = lowcase(clinical_impression);
if findw(clin_low,"duct") or code = 'D' then duct = 1;
else duct=0;
if findw(clin_low,"fluid") or code = 'D' then fluid = 1;
else fluid=0;
if findw(clin_low,"tail") then tail = 1;
else tail=0;
if findw(clin_low,"head") then head = 1;
else head=0;
if findw(clin_low,"cyst") then cyst = 1;
else cyst=0;
if findw(clin_low,"serum") then serum = 1;
else serum=0;
if findw(clin_low,"mucinous")then mucinous = 1;
else mucinous=0;
if findw(clin_low,"ipmn") or code ='Im' or code = 'Ib'
then ipmn = 1;
else ipmn=0;
if code = 'Ib' then IPMN_benign = 1;
else IPMN_benign=0;
if code = 'Im' then IPMN_malignant = 1;
else IPMN_malignant=0;
if code = 'Im' or findw(clin_low,"malignant")then malignant = 1;
else malignant=0;
if findw(clin_low,"body") then body = 1;
else body=0;
if code = 'S' or findw(clin_low,"serous cystadenoma") then
serous_cysta = 1;
else serous_cysta =0;
if code = 'M' or findw(clin_low,"mucinous cystadenoma")then
mucinous_cysta = 1;
else mucinous_cysta =0;
if findw(clin_low,"serous") or code = 'S' then serous = 1;
Page | 43 else serous=0;
if findw(clin_low,"benign") or code ='Ib' then benign = 1;
else benign=0;
if findw(clin_low,"cyst") then cyst = 1;
else cyst=0;
if findw(clin_low,"pseudocyst") then pseudocyst = 1;
else pseudocyst=0;
spec = specs;
drop specs;
run;
proc print data= specs.classify;
var spec code clin_low duct--pseudocyst;
run;
proc sort data= specs.clusters;
by spec;
run;
proc sort data= classify;
by spec;
run;
data clusters;
set specs.clusters;
if cluster = 1 then cluster_1 = 1; else cluster_1 = 0;
if cluster = 2 then cluster_2 = 1; else cluster_2 = 0;
if cluster = 3 then cluster_3 = 1; else cluster_3 = 0;
if cluster = 4 then cluster_4 = 1; else cluster_4 = 0;
if cluster = 5 then cluster_5 = 1; else cluster_5 = 0;
if cluster = 6 then cluster_6 = 1; else cluster_6 = 0;
run;
data specs.cluster_class;
merge clusters classify;
by spec;
run;
proc print data=specs.cluster_class;;
run;
proc freq data=specs.cluster_class;
tables (cluster_1-cluster_6)*(duct--pseudocyst)/crosslist fisher;
ods select crosslist fishersexact;
ods output fishersexact=fishersexact;
run;
proc sort data=fishersexact;
by cvalue1;
run;
proc print data=fishersexact;
where name1 ='XP2_FISH';
var table cvalue1;
run;
proc import replace
datafile="C:\Documents and
Settings\mathuser\Desktop\Regal\Junmin\Spec\March
11\June\specsclustered\peak.xls"
out=specs.peaking;
sheet='peaking';
run;
proc sort data=specs.peaking;
by spec;
run;
data specs.cluster_class;
Page | 44 merge specs.cluster_class specs.peaking;
by spec;
run;
proc contents data=cluster_class;
run;
proc format;
value yes_no_fmt 0='No' 1='Yes';
run;
options mprint;
goptions reset=all;
goptions hsize=3in vsize=5in;
symbol1 value=dot height=0.05in color=black;
%macro stepping(var);
* Stepwise;
proc stepdisc data=specs.cluster_class;
title "&var";
class &var;
var peak_2157 -- peak_19990;
*ods select none;
ods select bcorr summary variables counts;
run;
* Cross-validated Missclassification Rates;
proc discrim data=cluster_class crosslisterr;
class &var;
var &_stdvar;
* The selected peaks;
* ods select none;
run;
* Plotting;
proc candisc data=cluster_class out=outcan;
class &var;
var &_stdvar;
* ods select none;
run;
proc sort data=outcan;
by &var can1;
run;
* Move points so not on top of each other;
data outcan;
format &var.s yes_no_fmt.;
set outcan;
by &var can1;
prev_can1 = lag(can1);
if first.&var or (abs(can1 - first_can1) gt 0.1) then do;
&var.s = &var;
rep = 1;
first_can1 = can1;
end;
else do;
if &var = 0 then &var.s = &var + 0.01*rep;
else if &var = 1 then &var.s = &var - 0.005*rep;
rep = rep +1;
end;
retain rep first_can1;
run;
axis1 label=("&var") order= (0 to 1 by 1);
proc gplot data=outcan;
plot can1*&var.s/haxis=axis1;
Page | 45 run;
%mend;
ods pdf file= 'C:\Documents and
Settings\mathuser\Desktop\Regal\Junmin\Spec\March
11\June\specsclustered\Disc.pdf';
options nodate ;
%stepping(Duct);
%stepping(Benign);
%stepping(Body);
%stepping(Fluid);
%stepping(Tail);
%stepping(Cyst);
%stepping(Head);
%stepping(Serum);
%stepping(Mucinous);
%stepping(IPMN_Malignant);
%stepping(IPMN_Benign);
%stepping(Serous_Cysta);
%stepping(Malignant);
%stepping(Mucinous_Cysta);
%stepping(Serous);
%stepping(Cyst);
%stepping(Benign);
%stepping(Pseudocyst);
proc stepdisc data=cluster_class;
title "Cluster 4";
class cluster_4;
var peak_2157 -- peak_19990;
* ods select none;
ods select bcorr summary variables counts;
run;
proc discrim data=cluster_class crosslisterr;
class Cluster_4;
var &_stdvar;
* The selected peaks;
* ods select none;
run;
* Plotting;
proc candisc data=cluster_class out=outcan_cl;
class Cluster_4;
var &_stdvar;
* ods select none;
run;
proc sort data=outcan_cl;
by Cluster_4 can1;
run;
* Move points so not on top of each other;
data outcan_cl;
format Cluster_4s yes_no_fmt.;
set outcan_cl;
by Cluster_4 can1;
prev_can1 = lag(can1);
if first.Cluster_4 or (abs(can1 - first_can1) gt 0.1) then do;
Cluster_4s = Cluster_4;
rep = 1;
first_can1 = can1;
end;
else do;
Page | 46 if Cluster_4 = 0 then Cluster_4s = Cluster_4 + 0.01*rep;
else if Cluster_4 = 1 then Cluster_4s = Cluster_4 - 0.005*rep;
rep = rep +1;
end;
retain rep first_can1;
run;
axis1 label=("Cluster 4") order= (0 to 1 by 1);
proc gplot data=outcan_cl;
plot can1*cluster_4s/haxis=axis1;
run;
************************ ;
proc stepdisc data=cluster_class;
title 'Cluster 1';
class cluster_1;
var peak_2157 -- peak_19990;
* ods select none;
ods select bcorr summary variables counts;
run;
proc discrim data=cluster_class crosslisterr;
class Cluster_1;
var &_stdvar;
* The selected peaks;
* ods select none;
run;
* Plotting;
proc candisc data=cluster_class out=outcan_cl;
class Cluster_1;
var &_stdvar;
ods select none;
run;
ods select all; * ?????;
proc sort data=outcan_cl;
by Cluster_1 can1;
run;
* Move points so not on top of each other;
data outcan_cl;
format Cluster_1s yes_no_fmt.;
set outcan_cl;
by Cluster_1 can1;
prev_can1 = lag(can1);
if first.Cluster_1 or (abs(can1 - first_can1) gt 0.1) then do;
Cluster_1s = Cluster_1;
rep = 1;
first_can1 = can1;
end;
else do;
if Cluster_1 = 0 then Cluster_1s = Cluster_1 + 0.01*rep;
else if Cluster_1 = 1 then Cluster_1s = Cluster_1 - 0.005*rep;
rep = rep +1;
end;
retain rep first_can1;
run;
axis1 label=("Cluster 4") order= (0 to 1 by 1);
proc gplot data=outcan_cl;
plot can1*cluster_1s/haxis=axis1;
run;
******************;
ods pdf close;
Page | 47 10.3 Matlab codes Alignment by Icoshift %% Alignment of group1------------------------------------------------g1 = g1';
mz = g1(1,:);
%% iCOshift 1: aligns the whole spectra according to the mean
[g1_cos, intervals, indexes, target] = icoshift ('average', g1(2:4,:),
'whole', 'b', [2 1 1], mz);
%% iCOshift 2: splits the dataset in 100 regular intervals and aligns
each of them separately
[g1_cos, intervals, indexes, target] = icoshift ('average', g1(2:4,:),
50, 'b', [2 1 1], mz);
%% iCOshift 3: splits the dataset in regular intervals 500 points wide
and search for the "best" allowed shift for each of them separately
[g1_cos, intervals, indexes, target1] = icoshift ('average', g1(2:4,:),
'50', 'b', [2 1 1], mz);
%% Alignment of group2------------------------------------------------g2 = g2';
mz = g2(1,:);
%% iCOshift 1: aligns the whole spectra according to the mean
[g2_cos, intervals, indexes, target] = icoshift ('average', g2(2:6,:),
'whole', 'b', [2 1 1], mz);
%% iCOshift 2: splits the dataset in 100 regular intervals and aligns
each of them separately
[g2_cos, intervals, indexes, target] = icoshift ('average', g2(2:6,:),
50, 'b', [2 1 1], mz);
%% iCOshift 3: splits the dataset in regular intervals 500 points wide
and search for the "best" allowed shift for each of them separately
[g2_cos, intervals, indexes, target2] = icoshift ('average', g2(2:6,:),
'50', 'b', [2 1 1], mz);
%% Alignment of group3------------------------------------------------g3 = g3';
mz = g3(1,:);
%% iCOshift 1: aligns the whole spectra according to the mean
[g3_cos, intervals, indexes, target] = icoshift ('average', g3(2:16,:),
'whole', 'b', [2 1 1], mz);
%% iCOshift 2: splits the dataset in 100 regular intervals and aligns
each of them separately
[g3_cos, intervals, indexes, target] = icoshift ('average', g3(2:16,:),
50, 'b', [2 1 1], mz);
%% iCOshift 3: splits the dataset in regular intervals 500 points wide
and search for the "best" allowed shift for each of them separately
[g3_cos, intervals, indexes, target3] = icoshift ('average', g3(2:16,:),
'50', 'b', [2 1 1], mz);
%% plots for the purpose of comparison
subplot(2,2,1)
plot(mz,g3(2:16,:))
xlabel('m/z value'); ylabel('Intensity')
hold on
subplot(2,2,3)
plot(mz,g3_cos)
xlabel('m/z value'); ylabel('Intensity')
hold on
subplot(2,2,2)
Page | 48 plot(mz(8100:9200),g3(2:16, 8100:9200))
xlabel('m/z value'); ylabel('Intensity')
hold on
subplot(2,2,4)
plot(mz(8100:9200),g3_cos(:,8100:9200))
xlabel('m/z value'); ylabel('Intensity')
%% Alignment of group4------------------------------------------------g4 = g4';
mz = g4(1,:);
%% iCOshift 1: aligns the whole spectra according to the mean
[g4_cos, intervals, indexes, target] = icoshift ('average', g4(2:17,:),
'whole', 'b', [2 1 1], mz);
%% iCOshift 2: splits the dataset in 100 regular intervals and aligns
each of them separately
[g4_cos, intervals, indexes, target] = icoshift ('average', g4(2:17,:),
50, 'b', [2 1 1], mz);
%% iCOshift 3: splits the dataset in regular intervals 500 points wide
and search for the "best" allowed shift for each of them separately
[g4_cos, intervals, indexes, target4] = icoshift ('average', g4(2:17,:),
'50', 'b', [2 1 1], mz);
%% plots for the purpose of comparison
subplot(2,1,1)
plot(mz,g4(2:17,:))
xlabel('m/z value'); ylabel('Intensity')
hold on
subplot(2,1,2)
plot(mz,g4_cos)
xlabel('m/z value'); ylabel('Intensity')
%% Alignment of group5------------------------------------------------g5 = g5';
mz = g5(1,:);
%% iCOshift 1: aligns the whole spectra according to the mean
[g5_cos, intervals, indexes, target] = icoshift ('average', g5(2:19,:),
'whole', 'b', [2 1 1], mz);
%% iCOshift 2: splits the dataset in 100 regular intervals and aligns
each of them separately
[g5_cos, intervals, indexes, target] = icoshift ('average', g5(2:19,:),
50, 'b', [2 1 1], mz);
%% iCOshift 3: splits the dataset in regular intervals 500 points wide
and search for the "best" allowed shift for each of them separately
[g5_cos, intervals, indexes, target5] = icoshift ('average', g5(2:19,:),
'50', 'b', [2 1 1], mz);
%% plots for the purpose of comparison
subplot(2,1,1)
plot(mz,g5(2:19,:))
xlabel('m/z value'); ylabel('Intensity')
hold on
subplot(2,1,2)
plot(mz,g5_cos)
xlabel('m/z value'); ylabel('Intensity')
%% Alignment of group6
g6 = g6';
mz = g6(1,:);
%% iCOshift 1: aligns the whole spectra according to the mean
[g6_cos, intervals, indexes, target] = icoshift ('average', g6(2:40,:),
'whole', 'b', [2 1 1], mz);
Page | 49 %% iCOshift 2: splits the dataset in 100 regular intervals and aligns
each of them separately
[g6_cos, intervals, indexes, target] = icoshift ('average', g6(2:40,:),
40, 'b', [2 1 1], mz);
%% iCOshift 3: splits the dataset in regular intervals 500 points wide
and search for the "best" allowed shift for each of them separately
[g6_cos, intervals, indexes, target6] = icoshift ('average', g6(2:40,:),
'50', 'b', [2 1 1], mz);
%% plots for the purpose of comparison
subplot(2,1,1)
plot(mz,g6(2:40,:))
xlabel('m/z value'); ylabel('Intensity')
hold on
subplot(2,1,2)
plot(mz,g6_cos)
xlabel('m/z value'); ylabel('Intensity')
Alignment by COW %% Alignment of group1------------------------------------------------g1 = g1';
ref = g1(5,:);
%% Find the optimal segment length and slack size parameter
[optim_pars,OS,diagnos] = optim_cow(g1(2:5,:),[5 240 1 80],[1 3 50
0.15],ref);
%% Align the spectra using COW
[warping,Xw_cow1,diagnos] = cow(g1(5,:),g1(2:4,:),10,5,[0 1 0]);
%% Plot the warped results to visualize any apparent improvement
subplot(2,1,1)
plot(g1(1,:),g1(2,:),g1(1,:),g1(3,:),g1(1,:),g1(4,:),g1(1,:),g1(5,:))
xlabel('m/z value'); ylabel('Intensity')
legend('A1', 'B13', 'B17', 'Average')
hold on
subplot(2,1,2)
plot(g1(1,:),g1(5,:),g1(1,:),Xw_cow(1,:), g1(1,:),Xw_cow(2,:),
g1(1,:),Xw_cow(3,:))
xlabel('m/z value'); ylabel('Intensity')
legend('reference', 'aligned')
%% Alignment of group2------------------------------------------------g2 = g2';
ref = g2(7,:);
%% Find the optimal segment length and slack size parameter
[optim_pars,OS,diagnos] = optim_cow(g2(2:7,:),[5 240 1 80],[1 3 50
0.15],ref);
%% Align the spectra using COW
[warping,Xw_cow2,diagnos] = cow(g2(7,:),g2(2:6,:),18,1,[0 1 0]);
%% Alignment of group3------------------------------------------------g3 = g3';
ref = g3(17,:);
%% Find the optimal segment length and slack size parameter
[optim_pars,OS,diagnos] = optim_cow(g3(2:17,:),[5 240 1 80],[1 3 50
0.15],ref);
%% Align the spectra using COW
[warping,Xw_cow3,diagnos] = cow(g3(17,:),g3(2:16,:),24,9,[0 1 0]);
%% Alignment of group4-------------------------------------------------
Page | 50 g4 = g4';
ref = g4(18,:);
%% Find the optimal segment length and slack size parameter
[optim_pars,OS,diagnos] = optim_cow(g4(2:18,:),[5 240 1 80],[1 3 50
0.15],ref);
%% Align the spectra using COW
[warping,Xw_cow4,diagnos] = cow(g4(18,:),g4(2:17,:),18,1,[0 1 0]);
%% Alignment of group5------------------------------------------------g5 = g5';
ref = g5(20,:);
%% Find the optimal segment length and slack size parameter
[optim_pars,OS,diagnos] = optim_cow(g5(2:20,:),[5 240 1 80],[1 3 50
0.15],ref);
%% Align the spectra using COW
[warping,Xw_cow5,diagnos] = cow(g5(20,:),g5(2:19,:),11,5,[0 1 0]);
%% Alignment of group6------------------------------------------------g6 = g6';
ref = g6(41,:);
%% Find the optimal segment length and slack size parameter
[optim_pars,OS,diagnos] = optim_cow(g6(2:41,:),[5 240 1 80],[1 3 50
0.15],ref);
%% Align the spectra using COW
[warping,Xw_cow6,diagnos] = cow(g6(41,:),g6(2:40,:),11,4,[0 1 0]);
Calculations of correlation coefficient before and after alignment %% Comparison of correlation coefficients for COW
%group1---------------------------------------------------------------%%
%g1 = g1';
%g1_cos = g1_cos';
%target1 = target1';
rho1_bf = corr(g1(:,2:4),g1(:,5));
rho1_af = corr(g1_cos(:,2:4),g1_cos(:,5));
ratio_1 = (rho1_af.^2)./(rho1_bf.^2);
%group2---------------------------------------------------------------%%
%g2 = g2';
%g2_cos = g2_cos';
%target2 = target2';
rho2_bf = corr(g2(:,2:6),g2(:,7));
rho2_af = corr(g2_cos(:,2:6),g2_cos(:,7));
ratio_2 = (rho2_af.^2)./(rho2_bf.^2);
%group3---------------------------------------------------------------%%
%g3 = g3';
%g3_cos = g3_cos';
%target3 = target3';
rho3_bf = corr(g3(:,2:16),g3(:,17));
rho3_af = corr(g3_cos(:,2:16),g3_cos(:,17));
ratio_3 = (rho3_af.^2)./(rho3_bf.^2);
%group4---------------------------------------------------------------%%
%g4 = g4';
%g4_cos = g4_cos';
Page | 51 %target4 = target4';
rho4_bf = corr(g4(:,2:17),g4(:,18));
rho4_af = corr(g4_cos(1:20872,2:17),g4_cos(1:20872,18));
ratio_4 = (rho4_af.^2)./(rho4_bf.^2);
%group5---------------------------------------------------------------%%
%g5 = g5';
%g5_cos = g5_cos';
%target5 = target5';
rho5_bf = corr(g5(:,2:19),g5(:,20));
rho5_af = corr(g5_cos(:,2:19),g5_cos(:,20));
ratio_5 = (rho5_af.^2)./(rho5_bf.^2);
%group6---------------------------------------------------------------%%
%g6 = g6';
%g6_cos = g6_cos';
%target6 = target6';
rho6_bf = corr(g6(:,2:40),g6(:,41));
rho6_af = corr(g6_cos(:,2:40),g6_cos(:,41));
ratio_6 = (rho6_af.^2)./(rho6_bf.^2);
%put all r squared into one vector------------------------------------%%
r2_cow = [ratio_1;ratio_2;ratio_3;ratio_4;ratio_5;ratio_6];
%% plot the results
x = 1:96;
plot(x,r)
xlabel('Sample number'); ylabel('Ratio')
legend('COW', 'Icoshift', 'SpecAlign', 'PTW')
Fisher’s discriminant analysis g1_cow = peak(:,2:4);
g2_cow = peak(:,5:9);
g3_cow = peak(:,10:24);
g4_cow = peak(:,25:40);
g5_cow = peak(:,41:58);
g6_cow = peak(:,59:97);
%%
g1 = g1_cow';
g2 = g2_cow';
g3 = g3_cow';
g4 = g4_cow';
g5 = g5_cow';
g6 = g6_cow';
%% Calculate the sample mean vector and the covariance matrices
g1_mean = mean(g1);
g2_mean = mean(g2);
g3_mean = mean(g3);
g4_mean = mean(g4);
g5_mean = mean(g5);
g6_mean = mean(g6);
s1 = cov(g1);
s2 = cov(g2);
s3 = cov(g3);
s4 = cov(g4);
Page | 52 s5 = cov(g5);
s6 = cov(g6);
%% Define the overall average vector and pooled matrix of variance
total =
size(g1,1)+size(g2,1)+size(g3,1)+size(g4,1)+size(g5,1)+size(g6,1);
all_mean =
(size(g1,1)*g1_mean+size(g2,1)*g2_mean+size(g3,1)*g3_mean+ ...
size(g4,1)*g4_mean+size(g5,1)*g5_mean+size(g6,1)*g6_mean)/total;
s_pooled = ((size(g1,1)-1)/(total-5))*s1+((size(g2,1)-1)/(total5))*s2+...
((size(g3,1)-1)/(total-5))*s3+((size(g4,1)-1)/(total5))*s4+...
((size(g5,1)-1)/(total-5))*s5+((size(g6,1)-1)/(total-5))*s6;
%% Calculate the matrices B and W
w = (total-5)*s_pooled;
B = size(g1,1)*(g1_mean'-all_mean')*(g1_mean'-all_mean')'+ ...
size(g2,1)*(g2_mean'-all_mean')*(g2_mean'-all_mean')'+ ...
size(g3,1)*(g3_mean'-all_mean')*(g3_mean'-all_mean')'+ ...
size(g4,1)*(g4_mean'-all_mean')*(g4_mean'-all_mean')'+ ...
size(g5,1)*(g5_mean'-all_mean')*(g5_mean'-all_mean')'+ ...
size(g6,1)*(g6_mean'-all_mean')*(g6_mean'-all_mean')';
%% Calculate the eigenvelues and eigenvectors
[V1,D1] = eig(w^(-0.5)*B*w^(-0.5));
a = (w^(-0.5)*V1);
%%
y1 = a(:,1)'*peak(:,2:97);
y2 = a(:,2)'*peak(:,2:97);
%%
plot(y1(1,1:3),y2(1,1:3),'+b', 'MarkerSize',12)
hold on
plot(y1(1,4:8),y2(1,4:8),'*b','MarkerSize',12)
hold on
plot(y1(1,9:23),y2(1,9:23),'dg','MarkerSize',12)
hold on
plot(y1(1,24:39),y2(1,24:39),'xb','MarkerSize',12)
hold on
plot(y1(1,40:57),y2(1,40:57),'om','MarkerSize',12)
hold on
plot(y1(1,58:96),y2(1,58:96),'pr','MarkerSize',12)
legend('Group1','Group2', 'Group3', 'Group4', 'Group5', 'Group6')
xlabel('First discriminant'); ylabel('Second discriminant')
Page | 53 10.4 Proof Proof of Fisher’s Method for Discriminating among Several populations (Extended from Exercise 11.21 at Page 651 of Johnson et al., 2002) g
∑ (µ
iY
The ratio, − µY )
i =1
σ Y2
g
2
'
'
∑ (a µ − a µ )
2
'
i
=
i =1
a 'Σa
=
g
a [∑ ( µi − µ )( µi − µ )' ]a
i =1
a 'Σa
=
a ' Bµ a
a 'Σa
, measures the variability between the groups of Y-­‐values relative to the common variability within groups. The primary goal of Fisher’s discriminant analysis is to separate populations with its optimization reached as the ratio defined above is maximized for appropriate choices of a. Let u = Σ1/ 2 a , so u ' u = a ' Σ1/ 2Σ1/ 2 a = a ' Σa and u ' Σ −1/ 2 Bu Σ −1/ 2u = a ' Σ1/ 2Σ −1/ 2 Bu Σ −1/ 2Σ1/ 2 a = a ' Bu a . As a result, the problem reduces to u ' Σ−1/ 2 Bu Σ−1/ 2u
maximizing over u. From (2-­‐51), the maximum of this ratio is the largest u 'u
eigenvalue of Σ −1/ 2 Bu Σ −1/ 2 . This maximum occurs when u=e1, the normalized eigenvector associated with λ1 . Because e1=u= Σ1/ 2 a1 , or a1 = Σ −1/ 2 e1 , Var (a1' X ) = a1' Σa1 = e1' Σ −1/ 2ΣΣ −1/ 2e1 = e1' Σ −1/ 2Σ1/ 2Σ1/ 2Σ −1/ 2e1 = e1' e1 = 1. For the choice of e2 or a2, we have Var (a2' X ) = a2' Σa2 = e2' Σ −1/ 2ΣΣ −1/ 2 e2 = e2' Σ −1/ 2Σ1/ 2Σ1/ 2Σ −1/ 2e2 = e2' e2 = 1. and Cov(a2' X , a1' X ) = a2' Σa1 = e2' Σ −1/ 2 ΣΣ −1/ 2e1 = e2' e1 = 0. Note that if λ and e are an eigenvalue-­‐eigenvector pair of Σ −1/ 2 Bu Σ −1/ 2 , then Σ −1/ 2 Bu Σ −1/ 2 e = λ e and multiplication on the left by Σ−1/ 2 gives Σ −1/ 2 Σ −1/ 2 Bu Σ −1/ 2 e = λΣ −1/ 2 e or Σ −1 Bu (Σ −1/ 2 e) = λ (Σ −1/ 2 e) . Thus, Σ −1 Bu has the same eigenvalues as Σ −1/ 2 Bu Σ −1/ 2 , but the corresponding eigenvector is proportional to Σ −1/ 2 e = a . The linear combination a1' x is called the first discriminant. The choice a2 produces the second discriminant, a2' x , and continuing, we obtain the kth discriminant with k ≤ s (s ≤ min(g-­‐1,p)). Page | 54