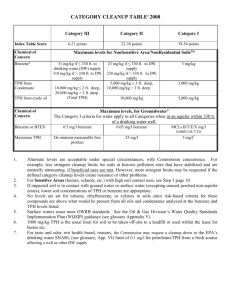
structure and function of tryptophan hydroxylase
advertisement

STRUCTURE AND FUNCTION OF TRYPTOPHAN HYDROXYLASE By GEORGE CHIH-THAI JIANG A Thesis Submitted to the Graduate Faculty of WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE in the Molecular Genetics Program of the Wake Forest University School of Medicine May 2003 Winston-Salem, North Carolina Approved by: Kent E. Vrana, Ph.D., Advisor _______________________ Examining Committee: Linda C. McPhail, Ph.D., Chairman _______________________ Mark O. Lively, Ph.D. _______________________ Leslie B. Poole, Ph.D. _______________________ William E. Sonntag, Ph.D. _______________________ DEDICATION I would like to dedicate this work to my parents, Allan Lii-Shang and Grace Mei-Chuon Jiang. Without your constant love and support throughout the turbulent times of my graduate career, this would not have been possible. Thank you for your constant words of wisdom and encouragement. ii ACKNOWLEDGMENTS First and foremost, I would like to thank Dr. Kent E. Vrana. I appreciate all the patience and guidance to achieve my career goals. My time at Wake Forest University has been filled with many ups and downs and I thank you for always supporting me, taking the time to further motivate and guide me in the right direction, and providing numerous opportunities for professional development. I would also like to acknowledge Dr. James Smith and the entire Department of Physiology and Pharmacology for providing the resources and supportive environment that have made my graduate career here so enjoyable. I would like to specifically thank all the members of the Vrana Lab, past and present, for their assistance and friendship in the laboratory. In particular, I would like to thank Dr. George J. Yohrling IV for his guidance and for serving as the model student-scientist which I have aspired to emulate. I would also like to thank Amanda Domer for all her assistance with protein expression, purification and enzymatic assays. Special thanks to Dean Melson, Dean Solano, Pearl Lawrence, and Beth Whitsett for their help in all aspects of the Graduate Student Association, and their support and assistance throughout my graduate career. I would also like to thank my committee members, Drs. Lively, McPhail, Poole, and Sonntag for their patience and guidance through the scientific process. Finally, but not least, I would like to thank all my classmates, friends, and colleagues. The support network you have provided has been remarkable and is greatly appreciated. iii TABLE OF CONTENTS LIST OF FIGURES AND TABLES.......................................................................vi ABBREVIATIONS.............................................................................................. viii ABSTRACT.......................................................................................................... x CHAPTER I - INTRODUCTION: STRUCTURAL AND FUNCTIONAL CHARACTERIZATION OF THE AROMATIC AMINO ACID HYDROXYLASES ..................................................................................... 1 Aromatic Amino Acid Hydroxylases...................................... 2 Tryptophan Hydroxylase and Serotonin Biosynthesis .......... 6 Serotonin in Human Health and Disease.............................. 7 AAAH Polymorphisms and Disease ..................................... 9 Protein Purification Studies ................................................ 13 X-Ray Crystallographic Studies.......................................... 17 References ......................................................................... 23 Statement of Purpose......................................................... 33 CHAPTER II - IDENTIFICATION OF SUBSTRATE ORIENTING AND PHOSPHORYLATION SITES WITHIN TRYPTOPHAN HYDROXYLASE USING HOMOLOGY-BASED MOLECULAR MODELING ..................... 35 This chapter is published in Journal of Molecular Biology 302:1005-1017 (2000). Abstract .............................................................................. 36 Introduction........................................................................ 38 Results .............................................................................. 44 Discussion ......................................................................... 60 Materials and Methods ....................................................... 71 References ........................................................................ 78 CHAPTER III – FUNCTIONAL ANALYSIS OF THE V177I CODING REGION POLYMORPHISM IN HUMAN TRYPTOPHAN HYDROXYLASE (TPH1) 82 In Preparation, Journal of Neurochemistry (2003) Abstract ............................................................................. 83 Introduction........................................................................ 84 Materials and Methods ...................................................... 88 iv Results .............................................................................. 94 Discussion ......................................................................... 96 References ...................................................................... 107 CHAPTER IV - SUMMARY AND CONCLUSIONS ......................................... 113 References ...................................................................... 121 APPENDIX - PHENYLKETONURIA …………………………………………..….. 124 This chapter is published in Encyclopedia of Life Sciences 14:150-154 (2000). Introduction...................................................................... 125 Clinical Signs and Symptoms ........................................... 126 Classical Phenylketonuria ............................................... 132 Nonclassical Phenylketonuria.......................................... 137 Future Treatments for Phenylketonuria ........................... 139 Summary ......................................................................... 143 Further Reading .............................................................. 144 CURRICULUM VITA ....................................................................................... 145 v LIST OF FIGURES AND TABLES CHAPTER I Figure 1: Biosynthetic pathway for catecholamines and indoleamines........... 5 Table I: Published TPH purification protocols ............................................ 13 Figure 2: Structural comparisons of the AAAHs ........................................... 22 CHAPTER II Table 1: Published TPH mutagenesis studies............................................. 41 Figure 1: Hypothetical model of human TPH (monomer) ............................. 46 Figure 2: Side by side comparison of TH, PAH, and hypothetical TPH ........ 48 Figure 3: Tetramer of hypothetical TPH ....................................................... 50 Table 2: Steady-state kinetic parameters of wild-type TPH and mutants .... 51 Figure 4: Phosphorylation and immunodetection of TPH ............................. 53 Figure 5: Specific activities of TPH active-site mutations ............................. 57 Figure 6: Lineweaver-Burk analysis of wild-type TPH and TPH Y235A ....... 59 Figure 7: Multiple sequence alignment ........................................................ 68 Figure 8: Sequence alignment of the aromatic amino acid hydroxylases..... 70 CHAPTER III Table I: Site-directed mutagenesis primers................................................ 90 Figure 1: Structural depictions of Val177 mutations ................................... 102 vi Table II: Purification data for wild-type and mutant enzymes .................... 103 Figure 2: Coomassie and immunoblots of purified proteins........................ 105 Table III: Steady-state kinetic parameters of wild-type TPH and mutants.. 106 CHAPTER IV Figure 1: Structural location of the functional residues of interest in the present work. .............................................................................. 118 APPENDIX Figure 1: Phenylalanine metabolism ......................................................... 128 Table 1: Genetic defects in phenylketonuria ............................................ 135 vii ABBREVIATIONS 5-HT 5-Hydroxytryptamine (Serotonin) 5-HTP 5-Hydroxytryptophan AAADC Aromatic Amino Acid Decarboxylase (EC 4.1.1.28) AAAH Aromatic Amino Acid Hydroxylase(s) BH4 Tetrahydrobiopterin ((6R)-5,6,7,8-Tetrahydro-L-biopterin) CaMPKII Calcium/Calmodulin-Dependent Protein Kinase II C∆/N∆ Carboxyl/Amino Terminal Deletion cDNA Complementary DNA CNS Central Nervous System DA Dopamine (3-hydroxydopamine) DNA Deoxyribonucleic Acid DOPA 3,4-Dihydroxyphenylalanine DTT Dithiothreitol EDTA Ethylenediaminetetraacetic Acid Fe Iron HEPES N-[2-Hydroxyethyl] Piperazine-N’-[2-Ethanesulfonic Acid] kb Kilobase kDa Kilodalton Km Michaelis-Menten Constant mRNA Messenger Ribonucleic Acid OCD Obsessive Compulsive Disorder viii PAH Phenylalanine Hydroxylase (phenylalanine-4-monooxygenase; EC 1.14.16.1) PD Parkinson’s Disease PKA Protein Kinase A; cAMP-Dependent Protein Kinase PMSF Phenylmethylsulfonyl Fluoride RMSD Root Mean Square Deviation SDS-PAGE Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis SEC Size-Exclusion Chromatography SN Substantia Nigra SNP Single Nucleotide Polymorphisms TH Tyrosine Hydroxylase (tyrosine-3-monooxygenase; EC 1.14.16.2) TPH Tryptophan Hydroxylase (tryptophan-5-monooxygenase; EC 1.14.16.4) Vmax Maximal Velocity ix ABSTRACT George C.-T. Jiang STRUCTURE AND FUNCTION OF TRYPTOPHAN HYDROXYLASE Thesis under the direction of Kent E. Vrana, Ph.D., Professor of Physiology and Pharmacology and Director of Graduate Studies Tryptophan hydroxylase (TPH) is the rate-limiting enzyme in the biosynthesis of serotonin, and is a member of a family of enzymes known as aromatic amino acid hydroxylases (AAAH). Studies into the regulation, structure, and function of TPH have lagged behind those of the other AAAH. In the absence of an experimentallydetermined crystal structure, we generated a hypothetical model of human TPH using multiple sequence alignment homology-based molecular modeling of the other AAAH. We then performed site-directed mutagenesis to identify functional domains within the TPH protein, and to validate the hypothetical model. Our studies demonstrated that tyrosine 235 plays a role in tryptophan substrate interactions in the active site, and that serine 58 and serine 260 are substrates for Ca2+/calmodulindependent protein kinase II. Additional studies analyzed a coding region polymorphism in human TPH, a subtle valine to isoleucine substitution at residue 177 (V177I). Our studies suggest that this residue is involved in the optimal x orientation of the tryptophan substrate binding pocket in the TPH active site. These studies have added to our understanding of the structure and function of TPH.Using this type of information, novel TPH-specific pharmacotherapies may be developed for use in the treatment of serotonergic disorders in human health and disease. xi CHAPTER I INTRODUCTION: STRUCTURAL AND FUNCTIONAL CHARACTERIZATION OF THE AROMATIC AMINO ACID HYDROXYLASES George C.-T. Jiang 1 2 AROMATIC AMINO ACID HYDROXYLASES Tryptophan hydroxylase (TPH; EC 1.14.16.4) is a member of a family of enzymes known as aromatic amino acid hydroxylases (AAAH). The other members of this family are tyrosine hydroxylase (TH; EC 1.14.16.2) and phenylalanine hydroxylase (PAH; EC 1.14.16.1). TPH catalyzes the rate-limiting step in the biosynthesis of the neurotransmitter serotonin (Grahame-Smith, 1964; Lovenberg et al., 1967), and is involved in melatonin biosynthesis (Chanut et al., 2002; Privat et al., 2002; reviewed in Martinez et al., 2001). TH is the rate-limiting enzyme in the synthesis of the catecholamines (dopamine, epinephrine, and norepinephrine), while PAH is rate-limiting in the synthesis of the amino acid tyrosine (for reviews, see Hufton et al., 1995; Kumer and Vrana, 1997) and the primary enzyme responsible for the catabolism of phenylalanine (reviewed in Martinez et al., 2001). All three enzymes share a high degree of amino acid identity and distinct structural and functional characteristics. Structurally, AAAH are composed of three regions - an amino terminal regulatory domain, a carboxyl catalytic domain, and an oligomeric binding domain at the extreme carboxyl terminus (Liu and Vrana, 1991; Wang et al., 2000; reviewed in Ftizpatrick, 1999). While the regulatory domains exhibit approximately 30% sequence identity, over 60% sequence identity is exhibited in the catalytic region. The junction delineating the regulatory and catalytic domains (Val-Pro-Trp-Phe-Pro) is completely conserved in all species of the hydroxylases, and marks the beginning of the conserved catalytic domain (Yang et al., 1994; Kumer et al., 1997a; Mockus et al., 1997b; Moran et al., 1998; reviewed in 3 Ftizpatrick 1999). Recent crystallographic data show that the proteins fold in a similar manner and are virtually superimposeable in their catalytic domains. It is generally accepted that the three hydroxylases evolved from a common progenitor (Grenett et al., 1987; Neckameyer and White, 1992). Evidence for this is demonstrated by the fruit fly, Drosophila melanogaster, which possesses only two AAAH – TH and an enzyme that hydroxylates both tryptophan and phenylalanine (Neckameyer and White, 1992). Functionally, all AAAHs require the same co-substrates – molecular oxygen (O2), tetrahydrobiopterin (BH4) – and a non-heme, ferrous iron co-factor to hydroxylate their respective amino acid (Figure 1). Another similarity revolves around the hydroxylation reaction itself, which produces water as a bi-product in all cases. The release of water serves as the basis of one method for determining enzyme activity. By utilizing a tritiated tryptophan substrate in the hydroxylation reaction, one can determine enzyme activity by measuring the tritiated water released (Beevers et al., 1983; Reinhard et al., 1986; Vrana et al., 1993). The other method for determining enzyme activity in vitro is based on a continuous fluorometric assay using the different spectral characteristics of tryptophan and 5hydroxytryptophan (Moran and Fitzpatrick, 1999). Using this assay, one directly detects the fluorescent yield of 5-hydroxytryptophan. 4 Figure 1: Biosynthetic pathway for catecholamines and indoleamines. TH is the rate-limiting enzyme in the synthesis of the catecholamines (DA, NE, and E), while TPH is the rate-limiting enzyme for serotonin formation. TPH also serves as a nonlimiting enzyme in the synthesis of melatonin. Both TH and TPH require the cosubstrates molecular oxygen (O2) and tetrahydrobiopterin (BH4), as well as the cofactor ferrous iron (Fe2+). Following hydroxylation of tryptophan at the 5' position, aromatic amino acid decarboxylase (AAADC) decarboxylates 5-hydroxytryptophan to form 5-hydroxytryptamine (serotonin). Serotonin is then acetylated in a ratelimiting fashion by N-acetyltransferase (NAT) to allow for melatonin formation (not shown). AAADC also serves to decarboxylate DOPA to form dopamine. Dopamine is further metabolized to norepinephrine (NE) and epinephrine (E) by dopamine-βhydroxylase (DBH) and phenylethanolamine N-methyltransferase (PNMT), respectively. 5 6 TRYPTOPHAN HYDROXYLASE AND SEROTONIN BIOSYNTHESIS TPH is the rate-limiting enzyme in the biosynthesis of the neurotransmitter serotonin (Grahame-Smith, 1964; Lovenberg et al., 1967). It is also involved in melatonin biosynthesis though is not rate-limiting in that reaction. Localization of cell bodies, as determined by in situ hydridization, illustrate that TPH mRNA-positive neurons can be found throughout the raphe nuclei and the pineal gland (Austin and O’Donnell, 1999). TPH is predominantly made in the central nervous system in these brain regions (Jequier et al., 1969; Joh et al., 1975). There is also evidence for peripheral synthesis of TPH, including cells of the enteric neurons of the gut, platelets, retina, and thyroid cells (reviewed in Mockus and Vrana, 1998). In mammalian species, serotonin has been detected in nearly all tissues of the body and plays a number of different roles (reviewed in Azmitia, 2001). Because of its important regulatory role, a number of processes have evolved to regulate serotonin biosynthesis. In the brain, rapid regulatory control is achieved by modulating TPH enzyme activity through phosphorylation. This mechanism of action allows for rapid and local regulation of 5-HT biosynthesis without requiring new enzymes to be transported from the cell body area to the synapse, which could take hours to days to accomplish (reviewed in Azmitia, 2001). 7 SEROTONIN IN HUMAN HEALTH AND DISEASE Serotonin (5-HT) is the neurotransmitter end-product following tryptophan hydroxylation by TPH and subsequent decarboxylation by AAADC. Serotonin is implicated in a variety of CNS functions such as temperature control, aggression, pain and memory (Kandasamy and Williams, 1984; reviewed in Oliver et al., 1995; Brentegani and Lico, 1982; and Cornwell-Jones et al., 1989). Aberrations in serotonin synthesis, regulation, and homeostasis have been associated with numerous psychiatric illnesses including depression, anxiety, obsessive-compulsive disorder (OCD), bulimia, migraines and drug abuse (reviewed in Mockus and Vrana, 1998). Moreover, 5-HT itself can be oxidized in vivo by superoxide to form the toxic metabolite, tryptamine-4,5-dione (T-4,5-D) which has been implicated in the degeneration of serotonergic neurons following methamphetamine exposure or cerebral ischemia reperfusion (Jiang et al., 1999). Recent data from the National Institute of Mental Health (NIMH) demonstrate that there is a severe socioeconomic impact of mental illness in the United States. An estimated 22.1% of Americans suffer from a diagnosable mental disorder in a given year (Regier et al., 1993). For example, it is estimated that 19 million adults in the United States (10% of the US population) suffer from depression, and only onethird of this population receives treatment. As mentioned earlier, depression is associated with aberrant serotonergic function. Previously, treatment regimen for depressive disorders involved tricyclic antidepressants (TCAs) which have a large number of side effects. Today, treatment predominantly involves selective serotonin 8 reuptake inhibitors (SSRIs) and/or monoamine oxidase inhibitors (MAOIs) that modulate neurotransmitter levels (including serotonin) with less side effects. In 2001, the pharmaceutical giants Eli Lilly and GlaxoSmithKline – producers of the leading antidepressant drugs, Prozac (fluoxetine) and Paxil (paroxetine) – reported $2.2 Billion and $6.2 Billion in US sales, respectively. Typically, the treatment for serotonin-related diseases is to modulate the serotonergic system, often by way of selective serotonin reuptake inhibitors, such as fluoxetine or sertraline as described above. Alternative treatments, using lithium or the former diet drug fenfluramine, work presynaptically to alter the serotonin system. However, the side effects are vast and long-term effects remain to be determined. TPH could contribute to the root of many of these serotonin-related disorders. Therefore, it is necessary to further understand the structure, function and regulation of this important enzyme to potentially provide an alternative therapeutic approach to treat these diseases. 9 AAAH POLYMORPHISMS AND DISEASE As the AAAHs are all rate-limiting enzymes in their respective metabolic pathways, it is not suprising that changes in enzyme structure could change enzyme function, and thereby manifest themselves, ultimately in abnormal clinical conditions. This is indeed the case for PAH and TH, and subtle hints of such polymorphisms resulting in human disease have been suggested for TPH. It has been demonstrated that a dysfunctional or compromised PAH will result in increased blood phenylalanine levels, or hyperphenylalaninemia (HPA) (Nowacki et al., 1997) due to the inability to catabolize the amino acid. More severe cases of HPA result in the condition commonly known as phenylketonuria (PKU; reviewed in Erlandsen et al., 2001; reviewed in Jiang et al., 2001a). Researchers have identified over 400 mutations in PAH that result in decreased enzymatic activity, and can be linked to PKU. Unfortunately, PKU results in severe mental retardation of patients, if left untreated. Numerous researchers have utilized PAH crystal structures (Erlandsen et al., 1997a; Fusetti et al., 1998; Kobe et al., 1999; Jennings et al., 2000; Miranda et al., 2002, reviewed in Erlandsen et al., 1997b) to explain the functional effect of many of these mutations on enzymatic activity. Often, these mutations map to important active site residues, but some map to amino acids in structural motifs, or in important hinge structures (Bjorgo et al., 2001; Jennings et al., 2001; Miranda et al., 2002; Horne et al., 2002). Polymorphisms in the coding region for TH have also been identified that result in clinical abnormalities. These point mutations in TH result in autosomal 10 recessive L-DOPA- responsive parkinsonism and dystonias (Ludecke et al., 1995, 1996, reviewed in Ichinose et al., 1999). Clinically, all of these polymorphisms manifest themselves as motor coordination symptoms in patients as they result in dopamine deficiency, yet without the neuronal death associated with juvenile parkinsonism or Parkinson’s Disease. To date, only a handful of TH polymorphisms have been described. A Glu381Lys (Q381K) change in human TH was first described (Knappskog et al., 1995; Ludecke et al., 1995) to result in decreased TH specific activity to about 15% of wild-type when expressed in vitro. Additionally, the Km for the tyrosine substrate of the mutant was 6-fold higher compared to wild-type enzyme (Knappskog et al., 1995). Similarly, a Leu205Pro (L205P) polymorphism was described that resulted in a recombinant enzyme that exhibits 1.5% of the activity of wild-type (Ludecke et al., 1996). Other TH polymorphisms that have been characterized include Arg233His (R233H) (van den Heuvel et al., 1998), Cys359Phe (C359F) (Bräutigam et al., 1999), Val112Met (V112M), and Asp498Gly (D498G) (Wevers et al., 1999). In all cases, the effect on enzyme function can be explained by the change in enzymatic structure, as rationalized using a crystal structure. Due to challenges in enzyme purification and retention of activity, research on the structure and regulation of TPH has fallen behind that of the other amino acid hydroxylases. Only recently has the TPH crystal structure been deciphered (Wang et al., 2002). As 5-HT plays a role in the etiology of a wide variety of neurological diseases, but only subtle phenotypic changes occur, it has been difficult to identify polymorphisms in the TPH gene that associate with a disease in a clear-cut manner. Nielsen and colleagues first described an intronic polymorphism in the human TPH 11 gene (intron 7) that is associated with suicidality and variable 5-hydroxyindolacetic acid concentrations in Finnish Caucasians (Nielsen et al., 1994). Another study has associated another intronic TPH gene polymorphism with aggression and angerrelated traits (Manuck et al., 1999). Recently, the first coding region polymorphism of TPH was described by Ramaekers and colleagues in a pediatric patient exhibiting neurodevelopment problems (Ramaekers et al., 2001). Analysis of cerebral spinal fluid (CSF) and serum from five pediatric patients exhibiting motor and neurodevelopment problems illustrated decreased serotonin metabolites, suggesting a deficiency in serotonin production. When treated with the end-product of TPH, 5hydroxytryptophan, the patients all regained normal motor and neurological function, further suggesting a potential problem with TPH. Sequencing of the TPH gene revealed a G529A polymorphism in the gene that would produce a subtle valine to isoleucine substitution at residue 177 (Val177Ile) in the protein. The role of this specific residue remains undetermined. However, a complicating factor is that the specific Val177Ile polymorphism only occurred in one of the five patients (Ramaekers et al., 2001). Interestingly, however, in all the TPH-associated clinical situations, structural and catalytic consequences of potential polymorphisms remain unknown. Through our work, we have developed a structural model for TPH to explain the effect of such polymorphisms, in conjunction with biochemical studies to assess the effect of such polymorphisms on TPH function. A fundamental hypothesis is that investigation of additional patient populations will reveal more TPH polymorphisms that could be the cause of the neurological disorders associated with aberrant 13 PROTEIN PURIFICATION STUDIES The first attempts to purify tryptophan hydroxylase started in the 1970s using a number of different sources and chromatographic techniques, though there were few publications when compared to the other AAAHs (reviewed in Cash, 1998). The difficulty in obtaining pure TPH protein can be attributed primarily to 1) the low quantities of TPH protein available in natural sources; and 2) the inherent sensitivity of the enzyme to proteolytic degradation and/or inactivation (reviewed in Cash, 1998). The initial work on tryptophan hydroxylase was focused on protein obtained from bovine (Jequier et al., 1969; Nukiwa et al., 1974), rabbit (Tong et al., 1975; Nakata et al., 1981; Banik et al., 1997; Moran et al., 1998), and rat (Jequier et al., 1969; Nakata et al., 1982; Cash et al., 1985; Cash, 1998) sources. However, TPH has been isolated from other mammalian species, including mouse (Hosada 1975; Nakata et al., 1982b; Fujisawa et al., 1987a; Park et al., 1994), pig (Youdim et al., 1974, 1975) and human samples (Yang et al., 1994; Kowlessur et al., 1999; Wang et al., 2002). Successful purification of TPH was difficult as the procedures required considerable time and extensive multi-step chromatography procedures. In fact, the enzyme frequently lost activity and the protocols typically produced small quantities of pure protein. Moreover, unlike more ubiquitous proteins, researchers found it challenging to obtain significant quantities of starting material. For example, the first purification of rat TPH used 17 rat brainstems and 9 grams of tissue yet only produced 13.6 mg of TPH (Jequier et al., 1969). More recent purification protocols 14 entail recombinant expression of a TPH cDNA and subsequent purification, which generates copious amounts of starting material at a very low cost. Table I also illustrates the different yields and fold purifications that have previously been achieved. The recent advances in protein chemistry technologies have facilitated the purification of greater amounts of protein, namely by the use of fusion proteins. The first use of an affinity tag for TPH purification was by Yang and Kaufman (1994), who produced recombinant full length and truncated human TPH fused to maltose binding protein in E. coli. Though this technique was very useful in obtaining pure protein, the concept of attaching a 49 kD maltose binding protein with a 2 kD linker to TPH, or any other protein for that matter, is not optimal. Banik and colleagues (Banik et al., 1997) utilized a much smaller fusion with an amino-terminal hexahistidine tag to express rabbit TPH in insect cells (Spodoptera frugiperda) for subsequent purification. These researchers removed the hexa-histidine tag after the purification procedures, unlike Hamdan and Ribeiro (Hamdan et al., 1999), who also used a hexa-histidine tag (amino terminal) to express and purify E. coli expressed rabbit TPH and TPH from the parasite Schistosoma mansoni (SmTPH). These latter researchers were able to achieve a 45-fold purification with a yield of 0.66-0.8 mg of purified SmTPH from 100ml of induced bacterial culture (with a specific activity of 170 nmol 5-HTP/min·mg). The recent crystallization of the catalytic core of hTPH was also done with TPH expressed as a hexa-hisitidine fusion enzyme, although the use of an alternative prokaryotic expression vector resulted in this enzyme being expressed with a carboxy-terminal hexa-hisitidine tag instead of an amino-terminal 15 fusion (Wang et al., 2002). The current work has also concurrently developed a purification system for human TPH, utilizing an amino-terminal hexa-histidine affinity tag for rapid purification as described in Chapter 3 of this thesis. 16 17 X-RAY CRYSTALLOGRAPHIC STUDIES Due to the inherent instability of TPH enzyme activity, large amounts of purified protein have been difficult to obtain (Kuhn et al., 1980; D’Sa et al., 1996a, Cash 1998). This posed a significant challenge for researchers attempting crystallization studies. As stated above, the recent advances in protein chemistry technologies have facilitated the purification of large amounts of protein and, thus, it was only recently that the first crystallographic structure of TPH was determined (Wang et al., 2002). Prior to the actual determination of the crystal structure of the enzyme, researchers had success in determining the crystal structures for the other members of the AAAH. In 1997, the structure for tyrosine hydroxylase was first resolved at 2.3Å (Goodwill et al., 1997). This structure was incomplete as it only represented the rat catalytic domain (residues 164-498), missing the entire amino terminal regulatory domain. This crystal structure validated the presence of a 40Å alpha helix consisting of a 4,3-hydrophobic repeat, that was first postulated by Liu and Vrana (1991) to be responsible for tetramerization through the interactions of anti-parallel coiled coils. The crystal structure also revealed potential intersubunit binding domains, such as a dimerization interface between two TH monomers formed by a salt bridge between Lys170 and Glu282 and adjacent hydrogen bonding interactions. This interaction is conserved in TPH and was studied by Yohrling and colleagues (Yohrling et al., 1999; Yohrling et al., 2000). Shortly following the report of the initial TH crystal structure, the second crystal structure of TH was resolved to 18 2.3Å by Goodwill and colleagues (1998). This new structure was also truncated, missing the entire regulatory domain (N∆1-159). Nonetheless, the structure did provide new insights to the enzyme, as it was resolved with iron and a pterin cosubstrate analogue in the active site (Goodwill et al., 1998). The number of published crystal structures for phenylalanine hydroxylase is significantly greater. The first PAH crystal structure was determined at about the same time as that for tyrosine hydroxylase in 1997. This crystal structure was a homodimer but truncated, only representing the catalytic domain of human PAH (residues 117-424, Erlandsen et al., 1997a). Soon after, the structure of the catalytic domain of human PAH (residues 118-452) in its tetrameric form was resolved (Fusetti et al., 1998). Additionally, researchers were able to determine the structure of the human PAH catalytic domain to 2.0Å (residues 117-424) with various catechol inhibitors bound (Erlandsen et al., 1998). These structures gave insight into residues in the active site that might play a role in substrate binding and catalysis. A more complete structure for PAH was resolved to 2.2Å with the crystallization of rat PAH (residues 1-429) that was missing only the carboxyl terminal tetramerization domain (Kobe et al., 1999). Interestingly, it was observed that phosphorylation did not result in dramatic structural changes, suggesting subtle allosteric regulatory mechanisms for PAH following phosphorylation. The structure for pterin-bound human PAH (residues 118-424) was determined by Erlandsen and colleagues in 2000 (Erlandsen et al., 2000). This provided a more detailed view into the active site of the enzyme, and the potential mechanism of catalysis. Soon thereafter, the structure of the human PAH catalytic 19 domain (N∆1-102/C∆428-452) with catalytically active Fe(II) and in a binary complex with the natural cosubstrate, BH4, was resolved (Andersen et al., 2001). This has been followed up with a report of the structure of a truncated human PAH enzyme (N∆1-102/C∆428-452) ternary complex with BH4 and 3-(2-thienyl)-L-phenylalanine (Andersen et al., 2002). Further insights into understanding PAH, and by extension, understanding the structure and function of TH and TPH, were provided by the comparison of bacterial versus human PAH (Erlandsen et al., 2002). These researchers observed similar folding in the catalytic domain, but different enzyme stability and reaction rates between the prokaryotic and eukaryotic enzymes. Recently, researchers have investigated the role of the amino terminus of PAH, which has provided no interpretable electron density in previous PAH crystallographic studies. This N-terminal sequence had been surmised to play a role in autoregulation of PAH, based on the crystal structure of PAH (Kobe et al., 1999) and deletion mutagenesis (Jennings et al., 2001, and Wang et al., 2001; Horne at al., 2002). Using NMR, Horne and colleagues demonstrated that the N-terminus is a mobile flexible region that associates with the folded core of the molecule after binding of the phenylalanine substrate, the association of which can be facilitated by phosphorylation (Horne at al., 2002). In spite of intensive efforts by several groups, no successful crystallization of TPH occurred until recently (Wang et al., 2002). In advance of such a structure, and given the high sequence homology between the AAAHs, the present work includes the construction of the first hypothetical molecular model of human TPH (Vrana, 1999; Jiang et al., 2000b; Figure 2) based upon the then available crystal structures 20 of both rat TH and rat PAH (Goodwill et al., 1997; Kobe et al., 1999). Due to the challenges involved in getting crystal formation with all domains present, the hypothetical model was, and still remains, the most comprehensive structure of any AAAH to date as it is nearly full length (missing only one amino acid residue). This model also has significant advantages over an automated model of TPH generated by SWISS-MODEL (Guex et al., 1997, 1999). This latter structure was truncated (containing residues 104-411), and not subject to rigorous investigator-interactive energy minimizations used in the present work. Potential functional motifs, as well as amino acids involved in substrate binding and phosphorylation, have been identified with the use of our hypothetical model. The recent crystallization of a truncated form of human TPH (N∆1-102/C∆402-444) at a resolution of 1.7Å and with bound cofactor analogue (7,8-dihydro-L-biopterin), validates the catalytic domain of this hypothetical model and provides new insights into tryptophan hydroxylation. An “iterative magic fit” of the TPH crystal structure and hypothetical model using Swiss-PdbViewer (Guex and Peitsch, 1997) indicate a root mean square deviation (R.M.S.D.) of 1.02 Å between the carbon backbones of the structures. 21 Figure 2: Structural Comparisons of the AAAHs. The crystal structures of PAH and TH are shown beside a predicted structure of the TPH catalytic domain. These structures only depict the catalytic domains of these AAAHs. The PAH structure (PDB entry 1PAH) depicts residues 117 to 424, thereby missing the entire regulatory domain and the extreme carboxy terminal. The TH structure (1TOH) depicts residues 164 to 498, and is also missing the entire regulatory domain. The predicted TPH catalytic domain was generated by homology-based modeling in the “Composer” module of Sybyl 6.5 (Tripos, St. Louis, MO). This figure was originally generated by Mr. Jiang and is reproduced from Vrana, 1999. 22 23 REFERENCES Andersen O.A., Flatmark T., and Hough E. (2001) High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J. Mol. Biol. 314, 270-291. Andersen O.A., Flatmark T., and Hough E. (2002) Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-L-alanine, and its implications for the mechanism of catalysis and substrate activation. J. Mol. Biol. 320, 10951108. Austin, M.C. and O’Donnell, S.M. (1999) Regional distribution and cellular expression of tryptophan hydroxylase messenger RNA in postmortem human brainstem and pineal gland. J. Neurochem. 72, 2065-2073. Azmitia E.C.G. (2001) Serotonin. Encyclopedia of Life Sciences 1-11. Banik U., Wang G.A., Wagner P.D., and Kaufman S. (1997) Interaction of phosphorylated tryptophan hydroxylase with 14-3-3 proteins. J. Biol. Chem. 272, 26219-25 Beevers, S.J., Knowles, R.G., and Pogson, C.I. (1983) A sensitive radiometric assay for tryptophan hydroxylase applicable to crude extracts. J. Neurochem. 40, 894-897. Boadle-Biber M.C. (1982) Blockade by haloperidol of the increase in tryptophan hydroxylase activity induced by incubation of slices of brain stem with dibutyryl cyclic AMP. Biochem. Pharmacol. 31, 2203-2207. Boadle-Biber M.C., and Phan T.H. (1987) Involvement of calmodulin-dependent phosphorylation of brainstem tryptophan hdydroxylase induced by depolarization of slices or other treatments that raise intracellular free calcium levels. Biochem Pharmacol. 36, 1174-1176. Bräutigam C., Steenbergen-Spanjers G.C.H., Hoffmann G.F., Dionisi-Vici C., van den Heuvel L.P.W.J., Smeitink J.A.M., and Wevers R.A. (1999). Biochemical and molecular genetic characteristics of the severe form of tyrosine hydroxylase deficiency. Clin. Chem. 45, 2073-2078. 24 Bjorgo E., de Carvalho R.M., and Flatmark T. (2001) A comparison of kinetic and regulatory properties of the tetrameric and dimeric forms of wild-type and Thr427->Pro mutant human phenylalanine hydroxylase: contribution of the flexible hinge region Asp425-Gln429 to the tetramerization and cooperative substrate binding. Eur. J. Biochem. 268, 997-1005. Brentegani, L.G. and Lico, M.C. (1982) Dental pain and the injection of agents into the trigeminal nerve of guinea pigs. Phys. and Behav. 28, 49-52. Cash C.D., Vayer P., Mandel P., and Maitre M. (1985) Tryptophan 5-hydroxylase. Rapid purification from whole rat brain and production of a specific antiserum. Eur. J. Biochem. 149, 239-245. Cash C.D. (1998) Why tryptophan hydroxylase is difficult to purify: a reactive oxygen-derived species-mediated phenomenon that may be implicated in human pathology. Gen. Pharmacol. 30, 569-574. Chanut E., Nguyen-Legros J., Labarthe B., Trouvin J.H., and Versaux-Botteri C. (2002) Serotonin synthesis and its light-dark variation in the rat retina. J. Neurochem. 83, 863-9. Cornwell-Jones, C.A., Decker, M.W., Chang, J.W., Cole, B., Goltz, K.M., Tran, T,, and McGaugh, J.L. (1989) Neonatal 6-hydroxydopa, but not DSP-4, elevates brainstem monoamines and impairs inhibitory avoidance learning in developing rats. Brain Res. 493, 258-68. Daubner S.C., Lohse D.L., and Fitzpatrick P.F. (1993) Expression and characterization of catalytic and regulatory domains of rat tyrosine hydroxylase. Protein Sci. 2, 1452-1460. Darmon M.C., Guibert B., Leviel V., Ehret M., Maitre M., and Mallet J. (1988) Sequence of two mRNAs encoding active rat tryptophan hydroxylase. J. Neurochem. 51, 312-316. D’Sa C.M., Arthur R.E. Jr., Jennings I., Cotton R.G.H., and Kuhn D.M. (1996a) Tryptophan hydroxylase: Purification by affinity chromatography on calmodulin-sepharose. J. Neurosci. Meth. 69, 149-153. D’Sa C.M., Arthur R.E.Jr., Kuhn D.M. (1996b) Expression and deletion mutagenesis of tryptophan hydroxylase fusion proteins: Delineation of the enzyme catalytic core. J. Neurochem. 67, 917-926. Erlandsen, H., Martinez A., Knappskog P.M., Haavik, J., Hough, E. and Flatmark, T. (1997a) Crystallization and preliminary diffraction analysis of a truncated homodimer of human phenylalanine hydroxylase. FEBS Letters. 406, 171174. 25 Erlandsen, H., Fusetti F., Martinez A., Hough E., Flatmark, T. and Stevens R.C. (1997b) Crystal structure of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nat. Struct. Biol. 4, 995-1000. Erlandsen H., Flatmark T., Stevens R.C., and Hough E. (1998) Crystallographic analysis of the human phenylalanine hydroxylase catalytic domain with bound catechol inhibitors at 2.0A resolution. Biochem. 37, 15638-15646. Erlandsen H., Bjorgo E., Flatmark T., and Stevens R.C. (2000) Crystal structure and site-specific mutagenesis of pterin-bound human phenylalanine hydroxylase. Biochem. 39, 2208-2217. Erlandsen H., and Stevens R.C. (2001). A structural hypothesis for BH4 responsiveness in patients with mild forms of hyperphenylalaninaemia and phenylketonuria. J. Inherit. Metab. Dis. 24, 213-30. Erlandsen H., Kim J.Y., Patch M.G., Han A., Volner A., Abu-Omar M.M., and Stevens R.C. (2002) Structural comparison of bacterial and human irondependent phenylalanine hydroxylases: Similar fold, different stability and reaction rates. J. Mol. Biol. 320, 645-661. Fisher, D.B. and Kaufman, S. (1973) The stimulation of rat liver phenylalanine hydroxylase by lysolecithin and chymotrypsin. J. Biol. Chem. 248, 43454353. Fitzpatrick P.F. (1999) Tetrahydrobiopterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 68, 355-381. Fujisawa H., and Nakata H. (1987a) Tryptophan 5-monooxygenase from mouse mastocytoma clone P815. Methods Enzymol. 142, 93-96. Fujisawa H., and Nakata H. (1987b) Tryptophan 5-monooxygenase from rat brain stem. Methods Enzymol. 142, 83-87. Fusetti F., Erlandsen H., Flatmark T., and Stevens R.C. (1998) Structure of tetrameric phenylalanine hydroxylase. J. Biol. Chem. 273, 16962-16967. Goodwill, K.E., Sabatier, C., Marks, C., Raag, R., Fitzpatrick, P.F., and Stevens, R.C. (1997) Crystal structure of tyrosine hydroxylase at 2.3 A and its implication for inherited neurodegenerative diseases. Nature Struct. Biol. 4, 578-585. Goodwill, K.E., Sabatier, C., and Stevens, R.C. (1998) Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3Å resolution: selfhydroxylation of Phe300 and pterin-binding site. Biochem. 37, 13437-13445. 26 Grahame-Smith, D.G. (1964) Tryptophan hydroxylation in brain. Biochem Biophys Res Commun 16, 586-592. Grenett H.E., Ledley F.D., Reed L.L., and Woo S.L.C. (1987) Full-length cDNA for rabbit tryptophan hydroxylase: Functional domains and evolution of aromatic amino acid hydroxylases. Proc. Natl. Acad. Sci. USA, 84, 5503-5534. Grima B., Lamouroux A., Blanot F., Faucon Biguet N., and Mallet J. (1985) Complete coding sequence of rat tyrosine hydroxylase mRNA. Proc. Natl. Acad. Sci. USA, 82, 617-621. Guex N., and Peitsch M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 27142723. Guex N., Diemand A., and Peitsch M.C. (1999) Protein modeling for all. Trends Biochem. 24, 364-367. Hamdan, F.F., and Ribeiro P. (1999) Characterization of a stable form of tryptophan hydroxylase from the human parasite Schistosoma mansoni. J. Biol. Chem. 274, 21746-21754. Horne J., Jennings I.G., The T., Gooley P.R., and Kobe B. (2002) Structural characterization of the N-terminal autoregulatory sequence of phenylalanine hydroxylase. Prot. Sci. 11, 2041-2047. Hosoda S. (1975) Further studies on tryptophan hydroxylase from neoplastic murine mast cells. Biochim. Biophys. Acta. 397, 58-68. Hufton S.E., Jennings I.G., and Cotton R.G.H. (1995) Structure and function of the aromatic amino acid hydroxylases. Biochem. J. 311, 353-366. Ichinose H., Suzuki T., Inagaki H., Ohye T., and Nagatsu T. (1999) Molecular genetics of Dopa-responsive dystonia. Biol. Chem. 380, 1355-1364. Jennings I.G., Cotton R.G., and Kobe B. (2000) Structural interpretation of mutations in phenylalanine hydroxylase protein aids in identifying genotypephenotype correlations in phenylketonuria. Eur. J. Hum. Genet. 8, 683-696. Jennings I.G., Teh T., and Kobe B. (2001) Essential role of the N-terminal autoregulatory sequence in the regulation of phenylalanine hydroxylase. FEBS Lett. 488,196-200. Jequier E., Robinson D.S., Lovenberg W., and Sjoerdsma A. (1969) Further studies on tryptophan hydroxylase in rat brain stem and beef pineal. Biochem. Pharmacol. 18, 1071-1081. 27 Jiang G.C.-T., Yohrling IV G.J., and Vrana K.E. (2000a). Phenylketonuria. In: Encyclopedia of Life Sciences. 14, 150-154. London: Nature-Scientific American Publishing Group. http://www.els.net (invited review). Jiang G.C.-T., Yohrling IV G.J., Schmitt J.D., and Vrana K.E. (2000b). Identification of substrate orienting and phosphorylation sites within tryptophan hydroxylase using homology-based molecular modeling. J. Mol. Biol. 302, 1005-1017. Jiang, X.R., Wrona, MZ., and Dryhurst, G. (1999) Tryptamine-4,5-dione, a putative endotoxic metabolite of the superoxide-mediated oxidation of serotonin, is a mitochondrial toxin: Possible implications in neurodegenerative brain disorders. Chem Res in Toxicol. 12, 429-436. Joh T.H., Shikimi T., Pickel V.M., and Reis D.J. (1975) Brain tryptophan hydroxylase: Purification of, production of antibodies to, and cellular and ultrastructural localization in serotonergic neurons of rat midbrain. Proc. Natl. Acad. Sci. USA 72, 3575-3579. Johansen P.A., Jennings I., Cotten R.G.H., and Kuhn D.M. (1995) Tryptophan hydroxylase is phosphorylated by protein kinase A. J. Neurochem. 65, 882888. Johansen P.A., Jennings I., Cotten R.G.H., and Kuhn D.M. (1996) Phosphorylation and activation of tryptophan hydroxylase by exogenous protein kinase A. J. Neurochem. 66, 817-823. Kandasamy, S.B. and Williams, B.A. (1984) Hypothermic and anti-pyretic effects of ACTH (1-24) and alpha-melanotropin in guinea pigs. Neuropharmacology 23, 49-53. Knappskog P.M., Flatmark T., Mallet J., Ludecke B., and Bartholome K. (1995) Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum. Mol. Genet. 4, 1209-1212. Kobe B, Jennings I.G., House, C.M., Feil, S.C., Michell, B.J., Tiganis, T., Parker, M.W., Cotton, R.G.H., and Kemp, B.E. (1997) Regulation and crystallization of phosphorylated and dephosphorylated forms of truncated dimeric phenylalanine hydroxylase. Protein Science, 6, 1352-1357. Kobe B, Jennings I.G., House, C.M., Michell, B.J., Goodwill, K.E., Santarsiero, B.D., Stevens, R.C., Cotton, R.G.H., and Kemp, B.E. (1999) Structural basis of auto regulation of phenylalanine hydroxylase. Nature Struct. Biol. 6, 442448. 28 Kowlessur D. and Kaufman S. (1999) Cloning and expression of recombinant human pineal tryptophan hydroxylase in Escherichia coli: purification and characterization of the cloned enzyme. Biochimica et Biophysica Acta. 1434, 317-330. Kuhn D.M., Rosenberg R.C., and Lovenberg W. (1979) Determination of some molecular parameters of tryptophan hydroxylase from rat midbrain and murine mast cells. J. Neurochem. 33, 15-21. Kuhn D.M., Ruskin B., and Lovenberg W. (1980) Tryptophan hydroxylase: The role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J. Biol. Chem. 255, 4137-4143. Kumer S.C., Mockus S.M., Rucker P.J., Vrana K.E. (1997a) Amino terminal deletion analysis of tryptophan hydroxylase: PKA phosphorylation occus at serine-58. J. Neurochem. 69, 1738-1745. Kumer S.C. and Vrana, K.E. (1997b) Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 67, 443-462. Ledley F.D., DiLella A.G., Kwok C.M., and Woo S.L.C. (1985) Homology between phenylalanine hydroxylase and tyrosine hydroxylase reveals common structural and functional domains. Biochemistry, 24, 3389-3394. Liu X., and Vrana K.E. (1991) leucine zippers and coiled-coils in the aromatic amino acid hydoxylases. Neurochem. Int. 18, 27-31. Lovenberg W., Jequier E., and Sjoerdsma A. (1967) Tryptophan hydroxylation: Measurement in pineal gland, brain stem and carcinoid tumor. Science 155, 217-219. Ludecke, B., Dworniczak, B., Bartholome, K. (1995) A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum. Genet. 95, 123-125. Ludecke, B., Knappskog P.M., Clayton P.T., Surtees R.A.H., Clelland J.D., Heales S.J.R., Brand M.P., Bartholome K., and Flatmark T. (1996) Recessively inherited L-dope responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum. Mol. Gen. 7, 10231028. Manuck, S.B., Flory, J.D., Ferrell, R.E., Dent, K.M., Mann, J.J., and Muldoon, M.F. (1999) Aggression and anger-related traits associated with a polymorphism of the tryptophan hydroxylase gene. Biol. Psych. 45, 603-614. 29 Martinez A., Knappskog P.M., and Haavik J. (2001) A structural approach into human tryptophan hydroxylase and its implications for the regulation of serotonin biosyntheis. Curr. Med. Chem. 8, 1077-1091. McKinney J., Teigen K., Froystein N.A., Salaun C., Knappskog P.M., Haavik J., and Martinez A. (2001) Conformation of the substrate and pterin cofactor bound to human tryptophan hydroxylase. Important role of Phe313 in substrate specificity. Biochemistry 40, 15591-601. Miranda FF, Teigen K, Thorolfsson M, Svebak RM, Knappskog PM, Flatmark T, and Martinez A. (2002) Phosphorylation and mutations of Ser(16) in human phenylalanine hydroxylase. Kinetic and structural effects. J. Biol. Chem. 277, 40937-40943. Mockus S.M., Kumer S.C., Vrana K.E. (1997a) Carboxyl terminal deletion analysis of tryptophan hydroxylase. Biochim. Biophys. Acta, 1342, 132-140. Mockus S.M., Kumer S.C., Vrana K.E. (1997b) A chimeric tyrosine/tryptophan hydroxylase: The tyrosine hydroxylase regulatory domain serves to stabilize enzyme activity. J. Mol. Neurosci. 9, 35-48. Mockus S.M., Yohrling IV, G.J., and Vrana K.E. (1998) Tyrosine hydroxylase and tryptophan hydroxylase do not form heterotetramers. 10, 45-51. Mockus S.M. and Vrana K.E. (1998) Advances in the molecular characterization of tryptophan hydroxylase. J. Mol. Neurosci. 10, 163-179. Moran G.R., Daubner S.C., and Fitzpatrick P.L. (1998) Expression and characterization of the catalytic core of tryptophan hydroxylase. J. Biol. Chem. 273, 12259-12266. Moran G.R., Fitzpatrick P.F. (1999). A continuous fluorescence assay for tryptophan hydroxylase. Anal. Biochem. 266,148-52. Nakata H, and Fujisawa H. (1981) Simple and rapid purification of tryptophan 5monooxygenase from rabbit brain by affinity chromatography. J. Biochem. 90, 567-569. Nakata H., and Fujisawa H. (1982a) Purification and properties of tryptophan 5monoxygenase from rat brainstem. Eur. J. Biochem. 122, 41-47. Nakata H, and Fujisawa H. (1982b) Tryptophan 5-monooxygenase from mouse mastocytoma P815. A simple purification and general properties. Eur. J. Biochem. 124, 595-601. 30 Neckameyer W.S., and White K. (1992) A single locus encodes both phenylalanine hydroxylase and tryptophan hydroxylase activities in Drosophila. J. Biol. Chem. 267, 4199-4206. Nielsen, D.A., Goldman, D., Virkkunen, M., Tokola, R., Rawlings, R., and Linnoila, M. (1994) Suicidality and 5-hydroxyindoleacetic acid concentration associated with a tryptophan hydroxylase polymorphism. Arch. Gen. Psych. 51, 34-38. Nowacki, P., Byck, S., Prevost, L., and Scriver, C.R. (1997) The PAH mutation analysis consortium database: update 1996. Nucleic Acids Res. 25, 139142. Nukiwa T., Toyama C., Okita C., Kataoka T., and Ichiyama A. (1974) Purification and some properties of bovine pineal tryptophan 5-monooxygenase. Biochem. Biophys. Res. Commun. 60, 1029-1035. Olivier, B., Mos, J., van Oorschot, R., and Hen, R. (1995) Serotonin receptors and animal models of aggressive behavior. Pharmacopsych. 28 Suppl 2, 80-90. Park D.H., Stone D.M., Kim K.S., and Joh T.H. (1994) Characterization of recombinant mouse tryptophan hydroxylase expressed in Escherichia coli. Mol. Cell. Neurosci. 5, 87-93. Privat K., Brisson C., Jouvet A., Chesneau D., Ravault J.P., and Fevre-Montange M. (2002) Evidence for implication of tryptophan hydroxylase in the regulation of melatonin synthesis in ovine pinealocytes in culture. Cell. Mol. Neurobiol. 22, 417-29. Ramaekers V.T., Senderek J., Hausler M., Haring M., Abeling N., Zerres K., Bergmann C., Heimann G., and Blau N. (2001) A novel neurodevelopmental syndrome responsive to 5-hydroxytryptophan and carbidopa. Mol. Genet. Metab. 73,179-87. Regier D.A., Narrow W.E., Rae D.S., Manderscheid R.W., Locke B.Z., and Goodwin F.K. (1993) The de facto mental and addictive disorders service system. Epidemiologic Catchment Area prospective 1-year prevalence rates of disorders and services. Archives of General Psychiatry 50, 85-94. Reinhard Jr J.F, Smith G.K., Nichol C.A. (1986) A rapid and sensitive assay for tyrosine-3-monooxygenase based upon the release of 3H2O and adsorption of [3H]-tyrosine by charcoal. Life Sci. 39, 2185-2189. Sawada M., and Nagatsu T. (1986) Stimulation of the serotonin autoreceptor prevents the calcium-calmodulin-dependent increase of serotonin biosynthesis in rat raphe slices. J. Neurochem. 46, 963-967. 31 Tong J.H., and Kaufman S. (1975) Tryptophan hydroxylase: purification and some properties of the enzyme from rabbit hindbrain. J. Biol. Chem. 250, 41524158. Van den Heuvel L.P., Luiten B., Smeitink J.A.M., de Rijk-van Andel J.F., Hyland K., Steenbergen-Spanjers G.C., Janssen R.J., and Wevers R.A. (1998). A common point mutation in the tyrosine hydroxylase gene in autosomal recessive L-DOPA-responsive dystonia in the Dutch population. Hum. Genet. 102, 644-646. Vrana K.E. (1999) How the regulatory and catalytic domains get together. Nat. Struct. Biol. 6, 401-402. Vrana, S.L., Dworkin, S.I., and Vrana, K.E. (1993) Radioenzymatic assay for tryptophan hydroxylase: [3H]H2O release assessed by charcoal adsorption. J. Neurosci. Meth. 48, 123-129. Walker S.J., Liu X., Roskoski R. Jr., and Vrana K.E. (1994) Catalytic core of rat tyrosine hydroxylase: Terminal deletion analysis of bacterially-expressed enzyme. Biochim. Biophys. Acta 1206, 113-119. Wang, G.A., Coon, S.L., and Kaufman, S. (1998) Alternative splicing at the 3'-cDNA of human tryptophan hydroxylase. J. Neurochem. 71, 1769-1762. Wang G.A., Gu P., and Kaufman S. (2001) Mutagenesis of the regulatory domain of phenylalanine hydroxylase. Proc. Natl. Acad. Sci. 98, 1537-1542. Wang L., Erlandsen H., Haavik J., Knappskog P.M., and Stevens R.C. (2002). Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochem. 41, 12569-12574. Wevers R.A., de Ruk-van Andel J.F., Bräutigam C., Guertz B., van den Heuvel L.P., Steenergen-Spanjers G.C., Smeitink J.A., Hoffman G.F., and Gabreels F.J. (1999). A review of biochemical and molecular genetic aspects of tyrosine hydroxylase deficiency including a novel mutation (291delC). J. Inherit. Metab. Dis. 22, 364-373. Widmer F., Mutus B., RamaMurthy J., Sniedkus V.A., and Viswanatha T. (1975) Partial purification of rabbit hind brain tryptophan hydroxylase by affinity chromatography. Life Sci. 17, 1297-1302. Yang X.J., and Kaufman S. (1994) High-level expression and deletion mutagenesis of human tryptophan hydroxylase. Proc. Natl. Acad. Sci. USA 91, 6659-6663. 32 Yohrling IV, G.J., Mockus, S.M., and Vrana, K.E. (1999) Identification of aminoterminal sequences contributing to tryptophan hydroxylase tetramer formation. J. Mol. Neurosci. 12, 23-34. Yohrling IV G.J., Jiang G.C., Mockus S.M., and Vrana K.E. (2000) Intersubunit binding domains within tyrosine hydroxylase and tryptophan hydroxylase. J. Neurosci. Res. 61,:313-20. Youdim M.B., Hamon M., and Bourgoin S. (1974) Purification of pig brainstem tryptophan hydroxylase and some of its properties. Adv. Biochem. Psychopharmacol. 11, 13-17. Youdim M.B., Hamon M., and Bourgoin S. (1975) Properties of partially purified pig brain stem tryptophan hydroxylase. J. Neurochem. 25, 407-414. 33 STATEMENT OF PURPOSE The goal of this dissertation research was to utilize recombinant DNA technologies as well as homology-based computer modeling to more fully understand the structure and function of human TPH. The studies presented in the next chapters are intended to advance our characterization of TPH, and the subtle role that mutations in the enzyme may play in disease. The first objective was to generate a hypothetical three-dimensional model of human TPH using homology-based molecular modeling in the absence of an experimentally-determined crystal structure. Previous in vitro structural data of TPH and the other AAAHs were mapped to this model. Our hypothetical model revealed information regarding the differential regulation and function of TPH when compared to previously determined structures for TH and PAH. Thereafter, we performed sitedirected mutagenesis to identify previously unidentified functional domains within the TPH protein, and to validate the hypothetical model. The second objective was to perform in vitro expression analysis of a naturally-occuring mutation in human TPH. The rationale for doing so is the same as those behind the analysis of PAH polymorphisms related to the metabolic condition PKU. That is, to confirm that a “disease-associated” mutation is truly pathogenic, to assess the severity of a mutation’s impact, and to elucidate the molecular mechanisms involved by understanding how a mutation exerts its deleterious effects. In the course of these experiments, we developed a bacterial expression system that permits the rapid purification of the human TPH enyme. 34 Overall, these studies have revealed that the structure of TPH closely resembles other members of the AAAH superfamily, and have illuminated new insights into the structural and functional aspects of the protein. Additionally, these studies indicate that the first described coding region polymorphism of TPH may have a minimal impact on enzyme activity. However, it has identified what is important new information on structural determinants of the TPH active site. CHAPTER II IDENTIFICATION OF SUBSTRATE ORIENTING AND PHOSPHORYLATION SITES WITHIN TRYPTOPHAN HYDROXYLASE USING HOMOLOGY-BASED MOLECULAR MODELING George C.-T. Jiang, George J. Yohrling IV, Jeffrey D. Schmitt, and Kent E. Vrana The following chapter is a paper published in Journal of Molecular Biology, volume 302, pages 1005-1017 (2000). The materials have been reprinted with permission. Stylistic variations are due to the requirements of the journal. The original proposal and concept for these studies were those of George Jiang, George Yohrling, and Kent Vrana. George Jiang and George Yohrling contributed equally to construction of the hypothetical model and preparation of the manuscript. Dr. Kent E. Vrana served in an advisory position on experimental design and manuscript preparation. Dr. Jeffrey D. Schmitt (Targacept Inc.) provided technical assistance and scientific advice with the molecular modeling software and aided with construction of the hypothetical model. All final energy minimizations of the model were performed by Dr. Schmitt. 35 36 Abstract Tryptophan hydroxylase (TPH) is the initial and rate-limiting enzyme in the biosynthesis of serotonin (5-HT). The inherent instability of TPH has prevented a crystallographic structure from being resolved. For this reason, multiple sequence alignment-based molecular modeling was utilized to generate a full-length model of human TPH. Previously determined crystal coordinates of two highly homologous proteins, phenylalanine hydroxylase (PAH) and tyrosine hydroxylase (TH), were used as templates. Analysis of the model aided rational mutagenesis studies to further dissect the regulation and catalysis of TPH. Using rational site-directed mutagenesis, it was determined that Tyr235 (Y235), within the active site of TPH, appears to be involved as a tryptophan substrate orienting residue. The mutants Y235A and Y235L displayed reduced specific activity compared to wild-type TPH (≈5% residual activity). The Km of tryptophan for the Y235A (564 µM) and Y235L (96 µM) mutant was significantly increased compared to wild-type TPH (42 µM). In addition, kinetic analyses were performed on wild-type TPH and a deletion construct that lacks the amino terminal autoregulatory sequence (TPH N ∆15). This sequence in phenylalanine hydroxylase (residues 19-33) has previously been proposed to act as a steric regulator of substrate accessibility to the active site. Changes in the steady-state kinetics for tetrahydrobiopterin (BH4) and tryptophan for TPH N ∆15 were not observed. Finally, it was demonstrated that both Ser58 and Ser260 are substrates for Ca2+/calmodulin-dependent protein kinase II. Additional analysis of this model will aid in deciphering the regulation and substrate specificity of TPH, as 37 well as providing a basis to understand as yet to be identified polymorphisms. 38 Introduction Tryptophan hydroxylase (TPH; EC 1.14.16.4) is a member of the aromatic amino acid hydroxylase superfamily (Fitzpatrick, 1999). The other members of this family include tyrosine hydroxylase (TH; EC 1.14.16.2) and phenylalanine hydroxylase (PAH; EC 1.14.16.1). The enzymes share approximately 50% amino acid sequence identity and are thought to have evolved from a common progenitor (Grenett et al., 1987; Neckameyer & White, 1992). TPH serves as the rate-limiting enzyme in the biosynthesis of the neurotransmitter serotonin (5-HT), and as a nonlimiting component of the melatonin synthetic pathway within the pineal gland. Recent evidence has shown TPH to be rate-limiting for melatonin biosynthesis in the retina (Thomas et al., 1998). Within the central nervous system, TPH is localized in the raphe nuclei, as well as in various peripheral locations such as the pineal gland, enteric neurons of the gut, and the enterochromaffin mast cells of the intestine and pancreas (Mockus & Vrana, 1998). Serotonin is an important monoamine that has been implicated in a wide variety of central nervous system functions including temperature control, aggression, pain, and memory (Kandasamy & Williams, 1984; Brentegani & Lico, 1982; Cornwell-Jones et al., 1989; Olivier at al., 1995). Aberrations in 5-HT synthesis and regulation have been linked to a diverse group of neurological and psychiatric disorders such as Parkinson’s disease, Gilles de la Tourette’s syndrome, multiple sclerosis, depression, and obsessive compulsive disorder (Volicier et al., 1985; Schauenburg & Dressler; 1992; Tabaddor et al., 1978; Mann et al., 1997; Delgado & Moreno, 1998). TPH could be a potential 39 pharmacotherapeutic target in the etiology of several of these neurological diseases. The hydroxylases are composed of an amino terminal regulatory domain and a carboxyl terminal catalytic domain (Grenett et al., 1987; Ledley et al., 1985; Darmon et al., 1988; Daubner et al., 1993). It is presumed that the regulatory domains of the enzymes act to modulate activity. Ser58, the one documented phosphorylation site in TPH resides in this regulatory domain (Kuhn et al., 1997; Kumer et al., 1997). The active sites reside within the catalytic domain and are responsible for the hydroxylation of their respective aromatic amino acid residues. Chimeric hydroxylases have been created to demonstrate that substrate specificity is controlled by the catalytic domain (Mockus et al., 1997b; Daubner et al., 1997). Table 1 summarizes all of the published recombinant TPH mutants that have been created to dissect the structure, function, and regulatory mechanisms of TPH. TPH is known to exist both in vivo and in vitro as a homotetramer (reviewed in Hufton et al., 1995). Recently, this laboratory has identified intersubunit binding domains in both the regulatory and catalytic domains of the rabbit TPH protein (Mockus et al., 1997a; Yohrling et al., 1999). These interactions are thought to be hydrophobic and necessary for the proper macromolecular assembly of TPH. Despite the high level of protein sequence identity in the catalytic domains of TH and TPH, they do not form heterotetramers (Mockus et al., 1998). It is hypothesized that there are additional structural domains within TPH that work in tandem with those already identified, to allow for its regulation. This is an additional reason that crystallization or theoretical modeling of TPH is of particular significance. Knowledge (or prediction) of a detailed quaternary structure will enable design of 40 potential pharmacotherapeutics to modulate TPH activity. Moreover, a detailed tertiary and quaternary structure of TPH will provide insight into the factors governing differential substrate specificity among the aromatic amino acid hydroxylases. 41 42 It has long been known that point mutations within the PAH gene lead to insufficient PAH activity and the build-up of the amino acid phenylalanine. This leads to the metabolic imbalances and mental retardation that are characteristic of phenylketonuria (PKU). There are in excess of 300 known distinct mutations in PAH that lead to PKU (Eisensmith & Woo, 1991). Recently, point mutations in the catalytic domain of TH have been linked to Segawa’s syndrome and L-dopa responsive Parkinsonism (Ludecke et al., 1995, 1996; Knappskog et al., 1995). In addition, an intronic polymorphism in the human TPH gene has been discovered that is associated with suicidality and variable 5-hydroxyindolacetic acid (5-HIAA) concentrations in Finnish Caucasians (Nielsen et al., 1992, 1994). To date, it is unclear what, if any, such an intronic polymorphism would have on TPH regulation. However, the genetic findings in PAH and TH suggest that point mutations in critical regulatory and structural domains of TPH will be discovered and that these mutations may explain selected neurological disorders associated with aberrant serotonergic levels. It is unfortunate that research advances on TPH have lagged behind that of the other aromatic amino acid hydroxylases. The extreme instability of TPH has precluded its purification in large quantities (Kuhn et al., 1980; D’Sa et al., 1996a,b; Cash, 1998; Moran et al., 1998), and without adequate purification, a complete crystallographic model for TPH will not be elucidated. For this reason, it has become important to apply the existing information on the related aromatic amino acid hydroxylases (PAH and TH) to aid in characterizing the structure and regulation of TPH. The recent crystallization of both PAH and TH provide much of this 43 information (PAH; Erlandsen et al., 1997a,b, 1998, 2000; TH; Goodwill et al., 1997, 1999). Here, we present a detailed structural model of human TPH, based on the existing structural coordinates for TH and PAH (1TOH and both 1PHZ and 1PAH, respectively; Research Collaboratory for Structural Bioinfomatics Protein Databank; RCSB-PDB) generated using multiple sequence alignment homology modeling techniques (Blundell et al., 1988; Sutcliffe et al., 1987). In addition to being stable relative to molecular mechanics, our model was utilized to successfully predict aspects of TPH assembly and function. 44 Results General topography of the hypothetical TPH model The fully minimized, hypothetical human TPH model is shown in Figure 1. The monomeric unit contains both the amino terminal regulatory domain and the carboxy terminal catalytic domain. The amino terminal end of the protein lacks an organized secondary structure. Previously determined structural and regulatory moieties are evident on the model. A Ramachandran plot (PROCHECK analysis at 2.0 Å) indicates that 84.7, 14.0, 1.0, and 0.3% of non-glycine and non-proline residues reside in most favorable, additionally allowed, generously allowed and disallowed regions respectively. Notably, the last category (disallowed) is composed of a single amino acid (Ile 2) at the disordered amino terminus. In Figure 2, the high degree of secondary and tertiary structural identity between TPH, PAH and TH is evident in a side-by-side comparison of all three structures. Slight structural variations are present between the regulatory domains of PAH and TPH. Figure 3 depicts a theoretical tetramer of human TPH. The tetramer is anchored by the Cterminal tetramerization domain that is composed of hydrophobic, coiled-coil interactions. The regulatory domain of a given monomer forms intersubunit contacts with both the tetramerization domain and regulatory domain of an adjacent subunit. 45 Figure 1: Hypothetical model of human TPH (monomer). A 3-D model of the monomeric subunit of human TPH is depicted. All amino acids were modeled except for the initial methionine residue. Secondary structure is identified by color: β-sheets (yellow), α-helix (pink), and random coil (white). The locations of Ser58, Ser260, and Tyr235 are indicated. The amino terminus of TPH begins with a nonstructured random coil formation termed the active-site gate. The carboxy terminal helix (tetramerization domain) and the amino terminal 4,3-repeat have previously been identified as structural motifs required for the proper macromolecular assembly of TPH. A proposed interface domain between the regulatory and catalytic domain of TPH is also indicated. The model was generated on a Silicon Graphics Workstation with the modeling program Sybyl 6.5 (Tripos Inc., St. Louis, MO). 46 47 Figure 2: Side by side comparison of TH, PAH, and hypothetical TPH. The previously resolved crystal structures for rat PAH (1PHZ) and rat TH (1TOH) are shown along with the full length human TPH model. The X-ray diffraction coordinates for both 1PHZ and 1TOH were used to perform homology based modeling. A high degree of secondary and tertiary structure identity among all the hydroxylase models is evident. 48 49 Figure 3: Tetramer of hypothetical TPH. Each monomer of TPH is shown in a different color. The tetramer is anchored by a carboxy terminal leucine zipper and additional intersubunit hydrophobic interactions between the regulatory domains. The model was created using an alignment based on the previously reported tetramer for TH (Goodwill et al., 1997). 50 51 TABLE 2. Steady-state kinetic parameters of wild-type TPH and mutants Enzyme BH4 Km (µM) L-tryptophan Km (µM) wt TPH 61 ± 19 30 ± 16 N ∆ 15 50 ± 6 41 ± 13 C ∆ 17 55 ± 4 30 ± 6 wt TPH 78 ± 0.4 42 ± 2 Y235A 34 ± 11 564 ± 85* Y235L 56 ± 9 96 ± 5* TPH activity was determined (see Materials and Methods) and the kinetic constants were calculated by non-linear regression analysis. The substrate concentrations were 100 µM BH4 (tryptophan variable) and 50 µM tryptophan (BH4 variable). The mean values for BH4 and tryptophan were compared independently of each other. * One-way ANOVA, followed by a Student’s Newman-Keuls test at 95% confidence indicated that the kinetic differences observed were statistically significant (p < 0.05). 52 Figure 4: Phosphorylation and immunodetection of TPH. (a) SDS-PAGE analysis of CaMPKII phosphorylated TPH proteins. Lane1: pET-21c negative control; lane 2: wild-type TPH; lane 3: S58A; lane 4: S260A; lane 5: S58A/S260A. A decrease in the amount of incorporated phosphate is observed in the double point mutant S58A/S260A. (b) Immunoblot of phosphorylated TPH. Immunoreactive proteins were detected with a monoclonal antibody (WH3) at a 1:1000 dilution followed by an anti-mouse IgG-HRP from sheep at a 1:1500 dilution. Each protein migrated with a molecular weight of 51 kDa. No immunoreactive protein was detected in the pET 21c control. 53 54 Kinetic analysis of autoregulatory sequence Amino terminal deletion of the proposed active-site guard (residues 1-15; N∆15) and subsequent kinetic analysis produced no change in the Km of TPH for tryptophan (41(±13) µM) when compared to wild-type TPH (30(±16) µM; Table 2). Likewise, no alteration in the Km for BH4 was observed in N ∆15 when compared to TPH. A similar analysis was performed on a TPH mutant that lacked the C-terminal tetramerization domain. Non-significant changes in the Km for both BH4 and tryptophan were observed for the truncated mutant enzyme (see Table 2). Phosphorylation of TPH by CaMPKII The hypothetical model indicates that Ser260 resides on the exterior of the TPH monomer. It has been postulated that Ser260 may serve as a substrate for phosphorylation by CaMPKII (56) because it is located within the consensus motif for this kinase (Pearson & Kemp, 1991). To determine whether Ser260 is phosphorylated by CaMPKII, a series of point mutations were created to identify the phosphoryl accepting residue. In Figure 4(a), a phosphorylated band at 51 kDa, corresponding to the molecular weight of TPH is observed for wild-type TPH, S58A, and S260A. However, little phosphorylation is apparent in the mutant S58A/S260A. An unknown bacterial protein (≈49 kDa) was phosphorylated (Figure 4(a)), but lacked immunoreactivity (Figure 4(b)). The data indicate that both Ser58 and Ser260 are substrates for CaMPKII. The corresponding western blot for the phosphorylation reaction is depicted in Figure 4(b) and demonstrates that comparable amounts of 55 TPH protein are present in all samples (even the unlabeled S58A/S260A double point mutation), and all TPH proteins migrated with a molecular weight of 51 kDa. Rational site-directed mutagenesis Analysis of the TPH model and a PAH structure with bound inhibitor (6PAH; Erlandsen et al., 1998) identified Tyr235 (Y235) in TPH as a potential tryptophan substrate orienting residue. It was determined that the side-chain of the corresponding active-site residue in 6PAH (L248) is 5.6 Å from the bound catechol inhibitor (Erlandsen et al., 1998). Site-directed mutagenesis of Y235 to an alanine residue (Y235A) and Y235 to a leucine residue (Y235L) significantly reduced TPH enzymatic activity when compared to wild-type TPH at near saturating substrate concentrations (p < 0.0005). Figure 5 shows the specific activities for wild-type TPH, Y235L, and Y235A. The values have been normalized for the amount of immunoreactive protein present via densitometric analysis (hence enzyme activity units of nmol hr-1 ROD-1). Steady-state kinetic analyses were then performed to investigate the nature of the reduction in activities for the Y235 mutations. The Km of Y235A (564(± 85) µM) and Y235L (96(± 5) µM) for tryptophan were both dramatically increased over wild-type TPH (42(± 2) µM). Moreover, the Km of the mutants, for BH4, were unchanged (wild-type TPH, 78(± 0.4) µM; Y235A, 34(± 11) µM; Y235L, 56(± 9) µM) (see Table 2). Tryptophan substrate inhibition was not observed in Y235A (Figure 6) or Y235L (data not shown). 56 Figure 5: Specific activities of TPH active-site mutations. Mutation of tyrosine 235 to an alanine residue (Y235A) and a leucine residue (Y235L) reduced the specific activity (nmol hr-1 ROD-1) of TPH by 94 and 95%, respectively. To determine these values, constant amounts of the three recombinant enzymes (as determined via a radioenzymatic activity assay) were applied to a SDS-PAGE gel and electroblotted. The blot was probed with a monoclonal anti-TPH antibody (WH3; Sigma) followed by a sheep anti-mouse IgG HRP-coupled secondary antibody (Amersham). The activity was then normalized for the amount of immunoreactive protein present in each sample by densitometric analyses (O’Neill et al., 1989). This method accounts for differences in bacterial expression and/or liberation of the recombinant proteins such that enzymatic activities can be directly compared. Shown here is the specific activity mean ± S.E.M. from three independent experiments. 57 58 Figure 6: Lineweaver-Burk analysis of wild-type TPH and TPH Y235A. Shown is a representative Lineweaver-Burk plot for wild-type TPH (solid circles z) and TPH Y235A (solid triangles ▼). Tryptophan concentrations ranged from 10 µM to 1mM. For wild-type TPH, regression analysis was performed after omitting the activity values recorded at 400 µM and 1mM due to significant TPH inhibition at these concentrations. For Y235A (this plot) and Y235L (data not shown), no inhibition of TPH activity was recorded at tryptophan concentrations of 400 µM or 1mM. In this particular experiment, TPH had a Km for tryptophan of 44 µM, while Y235A was 730 µM. 59 60 Discussion Our understanding of the aromatic amino acid hydroxylases has increased greatly in recent years. This progress culminated in the crystallization of the catalytic core of TH (Goodwil et al., 1997) and the first crystallization of a PAH regulatory domain (Kobe et al., 1999). Due to the extreme instability of TPH and the resulting inability to obtain significant amounts of purified protein, a crystal structure for TPH remains elusive. TPH shares a high degree of sequence identity with both TH and PAH (Grenett et al., 1987; Neckameyer & White, 1992). For this reason, we have utilized the crystal coordinates for both TH (1TOH) and PAH (1PHZ and 1PAH) to perform multiple sequence alignment-based molecular modeling of the human and rabbit TPH proteins. A Blast comparison (Altschul et al., 1990) of the rabbit and human protein sequences, yields 97% overall sequence identity. Only 11 out of the 444 amino acid residues are different and only four of these amino acid residues are non-conservative substitutions. With such a high degree of sequence identity, and no mutations in the active-site region which could cause putative changes in structure/function, only the human TPH model was subjected to further energy minimizations to make predictions with respect to aspects of catalysis and regulation. It is noteworthy to mention that Swiss-Model, the automated protein modeling server, (GlaxoWellcome Experimental Research Center in Geneva, Switzerland) has previously generated a model of TPH (Guex & Peitsch, 1997; Peitsch, 1995, 1996). This model is based on several phenylalanine hydroxylase structures (1PHZ, 2PHM, 61 1PAH, 3PAH, 4PAH), but is truncated at the carboxy terminus. Furthermore, it was generated without human intervention and interpretation. PROCHECK analysis indicates similar Ramachandran properties as our manually predicted human TPH model (85.1% most favored, 11.7% additionally allowed, 2.4% generously allowed, and 0.8% in disallowed regions) but with a greater number of residues in generously allowed and disallowed regions. In addition, the Swiss-Model structure has 46 bad contacts, a significantly greater number than the 2 that our model exhibits. The human 3-D TPH model presented here is the first full-length TPH structure to be reported. As expected from the modeling approach, the human TPH monomer has significant structural identity to the previously crystallized proteins, TH and PAH (Figure 2). The monomer of TPH has an amino terminal regulatory domain (residues 2-106), a carboxy terminal catalytic domain (residues 107-423), and a tetramerization domain (424-444). This regulatory and catalytic domain organization is consistent with experimental observations of the independent catalytic activity of the C-terminal portion of the enzyme (Kumer et al., 1997; D’Sa et al., 1996a; Yang & Kaufman, 1994). The tetramerization domain of TPH is composed of an extreme C-terminal 4,3-hydrophobic repeat. Interspersed within this repeat is a leucine zipper that was originally postulated by Liu and Vrana (1991) to be important for the tetramerization of the AAAH. Deletion mutagenesis of this domain resulted in dimers and monomers of TPH that retained activity (Mockus et al., 1997a; D’Sa et al., 1996b). Kinetic analysis of the mutant C ∆17 TPH, that lacks this tetramerization domain, failed to show a significant change in the Km of C ∆17 TPH for BH4 or tryptophan (Table 2). These data suggest that the tetramerization 62 domain of TPH is a structural motif that is not required for activity. The role of oligomerization in regulatory mechanisms of TPH and the other hydroxylases remain to be determined. Our human TPH model predicts an alternative regulatory domain orientation, relative to PAH and the Swiss-model predicted TPH structure. According to the model, the entire regulatory domain is rotated away from the catalytic domain. In PAH (1PHZ), three regions of interfacial contact are observed (46-65, 107-117, 412414 possessing 55%, 75%, and 67% sequence identity to TPH, respectively). Our findings suggest that the numerous interfacial domain contacts are more solvent exposed in TPH. The absence of these interface interactions in TPH may be a result of the multiple sequence alignment-based modeling. It was least successful in generating the former two regions where loop searching was required to fill the gaps (Figure 7). If this alternative N-terminal domain orientation proves to be correct, it could have implications for protein stability (Mockus et al., 1997b; Daubner et al., 1997) and/or regulation. Kobe et al. (1997a) have hypothesized that an amino terminal autoregulatory sequence is present in PAH. These amino acids in PAH (19 to 33) work to limit substrate accessibility into the active site. This sequence is homologous to amino acids 1 to 15 in TPH. Upon the deletion of the proposed active-site gate in TPH (as with the mutant N ∆15), the entrance proposed by Kobe and colleagues is no longer sterically inhibited. This should permit free substrate access and increase the chances of the slow step of BH4 binding to occur as previously described for TH (Fitzpatrick, 1988). However, such a mutation did not alter the steady-state kinetics 63 of TPH. Therefore, it is unlikely that an autoregulatory sequence, as observed in PAH functions in TPH. Instead, it appears as though access to the active site can be accomplished from the opposite side of the catalytic domain in addition to the previously described 12 Å active-site entrance for TH (Goodwill et al., 1997). This ready access may explain why no alteration in Michaelis-Menten constants was observed. However, more rigorous analyses are needed to verify this hypothesis. Similar studies in PAH must be performed to determine the exact function of its amino terminal autoregulatory sequence. Recent studies of the AAAH have identified several structural motifs important in macromolecular assembly. It has been proposed that a 4,3-hydrophobic repeat in the regulatory domain of TPH (residues 21-41) exists as an alpha helix to permit intersubunit hydrophobic interactions that support assembly (Yohrling et al., 1999). The present model verifies this earlier prediction that a portion of these amino acid residues exist as an alpha helix on the exterior of the subunit (Figure 1). In a similar way, the tetramerization domain (leucine zipper) that was hypothesized by Liu and Vrana (1991) to exist has been demonstrated for both TH (Goodwill et al., 1997) and PAH (Fusetti et al., 1998). The model also predicts that a parallel alpha helix within the regulatory domain (amino acid residues 76-85) could function as an intersubunit binding domain to allow for the tetramerization of TPH. A Blast comparison (Altschul et al., 1990) of the TH and TPH protein sequences yields only 30% overall sequence identity in this region. The structure of the homologous region in TH remains to be determined. However, such differences in amino acid identity could account for the 64 homotetramerization of TH and TPH, as could the different splice variants of human tyrosine hydroxylase. Numerous studies have shown TPH to be phosphorylated and/or activated by CaMPKII (Kuhn et al., 1980; Furukawa et al., 1993; Ichimura et al., 1987; Ehret et al., 1989; Yamauchi et al., 1981). However, TPH activation requires the presence of the activator protein 14-3-3 (Ichimura et al., 1987; Yamauchi et al., 1981). So far, no studies have identified the specific CaMPKII substrate residue(s) within TPH. The human TPH model and the initial rabbit TPH structure (data not shown) suggest that Ser260 could be a substrate for CaMPKII, due to its external orientation on the TPH monomer (Figure 1). The phosphorylation studies presented here verify these predictions by demonstrating that Ser58 and Ser260 are phosphorylated by CaMPKII. The double point mutant S58A/S260A appears to incorporate a minimal amount of phosphate. This may result from the partial phosphorylation of Ser443 in denatured TPH. Ser443 also resides in the midst of a consensus sequence for CaMPKII (Hufton et al., 1995). However, Ser443 is engaged in the strong hydrophobic interactions of the tetramerization domain. It is therefore unlikely that CaMPKII has ready access to this residue under non-denaturing conditions. The in vitro activation of recombinantly expressed TPH and its mutants, in the presence of both CaMPKII and 14-3-3 remains inconclusive (data not shown). The human TPH model presented here was successful in predicting an aspect of TPH active-site structure and specificity. Based on previous findings in TH and PAH, as well as primary sequence alignments, the predicted TPH active-site topology suggested that Y235 (corresponding to L294 in TH and L248 in PAH) could 65 serve to interact with and orient the tryptophan substrate. Mutagenesis of Y235 resulted in a significant reduction of TPH activity (≈95%). Kinetic analyses confirmed our hypothesis that Y235 is a tryptophan orienting residue. Both mutants (Y235A and Y235L) exhibit a dramatic increase in the Michaelis constant for tryptophan, but not BH4. Mutagenesis of this residue also eliminates the substrate inhibition of TPH observed at high levels (>200 µM) of tryptophan (Figure 6). The corresponding identity of Y235 in both TH and PAH is leucine. TPH Y235 is located in a region of 21 amino acid residues, at the entrance of the active site (active site loop and α6), that exhibits an extraordinary amount of amino acid identity (86%) for all aromatic amino acid hydroxylases (Figure 8). It is hypothesized that the longer side chain of leucine is required to hold the smaller amino acid substrates, phenylalanine and tyrosine, in the optimal orientation within the active sites of PAH and TH respectively. The aromatic ring of Tyr235 in TPH may form a π-stacking interaction with tryptophan. This type of interaction appears to play a significant role in mediating substrate specificity of TPH. In addition to the data presented here, the hypothetical model of human TPH will serve numerous other capacities. First, the molecular model will aid in the development of novel pharmacotherapeutics. The availability of a detailed active site conformation could help identify a new generation of highly specific inhibitors of TPH for the potential treatment of CNS disorders associated with excessive serotonergic activity (e.g., restless leg syndrome). Secondly, detailed modeling of TPH will assist biogenic amine research by identifying a list of potentially interesting residues that may be involved in the regulation and activity of TPH, thus assisting 66 rational site-directed mutagenesis studies. Third, the new model may function as the template on which as yet to be identified polymorphisms can be explained. Single nucleotide polymorphisms (SNPs) are known to play a pivotal role in human health and disease (Tomita-Mitchell et al., 1998; Uglialoro et al., 1998). This is particularly true of PAH where 330 independent polymorphisms have been reported that alter enzyme activity (Kobe et al., 1999). The present model provides a platform on which to base structural predictors for TPH following identification of the mutations. It is hoped that one day, clinical screening will be available to identify potentially deleterious polymorphisms in TPH to aid in the prevention and treatment of the numerous neurological diseases associated with aberrant 5-HT and TPH. 67 Figure 7: Multiple sequence alignment. A multiple sequence alignment of the template crystal structures 1PHZ, 1PAH and 1TOH and human TPH (1TPH). This alignment was generated by the Composer module of Sybyl 6.5 (Tripos Inc., St. Louis, MO) and used to construct the hypothetical human TPH model. Residues in red indicate where a gap resulted in the preliminary model, which were subsequently completed by the loop search function. Bolded residues indicate amino acids within the interface between the regulatory and catalytic domains as observed in PAH (1PHZ). Residue numbering is according to the human TPH amino acid sequence. 68 69 Figure 8: Sequence alignment of the aromatic amino acid hydroxylases. Amino acid alignments for human TPH (Accession number; P17752), rabbit TPH (P17290), rat TH (P04177), human TH (P07101), rat PAH (P04176), and human PAH (P00439) for a 21 amino acid stretch that spans the beginning of the active sites for each of the enzymes. 86% amino acid identity is observed in this region. The overall identity of the catalytic domains of the hydroxylases is significantly lower (≈50%). Tyrosine 235 (Y235) of rabbit TPH was mutated to both an alanine and a leucine residue. Above the amino acid residues, lines indicate the regions corresponding to the active-site loop and alpha helix 6 (α6). The active sites of the hydroxylases are thought to contain helices α6-α9 (Erlandsen et al., 1997a). 70 71 Materials and Methods Materials All materials were obtained from the Sigma Chemical Company (St. Louis, MO) with the following exceptions: restriction and DNA modifying enzymes were supplied by Promega Corporation (Madison, WI) and New England Biolabs (NEB; Beverly, MA); L-[5-3H] tryptophan and [γ-32P] ATP from Du Pont NEN Research Products (Boston, MA); IPTG, ampicillin, and activated charcoal (Darco G-60) from Fisher Scientific (Pittsburgh, PA). The pET-21c vector and BL21[DE3](F- OmpTrBmB-) competent cells were acquired from Novagen Corporation (Madison, WI). Reinforced nitrocellulose (Duralose-UVTM) and the QuikChangeTM site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). The Benchmark prestained protein ladder was purchased from Life Technologies (Gaithersburg, MD). Chemiluminescent reagents (RenaissanceTM) were purchased from NEN Research Products (Boston, MA). The mouse anti-TPH monoclonal antibody (WH3) was obtained from Sigma-RBI (St.Louis, MO). The sheep anti-mouse IgG HRP- coupled secondary antibody was purchased from Amersham Life Sciences (Arlington Heights, Il). Ca2+/Calmodulin-dependent protein kinase II α-subunit was purchased from Calbiochem (San Diego, CA). Finally, all sequencing was performed by the DNA Sequencing and Gene Analysis Facility of the Molecular Genetics Program (Wake Forest University School of Medicine) using a Perkin Elmer/Applied Biosystems 377 Prism automated DNA sequencer. 72 Molecular modeling The three-dimensional models of human and rabbit tryptophan hydroxylase (accession numbers NP_004170 and P17290 respectively) were generated using the multiple sequence alignment modeling approach in the “Composer” module of Sybyl 6.5 (Tripos Inc., St. Louis, MO). The experimentally-solved structures of phenylalanine hydroxylase (1PHZ and 1PAH) (Kobe et al., 1999; Erlandsen et al., 1997b) and tyrosine hydroxylase (1TOH) (Goodwill et al., 1997) were used as template molecules in a modular method to provide the most complete model. Initially, human TPH and rabbit TPH were predicted based on 1PHZ, 1PAH and 1TOH. This yielded initial structures with gaps at positions 1, 65 to 66, 100 to 101, and 411 to 444 (Figure 7). The gaps that resulted within the protein (residues 65 to 66, and 100 to 101) were bridged using the “Loop Search” functionality in the Sybyl biopolymer module. Before the loop search, each gap was enlarged by the removal of two amino acids flanking the gap. Loop candidates were visually evaluated and chosen on the basis of complementarity to the existing structure. In order to provide a complete model, human and rabbit TPH were separately predicted based solely on 1TOH and the carboxy terminus of the resulting structure (residues 411 to 444) was then merged with the ir respective continuous, but truncated TPH model based on 1PHZ, 1PAH, and 1TOH. The resulting full-length human TPH model (missing only the initiator methionine residue) was subjected to further refinement using the program PROCHECK to assess progress (Laskowski et al., 1993). First, polar hydrogen atoms were added, then proline and sidechain conformations were fixed and 73 relieved of bad contacts. Next, the peptide backbone was constrained and the model was subjected to convergent minimization, without electrostatics, in the Tripos forcefield (Clark et al., 1989). All the remaining valencies were hydrogen-filled and the model was again minimized to convergence. Backbone constraints were then removed and the model was minimized to a high level of convergence (gradient 0.005 kcal / mol⋅Å). Kollman charges (Weiner et al., 1984) were then added followed by five cycles of minimization, during which time the dielectric constant was gradually decreased from ε = 80 to ε = 20. The final minimization step was carried out to convergence at a high level (gradient 0.005 kcal / mol⋅Å). Bacterial expression of TPH A full length cDNA clone for rabbit tryptophan hydroxylase has previously been developed for prokaryotic expression (Vrana et al., 1993). The coding region of the parent clone (prbTRH479) (Grenett et al., 1987) was amplified by PCR to create a 5' NdeI site and a 3' BamHI site and subsequently subcloned into a pET21c prokaryotic expression vector. All recombinant proteins were expressed using the approach detailed by Yohrling et al. (1999). TPH deletion and point mutation constructs Polymerase chain reaction was utilized to create a number of different deletion mutations to examine TPH structure/function predictions from the human TPH model. For example, to generate the N∆15 mutant (N ∆ denotes an amino terminal deletion followed by the number of amino acid residues deleted from the 74 construct), a 5' primer was designed to introduce an NdeI consensus sequence (CATATG) followed by the sequences for amino acid residues 16 to 20. Details for the construction and characterization of the carboxy terminal mutant (C ∆17) are outlined in (Mockus et al., 1997a). In addition, the S58A mutant utilized in this report has been previously reported (Kumer et al., 1997). To elucidate a possible role for S260 as a substrate for phosphorylation by CaMPKII, the mutants S260A and S58A/S260A were created with the QuikChangeTM Site-Directed mutagenesis kit from Stratagene. Wild-type TPH and S58A (25 ng) were used as the respective DNA templates to create S260A and S58A/S260A. The conditions utilized are outlined in (Yohrling et al., 1999; Vrana et al., 1994). The primers used (but not previously reported) were as follows: TPH N ∆15: 5'- CTCAATAGTCCATATGGGAAGAGCAAGTCTC -3' TPH S260A Sense: 5'- GTGAGACACAGTGCAGACCCCTTCTATACC-3' Antisense: 5'- GGTATAGAAGGGGTCTGCACTGTGTCTCAC -3' TPH Y235A Sense: 5'- CGTCCTGTGGCTGGTGCCTTATCACCAAGAG -3' Antisense: 5'- CTCTTGGTGATAAGGCACCAGCCACAGGACG -3' TPH Y235L Sense: 5'- CGTCCTGTGGCTGGTCTCTTATCACCAAGAG-3' Antisense: 5'- CTCTTGGTGATAAGAGACCAGCCACAGGACG -3' The nucleotides underlined correspond to the NdeI recognition site. The bold nucleotides highlight the mutated codons for amino acid residues 260 and 235, respectively. Complete DNA sequencing was performed to verify the presence of the appropriate mutation in the coding sequences of all recombinant proteins. This also established that the PCR-based mutagenesis (N∆15) and non-PCR-based 75 mutagenesis (S260A, S58A/S260A, Y235A, and Y235L) did not introduce extraneous mutations. Kinetic analyses and TPH activity assay Michaelis-Menten constants for two of the cosubstrates, BH4 and tryptophan, were determined for wild-type TPH, TPH N ∆15, C ∆17, Y235A, and Y235L. First, bacterial pellets containing the various proteins were sonicated in a HEPES-based resuspension buffer and centrifuged at 50,000 x g to create a high speed supernatant (Yohrling et al., 1999). The Km values of each recombinant protein for BH4 were determined on comparable amounts of activity by performing a radioenzymatic TPH activity assay with BH4 concentrations ranging from 9 µM -220 µM (Beevers et al., 1983; Vrana et al., 1993). Likewise, the Km of each enzyme for tryptophan was calculated in the presence of tryptophan concentrations ranging from 20 µM-1 mM. The activity values recorded at 400 µM and 1 mM of tryptophan for wild-type TPH were not taken into account when determining the Michaelis constant because of substrate inhibition. Stock solutions of L-tryptophan were prepared in 200 mM Tris-HCl (pH 7.0). Activity values derived from each assay are expressed as nmol product ⋅ hr-1 ⋅ mg total protein-1. All kinetic data were fitted to the Michaelis-Menten equation using GraphPad Prism® version 3.0 (GraphPad Software, San Diego, CA, www.graphpad.com). 76 Phosphorylation of native and mutant TPH Bacterial supernatants of TPH and the mutants S58A, S260A, and S58A/S260A were subjected to phosphorylating conditions to ascertain whether S58A and/or S260 are phosphorylated by CaMPKII. Both amino acids reside in the consensus motif for CaMPKII (Arg-X-X-Ser), and are predicted, by the hypothetical model, to reside on the surface of the protein. Homogenates were incubated at 30°C for five minutes with 50 mM MgCl2, 3 mM CaCl2, 1µCi [γ-32P] ATP, 40 µM ATP, 0.75 µg calmodulin, and 400 units of CaMPKII. The reactions were terminated by adding 5 µl of 6X sample buffer [0.5M Tris-HCl (pH 6.8), 30% (v/v) glycerol, 10% (w/v) SDS, 0.6M DTT, and 0.012% bromophenol blue. The samples were immediately denatured at 100°C for ten minutes and then subjected to denaturing polyacrylamide gel electrophoresis (SDS-PAGE). TPH protein analysis and enzyme activity Denaturing polyacrylamide gel electrophoresis (10% acrylamide, 0.27% bisacrylamide) (Laemmli et al., 1970) was performed on phosphorylation reactions as outlined by Kumer et al. (1997). Following transfer of the proteins, radioactive proteins were visualized with a phosphoimager (Fuji Medical Systems). Immunoreactive TPH proteins were detected by probing with a 1:1000 dilution of affinity-purified mouse anti-TPH monoclonal antibody from mouse (WH3) followed by a 1:1500 dilution (1.5 µg / ml) of sheep anti-mouse IgG coupled with HRP. The incubation and wash conditions for western analysis are outlined in Mockus et al. (1997b). Immune complexes were visualized using enhanced chemiluminescence 77 (NEN Research Products, Boston, MA) and exposed to X-ray film (Kodak Biomax MR2) at exposure times (10 sec to 4 min) where the radioactivity signal was negligent. Acknowledgments We would like to thank both Dr. Mark O. Lively (Wake Forest University) and Dr. Susan M. Mockus (University of Washington) for their technical input. This work was supported by PHS Grant RO1 GM-38931 to K.E.V. 78 References Beevers, S.J., Knowles, R.G., and Pogson, C.I. (1983) J. Neurochem. 40, 894-897. Blundell, T.L., Carney, D.P., Gardner, S., Hayes, F.R.F., Howlin, B., Hubbard, T.J.P., Overington, J.P., Singh, D.A., Sibanda, B.L., and Sutcliffe, M.J. (1988) Eur. J. Biochem. 172, 513-520. Brentegani, L.G. and Lico, M.C. (1982) Phys. and Behav. 28, 49-52. Cash, C.D. (1998) Gen. Pharmacol. 30, 569-574. Clark, M., Cramer, III, R.D., Van Opdenbosch, N. (1989) J. Comp. Chem. 10, 9821012. Cornwell-Jones, C.A., Decker, M.W., Chang, J.W., Cole, B., Goltz, K.M., Tran, T,, and McGaugh, J.L. (1989) Brain Res. 493, 258-68. D’Sa, C.M., Arthur, R.E. Jr., Jennings, I.., Cotton, R.G.H., and Kuhn, D.M. (1996a) J. Neurosci. Meth. 69, 149-153. D’Sa, C.M., Arthur, R.E.Jr., and Kuhn, D.M. (1996b) J. Neurochem. 67, 917-926. Darmon, M.C., Guibert, B., Leviel, V., Ehret, M., Maitre, M., and Mallet, J. (1988) J. Neurochem. 51, 312-316. Daubner, S.C., Lohse, D.L., and Fitzpatrick P.F. (1993) Protein Sci. 2, 1452-1460. Daubner, S.C., Hillas, P.J., Fitzpatrick, P.F. (1997) Biochem. 36, 11574-11582. Delgado, P.L. and Moreno, F.A. (1998) J. Psychoactive Drugs 30, 359-366. Ehret, M., Cash, C.D., Hamon, M., and Maitre, M. (1989) J. Neurochem. 52, 18861891. Eisensmith, R.C. and Woo, S.L. (1991) Mol. Biol. and Med. 8 (1), 3-18. Erlandsen, H., Fusetti, F., Martinez, A., Hough, E., Flatmark, T., Stevens, R. C. (1997) Nature Struct. Biol. 4, 995-1000. Erlandsen, H., Martinez, A., Knappskog, P.M., Haavik, J., Hough, E., and Flatmark, T. (1997) FEBS Letters 406, 171-174. Erlandsen, H., Flatmark, T., Stevens, R.C., and Hough, E. (1998) Biochem. 37(45), 15638-15646. 79 Erlandsen, H., Bjorgo, E., Flatmark, T., and Stevens, R.C. (2000) Biochem. 39, 2208-2217. Fitzpatrick, P.F. (1988) J. Biol. Chem. 263 (31), 16058-16062. Fitzpatrick, P.F. (1999) Annu. Rev. Biochem. 68, 355-381. Furukawa, Y., Ikuta, N., Omata, S., Yamauchi, T., Isobe, T., and Ichimure, T. (1993) Biochem. Biophys. Res. Commun. 194, 144-149. Fusetti, F., Erlandsen, H., Flatmark, T., and Stevens, R.C. (1998) J. Biol. Chem. 273 (27),: 16962-16967. Goodwill, K.E., Sabatier, C., Marks, C., Raag, R., Fitzpatrick, P.F., and Stevens, R.C. (1997) Nature Struct. Biol. 4(7), 578-585. Goodwill, K.E., Sabatier, C., Stevens, R.C. (1998) Biochem, 37(39), 13437-13445. Grenett, H.E., Ledley, F.D., Reed, L.L., and Woo, S.L.C. (1987) Proc. Natl. Acad. Sci. USA, 84, 5503-5534. Guex, N. and Peitsch, M. C. (1997) Electrophoresis 18, 2714-2723. Hufton, S.E., Jennings, I.G., and Cotton, R.G.H. (1995) Biochem. J. 311, 353-366. Hufton, S.E., Jennings, I.G., and Cotton, R.G.H. (1998) Biochim. Biophys. Acta, 1382, 295-304. Ichimura, T., Isobe, T., Okuyama, T., Yamauchi, T., and Fujisawa, H. (1987) FEBS Lett. 219, 79-82. Kandasamy, S.B. and Williams, B.A. (1984) Neuropharmacology 23: 49-53. Knappskog, P.M., Flatmark, T., Mallet, J., Ludecke, B., and Bartholome, K. (1995) Hum. Mol. Genet. 4, 1209-1212. Kobe, B, Jennings, I.G., House, C.M., Feil, S.C., Michell, B.J., Tiganis, T., Parker, M.W., Cotton, R.G.H., and Kemp, B.E. (1997) Prot. Sci. 6, 1352-1357. Kobe, B, Jennings, I.G., House, C.M., Michell, B.J., Goodwill, K.E., Santarsiero, B.D., Stevens, R.C., Cotton, R.G.H., and Kemp, B.E. (1999) Nature Struct. Biol. 6(5), 442-448. Kuhn, D.M., Ruskin, B., and Lovenberg, W. (1980) J. Biol. Chem. 255, 4137-4143. Kuhn, D.M., Arthus, R., Jr., States, J.C. (1997) J. Neurochem. 68, 2220-2223. 80 Kumer, S.C., Mockus, S.M., Rucker, P.J., and Vrana K.E. (1997) J. Neurochem. 69, 1738-1745. Laemmli, U.K. (1970) Nature 227, 680-685. Laskowski, R.A., MacArthur, M.W., Moss, D.S. and Thornthon, J.M. (1993) J. Appl. Crystallogr. 26, 283-291. Ledley, F.D., DiLella, A.G., Kwok, C.M., and Woo, S.L.C. (1985) Biochemistry, 24, 3389-3394. Liu, X. and Vrana, K.E. (1991) Neurochem. Int. 18, 27-31. Ludecke, B., Dworniczak, B., and Bartholome, K. (1995) Hum. Genet. 95, 123-125. Ludecke, B., Knappskog, P.M., Clayton, P.T., Surtees, R.A.H., Clelland, J.D., Heales,S.J.R., Brand, M.P., Bartholome, K., and Flatmark. T. (1996) Hum. Mol. Genet. 5, 1023-1028. Mann, J.J., Malone, K.M., Nielsen, D.A., Goldman, D., Erdos, J., Gelernter, J. (1997) Amer. J. Psych. 154, 1451-1453. Mockus, S.M., Kumer, S.C., and Vrana, K.E. (1997a) Biochim. Biophys. Acta, 1342, 132-140. Mockus, S.M., Kumer, S.C., and Vrana K.E. (1997b) J. Mol. Neurosci. 9, 35-48. Mockus, S.M., Yohrling, IV, G.J., Vrana, K.E. (1998) J. Mol. Neurosci. 10, 45-51. Mockus, S.M. and Vrana, K.E. (1998) J. Mol. Neurosci. 10(3), 163-179. Moran, G.R., Daubner, S.C., and Fitzpatrick, P.L. (1998) J. Biol. Chem. 273, 1225912266. Neckameyer, W.S., and White, K. (1992) J. Biol. Chem. 267, 4199-4206. Nielsen, D.A., Dean, M., and Goldman, D. (1992) Amer. J. Hum. Genet. 51, 13661371. Nielsen, D.A., Goldman, D., Virkkunen, M., Tokola, R., Rawlings, R., and Linnoila, M. (1994) Arch. Gen. Psych. 51, 34-38. O’Neill, R.R., Mitchell, L.G., Merril, C.R., and Rasband, W.S. (1989) Appl. Theor. Electrophor. 1, 163-167. 81 Olivier, B., Mos, J., van Oorschot, R., and Hen, R. (1995) Pharmacopsych. 28 Suppl 2, 80-90. Pearson, R.B. and Kemp, B.E. (1991) Methods Enzymol. 200, 62-81. Peitsch, M.C. (1995) Bio/Technology 13, 658-660. Peitsch, M.C (1996) Biochem. Soc. Trans. 24, 274-279. Schauenburg, H. and Dressler, D. (1992) Nervearzt 63, 453-461. Sutcliffe, M.J., Haneef, I., Carney, D.P., and Blundell, T.L. (1987) Prot. Eng. 1, 377. Tabaddor, K., Wolfson, L.I., Sharpless, N.S. (1978) Neurology 28, 1249-1253. Thomas, K.B., Brown, A.D., and Iuvone, P.M., (1998) Neuroreport 9, 4041-4044. Tomita-Mitchell, A., Muniappan, B.P., Herrero-Jimenez, P., Zarbl, H., Thilly, W.G. (1998) Gene, 223, 381-391. Uglialoro, A.M., Turbay, D., Pesavento, P.A., Delgado, J.C., McKenzie, F.E., Gribben, J.G., Hartl, D., Yunis, E.J., Goldfeld, A.E. (1998) Tissue Antigens. 52, 359-67. Volicer, L., Direnfeld, L.K., Freedman, M., Albert, M., Langlias, P.J., Bird, E.D. (1985) Arch. of Neurol. 42, 127-29. Vrana, S.L., Dworkin, S.I., and Vrana, K.E. (1993) J. Neuorsci. Meth. 48, 123-129. Vrana, K.E., Rucker, P., and Kumer S.C. (1994a) Life Sci. 55, 1045-1052. Wang, G.A., Coon, S.L., Kaufman, S. (1998) J. Neurochem. 71, 1769-1772. Weiner, S.J., Kollman, P.A., Case, D.A., Singh, U.C., Ghio, C., Alagona, G., Profeta, S., Weiner, P. (1984) J. Amer. Chem. Soc. 106, 765-784. Yamauchi, T., Nakata, H., and Fujisawa H. (1981) J. Biol. Chem. 256, 5404-5409. Yang, X.J. and Kaufman, S. (1994) Proc. Natl. Acad. Sci. USA 91, 6659-6663. Yohrling, IV, G.J., Mockus, S.M., and Vrana, K.E. (1999) J. Mol. Neurosci. 12, 23-34. Yohrling, IV, GJ, Jiang, G.C-T., Mockus, S.M., and Vrana, K.E. (2000) J. Neurosci. Res. 61, 313-320. CHAPTER III FUNCTIONAL ANALYSIS OF THE V177I CODING REGION POLYMORPHISM IN HUMAN TRYPTOPHAN HYDROXYLASE (TPH1) George C-T. Jiang, Amanda C. Domer, and Kent E. Vrana The following manuscript is in preparation for submission to the Journal of Neurochemistry. Stylistic variations are due to the requirements of this journal. The original proposal and concept for these studies were those of George C.-T. Jiang. Dr. Kent E. Vrana served in an advisory position on experimental design and manuscript preparation. Amanda Domer assisted in the creation of point mutants, protein expression and purification, and TPH activity assays. 82 83 Abstract The aromatic amino acid hydroxylase (AAAH) superfamily consists of three important enzymes: phenylalanine hydroxylase (PH), tyrosine hydroxylase (TH) and tryptophan hydroxylase (TPH). These enzymes are rate-limiting in phenylalanine catabolism, catecholamine biosynthesis, and serotonin biosynthesis, respectively. Numerous polymorphisms in PH and TH have been described that have been associated with phenylketonuria and dystonias, respectively, while few polymorphisms have been described for TPH. Recently, the first human TPH coding region polymorphism has been described. This paper analyzes the functional implications of this polymorphism using site-directed mutagenesis, recombinant bacterial expression, and purified enzyme. The Michaelis-Menten constant for tetrahydrobiopterin was unchanged for V177I (Km, BH4 = 14.8 ± 0.9 µM), while the enzyme’s Michaelis constant for the tryptophan substrate (Km, Trp = 41.1 ± 6.5 µM) was increased 55% compared to wild-type enzyme (Km, BH4 = 15.8 ± 1.2 µM, Km, Trp = 26.5 ± 3.3 µM). Our results indicate that the subtle V177I mutation results in a somewhat less-efficient enzyme due to compromised tryptophan substrate binding. Additional mutational analysis of V177 indicates that the identity of this residue appears to be critical for proper enzyme function in that substitution of nearly any amino acid at this position (except alanine, V177A) produces enzyme with little, or no, activity. Therefore, while the specific polymorphism has a modest impact on enzyme activity, it highlights a region of considerable importance to enzyme structure and function. 84 Introduction The aromatic amino acid hydroxylase (AAAH) superfamily is a group of tetrahydrobiopterin-dependent monooxygenases with specificity for the benzeneand indole-containing amino acids. The members of this family include phenylalanine hydroxylase (PH; EC 1.14.16.1), tyrosine hydroxylase (TH; EC 1.14.16.2), and tryptophan hydroxylase (TPH; EC 1.14.16.4). These enzymes share approximately 50% amino acid sequence identity and are thought to have evolved from a common progenitor (Grenett et al., 1987; Neckameyer and White, 1992). Each AAAH enzyme catalyzes the hydroxylation reaction of its respective amino acid substrate in the presence of tetrahydrobiopterin (BH4) and molecular oxygen (O2) co-substrates, and a ferrous iron (Fe2+) cofactor. The catalytic mechanisms of the hydroxylases are believed to be sequential in nature with BH4 binding first and forming part of the amino acid substrate-binding pocket, followed by binding of either the molecular oxygen or the respective aromatic amino acid substrate (Fitzpatrick, 1991; Fitzpatrick, 1993; Andersen et al., 2002; Bassan et al., 2003). Recently, Volner et al. (2003) have conclusively established that the mechanism for bacterial PH is a fully-ordered ter-bi sequential mechanism. The AAAHs are all composed of an amino terminal regulatory domain and a carboxyl terminal catalytic domain (Grenett et al., 1987; Ledley et al., 1985; Darmon et al., 1988; Daubner et al., 1993; Walker et al., 1994; Kumer et al., 1997). The regulatory domains of the enzymes act to modulate activity. Documented phosphorylation sites in all AAAHs reside in this regulatory domain (Kuhn et al., 1997; Kumer et al., 1997; reviewed in Fitzpatrick, 2000). The active sites reside 85 within the catalytic domain and are responsible for the hydroxylation of their respective aromatic amino acids. Chimeric hydroxylases have been created to demonstrate that substrate specificity is conferred exclusively by the catalytic domain (Mockus et al., 1997; Kumer et al., 1997). Crystal structures are available for TH and PH (Goodwill et al., 1997; Goodwill et al., 1998; Erlandsen et al., 1997a; Erlandsen et al., 1997b; Erlandsen et al., 1998; Fusetti et al., 1998; Erlandsen et al., 2000; Erlandsen et al., 2002), while only one crystal structure is available for TPH (Wang et al., 2002). The TPH crystal structure (Wang et al., 2002), hypothetical AAAH models based on crystal structures (Jiang et al., 2000; McKinney et al., 2001; Andersen et al., 2002), and NMR studies with TPH and bound substrates by McKinney and colleagues (2001) predict to the residues involved in substrate binding in TPH, and confirm the hypothesis of two binding pockets in the active site – one for the aromatic amino acid substrate, and one for the tetrahydrobiopterin cosubstrate (BH4). These data are in agreement with the presumed catalytic mechanisms of the AAAHs. AAAHs have been implicated in several important clinical conditions. In the case of PH, over 400 polymorphisms have been described that result in phenylketonuria (PKU). A number of TH coding region polymorphisms have been described that are associated with L-DOPA responsive parkinsonism (L205P, Ludecke et al., 1996), dystonias (Q381K, Knappskog et al., 1995), and Segawa’s Disease (Ludecke et al., 1995). However, due to the complexity involved in diagnosing patients with serotonin aberrations, only one coding region polymorphism has been described for TPH (Ramaekers et al., 2001). This 86 polymorphism is a heterozygous missense mutation within exon 6 (G529A) of the TPH gene causing a substitution of isoleucine for valine at codon 177 (V177I). This has been interpreted as a rare DNA variant because the pedigree analysis of that individual did not provide direct genotype-phenotype correlation. This polymorphism was discovered in a single member of a patient population that exhibited novel neurodevelopment symptoms that were responsive to 5-hydroxytryptophan and carbidopa treatment. In the other four patients, the TPH gene analysis was normal. Notably, however, the individual exhibiting the TPH polymorphism also had another commonly found polymorphism (C677T heterozygous mutation) in the methylenetetrahydrofolate reductase (MTHFR) gene. The functional consequence of this V177I mutation on TPH remains unclear. Moreover, because of the lack of functional data, it is unclear if the serotonergic imbalances seen in the patients are due to compromised TPH enzymes, inactivated TPH enzymes, or the selective loss of serotonergic neurons. Structurally, we have localized the V177I polymorphism to the catalytic domain of TPH, at a position near the tryptophan substrate-binding pocket of the active site. Based on a TPH crystal structure (Wang et al., 2002) and hypothetical TPH model (Jiang et al., 2000), Val177 is located approximately 3-4 Å from Arg257 and approximately 5-6 Å from Ser336. These two residues are conserved among the hydroxylases and have been implicated, within the other AAAHs, in binding the alpha carboxyl group of the aromatic amino acid substrate (Moran et al., 1998) and decoupling oxygen-oxygen bond cleavage and tyrosine hydroxylation (Ellis et al., 2000), respectively. To examine the functional significance of the TPH polymorphism, we used 87 site-directed mutagenesis to introduce this mutation into a recombinant human TPH expression construct. The codon for valine 177 was altered to encode glycine, alanine, isoleucine, leucine, phenylalanine, serine, threonine, arginine, and glutamate. Mutant proteins were then expressed as hexa-histidine-tagged proteins, purified, and assayed for tryptophan hydroxylation in order to determine the biochemical consequences of the substitutions on TPH. 88 Materials and Methods Materials All materials were obtained from the Sigma Chemical Company (St. Louis, MO) with the following exceptions: restriction and DNA modifying enzymes were supplied by Promega Corporation (Madison, WI) and New England Biolabs (NEB; Beverly, MA); L-[5-3H] tryptophan, and Ni2+ chelating columns from Amersham Pharmacia Biotech (Piscataway, NJ); isopropyl-thio-β-D-galactopyranoside (IPTG), ampicillin, and activated charcoal (Darco G-60) from Fisher Scientific (Pittsburgh, PA). The pET-15b prokaryotic expression vector, and BL21(DE3)pLysS (F- OmpTrBmB-) competent cells were acquired from Novagen Corporation (Madison, WI). Reinforced nitrocellulose (Duralose-UVTM) and the QuickChangeTM Site-Directed Mutagenesis kit was purchased from Stratagene (La Jolla, CA). Benchmark Prestained Protein Ladder, Mark 12™ Unstained Standard and Colloidal Blue Staining Kit was purchased from Invitrogen (Carlsbad, CA). Chemiluminescence reagents (RenaissanceTM) were purchased from NEN Research Products (Boston, MA). The mouse anti-TPH monoclonal antibody (WH3) was obtained from SigmaRBI (St. Louis, MO). The sheep anti-mouse IgG HRP-coupled secondary antibody was purchased from Amersham Life Sciences (Arlington Heights, Il). Qiagen Miniprep and Hi-Speed Midiprep kits were purchased from Qiagen (Valencia, CA). Finally, all sequencing was performed by the DNA Sequencing and Gene Analysis Facility of the Molecular Genetics Program (Wake Forest University School of Medicine) using a Perkin Elmer/Applied Biosystems 377 Prism automated DNA 89 sequencer. TPH Point-Mutant Constructs The methods used for the construction of the various TPH point-mutants are similar to those outlined in Yohrling et al., 1998. The primers used are shown in Table I. Briefly, mutations were made with the Stratagene QuikChangeTM SiteDirected Mutagenesis kit using the pET-15b/hTPH plasmid as the template. Thermocycler conditions were set as follows: 20 cycles of 94°C, 30s, 45°C, 30s, 72°C, 12m; 25 cycles of 94°C, 30s, 55°C, 30s, 72°C, 12m; 4°C. 2 µl DpnI-treated products were used to transform supercompetent XL1-Blue cells. Random colonies were selected to determine if the desired mutation was present. Complete DNA sequencing was performed to verify the presence of the appropriate mutation in the coding sequences of all recombinant proteins and to discount the presence of spurious sequence mutations. 90 Table I – Site-directed mutagenesis primers h-V177Is 5’- G ACC TGG GGA GCC ATT TTC CAA GAG C -3’ h-V177Ia 5’- G CTC TTG GAA AAT GGC TCC CCA GGT C-3’ h-V177As 5’- G ACC TGG GGA GCC GCA TTC CAA GAG C -3’ h-V177Aa 5’- G CTC TTG GAA TGC GGC TCC CCA GGT C-3’ h-V177Rs 5’- G ACC TGG GGA GCC CGA TTC CAA GAG C -3’ h-V177Ra 5’- G CTC TTG GAA TCG GGC TCC CCA GGT C-3’ h-V177Es 5’- G ACC TGG GGA GCC GAA TTC CAA GAG C -3’ h-V177Ea 5’- G CTC TTG GAA TTC GGC TCC CCA GGT C-3’ h-V177Ls 5’- G ACC TGG GGA GCC CTT TTC CAA GAG C -3’ h-V177La 5’- G CTC TTG GAA AAG GGC TCC CCA GGT C-3’ h-V177Fs 5’- G ACC TGG GGA GCC TTT TTC CAA GAG C -3’ h-V177Fa 5’- G CTC TTG GAA AAA GGC TCC CCA GGT C-3’ h-V177Ga 5’- G ACC TGG GGA GCC GGT TTC CAA GAG C -3 h-V177Ga 5’- G CTC TTG GAA ACC GGC TCC CCA GGT C-3 h-V177Ss 5’- G ACC TGG GGA GCC TCT TTC CAA GAG C -3’ h-V177Sa 5’- G CTC TTG GAA AGA GGC TCC CCA GGT C-3’ h-V177Ts 5’- G ACC TGG GGA GCC ACT TTC CAA GAG C -3’ h-V177Ta 5’- G CTC TTG GAA AGT GGC TCC CCA GGT C-3’ Bolded nucleotides corresponding to the codon which yields an altered amino acid. 91 Protein Expression and Purification of Human TPH Human tryptophan hydroxylase (hTPH) cDNA was a generous gift of Dr. Jacques Mallet (Centre National de la Recherche Scientifique, Paris, France). Based on the recent report of Walther et al. (2003), this is the peripherallyexpressed isoform of the enzyme and might more accurately be described as TPH1. PCR primers integrating NdeI and BamHI sites were utilized to modify the respective 5’ and 3’ ends of the hTPH gene to enable subcloning into pET prokaryotic expression vectors (Novagen, Inc., Madison, WI). PCR products were digested with both restriction enzymes and then ligated into a similarly digested pET-15b expression vector that encodes an amino-terminal hexa-histidine tag. Clones were verified by complete DNA sequencing. This sequence analysis confirmed clone identity and eliminates any extraneous mutations that may have occurred during the PCR modification step. All recombinant proteins were expressed using the approach detailed in (Yohrling et al., 1999). Briefly, recombinant human TPH (hTPH) was expressed in bacteria by transforming the BL21(DE3)pLysS strain of Escherichia coli (E. coli). Bacteria were grown until they reached mid-log phase as determined by a spectrophotometer reading of OD600 = 0.4. Bacteria were then induced by the addition of isopropyl-thio-β-D-galactopyranoside (IPTG) to express hTPH for 2.5 hours at 30°C. Bacterial cells were harvested by low speed centrifugation. Recovery of recombinant protein was achieved by lysis of the cells via sonication in a HEPES-based resuspension buffer, centrifugation at 50,000 x g, and subsequent collection of the supernatant. 92 The clarified lysate (high speed supernatant) was then subject to a one-step metal chelate affinity chromatography procedure using an AktaFPLC automated workstation (Amersham Pharmacia Biotech, Piscataway, NJ) and an optimized protocol for the HisTrap Kit (Amerham Pharmacia Biotech, Piscataway, NJ). Our modifications to the purification protocol include the addition of 100mM imidazole in the start and wash buffers, and elution of the His-tagged protein in a buffer containing 300mM imidazole, all performed on a 5ml Ni2+-chelating column at a flow rate of 3ml/min. Purified protein was immediately assayed or stored at –80°C in a storage buffer consisting of 50mM HEPES (pH 7.5), 0.2M NaCl, 10% glycerol, 0.05% Tween 20, and 1mM dithiothreitol and protease inhibitors (Hamdan and Ribeiro, 1999). Kinetic Analyses and TPH activity assay TPH enzyme activity was determined using a radioenzymatic assay as previously described with substrate concentrations of 100 µM BH4 and 50 µM tryptophan (Beevers et al., 1983; Vrana et al., 1993; Jiang et al., 2000). MichaelisMenten constants for two of the co-substrates, BH4 and tryptophan, were determined for wild-type hTPH, and the hTPH V177 mutants. The Km values of each recombinant protein for BH4 were determined on comparable amounts of activity by performing a radioenzymatic TPH activity assay with BH4 concentrations ranging from 6-110 µM (at 50 µM tryptophan) (Beevers et al., 1983; Vrana et al., 1993; Jiang et al., 2000). Likewise, the Km of each enzyme for tryptophan was calculated in the presence of tryptophan concentrations ranging from 2µM-2mM (at 93 100 µM BH4) (Jiang et al., 2000). Activity values, derived from each assay, are expressed as nmol H2O product • min-1 • mg-1. Because 5-hydroxytryptophan production is equimolar with water production, the nmol of product are considered identical. All kinetic data were fitted to the Michaelis-Menten equation using GraphPad Prism version 3.03 (GraphPad Software, San Diego, CA, www.graphpad.com). 94 Results Expression and purification of wild-type and mutant enzymes was not significantly different (Table II and Figure 2). All active enzymes demonstrated similar half-lives and stability (activity retention) in storage buffer (data not shown), though initial baseline activity may have been different (Table II and III). The V177G, V177S, V177T, V177R, V177E, V177L and V177F mutants demonstrated significantly compromised enzyme activity compared to the wild-type enzyme, with the V177R and V177E mutants having no detectable activity. The experimental kinetic data for wild-type and active mutant enzymes are shown in Table III. Wild-type TPH exhibited a Michaelis-Menten constant for tetrahydrobiopterin (Km, BH4) of 15.8 ± 1.2 µM, and a Km, Trp = 26.5 ± 3.3 µM. The mutant V177I, which is the polymorphism described by Ramaekers and colleagues (2002), showed no change with respect to its Michaelis-Menten constant for tetrahydrobiopterin (Km, BH4 = 14.8 ± 0.9 µM), but demonstrated an apparent decrease in Vmax when assayed for its Km, BH4 at a fixed tryptophan concentration of 50 µM. This apparent decrease in Vmax was not observed when varying concentrations of BH4 were assayed with a higher tryptophan substrate concentration (500 µM). Conversely, the Michaelis-Menten constant for the actual tryptophan substrate increased (Km, Trp = 41.1 ± 6.5 µM) with no significant change in Vmax. Similarly, the V177A mutant did not exhibit a change in Km, BH4 (16.7 ± 2.5 µM) or in Km, Trp (31.6 ± 1.4 µM) with no significant changes in Vmax. Kinetic observations for V177G, V177S, V177T, V177L, V177F, V177R, and V177E were 95 unable to be determined as all of these mutants demonstrated little or no activitiy following purification (<5% of wild-type activity). 96 Discussion There is considerable similarity among the AAAH in terms of sequence identity, overall structure and catalytic reaction. Therefore, it has been suggested that the TPH active site is comprised of two distinct binding regions – one for tetrahydrobiopterin and one for tryptophan – and that TPH exhibits a comparable catalytic mechanism to the other AAAH. Namely, TPH would utilize an ordered terbi sequential binding order with tetrahydrobiopterin binding first followed by either oxygen or tryptophan (Fitzpatrick, 1991; Fitzpatrick, 1993; Andersen et al., 2002; Bassan et al., 2003; Volner et al., 2003). This mechanism, along with the hypothetical TPH models (Jiang et al., 2000; McKinney et al., 2001; Martinez et al., 2001) and TPH crystal structure (Wang et al., 2002), assist in interpreting the experimental data obtained with the V177 mutants. Specifically, our hypothetical model of TPH (Jiang et al., 2000), predictions based on NMR data (McKinney et al., 2001), and x-ray crystallography data (Wang et al., 2002) suggest a structural situation presented in Figure 1. Namely, that arginine 257 (R257) is actively engaged in orienting binding of the alpha-carboxyl of the tryptophan substrate, as in TH (Moran et al., 1998). While not directly involved in substrate binding, V177 is close enough to R257 (3-4 Å) to influence its orientation. Replacement of the valine residue with a slightly larger isoleucine residue mimics the coding region polymorphism of TPH (Ramaekers et al., 2001), and results in a decrease in Vmax (observed at lower fixed concentrations of tryptophan substrate) with no change in Km, BH4. However, this subtle mutation seems to hinder the binding and/or utilization of the tryptophan substrate as demonstrated by the 97 increase in Km, Trp with no change in Vmax. This change can be explained by the steric hindrance provided by the bulkier isoleucine residue to either the substrate itself or the position of the surrounding residues. Further support for this hypothesis is given by the data obtained from analysis of the valine to alanine substitution at residue 177 (V177A), which did not adversely affect the performance of the enzyme with respect to BH4 or tryptophan. However, a further reduction in side chain size and hydrophobicity, through construction of the V177G point mutant, is detrimental to enzymatic activity. In fact, this mutation nearly abolished enzyme activity. This can be explained by the lack of any steric hindrance, thus resulting in conformational flexibility that could dramatically perturb the size and shape of the active site pocket. Similarly, V177S and V177T, mutants exhibiting side chains similar in size to alanine and valine, respectively, but with hydrophilic properties, also exhibited little enzyme activity. Unexpectedly, the V177L mutant also had little enzyme activity. Our hypothesis was that this mutation would result in an enzyme that had activity with no change in Km, BH4, and an increase in the Km, Trp compared to V177I. Though typically considered a relatively conservative mutation, it is plausible that the addition of a methylene group could constrain the positioning of residue 177 and shift the orientation of surrounding residues (such as R257), thereby preventing optimal residue placement and decreasing normal enzyme function. In contrast, the lack of activity of the V177F mutant was expected as we hypothesized that the bulk and hydrophobicity of the phenylalanine side chain would perturb the local structure and/or proper binding of the tryptophan substrate to such a great extent that the 98 enzyme would not be able to function. Together, these mutagenesis experiments provide further support for a theory that this residue plays a critical, but indirect, role in the proper positioning of the tryptophan substrate. Replacement of the valine residue with charged residues, namely arginine and glutamate, resulted in point mutants that had no detectable activity. These results from V177R and V177E are not unexpected as these residues introduce a charge into the hydrophobic core of the enzyme, which could easily perturb local structure and thereby prevent proper binding and subsequent catalysis. Structural models of these mutants allowed for theoretical visualization of the residue at position 177 potentially interacting with the nearby arginine 257 residue (3-4Å). In the case of V177R, the close proximity of the side chains would result in Van der Waals repulsive forces, while a potential salt bridge could form between V177E and R257, all of which would perturb proper tryptophan substrate binding if R257 is indeed involved in coordinating the alpha carboxyl carbon of the amino acid substrate as in TH (Moran et al., 1998). Typically, a valine to isoleucine substitution is considered a subtle change. Our data indicate that this mutation at residue 177 (V177I) is not completely deleterious to TPH enzyme activity, and suggest that this polymorphism could exist in the population but remain undiagnosed as long as tryptophan substrate levels are high. Physiologically, such a mutation would exhibit itself only in the presence of an additional metabolism-related defect. This coincides with the findings of Ramaekers and colleagues, where only one out of the 5 patients that exhibited aberrant serotonin neurochemistry and the neurodevelopment problems possessed the TPH 99 polymorphism. Overall, our data give credence to the theory that the decrease in serotonin levels in these neurodevelopmentally-challenged patients results from a loss in serotonergic neurons rather than a decrease in serotonin levels due to a change in tryptophan hydroxylase activity. Complicating the explanation of the potential functional role of the V177I mutation is the recent discovery of a brain-specific isoform of TPH gene, Tph2 (Walther et al., 2003). This novel isoform, Tph2, encodes a TPH enzyme that predominates in the central nervous system, and resembles the TPH enzyme encoded by Tph1 (Boularand et al., 1990; Boularand et al., 1995), studied in Ramaekers et al. (2002), and reportedly to be located in the periphery (Walther et al., 2003). It is logical to assume that the neuronal-specific TPH (TPH2, Walther et al., 2003) plays the dominant role in the regulation of normal neurological development and function. Based on the fact that the V177I polymorphism was observed in only 1 out of 5 patients, coupled with the recent report that the TPH harboring this polymorphism is the peripheral enzyme, it is likely that V177I is coincidentally associated with the novel neurodevelopmental disorder. It is important to note, however, that the presence of this polymorphism highlighted a residue which has proven to be pivotally important to the structure of the active site. Without evidence of this polymorphism, there would have been no reason a priori to select valine 177 for mutational analysis. Fully understanding this neurodevelopmental disorder will have to await the characterization of TPH2 in these patients, as well as an understanding of its interrelationship with TPH1. It seems unlikely that a 100 polymorphism in the peripheral version of TPH would not play some role in a behavioral syndrome that is corrected with treatment with 5-hydroxytryptophan and carbidopa supplementation and is characterized by reduced serotonin metabolites. Acknowledgments We would like to thank Dr. Jacques Mallet (Centre National de la Recherche Scientifique, Paris, France) for the generous gift of human tryptophan hydroxylase (hTPH1). We would like to thank Dr. Linda C. McPhail, Dr. Mark O. Lively, Dr. Leslie B. Poole, and Dr. William E. Sonntag (Wake Forest University) for their technical input. This work was supported by PHS grant R01 GM-38931 (to K.E.V.). 101 Figure 1 – Structural depictions of Val177 mutations. A closeup view of the active site site pocket of the hypothetical model of human TPH is depicted (Jiang et al., 2000). Secondary structure is identified by color: β-sheets (yellow), α-helix (pink), and random coil (white). The locations of Tyr235, Arg257, Ser260, and Ser336 are indicated. A) The typical active site conformation and positioning of Val177; Predicted structural changes resulting from V177 substitutions are represented on the hypothetical TPH model in B) Val177Gly; C) Val177Ser; D) Val177Thr; E) Val177Ala; F) Val177Ile; G) Val177Leu; H) Val177Phe; I) Val177Arg; J) Val177Glu. These images were generated on a Silicon Graphics Workstation with the modeling program Sybyl 6.8 (Tripos Inc., St. Louis, MO). 102 A B C D E F G H I J 103 Table II - Purification data for wild-type and mutant enzymes Yield (µg) a Specific Activity b Fold purification c Wild-type 160 21.9 661 V177G 155 0.41 120 V177S 165 0.66 2.88 V177T 175 1.04 447 V177A 150 44.4 500 V177I 154 28.6 3920 V177L 180 0.99 102 V177F 171 ND - V177R 140 ND - V177E 106 ND - Protein a Yield of purified protein from 250ml of bacterial culture (average 25 mg total protein). b Units of specific activity were nmol 5-OH-Trp produced / min-mg. TPH enzymatic activity was determined (see Materials and Methods) using substrate concentrations of 100 µM BH4 and 50 µM tryptophan. c Fold purification was determined using specific activity values determined from enzyme activity in the crude bacterial extract and purified fraction. ND – No detectable activity. 104 Figure 2 - Coomassie and immunoblots of purified proteins. Human TPH was expressed in E. coli (BL21) as an amino-terminal oligohistidine fusion protein and purified to apparent homogeneity by affinity chromatography. 1 µg purified protein was subject to SDS-PAGE. (A) Colloidal Blue staining of purified TPH proteins. Lane L: Mark 12™ Unstained Standard; lane1: V177G; lane 2: V177S; lane 3: V177T; lane 4: V177A; lane 5: wild-type TPH; lane 6: V177I; lane 7: V177L; lane 8: V177F; lane 9: V177R; lane 10: V177E; lane 11: 1 µg BSA; lane 12: 5 µg BSA; lane 13: 10µg BSA. (B) Immunoblot of purified TPH proteins. Lane1: V177G; lane 2: V177S; lane 3: V177T; lane 4: V177A; lane 5: wild-type TPH; lane 6: V177I; lane 7: V177L; lane 8: V177F; lane 9: V177R; lane 10: V177E. Immunoreactive proteins were detected with a monoclonal antibody (WH3) at a 1:1000 dilution followed by an anti-mouse IgG-HRP from sheep at a 1:1500 dilution. Each protein migrated with an apparent molecular mass of 51 kDa. 105 106 Table III - Steady-state kinetic parameters of wild-type TPH and mutants Enzyme Km, BH4 (µM) Km, Trp (µM) Specific Activity Wild-type 15.8 ± 1.2 26.4 ± 3.3 21.9 V177A 16.7 ± 2.5 31.6 ± 1.4 44.4 V177I 14.8 ± 0.9 41.1 ± 6.5 * 28.6 V177G NR - 0.41 V177S NR - 0.66 V177T NR - 1.04 V177L - - 0.99 V177F - - 0.02 V177R - - - V177E - - - (nmol/min/mg) NR – Not Reliable. Activity levels were too low to provide accurate Km determinations, but values ranged from (17.5 µM to 55 µM). Cells denoted with “-“ provided no kinetic values. TPH activity was determined (see Materials and Methods) and the kinetic constants were calculated by direct fit to the Michaelis-Menten equation and non-linear regression analysis using GraphPad Prism version 3.03 (GraphPad Software, San Diego, CA, www.graphpad.com). Kinetic determinations used substrate concentrations at 100 µM BH4 (tryptophan variable) and 50 µM or 500 µM tryptophan (BH4 variable). The mean values for BH4 and tryptophan were compared independently of each other (n = 6 to 11). Specific activity was determined at substrate concentrations of 100 µM BH4 and 50 µM tryptophan. *One-way ANOVA, followed by a Student’s Newman-Keuls test at 95% confidence indicated that the kinetic differences observed were statistically significant (p = 0.004). 107 References Andersen O.A., Flatmark T., and Hough E. (2002) Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-L-alanine, and its implications for the mechanism of catalysis and substrate activation. J. Mol. Biol. 320, 10951108. Bassan A., Blomberg M.R., and Siegbahn P.E. (2003) Mechanism of dioxygen cleavage in tetrahydrobiopterin-dependent amino acid hydroxylases. Chemistry 9, 106-115. Beevers S.J., Knowles R.G., and Pogson C.I. 1983. A sensitive radiometric assay for tryptophan hydroxylase applicable to crude extracts. J. Neurochem. 40: 894-897. Boularand S., Darmon M.C., Ganem Y., Launay J.M., and Mallet J. (1990) Complete coding sequence of human tryptophan hydroxylase. Nucleic Acids Res. 18, 4257. Boularand S., Darmon M.C., and Mallet J. (1995) The human tryptophan hydroxylase gene. An unusual splicing complexity in the 5'-untranslated region. J. Biol. Chem. 270, 3748-56. Cornwell-Jones C.A., Decker M.W., Chang J.W., Cole B., Goltz K.M., Tran T., and McGaugh J.L. (1989) Neonatal 6-hydroxydopa, but not DSP-4, elevates brainstem monoamines and impairs inhibitory avoidance learning in developing rats. Brain Res. 493, 258-68. Daubner S.C., Lohse D.L., and Fitzpatrick P.F. (1993) Expression and characterization of catalytic and regulatory domains of rat tyrosine hydroxylase. Protein Sci. 2, 1452-1460. Darmon M.C., Guibert B., Leviel V., Ehret M., Maitre M., and Mallet J. (1988) Sequence of two mRNAs encoding active rat tryptophan hydroxylase. J. Neurochem. 51, 312-316. Delgado P.L. and Moreno F.A. (1998) Hallucinogens, serotonin and obsessive compulsive disorder. J. Psychoactive Drugs 30, 359-366. D’Sa C.M., Arthur R.E. Jr, Jennings I., Cotton R.G.H., and Kuhn D.M. (1996a) Tryptophan hydroxylase: Purification by affinity chromatography on calmodulin-Sepharose. J. Neurosci. Meth. 69, 149-153. 108 D’Sa C.M., Arthur R.E. Jr, and Kuhn D.M. (1996b) Expression and deletion mutagenesis of tryptophan hydroxylase fusion proteins: delineation of the enzyme catalytic core. J. Neurochem. 67, 917-926. Ellis H.R., Daubner S.C., and Fitzpatrick P.F. (2000) Mutation of serine 395 of tyrosine hydroxylase decouples oxygen-oxygen bond cleavage and tyrosine hydroxylation. Biochemistry. 39, 4174-4181. Erlandsen, H., Martinez A., Knappskog P.M., Haavik, J., Hough, E. and Flatmark, T. (1997a) Crystallization and preliminary diffraction analysis of a truncated homodimer of human phenylalanine hydroxylase. FEBS Letters. 406, 171174. Erlandsen, H., Fusetti F., Martinez A., Hough E., Flatmark, T. and Stevens R.C. (1997b) Crystal structure of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nat. Struct. Biol. 4, 995-1000. Erlandsen H., Flatmark T., Stevens R.C., and Hough E. (1998) Crystallographic analysis of the human phenylalanine hydroxylase catalytic domain with bound catechol inhibitors at 2.0A resolution. Biochem. 37, 15638-15646. Erlandsen H., Bjorgo E., Flatmark T., and Stevens R.C. (2000) Crystal structure and site-specific mutagenesis of pterin-bound human phenylalanine hydroxylase. Biochem. 39, 2208-2217. Erlandsen H., Kim J.Y., Patch M.G., Han A., Volner A., Abu-Omar M.M., and Stevens R.C. (2002) Structural comparison of bacterial and human irondependent phenylalanine hydroxylases: Similar fold, different stability and reaction rates. J. Mol. Biol. 320, 645-661. Fitzpatrick P.F. (1991) Steady-state kinetic mechanism of rat tyrosine hydroxylase. Biochem. 30, 3658-3662. Fitzpatrick P.F. (1993) Mechanistic studies of tyrosine hydroxylase. Adv. Exp. Med. Biol. 338, 81-86. Fitzpatrick P.F. (2000) The aromatic amino acid hydroxylases. Adv. Enzymol. Relat. Areas Mol. Biol. 74, 235-294. Fusetti F., Erlandsen H., Flatmark T., and Stevens R.C. (1998) Structure of tetrameric phenylalanine hydroxylase and its implications for phenylketonuria. J. Biol. Chem. 273, 16962-16967. Goodwill K.E., Sabatier C., Marks C., Raag R., Fitzpatrick P.F, and Stevens R.C. (1997) Crystal structure of tyrosine hydroxylase at 2.3 A and its implication for inherited neurodegenerative diseases. Nature Struct. Biol. 4, 578-585. 109 Goodwill K.E., Sabatier C., and Stevens R.C. (1998) Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3 A resolution: Selfhydroxylation of Phe300 and the pterin-binding site. Biochem. 37, 1343713445. Grenett H.E., Ledley F.D., Reed L.L., and Woo S.L.C. (1987) Full-length cDNA for rabbit tryptophan hydroxylase: Functional domains and evolution of aromatic amino acid hydroxylases. Proc. Natl. Acad. Sci. USA, 84, 5503-5534. Grima B., Lamouroux A., Blanot F., Biguet N.F., and Mallet J. (1985) Complete coding sequence of rat tyrosine hydroxylase cDNA. Proc. Natl. Acad. Sci. USA, 82, 617-621. Hamdan F.F., and Ribeiro P. (1999) Characterization of a stable form of tryptophan hydroxylase from the human parasite Schistosoma mansoni. J. Biol. Chem. 274, 21746-21754. Hufton S.E., Jennings I.G., and Cotton R.G.H. (1998) Structure/function analysis of the domains required for the multimerisation of phenylalanine hydroxylase. Biochim. Biophys. Acta., 1382, 295-304. Jiang G.C.-T., Yohrling IV G.J., Schmitt J.D., and Vrana K.E. (2000). Identification of substrate orienting and phosphorylation sites within tryptophan hydroxylase using homology-based molecular modeling. J. Mol. Biol. 302, 1005-1017. Kandasamy S.B. and Williams B.A. (1984) Hypothermic and anti-pyretic effects of ACTH (1-24) and alpha-melanotropin in guinea pigs. Neuropharmacology 23, 49-53. Knappskog P.M., Flatmark T., Mallet J., Ludecke B., and Bartholome K. (1995) Recessively inherited L-dopa-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Human Mol. Genet. 4, 12091212. Knappskog P.M., Flatmark T., Aarden J.M., Haavik J., and Martinez A. (1996) Structure/function relationships in human phenylalanine hydroxylase. Effect of terminal deletions on the oligomerization, activation and cooperativity of substrate binding to the enzyme. Eur. J. Biochem. 242, 813-821. Kobe B., Jennings I.G., House C.M., Feil S.C., Michell B.J., Tiganis T., Parker M.W., Cotton R.G.H., and Kemp B.E. (1997) Regulation and crystallization of phosphorylated and dephosphorylated forms of truncated dimeric phenylalanine hydroxylase. Protein Science 6, 1352-1357. 110 Kobe B., Jennings I.G., House C.M., Michell B.J., Goodwill K.E., Santarsiero B.D., Stevens R.C., Cotton R.G.H., and Kemp BE. (1999) Structural basis of auto regulation of phenylalanine hydroxylase. Nature Struct. Biol. 6, 442-448. Kuhn D.M., Ruskin B., and Lovenberg W. (1980) Tryptophan hydroxylase: the role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J. Biol. Chem. 255, 4137-4143. Kumer S.C., Mockus S.M., Rucker P.J., and Vrana K.E. (1997) Amino terminal deletion analysis of tryptophan hydroxylase: PKA phosphorylation occus at serine-58. J. Neurochem. 69,1738-1745. Kwok S.C., Ledley F.D., DiLella A.G., Robson K.J., and Woo S.L. (1985) Nucleotide sequence of a full-length complimentary DNA clone and amino acid sequence of human phenylalanine hydroxylase. Biochem. 24, 556-561. Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. Ledley F.D., DiLella A.G., Kwok C.M., and Woo S.L.C. (1985) Homology between phenylalanine hydroxylase and tyrosine hydroxylase reveals common structural and functional domains. Biochemistry, 24, 3389-3394. Liu X. and Vrana K.E. (1991) Leucine zippers and coiled-coils in the aromatic amino acid hydroxylases. Neurochem. Int. 18, 27-31. Lohse D.L., and Fitzpatrick P.F. (1993) Identification of the intersubunit binding region in rat tyrosine hydroxylase. Biochem. Biophys. Res. Commun. 197, 1543-1548. Ludecke B., Dworniczak B., and Bartholome K. (1995) A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum. Genet. 95, 123-125. Ludecke, B., Knappskog P.M., Clayton P.T., Surtees R.A.H., Clelland J.D., Heales S.J.R., Brand M.P., Bartholome K., and Flatmark T. (1996) Recessively inherited L-dope responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum. Mol. Gen. 5, 10231028. McKinney J., Teigen K., Froystein N.A., Salaun C., Knappskog P.M., Haavik J., and Martinez A. (2001) Conformation of the substrate and pterin cofactor bound to human tryptophan hydroxylase. Important role of Phe313 in substrate specificity. Biochemistry 40, 15591-601. 111 Mann J.J., Malone K.M., Nielsen D.A., Goldman D., Erdos J., and Gelernter J. (1997) Possible association of the tryptophan hydroxylase gene and suicidal behavior in depressed patients. Amer. J. Psych. 154, 1451-1453. Martinez A., Knappskog P.M., and Haavik J. (2001) A structural approach into human tryptophan hydroxylase and its implications for the regulation of serotonin biosyntheis. Curr. Med. Chem. 8, 1077-1091. Mockus S.M., Kumer S.C., and Vrana K.E. (1997a) Carboxyl terminal deletion analysis of tryptophan hydroxylase. Biochim. Biophys. Acta. 1342, 132-140. Mockus S.M., Kumer S.C., and Vrana K.E. (1997b) A chimeric tyrosine/tryptophan hydroxylase: The tyrosine hydroxylase regulatory domain serves to stabilize enzyme activity. J. Mol. Neurosci. 9, 35-48. Mockus S.M. and Vrana K.E. (1998) Advances in the molecular characterization of tryptophan hydroxylase. J. Mol. Neurosci. 10, 163-179. Moran G.R., Daubner S.C., and Fitzpatrick P.L. (1998) Expression and characterization of the catalytic core of tryptophan hydroxylase. J. Biol. Chem. 273, 12259-12266. Nielsen D.A., Dean M., and Goldman D. (1992) Genetic mapping of the human tryptophan hydroxylase gene on chromosome 11, using and intronic conformational polymorphism. Amer. J. Human Genet. 51, 1366-1371. Nielsen D.A., Goldman D., Virkkunen M., Tokola R., Rawlings R., and Linnoila M. (1994) Suicidality and 5-hydroxyindoleacetic acid concentration associated with a tryptophan hydroxylase polymorphism. Arch. Gen. Psych. 51, 34-38. Reinhard J.F., Smith G.K., and Nichol C.A. (1986) A rapid and sensitive assay for tyrosine-3-monooxygenase based upon the release of 3H2O and adsorption of [3H]-tyrosine by charcoal. Life. Sci. 39, 2185-2189. Schauenburg H. and Dressler D. (1992) Gilles de la Tourette Syndrome. Nervearzt 63, 453-461. Sindelar C.V., Hendsch Z.S., and Tidor B. (1998) Effects of salt bridges on protein structure and design. Protein Sci. 7, 1898-1914. Tabaddor K., Wolfson L.I., and Sharpless N.S. (1978) Ventricular fluid homovanillic acid and 5-hydroxyindolacetic acid concentrations in patients with movement disorders. Neurology 28, 1249-1253. 112 Volicer L., Direnfeld L.K., Freedman M., Albert M., Langlias P.J., Bird E.D. (1985) Serotonin and 5-hydroxyindolacetic acid in CSF. Difference in Parkinson’s disease and dementia of the Alzheimer’s type. Archives of Neurology 42, 127-29. Volner A., Zoidakis J., and Abu-Omar M.M. (2003) Order of substrate binding in bacterial phenylalanine hydroxylase and its mechanistic implication for pterindependent oxygenases. J. Biol. Inorg. Chem. 8, 121-128. Vrana S.L., Dworkin S.I., and Vrana K.E. (1993) Radioenzymatic assay for tryptophan hydroxylase: [3H2O] release assessed by charcoal adsorption. J. Neurosci. Meth. 48, 123-129. Vrana K.E., Rucker P., Kumer S.C. (1994a) Recombinant rabbit tryptophan hydroxylase is a substrate cAMP-dependent protein kinase. Life Sci. 55, 1045-1052. Vrana K.E., Walker S.J., Rucker P., and Liu X. (1994b) A carboxyl terminal leucine zipper is required for tyrosine hydroxylase tetramer formation. J. Neurochem. 63, 2014-2020. Walker S.J., Liu X., Roskoski R. Jr, and Vrana K.E. (1994) Catalytic core of rat tyrosine hydroxylase: Terminal deletion analysis of bacterially-expressed enzyme. Biochim. Biophys. Acta. 1206, 113-119. Walther D.J., Peter J.U., Bashammakh S., Hortnagl H., Voits M., Fink H., and Bader M. (2003) Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299, 76. Wang L., Erlandsen H., Haavik J., Knappskog P.M., and Stevens R.C. (2002). Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochem. 41, 12569-12574. Yohrling IV, G.J., Mockus S.M., and Vrana K.E. (1999) Identification of aminoterminal sequences contributing to tryptophan hydroxylase tetramer formation. J. Mol. Neurosci. 12, 23-34. CHAPTER IV SUMMARY AND CONCLUSIONS George C.-T. Jiang 113 114 Tryptophan hydroxylase (TPH) catalyzes the rate-limiting step in the formation of the neurotransmitter serotonin (5-HT, Grahame-Smith, 1964; Lovenberg et al., 1967), and also plays a role in the subsequent biosynthesis of melatonin (Chanut et al., 2002; Privat et al., 2002; reviewed in Martinez et al., 2001). TPH is localized predominantly within the raphe nuclei of the midbrain and is also peripherally located in the pineal gland, retina, and enteric neurons of the gut (Jequier et al., 1969; Joh et al., 1975; Austin and O’Donnell, 1999; reviewed in Mockus and Vrana, 1998). Recent studies reveal that this may be explained by the existence of two distinct isoforms of TPH that are differentially expressed in the CNS and periphery (Walther et al., 2003). Serotonin, and therefore TPH by inference, play important roles in a variety of physiological processes. Serotonin is implicated in a variety of CNS functions such as temperature control, aggression, pain and memory (Kandasamy and Williams, 1984; reviewed in Oliver et al., 1995; Brentegani and Lico, 1982; Cornwell-Jones et al., 1989). Aberrations in serotonin synthesis, regulation, and homeostasis have been associated with numerous psychiatric illnesses including depression, anxiety, obsessive-compulsive disorder (OCD), bulimia, migraines, and drug abuse (reviewed in Mockus and Vrana, 1998). The present work has encompassed the development of a hypothetical TPH model (Jiang et al., 2000), studies to validate the model and identify amino acid residues of importance (Jiang et al., 2000), and studies that address the significance of a TPH polymorphism found in one individual (Jiang et al., in preparation) (see Figure 1). The hypothetical model has served as an excellent tool to visualize structure and function relationships, and aided in mutagenesis studies, in lieu of a 115 TPH crystal structure that was only recently released (Wang et al., 2002). The recent publication and release of the TPH crystal structure definitively illustrates the molecular structure in the catalytic domain and further validates the hypothetical model, and the experimental studies conducted in this work. An “iterative magic fit” of the TPH crystal structure and hypothetical model using Swiss-PdbViewer (Guex and Peitsch, 1997) indicate a root mean square deviation (RMSD) of 1.02 Å between the carbon backbones of the structures. This demonstates the high degree of similarity between both structures considering rms deviations between models of homologous protein structures and real ones are typically between 2 and 4 Å, even in the best cases (Petsko, 2000). 116 Figure 1 – Structural location of the functional residues of interest in the present work. A view of the hypothetical model of human TPH is depicted (Jiang et al., 2000). Secondary structure is identified by color: β-sheets (yellow), α-helix (pink), and random coil (white). Residues of note include Ser58, Val177, Tyr235, Ser260, and Ser443. This image was generated on a Silicon Graphics Workstation with the modeling program Sybyl 6.8 (Tripos Inc., St. Louis, MO). 117 118 The present work has identified CaMPKII phosphorylation sites in TPH. The substrate amino acid residues include Ser58, Ser260, and Ser443. Mutagenesis studies with Ser58Ala and Ser260Ala mutants indicate that phosphorylation can occur at either residue and only minimally at Ser443. We surmise that Ser443 is only a potential CaMPKII phosphorylation site, and probably plays a minimal role in in vivo regulation of TPH as it is located in the extreme carboxy terminus tetramerization domain of the enzyme and would therefore normally be buried deep within the protein. The small amount of phosphorylation that was observed in the double point mutant (Ser58Ala/Ser260Ala) was, in all probability, a result of the presence of monomeric or denatured TPH in solution. The development of a hypothetical TPH structure has been useful in further understanding structure/function relationships in this important biosynthetic enzyme by assisting in the selection of amino acid residues for mutagenesis studies. Mutagenesis experiments and subsequent characterization have identified and confirmed the importance of Val177 and Tyr235 in the catalytic domain of TPH. Site-directed mutagenesis of Val177 allowed the correlation of functional data for a naturally-occurring coding region polymorphism associated with a developmentallyimpaired patient (Ramaekers et al., 2001). Our in vitro studies with recombinantlyexpressed human TPH indicate that Val177 plays a role in tryptophan substrate utilization and the identity of the residue is critical for proper enzyme structure and function (Jiang et al., in preparation). Similarly, mutagenesis studies on Tyr235 reveal that the residue plays an integral role in the BH4 binding pocket that 119 dramatically affects subsequent tryptophan hydroxylation (Jiang et al., 2000; McKinney et al., 2001; Wang et al., 2002; reviewed in Martinez et al., 2001). Recently, a genetic knock-out of the known TPH gene was created in mice (Walther et al., 2003). Interestingly, the knock-out mice (Tph-/-) still had normal serotonin (5-HT) levels in classically serotonergic brain regions in the central nervous system, while they lacked 5-HT in the periphery. This led the authors to identify a new isoform of TPH (Tph2) in mice, and subsequently in rat and human species. The knockout mice (Tph1-/-) exhibited no significant behavioral differences in elevated plus maze and hole board tests, which are indicative of 5-HT-related behaviors. These data from mice are different from the C. elegans TPH knockout that was previously generated (Sze et al., 2000). These TPH deletion mutant worms were viable, but did not synthesize serotonin, and exhibited alterations in their behavior and metabolism, including decreased feeding and egg laying rates, and an increase in fat storage (Sze et al., 2000). Comparisons between the amino acid sequences of Tph1, Tph2, and C. elegans Tph reveal that the bulk of the differences lie in the amino terminal regulatory domain, but do not identify the C. elegans TPH gene more closely with either of the mammalian genes. The recent identification of the new TPH isoform in vertebrates, which predominates in the central nervous system, raises interesting new questions regarding the regulation of serotonin biosynthesis in animals, and the possibility for potential intervention with novel pharmacotherapies specific for CNS TPH or peripheral TPH. This could have dramatic implications for future treatment of serotonergic disorders. One can envision using genetic therapies by engineering the 120 necessary TPH gene to either increase or decrease the protein’s inherent enzyme stability or perhaps its basal activity. The full-length human TPH model and crystal structure have been useful in filling some of the gaps in our knowledge of the structure, function, and regulation of TPH, and will continue to do so in the fuure. In conclusion, the AAAH are highly homologous proteins, but subtle nuances exist between the enzymes that determine substrate specificity, stability, and enzyme regulation. Though the short half-life of TPH and minute quantity available in a single animal have historically impeded progress on the research of TPH, these challenges are slowly being conquered. The recent advances in protein expression technologies has helped to overcome problems with enzyme purification, while the identification of a stable form of TPH in Schistosoma mansoni (Hamdan and Ribeiro, 1999) has provided new insights to TPH enzyme stability and regulation. Moreover, the development of a hypothetical TPH structure and the subsequent crystallization provide new tools to further expand the knowledge base regarding TPH structure and function. It is hoped that information from this thesis, and future studies using the contemporary tools and approaches available will assist in addressing the important issues and intricate details that remain about serotonin biosynthesis and TPH regulation in human health and disease. 121 REFERENCES Austin, M.C. and O’Donnell, S.M. (1999) Regional distribution and cellular expression of tryptophan hydroxylase messenger RNA in postmortem human brainstem and pineal gland. J. Neurochem. 72, 2065-2073. Brentegani, L.G. and Lico, M.C. (1982) Dental pain and the injection of agents into the trigeminal nerve of guinea pigs. Phys. and Behav. 28, 49-52. Chanut E., Nguyen-Legros J., Labarthe B., Trouvin J.H., and Versaux-Botteri C. (2002) Serotonin synthesis and its light-dark variation in the rat retina. J. Neurochem. 83, 863-869. Cornwell-Jones, C.A., Decker, M.W., Chang, J.W., Cole, B., Goltz, K.M., Tran, T,, and McGaugh, J.L. (1989) Neonatal 6-hydroxydopa, but not DSP-4, elevates brainstem monoamines and impairs inhibitory avoidance learning in developing rats. Brain Res. 493, 258-68. Grahame-Smith, D.G. (1964) Tryptophan hydroxylation in brain. Biochem Biophys Res Commun 16, 586-592. Guex, N. and Peitsch, M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18, 27142723. Hamdan, F.F., and Ribeiro P. (1999) Characterization of a stable form of tryptophan hydroxylase from the human parasite Schistosoma mansoni. J. Biol. Chem. 274, 21746-21754. Jequier E., Robinson D.S., Lovenberg W., and Sjoerdsma A. (1969) Further studies on tryptophan hydroxylase in rat brain stem and beef pineal. Biochem. Pharmacol. 18, 1071-1081. Jiang G.C.-T., Yohrling IV G.J., Schmitt J.D., and Vrana K.E. (2000). Identification of substrate orienting and phosphorylation sites within tryptophan hydroxylase using homology-based molecular modeling. J. Mol. Biol. 302, 1005-1017. Joh, T.H., Shikimi, T., Pickel, V.M., and Reis, D.J. (1975) Brain tryptophan hydroxylase: Purification of, production of antibodies to, and cellular and ultrastructural localization in serotonergic neurons of rat midbrain. Proc. Natl. Acad. Sci. 72, 3575-3579. 122 Kandasamy, S.B. and Williams, B.A. (1984) Hypothermic and anti-pyretic effects of ACTH (1-24) and alpha-melanotropin in guinea pigs. Neuropharmacology 23, 49-53. Lovenberg W., Jequier E., and Sjoerdsma A. (1967) Tryptophan hydroxylation: Measurement in pineal gland, brain stem and carcinoid tumor. Science 155, 217-219. Ludecke, B., Dworniczak, B., Bartholome, K. (1995) A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum. Genet. 95, 123-125. Martinez A., Knappskog P.M., and Haavik J. (2001) A structural approach into human tryptophan hydroxylase and its implications for the regulation of serotonin biosyntheis. Curr. Med. Chem. 8, 1077-1091. McKinney J., Teigen K., Froystein N.A., Salaun C., Knappskog P.M., Haavik J., and Martinez A. (2001) Conformation of the substrate and pterin cofactor bound to human tryptophan hydroxylase. Important role of Phe313 in substrate specificity. Biochemistry 40, 15591-601. Mockus S.M. and Vrana K.E. (1998) Advances in the molecular characterization of tryptophan hydroxylase. J. Mol. Neurosci. 10, 163-179. Olivier, B., Mos, J., van Oorschot, R., and Hen, R. (1995) Serotonin receptors and animal models of aggressive behavior. Pharmacopsych. 28 Suppl 2, 80-90. Petsko G.A. (2000) The Grail problem. Genome Biol. 1, comment002 Privat K., Brisson C., Jouvet A., Chesneau D., Ravault J.P., and Fevre-Montange M. (2002) Evidence for implication of tryptophan hydroxylase in the regulation of melatonin synthesis in ovine pinealocytes in culture. Cell. Mol. Neurobiol. 22, 417-29. Ramaekers V.T., Senderek J., Hausler M., Haring M., Abeling N., Zerres K., Bergmann C., Heimann G., and Blau N. (2001) A novel neurodevelopmental syndrome responsive to 5-hydroxytryptophan and carbidopa. Mol. Genet. Metab. 73,179-87. Sze J.Y., Victor M., Loer C., Shi Y., and Ruvkun G. (2000) Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature 403, 560-564. Walther D.J., Peter J.U., Bashammakh S., Hortnagl H., Voits M., Fink H., and Bader M. (2003) Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299, 76. 123 Wang L., Erlandsen H., Haavik J., Knappskog P.M., and Stevens R.C. (2002). Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochem. 41, 12569-12574. APPENDIX PHENYLKETONURIA George C-T. Jiang, George J. Yohrling IV, and Kent E. Vrana The following educational review article is in press for publication in the Encyclopedia of Life Sciences and is reprinted with permission from the Nature Publishing Group, www.els.net. Stylistic variations are due to the requirements of this journal. The manuscript was prepared and edited by George C.-T. Jiang. Authorship is shared with George J. Yohrling IV and Dr. Kent E. Vrana. George J. Yohrling IV wrote the initial draft of the Future Treatments for Phenylketonuria section. Dr. Kent E. Vrana served as a scientific advisor/editor for this project. 124 125 Introduction Metabolic disorders can adversely affect the body’s natural homeostatic or steady state and lead to chemical imbalances and severe pathological conditions. Phenylketonuria is such an example in which the normal conversion of the dietary amino acid phenylalanine to tyrosine is blocked. The resulting build-up of phenylalanine and its metabolites in young patients produces a number of severe side effects including intellectual impairment and cutaneous changes. This article reviews the autosomal recessive metabolic disorder of phenylketonuria, covering clinical diagnoses, typical and atypical causes, as well as current and potential treatments. Phenylketonuria, also known as PKU, is a disorder that affects 1 in 12,000– 15,000 births. In most cases, the cause is a deficiency in the enzyme phenylalanine hydroxylase (PH). This genetic condition results in numerous complications due to the incomplete metabolism of the essential amino acid phenylalanine. Clinical manifestations, if the disease is left untreated, include developmental delay and severe intellectual impairment, seizures, autism, eczema, hyperactivity, aggressive behaviour and scleroderma-like skin changes with dilution of hair and skin colour. PKU is implicated in about 0.64% of institutionalized patients worldwide. Early diagnosis after birth is critical and treatment with a phenylalanine-restricted diet will prevent onset of intellectual impairment and neurological abnormalities. Routine paediatric screening for PKU began in the United States in 1961, and the success of screening has spurred the development of similar procedures for other genetic metabolic disorders. 126 Clinical Signs and Conditions PKU is caused by a deficiency in the enzyme PH or reduction in the synthesis or regeneration of a critical co-substrate for the enzyme. In patients afflicted with PKU, this results in a build-up of phenylalanine, as it is not converted to tyrosine. Collectively, the various forms of PKU are also referred to as hyperphenylalaninemia owing to the increased circulating levels of the amino acid. The excess phenylalanine is metabolized to abnormal breakdown products (the phenylketones) accounting for the common name of the disorder, phenylketonuria. Increased levels of phenylalanine and its metabolites cause irreparable damage to the developing central nervous system unless the condition is diagnosed and treated within the first 3-6 weeks of life. Furthermore, this biochemical defect can result in a variety of cutaneous abnormalities, including diffuse hypopigmentation, eczema, and photosensitivity. These cutaneous changes may result from the toxic effects of phenylalanine and its decomposition products in the skin (Figure 1). 128 129 Normally, the essential amino acid phenylalanine is converted to tyrosine by the enzyme PH. The resulting tyrosine can then be converted, via independent pathways, to catecholamines (the neurotransmitters dopamine, noradrenaline (norepinephrine) and adrenaline (epinephrine)), melanin, or thyroid hormone. In addition, tyrosine can be incorporated, as a fundamental building block, into new proteins. Because tyrosine is itself a nonessential amino acid, PKU is not fatal since a diet high in tyrosine can make up for the lack of PH activity. Reduced synthesis of the cosubstrate tetrahydrobiopterin, or a deficiency in the regenerating enzyme dihydropteridine reductase may also result in phenylketonuria. However, such cases are rarer and are referred to as non-classical PKU. With the aid of routine paediatric screening, a diagnosis of PKU is usually established soon after birth. The newborn screening test that is performed involves the measurement of blood phenylalanine levels by the Guthrie test, a bacterial inhibition assay. Normally, blood phenylalanine levels are below 2mg/dL-1. A blood phenylalanine concentration of greater than 20mg/dL-1 with a normal or reduced concentration of tyrosine is diagnostic of classical PKU. For the confirmation of PKU, all amino acid levels must be measured by a quantitative technique and screening programmes often request repeat specimens in any infant with a blood phenylalanine level greater than normal. The essential neurological symptoms are intellectual impairment, which may cause corresponding changes in electroencephalogram, disturbed gait, ataxia, extrapyramidal symptoms, focalized or generalized epileptiform seizures. In general, if treatment of PKU is instituted by 3 weeks of age, prevention of intellectual 130 impairment and neurologic abnormalities is seen. However, learning disabilities have been reported in even well-treated patients. Today, it is recommended that patients with PKU continue a phenylalanine-restricted diet indefinitely because of several reports of decreased intellectual ability in children who were taken off the diet at 5 or 6 years of age. Continuation of the diet also aids in prevention of complications that are seen in genetically normal infants of women with PKU (maternal PKU). These complications include microcephaly, congenital heart disease and intrauterine growth delay. In patients with PKU, cutaneous changes can result from the toxic effects of phenylalanine and its decomposition products in the skin. Changes include reduction of skin, hair, and eye colour with increased photosensitivity, although photosensitivity is not universal. Dermatitis is also seen in up to 50% of patients. This dermatitis is virtually indistinguishable from the true atopic dermatitis, a chronic skin condition characterised by red itchy skin. Some are indeed true cases, but others only mimic such lesions. Hair and skin darkening and other skin change reversals may occur with dietary restriction of phenylalanine intake. However, when a diet with normal levels of phenylalanine is resumed, facial eczema may reappear within 24 h. The current treatment for patients afflicted with PKU is a strict dietary regimen that is restricted in phenylalanine intake yet provides all the other amino acids. The earlier the treatment is started, the more beneficial the effects. While on dietary treatment, patients with PKU need to be wary of products that contain the artificial sweetener aspartame (Nutrasweet). Aspartame, chemically known as N- 131 aspartylphenylalanine methyl ester, is commonly used in sweetened drinks, and the amount in a litre of sweetened drink can be equivalent to the amount of phenylalanine normally obtained from the daily diet. Uninformed patients may therefore be at risk for contradicting their phenylalanine-reduced diet. 132 Classical Phenylketonuria Typically, the term classical PKU refers to hyperphenylalaninemias resulting from a deficient PH enzyme. If left untreated, classical PKU can lead to severe intellectual impairment. As stated previously, PH is the mixed function oxidase which, in its central reaction mechanism, converts the essential amino acid Lphenylalanine to L-tyrosine in the presence of the co-substrates tetrahydrobiopterin and molecular oxygen. The recognition that PH was the cause of the hyperphenylalaninemia was not made until the late 1940s, over 20 years after classical PKU was initially characterised by Asbjörn Fölling in 1934. It is important to note that PKU does not necessarily result from a lack of the entire PH gene. Assays to detect the PH protein show that patients with PKU can produce the enzyme and kinetic studies provide evidence for enzymatic deficiencies. The human PH gene was first cloned in 1985; since then many researchers have studied it to determine the molecular basis for PKU. The PH gene comprises 13 exons and spans approximately 90 kilobases on the human chromosomes. Many different defects in the PH gene have been observed and reported for patients with PKU. These polymorphisms include single nucleotide base changes or point mutations, deletions, splice variants, premature stop signals and insertions. Most of the identified polymorphisms are single nucleotide changes that result in a protein with one amino acid substituted by another. These missense mutations constitute approximately 60% of all the known mutations that have been characterized. Splicing mutations and deletions each constitute about 13% of all the mutations. In these cases, a mutation results in defective ribonucleic acid processing (splicing). 133 This can produce an absence of PH or a defective form of the enzyme. Nonsense mutations (inappropriate protein synthesis stop signals) and insertions make up the remainder of the polymorphisms. The majority of the mutations (approximately 80%) lie in the central catalytic domain of the protein between exons 5 and 12, but mutations have been seen in all the different exons. Table 1 lists the various causes of PKU. 134 Table 1 Genetic defects and substrate deficiencies in phenylketonuria. 135 136 The frequencies at which different populations exhibit distinct polymorphisms vary greatly. Caucasians tend to have a mutation in the splice site of intron 12 where an adenosine is substituted for a guanosine (G→A), resulting in a truncated form of PH that is missing the last 52 amino acids. Asians, on the other hand, do not have such a general genotype. Based on these types of geographic localizations, it is clear that PKU has evolved from numerous independent mutations. 137 Nonclassical Phenylketonuria In addition to the classical forms of PKU involving mutations in the PH gene, there are a number of examples of the disease (1-3% of the PKU cases) where the underlying defect is in other systems. Notably, these involve mutations in the dihydropteridine reductase (DHPR), or in the biosynthetic enzymes responsible for biopterin synthesis (see Figure 1). For this reason, neonatal patients exhibiting hyperphenylalaninemia must also be tested for urinary pteridines, blood DHPR, and additional biogenic amines. tetrahydrobiopterin (BH4) This last test is necessary because defects in synthesis or regeneration (from quinonoid dihydrobiopterin; q-BH2) will affect other key biosynthetic enzymes, including tyrosine hydroxylase (rate-limiting enzyme in dopamine, noradrenaline and adrenaline biosynthesis) and tryptophan hydroxylase (rate-limiting enzyme in serotonin biosynthesis). Unfortunately, these patients are subject to a much more severe prognosis than those with classical PKU. Disruption of these other neurotransmitter systems produces mental health problems and motor problems (similar to Parkinson disease). Infants with nonclassical PKU (pterin cosubstrate deficiencies) will require additional therapeutic interventions. First, they require limitations in dietary phenylalanine to prevent the build-up of the amino acid and its phenylketone metabolites. However, this will further exacerbate the deficiency in the catecholamine neurotransmitters (dopamine, noradrenaline and adrenaline). Therefore, patients with biopterin defects are also treated with precursors to the catecholamines that bypass the tyrosine hydroxylase step (L-dopamine; the same 139 Future Treatments for Phenylketonuria As stated above, the current therapeutic regimen for the majority of the patients with PKU is the dietary restriction of the amino acid, phenylalanine. This can reduce the blood levels of phenylalanine and help prevent the severe intellectual impairment common with this disease. Regardless of the successes with this intervention, there are problems with this approach. Dietary restriction is often difficult and lends itself to non-compliance. This is particularly true in those cases where dietary phenylalanine restriction is recommended for life. It has been reported, for instance, that a decline in intellectual function and behavioural performance is evident in adults with PKU who have not maintained a low phenylalanine diet. Dietary management is particularly important in pregnant women with classical PKU who have previously curtailed phenylalanine restriction. Dietary noncompliance during pregnancy can produce PKU-like symptoms in their genetically unaffected (heterozygous) offspring. This syndrome is referred to as “maternal PKU”. In addition to the intellectual impairments of their offspring, low birth weight and congenital heart disease are often present. To prevent this, dietary treatment must be commenced before the pregnancy even begins, and rigidly maintained for the duration of the pregnancy. While dietary treatment for PKU is effective and non-hazardous, additional developments in the field are needed in order to find a “cure” for PKU. There are several different mechanisms that are amenable to clinical intervention. For example, drugs could be designed that would prevent phenylalanine absorption from 140 the gut. This would cause ingested phenylalanine to be excreted and may prevent its damaging effects. Enzyme therapies may also be utilized. Insertion of active PH into a “fatty” liposome carrier or administration of regular enzyme injections may serve potential roles in the treatment of PKU. Additional attention needs to be paid to PH, the primary enzyme in the metabolism of phenylalanine. As stated above, PH hydroxylates phenylalanine in the presence of the necessary substrates to form the amino acid tyrosine. Numerous defects in the gene encoding PH lead to an inactive enzyme and the systemic build-up of phenylalanine in the body. The recent X-ray crystallographic analysis of PH structure has supplied valuable information regarding the structure of the protein. These data may assist researchers in the design of pharmacotherapeutic interventions to treat PKU by accelerating the metabolism of phenylalanine to tyrosine. Further structural and functional studies, such as these, are necessary if we hope to eradicate PKU completely. Recently, researchers using rodent models found that use of oral enzyme therapy, with the enzyme phenylalanine ammonia lyase (PAL), combined with a low protein diet has the potential to replace the traditional phenylalanine-restricted dietary regimen. PAL is an enzyme that does not require a cofactor to convert phenylalanine to trans-cinnamic acid and insignificant amounts of ammonia. Transcinnamic acid is a harmless metabolite that is further converted to benzoic acid and then to hippurate which is excreted in urine. In theory, patients on PAL could break down phenylalanine such that phenylalanine ingestion could be tolerated. In rodent models, it was found that oral administration of PAL attenuated PKU. When 141 humans were used as test subjects, similar results were observed. However, only limited data are available from the studies because of the costs necessary to obtain sufficient amounts of PAL. Further research must be performed before the general population can use PAL, but these studies indicate that oral administration of PAL appears to have great potential as a future treatment for PKU. The costs of acquiring PAL must decrease however. Research indicates that PKU is primarily the result of single gene malfunctions. Therefore, a promising alternative to dietary restriction of phenylalanine in the management of PKU is somatic gene therapy. This would involve the insertion of a normal PH gene into a chromosomal location in the nucleus of liver cells (hepatocytes). The normal PH would then be expressed in place of, or in addition to, the mutated PH. Hepatocytes would be the appropriate targets for gene therapy since PH is known to be expressed predominantly within the liver. In order to get the normal gene to its proper target, the deoxyribonucleic acid (DNA) needs to be inserted into a vector or “carrier” system. To date, the use of three different vector systems has been explored. They include recombinant retroviral vectors, recombinant adenoviral vectors, and DNA-protein complexes. Numerous studies have shown these gene carriers to be successful in lowering blood phenylalanine levels. However, all have serious limitations, such as low transduction efficiencies and the inability to integrate into non-dividing cells. Additionally, these vectors may eventually lead to immune responses, or become inactive, which limits long-term efficacy. All of these limitations must first be 142 overcome if genetic therapy for PKU is to become a reality. 143 Summary Phenylketonuria (hyperphenylalaninemia) is one of the more common genetic disorders of intermediary metabolism. Disruption of the normal metabolic conversion of phenylalanine to tyrosine results in increased circulating levels of phenylalanine and its metabolic breakdown products. In the developing neonate, these metabolic imbalances produce dramatic clinical problems including severe intellectual impairment. In most cases, the genetic culprit in this problem is a defect in the biosynthetic enzyme PH. In a minority of the cases, there is an alternative problem in the biosynthesis or regeneration of a pivotal co-substrate of the PH reaction (BH4). Luckily, if properly diagnosed soon after birth, PKU can be managed through dietary interventions that decrease the levels of circulating phenylalanine. Knowing the molecular biology of the disorder provides a long-term hope of “curing” the disease through gene replacement strategies. These approaches may well become routinely available in the future. 144 Further Reading Bremer, H.J., Duran, D., Kamerling, J.P., Przyrembel, H., Wadman, S.K. (1981) Clinical Chemistry and Diagnosis of Inherited Diseases. Disturbances of Amino Acid Metabolism: Clinical Chemistry and Diagnosis. pp. 307-327. Baltimore: Urban & Scwharzenberg. [Chapter on clinical chemistry and diagnosis of inherited diseases.] Eisensmith R.C. and Woo, S.L.C. (1995) Molecular Genetics of Phenylketonuria: From Molecular Anthropology to Gene Therapy. Advances in Genetics 32, 199-271. Hoang L., Byck S., Prevost L., Scriver C.R. (1996) PAH Mutation Analysis Consortium Database: a database for disease-producing and other allelic variation at the human PAH locus. Nucleic Acids Research, 24(1):127-131. Kaufman, S. (1997) Tetrahydrobiopterin. pp. 262-322. Baltimore: John Hopkins University Press. [Chapter on phenylketonuria and its variants.] Nowacki P.M., Byck S., Prevost L., Scriver C.R. (1997) The PAH Mutation Analysis Consortium Database Update 1996. Nucleic Acids Research 25(1):139-142. Wurtman, R. and Ritter-Walker, E. (1988) Dietary Phenylalanine and Brain Function. Boston, MA: Birkhäuser. CURRICULUM VITA George Chih-Thai Jiang BORN: June 12, 1978 Bangkok, Thailand EDUCATION: Wake Forest University School of Medicine Winston-Salem, North Carolina M.S., Molecular Genetics, 2003 Wake Forest University Winston-Salem, North Carolina M.B.A., 2003 University of Maryland, College Park College Park, Maryland B.S., General Biology, 1998 SCHOLASTIC AND PROFESSIONAL EXPERIENCE: Howard Hughes Undergraduate Research Fellow, Department of Microbiology and Imuunology, University of Maryland, College Park, College Park, MD, 1997-1998. Advisor: Dr. Elisabeth Gantt Research Assistant, Department of Plant Biology, University of Maryland, College Park, College Park, MD, 1994-1997. Advisor: Dr. Elisabeth Gantt Research Assistant, Department of Gastroenterology, Walter Reed Army Institute of Research, Washington, D.C., 1994-1995. Advisor: Dr Yuan-Heng Tai 145 146 HONORS and AWARDS: Dean’s Fellowship, Wake Forest University Graduate School of Arts and Sciences, 1998-1999. Bacock Scholarship, Wake Forest University Babcock School of Management, 2001-2002. 2nd Place Graduate Student Poster Award, Western North Carolina Society for Neuroscience, 2001. PROFESSIONAL APPOINTMENTS AND ACTIVITIES: Graduate School Activities Volunteer, Community Public Affairs Committee, 1999-present Representative, Graduate Student Association, 1999present President, Graduate Student Association, 2001-2002 Member, Wake Forest University Graduate Council, 2001-2002 Graduate Teaching Experience Laboratory Instructor – Recombinant DNA Technologies, 1999, 2000 Course Coordinator – Drug Discovery and Development, 2002 Ad hoc reviewer: Journal of Biological Chemistry Journal of Genetics and Development Journal of Neurochemistry Nature Structural Biology Proceedings of the National Academy of Sciences Structure PROFESSIONAL MEMBERSHIPS: Society for Neuroscience, 1999-present. Western North Carolina Society for Neuroscience, 2000-present. American Society for Biochemistry and Molecular Biology, 2000-present. National Association of Graduate Professional Students, 2001-present. 147 BIBLIOGRAPHY: JOURNAL ARTICLES: Yohrling IV GJ, Jiang GC-T, Mockus SM, and Vrana KE (2000). Intersubunit binding domains within tyrosine hydroxylase and tryptophan hydroxylase. J. Neurosci. Res. 61(3): 313-320. Jiang GC-T, Yohrling IV GJ, Schmitt JD, and Vrana KE (2000). Identification of substrate orienting and phosphorylation sites within tryptophan hydroxylase using homology-based molecular modeling. J. Mol. Biol. 302(4): 1005-1017. Blanchard-Fillion B, Souza JM, Friel T, Jiang GC-T, Vrana KE, Sharov V, Barón L, Schöneich C, Quijano C, Alvarez B, Radi R, Przedborski S, Fernando GS, Horwitz J, and Ischiropoulos H (2001). Nitration and Inactivation of Tyrosine Hydroxylase by Peroxynitrite. J. Biol. Chem. 276: 46017-46023. Yohrling IV GJ, Jiang GC-T, DeJohn MM, Robertson DJ, Vrana KE, Cha JH-J (2002). Inhibition of Tryptophan Hydroxylase Activity and Decreased 5-HT1A Receptor Binding in a Mouse Model of Huntington’s Disease. J. Neurochem. 82:1416-1423. Stredrick DL, Jiang GC-T, Suber L, Mockus S, Aschner M, and Vrana KE. Manganese-Mediated Inhibition of Tyrosine Hydroxylase in CATH.a Cells. Mol. Brain Res. (in revision). Yohrling IV GJ, Jiang GC-T, DeJohn MM, Miller DW, Young AB, Vrana KE, Cha JHJ. Analysis of Cellular, Transgenic and Human Models of Huntington’s Disease Reveals Tyrosine Hydroxylase Alterations in Substantia Nigra Neuropathology. J. Neurosci. (submitted). Jiang GC-T, Domer AC, and Vrana KE. Functional Analysis of the V177I Coding Region Polymorphism in Human Tryptophan Hydroxylase (TPH1). (in preparation). BOOK CHAPTERS: Jiang GC-T, Yohrling IV GJ, and Vrana KE. (March 2000). Phenylketonuria. In: Encyclopedia of Life Sciences. Vol. 14, pp. 150-154. London: NatureScientific American Publishing Group. http://www.els.net (invited review). 148 Yohrling IV GJ, Jiang GC-T, and Vrana KE. (August 2000). Control of Enzymatic Activity. In: Encyclopedia of Life Sciences. London: Nature-Scientific American Publishing Group. http://www.els.net (invited review). ABSTRACTS: Yohrling IV GJ, Jiang GC-T, Mockus SM, and Vrana KE. Site-directed mutagenesis of tryptophan hydroxylase reveals K111-E223 to be an intersubunit dimerization bridge. Soc. Neurosci. Abst. 25: 178, 1999. Jiang GC-T, Yohrling IV GJ, Schmitt JD, and Vrana KE. Molecular modeling of human tryptophan hydroxylase: Implications for subunit assembly and drug development. FASEB J. 14(8): A1453, 2000. Yohrling IV GJ, Jiang, GC-T, Schmitt, JD, and Vrana KE. Identification of a tryptophan orienting residue in the active site of tryptophan hydroxylase. Soc. Neurosci. Abst. 26: 2000. Robertson DJ, Allen JW, Freeman WM, Jiang GC-T, Walker SJ, Hyyta P, Kiianmaa K, Sampson HH and Vrana KE. Changes in VTA gene expression following ethanol self-administration in AA/ANA rats. RSA Abst. 2000. Yohrling IV GJ, Jiang GC-T, Schmitt JD, and Vrana KE. Identification of a tryptophan orienting residue in the active site of tryptophan hydroxylase. Soc. Neurosci. Abst. 26:400, 2000. Jiang GC-T, Freeman WM, Vrana KE, Caron MG, and Jones SR. Functional neurogenomics and biochemical analyses of enzymatic differences in dopamine transporter knockout mice. Soc. Neurosci. Abst. 27, 2001. Yohrling IV GJ, Jiang GC-T, Robertson DJ, Vrana KE, and Cha J-HJ. Biochemical Analyses of Tyrosine and Tryptophan Hydroxylase in the R6/2 Mouse Model of Huntington’s Disease. Soc. Neurosci. Abst. 27, 2001. Jiang GC-T, Yohrling IV GJ, Miller DW, DeJohn MM, Vrana KE, and Cha J-HJ. Tyrosine hydroxylase activity and mRNA changes in Huntington’s disease. Soc. Neurosci. Abst. 28, 2002.