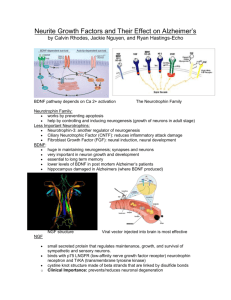
The Trk A, B, C`s of Neurotrophins in the Cochlea
advertisement

THE ANATOMICAL RECORD 295:1877–1895 (2012) The Trk A, B, C’s of Neurotrophins in the Cochlea STEVEN H. GREEN,1,2* ERIN BAILEY,1 QIONG WANG,1 AND ROBIN L. DAVIS3 1 Department of Biology, University of Iowa, Iowa City, Iowa 2 Department of Otolaryngology, University of Iowa, Iowa City, Iowa 3 Department of Cell Biology and Neuroscience, Rutgers University, Piscataway, New Jersey ABSTRACT The spiral ganglion neurons (SGNs) are the afferent neurons of the cochlea, connecting the auditory sensory cells—hair cells—to the brainstem cochlear nuclei. The neurotrophins neurotrophin-3 (NT-3) and brainderived neurotrophic factor (BDNF) are expressed in the cochlea and both support SGN survival during development. These neurotrophins remain expressed in the postnatal cochlea and continue to play additional roles for SGNs, contributing to maintenance of hair cell-SGN synapses and regulating expression of ion channels, presynaptic and postsynaptic proteins, and SGN membrane electrical properties in a physiologically important spatial pattern. Remarkably, NT-3 and BDNF have different, even opposing, effects on SGN physiology despite the close similarity of their receptors TrkB and TrkC. Recent studies have also raised the possibility that precursor proneurotrophin forms of the neurotrophins play a role in responses to trauma in the cochlea, signaling through the proneurotrophin receptor p75NTR. Here, we review expression and function of neurotrophins and their p75NTR and Trk-family receptors in the cochlea. We focus, in particular, on neurotrophin functions other than support of SGN survival, including regulation of SGN neurite growth, synaptic and membrane physiology. These functions, unlike survival, are ones for which BDNF and NT-3 substantially differ in their effects. Signal transduction mechanisms of p75NTR and of Trk-family receptors are discussed, indicating how these lead to different responses, and we speculate on how BDNF and NT-3 can cause different phenotypic changes in SGNs. Because these complex signaling interactions remain incompletely understood, use of neurotrophins as therapeutic agents in the cochlea should be approached with caution. Anat C 2012 Wiley Periodicals, Inc. Rec, 295:1877–1895, 2012. V Key words: neurotrophins; spiral ganglion neurons; phenotypic changes; neurotrophic factors; signal transduction; neurotrophin receptors; expression patterns; hearing; cochlea INTRODUCTION During neuronal development, neurotrophic factors (NTFs) play an essential role in establishment of neuron number through their control of cell survival/death (Huang and Reichardt, 2001). This is certainly the case in the developing peripheral auditory system: the cochlea. The afferent neurons of the cochlea, termed spiral ganglion neurons (SGNs), require neurotrophic support for their survival during development. This subject has been extensively reviewed (Fritzsch et al., 2004), C 2012 WILEY PERIODICALS, INC. V Grant sponsor: NIH/NIDCD; Grant numbers: RO1 DC01856 (to R.L.D.), R01 DC002961 (to S.H.G.), R01 DC009405 (to S.H.G.), P30 DC010362, F31 DC011680 (to E.B.); Grant sponsor: American Hearing Research Foundation (to Q.W.). *Correspondence to: Steven Green, Department of Biology, University of Iowa, 143 Biology Building, Iowa City, IA 52242-1324. E-mail: steven-green@uiowa.edu Received 24 July 2012; Accepted 24 July 2012. DOI 10.1002/ar.22587 Published online 8 October 2012 in Wiley Online Library (wileyonlinelibrary.com). 1878 GREEN ET AL. particularly with regard to one family of NTFs, the neurotrophins, and their receptors. Here, we review roles for neurotrophins other than control of developmental programmed cell death, with a focus on the postnatal cochlea and distinct functions of BDNF and NT-3. THE SENSORINEURAL CELLS OF THE COCHLEA The anatomy and physiology of the cochlea have been described elsewhere in detail (Slepecky, 1996) and is summarized here and in Fig. 1. The cochlea is structured so that tuning to sound frequency is graded from the cochlea base, which responds to the tones at the high end of the organism’s frequency range, to the apex, which responds to the lowest tones of the frequency range. This defines a ‘‘tonotopic axis’’ of the cochlea. The middle of the cochlea responds to midrange tones and is also the region with the lowest threshold (highest sensitivity) for sound. This physiological gradient along the cochlea largely results from base to apex changes in physical properties (e.g., width and stiffness) of the ‘‘basilar membrane,’’ that is, the structure that conducts sound vibrations in the cochlea. They are also reflected in physiological characteristics of the neural elements, as has been previously reviewed (Davis and Liu, 2011). The sensory elements of the cochlea reside in the ‘‘organ of Corti,’’ a long narrow structure lying on the basilar membrane that extends from the base of the cochlea to its apex. Parallel to the length of the organ of Corti are one row of inner hair cells (IHCs) and three rows of outer hair cells (OHCs), with the fluid-filled tunnel of Corti between the IHCs and OHCs. In the rat cochlea, there are 1,000 IHCs and 3,400 OHCs (Keithley and Feldman, 1982). The cochlear hair cells are the auditory sensory cells. The OHCs act primarily, as mechanical amplifiers and the IHCs are the primary sensory transducers. Correspondingly, the IHCs are densely innervated by SGNs and the OHCs only sparsely innervated. In addition to the hair cells, the organ of Corti contains well-defined rows of stereotyped specialized ‘‘supporting cells,’’ which are essential to the structure and function of the organ. Bordering the row of IHCs on the inner or modiolar side of the organ of Corti is a row of ‘‘border cells’’; on the other side of the IHC row is a row of ‘‘inner pillar cells,’’ which are also the inner side of the tunnel of Corti. The OHCs are bordered on the modiolar side by the ‘‘outer pillar cells,’’ which are also the outer side of the tunnel of Corti; on the outer side of the OHC rows are ‘‘Hensen’s cells.’’ Directly associated with these hair cells are the ‘‘inner and outer phalangeal cells’’ (the latter usually called ‘‘Deiter’s cells’’), with a row of inner phalangeal cells in contact with the IHC and three rows of Deiter’s cells supporting the OHCs. The spiral ganglion extends along the organ of Corti and is modiolar to it. The SGNs are bipolar neurons. Their central axons enter the modiolus (the central core of the cochlear spiral) and collect to form the auditory portion of the VIIIth nerve that projects to the cochlear nuclei in the brainstem. The peripheral axons of the SGNs extend radially to the organ of Corti. The type I SGNs that innervate the IHCs comprise 95% of the total SGN population. Each type I SGN makes a synaptic contact with just a single IHC. Because there are >18,000 SGNs in the rat (Rueda et al., 1987), this means that each IHC is presynaptic to many type I SGNs. In the rat and mouse, there are 20 afferent synapses per IHC in the middle of the cochlea, the region of lowest auditory threshold (greatest sensitivity), but the number of synapses/IHC declines toward the apex and base (Francis et al., 2004, 2006; Kujawa and Liberman, 2009; Meyer et al., 2009). The synapses are excitatory and glutamatergic. Type I SGNs are myelinated except at the distal end of the peripheral axon near the synapse with the hair cell. The type II SGN axons, which are entirely unmyelinated, extend past the IHC row and then turn in a basal direction to run parallel to an OHC row and make en passant synapses with many OHCs. NTFs AND THEIR RECEPTORS The neurotrophin family (Fig. 2) has four members, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin4/5 (NT-4/5; not shown in the figure). These are encoded, respectively, by the Ngf, Bdnf, Ntf3, and Ntf4 genes in the rat; Ngf, Bdnf, Ntf3, and Ntf5 genes in the mouse; NGF, BDNF, NTF3, and NTF4 genes in human genomic nomenclature. Neurotrophins assemble into tightly associated dimers, a structural feature crucial for their function, as will be discussed later. As is the case for other peptide growth factors and hormones, the neurotrophins are proteolytically cleaved from larger precursors. Neurotrophins are distinctive in that the proneurotrophin precursors are released and bind to their own receptor providing a signaling pathway divergent from that of the processed neurotrophins. Conversion from proneurotrophins to mature neurotrophins can be carried out after release by proteases in the extracellular matrix, and this can happen in an activitydependent manner (Nagappan et al., 2009; Yang et al., 2009). Another distinctive aspect of neurotrophin signaling is that release of neurotrophins can occur constitutively, as is typical for peptide growth factors and hormones or can occur in an activity-dependent manner (Kohara et al., 2001). Thus, regulation of neurotrophin signaling can occur at any or all the levels of transcription, translation, intracellular trafficking, intracellular proteolytic processing, secretion, and extracellular processing. As summarized in Fig. 2, neurotrophins and proneurotrophins signal through two different types of receptors (Reichardt, 2006). Trk family receptor protein-tyrosine kinases are high-affinity receptors for neurotrophins. There are three members of the Trk family: TrkA, TrkB, and TrkC, encoded, respectively, by the genes Ntrk1, Ntrk2, and Ntrk3 in rat or mouse genomic nomenclature, by NTRK1, NTRK2, and NTRK3 in human genomic nomenclature. TrkA is the high-affinity receptor for NGF, TrkB for BDNF and NT-4, and TrkC is the high-affinity receptor for NT-3. Also, NT-3 binds with a lower affinity to both TrkA and TrkB. Signaling through these receptor protein-tyrosine kinases is generally hypertrophic and increases neuronal metabolism, specifically, promoting neuronal survival, stimulating neurite growth, promoting synaptogenesis, and potentiating synaptic strength (Huang and Reichardt, 2003; Reichardt, 2006). The neurotrophin receptor p75NTR binds all neurotrophins with low affinity but, in complex NEUROTROPHINS IN THE COCHLEA 1879 Fig. 1. Basic neuroanatomy of the cochlea. A. Image showing innervation pattern of hair cells. This is the middle region of a neonatal rat cochlea dissected and placed in culture (Wang and Green, 2011). The hair cells (blue) are labeled with anti-myosin VI antibody. The SGNs (red) are labeled with anti-high molecular weight neurofilament antibody. The three rows of OHCs and the single row of IHCs are shown. The former are innervated by spiral bundles of type II SGN axons that turn to run parallel to the organ of Corti spiral. The IHCs are densely innervated by radial SGN axons. Scale bar ¼ 50 lm. B. Diagrammatic representation of cochlear innervation: the pattern of radial and spiral innervation (red) of, respectively, IHCs and OHCs (blue) is qualitatively represented, as is the increased density of innervation in the middle of the cochlea. The representation is not intended to be quantitatively accurate. C. Image showing innervation of hair cells. This is similar to (A) but at higher magnification and with synapses (green) labeled with anti-PSD95 antibody. The dense innervation of IHCs by type I SGN axons can be visualized by the number of radial fibers and number of synapses on each IHC. Type II fibers, which form the spiral bundles, can be seen extending past the IHC row. Scale bar ¼ 20 lm. D. Diagrammatic representation of cochlear innervation (red) and synapses (green) on IHCs and OHCs (respectively, dark blue and light blue). As for (B), the representation is intended to be accurate qualitatively but not quantitatively. with the ubiquitous protein sortilin, makes a high-affinity receptor for proneurotrophins (Teng et al., 2010). p75NTR is not a receptor protein-tyrosine kinase and recruits intracellular signaling different from that activated by Trks (Teng et al., 2010). p75NTR signaling is generally atrophic, promoting apoptosis, inhibiting neurite growth, and depressing synaptic strength (Teng et al., 2010). Unlike Trks, p75NTR is expressed on glial cells as well as on neurons. In the peripheral nervous system, p75NTR is expressed on Schwann cells after axotomy (Johnson et al., 1988). Note that because proneurotrophin cleavage and conversion from proneurotrophin to neurotrophin is regulated by activity and, possibly, other stimuli, this amounts to an activity-dependent regulation of the ratio of Trk to p75NTR signaling. Because these receptors elicit dramatically 1880 GREEN ET AL. Fig. 2. Neurotrophins and their receptors. The diagram summarizes relationships among proneurotrophins, neurotrophins, and their respective high-affinity signaling receptors p75NTR and Trks. Expression of BDNF, NT-3, and, after trauma, proBDNF, has been shown in the cochlea. SGNs express TrkB and TrkC. Schwann cells or organ of Corti supporting cells appear to express p75NTR after trauma. See text for details. different responses, this regulation has crucially important consequences for the cell. NEUROTROPHIN EXPRESSION IN THE COCHLEA As summarized in Fig. 2, neither NGF nor TrkA is expressed in the cochlea during development (Schecterson and Bothwell, 1994) and is not expressed in the cochlea postnatally (Qiong Wang, Erin Bailey, Steven H. Green, unpublished observations). All SGNs do express both TrkB and TrkC (Ylikoski et al., 1993; Pirvola et al., 1994; Mou et al., 1997), and SGN survival can be supported in vivo and in vitro by either BDNF or NT-3 (Lefebvre et al., 1994; Malgrange et al., 1996; Hegarty et al., 1997; Mou et al., 1997). Thus, we focus here on the roles of NT-3 and BDNF in the cochlea. Expression of NT-3 and BDNF in the organ of Corti is summarized in Fig. 3. NT-3 expression has been investigated by use of in situ hybridization in rats and mice (Pirvola et al., 1992b, 1994; Ylikoski et al., 1993; Schecterson and Bothwell, 1994; Wheeler et al., 1994) as well as by b-galactosidase labeling in mice in which the LacZ coding sequence replaces that of Ntf3 (Fari~ nas et al., 2001; Sugawara et al., 2005, 2007). During development, NT-3 expression appears to be primarily in supporting cells of the cochlear sensory epithelium rather than in the hair cells themselves (Fari~ nas et al., 2001). Postnatally, NT-3 expression is highest in the hair cells, but it is also abundantly expressed in adjacent supporting cells, for example, phalangeal cells, in the organ of Corti (Sugawara et al., 2007). These studies identified two spatial gradients of NT-3 expression in the neonatal and postnatal organ of Corti. The first is along the tonotopic axis: NT-3 expression is highest in the cochlear apex and lowest in the base. Second, NT-3 expression is higher on the modiolar side of the organ of Corti, that is, higher in the IHC region than in the OHC region. As described ‘‘BDNF and NT-3 have distinct actions in regulation of SGN electrophysiological and synaptic phenotype’’ section, the spatial gradient of NT-3 expression plays an important role in specifying position-dependent features of SGNs. For example, exposure to NT-3 in vitro causes SGNs to adopt an apical phenotype (Adamson et al., 2002a). NT-3 expression declines postnatally, but some expression remains in hair cells and immediately adjacent sensory cells even in older mice, primarily in the IHCs. There is also evidence that NT-3 is expressed in SGNs and glia in the postnatal cochlea (Hansen et al., 2001a,b), but there does not appear to be a spatial gradient in the spiral ganglion (Bailey et al., 2012). The spatial expression pattern of BDNF has been analyzed parallel to that of NT-3, in rats and mice, using the same methods: in situ hybridization and LacZ ‘‘knock-in’’ (Pirvola et al., 1992b, 1994; Ylikoski et al., 1993; Schecterson and Bothwell, 1994; Fari~ nas et al., 2001). These studies have shown that, during embryonic development, BDNF is expressed in the organ of Corti NEUROTROPHINS IN THE COCHLEA 1881 Fig. 3. Correlation of neurotrophin expression and SGN physiological phenotype. NT-3 is expressed in an apex to base gradient in the organ of Corti, whereas there is an opposite base to apex gradient of cochlear BDNF expression. This is correlated with and causal to apex to base differences in SGN membrane electrical properties and expression of ion channels and synaptic proteins, summarized in the text. and, like NT-3, in an apex to base gradient, highest in the apex and lowest in the base. Unlike NT-3, BDNF expression is restricted to hair cells in the cochlear base with expression at approximately equal levels in IHCs and OHCs. While always higher in the apex, BDNF expression declines in the prenatal period throughout the organ of Corti, so that BDNF mRNA is at very low levels by postnatal day 1 (P1). BDNF expression transiently increases in the rodent organ of Corti postnatally between approximately P4 and P9 (Wiechers et al., 1999) in a base to apex gradient (Tan and Shepherd, 2006; Flores-Otero and Davis, 2011). Although BDNF mRNA may be at low levels (Wiechers et al., 1999; Bailey et al., 2011), BDNF immunoreactivity has been detected in organ of Corti hair cells in the adult (Tan and Shepherd, 2006; Flores-Otero and Davis, 2011). The spatially patterned BDNF expression observed in the developing and postnatal organ of Corti plays important roles in position-specific SGN differentiation (see ‘‘BDNF and NT-3 have distinct actions in regulation of SGN electrophysiological and synaptic phenotype’’ section), for example, basal portions of the organ of Corti cocultured with SGNs induce a basal SGN phenotype in a BDNF-dependent manner (Flores-Otero et al., 2007). Moreover, BDNF is expressed in the postnatal spiral ganglion in rats and mice (Wiechers et al., 1999; Hansen et al., 2001b; Zha et al., 2001; Schimmang et al., 2003; Ruttiger et al., 2007; Singer et al., 2008), and one study has shown this expression to be in a base to apex gradient (Schimmang et al., 2003). Quantitative real-time PCR (qPCR) analysis (Stankovic and Corfas, 2003) has been used to quantify postnatal expression of BDNF and NT-3 in the cochlea and vestibule although, because the entire organs were assayed, localization of expression to specific cell types within the inner ear could not be determined. The study does confirm that both NT-3 and BDNF are expressed in the mature inner ear, although BDNF levels are very low in the cochlea relative to the vestibule. NT-3 expression in the cochlea appears to require the neuregulin signaling pathway. The neuregulins are a family of intercellular signals, related to epidermal growth factor, that signal via receptor protein-tyrosine kinases of the ErbB family (Burden and Yarden, 1997). In some cell types, the active receptor is an ErbB4 homodimer; in others, it is an ErbB2–ErbB3 heterodimer (Burden and Yarden, 1997). In the cochlea, organ of Corti supporting cells (Stankovic et al., 2004) and spiral ganglion Schwann cells (Hansen et al., 2001a) express ErbB2 and ErbB3, so are capable of responding to neuregulin. SGNs themselves express neuregulin (Hansen et al., 2001a; Stankovic et al., 2004) so are, reciprocally, capable of signaling to these Erb2/3-expressing cells. In developing sympathetic ganglia, Verdi et al. (1996) showed a reciprocal relationship between neuroblasts and non-neuronal cells in which the former express TrkC and are supported by NT-3 produced by non-neuronal cells. The latter express both ErbB2 and ErbB3 and are supported by neuroblast-derived neuregulin. A similar reciprocal trophic relationship may exist between SGNs and organ of Corti supporting cells in the cochlea. Inhibition of ErbB function in supporting cells reduces NT-3 expression in the cochlea and results in death of most SGNs. Therefore, SGNs appear to be supported by NT-3 derived from the organ of Corti and, in turn, induce NT-3 expression in these cells via neuregulin signaling (Stankovic et al., 2004). Moreover, synaptogenesis in the vestibule appears to depend on BDNF produced in supporting cells, again in an ErbB-dependent manner (Gomez-Casati et al., 2010). In summary, both NT-3 and BDNF are expressed in the developing cochlea in apex to base gradients. NT-3 continues to be expressed in an apex to base gradient in the postnatal organ of Corti, although at lower levels than prenatally. BDNF expression is low in the postnatal organ of Corti. Both BDNF and NT-3 are expressed postnatally in the spiral ganglion in neurons and/or glial cells. Thus, BDNF and NT-3 can mediate 1882 GREEN ET AL. trophic or tropic signaling from the organ of Corti to SGNs and can provide autocrine or paracrine trophic or tropic signaling within the spiral ganglion. Consistent with this, either BDNF or NT-3 can maintain survival of SGNs in culture in the absence of other trophic factors (Malgrange et al., 1996; Hegarty et al., 1997; Mou et al., 1997). Interactions among neurons and non-neuronal cells are likely to be important in inducing expression of neurotrophins, although it is not known how the neurotrophin expression gradients along the tonotopic axis are generated and maintained. COCHLEAR EXPRESSION OF NTFs OTHER THAN NEUROTROPHINS There is evidence for NTFs other than neurotrophins in the postnatal cochlea. Studies of rat cochleae (Ylikoski et al., 1998) show glial cell line-derived neurotrophic factor (GDNF) mRNA present starting at postnatal day 7 (P7), just prior to hearing onset. In situ hybridization shows that GDNF expression is in hair cells, becoming restricted to IHCs over time (Ylikoski et al., 1998). Quantitation with qPCR shows GDNF levels greater than those of NT-3 in the cochlea of the mature mouse (Stankovic and Corfas, 2003). Transcripts of other members of the GDNF family—neurturin, artemin, and persephin—have also been found by reverse transcriptase-polymerase chain reaction (RT-PCR) in the cochlea in a tissue isolate containing the organ of Corti and in a modiolar tissue isolate containing the spiral ganglion (St€over et al., 2000), although localization on the cellular level within these regions has not yet been reported. GDNF supports survival of cultured SGNs (Ylikoski et al., 1998) indicating that SGNs must express a functional GDNF receptor complex. The canonical highaffinity receptor for GDNF family members contains the receptor protein-tyrosine kinase Ret and one of the members of the GDNF family receptor alpha (GFRa) family. The former appears to be the signal-transducing receptor, while the GFRa family members, which consist only of an extracellular domain glycosylphosphatidylinositol (GPI)-linked to the plasma membrane, serve to confer high-affinity binding to Ret and not to be signal transducers (Airaksinen and Saarma, 2002). With regard to the latter, several studies (Ylikoski et al., 1998; St€over et al., 2000, 2001) have shown expression of GFRa1 in the cochlea, specifically localized to SGNs (St€over et al., 2001). St€over et al. (2000) have provided evidence for expression of the other two members of the GFRa family, GFRa2 and GFRa3, in the cochlea. Microarray gene expression profiling of P32 and P60 rat spiral ganglia (Bailey et al., 2012) confirms expression of GFRa1 and GFRa2, although GFRa3 was not detected. Thus, coreceptors are potentially available for high-affinity binding of the members of the GDNF family of NTFs. The presence of the signal-transducing receptor Ret in the spiral ganglion is still uncertain. Although two studies raised a possibility of Ret expression (St€over et al., 2000, 2001), others have failed to detect Ret expression (Ylikoski et al., 1998; Hashino et al., 1999). The previously mentioned microarray gene expression profiling indicated only a low level of Ret expression in the mature rat spiral ganglion, but this same study did show expression of neural cell adhesion molecule (NCAM) in the spiral ganglion. Because NCAM can sub- stitute for Ret in GDNF signal transduction (Paratcha et al., 2003), the fact that SGN survival and neurite growth are promoted by GDNF (see later) is not incompatible with absence of Ret. Finally, there is evidence that ciliary neurotrophic factor (CNTF) and its receptors are expressed in the cochlea (Malgrange et al., 1998). Microarray gene expression profiling (Bailey et al., 2012) has confirmed expression of CNTF and CNTF receptors in the spiral ganglion. CNTF promotes survival of cultured SGNs (Hartnick et al., 1996; Whitlon et al., 2007) and has a strong synergistic survival-promoting effect in combination with NT-3 (Staecker et al., 1995; Hartnick et al., 1996). NEUROTROPHIN KNOCKOUTS REDUCE SGN SURVIVAL IN THE DEVELOPING COCHLEA The effects of knockouts of NT-3, of BDNF, or of their receptors have been previously reviewed (Fritzsch et al., 2004). Briefly, mice with knockouts (KOs) of the NT-3 gene or of the BDNF gene show that either of these NTFs can support SGN survival during development but that presence of at least one is necessary for SGN survival. Double knockout of both BDNF and NT-3 results in complete loss of SGNs (Ernfors et al., 1995). Similarly, double knockout of TrkB and TrkC results in complete loss of SGNs (Fritzsch et al., 1995). Evidently, any other NTFs expressed in the developing cochlea do not suffice to support survival. Knockout of BDNF alone has a relatively modest effect on cochlear innervation. In contrast, vestibular neuron survival in BDNF KO mice is severely impaired (Ernfors et al., 1994, 1995; Jones et al., 1994) indicating that BDNF is an essential NTF for vestibular neurons. This is consistent with the mature expression pattern in which BDNF is expressed at much higher levels in the vestibule than in the cochlea (Stankovic and Corfas, 2003). In the cochlea, TrkB or BDNF KO reduces innervation of the OHCs (Ernfors et al., 1995; Fritzsch et al., 1998). This may be a consequence of lower levels of NT-3 expression in OHCs relative to IHCs, which could make OHC innervation more dependent on BDNF. NT-3 or TrkC KO results in loss of most SGNs (Ernfors et al., 1995; Fritzsch et al., 1997, 1998; Fari~ nas et al., 2001) although SGNs do survive in the apex. SGN death in NT-3 KOs occurs during the period embryonic day (E) 13.5–15.5, just after innervation of hair cells. At this time, BDNF expression is declining in the organ of Corti, and BDNF is present in the apex but not in the base (Fari~ nas et al., 2001). Therefore, survival of SGNs in the apex in NT-3 KO mice is likely to be due to support of SGN survival by BDNF. Indeed, in a combined BDNF and NT-3 double KO or a combined TrkB and TrkC double KO, neither SGN survival is observed in the cochlea nor vestibular neurons survival is observed in Scarpa’s ganglion (Ernfors et al., 1995; Fritzsch et al., 1995; Silos-Santiago et al., 1997). However, genetic replacement of NT-3 by BDNF completely rescues SGN survival in mice (Fari~ nas et al., 2001). These data imply that BDNF and NT-3 are essentially equivalent in their trophic action on SGNs during development. Further support for this comes from observations of cultured SGNs, which show that SGN survival can be supported by adding either BDNF or NT-3 or NT-4/5 to the culture medium (Zheng et al., 1995; Malgrange et al., 1996; NEUROTROPHINS IN THE COCHLEA Hegarty et al., 1997; Mou et al., 1997). Although NT-4/5 is not expressed in the cochlea, it is a TrkB ligand comparable with BDNF in ability to signal through TrkB. NEUROTROPHINS SUPPORT SURVIVAL OF DEAFFERENTED SGNs IN THE MATURE COCHLEA If hair cells are killed by certain means, for example, by exposure to aminoglycoside antibiotics, SGNs gradually die but this SGN death can be reduced by intracochlear infusion of NTFs or of viral vectors that drive expression of NTFs; a subject that has been previously reviewed (Roehm and Hansen, 2005; Green et al., 2008) and recently reviewed in detail in Budenz et al. (in this issue). Briefly, in a paradigm that has been applied with comparable results using cats, guinea pigs, or rats, the animals are deafened by injection of an ototoxic aminoglycoside, in some cases combined with a loop diuretic, that rapidly kills hair cells. SGNs die after deafferentation although, unlike the very rapid death that occurs after NT-3 KO in embryonic mice, the death of SGNs in adult animals is slow. In deafened rats and guinea pigs, SGN death occurs over a period of a few months (Webster and Webster, 1981; Koitchev et al., 1982; Bichler et al., 1983; Alam et al., 2007); in deafened cats, SGN death occurs over a period of months to years (Leake and Hradek, 1988). Observations of human postmortem tissue indicate that SGNs are capable of surviving for many years in the absence of hair cells (Nadol et al., 1989; Nadol, 1990, 1997). If hair cell-derived NTFs were the sole source of neurotrophic support to SGNs, one would expect the SGNs to die rapidly after loss of hair cells. However, the loss of SGNs postdeafferentation is slow and gradual. Moreover, elimination of hair cells by some means, for example, thiamine deprivation, does not result in SGN death. These data suggest, rather, that NTFs from sources other than hair cells can provide neurotrophic support to SGNs. Such sources include organ of Corti supporting cells (Sugawara et al., 2005), neuronal and glial sources within the spiral ganglion (Hansen et al., 2001a,b; Bailey et al., 2011), and the cochlear nuclei, which produce NTFs (Bailey et al., 2011) and are necessary for SGN survival (Spoendlin, 1971). The neurotrophic support provided by these sources is not restricted to neurotrophins, BDNF and NT-3, but includes other NTFs, including those summarized earlier (Bailey et al., 2011). Nevertheless, as described by Budenz et al. (in this issue), when SGN death would occur following hair cell loss, it is reduced by supplying NTFs to the cochlea during the period when SGNs are dying. To take just a few of many examples, intracochlear infusion of NT-3 from an implanted minipump and cannula has been shown to reduce SGN death in deafened guinea pigs (Ernfors et al., 1996; Staecker et al., 1996). Similarly, intracochlear infusion of BDNF has been shown to reduce SGN death in deafened guinea pigs (Staecker et al., 1996; Miller et al., 1997; Shinohara et al., 2002), rats (McGuinness and Shepherd, 2005), or cats (Leake et al., 2011). Reduced SGN death was also observed in deafened guinea pigs with intracochlear expression of BDNF from intracochlear injection of viral expression vectors (Nakaizumi et al., 2004) or fibroblasts genetically engineered to express BDNF (Rejali et al., 2007). Aside from 1883 similar effects on survival of SGNs, experiments using cultured SGNs show that NT-3 and BDNF are both able to elicit neurite growth from SGNs (Malgrange et al., 1996; Hegarty et al., 1997). NTFs other than neurotrophins that promote SGN survival in vitro also reduce SGN death in deafened animals. Intracochlear infusion of GDNF (Ylikoski et al., 1998), or artemin (Warnecke et al., 2010) reduces SGN death in deafened guinea pigs. Also, intracochlear injection of GDNF (Yagi et al., 2000; Kanzaki et al., 2002) or CNTF (Nakaizumi et al., 2004) viral expression vectors into deafened guinea pigs reduces SGN death. While these experiments show that BDNF, NT-3, and other NTFs are sufficient to maintain survival of SGNs after deafferentation, it is important to note that these experiments do not show that NT-3 or any particular NTF is necessary for survival of SGNs in the postnatal cochlea. In contrast to the situation in NT-3 KO mice, in which SGNs die rapidly, SGN death after deafferentation is slow. Thus, it is possible that NT-3 from sources other than hair cells can support SGNs after hair cell death. Organ of Corti supporting cells have been suggested as a source of NT-3 crucial to SGN survival (Sugawara et al., 2005). NT-3 and BDNF may be expressed in the ganglion itself (Hansen et al., 2001a,b; Schimmang et al., 2003; Bailey et al., 2011) and provide autocrine or paracrine trophic support to SGNs (Hansen et al., 2001b). Another possibility is that SGNs may become independent of support by NT-3 alone during postnatal maturation and acquire the ability to have their survival supported by other NTFs in the cochlea. (For example, as noted above, GDNF is expressed in the postnatal cochlea, can support SGNs in vitro, and can support SGN survival after hair cell death.) These possibilities cannot be distinguished without experiments in which NT-3 signaling is normal during development but disrupted postnatally in hair cells or supporting cells or cells in the spiral ganglion. NEUROTROPHIN REGULATION OF SGN MORPHOLOGICAL PHENOTYPE Because type I neurons comprise approximately 95% of the ganglion, we will examine spiral ganglion morphological phenotype in the context of this class of neurons. Similar to olfactory neurons and in distinction to those in the dorsal root ganglion, most type I SGNs elaborate a classical bipolar morphology. Each cell soma is surrounded by its axonal-like distal and proximal myelinated processes that convey electrical signals into the CNS. Thus, once the electrical signal is generated in the initial segment (IS) in the cochlea, extending from the postsynaptic contact to the hair cell to the heminode adjacent to the foramina nervosa (Hossain et al., 2005), it then travels via saltatory conduction through a distal peripheral (DP) axonal process, through the cell soma, and into a proximal peripheral (PP) axonal process. The entire extent of the peripheral regions of the neuron, aside from the IS, is myelinated by Schwann cells; compact myelin ensheaths the axonal processes, whereas a unique form of loose myelin surrounds the cell soma (Rosenbluth, 1962). The axon then enters the CNS through the internal auditory meatus, assuming the features of a central process (CP), myelinated by 1884 GREEN ET AL. oligodendrocytes before bifurcating and extending to form distinct tonotopic maps within the cochlear nucleus (Lorente de No, 1933; Luo et al., 2009). Neurite Outgrowth The axonal details elaborated above are important to keep in mind, because the lengths of specific regions of the peripheral and central axonal compartments, like the cell soma, vary tonotopically. Although the total length of SGNs from their synapse on a hair cell to their prominent bifurcation in the CNS is relatively uniform, distinct axonal regions vary in length along the tonotopic axis (Fekete et al., 1984). Because the basilar membrane is narrow in the high-frequency region and the somata of basal neurons are closer to the internal auditory meatus, the basal neuron IS, DP, and PP are shorter compared to those of apical neurons (Ryugo, 1992). And because the high-frequency regions of the cochlear nucleus tonotopic maps are further from the internal auditory meatus than are the low frequency regions, the CP is longer in basal neurons compared to apical neurons (Lorente de No, 1933). Observations of neurite outgrowth are central to the original observations of neurotrophin effects on the primary auditory afferents and may require precise controls because of the tonotopically associated differences in neurite length. In addition to other factors that play a role in neurite regeneration, such as FGF-1, FGF2, LIF, and depolarization (Hegarty et al., 1997; Gillespie et al., 2001; Hansen et al., 2001b; Hossain et al., 2002; Aletsee et al., 2003; Whitlon et al., 2007), both BDNF and NT-3 have also been shown to enhance spiral ganglion neurite outgrowth in vitro (Malgrange et al., 1996; Hegarty et al., 1997). A comparison of the effects of BDNF and NT-3 on early embryonic (E11) spiral ganglia showed that, at the concentrations used, BDNF was more efficacious in promoting spiral ganglion neurite outgrowth than was NT-3 (Pirvola et al., 1992a). Observations were made on total explants, thus, only an overall assessment, rather than specific evaluations, can be made from these data. When examined in vivo, the effects of BDNF and NT-3 on the cochlea are clearly complex (Yang et al., 2011). In accord with in vitro studies, addition of BDNF, either alone or in combination with other trophic factors, has been shown to increase SGN sprouting in vivo (Wise et al., 2005; Glueckert et al., 2008; Leake et al., 2011). In addition to promoting neuritogenesis, BDNF has also been shown to have the potential to provide a cue for targeting. In a study in which adenoviral vectors expressing either BDNF or NT-3 were injected into the scala tympani or scala media (Wise et al., 2010), SGNs showed more organized outgrowth toward the injection site. This approach could be highly useful when attempting to distinguish neurotrophin effects on neuronal survival from a role in axon guidance and synapse formation. Soma Size Another aspect of the phenotypic profile of SGNs that varies along the tonotopic contour is soma size, with neuron somata in the apex being smaller than those in the base (Liberman and Oliver, 1984; Nadol et al., 1990; Echteler and Nofsinger, 2000). Because the soma of these bipolar neurons is interposed within the electrical trajectory from sensory receptor to postsynaptic target, soma size, which affects the amount of membrane to charge and the internal resistance, can have a profound impact on the timing and filtering characteristics of signal transmission (Robertson, 1976). Thus, in a system in which action potential timing is critical to convey accurate information about sound frequency, one might hypothesize that the feature of soma size must also be highly regulated. Further, because neurotrophins are tonotopically expressed and have the ability to alter aspects of the electrophysiological phenotype of SGNs, this opens the possibility that this morphological feature is regulated in tandem with the electrophysiological phenotype. Studies examining the effects of neurotrophins infused in damaged cochleae in vivo, clearly show that in addition to promoting increased survival BDNF can maintain (Leake et al., 2011) or increase the original soma area, particularly in the basal region of the cochlea (McGuinness and Shepherd, 2005; Shepherd et al., 2005; Agterberg et al., 2008). A possible explanation for this observation is that the site of BNDF delivery is the round window, but based on the concentrations needed to produce an effect on survival, it has been argued that this is unlikely (Agterberg et al., 2008). An alternative explanation is that the basal cochlea is more BDNF-responsive than the apex, a result consistent with other actions of this neurotrophin on the properties of the SGNs. A complication to the interpretation of the observations described earlier is that regulated enlargement of neuronal soma may be difficult to distinguish from the possibility that a cell with appropriate trophic support may be larger and, as a result, ‘‘healthier’’ than one devoid of it. One way to examine this is to determine whether a neurotrophin can predictably decrease soma area while simultaneously promoting survival. This type of mechanism might be predicted to regulate the smaller size of apical spiral ganglion. Interestingly, this has been observed in experiments carried out in vitro. In an NT-3 concentration series that was found to increase the survival of apical SGNs, significant decreases in soma size were noted (Smith and Davis, 2012). Taken together, from the studies carried out to date, there are strong indications that soma size, in addition to electrophysiological phenotype, is likely to be differentially affected by BDNF and NT-3. Additional experiments, however, are required to determine how neurotrophin regulation of soma size compares to that of ion channel or synaptic protein regulatory mechanisms. Overview Additional investigation is required to explain the signaling mechanisms that underlie observations of differential BDNF and NT-3 actions and how they intersect with other processes and factors to control the overall characteristics of SGNs. Our current understanding does, however, indicate that, like other neurons in the peripheral and central nervous systems, the primary auditory afferents require complex and overlapping neurotrophin signaling mechanisms to attain their final form. Furthermore, understanding how the tonotopically distributed morphology, synaptic connections, and NEUROTROPHINS IN THE COCHLEA electrical transmission properties are achieved presents a future challenge required to define the unique neurons that compose the spiral ganglion. BDNF AND NT-3 HAVE DISTINCTIVE ACTIONS IN COCHLEAR INNERVATION The above experiments imply that BDNF and NT-3 are approximately equivalent in their ability to promote SGN survival or elicit neurite growth from SGNs. Indeed, experiments in which the NT-3 gene is replaced by a BDNF gene show that BDNF can rescue SGN survival in the absence of NT-3 (Coppola et al., 2001). However, these same experiments show that BDNF and NT-3 have distinctive actions in directing innervation of the cochlea. While SGN number is normal or nearly so in mice in which BDNF has replaced NT-3, the innervation of the organ of Corti is abnormal (Coppola et al., 2001; Tessarollo et al., 2004; Yang et al., 2011). This is due, in part, to invasion of the cochlea base by vestibular neuron fibers (Tessarollo et al., 2004), which are BDNFdependent, but the spiral ganglion projection itself is abnormal, particularly the projection to the OHCs, which is diminished (Yang et al., 2011). Synapses between IHCs and SGNs are susceptible to excitotoxic trauma: exposure to agonists of non-Nmethyl-D-aspartate (NMDA)-type ionotropic glutamate receptors results in destruction of these synapses in vivo (Puel et al., 1995) and in vitro (Wang and Green, 2011). There is some limited synapse regeneration after excitotoxic trauma and the number of regenerated synapses is increased by addition of either BDNF or NT-3, with the two neurotrophins being equally effective (Wang and Green, 2011). However, blockade of NT-3 signaling significantly reduces synapse regeneration, even in the presence of added BDNF (Wang and Green, 2011). One interpretation of these results is that NT-3 and BDNF are not equivalent in their effect on synapse regeneration in this system and that BDNF cannot completely replace NT-3. BDNF AND NT-3 HAVE DISTINCT ACTIONS IN REGULATION OF SGN ELECTROPHYSIOLOGICAL AND SYNAPTIC PHENOTYPE Primary auditory afferents possess both TrkB and TrkC receptors, making them capable of responding to multiple neurotrophins, and thus enabling a more complex and rich response than neurons that possess only a single Trk receptor type. Fortunately, the functional effects of both BDNF and NT-3 can be effectively explored within the spiral ganglion because of its simple and highly ordered organization. As mentioned earlier, the ganglion is composed of a relatively uniform population of cells, 95% of which are classified as type I neurons, which are predominately bipolar neurons that make one-to-one synaptic connections with IHCs (Spoendlin, 1973). Further, peripheral innervation patterns of type I neurons are organized into an array that is systematically graded from the base to the apex of the cochlea reiterating the tonotopic map of sound extending from high to low frequencies, respectively. Thus, the spiral ganglion, with its highly regular peripheral synaptic connections, follows the tonotopic map of sound that 1885 originates from mechanical and cellular cochlea specializations (Rubel and Fritzsch, 2002). Commensurate with this, the intrinsic membrane properties of early postnatal SGNs separated from their synaptic connections in vitro display tonotopic kinetic features (summarized in Fig. 3). Neurons that innervate high-frequency regions display fast firing properties, in response to constant current injection, such as rapid accommodation and abbreviated action potential duration and latency. Conversely, neurons that innervate low-frequency regions display slower firing properties, such as rapid to slow accommodation and prolonged action potential duration and latency (Adamson et al., 2002b). Voltage-Gated Ion Channels The graded kinetic membrane properties of SGNs are largely orchestrated by the relative density and types of voltage-gated ion channels, although structural features of the neurons (see earlier) may also have a significant impact. The specific ion channel types studied in this context have been Kv1.1, Kv3.1, Kv3.3, Kv4.2, and large conductance Ca2þ-activated Kþ (BK) channels (Adamson et al., 2002b; Chen and Davis, 2006). Neurons capable of rapid firing in the basal cochlea would be expected to possess rapidly activating, high-voltage activated Kþ channels, such as Kv3.1 and Kv3.3, voltage and Ca2þactivated Kþ channels, such as BK channels, and lowvoltage activated Kþ channels, such as Kv1.1, because, in combination, these would limit action potential duration and increase accommodation (Wang et al., 1998; Brew et al., 2003; Edgerton and Reinhart, 2003). Interestingly, as a consequence of the presence of these Kþ channels, there would also be an elevation of action potential firing threshold, but this is offset by the high density of 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA)-type receptors also found in this neuronal population (see later). In contrast, neurons with slow firing features in the apical cochlea would be expected to possess low-voltage activated, inactivating ion channels, such as Kv4.2, which are capable of delaying action potential responses to stimulus onset (Baranauskas, 2007). Thus, the distribution pattern of these ion channel types within the ganglion is consistent with their physiological role enabling rapid changes in membrane potential for high-frequency sounds and following slower changes in voltage for low-frequency sounds. Expectedly, ion channel types that contribute to fast firing characteristics (Kv1.1, Kv3.1, and BK) are found in greater abundance in basal, high-frequency coding SGNs, whereas the ion channel type that contributes to slow firing characteristics (Kv4.2) is enriched in the apex, low-frequency coding SGNs (Adamson et al., 2002b). One might hypothesize that these features must be precisely regulated to achieve the frequency-associated kinetic differences that have been described earlier. Indeed, ion channel composition and firing features, evaluated with immunocytochemistry and whole-cell patch clamp electrophysiology, can be altered by both BDNF and NT-3 and, in some cases, these two neurotrophins have opposing actions. This is the case for the assessment of two ion channel types responsible for high-frequency firing: Kv3.1 and BK. BDNF increases the abundance of both ion channel types in apical 1886 GREEN ET AL. neurons, whereas NT-3 decreases their abundance in basal neurons (Adamson et al., 2002a). Interestingly, a channel type that has a major impact on neuronal sensitivity, Kv1.1, that is enriched in the basal neurons (Adamson et al., 2002b), is not oppositely regulated by BDNF and NT-3. Its level is enhanced by BDNF, with NT-3 having little or no effect. Similarly, yet in distinction to this, the ion channel associated with lowfrequency firing, Kv4.2, shows increased levels following exposure to NT-3, with exposure to BDNF having little or no effect (Adamson et al., 2002a). These observations demonstrate two broad mechanisms of regulation within the classes of ion channels identified in the spiral ganglion. First, BDNF and NT-3 have opposite regulatory effects on a specific set of ion channel types; second, each neurotrophin acts separately to up-regulate defined ion channels. If one assumes in each of these examples that BDNF and NT-3 are working primarily through their cognate high-affinity receptors, TrkB and TrkC, respectively, then these complex patterns of ion channel regulation indicate that their signaling pathways are necessarily different. In the first example cited above for Kv3.1 and BK, one hypothesis could be that a common TrkB/TrkC signaling pathway may be activated, yet in one Trk-evoked pathway the sign of the response is inverted to mediate a reversed outcome. In the second example cited above for Kv1.1 and Kv4.2, one hypothesis might be that distinct TrkB/TrkC signaling pathways are utilized, each having exclusive regulatory effects on different ion channel types. Although such fine distinctions have not yet been revealed in different Trk signaling pathways, these types of observations are valuable for formulating models that can be tested in appropriate systems, such as the spiral ganglion (Huang and Reichardt, 2003). Synaptic Proteins Because the function of primary afferents is to deliver reliably the electrical output of sensory receptor generator potentials to higher order neural centers, it is reasonable to suppose that the synaptic proteins that comprise both the presynaptic and postsynaptic machinery of SGNs may also be highly regulated in a manner commensurate with that seen for voltage-gated ion channels. Indeed, studies have shown that this is the case. Superimposed upon the fine structure of the tonotopic distribution of synapse number (Meyer et al., 2009) and their structural complexities characterized around each IHC circumference (Liberman et al., 2011), observations of overall synaptic proteins levels throughout the ganglion have also been shown to be neurotrophindependent. It is already known that apical SGNs form notably larger end bulbs of Held with bushy cells in the cochlear nucleus (Rouiller et al., 1986) and also show graded levels of presynaptic proteins compared to their basal counterparts, as assessed by immunocytochemical analysis of anti-SNAP-25 and antisynaptophysin antibody immunolabeling (Flores-Otero and Davis, 2011). In contrast to this, basal SGNs, which have substantially higher voltage thresholds and lower resting membrane potentials than apical neurons, show higher levels of postsynaptic AMPA receptors compared to their apical counterparts, as assessed by immunocytochemical analy- sis of anti-GluR2, anti-GluR3, and anti-GluR2/3 antibody immunolabeling (Flores-Otero and Davis, 2011). What is notable about both of these distributions is that the assessed presynaptic and postsynaptic proteins are regulated by neurotrophins similar to that described for Kv3.1 and BK, such that BDNF and NT-3 are capable of working together to regulate synaptic proteins reciprocally. AMPA receptors are up-regulated by BDNF and down-regulated by NT-3. In contrast, SNAP25 and synaptophysin are down-regulated by BDNF and up-regulated by NT-3. Thus, the presynaptic proteins assessed in the spiral ganglion were oppositely regulated by BDNF and NT-3, a mechanism that was also observed for postsynaptic proteins, but as a mirror image (Flores-Otero et al., 2007). These results are supported by experiments using an in vitro preparation in which microisolates removed from different tonotopic areas of the organ of Corti were cocultured with apical or basal SGNs (Flores-Otero et al., 2007). Basal SGNs were altered to a more apicallike phenotype when cocultured with apical organ of Corti microisolates, and this effect was inhibited by an anti-NT-3 function-blocking antibody. Conversely, apical SGNs were altered to a more basal-like phenotype when cocultured with basal microisolates; these characteristics were significantly decreased by the additional of an antiBDNF function-blocking antibody. This is consistent with the hypothesis that a high ratio of NT-3 to BDNF in the apex results in enhanced NT-3 secretion from the apical regions of the organ of Corti relative to the base, and this induces apical characteristics in SGNs. The inverse is the case in the base, a low ratio of NT-3 to BDNF results in enhanced BDNF secretion from basal regions of the organ of Corti compared to apical regions and BDNF derived from the basal organ of Corti induces basal characteristics in SGNs. The observations above indicate that the overall electrophysiological signature of a SGN is the outcome of complex regulatory mechanisms, in which distinctive signaling by BDNF and NT-3 plays a crucial role. This unique pattern of regulation provides a platform to explore interactions between TrkB and TrkC signaling pathways that appear to be substantially different, yet highly interdependent. Observations from other neuronal systems and functional contexts suggest that yin and yang interactions are a general principle of neurotrophin signaling (Lu et al., 2005). For example, it has recently been observed that phenotypes as disparate as survival/apoptosis (Hempstead, 2006) and long-term potentiation (LTP) and long-term depression (LTD; Minichiello, 2009) are regulated by BDNF by novel mechanisms associated with differential signaling through p75NTR versus TrkB. Depending on the extent to which proBDNF is cleaved to mature BDNF, respectively, p75NTR or BDNF will be preferentially bound, with very different intracellular signaling pathways and outcomes (see later) as a result. Although one observes a yin and yang pattern of voltage-gated ion channel and synaptic protein regulation in SGNs (Davis and Liu, 2011), the mechanism is unlikely to involve p75NTR, which does not appear to be expressed on SGNs. Rather, two similar receptor types, TrkB and TrkC, are preferentially bound by two different ligands. From studies evaluating neurotrophin levels reviewed earlier, NT-3 concentrations are expected to be NEUROTROPHINS IN THE COCHLEA higher in the apical compared to the basal regions of the cochlea, which is consistent with its phenotypic actions on the SGNs. This poses an interesting issue, whether at the highest concentrations NT-3, with its known promiscuous binding, also interacts with TrkB receptors, thus increasing the potential complexity of neurotrophin interactions within the spiral ganglion. Observations consistent with this type of interaction have been reported (Hansen et al., 2001b; Zhou et al., 2005), however, it is currently unclear whether this mechanism contributes to the final functional regulation within the spiral ganglion. A significant limitation to our ability to explain the complex pattern of divergence and convergence in neurotrophin effects on SGNs is that kinetics of neurotrophin binding to SGNs has never been assessed. Thus, the actual affinities of SGNs for NT-3 and BDNF are not known. The role of BDNF in this process is even less clear. In addition to the open question of whether or not this neurotrophin is produced and secreted by cochlear hair cells postnatally, it is currently unresolved as to whether, as would be consistent with functional data, BDNF concentrations are higher in basal regions of the cochlea (see earlier). Furthermore, the presence of BDNF in the spiral ganglion, which is higher in basal than apical neurons (Schimmang et al., 2003) opens up the question as to whether BDNF has an autocrine or paracrine role in phenotypic regulation, and how that compares with neuronal survival (Hansen et al., 2001b). In this case, the mechanisms of secretion and neurotrophin processing may be key factors to consider. Thus, we anticipate that novel processes may underlie the multiple regulatory mechanisms that have been observed in SGNs. One might imagine that in addition to specificity of neurotrophin concentrations and secretion mechanisms, that processing of the neurotrophin proforms, novel receptor binding, or alternative signaling pathways, are also involved. POSSIBLE FUNCTIONS OF p75NTR IN THE COCHLEA Neurons that express p75NTR also express at least one of the Trks and Trk-p75NTR association increases specificity of the Trk for its corresponding neurotrophin(s) and increases binding affinity. However, p75NTR also has its own distinct signaling very different from that of Trk. Signaling by p75NTR has been recently reviewed in detail (Reichardt, 2006; Schweigreiter, 2006; Teng et al., 2010) so will be only briefly summarized here. p75NTR is expressed in neurons with at least one member of the Trk family but may also be expressed in non-neuronal cells, including central and peripheral glia. As diagrammed in Fig. 2, p75NTR alone binds neurotrophins with low affinity but, with the ubiquitous protein sortilin as a coreceptor, is a high-affinity receptor for proneurotrophins (Teng and Hempstead, 2004; Schweigreiter, 2006). In either case, p75NTR signaling is generally, although not necessarily, proapoptotic (Reichardt, 2006; Teng et al., 2010) and, in neurons, also inhibits neurite growth (Teng et al., 2010) and depresses synaptic activity (Lu, 2003). Thus, for a cellular decision of survival versus apoptosis, the presence of specific receptors and ligands coordinate distinct functional outcomes. Cells can be preferentially triggered to undergo apoptosis in 1887 the presence of pro-BDNF, which can bind with high affinity to a p75NTR/sortilin receptor complex. Yet, these same cells preferentially survive when mature BDNF binds to TrkB (Lee et al., 2001). In another example, LTP and LTD are differentially induced in hippocampal neurons via, respectively, TrkB or p75NTR, depending on whether proBDNF is processed to BDNF (Figurov et al., 1996; Woo et al., 2005). Proapoptotic signaling by p75NTR is due, in part, to activation of the proapoptotic mitogen-activated protein (MAP) kinase c-Jun N-terminal kinase (JNK) signaling pathway (Harrington et al., 2002; Bhakar et al., 2003; Kenchappa et al., 2010) and activation of the tumor necrosis factor-alpha-converting enzyme/ADAM17 (Kenchappa et al., 2010). At least one other p75NTR-associated protein, neurotrophin receptor-interacting MAGE homolog (NRAGE), may contribute to p75NTR-induced apoptosis via a separate signaling pathway (Bertrand et al., 2008) and also contribute to inhibition of neurite growth (Feng et al., 2010). The intracellular domain (ICD) of p75NTR can be cleaved and released by the protease c-secretase, which allows translocation of the ICD to the nucleus (Parkhurst et al., 2010). This results in increased transcription of at least one gene, cyclin E1 (Parkhurst et al., 2010). In addition, nuclear translocation of the p75NTR ICD brings p75NTR-associated proteins including the zinc-finger transcription factor neurotrophin receptor interacting factor (NRIF) (Linggi et al., 2005) to the nucleus. This event also contributes to apoptosis (Kenchappa et al., 2006), although the specific genes so regulated have not yet been identified. Other intracellular signals activated by this versatile receptor include RhoA, which contributes to antagonism of axon growth by p75NTR (Yamashita et al., 1999), and NF-jB, which contributes to cell differentiation and cell survival responses to p75NTR observed in some contexts (Hamanoue et al., 1999). p75NTR signaling can be antagonistic to neurotrophic signaling without necessarily being proapoptotic (Greferath et al., 2012). In the cochlea, p75NTR is clearly expressed in the embryo (Abe et al., 1991; Despres et al., 1991), but p75NTR expression declines to undetectable levels in the first postnatal week and remains undetectable in the postnatal and mature cochlea. In the organ of Corti, p75NTR expression is restricted to the inner pillar cells in the embryo and early neonate, but this expression disappears postnatally (von Bartheld et al., 1991; Sato et al., 2006). In the spiral ganglion, p75NTR expression is evident in embryonic and neonatal SGNs (von Bartheld et al., 1991; Schecterson and Bothwell, 1994; Sato et al., 2006) but, as in the organ of Corti, p75NTR expression in the spiral ganglion disappears after birth. Although Sato et al. (2006) showed p75NTR immunoreactivity in SGNs in 1-month-old C57BL/6 mice, several studies (von Bartheld et al., 1991; Tan and Shepherd, 2006; Provenzano et al., 2011) showed no p75NTR immunoreactivity in SGN somata in 1-month-old normal hearing rats. In this respect, SGNs are highly unusual neurons in expressing Trks (specifically, TrkB and TrkC) but not p75NTR. Given the known functions of p75NTR in Trk-expressing neurons, this suggests that affinity of SGNs for neurotrophins may be lower than in most neurons, that is, relatively high concentrations of neurotrophins may be needed to elicit trophic or tropic effects on SGNs. 1888 GREEN ET AL. Nearly all published studies in which neurotrophins have been used to support SGN survival in vitro or in vivo have used >1 nM concentrations that greatly exceed the high-affinity Kd of neurotrophin binding, 0.01 nM (Hempstead et al., 1991), so it is not known whether SGNs can bind or respond to neurotrophins with high-affinity. As noted earlier, neurotrophin binding kinetics have never been assessed for SGNs. Moreover, because of the lack of p75NTR on SGNs, it is conceivable that TrkB and TrkC receptors on SGNs may bind neurotrophins with less specificity than these receptors on most other neurons, which could allow greater cross-talk between neurotrophins and Trks. Remarkably, in deafened rats, p75NTR and its high-affinity ligand proBDNF are expressed in the spiral ganglion (Tan and Shepherd, 2006; Provenzano et al., 2011), although the expression is largely restricted to spiral ganglion satellite and Schwann cells and not the SGNs themselves (Provenzano et al., 2011). p75NTR knockout mice have normal hearing onset and young p75NTR knockout mice have normal hearing thresholds (Sato et al., 2006; Brors et al., 2008), implying that p75NTR is not necessary for normal hearing development. However, Sato et al. (2006) show that p75NTR KO mice have an accelerated loss of SGNs in aging and SGN death after deafening is accelerated in p75NTR KO mice (Tan et al., 2010). These studies imply a protective role for p75NTR in the cochlea. This is remarkable given that p75NTR is typically thought of as proapoptotic. Nevertheless, the available data suggest that, after trauma, p75NTR, which is otherwise not expressed in the mature cochlea, is upregulated in non-neuronal cells and this, in turn, reduces the death of SGNs. While the mechanism is not known, Provenzano et al. (2011) have shown that nuclear translocation of the p75NTR ICD occurs in spiral ganglion glia after deafening and that this is associated with entry to the cell cycle. Because spiral ganglion glia produce NTFs, including NT-3 (Hansen et al., 2001a), p75NTR may be exerting a protective effect on neurons indirectly, by maintaining glial number in the degenerating ganglion, allowing the glia to provide trophic support to the neurons. More direct evidence for this has come from use of a previously mentioned model for study of trauma in the cochlea, that of excitotoxic trauma to IHC-SGN synapses in vitro (Wang and Green, 2011). Following trauma, p75NTR is expressed in the organ of Corti supporting cells, and this has a protective role in that it promotes synapse regeneration in a NT-3-dependent manner (Wang et al., 2009). SIGNAL TRANSDUCTION BY Trk-FAMILY RECEPTORS While signal transduction by Trk (summarized in Fig. 4) has been extensively studied and reviewed in detail (Huang and Reichardt, 2003; Reichardt, 2006; Schweigreiter, 2006), most studies have been of TrkA, the firstdiscovered member of the family; TrkB and TrkC have not been extensively investigated. Nevertheless, similarity of key sequences in the ICD of all three Trks suggests that the mechanisms revealed by study of TrkA and other receptor protein-tyrosine kinases apply also to TrkB and TrkC. For any receptor protein-tyrosine kinase, ligands are divalent or multivalent. In the case of neurotrophin signaling, all neurotrophins are noncovalently but tightly linked dimers. Binding of ligand to the receptor extracellularly results in receptor dimerization, cross-phosphorylation on certain tyrosine residues in the intracellular portion of the dimerized protein-tyrosine kinases. This ‘‘autophosphorylation’’ by protein-tyrosine kinases is crucial, as it causes recruitment to the receptor of effector or adaptor proteins that contain phosphotyrosine-binding motifs. The effectors then initiate intracellular signaling pathways; adaptor proteins recruit other effectors that initiate intracellular signaling pathways. Thus, receptor-tyrosine kinases simultaneously activate several intracellular signaling pathways after binding ligand. Multiple effector and adaptor proteins bind Trk receptors, including phosphatidylinositol-3-kinase, Shc, Grb2, c-Abl, Frs2, phospholipase C-c, and ARMS (Reichardt, 2006). These adaptors, in turn, recruit and activate protein kinases such as protein kinase B (PKB/Akt) and MAP kinases including extracellular signal-regulated kinase (ERK)1/2, ERK5, p38, Src-family protein-tyrosine kinases, and others. By targeting cytoplasmic proteins and nuclear transcription factors, these kinases promote survival, neuronal differentiation, neurite growth, synapse potentiation, and most of the other Trk-mediated effects of neurotrophins. Small GTPases, Ras and Rap1, participate in linking Trk to activation of the ERK MAP kinases but Trk also activates other small GTPases— Rho family members Rac and Cdc42—that directly affect organization of the actin cytoskeleton and so affect membrane, cell, and growth cone motility and guidance of axon growth. Neuronal Survival The ability of trophic factors to promote survival via protein-tyrosine kinase signaling depends crucially on the protein kinase PKB/Akt (Matheny and Adamo, 2009). This general concept also applies specifically to SGNs: inhibition of Akt prevents BDNF or NT-3 from promoting SGN survival (Hansen et al., 2001b). Akt is a multifunctional protein kinase, targeting multiple effectors to inhibit apoptosis. For example, Akt phosphorylates and inactivates proapoptotic transcription factors of the Forkhead/FoxO family (Brunet et al., 1999) and p53 (Ogawara et al., 2002) and Akt phosphorylates and inactivates the proapoptotic Bcl-2 family protein Bad (Downward, 1999). Activation of the MAP kinase JNK—which mediates proapoptotic effects of NTF withdrawal (Maroney et al., 1999), cellular stress (Lin, 2003) and of p75NTR (Teng et al., 2010)—is also inhibited by Akt (Barthwal et al., 2003; Widenmaier et al., 2009) thereby providing a mechanism by which Trk signaling can abrogate proapoptotic p75NTR signaling. Suppression of JNK signaling may be relevant also to SGN survival in vivo, because SGN apoptosis following loss of hair cells is accompanied by JNK activation (Alam et al., 2007). Neurite Growth Neurotrophins can stimulate neurite growth via Trk. With all of the factors and mechanisms that underlie neurite outgrowth, it is not surprising to find that multiple, yet specific, signaling mechanisms, including ERK NEUROTROPHINS IN THE COCHLEA Fig. 4. Signal transduction by Trk-family receptor protein-tyrosine kinases. Trks are dimerized by divalent neurotrophins allowing autophosphorylation of certain tyrosine residues, some of which are diagrammatically illustrated by small blue circles in the intracellular portion of Trk. On the left side, some effectors activated by Trk after ligand binding are shown. The phosphotyrosine in the juxtamembrane domain (Tyr515 in mouse TrkB and Tyr516 in mouse TrkC) is the primary site for recruitment of ERK MAP kinases and PKB/Akt. Trk shares with other protein-tyrosine kinases the ability to assemble an adaptor protein complex that strongly but transiently activate ERK MAP kinases and other effectors at the plasma membrane. In addition, Trk has an atypical ability to assemble, on the juxtamembrane MAP kinases, Akt, and small GTPases, underlie this process. Interestingly, different aspects of axon growth may be modulated by different signaling pathways with, for example, ERK signaling increasing length and Akt signaling increasing axon caliber and branching in sensory neurons (Markus et al., 2002). NT-3-evoked extension of SGN neurites requires the Ras/ERK pathway (Aletsee et al., 2001) and JNK activity (Bodmer et al., 2002; Atkinson et al., 2011) but is independent of p38 (Aletsee et al., 2001) and is inhibited by Rho kinase (Lie et al., 2010). Of depolarization-activated signals, CaMKII, but not CaMKIV, inhibits SGN neurite growth, although both kinases converge to promote survival in these same cells (Hansen et al., 2003). To date, signaling mechanisms involved specifically in BDNF-evoked neurite extension are unknown, limiting our ability to make comparisons. Nevertheless, studies reported to date have shown that inroads are being made to understand better the diverse signaling contributions to spiral ganglion neurite outgrowth. 1889 domain phosphotyrosine, an adaptor protein complex that causes a sustained activation of ERK MAP kinases. This allows retrograde signaling from axon terminals to the soma, crucial for neurotrophic gene regulation. As indicated on the right side, phosphorylation of this juxtamembrane tyrosine is necessary for full enzymatic activation and autophosphorylation in TrkB, but it is not necessary for this in TrkC (Postigo et al., 2002). Additional phosphotyrosine residues within the kinase domain contribute to increased enzymatic activity and may serve as alternative sites for effector recruitment. Finally, a phosphotyrosine residue at the C-terminal recruits the signaling enzyme phospholipase C-c. Distinctive Temporal and Spatial Signaling by Trk Receptors The ERK family of MAP kinases (Johnson and Lapadat, 2002) are crucial effectors of receptor proteintyrosine kinases and are largely responsible for the mitogenic effect of these receptors. In neurons, which do not undergo cell division, ERKs function in neuronal differentiation, neurite growth, neuronal plasticity, and other roles. This involves a distinctive mode of ERK activation by Trk, not shared by mitogenic signal transduction. Trk engages two distinct mechanisms for ERK activation with different temporal and spatial characteristics. The adaptor proteins Shc and Grb2 recruit a Ras activator, and activated Ras initiates a kinase cascade that results in a rapid, strong, but transient Erk activation at the plasma membrane. This mechanism shared by all receptor protein-tyrosine kinases and is a canonical mechanism for triggering cell cycle entry and mitosis (Davis, 1993). 1890 GREEN ET AL. A second mechanism, requiring adaptor proteins Frs2 or ARMS, leads instead to recruitment of the small GTPase Rap1 and prolonged Erk activity (Kao et al., 2001; Arevalo et al., 2004). This mechanism is distinctive of Trk and, possibly, other NTF receptors, and is not used by mitogenic receptor protein-tyrosine kinases. This latter mechanism occurs in neurons that are postmitotic and, rather than triggering mitosis, the prolonged Erk activity promotes neuronal differentiation (Qiu and Green, 1992). Moreover, this mechanism operates on endosomes, as opposed to the plasma membrane, the site of Ras-mediated ERK activation (Wu et al., 2001). Activation of Trk by neurotrophin binding causes Trk to be internalized in endosomes. Endosomes can be retrogradely transported from axon terminals, where neurons encounter target-derived neurotrophins, to the soma. This allows activated internalized Trk to convey persistent Erk signaling from the terminal to the soma to effect cell survival and regulate gene expression (Ginty and Segal, 2002). Even this property of Trk receptors may vary with different ligand–receptor interactions. For example, although TrkA can bind both NGF and NT-3, only NGF can elicit TrkA internalization and retrograde signaling (Kuruvilla et al., 2004). Thus, Trk can activate ERK via two different signaling pathways. The Ras-dependent pathway, common to all receptor protein-tyrosine kinases, is transient and activates ERKs locally at the plasma membrane and is important for mitogenic actions of ERKs as well as for local effects of ERKs on cell metabolism and motility, including promotion of growth cone motility and axon growth. The Rap1-dependent pathway is persistent and activates ERKs on endosomes allowing retrograde signaling to the neuronal soma for promotion of cell survival and regulation of gene expression. This brief summary glosses over some critical details of Trk signal transduction, reviewed at length by Reichardt (2006) that may be crucial to consideration of how TrkB and TrkC might elicit distinct responses. One such detail, previously mentioned, is that TrkC cannot bind BDNF but TrkB can bind NT-3, albeit at reduced affinity. Thus, NT-3 can elicit signaling through TrkB as well as TrkC and may consequently elicit a higher level of Trk signaling in SGNs than can BDNF. Another such detail is that there is generally not an exclusive relationship between a particular adaptorbinding site on Trk and a protein kinase. Rather, the same or different adaptors binding to different sites on activated Trk are able to activate the protein kinases Erks and/or Akt, although the level or duration of activity may vary. Thus, because of such cross-talk, deletion of a single adaptor-binding site on Trk may have subtle quantitative but not necessarily drastic qualitative effects on responses to NTFs. Similarly, subtle differences among Trks in relative affinity of specific adaptorbinding sites for their respective adaptors could result in quantitative differences in intracellular signaling generated by the Trks. Even quantitative differences in intracellular signaling may yield substantial differences in outcome on the cellular level. For example, difference in duration of activity of a particular intracellular signal, the Erk MAP kinase, means a difference between mitogenic signaling and induction of neuronal differentiation. DISTINCT FUNCTION OF TrkB AND TrkC RECEPTORS IN THE COCHLEA As discussed earlier, SGN express TrkB and TrkC so are capable of responding to both BDNF and NT-3. Indeed, BDNF and NT-3 can elicit many of the same responses in SGNs in vivo or in vitro, for example, both promote survival and neurite growth to approximately the same degree. However, there are also instances where BDNF and NT-3 elicit different or opposing effects in neurons, for example, different turning responses in spinal neuron growth cones (Song and Poo, 1999) and different or opposing effects on dendrite growth in cortical pyramidal neurons (McAllister et al., 1997). In SGNs, BDNF and NT-3 clearly can elicit different or opposing effects, for example, regulation of membrane physiology and expression of channels, receptors and other synaptic proteins (Adamson et al., 2002a), as discussed earlier. This implies that TrkB and TrkC intracellular signaling share some similarities but also differ in important respects. Although TrkA, TrkB, and TrkC appear to activate the same major intracellular signals—Erk1/2 and Erk5 MAP kinases, PKB/Akt, phospholipase C-c, p38, Rac, and so forth—and similarly promote neuronal differentiation, survival, neurite growth, synaptogenesis and increased synaptic activity (Reichardt, 2006), it is possible that they can, nevertheless, direct some dissimilar neuronal responses via nuanced quantitative differences in the signaling generated. Such differences in signaling might underlie different responses of SGNs to BDNF and to NT-3. For example, as noted above, NT-3 is better able than BDNF to promote proper synaptogenesis on hair cells. This is evidenced by reduced synapse regeneration on IHCs after excitotoxic disruption in vitro when NT-3 signaling is blocked, even in the presence of added BDNF (Wang and Green, 2011). Moreover, genetic replacement of NT-3 by BDNF in vivo (Tessarollo et al., 2004) results in no significant reduction in SGN number, consistent with similar ability of BDNF and NT-3 to promote SGN survival, but innervation of the organ of Corti is disorganized. Thus, despite being equivalent in ability to promote SGN survival, BDNF is not equivalent to NT3 in ability to promote and organize innervation of the organ of Corti. As noted earlier, distinctive effects of BDNF vs. NT-3 have been observed in CNS neurons that express both TrkB and TrkC. However, the relative simplicity of cochlear innervation and well-defined effects of the neurotrophins on SGNs makes these neurons a particularly favorable system for studying the mechanisms by which TrkB and TrkC can elicit different responses. An analysis by Postigo et al. (2002) of the role of the important juxtamembrane domain adaptor binding site—tyrosine 516 in TrkC—is consistent with a hypothesis that differences in the relative significance of such sites in Trk proteins could underlie differences in the ability of NT-3 and BDNF to support innervation of the cochlea. Mutation of this site prevents association of critical adaptors, including Shc and Frs2, with activated TrkC. This should and does compromise activation of Erks and PKB/Akt—key effectors of neuronal survival and neurite growth—by TrkC. However, because other adaptors that can activate these kinase signaling pathways are able to bind TrkC at other sites, complete 1891 NEUROTROPHINS IN THE COCHLEA blockade of kinase activation would be unlikely and, in fact, was not observed (Postigo et al., 2002). Consistent with this, mice homozygous for this mutation of the TrkC Shc-binding site (trkCshc/shc mice) are viable postnatally, unlike trkC knockout (trkC/) mice. trkCshc/shc mice had a decreased number of SGNs but the decrease, only 25%, was modest compared to that in trkC–/– mice. The corresponding mutation in TrkB had similar consequences for vestibular neurons. However, while the trkBshc mutation significantly affected peripheral innervation by vestibular neurons, the trkCshc mutation had little apparent effect on peripheral innervation by SGNs. These data imply that TrkB and TrkC are similar in their reliance on this adaptor site for promoting survival but differ significantly their reliance on this adaptor site for directing axon growth and synaptogenesis. What can account for this on the molecular level? Postigo et al. observed that mutation of tyrosine 516 in the TrkC juxtamembrane domain, while markedly reducing activation of ERKs and PKB, does not reduce TrkC autophosphorylation. That is, TrkC remains enzymatically active and the remaining phosphotyrosine residues are, presumably, capable of some recruitment of intracellular signaling. In contrast, mutation of the corresponding juxtamembrane phosphorylatable tyrosine in TrkB (Tyr515) results in greatly diminished autophosphorylation. Presumably, in TrkB, full enzymatic activation requires phosphorylation of Tyr515 while in TrkC enzymatic activity is largely independent. While it is not clear how this mechanistic difference between TrkB and TrkC can account for different responses to BDNF and NT-3 in SGNs, it is consistent with a hypothesis that lack of equivalence in TrkB and TrkC signal transduction can account for these different responses. SUMMARY AND CONCLUSIONS Two types of receptors, p75NTR and Trk-family receptor protein-tyrosine kinases, can bind neurotrophins and mediate responses to these important NTFs. The highaffinity proneurotrophin receptor p75NTR is expressed in the developing cochlea and, after trauma, in the mature cochlea, where it may have a protective role in response to the trauma. SGNs express TrkB and TrkC so can respond to both BDNF and NT-3. Both of these neurotrophins can promote SGN survival and stimulate neurite growth. Nevertheless, there are significant differences in SGN responses to BDNF and NT-3. For example, NT-3 may promote and properly organize synapse formation in the organ of Corti in a way that BDNF is unable to accomplish. BDNF induces physiological properties in SGNs characteristic of SGNs in the basal cochlea, whereas NT-3 induces physiological properties in SGNs characteristic of those in the apical cochlea. These differences presumably derive from differences in association of BDNF and NT-3 with TrkB and TrkC and differences in the association of TrkB and TrkC with diverse intracellular signaling pathways. However, such differences are largely unknown and may involve relatively subtle quantitative aspects of signal transduction. As described by Budenz et al. (in this issue), significant effort has been expended in determining the extent to which NTFs, especially BDNF, NT-3, and GDNF, can maintain SGN survival in vitro or in vivo in the absence of normal afferent input. Because these factors are indeed able to promote SGN survival in animal studies, they have been proposed as therapeutic agents in humans with sensorineural deafness due to loss of hair cells for the purpose of maintaining SGN survival to allow long-term efficacy of cochlear implants and for the purpose of attracting SGN neurite growth toward electrodes to allow reduced stimulating current (Roehm and Hansen, 2005; Shibata et al., 2011). Should it become possible to regenerate lost hair cells, presumably there will be proposals to use NTFs to maintain SGN survival and promote synaptogenesis on regenerated hair cells. While these are potentially beneficial uses of NTFs, they should be approached with caution. As discussed earlier, NTFs have many diverse effects on SGNs and non-neuronal cells of the cochlea and may use multiple signal transduction mechanisms in rather subtle ways to achieve these effects. Some of these effects may be desirable therapeutic outcomes and some not. Considerable investigation remains to be done on the effects of NTFs on SGNs before we can reliably predict the outcome of NTF therapy on SGNs and choose an appropriate regimen of NTFs to optimize physiological and structural characteristics of surviving SGNs. LITERATURE CITED Abe H, Wataya H, Amano O, Kondo H. 1991. Localization of nerve growth factor receptor in developing inner ear of rats. Acta Oto-Laryngol 111:691–698. Adamson CL, Reid MA, Davis RL. 2002a. Opposite actions of brainderived neurotrophic factor and neurotrophin-3 on firing features and ion channel composition of murine spiral ganglion neurons. J Neurosci 22:1385–1396. Adamson CL, Reid MA, Mo ZL, Bowne-English J, Davis RL. 2002b. Firing features and potassium channel content of murine spiral ganglion neurons vary with cochlear location. J Comp Neurol 447: 331–350. Agterberg MJ, Versnel H, de Groot JC, Smoorenburg GF, Albers FW, Klis SF. 2008. Morphological changes in spiral ganglion cells after intracochlear application of brain-derived neurotrophic factor in deafened guinea pigs. Hear Res 244:25–34. Airaksinen MS, Saarma M. 2002. The GDNF family: signalling, biological functions and therapeutic value. Nature Rev Neurosci 3: 383–394. Alam S, Robinson BK, Huang J, Green SH. 2007. Prosurvival and proapoptotic intracellular signaling in rat spiral ganglion neurons in vivo after the loss of hair cells. J Comp Neurol 503:832–852. Aletsee C, Beros A, Mullen L, Palacios S, Pak K, Dazert S, Ryan AF. 2001. Ras/MEK but not p38 signaling mediates NT-3-induced neurite extension from spiral ganglion neurons. J Assoc Res Otolaryngol 2:377–387. Aletsee C, Brors D, Mlynski R, Ryan AF, Dazert S. 2003. Branching of spiral ganglion neurites is induced by focal application of fibroblast growth factor-1. Laryngoscope 113:791–796. Arevalo JC, Yano H, Teng KK, Chao MV. 2004. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. EMBO J 23:2358–2368. Atkinson PJ, Cho CH, Hansen MR, Green SH. 2011. Activity of all JNK isoforms contributes to neurite growth in spiral ganglion neurons. Hear Res 278:77–85. Bailey E, Galindo, R, Green SH. 2012. Neurotrophic support of spiral ganglion neurons (SGNs): a perspective from gene expression profiling. In: Presented at the 35th Annual Midwinter Meeting of the Association for Research in Otolaryngology, San Diego, CA. Bailey EM, Kopelovich JC, Green SH. 2011. Neurotrophic factor expression in the rat organ of Corti (OC), cochlear nucleus (CN), and spiral ganglion after deafening. In: Presented at the 34th 1892 GREEN ET AL. Annual Midwinter Meeting of the Association for Research in Otolaryngology, Baltimore, MD. Baranauskas G. 2007. Ionic channel function in action potential generation: current perspective. Mol Neurobiol 35:129–150. Barthwal MK, Sathyanarayana P, Kundu CN, Rana B, Pradeep A, Sharma C, Woodgett JR, Rana A. 2003. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem 278:3897–3902. Bertrand MJ, Kenchappa RS, Andrieu D, Leclercq-Smekens M, Nguyen HN, Carter BD, Muscatelli F, Barker PA, De Backer O. 2008. NRAGE, a p75NTR adaptor protein, is required for developmental apoptosis in vivo. Cell Death Differ 15:1921–1929. Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EB, Said F, Bonni A, Barker PA. 2003. Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J Neurosci 23:11373–11381. Bichler E, Spoendlin H, Rauchegger H. 1983. Degeneration of cochlear neurons after amikacin intoxication in the rat. Arch Otorhinolaryngol 237:201–208. Bodmer D, Gloddek B, Ryan AF, Huverstuhl J, Brors D. 2002. Inhibition of the c-Jun N-terminal kinase signaling pathway influences neurite outgrowth of spiral ganglion neurons in vitro. Laryngoscope 112:2057–2061. Brew HM, Hallows JL, Tempel BL. 2003. Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J Physiol 548:1–20. Brors D, Hansen S, Mlynski R, Volkenstein S, Aletsee C, Sendtner M, Ryan AF, Dazert S. 2008. Spiral ganglion outgrowth and hearing development in p75-deficient mice. Audiol Neurootol 13: 388–395. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 96:857–868. Burden S, Yarden Y. 1997. Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron 18:847–855. Chen WC, Davis RL. 2006. Voltage-gated and two-pore-domain potassium channels in murine spiral ganglion neurons. Hear Res 222:89–99. Coppola V, Kucera J, Palko ME, Martinez-De Velasco J, Lyons WE, Fritzsch B, Tessarollo L. 2001. Dissection of NT-3 functions in vivo by gene replacement strategy. Development 128:4315–4327. Davis RJ. 1993. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem 268:14553–14556. Davis RL, Liu Q. 2011. Complex primary afferents: what the distribution of electrophysiologically-relevant phenotypes within the spiral ganglion tells us about peripheral neural coding. Hear Res 276:34–43. Despres G, Hafidi A, Romand R. 1991. Immunohistochemical localization of nerve growth factor receptor in the cochlea and in the brainstem of the perinatal rat. Hear Res 52:157–165. Downward J. 1999. How BAD phosphorylation is good for survival. Nat Cell Biol 1:E33–E35. Echteler SM, Nofsinger YC. 2000. Development of ganglion cell topography in the postnatal cochlea. J Comp Neurol 425:436–446. Edgerton JR, Reinhart PH. 2003. Distinct contributions of small and large conductance Ca2þ-activated Kþ channels to rat Purkinje neuron function. J Physiol 548:53–69. Ernfors P, Duan ML, ElShamy WM, Canlon B. 1996. Protection of auditory neurons from aminoglycoside toxicity by neurotrophin-3. Nature Med 2:463–467. Ernfors P, Lee KF, Jaenisch R. 1994. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368: 147–150. Ernfors P, Van De Water T, Loring J, Jaenisch R. 1995. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron 14:1153–1164. Fari~ nas I, Jones KR, Tessarollo L, Vigers AJ, Huang E, Kirstein M, de Caprona DC, Coppola V, Backus C, Reichardt LF, Fritzsch B. 2001. Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. J Neurosci 21:6170–6180. Fekete DM, Rouiller EM, Liberman MC, Ryugo DK. 1984. The central projections of intracellularly labeled auditory nerve fibers in cats. J Comp Neurol 229:432–450. Feng Z, Li K, Liu M, Wen C. 2010. NRAGE is a negative regulator of nerve growth factor-stimulated neurite outgrowth in PC12 cells mediated through TrkA-ERK signaling. J Neurosci Res 88: 1822–1828. Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. 1996. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381:706–709. Flores-Otero J, Davis RL. 2011. Synaptic proteins are tonotopically graded in postnatal and adult type I and type II spiral ganglion neurons. J Comp Neurol 519:1455–1475. Flores-Otero J, Xue HZ, Davis RL. 2007. Reciprocal regulation of presynaptic and postsynaptic proteins in bipolar spiral ganglion neurons by neurotrophins. J Neurosci 27:14023–14034. Francis HW, Rivas A, Lehar M, Ryugo DK. 2004. Two types of afferent terminals innervate cochlear inner hair cells in C57BL/6J mice. Brain Res 1016:182–194. Francis HW, Rivas A, Lehar M, Saito Y, Mouton PR, Ryugo DK. 2006. Efficient quantification of afferent cochlear ultrastructure using design-based stereology. J Neurosci Methods 150:150–158. Fritzsch B, Barbacid M, Silos-Santiago I. 1998. The combined effects of trkB and trkC mutations on the innervation of the inner ear. Int J Dev Neurosci 16:493–505. Fritzsch B, Fari~ nas I, Reichardt LF. 1997. Lack of neurotrophin 3 causes losses of both classes of spiral ganglion neurons in the cochlea in a region-specific fashion. J Neurosci 17:6213–6225. Fritzsch B, Silos-Santiago I, Smeyne R, Fagan AM, Barbacid M. 1995. Reduction and loss of inner ear innervation in trkB and trkC receptor knockout mice: a whole mount DiI and scanning electron microscopic analysis. Audit Neurosci 1:401–417. Fritzsch B, Tessarollo L, Coppola E, Reichardt LF. 2004. Neurotrophins in the ear: their roles in sensory neuron survival and fiber guidance. Prog Brain Res 146:265–278. Gillespie LN, Clark GM, Bartlett PF, Marzella PL. 2001. LIF is more potent than BDNF in promoting neurite outgrowth of mammalian auditory neurons in vitro. Neuroreport 12:275–279. Ginty DD, Segal RA. 2002. Retrograde neurotrophin signaling: Trking along the axon. Curr Opin Neurobiol 12:268–274. Glueckert R, Bitsche M, Miller JM, Zhu Y, Prieskorn DM, Altschuler RA, Schrott-Fischer A. 2008. Deafferentation-associated changes in afferent and efferent processes in the guinea pig cochlea and afferent regeneration with chronic intrascalar brainderived neurotrophic factor and acidic fibroblast growth factor. J Comp Neurol 507:1602–1621. Gomez-Casati ME, Murtie JC, Rio C, Stankovic K, Liberman MC, Corfas G. 2010. Nonneuronal cells regulate synapse formation in the vestibular sensory epithelium via erbB-dependent BDNF expression. Proc Natl Acad Sci USA 107:17005–17010. Green SH, Altschuler RA, Miller JM. 2008. Cell death and cochlear protection. In: Schacht J, Popper AN, Fay RR, editors. Auditory trauma, protection and repair. New York: Springer-Verlag. pp. 275–319. Greferath U, Trieu J, Barrett GL. 2012. The p75 neurotrophin receptor has nonapoptotic antineurotrophic actions in the basal forebrain. J Neurosci Res 90:278–287. Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. 1999. p75-mediated NF-jB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci 14:28–40. Hansen MR, Bok J, Devaiah AK, Zha XM, Green SH. 2003. Ca2þ/ calmodulin-dependent protein kinases II and IV both promote survival but differ in their effects on axon growth in spiral ganglion neurons. J Neurosci Res 72:169–184. Hansen MR, Vijapurkar U, Koland JG, Green SH. 2001a. Reciprocal signaling between spiral ganglion neurons and Schwann cells involves neuregulin and neurotrophins. Hear Res 161:87–98. Hansen MR, Zha X-M, Bok J, Green SH. 2001b. Multiple distinct signal pathways, including an autocrine neurotrophic mechanism, contribute to the survival-promoting effect of depolarization on spiral ganglion neurons. J Neurosci 21:2256–2267. NEUROTROPHINS IN THE COCHLEA Harrington AW, Kim JY, Yoon SO. 2002. Activation of Rac GTPase by p75 is necessary for c-Jun N-terminal kinase-mediated apoptosis. J Neurosci 22:156–166. Hartnick C, Staecker H, Malgrange B, Lefebvre P, Liu W, Moonen G, Van de Water TR. 1996. Neurotrophic effects of BDNF and CNTF, alone and in combination, on postnatal day 5 rat acoustic ganglion neurons. J Neurobiol 30:246–254. Hashino E, Johnson EM, Jr., Milbrandt J, Shero M, Salvi RJ, Cohan CS. 1999. Multiple actions of neurturin correlate with spatiotemporal patterns of ret expression in developing chick cranial ganglion neurons. J Neurosci 19:8476–8486. Hegarty JL, Kay AR, Green SH. 1997. Trophic support of cultured spiral ganglion neurons by depolarization exceeds and is additive with that by neurotrophins or cyclic AMP, and requires elevation of [Ca2þ]i within a set range. J Neurosci 17:1959–1970. Hempstead BL. 2006. Dissecting the diverse actions of pro- and mature neurotrophins. Curr Alzheimer Res 3:19–24. Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. 1991. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350: 678–683. Hossain WA, Antic SD, Yang Y, Rasband MN, Morest DK. 2005. Where is the spike generator of the cochlear nerve? Voltage-gated sodium channels in the mouse cochlea. J Neurosci 25:6857–6868. Hossain WA, Brumwell CL, Morest DK. 2002. Sequential interactions of fibroblast growth factor-2, brain-derived neurotrophic factor, neurotrophin-3, and their receptors define critical periods in the development of cochlear ganglion cells. Exp Neurol 175: 138–151. Huang EJ, Reichardt LF. 2001. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. Huang EJ, Reichardt LF. 2003. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609–642. Johnson EM, Jr., Taniuchi M, DiStefano PS. 1988. Expression and possible function of nerve growth factor receptors on Schwann cells. Trends Neurosci 11:299–304. Johnson GL, Lapadat R. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298:1911–1912. Jones KR, Fari~ nas I, Backus C, Reichardt LF. 1994. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989–999. Kanzaki S, Stover T, Kawamoto K, Prieskorn DM, Altschuler RA, Miller JM, Raphael Y. 2002. Glial cell line-derived neurotrophic factor and chronic electrical stimulation prevent VIII cranial nerve degeneration following denervation. J Comp Neurol 454: 350–360. Kao S, Jaiswal RK, Kolch W, Landreth GE. 2001. Identification of the mechanisms regulating the differential activation of the MAPK cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem 276:18169–18177. Keithley EM, Feldman ML. 1982. Hair cell counts in an age-graded series of rat cochleas. Hear Res 8:249–262. Kenchappa RS, Tep C, Korade Z, Urra S, Bronfman FC, Yoon SO, Carter BD. 2010. p75 neurotrophin receptor-mediated apoptosis in sympathetic neurons involves a biphasic activation of JNK and up-regulation of tumor necrosis factor-alpha-converting enzyme/ ADAM17. J Biol Chem 285:20358–20368. Kenchappa RS, Zampieri N, Chao MV, Barker PA, Teng HK, Hempstead BL, Carter BD. 2006. Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 50:219–232. Kohara K, Kitamura A, Morishima M, Tsumoto T. 2001. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291:2419–2423. Koitchev K, Guilhaume A, Cazals Y, Aran J-M. 1982. Spiral ganglion changes after massive aminoglycoside treatment in the guinea pig. Counts and ultrastructure. Acta Otolaryngol 94: 431–438. Kujawa SG, Liberman MC. 2009. Adding insult to injury: cochlear nerve degeneration after temporary noise-induced hearing loss. J Neurosci 29:14077–14085. 1893 Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. 2004. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 118:243–255. Leake PA, Hradek GT. 1988. Cochlear pathology of long term neomycin induced deafness in cats. Hear Res 33:11–33. Leake PA, Hradek GT, Hetherington AM, Stakhovskaya O. 2011. Brain-derived neurotrophic factor promotes cochlear spiral ganglion cell survival and function in deafened, developing cats. J Comp Neurol 519:1526–1545. Lee R, Kermani P, Teng KK, Hempstead BL. 2001. Regulation of cell survival by secreted proneurotrophins. Science 294: 1945–1948. Lefebvre PP, Malgrange B, Staecker H, Moghadass M, Van De Water TR, Moonen G. 1994. Neurotrophins affect survival and neuritogenesis by adult injured auditory neurons in vitro. NeuroReports 5:865–868. Liberman LD, Wang H, Liberman MC. 2011. Opposing gradients of ribbon size and AMPA receptor expression underlie sensitivity differences among cochlear-nerve/hair-cell synapses. J Neurosci 31: 801–808. Liberman MC, Oliver ME. 1984. Morphometry of intracellularly labeled neurons of the auditory nerve: correlations with functional properties. J Comp Neurol 223:163–176. Lie M, Grover M, Whitlon DS. 2010. Accelerated neurite growth from spiral ganglion neurons exposed to the Rho kinase inhibitor H-1152. Neuroscience 169:855–862. Lin A. 2003. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25:17–24. Linggi MS, Burke TL, Williams BB, Harrington A, Kraemer R, Hempstead BL, Yoon SO, Carter BD. 2005. Neurotrophin receptor interacting factor (NRIF) is an essential mediator of apoptotic signaling by the p75 neurotrophin receptor. J Biol Chem 280: 13801–13808. Lorente de N o R. 1933. Anatomy of the eighth nerve. I. The central projection of the nerve endings of the internal ear. Laryngoscope 43:1–38. Lu B. 2003. Pro-region of neurotrophins: role in synaptic modulation. Neuron 39:735–738. Lu B, Pang PT, Woo NH. 2005. The yin and yang of neurotrophin action. Nat Rev Neurosci 6:603–614. Luo F, Wang Q, Farid N, Liu X, Yan J. 2009. Three-dimensional tonotopic organization of the C57 mouse cochlear nucleus. Hear Res 257:75–82. Malgrange B, Lefebvre P, Van de Water TR, Staecker H, Moonen G. 1996. Effects of neurotrophins on early auditory neurones in cell culture. Neuroreport 7:913–917. Malgrange B, Rogister B, Lefebvre PP, Mazy-Servais C, Welcher AA, Bonnet C, Hsu RY, Rigo JM, Van De Water TR, Moonen G. 1998. Expression of growth factors and their receptors in the postnatal rat cochlea. Neurochem Res 23:1133–1138. Markus A, Zhong J, Snider WD. 2002. Raf and Akt mediate distinct aspects of sensory axon growth. Neuron 35:65–76. Maroney AC, Finn JP, Bozyczko-Coyne D, O’Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA. 1999. CEP1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem 73: 1901–1912. Matheny RW, Jr., Adamo ML. 2009. Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med 234:1264–1270. McAllister AK, Katz LC, Lo DC. 1997. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron 18:767–778. McGuinness SL, Shepherd RK. 2005. Exogenous BDNF rescues rat spiral ganglion neurons in vivo. Otol Neurotol 26:1064–1072. Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin YM, Harke B, Hell SW, Egner A, Moser T. 2009. Tuning of synapse number, structure and function in the cochlea. Nat Neurosci 12:444–453. Miller JM, Chi DH, O’Keeffe LJ, Kruszka P, Raphael Y, Altschuler RA. 1997. Neurotrophins can enhance spiral ganglion 1894 GREEN ET AL. cell survival after inner hair cell loss. Int J Dev Neurosci 15: 631–643. Minichiello L. 2009. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10:850–860. Mou K, Hunsberger CL, Cleary JM, Davis RL. 1997. Synergistic effects of BDNF and NT-3 on postnatal spiral ganglion neurons. J Comp Neurol 386:529–539. Nadol JB, Jr. 1990. Degeneration of cochlear neurons as seen in the spiral ganglion of man. Hear Res 49:141–154. Nadol JB, Jr. 1997. Patterns of neural degeneration in the human cochlea and auditory nerve: implications for cochlear implantation. Otolaryngol Head Neck Surg 117:220–228. Nadol JB, Jr., Burgess BJ, Reisser C. 1990. Morphometric analysis of normal human spiral ganglion cells. Ann Otol Rhinol Laryngol 99:340–348. Nadol JB, Jr., Young YS, Glynn RJ. 1989. Survival of spiral ganglion cells in profound sensorineural hearing loss: implications for cochlear implantation. Ann Otol Rhinol Laryngol 98:411–416. Nagappan G, Zaitsev E, Senatorov VV, Jr., Yang J, Hempstead BL, Lu B. 2009. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc Natl Acad Sci USA 106: 1267–1272. Nakaizumi T, Kawamoto K, Minoda R, Raphael Y. 2004. Adenovirus-mediated expression of brain-derived neurotrophic factor protects spiral ganglion neurons from ototoxic damage. Audiol Neurootol 9:135–143. Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. 2002. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem 277:21843–21850. ~ ez CF. 2003. The neural cell adhesion Paratcha G, Ledda F, Ib an molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 113:867–879. Parkhurst CN, Zampieri N, Chao MV. 2010. Nuclear localization of the p75 neurotrophin receptor intracellular domain. J Biol Chem 285:5361–5368. Pirvola U, Arumae U, Moshnyakov M, Palgi J, Saarma M, Ylikoski J. 1994. Coordinated expression and function of neurotrophins and their receptors in the rat inner ear during target innervation. Hear Res 75:131–144. Pirvola U, Ylikoski J, Palgi J, Lehtonen E, Arumae U, Saarma M. 1992a. Brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the peripheral target fields of developing inner ear ganglia. Proc Natl Acad Sci USA 89:9915–9919. Pirvola U, Ylikoski J, Palgi J, Lehtonen E, Arumae U, Saarma M. 1992b. Brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the peripheral target fields of developing inner ear ganglia. Proc Natl Acad Sci USA 89:9915–9919. Postigo A, Calella AM, Fritzsch B, Knipper M, Katz D, Eilers A, Schimmang T, Lewin GR, Klein R, Minichiello L. 2002. Distinct requirements for TrkB and TrkC signaling in target innervation by sensory neurons. Genes Dev 16:633–645. Provenzano MJ, Minner SA, Zander K, Clark JJ, Kane CJ, Green SH, Hansen MR. 2011. p75NTR expression and nuclear localization of p75NTR intracellular domain in spiral ganglion Schwann cells following deafness correlates with cell proliferation. Mol Cell Neurosci 47:306–315. Puel JL, Saffiedine S, Gervais d’Aldin C, Eybalin M, Pujol R. 1995. Synaptic regeneration and functional recovery after excitotoxic injury in the guinea pig cochlea. C R Acad Sci III—Life Sci 318: 67–75. Qiu M-S, Green SH. 1992. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged tyrosine phosphorylation of ERKs. Neuron 9:705–717. Reichardt LF. 2006. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond 361:1545–1564. Rejali D, Lee VA, Abrashkin KA, Humayun N, Swiderski DL, Raphael Y. 2007. Cochlear implants and ex vivo BDNF gene therapy protect spiral ganglion neurons. Hear Res 228:180–187. Robertson D. 1976. Possible relation between structure and spike shapes of neurones in guinea pig cochlear ganglion. Brain Res 109:487–496. Roehm PC, Hansen MR. 2005. Strategies to preserve or regenerate spiral ganglion neurons. Curr Opin Otolaryngol Head Neck Surg 13:294–300. Rosenbluth J. 1962. The fine structure of acoustic ganglia in the rat. J Cell Biol 12:329–359. Rouiller EM, Cronin-Schreiber R, Fekete DM, Ryugo DK. 1986. The central projections of intracellularly labeled auditory nerve fibers in cats: an analysis of terminal morphology. J Comp Neurol 249: 261–278. Rubel EW, Fritzsch B. 2002. Auditory system development: primary auditory neurons and their targets. Annu Rev Neurosci 25: 51–101. Rueda J, De La Sen C, Juiz JM, Merch an JA. 1987. Neuronal loss in the spiral ganglion of young rats. Acta Otolaryngol 104:417–421. Ruttiger L, Panford-Walsh R, Schimmang T, Tan J, Zimmermann U, Rohbock K, Kopschall I, Limberger A, Muller M, Fraenzer JT, Cimerman J, Knipper M. 2007. BDNF mRNA expression and protein localization are changed in age-related hearing loss. Neurobiol Aging 28:586–601. Ryugo DK. 1992. The auditory nerve: peripheral innervation, cell body morphology, and central projections. In: Webster DB, Popper AN, Fay RR, editors. The mammalian auditory pathway: neuroanatomy. New York: Springer-Verlag. p34–93. Sato T, Doi K, Taniguchi M, Yamashita T, Kubo T, Tohyama M. 2006. Progressive hearing loss in mice carrying a mutation in the p75 gene. Brain Res 1091:224–234. Schecterson LC, Bothwell M. 1994. Neurotrophin and neurotrophin receptor mRNA expression in developing inner ear. Hear Res 73: 92–100. Schimmang T, Tan J, Muller M, Zimmermann U, Rohbock K, Kopschall I, Limberger A, Minichiello L, Knipper M. 2003. Lack of BDNF and TrkB signalling in the postnatal cochlea leads to a spatial reshaping of innervation along the tonotopic axis and hearing loss. Development 130:4741–4750. Schweigreiter R. 2006. The dual nature of neurotrophins. Bioessays 28:583–594. Shepherd RK, Coco A, Epp SB, Crook JM. 2005. Chronic depolarization enhances the trophic effects of brain-derived neurotrophic factor in rescuing auditory neurons following a sensorineural hearing loss. J Comp Neurol 486:145–158. Shibata SB, Budenz CL, Bowling SA, Pfingst BE, Raphael Y. 2011. Nerve maintenance and regeneration in the damaged cochlea. Hear Res 281:56–64. Shinohara T, Bredberg G, Ulfendahl M, Pyykko I, Olivius NP, Kaksonen R, Lindstrom B, Altschuler R, Miller JM. 2002. Neurotrophic factor intervention restores auditory function in deafened animals. Proc Natl Acad Sci USA 99:1657–1660. Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M. 1997. Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur J Neurosci 9:2045–2056. Singer W, Panford-Walsh R, Watermann D, Hendrich O, Zimmermann U, Kopschall I, Rohbock K, Knipper M. 2008. Salicylate alters the expression of calcium response transcription factor 1 in the cochlea: implications for brain-derived neurotrophic factor transcriptional regulation. Mol Pharmacol 73:1085–1091. Slepecky NB. 1996. Structure of the mammalian cochlea. In: Dallos P, Popper AN, Fay RR, editors. The cochlea. New York: SpringerVerlag. p44–129. Smith FL, Davis RL. 2012. Neurotrophins can differentially alter the soma area and survival of apical and basal spiral ganglion neurons in vitro. In: Presented at the 35th Annual Midwinter Meeting of the Association for Research in Otolaryngology, San Diego, CA. Song HJ, Poo MM. 1999. Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol 9: 355–363. Spoendlin H. 1971. Degeneration behavior of the cochlear nerve. Archiv Klin Exp Ohren Nasen Kehlkopfheilk 200:275–291. Spoendlin H. 1973. The innervation of the cochlear receptor. In: Møller AR, Boston P, editors. Basic mechanisms in hearing. New York: Academic Press. p185–234. NEUROTROPHINS IN THE COCHLEA Staecker H, Kopke R, Malgrange B, Lefebvre P, Van de Water TR. 1996. NT-3 and/or BDNF therapy prevents loss of auditory neurons following loss of hair cells. Neuroreport 7:889–894. Staecker H, Liu W, Hartnick C, Lefebvre P, Malgrange B, Moonen G, Van de Water TR. 1995. NT-3 combined with CNTF promotes survival of neurons in modiolus-spiral ganglion explants. Neuroreport 6:1533–1537. Stankovic K, Rio C, Xia A, Sugawara M, Adams JC, Liberman MC, Corfas G. 2004. Survival of adult spiral ganglion neurons requires erbB receptor signaling in the inner ear. J Neurosci 24: 8651–8661. Stankovic KM, Corfas G. 2003. Real-time quantitative RT-PCR for low-abundance transcripts in the inner ear: analysis of neurotrophic factor expression. Hear Res 185:97–108. St€over T, Gong TL, Cho Y, Altschuler RA, Lomax MI. 2000. Expression of the GDNF family members and their receptors in the mature rat cochlea. Brain Res Mol Brain Res 76:25–35. St€over T, Nam Y, Gong TL, Lomax MI, Altschuler RA. 2001. Glial cell line-derived neurotrophic factor (GDNF) and its receptor complex are expressed in the auditory nerve of the mature rat cochlea. Hear Res 155:143–151. Sugawara M, Corfas G, Liberman MC. 2005. Influence of supporting cells on neurodegeneration after hair cell loss. J Assoc Res Otolaryngol 6:136–147. Sugawara M, Murtie JC, Stankovic KM, Liberman MC, Corfas G. 2007. Dynamic patterns of neurotrophin 3 expression in the postnatal mouse inner ear. J Comp Neurol 501:30–37. Tan J, Clarke M, Barrett G, Millard R. 2010. The p75 neurotrophin receptor protects primary auditory neurons against acoustic trauma in mice. Hear Res 268:46–59. Tan J, Shepherd RK. 2006. Aminoglycoside-induced degeneration of adult spiral ganglion neurons involves differential modulation of Tyrosine Kinase B and p75 neurotrophin receptor signaling. Am J Pathol 169:528–543. Teng KK, Felice S, Kim T, Hempstead BL. 2010. Understanding proneurotrophin actions: recent advances and challenges. Dev Neurobiol 70:350–359. Teng KK, Hempstead BL. 2004. Neurotrophins and their receptors: signaling trios in complex biological systems. Cell Mol Life Sci 61: 35–48. Tessarollo L, Coppola V, Fritzsch B. 2004. NT-3 replacement with brain-derived neurotrophic factor redirects vestibular nerve fibers to the cochlea. J Neurosci 24:2575–2584. Verdi JM, Groves AK, Fari~ nas I, Jones K, Marchionni MA, Reichardt LF, Anderson DJ. 1996. A reciprocal cell-cell interaction mediated by NT-3 and neuregulins controls the early survival and development of sympathetic neuroblasts. Neuron 16:515–527. von Bartheld CS, Patterson SL, Heuer JG, Wheeler EF, Bothwell M, Rubel EW. 1991. Expression of nerve growth factor (NGF) receptors in the developing inner ear of chick and rat. Development 113:455–470. Wang LY, Gan L, Forsythe ID, Kaczmarek LK. 1998. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol 509 (Pt 1):183–194. Wang Q, Green SH. 2011. Functional role of neurotrophin-3 in synapse regeneration by spiral ganglion neurons on inner hair cells after excitotoxic trauma in vitro. J Neurosci 31:7938–7949. Wang Q, Liang I-C, Kane CJ, Green SH. 2009. Inner hair cell (IHC)-spiral ganglion neuron (SGN) synapse regeneration following excitotoxic trauma is deficient in mice lacking P75NTR. In: Presented at the 32nd Annual Midwinter Meeting of the Association for Research in Otolaryngology, Baltimore, MD. Warnecke A, Scheper V, Buhr I, Wenzel GI, Wissel K, Paasche G, Berkingali N, Jorgensen JR, Lenarz T, Stover T. 2010. Artemin improves survival of spiral ganglion neurons in vivo and in vitro. Neuroreport 21:517–521. 1895 Webster M, Webster DB. 1981. Spiral ganglion neuron loss following organ of Corti loss: a quantitative study. Brain Res 212:17–30. Wheeler EF, Bothwell M, Schecterson LC, von Bartheld CS. 1994. Expression of BDNF and NT-3 mRNA in hair cells of the organ of Corti: quantitative analysis in developing rats. Hear Res 73: 46–56. Whitlon DS, Grover M, Tristano J, Williams T, Coulson MT. 2007. Culture conditions determine the prevalence of bipolar and monopolar neurons in cultures of dissociated spiral ganglion. Neuroscience 146:833–840. Widenmaier SB, Ao Z, Kim SJ, Warnock G, McIntosh CH. 2009. Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J Biol Chem 284:30372–30382. Wiechers B, Gestwa G, Mack A, Carroll P, Zenner HP, Knipper M. 1999. A changing pattern of brain-derived neurotrophic factor expression correlates with the rearrangement of fibers during cochlear development of rats and mice. J Neurosci 19:3033–3042. Wise AK, Hume CR, Flynn BO, Jeelall YS, Suhr CL, Sgro BE, O’Leary SJ, Shepherd RK, Richardson RT. 2010. Effects of localized neurotrophin gene expression on spiral ganglion neuron resprouting in the deafened cochlea. Mol Ther 18:1111–1122. Wise AK, Richardson R, Hardman J, Clark G, O’Leary S. 2005. Resprouting and survival of guinea pig cochlear neurons in response to the administration of the neurotrophins brain-derived neurotrophic factor and neurotrophin-3. J Comp Neurol 487: 147–165. Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B. 2005. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8: 1069–1077. Wu C, Lai CF, Mobley WC. 2001. Nerve growth factor activates persistent Rap1 signaling in endosomes. J Neurosci 21:5406–5416. Yagi M, Kanzaki S, Kawamoto K, Shin B, Shah PP, Magal E, Sheng J, Raphael Y. 2000. Spiral ganglion neurons are protected from degeneration by GDNF gene therapy. J Assoc Res Otolaryngol 1: 315–325. Yamashita T, Tucker KL, Barde YA. 1999. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24:585–593. Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. 2009. Neuronal release of proBDNF. Nat Neurosci 12:113–115. Yang T, Kersigo J, Jahan I, Pan N, Fritzsch B. 2011. The molecular basis of making spiral ganglion neurons and connecting them to hair cells of the organ of Corti. Hear Res 278:21–33. Ylikoski J, Pirvola U, Moshnyakov M, Palgi J, Arum€ ae U, Saarma M. 1993. Expression patterns of neurotrophin and their receptor mRNAs in the rat inner ear. Hear Res 65:69–78. Ylikoski J, Pirvola U, Virkkala J, Suvanto P, Liang XQ, Magal E, Altschuler R, Miller JM, Saarma M. 1998. Guinea pig auditory neurons are protected by glial cell line-derived growth factor from degeneration after noise trauma. Hear Res 124:17–26. Zha X-M, Bishop JF, Hansen MR, Victoria L, Abbas PJ, Mouradian MM, Green SH. 2001. BDNF synthesis in spiral gangion neurons is constitutive and CREB-dependent. Hear Res 156:53–68. Zheng JL, Stewart RR, Gao W-Q. 1995. Neurotrophin-4/5 enhances survival of cultured spiral ganglion neurons and protects them from cisplatin neurotoxicity. J Neurosci 15:5079–5087. Zhou Z, Liu Q, Davis RL. 2005. Complex regulation of spiral ganglion neuron firing patterns by neurotrophin-3. J Neurosci 25: 7558–7566. Zilberstein Y, Liberman MC, Corfas G. 2012. Inner hair cells are not required for survival of spiral ganglion neurons in the adult cochlea. J Neurosci 32:405–410.