- Wiley Online Library
advertisement

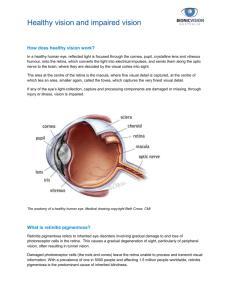
Ocular perfusion and age-related macular degeneration Thomas A. Ciulla, Alon Harris and Bruce J. Martin Indiana University School of Medicine, Indianapolis, Indiana, USA ABSTRACT. Purpose: To review the role of ocular perfusion in the pathophysiology of agerelated macular degeneration (AMD), the leading cause of irreversible blindness in the industrialized world. Methods: Medline search of the literature published in English or with English abstracts from 1966 to 2000 was performed using various combinations of relevant key words. Results: Vascular defects have been identified in both nonexudative and exudative AMD patients using fluorescein angiographic methods, laser Doppler flowmetry, indocyanine green angiography, and color Doppler imaging. Conclusion: Although these studies lend some support to the vascular pathogenesis of AMD, it is not possible to determine if the choroidal perfusion abnormalities play a causative role in nonexudative AMD, if they are simply an association with another primary alteration, such as a primary RPE defect or a genetic defect at the photoreceptor level, or if they are more strongly associated with one particular form of this heterogeneous disease. Further study is warranted. Key words: age-related macular degeneration – ocular perfusion – blood flow – choriod – choriocapiIlaris – retinal circulation. Acta Ophthalmol. Scand. 2001: 79: 108–115 Copyright c Acta Ophthalmol Scand 2001. ISSN 1395-3907 A ge related macular degeneration (AMD) is the leading cause of irreversible visual loss in the United States, occurring in over 10% of the population aged 65 to 74 years and over 25% of the population over the age of 74 years (Leibowitz et al. 1980). There are two types of macular degeneration. The nonexudative form involves atrophic and hypertrophic changes in the retinal pigment epithelium (RPE) underlying the central retina or macula, drusen. Patients with nonexudative AMD can progress to the exudative form of AMD, in which choroidal neovascular membranes (CNVM) develop under the retina, leak fluid and blood, and ultimately cause a blinding ‘‘disciform’’ scar in and under the retina. Nonexudative AMD occurs in approximately 27% of patients over 75 years, and exudative AMD occurs in nearly 5% of this group (Klein et al. 1992). Overall, ap- 108 proximately 10–20% of patients with nonexudative AMD progress to the exudative form, which is responsible for most of the estimated 1.2 million cases of severe visual loss from AMD (Hyman et al. 1993; Tielsch et al. 1995). Also, it should be noted that AMD is a bilateral disorder; CNVM develop in over onefourth (26%) of fellow eyes that are initially free of exudative AMD over a fiveyear period (MPS 1993). As the population in the United States ages, visual loss from AMD will become even more prevalent. Nonexudative AMD is much more common than exudative AMD and is usually a precursor of exudative AMD. The nonexudative form involves a variety of presentations including hard drusen, soft drusen and geographic (areolar) atrophy of the RPE. Hard drusen are associated with localized dysfunction of the RPE, while soft drusen are associated with diffuse dysfunction of the RPE (Bressler et al. 1988). Although more typically associated with exudative AMD, severe visual loss can occur from nonexudative AMD, particularly when geographic atrophy of the RPE develops in the fovea to cause a central scotoma (Ciulla et al. 1998). Unfortunately, there is no widely accepted treatment modality for this devastating form of AMD, although some investigators have suggested a protective effect from various micronutrients and vitamins and other investigators are studying laser treatment of drusen (Ciulla et al. 1998). Although the exudative form is treatable, treatment efficacy is currently low. Currently, the only well-studied and widely-accepted method of treatment is laser photocoagulation of the CNVM as demonstrated in several well designed clinical trials, including the Macular Photocoagulation Study (MPS), a group of multicenter, randomized, controlled clinical trials of laser photocoagulation of choroidal neovascularization in patients with AMD, the presumed ocular histoplasmosis syndrome, or in idiopathic cases. The MPS showed that photocoagulation effectively prevented large decreases in visual acuity compared to observation. However, only 13–26% of patients with exudative AMD show welldemarcated ‘‘classic’’ CNVM (recognized on fluorescein angiography as discreet early hyperfluorescence with late leakage) amenable to laser treatment and at least half of these patients suffer from persistent or recurrent CNVM formation within two years. Patients with poorly demarcated (due to subretinal hemorrhage, for example) or ‘‘occult’’ CNVM (recognized on fluorescein angiography as a fibrovascular pigment epithelial detachment or late leakage of undetermined source) make up the majority of patients with exudative AMD and are not eligible for Fig. 1. According to the traditonal theory, primary genetic, dietary, or photo-oxidative stress leads to RPE dysfunction, which consequently leads to choriocapillaris atrophy and choroidal perfusion defects. laser photocoagulation (Freund et al. 1993; Moisseiev et al. 1995; MPS 1982; MPS 1986; MPS 1991). Pathogenesis of AMD Traditional theory Several theories of pathogenesis have been proposed and these include primary RPE and Bruch’s membrane senescence, primary genetic defects, and primary ocular perfusion abnormalities. Oxidative insults have also been proposed as a contributing factor. Traditionally, investigators have felt that senescence of the RPE, which metabolically supports and maintains the photoreceptors, leads to AMD (Eagle 1984; Young 1987). It has been felt that the senescent RPE accumulates metabolic debris as remnants of incomplete degradation from phagocytosed rod and cone membranes and that progressive engorgement of these RPE cells leads to drusen formation with subsequent progressive further dysfunction of the remaining RPE (Eagle 1984; Young 1987). (Figs. 1 & 2). Bruch’s membrane, thickened with drusen, could be predisposed to crack formation (Green et al. 1985; Sarks 1976). Calcification and fragmentation of Bruch’s membrane is more prominent in eyes with exudative AMD, and it is thought that these defects in Bruch’s membrane could facilitate development of CNVM (Spraul & Grossniklaus 1997). This theory is supported by findings in Fig. 2. According to the traditional theory, senescent RPE is susceptible to genetic, dietary, or photo-oxidative stress, which in turn leads to failure to metabolize photoreceptor outer segments that are constantly being shed. This leads to drusen formation. In addition, RPE dysfunction or atrophy leads to choriocapillaris atrophy. myopic degeneration and angioid streaks in which CNVM develop through breaks in Bruch’s membrane. The well-known primate-laser model, developed by Ryan, may also support this theory for CNVM (Ishibashi et al. 1995, 1985, 1987; Miller et al. 1990; Nishimura et al. 1990; Ohkuma & Ryan 1982, 1983; Ryan 1979, 1982). In this model, high intensity laser burns are used to create ruptures in Bruch’s membrane/RPE complex to initiate a repair process in the fundus that results in the development of subretinal neovascularization (Miller et al. l990). The exact stimulus for CNVM formation is unclear; it is possible that macrophages involved in the initial response to Bruch’s membrane injury secrete angiogenic growth factors (Ishibashi et al. 1985; Nishimura et al. 1990). Another potential mechanism by which CNVM could develop in response to fragmentation of Bruch’s membrane could relate to matrix metalloproteinases (MMP) which are extracellular matrix degrading enzymes that may play a key role in angiogenesis and CNVM formation (Steen et al. 1998). Bruch’s membrane has been shown to contain tissue inhibitor of metalloproteinases (TlMP)-3 (Faris et al. 1997) and eyes with AMD have been shown to have abnormal levels of TIMP-3 compared to normal agematched control eyes (Kamei & Holleyfield 1999); calcification and fragmentation observed in Bruch’s membrane may represent a breach in this antiangiogenic barrier, facilitating CNVM devel- opment. Whatever the initial stimulus for CNVM formation, it is clear that angiogenic growth factors are ultimately involved. Surgically excised and post mortem CNVM tissue, as well RPE cells, have been shown to be immunoreactive for various growth factors thought to be angiogenic, including vascular endothelial growth factor (VEGF), transforming growth factor-beta (TGF-b), platelet derived growth factor (PDGF) and basic fibroblast growth factor (FGF) (Amin et al. 1994: Kvanta 1995; Lopez et al. 1996; Reddy et al. 1995). Genetic Defects Yet another theory involves genetic defects. A variety of genes have been suggested. For example, some investigators recently reported that 16% of AMD patients in their study had a genetic defect in a gene encoding a retinal rod protein, the ABCR gene, which has also been found to be defective in Stargardt’s disease (Allikmets et al. 1997). However, there have been two more recent publications suggesting that the ABCR mutations might not be linked to AMD (De La Paz et al. 1999; Stone et al. 1998). There have also been recent reports of a genetic association between AMD and apolipoprotein E, a protein that plays a role in central nervous system lipid homeostasis (Klaver et al. 1998; Souied et al. 1998). Investigators are also studying other hereditary dystrophies with some 109 features similar to AMD, such as Malattia Leventinese and Doyne’s Honeycomb retinal dystrophy, which are characterized by drusen and have been associated with a single mutation in the gene for EGFcontaining fibrillin-like extracellular matrix protein 1 (Stone et al. 1999). Sorsby’s dystrophy, an autosomal dominant disorder that presents in the fifth decade, shows several similarities to AMD, including submacular exudates and hemorrhages initially followed by cicatrization, and a point mutation on the TIMP-3 gene has been reported (Weber et al. 1994). Another group, however, found no evidence of linkage or association between the TIMP-3 gene in 38 multiplex families with AMD (De La Paz et al. 1997). group of investigators showed correlation between the regions of sparring in annular maculopathy and the spatial distribution of macular pigments (Weiter et al. 1988). Furthermore, factors known to decrease macular pigment optical density (MPOD) levels, such as cigarette smoking (Hammond et al. 1996), light iris color (Ciulla et al. 2000; Hammond et al. 1996), and female gender (Hammond et al. 1996), have also been implicated to increase the risk of AMD in epidemiologic studies; this parallel is directional, with factors that decrease MPOD leading to increased risk of AMD and factors that increase MPOD leading to decreased risk of AMD, consistent with a potential protective role of macular pigments in AMD. Oxidative Insults Ocular Perfusion Defects in AMD Oxidative insults have also been proposed as a contributing factor and this may involve the macular pigments, lutein and zeaxanthin, which are primarily obtained from dark green, leafy vegetables and account for the yellow pigmentation of the macula lutea (Bone et al. 1993, 1985). Macular pigment is of dietary origin as humans and non-human primates do not synthesize carotenoids de novo (Malinow et al. 1980; Neuringer et al. 1999). Macular pigment has been hypothesized to play a protective role against the development of AMD through the limitation of oxidative insults (Katz et al. 1982; Schalch 1992; Seddon & Hennekens 1994; Snodderly et al. 1984) by filtering out harmful wavelengths of light (Bone et al. 1985) or by the antioxidant properties of its components (Katz et al. 1982; Schalch 1999). A recent study showed that primates raised on carotenoid-depleted diets had a significantly increased incidence of angiographic transmission defects in the macular regions (Neuringer et al. 1999). Previous studies have shown that a higher dietary intake of L and Z has been associated with a lower risk for AMD (Seddon et al. 1994; West et al. 1994). Another link between macular pigment and AMD derives from observations of atrophic AMD. The central fovea, which contains the highest concentrations of macular pigment, is often spared from atrophy until late in progression, causing some investigators to postulate that macular pigments account for the relative resistance of this area to degenerative change; one 110 With regard to the role of choroidal perfusion defects, it is well known that the choriocapillaris supplies the metabolic needs of the retinal pigment epithelium and the outer retina; a primary perfusion defect in the choriocapillaris could account for some of the physiologic and pathologic changes in AMD. For example, both acute ischemia and the subsequent reperfusion of the brain are associated with CNS cell death (Chen et al. 1997; Gillardon et al. 1996; Leib et al. 1996; Macaya 1996; Nickells 1996; Quigley et al. 1995). Cell death after ischemia occurs primarily by apoptosis, especially when the insult is mild (Chen et al. 1997; Gillardon et al. 1996; Leib et al. 1996; Macaya 1996). Mild ischemia is postulated to provoke precisely such a cellular event in retinal ganglion cells in glaucoma (Chen et al. 1997; Gillardon et al. 1996; Harris et al. 2000; Leib et al. 1996; Macaya 1996; Nickells 1996; Quigley et al. 1995), which is characterized by slow loss of these cells over years, and some forms of AMD (such as geographic atrophy of the RPE characterized by progressive loss of RPE cells) could have a similar pathogenesis to glaucoma. In the vascular model for AMD pathogenesis proposed by Friedman, it is theorized that lipid deposition in sclera and Bruch’s membrane leads to scleral stiffening and impaired choroidal perfusion, which would in turn adversely affect metabolic transport function of the retinal pigment epithelium (Friedman 1997; Friedman et al. 1995). The im- paired RPE cannot metabolize and transport material shed from the photoreceptors, leading to accumulation of metabolic debris and drusen (Friedman 1997; Friedman et al. 1995). (Figs. 3–5). This theory is supported by studies demonstrating an association between increased scleral rigidity and AMD (Friedman et al. 1989). In addition, AMD, with its widely varying clinical presentations, may actually represent several distinct disorders that have yet to be more clearly differentiated on a pathogenic basis; it is possible that RPE senescence represents the primary derangement in some subforms, choroidal perfusion defects represent the primary derangement in other subforms, and perhaps specific genetic defects represent the primary defect in yet other subforms. Epidemiological risk factors, such as tobacco use (Christen et al. 1996; Seddon et al. 1996), blue light or sunlight exposure (Cruickshanks et al. 1993) and nutritional factors (MaresPerlman et al. 1995; Seddon et al. 1994) could represent environmental influences that exert a detrimental secondary effect on individuals with any of the underlying primary derangements noted above. Of note, many of the risk factors for AMD are similar to those for cardiovascular disease, including tobacco use, hypertension, and nutritional factors, lending further support for a vascular role in AMD. The vascular theory is supported by studies demonstrating delayed choroidal filling in AMD using conventional angiographic techniques (Boker et al. 1993; Remulla et al. 1995; Zhao et al. 1995). Delayed choroidal filling may have histologic significance as it may correlate with diffuse thickening of Bruch’s membrane (Pauleikhoff et al. 1990). This sign also has functional significance as eyes with this sign harbor discrete areas of increased threshold on static perimetry (Chen et al. 1992) and are at risk for loss of vision (Piguet et al. 1992). Specifically, in one study, 38% of 32 eyes with this sign lost two or more lines of vision by two years compared to 14% of 64 eyes without this sign (Piguet et al. 1992). This difference was related to the greater incidence of geographic atrophy in the patients with delayed choroidal filling; significantly, the incidence of CNVM was similar in each group of patients (Piguet et al. 1992). It is very difficult to quantify choroidal blood flow angiographically given the overlying retinal circulation and multilay- Fig. 3. According to the vascular theory, primary choroidal perfusion abnormalities lead to RPE dysfunction and then to AMD. Fig. 5. Decreased choroidal perfusion impairs RPE transport function as the RPE must pump against a higher osmotic gradient. The photoreceptor outer segments, which are constantly being shed, are not properly metabolized by the RPE, which leads to drusen formation. Fig. 4. Choroidal perfusion abnormalities arise as lipid is deposited in the sclera and Bruch’s membrane, which then leads to collagen and elastic degeneration, causing scleral stiffening. The increase in scleral rigidity leads to decreased compliance and increased resistance in the choroid, which then leads to decreased choroidal perfusion. Fig. 6. According to the vascular model as proposed by Friedman (Friedman 1997; Freidman et al. 1995), there is a generalized stiffening and increase in resistance, not only in the choroidal vasculature, but also in the cerebral vasculature. If the choroidal resistance increases more that the cerebral vascular resistance, there is a decrease in choroidal perfusion with an increase in the osmotic gradient against which the RPE must pump, leading to an accumulation of metabolic debris in the form of drusen. If the choroidal resistance increases less than the cereoral vascular resistance, there is higher choroidal perfusion pressure, which facilitates CNVM development. ered choroidal circulation that complicates analysis. Recently, however, new technologies have been employed to corroborate the existence of choroidal perfusion anomalies in AMD, including ICG angiography, laser Doppler flowmetry, and color Doppler imaging (Harris et al. 1999). One group recently used a new analysis technique based on indocyanine green angiography to compare the choroidal circulation in patients with AMD to a control group. ICG facilitates study of the choroidal circulation for several reasons. First, ICG better delineates the choroidal circulation than fluorescein because the near-infrared light absorbed by ICG penetrates the retina pigment epithelium better than the shorter wavelength absorbed by fluorescein. Also, unlike fluorescein, ICG is strongly bound to plasma proteins, which prevents diffusion of the compound through the fenestrated choroidal capillaries, and permits better delineation of choroidal details. This study showed a statistically significant increased frequency of presumed macular watershed filling (PMWF), which was described as ‘‘characteristic vertical, angled, or stellate-shaped zones of early-phase indocyanine green videoangiographic hypofluorescence, assumed to be hypoperfusion, which disappeared in the early phase of the angiogram’’ (Ross et al. l998). The investigators noted that 55.4% of 74 patients with AMD versus 15.0% 111 of 20 normal control patients exhibited PMWF. They also note that 59.0% of the 61 patients with AMD-associated choroidal neovascularization exhibited PMWF and that the CNVM arose from the PMWF zone in 91.7% of these cases. This analysis approach provides valuable insight into abnormalities of choroidal circulation in AMD, although this method requires subjective assessment for the presence and location of PMWF. Another more objective approach has been employed using a new, area dilution analysis technique applied to ICG angiography (Ciulla et al. 1998; Harris et al. 1998). Scanning laser ophthalmoscope angiograms in 21 nonexudative AMD subjects were compared to 21 agematched control subjects (Ciulla et al. 2000). After correction for eye movements, the digital image analysis system recorded mean intensity levels at each area over time. Four areas around the macula and two areas in the temporal peripapillary retina were evaluated. Fluorescence density in each block was averaged over time, graphed, and analyzed for dye appearance time and maximal fluorescence. In addition, intensity curves were analyzed by quantifying the slope of the filling portion of the curves, the amount of time required to rise 10% and 63% above baseline by graphing intensity of fluorescence of each area over time. Although the exact concentration of ICG in each region cannot be determined, simultaneous acquisition of dye dilution curves from these regions within the choroid facilitates comparison of relative concentrations between these regions. Since the six analysis regions are identically positioned on each subjects angiogram, the resulting analysis represents a very objective evaluation of choroidal perfusion characteristics and does not rely on subjective assessment. This technique showed statistically significant delayed and heterogeneous filling within the choriocapillaris of nonexudative AMD patients when compared to normal age-matched controls, and these changes showed some macular-region specificity (Ciulla et al. 2000). In addition, the localized choroidal perfusion defects correlated in a statistically significant fashion with visual acuity and severity of non-exudative agerelated macular degeneration. Another group used a new technique called laser Doppler flowmetry in subjects with nonexudative AMD to show that the choroidal blood flow was decreased at the center of the fovea com- 112 pared to a control group (Grunwald et al. 1998). This technique cannot be readily applied outside the foveal center as the overlying retinal circulation would Doppler shift the reflected light from the laser and prevent analysis of the choroid. Nevertheless, this study and the ICG angiogram studies were consistent in that this prior study showed alterations in choroidal flow in the foveal center and the ICG angiogram studies confirmed alterations with some macular-region specificity, although the foveal center itself was not measured. Color Doppler imaging has been used to evaluate the retrobulbar vasculature in AMD; two groups have found statistically significant differences in the central retinal and posterior ciliary arteries in patients with AMD compared to controls (Ciulla et al. 1999; Friedman 1995, 1997). Critics of the vascular theory point to literature that suggests that healthy RPE necessary for maintenance of the choroid and choriocapillaris (Del Priore et al. 1995, 1996; Henkind & Gartner 1983; Korte et al. 1984; Nasir et al. 1997; Pollack et al. 1996; Takeuchi et al. 1993) and that the vascular changes found in AMD are secondary to primary RPE dysfunction. In primate eyes, in which the RPE was selectively damaged by intravitreal ornithine (Takeuchi et al. 1993) and in porcine eyes in which the RPE was debrided surgically, the choriocapillaris degenerated by two months and one week, respectively (Del Priore et al. 1995, 1996). Another group obtained a similar result in a rabbit model in which the RPE was damaged by intravenous sodium iodate, and they also observed absence of the choriocapillaris histopathologically in human eyes with retinitis pigmentosa; they postulated the existence of a diffusable vascular modulation factor, produced by the RPE and responsible for choriocapillaris fenestrae formation and maintenance (Henkind & Gartner 1983; Korte et al. 1984). Some reports suggest that surgical removal of the RPE in humans (during removal of choroidal neovascular membranes) leads to abnormal perfusion of the choriocapillaris, although it is not possible to rule out preexisting atrophy or intraoperative damage (Nasir et al. 1997; Pollack et al. 1996). Although it may not be possible to definitively determine it the vascular derangements found in AMD are primary or secondary, findings from color Doppler imaging may support primary vascular dysfunction. Specifically, it is intriguing to elicit perfusion deficits in the central retinal artery of AMD subjects, as one would not necessarily anticipate a vascular derangement beyond the posterior ciliary arteries, which supply the choroid. These results suggest that there may be a more generalized perfusion defect beyond the choroid in patients with AMD. In addition, the vascular model could account for development of both the nonexudative and exudative forms of AMD. According to the vascular model, there is a generalized stiffening and increase in resistance, not only in the choroidal vasculature, but also in the cerebral vasculature (Friedman 1997; Friedman et al. 1995). If the choroidal resistance increases more than the cerebral vascular resistance, there is a decrease in choroidal perfusion with an increase in the osmotic gradient against which the RPE must pump, leading to an accumulation of metabolic debris in the form of drusen. If the choroidal resistance increases less than the cerebral vascular resistance, there is higher choroidal perfusion pressure, which facilitates CNVM development. This mechanism could partially account for the development of CNVM in the presence of Bruch’s membrane senescence or cracks. Therapies Based on Blood Flow It is unclear if improving macular perfusion would lead to improved function in nonexudative AMD or delayed disease progression. Friedman originally proposed a clinical study that involves lamellar scleral resection as a means of decreasing scleral rigidity to enhance choroidal perfusion and in turn arrest the progression of the disease. However, this proposition generated significant controversy, given the invasive nature of this procedure and the unsettled role of ocular perfusion defects in the many presentations and types of AMD. Nevertheless, several currently available topical ophthalmic and systemic pharmaceutical agents have been shown to enhance ocular blood flow in normal and glaucomatous eyes and these agents could have a potential role in AMD. In healthy individuals, for example, three different topical beta-blockers, including both Beta1selective and nonselective agents (levobunolol, betaxolol, and timolol) each re- duced the intraocular pressure (IOP), decreased the retinal arteriovenous passage time, and increased macular capillary and epipapillary passage time, without altering superior temporal arterial or venous diameters (Harris et al. 1995). These results suggest enhanced perfusion of the retina. In a subsequent comparison of timolol and betaxolol in normal tension glaucoma, there was a dissociation between IOP alterations and blood flow changes, with timolol (but not betaxolol) significantly lowering IOP, while betaxolol (but not timolol) increasing end-diastolic velocities and reducing resistance indices in the average of four different retrobulbar vessels (Harris et al. 1995). These results suggest that betaxolol can enhance ocular perfusion via a non-IOP lowering mechanism. Another pharmaceutical agent that can enhance ocular perfusion is topical dorzolamide. In 11 healthy subjects, dorzolamide reduced IOP decreased arteriovenous passage time, and increased macular and superficial optic nerve head capillary transit velocities, without altering the diameter or the superior temporal artery or vein. It did not alter flow velocities or resistance indices in four retrobulbar vessels on color Doppler imaging (Harris et al. 1996). Similar results have been obtained in studies involving subjects with normal tension glaucoma, with a 24% decrease in retinal arteriovenous passage time after dorzolamide treatment despite no significant alteration in retrobulbar flow velocities (Harris et al. 1996). Another category of agents includes calcium channel blockers. In one study, there was a correlation between the mean visual field defect and central retinal artery resistance index in 14 subjects with normal tension glaucoma who were treated with calcium channel blockers (Pillanat et al. 1994, 1995). This study also showed that those patients with the greatest initial central retinal artery resistance index experienced the greatest improvement in mean visual field defect (Pillanat et al. 1994, 1995). Another study showed an improvement in mean contrast sensitivity and alterations in ophthalmic artery peak systolic and enddiastolic velocities in 16 of 21 normal tension glaucoma patients treated with oral nifedipine (Harris et al. 1997). These findings corroborate the idea that ocular perfusion and ophthalmic visual function are correlated. Consequently, it is apparent that modu- lation of ocular blood flow is possible using currently available topical ophthalmics (glaucoma therapy) and systemic agents (calcium channel antagonists). These agents may improve visual function as measured by contrast sensitivity and visual field assessment. Further study of these agents in nonexudative age-related macular degeneration is warranted. Conclusion In summary, there appears to be a specific derangement in the choroidal circulation in patients with AMD. Specifically, vascular defects have been identified in both nonexudative and exudative AMD patients using fluorescein angiographic methods, laser Doppler flowmetry, indocyanine green angiography, and color Doppler imaging. Although these studies lend some support to the vascular pathogenesis of AMD, it is not possible to determine if the choroidal perfusion abnormalities play a causative role in nonexudative AMD, if they are simply an association with another primary alteration, such as a primary RPE defect or a genetic defect at the photoreceptor level, or if they are more strongly associated with one particular form of this heterogeneous disease. Specific issues to be addressed in future studies include correlation of perfusion defects with various subtypes of nonexudative AMD, further correlation of perfusion defects with severity of the disease process within these subtypes, correlation of perfusion defects with disease progression, and assessment of currently-available vasoactive pharmaceutics in improving macular perfusion and function in nonexudative AMD or to slow disease progression in this disorder. Acknowledgement Supported, in part, by grants from Research to Prevent Blindness, Inc.; and N.I.H. grant EY10801. Dr. Ciulla is a recipient af a career development award from Research to Prevent Blindness, Inc., New York. References Allikmets R, Shroyer N, Singh N, Seddon J, Lewis R, Bernstein P, Peiffer A, Zabriskie N, Li Y, Hutchinson A, Dean M, Lupski J & Leppert M (1997): Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 277: 1805– 1807. Amin R, Puklin J & Frank R (1994): Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest Ophthalmol Vis Sci 35: 3178–3188. Boker T, Fang T & Steinmetz R (1993). Refractive error and choroidal perfusion characteristics in patients with choroidal neovascularization and age-related macular degeneration. Ger J Ophthalmol 2: 10–13. Bone RA, Landrum JT, Hime GW, Cains A & Zamor J (1993): Stereochemistry of the human macular carotenoids. Invest Ophthalmol Vis Sci 34: 2033–2040. Bone RA, Landrum JT & Tarsis SL (1985): Preliminary identification of the human macular pigment. Vision Res 25: 1521–1525. Bressler N, Bressler S & Fine S (1988): Agerelated macular degeneration. Surv Ophthalmol 32: 375–413. Chen J, Fitzke F, Pauleikhoff D & Bird A (1992): Functional loss in age-related Bruch’s membrane change with choroidal perfusion defect. Invest Ophthalmol Vis Sci 33: 334–340. Chen J, Graham S, Nakayama M, Zhu R, Jin K, Stetler R & Simon R (1997): Apoptosis repressor genes Bcl-2 and Bcl-x-long are expressed in the rat brain following global ischemia. J Cereb Blood Flow Metab 98: 2– 10. Christen W, Glynn R, Manson J, Ajani U & Buring J (1996): A prospective study of cigarette smoking and risk in age-related macular degeneration in men. JAMA 276: 1147– 1151. Ciulla TA, Curran-Celentano J, Cooper DA, Hammond BR Jr., Danis RP, Pratt LM, Riccardi KA & Filloon TG (2000): Macular pigment optical density in a Midwestern sample. Ophthalmology (in press). Ciulla TA, Danis RP & Harris A (1998): Agerelated macular degeneration: a review of experimental treatments. Surv Ophthalmol 43: 134–146. Ciulla TA, Harris A, Chung HS, Danis RP, Kagemann L, Martin BJ & Garzozi H (2000): Choroidal perfusion is delayed in non-exudative age-related macular degeneration. Ophthalmology (in press). Ciulla TA, Harris A, Chung HS, Danis RP, Kagemann L, McNulty L, Pratt LM & Martin BJ (1999): Color Doppler imaging discloses reduced ocular blood flow velocities in nonexudative age-related macular degeneration. Am J Ophthalmol 128: 75–80. Ciulla TA, Harris A & Danis RP (1998): Presumed macular choroidal watershed vascular filling, choroidal neovascularization, and systemic vascular disease in patients with age-related macular degeneration [letter; comment]. Am J Ophthalmol 126: 153–155. Cruickshanks K, Klein R & Klein B (1993): Sunlight and age-related macular degeneration. The Beaver Dam Eye Study. Arch Ophthalmol 111: 514–518. 113 De La Paz M, Guy V, Abou-Donia S, Heinis R, Bracken B, Vance J, Gilbert J, Gass J, Haines J & Pericak-Vance M (1999): Analysis of the Stargardt disease gene (ABCR) in age-related macular degeneration. Ophthalmology 106: 1531–1536. De La Paz MA, Pericak-Vance MA, Lennon F, Haines JL & Seddon JM (1997): Exclusion of TIMP3 as a candidate locus in age-related macular degeneration. Invest Ophthalmol Vis Sci 38: 1060–1065. Del Priore L, Hornbeck R, Kaplan H, Jores Z, Valentino T, Mosinger-Ogilvie J & Swinn M (1995): Debridement of the pig retinal pigment epithelium in vivo. Arch Ophthalmol 113: 939–944. Del Priore L, Kaplan H, Hornbeck R, Jones Z & Swinn M (1996): Retinal pigment epithelial debridement as a model for the pathogenesis and treatment of macular degeneration. Am J Ophthalmol 122: 629–643. Eagle RJ (1984): Mechanisms of maculopathy. Ophthalmology 91: 613–625. Faris R, Apte S, Olsen B, Iwata K & Milam A (1997): Tissue inhibitor of metalloproteinases-3 is a component of Bruch’s membrane of the eye. Am J Pathol 150: 323–328. Freund K, Yannuzzi L & Sorenson J (1993): Age-related macular degeneration and choroidal neovascularization. Am J Ophthalmol 115: 786–791. Friedman E (1997): A hemodynamic model of the pathogenesis of age-related macular degeneration. Am J Ophthalmol 124: 677–682. Friedman E, Ivry M, Ebert E, Glynn R, Gragoudas ES & Seddon J (1989): Increased scleral rigidity and age-related macular degeneration. Surv Ophthalmol 96: 104–108. Friedman E, Krupsky S, Lane A, Oak S, Friedman E, Egan K & Gragoudas E (1995): Ocular blood flow velocity in age-related macular degeneration. Ophthalmology 102: 640–646. Gillardon F, Lenz C, Waschke K, Krajewski S, Reed J, Zimmerman M & Kuschinsky W (1996): Altered expression of Bcl-2, Bcl-x, Bax, and c-Fos colonizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Brain Res Mol Brain Res 40: 254–260. Green W, McDonnell P & Yeo J (1985): Pathologic features of senile macular degeneration. Ophthalmology 92: 615–627. Grunwald J, Hariprasad S, DuPont J, Maguire M, Fine S, Brucker A, Maguire A & Ho A (1998): Foveolar choroidal blood flow in age-related macular degeneration. Invest Ophthalmol Vis Sci 1998: 385–390. Hammond BR Jr., Curran-Celentano J, Judd S, Fuld K, Krinsky NI, Wooten BR & Snodderly DM (1996): Sex differences in macular pigment optical density: relation to plasma carotenoid concentrations and dietary patterns. Vision Res 36: 2001–2012. Hammond BR Jr., Fuld K & Snodderly DM (1996): Iris color and macular pigment optical density. Exp Eye Res 62: 293–297. Hammond BR Jr., Wooten BR & Snodderly 114 DM (1996): Cigarette smoking and retinal carotenoids: implications for age-related macular degeneration. Vision Res 36: 3003– 3009. Harris A, Arend O, Arend S & Martin B (1996): Effects of topical dorzolamide on retinal and retrobulbar hemodynamics. Acta Ophthalmol Scand 74: 569–572. Harris A, Chung HS, Ciulla TA & Kagemann L (1999): Progress in measurement of ocular blood flow and relevance to our understanding of glaucoma and age-related macular degeneration. Prog Retin Eye Res 18: 669– 687. Harris A, Ciulla TA, Kagemann L, Zarfait D & Martin B (2000): Vasoprotection as neuroprotection for the optic nerve. Eye 14: 473–475. Harris A, Evans D, Cantor L & Martin B (1997): Hemodynamic and visual function effects of oral nifedipine in normal tension glaucoma. Am J Ophthalmol 124: 296–302. Harris A, Kagemann L, Chung H & Ciulla T (1998): Ophthalmic Imaging and Diagnostics: The use of dye dilution curve analysis in the quantification of indocyanine green angiograms of the human choroid. Ophthalmology Clinics of North America, St Louis, W. B. Saunders. Harris A, Shoemaker J, Burgoyne J, Weinland M, Sponsel W & Cantor L (1995): The acute effect of topical beta-adrenergic antagonists on normal perimacular hemodynamics. J Glaucoma 4: 36–40. Harris A, Spaeth G, Sergott R, Katz L, Cantor L & Martin B (1995): Retrobulbar arterial hemodynamic effects of betaxolol and timolol in normal-tension glaucoma. Am J Ophthalmol 120: 168–175. Henkind P & Gartner S (1983): The relationship between retinal pigment epithelium and the choriocapillaris. Trans Ophthalmol Soc UK 103: 444–447. Hyman L, He O, Grimson R, Oden N, Schachat A, Leske M & a T A-r MPS Group (1992): Risk factors for age-related maculopathy. Invest Ophthalmol Vis Sci (Suppl) 33: 548. Ishibashi T, Inomata H, Sakamoto T & Ryan S (1995): Pericytes of newly formed vessels in experimental subretinal neovascularization. Arch Ophthalmol 113: 227–231. Ishibashi T, Miki K, Sorgente N, Patterson R & Ryan S (1985): Effects of intravitreal administration of steroids on experimental subretinal neovascularizalion in the subhuman primate. Arch Ophthalmol 103: 708– 711. Ishibashi T, Miller H, Orr G, Sorgente N & Ryan S (1987): Morphologic observations on experimental subretinal neovascularization in the monkey. Invest Ophthalmol Vis Sci 28: 1116–1130. Kamei M & Holleyfield J (1999): TIMP-3 in Bruch’s membrane: change during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci 40: 2367–2375. Katz M, Parker K, Handelman G, Bramel T & Ea D (1982): Effects of antioxidant nutrient deficiency on the retina and retinal pigment epithelium of albino rats: A light and electron microscopic study. Exp Eye Res 34: 339–369. Klaver C, Kliffen M, van Duijn C, Hofman A, Cruts M, Grobbee D, van Broeckhoven C & de Jong P (1998): Genetic association of apoliprotein E with age-related macular degeneration. Am J Hum Genet 63: 200–206. Klein R, Klein B & Linton K (1992): Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology 99: 933–943. Korte G, Reppucci V & Henkind P (1984): RPE destruction cause choriocapillary atrophy. Invest Ophthalmol Vis Sci 25: 1135– 1145. Kvanta A (1995): Expression and regulation of vascular endothelial growth factor in choroidal fibroblasts. Curr Eye Res 14: 1015– 1020. Leib S, Kim Y, Chow L, Sheldon T & Tauber M (1996): Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest 98: 2632–2639. Leibowitz H, Krueger D, Maunder L, Milton R, Kini M, Kahn H, Nickerson R, Pool J, Colton T, Ganley J, Loewenstein J & Dawber T (1980): The Framingham Eye Study monograph: An ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol 24: 335–610. Lopez PF, Sippy BD, Lambert HM, Thach AB & Hinton DR (1996): Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci 37: 855–868. Macaya A (1996): Apoptosis in the nervous system. Rev Neurol 24: 1356–1360. Malinow MR, Feeney-Burns L, Peterson LH, Klein ML & Neuringer M (1980): Diet-related macular anomalies in monkeys. Invest Ophthalmol Vis Sci 19: 857–863. Mares-Perlman J, Brady W, Klein R, Klein B, Bowen P, Stacewicz-Sapuntzakis M & Palta M (1995): Serum antioxidants and age-related macular degeneration in a populationbased case-control study. Arch Ophthalmol 113: 1518–1523. Miller H, Miller B, Ishibashi T & Ryan S (1990): Pathogenesis of laser-induced choroidal subretinal neovascularization. Invest Ophthalmol Vis Sci 31: 899–908. Moisseiev J, Alhalel A, Masuri R & Treister G (1995): The impact of the macular photocoagulation study results on the treatment of exudative age-related macular degeneration. Arch Ophthalmol 113: 185–189. MPS (1982): Argon laser photocoagulation for senile macular degeneration. Results of a randomized clinical trial. Arch Ophthalmol 100: 912–918. MPS (1986): Argon laser photocoagulation for neovascular maculopathy. Three-year results from randomized clinical trials. Arch Ophthalmol 104: 694–701. MPS (1991): Argon laser photocoagulation for neovascular maculopathy. Five-year results from randomized clinical trials. Arch Ophthalmol 109: 1109–1114. MPS (1993): Five-year follow-up of fellow eyes of patients with age-related macular degeneration and unilateral extrafoveal choroidal neovascularization. Arch Ophthalmol 111: 1189–1199. Nasir M, Sugino I & Zarbin M (1997): Decreased choriocapillaris perfusion following surgical excision of choroidal neovascular membranes in age-related macular degeneration. Br J Ophthalmol 81: 481–489. Neuringer M, Klein M, Snodderly M, Hohnson E, Feeney-Burns L & Kittleson C (1999): Effects of carotenoid depletion and N-3 fatty acid status on macular defects in Rhesus monkeys (ARVO abstract). Invest Ophthalmol Vis Sci (suppl) 40: 164. Nickells R (1996): Retinal ganglion cell death in glaucoma: the how, the why, and the maybe. J Glaucoma 5: 345–356. Nishimura T, Goodnight R, Prendergast R & Ryan S (1990): Activated macrophages in experimental subretinal neovascularization. Ophthalmologica 200: 39–44. Ohkuma H & Ryan S (1982): Vascular casts of experimental subretinal neovascularization in monkeys: a preliminary report. Jpn J Ophthalmol 26: 150–158. Ohkuma H & Ryan S (1983): Experimental subretinal neovascularization in the monkey. Permeability of new vessel. Arch Ophthalmol 101: 1102–1110. Ohkuma H & Ryan S (1983): Vascular casts of experimental subretinal neovascularization in monkeys. Invest Ophthalmol Vis Sci 24: 481–490. Pauleikhoff D, Chen J, Chisholm I & Bird A (1990): Choroidal perfusion abnormality with age-related Bruch’s membrane change. Am J Ophthalmol 109: 211–217. Piguet B, Palmvang I, Chisholm I, Minassian D & Bird A (1992): Evolution of age-related macular degeneration with choroidal perfusion abnormality. Am J Ophthalmol 113: 657–663. Pillunat L, Lang G & Harris A (1994): The visual response to increased ocular blood flow in normal pressure glaucoma. Surv Ophthalmol 38: S139-S145. Pillunat L, Lang G & Harris A (1995): Ocular carbon dioxide reactivity and calcium channel blockers in normal tension glaucoma. Glaucoma, Ocular Blood Flow and Drug Treatment. S. M. Drance. Amsterdam, Kugler: 67–71. Pollack J, Del Priore L, Smith M, Feiner M & Kaplan H (1996): Postoperative abnormalities of the choriocapillaris in exudative age- related macular degeneration. Br J Ophthalmol 80: 314–318. Quigley H, Nickells R, Kerrigan L, Pease M, Thibault D & Zack D (1995): Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci 36: 774–786. Reddy VM, Zamora RL & Kaplan HJ (1995): Distribution of growth factors in subfoveal neovascular membranes in age-related macular degeneration and presumed ocular histoplasmosis syndrome. Am J Ophthalmol 120: 291–301. Remulla J, Gaudio A, Miller S & Sandberg M (1995): Foveal electroretinograms and choroidal perfusion characteristics in fellow eyes of patients with unilateral neovascular agerelated macular degeneration. Br J Ophthalmol 79: 558–561. Ross R, Barofsky J, Cohen G, Baber W, Palao S & Gitter K (1998): Presumed macular choroidal watershed vascular filling, choroidal neovascularization, and systemic vascular disease in patients with age-related macular degeneration. Am J Ophthalmol 125: 71–80. Ryan S (1979): The development of an experimental model of subretinal neovascularization in disciform macular degeneration. Trans Am Ophthalmol Soc 77: 707–745. Ryan S (1980): Subretinal neovascularization. Natural history of an experimental model. Arch Ophthalmol 100: 1804–1809. Sarks SH (1976): Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol 60: 324–341. Schalch W (1992): Carotenoids in the retina – a review of their possible role to prevent or limit damage caused by light and oxygen. Free Radicals and Aging 62: 280–298. Seddon J, Ajani U, Sperduto R, Hiller R, Blair N, Burton T, Farber M, Gragoudas E, Haller J, Miller D et al. (1994): Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group [see comments] [published erratum appears in JAMA 1995 Feb 22;273(8):622]. JAMA 272: 1413– 1420. Seddon J & Hennekens C (1994): Vitamins, minerals, and macular degeneration: Promising but unproven hypotheses. Arch Ophthalmol 112: 176–178. Seddon J, Willet W, Speizer F & Hankinson S (1996): A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA 276: 1141–1146. Snodderly DM, Auran JD & Delori FC (1984): The macular pigment. II. Spatial distribution in primate retinas. Invest Ophthalmol Vis Sci 25: 674–685. Souied E, Benlian P, Amouyel P, Feingold J, Lagarde J, Munnich A, Kaplan J, Coscas G & Soubrane G (1998): The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am J Ophthalmol 125: 353–359. Spraul C & Grossniklaus H (1997): Character- istics of drusen and Bruch’s membrane in postmortem eyes with age-related macular degeneration. Arch Ophthalmol 115: 267– 273. Steen B, Sejersen S, Berglin L, Seregard S & Kvanta A (1998): Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Invest Ophthalmol Vis Sci 39: 2194–2200. Stone E, Lotery A, Munier F, Heon E, Piguet B, Guymer R, Vandenburgh K, Cousin P, Nishimura D, Swiderski R, Silvestri G, Mackey D, Hageman G, Bird A, Sheffield V & Schorderet D (1999): A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet 22: 199–202. Stone E, Webster A, Vandenburgh K, Streb L, Hockey R, Lotery A & Sheffield V (1998): Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration (letter). Nat Genet 20: 328– 329. Takeuchi M, Itagaki T, Takahashi K, Ohkuma H & Uyama M (1993): Changes in the intermediate stage of retinal degeneration after intravitreal injection of ornithine. Nippon Ganka Gakkai Zasshi 1: 17–28. Tielsch J, Javitt J, Coleman A, Katz J & Sommer A (1995): The prevalence of blindness and visual impairment among nursing home residents in Baltimore [see comments]. N Engl J Med 332: 1205–1209. Weber B, Vogt G, Pruett R, Stohr H & Felbor U (1994): Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet 4: 352–356. Weber B, Vogt G, Wolz W, Ives E & Ewing C (1994): Sorsby’s fundus dystrophy is genetically linked to chromosome 22q13-qter. Nat Genet 7: 158–161. Weiter JJ, Delori F & Dorey CK (1988): Central sparing in annular macular degeneration. Am J Ophthalmol 106: 286–292. West S, Vitale S, Hallfrisch J (all names, please) (1994): Antioxidants and supplements: Protective for age-related macular degeneration? Arch Ophthalmol 112: 222–227. Young R (1987): Pathophysiology of age-related macular degeneration. Surv Ophthalmol 31: 291–306. Zhao J, Frambach D, Lee P, Lee M & Lopez P (1995): Delayed macular choriocapillary circulation in age-related macular degeneration. Int Ophthalmol 19: 1–12. Received on August 18th, 2000. Accepted on August 25th, 2000. Corresponding author: Alon Harris, Ph.D. 702 Rotary Circle, IUMC Indianapolis, IN, 46260 USA Phone: 317 278 0134 115