International Journal of Inorganic Materials 2 (2000) 123–129
XRD study of ZrW2 O 8 versus temperature and pressure
´ a,
Jose´ Manuel Gallardo-Amores a , Ulises Amador b , *, Emilio Moran
´
Miguel Angel
Alario-Franco a
a
´
Lab. de Altas Presiones, Fac. de Ciencias Quımicas
, Universidad Complutense, 28040 -Ciudad Universitaria-Madrid, Madrid, Spain
b
´
´
´
´
Inorganica
y Materiales, F.C.C. Experimentales y Tecnicas
, Universidad San Pablo CEU, Urb. Monteprıncipe
,
Dep. Quımica
28668 -Boadilla del Monte, Madrid, Spain
Received 15 December 1999; accepted 27 December 1999
Abstract
ZrW2 O 8 samples have been prepared from ZrO 2 and WO 3 precursors by a ceramic method. Different portions of this material were
subjected to non cumulative pressure treatments within the 2–65 Kbar range; the metastable high-pressure phase, g-ZrW2 O 8 , has been
isolated by quenching from 6 Kbar. Further pressure increases up to 10 Kbar produced the volume reduction of the g-phase; a minimum
volume was found even after having released the pressure. Otherwise, heating above 6008C, followed by quenching at 77 K leads to the
high-temperature phase, b-ZrW2 O 8 , which is stable at room temperature. Finally, when g-ZrW2 O 8 was subjected to pressure and
temperature, g-ZrW2 O 8 could also be quenched with a still larger volume reduction at 6 Kbar and 6008C. 2000 Elsevier Science Ltd.
All rights reserved.
Keywords: A. ceramics; C. high pressure; C. X-ray diffraction; D. thermal expansion
1. Introduction
Most solids expand when heated; this is a direct effect
of the increased amplitude of thermal vibration at a higher
temperature. Nevertheless, there is increasing evidence of
thermal contraction in solids as reported by different
authors: for example, the ZrV22x Px O 7 solid solution [1–3],
compounds with the general formula A 2 M 3 O 12 (where M
is Mo or W and A can be Y and Sc) and materials in the
Ta 2 O 5 –WO 3 system [4–6]. These systems present an
isotropic thermal contraction, that is, all cell parameters
decrease under heating.
In more recent studies on aluminosilicates, such as
cordierite (Mg 2 Al 2 Si 5 O 18 ) and phosphates such as NZP
(NaZr 2 (PO 4 ) 3 ), an anisotropic variation of the cell parameters has been reported, giving rise to a light expansion of
the cell volume. In these structures, thermal increase leads
to the normal expansion in one or two dimensions causing
the polyhedra to rotate in such a way that it results in
contraction in another direction [7]. In other cases, as for
*Corresponding author. Tel.: 134-1352-0144 ext.229; fax: 134-13510475.
E-mail addresses: amores@eucmos.sim.ucm.es (J.M. GallardoAmores), uamador@ceu.es (U. Amador)
the zeolite b-eucryptite, the interstitial cations, when
heated, may alter their position changing to other available
sites, and thus giving rise to the increase of one or two cell
parameters [8,9].
Other families of compounds such as MeW2 O 8 (Me5
Hf, Mo, . . . ) and different solid solutions of these show a
similar behaviour [10,11] but, in this case the thermal
contraction is greater than in those previously mentioned;
it is also remarkable that the contraction takes place in the
entire stability range, 0.3–1003 K. These crystals are
constituted by linked polyhedra which present, when
heated, a balance of forces between the static bending and
the dynamic thermal vibration of the oxygen atoms [12].
The result is a whole cell volume reduction. It is obviously
of much interest to see what influence pressure has on
these types of materials. Evans et al. [10] have shown that
a phase transition at about 6 Kbar preceeds the amorphization that takes place at higher pressure and temperature.
Meanwhile, when only heated above 473 K, a new phase is
obtained, b-ZrW2 O 8 , changing the space group, from P2 1 3
to a centric one Pa-3 [13]. More recently, a new trigonal
symmetry polymorph of ZrW2 O 8 has been prepared using
a non-hydrolitic sol–gel method [14].
Moreover, ZrW2 O 8 presents a metastable character when
synthesised at room pressure, partially decomposing if
1466-6049 / 00 / $ – see front matter 2000 Elsevier Science Ltd. All rights reserved.
PII: S1466-6049( 00 )00004-0
124
J.M. Gallardo-Amores et al. / International Journal of Inorganic Materials 2 (2000) 123 – 129
careful quenching is not performed. In recent works, a new
synthesis at low temperature has been proposed to avoid
the traditional ceramic method [15]. In order to obtain
further insight into the behaviour of this material we have
performed a detailed study of the influence of pressure
both at room and higher temperatures in some of these
materials. We report here some results concerning ZrW2 O 8 .
2. Experimental
2.1. Synthesis
ZrW2 O 8 powders were synthesised by a conventional
ceramic method. Zirconium and tungsten oxides, supplied
by Aldrich (99%), were calcined at 6008C for 2 h, mixed
and well ground in an agate mortar for 1 h. Then, they
were pressed giving rise to pellets of about 2 g and finally,
they were calcined at between 1200 and 14008C for 5 h.
To prevent decomposition back to the precursors due to the
metastable character of ZrW2 O 8 , different procedures were
carried out: (i) a continuous oxygen flow was passed
during the reaction, (ii) time calcination was increased,
(iii) temperature calcination was varied in the 1200–1400
range and (iv) samples were quenched either at room
temperature or at 77 K.
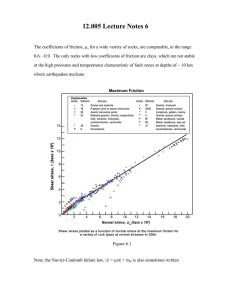
In Fig. 1, some X-ray diffraction patterns of different
preparations are compared: (a) the precursors were calcined at 12508C for 15 h in a continuous oxygen flow
followed by quenching at room temperature, (b) the same
method, except that the temperature was raised up to
13508C and (c) the precursors were calcined at 12008C for
8 h followed by quenching at 77 K. In Table 1 their cell
parameters and phase percentages are shown. All ZrW2 O 8
powders prepared in the 1200–13508C temperature range
show XRD patterns corresponding to a cubic phase (cell
˚ identified as a-ZrW2 O 8 (IDDE no.
parameter59.1477 A),
13-557) with little traces of ZrO 2 (baddeleyite, IDDE no.
72-1669) and WO 3 (IDDE no. 20-1323). The best results
were obtained at 12008C for 8 h without oxygen flow and
quenched at 77 K, obtaining a light green powder, with
|4% WO 3 present as an impurity.
Several portions were pressed at room temperature in a
2–65 Kbar range in a non-cumulative way in a belt-type
apparatus or a piston-cylinder press; samples were also
calcined in a 20–10008C range, and some of them were
also treated under the combined effect of pressure and
temperature.
2.2. Powder X-ray diffraction
Powder X-ray diffraction patterns were recorded for all
samples using a Siemens D-500 diffractometer at 35 KV
and 35 mA. This instrument is equipped with a CuK a
radiation source and a Ni-filter. For all of the reported data
sets, a step-size of 0.038 and step-time of 5 s were
employed in the entire range of analysis. All samples were
analysed after their respective treatments. The X-ray
diffraction data were analysed using the program FULLPROF
[16] to determine the cell parameters and weight ratios of
the phases present in the samples. The compositions of the
samples were obtained from the scale factors corre-
Fig. 1. X-ray patterns of several ZrW2 O 8 samples synthesised by: (a) calcination at 12508C for 15 h in continuous oxygen flow followed by quenching at
room temperature, (b) method (a) except that the temperature was varied up to 13508C and (c) calcination at 12008C for 8 h followed by quenching at 77 K.
J.M. Gallardo-Amores et al. / International Journal of Inorganic Materials 2 (2000) 123 – 129
125
Table 1
Cell parameters, volume and phase percentage for samples treated under pressure
P (Kbar)
1 atm (a)
1 atm (b)
1 atm (c)
2
4
6
8
9
10
12
..12
a
˚
a (A)
˚
b (A)
91431(4)
9.1443(7)
9.1477(3)
9.1220(8)
9.0495(9)
26.873(3)
9.1464(7)
9.0808(2)
27.065(9)
9.1139(13)
26.995(4)
9.0767(12)
27.035(4)
9.0927(11)
26.981(4)
9.0719 (15)
26.987(5)
9.0825(13)
27.041(4)
Strong increase of amorphization
3
˚
c (A)
8.8763(8)
8.929(3)
8.9258(9)
8.9216(12)
8.9217(12)
8.9119(16)
8.9235(15)
˚ )
V (A
Phase (%)
764.3
764.6
765.5
759.1
2159
765
2195
2196
2189
2187
2182
2190
a(91)–Z(3)–W(6)
a(89)–Z(5)–W(6)
a(96)–W(4)
a-ZrW2 O 8 (61)
g-ZrW2 O 8 (34)
a-ZrW2 O 8 (45)
g-ZrW2 O 8 (50)
g-ZrW2 O 8
g-ZrW2 O 8
g-ZrW2 O 8
g-ZrW2 O 8
g-ZrW2 O 8
a-ZrW2 O 8
a
a5a-ZrW2 O 8 , Z5ZrO 2 , W5WO 3 .
sponding to the different phases, assuming a similar
crystallinity.
2.3. XDS measurements
air up to 1173 K using a Seiko 320U system at a heating
rate of 10 K / min.
3. Results
The composition of some of the phases obtained was
confirmed by energy dispersive X-ray (XDS) analysis
using a Link scanning electron microscope equipped with a
Link energy-dispersive X-ray detector.
2.4. Thermal analysis
Differential thermal analyses (DTA) were performed on
3.1. Behaviour upon the increase of pressure
The X-ray patterns of a a-ZrW2 O 8 sample as prepared
by method (c) (see Section 2.1) and treated at different
pressures (within a 2–12 Kbar range) are compared in Fig.
2. When the pressure is increased, a-ZrW2 O 8 (the cubic
phase) transforms progressively into g-ZrW2 O 8 (the tetragonal phase, P2 1 2 1 2 1 space group) reaching a g:a ratio of
Fig. 2. X-ray patterns of a-ZrW2 O 8 after treated at non-cumulative different pressures.
126
J.M. Gallardo-Amores et al. / International Journal of Inorganic Materials 2 (2000) 123 – 129
about 34% at 2 Kbar with a volume contraction for both
phases, this is greater for g-ZrW2 O 8 , with about a 21.7%
of volume reduction. The orthorhombic phase proportion
becomes |50% at 4 Kbar showing cell parameters similar
to those of the initial phases; however, the g-phase appears
pure at 6 Kbar. Until this pressure, there are not significant
variations in the amount of WO 3 and ZrO 2 by-products,
indicating that at high-pressure the g-phase does not
decompose. These data are in agreement with those
˚ 3 for
reported elsewhere [17], where a volume of 2186 A
the g-phase was obtained.
Subsequently, at higher pressures the g-phase tends to
reduce the volume notably until reaching a minimum value
˚ 3 at 10 Kbar. It means a volume contraction of
of 2182 A
about 0.6% with respect to that one at 6 Kbar and a
significant amount of amorphous material appears. Further
pressure increases show the metastable character of this
structure leading to a significant volume increase up to
˚ 3 . These results are in agreement with a previous
2190 A
work [17], where the g-phase compressibility was studied
under pressure; our data refer to the quenched phases after
releasing the pressure. From 13 Kbar onwards, a very
strong amorphization process is observed and only the
main reflections of a-ZrW2 O 8 , which are progressively
weakened, can be detected in the XRD patterns (Fig. 2).
The diffraction peaks become broader above 18 Kbar and
at 40 Kbar they could not be distinguished anymore,
indicating that the samples had undergone full amorphization. No evidence of new transformations was observed up
to 65 Kbar. After releasing pressure, the high-pressure
amorphous phase was retained at room temperature. According to Perottoni et al. [18], this behaviour could be the
result of a large number of overlapped Bragg peaks from a
low-symmetry structure, which were not resolved due to
the intrinsically low resolution of the experimental technique.
On the basis of these results, it can be concluded that
this structure shows a very flexible framework. There is a
minimum volume for g-ZrW2 O 8 when subjected to a
pressure of 10 Kbar after which the structure is very
unstable and tends to transforms into a-ZrW2 O 8 and
finally, it suffers pressure-induced amorphization.
3.2. Behaviour upon the increase of temperature
In Fig. 3, X-ray patterns of a-ZrW2 O 8 powders calcined
at different temperatures are shown. It is well-known that
temperature increase produces a volume contraction of
a-ZrW2 O 8 , an effect usually known as negative thermal
expansion (NTE), which is in fact a thermal contraction
[7] but, in all cases, smaller than that reached by pressure.
Moreover, when a-ZrW2 O 8 is heated in air, an endothermic peak at 2108C together with negligible weight
losses is observed in the DTA experiments (Fig. 4). It
corresponds to the conversion of a- to b-ZrW2 O 8 with a
centric space group Pa-3. This phase was up to now only
detected by X-ray diffraction experiments during the
heating. Basically, it is characterised by the progressive
intensity decrease of the peaks 111, 221 and 310. These
are practically absent at 2408C. Otherwise, the material is
stable up to temperatures lower than 8008C, then it
decomposes in monoclinic ZrO 2 and triclinic WO 3 . If the
X-ray diffraction experiment is carried out during the
heating the tetragonal ZrO 2 (IDDE no. 71-1282) is identified, instead. We have prepared a b-ZrW2 O 8 sample by
calcining a-ZrW2 O 8 at 6008C for 4 h and quenching at 77
K (even at 3008C there is unequivocal evidence of its
stabilisation, appearing as a a1b-phase mixture). As
Fig. 3. X-ray patterns of a-ZrW2 O 8 calcined at several temperatures followed by quenching at 77 K.
J.M. Gallardo-Amores et al. / International Journal of Inorganic Materials 2 (2000) 123 – 129
127
shown in Fig. 3, there are variations in the intensities of
the above peaks named before. In spite of the fact that
these peaks should be absent at 6008C, we find a better
fitting for the model based in the space group Pa-3 (Table
2). In fact, these data can be explained taking into account
the small differences between both space groups (a symmetry centre) linked to the quenching effect which can
retain the symmetry of the local site within the average
one. Due to the well-known thermal properties of this
phase, the cell parameter is increased with respect to that
˚
calculated from the XRD pattern at 6008C to 9.1269 A
[13].
3.3. Treatments under pressure and temperature
Fig. 4. Thermal analysis (DTA) of a ZrW2 O 8 powder.
Table 2
Cell parameters, volume and phase percentage for samples treated under
temperature a
T (8C)
˚
a (A)
25 (c)
120, 2h
300, 2 h, Q
9.1477(3)
9.1471(2)
9.1475(3)
9.1439(5)
9.1468(3)
Decomposition in WO 3 (t) and
600, 2 h, Q
800, 2 h, Q
a
˚
b (A)
˚
c (A)
˚ 3)
V (A
Phase (%)
765.5
765.3
765.4
764.5
765.2
ZrO 2 (m)
a-ZrW2 O 8
a-ZrW2 O 8
a-ZrW2 O 8 (80)
b-ZrW2 O 8 (11)
b-ZrW2 O 8
t5Triclinic, m5monoclinic, Q5quenched phase and (c)5as in Fig. 1.
After studying the separate effects of pressure and
temperature on a-ZrW2 O 8 , the combined effect of both
was studied. In Fig. 5, the X-ray diffraction patterns of
samples treated simultaneously under pressure and temperature are compared and their structural features are
listed in Table 3. Samples treated at 6 Kbar and 6008C are
constituted mainly by |72% g-ZrW2 O 8 , 6% a-ZrW2 O 8 , as
well as a 9% monoclinic ZrO 2 and 13% triclinic WO 3 as
by-products with a DV|20.7% for the g-phase. These data
show the g-ZrW2 O 8 tendency to a reduction volume when
the temperature is increased, in good agreement with
Sleight et al. [17]; however, in this case, the pressure
allows the structure to be quenched and even at room
temperature. Otherwise, the increase in the amount of the
by-products and the appearance of a-ZrW2 O 3 indicate that
the g high-pressure phase is metastable and starts to
decompose at room temperature and pressure. This is
indeed confirmed by increasing the temperature by 2008C,
Fig. 5. X-ray patterns of a-ZrW2 O 8 after subjected to the combined effects of pressure and temperature.
J.M. Gallardo-Amores et al. / International Journal of Inorganic Materials 2 (2000) 123 – 129
128
Table 3
Cell parameters, volume and phase percentage for samples treated under pressure and temperature a
Conditions
6 Kbar16008C
6 Kbar18008C
10 Kbar16008C
a
˚
a (A)
˚
b (A)
˚
c (A)
˚ 3)
V (A
Phase (%)
26.968(16)
8.932(5)
759
2180
5.2334(7)
b 5101.7(1)
7.3717(8)
7.5092(7)
a 589.27(1)
b 591.17(1)
Pressure-induced amorphization
5.368(1)
134
146
a-ZrW2 O 8 (6)
g-ZrW2 O 8 (72)
Z(9)–W(13)
ZrO 2 (c,8)
ZrO 2 (m,1)
425
WO 3 (t,91)
9.123(4)
9.053(5)
5.127 (5)
5.315(1)
7.6784(7)
g 591.08(1)
a-ZrW2 O 8
c5Cubic, m5monoclinic, t5triclinic, Z5ZrO 2 , W5WO 3 .
where we did find a material constituted by ZrO 2 (cubic,
IDDE no. 27-0997), ZrO 2 (monoclinic, IDDE no. 371484) and WO 3 (triclinic, no. 83-0951) (Fig. 5). In fact,
the ZrO 2 cubic phase is unstable at room temperature, but
in this sample, the cell parameter is notably enhanced from
˚ (IDDE no. 27-0997) to a55.1223(3) A,
˚
a55.0900 A
which could be indicative of some tungsten / zirconium
replacement. Nevertheless, the percentage for this phase is
below 10%.
Experiments carried out at 10 Kbar and 6008C give rise
to almost completely amorphous materials comparable to
those obtained at 45 Kbar.
g-ZrW2 O 8 . The X-ray diffraction patterns of a sample
subjected to 6 Kbar after several treatments are compared
in Fig. 6 and the cell parameters and percentages of all
phases are listed in Table 4. After 7 days at room
temperature, the sample remarkably changes (Fig. 6),
converting to a phase mixture with a a-ZrW2 O 8 :g-ZrW2 O 8
ratio of 30:64, but if pressure again is applied, it tends to
transform back into the g-phase. In this case, the sample
reaches about 84% of g-ZrW2 O 8 after being treated at 6
Kbar for 1 h. This fact is indicative that the a→g phase
transition of ZrW2 O 8 is reversible and that g-ZrW2 O 8
shows a metastable behaviour [17].
3.4. Relaxation of the sample with time after the effect
of pressure
4. Conclusions
A study was performed with some samples that seem to
decompose with time after releasing the pressure. We show
the results obtained for the sample at 6 Kbar formed by
• a-ZrW2 O 8 shows thermal contraction in the range of
25–8008C and transforms into the b-phase at about
2108C, as detected by an endothermic peak in the
thermal analysis and the extinction of several reflec-
Fig. 6. X-ray pattern evolution of g-ZrW2 O 8 with time and subsequent treatment upon pressure at 6 Kbar.
J.M. Gallardo-Amores et al. / International Journal of Inorganic Materials 2 (2000) 123 – 129
129
Table 4
Cell parameters, volume and phase percentage for a sample treated at 6 Kbar, its evolution with time and newly treated at 6 Kbar
Conditions
˚
a (A)
˚
b (A)
˚
c (A)
˚ 3)
V (A
Phase (%)
6 Kbar
9.1139(13)
26.995(4)
8.9258(9)
2196
g-ZrW2 O 8
6 Kbar17 days
9.1465(5)
9.0694(12)
27.025(4)
8.9118(12)
765
2184
a-ZrW2 O 8 (30)
g-ZrW2 O 8 (64)
9.1396(13)
9.0686(18)
27.029(6)
8.9112(18)
763.5
2184
a-ZrW2 O 8 (13)
g-ZrW2 O 8 (81)
6 Kbar1 7 days
1 6 Kbar
tions in the X-ray diffraction pattern. b-ZrW2 O 8 was
synthesised by calcining a-ZrW2 O 8 at 6008C followed by quenching at 77 K.
• g-ZrW2 O 8 can be compressed up to 10 Kbar, where a
volume reduction even after releasing the pressure
was found. From 12 Kbar onwards, a strong pressure-induced amorphization is seen. g-ZrW2 O 8 , transforms in a g1a-phase mixture after 7 days of
releasing the pressure and tends to revert to gZrW2 O 8 if the pressure is again applied, showing the
reversible character of this transition.
• When pressure and temperature are simultaneously
applied to a-ZrW2 O 8 , g-ZrW2 O 8 is obtained with a
˚ 3 ) than that observed
larger volume reduction (2180 A
when only pressure is applied. Thus, the pressure acts
as a stabilising factor for this metastable phase.
Acknowledgements
Authors would like to thank CICYT, Project MAT
98-1053-V04-01, for financial support.
References
[1] Taylor D. J Br Ceram Trans 1984;83:5.
[2] Korthius V, Khrosrovani N, Sleight AW, Roberts N, Dupree R,
Warren Jr. WW. Chem Mater 1995;7:412–7.
[3] Khrosrovani N, Korthius V, Sleight AW. Inorg Chem 1996;35:485.
[4] Foster PM, Sleight AW. J Inter Inorg Mater 1999;1:123–7.
[5] Evans JSO, Mary TA, Sleight AW. J Solid State Chem
1998;137:148–60.
[6] Smith DD, Holcombe CE. J Am Ceram Soc 1978;61:163.
[7] Sleight AW. Endeavour 1995;64:19.
˜
´
[8] Woodcock DA, Lightfoot P, Villaescusa LA, Dıaz-Cabanas
MJ,
Camblor MA, Engberg D. Chem Mater 1999;11:2508–14.
[9] Swainson IP, Dove MT. Phys Chem Minerals 1995;22:61–5.
[10] Mary TA, Evans JSO, Vogt T, Sleight AW. Science 1996;272:90–2.
[11] Seo DK, Whangbo MH. J Solid State Chem 1997;129:160–3.
[12] Cahn RW. Nature 1997;386:22–3.
[13] Evans JSO, Mary TA, Vogt T, Subramanian MA, Sleight AW. Chem
Mater 1996;8:2809–23.
[14] Wilkinson AP, Lind C, Pattanaik S. Chem Mater 1999;11:101–8.
[15] Closmann C, Sleight AW, Haygarth JC. J Solid State Chem
1998;139:424–6.
´
[16] Rodrıguez-Carvajal
J. FULLPROF 2.4.2, ILL, December 1993.
[17] Evans JSO, Hu Z, Jorgensen JD, Argyriou DN, Short S, Sleight AW.
Science 1997;275:61–5.
[18] Perottoni CA, da Jornada JAH. Science 1998;280:886.