Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
C The Electrochemical Society
0013-4651/2011/158(8)/B1029/13/$28.00 V
B1029
Oxygen Reduction Reaction Electrocatalytic Activity of
Glancing Angle Deposited Platinum Nanorod Arrays
Wisam J. Khudhayer,a,*,z Nancy N. Kariuki,b Xiaoping Wang,b Deborah J. Myers,b
Ali U. Shaikh,c and Tansel Karabacakd
a
Department of Systems Engineering, cDepartment of Chemistry, and dDepartment of Applied Science,
University of Arkansas at Little Rock, Little Rock, Arkansas 72204, USA
b
Chemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, Illinois 60439-4837, USA
The electrocatalytic oxygen reduction reaction (ORR) activity of vertically-aligned Pt nanorods has been evaluated utilizing cyclic
voltammetry (CV) and rotating-disk electrode (RDE) techniques in a 0.1 M HClO4 solution at temperatures ranging from 20 to
60 C. A glancing angle deposition (GLAD) technique was used to fabricate Pt nanorod arrays on glassy carbon (GC) electrodes.
GLAD catalyst nanorods, without any carbon support, have been produced at different lengths varying between 50 and 400 nm,
corresponding to 0.04–0.32 mg/cm2 Pt loadings, with diameter and spacing values ranging from about 5 up to 100 nm. The scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray diffraction (XRD) results reveal that Pt
nanorods are well-isolated, vertically aligned, and single-crystal. Crystal orientation analysis demonstrates that large surface area
Pt nanorod sidewalls are mainly dominated by Pt(110) planes, which is known to be the most active crystal plane of Pt for the
ORR. Compared to a commercial high-surface-area-supported Pt (Pt/C) catalyst, the CV results show that the Pt-nanorod electrocatalyst exhibits a more positive oxide reduction peak potential, indicating that GLAD Pt nanorods are less oxophilic. Moreover,
the nanorods exhibit enhanced stability against loss of electrochemically-active surface area as a result of potential cycling in
acidic electrolyte as compared to the Pt/C catalyst. Specific ORR activities determined by the RDE technique for GLAD Pt nanorods of different lengths are analyzed and compared to literature values for polycrystalline Pt, nano-structured thin film Pt (3M
NSTF Pt), and to those measured for Pt/C. RDE results reveal that Pt-nanorod electrocatalysts exhibit higher area-specific activity,
higher electron-transfer rate constant, and comparable activation energy for ORR than those of Pt/C due to their larger crystallite
size, single-crystal property, and dominance of the preferred crystal orientations for ORR. However, Pt nanorods show lower mass
specific activity than that of Pt/C electrocatalyst due to the large diameter of nanorods.
C 2011 The Electrochemical Society.
V
[DOI: 10.1149/1.3599901] All rights reserved.
Manuscript submitted July 23, 2010; revised manuscript received April 29, 2011. Published June 22, 2011.
Fuel cells are attractive power sources for use in portable, stationary, and transportation applications.1,2 Polymer electrolyte
membrane (PEM) fuel cells, among the various types of fuel cells,
are expected to be a high impact technology because they operate at
relatively low temperatures (80 C), have high power density, can
vary their output quickly to meet changes in power demand, and are
suited for automotive applications where quick startup is required.2–4
However, the high cost of Pt electrocatalysts is one of the major barriers to widespread implementation of fuel cell power sources in a
variety of applications. The oxygen reduction reaction (ORR) at the
cathode is the major contributor to single cell efficiency losses due
to the slow kinetics, unlike the anodic hydrogen oxidation reaction
(HOR), necessitating high Pt loadings at the cathode to reduce these
losses. Typical anode and cathode electrocatalysts are comprised of
platinum nanoparticles (3–5 nm in size) supported on carbon black.3
In addition to the high cost of the typical Pt electrocatalysts, the carbon support causes additional challenges. These challenges include
oxidation of the carbon support resulting in loss of Pt nanoparticles,5
promotion of the formation of peroxide species by the carbon support leading to degradation of the membrane polymer,6 and separation of carbon from the ionomer in the electrode structure leading to
loss of catalyst electrochemically-active surface area.5 Due to these
issues, extensive efforts are underway to develop high-performance,
durable, carbon-free, and low cost (low Pt loading) electrocatalyst
materials.4
Carbon-free unsupported Pt nanoparticles (Pt-black) can avoid
the problems associated with carbon, however, the large particle size
of Pt-black translates to high cathode Pt loadings required to achieve
adequate electrochemically-active surface area (approximately 2 mg
Pt/cm2 compared to < 0.4 mg Pt/cm2 for carbon-supported Pt) and
electronic conductivity.3,4 In recent years, carbon-free catalyst electrode approaches have been proposed, such as coating of Pt films of
sub-micron thickness directly on the membrane, gas diffusion layer
(GDL),7–13 columnar Ti and CrN supports,14,15 or on an organic
* Electrochemical Society Student Member.
z
E-mail: wjkhudhayer@ualr.edu
whisker support layer.16 Using these approaches, the carbon-related
problems mentioned above can be avoided and promising results
have been demonstrated. With the exception of the organic whisker
approach, however, the fuel cell performance of such continuous Pt
film approaches7–13 generally falls below that of conventional fuel
cells with higher loadings (typically 0.4 mg Pt/cm2) due to the
continuous, non-porous, and polycrystalline structure of the Pt thin
film.
Recently, 3M has demonstrated the improved durability and
area-specific activity of nano-structured thin film Pt (3M NSTF Pt)
electrocatalyst layers consisting of large-grained polycrystalline Pt
thin films deposited on and encapsulating oriented crystalline
whiskers of an organic pigment material.16 However, the polycrystalline nature of Pt thin films has the important disadvantages of
being prone to oxidation at the grain boundaries17 and comprised of
a mixture of low index planes with varying ORR activities. The
(111) plane is the energetically-favorable growth plane of Pt films.
Single-crystal data of Markovic et al.18 show that the ORR activity
in perchloric acid electrolyte for different crystal planes of platinum
follows the trend Pt(110) > Pt(111) > Pt(100), with relative ORR
activities of approximately 1.5, 1.2, and 0.6 as compared to polycrystalline Pt.19 In sulfuric acid electrolyte, the activity trend was
found to be Pt(110) > Pt(100) >> Pt(111)17. Kadiri et al.,20 employing the same method, found that the ORR in perchloric acid (a nonadsorbing anion) is relatively insensitive to the Pt surface structure,
but is structure-sensitive in solutions containing adsorbing anions
like bisulfate, phosphate, or chloride, which impede the reaction.
Recently, Subbaraman et al.21,22 have shown that the sulfonate ion
of the perfluorosulfonic acid fuel cell electrolyte (e.g., Nafion) cannot be considered non-adsorbing and that the sensitivity of the crystal face to deactivation of ORR by adsorbed sulfonate is the same as
that of the [bi]sulfate anion. In the presence of perfluorosulfonic
acid ionomer in the fuel cell electrode, it is therefore beneficial to
decrease the ORR inhibiting effect of adsorbed sulfonate by maximizing the fraction of exposed crystal faces with weak sulfonate
adsorption (e.g., (110)). In another study, Chen et al.23 reported that
enhanced ORR electrocatalytic activity of Pt nanotubes in 0.5 M
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
B1030
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
H2SO4 might be attributed to preferential exposure of certain crystal
facets of nanotubes. Therefore, controlling the electrode roughness
factor (electrochemically-active surface area/geometric surface
area), promoting a single-crystal property, and controlling the crystal orientation by promoting the Pt[110] orientation over the Pt[111]
orientation may further enhance the electrochemical activity of
PEM fuel cell cathodes.
The aim of this study is to investigate the morphology, crystal
property, electrochemical activity, and ORR kinetics of verticallyaligned Pt nanorod arrays fabricated by the glancing angle deposition (GLAD) technique. The GLAD technique provides the novel
capability of growing 3D nanostructure arrays with interesting
material properties.24–31 It offers a simple, single-step, cost- and
time-efficient method for fabricating nano-structured arrays of various elemental materials as well as alloys and oxides. The GLAD
technique uses the “shadowing effect,” which is a “physical selfassembly” process through which obliquely incident atoms/molecules
can only deposit on the tops of higher surface points, such as the
tips of a nano-structured array or the highest points of a rough or
patterned substrate. Recently, Gasda et al.32 compared the fuel cell
performance of conventional polycrystalline Pt thin films and Pt
particles produced by GLAD and demonstrated that the particleelectrodes exhibit a higher mass activity than continuous-layer film
and conventional Teflon-bonded Pt-black (TBPBE) electrodes at
high current densities. The results of Gasda et al. suggest that the
GLAD technique combines the advantages of thin film electrodes
with the ability to fabricate porous electrodes allowing efficient
reactant flow especially for high-current density operations.
Scanning electron microscopy (SEM), transmission electron microscopy (TEM), h–2h scan and pole-figure X-ray diffraction
(XRD) have been utilized to study the morphology and crystallographic structure of the Pt nanorods. Cyclic voltammetry (CV) and
rotating-disk electrode experiments (RDE) were performed at room
temperature to characterize the ORR activity and activity stability
of the Pt nanorods and of the conventional Pt/C PEM fuel cell cathode electrocatalyst for comparison. Finally, the temperature dependence of the ORR kinetics was studied at temperatures between 20
and 60 C to calculate the apparent rate constant and the activation
energy for ORR.
Experimental
Fabrication and material characterization of Pt nanorods— We
employed a DC magnetron sputter GLAD technique (Excel Instru-
ments, India) to fabricate vertically-aligned Pt nanorods. A growth
time of 8, 30, and 60 min resulted in 50, 200, and 400 nm long nanorod arrays. The schematic of the GLAD experimental setup is shown
in Fig. 1. Pt nanorods were deposited at a glancing angle of 85
(with respect to substrate normal) on glassy carbon disk inserts (5
mm OD 4 mm thick, Pine Instrument), using a 99.99% pure Pt
cathode (diameter about 2.54 cm). The substrate (glassy carbon disk
insert), located at a distance of about 12 cm from the cathode, was
mounted on a sample holder which was attached to a stepper motor
and rotated at a speed of 2 rpm around the substrate normal axis.
The depositions were performed under a base pressure of 2.4 106
Torr that was achieved using a turbo-molecular pump backed by a
mechanical pump. During Pt deposition, the sputter power was
150 W with an ultrapure Ar working gas pressure of 2.4 mTorr. The
Pt nanorod deposition rate (i.e., nanorod length per growth time)
was approximately 7.5 nm/min as measured by the analysis of
cross-sectional SEM images and confirmed by quartz crystal microbalance measurements (QCM, Inficon Q-pod QCM monitor, crystal:
6 MHz gold coated standard quartz). For the QCM measurements,
the Pt nanorods were deposited directly on quartz crystals and the
loading values were determined by comparing the oscillation frequencies of the blank and the coated crystal.33
The surface morphology of Pt nanorods was investigated using
SEM analysis (FESEM-6330F, JEOL Ltd, Tokyo, Japan). The Crystallographic structure of the nanorods was determined using TEM
analysis (JEOL JEM 2100F, Ltd, Tokyo, Japan), h–2h XRD (Bruker
D8 discover), and pole-figure XRD. For SEM and XRD analysis, Pt
nanorods were deposited on Si(100) wafer pieces which were
mounted on the substrate holder in the same Pt deposition experiments carried out on the glassy carbon samples. For TEM analysis,
Pt nanorods deposited on silicon substrates were scraped off onto
holey carbon grids. The h–2h XRD scan method is a versatile technique for the study of crystal orientations perpendicular to the
substrate plane (i.e., orientation of crystal planes parallel to the substrate surface or, in other words, “texture”) of thin films and nanostructured coatings. On the other hand, XRD pole-figure analysis
provides information on the angular dependence of a specific crystal
orientation over angles ranging from 0 (perpendicular to the substrate plane) to about 90 (parallel to the substrate plane). In our
study, XRD pole-figures of Pt(100), Pt(110), and Pt(111) were performed using a step size of the azimuth angle D/ ¼ 3 (/ ¼ 0–360 )
and 1 for the range of v values of 0–50 and 60–90 , respectively,
with a step size of 10 . For example, a pole-figure peak observed at
v ¼ 0 corresponds to a crystal plane oriented perpendicular to the
Figure 1. (Color online) Schematic of the
glancing angle deposition (GLAD) technique used for the fabrication of nanorod
arrays.
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
substrate (i.e., along the nanorod axis), while pole-figure peaks or a
ring-shaped intensity pattern located at v 90 indicates the existence of crystal orientations parallel to the substrate plane (i.e., crystal planes on the sidewalls of nanorods).
Electrode preparation and electrochemical characterization—
Eelectrochemical measurements (CV and RDE) were performed at
different temperatures to characterize the ORR activity and kinetics
of the Pt nanorod and Pt/C catalysts using a three-electrode cell and
a potentiostat (Pine Instrument, CH Instruments bipotentiostat). The
measurement setup was a typical three-electrode system, consisting
of a working electrode (Pt nanorods or Pt/C on glassy carbon), a Pt
wire counter electrode located in a separate fritted compartment,
and a mercury/mercurous sulfate (Hg/Hg2SO4) reference electrode
with a filling solution of 0.5 M H2SO4. The measured potential of
this reference electrode versus the reversible hydrogen electrode
(RHE) was used to convert all potentials, given in the remainder of
the paper, to the RHE scale. The electrolyte was deaerated or oxygen (O2)-saturated 0.1 M HClO4 (GFS Chemical, Inc., veritas double-distilled in >18 MX-cm1 Millipore water). Both the argon (Ar,
for deaeration) and O2 gases used were ultrahigh purity (99.999%
Ar, 99.99% O2, Linde). An interchangeable, removable glassy carbon disk (5 mm OD 4 mm thick) of a ring-disk electrode (RDE,
6.5 mm ID, 7.5 mm OD, Pine Instruments) was used as a substrate
for preparing the Pt nanorod electrodes. This interchangeable electrode allowed us to use the same ring assembly with different disk
inserts. The geometric area of the glassy carbon disk working electrode was 0.196 cm2. After sealing in the ring-disk housing and
immersion in the electrolyte, the glassy carbon/Pt nanorod working
electrodes were scanned from 0.05 to 1.05 V at a scan rate of 10
mV/s. The samples were also scanned numerous times in the 0.6 to
1.0 V range at 50 mV/s to establish the stability of the Pt-nanorod
samples in the acidic environment. RDE measurements were performed at room temperature in an oxygen-saturated 0.1 M HClO4
solution at scan rates of 10, 20, and 50 mV/s at rotation rates of
1600, 2025, and 2500 rpm, and at elevated temperatures at 20 mV/s
and a rotation rate of 1600 rpm.
The conventional carbon-supported Pt nanoparticle electrode
(Pt/C 20 wt % Pt on carbon-black, Clean Fuel Cell Energy, LLC)
was prepared as described by Gasteiger et al.4 and Garsany et al.34
Briefly, a stock solution of 20% isopropanol and 0.02% Nafion ionomer was prepared by mixing 20 ml of isopropanol with 79.6 ml of
DI water, and 0.4 ml of 5% Nafion ionomer solution (Clean Fuel
Cell Energy, LLC). Ten milligrams of the Pt/C catalyst was mixed
with 5 ml of the stock isopropanol/Nafion solution using an ultrasonicator. A 10 L droplet of the well-dispersed catalyst ink was
pipetted onto the clean, polished glassy carbon rotating disk electrode, leading to a Pt loading of 20 g Pt/cm2. In order to get smooth
and crack-free films covering the entire surface of glassy carbon
electrode, solvent evaporation was slowed by covering the catalyst
ink-coated electrode with a beaker. The Nafion film thickness based
on the loading on the glassy carbon electrode was estimated to be
less than 0.1 m, which is in the range where diffusion effects
within the thin film are considered to be negligible.35 After preparation, the electrodes were immersed in deaerated 0.1 M HClO4 under
potential control of 0.5 V. The potential was then cycled between 0
and 1.0 V in order to produce an electrochemically-clean electrode
surface.
Results and Discussion
Surface morphology and Pt loadings— Top view and crosssection SEM images reveal that Pt nanorod arrays deposited by
GLAD have an isolated columnar morphology (Fig. 2). In the initial
stages of GLAD growth, the number density of the rods is large, and
the rods have diameters ranging from 5–35 nm. However, as the
rods get longer, they also grow in the lateral direction to diameters
up to about 100 nm. During this stage, the average gap between the
rods also changes with increasing length from an initial gap of 5–10
B1031
to 50–100 nm. When a critical length is reached close to about 300
nm, some of the rods cease to grow in the vertical direction, and the
rest of them continue to grow upwards with almost constant diameter and gap values, leading to a straighter rod-like geometry. By
utilizing image processing software (SPIP, version 3.3.5.0), we
measured the number density and average diameter of the nanorods
using the top view SEM images shown in Fig. 2. The number density of the 50, 200, and 400 nm long Pt nanorods were calculated to
be approximately 5.4 1010, 1.4 1010, and 3 109 nanorods/cm2,
and their average diameters were found to be approximately 15, 50,
and 100 nm, respectively. The average diameter and gap between
GLAD nanorods can be changed through control of the deposition
parameters such as deposition angle, sputter power, substrate temperature, and surface pattern/roughness of the substrate. The isolated
nature of the rods in lateral directions leads to a channeled porosity
aligned in the vertical direction to the substrate surface. This novel
geometry can greatly help the effective transport of hydrogen molecules to the catalyst sites in the anode and, more importantly, proton
and oxygen transport in the cathode layer. Moreover, high resolution
top view SEM analysis of the Pt nanorods (Fig. 2) reveals that some
of the rods have 6-fold symmetric faceted tips indicating that the
individual nanorods potentially have a single-crystal structure,
which was proven to be correct in the following section. The tendency for individual metallic nanorods fabricated by GLAD to be single-crystal has also been noted in previous studies.36–38
The weight loading of the 50, 200, and 400 nm Pt nanorod
deposits, as determined by QCM, was approximately 0.04, 0.16, and
0.32 mg Pt/cm2, respectively. Based on these QCM measurements,
the weight loading of Pt nanorods was calculated to be as low as 0.8
g Pt/cm2 per nanometer of rod length.
Crystal structure and orientation analysis— Figures 3a and 3b
show the bright-field TEM image and the corresponding diffraction
pattern, respectively, of an individual vertical Pt nanorod. The diffraction spots are perfectly matched by the theoretical diffraction
pattern for a face-centered cubic lattice. The presence of only a single set of diffraction spots indicates that the individual GLAD Pt
nanorods are single crystals and thus do not have interior grain
boundaries (a polycrystalline material would give a TEM diffraction
pattern with rings of different crystal orientations). Grain boundaries
in crystalline materials have been shown to be easy diffusion pathways for oxygen17 and therefore can act as initiation sites for both
surface and bulk oxide formation and oxidation-related degradation.39
It has been shown that oxidation can significantly inhibit the ORR,
and even lead to Pt dissolution, leading to loss of electrochemicallyactive surface area.16 Lack of grain boundaries in single-crystal Pt
nanorods is therefore expected to lead to enhanced electrochemical
activity and increased resistance to oxidation-related degradation of
the electrode layer in the fuel cell environment.
The XRD profile of a GLAD Pt nanorod array (Fig. 4) shows
that the Pt nanorods are mainly oriented in the [200] direction
(equivalent to the [100] direction) perpendicular to the substrate.
The (100) texture of Pt nanorods obtained from h–2h XRD scans
indicates the dominant crystal plane in the bulk of the rods parallel
to the substrate and cannot provide any direct information on the
crystal planes on the nanorod surface (tip-facets and sidewalls). The
surface crystal planes, which are those involved in the electrochemical reactions, were investigated using crystal simulation and XRD
pole-figure analysis described later in the discussion. It should also
be noted that (111) is the energetically preferred growth plane of
conventional polycrystalline Pt films.22 The formation of energetically unfavorable texture during glancing angle deposition is
believed to be due to lower adatom mobility and faster vertical
growth rates of some crystal orientations.40,41 The islands of these
orientations can grow longer, thereby shadowing others, and giving
rise to the novel texture of the GLAD Pt nanorods.
In our previous study,42 we showed that GLAD Pt nanorods are
highly hydrophilic enabling penetration of aqueous electrolyte and/
or ionomer into the nanorod arrays, and interaction of the electrolyte
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
B1032
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
Figure 2. Top and cross-section scanning
electron microscopy (SEM) views of
GLAD Pt nanorod arrays grown at lengths
of (a) 50, (b) 200, and (c) 400 nm.
with not only the tips, but also with the high-surface-area nanorod
side walls as well. Therefore, we have investigated the possible
crystal orientations of the Pt nanorod tip facets and side walls by
calculating the angles between the different planes of the Pt nanorods utilizing Crystal-Maker software (CrystalMaker Software
Ltd.). Pt has a face-centered cubic (fcc) crystal structure and has
several equivalent crystal planes due to the symmetry of the fcc lattice. For example, the (100), (010), and (001) planes of Pt are all
identical, and in the following sections we will call them (100)-type
planes (one can also use the notation {100} to represent the family
of (100) planes). Similarly, (110), (101), and (011) planes of Pt have
the same atomic configuration and we will call them (110)-type
planes. Individual single-crystal GLAD Pt nanorods are mainly oriented with the [100] direction perpendicular to the substrate plane
(Fig. 4) and it is possible for (100) planes to make angles of 90, 90,
55, 45, 45, and 90 with the crystal planes (001), (010), (111), (110),
(101), and (011), respectively (Note: This method of crystal orientation calculation cannot be used for a polycrystalline material due to
the randomly oriented nature of crystal grains). This implies that the
nanorod sidewalls are most likely oriented in the [001] and [010]
directions, which are identical to [100] orientation. The (011) plane,
which is a (110)-type plane, also makes a 90 angle with the (100)
plane. Hence, the side walls of Pt nanorods are expected to have
(100) and (110)-type crystal planes, as shown in Fig. 5. For the facets observed at the tips of the nanorods (Fig. 2), the tilt angle a of
the facets, measured from the line parallel to the nanorod axis utilizing cross-sectional SEM images, was found to range from 54 to 45 ,
indicating that the facets seen on the tips are likely to be oriented in
the [111], [110], and [101] directions (Fig. 5). Therefore, the facets
of the tips of the nanorods are expected to have (111) and (110)type planes.
The theoretical calculation above has been confirmed by XRD
pole-figure measurements on Pt nanorods, as shown in Fig. 6. Polefigure results indicate that the sidewalls of the nanorods, which
dominate the overall surface area of the rods, mainly involve (110)
planes (Fig. 6c) as they are seen to form a strong ring pattern located
at approximately 80 from the surface normal. A weak and incomplete ring, at about 80 , has also been observed for the (100) planes
at the sidewalls of the nanorods (Fig. 6b). This indicates that some
of the sidewalls are oriented along [100] directions, consistent with
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
B1033
presence of the (100) plane at the tips of the nanorods. This might
be attributed to the presence of some nanorods which are oriented in
the [110] direction perpendicular to the substrate (i.e., weak Pt(220)
peaks observed in XRD results shown in Fig. 4), thus having [100]
planes at the tips of these nanorods. Therefore, especially for long
nanorods (i.e., length > tip diameter), the nanorods are expected to
have a large fraction of Pt(110) planes because these are the dominant planes at the high-surface-area nanorod sidewalls. We believe
that the dominant exposed crystal plane of the nanorods is Pt(110),
the plane that is the most active for the ORR.18–22
Electrochemical characterization— Electrochemical active surface area measurements and surface area stability— CV and RDE
measurements were performed on 50, 200, and 400 nm long Pt
nanorods which correspond to Pt loadings of 0.04, 0.16, and 0.32
mg/cm2, respectively. For comparison, these measurements were
also performed on Pt/C with a Pt loading 0.02 mg/cm2. During CV
measurements, the GLAD Pt nanorod array electrodes were scanned
between 0 and 1.0 V in O2-free 0.1 M HClO4 at a scan rate of 10
mV/s. As shown in the CVs for the Pt nanorods in O2-free electrolyte (Figs. 7a and 7b), the Pt nanorods have an oxide reduction peak
potential of 0.83 V. This potential is 50 mV more positive than the
oxide reduction peak observed for Pt/C (Fig. 7b) indicating that the
GLAD Pt nanorods are less oxophilic than Pt/C.43 As shown in
Fig. 7b, the peak potential for oxide reduction is nearly identical to
that of polycrystalline Pt.
The electrochemically-active surface area (ECSA) of a high surface area catalyst is a critical parameter for defining the Pt loadings
necessary in the fuel cell application. The ECSAs of the Pt/C and
GLAD Pt nanorods were estimated using the charge for hydrogen
adsorption derived from the room temperature CVs. These were
obtained by integrating the charge between the double layer region
and the onset of hydrogen evolution, after subtracting the double
layer charging current, and using the following equation to convert
this charge to ECSAs44)
Figure 3. (a) Bright-field transmssion electron microscopy (TEM) image of
an individual Pt nanorod and (b) the correspoding selective area electron diffraction (SAED) pattern. Scale bar shown is 50 nm. The single set of diffraction spots in the SAED pattern in (b) shows that the individual Pt nanorod
has a single crystal structure.
the theoretical predictions explained above (Fig. 5). From Figs. 6a,
6b, and 6c, it can be clearly seen that the facets of the nanorod tips
have [111], [100], and [110] orientations as they form rings at about
58, 55, and 44 , respectively. These pole-figure results are also consistent with the simulation results (Fig. 5) with the exception of the
Figure 4. X-ray diffraction (XRD) profiles of 50, 200, and 400 nm long
GLAD Pt nanorod arrays. Data is offset for clarity.
ECSAðcm2 Pt=gPtÞ ¼
ChargeðC=cm2 Þ
210ðC=cm2 ÞPtLoading ðgPt=cm2 Þ
[1]
The results show that the ECSAs of 50, 200, and 400 nm long Pt
nanorods and of Pt/C with Pt loadings of 0.04, 0.16, 0.32 and 0.02
mg/cm2 are 13, 12, 6.7, and 60 m2/g Pt, respectively. The ECSA
value of Pt/C electrocatalyst is in good agreement with values
Figure 5. Top-view of a single crystal Pt nanorod oriented in the Pt[100]
direction (with respect to surface normal along the x-direction out of the
page) showing the simulated results of possible crystal orientations at its
sidewalls and at the facets of the nanorod tip.
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
B1034
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
Figure 6. (Color online) XRD pole-figure
results for the Pt(111), Pt(100), and
Pt(110) planes for glancing angle deposited (GLAD) Pt nanorods. The Pt(110)
plane are mainly oriented at about 80
from the surface normal, which corresponds to the nanorod sidewalls.
reported in the literature.4 The decreasing ECSA with increasing
nanorod length may be attributed to the increase in nanorod diameter concurrent with the increase in length. Larger diameter decreases
the surface area to volume ratio of the rods. In addition, incomplete
wetting of the nanorods with increasing nanorod length can further
reduce the measured ECSA. Based on previously reported BET
measurements on GLAD carbon nanorods,45 Pt nanorods of 500 nm
length are expected to have an effective surface area enhancement
factor (SAEF, ratio of real surface area to substrate geometric area)
of 40. Due to the similar number density and diameters of 400 and
500 nm long nanorods, the estimated SAEF of 400 nm Pt nanorods
is calculated to be approximately 32 (40 400 nm/500 nm). However, as can be seen from the SEM images shown in Fig. 2, Pt nanorods that are shorter than about 300 nm do not have uniform heights
due to the competitive growth mechanism during the initial stages
of deposition. This causes a change of number density as a function
of nanorod length as the growth progresses until they reach a satura-
tion thickness at about 300 nm. At these initial stages, the diameters
of nanorods also increase with their length. Therefore, below 300
nm, the total surface area of the Pt nanorods is not expected to linearly depend on their length. However, we can assume that the surface area of the nanorods is dominated by the sidewall area, which
is proportional to the nanorod length, diameter, and number density.
By comparing the length, average diameter, and number density of
200 and 400 nm long rods, we estimated the SAEF of 200 nm Pt
nanorods as 25 [ ¼ 32 (200/400) (50/100) (14 109/
3 109)]. Similarly, by comparing 50 and 200 nm long rods, we calculated the SAEF of the 50 nm Pt nanorods to be 7 [ ¼ 25 (50/
200) (15/50) (54 109/14 109)]. Dividing these the SAEFs by
the measured Pt loading values, the effective surface areas for the
50, 200, and 400 nm long Pt nanorods were calculated to be approximately 17, 15, and 10 m2/g Pt, respectively. Based on our electrochemical measurements the 50, 200, and 400 nm long Pt nanorods
have ECSAs of 13, 12, and 6.7 m2/g Pt, respectively. Thus, the
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
B1035
platinum particles sizes with cycling caused by ripening processes,
as recently summarized by Shao-Horn et al.46 This behavior has
been also been observed by Debe et al.16
Figure 7. (Color online) (a) Room temperature CVs in O2-free 0.1 M
HClO4 at a scan rate 10 mV/s of 50, 200, and 400 nm long Pt nanorods,
which correspond to Pt loadings of 0.04, 0.16, and 0.32 mg/cm2, respectively, and (b) Comparison of CVs of Poly-Pt, Pt/C, and 200 nm long Pt
nanorods in O2-free 0.1 M HClO4 at a scan rate 10 mV/s. Data in (b) were
normalized to measured Pt ECSA.
utilization factor of 50, 200, and 400 nm long Pt nanorods were estimated to be approximately 76% (13/17 100), 80% (12/15 100)
and 67% (6.7/10 100), respectively. Higher utilization of shorter
nanorods is believed to be due to the more complete wetting of the
nanorod sidewalls by the electrolyte solution.
The 50, 200, and 400 nm long Pt nanorods samples were scanned
multiple times in the 0.6 to 1.0 V potential range at a scan rate of 50
mV/s to establish their stability in an acidic environment. Figure 8
shows a series of CVs, taken at room temperature, from 50, 200,
and 400 nm Pt nanorods over 250 and 4500 cycles. For comparison,
the results for the Pt/C are shown in Fig. 9a. ECSA analysis shows
that only 60% of the initial ECSA of the Pt/C sample remained after
1000 potential cycles, while after 4500 cycles the 50, 200, and 400
nm Pt nanorods retained 59, 63, and 82% of their initial ECSAs,
respectively (Fig. 8). These results indicate that the Pt nanorods are
more stable than Pt/C with potential cycling over a potential region
and in an acidic environment relevant to the PEM fuel cell cathode
environment. A shift in the final Pt/C oxide formation and reduction
peak potentials (Fig.9a) toward those of the Pt nanorods may be
attributed to a decrease in Pt oxophilicity due to an increase in Pt/C
ORR kinetic analysis— The RDE CV traces taken at a rotation rate
of 1600 rpm were used to evaluate and compare the ORR kinetic
current densities of the Pt nanorod and Pt/C catalysts. Figures 9b
and 10a show the ORR current densities, as a function of potential,
obtained from RDE tests in oxygen-saturated 0.1 M HClO4 at 20 C
and 1600 rpm, using Pt/C (20 wt % Pt) catalyst at a Pt loading of
0.02 mg/cm2, and 50, 200, and 400 nm long Pt nanorods with Pt
loadings of 0.04–0.32 mg/cm2. The scan rates were 10 and 20 mV/s
for Pt nanorods and Pt/C catalysts, respectively. The effects of
pseudo-capacitive currents on the calculated ORR activities were
eliminated by subtracting the background currents in CVs taken in
argon-purged electrolyte from the currents in CVs taken in oxygensaturated electrolyte. As shown in Figs. 9b and 10a, both Pt/C and
the Pt nanorods exhibit the typical mixed kinetic-diffusion controlled region from approximately 0.75 to 1.00 V and diffusion-limited current region from 0.2 to 0.7 V. The diffusion-limited current
densities normalized to geometric surface area are well-defined,
similar for the Pt nanorod catalyst and for the Pt/C, and are within
10% of the theoretical diffusion limiting current expected for a rotation rate of 1600 rpm and room temperature (5.7 mA/cm2).4,47,48
This agreement clearly indicates adequate coverage of the glassy
carbon electrodes by the Pt/C film and by the nanorods and minimal
effects of oxygen diffusion within the electrode films.4,7,47 Slight
differences in diffusion-limiting current were observed for the 50,
200, and 400 nm nanorod samples; these differences will be discussed later. The slight decrease in the limiting current of Pt/C at
potentials less than 0.20 V (Fig. 9b), in the region of hydrogen
adsorption, indicates the formation of H2O2 and a change in the
ORR pathway from a four- to a two-electron mechanism.18 This
change in the mechanism is less pronounced in the case of Pt nanorods (i.e., hydrogen peroxide production rate on Pt nanorods is
lower than that of Pt/C) as can be seen in Fig. 10a, indicating that
the contribution from the two-electron process at <0.2 V is lower
for the nanorods. This observation is similar to what was seen for
single-crystal Pt(110) electrodes.18
The electrocatalytic activities of Pt nanorods for the ORR were
extracted from the background-corrected RDE CVs using the wellknown mass-transport correction for RDE measurements4,47,48
ik ¼
ðilim iÞ
ðilim iÞ
[2]
where i is the measured current at a specified potential, ilim is the
measured limiting current, and ik is the kinetic current. The assumptions for extracting ik from the RDE data using Eq. (2) are valid
over the current range of 0.1 ilim < i < 0.8 ilim.47 Specific activities
can be determined by calculation of ik using Eq. (2) and normalization to measured Pt ECSA and Pt loading. The area-specific activity
provides a measure of the electrocatalytic activity of Pt atoms at the
surface, thus, it is specifically meaningful for structure-sensitive
reactions. The mass-specific activity has practical implications for
fuel cells in that it can be used to calculate catalyst cost per unit
power output. The ORR area-specific activities and mass-specific
activities for the Pt nanorod and Pt/C catalysts are shown in
Figs. 10b-d. Equation (2) has been shown to provide accurate
assessment of the electrocatalytic activity of catalysts in thin film
RDE electrodes for film thicknesses of <0.1 m.4,7 Mass transport
within the electrode structure can have a significant influence on the
thin film RDE data for thicker films.7
To verify the assumption of negligible effects of mass transport
of oxygen within the nanorod electrodes, an analysis of the rotation
rate dependence of the RDE ORR data for the thickest electrode
comprised of the 400 nm Pt nanorods was performed. Figure 11a
shows the RDE profiles of 400 nm long Pt nanorods at different
rotation speeds of 1600, 2025, and 2500 rpm in oxygen-saturated
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
B1036
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
Figure 8. (Color online) Room temperature CVs of (a) 50, (b) 200, and (c)
400 nm long Pt nanorods over first cycle, 250, and 4500 cycles (cycling
between 0.6 and 1.0 V at 50 mV/s). Data was recorded in O2-free 0.1 M
HClO4.
Figure 9. (Color online) (a) Room temperature CV of Pt/C in O2-free 0.1 M
HClO4 at sweep rate 50 mV/s; first cycle and after 1000 cycles, (b) Positivegoing RDE plot of Pt/C in oxygen-saturated 0.1 M HClO4 at sweep rate 20
mV/s and rotation rate 1600 rpm, and (c) ORR polarization curve of Pt/C
with Pt loading of 0.02 mg/cm2.
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
B1037
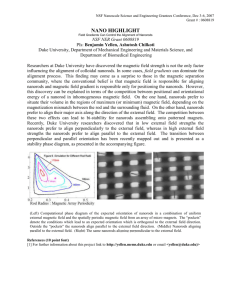
Figure 10. (Color online) (a) Room temperature CVs of 50, 200, 400 nm long Pt nanorods in oxygen-saturated 0.1 M HClO4 at a scan rate of 10 mV/s and rotation speed of 1600 rpm, (b,c) Calculated ORR area- and mass-specific activities as a function of potential on 50, 200, and 400 nm long Pt nanorods, (d) dependence of area- and mass-specific activities on the ECSA of Pt nanorods of different lengths (different Pt loadings), and comparison of the ORR specific activity
from polycrystalline Pt, Pt/C, NSTF Pt (3M), and Pt nanorods of different lengths. Data are plotted for the positive-going sweep. The data for polycrystalline Pt
and NSTF Pt (3M) catalysts were taken from Mayrhofer et al.47 Based on the current range (0.1 ilim < i < 0.8 ilim) used for extracting kinetic currents, (b) and (c)
were restricted to potential ranges of 0.81–0.96, 0.89–0.98, and 0.90–1.00 V for 50, 200, and 400 nm Pt nanorods, respectively.
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
B1038
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
0.1 M HClO4 at a scan rate of 10 mV/s. Equation 2 can be expanded
to take into account the effects of diffusion within a film electrode49
1 1 1 1
1 1
1
¼ þ þ ¼ þ þ
i ik if id ik if Kx12
[3]
where if represents the film-diffusion-limited current or current density controlled by reactant diffusion within the electrode film, K is a
constant dependent on oxygen concentration and diffusion coefficient of oxygen in the electrolyte and on its kinematic viscosity, x is
the angular rotation velocity of the RDE, and the other terms have
the same meaning as in Eq. (2). A Koutecky-Levich plot,50 (1/i) vs.
1/x1/2, for a fixed potential should show a linear relationship with a
slope of 1/K and a y-intercept of 1/ik þ 1/if. The Koutecky-Levich
plots for the 400 nm Pt nanorod RDE ORR data at rotation rates of
1600, 2025, and 2500 rpm are shown in Fig. 11b for the diffusionlimited current (current at 0.4 V) and for the currents at 0.90 and
0.95 V. These plots are linear and parallel, as expected. To determine if film diffusion is affecting the kinetic currents obtained from
RDE measurements and to correct for its effect, the y-intercept of
the Koutecky-Levich plot for i ¼ id is subtracted from the y-intercept
for the same plot at the potential of interest (e.g., 0.90 V). Comparison of the kinetic currents derived directly from the y-intercepts of
the Koutecky-Levich plots (ik) to those corrected for film diffusion
(ik’) indicates the extent of the influence of film diffusion on the
RDE measurements. This comparison for the 400 nm Pt nanorod
RDE data shows that film diffusion has minimal effect on the currents at 0.90 and 0.95 V, with <5% difference between ik and ik’.
Since all the other films tested in this study were thinner than the
400 nm film, it is expected that film diffusion has minimal influence
on the kinetic currents at 0.90 and 0.95 V for these films as well.
Based on the absence of film diffusion effects at 0.90 and 0.95
V, Eq. (2) was used to extract kinetic currents from the oxygen-saturated electrolyte RDE CVs (Figs. 9b and 10a). The effect of different Pt loadings (i.e., nanorod length and diameter) for GLAD Pt
nanorods is demonstrated in Figs. 10a, 10b and 10c. Based on the
current range (0.1 ilim < i < 0.8 ilim) used for extracting kinetic currents, the polarization curves in Figs. 10b and 10c for 50, 200, and
400 nm long Pt nanorods were restricted to potential ranges of 0.81–
0.96, 0.89–0.98, and 0.90–1.00 V, respectively. As shown in Fig.
10b, the 200 and 400 nm long nanorods have nearly identical areaspecific ORR activities at 0.90 V, while the 50 nm nanorods have
relatively low activity. Two factors that may contribute to the lower
relative ORR activity of the 50 nm nanorods are: (1) the much lower
Pt loading (0.04 versus 0.16 and 0.32 mg Pt/cm2) leading to reduced
coverage of the glassy carbon disk with nanorods, evident in the
slightly lower diffusion-limited current for this sample (Fig. 10a)
and (2) the 50 nm nanorods do not have well-developed sidewalls
and have almost a particle-like structure as compared to the 200 and
400 nm long nanorods (Fig. 2). This means that the electrolyte is in
contact mainly with the facets at the tips of the nanorods which are
oriented in the [111], [100], and [110] directions (Figs. 4, 5, and 6).
The lower electrochemical activity of the (111) and (100) faces
compared to that of the (110) face will result in lower ORR activity
of the 50 nm long Pt nanorods compared to the activity of the 200
and 400 nm long nanorods that have well-developed (110) sidewalls
in addition to the tip and facet orientations. Figure 10c also shows
that the 400 nm long nanorod sample has lower mass-specific activity at 0.9 V than that of the 50 and 200 nm long nanorod samples
due to larger diameters and incomplete wetting compared to shorter
nanorods. This observation is also supported by the utilization calculation shown in the previous section.
The area- and mass-specific activities for Pt/C and Pt nanorods at
0.9 and 0.95 V are reported in Table I. The reported values of areaand mass-specific activities at 0.9 V taken from the literature4,16,47,49
for polycrystalline Pt and Pt/C are also given in Table I for comparison. Based on the reported area-specific activity and ECSA of 8 m2/
g for NSTF,47 the mass-specific activity was calculated and is also
reported in Table I. The area- and mass-specific activities measured
Figure 11. (Color online) (a) Room temperature positive-going RDE curves
for 400 nm long Pt nanorods in oxygen-saturated 0.1 M HClO4 at scan rate
10 mV/s and different rotation speeds (1600, 2025, and 2500 rpm) and (b)
Koutecky-Levich plots for 400 nm long Pt nanorods extracted from (a) at
0.4, 0.90, and 0.95 V.
for Pt/C in this study are in good agreement with the values reported
by Gasteiger et al.4 and by Paulus et al.48 and slightly lower than
those reported by Garsany et al.34 As discussed by Garsany et al.,34
the higher SA and MA values reported in their study might be attributed to optimization of the catalyst ink formulations. Figure 10d
shows the dependence of area- and mass-specific ORR activity at
0.90 V on the length and ECSA of Pt nanorods. Of the three nanorod
lengths studied, the 200 nm length exhibited the highest mass activity and area-specific activity nearly identical to that of the 400 nm
sample. As discussed above, the 200 nm long nanorods are long
enough to have well-developed sidewalls with the preferred [110]
orientation but, unlike the 400 nm long nanorods, they have smaller
diameters and better wetting. As shown in Table I and Fig. 10e, the
area-specific ORR activity of the 50 nm long Pt nanorods is higher
than that of Pt/C, while the 200 nm and 400 nm long nanorods have
area-specific activities that are higher than the Pt NSTF catalyst and
comparable to that of polycrystalline Pt. As explained in the introduction, electrochemical activity in 0.1 M HClO4 electrolyte follows
the trend Pt(110) > Pt(111) > polycrystalline Pt > Pt(100).18,19
Because the sidewalls of the Pt nanorods are dominated by (110)
planes, one would expect a higher area-specific ORR activity for
GLAD nanorods compared to polycrystalline Pt. Polycrystalline Ptlike ORR activities may be due to the existence of Pt(100) planes at
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
B1039
Table I. Summary of the evaluated ORR electrocatalytic activity (area-specific and mass-specific activities at 0.90 and 0.95 V) of Pt-nanorod
array and Pt/C catalysts in 0.1 M HClO4 and comparison to the literature ORR activity values for various Pt catalysts.
Method
Sample
RDE
RDE
RDE
TF-RDE
RDE
TF-RDE
TF-RDE
TF-RDE
TF-RDE
TF-RDE
TF-RDE
50 nm long Pt nanorods
200 nm long Pt nanorods
400 nm long Pt nanorods
20% Pt/C
Bulk Poly-Pt
NSTF Pt
Pt/C (TKK)
Pt/C (TKK)
PT/C (TKK)
46% Pt/HSC (TKK)
20% Pt/C
Pt
loading
(mg/cm2)
ECSA
(m2/g)
—
0.042
0.014 (d ¼ 1–1.5 nm)
0.014 (d ¼ 2–3 nm)
0.014 (d ¼ 5 nm)
0.017
0.014
13
12
6.7
60
—
8
>60
>60
>60
132
66
0.040
0.160
0.320
0.020
SA(A/cm2)
MA(A/mg)
T
( C)
Scan
rate
(mV/s)
0.90(V)
0.95(V)
0.90(V)
0.95(V)
References
20
20
20
20
20
20
20
20
20
25
30
10
10
10
20
20
20
20
20
20
10
20
632
1080
1194
288
1200
750
100
200
350
292
305
166
250
266
76
—
—
—
—
—
—
—
0.08
0.13
0.08
0.18
—
0.06
—
—
—
0.27
0.21
0.02
0.03
0.02
0.05
—
—
—
—
—
—
—
This work
This work
This work
This work
Ref. 47
Ref. 47
Ref. 47
Ref. 47
Ref. 47
Ref. 51
Ref. 34
the sidewalls and tips of some of the nanorods and to the presence of
low coordination sites at the edges of the low index planes, which
can reduce the electrochemical activity compared to that of ideal
pure Pt(110) in perchloric acid electrolyte. It should be noted that
the existence of Pt(100) planes is expected to improve ORR electrocatalytic activity if adsorbing electrolytes such as sulfuric acid or
perfluorosulfonic acid fuel cell electrolyte (e.g., Nafion) are used
instead of non-adsorbing perchloric acid electrolyte,21,22 as
explained in the introduction.
The higher area-specific activity of the Pt NSTF catalysts compared to Pt/C catalysts has been attributed to the large Pt crystallite
size resulting in average coordination numbers of surface Pt and
corresponding surface electronic properties approaching those of
polycrystalline Pt.47 As shown in Fig. 2a, the 50 nm long nanorods
have diameters ranging from 5 to 20 nm and thus, due to the singlecrystal nature, their crystallite dimensions are expected to be in the
range of 5–50 nm (i.e., diameter-length). The comparable area-specific activities of the 50 nm Pt nanorod and the Pt NSTF catalysts is
believed to be due to the similar Pt crystallite dimensions of these
catalysts. In addition to the effects of electrochemically active
Pt(110) planes of Pt nanorods discussed above, the higher area-specific activity of the 200 and 400 nm nanorods can likewise be attributed in part to their larger crystallite dimensions compared to the 50
nm nanorods, Pt/C, and to the Pt NSTF. However, the large crystallite size of the Pt nanorod and the Pt NSTF catalysts result in a
lower ratio of surface to bulk Pt atoms and a concomitantly lower
mass activity than Pt/C, as shown in Fig. 10e and Table I
Temperature-dependence study— Hydrodynamic voltammograms of
the Pt nanorods and the Pt/C catalyst were obtained as a function of
temperature between 20 and 60 C in oxygen-saturated 0.1 M HClO4
at 20 mV/s scan rate and 1600 rpm electrode rotation speed. Figure
12 shows an increase in the limiting current in the temperature range
of 20–40 C. The increase in current density can be attributed to an
increase in the oxygen diffusion rates with increasing temperature
due to a decrease in electrolyte viscosity. At a temperature of 60 C,
a slight decrease in the limiting current density was observed (Fig.
12), indicating a decrease in both the oxygen concentration and density of the electrolyte at elevated temperature due to a drastic
increase in the vapor pressure of water. This observation was confirmed by Paulus et al.48 who derived an expression which represents the temperature-dependent limiting current density using the
physical-chemical properties of pure water.
Since ik depends on the oxygen concentration in the electrolyte,
which decreases with increasing the operating temperature,48,52,53 it
is most appropriate to define the kinetics of the reaction in terms of
a concentration-independent apparent rate constant, kapp.52 The
apparent rate constants of ORR can be calculated from Eq. (4) at a
constant overpotential52
Figure 12. (Color online) RDE plots at different temperatures varying
between 20 and 60 C of (a) Pt/C and (b) 200 nm Pt nanorods in oxygen-saturated 0.1 M HClO4 at sweep rate 20 mV/s and rotation rate 1600 rpm.
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
B1040
kapp ¼
ik
nFAreal ½O2 ½H þ [4]
where kapp is the apparent rate constant, ik is the kinetic current calculated from Eq. (2), n is the number of electrons involved in the
reaction, F is the Faraday constant, Areal is the electrochemicallyactive surface area, [O2] is the oxygen concentration in the bulk of
the electrolyte solution, and [Hþ] is the bulk concentration of protons (0.1 M). The oxygen concentration [O2] values at different
temperatures were taken from the values reported by Wakabayashi
et al.52 in which [O2] values were calculated based on Henry’s law.
The apparent rate constants for ORR on Pt/C and Pt nanorods were
calculated at 0.93 and 0.95 V, where the rate of electron transfer is
very slow and clearly the rate-determining step. The apparent rate
constant values were calculated at potentials higher than 0.90 V
because the RDE current of Pt nanorods at 0.90 V and 60 C is in the
diffusion-limited region, as shown in Fig. 12b. Table II shows that
the apparent ORR rate constant on Pt nanorods (5 cm4/mol s) is
higher than that of the conventional catalysts (2 cm4/mol s) indicating that the ORR rate on Pt nanorods is larger than on Pt/C. The
Arrhenius equation48,53 was utilized to calculate the activation energies for ORR on both Pt/C and Pt nanorods
ln kapp ¼
Ea 1
þ ln A
R T
Figure 13. (Color online) Arrhenius plots for the apparent rate constant kapp
on Pt/C and GLAD Pt nanorods at 0.93 and 0.95 V vs. RHE.
[5]
where Ea is the activation energy, A is the pre-exponential factor, T
is the operating temperature, and R is the gas constant. Figure 13
shows the Arrhenius plots of the value of kapp evaluated at 0.93 and
0.95 V from Eq. (4) versus (1/T) which gives straight lines. The
ORR activation energies for Pt/C and Pt nanorods, determined from
the slopes of these lines, are listed in Table II. It should be noted
that there are contradictory reports about the activation energy of Pt/
C in the literature. Grgur et al.54 calculated the activation energy to
be 42 kJ/mol on all three low index Pt single crystal surfaces in 0.05
M H2SO4 at 0.8 V. Paulus et al.48 reported lower activation energies
of 26 and 28 kJ/mol on Pt/C in 0.5 M H2SO4 and 0.5 M HClO4,
respectively. It was found that these values are in good agreement
with the values reported for polycrystalline Pt in sulfuric acid with
pH ¼ 1 at 0.8 V,55 and in perchloric acid with pH ¼ 1.9 at 0.8 V,56
which are 25 and 20 kJ/mol, respectively. Recently, Kongkanand et
al.53 reported an activation energy value of 25 kJ/mol at 0.575 and
0.625 V (vs. SCE) on Pt/C and single-wall carbon nanotube-supported Pt nanoparticles, while the ORR rate constant is 2-fold higher
for the Pt/SWCNT electrode than that of Pt/C. On the other hand,
significantly larger activation energy of 40 kJ/mol was observed
by Yano et al.57 in 0.1 M HClO4 at 0.8 V on Nafion-coated film
electrodes of bulk Pt and Pt nanoparticles dispersed on carbon black.
This value is comparable to those on the bulk Pt electrode measured
by Wakabayashi et al.52 In summary, activation energy values of Pt/
C electrode reported in the literature vary within the range of 20–40
kJ/mol in liquid electrolytes, and can get as large as 60 kJ/mol in
operating PEM fuel cells.58–60 Neyerlin et al.61 determined the apparent activation energies of Pt/C electrode in operating PEM fuel
cells at zero overpotential, constant overpotentials of 0.3 and 0.35
V, and 0.9 V iR-free cell voltage to be 67, 38 and 33, and 10 kJ/mol,
respectively. It was suggested that scattered activation energy data
in the literature is mainly due to the use of different definition of the
terms and conditions utilized to extract the kinetics data.61 Activation energy value of 30 kJ/mol calculated from our experiments on
Pt/C is in good agreement with majority of the reported values
which are in the range of 20–40 kJ/mol.48,53,55,56,61 Moreover, we
note that the activation energy of 25.8 kJ/mol for the Pt nanorod catalyst falls in the lower limit of the values reported in the literature
(i.e., in the range of 20–40 kJ/mol) for Pt/C and polycrystalline Pt.
Conclusion
The glancing angle deposition technique was utilized to fabricate
vertically-aligned Pt nanorod arrays on glassy carbon substrates.
The ORR activity of the nanorod arrays was investigated using
cyclic volammtery and rotating disk electrode techniques at room
temperature and as a function of temperature up to 60 C. It was
found that GLAD Pt nanorods exhibit higher area-specific activity,
higher reaction rate constant, comparable activation energy, and
greater stability against electrochemically-active surface area loss
compared to conventional Pt/C electrode. The enhanced ORR activity of Pt nanorods was attributed to their larger crystallite size and
the dominance of Pt(110) crystal planes on the nanorod sidewalls,
which was reported to be the most active plane for ORR. The massspecific ORR activity of the Pt nanorods was found to be lower than
that of the Pt/C catalyst, mainly due to the large diameter of nanorods. Decreasing the diameter of the nanorods, thereby increasing
the surface/bulk atom ratio, or using alloyed Pt nanorods, platinum
mass-specific activity can be potentially improved. These and similar surface engineering and chemical modification approaches may
open the way for utilizing GLAD nanorods as cathode catalysts in
PEM fuel cells.
Acknowledgments
Table II. Calculated apparent reaction rate constant and activation energy for the ORR on Pt/C and GLAD Pt nanorods at
0.95 V.
Sample
ECSA
(m2/g Pt)
kapp at 0.95 V
and 20 C
(cm4/mol s)
Ea (kJ/mol)
at 0.95 V
20% Pt/C
200 nm long Pt nanorods
60
12
2
5
30.0 6 0.5
25.8 6 0.5
The authors would like to thank the UALR Nanotechnology
Center and Dr. Fumiya Watanabe for his valuable support and discussions during SEM, XRD, and TEM measurements. A portion of
this research was conducted at Argonne National Laboratory, a U.S.
Department of Energy, Office of Science Laboratory, operated
by UChicago Argonne, LLC, under contract no. DE-AC0206CH11357. The Argonne National Laboratory authors would like
to acknowledge the support of the U.S. Department of Energy,
Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Program, program manager Nancy Garland.
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp
Journal of The Electrochemical Society, 158 (8) B1029-B1041 (2011)
References
1. Report of the DOE Basic Energy Sciences Workshop on Hydrogen Production,
Storage, and Use (May 2003), online copy available at http://www.sc.doe.gov/bes/
hydrogen.pdf.
2. Fuel Cell Handbook, EG&G Technical Services Inc, 7th Ed. (U.S. Department of
Energy, Office of Fossil Energy, National Energy Technology Laboratory, 2004),
online copy available at http://www.netl.doe.gov/technologies/coalpower/fuelcells/
seca/pubs/FCHandbook7.pdf.
3. J. Zhang, PEM fuel cell electrocatalysts and catalyst layer: fundamentals and
applications, Springer-Verlag, London (2008).
4. H. A. Gasteiger, S. S. Kocha, B. Sompalli, and F. T. Wagner, Appl. Catal., B, 56, 9
(2005).
5. H. Tang, Z.G. Qi, M. Ramani, and J. F. Elter, J. Power Sources, 158, 1306 (2006).
6. J. L. Qiao, M. Saito, K. Hayamizu, and T. J. Okada, J. Electrochem. Soc., 153,
A967 (2006).
7. U. A. Paulus, T. J. Schmidt, H. A. Gasteiger, and R. J. Behm, J. Electroanal.
Chem., 495, 134 (2001).
8. D. Gruber, N. Ponath, J. Muller, and F. Lindstaedt, J. Power Sources, 150, 67
(2005).
9. M. K. Debe, A. K. Schomoeckel, G. D. Vernstrom, and R. Atanasoski, J. Power
Sources, 161, 1002 (2006).
10. D. Gurber and J. Muller, J. Power Sources, 171, 294 (2007).
11. A. Caillard, C. Coutanceau, P. Braulta, J. Mathias, and J.-M. Léger, J. Power Sources, 162, 66 (2006).
12. M. Gustavsson, H. Ekstrom, P. Hanarpa, L. Eureniusa, G. Lindbergh, E. Olsson,
and B. Kasemoa, J. Power Sources, 163, 671 (2007).
13. C. Chien and K. Jeng, Mater. Chem. Phys., 103, 400 (2007).
14. A. Bonakdarpour, M. D. Fleischauer, M.J. Brett, and J. R. Dahn, Appl. Catal., A,
349, 110 (2008).
15. M. D. Gasda, G. A. Eisman, and D. Gall, J. Electrochem. Soc., 157, B71 (2010).
16. M. K. Debe, A. K. Schmoeckel, S. M. Hendricks, G. D. Vernstrom, G. M. Haugen,
and R. T. Atanasoski, ECS Trans., 1(8), 51 (2006).
17. A. Atkinson, R. I. Taylor, and A. E. Hughes, Philos. Mag. A, 45(5), 823 (1982).
18. N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 45, 121 (2002).
19. V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas, and
N. M. Marković, Science, 315, 493 (2007).
20. F. El Kadiri, R. Faure, and R. Durand, J. Electroanal. Chem., 301, 177 (1999).
21. R. Subbaraman, D. Strmcnik, V. Stamenkovic, and N. M. Markovic, J. Phys.
Chem. C, 114, 8414 (2010).
22. R. Subbaraman, D. Strmcnik, A. P. Paulikas, V. R. Stamenkovic, and N. M. Markovic, ChemPhysChem., 11, 2825 (2010).
23. Z. Chen, M. Waje, W. Li, and Y. Yan, Angew. Chem., Int. Ed., 46, 4060 (2007).
24. S. Antonino, B. Peter, S. Bruno, T. Jean-Marie, S. Walter van, Nature Mater., 4,
366 (2005).
25. T. Karabacak and T.-M. Lu, in Handbook of Theoretical and Computational Nanotechnology, M. Rieth and W. Schommers, Editors, Chap. 69, p. 729, American.
Scientific, Stevenson Ranch, CA (2005).
26. T. Karabacak, G.-C. Wang, and T.-M. Lu, J. Vac. Sci. Technol. A, 22, 1778 (2004).
27. F. Tang, T. Karabacak, L. Li, M. Pelliccione, G.-C. Wang, and T.-M. Lu, J. Vac.
Sci. Technol. A, 25, 160 (2007).
28. S. Kim, N. Koratkar, T. Karabacak, and T.-M. Lu, Appl. Phys. Lett., 88, 263106
(2006).
29. P. Morrow, F. Tang, T. Karabacak, P.-I. Wang, D.-X. Ye, G.-C. Wang, and T.-M.
Lu, J. Vac. Sci. Technol. A, 24, 205 (2006).
B1041
30. F. Tang, T. Karabacak, P. Morrow, C. Gaire, G.-C. Wang, and T.-M. Lu, Phys.
Rev. B, 72, 165402 (2005).
31. E. Main, T. Karabacak, and T.-M. Lu, J. Appl. Phys., 95, 4346 (2004).
32. M. D. Gasda, R. Teki, T.-M. Lu, N. Koratkar, G. A. Eisman, and D. Gall, J. Electrochem. Soc., 156, B614 (2009).
33. W. J. Khudhayer, A. U. Shaikh, and T. Karabacak, Advanced Science Letters, 4,
1-9 (2011)
34. Y. Garsany, O. A. Baturina, K. E. Swider-Lyons, and S. S. Kocha, Anal. Chem.,
82, 6321 (2010).
35. E. Higuchi, H. Uchida, and M. Watanabe, J. Electroanal. Chem., 583, 69 (2005).
36. T. Karabacak, J. S. DeLuca, D. Ye, P.-I Wang, G.-C. Wang, and T.M. Lu, J. Appl.
Phys., 99, 064304 (2006).
37. T. Karabacak, P.-I. Wang, G.-C. Wang, and T.-M. Lu, Thin Solid Films, 493, 293
(2005).
38. T. Karabacak, P.-I. Wang, G.-C. Wang, and T.-M. Lu, Mat. Res. Soc. Symp. Proc.,
788, 75 (2004).
39. V. Komanicky, K. C. Chang, A. Menzel, N. M. Markovic, H. You, X. Wang, and
D. Myers, J. Electrochem. Soc., 153, B446 (2006).
40. P. Morrow, F. Tang, T. Karabacak, P.-I. Wang, D.-X. Ye, G.-C. Wang, and T.-M.
Lu, J. Vac. Sci. Technol. A, 24, 205 (2006).
41. F. Tang, T. Karabacak, P. Morrow, C. Gaire, G.-C. Wang, and T.-M. Lu, Phys.
Rev. B, 72, 165402 (2005).
42. W. J. Khudhayer, R. Sharma, and T. Karabacak, Nanotechnology, 20, 275302
(2009).
43. N. Markovic, H. Gasteiger, and P. N. Ross, J. Electrochem. Soc., 144, 1591
(1997).
44. T. R. Ralph, G. A. Hards, J. E. Keating, S. A. Campbell, D. P. Wilkinson, M.
Davis, J. St-Pierre, and M. C. Johnson, J. Electrochem. Soc., 144, 3845 (1997).
45. G. K. Klema and M. J. Brett, J. Electrochem. Soc., 150, E342 (2003).
46. Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby, and D. Morgan,
Top. Catal., 46, 285 (2007).
47. K. J. J. Mayrhofer, D. Strmcink, B. B. Blizanac, V. Stamenkovic, M. Arenz, and
N. M. Markovic, Electrochim Acta, 53, 3181 (2008).
48. U. A. Paulus, T. J. Schmidt, H. A. Gasteiger, and R. J. Behm, J. Electroanal.
Chem., 495, 134, (2001).
49. D. R. Lawson, L. U. Whiteley, C. R. Martin, M. N. Szentimay, and J. I. Song,
J. Electrochem. Soc., 135, 2247 (1988).
50. A. J. Bard and L. R. Faulkner, Electrochemical Methods, John Wiley & Sons, New
York (1980).
51. I. Takahashi and S. S. Kocha, J. Power Sources, 195, 6312 (2010).
52. N. Wakabayashi, M. Takeichi, M. Itagaki, H. Uchida, and M. Watanabe, J. Electroanal. Chem., 574, 339 (2005).
53. A. Kongkanand, S. Kuwabata, G. Girishkumar, and P. Kamat, Langmuir, 22, 2392,
(2006).
54. B. N. Grgur, N. M. Markovic, and P. N. Ross, Can. J. Chem., 75, 1465 (1997).
55. A. Damjanovic and D. B. Sepa, Electrochim. Acta, 35, 1157 (1990).
56. D. B. Sepa, M. V. Vojnovic, L. Vracar, and A. Damjanovic, Electrochim. Acta, 31,
91 (1986).
57. H. Yano, E. Higuchi, H. Uchida, and M. Watanabe, J. Phys. Chem. B, 110, 16544
(2006).
58. S. Mukerjee, S. Srinivasan, and M. P. Soriaga, J. Phys. Chem., 99, 4577 (1995).
59. P. S. Beattie, V. I. Basura, and S. Holdcroft, J. Electroanal. Chem., 468, 180 (1999).
60. A. J. Appleby, J. Electrochem. Soc., 117, 328 (1970).
61. K. C. Neyerlin, W. Gu, J. Jorne, and H. A. Gasteiger, J. Electrochem. Soc., 153,
A1955 (2006).
Downloaded 29 Jun 2011 to 144.167.57.133. Redistribution subject to ECS license or copyright; see http://www.ecsdl.org/terms_use.jsp