Advanced Drug Delivery Reviews 63 (2011) 1186–1209
Contents lists available at SciVerse ScienceDirect
Advanced Drug Delivery Reviews
j o u r n a l h o m e p a g e : w w w. e l s ev i e r. c o m / l o c a t e / a d d r
Multiple aspects of the interaction of biomacromolecules with inorganic surfaces☆
Ivana Fenoglio a, b,⁎, Bice Fubini a, b, Elena M. Ghibaudi a, Francesco Turci a, b
a
Dipartimento di Chimica Inorganica, Chimica Fisica e Chimica dei Materiali, University of Torino, via Pietro Giuria 7, 10125 Torino, Italy
“G. Scansetti” Interdepartmental Center for Studies on Asbestos and other Toxic Particulates and NIS — Interdepartmental Center for Nanostructured Interfaces and Surfaces,
University of Torino, via Pietro Giuria 7, 10125 Torino, Italy
b
a r t i c l e
i n f o
Article history:
Received 5 April 2011
Accepted 2 August 2011
Available online 16 August 2011
Keywords:
Protein corona
DNA
Oxidative damage
Free radicals
Silica
Titania
Gold nanoparticles
Carbon nanotubes
Conformation
Adsorption
a b s t r a c t
The understanding of the mechanisms involved in the interaction of biological systems with inorganic
materials is of interest in both fundamental and applied disciplines. The adsorption of proteins modulates the
formation of biofilms onto surfaces, a process important in infections associated to medical implants, in dental
caries, in environmental technologies. The interaction with biomacromolecules is crucial to determine the
beneficial/adverse response of cells to foreign inorganic materials as implants, engineered or accidentally
produced inorganic nanoparticles. A detailed knowledge of the surface/biological fluids interface processes is
needed for the design of new biocompatible materials. Researchers involved in the different disciplines face
up with similar difficulties in describing and predicting phenomena occurring at the interface between solid
phases and biological fluids. This review represents an attempt to integrate the knowledge from different
research areas by focussing on the search for determinants driving the interaction of inorganic surfaces with
biological matter.
© 2011 Elsevier B.V. All rights reserved.
Contents
1.
2.
3.
Introduction
. . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Properties of inorganic surfaces
. . . . . . . . . . . . . . . . . . . .
2.1.
Hydrophobicity/hydrophilicity . . . . . . . . . . . . . . . . . .
2.2.
Surface charge . . . . . . . . . . . . . . . . . . . . . . . . .
2.3.
Nanotopography . . . . . . . . . . . . . . . . . . . . . . . .
2.4.
Curvature . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.5.
Reactive sites . . . . . . . . . . . . . . . . . . . . . . . . . .
2.6.
Chirality . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.7.
Dissolution/re-precipitation equilibria . . . . . . . . . . . . . .
2.8.
Techniques to evaluate physico-chemical surface properties . . . .
Adsorption of proteins . . . . . . . . . . . . . . . . . . . . . . . . .
3.1.
Alteration of the surface properties of the solid . . . . . . . . . .
3.2.
Structural modifications induced by the adsorption on the protein fold
3.2.1.
The intrinsic protein stability . . . . . . . . . . . . . .
3.2.2.
The characteristics of the sorbent surface . . . . . . . .
3.2.3.
The surface curvature
. . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
1187
1187
1188
1188
1189
1189
1190
1191
1191
1192
1194
1194
1196
1196
1197
1197
Abbreviations: AFM, atomic force microscopy; ANS, 1-anilino-8-naphthalenesulfonate; BSA, bovine serum albumin; CNT, carbon nanotubes; DC, circular dichroism; DSC,
differential scanning calorimetry; ESR, electron spin resonance; FTIR, Fourier transform infrared spectroscopy; HEL, hen egg lysozyme; IEP, isoelectric point; MWCNT, multi-walled
carbon nanotubes; NMR, nuclear magnetic resonance; PZC, point of zero-charge; ROS, reactive oxygen species; SDS, sodium dodecyl sulfate; SDSL, site-directed spin-labeling; SEM,
scanning electron microscopy; SERS, surface-enhanced Raman scattering; SWCNT, single-walled carbon nanotubes; TEM, transmission electron microscopy; TIRFM, total internal
reflection fluorescence microscopy; Trp, tryptophan; UV, ultraviolet.
☆ This review is part of the Advanced Drug Delivery Reviews theme issue on “Formulating Biomolecules: Mechanistics Insights in Molecular Interactions”.
⁎ Corresponding author at: Dip. di Chimica IFM, University of Torino, via Pietro Giuria 7, 10125 Torino, Italy. Tel.: +39 011 670 75 06; fax: +39 011 670 75 77.
E-mail address: ivana.fenoglio@unito.it (I. Fenoglio).
0169-409X/$ – see front matter © 2011 Elsevier B.V. All rights reserved.
doi:10.1016/j.addr.2011.08.001
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
3.2.4.
The protein concentration at the surface . . . . . . . . . . . . . . . . . . .
3.2.5.
The pH and ionic strength . . . . . . . . . . . . . . . . . . . . . . . . . .
3.3.
Changes of protein activity related with structural modifications or selective orientation of
3.4.
Techniques to evaluate protein adsorption, unfolding and orientation . . . . . . . . . . .
4.
Adsorption of nucleic acids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
5.
Surface driven oxidative damage to biomolecules . . . . . . . . . . . . . . . . . . . . . . .
6.
Role of the interactions with biomacromolecules in the in vivo response to nanoparticles . . . .
7.
Conclusions
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1. Introduction
The phenomena occurring when an inorganic material meets a
biofluid are extremely complex. Dissolution/precipitation processes,
reconstruction of the surface, adsorption of ions, small molecules and
macromolecules, and redox reactions may occur at once [1].
The comprehension of the molecular mechanisms of such
processes is an intriguing challenge which rises interest in several
different fields such as astrobiology, ecology, biology, biotechnology,
engineering, and medicine [1–4]. In medicine a deep knowledge of
the surface/biological fluid interface processes is needed for the
design of new biocompatible materials for implants, drug delivery and
nanodevices for diagnosis and therapy.
One of the main problems with implantable medical devices is to
prevent adverse reactions as inflammation or infections. For this reason
effort has been placed on creating surfaces which should promote the
attachment of target tissue cells but preventing bacterial adhesion. The
mechanisms of interaction of surfaces with tissues at a molecular level
are however largely unknown [5], thus a predictive approach to the
development of new materials is currently difficult to achieve.
The development of methods to manipulate matter at the nanolevel
has recently lead to the production of several different kinds of inorganic
nanoparticles (NPs) which may possibly find medical application in the
next future. At the same time the rapid diffusion of nanotechnological
products opens new concerns on the possible adverse effects following
the direct or indirect (i.e. through leakage in the environment) exposure
of humans to NPs. The interaction of NPs with blood proteins is
considered the most critical step determining the NPs biodistribution,
toxicity and/or efficacy. Interaction of NPs with blood proteins has been
in fact associated to thrombosis [6] or adverse responses of the immune
system, macrophage uptake and elimination [7].
The definition of common determinants in cell/surface interface
processes is difficult to achieve since inorganic surfaces and biomolecules,
as well as the fluids wherein they interact are extremely variable entities.
Because of the specificity of its complex structure each single
protein or nucleic acid molecules exhibits its own “personality” [8]
and therefore interacts differently with surfaces. On the other hand,
inorganic surfaces may expose to the solvent distinct functionalities or
defects that may react or interact with solvent, solutes, biomolecules
and cells. Such properties vary in abundance and typology not only in
a way that depends on the kind of material, but also within materials
having the same elemental composition but different structure or
origin [9,10].
Large differences in chemical composition, ionic strength and
acidity of biological fluids exist depending on tissues and cellular
compartments [11]. Furthermore the composition of fluids varies as it
depends on the cell responses to environmental stimuli. Biological
compartments are open to matter and energy exchanges and therefore
they never reach an equilibrium state. Kinetics are therefore relevant
in determining which process prevails.
When the surface of a foreign body comes in contact with a
biological fluid it is quickly covered by the medium components —
particularly proteins — forming a complex layer which has been
defined by Dawson and co-workers as “protein corona” in the case of
. . . . . .
. . . . . .
the proteins
. . . . . .
. . . . . .
. . . . . .
. . . . . .
. . . . . .
. . . . . .
1187
. . . . . . . .
. . . . . . . .
onto the surface
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . .
. . .
. .
. . .
. . .
. . .
. . .
. . .
. . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
1197
1198
1199
1200
1202
1203
1204
1204
1205
particles. Such corona is a dual system, composed by a core of strongly
bound proteins and an outer layer of fast exchanging molecules
[12,13].
The establishment of such a corona is a competitive process driven by
thermodynamic and kinetic factors, including the stability of the NP–
protein adduct, the relative concentrations of proteins in the medium, the
charge distribution on the NP as well as on the protein surface (which are
both pH-dependent), the chemical nature and reactivity of the NP surface,
the NPs abundance, the temperature [12].
Moreover the corona is expected to change depending on the nature
and rate of “relaxation processes” as particles redistribute from one
compartment or organ to another, as well as upon receptor-mediated
endocytosis from the extracellular environment into the primary
endosomal cavity, or from the cytosol to the nucleus [13].
Dawson has very clearly highlighted that the interaction of the cell
with NPs is strongly driven by the protein corona, rather than by the NP
itself because the cell “sees” protein-coated NPs rather than the bare NP
surface. On the other hand, the formation of such a corona is heavily
influenced by nature, size and shape of the NP, as well as by the
composition of the medium [12]. Consequently, in order to foresee the
cell reaction towards the protein-coated NP, it is essential to clarify the
mechanisms that lead to the formation of the protein corona. On one
side, this implies the need for a standardized multi-techniques approach
for the characterization of the protein corona. On the other one, an
accurate knowledge of the structural and chemical nature of the NP (and
especially the NP surface) influencing the adsorption processes is
needed as well [12].
A large number of coatings and surface decorations have been
proposed to improve the biocompatibility of materials used in
prosthesis and of NPs used in therapy and diagnosis. The description
of the properties of these modified materials is beyond the scope of
the present paper and we refer to other reviews for an extensive
discussion of this kind of modified materials [5,14,15]. Here, we
mainly focus on the interaction with bare inorganic surfaces. Note that
coatings may be instable in fluids [16,17] and the long permanence in
the body of functionalized surfaces may result in coating degradation
[18,19]. Exposure of the internal inorganic core is therefore a realistic
event at some stages.
This review tries to merge the information coming from different
scientific areas in order to summarize the current knowledge
concerning the behavior of proteins and nucleic acids approaching
an inorganic surface as well as the physico-chemical determinants
that drive such processes. In particular we will focus on those
inorganic materials that have been proposed — or are currently used —
as nanovectors or as excipients in pharmaceutical preparations.
2. Properties of inorganic surfaces
The importance of the surface in driving and modulating the
biochemical fate of inorganic particles is now a solid and wellassessed key fact. The physico-chemical properties of inorganic
surfaces play a crucial role in determining the interaction of a solid
with biomolecules and hence with living matter [1,5,20–22]. Among
the recent revolutionary advances in the field of surface science the
1188
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
most fundamental discovery that has been made is that — in contrast
to bulk properties which are derived from both chemical composition
and structure — the properties of a surface depend on its own surfacespecific composition and structure — which largely differ from bulk —
and on nanotopography. Outside the fields of solid-state chemistry
and engineering sciences, the fact that the bulk and the surface of a
material are very distinct — though related — entities is not always
enough stressed out. This consideration is indeed central in the
study of all the complex physico-chemical aspects of the surface. The
last atomic layer of a solid is a discontinuity point, an interface
between the bulk structure (either crystalline or amorphous) and
the surrounding medium (air, water, solvent, complex media like
serum or blood). The surface of a solid is a complex and dynamic
entity, that can actively interact with surrounding medium, e.g.
undergoing protonation/ deprotonation, adsorbing/desorbing moieties and biomolecules, being dissolved or assisting the precipitation
of compounds.
Here we will focus on those surface features strictly ascribed to the
chemical properties which are known to modulate the interaction
with biomolecules: i) the hydrophobic or hydrophilic behavior; ii) the
surface charge; iii) the sub-micrometric topography of the surface
(nanotopography); iv) the surface curvature; v) the occurrence and
nature of reactive sites; vi) the chirality; and vii) the dissolution/reprecipitation equilibrium.
2.1. Hydrophobicity/hydrophilicity
Defining the hydrophobicity and the hydrophilicity of a surface from
a chemical point of view is not trivial. Macroscopically and qualitatively,
the wettability of a material defines its hydrophilicity that can be
measured evaluating its mirrored effect such as the contact angle of a
water droplet. The more hydrophilic the material, the higher the
wettability, the lower the contact angle. The contact angle is the
resultant between adhesive (droplet-surface) and cohesive (droplet–
droplet) forces. In other words, the tendency of a drop to spread out over
a flat, solid surface (wettability) increases as the contact angle decreases.
Down to the atomic scale, hydrophilicity originates in polar surface
chemical functionalities (e.g. Si–OH, Ti–OH) or under-coordinated metal
ions at the surface (Ti3+, Al3+, Fe 2+/3+) [23,24]. The abundance of such
sites determines the degree of hydrophilicity/hydrophobicity, which is
one of the determinants for biomolecules adsorption. The very same
bulk material may show dramatic changes in hydrophilicity upon
heating or surface alteration/functionalisation [25]. In general at the
atomic scale there is a marked heterogeneity also in hydrophilicity,
which can be evaluated from the strength of the interaction of each site
with water molecules. The interaction of surface sites with water
molecules is obtained by measuring the adsorption enthalpy as a
function of coverage, whose chemical nature may be assessed by
integrating data from microcalorimetry and infrared spectroscopy [23].
For example, detailed studies on the hydrophilicity of several quartz
samples have been performed in the past ([23] and ref therein) some of
which have shown a large variation in cytotoxicity upon variation in
hydrophilicity [26]. The occurrence of interstitial or lattice substituting
ions such as Al3+ or Fe3+ largely modifies the hydrophilic behavior of
SiO2 surface.
Carbon-based nanoparticles, CNTs and fullerenes are highly
hydrophobic materials. However, they may be easily functionalized
by inserting surface functionalities as hydroxyl (–OH) or carboxyl
(–COOH) groups which impart to the surface a hydrophilic character.
2.2. Surface charge
We have previously clarified that bulk and surface of a material
are very distinct entities. This consideration is central in the study of
the surface charge. The chemical nature of the surface and the
occurrence of polarized or charged moieties determine the electric
nature of the surface. When any object with a charged surface is
placed into an aqueous solution an electrical double layer — two
parallel layers of charge surrounding the object — is formed on the
surface. The first layer is made of ions adsorbed directly onto the
surface due to specific chemical interactions with the surface, e.g.
electrostatic forces, hydrogen bond, coordinative bond, and van der
Waals interactions. The second layer loosely associated with the
surface via the weak electrostatic forces, is composed of free ions
which move in the fluid under the influence of electric attraction and
thermal motion rather than being firmly anchored to the surface. This
second layer is thus called the diffuse layer. The potential drop across
the mobile part of the double layer responsible for electrokinetic
phenomena is called ζ-potential [27]. Conventionally, the sign of this
potential is reported accordingly to the charge of the surface
generating it, thus being positive or negative for positively or
negatively charged surfaces respectively. Being an interface measure
the ζ-potential of a solid surface largely depends on the chemical
nature of the solution interfacing with it. Thus a ζ-potential value
reported without defining the solution conditions is a virtually
meaningless number. In aqueous media, the pH and the ionic
strength of the solution are the most important factors that affect
ζ-potential and should always be explicitly indicated.
In colloid science the determination of ζ-potential is commonly
used to evaluate the stability of a suspension. A potential of 25–30 mV
(positive or negative) can be taken as the arbitrary value that separates
low-charged surfaces from highly-charged surfaces, hence instable
from stable colloids [28]. The condition when the electrical charge
density of a surface becomes zero is called point of zero charge (PZC).
This point is usually experimentally determined by acid–base
titrations while monitoring the electrophoretic mobility of the
particles and the pH of the suspension (see below). From an
electrokinetic perspective, the iso-electric point (IEP) and the PZC,
i.e. the pH at which electrophoretic mobility is null, are generally
considered equivalent. However, PZC and IEP have different meanings.
According to Jolivet [29], when the surface is not charged, i.e. there are
neither positive nor negative charges, the surface is best described by
the PZC. When an equal amount of positive and negative charges are
present, the IEP should be used instead.
When in the nanoscale, particles likely form agglomerates or are
found in chemically stable aggregates. Under these circumstances the
agglomeration and/or aggregation both in liquids or in air determine
the actual particle size of NPs [30]. The role of important factors such
as ionic strength, pH and particle surface chemistry which control
dispersion of NPs was recently examined in a thorough work on some
commercial and lab-synthesized titanium dioxide (TiO2 ) and
quantum dot samples [31]. In this study the difference between
aggregation and agglomeration is discussed. Aggregated NPs are
bound with hard, likely covalent bonds between primary particles.
These sintering processes are likely to occur during NP preparation
and cannot be avoided. Conversely agglomerated particles that are
held together by van der Waals forces can be separated by overcoming these weaker interactions by several methodologies. The
measurement of the ζ-potential can be of great usefulness in
selecting the appropriate condition for manipulating a NP promoting
or favoring aggregation to achieve different results. For instance, the
dispersion of hydrophobic carbon-based nanomaterials in water may
be largely improved by introducing a sufficient number of charged
functionalities at the surface to generate repulsion among particles
[32–34].
Besides being a well-know tool for the investigation of colloid
stability the ζ-potential of inorganic solids is rapidly becoming an
indispensable surface measurement to characterize NPs and to predict
their behavior in terms of adsorption of biomolecules ([35] and ref
therein). Recently ζ-potential measurements were also adapted to
investigate the stability and the partial vs. full coverage of proteins
adsorbed on different silica NPs [36].
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
The ζ-potential of a solid measured during the acid–base titration of
the surface functionalities reported as function of pH, i.e. the ζ-plot, is a
straightforward tool to characterize different NPs and to explain and
predict their behavior in a biological fluid. Si, Ti and Fe oxide NPs of
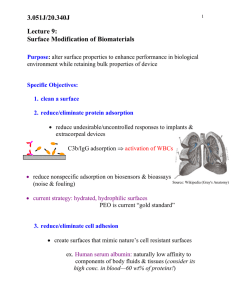
similar size exhibit largely different ζ-plots (Fig. 1). PZCs are in the order
silica b titania b hematite, clearly indicating the order of acidic strength
of the hydroxylated surface functions (Si–OHN Ti–OHN Fe–OH). The
value of ζ-potential for biologically relevant pH values is highlighted. At
physiological pH silica and titania are negatively charged, while
hematite is positive (close to PZC). On the other hand at acidic pH —
e.g. 4.5 inside phagolysosomial vesicles — silica remains negatively
charged, while titania and hematite exhibit positively charged surfaces.
ζ-potential is not only related to the type of material: Kosmulski
dedicated a recent review [37] to remark that the ζ-potential of a solid
depends upon a number of material-specific features such as the
chemical and crystallographic composition (commercial samples of
the same material may largely differ in such characteristics), particle
size, crystal lattice and defects, impurities, and the presence of
thermodynamically unstable phases.
In a recent work specifically devoted to investigate the effect of
some TiO2 surface features on ζ-potential [38] PZC was found to be a
function of primary particle size. When primary particle size of labsynthesized TiO2 anatase NPs increased from 6 to 104 nm, the PZC
decreased from 6.0 to 3.8 pH unit. At the same pH and ionic strength
conditions an increase in size determines a decrease of ζ-potential
throughout the potential curve (at neutral pH, ζ-potential decreases
from −20 to −40 when particle size increases from 6 nm to 104 nm).
This size-dependent mechanism is likely due to the differences in
terms of acidity of surface Ti–OH. When the size of a NP increases, the
acidity of surface hydroxyl increases, as indicated by the more acidic
PZC reported for TiO2. At the same time, the larger particles, being
more acidic, will be more likely deprotonated in neutral or alkaline
solutions, thus showing a more negative ζ-potential. The opposite is
expected to occur at acidic pH where positively charged species may
account for this behavior. The chemical nature and the electrostatic
charge of both rutile and anatase TiO2 surface functionalities
accounting for ζ-potential and PZC have been investigated with
theoretical considerations and experimental measurements by Panagiotou and co-workers [39].
It is very hard, if possible at all, to draw general conclusions on the
ζ-potential trend with respect to surface and bulk features of NPs.
There are however some well-assessed general rules that still apply to
the PZC of metal oxides [40]: i) anionic impurities shift the PZC to
more acid values, cationic impurities to more basic values or toward
the PZC of the impurity oxide; ii) even a partial oxidation and
60
ζ Potential (mV)
40
0
1
2
3
4
5
6
7
8
9
10
11
12
pH
-20
Fe2O3
TiO2
-60
reduction of the material may shift the PZC toward that characteristic
of the oxidation state produced, i.e. oxides at lower oxidation states
have higher PZC; iii) an increase of the ionic strength of the solution
produces a decrease of the ζ-potential of the suspended particles.
2.3. Nanotopography
A smooth surface at the macroscopic level may be very irregular at
the molecular level. Edges, kinks and steps are present to some extent
in all crystals, and many solids have indented edges, irrespective of
whether they are crystalline or amorphous. Exposed atoms or ions at
these positions are more reactive, so that, in general, irregular surfaces
behave differently from smooth ones, with the latter being more inert.
Surface topographic parameters may play thus an important role to
obtain effective material with high biocompatibility and good tissue
interaction. Often the clinical success of an implant is related to an
early osseointegration, i.e. the creation of a direct response is related
to direct bone–implant relationship without the formation of an
intervening connective tissue layer. Geometry and surface topography
are crucial for the short- and long-term success of implants. Kim and
Ramaswamy [41] recently reviewed the latest electrochemical
modifications performed on the native surface of metallic titanium.
Titanium and titanium based alloys are commonly used for implantation in bone contact because of their mechanical properties as well
as their excellent corrosion stability and osseointegration, both
ascribed to the air-formed passive layer always covering the metal
surface. Aluminum oxide is also used for implant due to excellent
bioinertness and mechanical properties. However, all biomaterials
require surface modifications to increase the biocompatibility and
improve implant stability. Anodic oxidation is a common technique to
obtain desired roughness, porosity and chemical composition of the
oxide. Self-organized nano-titanium dioxide tubes are one of the most
promising modifications, because the tubular structure acts as better
scaffold for cell adhesion than the conventional flat surface. Cathodic
electrochemical techniques, such as electrophoretic and cathodic
depositions, are the other electrochemical pathway to surface modification. Deposition of nano-grained hydroxyapatite on titanium
implant makes the implant surface a better substrate for cell interaction than the more coarse-grained [42]. Denis and co-workers [43]
studied collagen adsorption as function of surface nanotopography
and hydrophilicity. They found that the adsorbed amounts on rough
substrata were similar to those found on smooth substrata, but the
protein film morphology clearly depended on the substratum
topography with elongated supramolecular assemblages formed on
smooth but not rough substrata. The authors suggested that
differences in the protein mobility may account for supramolecular
rearrangement of protein where the collagen molecules would be
relatively free to move and assemble on smooth surfaces while the
nanoscale protrusions of rough substrata would inhibit the collagen
mobility.
2.4. Curvature
20
-40
1189
SiO2
Fig. 1. Dependence of surface charge on pH. Variation of ζ-potential as a function of pH
in three different metal oxides. Blue and pink bars indicate the pH in macrophages
cytoplasm and phagolysosomial vesicles respectively. Adapted from Ref. [35].
It is now well assessed that size is a key factor in determining
the interaction of inorganic particles with biological systems,
thereof a cell-specific optimal diameter is relevant for promoting or
avoiding cellular uptake [1]. However, when one refers to surface–
biomolecules interaction the size of an object becomes relevant in
terms of surface curvature. A large particle exposes to biomolecules a
quasi-flat surface, while smaller ones exposing a surface with a high
curvature are reported being able to host a larger amount of adsorbed
molecules per unit surface area [44].
The curvature of the particles is reported to be a dominant factor
for conformational changes occurring to proteins during adsorption
[45]. The surface curvature can affect two distinct surface properties,
the surface geometry and the type of surface functional groups. The
1190
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
two effects will be discussed separately, even if they are mutually
related and only well designed experimental settings can discriminate
between them.
In the paper by Lundqvist and coworkers [46] large amorphous
silica NPs (diameter 15 nm) affect the secondary structure of a
carbonic anhydrase (HCA I) more than smaller ones (6 nm). Under
these circumstances geometrical reasons account for the observed
conformational changes, whereas the interaction energy is higher for
a large interaction area as the sum of the interactions established with
the solid surface is stronger than the energy associated to the
conformational structure of the protein.
The effect of surface curvature on SiO2 NPs in the range 5–100 nm of
diameter was tested on the main phase transition of phosphotidylcholine
bilayers of different chain length around the particles [47]. The data
reported by Ahmed and Wunder are based upon a phosphotidylcholine
bilayer model in which as the SiO2 NP size decreases the difference in the
curvature of the inner and outer leaflets of the bilayer increases. Flat
surfaces and large particles (low curvature) adsorb lipids in a similar way.
On highly curved surfaces (small particles) the lipids form bilayers that
adopt morphology with outer polar heads being widely separated from
each other. To compensate the high free volume and the increased
headgroup spacing, more chains are interlocked with the outer layer of
adsorbed lipids.
A similar conclusion is reported in the study by Cederquist and
Keating [44] on the impact of the diameter of gold nanosphere on DNA
adsorption. The experimental study demonstrates that the particle
curvature controls the surface density of DNA with large particles
more densely coated than small ones. They also propose a model that
can be used to predict DNA packing on nonspherical particles.
On a very different system, poly(methyl methacrylate) adsorbed on
aluminum oxide (Al2O3) clusters, Hershkovits et al. [48] came to similar
conclusions. In this well-understood system, the mechanism of adsorption
results in the formation of a coordination bond (chemisorption) between
the methyl methacrylate (MMA) segments and Al3+ sites on the surface of
the oxide. Three different regimes of adsorption of poly(methyl
methacrylate) (PMMA) of various molecular weights and length on
Al2O3 particles of different sizes have been proposed: (a) when the
alumina cluster radius is much larger than the polymer length, the
adsorption is similar to adsorption on a flat surface and the proportion of
polymer segments that experience conformational changes is indicative of
the bonding process; (b) when the cluster size is on the order of
magnitude of the polymer, geometrical constraints force the scaling of the
adsorbed monomer densities with the curvature. In this case, the
thickness of the adsorbed polymer layer is similar to the thickness of
the adsorbed layer on a flat surface; (c) when the cluster size is on the
same order of magnitude as the thickness of the adsorbed layer, the
curvature is too high to support an energy gradient between the adsorbed
layer and the bulk. As a result, the adsorbed polymer chains will extend
outwardly from the surface of the cluster in order to decrease the density
gradient and the clusters are enveloped by a mixture of adsorbed polymer
chains that extend outward into the solution and entangled polymer
chains from the solution.
Beside geometrical considerations the physico-chemical properties of
the surface may be highly dependent on surface curvature. Curvatureinduced surface modification reflects upon many other biologically
relevant macroscopic features, such as hydrophilicity, ζ-potential,
strength of hydrogen bonding. As reported above the silica surface
terminates with hydroxylated functions attached to Si atoms named
silanols (≡Si–OH). Silanols can be broadly classified as i) isolated,
ii) interacting, and iii) geminal. Siloxane bridges (Si–O–Si) are formed
upon condensation of two adjacent silanols [49]. The presence of surface
silanols and siloxanes and their nature (structure) are determined via
diffuse and reflected FT-IR and FT-NIR analyses. In a study on amorphous
silica powders with different diameters in the range 8–260 nm, the
amount of isolated silanol (IR adsorption band at 3750 cm− 1) was shown
to decrease as the particle diameter increases, almost disappearing in the
case of the largest NP (260 nm). In contrast, the ratio of absorption of the
hydrogen-bonded silanol IR band to that of the isolated group increases
as the particle diameter increases [50]. Generally speaking, the curvature
of silica NPs determines the relative abundance of isolated vs. interacting
silanols and thus influences the dispersibility of silica in water
suspension.
Theoretical considerations on the adhesion of an organic polymer
(oligothiophene) on large-scale atomistic models of nanostructured
titania surfaces were recently carried out by Melis [51]. Adhesion of
the polymer appears to depend upon the local curvature and
roughness. The interaction between titania and the polymer was
reported to be dominated by electrostatic interactions. The adhesion
forces on the curved surface were very different from the case of a
planar surface. Better adhesion was achieved on flat or poorly curved
surfaces and the molecular strain induced by a highly curved surface
had a detrimental effect on adhesion. The authors discuss the binding
force on flat surface, which is dominated by the Coulomb contribution
[52] and report that, at the surface of nanostructured titania, a
covalent contribution larger than that on a flat surface is expected.
2.5. Reactive sites
The surfaces of both covalent and ionic solids exhibit defective
sites generated by the interruption of the bulk structure of the solid.
Freshly ground, abraded, indented, and defective surfaces may expose
surface charges, dangling bonds (i.e. surface-bound radicals) and
poorly coordinated ions which may react with biological molecules. In
some cases such reactions may cause detrimental effects on cells ([9]
and ref therein) as discussed in Section 5.
Crystalline — but not amorphous — silica dusts are known to be
inflammatory and fibrogenic when inhaled [53]. Silica-derived reactive
oxygen species (ROS) that are generated at the particle surface are deeply
involved in the mechanism of toxicity of quartz ([54] and ref therein).
Two cooperative particle-derived ROS-generating mechanisms are
commonly reported. One is mediated by the presence of dangling
bonds on the quartz surface, the other is related to the occurrence of
redox-reactive transition metals exposed at the surface. The first
mechanism involves the presence of homolytically (Si•, SiO•) and
heterolytically generated (Si+, SiO−) reactive sites, which originate
from the grinding processes used to produce quartz dusts, that promptly
react with molecular oxygen originating several surface radical species,
i.e. SiO2•, SiO3•, Si+–O2−•. The second mechanism involves the presence of
transition metal traces, typically iron [55], at the particle surface. Both
dangling bonds and metal impurities are not present on synthetic
amorphous silicas (precipitated or pyrogenic silicas) which appear
therefore completely inactive in generating free radicals [56]. Metals
exposed to the surface may react via radical mechanisms or electron
transfer reactions with biological relevant molecules, such as H2O2, C–H
bonds or thiol groups in proteins, and nucleic acids. Iron is a common
contaminant also in many natural specimens and can substitute for
structural ions (e.g. Mg2+) in silicates. The reactivity of iron is involved in
fact in the pathogenic mechanism of asbestos. An asbestos fiber that does
not expose iron has been reported to be non-reactive in terms of ROS
generation [57] and resulted inactive in altering cellular metabolism [58].
ROS-generating iron ions may also be acquired by the environment
surrounding the surface. Inhaled asbestos fibers, for example, are often
found to be coated with iron oxyhydroxides (asbestos bodies) which may
originate from the adsorption of endogenous ferritin. Mammalian ferritin
was in fact found to adsorb strongly, from aqueous solutions, onto the
asbestos minerals amosite and crocidolite [59]. The fibers containing
adsorbed ferritin were reported to show high free-radical activity leading
to radical damage to DNA.
Generally speaking, it is now well-documented that iron-related
radical release from solid surfaces is not related to iron abundance but to
a well-defined redox and coordination state of the iron ions at the
surface [57,60–64]. Pioneering work by Fubini's group [65] measured
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
the free radical release by means of the spin trapping technique on three
kinds of iron containing particulate: two asbestos fibers (chrysotile and
crocidolite, Fe ca. 5% and 27% respectively); an iron-exchanged zeolite
(Fe ca. 4%) and two iron oxides (magnetite and hematite). The authors
give evidence for three different kinds of iron sites: one acting as
hydrogen atoms abstractor, the other as a heterogeneous catalyst for
hydroxyl radical release, the third one related to catalysis of hydrogen
peroxide disproportionation. In both mechanisms of free radical release,
the Fe-exchanged zeolite mimics the behavior of asbestos whereas the
two oxides are mostly inert. Conversely magnetite turns out to be an
excellent catalyst for hydrogen peroxide disproportionation while
hematite is inactive also in this reaction.
Even if iron contamination is involved in many detrimental
reaction of mineral surface, a recent study clarified that the high
toxicity of some very pure quartz sample (DQ12) is not related to
metal contamination but arises from the presence of undissociated
silanols with potential to form strong hydrogen bonds, rather than
being due to radical surface reactions. The reactivity of these surfaces
is believed to interact in such a strong manner with biomolecules to
bind and disrupt cell membranes and/or proteins [64].
Reactivity mechanisms also originated by metal impurities are
partially involved in the biological detrimental reactivity of carbon
nanotubes. Most commercial CNTs contain ultrafine metal particles
(e.g. Fe, Ni, Y, Co, and/or Mo) derived from the original growth catalyst
or support. It has been reported that carbon nanotubes containing
iron as impurity were able to induce intracellular ROS. When acid
treated to remove metal contaminants, the cellular ROS generation
did not occur, suggesting that the metal catalyst contaminants are
responsible for the ROS generation [66]. On the other hand, cell-free
experiments using multiwall carbon nanotubes (MWCNT) in aqueous
suspension determined that MWCNT do not directly generate either
oxygen or carbon-centered free radicals whereas are able to quench
such reactive species [67–69].
In the case of semiconducting materials, reactivity may arise
following the charge separation occurring in the bulk of the oxide
leading to the promotion of an electron in the conduction band and to
the formation of a hole in the valence band. When the charge carriers
reach the surface of the solid they entail reduction (e −) and oxidation
(h +) reactions with the surrounding medium [70].
TiO2 is a well-known photocatalyst. Under UV illumination it
produces highly reactive radical species such as O2−• (e − transfer to
O2) and HO• (hole interaction with water) [71].
Ti
3þ
þ O2 → Ti
4þ
•
þ O−
2
þ
h þ OH− → ΗO•
Superoxide radicals may be oxidized to singlet oxygen by electron
holes [72].
For TiO2 NPs entering inside the body no reactivity is expected in
the absence of illumination. However it was recently reported that the
generation of free radicals from uncoated rutile and anatase surfaces
also occurs in the dark in the presence of hydrogen peroxide or
organic molecules [73]. In the case of titania, the crystalline phase
largely modulates the surface reactivity, anatase being more reactive
than rutile under illumination [73,74]. Photoreactivity of titania also
depends upon the size of particles and method of synthesis [75].
Other materials (e.g., quantum dots or fullerenes) [76,77] have
been reported to generate free radicals under UV illumination.
A size effect is generally observed for particles in the nanometric
range. By reducing the particle size down to nanometers the disorder
of the structure increases and the generation of defects affects the
physical properties of the material and possibly (e.g. Au and Fe NPs)
surface reactivity [20,78]. However, this is not a rule since it strictly
depends upon the type of material.
1191
2.6. Chirality
Biomolecules generally rely on chirality to induce site-specific
molecular recognition as the basis for their correct function. Proteins
have mostly evolved to recognize specific substrates, or be recognized
by specific receptors and is not surprising that asymmetry plays a
significant role in recognition processes. Addadi's and her coworkers'
work has over the years elegantly clarified that the asymmetry of
organized surfaces, such as chiral faces of calcium tartrate crystals, is
selectively recognized by some proteins, antibodies and eventually
differentiated in terms of cell adhesion [79]. Experimental studies have
been devoted to the adsorption of organic molecules on right- vs. lefthanded quartz, which is the most common acentric mineral [80].
Although the chirality of quartz crystal seems not to account for
homochirality of living matter [81], intrinsically chiral mineral and
metal surfaces have been reported to act as enantioselective agents in
several reactions [82,83]. Surfaces of metals with achiral bulk lattice
symmetry can become chiral by cutting a single crystal along specific
high Miller index planes. Intrinsically chiral surfaces of some common
minerals with both chiral and achiral bulk symmetry, such as α-quartz
or calcite respectively, have been investigated with regard to the
designing of catalysts with high yield in terms of enantiomeric excesses.
Many more details on this topic can be found in the Mallat, Orglmeister
and Baiker extensive review on the asymmetric catalysis at chiral metal
surfaces [84].
Chirality may also be observed in engineered single-walled carbon
nanotubes (SWCNT). SWCNT are made of a single graphene sheet rolled
up to form tubes which may have distinct structures [85]. Depending
upon the method of synthesis chiral helical tubes may be formed albeit a
mixture of SWCNT having different diameter and chirality are generally
obtained [86]. The preparation of enantiomerically pure SWCNTs has
raised interest in the past years since enatiomers exhibit distinct
electronic and optical properties [85,86].
2.7. Dissolution/re-precipitation equilibria
Solid materials may be constituted by atoms bound through i) polar
or apolar covalent bonds (e.g. elemental carbon, silicon dioxide or
titanium oxide); ii) ionic bonds (typically alkaline, alkaline earth, e.g.
NaCl, CaCO3); and iii) metallic bond (e.g. Au, Ag). When an ionic or
covalent material is immersed in water, the solubility depends on the
balance between reticular free energy and energy of solvatation of the
ions and related entropy changes. In the case of metals in the
elementary form both the tendency to be oxidized in aqueous media
and the solubility of the oxidized form have to be considered.
Biological fluids contain several chemical species which can form
stable coordination compounds. Therefore the solubility in biological
fluids of metal-containing materials is generally higher than in pure
water [87,88]. A similar effect has been reported also for silica [89].
The dissolution of a solid in biological fluids is relevant for at least
two reasons: a) metal ions in the solid may be progressively released
and become involved in many reactions and b) the chemical nature of
the surface and sub-surface layers are progressively modified. The
metal mobilized may then be a direct or indirect source of damage for
biomolecules, such as lipids, proteins or DNA [1,88]. The ion-depleted
material may in turn re-acquire the same ions, or other ions which are
similar in size and charge, from the surrounding solution. The structure
and composition of the surface inorganic solids may therefore largely
be modified during the permanence in biological fluids.
The crystal structure and surface morphology determine to what
extent ion depletion occurs, and which are the chelators more
appropriate to extract a given ion from a particular solid. Many cases
of detrimental interactions with cells induced by solubilised ions
instead of surface-driven mechanism have been reported, e.g. for iron
oxides, Ag and ZnO NPs ([1] and ref therein).
1192
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
Different bulk structure (crystalline vs. amorphous, type of
crystalline phase or insertion of doping elements) may induce large
differences in terms of solubility or rate of dissolution of an inorganic
compound. As an example, the dissolution rate of amorphous silica is
much greater than quartz in similar conditions. A large variability
exists among the solubility of crystalline silica particles with different
lattice symmetry [49]. The higher solubility of amorphous silica by
respect to crystalline forms is one of the various factors claimed to
play a role in the much lower toxic response elicited [90–93].
Finally, as far as nanometric particles are less thermodynamically
stable than their larger counterparts and expose to the fluids a larger
surface area per unit mass, the dissolution kinetic is expected to be
much faster than that of bulk material [78].
2.8. Techniques to evaluate physico-chemical surface properties
The surface chemistry of a solid and particularly the properties that
govern the response to bio-interactions, can be studied by several
techniques, many of which are now virtually able to give a response at
the atomic resolution. Among the many possible ways to organize
these techniques, we chose to discuss them with regard to their direct
or indirect interaction with the surface. Direct tools include imaging
techniques, such as scanning and transmission electron microscopy
(SEM and TEM), atomic force microscopy (AFM), and elemental
techniques, such as X-ray photoelectron and Auger electron spectroscopy (XPS and AES) and secondary ion mass spectrometry (SIMS),
just to name a few. Indirect methods gather information about the
surface analyzing the broader effect of some relevant surface features
(e.g. ζ-potential/electrophoretic mobility) or the interaction with a
molecular probe for specific surface sites (e.g., adsorption enthalpy
measure — calorimetry; ESR spectroscopy coupled with spin-trapping
technique; FTIR and Raman detection of site-specific vibrational
probes).
Scanning electron microscopy (SEM) is largely the most used
technique to study surface topography. When a high-energy electron
beam (1–30 keV) is spotted across a sample surface, low-energy
secondary electrons are generated. Due to their very low energy
(b50 eV), secondary electrons may escape only from the uppermost
surface layers (ca. 5 nm) of an object and are hence able to provide
high spatial resolution (up to 0.5 nm) and impressive long-depth-offield images of the sample surface. The main difficulties that arose in
SEM imaging are due to sample preparation. The sample has to be
conductive per se or coated with a conductive thin film (usually C, Au,
Pt). Normally SEM microscopes operate under high vacuum condition
(ca. 10 − 6 Torr) to prevent electron beam to be deviated by gas
molecules. These operating conditions pose particular difficulties
when analyzing biological samples generally electrically insulators
and water-rich. Variable pressure SEM microscopes overcome to
these limitations by allowing non-conductive and partially hydrated
samples to be imaged but a general loss in spatial resolution is
observed. Very low-energy electron emission source (Field emission
gun) can be operated to achieve better performance on poorly
conductive samples.
When an electron beam interacts with an element, X-rays are also
emitted. For this reason, SEM are often coupled with X-ray analyzers
(energy-dispersive or wavelength dispersive X-ray spectroscopy, EDS
or WDS) that allow to obtain information on the elemental
composition of the sample. However, opposite to secondary electron,
X-rays have high-energy (in the keV range) and can be produced
several microns below the surface. Thus the information provided
describes the bulk composition of the material rather than its surface,
which has to be investigated with surface specific techniques (see
below). The spatial resolution of the X-ray spectroscopy coupled with
SEM is however high enough to allow the elemental mapping of a
surface with a submicron resolution.
Similar to SEM, TEM uses an electron beam to create an image of the
sample. However, in TEM the electrons are forced to pass through the
sample which has to be thin or ultra-thin. TEM samples will have a
thickness that is comparable to the mean free path of the electrons that
travel through the samples, which may be only a few tens of
nanometres. TEM may operate in bright field or in dark field. The bright
field imaging mode is the most common mode of operation for a TEM. In
this mode the image, thus the contrast, is formed directly by occlusion
and absorption of electrons in the sample. Denser regions will appear
darker, while regions with less density (both in terms of crystal lattice or
atomic number) or with no sample in the beam path will appear
brighter. The bright field image is virtually a two dimensional projection
of the sample down the microscope optic axis. The very short
wavelength of electrons used in this case as beam of electromagnetic
radiation makes it possible to achieve a very high spatial resolution
which makes TEM capable to attain an atomic resolution. The dark field
imaging mode uses the electrons diffracted by one or more crystallographic planes of the sample. In this mode, the image is formed by the
scattering of specific crystal planes and the zones where there is no
sample remaining dark. Bright and dark field images can be combined
obtaining a so called high-resolution image (HR-TEM). Although TEM is
intrinsically a bulk technique, the necessary preparation of thin crosssections of massive samples often permit to obtain fundamental
information about many surface characteristics, in particular elemental
composition, contamination, defects, and crystal lattice modification, as
well as surface amorphization.
The most recently developed but today widely used imaging
technique with atomic resolution is the atomic force microscopy
(AFM). AFM is a scanning probe microscopy (SPM), a branch of
microscopy that forms images of surfaces using a physical probe that
scans the specimen. The AFM probe consists of an oscillating silicon
cantilever with a sharp tip (few nanometers wide) at its end. When
the tip is brought into proximity of a sample surface, several different
forces are established between the tip and the sample and the
cantilever is deflected. The cantilever deflection or the modification of
its natural oscillation frequency can be followed with a laser spot
reflected from the top surface of the cantilever on an array of
photodiodes. The AFM can be operated in a number of modes,
depending on the application. Topographical analysis can be readily
performed by scanning the surface maintaining the tip at a constant
height, thus exerting a constant force between the tip and the sample.
This is obtained with the aid of a piezoelectric tube that can move the
sample in the z direction for maintaining a constant force, as well as
along x and y axes. The resolution along z can be sub-nanometric,
whereas AFM lateral resolution is typically of the order of 1 nm.
Besides static mode (also called contact), a variety of dynamic (or
non-contact) modes where the cantilever is vibrated exist. Although
the lateral resolution of AFM is comparable with SEM and TEM
imaging technique, the extraordinary resolution on z and the
possibility to operate in complex media (water, but also buffers or
solvent) make AFM often the most suitable technique for surface
imaging. Furthermore, the silicon probe can be variously functionalized making AFM an even more powerful tool to characterize surface
properties [94,95]. Many reviews exist on the topic and can be used
for further reading [96,97].
X-ray photoelectron spectroscopy, also known as electron
spectroscopy for chemical analysis (ESCA), is used to determine
quantitatively the atomic composition of a material and the chemical
properties of its elements. During XPS experiment, the sample is
irradiated with an X-ray beam that interacts with the electronic shell
of the material atoms determining the emission of some electrons,
thus called photoelectrons. The kinetic energy of these electrons,
subtracted by the X-ray photon energy and some instrumental
parameters (the work function), corresponds to the binding energy of
the electron with the nucleus and is therefore directly representative
of the atomic number of the element (Z). Furthermore, element in
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
different oxidation state (e.g., Fe 2+ and Fe 3+ or S 2− and SO42−)
exhibit different electronic shell and produce differentiable XPS
signals. The low energy of photoemitted electrons makes them
impossible to escape from inner material layers, thus the XPS signal is
representative of the first 10–100 Å. One of the most interesting
features of XPS spectroscopy is the large spectrum of elements
detected, when compared to other electronic spectroscopies. Virtually
only H and He are not emitting photoelectrons in a sufficient amount
to allow determination. Elements from Li (Z = 3) and above can be
quantitatively analyzed by means of XPS. Unfortunately, the limit of
detection is in the order of part per thousands. Detection limits of
parts per million (ppm) are possible, but require special experimental
conditions. As for the other electron involving techniques, ultra high
vacuum conditions are required. This is the primary drawback for
analyzing biological samples. For most applications, XPS is in effect a
non-destructive technique that measures the surface chemistry of
any material.
Complementary to XPS measures many quasi-surface analytical
techniques exist that involve either X-rays or electron as excitation or
information source. Among them, Auger electron spectroscopy is of
particular interest for surface analysis. Auger electrons in fact are
generated when a high-energy electron beam is focused on a material
surface. As for secondary electrons used in SEM imaging, the low
energy of Auger electrons (50 eV–3 keV) has a short free path in a
solid and is therefore related to the uppermost surface or quasisurface atomic layers (few nanometres). For this reason, however, AES
has to be operated under ultra-high vacuum conditions. As in XPS, AES
measures the kinetic energy of the emitted Auger electrons, which is
characteristic of the element present at the surface. Some modern
electron scanning microscopes have been specifically designed for
coupling with Auger spectrometer; these scanning Auger microscopes
(SAM) can produce high resolution, spatially resolved surface
chemical images, since Auger electrons can be discriminated from
secondary electrons on a kinetic basis, being Auger more energetic
than secondary electrons. Besides surface information, depth profiles
are often obtained with AES coupled with a sputtering device able
to dig into the surface with a subnanometric resolution. Opposite
to quantitative elemental methods using X-rays (such as EDS, WDS or
X-ray fluorescence), AES is sensitive to the lighter elements and Auger
peaks can be detected for elements as light as lithium (Z = 3), which is
the lower limit for AES sensitivity. Neither H nor He can be detected
with this technique. The very low Auger yield for elements with Z N 50
strongly limits AES to identify heavier elements. Beside atomic
number, there are several factors that can limit AES applicability.
The most common limitation is often due to charging effects in nonconducting or poorly-conducting samples. Since excitation source of
AES is conceptually identical to SEM, poorly conducting sample will
gain a net polarity at the surface during the experiment. However, in
this case, a conductive surface coating is obviously not applicable.
Both positive and negative surface charges severely alter the yield of
electrons emitted from the sample and hence distort the measured
Auger peaks.
In this brief overview of surface techniques, it is worth mentioning
the currently most sensitive surface analysis technique, secondary ion
mass spectrometry (SIMS), which is able to detect elements present in
the parts per billion range. When a solid surface is bombarded with
high energy ions (primary ions), new ions from the sample are formed
(secondary ions) and are sputtered away from the sample surface. The
secondary ions can thus be detected and analyzed by means of a mass
spectrometer, such as a time-of-flight (TOF) or a quadrupole mass
analyzer. The erosion of the sample by an accessory ion beam can also
provide a depth profile of the sample, and the ion beam, opportunely
focused up to a 50 nm lateral resolution, can be scanned over the
surface to provide an elemental image. SIMS is of particular interest
when high sensitivity measures for dopants and impurities are
required. It is also the reference choice for depth profiles with
1193
simultaneously excellent detection limits and depth resolution
(subnanometric). Using mass detectors, SIMS can analyze virtually
any element of the periodic table, and can be fine-tuned to
discriminate among isotopes, including H.
The most external atomic layer of a solid is a discontinuity point, an
interface between the outlying bulk structure and the surrounding
medium. Structural ligands of surface atoms are generally replaced by
molecular water or hydroxyl groups. The degree of ligand loss, and
thus of coordination, varies with the location of the ions at the surface
(e.g., extended surfaces, edge or corner positions). It is well-known
that such poorly coordinated ions play a determinant role in the
surface reactivity. Nature and abundance of the surface active sites
may be evaluated by adsorption of suitable probe molecules from the
gas phase on the material surfaces deprived of the adsorbed
adventitious molecules by a standard treatment in vacuum [98–102].
The coordinative unsaturation created in such way can then be filled
by a suitable probe molecule. Both the adsorption/ desorption
enthalpies and the vibrational features of the probe depend on the
characteristics of the surface centers where it is adsorbed, while the
total amount adsorbed evaluates the abundance of surface active ions.
The vibrational features of adsorbed probe are practically monitored by
means of FTIR spectroscopy and the stepwise adsorption/desorption
enthalpies of the process can be followed by a conventional
adsorption microcalorimetry.
Many well-established surface site-probe couples have been
thoroughly investigated: Fe–NO, Al–CO, Cu–CO, Ca–CO2, Brønsted
acid-NH3 or pyridine, noble metal (Au, Pt)-CO [57,61,62,64,98,
103–106].
The information which can be obtained from FTIR spectra and
microcalorimetry using molecular probes concerns: (i) the abundance
and nature of Brønsted and Lewis acidic or basic groups; (ii) the
occurrence structural defects; (iii) and the nature and location of
framework cations.
Among indirect surface properties measurements, ζ-potential is
becoming more and more important in understanding and prediction
of solid surface-biomolecule interaction [37], with a particular regard
for NP studies [31]. ζ-potential can be measured using several
complementary techniques. A complete report on such techniques is
beyond the purposes of this review and only the most commonly
applied electrophoretic light scattering (ELS) and electroacoustic
phenomena measurements will be discussed. When an electric field is
applied across a dispersion, charged particles move toward the
electrode of opposite polarity. This phenomenon is called electrophoresis. If a laser beam is passed through the sample undergoing
electrophoresis, the scattered light from the moving particles will be
frequency shifted. By measuring the frequency shift, the electrophoretic mobility can be determined given the laser wavelength and the
scattering angle. ζ-potential can then be calculated from the
electrophoretic mobility. It should be noted that in the surrounding
electrical double layer there is a notional boundary (slipping plane),
within which the liquid moves together with particles. The measured
ζ-potential is the potential at this slipping plane. ζ-potential is not
exactly the surface potential (surface charge), but is the potential of
practical interest in dispersion stability because it determines the
interparticle forces [37,107]. Electroacoustic phenomena measurements are based on the generation of and electric signal following the
propagation of ultrasound through a heterogeneous fluid, such as
dispersions or emulsions. This electric signal is called colloid vibration
potential/current (CVI) and can be used for characterizing the ζpotential of various dispersions and emulsions. At the opposite, when
an electric field propagates through a suspension an ultrasonic
acoustic wave arises. The so called Electric Sonic Amplitude (ESA),
the inverse of CVI effect, can be detected by an acoustic transducer
behind the electrode. The generated sound wave is at the same
frequency as the applied electric field and its amplitude simply
correlates with the dynamic electrophoretic mobility of spherical
1194
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
particles hence with its ζ-potential. A review of modern electroacoustic methods for measuring ζ-potential in non-diluted samples is
available [108].
The nature and occurrence of reactive surface centers can be
investigated by provoking specific chemical reactions with suitable
target molecules. Fenton-like reactivity using hydrogen peroxide as a
probe has been extensively applied for the characterization of reactivity
of iron-bearing mineral surfaces [57,65]. Formic, ascorbic, linoleic acid,
glutathione, and cysteine were also used to investigate surface reactivity
of various quartz samples, TiO2, cobalt–tungsten carbide, indium–tin
oxide [68,73,109–111] The surface-driven reaction on a specific target
molecule unveils very informative details on the coordinative and
oxidative state of reactive centers, as well as their abundance. Different
methods have been proposed to evaluate the oxidative potential of
particulates [112]. Among the various methods, spin-trapping technique,
coupled with electron spin resonance (ESR) spectroscopy, is probably the
most informative procedure to assess such relevant surface features
which impart radical reactivity to a material or particulate. In a spin
trapping experiment [113], the radicals generated from a target molecule
is stabilized by a covalent bond with a “spin-trapping” molecule. The
stable radical adduct generated is analyzed by ESR. The intensity of the
ESR signal is proportional to the amount of the radical species in solution.
Among the different probes that have been developed to evaluate the
oxidative potential of particles [58,112,114,115] ESR is the only available
technique that allows to unveil the chemical nature of the radical formed
and to discriminate among different radicals simultaneously generated.
The ESR signal can be fitted with appropriate simulation software and the
spectroscopic resonant parameters (the hyperfine splitting-constants)
may be usefully used to identify the chemical nature of the radical adduct
formed.
3. Adsorption of proteins
Among the different processes occurring at the interface between
surfaces and biological fluids the interaction with proteins is the most
relevant in the response of the tissues/cells to the xenobiotic entity. At
the same time this process is the most complex to describe mainly
because of its dynamic nature. In the case of nanoparticles the surface/
biofluid interface may be described as a protein corona, i.e. a layer
characterized by a slowly established core, with relatively stable
composition, and an outer layer made of fast-exchanging proteins,
whose composition is strongly influenced by the biological environment
and is subjected to relaxation processes. Such corona is time-dependent
[1,116], as Leo Vroman showed in 1962 with his work on the adsorption
of blood serum proteins onto an inorganic surface, and it is the result of
both thermodynamic and kinetic factors, as it implies competition
between proteins for the adsorption sites. The proteins with higher
mobility absorb first and are subsequently replaced by less motile
proteins with a higher affinity for the surface. The process may take
several hours to get to equilibrium [116] and the outcome is influenced
by the environment which may change significantly whenever the
particle moves from one biological compartment to another.
The two main factors that drive protein competition for the solid
surface are: i) the relative concentration of the proteins in the
biological environment; ii) the stability of the protein–particle adduct.
The adduct stability ultimately depends on the intermolecular forces
that dominate in the mutual interaction and are influenced by the
type of proteins involved [117] and by the chemical properties of the
solid surface [46,118].
The adduct stability reflects on the entropy vs. enthalpy balance.
Enthalpic changes are usually related with the formation/disruption of a
number of chemical bonds either between protein and surface or within
the protein molecule after adsorption or following the redistribution of
charged groups (ions) when the electrical double layers around the
protein molecules and the sorbent surface overlap; conversely, entropic
changes are usually related with the release of bound water from the solid
surface or with the presence of denaturation/structural rearrangement
phenomena within the protein molecule [8].
A significant factor that may influence the final composition and
aspect of the protein corona is the size of NPs with respect to the size
of proteins. This may influence the molecular crowding on the surface
as well as the establishment of protein–protein interactions that may
affect the conformation of the adsorbed proteins.
Despite the amplitude of factors that influence the process of
formation and the composition of the protein corona and the
consequent difficulties in predicting the outcome of the interactions
between NPs and biological fluids it's worth to mention that several
attempts of building models for such process have been already made.
Dell'Orco et al. [119] propose a dynamic model for the prediction of
time evolution and equilibrium composition of the protein corona
that forms when polymer NPs come into contact with human plasma.
The model is based on affinity, stoichiometry and rate constants.
Similarly, Raffaini et al. [120] employ computational methods to
predict the behavior of albumin in the presence of carbon nanotubes
or C60 fullerenes. Other theoretical studies on proteins interacting
with different surfaces have been reviewed by Gray [121].
In this section we will focus on three phenomena that occur after
protein adsorption on the solid surface and that may heavily influence
the cell response: i) the alteration of surface properties of the solid
due to protein coverage; ii) the structural modifications induced by
the adsorption on the protein fold; iii) the changes of protein activity
related with structural modifications or selective orientation of the
proteins onto the NP surface. Fig. 2 reports a summary of the events
that may occur after protein adsorption.
A number of studies have been devoted to the different aspects of
the subject. Our aim is to provide an overview on the kind of
modifications that may occur as well as a survey of the available
methods for characterizing such changes.
3.1. Alteration of the surface properties of the solid
The first consequence of the interaction of a solid surface with
biological fluids is the alteration of surface properties of the solid.
The coverage of a solid surface by proteins implies important
changes in the surface charge distribution, in the ζ-potential (in case
of NPs) and in the accessibility of a number of chemical functions that
affects the ability of the solid to establish H-bonds, electrostatic or
hydrophobic interactions, etc., that is to say the ability of the surface to
interact with the chemical environment.
A progressive modification in the surface charge of silica NPs as a result
of increasing protein coverage of the surface has been reported by various
research groups [122–124] and recently by some of us [36]. Fig. 3A
illustrates some data reported in this latter study on the behavior of hen
egg lysozyme (HEL) at the surface of silica NPs. By increasing the protein
concentration in the supernatant, the amount of HEL adsorbed onto the
silica surface gradually reached the theoretical monolayer (Fig. 3A, B). A
high degree of coverage of the surface was confirmed by the ζ potential
values which showed a gradual decrease of the negative surface charge of
silica, which became even positive at the highest degree of coverage
(Fig. 3B). Note however that HEL would hardly completely mask the silica
surface in complex media because the adsorption of HEL on silica is
partially reversible [36] thus HEL is likely to be displaced in vivo by any
other protein with a higher affinity for the surface.
Any modification of surface charge is particularly relevant in NPs
studies as it reflects on both the stability of the colloidal suspension
and the biological response.
Colloidal instability may concern both NPs (e.g. particle aggregation, flocculation, precipitation, etc.) and proteins (through protein
aggregation, clustering, fibrillation, etc.) and heavily influences the
initial biological response to NPs [13].
Experimental evidence suggests that formation of a protein corona
improves colloidal stability. A study on polysterene NPs by Walczyk
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
1195
proteins
nanoparticles
particle-induced
protein fibrillation
a
f
b
colloidal
stabilization
conformational
changes
e
c
proteins-mediated
agglomeration
modification of
protein activity
d
masking of particles
charge
and reactivity
orientation of proteins
on the surface
e
Fig. 2. Summary of the events which may follow protein adsorption on particles. When particles come into contact with a pool of proteins in a biological fluid a protein corona is
rapidly formed (a). The protein corona composition depends on the relative abundance and type of proteins in the fluids, pH and ionic strength and chemical nature of the sorbent
surface. Protein corona may evolve and the following events may occur: stabilization of NPs suspension (b) or proteins-mediated agglomeration (c); masking of particles charge and
reactivity (d); modification of proteins activity (e) or particle-induced protein fibrillation (f).
et al. [12] remarks that — on the only base of the low surface charge —
the protein-coated NPs would not be expected to be colloidal and
assigns the observed stability (which is comparable to that of bare
particles) to the presence of the protein corona. Similarly, a study on
gold NPs by Casals et al. [125] shows that AuNPs instantaneously
aggregate when dispersed in protein-free cell culture medium, while
they are stable upon addition of serum to the solution. This is also
taken as an indication that NPs protein coating is faster than NPs
aggregation in those experimental conditions [125].
This protein-mediated ability to stabilize nanomaterials has even
been exploited for NPs assembly [126] as is the case of biotin–
streptavidin [127] and antigen–antibody [128] interactions. Anisotropic particles such as nanowires, nanotubes and M13 viruses have
been also assembled through biomolecular recognition mechanisms
[129]. Interestingly, even the structural stability and instability of
proteins upon binding may be exploited to modulate NPs spacing, as
reported by the group of Rotello [130] in their study on chymotrypsin
(which denatured upon binding) and cytochrome c (which retained
its native structure).
Deguchi et al. [131] show that fullerenes C60 can be stabilized by
adsorption of proteins in the physiological environment. They found
that the salt-induced coagulation of C60 was suppressed by the presence
of human serum albumin (HSA) in a molar ratio-dependent mode.
Similarly, CNTs aqueous suspensions are stabilized by proteins [132].
Schulze et al. [133] report that ZrO2, CeO2 and TiO2 particles exposed to Dulbecco's Modified Eagle's Medium (DMEM) were strongly
agglomerated. Nevertheless a deagglomeration effect was found with
ZrO2, CeO2 upon enrichment of the medium with FCS (Fetal Calf
Serum) where deagglomeration was scaled with the protein content.
In some cases interaction of particles with proteins lead to a destabilization of the suspension. Agglomeration was observed in NP
suspensions following interaction with fibrinogen [134]. In this case,
bridges of proteins between NPs were observed by SEM. Similarly, Xu
and coworkers [135] observe that cytochrome c, DNase II or
hemoglobin tend to form coralloids with SiO2 NPs by bridging
between them.
The impact of the interaction with a solid phase on protein stability
will be discussed in a further section. Nevertheless in discussing the
alteration of surface properties of the protein–solid particle system is
worth to mention here that NPs have been reported to influence
protein fibrillation processes [136–138]. This is likely related with the
conformational perturbations associated with protein binding to solid
surfaces, whose effect can be to bring protein domains that are
normally buried inside the protein towards the surface. It's worth to
mention that formation of protein fibrils as a result of conformational
changes is a relevant phenomenon for a number of diseases, such as
amyloidosis, Alzheimer, etc.
Lynch et al. [136] show that the fibrillation rate of β-2-microglobulin —
an amyloidogenic protein — is increased in the presence of polymer NPs.
As the resulting fibrils are not associated with the NP, a mechanism
implying pre-concentration of protein on the NP surface and subsequent
detachment of the pre-fibrillar aggregate, which migrates in the solution
phase, has been proposed. Similar findings have been reported by Bellezza
and coworkers [137] on myoglobin (Mb) with phosphate-grafted zirconia
NPs. Conversely, C60 hydrated fullerene has been reported to display
anti-amyloidogenic ability as it inhibits fibrillation of the amyloid β2535-peptide [139]. If confirmed this finding may have important
implications in the development of NP-based therapies against neurodegenerative diseases.
Another important effect which may follow protein adsorption is
the inhibition of the ability of inorganic surfaces to react with
biomolecules or to generate free radicals.
1196
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
A
B
1.2
25
20
15
0.8
10
Potential ζ , mV
θ, nmol ads / nmol monolayer
side-on theoretical monolayer
1.0
0.6
0.4
0.2
IP-HEL
5
0
-5
1
-10
2
3
4
5
6
7
8
9
10
11
12
pH
silica
-15
-20
0.0
0
100
200
300
400
-25
500
[protein]free, μM
-30
-35
C
D
extracellular
proteins adsorb onto
particle surface
a
phagocytosis
surface
coverage
b
particle
c
protein
digestion
3320
3340
3360
enzymes
in phagolysosome
remove proteins
macrophage
3380
Field (G)
Fig. 3. Modification of surface charge and surface reactivity following coverage with proteins. A) Isotherm of adsorption of lysozyme (HEL) on silica NPs; B) shift of ζ-potential
following HEL adsorption; C) ESR spectra recorded on a suspension of a commercial quartz powder in a buffered solution of sodium formate in the presence of a spin trap (DMPO):
a) quartz powder; b) quartz powder coated with bovine serum albumin; c) quartz powder after digestion of albumin with a protease.
Ground quartz dusts are highly reactive surfaces and are able to
generate free radicals or to directly react with organic molecules (see
paragraph 2). A useful method to monitor such reactivity consists in
evaluating the amount of carbon-centered radicals generated by a
definite amount of powder in an aqueous suspension of a model protein
by means of the ESR/spin trapping technique [89,110]. In Fig. 3C the ESR
signal obtained by a commercial quartz dust is reported (spectrum a).
When pre-incubated with a solution of bovine serum albumin (BSA),
which rapidly adsorbs onto silica surfaces in an irreversible mode, the
signal is completely suppressed (spectrum b) suggesting that the
reactive sites have been completely covered by protein molecules.
However upon removal of the adsorbed BSA by enzymatic degradation
the ability to generate radicals was restored (spectrum c). This
experiment recalls what may happen when a particle comes into
contact with proteins in extracellular fluids and is subsequently
phagocytised by macrophages. Proteinases inside the cell may degrade
the protein corona and hence restore the original reactivity of the dust.
In panel D, Fig. 3 a scheme of such process is depicted.
3.2. Structural modifications induced by the adsorption on the protein fold
Adsorption onto a surface may bring about important structural
changes in the protein. These are strictly dependent on the mode of
interaction of the protein with the surface which, in turn, is influenced
by the physico-chemical nature of the solid surface and by the intrinsic
properties of the protein [46]. The final outcome results from a balance
between the extrinsic and intrinsic forces involved in the process.
The degree of conformational change and the rate at which the
proteins undergo conformational changes after adsorption on a solid
surface are influenced by a number of parameters:
i.
ii.
iii.
iv.
v.
The intrinsic protein stability
The characteristic of the sorbent surface
The surface curvature
The protein concentration on the surface
The pH and the ionic strength
In the following lines, we will try to discuss each one of these
factors, through meaningful examples taken from the literature.
3.2.1. The intrinsic protein stability
This parameter deals with the intrinsic resistance of the protein to
undergo conformational changes in the presence of a solid surface.
Following a classification by Norde [8] proteins may be divided in two
groups: hard, which designates relatively rigid structures that tend to
maintain their original conformation and soft, which indicates those
that readily undergo conformational changes or deformation
[140,141].
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
The hardness and softness character of a protein is particularly
relevant as one considers that adsorption of proteins on a solid surface
normally results in conformational heterogeneity of the surface-bound
proteins that implies the presence of a population of protein structures
with different degrees of perturbation [46,142,143]. Wu et al. [143] while
commenting on the extent of unfolding of globular proteins, underline
that highly flexible proteins (e.g. β-casein) adsorb much faster and
spread at the interface to a greater extent than very rigid and compact
ones [144] whose tertiary structure should undergo conformational
changes in order to spread at the interface.
Protein hardness and softness [46,145] depends on structural
determinants (such as the number of disulfide covalent bridges within
the protein, the presence and distribution of salt bridges in the protein
structure, the presence of stabilizing sites such as metal-coordination
sites that may both influence the spatial arrangement of the protein
domains and exert a charge stabilization effect, etc.) and on thermodynamic factors (the balance between enthalpic and entropic contributions,
that finally defines the amplitude of conformational heterogeneity).
Larsericsdotter [145] reports that the structural heterogeneity induced by
the adsorption of RNAse and lysozyme on silica particles is decreased by
calcium ions; in fact, co-adsorption of bivalent cations has been reported
to prevent effectively charge accumulation in the adsorbed protein layers
which seems to be a main reason for structural heterogeneity. Similarly
Billsten et al. [146] have shown that the protein's structural stability is a
key factor in determining the rate of conformational change for different
mutants of human carbonic anhydrase II. Further, Wu et al. [117] report
that the extent of unfolding for lysozyme (positively charged) is comparable to the values for β lactoglobulin (negatively charged) [143] in
spite of favorable electrostatic interaction with the silica surface and
assign this peculiar behavior to the presence of disulfide bonds stabilizing
the tertiary structure of both proteins.
Albeit the overall structural stability of proteins is definitely an
important determinant in the adsorption of proteins, differences in
the resistance to conformational changes among the different
domains of proteins should be considered in describing the process
at a molecular level.
3.2.2. The characteristics of the sorbent surface
A huge number of papers show that the chemical nature of the
sorbent surface has a strong influence on the adsorption outcome, in
terms of affinity and structural distortion of the protein.
Zoungrana [147] reports that adsorption of α-chymotrypsin on
positively or negatively-charged polystyrene surfaces decreases the
enzymatic activity at a different extent suggesting that electrostatic
interactions between the protein residues and the solid surface play a
key role in determining the degree of unfolding.
Electrostatic interactions appear to be important also in the case of
silica particles. Xu et al. [135] report that the stability constant of
adducts of negatively charged silica particles with cytochrome c,
deoxyribonuclease and hemoglobin increases with increasing numbers
of positively charged Lys and Arg residues. The role of these residues in
the interaction with the surface was confirmed by the involvement of
the polar residues of cytochrome c located near positively charged
residues in H-bonding to silanols in silica. Similarly, a study on the
adsorption of lysozyme onto silica [148,149] shows that the amount
adsorbed decreases as the pH decreases. This posits in favor of a
mechanism of adsorption dominated by coulombic interactions [148].
It is generally accepted that the hydrophobicity/hydrophilicity degree
of the surface plays an important role in the affinity of proteins and in
the extent of surface-driven unfolding. Irreversibility is strictly related
to consistent surface-driven conformational changes that occur mainly
when protein adsorbs through hydrophobic interactions [8,142].
Adsorption of lysozyme onto silica NP was shown to result in a
marked loss of α-helix content for larger surface coverage [148]. The
same protein adsorbed onto polytetrafluoroethylene (PTFE) surfaces
1197
underwent a biphasic conversion of α-helix to β-sheet, with an initial
quick phase followed by a much slower conversion [150].
Interestingly, Norde [8] highlights that apolar surfaces may induce a
higher degree of ordered structure in the adsorbed proteins, in that they
may promote H-bonding between peptide units in the protein–surface
contact region, although this cannot be taken as a general rule.
3.2.3. The surface curvature
The surface curvature has been shown to influence both the
composition of the protein corona and the conformational state of the
adsorbed protein.
Lynch et al. [136] underline that — departing from the simple
limiting case of flat surfaces — as the particles become smaller and
approaches the size of the adsorbed proteins, the composition and
organization of the protein layer changes dramatically. According to
Lundqvist [46] this is related with the fact that a large interaction area
between the protein and particle (typical of larger particles) affects
the secondary structure more than a small one, because of the higher
interaction energy. Nevertheless foreseeing the trend of such changes
is not straightforward, as the effect of curvature is also strongly
dependent on the nature of the adsorbed protein.
Several examples of opposite behaviors that are paradigmatic of such
dependence are available in the literature. Vertegel et al. [148] and
Lundqvist et al. [46], in their work on the adsorption of lysozyme and
human carbonic anhydrase on silica NPs show that these proteins
retained more native-like structure on smaller particles. Oppositely, a
higher curvature results in a more destabilized protein in the case of
RNase adsorbed onto silica [151]. A similar behavior has been reported
for albumin adsorbed on silica particles, whereas fibrinogen follows the
opposite trend. Roach et al. report that, although both proteins showed
similar trends in binding affinity and saturation, the loss of secondary
structure of fibrinogen is more important in the presence of particles
with higher surface curvature [45]. Besides, Karajanagi et al. [152] report
that soybean peroxidase on single-walled carbon nanotubes retained
more of its native structure and activity than chymotrypsin, which
exhibited a nearly complete loss in activity and structure. In conclusion
the surface curvature modulates the degree of unfolding of proteins,
however, a common trend may not be found since the structure of an
adsorbed protein is not independent from the nature of the protein
itself.
A protein stabilization effect exerted by highly curved surfaces has
been reported by Asuri et al. [126]. They report that single-walled
carbon nanotubes (SWNTs) may stabilize proteins in strongly
denaturing environments to a greater extent than flat supports. The
provided explanation is that lateral interactions between adjacent
adsorbed proteins contribute to protein deactivation in harsh
environments and that these unfavorable interactions are suppressed
on highly curved supports such as SWNTs relative to flat surfaces
[126]. Similarly, the highly curved surface of C60 fullerenes have also
been reported to enhance enzyme stability [136].
3.2.4. The protein concentration at the surface
Molecular crowding, which depends on the abundance of proteins
approaching the surface, is another factor that seems to have an
influence on both the extent and the kinetic of unfolding of adsorbed
proteins. Wu et al. [143] in their investigation on β-lactoglobulin
adsorbed on silica particles remark that the protein unfolded to a greater
extent at low surface concentration; in addition, as compared to higher
surface concentration, the process was faster and kinetic curves were
different. The energy barrier for unfolding in a crowed surface seems to
be higher; the rationale behind this finding lies in protein–protein
interactions that would predominate in these conditions, whereas, at
low surface concentration, proteins mainly interact with the solid
surface. At high surface concentration the unfolding behavior is affected
by the interaction between protein molecules, due to limitation of the
physical space and to the energy barriers that arise from the interaction
1198
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
with other nearby protein molecules; such barrier also decreases the
unfolding rate.
A similar behavior has also been reported in the case of lysozyme
adsorption onto silica nanoparticle [117].
3.2.5. The pH and ionic strength
As discussed in the Introduction section, pH and ionic strength of
biological fluids may largely vary in the body [11] and this is highly
relevant for protein adsorption phenomena onto solid surfaces.
Both the pH and ionic strength of the medium have been reported
to display an effect on the folding state of adsorbed proteins. pH can
modulate the protonation state of all the ionisable groups found in the
protein–solid dual system. So it may influence the surface charge
distribution of the solid phase and the protonation state of aminoacids
involved in H-bonding, columbic interactions, etc. It is well known
that pH has a strong effect on the H-bonding network of proteins and
this may result in protein destabilization [153]. Changes of ionic
strength are known to influence protein stability and may result in
aggregation or protein precipitation.
A strong effect of pH on the secondary structure of lysozyme
adsorbed on silica NPs is reported by Vertegel et al. [148]. They report
that the α-helix content decreases from 40% (in the free enzyme in
solution) to respectively 8 and 5% for lysozyme adsorbed on 100-nm
particles at pH 6.9 and 5.0.
As for the ionic strength, higher values of this parameter were
found to induce a much higher extent of unfolding at higher surface
concentration, in the case of the adsorption of β-lactoglobulin onto
silica NPs [143]. According to Wu et al. [117] at higher ionic strength
the electrical double layer in the vicinity of the adsorbed protein
molecule is compressed. As a result, the electrostatic interaction
between adsorbed protein molecules is reduced and this may improve
unfolding. Interestingly, such an effect of ionic strength was strongly
dependent on the surface concentration and it was not significant at
lower concentration, where the average distance between adsorbed
protein molecules is far greater than the interaction range of the
double layers of different protein molecules [117].
Another important issue concerns the dependence of the amount
of adsorbed protein on ionic strength, as it may provide information
on the nature of interaction forces that dominate the establishment of
the protein–NP system. In a study on the adsorption of CytC, DNAse II
and Hb to silica NPs, Xu et al. [135] find that the amount of bound
protein decreased sharply with increasing ionic strength. This is taken
as evidence that electrostatic attraction is the main interaction force
between the protein and the nano-SiO2 particle. The double electric
layer on the nano-SiO2 particles adsorbed Na + so the positively
charged side groups of proteins were repelled. The induction,
orientation and dispersion forces between proteins and nano-SiO2
become stronger in a high salt medium owing to polarization of the
double electric layer. However, once again, this mechanism cannot be
generalized as a different behavior is found in the case of the
adsorption of HSA and nano-TiO2 [154].
The understanding of the conformational changes undergone by
proteins upon binding to a surface implies the necessity of grasping
information on two different aspects: i) to get a detailed description of
such structural changes at a molecular level of resolution; and ii) to
clarify the mechanism that underlies such changes.
In order to face these problems, both traditional and new investigation
approaches needs to be used.
Traditional approaches include the techniques normally used in
protein stability and conformational studies, such as Trp fluorescence,
circular dichroism (CD), differential scanning calorimetry (DSC), etc.
Nevertheless, these approaches need to be somehow refreshed as
soon as they are applied to heterogeneous systems (solid plus protein
dispersed in liquid media) instead of homogeneous media.
Unraveling the molecular details and the behavior of such heterogeneous systems requires the development of synergistic approaches in
terms of characterization techniques. A multi-techniques and multidisciplinary approach is required, able to put together two distinct fields
of expertise (protein structure and function, on one side; the characterization of solid surfaces and particles, on the other side), to combine
their experimental strategies and to merge their disciplinary viewpoints.
This strong challenge will influence the future perspectives for the
employment of NPs in health sciences.
A number of studies have been performed on different proteins and
different materials with the aim of highlighting the influence of
adsorption on protein conformation. As expected, a huge variety of
cases has been collected, due to the number of factors that influence
these complex phenomena and to the peculiar individual character of
each protein. It is almost impossible to extrapolate a common trend and
a systematic exploration of different combinations of proteins and
materials appears necessary, in order to build a huge reference database.
Conformational changes induced by protein adsorption are often a
stepwise process. This is clearly shown by Karlsson et al. [155] in a study
on the kinetics of conformational changes of different destabilized
mutants of human carbonic anhydrase II (HCAII) adsorbed on silica NPs.
By using a combination of circular dichroism (CD), Trp fluorescence and
ANS (1-anilino-8-naphthalenesulfonate) fluorescence spectroscopy,
Karlsson et al. show that the process takes place in a progressive
mode. At first, HCAII binds to the solid surface and structural changes
occur afterwards. The disruption of the active site occurs first and it is
followed by the rest of the tertiary structure; half-lives are reported for
each step. The stability of HCAII mutants affects the rate of conformational rearrangements (i.e. the kinetic of denaturation). The less stable
the structure, the more firmly the protein is associated with the surface
and the more quickly the structure is unfolded. Interestingly the extent
of conformational changes is not influenced by protein stability as all
mutants unfold to the same extent, i.e. a molten globule state. According
to the authors this is taken as an indication that surface-driven
denaturation differs from chemical denaturation, as the first deals
with stability at the surface and the second with global stability.
According to Karlsson, this is because the protein interacts in a specific
orientation with surfaces, while in chemical denaturation the protein is
immersed in the denaturing medium.
A study on the adsorption of Cyt c, DNAse II and hemoglobin onto
silica NPs [135] reports distinct behaviors. In Cyt c the fractions of
α-helix and β-turn decreased, whereas the β-sheet fraction increased
with increasing SiO2 particle number. Conversely, the fractions of
α-helix and β-turn in DNase II and Hb increased with increasing SiO2
particle number and a decrease of the β-sheet fraction was found.
According to Xu et al. [135] the rationale behind this finding is that the
β-pleated sheet areas of DNase II and Hb bound directly to the SiO2
particles and the press-and-pull interactions twisted the sheets, thus
inducing the emergence of a α-helix-like structure.
When protein is adsorbed on a surface, rearrangement phenomena
may occur: these may be associated with partial protein refolding. This
is what Wu et al. report in the case of lysozyme and β-lactoglobulin
adsorbed onto silica NPs [117]. These authors found that a comparison of
the kinetics of tertiary conformational changes of the two proteins
indicates that, the structural perturbation is faster and more extended in
lysozyme as compared to β-lactoglobulin during the first phase of
adsorption. Nevertheless, in a further step lysozyme was able to
undergo a refolding process that was absent for β-lactoglobulin.
The initial rapid unfolding of lysozyme upon adsorption seems to be
associated with the rapid decoupling of the α and β domains of lysozyme,
with consequent disruption of the tertiary structure, loss of α-helix and,
increase in β-turn and random coil [150]. This results in a ‘molten
globule-like’ structure in which α-helix is transformed into β-sheet,
thereby resulting in a refolding phase [117]. Thus at least for lysozyme
the adsorption process occurs in three phases: an initial unfolding phase,
that occurs as the proteins interact with the surface, followed by partial
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
renaturation (second phase) likely associated with protein reorientation
on the surface. The third phase is characterized by a further slow
unfolding step, which drives the system towards the equilibrium.
Another aspect of conformational changes is the degree of
reversibility of the induced modifications upon protein desorption
from the surface.
The case of BSA interacting with synthetic chrysotile nanocrystals,
investigated by Sabatino et al. [156], is a typical example. This study
shows that BSA modifications are driven by surface interaction with
chrysotile. Once BSA is desorbed back into the solution its structure
rearranges, without getting back to the native conformation. A
significant amount of β-structures is found in the desorbed molecules.
According to Sabatino, β-structures are more involved in the surface
adhesion process adsorption and their increase is related to the extent of
protein unfolding that exposes to the solvent protein regions that are
normally buried in the interior. Sabatino et al. [156] suggest that this
change in secondary structure is driven by the formation of surfacemediated hydrogen bonds with the polar aminoacidic side-chain groups
pointing outwards and serving as sites for molecular recognition with
the hydroxylic substrate groups.
3.3. Changes of protein activity related with structural modifications or
selective orientation of the proteins onto the surface
The issue of protein conformation is important as it reflects on the
protein behavior. This is especially dramatic in the case of enzymes,
whose catalytic activity is strongly dependent on a correct protein
folding and hence may be seriously influenced — even completely
disrupted — as an effect of the interaction with a surface. It may also be
relevant for proteins involved in molecular recognition processes or in
the interaction with receptors. Such kind of processes requires that the
protein or specific protein domains maintain their conformation in
order to bring about the expected effect. Even when the general fold of
the protein and the conformation of the active site is not compromised
by the adsorption process there is still another issue that may influence
protein functionality, namely the orientation of the molecule onto the
solid surface. In the case of enzyme, the interaction with the surface may
hinder the entrance of the substrate channel or interfere with the
diffusion of substrates within the active site. In addition, if a protein
involved in molecular recognition processes exposes to the solid surface
the epitope that needs to be recognized by a receptor or a molecular
partner, the related process will be impeached [13].
As in the case of conformational changes the effects of adsorption
on the functional behavior of proteins are extremely varied.
Foreseeing a general common trend is unfeasible as too many factors
concur in determining the final exit.
Important conformational changes recorded upon adsorption of
lysozyme onto silica NPs reflect on the enzyme activity: about 60% of
the native lysozyme activity is retained when the α-helix content is
reduced to ~ 30% of the initial value. Further loss of secondary
structure results in a much sharper activity decrease [148]. As for the
orientation of the protein [157] it has been shown that lysozyme
adsorbed onto flat silica surfaces exposes the largest positively
charged patch on its surface (which contains 7 of the total 17 basic
aminoacid residues of lysozyme) towards the negatively charged
silica surface. As the active site of lysozyme lies on the opposite side
[158] one can speculate that the moderate loss in activity is due to the
distance existing between the structurally perturbed region and the
catalytic site. Nevertheless further activity loss is found, as the
perturbation spreads over the whole structure. Conversely, both
cytochrome c and DNase II were reported to be inactivated up to 75%
upon adsorption on aggregated nanometric SiO2 [135].
Activity measurements performed on RNase [151] show that the
enzyme retained 90% of its intrinsic activity upon adsorption on 4 nm
silica NPs, whereas a net 30% loss was found with 15 nm particles. This
1199
is clearly related with the higher degree of conformational integrity of
protein molecules associated with smaller particles (see Section 3.2).
The degree of hydrophilicity/hydrophobicity of the solid surface
influences the functional perturbation of some proteins, as it reflects
on the kind of interactions that a protein can establish with the solid
phase which, in turn, may affect its conformation. Noinville et al. [159]
report that the activity of α-chymotrypsin is more affected when the
protein interacts with hydrophobic surfaces because of greater driving
force for unfolding.
In addition, as protein concentration at the surface has been shown to
influence the kinetic of unfolding, an effect on their activity is expected
as well. Wu et al. [117] in their investigation on lysozyme adsorbed onto
silica particles found that the immobilized enzyme retains its activity
only at high packing densities.
Although this review does not deal with coated-particles, it is worth
to mention the study by Rotello and coworkers [130,160] who showed
that, by controlling the surface chemistry, it was possible to achieve
distinct levels of interaction of chymotrypsin with CdSe NPs, implying
enzyme inhibition with denaturation or with retention of structure [130].
In the case of enzymes with dual catalytic functions, adsorption may
modulate the balance between them. A study by Shang et al. [161] on Cyt
c shows that the heme environment is perturbed by the adsorption of
the protein onto a solid surface and this increases solvent accessibility of
the heme. This reflects on the peroxidatic activity, which is usually weak
in native Cyt c: a size-dependent increase of peroxidase activity was
found in Cyt c adsorbed on silica NPs of different size.
As previously mentioned, a loss of activity upon protein adsorption
has not always to be expected. Several reports have shown that using
some NPs as solid supports does not necessarily imply important
activity loss [143]. This is especially true in the case of biochemically
functionalized silica NPs with enzymes, that have been successfully
employed for cell membrane staining and other applications [143,146].
As far as the issue of protein orientation is concerned, it is worth to
mention the potentialities of labeling techniques, that allows collecting
topological information on the interaction between the protein and the
surface.
Site-directed spin-labeling (SDSL) is a labeling technique developed
by Hubbell et al. [162] that relies on the conjugation of a paramagnetic
spin-label on specific sites of a protein, usually exploiting Cys residues as
the anchor-point. Cys may be introduced by site-directed mutagenesis.
Such approach is useful to investigate protein dynamics at a level of
molecular details [163,164]: changes of the ESR spectrum as a
consequence of conformational transitions of the labeled protein allow
evaluating a number of parameters related with the anisotropy degree of
the paramagnetic probe motion and to measure interatomic distances.
This approach has been applied by Jacobsen et al. [165] on a series
of T4 lysozyme mutants adsorbed on quartz surfaces, with the aim of
evaluating changes of the local environment of distinctly labeled sites,
spread over the whole lysozyme structure, upon protein adsorption.
Based on the comparison of ESR spectra, the authors were able to
show that the C-terminal part of the protein is actively involved in the
adsorption process.
An ESR study by Nicholov et al. [166] on human serum albumin
labeled on the Cys34 residue allowed establishing the degree of
conformational distortion of the protein environment around the
labeled site, thus providing information on the orientation of HSA on
three distinct solid supports.
In a recent study [36] we employed SDSL to investigate the
interaction of lysozyme and ribonuclease with nanostructured silica
particles. We were able to conclude that the lysozyme region containing
the spin-labeling site adhered quite flatly to the silica surface, whereas
RNase adsorption resulted in a label-containing cavity that was almost
inaccessible from the outside, but still allowed a certain degree of
mobility to the spin-label trapped inside. These details allowed
concluding that both proteins adsorb preferably on one side of the
molecule but each enzyme exhibited a peculiar adsorption mode.
1200
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
3.4. Techniques to evaluate protein adsorption, unfolding and orientation
A number of techniques and experimental approaches have been
applied either separately or in synergy to investigate NPs contact with
proteins.
A thorough characterization of the protein–NP system needs
distinct approaches, depending on the aspect of the system that one
aims to focus on. Different techniques are needed for characterizing
the analytical composition of the protein corona and the dynamic
aspects related with its formation (Table 1), as compared to the
changes in protein fold brought about by protein adsorption as well as
the orientation of proteins onto the solid surface (Table 2).
In this section we will try to report a schematic overview of the
available techniques and the kind of information they can provide.
Size exclusion chromatography allows separating NP-associated
proteins from complex mixtures such as plasma and other biological
fluids. It allows estimating the exchange-rate of proteins adsorbed on
NPs because only slow-exchanging proteins, with a residence time
several times longer than the separation time, are recovered in the
same final fraction as NPs. Conversely, very fast exchanging proteins
elute at the same time as isolated proteins. A study by Cedervall et al.
on the adsorption of plasma proteins to N-isopropylacrylamide
(NIPAM)/N-tert-butylacrylamide NPs shows a dependence of the
exchange rates on NPs' hydrophobicity, as apolipoprotein A-I coeluted
with the particles, whereas HSA and fibrinogen eluted much later
[118]. According to Cedervall et al. size-exclusion chromatography is
potentially less perturbing of protein–particle complexes than
centrifugation, because the outcome of a centrifugation assay may
be affected by the duration of washing steps and the solution volumes
used in these steps [13]. As a consequence Lynch et al. underline that
highly abundant proteins may be retrieved either due to insufficient
washings or because they are truly associated with the NP.
Sedimentation of large proteins, protein aggregates, and co-precipitation have also to be taken into account. Nevertheless, centrifugation
assays conducted with care and associated with other methods still
allow to retrieve enough proteins for safe identification of proteins by
mass spectrometry [13].
Aggarwal et al. report that [167] a combination of SDS-PAGE and
Western blotting may provide general information concerning the
proteins adsorbed onto NPs, although quantitative results are difficult
to obtain and these methods may be used mainly for comparison
purposes. SDS performed on fluorescent dye-labeled protein separates
the proteins based on molecular weight [168] and allows measuring
protein concentration based on fluorescence measurements. The same
authors remind that another possibility for spotting the corona's
proteins, once they are separated from NPs is two-dimensional
polyacrylamide gel electrophoresis (2D-PAGE). This can be used in
association with N-terminal sequencing and/or mass spectrometry,
applied on individual excised protein spots from the 2D gel and
compared to a known database of proteins. Immune-blotting and
Western blotting may also help in identifying specific proteins [167].
Stoichiometry, affinity and enthalpy of protein–NP interaction
may be assessed by isothermal titration calorimetry (ITC) [13,118].
Kinetics of adsorption may be studied by means of surface plasmon
resonance (SPR) studies which may provide information on protein–
protein interaction kinetics as well as on protein association to and
dissociation from NPs [13].
Dynamic light scattering (DLS) has been used to determine the
thickness of the protein layer on NPs [169]. A study by Casals et al.
[125] provides an example of how DLS may be used to follow the
evolution of NPs coating in metallic Au NPs exposed to protein-rich
media.
The ζ-potential and the isoelectric point correlate with the degree
of coverage of the particle surface and provide indirect information on
the changes in surface net charge of the NP after protein adsorption
[36]. This technique may be also exploited to obtain information on
the degree of reversibility of the adsorption process by measuring the
variation of ζ-potential after applying negative gradients at the
surface of the NPs–protein conjugate [36].
Scanning electron microscopy (SEM) and transmission electron
microscopy (TEM) may help in grasping information on the morphological changes occurring at the NP's surface after protein adsorption.
These techniques have also been employed to highlight association of
NPs, NP-induced protein fibrillation, etc. [136]. Aberration corrected
Table 1
Techniques for characterizing the protein composition of the NPs' corona and its evolution with time.
Technique
Issue
Information provided
References
Size-exclusion chromatography
(gel filtration)
Centrifugation associated with MS
Chemical composition of the protein corona
[13,118,136]
Isothermal titration calorimetry (ITC)
Chemical composition of the protein corona
Thermodynamic stability
Kinetic evolution of the protein corona
Identity of the proteins on nanoparticles
Rates of exchange with proteins in the medium
In association with MS, identification of proteins
in the corona.
Used to assess the stoichiometry, affinity and
enthalpy of protein binding to NPs
Protein–protein interaction kinetics of protein
association to and dissociation from nanoparticles
Changes in NPs diameter due to protein adsorption
Changes in surface charge distribution of NPs due
to protein adsorption, reversibility of adsorption
In association with Western blotting or performed
on fluorescent dye-labeled proteins. Used to monitor
the kinetic of protein adsorption
Associated with N-terminal protein sequencing or
MS or with immunoblotting and Western blotting;
identification of proteins in the corona.
Provides information on the morphology of the
NP–protein surface
Provides information on the morphology of the
NP–protein surface
Allows to observe single molecule fluorescence
at surfaces
Allow to follow high resolution imaging of movement
through intracellular environments
Measures the enhanced Raman scattering of molecules
adsorbed on metal surfaces
Surface plasmon resonance (SPR)
Dynamic light scattering
Electrophoretic light scattering
Chemical composition of the protein corona
SDS-PAGE
Size of the protein–NP system
Degree of surface coverage by proteins;
colloidal stability
Kinetic evolution of the protein corona
2D-PAGE electrophoresis
Chemical composition of the protein corona
TEM
Modifications of the solid surface
SEM
Modifications of the solid surface
Total internal reflection fluorescence
microscopy (TIRFM)
Live cell confocal microscopy
Biodistribution
Surface-enhanced Raman scattering (SERS)
Biodistribution
Aggregation state of the particles
Biodistribution
[13,118]
[13,118]
[13,118]
[125,169]
[36]
[168,248]
[167]
[1]
[1,135]
[172]
[173]
[174]
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
1201
Table 2
Techniques for characterizing the changes in protein fold and activity and the orientation of proteins adsorbed onto solid surfaces.
Technique
Issue
Information provided
References
Circular dichroism (CD)
Conformational state of proteins
[146]
Trp fluorescence
Conformational state of proteins
ANS binding
Conformational state of proteins
Infrared (IR) and Raman spectroscopy
Conformational state of proteins
Nuclear magnetic resonance (NMR)
Conformational state of proteins
Neutron reflectivity (NR)
Conformational state of proteins
Atomic force microscopy
Topography of protein adsorption
ESR-SDSL
Topology and orientation
SERS
Protein orientation
Analytical ultracentrifugation and gel
permeation chromatography
Activity assays
Kinetic of conformational changes
Allows to monitor secondary and tertiary structure changes
of proteins and peptides; unfolding processes at the surface
Used to monitor conformational changes and unfolding
processes, based on the change of fluorescence emission of
Trp residues
Allows to detect molten globule states or to highlight the
exposure of hydrophobic patches to solvent
Analysis of protein secondary structure, in combination with
other techniques
Used to probe conformational changes in proteins and peptides
adsorbed onto solid surfaces at a level of molecular details and
to identify protein–NP sites of interaction
Used to determine conformations of peptide and protein layers
adsorbed onto solid surfaces at a level of molecular details
Imaging of proteins adsorbed onto solid surfaces; determination
of the strength of the molecular interactions between the protein
and the surface
Monitoring secondary and tertiary structure changes of proteins
and peptides
Molecular recognition processes; unfolding processes at the surface
Provides vibrational spectra of biomolecules adsorbed on surfaces,
useful for assessing protein orientation.
May provide information on the rate of protein conformational
transition occurring upon adsorption
Allows to correlate the conformational state of the adsorbed
protein to activity changes
Functional state of the adsorbed protein
TEM instruments with improved resolution are now available: they
allow imaging the volumes and surface edge atomic structures of NPs
using both transmission and scanning transmission modes [170,171].
Finally total internal reflection fluorescence microscopy (TIRFM) —
which allows to observe single molecule fluorescence at surfaces
[172] and live-cell confocal microscopy [173] — which allows high
resolution imaging of movement through intracellular environments
— are helpful to follow the particle biodistribution. The same objective
may be achieved by using surface-enhanced Raman scattering (SERS),
that measures the enhanced Raman scattering of molecules adsorbed
on metal surfaces [1]. The same techniques also provide information
on the state of aggregation of NPs [174].
An accurate monitoring of the conformational changes underwent
by the adsorbed proteins may rely on several techniques.
• Far UV-circular dichroism (CD) is useful for monitoring the
secondary structure changes of proteins and peptides; near UV-CD
provides information on the tertiary structure. Both are used to
monitor unfolding processes.
• Trp fluorescence is employed for monitoring conformational changes
and unfolding processes, based on the change of fluorescence
emission of Trp residues, that is strongly influenced by the
hydrophobicity of its chemical environment [175]. In particular, the
changes in tertiary conformation may be correlated to the extent of
blue shift of the emission spectrum.
• ANS binding allows to detect molten globule states or to highlight the
exposure of hydrophobic patches to solvent. These three techniques are
often used together, in order to get an exhaustive picture of the
modifications ongoing in the protein structure during unfolding [164].
• Fourier transform infrared spectroscopy (FTIR) [150,159,176] and
Raman spectroscopy are well established methods for the analysis of
protein secondary structure, in solution and when adsorbed to
surfaces. The frequency shift of mode of vibration of some peculiar
bonds, such as the amide I [177] may provide information on the
degree of conformational integrity of the adsorbed protein. In fact the
presence of a number of Amide I band frequencies has been
correlated with the presence of α-helical, antiparallel and parallel
•
•
•
•
•
•
[1,117,136,167]
[146]
[150,159,176,177]
[178,181]
[179]
[182,183]
[36,165]
[184,249]
[46]
[135,143,148]
β-sheets and random coil structures. FTIR can be a very powerful tool
when used in combination with other techniques, like CD.
Nuclear magnetic resonance (NMR) [178,179] is useful to probe
conformational changes through modification of dipolar interactions
between distinct parts of the protein. Localized conformational
information has been obtained for some adsorbed peptides by using
solid-state NMR [180]. A recent study by Calzolai et al. [181] reports
that NMR measurements, chemical shift perturbation analysis and
dynamic light scattering, allowed to identify the interaction site of
ubiquitin with gold NPs at aminoacid scale, in solution.
AFM has the ability to image adsorbed proteins near native
conditions. Silva reports that [182] high resolution topographs of
ribonuclease A adsorbed on mica surface have shown the presence
of well-defined monolayer two-dimensional structural arrays of
oligomers. Determination of the force vs. distance curves by AFM
provides a direct understanding of the strength of the molecular
interactions between the protein and the surface [183].
SDSL coupled to ESR spectroscopy (either CW or pulsed) may be
successfully employed to get information on the folding state of the
protein and on conformational transitions. It allows measuring
spin–spin distances and represents a powerful tool for following the
mechanism of conformational perturbation at a level of aminoacid
residues detail. In addition, it has been employed to grasp
information on the orientation of proteins on the surface [36,165].
A study by Yu et al. [184] on cytochrome c adsorbed on chemically
modified gold nanohole arrays demonstrates that SERS may be
successfully employed for studying the orientation of proteins
adsorbed on nano-patterned metal surfaces.
Analytical ultracentrifugation and gel permeation chromatography
may provide information on the rate of conformational transition
occurring in proteins upon adsorption on solid surfaces. In a study on
the adsorption of human carbonic anhydrase to silica surfaces [46],
analytical ultracentrifugation and gel permeation chromatography
data allow to conclude that conversion of the adsorbed protein from
the native to non native conformation is slow or readily reversible.
Activity assays are a useful tool for grasping indirect information on
the folding state of a protein and, in particular, of the active site of
1202
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
adsorbed proteins, as the enzyme activity is strictly dependent on
protein conformation and changes in protein folding may result in
loss of activity.
4. Adsorption of nucleic acids
Similar to proteins nucleic acids exhibit a high tendency to adsorb
onto surfaces through electrostatic interactions and coordinative
bonds, hydrogen bonds or hydrophobic interactions. The presence of
binding sites at well defined distances at the surface of crystalline
solids may lead to specific adsorption nucleic acids and, in some cases,
to a molecular recognition. Like for proteins, adsorption may lead to
modification of the activity of nucleic acid. However, in the case of
DNA and RNA, more severe adverse effects on cell functions are
expected if irreversible adsorption or even unfolding occurs.
Direct interaction with DNA (primary direct genotoxicity) is one of
the possible modes of action of genotoxic molecular substances as
well as particulates [18,185–187].
To get in contact with DNA, particles need to enter cells and to
cross the nuclear membrane. However, DNA aberrations may also
arise from the contact of particles with DNA during mitosis or,
alternatively, as a result of interferences with mitotic spindle.
A growing number of studies devoted to the evaluation of
genotoxicity of engineered NPs are currently published [18,187,188]
since the evaluation of carcinogenic and mutagenic potential is a key
area in the risk assessment of new materials.
Not many examples of genotoxicity triggered by the direct
interaction of inorganic NPs with nucleic acids are available (Table 3).
However, reports of specific interaction may be found in the literature
from other scientific area.
The interaction of RNA with solid surfaces is relevant to studies
concerning the origin of life. In fact, one of the leading theories is based
on the assumption that life started in an RNA-based world. The
interaction of RNA with minerals, as catalytic surfaces, might have
played a role in the insurgence of the first self-organized systems
capable of self-replication. The idea of an origin of life mediated by the
surface of minerals was first proposed by Bernal in 1949 [189] and
supported by several authors. Studies hunting for a specific interaction
between RNA and widespread minerals have been published. Clays
were reported to catalyze the formation of RNA oligomers in a
regioselective mode [190,191] and to confine and conformationally
restrict RNA, similarly to what was observed within the ribosome of
living cells [191]. RNA adsorbed on clays was also reported to be less
susceptible of degradation by RNase but still able to act as template for
molecular replication [192]. Both clays, which are aluminosilicates, and
RNA strands are negatively charged and therefore is unlikely that
electrostatic interactions could be the driving forces for adsorption. It
has been proposed that soluble cations as Ca 2+ than Na + could
potentially mediate such interaction by screening the negative charge
with monovalent cations, or by making divalent cation bridges between
the two layers of negative charge formed by the nucleic acid and the
mineral surface [191]. Theoretical calculations strengthen the role of
Na+ ions in the interaction of DNA and RNA nucleobases with the clay
surface [193]. Adsorption of DNA against electrostatic repulsion was also
observed on silica. In this case the process was suggested to be driven by
dehydration of both the DNA molecules and the silica surface as well as
by the formation of intramolecular hydrogen bonds [194]. The possible
interaction of clays with DNA is relevant in nanotoxicology. Nanometric
clays (nanoclays) are currently used as filler to improve mechanical and
thermal resistance of polymers. Exposure of humans to these
nanomaterials is therefore likely to occur. Only few toxicity studies
have been published on this kind of materials [195–198]. Two of them
explore the possible genotoxicity of different type of nanoclays and both
report no evidence of adverse effects [196,197].
Stabilization of nucleic acids does not occur at the surface of all
minerals. Specificity was not observed on pyrite, a very common
mineral composed by iron sulfide (FeS2). In this case DNA-like
polymers were shown to adsorb onto pyrite surface through N–Fe
interaction involving nitrogen groups of the nucleobases. However,
the adsorption was aspecific and inhibited the hybridization process
with complementary nucleic acid [199].
DNA molecules are chiral and may interact stereospecifically with
chiral surfaces. Tang and co-workers nicely demonstrated a stereoselective interaction between DNA and a chiral surface possibly
Table 3
Summary of studies reporting a primary direct genotoxicity of inorganic particles.
NP type
Cell type/model
Localization of
particles
Genotoxic effects observed
Mechanism of interaction
proposed
Techniques used to
evaluate particle-DNA
interaction
References
TiO2 anatase (5 nm)
ICR-mice
Accumulation in
liver DNA
Alteration of the secondary
structure of DNA, cleavage
at the higher dose
Insertion into DNA base
pairs and binding to DNA
nucleotide through Ti–O
or Ti–N bonds
UV/Vis, ICP-MS , EXAFS,
CD, agarose gel
electrophoresis
[250]
Au nanoparticles
(30 nm), nuclear
targeting
Carbon nanoparticles
Cancer cells
Nucleus
Cytokinesis arrest, failure
cell division, apoptosis
Escherichia coli
Extracted DNA
Induction of DNA
aggregation
Quartz particles
Alveolar epithelial cells
Localization of quartz
particles in the nuclei
and mitotic spindles
Asbestos fibers
Different cell lines
Nucleus
Induction of DNA strand
breaks and induction of
(8-OHdG)
SiO2 NPs (50–100 nm)
HEp-2 and RPMI 2650 cells
Nucleus
Ag NPs (6–20 nm)
Human lung fibroblast cells
(IMR-90) and human
glioblastoma cells (U251).
Nucleus and
mitochondria
Inhibition of replication,
transcription, and cell
proliferation but not cell
viability
DNA damage, cell cycle
arrest in G2/M phase
[251]
Van der Waals forces
AFM
[252]
Hydrogen bonding
between surface silanol
groups and the
phosphate-sugar
backbone of DNA
Direct oxidative damage
to DNA
Fourier transform
infrared spectroscopy
[253]
Agarose gel
electrophoresis of
plasmid DNA after
incubation with fibers
Immunofluorescence
[107]
Indirect, formation of
intranuclear protein
aggregates of
topoisomerases
Direct (Ag+ ions) or
indirect (oxidative stress
via disruption of the
mitochondrial respiratory
chain)
[254]
[255]
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
through hydrogen bonds [200] giving insights into the stereospecific
interaction of cells with surfaces [201].
The immobilization of DNA on different solid supports is an
important issue in different fields ranging from medicine to analytical
chemistry and molecular electronics. Immobilization may be performed
by simple adsorption or by covalent bonding. In such applications DNA
strands need to preserve their conformational structure for hybridization with complementary chains and therefore organic linkers are often
used to minimize the interaction with the surface.
Au/DNA conjugates find application in biosensing and nanobiotechnological applications [202]. DNA thiolated strands are generally used to
synthesize Au/DNA NPs since thiol groups are known to form stable
bonds with gold surfaces. DNA unfolding was not observed on Au NPs at
low coverage suggesting that intramolecular hydrogen bonds and base
stacking which stabilize the double DNA strand are stronger than
nonspecific interactions between the bases and the gold surface [203].
Recently a large interest has been focused on DNA/carbon nanotubes
conjugates. The remarkable sensitivity of CNT conductivity to the surface
adsorbates allows using CNTs as highly sensitive nanoscale sensors [204].
CNTs are also of large interest in medicine as gene nanocarriers [205,206].
Interaction of DNA with non-functionalized single-walled CNTs (with
diameter ranging from 1.2 to 1.6 nm) resulted in DNA wrapping around
the tube and structural changes [207,208] similar to those observed in
chromosomes during histones-mediated DNA assembly. The same
authors studied the interaction of SWCNT with poly-A fragment and
found that the interaction occurs through adenine and phosphate groups
[209]. Interestingly, some authors recently report that single-walled CNT
may recognize particular sequences of single stranded DNA depending
upon the chirality of their graphenic structure [210].
Adsorption of nucleic acid on inorganic particles may occur only
when the size of particles is larger than the nucleic acid molecule's
size. When particles become very small (few nanometers), the
interaction with DNA helixes resembles that reported for soluble
metals (such as Cr(VI) or Cd) as they fit into the DNA grooves through
very size-dependent specific interactions, as demonstrated for Au NPs
[211] and for C60 fullerenol [212].
Finally, when an inorganic particle approaching nucleic acids
exhibits reactive sites at the surface which may generate radical
species, oxidative damage of nucleic acid may occur. This type of
interaction will be discussed in the next session.
5. Surface driven oxidative damage to biomolecules
Reactive oxygen species (ROS•) is a term that includes oxygen-based
free radicals such as superoxide anion (O2•−), hydroxyl (HO•) and
biomolecules-derived species as alkylperoxyl (RO2•) and alkoxyl (RO•)
radicals. Reactive non-radical species such as hydrogen peroxide (H2O2)
and singlet oxygen (1O2) are also considered ROS [213]. These reactive
species are physiologically produced in cells from several different
sources and cover many important physiological functions [214].
ROS have been demonstrated to be involved in many signal
transduction pathways. However, increased ROS may also be
detrimental leading to cell death or to acceleration in aging and
age-related diseases. Increased ROS levels cause random damage to
proteins, lipids and DNA. In addition to these effects, a rise in ROS
levels may also represent a stress signal that activates specific redoxsensitive signaling pathways [214].
Oxidative stress, resulting from an imbalance between production
of reactive oxygen species (ROS) or reactive nitrogen species (RNS)
and antioxidant defenses, is considered an important mechanism of
particle-induced health effects [215,216].
Particles may generate reactive oxygen species by a direct
mechanism (surface-derived ROS), or by an indirect one relying on
the alteration of mitochondrial functions or the activation of cells of
the immune systems (cell-derived ROS) [54,112,217]. Oxidative stress
may also be a consequence of leaching of redox-active ions like iron
1203
[218] or of the impairment of ROS homeostasis by depletion of
endogenous antioxidants [89,110,112,219,220].
Finally, damage may follow the direct reaction of biomolecules
with the surface. In Fig. 4 the different paths which may lead to
particle-induced oxidative damage are summarized.
Internalization and localization of NPs inside cells play a key role in
the particle-derived oxidative stress since free radicals and singlet
oxygen are extremely unstable. Therefore in order to trigger oxidative
damage they need to be generated close to the target molecule.
Both generation of ROS and direct damage to biomolecules are
related to the existence of sites accessible to the fluid and able to
undergo redox cycling. The chemical nature of these reactive sites
depends upon the type of solid (see Section 2.5) [9,20,54,221].
Covalent solids as silica or carbon nanotubes may exhibit radical
defects (dangling bonds) generated by the homolytic rupture of the
covalent bonds when ground. However, while in crystalline silica forms,
e.g. quartz, such radical species are known to promote the generation of
free radicals in biological fluids [54] and to cause oxidative damage to
biomolecules [222], in carbon nanotubes they have been related to a
scavenging activity toward free radicals [68,69]. Radical defects are not
present on amorphous silica forms which are therefore essentially
unable to cause direct oxidative damage.
Direct oxidative damage may also derive from redox reactions
involving redox-active metal ions exposed at the surface. Such ions
may be present as contaminants [64,217], may derive by adsorption of
ions from fluids, or may be present in the structure of the solids, e.g. in
asbestos, pyrite or iron oxides [57,61,65,221,223]. As discussed in
Section 2.5, the oxidative potential depends upon the coordinative
and oxidative states of exposed ions. In some cases a strong reactivity
has been observed. For example asbestos fibers were shown to induce
strand breaks in supercoiled plasmid DNA [88,107,224–226] and lipid
peroxidation [227]. Similar reactivity toward DNA was observed in
pyrite [223]. Alternatively oxidative stress may derive from metals
leached from the surface by endogenous chelators [228].
Semiconducting materials are intrinsically able to exchange
electrons with molecular entities which come into contact with the
surface, if activated by light or heat.
One of the most common semiconductors is TiO2. As described in
the Section 2.5 under sunlight illumination TiO2, when in contact with
oxygen or water, generates large amounts of radical species such as
O2• −, HO• and 1O2. As these species are highly aggressive, they may
efficiently degrade organic molecules or act as bactericidal agents.
Therefore, TiO2 has been proposed as catalyst for the degradation of
pollutants in waste waters [229,230] or as antibacterial and selfcleaning components in coatings and textiles [231,232]. At the same
time, direct DNA and RNA damage [233,234] and lipid peroxidation
[16,17,109] caused by UV-irradiated TiO2 have been reported.
Depending upon the method of synthesis TiO2 particles may
expose at the surface few persistent active centers which are still
reactive in the dark. Recently, some of us reported the ability of TiO2
powders to generate free radicals form organic molecules in the
absence of illumination [73]. This reactivity was related to the
presence of persistent Ti 3+ species and electron holes.
The capacity of CdSe quantum dots to induce oxidative damage to
DNA was also reported under photoactivation or in the dark [76].
Conductive indium-doped tin oxide (ITO) powders was also
reported to generate free radicals [111]. However the nature of the
reactive sites has not been clarified.
Carbon-based NPs and multiwalled carbon nanotubes are intrinsically
unable to generate free radicals. At the opposite they may act as
antioxidant by scavenging free radicals [67–69,235] and therefore may
potentially protect biomolecules from oxidative damage.
C60 are known to quench various free radicals, behaving as a “free
radical sponge” C60 [77,236]. Exploiting this properties, fullerenes
have been proposed in antioxidant therapy as neuroprotective agents
[237]. However, if excited with UV light they may be promoted to a to
1204
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
Fig. 4. Possible mechanisms of oxidative damage induced by inorganic particles. Inorganic particles may induce oxidative damage to cells through different path. The first path (direct
oxidative damage) may be a consequence of the presence of: i. reactive sites at the surface which cause the generation of reactive oxygen species (particle-derived ROS) e.g.,
hydroxyl radicals (HO•) from endogenous hydrogen peroxide (H2O2), superoxide ions (O2−•) and singlet oxygen (1O2) from oxygen dissolved in fluids, or ii. reactive sites which
directly damage biomolecules. Alternatively, the release of redox-active metals (Mn+) may contribute to increase the particle-derived oxidative stress. The impairment of ROS
homeostasis by depletion endogenous antioxidants may contribute to the overall process. A second path (indirect oxidative damage) consists in the generation of ROS (cell-derived
ROS) indirectly by altering mitochondrial function or activating cells of the immune systems.
triplet state which may transfer energy to oxygen to generate singlet
oxygen ( 1O2) [77] and for this reason they have been proposed in
photodynamic therapy of tumors.
6. Role of the interactions with biomacromolecules in the in vivo
response to nanoparticles
The complexity of the processes occurring at the solid/biofluid
interface is particularly high in the case of nanoparticles. NPs are in
fact potentially able to distribute through the organs and in the
various cellular and extracellular compartments, thus implying that
they may experience different conditions in terms of pH, ionic
strength and fluid composition. At the same time, NPs may come into
contact with macrobiomolecules as nucleic acids unaccessible to bulk
inorganic materials and even micrometric particles.
The fate of a NP in the body and the adverse/beneficial responses to
them is largely related to the events occurring at the interfaces [136]. In
particular the following events need to be considered: i. modification of
the surface properties affecting biodistribution; ii. perturbation of the
composition of biological fluids; iii. perturbation of the biomacromolecule
physiological functions; iv. generation of bioactive protein/NP adducts.
Physico-chemical properties as size, charge and hydrophilic degree
have been reported to modulate biodistribution, bioaccumulation and
biocompatibility of NPs [22,238–240]. According to Aggarwal et al.
[167] and Lynch et al. [136], it is the NP–protein corona, whose
composition is largely determined by the physico-chemical properties
of NPs, that actually determines both biodistribution and the final
subcellular location of a specific NP.
An aspect that deserves attention, as far as protein adsorption on
NPs is concerned, it is the effect of protein depletion in fluids following
adsorption. Several in vitro studies report changes in the biological
activity as a consequence of the modified composition of cellular
media interacting with NPs. Data are available for metal oxide,
polystyrene NPs and carbon nanotubes [35,131,241–244]. Similarly,
depletion of proteins may occur in biofluid. For examples, selective
binding of supernatant proteins from bronchoalveolar lavage fluid by
double-walled nanotubes has been reported by Salvador-Morales
et al. [245]. The authors show that a chronic level of exposure to
carbon nanotubes may result in sequestration of surfactant proteins A
and D, with consequently higher probability for the insurgence of
pathological states.
In the previous chapters the occurrence of modification of the
enzymatic activity of enzymes or perturbation of the activity of nucleic
acids following adsorption (Sections 3.3 and 4) and of oxidative
damage (Section 5) has been discussed. How such modifications
overcome the capacity of cells to re-establish their physiological (or
pathological) state needs to be clarified.
Finally, as far as the adverse effects that may arise from exposure to
NPs are concerned, it is worth to remember that the response of the
immune system to NPs has been related to the generation of bioactive
proteins/NP adducts. Protein adsorption may in fact lead to the
exposure of receptors involved in the various paths driving to
inflammation [246]. At the same time NPs may improve antigenicity
of conjugated weak antigens thus serving as adjuvants [247].
7. Conclusions
A deep understanding of the mechanisms driving the interaction
between biological fluids or cells constituents and surfaces is
instrumental for designing strategies apt to prevent the toxicity and
premature clearance of NPs used in diagnosis and therapy, and for
avoiding adverse reactions to materials used as implants or toxic
effects that may follow the accidental exposure of organisms to
nanomaterials.
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
1205
Table 4
Conclusive perspective of the effects induced on macrobiomolecules by the most relevant physico-chemical surface features.
Effect on macrobiomolecules
Type of interaction force
Physico–chemical surface feature
Hydrophobic
interaction
Affinity changes?
Conformational
changes?
Selective orientation/
interaction?
Modification of activity?
Increase with protein
softness
Correlate with protein
softness
Only for proteins exposing
hydrophobic patches
May occur following
unfolding
Hydrogen
bonding
Surface
charges
Secondary structure
changes may occur
Driven by coulombic
interactions
Correlate with protein
charge distribution
May result from secondary
changes
Only for proteins having a
non–homogeneous surface
charge distribution
May occur following
selective orientation
Coordinative
bonds
May occur following
stabilization/destabilization
of nucleic acids
Reactive sites
Possible, following direct
or ROS–mediated oxidative
damage
Size/
curvature
Higher curvatures may
prevent efficient close
packing of the proteins
Generally induced by lower
curvature. Some protein–
dependent exceptions reported
Chirality
The physico-chemical properties of the surface definitely play a
pivotal role in modulating the various possible processes at the
interface between biological fluids and solid surfaces; the peculiar
features and behavior of biomacromolecules further complicate the
picture. In Table 4 we have tried to summarize the main effects played
by the most relevant surface properties on biomacromolecules
adsorbed onto solids. These evidences come mainly from studies on
model systems; it has to be born in mind that — in real systems — the
different effects are difficult to disentangle.
In the light of these considerations, as well as of the available
literature data, we strongly believe that a full comprehension of
the physico-chemical determinants of the solid/biofluid interface
processes will only be achieved by assembling experimental evidences
from both real and model systems (where well characterized surfaces
interact with proteins with well-defined properties) with the contribution of theoretical calculations.
References
[1] A.E. Nel, L. Madler, D. Velegol, T. Xia, E.M.V. Hoek, P. Somasundaran, F. Klaessig, V.
Castranova, M. Thompson, Understanding biophysicochemical interactions at
the nano-bio interface, Nature Materials 8 (2009) 543–557.
[2] O. Cohavi, S. Corni, F. De Rienzo, R. Di Felice, K.E. Gottschalk, M. Hoefling, D. Kokh,
E. Molinari, G. Schreiber, A. Vaskevich, R.C. Wade, Protein–surface interactions:
challenging experiments and computations, Journal of Molecular Recognition 23
(2010) 259–262.
[3] H. Chen, L. Yuan, W. Song, Z.K. Wu, D. Li, Biocompatible polymer materials: role
of protein–surface interactions, Progress in Polymer Science 33 (2008)
1059–1087.
[4] M. Corno, A. Rimola, V. Bolis, P. Ugliengo, Hydroxyapatite as a key biomaterial:
quantum-mechanical simulation of its surfaces in interaction with biomolecules,
Physical Chemistry Chemical Physics 12 (2010) 6309–6329.
[5] P. Thevenot, W.J. Hu, L.P. Tang, Surface chemistry influences implant biocompatibility, Current Topics in Medicinal Chemistry 8 (2008) 270–280.
[6] A. Khandoga, T. Stoeger, A.G. Khandoga, P. Bihari, E. Karg, D. Ettehadieh, S.
Lakatos, J. Fent, H. Schulz, F. Krombach, Platelet adhesion and fibrinogen
deposition in murine microvessels upon inhalation of nanosized carbon
particles, Journal of Thrombosis and Haemostasis 8 (2010) 1632–1640.
Larger particles induce
higher activity loss
Stereoselective molecular
recognition of protein and
nucleic acids
[7] P.P. Karmali, D. Simberg, Interactions of nanoparticles with plasma proteins:
implication on clearance and toxicity of drug delivery systems, Expert Opinion
on Drug Delivery 8 (2011) 343–357.
[8] W. Norde, My voyage of discovery to proteins in flatland … and beyond, Colloids
and Surfaces. B, Biointerfaces 61 (2008) 1–9.
[9] B. Fubini, I. Fenoglio, Toxic potential of mineral dusts, Elements 3 (2007)
407–414.
[10] B. Fubini, I. Fenoglio, M. Tomatis, Physico-chemical Characteristics of Nanoparticles which Determine their Potential Toxicity, Informa Healthcare, New York,
2007.
[11] G.S. Plumlee, S.A. Morman, T.L. Ziegler, The toxicological geochemistry of
earth materials: an overview of processes and the interdisciplinary methods
used to understand them, Medical Mineralogy and Geochemistry 64 (2006)
5–57.
[12] D. Walczyk, F.B. Bombelli, M.P. Monopoli, I. Lynch, K.A. Dawson, What the cell
“sees” in bionanoscience, Journal of the American Chemical Society 132 (2010)
5761–5768.
[13] I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse, K.A. Dawson, The
nanoparticle–protein complex as a biological entity; a complex fluids and
surface science challenge for the 21st century, Advances in Colloid and Interface
Science 134–35 (2007) 167–174.
[14] C.M. Niemeyer, Nanoparticles, proteins, and nucleic acids: biotechnology meets
materials science, Angewandte Chemie, International Edition 40 (2001)
4128–4158.
[15] Y.Y. Liu, H. Miyoshi, M. Nakamura, Nanomedicine for drug delivery and imaging:
a promising avenue for cancer therapy and diagnosis using targeted functional
nanoparticles, International Journal of Cancer 120 (2007) 2527–2537.
[16] M.E. Carlotti, E. Ugazio, S. Sapino, I. Fenoglio, G. Greco, B. Fubini, Role of particle
coating in controlling skin damage photoinduced by titania nanoparticles, Free
Radical Research 43 (2009) 312–322.
[17] V. Brezova, S. Gabcova, D. Dvoranova, A. Stasko, Reactive oxygen species
produced upon photoexcitation of sunscreens containing titanium dioxide (an
EPR study), Journal of Photochemistry and Photobiology B, Biology 79 (2005)
121–134.
[18] N. Singh, B. Manshian, G.J.S. Jenkins, S.M. Griffiths, P.M. Williams, T.G.G. Maffeis,
C.J. Wright, S.H. Doak, NanoGenotoxicology: the DNA damaging potential of
engineered nanomaterials, Biomaterials 30 (2009) 3891–3914.
[19] S.J. Cho, D. Maysinger, M. Jain, B. Roder, S. Hackbarth, F.M. Winnik, Long-term
exposure to CdTe quantum dots causes functional impairments in live cells,
Langmuir 23 (2007) 1974–1980.
[20] A. Nel, T. Xia, L. Madler, N. Li, Toxic potential of materials at the nanolevel,
Science 311 (2006) 622–627.
[21] D.B. Warheit, How meaningful are the results of nanotoxicity studies in the
absence of adequate material characterization? Toxicological Sciences 101
(2008) 183–185.
1206
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
[22] P. Rivera Gil, G. Oberdorster, A. Elder, V. Puntes, W.J. Parak, Correlating physicochemical with toxicological properties of nanoparticles: the present and the
future, ACS Nano 4 (2010) 5527–5531.
[23] B. Fubini, I. Fenoglio, R. Ceschino, M. Ghiazza, G. Martra, M. Tomatis, P. Borm, R.
Schins, J. Bruch, Relationship between the state of the surface of four commercial
quartz flours and their biological activity in vitro and in vivo, International
Journal of Hygiene and Environmental Health 207 (2004) 89–104.
[24] P. Maksymovych, S. Mezhenny, J.T. Yates, STM study of water adsorption on the
TiO2(110)-(1 × 2) surface, Chemical Physics Letters 382 (2003) 270–276.
[25] K. Liu, X. Yao, L. Jiang, Recent developments in bio-inspired special wettability,
Chemical Society Reviews 39 (2010) 3240–3255.
[26] B. Fubini, G. Zanetti, S. Altilia, R. Tiozzo, D. Lison, U. Saffiotti, Relationship
between surface properties and cellular responses to crystalline silica: studies
with heat-treated cristobalite, Chemical Research in Toxicology 12 (1999)
737–745.
[27] IUPAC, Compendium of Chemical Terminology (the “Gold Book”). Compiled by
A. D. McNaught and A. Wilkinson, 2nd ed. Blackwell Scientific Publications,
Oxford, 2006.
[28] J. Lyklema, Fundamentals of Interface and Colloid Science, Academic Press,
London, Sidney, 1991.
[29] J.P. Jolivet, Metal Oxide Chemistry and Synthesis. From Solution to Solid State,
John Wiley & Sons, Hoboken, NJ, 2000.
[30] G. Oberdorster, Safety assessment for nanotechnology and nanomedicine:
concepts of nanotoxicology, Journal of Internal Medicine 267 (2010) 89–105.
[31] J. Jiang, G. Oberdorster, P. Biswas, Characterization of size, surface charge, and
agglomeration state of nanoparticle dispersions for toxicological studies, Journal
of Nanoparticle Research 11 (2009) 77–89.
[32] S. Banerjee, T. Hemraj-Benny, S.S. Wong, Covalent surface chemistry of singlewalled carbon nanotubes, Advanced Materials 17 (2005) 17–29.
[33] D. Tasis, N. Tagmatarchis, A. Bianco, M. Prato, Chemistry of carbon nanotubes,
Chemical Reviews 106 (2006) 1105–1136.
[34] A. Hirsch, O. Vostrowsky, Functionalization of carbon nanotubes, Functional
Molecular Nanostructures, Springer-Verlag Berlin, Berlin, 2005, pp. 193–237.
[35] B. Fubini, M. Ghiazza, I. Fenoglio, Physico-chemical features of engineered
nanoparticles relevant to their toxicity, Nanotoxicology 4 (2010) 347–363.
[36] F. Turci, E. Ghibaudi, M. Colonna, B. Boscolo, I. Fenoglio, B. Fubini, An integrated
approach to the study of the interaction between proteins and nanoparticles,
Langmuir 26 (2010) 8336–8346.
[37] M. Kosmulski, Surface Charging and Points of Zero Charge, CRC Press, 2009.
[38] K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul, B.
Prati, Role of surface area, primary particle size, and crystal phase on
titanium dioxide nanoparticle dispersion properties, Nanoscale Research Letters
6 (2011).
[39] G.D. Panagiotou, T. Petsi, K. Bourikas, C.S. Garoufalis, A. Tsevis, N. Spanos, C.
Kordulis, A. Lycourghiotis, Mapping the surface (hydr)oxo-groups of titanium
oxide and its interface with an aqueous solution: the state of the art and a new
approach, Advances in Colloid and Interface Science 142 (2008) 20–42.
[40] G.A. Parks, The isoelectric points of solid oxides, solid hydroxides, and aqueous
hydroxo complex systems, Chemical Reviews 65 (1965) 177–198.
[41] K.H. Kim, N. Ramaswamy, Electrochemical surface modification of titanium in
dentistry, Dental Materials Journal 28 (2009) 20–36.
[42] C. Ergun, H. Liu, J.W. Halloran, T.J. Webster, Increased osteoblast adhesion on
nanograined hydroxyapatite and tricalcium phosphate containing calcium
titanate, Journal of Biomedical Materials Research. Part A 80A (2007) 990–997.
[43] F.A. Denis, A. Pallandre, B. Nysten, A.M. Jonas, C.C. Dupont-Gillain, Alignment and
assembly of adsorbed collagen molecules induced by anisotropic chemical
nanopatterns, Small 1 (2005) 984–991.
[44] K.B. Cederquist, C.D. Keating, Curvature effects in DNA:Au nanoparticle
conjugates, ACS Nano 3 (2009) 256–260.
[45] P. Roach, D. Farrar, C.C. Perry, Surface tailoring for controlled protein adsorption:
effect of topography at the nanometer scale and chemistry, Journal of the
American Chemical Society 128 (2006) 3939–3945.
[46] M. Lundqvist, I. Sethson, B.H. Jonsson, Protein adsorption onto silica nanoparticles: conformational changes depend on the particles' curvature and the
protein stability, Langmuir 20 (2004) 10639–10647.
[47] S. Ahmed, S.L. Wunder, Effect of high surface curvature on the main phase
transition of supported phospholipid bilayers on SiO2 nanoparticles, Langmuir
25 (2009) 3682–3691.
[48] E. Hershkovits, A. Tannenbaum, R. Tannenbaum, Polymer adsorption on curved
surfaces: a geometric approach, Journal of Physical Chemistry C 111 (2007)
12369–12375.
[49] R.K. Iler, The Chemistry of Silica, Wiley and Sons, New York, 1979.
[50] H. Kamiya, M. Mitsui, H. Takano, S. Miyazawa, Influence of particle diameter on
surface silanol structure, hydration forces, and aggregation behavior of alkoxidederived silica particles, Journal of the American Ceramic Society 83 (2000)
287–293.
[51] C. Melis, A. Mattoni, L. Colombo, Atomistic investigation of poly(3-hexylthiophene) adhesion on nanostructured titania, Journal of Physical Chemistry C 114
(2010) 3401–3406.
[52] S. Reiss, H. Krumm, A. Niklewski, V. Staemmler, C. Woll, The adsorption of acenes
on rutile TiO2(110): a multi-technique investigation, Journal of Chemical Physics
116 (2002) 7704–7713.
[53] K. Straif, L. Benbrahim-Tallaa, R. Baan, Y. Grosse, B. Secretan, F. El Ghissassi, V.
Bouvard, N. Guha, C. Freeman, L. Galichet, V. Cogliano, A review of human
carcinogens Part C: metals, arsenic, dusts, and fibres, The Lancet Oncology 10
(2009) 453–454.
[54] B. Fubini, A. Hubbard, Reactive oxygen species (ROS) and reactive nitrogen
species (RNS) generation by silica in inflammation and fibrosis, Free Radical
Biology & Medicine 34 (2003) 1507–1516.
[55] V. Castranova, V. Vallyathan, D.M. Ramsey, J.L. McLaurin, D. Pack, S. Leonard,
M.W. Barger, J.Y.C. Ma, N.S. Dalal, A. Teass, Augmentation of pulmonary reactions
to quartz inhalation by trace amounts of iron-containing particles, Environmental
Health Perspectives 105 (1997) 1319–1324.
[56] M. Ghiazza, M. Polimeni, I. Fenoglio, E. Gazzano, D. Ghigo, B. Fubini, Does vitreous
silica contradict the toxicity of the crystalline silica paradigm? Chemical
Research in Toxicology 23 (2010) 620–629.
[57] F. Turci, M. Tomatis, I.G. Lesci, N. Roveri, B. Fubini, The iron-related molecular
toxicity mechanism of synthetic asbestos nanofibres: a model study for highaspect-ratio nanoparticles, Chemistry-A European Journal 17 (2011) 350–358.
[58] E. Gazzano, F. Turci, E. Foresti, M.G. Putzu, E. Aldieri, F. Silvagno, I.G. Lesci, M.
Tomatis, C. Riganti, C. Romano, B. Fubini, N. Roveri, D. Ghigo, Iron-loaded
synthetic chrysotile: a new model solid for studying the role of iron in asbestos
toxicity, Chemical Research in Toxicology 20 (2007) 380–387.
[59] C. Otero-Areán, F. Barcelo, Free radical activity of mineral fibres containing
adsorbed ferritin: detection using supercoiled DNA, Research on Chemical
Intermediates 25 (1999) 177–185.
[60] B. Fubini, L. Mollo, Role of iron in the reactivity of mineral fibers, Toxicology
Letters 82–3 (1995) 951–960.
[61] G. Martra, E. Chiardola, S. Coluccia, L. Marchese, M. Tomatis, B. Fubini, Reactive
sites at the surface of crocidolite asbestos, Langmuir 15 (1999) 5742–5752.
[62] G. Martra, M. Tomatis, I. Fenoglio, S. Coluccia, B. Fubini, Ascorbic acid modifies
the surface of asbestos: possible implications in the molecular mechanisms of
toxicity, Chemical Research in Toxicology 16 (2003) 328–335.
[63] F. Turci, M. Tomatis, S. Mantegna, G. Cravotto, B. Fubini, The combination of
oxalic acid with power ultrasound fully degrades chrysotile asbestos fibres,
Journal of Environmental Monitoring 9 (2007) 1064–1066.
[64] M. Ghiazza, A.M. Scherbart, I. Fenoglio, F. Grendene, F. Turci, G. Martra, C.
Albrecht, R.P.F. Schins, B. Fubini, Surface iron inhibits quartz-induced cytotoxic
and inflammatory responses in alveolar macrophages, Chemical Research in
Toxicology 24 (2011) 99–110.
[65] B. Fubini, L. Mollo, E. Giamello, Free-radical generation at the solid/liquid
interface in iron-containing minerals, Free Radical Research 23 (1995)
593–614.
[66] K. Pulskamp, S. Diabate, H.F. Krug, Carbon nanotubes show no sign of acute
toxicity but induce intracellular reactive oxygen species in dependence on
contaminants, Toxicology Letters 168 (2007) 58–74.
[67] I. Fenoglio, M. Tomatis, D. Lison, J. Muller, A. Fonseca, J.B. Nagy, B. Fubini,
Reactivity of carbon nanotubes: free radical generation or scavenging activity?
Free Radical Biology & Medicine 40 (2006) 1227–1233.
[68] I. Fenoglio, G. Greco, M. Tornatis, J. Muller, E. Rayrnundo-Pinero, F. Beguin, A.
Fonseca, J.B. Nagy, D. Lison, B. Fubini, Structural defects play a major role in the
acute lung toxicity of multiwall carbon nanotubes: physicochemical aspects,
Chemical Research in Toxicology 21 (2008) 1690–1697.
[69] A. Galano, M. Francisco-Marquez, A. Martinez, Influence of point defects on the
free-radical scavenging capability of single-walled carbon nanotubes, Journal of
Physical Chemistry C 114 (2010) 8302–8308.
[70] J.T. Yates Jr., Photochemistry on TiO(2): mechanisms behind the surface
chemistry, Surface Science 603 (2009) 1605–1612.
[71] A. Fujishima, X. Zhang, D.A. Tryk, TiO(2) photocatalysis and related surface
phenomena, Surface Science Reports 63 (2008) 515–582.
[72] T. Daimon, T. Hirakawa, M. Kitazawa, J. Suetake, Y. Nosaka, Formation of singlet
molecular oxygen associated with the formation of superoxide radicals in
aqueous suspensions of TiO2 photocatalysts, Applied Catalysis A: General 340
(2008) 169–175.
[73] I. Fenoglio, G. Greco, S. Livraghi, B. Fubini, Non-UV-induced radical reactions at
the surface of TiO2 nanoparticles that may trigger toxic responses, Chemistry-A
European Journal 15 (2009) 4614–4621.
[74] T. Hirakawa, K. Yawata, Y. Nosaka, Photocatalytic reactivity for O−2(center
dot−) and OH center dot radical formation in anatase and rutile TiO2
suspension as the effect of H2O2 addition, Applied Catalysis A: General 325
(2007) 105–111.
[75] C.B. Almquist, P. Biswas, Role of synthesis method and particle size of nanostructured TiO2 on its photoactivity, Journal of Catalysis 212 (2002) 145–156.
[76] E.J. Petersen, B.C. Nelson, Mechanisms and measurements of nanomaterialinduced oxidative damage to DNA, Analytical and Bioanalytical Chemistry 398
(2010) 613–650.
[77] Z. Markovic, V. Trajkovic, Biomedical potential of the reactive oxygen species
generation and quenching by fullerenes (C-60), Biomaterials 29 (2008)
3561–3573.
[78] M. Auffan, J. Rose, J.Y. Bottero, G.V. Lowry, J.P. Jolivet, M.R. Wiesner, Towards a
definition of inorganic nanoparticles from an environmental, health and safety
perspective, Nature Nanotechnology 4 (2009) 634–641.
[79] L. Addadi, M. Geva, Molecular recognition at the interface between crystals and
biology: generation, manifestation and detection of chirality at crystal surfaces,
CrystEngComm (2003) 140–146.
[80] K. Soai, S. Osanai, K. Kadowaki, S. Yonekubo, T. Shibata, I. Sato, d- and l-quartzpromoted highly enantioselective synthesis of a chiral organic compound,
Journal of the American Chemical Society 121 (1999) 11235–11236.
[81] E.I. Klabunovskii, Can enantiomorphic crystals like quartz play a role in the origin
of homochirality on earth? Astrobiology 1 (2001) 127–131.
[82] C.F. McFadden, P.S. Cremer, A.J. Gellman, Adsorption of chiral alcohols on “chiral”
metal surfaces, Langmuir 12 (1996) 2483–2487.
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
[83] G.A. Attard, Electrochemical studies of enantioselectivity at chiral metal surfaces,
The Journal of Physical Chemistry. B 105 (2001) 3158–3167.
[84] T. Mallat, E. Orglmeister, A. Baiker, Asymmetric catalysis at chiral metal surfaces,
Chemical Reviews 107 (2007) 4863–4890.
[85] C.H. Liu, H.L. Zhang, Chemical approaches towards single-species single-walled
carbon nanotubes, Nanoscale 2 (2010) 1901–1918.
[86] Y.N. Zhang, L.X. Zheng, Towards chirality-pure carbon nanotubes, Nanoscale 2
(2010) 1919–1929.
[87] M. Blesa, P.J. Morando, A.E. Regazzoni, Chemical Dissolution of Metal Oxides, CRC
Press, Boca Raton, FL, USA, 1994.
[88] J.A. Hardy, A.E. Aust, The effect of iron-binding on the ability of crocidolite
asbestos to catalyze DNA single-strand breaks, Carcinogenesis 16 (1995)
319–325.
[89] I. Fenoglio, G. Martra, S. Coluccia, B. Fubini, Possible role of ascorbic acid in the
oxidative damage induced by inhaled crystalline silica particles, Chemical
Research in Toxicology 13 (2000) 971–975.
[90] D.B. Warheit, T.A. McHugh, M.A. Hartsky, Differential pulmonary responses in
rats inhaling crystalline, colloidal or amorphous silica dusts, Scandinavian
Journal of Work, Environment & Health 21 (1995) 19–21.
[91] I.S. Yuen, M.A. Hartsky, S.I. Snajdr, D.B. Warheit, Time course of chemotactic
factor generation and neutrophil recruitment in the lungs of dust-exposed rats,
American Journal of Respiratory Cell and Molecular Biology 15 (1996) 268–274.
[92] P.G.J. Reuzel, J.P. Bruijntjes, V.J. Feron, R.A. Woutersen, Subchronic inhalation
toxicity of amorphous silicas and quartz dust in rats, Food and Chemical
Toxicology 29 (1991) 341–354.
[93] J.K. McLaughlin, W.H. Chow, L.S. Levy, Amorphous silica: a review of health
effects from inhalation exposure with particular reference to cancer, Journal of
Toxicology and Environmental Health 50 (1997) 553–566.
[94] S.K. Lower, M.F. Hochella, T.J. Beveridge, Bacterial recognition of mineral
surfaces: nanoscale interactions between Shewanella and alpha-FeOOH, Science
292 (2001) 1360–1363.
[95] P. Hinterdorfer, Y.F. Dufrene, Detection and localization of single molecular recognition
events using atomic force microscopy, Nature Methods 3 (2006) 347–355.
[96] D.J. Mueller, Y.F. Dufrene, Atomic force microscopy as a multifunctional molecular
toolbox in nanobiotechnology, Nature Nanotechnology 3 (2008) 261–269.
[97] B.J. Albers, T.C. Schwendemann, M.Z. Baykara, N. Pilet, M. Liebmann, E.I. Altman,
U.D. Schwarz, Three-dimensional imaging of short-range chemical forces with
picometre resolution, Nature Nanotechnology 4 (2009) 307–310.
[98] A. Zecchina, C.O. Arean, Diatomic molecular probes for mid-IR studies of zeolites,
Chemical Society Reviews 25 (1996) 187–197.
[99] V. Bolis, B. Fubini, L. Marchese, G. Martra, D. Costa, Hydrophilic and hydrophobic
sites on dehydrated crystalline and amorphous silicas, Journal of the Chemical
Society, Faraday Transactions 87 (1991) 497–505.
[100] V. Bolis, A. Cavenago, B. Fubini, Surface heterogeneity on hydrophilic and
hydrophobic silicas: water and alcohols as probes for H-bonding and dispersion
forces, Langmuir 13 (1997) 895–902.
[101] V. Bolis, C. Morterra, B. Fubini, P. Ugliengo, E. Garrone, Temkin-type model for the
description of induced heterogeneity — co adsorption on group-4 transitionmetal dioxides, Langmuir 9 (1993) 1521–1528.
[102] B. Fubini, V. Bolis, A. Cavenago, E. Garrone, P. Ugliengo, Structural and induced
heterogeneity at the surface of some SiO2 polymorphs from the enthalpy of
adsorption of various molecules, Langmuir 9 (1993) 2712–2720.
[103] M. Cerruti, G. Magnacca, V. Bolis, C. Morterra, Characterization of sol–gel
bioglasses with the use of simple model systems: a surface-chemistry approach,
Journal of Materials Chemistry 13 (2003) 1279–1286.
[104] E. Giamello, D. Murphy, G. Magnacca, C. Morterra, Y. Shioya, T. Nomura, M. Anpo,
The interaction of NO with copper ions in ZSM5 — an EPR and IR investigation,
Journal of Catalysis 136 (1992) 510–520.
[105] C. Morterra, V. Bolis, G. Magnacca, IR spectroscopic and microcalorimetric
characterization of Lewis-acid sites on (transition phase) Al2O3 using adsorbed
CO, Langmuir 10 (1994) 1812–1824.
[106] C. Morterra, G. Magnacca, A case study: surface chemistry and surface structure
of catalytic aluminas, as studied by vibrational spectroscopy of adsorbed species,
Catalysis Today 27 (1996) 497–532.
[107] L. Jiang, H. Nagai, H. Ohara, S. Hara, M. Tachibana, S. Hirano, Y. Shinohara, N.
Kohyama, S. Akatsuka, S. Toyokuni, Characteristics and modifying factors of
asbestos-induced oxidative DNA damage, Cancer Science 99 (2008) 2142–2151.
[108] R. Greenwood, Review of the measurement of zeta potentials in concentrated
aqueous suspensions using electroacoustics, Advances in Colloid and Interface
Science 106 (2003) 55–81.
[109] S. Livraghi, I. Corazzari, M.C. Paganini, G. Ceccone, E. Giamello, B. Fubini, I.
Fenoglio, Decreasing the oxidative potential of TiO2 nanoparticles through
modification of the surface with carbon: a new strategy for the production of safe
UV filters, Chemical Communications 46 (2010) 8478–8480.
[110] I. Fenoglio, S. Fonsato, B. Fubini, Reaction of cysteine and glutathione (GSH) at
the freshly fractured quartz surface: a possible role in silica-related diseases?
Free Radical Biology & Medicine 35 (2003) 752–762.
[111] D. Lison, J. Laloy, I. Corazzari, J. Muller, V. Rabolli, N. Panin, F. Huaux, I. Fenoglio, B.
Fubini, Sintered indium-tin-oxide (ITO) particles: a new pneumotoxic entity,
Toxicological Sciences 108 (2009) 472–481.
[112] J.G. Ayres, P. Borm, F.R. Cassee, V. Castranova, K. Donaldson, A. Ghio, R.M.
Harrison, R. Hider, F. Kelly, I.M. Kooter, F. Marano, R.L. Maynard, I. Mudway, A.
Nel, C. Sioutas, S. Smith, A. Baeza-Squiban, A. Cho, S. Duggan, J. Froines,
Evaluating the toxicity of airborne particulate matter and nanoparticles by
measuring oxidative stress potential — a workshop report and consensus
statement, Inhalation Toxicology 20 (2008) 75–99.
1207
[113] M.A.A. Shoonen, C.A. Cohn, E. Roemer, R. Laffers, S.R. Simon, T. O'Riordan,
Mineral-induced formation of reactive oxygen species, in: N. Shai, M.A.A.
Schoonen (Eds.), Medical Mineralogy and Geochamistry, The Mineralogical
Society of America, Chantilly, Virginia, U.S.A, 2006, pp. 179–222.
[114] N.V. Blough, D.J. Simpson, Chemically mediated fluorescence yield switching in
nitroxide-fluorophore adducts: optical sensors of radical/redox reactions,
Journal of the American Chemical Society 110 (1988) 1915–1917.
[115] K. Setsukinai, Y. Urano, K. Kakinuma, H.J. Majima, T. Nagano, Development of
novel fluorescence probes that can reliably detect reactive oxygen species and
distinguish specific species, The Journal of Biological Chemistry 278 (2003)
3170–3175.
[116] L. Vroman, Effect of adsorbed proteins on the wettability of hydrophilic and
hydrophobic solids, Nature 196 (1962) 476–477.
[117] X.Y. Wu, G. Narsimhan, Effect of surface concentration on secondary and tertiary
conformational changes of lysozyme adsorbed on silica nanoparticles, Biochimica et Biophysica Acta-Proteins and Proteomics 1784 (2008) 1694–1701.
[118] T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K.A. Dawson,
S. Linse, Understanding the nanoparticle–protein corona using methods to
quantify exchange rates and affinities of proteins for nanoparticles, Proceedings
of the National Academy of Sciences of the United States of America 104 (2007)
2050–2055.
[119] D. Dell'Orco, M. Lundqvist, C. Oslakovic, T. Cedervall, S. Linse, Modeling the
time evolution of the nanoparticle–protein corona in a body fluid, PloS One 5
(2010).
[120] G. Raffaini, F. Ganazzoli, Understanding the performance of biomaterials through
molecular modeling: crossing the bridge between their intrinsic properties and
the surface adsorption of proteins, Macromolecular Bioscience 7 (2007)
552–566.
[121] J.J. Gray, The interaction of proteins with solid surfaces, Current Opinion in
Structural Biology 14 (2004) 110–115.
[122] F. Galisteo, W. Norde, Adsorption of lysozyme and alpha-lactalbumin on poly
(styrenesulphonate) latices .1. Adsorption and desorption behavior, Colloids and
Surfaces. B, Biointerfaces 4 (1995) 375–387.
[123] J. Etheve, P. Dejardin, Adsorption kinetics of lysozyme on silica at pH 7.4:
correlation between streaming potential and adsorbed amount, Langmuir 18
(2002) 1777–1785.
[124] K. Rezwan, L.P. Meier, M. Rezwan, J. Voros, M. Textor, L.J. Gauckler, Bovine serum
albumin adsorption onto colloidal Al2O3 particles: a new model based on zeta
potential and UV–vis measurements, Langmuir 20 (2004) 10055–10061.
[125] E. Casals, T. Pfaller, A. Duschl, G.J. Oostingh, V. Puntes, Time evolution of the
nanoparticle protein corona, ACS Nano 4 (2010) 3623–3632.
[126] P. Asuri, S.S. Karajanagi, H.C. Yang, T.J. Yim, R.S. Kane, J.S. Dordick, Increasing
protein stability through control of the nanoscale environment, Langmuir 22
(2006) 5833–5836.
[127] S.J. Park, A.A. Lazarides, C.A. Mirkin, R.L. Letsinger, Directed assembly of periodic
materials from protein and oligonucleotide-modified nanoparticle building
blocks, Angewandte Chemie, International Edition 40 (2001) 2909–2912.
[128] W. Shenton, S.A. Davis, S. Mann, Directed self-assembly of nanoparticles into
macroscopic materials using antibody–antigen recognition, Advanced Materials
11 (1999) 449-+.
[129] P. Asuri, S.S. Bale, S.S. Karajanagi, R.S. Kane, The protein–nanomaterial interface,
Current Opinion in Biotechnology 17 (2006) 562–568.
[130] R. Hong, N.O. Fischer, A. Verma, C.M. Goodman, T. Emrick, V.M. Rotello, Control of
protein structure and function through surface recognition by tailored
nanoparticle scaffolds, Journal of the American Chemical Society 126 (2004)
739–743.
[131] S. Deguchi, T. Yamazaki, S. Mukai, R. Usami, K. Horikoshi, Stabilization of C-60
nanoparticles by protein adsorption and its implications for toxicity studies,
Chemical Research in Toxicology 20 (2007) 854–858.
[132] D. Elgrabli, S. Abella-Gallart, O. Aguerre-Chariol, F. Robidel, F. Rogerieux, J.
Boczkowski, G. Lacroix, Effect of BSA on carbon nanotube dispersion for in vivo
and in vitro studies, Nanotoxicology 1 (2007) 266–278.
[133] C. Schulze, A. Kroll, C.M. Lehr, U.F. Schafer, K. Becker, J. Schnekenburger, C.S.
Isfort, R. Landsiedel, W. Wohlleben, Not ready to use — overcoming pitfalls when
dispersing nanoparticles in physiological media, Nanotoxicology 2 (2008)
51–61.
[134] M. Kendall, P. Ding, K. Kendall, Particle and nanoparticle interactions with
fibrinogen: the importance of aggregation in nanotoxicology, Nanotoxicology 5
(2011) 55–65.
[135] Z. Xu, S.L. Wang, H.W. Gao, Effects of nano-sized silicon dioxide on the structures
and activities of three functional proteins, Journal of Hazardous Materials 180
(2010) 375–383.
[136] I. Lynch, K.A. Dawson, Protein–nanoparticle interactions, Nano Today 3 (2008)
40–47.
[137] F. Bellezza, A. Cipiciani, M.A. Quotadarno, S. Cinelli, G. Onori, S. Tacchi, Structure,
stability, and activity of myoglobin adsorbed onto phosphate-grafted zirconia
nanoparticles, Langmuir 23 (2007) 13007–13012.
[138] S. Linse, C. Cabaleiro-Lago, W.F. Xue, I. Lynch, S. Lindman, E. Thulin, S.E. Radford,
K.A. Dawson, Nucleation of protein fibrillation by nanoparticles, Proceedings of
the National Academy of Sciences of the United States of America 104 (2007)
8691–8696.
[139] I.Y. Podolski, Z.A. Podlubnaya, E.A. Kosenko, E.A. Mugantseva, E.G. Makarova, L.G.
Marsagishvili, M.D. Shpagina, Y.G. Kaminsky, G.V. Andrievsky, V.K. Klochkov,
Effects of hydrated forms of C-60 fullerene on amyloid beta-peptide fibrillization
in vitro and performance of the cognitive task, Journal of Nanoscience and
Nanotechnology 7 (2007) 1479–1485.
1208
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
[140] S. Duinhoven, R. Poort, G. Vandervoet, W.G.M. Agterof, W. Norde, J. Lyklema, Driving
forces for enzyme adsorption at solid–liquid interfaces.1. The serine-protease
savinase, Journal of Colloid and Interface Science 170 (1995) 340–350.
[141] T. Arai, W. Norde, The behavior of some model proteins at solid liquid
interfaces.1. Adsorption from single protein solutions, Colloids and Surfaces 51
(1990) 1–15.
[142] W. Norde, C.A. Haynes, Reversibility and the mechanism of protein adsorption,
in: T.A. Horbett, J.L. Brash (Eds.), Proteins at Interfaces II — Fundamentals and
Applications, ACS, Washington, DC — USA, 1995, pp. 26–40.
[143] X. Wu, G. Narsimhan, Characterization of secondary and tertiary conformational
changes of beta-lactoglobulin adsorbed on silica nanoparticle surfaces, Langmuir
24 (2008) 4989–4998.
[144] D.E. Graham, M.C. Phillips, Proteins at liquid interfaces: I. Kinetics of adsorption and
surface denaturation, Journal of Colloid and Interface Science 70 (1979) 403–414.
[145] H. Larsericsdotter, S. Oscarsson, J. Buijs, Thermodynamic analysis of proteins
adsorbed on silica particles: electrostatic effects, Journal of Colloid and Interface
Science 237 (2001) 98–103.
[146] P. Billsten, P.O. Freskgard, U. Carlsson, B.H. Jonsson, H. Elwing, Adsorption to
silica nanoparticles of human carbonic anhydrase II and truncated forms induce
a molten-globule-like structure, FEBS Letters 402 (1997) 67–72.
[147] T. Zoungrana, W. Norde, Thermal stability and enzymatic activity of alphachymotrypsin adsorbed on polystyrene surfaces, Colloids and Surfaces. B,
Biointerfaces 9 (1997) 157–167.
[148] A.A. Vertegel, R.W. Siegel, J.S. Dordick, Silica nanoparticle size influences the
structure and enzymatic activity of adsorbed lysozyme, Langmuir 20 (2004)
6800–6807.
[149] T.J. Su, J.R. Lu, R.K. Thomas, Z.F. Cui, J. Penfold, The effect of solution pH on the
structure of lysozyme layers adsorbed at the silica–water interface studied by
neutron reflection, Langmuir 14 (1998) 438–445.
[150] A. Sethuraman, G. Vedantham, T. Imoto, T. Przybycien, G. Belfort, Protein
unfolding at interfaces: slow dynamics of alpha-helix to beta-sheet transition,
Proteins: Structure Function and Bioinformatics 56 (2004) 669–678.
[151] W. Shang, J.H. Nuffer, J.S. Dordick, R.W. Siegel, Unfolding of ribonuclease A on
silica nanoparticle surfaces, Nano Letters 7 (2007) 1991–1995.
[152] S.S. Karajanagi, A.A. Vertegel, R.S. Kane, J.S. Dordick, Structure and function of
enzymes adsorbed onto single-walled carbon nanotubes, Langmuir 20 (2004)
11594–11599.
[153] B. Boscolo, S.S. Leal, C.A. Salgueiro, E.M. Ghibaudi, C.M. Gomes, The prominent
conformational plasticity of lactoperoxidase: a chemical and pH stability
analysis, Biochimica et Biophysica Acta-Proteins and Proteomics 1794 (2009)
1041–1048.
[154] F.Y. Oliva, L.B. Avalle, O.R. Camara, C.P. De Pauli, Adsorption of human serum
albumin (HSA) onto colloidal TiO2 particles, part I, Journal of Colloid and
Interface Science 261 (2003) 299–311.
[155] M. Karlsson, L.G. Martensson, B.H. Jonsson, U. Carlsson, Adsorption of human
carbonic anhydrase II variants to silica nanoparticles occur stepwise: binding is
followed by successive conformational changes to a molten-globule-like state,
Langmuir 16 (2000) 8470–8479.
[156] P. Sabatino, L. Casella, A. Granata, M. Iafisco, I.G. Lesci, E. Monzani, N. Roveri,
Synthetic chrysotile nanocrystals as a reference standard to investigate surfaceinduced serum albumin structural modifications, Journal of Colloid and Interface
Science 314 (2007) 389–397.
[157] J.L. Robeson, R.D. Tilton, Spontaneous reconfiguration of adsorbed lysozyme
layers observed by total internal reflection fluorescence with a pH-sensitive
fluorophore, Langmuir 12 (1996) 6104–6113.
[158] S.M. Daly, T.M. Przybycien, R.D. Tilton, Coverage-dependent orientation of
lysozyme adsorbed on silica, Langmuir 19 (2003) 3848–3857.
[159] S. Noinville, M. Revault, M.H. Baron, Conformational changes of enzymes
adsorbed at liquid–solid interface: relevance to enzymatic activity, Biopolymers
67 (2002) 323–326.
[160] C.C. You, M. De, G. Han, V.M. Rotello, Tunable inhibition and denaturation of
alpha-chymotrypsin with amino acid-functionalized gold nanoparticles, Journal
of the American Chemical Society 127 (2005) 12873–12881.
[161] W. Shang, J.H. Nuffer, V.A. Muniz-Papandrea, W. Colon, R.W. Siegel, J.S. Dordick,
Cytochrome c on silica nanoparticles: influence of nanoparticle size on protein
structure, stability, and activity, Small 5 (2009) 470–476.
[162] W.L. Hubbell, D.S. Cafiso, C. Altenbach, Identifying conformational changes with
site-directed spin labeling, Nature Structural Biology 7 (2000) 735–739.
[163] W.L. Hubbell, H.S. McHaourab, C. Altenbach, M.A. Lietzow, Watching proteins
move using site-directed spin labeling, Structure 4 (1996) 779–783.
[164] B. Boscolo, S.S. Leal, E.M. Ghibaudi, C.M. Gomes, Lactoperoxidase folding and
catalysis relies on the stabilization of the alpha-helix rich core domain: a thermal
unfolding study, Biochimica et Biophysica Acta-Proteins and Proteomics 1774
(2007) 1164–1172.
[165] K. Jacobsen, W.L. Hubbell, O.P. Ernst, T. Risse, Details of the partial unfolding of T4
lysozyme on quartz using site-directed spin labeling, Angewandte Chemie,
International Edition 45 (2006) 3874–3877.
[166] R. Nicholov, N. Lum, R.P.N. Veregin, F. DoCosmo, Human serum albumin
adsorption at solid–liquid interface monitored by electron spin resonance
spectroscopy, in: T.A. Horbett, J.L. Brash (Eds.), Proteins at Interfaces II:
Fundamental and Applications, American Chemical Society, Washington, USA,
1995, pp. 280–295.
[167] P. Aggarwal, J.B. Hall, C.B. McLeland, M.A. Dobrovolskaia, S.E. McNeil,
Nanoparticle interaction with plasma proteins as it relates to particle
biodistribution, biocompatibility and therapeutic efficacy, Advanced Drug
Delivery Reviews 61 (2009) 428–437.
[168] H.R. Kim, K. Andrieux, C. Delomenie, H. Chacun, M. Appel, D. Desmaele, F. Taran,
D. Georgin, P. Couvreur, M. Taverna, Analysis of plasma protein adsorption onto
PEGylated nanoparticles by complementary methods: 2-DE, CE and protein Labon-chip((R)) system, Electrophoresis 28 (2007) 2252–2261.
[169] H. Kato, M. Suzuki, K. Fujita, M. Horie, S. Endoh, Y. Yoshida, H. Iwahashi, K.
Takahashi, A. Nakamura, S. Kinugasa, Reliable size determination of nanoparticles using dynamic light scattering method for in vitro toxicology assessment,
Toxicology In Vitro 23 (2009) 927–934.
[170] B. Carragher, D. Fellmann, F. Guerra, R.A. Milligan, F. Mouche, J. Pulokas, B. Sheehan,
J. Quispe, C. Suloway, Y.X. Zhu, C.S. Potter, Rapid, routine structure determination
of macromolecular assemblies using electron microscopy: current progress and
further challenges, Journal of Synchrotron Radiation 11 (2004) 83–85.
[171] K. Kaneko, K. Inoke, B. Freitag, A.B. Hungria, P.A. Midgley, T.W. Hansen, J. Zhang,
S. Ohara, T. Adschiri, Structural and morphological characterization of cerium
oxide nanocrystals prepared by hydrothermal synthesis, Nano Letters 7 (2007)
421–425.
[172] S. Gon, M. Bendersky, J.L. Ross, M.M. Santore, Manipulating protein adsorption
using a patchy protein-resistant brush, Langmuir 26 (2010) 12147–12154.
[173] D.J. Stephens, V.J. Allan, Light microscopy techniques for live cell imaging,
Science 300 (2003) 82–86.
[174] C.I. Brady, N.H. Mack, L.O. Brown, S.K. Doorn, Self-assembly approach to
multiplexed surface-enhanced Raman spectral-encoder beads, Analytical Chemistry 81 (2009) 7181–7188.
[175] V.K. Sharma, D.S. Kalonia, Steady-state tryptophan fluorescence spectroscopy
study to probe tertiary structure of proteins in solid powders, Journal of
Pharmaceutical Sciences 92 (2003) 890–899.
[176] M.H. Baron, M. Revault, S. Servagent-Noinville, J. Abadie, H. Quiquampoix,
Chymotrypsin adsorption on montmorillonite: enzymatic activity and kinetic
FTIR structural analysis, Journal of Colloid and Interface Science 214 (1999)
319–332.
[177] Y.I. Tarasevich, L.I. Monakhova, Interaction between globular proteins and silica
surfaces, Colloid Journal 64 (2002) 482–487.
[178] M.J. Read, S.L. Burkett, Asymmetric alpha-helicity loss within a peptide adsorbed
onto charged colloidal substrates, Journal of Colloid and Interface Science 261
(2003) 255–263.
[179] J.R. Lu, S. Perumal, I. Hopkinson, J.R.P. Webster, J. Penfold, W. Hwang, S.G. Zhang,
Interfacial nano-structuring of designed peptides regulated by solution pH,
Journal of the American Chemical Society 126 (2004) 8940–8947.
[180] P. Billsten, M. Wahlgren, T. Arnebrant, J. McGuire, H. Elwing, Structural-changes
of T4 lysozyme upon adsorption to silica nanoparticles measured by circulardichroism, Journal of Colloid and Interface Science 175 (1995) 77–82.
[181] L. Calzolai, F. Franchini, D. Gilliland, F. Rossi, Protein–nanoparticle interaction:
identification of the ubiquitin–gold nanoparticle interaction site, Nano Letters 10
(2010) 3101–3105.
[182] L.P. Silva, Imaging proteins with atomic force microscopy: an overview, Current
Protein & Peptide Science 6 (2005) 387–395.
[183] S. Kidoaki, T. Matsuda, Mechanistic aspects of protein/material interactions
probed by atomic force microscopy, Colloids and Surfaces. B, Biointerfaces 23
(2002) 153–163.
[184] Q.M. Yu, G. Golden, Probing the protein orientation on charged self-assembled
monolayers on gold nanohole arrays by SERS, Langmuir 23 (2007) 8659–8662.
[185] M.C. Jaurand, Mechanisms of fiber-induced genotoxicity, Environmental Health
Perspectives 105 (1997) 1073–1084.
[186] R.P.F. Schins, Mechanisms of genotoxicity of particles and fibers, Inhalation
Toxicology 14 (2002) 57–78.
[187] L. Gonzalez, D. Lison, M. Kirsch-Volders, Genotoxicity of engineered nanomaterials: a critical review, Nanotoxicology 2 (2008) 252–273.
[188] K. Donaldson, C.A. Poland, R.P.F. Schins, Possible genotoxic mechanisms of
nanoparticles: criteria for improved test strategies, Nanotoxicology 4 (2010)
414–420.
[189] J.D. Bernal, The physical basis of life, The Proceedings of the Physical Society 62
(1949) 537–558.
[190] W.H. Huang, J.P. Ferris, One-step, regioselective synthesis of up to 50-mers of
RNA oligomers by montmorillonite catalysis, Journal of the American Chemical
Society 128 (2006) 8914–8919.
[191] J.B. Swadling, P.V. Coveney, H.C. Greenwell, Clay minerals mediate folding and
regioselective interactions of RNA: a large-scale atomistic simulation study,
Journal of the American Chemical Society 132 (2010) 13750–13764.
[192] M. Franchi, E. Gallori, A surface-mediated origin of the RNA world: biogenic
activities of clay-adsorbed RNA molecules, Gene 346 (2005) 205–214.
[193] P. Mignon, P. Ugliengo, M. Sodupe, Theoretical study of the adsorption of
RNA/DNA bases on the external surfaces of Na+-montmorillonite, Journal of
Physical Chemistry C 113 (2009) 13741–13749.
[194] K.A. Melzak, C.S. Sherwood, R.F.B. Turner, C.A. Haynes, Driving forces for DNA
adsorption to silica in perchlorate solutions, Journal of Colloid and Interface
Science 181 (1996) 635–644.
[195] D.B. Warheit, C.M. Sayes, S.R. Frame, K.L. Reed, Pulmonary exposures to Sepiolite
nanoclay particulates in rats: resolution following multinucleate giant cell
formation, Toxicology Letters 192 (2010) 286–293.
[196] A.K. Sharma, B. Schmidt, H. Frandsen, N.R. Jacobsen, E.H. Larsen, M.L. Binderup,
Genotoxicity of unmodified and organo-modified montmorillonite, Mutation
Research, Genetic Toxicology and Environmental Mutagenesis 700 (2010)
18–25.
[197] P.R. Li, J.C. Wei, Y.F. Chiu, H.L. Su, F.C. Peng, J.J. Lin, Evaluation on cytotoxicity and
genotoxicity of the exfoliated silicate nanoclay, ACS Applied Materials &
Interfaces 2 (2010) 1608–1613.
I. Fenoglio et al. / Advanced Drug Delivery Reviews 63 (2011) 1186–1209
[198] S. Lordan, J.E. Kennedy, C.L. Higginbotham, Cytotoxic effects induced by
unmodified and organically modified nanoclays in the human hepatic HepG2
cell line, Journal of Applied Toxicology 31 (2011) 27–35.
[199] E. Mateo-Marti, C. Briones, C. Rogero, C. Gomez-Navarro, C. Methivier, C.M.
Pradier, J.A. Martin-Gago, Nucleic acid interactions with pyrite surfaces,
Chemical Physics 352 (2008) 11–18.
[200] K.J. Tang, H. Gan, Y. Li, L.F. Chi, T.L. Sun, H. Fuchs, Stereoselective interaction
between DNA and chiral surfaces, Journal of the American Chemical Society 130
(2008) 11284-+.
[201] D. Hanein, B. Geiger, L. Addadi, Differential adhesion of cells to enantiomorphous
crystal-surfaces, Science 263 (1994) 1413–1416.
[202] P. Alivisatos, The use of nanocrystals in biological detection, Nature Biotechnology 22 (2004) 47–52.
[203] D. Peled, R. Naaman, S.S. Daube, Packed DNA denatures on gold nanoparticles,
The Journal of Physical Chemistry. B 114 (2010) 8581–8584.
[204] A. Qureshi, W.P. Kang, J.L. Davidson, Y. Gurbuz, Review on carbon-derived, solidstate, micro and nano sensors for electrochemical sensing applications, Diamond
and Related Materials 18 (2009) 1401–1420.
[205] G. Liu, M. Swierczewska, S. Lee, X.Y. Chen, Functional nanoparticles for molecular
imaging guided gene delivery, Nano Today 5 (2010) 524–539.
[206] Y. Wang, Z.G. Li, Y. Han, L.H. Liang, A.M. Ji, Nanoparticle-based delivery system
for application of siRNA in vivo, Current Drug Metabolism 11 (2010) 182–196.
[207] G.I. Dovbeshko, O.P. Repnytska, E.D. Obraztsova, Y.V. Shtogun, DNA interaction
with single-walled carbon nanotubes: a SEIRA study, Chemical Physics Letters
372 (2003) 432–437.
[208] G.I. Dovbeshko, O.M. Fesenko, E.D. Obraztsova, K.R. Allakhverdiev, A.E. Kaja,
Conformation analysis of nucleic acids and proteins adsorbed on single-shell
carbon nanotubes, Journal of Structural Chemistry 50 (2009) 954–961.
[209] G. Dovbeshko, O. Fesenko, K. Yakovkin, S. Bertarione, A. Damin, D. Scarano, A.
Zeechina, E. Obraztsova, The poly-A interaction and interfaces with carbon
nanotubes, Molecular Crystals and Liquid Crystals 496 (2008).
[210] X.M. Tu, S. Manohar, A. Jagota, M. Zheng, DNA sequence motifs for structurespecific recognition and separation of carbon nanotubes, Nature 460 (2009)
250–253.
[211] G. Schmid, The relevance of shape and size of Au-55 clusters, Chemical Society
Reviews 37 (2008) 1909–1930.
[212] M. Pinteala, A. Dascalu, C. Ungurenasu, Binding fullerenol C-60(OH)(24) to
dsDNA, International Journal of Nanomedicine 4 (2009) 193–199.
[213] M.L. Circu, T.Y. Aw, Reactive oxygen species, cellular redox systems, and
apoptosis, Free Radical Biology & Medicine 48 (2010) 749–762.
[214] T. Finkel, N.J. Holbrook, Oxidants, oxidative stress and the biology of ageing,
Nature 408 (2000) 239–247.
[215] G. Oberdorster, A. Maynard, K. Donaldson, V. Castranova, J. Fitzpatrick, K.
Ausman, J. Carter, B. Karn, W. Kreyling, D. Lai, S. Olin, N. Monteiro-Riviere, D.
Warheit, H. Yang, Principles for characterizing the potential human health
effects from exposure to nanomaterials: elements of a screening strategy,
Particle and Fibre Toxicology 2 (2005) 8.
[216] T. Xia, M. Kovochich, J. Brant, M. Hotze, J. Sempf, T. Oberley, C. Sioutas, J.I. Yeh,
M.R. Wiesner, A.E. Nel, Comparison of the abilities of ambient and manufactured
nanoparticles to induce cellular toxicity according to an oxidative stress
paradigm, Nano Letters 6 (2006) 1794–1807.
[217] N. Li, T. Xia, A.E. Nel, The role of oxidative stress in ambient particulate matterinduced lung diseases and its implications in the toxicity of engineered
nanoparticles, Free Radical Biology & Medicine 44 (2008) 1689–1699.
[218] K.D. Welch, T.Z. Davis, M.E. Van Eden, S.D. Aust, Deleterious iron-mediated
oxidation of biomolecules, Free Radical Biology & Medicine 32 (2002) 577–583.
[219] H. Zielinski, I.S. Mudway, K.A. Berube, S. Murphy, R. Richards, F.J. Kelly, Modeling
the interactions of particulates with epithelial lining fluid antioxidants,
American Journal of Physiology. Lung Cellular and Molecular Physiology 277
(1999) L719–L726.
[220] P. Moller, N.R. Jacobsen, J.K. Folkmann, P.H. Danielsen, L. Mikkelsen, J.G.
Hemmingsen, L.K. Vesterdal, L. Forchhammer, H. Wallin, S. Loft, Role of oxidative
damage in toxicity of particulates, Free Radical Research 44 (2010) 1–46.
[221] M.A.A. Schoonen, C.A. Cohn, E. Roemer, R. Laffers, S.R. Simon, T. O'Riordan,
Mineral-induced formation of reactive oxygen species, Medical Mineraology and
Geochemistry, 2006, pp. 179–221.
[222] X.L. Shi, Y. Mao, L.N. Daniel, U. Saffiotti, N.S. Dalal, V. Vallyathan, Silica radicalinduced DNA-damage and lipid-peroxidation, Environmental Health Perspectives 102 (1994) 149–154.
[223] C.A. Cohn, R. Laffers, M.A.A. Schoonen, Using yeast RNA as a probe for generation
of hydroxyl radicals by earth materials, Environmental Science & Technology 40
(2006) 2838–2843.
[224] P.S. Gilmour, P.H. Beswick, D.M. Brown, K. Donaldson, Detection of surface free
radical activity of respirable industrial fibres using supercoiled phi X174 RF1
plasmid DNA, Carcinogenesis 16 (1995) 2973–2979.
[225] L.G. Lund, M.G. Williams, R.F. Dodson, A.E. Aust, Iron associated with asbestos
bodies is responsible for the formation of single-strand breaks in phi X174 RFI
DNA, Occupational and Environmental Medicine 51 (1994) 200–204.
[226] C. Otero-Areán, F. Barcelo, I. Fenoglio, B. Fubini, F. Xamena, M. Tomatis, Free
radical activity of natural and heat treated amphibole asbestos, Journal of
Inorganic Biochemistry 83 (2001) 211–216.
[227] M. Gulumian, F. Sardianos, T. Kilroe-Smith, G. Ockerse, Lipid peroxidation in
microsomes induced by crocidolite fibres, Chemico-Biological Interactions 44
(1983) 111–118.
1209
[228] L.G. Lund, A.E. Aust, Iron mobilization from asbestos by chelators and ascorbicacid, Archives of Biochemistry and Biophysics 278 (1990) 60–64.
[229] A.G. Agrios, P. Pichat, State of the art and perspectives on materials and
applications of photocatalysis over TiO2, Journal of Applied Electrochemistry 35
(2005) 655–663.
[230] T.L. Thompson, J.T. Yates, Surface science studies of the photoactivation of TiO2 —
new photochemical processes, Chemical Reviews 106 (2006) 4428–4453.
[231] H. Yaghoubi, N. Taghavinia, E.K. Alamdari, Self cleaning TiO2 coating on
polycarbonate: surface treatment, photocatalytic and nanomechanical properties, Surface and Coatings Technology 204 (2010) 1562–1568.
[232] N. Abidi, L. Cabrales, E. Hequet, Functionalization of a cotton fabric surface with
titania nanosols: applications for self-cleaning and UV-protection properties,
ACS Applied Materials & Interfaces 1 (2009) 2141–2146.
[233] K. Hirakawa, M. Mori, M. Yoshida, S. Oikawa, S. Kawanishi, Photo-irradiated
titanium dioxide catalyzes site specific DNA damage via generation of hydrogen
peroxide, Free Radical Research 38 (2004) 439–447.
[234] R. Dunford, A. Salinaro, L.Z. Cai, N. Serpone, S. Horikoshi, H. Hidaka, J. Knowland,
Chemical oxidation and DNA damage catalysed by inorganic sunscreen
ingredients, FEBS Letters 418 (1997) 87–90.
[235] D. Crouzier, S. Follot, E. Gentilhomme, E. Flahaut, R. Arnaud, V. Dabouis, C.
Castellarin, J.C. Debouzy, Carbon nanotubes induce inflammation but decrease
the production of reactive oxygen species in lung, Toxicology 272 (2010) 39–45.
[236] P.J. Krusic, E. Wasserman, P.N. Keizer, J.R. Morton, K.F. Preston, Radical reactions
of C60, Science 254 (1991) 1183–1185.
[237] L.L. Dugan, D.M. Turetsky, C. Du, D. Lobner, M. Wheeler, C.R. Almli, C.K.F. Shen,
T.Y. Luh, D.W. Choi, T.S. Lin, Carboxyfullerenes as neuroprotective agents,
Proceedings of the National Academy of Sciences of the United States of America
94 (1997) 9434–9439.
[238] T. Xia, L. Rome, A. Nel, Nanobiology — particles slip cell security, Nature Materials
7 (2008) 519–520.
[239] H.S. Choi, J.V. Frangioni, Nanoparticles for biomedical imaging: fundamentals of
clinical translation, Molecular Imaging 9 (2010) 291–310.
[240] K.L. Aillon, Y. Xie, N. El-Gendy, C.J. Berkland, M.L. Forrest, Effects of nanomaterial
physicochemical properties on in vivo toxicity, Advanced Drug Delivery Reviews
61 (2009) 457–466.
[241] D. Dutta, S.K. Sundaram, J.G. Teeguarden, B.J. Riley, L.S. Fifield, J.M. Jacobs, S.R.
Addleman, G.A. Kaysen, B.M. Moudgil, T.J. Weber, Adsorbed proteins influence
the biological activity and molecular targeting of nanomaterials, Toxicological
Sciences 100 (2007) 303–315.
[242] M.S. Lord, B.G. Cousins, P.J. Doherty, J.M. Whitelock, A. Simmons, R.L. Williams,
B.K. Milthorpe, The effect of silica nanoparticulate coatings on serum protein
adsorption and cellular response, Biomaterials 27 (2006) 4856–4862.
[243] M. Horie, K. Nishio, K. Fujita, S. Endoh, A. Miyauchi, Y. Saito, H. Iwahashi, K.
Yamamoto, H. Murayama, H. Nakano, N. Nanashima, E. Niki, Y. Yoshida, Protein
adsorption of ultrafine metal oxide and its influence on cytotoxicity toward
cultured cells, Chemical Research in Toxicology 22 (2009) 543–553.
[244] M.S. Ehrenberg, A.E. Friedman, J.N. Finkelstein, G. Oberdorster, J.L. McGrath, The
influence of protein adsorption on nanoparticle association with cultured
endothelial cells, Biomaterials 30 (2009) 603–610.
[245] C. Salvador-Morales, P. Townsend, E. Flahaut, C. Venien-Bryan, A. Vlandas, M.L.H.
Green, R.B. Sim, Binding of pulmonary surfactant proteins to carbon nanotubes;
potential for damage to lung immune defense mechanisms, Carbon 45 (2007)
607–617.
[246] Z.J. Deng, M. Liang, M. Monteiro, I. Toth, R.F. Minchin, Nanoparticle-induced
unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation,
Nature Nanotechnology 6 (2011) 39–44.
[247] B.S. Zolnik, A. Gonzalez-Fernandez, N. Sadrieh, M.A. Dobrovolskaia, Minireview:
nanoparticles and the immune system, Endocrinology 151 (2010) 458–465.
[248] S. Nagayama, K. Ogawara, Y. Fukuoka, K. Higaki, T. Kimura, Time-dependent
changes in opsonin amount associated on nanoparticles alter their hepatic
uptake characteristics, International Journal of Pharmaceutics 342 (2007)
215–221.
[249] L.A. Dick, A.J. Haes, R.P. Van Duyne, Distance and orientation dependence of
heterogeneous electron transfer: a surface-enhanced resonance Raman scattering study of cytochrome c bound to carboxylic acid terminated alkanethiols
adsorbed on silver electrodes, The Journal of Physical Chemistry. B 104 (2000)
11752–11762.
[250] N. Li, L.L. Ma, J. Wang, L. Zheng, J. Liu, Y.M. Duan, H.T. Liu, X.Y. Zhao, S.S. Wang, H.
Wang, F.S. Hong, Y.N. Xie, Interaction between nano-anatase TiO2 and liver DNA
from mice in vivo, Nanoscale Research Letters 5 (2010) 108–115.
[251] B. Kang, M.A. Mackey, M.A. El-Sayed, Nuclear targeting of gold nanoparticles in
cancer cells induces DNA damage, causing cytokinesis arrest and apoptosis,
Journal of the American Chemical Society 132 (2010).
[252] H.J. An, Q.D. Liu, Q.L. Ji, B. Jin, DNA binding and aggregation by carbon
nanoparticles, Biochemical and Biophysical Research Communications 393
(2010) 571–576.
[253] L.N. Daniel, Y. Mao, A.O. Williams, U. Saffiotti, Direct interaction between
crystalline silica and DNA — a proposed model for silica carcinogenesis,
Scandinavian Journal of Work, Environment & Health 21 (1995) 22–26.
[254] M. Chen, A. von Mikecz, Formation of nucleoplasmic protein aggregates impairs
nuclear function in response to SiO2 nanoparticles, Experimental Cell Research
305 (2005) 51–62.
[255] P.V. AshaRani, G.L.K. Mun, M.P. Hande, S. Valiyaveettil, Cytotoxicity and
genotoxicity of silver nanoparticles in human cells, ACS Nano 3 (2009) 279–290.