Copyright by Tingji Tang August 2006
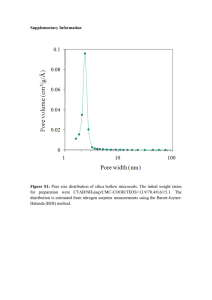
advertisement

Copyright by Tingji Tang August 2006 The Dissertation Committee for Tingji Tang certifies that this is the approved version of the following dissertation: Photophysics of Perylene Diimides in Solutions and Self-Assembled Films Committee: David A. Vanden Bout, Supervisor Peter J. Rossky Keith J. Stevenson Alan Campion Benny D. Freeman Stephen E. Webber Photophysics of Perylene Diimides in Solutions and Self-Assembled Films by Tingji Tang, B.E.; M.S. Dissertation Presented to the Faculty of the Graduate School of The University of Texas at Austin in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy The University of Texas at Austin August 2006 This work is for my wife Yanbo Wang, our son, Bryan Benshuo Tang, my daddy Jiaen Tang, Mommy Chengfeng Lu and my brother Tingdong Tang. Acknowledgements I want to express my timeless gratitude to my research supervising Professor Dr. Steve Webber, for his unselfish dedication to helping me through my studies and research. I could not even begin to quantify the aid that he has given me. Both of his moral and professional support has inspired me to be good scientist instead of a technician. I am also very grateful to Dr. Vanden Bout for his generosity serving as my official supervisor since Dr. Webber’s retirement. I really appreciate the collaboration with Professor Klaus Müllen from Max-Planck-Institute for Polymer Research for his wonderful compounds. I would like to show my appreciation to various people who helped me in many ways during my graduate school years. They are Dr. Mehdi Moini for his suggestions concerning Mass Spectrscopy techniques; Dr. Yangming Sun for SEM traning and use; Ms. Lynn Rozanski for her help in AFM measurements; Mr. Li-hsin Han for his collaboration with me on the azo-related LBL study; Dr. Ti Cao for his intellectual discussion of my research and fluorescence techniques; Dr. Jungseok Hahn and Mr. Ye Hu for helping me in various ways in the labs; I particularly want to thank my family: my parents, Jiaen Tang and Chengfeng Lu and brother, Tingdong Tang. Thank you all for bringing me up with happiness and love, which could not be more important. Lastly, by no means least, I wish to thank Yanbo Wang for her endless love, sacrifice, understanding and encouragement. Bryan Tang has become part of my life and happiness. I really enjoy the time we spent together, not only past, but now, future and forever. v Photophysics of Perylene Diimides in Solutions and Self-Assembled Films Publication No._____________ Tingji Tang, Ph.D The University of Texas at Austin, 2006 Supervisor: David A. Vanden Bout New perylene diimide based dendrimers were synthesized by the divergent synthetic method. Diamines with different lengths and methyl acrylate (or t-butyl acrylate) were used as the extension regents for the dendrimer growth steps. The photophysics of these synthesized PDIs was investigated with and without adding TFA. Intramolecular fluorescence quenching was observed by comparing the fluorescence intensity and lifetime before and after adding TFA, demonstrating a stronger quenching process for those PDIs having 2 or 3 carbons between the two nitrogens of the diamines. A 6 or 7 member ring mechanism for intramolecular electron transfer was presented. Photophysics of new water soluble perylene diimide, anionic n-PDI, CAS [694438-88-5] and cationic p-PDI, CAS [817207-4-7], was investigated in different media. The absorption spectra and quantum yield were modified by the presence of these reagents in a manner that suggests that weakly interacting complexes (such as H- or Jaggregates) are present in pure water and that complex formation can be enhanced by vi adding NaCl and polyelectrolytes or diminished by interacting with surfactants such as SDS and DTAC. A molecular layer-by-layer (MLBL) deposition process has been carried out for n-PDI and p-PDI through the strong π−π and electrostatic interactions. The fluorescence of these films shows an alternation of intensity according to which perylene species is on the outer layer, which is interpreted as the effect of facile energy transfer between the perylenes. Energy transfer in these films was also characterized by studying fluorescence quenching produced by the deposition of a single energy trapping layer (n-TDI, CAS[862852-56-0]). The MLBL technique can be utilized for other multiply-charged water soluble conjugated dye molecules such as n-PSA (CAS[59572-10-0]) and n-TDI, thereby demonstrating considerable potential for making organic semiconductor films. Polyelectrolyte films containing n-PDI and p-PDI were prepared from PSS and PDAC using the layer-by-layer (LBL) methodology. The optical density and fluorescence intensity of the LBL films grows linearly with the number of layers and we do not observe extraction of the PDI by subsequent polyelectrolyte deposition in the presence of 0.5M NaCl. The PDI fluorescence is substantially quenched in these films which we interpret as a self-quenching effect, enhanced by inter- and intra-layer energy transfer. vii Table of Contents List of Tables……………………………………………………………………..…….xiii Lsit of Figures……………………………………………………………………..........xiv Chapter 1 Introduction………………………………………………………………….1 1.1 Peryelne Diimides…………………………………………………………1 1.1.1 Synthesis of Perylene Diimides…………………………………………..2 1.1.2 Applications of PDIs in Nanoscience and Nanotechnology……………...3 1.1.2.1 Organic Thin Film Transistors………………………………………5 1.1.2.2 Organic Light Emitting Diodes……………………………………...6 1.1.2.3 Photovoltaics………………………………………………………...7 1.1.2.4 Photoharvesting Arrays based on PDI dyes…………………………8 1.1.2.5 Supramolecular Self-Assembled Nanostructures……………………9 1.2 Layer-by-Layer Self-Assembly (LBL)………………………………………...10 1.3 References……………………………………………………………………..12 Chapter 2 Synthesis and Photophysics Study of 3, 4, 9, 10-Perylene Tetracarboxylic Diimide Dendrimers…………………………………………22 2.1 Introduction………………………………………………………………..…..22 2.1.1 General Introduction…………………………………………………..….22 2.1.2 Nomenclature…………………………………………………………..…24 2.2 Experimental Section……………………………………………….................26 2.2.1 Materials……………………………………………………………..…...26 2.2.2 Instruments and Measurements………………………………………..….27 2.2.3 Synthesis of PDIs…………………………………………………………27 2.2.3.1 Synthesis of PDI dendrimers by Divergent method……………........27 2.3.1.1.1 Condensation of PDA with diamines…………………………...28 2.3.1.1.2 Michael Addition of PDI (0.0) with acrylates………………..…29 2.2.3.2 Synthesis of other PDIs by one step condensation…………………..30 2.3 Results and Discussions……………………………………………………….31 2.3.1 Absorption and Emission spectra of PDIs………………………………..32 viii 2.3.1.1 Molar Extinction Coefficient for PDIs…………………………..…..32 2.3.1.2 Emission Spectra of PDIs and Quantum Yield measurements…........33 2.3.2 Lifetime measurements for PDIs………………………………………….35 2.3.3 Photophysical behaviors of PDIs upon adding TFA…………...................35 2.3.3.1 Protocol for studies of the effect of added TFA…………………...…36 2.3.3.2 Photophysical properties of PDI compounds in the presence of TFA.36 2.3.3.2.1 Effect of TFA on the absorption spectra of PDIs……………...36 2.3.3.2.2 Effect of TFA on the fluorescence spectra of PDIs……………37 2.3.3.2.3 Lifetimes of PDIs in the presence of TFA……………………..39 2.4 Summary…………………………....................................................................39 2.5 References……………………………………………………………………..40 Chapter 3 Solution Photophysics of Water Soluble Perylene Diimides…………….68 3.1 Introduction……………………………………………………………………68 3.2 Experimental Section………………………………………………………….69 3.2.1 Materials………………………………………………………………….69 3.2.2 Instruments and Measurements…………………………………………...69 3.3 Results and Discussion……………………………………………...................70 3.3.1 Absorption and Emission Spectra of p-PDI and n-PDI in Solutions……..70 3.3.2 Photophysics of n-PDI and p-PDI in Surfactant Solutions.........................71 3.3.2.1 Photophysics Study upon complexing between PDI and appositively charged surfactant (n-PDI/DTAC or p-PDI/SDS)……………………..74 3.3.2.2 Photophysics Study upon complexing between PDI and same charged surfactant………………………………………………………………75 3.3.2.3 Photophysics Study upon Mixing the PDI/Surfactant complexes…….77 3.3.2.4 Photophysics study of n-PDI titrated by DTAC…………....................77 3.3.3 Photophysics study of n-TDI upon complexing with DTAC surfactant solutions…………………………………………………………………….78 3.3.4 Effect of NaCl on the n-PDI and p-PDI Solutions…………………………78 3.3.5 Effect of PSS-Na+, PDAC+Cl- Polyelectrolytes on n-PDI and p-PDI Solutions………………………………………..........................................79 ix 3.3.6 Molecular Dynamics Simulation of p-PDI Dimer…………………………81 3.4 Summary………………………………………………………………............82 3.5 References……………………………………………………………………..83 Chapter 4 Molecular Layer-by-layer Self-assembly of Water Soluble Dyes through the π-π and Electrostatic Interactions…………………...............103 4.1 Introduction…………………………………………......................................103 4.2 Experimental Section……………………………………………...................104 4.2.1 Materials……………………………………………………………........104 4.2.2 Molecular Layer-by-Layer procedures…………………………………..105 4.2.3 Instruments and measurements…………………………………………..105 4.3 Results and Discussions………………………………...................................106 4.3.1 Molecular Layer-by-Layer of p-PDI and n-PDI………………………....106 4.3.1.1 Absorption spectra of (p-PDI/n-PDI)n……………………………....106 4.3.1.2 Characterization of (p-PDI/n-PDI)n…………………………….…...107 4.3.1.3 Emission spectra of (p-PDI/n-PDI)n………………………………...108 4.3.2 Molecular Layer-by-Layer of p-PDI and n-PSA………………………...109 4.3.2.1 Absorption spectra of (p-PDI/n-PSA)n……………………………...110 4.3.2.2 Emission spectra of (p-PDI/n-PSA)n………………………………..110 4.3.3 Molecular Layer-by-Layer of (p-PDI/n-TDI)n…………………………..111 4.3.3.1 Absorption spectra of (p-PDI/n-TDI)n………………………………111 4.3.3.2 Emission spectra of (p-PDI/n-TDI)n…………………………...........112 4.3.4 Molecular Layer-by-Layer of (p-PDI/(n-TDI:n-PSA))n…………………112 4.3.5 Energy Transfer for TDI/(p-PDI/n-PDI)n..................................................113 4.3.5.1 Absorption spectra for n-TDI/(p-PDI/n-PDI)n………………….......114 4.3.5.2 Energy Transfer for n-TDI/(p-PDI/n-PDI)n........................................114 4.4 Summary……………………………………..................................................116 4.5 References……………………………………………………………………118 Chapter 5 Incorporation of Water Soluble Perylene Diimides into Poylelectrolyte Films by the Layer-by-Layer Method………………….133 5.1 Introduction…………………………………………………………………..133 x 5.2 Experimental Section………………………………………………………...134 5.2.1 Materials…………………………………………………………………134 5.2.2 Layer-by-Layer Procedures……………………………………………...134 5.2.3 Spin-Assisted Layer-by-Layer (SA-LBL)……………………………….135 5.2.4 UV-Vis and Fluorescence Spectroscopy Measurements……...................135 5.2.5 Atomic Force Microscopy (AFM) Measurements…………....................136 5.2.6 Förster Resonance Energy Transfer (FRET) Study……………………..136 5.3 Results and Discussions…………………………………………...................137 5.3.1 Dye Penetration into PEI/(PSS/PDAC)n LBL Films……………………137 5.3.2 LBL of n-PDI or p-PDI with PSS and PDAC…………………………...138 5.3.2.1 LBL with 0.5M NaCl in Polyelectrolyte Solution……………...…..138 5.3.2.2 LBL with no NaCl in Polyelectrolyte Solution……………………..141 5.3.2.3 Characterization of p-PDI or n-PDI in the LBL films………………143 5.3.3 Energy Transfer Study Between PDI films and BG……………………..144 5.4 Summary……………………..........................................................................146 5.5 References……………………………………………………………………148 Chapter 6 Some Preliminary Results and Suggestion for Future Work…………..166 6.1 Synthesis of n-type and p-type PDI materials………………………………..166 6.1.1 Introduction……………………………………………………………...167 6.1.2 Synthesis…………………………………………………………………167 6.1.2.1 Bromination of Perylene Dianhydride(PDA)……………………..167 6.1.2.2 Imidization of crude Bromination Product………………………..168 6.1.3 Photophysics of the synthesized PDIs………….......................................169 6.1.4. Future work……………………………………………………………..170 6.2 Self-assembly of n-type semiconductor PDIs………………………………..170 6.2.1 Introduction…….......................................................................................170 6.2.2 Self-assembly of PDIs…….......................................................................171 6.2.2.1 Chemical structures of PDIs………………………………………...171 6.2.2.2 Solution based Self-assemblying……………………………………171 6.2.3 Future of Self-assembly of PDIs………………………………………...173 xi 6.3 References……………………………………………………………………173 Bibliography……………………………………………………………...……………185 Vita……………………………………………………………………………………..187 xii List of Tables Table 2.1 Characterization of synthesized PDIs………………………………………....66 Table 2.2 Spectroscopic characteristic of PDI compounds with no TFA added…….......67 Table 2.3 Spectroscopic characteristics of PDI compounds with TFA added…………..68 Table 3.1 Photophysical parameters for n-PDI and p-PDI in different solutions………102 Table 5.1 FRET parameters for D-A pairs of p-PDI, n-PDI and BG…………………..164 Table 5.2 Characteristics of UV-Vis and fluorescence spectra of p-PDI and n-PDI LBL films with 1, 3, and 5 SLs with and without using NaCl……………………………….165 xiii List of Figures Figure 1.1 Chemical Structures of Perylene and Peryelne derivatives………………....18 Figure 1.2 HOMO (top) and LUMO(bottom) of perylene diimides…………………....19 Figure 1.3 Procedure of synthesizing non symmetric PDIs……………………………..20 Figure 1.4 Schematic representation of Layer-by-Layer deposition process……………21 Figure 2.1 Schematic representation of Divergent and Covergent approach for dendrimer synthesis………………………………………………………………………………….41 Figure 2.2 Scheme for the PDI compounds synthesis…………………………………...42 Figure 2.3 Mass spectrum of PDI(0.0)-16 by CI+………………………………………..43 Figure 2.4 Mass Spectra of PDI0.5MA-12 by CI+ and PDI0.5MA-13 by CI-…………...44 Figure 2.5 Mass Spectra of PDI0.5MA-2213 by CI (a) and PDI0.5tBA-12 by CI+ (b)....45 Figure 2.6 Mass Spectra of PDI0.5MA-14 (a) and PDI0.5MA-17(b) by CI-……………46 Figure 2.7 Mass Spectra of PDI0.5MA-112 by CI+……………………………………..47 Figure 2.8 Mass Spectra of PDI-DBPA (a) and PDI-18 (b) by CI+……………………..48 Figure 2.9 Chemical Structures of PDI compounds…………………………………….49 Figure 2.10 (a) Absorption spectra of PDI0.5MA-12 as a function of concentration. (b) Absorbance at 526nm (●) and 490nm (▲) as a function of concentration for deriving the molar extinction coefficient showing as the y value in the figure………………………51 Figure 2.11 (a) and (b) Normalized absorption (solid line) and emission (dashed line) spectra for PDI0.5MA-112 and PDI-DBPA, respectively………………………………52 xiv Figure 2.12 (a) Fluorescence quenching by the electron transfer mechanism from tertiary amine. (b) Fluorescence electron transfer quenching mechanism suppressed by combining with TFA…………………………………………………………………….53 Figure 2.13 6-member ring and 7-member ring quenching mechanism by electron transfer from nitrogen to carbonyl group for PDI0.5-12, PDI0.5MA-13 and PDI0.5MA-2213, respectively………………………………………………………………………………54 Figure 2.14 Fluorescence decay curve for PDI0.5MA-2214 before and after adding TFA. Black: No TFA added, Red: adding TFA at [TFA]/[PDI]=106………………………….55 Figure 2.15 Absorption spectra responding to the addition of TFA for different PDI compounds……………………………………………………………………………….56 Figure 2.16 Normalized emission spectra of PDI compounds at different [TFA]/[PDI] ratios……………………………………………………………………………………...57 Figure 2.17 Ratio of fluorescence intensity to the absorbance at 490nm as a function of Log ([TFA]/[PDI]). The red triangle represents the ratio where no TFA was added……62 Figure 2.18 The protonation of tertiary amine by TFA………………………………….63 Figure 3.1 Chemical Structures of n-PDI, p-PDI and n-TDI…………………………….87 Figure 3.2 Synthetic Scheme for n-PDI and p-PDI……………………………………...88 Figure 3.3 (a) and (b) Absorption and emission spectra of 1.5 x 10-6 M n-PDI and p-PDI in water (solid line) and DMSO (dotted line), respectively……………………………..89 Figure 3.4 Absorption (left) and fluorescence emission (right) spectra in water for n-PDI (black line), p-PDI (dark blue line), p-PDI/n-PDI mixed in a 1:1 ratio, (red line) and the sum of the p-PDI and n-PDI spectra (light blue line)…………………………………..90 xv Figure 3.5 Absorption spectra of n-PDI with DTAC (a) and p-PDI with SDS (b) at different concentrations. …………………………………………..…………………….91 Figure 3.6 Fluorescence emission spectra of n-PDI (a) and p-PDI (b) mixing with SDS and DTAC surfactant solutions at different concentrations, respectively……………….92 Figure 3.7 (a) and (b) Absorption and emission spectra of n-PDI upon mixing with SDS solutions at different concentrations. Ex=540nm. ………………………………………93 Figure 3.8 (a) and (b) Absorption and emission spectra of p-PDI upon mixing with DTAC solutions at different concentrations. Ex=540nm. ………………………………94 Figure 3.9 (a) Absorption and emission spectra of p-PDI/SDS mixing with n-PDI/SDS at [SDS]=10mM (Red) and the average absorption spectrum of individual p-PDI/SDS and n-PDI/SDS at [SDS]=10mM(black). (b) Fluorescence intensity upon mixing p-PDI/SDS and n-PDI/SDS (red circle) and averaging the individual p-PDI/SDS and n-PDI/SDS (black triangle) as a function of SDS concentration……………………………………..95 Figure 3.10 (a) and (b) Absorption and emission spectra of n-PDI titrated by concentrated DTAC DMF solution. …..…………………………………………………96 Figure 3.11 (a) and (b) Absorption and emission spectra of n-TDI upon mixing with DTAC solutions at different concentrations. Ex=640nm. ………………………………97 Figure 3.12 (a) Absorption spectra of n-PDI and (b) p-PDI as a function of NaCl concentration, with insets showing the S-V fluorescence quenching curves……………98 Figure 3.13 Absorption spectra for n-PDI (a) and p-PDI (b) with the addition of PDAC and PSS solutions.………………………………………………………………..………99 Figure 3.14 (a) Absorption and fluorescence spectra of n-PDI before (black) and after (red) mixing with 1 mg/ml PDAC solution, demonstrating fluorescence quenching under xvi these conditions (b) Absorption and fluorescence spectra of p-PDI before (black) and after (red) mixing with 1 mg/ml PSS solution, demonstrating fluorescence enhancement under these conditions…………………………………………………………………..100 Figure 3.15 MD structure for p-PDI dimer in pure water………………………………101 Figure 4.1 (a) UV-Vis absorption spectra of the monolayer of p-PDI (Black) and n-PDI (Red). (b) The peak absorbance around 575 nm as a function of monolayer PDI with pPDI as the first layer. (c) The peak absorbance around 284nm as a function of monolayer PDI with p-PDI as the first layer……………………………………………………….120 Figure 4.2 The absorption spectra of p-PDI dipping cycles up to 10. ……..…………..121 Figure 4.3 (a) The TM-AFM image of (p-PDI/n-PDI)10 annealed at 80ºC overnight. (b) Ellipsometric film thickness as a function of PDI monolayers………………………...122 Figure 4.4 (a) Fluorescence spectra from the monolayer of p-PDI/n-PDI with p-PDI as the first layer (b) Fluorescence intensity as a function of double layer p-PDI/n-PDI….123 Figure 4.5 (a) Absorption and emission spectra of n-PSA in water with the inset showing n-PSA’s chemical structure. Ex=360nm. (b) Absorption spectra of monolayer of p-PDI and n-PSA starting with p-PDI layer. (c) Peak absorbance as a function of double layer (p-PDI/n-PSA) for p-PDI at 575nm (circles) and n-PSA at 379nm (diamonds) after correcting from the absorbance at 379nm for p-PDI…………………………………...124 Figure 4.6 (a) Emission spectra of 1st p-PDI (Red) monolayer and (p-PDI/n-PSA)1 (Pink). Black curve is the subtraction of 1st p-PDI and (p-PDI/n-PSA)1. Ex=360nm. (b) Emission spectra of (p-PDI/n-PSA)n-1/p-PDI with Ex=540nm. (c) Electron transfer mechanism from the HOMO of PSA to p-PDI exciton……………………………………………..125 xvii Figure 4.7 (a) UV-Vis absorption spectra of the monolayer of p-PDI (Black) and n-TDI (Red). (b) The peak absorbance around 575 nm for p-PDI (black circles) and 700nm for n-TDI (red diamonds) as a function of double layers. (c) The peak absorbance around 284nm as a function of deposition layers with p-PDI as the first layer………………...126 Figure 4.8 Emission spectra of (p-PDI/n-TDI)n with Ex=540nm………………………127 Figure 4.9 (a) UV-Vis absorption spectra of the monolayer of p-PDI (Black) and nTDI:n-PSA (Red). (b) The peak absorbance around 575 nm for p-PDI (black circles) and 700nm for n-TDI (red diamonds) as a function of the number of double layers……….128 Figure 4.10 Emission spectrum of (p-PDI/n-PDI)10(Red) and absorption spectrum of nTDI (Green)…………………………………………………………………………….129 Figure 4.11 (a) UV-Vis absorption spectra of n-TDI/(p-PDI/n-PDI)n. Green: n-TDI, Black: p-PDI and Red: n-PDI. (b) Absorption spectra of n-TDI/(p-PDI/n-PDI)n upon subtracting the absorption spectrum from n-TDI foundation layer. Black: p-PDI and Red: n-PDI. (c) The peak absorbance around 579 nm as a function of monolayer PDI with pPDI as the first layer. …………………………………………………………………..130 Figure 4.12 (a) Schematic representation of deriving the average distance from (p-PDI/nPDI)4/p-PDI to the center of n-TDI layer. (b) Fluorescence emission intensity for (pPDI/n-PDI)n and n-TDI/(p-PDI/n-PDI)n as a function of PDI double layers. (c) Plot of Log[(I0/Id)-1] versus Log(d), yielding a slope of 2.02 and intercept 3.61……………...131 Figure 4.13 (a) Energy transfer efficiency as a function of number of p-PDI layers. (b) pPDI fluorescence dependence as a function of number of p-PDI layers……………….132 Figure 5.1 (a) Absorbance at 580nm and (b) fluorescence intensity of n-PDI as a function of dipping time for “precursor” LBL films of PEI/(PSS/PDAC)n (n=1, 2, and 3)……..133 xviii Figure 5.2 (a) and (b) Absorption and fluorescence emission spectra of n-PDI dipping into the preassembled PEI/(PSS/PDAC)6 as a function of immersing time……………151 Figure 5.3 (a) Absorbance at 587nm and (b) fluorescence intensity of p-PDI as a function of dipping time for “precursor” LBL films of PEI/(PSS/PDAC)n-1/PSS……………….152 Figure 5.4 (a) and (b) Absorption spectra of n-PDI and p-PDI layer-by-layer assembly with PSS and PDAC solutions made in 0.5 M NaCl and 3 SLs, respectively………….153 Figure 5.5 (a) and (b) Fluorescence emission spectra of n-PDI and p-PDI with PSS and PDAC (0.5M NaCl) LBL films………………………………………………………...154 Figure 5.6 (a) Fluorescence intensity as a function of sequential deposition of polyelectrolyte layers following the third p-PDI deposition (b) Fluorescence intensity as a function of sequential deposition of polyelectrolyte layers following the third n-PDI deposition………………………………………………………………………………155 Figure 5.7 (a) and (b) Absorption spectra of n-PDI and p-PDI with PSS and PDAC (3SLs) in the LBL films (with no NaCl in the polyelectrolyte solution)………………156 Figure 5.8 (a) and (b) fluorescence spectra for n-PDI and p-PDI……………………...157 Figure 5.9 Absorption vs. the number of n-PDI layers deposited (●) and the normalized fluorescence intensity (■) for each layer. The number of separation layers are indicated between each n-PDI deposition (SL=1, 3, 5, 7, 9)…………………………………….158 Figure 5.10 TM-AFM angle view images of (a) before p-PDI deposition and (b) after pPDI depositions………………………………………………………………………...159 Figure 5.11 Schematic representation of p-PDI in LBL films with 3SLs along with showing the intralayer and interlayer FRET process………………………………….160 xix Figure 5.12 (a) and (b) Energy transfer efficiency as a function of spin-casting BG concentration for 4 n-PDI and p-PDI LBL films with 3SLs……………………………161 Figure 5.13 (a) Energy transfer efficiency as a function of number of n-PDI layers at BG concentration of 1.3x10-3M. (b) Energy transfer efficiency as a function of number of p- PDI layers at BG concentration of 8.3x10-6M………………………………………….162 Figure 5.14 Schematic representation of BG distribution in p-PDI and n-PDI LBL films with 3SLs……………………………………………………………………………….163 Figure 6.1 Synthetic routes to Compounds PDI-NP1, PDI-NP2, and PDI-PN9……….176 Figure 6.2 Mass Spectrum of PDI-NP1 by CI+ (a) and H-NMR in CDCl3 (b)………...177 Figure 6.3 Mass Spectrum of PDI-NP2 by CI- (a) and H-NMR in CDCl3 (b)…………178 Figure 6.4. Mass Spectrum of PDI-PN9 by CI+………………………………………...179 Figure 6.5 Chemical Structures of Synthesized PDIs…………………………………..180 Figure 6.6 Normalized absorption (a) and emission spectra (b and c) of PDI-NP1 (Red), PDI-NP-2(pink) and PDI-PN9 (Blue) in DCE………………………………………….181 Figure 6.7 SEM images of PDI-DEAPA……………………………………………….182 Figure 6.8 Large area SEM images of PDI-DMPA…………………………………….183 Figure 6.9 SEMs images of PDI-DMEA……………………………………………….184 xx Chapter 1 Introduction 1.1 Perylene Diimides (PDIs) Perylene is a conjugated planar molecule with 5 fused phenyl rings leading to very strong intermolecular π-π interactions (see Figure 1.1a). Perylene dyes were first discovered by Kardos in 1913 and have been used for a variety of applications as pigments, including textiles, automobiles and general paints and plastics. Because of their poor solubility due to strong π-π stacking interactions, the highly fluorescent nature of perylenes diimides was not recognized until 1959. Since then, a variety of synthetic schemes has been applied to modify perylene and perylene derivatives in order to improve their solubilities and processibilities. 1 However, the synthesis of perylene derivatives, such as perylene tetracarboxylic acid diimide (in short perylene diimide, PDI), was not very successful until the discovery of 3,4,9,10-perylne tetracarboxylic dianhydride (PDA) as shown in Figure 1.1b. Since then, PDIs have received considerable attention in academic as well as industrial dye and pigment research. 2 Perylene diimides were initially applied for industrial purposes as red vat dyes. After 1950, some PDIs were found to be high grade pigments (especially in automotive finishes) due to their favorable combination of insolubility and migrational stability, light- and weather-fastness, thermal stability and chemical inertness as well as high tinctorial strength with hues ranging from red to violet, and even black shades. 3 Nowadays, PDIs have become very promising organic semiconductor materials for various applications in novel high technologies such as photoconductors, filed effect transistors, or photovoltaic devices. 1 1.1.1 Synthesis of Perylene Diimides (PDIs) From the starting material of PDA, two synthetic routes have proved to be successful for making perylene diimides (PDIs) derivatives (see Figure 1.1c) with high solubility and functionality. Approach 1, discovered by Langhals et al in 1959, is to introduce long chain alkyl groups at the imide nitrogen (head and tail positions) which can dramatically increase the solubility of PDIs (see Figure 1.1c ). PDIs obtained by this approach exhibit essentially indistinguishable absorption and emission properties because the nodes in the HOMO and LUMO at the imide nitrogen reduce the coupling between the perylene core and imide substitutents to a minimum, as shown in Figure 1. 2. 4 In other words, PDIs can be regarded as a closed chromophoric system with an S0-S1 transition (polarized along the long molecular axis) whose intensity and position remain unaltered by the specific imide substituents. Therefore, introducing long alkyl chains at the head and tail positions does not affect the conjugation plane of the perylene core, so that substituted PDIs usually have a high fluorescence yield and small Stokes Shift along with well-resolved vibrational structures in both the absorption and emission spectra. However, the ability to increase PDI’s solubility is limited due to the very strong π-π interactions in solutions and solid films. Würthner et al at Universität Würzburg have shown great success in synthesizing new functional materials via receptor substitutions at the head and tail positions, which are then used as supermolecular assembly blocks. 5 The second more elaborate synthetic strategy was to introduce substituents in the carbocyclic scaffold in the so called bay-area (positions such 1, 6, 7, 12 in Figure 1.1c). This approach was first discovered by Seybold and coworkers at BASF. They successfully incorporated four phenoxy groups in high yields by nucleophilic displacement of chlorine 2 constituents, although the fourfold chlorination of PDA was contaminated by the threeand five- fold chlorination products. 6 Other nucleophilic displacements were not successful for PDA chlorides. In 1997, di-substituent PDI derivatives were discovered with the di-bromination of PDA. 7 However, the product mixture obtained by bromination was even worse than in the case of chlorination because three-fold bromination products and significant amounts of a second dibromo regioisomer are formed which are only detectable in high field (>400MHz) H-NMR spectroscopy. Only very recently was it discovered that the di-bromination of PDA was achieved in high yields(>70%) with the addition of iodine at 80°C. 8 Even four-fold bromination of PDA was obtained in a reasonable yield (30%) by adding additional iodine during reflux and increasing the temperature and reaction time. 9 Gratifyingly, the displacement of bromine substituents in PDA is very straightforward. Therefore, carbon, cyano, oxygen, oligothiophene and nitrogen nucleophiles could be coupled to perylene core leading to novel perylene diimides with unique properties. 10 In this case, the two naphthalene planes that make up the perylene structure were twisted about 20-30 º depending on the size and number of substituent groups. 5 Because of the easy control of the substituent at the 1, 6, 7, and 12 positions, tetra- or di-substituted materials have been widely synthesized. The introduction of polyphenylene dendrons at bay areas was systematically carried out by Müllen et al at MPI. 11 The higher generation of polyphenylene dendrons leads to diminished aggregation of PDIs in solutions and films due to their steric hindrance. These dendrons can efficiently block the formation of PDI excimers and exciplexes. Instead of small substituents at the bay positions, some star polymers with perylene as the core have been synthesized and provide some unique mechanical 3 properties. 12 Therefore, bay-substitution is very versatile approach to control PDI chemical structures. However, in all these cases, the absorption and emission spectra were broadened and Stokes shift increases along with the decrease of quantum yield. However, the solubility can be dramatically increased upon adding larger substituent groups in the bay positions, due to the diminished π-π interactions. While synthesizing symmetric PDIs which have the same groups at the nitrogen positions is relatively straightforward by reaction with an excess of primary amine, nonsymmetric PDIs with different groups at the imide positions can not be prepared by a stepwise condensation of primary amines with PDA. In some cases, a simultaneous condensation of PDA with a mixture of primary amines has been carried out, but the reactivity of the two amines must be similar and the reaction mixtures so obtained are difficult to separate. Nagao, Misono, and coworkers have found a synthetic route by a partial acid saponification of symmetric PDIs in conc. sulfuric acid at 180-200°C. 13 Simple aliphatic substituents R=methyl, ethyl, and butyl have been used. Problems occur with aromatic substituents which can be sulfonated. An elegant synthesis has been found by Tröster, which is shown in Figure 1. 3. 14 The insoluble PDA is converted to the tetra potassium salt 2 which is readily soluble in water. The monoanhydride monopotassium salt 3 is precipitated by a moderate acidification with orthophosphoric acid. The procedure can be further optimized by the application of acetic acid, 15 because traces of the latter can be more easily removed. The driving force for the formation of 3 is its extraordinarily high lattice energy-the substance is absolutely insoluble in any solvent, even at high temperature-which removes it from the protonation equilibria. 3 can be condensed to nonsymmetric PDIs with primary amines in water. The necessity of an 4 aqueous medium, however, limits the scope of the condensation reaction. The yield is very high for short chain water soluble amines, moderate with less soluble amines and low with highly hydrophobic amines. However, the condensation of the latter is important for the preparation of PDIs with a high solubility in organic solvents. 1 1.1.2 Applications of PDIs in Nanoscience and Nanotechnology Molecules of PDIs form a unique class of n-type organic semiconductors exhibiting relatively high electron affinity, brilliant colors, strong absorption and fluorescence, and outstanding chemical, thermal and photochemical stability, in comparison with the more common p-type counterpart in organic semiconductors. Bulk phase materials have been widely used in various optoelectronic devices. Typical examples include organic thin film transistors, photovoltaics, and organic light emitting diodes (OLED). Due to their high thermal and photostability, PDIs have also attracted increasing interest in fabrication of single molecule devices such as fluorescence switches, sensors, molecular wires and transistors. Besides their applications as semiconductors, they have been widely used in building blocks for making supramolecular assemblies, nanobelts, and even photoharvesting systems through their strong π-π intermolecular interactions. 1.1.2.1 Organic Thin Film Transistors (OTFTs) Field-effect transistors (FET) devices, which can be prepared by both bottom-up and top down methodologies, have been fabricated with thin films of perylene on SiO2 and polyimide gate insulators and p-channel FET properties have been found in both FET devices, like the FET properties of pentacene and oligothiophenes. The hole mobility was measured to be on the order of ~10-3 cm2V-1S-1 and current on/off ratios >105 were 5 obtained. 16 The highest hole carrier mobility achieved in p-type OTFTs was 1.5 cm2V-1S1 for pentacene transistors, which implies band transport of hole carriers. 17 Therefore, the OTFTs based on n-type materials, in which electrons are the majority carrier, are also desired since they will enable the fabrication of complimentary circuits. The “bottom-up” approach requires the deposition of organic thin films on the pre-made substrates with gate, source and drain electrodes on it while the " top-down” approach is to deposit the organic thin films onto the gate substrate and then source and drain electrodes are coated on top of the organic films. Malenfant et al. reported that the OTFTs of N,N’-dioctyl3,4,9,10-perylene diimide (PDI-C8) can reach an electron mobility up to 0.6 cm2V-1S-1 and current on/off ratios >105 using the bottom-up configuration. 18 Using Top-down configuration, electron mobility of PDI-C8 OFETs made under higher vacuum (10-7 Torr) can reach to 1.7 cm2V-1S-1 with on/off ratios around 107, agreeing with the trap-limited band transport. 19 1.1.2.2 Organic Light Emitting Diodes (OLEDs) Due to their high fluorescence quantum yield and excellent chemical and thermal stability, PDIs are considered to very good materials for making OLEDs as the emitting/electron transporting layer in OLED fabrications. For example, Jiang et al in 1997 reported the multilayered N, N’-bis(naphthyl)-3,4,9,10-perylene diimides based OLED showing a maximum brightness of 26 cd/m2. 20 However, it exhibited self quenching and strong interaction between the PDI layer and cathode metals. Since then, the application of PDIs in OLEDs has suffered from two problems which reduce their utility in such devices. First, many PDIs have low solubility, and therefore the preparation of thin films layers has to be carried out by thermal vapor deposition, which 6 is very disadvantageous for fabricating large area films compared to spin-coating due to its higher cost. Second, in the thin film solid state, PDIs have a very strong tendency to form aggregates and even crystals, originating from the strong π-π intermolecular interactions, which can lead to the decrease of emission efficiency due to the enhancement of nonradiative decay pathways or red shifts in the emission. J. Qu, et al. proposed an attractive way to solve both these problems simultaneously by incorporating the PDI core within a polyphenylene dendrimer (one and second generation respectively) shield (referred to as dendronized PDIs). 11 Polyphenylene dendrimers have the advantage of being rigid and shape persistent and have been shown to have high chemical and thermal stability. By connecting the polyphenylene dendrons of different generations to a PDI core, the size of the resulting nanostructure can be controlled, and the PDI core is separated from the surrounding media. The incorporation of polyphenylene dendrimers at the bay positions is more efficient in shielding the π-π interactions especially at the solid film states, also possessing excellent film formation properties. These dendronized PDIs showed strong red-orange photoluminescence. However, single layer OLEDs of dendronized PDIs showed low efficiencies and brightness and current density (0.14cd/m2 and 3.66mA/cm2 at 10V), especially with the second generation dendrimers, which is attributed to the poor charge transport, presumably because the perylene diimide cores are now too far apart. 11 1.1.2.3 Photovoltaics Organic photovoltaic devices are potentially one of the most important emerging new technologies, since the harvesting of energy by solar cells promises to be of increasing necessity as our reserves of fossil fuels dwindle. The most widely used design 7 of organic solar cells are so-called bulk-heterojunction devices in which the active layer consists of a blend of an electron-donating(p-type) and an electron-accepting (n-type) material. Soluble polythiophenes and polyphenylenevinylene are widely used as the ptype materials. Employing PDIs as n-type materials in photovoltaics has gained much attention due to their high electron affinity, large molar absorption coefficients and possible generation of a highly conducting direction along the π-π stacking axis, in addition to their other merits such as chemical robustness, thermal stability and low cost. Tang C.W. at Kodak did the pioneering work of using PDIs as the active layer in solar cells. 21 Solar cells fabricated with the bulk heterojunction from poly(2,7-carbazole) as the p-type materials and PDI as the n-type materials showed a high external quantum efficiency (EQE) of 16% at 490nm, FF around 0.4 and a power conversion efficiency of 0.6% illuminated with solar light. 22 The introduction of an electron donating group such as pyrolidine at the bay positions of PDIs can lead to a 160nm bathochromic shift with emission in the infrared region, which results in increasing the open circuit voltage (Voc) and hence the current density due to the improved absorption overlap with the solar. 23 1.1.2.4 Photoharvesting Arrays based on PDI dyes The development of efficient artificial systems for solar energy conversion is important for sustainable energy utilization. Natural photosynthesis relies upon monovalent interactions between similar chromophores to regulate energy and electron flow. The π-π stacking in both solutions and the solid state can lead to the strong electron wavefunction overlap between the adjacent PDIs, facilitating the energy and electron flow upon photoexcitation. PDI based light harvesting arrays have been extensively studied by Wasielewski in the last decade. 24 8 Hippius et al. have recently synthesized a calix[4]arene-based light harvesting arrays containing up to 5 PDI dyes (emitting in orange, violet and green colors) in a cofacial arrangement. Efficient energy transfer could be observed even from initially nonfluorescent PDI to the green PDI moiety in the molecule (fluorescence was quenched by photoinduced electron transfer). 25 1.1.2.5 Supramolecular Self-Assembled Nanostructures Through precise molecular design one can obtain the spatial organization of PDIs by molecular-recognition-directed self assembly. Such self-organization of functional dyes should be of great utility to tailor defined multichromophoric objects or bulk solid state materials with novel optical and electronic functionalities. π-π interactions, metalligand coordination, hydrogen-bonding and even ionic interaction can be combined to direct the self-assembly of the individual PDI blocks in a defined manner. Würthner et al. have done much of the research in the supramolecular chemistry in both solution and solid state for the PDIs. 5 Although some organized structures (e.g., particles, liquid crystals, networks) have been fabricated, 26 the 1D self-assembly (e.g. nanowires) of PDI molecules remains challenging. Very recently, Zang Ling et al. used “phase transfer” self-assembly between good and poor solvents and a surface annealing based self-assembly method for making organized nanobelts of PDIs by controlling the π-π stacking and the hydrophobic interactions between the side chains linked at the imide positions. In other words, the 1D self-assembly of PDIs represents a balance between molecular stacking and solubility. 27 1.2 Layer-by-Layer Self-Assembly (LBL) The preparation of organic thin films from the spontaneous assembly of functional materials has been dramatically sped up by the introduction of the layer-by-layer (LBL) 9 technique introduced by Decher et al., based on the electrostatic interactions between the polycations and polyanions. 28 The procedure is shown in Figure 1. 4 in which the substrate is dipped alternatively into polycation and polyanion solutions. Two processes for each polyelectrolyte dipping step are essential. The first is the charge compensation and the other is the charge over-compensation which is crucial for the next polyelectrolyte layer deposition. 29 LBL film fabrication can also be influenced by secondary interactions such as hydrogen bonding, donor and acceptor interactions and ππ interactions. 30 Because of its simplicity, reproducibility and film uniformity over a large area the LBL technique has been widely used in fabricating all kinds of heterostructural films incorporating dendrimers, quantum dots, polymer micelles, proteins and even carbon nanotubes in support of applications such as chemical sensors, fuel cells, nano- or ultra filtration, OLED etc. 29, 30, 31, 32 Besides producing flat films, the LBL process can be conducted on substrates with other geometries such as polystyrene latexes, optical fibers and even nanotubes. Free standing LBL films, hollow shells and hollow nanotubes based on the LBL films can be obtained by the subsequent removal of the substrates. 33 The mechanical properties of free standing LBL films were also investigated with high modulus larger than 1GPa.c 34, 35 The recently discovered spray assisted LBL process by Decher et al. can increase the time efficiency for making films with desired compositions and thickness by 100 times. Spin-assisted LBL approach was also discovered for making films more efficiently. 36 The film build up process can be monitored by all kinds of techniques such as UV-Vis spectroscopy, ellipsometry, quartz crystal microbalance (QCM), Zeta potential, neutron/X-ray scattering etc. Neutron reflectometry reveals the strong interpenetration of 10 the polyelectrolyte in the films up to 5 layers. 37 This also means that the polyelectrolyte is not confined in a strict single layer fashion. Fluorescence Resonance Energy Transfer (FRET) was also used to elucidate the nature of polyelectrolyte entanglement in the LBL films. 38 A Free Energy Model for the LBL process has been developed by Park. et al allowing for the predication of cumulative thickness as a function of number of deposited layer as well as hydrated layer thickness and concentration of each absorbed layer within the LBL films. 39 In addition to the use of polymeric molecules as the building blocks for functional films, small dye molecules can be incorporated into LBL films, which enhances their potential applications to OLEDs, photovoltaics and sensors. Furthermore, incorporation of dye molecules into the films also enables one to use them as a probe to obtain detailed information about molecular level local properties such as polarity or molecular mobility. For example, pyrene molecules have been widely employed as the molecular probes to sense the microenvironment polarity and viscosity. 40 A pyrene derivative, 1,3,6,8-pyrene tetrasulfonic acid sodium salt, has been used in a LBL study of both flat substrates and colloid particles. 41 Previous studies that applied the LBL process to small charged organic dye molecules have shown that regular LBL film formation depends critically on several factors: polymer concentration, ionic strength of the polyelectrolyte solution and dye molecular shape and dimension. 42, 43 More recently, LBL films combining small organic molecules with polyelectrolytes have been used as a model system for drug release in which the assembled LBL films will decompose in response to temperature or pH stimuli, thereby releasing the incorporated molecules. 44 The locally high density of small molecules in the LBL films has inspired researchers to 11 design the photo-harvesting architectures mimicking photosynthetic process. 45 However it is often observed that the dye molecules are extracted by the next polyelectrolyte layer (of opposite charge), which significantly impedes the loading efficiency of small molecules into the films, affects the films’ integrity and stability and also compromises the polyelectrolyte dipping solution. In the dissertation, we will demonstrate that this extraction problem is minimal for our PDI molecules. 1.3 Reference: 1. Langhals, H. Heterocycles 1995, 40(1), 477-500. 2. a) Zollinger, H. Color Chemistry: Synthesis, Properties and Applications of Organic Dyes and Pigments, 2nd, Rev. Ed. VCH: Weinheim, 1991. (b) Law, K. Y. Chem. Rev. 1993, 93, 449. 3. Herbst, W. and K. Hunger, K. Industrial Organic Pigments: Production, Properties, Applications, 2nd edn., WILEY-VCH, Weinheim, 1997. 4. H. Langals Spectrachim. Acta. 1988, 44A, 1189. 5. Würthner, F. Chem. Comm. 2004, 1564-1579. 6. (a) G. Seybold, G. and G. Wagenblast, G. Dyes Pigs. 1989, 11, 303-317. (b) Sandrai, M.; Hadel, L.; Sauers, R. R.; Husain, S.; Krogh-Jespersen, K.; Westbrook, J.D.; Bird, G.R. J. Phys. Chem. 1992, 96, 7988-7996. 7. Böhm, A.; Arms, H.; Henning, G.; Blaschka, P. (BASF AG), Ger. Pat. Appl., DE 19547209 Al, 1997. 8. Würthner, F., Stepanenko, V., Chen, Z., Saha-Möller, C. R., Kocher, N., Stalker, D. J. Org. Chem. 2004, 69, 7933-7939. 9. Fan, L.; Xu, Y., He, T. Tetrahedron. Lett. 2005, 46, 4443-4447. 12 10. (a) Chao, C-C.; Leung, M-K., Su, Y.O., Chiu, K-Y.; Lin, T-H., Shieh, S-J., Lin, S-C. J. Org. Chem. 2005, 70, 4323-4331. (b) Ahrens, A. H., Fuller, M. J., Wasielewski, M. R. Chem. Mater. 2003, 15, 2684-2686. (c) Zhao, Y., Wasielewski, M. R. Tetrahedron. Lett. 1999, 40, 7047-7050. (d) Lukas, A. S., Zhao, Y., Miller, S. E., Wasielewski, M. R. J. Phys. Chem. B. 2002, 106, 12991306. (e) Shen, S., Liu, Y., Qiu, W., Sun, X., Ma, Y., Zhu, D. Chem. Mater. 2005, 17, 2208-2215. 11. Qu, J., Zhang, J., Drimsdale, A. C., Müllen, K., Jaiser, F., Yang, X., Neher, D. Macromolecules 2004, 37, 8197-8306. 12. Klok, H.A.; Becker, S.; Schuch, F.; Pakula, T.; Müllen, K. Macromol. Chem. Phys. 2002, 203, 1106-1113. 13. Nagao, Y.; Misono, T. Bull. Chem. Soc. Jpn. 1981, 54, 1269. 14. Tröster H. Dyes Pigm. 1983, 4, 171. 15. Kaiser, H.; Linder, J.; Langhals, H. Chem. Ber. 1991, 124, 529. 16. Ohta, T.; Nagano, T.; Ochi, K.; Kubozono, Y.; Fujiwara, A. Appl. Phys. Lett. 2006, 88, 103506. 17. Lin, Y-Y.; Gundlach, D. J.; Nelson, S.F.; Jackson, T.N. IEEE Electron Device Lett. 1997, 18, 606. 18. Malenfant, P. R.; Dimitrakopouls, C.D.; Gelorme, J. D.; Kosbar, L.; Graham, T. O; Curioni, A.; Andreoni, W. Appl. Phys. Lett. 2002, 80, 2517. 19. Chesterfield, R.J.; Mckeen, J. C., Newman, C. R., Ewbank, P. C., da Silva Filho, D. A., Brédas, J-L., Miller, L. L., Mann, K. R., Frisbie, C. D. J. Phys. Chem.B 2004, 108, 19281-19292. 13 20. Jiang, X. Z., Liu, Y. Q., Liu, X. G., Qiu, W. F., Song, X. Q., Zhu. D. B. Synth. Met. 1997, 91, 253. 21. Tang, C.W. Appl. Phys. Lett. 1986, 48, 183-185. 22. Li. J.; Dierschke, F.; Wu, J. Grimsdale, A.C.; Müllen, K. J. Mater. Chem. 2006, 16, 96-100. 23. (a) Shin, W.S.; Jeong, H.-H.; Kim, M.-K.; Jin, S.-H.; Kim, M.-R.; Lee, J.-K.; Lee, J. W.; Gal, Y.S. J. Mater. Chem. 2006, 16, 384-390. (b) Dinçalp, H., Içli, S. Solar. Energy 2006, 80, 332-346. 24. (a) O’Neil, M. P., Niemaczyk M. P., Svec, W. A., Gosztola, D., Gaines III, G. L., Wasielewski, M. R. Science 1992, 257, 63-65. (b) Rybtchinski, B., Sinks, L. E., Wasielewski, M. R. J. Am. Chem. Soc. 2004, 126, 12268-12269. (c) Ahrens, M. J., Sinks, L. E., Rybtchinski, B. Liu, W., Jones, B. A., Giaimo, J. M., Gusev, A. V., Goshe, A. J., Tiede, D. M., Wasielewski, M. R. J. Am. Chem. Soc. 2004, 126, 8284-8294. 25. Hippius, C.; Schlosser, F.; Vysotsky, M.O.; Böhmer, V.; Würthner, F. J. Am. Chem. Soc. 2006, 128, 3870-3871. 26. (a) Wang, W.; Li, L.-S.; Helms, G.; Zhou, H.-H.; Li, A.D.Q. J. Am. Chem. Soc. 2003, 125, 1120-1121. (b) Van, Herrikhuyzen, J.; Syamakumari, A.; Schenning, A.P.; Weiss, E.A.; Wasielewski, M.R. J. Am. Chem. Soc. 2002, 124, 9582-9590. (c) Sugiyasu, K.; Fujita, N.; Shinkai, S. Angew. Chem. Int. Ed. 2004, 43, 12291233. 27. (a) Balakrishnan, K., Datar, A., Naddo, T., Huang, J., Oikter, R., Yen, M., Zhao, J., Zang, L. J. Am. Chem. Soc. 2006, ASAP. (b) Balakrishnan, K., Datar, A., 14 Oikter, R., Chen, H., Zuo, J., Zang, L. J. Am. Chem. Soc. 2005, 127, 1049610497. (c) Datar, A., Oikter, R., Zang, L. Chem. Comm. 2006, 1649-1651. 28. (a) Decher, G.; Hong, J. Makromol. Chem. Symp. 1991, 46, 321. (b) Decher, G. Science 1997, 277, 1232. 29. Tripathy, S., Kumar, J., Nalwa, H. S. Handbook of Polyelectrolytes and Their Applications (V1): Polyelectrolyte-Based Multilayers, Self-Assemblies and Nanostructures, 2002, American Scientific Publishers, California. 30. (a) Schmitt, J.; Decher, G.; Dressik, W. J.; Brandow, S. L.; Geer, R. E.; Shashidbar, R.; Calvert, J. M. AdV. Mater. 1997, 9, 61. (b) Gao, M. Y.; Richter, B.; Kirstein, S.; Möhwald, H. J. Phys. Chem. B 1998, 102, 4096. (c) Ariga, K.; Lvov, Y.; Kunitake, T. J. Am. Chem. Soc. 1997, 119, 2224. (d) Lvov, Y.; Ariga, K.; Ichinose, I.; Kunitake, T. J. Am. Chem. Soc. 1995, 117, 6117. (e) Caruso, F.; Möhwald, H. J. Am. Chem. Soc. 1999, 121, 6039. (f) Tang, T.J.; Qu, J. Q.; Klaus, M.; Webber, S. E. Langmuir 2006, 22; 26-28. 31. Decher, G., Schlenoff, J. B. Multilayer Thin Films, 2002, Wiley-VCH. 32. (a) Wang, Y.; Tang, Z. Y.; Correa-Duarte, M. A.; Liz-Marza’n, L. M.; Kotov, N. A. J. Am. Chem. Soc. 2003, 125, 2830. (b) Wu, A.; Yoo, J.K.; Rubner, M.F. J. Am. Chem. Soc. 1999, 121, 4883. (c) Hiller, L.; Mendelsohn, J. D.; Rubner, M. F. Nat. Mater. 2002, 1, 59. (d) Tang, Z.; Wang, Y.; Kotov, N. A. Langmuir 2002, 18, 7035. (e) Park, J.; Hammond, P.T.; Adv. Mater. 2004, 6, 520. (f) Caruso, F.; Caruso, R. A.; Möhwald, H. Chem. Mater. 2001, 13, 1076. (g) Harris, J.L.; Stair, J.L.; Bruening, M.L. Chem. Mater. 2000, 12, 1941. 15 33. (a) Liang, Z., Susha, A. S., Yu, A., Caruso, F. Adv. Mater. 2003, 15, 1849-1853. (b) Ai S., Lu, G., He, Q., Li, J. J. Am. Chem. Soc. 2003, 125, 11140-11141. (c) Déjunat, C., Sukhorukov, G. B. Langmuir 2004, 20, 7265-7269. (d) Caruso, F. Adv. Mater. 2001, 13, 11-22. (e) Li, Q., Quinn, J. F., Caruso, F. Adv. Mater. 2005, 17, 2058-2062. 34. (a) Markutsya, S., Jiang, C., Tsukruk, V. V. Adv. Funct. Mater. 2005, 15, 771780. (b) Jiang, C., Markutsya, S., Pikus, Y., Tsukruk, V. V. Nature Materials 2004, 3, 728. (c) Jiang, C., Markutsya, S., Tsukruk, V. V. Adv. Mater. 2004, 16, 157-161. (d) Mamedov, A. A., Kotov, N. A. Langmuir 2000, 16, 5530-5533. (e) Ono, S. S., Decher, G. Nano. Lett. 2006, 6, 592-598. 35. (a) Nolte, A. J., Cohen, R. E., Rubner, M. F. Macromolecules, 2006, ASAP. (b) Jaber, J. A., Schlenoof, J. B. J. Am. Chem. Soc. 2006, 128, 2940-2947. 36. (a) Izquierdo, A.; Ono, S. S., Voegel, J. C., Schaaf, P., Decher, G. Langmuir 2005, 21, 7558-7567. (b) Lee, S-S., Hong, J-D., Kim, C. H., Kim, K., Koo, J. P., Lee, K-B. Macromolecules 2001, 34, 5358-5360. 37. (a) Kellog, G. J., Mayes, A. M., Stockton, W.B., Ferreira, M., Rubner, M. F. Langmuir 1996, 12, 5109. (b) Lösche, M., Schmitt, J., Decher, G. Bouwman, W. G., Kajer, K. Macromolecules 1998, 31, 8893. (c) Tarabia, M., Hong, H., Davidov, D., Kirstein, S., Steitz, R., Neumann, R., Avny, Y. Appl. Phys. Lett. 1998, 83, 725. 38. (a) Kerimo, J., Adams, D. M., Barbara, P. F., Kaschak, D. M., Mallouk, T. E. J. Phys. Chem. B 1998, 102, 9451. (b) Baur, J. W., Rubner, M. F., Reynolds, J. R., Kim, S. Langmuir 1999, 15, 6460. (c) Richter, B., Kirstein, S. J. Chem. Phys. 16 1999, 111, 5191. (d) Lowman, G. M., Daoud, N., Case, R. M., Carson, P. J., Buratto, S. K. Nano. Lett. 2001, 1, 677-682. 39. (a) Park, S. Y., Rubner, M. F., Mayes, A. M. Langmuir 2002, 18, 9600-9604. (b) Schlenoff, J. B., Dubas, S. T. Macromolecules 2001, 34, 592-598. 40. Winnik, F.M. Chem. Rev. 1993, 93, 587. 41. (a) Tedeschi, C.; Caruso, F.; Möhwald, H.; Kirstein, S. J. Am. Chem. Soc. 2001, 122, 5841. (b) Caruso, F.; Lichtenfeld, H.; Donath, E.; Möhwald, H. Macromolecules 1999, 32, 2317. 42. (a) Araki, K.; Wegner, M.J; Wrighton, M.S. Langmuir 1996, 12, 5393.; (b) Cooper, T.; Campbell, A.; Crane, R. Lamgmuir 1995 11, 2713. 43. (a) Ariga, K.; Lvov, Y.; Kunitake, T. J. Am. Chem. Soc. 1997, 119, 2224.; (b) Salomäki, M.; Tervasmäki, P.; Areva, S.; Kankare, J. Langmuir, 2004, 20, 3679. (c) Linford, M.R.; Auch, M.; Möhwald, H. J. Am. Chem. Soc. 1998, 120, 178. (d) Quin, J. F.; Caruso, F. Langmuir 2004, 20, 20. 44. (a) Wood, K.C.; Boedicker, J.Q.; Lynn, D.M.; Hammond, P.T. Langmuir 2005, 21, 1603-1609. (b) Yoo, P. J., Nam, K. T., Qi, J., Lee, S-K., Park, J., Belcher, A. M. Hammond, P. T. Nature Materials 2006, 5, 234-240. 45. Dai, Z.; Dähne, L.; Donath, E.; Möhwald, H. J. Phys. Chem. B. 2002, 106, 11501. 17 R O O O 3 3 5 1 3 5 1 6 12 7 11 8 10 9 (a) 4 2 5 1 6 Bay Bay 7 12 6 12 O 4 2 4 2 O Head N 7 10 O 10 8 11 9 O O O 8 11 9 N R (c) (b) Figure 1.1 Chemical Structures of perylene and its derivatives 18 O Tail Figure 1. 2 HOMO (top) and LUMO(bottom) of perylene diimides.(Reproduced from Würthner, F. 5) 19 O O O O CO2K CO2K O O O O O N O R-NH2 O O O CO2K CO2K CO2K CO2K O R 1 2 3 Figure 1. 3 Procedure of synthesizing non symmetric PDIs. . 20 4 Figure 1.4 Schematic representation of Layer-by-Layer deposition process.(Reproduced from Decher, G. et al.28b) 21 Chapter 2 Synthesis and Photophysics Study of 3, 4, 9, 10-Perylene Tetracarboxylic Diimide Dendrimers 2.1 INTRODUCTION 2.1.1 General Introduction Starburst dendrimers are monodisperse, hyperbranched polymers, which are roughly spherical in shape and highly functionalized on the exterior. Because of their well-defined, unique macromolecular structure, dendrimers are attractive scaffolds for a variety of high end applications such as drug or gene delivery, light-harvesting, sensors, and amplifications functions. 1-3 The synthesis of dendrimers has been achieved by the divergent and convergent approaches, which are shown in Figure 2.1. Tomalia et al. disclosed the synthesis and characterization of family of dendrimers in 1984-1985, using the so-called divergent method. The synthesis was carried out by the Michael addition of a “core” molecule of ammonia to three molecules of methyl acrylate, followed by exhaustive amidation of the triester adduct that generates a molecule with six terminal amine groups. Iterative growth is then continued using alternating Michael addition and amidation steps with the appropriate excess of reagents.1 Since then, Newkome reported another family of trisbranched polyamide dendrimers.2 Although dendrimers obtained by this approach can be obtained on a commercial scale with molecular weight over 25,000, the repetition of the coupling and activations steps leads to an exponential increase in the number of reactions at the periphery (Figure 2.1a), which requires a large excess of reagents for each step. Therefore, the likelihood of incomplete coupling or activation steps increases exponentially as well. Although the removal of the monomer may be straightforward by a 22 simple distillation, precipitation or ultrafiltration, any flawed molecules resulting from cyclization or incomplete reactions cannot easily be removed because of their structural similarity to the intended product. In 1989-1990, Hawker and Fréchet introduced the convergent growth approach to dendrimers with a more versatile and elegant control of the dendrimer growth.3 This approach initiates growth from what will eventually become the exterior of the molecule (Figure 2.1b), and progresses inward by coupling end groups to each branch of the monomer (Figure 2.1b: coupling step). After completion of the coupling, the single functional group located at the focal point of the wedge-shaped dendron can be activated (Figure 2.1b: activation step). Coupling of this activated dendron to each of the complementary functionalities on an additional monomer units affords a higher generation dendron. After sufficient repetition of this process, these dendrons can be attached to a multifunctional core through their focal point to form a globular multidendron dendrimer. Although again an iterative synthesis, the convergent route contrasts strongly with its divergent counterpart since it involves only a small number of reactions per molecule during the coupling and activation steps. The contribution of monomer to the mass of the product decreases exponentially as the generation number increases. As a result, the chromatographic purification can be carried out to produce the purest synthetic macromolecules prepared to date due to the large dissimilarity between the monomers and dendrons. This approach provides greater structure control than the divergent approach due to its relatively low number of coupling reactions at each growth step, allowing access to dendritic products of high purity and functionalities. However, the commercialization is far more limited, because of it is less readily scaled up than the divergent synthesis. 23 When compared with to linear analogues, dendrimers demonstrate significantly increased solubility that can be readily tuned by the peripheral groups, and they also exhibit very low intrinsic viscosities compared to linear polymers of comparable molecular weight. As an alternative strategy to improve solubility and provide additional chemical functionality for PDI’s, poly (amidoamine) (PAMAM) functionalized PDI had been synthesized in our lab using PDA as the core through the divergent approach.4 From these initial studies, it was found that there was obvious fluorescence quenching for all these PDI dendrimers even in dilute solutions, probably the result of electron transfer quenching from the tertiary amines in the PDI dendrimers. In this study, half-generation PDI dendrimers with different length of alkyl chains in PDI-diimidediamine core were prepared using the synthetic steps described in Figure 2.2. The photophysics of these synthesized PDI dendrimers was studied in the presence trifluoroacetic acid (TFA), which protonates the tertiary amine and prevents electron-transfer quenching. It is our hope that the variety of our synthesized PDI dendrimers makes it possible to elucidate the exact mechanism for the fluorescence quenching behavior. 2.1.2 Nomenclature PDA=3, 4, 9, 10-perylene tetracarboxylic dianhydride. PDI=3, 4, 9, 10-perylene tetracarboxylic diimide. PDI (0.0)-1n=3, 4, 9, 10-perylene tetracarboxylic diimide core free amine (1,nalkyl diamine). PDI0.5MA-12=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 1,2-ethylenediamine as Michael addition and diimide condensation reagents, respectively. 24 PDI0.5tBA-12=3, 4, 9 ,10-perylene tetracarboxylic diimide dendrimer using tbutyl acrylate and 1,2-ethylenediamine as Michael addition and diimide condensation reagents, respectively. PDI0.5MA-13=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 1,3-propanediamine as Michael addition and diimide condensation reagents, respectively. PDI0.5MA-2213=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 2,2-dimethyl-1,3-propanediamine as Michael addition and diimide condensation reagents, respectively. PDI0.5MA-14=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 1,4-butanediamine as Michael addition and diimide condensation reagents, respectively. PDI0.5MA-16=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 1,6-hexanediamine as Michael addition and diimide condensation reagents, respectively. PDI0.5MA-17=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 1,7-heptanediamine as Michael addition and diimide condensation reagents, respectively. PDI0.5MA-112=3, 4, 9, 10-perylene tetracarboxylic diimide dendrimer using methyl acrylate and 1,12-dodecanediamine as Michael addition and diimide condensation reagents, respectively. PDI18= N,N’-bis(1-aminooctadecane)-3, 4, 9, 10-perylene tetracarboxylic diimide. 25 PDI-DBPA=N,N’-bis(N,N-dibutyl-3-aminopropyl)-3, 4, 9, 10-perylene 4, 9, 10-perylene tetracarboxylic diimide. PDI-DEPA=N,N’-bis(N,N-diethyl-3-aminopropyl)-3, tetracarboxylic diimide. PDI-DMPA=N,N’-bis(N,N-dimethyl-3-aminopropyl)-3, 4, 9, 10-perylene 9, 10-perylene tetracarboxylic diimide. PDI-DMEA=N,N’-bis(N,N-dimethyl-2-aminoethyl)-3, 4, tetracarboxylic diimide. PDI-DBP=Model Compound= N,N’-bis(2,5-ditert-butylephenyl)-3, 4, 9, 10perylene tetracarboxylic diimide. 2.2 Experimental Section 2.2.1 Materials 3,4,9,10-perylene tetracarboxylic dianhydride (PDA, 97% purity) was purchased from Aldrich and used as received. All amines were purchased from Aldrich and used without further purification. These amines were ethylene diamine, 1, 3-propanediamine, 2,2-dimethyl-1,3-propanediamine, 1,4-butanediamine, 1,6-hexanediamine, 1,7- heptanediamine, 1,12-dodecanediamine and octadecylamine all with purity greater than 97%. Both methyl acrylate (MA) and t-butyl acrylate (t-BA) were obtained from Aldrich and they were also used as received. PDI-DBP was obtained from Aldrich and purified through flash chromatography by dichloromethane as the eluent. Trifluoroacetic acid (TFA) was purchased from Fisher and used as received. Spectroscopy grade 1, 2dicholorethane (DCE) from Fisher was used for preparing PDI samples for the 26 photophysics study. The TLC plates we used were from J.T. Baker Baker-Flex Silica Gel IB-F. Silicon gel was bought from Sorbent technology. 2.2.2 Instruments and Measurements. Mass spectroscopy was taken on the ZAB2-E instrument by either negative or positive chemical ionization. UV-Vis absorption spectra were obtained with HP-8453 diode array spectrometer. The fluorimeter used was a SPEX Fluorolog-τ2 equipped with a 450 W xenon light source, Czerny-Turner double grating excitation and emission monochromators. A photomultiplier voltage of 950V was typically used and the excitation and emission slit widths were set at 0.5/0.5/1/1mm for solution samples. Emission spectra was collected in front face mode with λex=490nm. Time-correlated single-photon counting system (TCSPC) was used for the life time measurements. A Mira 900-D Ti-Sapphire Mode-locked femtosecond oscillator cavity with Optima Controller serves as the light source, whose tunable wavelength is in the range of 700-980nm with pulse width less than 200fs. An optical frequency doubler was coupled to extend the wavelength range from 350 to 490nm. The excitation wavelength was adjusted at 463nm. The right angle mode was used for data collection. The optical densities for all the samples were adjusted around 0.09. The data were analyzed with the PicoQuant FluoFit Version4.0. 2.2.3 Synthesis of PDIs 2.2.3.1 Synthesis of PDI dendrimers by Divergent method The general procedure of synthesizing the PDI dendrimers was described in Figure 2.2. The first step is the imide condensation between PDA and diamines at high 27 temperature. The second step involves of Michael addition of either methyl acrylate or tbutyl acrylate. 2.3.1.1.1 Condensation of PDA with diamines The synthesis was executed according to the procedures described in Dr. Cohen’s dissertation 20 but with some improvements to making the synthesizing process easier and more efficient. The general procedure was described in Figure 2.2. According to the solubility of diamines, the condensation of different diamines with PDA can be divided into in two categories. For example, 1,2-ethylenediamine, 1,3-propanediamine, 2,2dimethyl-1,3-propanediamine and 1,4-butanediamine are water soluble, therefore, the purification of their condensation product with PDA can be achieved by precipitating the reaction mixtures into water. The amines with alkyl chains longer than 6 are not soluble in water and also the amine reactivity is very low. Therefore, the reaction was carried out in sealed tube (with three cycles of vacuum-melting-vacuum degassing) at the amine refluxing temperature. After centrifuging the product solution, chloroform was used to wash the solid until the pH paper did not turn blue indicating the complete remove of free amines. Protocol 1 for condensation of water soluble diamines with PDA. PDA (0.102 g, 0.26mmol) was mixed with 30 mL EDA (26g, 0.43mol) in a 250 mL flask with the aid of 35 mL pyridine. Before the flask was put into 100°C oil bath for refluxing under N2 overnight, the mixture suspension was ultrasonicated for 10 minutes. The final mixture turned the characteristic deep purple color of the PDI compounds. The final solution was centrifuged to remove the un-reacted PDA, and then poured into 350mL of water for PDI (0.0) precipitation. The precipitation can be completely finished by leaving the mixture 28 overnight. Then the precipitate was collected by vacuum filtration using G4 filter funnel. The product was dried overnight under high vacuum (~50milltorr). 0.1g PDI (0.0)-12 was obtained with a yield of 95%. The product was verified by the CI(-)-MS.4b This product was almost insoluble in any organic solvent. The 1H-NMR was performed on the formic acid salt in D2O and showed the expected ratio of aromatic to aliphatic peaks.4 Protocol 2 for condensation of longer alkyl chain diamines with PDA. PDA (200mg, 0.51mmol) was added into the tube and then 1,6-hexanediamine (0.6g, 5.17mmol) was added into it with the aid of heating. After three cycles of freezingmelting under vacuum, the tube was sealed under vacuum. The sealed tube was put into 120°C oil bath for 12 hrs. After 3 hours, the solution became dark purple color. High vacuum was used to remove the excess 1, 6-hexanediamine with the liquid nitrogen trap. Then chloroform was employed to wash the dark red solid until pH paper exposed to the supernatant did not turn blue. The MS showed that this step was very successful without byproduct, since there was only one strong peak at 589(m/Z, CI+), illustrated in Figure 2.3. This condensation was pretty successful (yield up to 94%). By utilizing either protocol 1 or protocol 2, the PDI(0.0)s for different diamines were synthesized and characterized by mass spectroscopy. 2.3.1.1.2 Michael Addition of PDI (0.0) with acrylates According to Cohen’s initial study, the PDI (0.5) product has a very good solubility in the co-solvent of CHCl3/CH3OH (2/1 by v/v).4 Therefore, the co-solvent of CHCl3/CH3OH was used as the reaction media in order to make the reaction equilibrium move to the product side. The Michael addition was carried out in a 100mL flask by dissolving 50 mg PDI(0.0)-12 in 30 mL methyl acrylate (MA) with the aid of 15 mL 29 methanol and 30mL chloroform. Then the solution was refluxed at 60 °C under N2. TLC was used to monitor the reaction process and the reaction was stopped after 3 days. The final solution became deep red. After centrifuging, the methyl acrylate was removed by rotavaporation with the aid of methanol. The yield for this process is less than 40%, due to the incomplete Michael Additions for two-arm or 3-arm byproducts. The purified product was obtained by silica-gel column chromography in CH2Cl2/CH3OH (95/05) until only one point was observed on the TLC plate under UV-365nm exposure. Rf was ~0.42 in CH2Cl2/CH3OH (95/05). Only one strong peak (821 m/z, CI+) was found in the CI-MS. Its mass spectrum was shown in Figure 2.4. Substituting t-butyl acrylate for MA, PDI(0.5)t-BA-12 was then synthesized and purified by liquid chromatography following the same steps as described above. Its mass spectrum by chemical ionization was shown in Figure 2.5. Following the above process, the other PDI(0.5) dendrimers were synthesized and purified by liquid chromatography using the co-solvent CH2Cl2/CH3OH (95/05) as eluent. Following the same protocol, PDI0.5MA-13, PDI0.5MA-2213, PDI0.5MA-14, PDI0.5MA-17, and PDI0.5MA-113 were synthesized with yields between 10% and 40%, whose mass spectra were shown in Figure 2.5 and Figure 2.6 and Figure 2.7. All these compounds were characterized by TLC and CI-MS spectroscopy. 2.2.3.2 Synthesis of other PDIs by one step condensation All other PDI compounds such as PDI18, PDI-DBPA, PDI-DEPA, PDI-DMPA and PDI-DMEA were synthesized by the one step condensation between PDA and primary amines. 30 For PDI-DBPA, PDA (0.3g, 0.76mmol) was added into 30mL pyridine. The suspension was then refluxed in N2 for 20 minutes before 0.7mL 3-(dibutylamino)propylamine was added into it. The whole mixture was refluxed for 5 hrs, becoming deep red eventually. After the reaction was stopped, the whole solution was precipitated into acetone. After vacuum filtration, the dark red solid was obtained, which was weighed at 0.98g (yield about 92%). Its CI-MS is shown in Figure 2.7. The other PDIs were synthesized following the same protocol and their MS is shown in Figure 2.8. The molecular ion peak position, molecular weight and product yield of the synthesized PDIs are listed in Table 2.1. As can be clearly seen, PDIs synthesized from the one step condensation had higher yields. The Michael addition of acrylate to the amine was not so effective because the strong π-π interactions from PDI (0.0) and intermolecular cyclization etc.4 The chemical structures of all our PDI compounds are shown in Figure 2.9. The differences in the chemical structure can allow us to study the structure-spectroscopy relation of PDIs. Some PDIs have the different alkyl lengths in the spacer diamines, such as ethylenediamine, propylenediamine, 1,4-butanediamine, 1,7-heptanediamine, and 1,12-dodecanediamine. PDI0.5MA-12 and PDI0.5tBA-12 have differences only in the ester substituted group. The t-butyl groups are expected to have more steric hindrance for more compact conformations. The branched structure in a diamine such as PDI0.5MA2213 was also investigated. The oxygen atom in the ester group from PDI0.5MA-13 will also be studied compared with the similar PDI compound PDI-DBPA. 2.3 Results and Discussions 2.3.1 Absorption and Emission spectra of PDIs 31 2.3.1.1 Molar Extinction Coefficient for PDIs Stock solutions were prepared in a 50mL volumetric flask at room temperature using PDIs that were dried in vacuum at 70ºC overnight. For example, 155.91mg PDI0.5MA-12 was dissolved in the 50mL dichloroethane (DCE) (molar concentration 1.9013x10-4 mol/L). This stock solution was left overnight before it was diluted into five solutions with different concentrations Then the UV-Vis absorption spectrum of these solutions was monitored, washing the UV cell by acetone between measurements. The UV-Vis absorption spectra over a large range was monitored as shown in Figure 2.10(a) and the absorbance at 526 nm and 490 nm were plotted against the PDI0.5MA-12 concentration thereby deriving the molar extinction coefficients (ε). The absorption spectra in DCE for PDI0.5MA-12 have a similar shape as the spectra for all other PDIs in organic solvents. The broad S0-S1 transition along the long axis is located between 450 nm and 540nm and the weak S0-S2 transition along the short axis shows a second peak between 360nm and 450nm. A strongly pronounced vibronic structure was observed with an absorption maximum at 526 nm which belongs to the electronic S0,0-S1,0 transition. The absorption has neither band shifting nor band broadening over our test concentration range, which demonstrates that there is no strong intermolecular interactions in this concentration range for PDI0.5MA-12. It is expected that the branched structures in our PDIs impede the close contact of the inner perylene diimide cores of adjacent molecules, preventing π-π stacking in the solution. The very strict linearity between the absorbance for the two highest peaks and concentration suggests that this expectation is correct. The molar extinction coefficients for the two highest bands were found to be 76,000 and 48000 M-1cm-1 at 526 and 490nm, respectively. The fitting curves 32 were shown in Figure 2.10 (b). This is a little less than the reported value of 90,000 M1 cm-1for the model compound PDI-DBP. Similar absorption spectra were obtained for all other synthesized compounds. The highest two peak positions and their corresponding molar extinction coefficients are listed in Table 2.2. As can be clearly see from Table 2.2, the peak positions are independent of the chemical structure of all our synthesized PDI compounds and essentially identical to the model PDI compound. This lack of spectral sensitivity is due to the nodes present at the imide nitrogen positions (according to the MO calculations) which make the electron coupling between the substituents at the head and tail positions and the perylene core extremely weak.5 The molar extinction coefficient for all the compounds lies in the range of 60,000 to 80,000 M-1cm-1 for the highest peak. 2.3.1.2 Emission Spectra of PDIs and Quantum Yield measurements The normalized emission spectra reflect very good mirror images with respect to the normalized absorption spectra for PDI0.5MA-112 and PDI-DBPA, as can be clearly seen from Figure 2.11(a) and (b) respectively. The Stokes shifts for PDI0.5MA-112 and PDI-DBPA are 12 and 11nm respectively. The Stokes shifts for other PDIs are listed in Table 2.2. The relatively small Stokes shift indicates that the configurations and interaction with the solvent of the excited and the ground state are very similar. The quantum yields for all the synthesized PDIs compounds were measured in DCE using PDI-DBP (QY=1) as the reference. The quantum yields for all the PDI compounds are listed in Table 2.2. The quantum yields for PDI0.5MA-12, PDI0.5MAtBA-12, and PDI0.5MA-2213 were 0.5 or less. All the other PDI compounds have QYs higher than 0.7. PDI0.5-112 has a very high QY of 0.93 and PDI-18 has a QY of unity. 33 PDI0.5MA-2213 is anomalous in this series of compounds as it is the only compounds with a three-methylene spacer that shows a relatively low fluorescence quantum yield, and as we will see below, a strong enhancement of fluorescence in the presence of TFA. Because it is an electron donor the tertiary amine nitrogen atom is good fluorescence quencher by the electron transfer process shown in Figure 2.12. Due to the electron donating properties of the alkyl chains in diamines, the tertiary nitrogen in the PDI dendrimer is more prone to quenching the perylene diimide fluorescence. This is why the QY of PDI18, without any tertiary amines, is unity. PDI0.5MA-112 has a much higher QY than the other PDI0.5MA dendrimers, so we conclude that long distance electron transfer along the alkyl chain is not possible nor is there a significant folding back of the dendrimer “arms” onto the perylene core. Therefore we propose a quenching mechanism based on the formation of a 6 or 7 member ring between the nitrogen atom and carbonyl group in the PDI core, as represented in Figure 2.13. The higher electron affinity for oxygen atom facilitates the electron transfer from electron rich nitrogen atom to the electron deficient perylene core. From the standpoint view of thermodynamic and conformational analysis, 6 and 7 member rings are the most stable ones.6 In other words, PDIs made from ethylenediamine and propylenediamide should have lower QYs due to the ease of formation of 6 and 7 member rings. Even a bulky group like tert-butyl, as in the case of PDI0.5tBA-12, cannot prevent the formation of rings. Evidently the branched structure in diamines such as in PDI0.5MA-2213 cannot prevent the formation of quenching rings either, as a very low quantum yield of 24% was found for PDI0.5MA2213. We presume this is due to the additional electron donating groups in the 2,2dimethyl-1,3-propylenediamine, which compensates for the larger N-O separation (i.e. 34 compare PDI0.5MA-13 and PDI0.5MA-2213). For other longer diamines the tertiary amine in the PDI structure is separated from the perylene core by a larger distance and cannot very easily form the required ring structure illustrated in Figure 2.13 although thermodynamic fluctuations can bring the tertiary amine into closer proximity to the perylene diimide chromophore. 2.3.2 Lifetime measurements for PDIs The lifetimes for these PDI compounds in DCE are listed in Table 2.3. Only monoexponential fluorescence decay was observed for PDI0.5MA-14, PDI0.5MA-17, PDI0.5MA-112 and PDI-18 with a lifetime around 4ns, which is the typical lifetime for PDIs.7 All the fluorescence decay curves were fitted with Σ2 less than 2.5 For the other PDI compounds from the reaction with ethylenediamine or propylenediamine, a double exponential fluorescence decay was observed with a fast decay component of approximately 1 ns, which we assume corresponds to the electron transfer quenching process from tertiary amine. For PDI0.5MA-2213, a more obvious fast component with a decay lifetime of 0.23ns was obtained, which might be tentatively attributed to the high electron density of amines and perhaps static quenching (discussed later). The fluorescence decay curve of PDI0.5MA-2213 in DCE is shown in Figure 2.14. Two longer lifetimes at 5.35 and 2.91ns were observed for PDI0.5tBA-12, which can be attributed to the bulky structure of tertiary butyl groups in the exterior which makes excited state relaxation more difficult. 2.3.3 Photophysical behaviors of PDIs upon adding TFA It is well known that PDIs form aggregates easily in solution due to the π-π interaction which leads to a diminished fluorescent quantum yield. The addition of an 35 acid such as TFA can potentially break up these aggregates and make the fluorescence yield increase.8 Additionally, as a strong organic acid, TFA has very strong tendency to protonate the tertiary amines which will greatly diminish electron-transfer quenching, as illustrated in Figure 2.12(b). Thus the TFA effect on the fluorescence spectroscopy of these PDI compounds was investigated and DCE was selected as the solvent because of its low volatility. 2.3.3.1 Protocol for studies of the effect of added TFA The TFA effect on the fluorescence spectra of PDIs in DCE was carried out using the following process: Solutions of PDIs in DCE were prepared with a concentration of approximately 10-6 M so that the optical density of the second highest was less than 0.1. These PDI solutions were used to adjust the [TFA]/[PDI] ratio by progressive dilutions. The molar concentration for pure TFA was calculated at 13.463 M. For each PDI solution, a small amount of TFA was initially added into the above prepared PDI solution so that [TFA]/[PDI] was 2x105. Then the prepared stock PDI solution was used to dilute the above solution to make the second solution with [TFA]/[PDI] at 104. A third solution was prepared by diluting the second solution to make [TFA]/[PDI] at 103 and so on until the solution having [TFA]/[PDI] = 10-2 was prepared. Thus a series of PDI-TFA solutions with the following ratios of [TFA]/[PDI] were obtained: 10-2, 102, 103, 104, 10-1, 100, 101, 2x105. All these solutions were sealed and placed in the dark overnight before measuring their UV and fluorescence spectra. 2.3.3.2 Photophysical properties of PDI compounds in the presence of TFA 2.3.3.2.1 Effect of TFA on the absorption spectra of PDIs 36 Figure 2.15 demonstrates the change in the absorption spectra for some PDI compounds upon adding TFA. As we can see, the absorption initially decreased to some extent, then increased until the [TFA]/[PDI] exceeded 103. The decrease of molar extinction coefficient for PDI0.5MA-12, PDI0.5MA-13, and PDI0.5MA-2213 is dramatic compared with the other PDI compounds. For PDI0.5MA-112 and PDI18 there is hardly any change in the molar extinction coefficients by the addition of TFA. In the literature, a decrease in the molar extinction coefficient upon molecular aggregation is known as the “hypochromic effect”.9 It is possible that the TFA association with the amines of the PDIs might slightly perturb the PDI molecular orbital and therefore modify the extinction coefficient also affected by the changing the solvent polarity and acidity. This can explain the fact that no molar extinction change was observed for PDI-18 (which contains no amine group) during the TFA titration. Clearly there is very strong red shift (~45nm) at the highest TFA concentration examined. This is due to the fact that the energy of excited state can be lowered by increasing the solvent polarity. However, this effect is not obvious when the TFA amount is much smaller compared to the DCE solvent. This results in the strong red shift only at very high TFA amount. The red shift for absorption spectra at [TFA]/ [PDI] =2x105 is presented in Table 2.3. 2.3.3.2.2 Effect of TFA on the fluorescence spectra of PDIs The normalized emission spectra at 4 different TFA concentrations for the PDI compounds are shown in Figure 2.16. For each PDI compound a very structured emission spectrum was observed with three major peaks corresponding to the transition from S1>S0, a mirror image of their absorption spectra. Similarly, a strong spectral red shift was shown at a very high TFA concentration for each PDI. For a PDI compound such as 37 PDI0.5MA-12 (Figure 2.16a), PDI0.5MA-2213 (Figure 2.16b), PDI0.5tBA-12 (Figure 2.16c), PDI0.5MA-13 (not shown here), and PDI-DBPA (Figure 2.16f), there is a progressive red shift with the increasing of TFA concentration. For PDI0.5MA-14 (Figure 2.16d), PDI0.5MA-17 (not shown here), PDI0.5MA-112 (not shown here) and PDI-18 (Figure 2.16e), there is hardly any red shift before a very high TFA concentration was reached. We presume that the strong red shift at very high concentration is from the high polarity of TFA. The volume fraction at [TFA]/[PDI]=2x105 is about 0.1. In order to compare the exact effect of TFA on the fluorescence QY of PDI, the normalized ratio of fluorescence intensity to the optical density at the excited wavelength was plotted against the ratio [TFA]/[PDI], as shown in Figure 2.17 with the red point indicating the value in the absence of TFA. The ratio increases much more dramatically for PDI0.5MA-12, PDI0.5MA-13, PDI0.5MA-2213, PDI0.5tBA-12 and PDI-DBPA, whose QYs reached over 80% at [TFA]/[PDI]=4, compared with the other PDI cases. For PDI0.5MA-14, PDI0.5MA-17 and PDI0.5MA-112, there is little change for the ratio after a small jump with the initial addition of TFA (resulting in a QY higher than 90%). Once again, at a very high TFA concentration, the QY started to drop. The smaller effect of TFA on the QY for all these PDIs indicates that conformation fluctuations that can bring the tertiary amine into closer proximity to the perylene diimide core are rare. For PDI18, there is no change in the ratio until it begins to decrease at very high [TFA]/[PDI] ratio. Upon the total protonation of the tertiary amines by TFA electron transfer from nitrogen to perylene diimide through the carbonyls was suppressed (illustrated in Figure 2.18), which leads to a larger jump around [TFA]/[PDI]=4 for the QYs of PDIs such as PDI0.5MA-12, PDI0.5MA-13, PDI0.5MA-2213, PDI0.5tBA-12 and PDI-DBPA. For 38 other PDI compounds like PDI0.5MA-14, PDI0.5MA-17 and PDI0.5MA-112, there is hardly any change in the ratio after the initial addition of TFA, suggesting that there is no electron transfer quenching in DCE solution. 2.3.3.2.3 Lifetimes of PDIs in the presence of TFA The lifetimes of PDIs at very high TFA concentration (TFA/PDI~106) were measured and their values are listed in Table 2.2. A mono-exponentional decay of fluorescence was detected for every PDI compound and all were fit by a lifetime around 4ns. The fluorescence decay curve for PDI0.5MA-2213 at a high TFA concentration is shown in Figure 2.14 as the red curve. The simple exponential decay suggests that all intramolecular quenching, including static quenching, has been suppressed. The lifetimes for PDI0.5MA-14, PDI0.5MA-17 and PDI0.5MA-112 and PDI-18 showed not much change before and after adding TFA. The lifetimes for PDI0.5MA-12, PDI0.5MA-13, PDI-DBPA and PDI0.5MA-2213 became a little longer due to the suppressed quenching from tertiary amine upon adding TFA. After adding TFA (especially at very high concentration), the photophysics of the excited state of perylene core is about the same for every compound and exhibit the same photophysics of relaxation to the ground state. By comparing the two ratios of ΦTFA/ Φ0 and τ(TFA)/τ(DCE) (see Table 2.3), we found that ΦTFA/ Φ0 is larger than τ(TFA)/τ(DCE) for PDI0.5MA-12, PDI0.5MA-2213 and PDI0.5tBA-12, which means the presence of the static quenching for these compounds. For all other compounds even for PDI0.5MA-13 and PDI-DBPA, these two values are very close to each other indicating no static quenching. These static quenching can further change the lifetime behavior of these compounds. 2.4 Summary 39 New perylene diimide based dendrimers were synthesized with an acceptable yield, following the divergent synthetic method. Diamines with different lengths and methyl acrylate (or t-butyl acrylate in one case) were used as the extension regents for the dendrimer growth steps, leading to dendrimers with different spacers between the tertiary amine and perylene diimide core. The photophysics of these synthesized PDIs was investigated with and without adding TFA. The (presumed) intramolecular fluorescence quenching was observed by comparing the fluorescence intensity and lifetime before and after adding TFA, demonstrating a stronger quenching process for those PDIs that have 2 or 3 carbons between the two nitrogens of the diamines. The branched structure in the diamine (PDI0.5MA-2213) leads to a more obvious quenching of fluorescence, preseumably due to the higher electron density in the amine. Moreover, the static quenching was observed for the compounds of PDI0.5MA-12, PDI0.5MA-2213 and PDI0.5tBA-12 by comparing the ratios of ΦTFA/ Φ0 and τ(TFA)/τ(DCE). Upon protonating the tertiary amine in the PDIs, all the PDIs show a similar single fluorescence lifetime. By comparing the chemical structures of these synthesized PDIs (such as PDI0.5MA-12 with PDI0.5tBA-12 and PDI0.5MA-13 with PDI-DBPA), we can conclude that the ester groups in the PDI dendrimers did not have much effect on their fluorescence behaviors. With the exception of PDI0.5-MA-2213, a spacer longer than two –CH2- groups essentially eliminated intramolecular fluorescence quenching, a result consistant with the ring-closure mechanism illustrated in Figures 2.13 and 2.18. 2.5 References: 1. Tomalia, D.A.; baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Poly. J. 1985, 17, 117-132. 40 2. Newkome, G. R.; Yao, Z.; Baker, G. R.; Gupta, V.K. J. Org. Chem. 1985, 50, 2003-2004. 3. (a) Hawker, C.J.; Fréchet, J. M. J. J. Am. Chem. Soc. 1990, 112, 7638-7647. (b) Grayson, S.M.; Fréchet, J. M. J. Chem. Rev. 2001, 101, 3819-3867. 4. (a) Cohen, T.S.; Parson, W.S.; Webber, S.E. Poly. Mater. Sci. Eng. 1997, 77, 228229. (b) Cohen, T. S. Syntheis and Photophysics of 3, 4, 9, 10-perylene Tetracarboxylic Diimide Dendrimers, Ph.D. Dissertation, 2000. (b) Parsons, W.S. Photophysics of Polymers in Solutions, The University of Texas at Austin, Ph.D. dissertation, 1994. 5. H. Langals Spectrachim. Acta. 1988, 44A, 1189. 6. Organic Chemistry, 4th ed. John McMurry, Brooks/Cole Publishing, 1996. 7. Hofkens, J., Vosch, T., Maus, M., Köhn, F., Cotlet, M., Weil, T., Herrmann, A., Müllen, K., De Schryver, F. C. Chem. Phys. Lett. 2001, 333, 255. 8. Gvishi, R.; Reisfel, R.; Burshtein, Z. Chem. Phys. Lett. 1993, 213, 338. 9. El-Daly, S.A. Spectrochim. Acta A., 1999, 143. 41 Figure 2.1 Schematic representation of Divergent and Covergent approach for dendrimer synthesis. (reproduced from Fréchet, J. M. J. 3b) 42 O O O O O O R-NH2 i R-N O PDI H2N-R-NH2 O O H2N-R-N N-R-NH2 O O PDI0.0 ii N-R O O PDA i O MA or t-BA O PDI0.5MA-12 PDI0.5tBA-12 PDI0.5MA-13 PDI0.5MA-2213 PDI0.5MA-14 PDI0.5MA-16 PDI0.5MA-17 PDI0.5MA-112 PDI-18 PDI-DBPA PDI-DEPA PDI-DMPA PDI-DMEA O R'OOCH2CH2C CH2CH2COOR' N-R-N N-R-N R'OOCH2CH2C O CH2CH2COOR' O PDI0.5 Figure 2.2 Scheme for the PDI compounds synthesis. 43 R=1,2-ethylene, R'=methyl R=1,2-ethylene, R'=t-butyl R=1,3-propylene, R'=Methyl R=2,2-dimethyl-1,3-propylene R=1,4-butyl, R'=Methyl R=1,6-hexane, R'=Methyl R=1,7-heptane, R'=Methyl R=1,12-dodecane, R'=Methyl R=octadecyl R=N,N-dibutyl-3-aminopropyl R=N,N-diethyl-3-aminopropyl R=N,N-dimethyl-3-aminopropyl R=N,N-dimethyl-2-aminoethyl Figure 2.3 Mass spectrum of PDI(0.0)-16 by CI+. 44 a b Figure 2.4 Mass Spectra of PDI0.5MA-12 by CI+ and PDI0.5MA-13 by CI-. 45 a b Figure 2.5 Mass Spectra of PDI0.5MA-2213 by CI- (a) and PDI0.5tBA-12 by CI+ (b). 46 a b Figure 2.6 Mass Spectra of PDI0.5MA-14 (a) and PDI0.5MA-17(b) by CI-. 47 Figure 2.7 Mass Spectra of PDI0.5MA-112 by CI+. 48 a b Figure 2.8 Mass Spectra of PDI-DBPA (a) and PDI-18 (b) by CI+. 49 O H3COOCH2CH2C H3COOCH2CH2C O CH2CH2COOCH3 NH2CH2C N N CH2CH2N O CH2CH2COOCH3 O PDI0.5MA-12 O (H3C)3COOCH2CH2C O NH2CH2C N (H3C)3COOCH2CH2C CH2CH2COOC(CH3)3 N CH2CH2N O CH2CH2COOC(CH3)3 O PDI0.5tBA-12 O H3COOCH2CH2C O NH2CH2CH2C N H3COOCH2CH2C CH2CH2COOCH3 N CH2CH2CH2N CH2CH2COOCH3 O O PDI0.5MA-13 H3COOCH2CH2C H3COOCH2CH2C H3C O O NH2CCH2C N H3C CH3 N CH2CCH2N O O CH3 CH2CH2COOCH3 CH2CH2COOCH3 PDI0.5MA-2213 H3COOCH2CH2C O O N(H2CH2C)2 N H3COOCH2CH2C CH2CH2COOCH3 N (CH2CH2)2N CH2CH2COOCH3 O O PDI0.5MA-14 50 O H3COOCH2CH2C H3COOCH2CH2C NH2C(H2CH2C)3 O N N O (CH2CH2)3CH2N CH2CH2COOCH3 CH2CH2COOCH3 O PDI0.5MA-17 O H3COOCH2CH2C O N(H2CH2C)12 N H3COOCH2CH2C CH2CH2COOCH3 N (CH2CH2)12N CH2CH2COOCH3 O O PDI0.5MA-112 O O H3CH2CH2CH2C CH2CH2CH2CH3 NH2CH2CH2CH2C N H3CH2CH2CH2C N CH2CH2CH2N CH2CH2CH2CH3 O O PDI-DBPA O H3CH2C(H2CH2C)8 O N N O (CH2CH2)8CH2CH3 O PDI-18 O O N N O PDI-DBP Figure 2.9 Chemical Structures of PDI compounds. 51 O Absorbance 2.5 526nm a 2 490nm 1.5 1 457nm 0.5 0 300 360 420 480 540 600 Wavelength(nm) 660 720 3.5 Absorbance 3 b 2.5 2 y = 75698x 1.5 R = 0.9997 2 y = 47834x 1 2 R = 0.9997 0.5 0 0.0E+00 1.0E-05 2.0E-05 3.0E-05 4.0E-05 PDI0.5MA-12 Concentration/M Figure 2.10 (a) Absorption spectra of PDI0.5MA-12 as a function of concentration. (b) Absorbance at 526nm (●) and 490nm (▲) as a function of concentration for deriving the molar extinction coefficient showing as the y value in the figure. 52 1.2 a Norm. Abs. 1 1 Norm. Fluo. Inten. 1.2 0.8 0.8 0.6 0.6 0.4 0.4 0.2 0.2 0 350 400 450 500 550 600 650 0 700 Wavelength(nm) 1.2 b Norm. Abs. 1 1 0.8 0.8 0.6 0.6 0.4 0.4 0.2 0.2 0 350 400 450 500 550 Wavelength(nm) 600 650 Norm. Fluo. Inten. 1.2 0 700 Figure 2.11 (a) and (b) Normalized absorption (solid line) and emission (dashed line) spectra for PDI0.5MA-112 and PDI-DBPA, respectively. 53 P∗ + NR 3 → + P− + → NR 3 P + NR 3 P− e− e− → → (a) P∗ + NR 3 ∗ + TFA → P + H − NR 3 → hν → P + H − + NR 3 + hν → (b) Figure 2.12 (a) Fluorescence quenching by the electron transfer mechanism from tertiary amine. (b) Fluorescence electron transfer quenching mechanism suppressed by combining with TFA. 54 R e O O N N R N O e R O N R (a) e R N R N O O N N O O R R e (b) e O O R N R N N O O R N R e (c) Figure 2.13 6-member ring and 7-member ring quenching mechanism by electron transfer from nitrogen to carbonyl group for PDI0.5-12, PDI0.5MA-13 and PDI0.5MA-2213, respectively. 55 Log[Intensity(counts)] 4 3 2 1 2 4 6 8 10 12 14 16 18 Time(ns) 20 22 24 26 28 Figure 2.14 Fluorescence decay curve for PDI0.5MA-2214 before and after adding TFA. Black: No TFA added, Red: adding TFA at [TFA]/[PDI]=106. 56 0.12 0.12 0.11 a 0.1 0.09 Absorbance 0.1 0.08 0.08 0.07 0.06 0.06 -3 0.04 -2 -1 0.02 0 340 4 5 6 PDI0.5MA-12 400 460 0.12 520 580 640 700 Wavelength(nm) 760 820 880 0.11 b 0.1 Absorbance 0 1 2 3 Log([TFA]/[PDI]) 0.09 0.08 0.07 0.06 0.05 -3 0.04 0.02 -2 -1 0 1 2 3 Log([TFA]/[PDI]) 4 5 6 PDI0.5MA-13 0 340 400 460 520 580 640 700 760 820 880 Wavelength(nm) 0.1 c Absorbance 0.08 0.06 PDI0.5MA-17 0.04 0.02 0 340 400 460 520 580 Wavelength(nm) 57 640 700 Absorbance 0.1 d 0.08 0.06 PDI0.5MA-112 0.04 0.02 0 340 Absorbance 0.06 400 460 520 580 Wavelength(nm) 640 700 e 0.04 PDI-18 0.02 0 340 400 460 520 580 Wavelength(nm) 640 700 Figure 2.15 Absorption spectra responding to the addition of TFA for different PDI compounds. 58 Norm. Fluo. Inten. 1 a 0.8 0.6 PDI0.5MA-12 0.4 0.2 Norm. Fluo. Inten. 0 500 1 550 600 Wavlength(nm) 650 700 b 0.8 0.6 PDI0.5MA-2213 0.4 0.2 0 500 Norm. Fluo. Inten. 1 550 600 Wavelength(nm) 650 700 c 0.8 0.6 PDI0.5tBA-12 0.4 0.2 0 500 550 600 Wavelength(nm) 59 650 700 Norm. Fluo. Inten. 1 d 0.8 0.6 PDI0.5MA-14 0.4 0.2 0 500 Norm. Fluo. Inten. 1 550 600 Wavelength(nm) 650 700 e 0.8 0.6 PDI-18 0.4 0.2 0 Norm. Fluo. Inten. 500 1 550 600 Wavelength(nm) 650 700 f 0.8 PDI-DBAPA 0.6 0.4 0.2 0 500 550 600 Wavelength(nm) 650 700 Figure 2.16 Normalized emission spectra of PDI compounds at different [TFA]/[PDI] ratios. Red: 0; Pink: 10; Black: 1000; Blue: 2x105. 60 Fluo. Inten./Abs(490nm) 1.4E+08 a 1.2E+08 1.0E+08 PDI0.5MA-12 8.0E+07 6.0E+07 -3 -2 -1 0 1 2 3 4 5 6 Fluo. Inten./Abs(490nm) Log([TFA]/[PDI]) 1.6E+08 b 1.3E+08 1.0E+08 PDI0.5tBA-12 7.0E+07 4.0E+07 Fluo. Inten./Abs(490nm) -3 -2 -1 0 1 2 3 Log([TFA]/[PDI]) 4 5 6 c 1.4E+08 1.3E+08 PDI0.5MA-13 1.2E+08 1.1E+08 -3 -2 -1 0 1 2 Log([TFA]/[PDI]) 61 3 4 5 6 Fluo. Inten./Absorbance 1.6E+08 d 1.2E+08 8.0E+07 4.0E+07 PDI0.5MA-2213 0.0E+00 Fluo. Inten./Abs(490nm) -3 -2 -1 0 1 2 3 Log([TFA]/[PDI]) 4 5 6 1.6E+08 e 1.4E+08 1.2E+08 PDI-DBAPA 1.0E+08 -3 -2 -1 0 1 2 3 4 5 6 4 5 6 Fluo. Inten./Abs(490nm) Log([TFA]/[PDI]) 1.4E+08 f 1.2E+08 1.0E+08 PDI0.5MA-14 8.0E+07 -3 -2 -1 0 1 2 Log([TFA]/[PDI]) 62 3 Fluo. Inten./Absorbance 2.0E+08 g 1.6E+08 1.2E+08 PDI0.5MA-17 8.0E+07 4.0E+07 Fluo. Inten./Abs(490nm) -3 -2 0 1 2 3 Log([TFA]/[PDI]) 4 5 6 4 5 6 4 5 6 h 1.7E+08 1.5E+08 1.3E+08 PDI0.5MA-112 1.1E+08 9.0E+07 -3 Fluo. Inten./Abs(490nm) -1 1.9E+08 -2 -1 0 1 2 3 Log([TFA]/[PDI]) i 1.6E+08 1.3E+08 PDI-18 1.0E+08 -3 -2 -1 0 1 2 Log([TFA]/[PDI]) 3 Figure 2.17 Ratio of fluorescence intensity to the absorbance at 490nm as a function of Log ([TFA]/[PDI]). The red triangle represents the ratio where no TFA was added. 63 R O HN O N N R NH O R O R (a) R O O R NH N N O O NH R R (b) R O O R NH N N O O HN R R (c) Figure 2.18 The protonation of tertiary amine by TFA 64 Table 2.1 Characterization of synthesized PDIs. PDI0.5MA-12 PDI0.5tBA-12 PDI0.5MA-13 PDI0.5MA-2213 PDI0.5MA-14 PDI0.5MA-17 PDI0.5MA-112 PDI-DBPA PDI-18 Yield 35% 28% 32% 16% 40% 12% 16% 92% 87% MW(g/mol) 820 988 848 904 876 960 1101 728 895 65 MS Mode CI+ CI+ CICICICICI+ CICI+ M+ 821 990 848 904 878 960 1102 729 896 Table 2.2 Spectroscopic characteristic of PDI compounds with no TFA added PDI0.5MA-12 PDI0.5tBA-12 PDI0.5MA-13 PDI0.5MA-2213 PDI0.5MA-14 PDI0.5MA-17 PDI0.5MA-112 PDI-DBPA PDI-18 λ0,0-1,0 (nm) 526 528 527 524 524 524 524 524 524 ε0-0 (M-1cm-1) 76000 59000 60000 73000 62000 76000 74000 71000 90000a λ0,0-1,1 (nm) 490 492 490 488 488 487 487 488 488 a ε0-1 ( M-1cm-1) 48000 42000 46000 49000 42000 48000 46000 45000 N/A λ1,0-0,0 (nm) 533 534 533 535 532 532 531 533 531 Stokes Shift(nm) 7 6 6 11 8 8 7 9 7 : Due to it low solubility, so it was assumed to have the same molar extinction as PDI-DBP. 66 Table 2.3 Spectroscopic characteristics of PDI compounds with TFA added PDI0.5MA-12 PDI0.5tBA-12 PDI0.5MA-13 PDI0.5MA-2213 PDI0.5MA-14 PDI0.5MA-17 PDI0.5MA-112 PDI-DBPA PDI-18 Red Shift a 9nm 7nm 9nm 12nm 12nm 12nm 11nm 13nm 11nm Red shift b 18nm 15nm 16nm 14nm 18nm 18nm 18nm 18nm 17nm Φ0 c 0.83 0.88 0.86 0.86 0.82 0.98 1.0 0.98 1.0 ΦTFAd 0.50 0.40 0.72 0.25 0.73 0.85 0.93 0.76 1.0 ΦTFA/ Φ0 1.66 2.2 1.19 3.44 1.12 1.14 1.07 1.29 1 a : Ref shift of λ0,0-1,0 with adding TFA at [TFA]/[PDI]=2x105. : Ref shift of λ1,0-0,0 with adding TFA at [TFA]/[PDI]=2x105. c : Quantum yiled of PDI with adding TFA at [TFA]/[PDI]=2x105. d : Quantum yiled of PDI without TFA. e : lifetime of PDI without adding TFA in DCE. f : lifetime of PDI with adding TFA at [TFA]/[PDI]=2x105. g : The average lifetime was taken for calculation. h : The number in the parenthesis is the pre-exponential factor. b 67 τ(DCE)e 3.89(0.70h),1.24(0.30) 5.35(0.31), 291(0.69) 3.86(0.85), 1.24(0.15) 3.83(0.46), 0.23(0.52) 3.81 4.01 4.04 3.71(0.84), 0.92(0.16) 4.07 τ(TFA)f 4.07 4.16 4.20 4.15 4.18 4.12 4.22 4.17 3.97 τ(TFA)/τ(DCE)g 1.31 1.13 1.21 2.21 1.10 1.04 1.04 1.28 0.98 Chapter 3 Solution Photophysics of Water Soluble Perylene Diimides 3.1 Introduction In recent years, different attempts were made to improve the photophysical/photochemical properties of the water-soluble fluorescent dyes as labels for cells, antibodies, and DNA, which has gained additional momentum owing to the use of single molecular spectroscopy to gain deep insight into biological systems.1 Although there are a large variety of water-soluble chromophores commercially available today, most of them exhibit relatively low fluorescence quantum yield and/or photochemical instabilities. Cyanine dyes, for example, are highly unstable towards oxygen and light, while xanthene dyes tend to aggregate in aqueous medium. 1a, 2 The ideal dye should have the following characteristics: 1) good water solubility, 2) high fluorescence quantum yield, 3) high chemical and photostability, 4) nontoxity, 5) good biocompatibility, as well as 6) possible commercial viability and scalable production. The highly fluorescent 3, 4, 9, 10-perylene tetracarboxylic diimide (PDI) chromophore is widely used as a commercial dye and pigment due to its outstanding chemical, thermal and photochemical stability. Owing to their optical properties PDIs should be excellent biological probes.3, 4 However, water soluble PDIs with functionalities at their imide sites showed poor water solubility and very weak fluorescence in water due to the strong π-π stacking.5 It is well known that the solubility of PDIs in organic solvents can be dramatically increased when the substituents are attached in the bay region.6 It is expected that water solubility can be achieved by introducing charged groups in the bay region which also results in a twist within the perylene core leading to less tendency for π-π stacking. Following this strategy, Qu J. et al. synthesized water soluble PDIs with moderate to high fluorescence quantum yields 68 which have demonstrated their promise as the biological labels for living cells.4,6 However, their solution photophysics of these water soluble PDIS have not been systematically studied. In this chapter, the photophysics of water soluble PDIs in different media was investigated. 3.2 Experimental Section 3.2.1 Materials Positively and negatively charged water soluble p-PDI (CAS [817207-4-7]) and nPDI (CAS [694438-88-5]) (Figure 3.1, with dimensions estimated from MD simulations of 2.13x1.33x1.89 nm and 2.13x1.15x1.49 nm (LWH), respectively 7) were synthesized by Qu, J. et al with synthetic scheme presented in Figure 3.2. Poly(diallydimethylammonium chloride) (PDAC, Mw = 25,000g/mol), and poly(styrene sulfonate) (PSS, Mw = 70,000g/mol) were purchased from Sigma-Aldrich and used without further purification. Sodium dodecyl sulfate (SDS) was purchased from Aldrich and crystallized in ethanol before use. Dodecyltrimethylammonium chloride (DTAC, puriss grade ≥99%) was purchased from Fluka and used as received. The anionic and cationic surfactants SDS and DTAC were employed in these studies (their corresponding CMC are 8 and 20mM in neutral water, respectively). The effects of surfactants such as SDS and DTAC or NaCl on the absorption and fluorescence emission spectra of p-PDI and n-PDI were studied by diluting concentrated p-PDI and n-PDI aqueous solutions with the SDS and DTAC solutions of different concentrations. All samples were stirred overnight before taking their spectra. 3.2.2 Instruments and Measurements Same UV-Vis spectrometer and Spex τ2 were used for measurements except that the Ex/Em slit width used in these experiments were 1/1/2/2 mm. 69 Same TCSPC instruments were used to get the lifetime of p-PDI and n-PDI in different medium except that excitation light source at 463nm was used instead at 422nm. 3.3 Results and Discussion 3.3.1 Absorption and Emission Spectra of p-PDI and n-PDI in Solutions. Due to strong intermolecular forces, such as π-π interaction, self-association of dyes in aqueous solution is often encountered and similar behavior would be expected for n- and p-PDI. It is to be expected that in DMSO water soluble PDIs will remain in monomeric form, as has been found for larger π-electron system such as water soluble terylenediimdes.8 In Figure 3.3 is shown the absorption and emission spectra of p-PDI and n-PDI in water and DMSO, respectively. Their absorption spectra in water have a similar shape as the spectra for other PDIs in organic solvents.9 The broad S0-S1 transition along the long axis is located between 480 nm and 630nm and the weak S0-S2 transition along the short axis shows a second peak between 360nm and 480nm. For n-PDI in water the S0-S1 transition has a maxima peak at 568nm (λ0-0; ε=2.97 x 104 M-1cm-1) and a shoulder at 540nm, and its emission spectrum is composed of only one peak at 618nm with a quantum yield of about 0.54 and a large Stokes shift of 50nm, as in Figure 3.3a. When n-PDI was dissolved in DMSO the absorption spectra was very similar to that reported for other PDIs in the monomeric form, 21 showing a better-resolved vibrational structure with maxima at 577nm (λ0-0; ε=3.44 x 104 M-1cm-1) and 540nm (λ0-1, ε=2.26 x 104 M-1cm-1). The increase of the molar extinction coefficient in DMSO is attributed to breaking up of n-PDI aggregates.10 The emission peak in DMSO was centered at 611nm with the quantum yield increasing from 0.54 in water to 0.82 in DMSO. The Stokes shift was decreased to 34nm in DMSO. 70 A similar trend was observed for p-PDI in water and DMSO, except that both absorption and fluorescence spectra are red shifted in water (λ0-0 was red shifted from 581nm (3.85 x 104 M-1cm-1) in DMSO to 587nm (3.28 x 104 M-1cm-1) in water) suggesting the formation of aggregates in water, as shown in Figure 3.3b. 5,7,11 The quantum yield in DMSO was three times larger than in water. Again, the increases in both quantum yields and molar extinction coefficient for p-PDI in DMSO suggest the presence of monomeric form of p-PDI in DMSO. The Stokes shift was about the same as in these two solvents. Spectral data are collected in Table 3.1. According to Molecular Exciton Theory, two main types of dye aggregates are distinguished as H- and J- aggregates.11 H-aggregates are formed with the face-to-face or card-pack stacking configuration, leading to the absorption blue shift in respect to the isolated chromophore and a total loss of fluorescence if two PDI’s transistion dipoles are strictly parallel to each other. However, J-aggregates with the head-to-tail configuration usually shows fluorescence featured by the red shifts of both absorption and fluorescence emission spectra peaks.11 Comparing these cases with our observations (see Table 3.1), we conclude that p-PDI forms H-aggregates in water. However, the orientation of dipole moments from the adjacent p-PDI molecules are most likely not strictly parallel to each other, according to MD simulations of the p-PDI dimer. For n-PDI, although it had a blue shift of absorption peak in water by 9nm relative to that in DMSO, it has a very strong emission from water with a red shift of about 7nm. Therefore, we assume that the n-PDI forms aggregates whose configuration lies between H- aggregates and J-aggregates. Lifetimes of n-PDI and p-PDI in both water and DMSO are listed in Table 3.1. All the lifetimes were fit with Σ less than 2. As can be seen from Table 3.1, fluorescence decays for n-PDI was monoexponential with a lifetime of 4.78 and two lifetimes were 71 obtained for p-PDI in water (6.35 and 0.13 ns) showing that there probably exist rather complex structures of p-PDI in solution. The very short lifetime species might be attributed to the H-type like aggregate of p-PDI in water (see above or later simulation). Slightly longer lifetimes of n-PDI and p-PDI at DMSO, following mono-exponential decay, were observed at 5.49ns and 5.41 ns, indicating the dissociation of n-PDI and pPDI aggregates in water. The fact that the fluorescence lifetimes in water and DMSO are fairly similar but there is an appreciable difference in fluorescence quantum yield is consistent with the presume of dimers or aggregates and static quenching of fluorescence for p-PDI. The following equations are usually applied to extract the the fluorescence rate constant (kf ) and nonradiative rate constant (knr): φf = τ= kf (1) k f + k nr 1 k f + k nr (2) For example, n-PDI in DMSO has a lifetime of 5.49ns and quantum yield of 82%, we obtain kf=1.49x108s-1 and knr=3.31x107s-1. Hence, kf>knr. The other kf and knr values are listed in Table 3.1. A higher fluorescence rate constant was obtained for n-PDI and a higher nonradiative rate constant for p-PDI. In order to examine the complexation between the p-PDI and n-PDI, experiments in solution were conducted. Both the absorption and fluorescence spectra were shown in Figure 3.4 for p-PDI, n-PDI, and the p-PDI/n-PDI mixture (1/1 mol/mol), respectively in water at pH=6.5. The simple sum of the p-PDI and n-PDI UV individual absorption and fluorescence spectra are also displayed in Figure 3.4 labeled as “p-PDI + n-PDI”. UV spectra for p-PDI/n-PDI (1/1) and p-PDI + n-PDI are not identical to each other, which suggests that a complex is formed between the p-PDI and n-PDI upon mixing. 72 However, the absorption spectrum of the 1:1 complex is only slightly modified from the individual species, unlike most examples of PDI aggregates.12 The oppositely charged PDI molecules favor the formation of p-PDI and n-PDI complexation through both the electrostatic and π-π interaction. The fluorescence from the 1/1 PDI mixture is much lower than p-PDI, n-PDI or p-PDI + n-PDI due to the formation of a complex (presumably via π-π stacking) between the p-PDI and n-PDI. The complexation of p-PDI and n-PDI with a 1:1 ratio have the following optical characteristics: absorption peaks at 450 nm, 545 nm and 577 nm and an emission peak at 614 nm. At the same time, the emission from p-PDI/n-PDI is about 7.6% of that of p-PDI, or a quenching efficiency of approximately 13. 3.3.2 Photophysics of n-PDI and p-PDI in Surfactant Solutions. The Ionic Self-Assembly (ISA) process utilizes the electrostatic interactions between charged surfactants and oppositely charged oligoelectrolytic species as the primary interaction.13 Other noncovalent interactions such as H-bonding, π-π and hydrophobic interactions can also promote this self-assembly process.14 By selecting different functional moieties, ISA materials can be tailored to show desired properties such as switchable luminescent and reversibly switchable conducting properties.15 Only a few papers have reported on the application of ISA process for the water soluble PDIs up to now. 16 As discussed above, our studied n-PDI and p-PDI molecules showed a strong tendency to form aggregates in water due to their strong π-π interaction and hydrophobic core. Surfactants form micelles above their CMC in water with a hydrophobic core and hydrophilic corona. Hence, besides the electrostatic interactions between the PDI and surfactant, the hydrophobic nature of n-PDI or p-PDI (p-PDI is more hydrophobic than n- 73 PDI) will serve as the secondary motifs by interacting with the micelle core to promote and stabilize the combined surfactant/PDI micelle complex. 3.3.2.1 Photophysics Study upon complexing between PDI and appositively charged surfactant (n-PDI/DTAC or p-PDI/SDS) SDS or DTAC solutions were made at different concentrations and then added to dilute the PDI solutions to vary the PDI/surfactant ratio. The effects were quite dramatic. Adding DTAC to n-PDI (abbreviated as n-PDI/DTAC) or SDS to p-PDI (p-PDI/SDS) resulted in an increase in the extinction coefficient, a well-resolved absorption spectra and a significant increase in the fluorescence intensity above the surfactant CMC (see the insets in Figure 3.5(a) and (b) and also Table 3.1). n-PDI/DTAC, at the highest DTAC concentration (35 mM) had an absorption spectrum very similar to n-PDI in DMSO (λ0-0 at 581 nm; ε=3.71 x 104 M-1cm-1 and λ0-1 at 540nm ε=2.28 x 104 M-1cm-1 and a shoulder at 500nm). Fluorescence emission spectra for n-PDI/SDS showed a blue shift to 606nm from 616nm in water shown in Figure 3.6b. For n-PDI/DTAC there was an initial decrease in the fluorescence intensity with the addition of DTAC followed by an enhancement by a factor of ca. 1.8 above the CMC (quantum yield approximately 0.96). The strong fluorescence decrease at low DTAC concentration is due to the formation of non-fluorescent premicellar aggregates.17 It is well known that quaternary ammonium groups in DTAC could help in stabilizing H-aggregates by forming ion pairs with sulfate groups.19 This is the highest quantum yield reported for a water soluble PDIs so far.4,6 The blue shift of emission from 616 in water to 606 in the n-PDI/DTAC complex, as shown in Figure 3.6a, suggests a less polar micelle environment. All results imply that the n-PDI was monomerically dissolved in the DTAC micelles. It also has a lifetime of 6.91ns, longer compared with n-PDI in water and DMSO indicating the dissociation of n- 74 PDI aggregates when solvated by DTAC micelles. A very high fluorescence rate constant was obtained for n-PDI in DMSO, compared with its non-radiative rate constant. For p-PDI/SDS, similar structured absorption spectra were observed (λ0-0 at 592 nm, ε=4.97 x 104 M-1cm-1 and λ0-1 at 547nm, ε=2.71 x 104 M-1cm-1 and a shoulder around 500nm). The plot of fluorescence intensity versus [SDS] in Figure 3.5b shows a monotonic increase, ultimately leveling off above 10 mM with an increase by a factor of ca. 4 (quantum yield about 0.42 after correcting for the OD change at 540nm). The fluorescence emission spectra showed a blue shift of 14nm relative to that in water, as shown in Figure 3.6b. The structured absorption spectra and blue shift of the fluorescence emission spectra indicates the incorporation of single PDI molecules into the SDS micelle core. Again we observed a double exponential fluorescence decay curve for p-PDI/SDS with lifetimes of 5.20 and 0.54ns. Very short average lifetime was obtained for p-PDI at 0.72ns. Very high fluorescence rate and non-radiative rate constants are obtained. 3.3.2.2 Photophysics Study upon complexing between PDI and same charged surfactant (n-PDI/SDS or p-PDI/DTAC) Even for surfactants with the same charge as the PDI moiety (e.g. p-PDI/DTAC or n-PDI/SDS), a red shift in absorption spectra and blue shift in fluorescence emission spectra could also be observed along with an increase in the fluorescence quantum yield (Table 3.1). The absorption spectra for n-PDI/SDS changed very little with a 12nm red shift and almost no shift in the fluorescence spectrum compared to water (Figure 3.7a and b). The fluorescence intensity increased very little over the investigated concentration range, with a change in the quantum yield from 0.54 in pure water to 0.59 in SDS. A monoexponential fluorescence decay with a lifetime of 6.09ns was observed. These results suggest that the incorporation of n-PDI into the SDS micelle is possible but the 75 environment is not appreciably different than in pure water (hence dimers or small aggregates may be incorporated into the micelles). In Figure 3.8 is given the absorption and emission spectra of p-PDI upon mixing with DTAC, which is totally different from the case of n-PDI/SDS. Structured absorption spectra were observed, similar to p-PDI in DMSO except for the 10nm red shift. The new peak around 540nm was attributed to the strong hydrophobic interaction between p-PDI and DTAC core. It is more obvious that p-PDI is incorporated into the DTAC micelle core based on the 8nm blue shift of emission spectra (Figure 3.8b) and increased fluorescence quantum yield (Table 3.1). Interestingly, the fluorescence increase in this case is even higher than that for p-PDI/SDS complexes. The maximum quantum yield was 0.54, similar to that of p-PDI in DMSO. Again two fluorescence lifetimes were observed for p-PDI/DTAC, suggesting different local environments for individual p-PDI moieties. 3.3.2.3 Photophysics Study upon Mixing the PDI/Surfactant complexes Based on the above experiments we believe that p-PDI and n-PDI were incorporated into the core of SDS and DTAC micelles, respectively. Therefore, experiments consisting of mixing p-PDI/SDS and n-PDI/SDS complexes or n-PDI/DTAC and p-PDI/DTAC complexes were conducted to test this argument. As expected, the PDI moiety is “protected” by the micelle and there is no fluorescence quenching upon mixing p-PDI/SDS and n-PDI/SDS. The absorption spectrum upon mixing p-PDI/SDS and nPDI/SDS at 10mM is identical with the average absorption spectrum in Figure 3.9a, which is also the same result as all other SDS concentrations from 2 to14mM. The fluorescence intensity as a function of SDS concentration upon mixing the two complexes agrees very well with the average of fluorescence intensity from the individual 76 complexes. All these results imply that the solubilized p-PDI or n-PDI molecules in surfactants are confined to the core of SDS or DTAC micelles, and subsequently unable to complex with each other upon further mixing, unlike the situation in pure water.7 3.3.2.4 Photophysics study of n-PDI titrated by DTAC. The titration of n-PDI solution by DTAC concentrated solution can provide further information about the interactions between PDI and surfactant molecules. Concentrated DTAC solution was prepared in DMF at 0.65M. A very similar absorption of n-PDI was obtained with the titration of DTAC as for the n-PDI/DTAC complexes with a new peak emerging at 540nm originating from the interaction between n-PDI and DTAC molecules (see Figure 3.10a and Figure 3.3a). The fluorescence intensity also showed a decrease with the initial addition of DTAC and then increased after reaching the CMC of DTAC (20mM), the same as for n-PDI/DTAC. This indicates that the presence of n-PDI did not change the CMC of DTAC due to its very low concentration (about 10-3mM). The quantum yield of n-PDI can reach 95% with [DTAC] ≥ 20mM. Compared with the 10nm blue shift for n-PDI/DTAC complexes, a blue shift of 12nm was observed for the n-PDI emission spectra by DTAC, suggesting a less polar environment the n-PDI molecules. 3.3.3 Photophysics study of n-TDI upon complexing with DTAC surfactant solutions (n-TDI/DTAC). Due to very strong π-π interactions, n-TDI is nonfluorescent in water. However, complexing n-TDI with DTAC surfactant solutions can make n-TDI fluorescent. A pronounced reduction of intensity of absorption peak near 630nm in favor of band at longer wavelength of 690nm (λ0,0-1,0) is observed with increasing DTAC concentration (Figure 3.11a). The structured absorption spectra indicate the monomeric dispersion of 77 n-TDI into the DTAC micelles. A fluorescence peak at 724nm emerged with a DTAC concentration higher than 20mM, corresponding to the CMC of DTAC. The fluorescence intensity saturated after 25mM. At a concentration of 35mM of DTAC the quantum yield of n-TDI can reach to 3%. The fast nonraditive rate constant (109s-1) has been reported for a similar n-TDI compounds which leads to the low quantum yield of n-TDI in medium.8 Mixing n-TDI/DTAC and p-PDI/DTAC complexes above 20mM of DTAC showed no fluorescence quenching for either p-PDI or TDI and indicates that the n-TDI is isolated in the DTAC micelle cores. 3.3.4 Effect of NaCl on the n-PDI and p-PDI Solutions. The effect on the absorption spectrum of adding NaCl to p-PDI or n-PDI solutions is shown in Figure 3.12a and b. As can be seen the OD is reduced by about 20% over the full range of NaCl concentration. We interpret this as a hypochromic effect10 arising from NaCl-induced further aggregation. The magnitude of this effect is similar for both n-PDI and p-PDI but the accompanying decrease in fluorescence is much stronger for pPDI due to the more hydrophobic nature of p-PDI. The fluorescence “quenching” by NaCl yields a linear Stern-Volmer curve with slopes 6.23 M-1 and 42.8 M-1 for n-PDI and p-PDI respectively. As neither the Na+ nor the Cl- ion is expected to quench the fluorescence by any direct mechanism (e.g. electron transfer or external heavy atom effects) this result can only be rationalized as the effect of NaCl-induced aggregation. If we assume that the extent of aggregation is similar for both species (based on the OD changes) then these results suggest that p-PDI is more prone to self-quenching than nPDI, because of its more hydrophobic nature.6 3.3.5 Effect of PSS-Na+, PDAC+Cl- Polyelectrolytes on n-PDI and p-PDI Solutions. 78 Zimerman et al. have studied the interaction between the water soluble pyrenetetrasulfonic acid chromophore and cationic polyelectrolytes.19 When the negatively charged pyrene molecules are mixed with a cationic polyelectrolyte the excimer fluorescence band of the pyrene is enhanced, presumably because the pyrene moieties are sequestered within the polyelectrolyte with a high local concentration. Thus it is reasonable to expect that addition of our charged PDI species to polyelectrolyte of the opposite charge may result in PDI aggregation. Xie et al. examined the complex formed between an unsymmetric cationic PDI species with anionic poly(acrylates) and found considerable broadening of the absorption and fluorescence spectra, which was interpreted as the result of PDI excimers (the magnitude of the fluorescence quenching was not reported).20 Absorption spectra of n-PDI and p-PDI titrated with the oppositely charged polyelectrolyte are shown in Figure 3.13a and b with the insets showing the fluorescence quenching curves (log (F0/F) and F0/F respectively). The slow addition of polyelectrolyte to either PDI solution results in a decrease in the absorbance, accompanied by a slight change in the spectral shape and fluorescence quenching, especially for n-PDI and PDAC, as is seen from the inset in Figure 3.13a. A monomer unit concentration of PDAC of only 2x10-5 M (in polymer repeating units) will decrease the fluorescence by a factor of ~220 (approximately a 10:1 ratio of PDAC monomer units to n-PDI). The sequestering of n-PDI by PDAC will lead to an increase of n-PDI’s local concentration and we presume that the fluorescence quenching is due to the formation of larger aggregates, even though the spectral changes are relatively minor (but consistent with hypochromism10). The excited state of n-PDI is expected to be a good electron acceptor so we would not expect the quaternized amine of PDAC to be an effective fluorescence 79 quencher by an electron transfer mechanism. The quenching of the p-PDI fluorescence by the slow addition of PSS solution shown in Figure 13b is much smaller compared to nPDI and PDAC, but still substantial, a factor of about 12. Another set of experiments was carried out in which equal volumes of PDI solution were diluted all at once with a 1 mg/mL solution of the oppositely charged polyelectrolyte. Once again a very strong quenching of n-PDI by PDAC was observed (see Figure 3.13a) with similar spectral changes as illustrated in Figure 3.14a. However, for p-PDI and PSS the absorption increased and became much more structured (see Figure 3.14b) and a fluorescence enhancement by a factor of 5 was observed (when corrected for the increased absorption the fluorescence increase is approximately a factor of 4), which is similar with that for p-PDI/SDS complexes. For this experiment the ratio of styrene sulfonate group to p-PDI is approximately 6000:1. If a slowly titrated pPDI/PSS solution is added all at once to excess PSS a fluorescence enhancement is once again observed, demonstrating that the p-PDI/PSS complex is not kinetically frozen. In both cases no spectral shift is observed for the fluorescence,. We assume that all these polyelectrolyte-induced phenomena are related to the formation of aggregates of the PDI when sequestered within a polyelectrolyte and furthermore, at least in the case of p-PDI/PSS, the particular nature of the aggregates is strongly dependent on the p-PDI/PSS ratio. Thus when the PSS is present in large excess the p-PDI is more isolated from other p-PDI moieties and this is reflected in a sharpening of the absorption spectrum and an increase in the fluorescence quantum yield. Thus one can conclude that the p-PDIs form aggregates in pure water, perhaps to a greater extent than n-PDI, which is why p-PDI has a substantially lower quantum yield than n-PDI. When PSS is added slowly at a lower PSS/p-PDI ratio, the local 80 concentration of p-PDI is actually increased, as reflected in the decrease of OD and fluorescence intensity. The reason PSS is able to break up the p-PDI aggregate is attributed to the amphiphilic nature of PSS chain. Following the same logic, PDAC always strongly enhances the aggregation of n-PDI regardless of the n-PDI/PDAC ratio. 3.3.6 Molecular Dynamics Simulation of p-PDI Dimer Computer simulation of the structure of a possible p-PDI dimer was carried out using the IMPAC software (with periodic boundary conditions) from the Schroedinger Corporation.21 For these calculations two p-PDI molecules were placed in the vicinity of each other with Cl- counterions (for simplicity) and the remaining volume filled with H2O molecules. After equilibration of this starting structure the H2O molecules were removed and then reinserted into the available volume and the structure re-equilibrated (see Figure 3.15). Then the atomic coordinates from this final structure were used to calculate the molecular center-to-center distance (7.76 Å) and the value of κ2 = 0.2804, which is the orientation factor that governs the strength of the dipole-dipole interaction (see discussion of Förster energy transfer, presented in Chapter 5.2.6). The transition dipole is assumed to line along the long molecular axis (parallel to the vector joining the two imide nitrogen atoms). (µ1· R12)( µ2· R12). The unit vector along this direction is µi. and κ = µ1· µ2 – 3 From the coordinates we find µ1· µ2 = -0.99792, (µ1·R12) =- 0.40077 (µ2· R12) = 0.38961) and κ = 0.2804. An analogous simulation was carried out for a n-PDI dimer (structure not shown) with a similar center-to-center distance (7.49 Å) but a much smaller value of κ2 (0.0574). The center-to-center distances are larger than one would expect for classical excimer formation, but for this distance and mutual orientation of the transition dipoles it is plausible that there could be a significant interaction between the p-PDIs in the excited state, but a much weaker one for the n-PDI 81 dimer. As can be seen from Figure 3.15, the two molecular planes are approximately parallel (and the twisting of the perylene out of planarity is also easily seen), indicating a H-aggregate like molecular aggregates. The structure of the n-PDI dimer is qualitatively similar. 3.4 Summary The photophysics of these PDI compounds in water were complicated by the formation of aggregates, which are evidently fairly weakly interacting (at least with respect to π−π stacking) given their significant fluorescence quantum yield and the absence of strongly perturbed absorption or fluorescence spectra. Some aggregates similar to H-aggregates are formed for p-PDI in water and J-aggregates are formed for nPDI in water. We found that adding classical surfactants (SDS and DTAC) to aqueous solutions of the PDIs sharpened the absorption spectrum, increased the extinction coefficient and the fluorescence yield, indicating the breaking up of aggregates of p-PDI or n-PDI by interacting with surfactant solutions. We also observed that the extinction coefficient and fluorescence quantum yield of both n- and p-PDI could be diminished by the addition of NaCl or low concentrations of polyelectrolytes (PSS or PDAC) to their aqueous solution. One would expect NaCl to encourage aggregation by electrostatic screening and if the PDI compound is sequestered onto a polyelectrolyte of opposite charge then it is reasonable to think that there could be a high local concentration of the PDI with concomitant changes in the absorption spectrum and fluorescence quantum yield. However mixing of p-PDI with PSS and n-PDI with PDAC at high polyelectrolyte concentrations provides an interesting contrast. For p-PDI and a large excess of PSS the absorption spectrum becomes more structured, the extinction coefficient increases and the fluorescence yield increases by a factor of approximately 82 three. Thus in this case we suggest that any p-PDI aggregates in aqueous solution are broken up and the p-PDI moieties are distributed as isolated monomers along the PSS chain. For n-PDI and a large excess of PDAC is this not the case and essentially the same changes in the absorption spectrum and fluorescence yield are observed as for low PDAC concentrations. We do not have a good explanation for this difference – our LBL experiments to not indicate that PDAC is an intrinsic quencher for n-PDI (see Chapter 5). Given this evidence for some kind of “weakly bound aggregate” some molecular simulations were carried out for two p-PDI molecules interacting in water. According to these calculations the perylene aromatic rings are approximately co-parallel but offset from each other with a center-to-center separation on the order of 8 Å, i.e. larger than for classical excimer formation but close enough for a significant dipole-dipole interaction in the excited state. 3.5 References 1. (a) Haugland, P. R. Handbook of Fluorescenct Probes and Research Products, 9th ed., Molecular Probes, Eugene, 2002. (b) Seisenberger, G., Ried, M. U., Endress, T., Buning, H., Hallek, M., Brächle, C. Science, 2001, 294, 1929. (c) Cotlet, M., Hofkens, J., Maus, M., Gensch, T., Van der Auweraer, M., Michiels, J., Dirix, G., Van Guyse, M., Vanderleyden, J., Visser, A. J. W. G., De Schryver, F. C. J. Phys. Chem. 2001, 105, 4999. 2. Fischer, M., Georges, J. Chem. Phys. Lett. 1996, 260, 115. 3. Margineanu, A., Holfkens, J., Cotlet, M.; Habuchi, S., Stefan, A., Qu, J., Kohl, C., , Vercammen, J., Engelborghs, Y., Gensch, T., De Schryver, F. C. J. Phys. Chem. B. 2004, 108, 12242. 4. Qu, J., Kohl, C., Pottek, M., Müllen, K. Angew. Che. Int. Ed. 2004, 43, 1528. 83 5. (a) Schnupfeil, G., Stark, J., Wöhrle. D. Dyes Pigm. 1995, 27, 339. (b) Langhals, H., Jona, W., Einsiedl, F., Wohnlich, S. Adv. Mater. 1998, 10, 1022. 6. Kohl, C., Weil, T., Qu, J., Müllen, K. Chem. Eur. J. 2004, 10, 5297. 7. Tang, T. Qu, J. Müllen, K., Webber, S. E. Langmuir 2006, 22, 26. 8. Jung, C., Müller, B., Lamb, D.C., Nolde, F., Müllen, K., Bräuchle, C. J. Am. Chem. Soc. 2006, 128, 5283. 9. (a) Hofkens, J., Vosch, T., Maus, M., Köhn, F., Cotlet, M., Weil, T., Herrmann, A., Grebel-Koether, D., Qu, J., Müllen, K., De Schryver, F.C. Chem. Phys. Lett. 2001, 333, 255. (b) Liu, D., De Feyter, S., Cotlet, U.-M., Weil, T., Herrmann, A., Grebel-Koether, D., Qu.J., Müllen, K.; De Schryver, F. C. Macromolecules, 2003, 36, 5918. 10. Rhodes, W., Chase, M. Review of Modern Physics 1967, 39, 348. 11. Ford, W. E. J. Photochem. 1987, 37, 189. 12. Würthner, F. Chem. Comm. 2001, 1564. 13. Ford, W. E. J. Photochem. 1987, 37, 189. 14. Faul, C.F.J.; Antonietti, M. Adv. Mater. 2003, 15, 673. 15. (a) Faul, C.F.J.; Antonietti, M. Chem. Eur. J. 2002, 8, 2764. (b) Wei, Z., Laitinen, T., Smarsly, B., Ikkala, O., Faul, C. F. J. Angew. Chem. Int. Ed. 2005, 44, 751. 16. (a) Franke, D., Vos, M., Antonietti, M., Sommerdijk, N. A. J. M., Faul, C. F. J. Chem. Mater. 2006, 18, 1839. (b) Marcon, R. O., dos Santos, J. G., Figueiredo, K. M., Brochsztain, S. Langmuir 2006, 22, 1680. 17. Fendler, J. H. Membrane Minmetic Chemistry John Wiley & Sons, New York, 1982. 84 18. Rosen, M.J. Surfactants and Interfacial Phenomena 2nd ed., Wiley & Sons, New York, 1989. 19. (a) Zimerman,O.; Cosa, J.J.; Previtail, C.M. J.M.S.-Pure Appl. Chem. 1994, A31, 859. (b) Caruso, F.; Donath, E.; Möhwald, H.; Georgieva, R. Macromolecules, 1998, 31, 7365. 20. Xie, A. F., Liu, B., Hall, J. E., Barron, S. L., Higgins, D.A. Langmuir 2005, 21, 4149. 21. These calculations were carried out by Jessica Kingsberg in the laboratory of Prof. E. Dormidontova, Department of Macromolecular Science and Engineering, Case Western Reserve University, Cleveland, Ohio. 85 a b O N O O N HO3S SO3H O O N+ N O O HO3S O O SO3H O N O + O O N+ N+ O O N c O N O NaO3S SO3Na O O O O NaO3S SO3Na O N O Figure 3.1 Chemical Structures of n-PDI, p-PDI and n-TDI. 86 O 4MeSO3- O N O O N O HO3S O O ii:94% O O SO3H O O O O HO3S O N HO O O N SO3H O O N O i: 89% Cl Cl Cl Cl O N n-PDI HO O N iii: 79% O N O N O N O O O O N O N O O N+ O O iv:94% N N N+ O O 4MeSO3- N+ N+ O N O p-PDI Figure 3.2 Synthetic Scheme for n-PDI and p-PDI. i) Phenol, N-methyl-2 pyrrolidinone(NMP), K2CO3, 80ºC, 15hrs, 89%. ii) Conc. Sulfuric acid r.t. 15hrs, 93%. iii) 4-[2-(dimethylamino)ethyl]phenol, NMP, K2CO3, 80ºC, 15hrs, 79%. iv) silver methanesulfonate, r.t., 5hrs, 95%. 87 a 1 n-PDI Fluo. Inten.(a.u.) Absorbance 0.06 0.8 0.04 0.6 0.4 0.02 0.2 0 0 b 0.06 Absorbance 400 460 520 580 640 Wavelength(nm) 700 760 1 p-PDI 0.8 0.04 0.6 0.4 0.02 Fluo. Inten.(a.u) 340 0.2 0 0 340 Figure 3.3 400 460 520 580 640 Wavelength(nm) 700 760 (a) and (b) Absorption and emission spectra of 1.5 x 10-6 M n-PDI and pPDI in water (solid line) and DMSO (dotted line), respectively. 88 UV n-PDI UV p-PDI UV p-PDI/n-PDI UV p-PDI+n-PDI FL n-PDI FL p-PDI FL p-PDI/n-PDI FL p-PDI+n-PDI Absorbance 0.08 0.06 0.04 0.02 Fluo.Inten.(a.u.) 0.1 0 340 Figure 3.4 400 460 520 580 640 700 Wavelength(nm) 760 820 880 Absorption (left) and fluorescence emission (right) spectra in water for nPDI (black line), p-PDI (dark blue line), p-PDI/n-PDI mixed in a 1:1 ratio, (red line) and the sum of the p-PDI and n-PDI spectra (light blue line). 89 2 a 0.06 Fluo. Inten. Absorbance 0.08 n-PDI 1.5 1 0.5 0 0.04 0 5 10 15 20 25 [DTAC]/mM 30 35 40 0.02 0 400 500 600 700 Wavelength(nm) 900 6 0.08 Fluo. Inten. b 0.06 Absorbance 800 4 2 0 0.04 0 0.02 2 4 6 8 [SDS]/mM 10 12 14 p-PDI 0 400 500 600 700 Wavelength(nm) 800 900 Figure 3.5 Absorption spectra of n-PDI with DTAC (a) and p-PDI with SDS (b) at different concentrations. Insets show the fluorescence intensity as a function of surfactant concentration. Arrow indicates increasing surfactant concentration. 90 a n-PDI 5mMDTAC 10mMDTAC 15mMDTAC 20mMDTAC 25mMDTAC 30mMDTAC 35mMDTAC Fluo. Inten. n-PDI/DTAC 560 600 640 680 Wavelength(nm) 720 760 800 p-PDI b Fluo. inten. p-PDI/SDS 2mMSDS 4mMSDS 6mMSDS 8mMSDS 10mMSDS 12mMSDS 14mMSDS 560 600 640 680 720 Wavelength(nm) 760 800 Figure 3.6 Fluorescence emission spectra of n-PDI (a) and p-PDI (b) mixing with SDS and DTAC surfactant solutions at different concentrations, respectively. 91 0.1 a Absorbance 0.08 0.06 0.04 0.02 0 300 400 500 600 Wavelength(nm) 700 800 1.4 Fluo. Inten. Fluo. Inten. b 1.2 1 0.8 0 560 600 2 640 680 Wavelength(nm) 4 720 6 8 10 [SDS]/mM 760 12 14 800 Figure 3.7 (a) and (b) Absorption and emission spectra of n-PDI upon mixing with SDS solutions at different concentrations. Ex=540nm. Inset in (a) showing the fluorescence intensity (corrected to the abs) as a function of SDS concentrations. Absorption and emission spectra of n-PDI in water are shown as the red line. The arrow indicates the concentration increasing of SDS. 92 0.06 p-PDI a 0.05 5mMDTAC Absorbance 10mMDTAC 0.04 15mMDTAC 20mMDTAC 0.03 25mMDTAC 30mMDTAC 0.02 35mMDTAC 0.01 0 340 420 500 580 660 Wavelength(nm) 740 820 900 Fluo. Inten. 6 Fluo. Inten. d 4 2 0 0 560 600 5 640 680 Wavelength(nm) 10 15 20 [DTAC ]/mM 720 25 760 30 35 800 Figure 3.8 (a) and (b) Absorption and emission spectra of p-PDI upon mixing with DTAC solutions at different concentrations. Ex=540nm. Inset in (b) showing the fluorescence intensity as a function of DTAC concentration. The arrow indicates the concentration increasing of DTAC. 93 a 0.08 Fluo. Inten. Absorbance 0.1 0.06 0.04 0.02 0 360 420 480 540 600 660 Wavelength(nm) 720 780 Fluo. Inten. b 2 Figure 3.9 4 6 8 10 [SDS]/M 12 14 16 (a) Absorption and emission spectra of p-PDI/SDS mixing with nPDI/SDS at [SDS]=10mM (Red) and the average absorption spectrum of individual p-PDI/SDS and n-PDI/SDS at [SDS]=10mM(black). (b) Fluorescence intensity upon mixing p-PDI/SDS and n-PDI/SDS (red circle) and averaging the individual p-PDI/SDS and n-PDI/SDS (black triangle) as a function of SDS concentration. 94 0.03 Absorbance a 0.02 0.01 0 340 400 460 520 580 640 Wavelength(nm) 700 760 Fluo. Inten. b 0 560 600 640 5 10 15 20 25 DTAC Concentration/mM 680 720 Wavelength(nm) 760 30 800 Figure 3.10 (a) and (b) Absorption and emission spectra of n-PDI titrated by concentrated DTAC DMF solution. Red curves represent the spectra of nPDI in water. Inset shows the fluorescence versus DTAC concentration and the arrow indicates increasing DTAC concentration. 95 0.06 Absorbance a 0.04 0.02 0 400 460 520 580 640 700 760 Wavelength(nm) 1 b Fluo. Inten. Fluo. Inten. 0.8 0.6 0.4 0.2 0 0 660 700 740 5 780 820 Wavelength(nm) 10 15 20 25 DTAC Concentration/mM 860 900 30 35 940 Figure 3.11 (a) and (b) Absorption and emission spectra of n-TDI upon mixing with DTAC solutions at different concentrations. Ex=640nm. Inset in (a) showing the fluorescence intensity as a function of DTAC concentrations. Absorption and emission spectra of n-TDI in water are shown as the red line. The arrow indicates the concentration increasing of DTAC. 96 3 a 2 NaCl Increases 0.06 F0/F Absorbance 0.08 2.5 1.5 1 0.5 0.04 0 0 0.04 0.08 0.12 0.16 [NaCl](M) 0.2 0.24 0.02 0 400 460 520 580 640 700 Wavelength(nm) 0.12 820 880 7 0.08 6 5 NaCl increases 0.06 F0/F b 0.1 Absorbance 760 4 3 2 1 0 0 0.04 0.02 0.04 0.06 0.08 [NaCl]/M 0.1 0.12 0.02 0 400 460 520 580 640 700 760 Wavelength(nm) 820 880 Figure 3.12(a) Absorption spectra of n-PDI and (b) p-PDI as a function of NaCl concentration, with insets showing the S-V fluorescence quenching curves. The arrow indicates increasing NaCl concentration. 97 a Absorbance 0.1 PDAC Increase 0.08 Log[F0/F]d 0.12 2 1 0 0.06 0.0E+00 0.04 1.0E-05 2.0E-05 3.0E-05 monomer Comcentration/M 4.0E-05 0.02 0 Absorbance 0.08 460 520 760 820 880 10 b PSS increases 0.06 580 640 700 Wavelength(nm) 0.04 F0/F 400 5 0 0.0E+00 3.0E-06 6.0E-06 9.0E-06 Monomer Concentration(M) 0.02 0 400 500 600 700 Wavelength(nm) 800 900 Figure 3.13 Absorption spectra for n-PDI (a) and p-PDI (b) with the addition of PDAC and PSS solutions with insets showing the fluorescence ratio log(F0/F) or F0/F, respectively. Arrow indicates increasing PDAC and PSS concentration. Red curve represents the spectrum in water. 98 0.08 a n-PDI Fluo. Inten. Absorbance 0.06 0.04 0.02 0 300 400 500 600 700 800 Wavelength(nm) 0.08 b p-PDI Fluo. Inten. Absorbance 0.06 0.04 0.02 0 400 450 500 550 600 650 Wavelength(nm) 700 750 800 Figure 3.14 (a) Absorption and fluorescence spectra of n-PDI before (black) and after (red) mixing with 1 mg/ml PDAC solution, demonstrating fluorescence quenching under these conditions (b) Absorption and fluorescence spectra of p-PDI before (black) and after (red) mixing with 1 mg/ml PSS solution, demonstrating fluorescence enhancement under these conditions. 99 Figure 3.15 MD structure for p-PDI dimer in pure water 100 Table 3.1 Photophysical parameters for n-PDI and p-PDI in different solutions Media n-PDI p-PDI Water DMSO DTAC SDS Water DMSO DTAC SDS λ0,0-0,1 (nm) 568 577 581 580 587 581 593 592 λ1,0-0,0 (nm) 618 611 606 617 628 612 622 614 εmax (M-1cm-1) 29700 34400 37100 30200 32800 38500 62900 49700 ΦFa τb (ns) 4.78 5.49 6.91 6.03 6.35(0.52); 0.13(0.48) [3.36]c 5.41 5.61(0.75); 0.78(0.25) [4.40]c 5.20(0.04); 0.54(0.96) [0.72]c 0.54 0.82 0.96 0.59 0.14 0.55 0.52 0.42 kf (x10-8s-1) 1.13 1.49 1.39 0.98 0.26 1.02 1.18 5.83 knr ( x10-8s-1) 0.96 0.33 0.059 0.68 1.59 0.83 1.09 8.06 . a. The quantum yield (ΦF) was measured relative to the standard PDI compound N,N’-bis(2,6-dimethylphenyl)-3,4,9,10perylenetetracarboxylic diimide in dichloroethane, for which ΦF = 1 is assumed. b.Values in ( ) are the pre-exponential factors in a bi-exponential fit. c. The average fluorescence lifetime by weighing the pre-exponential factors. 101 Chapter 4 Molecular Layer-by-Layer Self-assembly of Water Soluble Dyes through the π-π and Electrostatic Interactions 4.1 Introduction Conventionally, the Layer-by-Layer (LBL) process is based on the sequential adsorption of polycations and polyanions from dilute aqueous solution onto a solid support with film formation a consequence of the electrostatic interaction and complex formation between the oppositely charged polyelectrolytes.1 Functional films for making thin-film filed-effect transistiors and photovoltaics require the preparation of well defined films composed of molecules with appropriate properties in a specified geometrical arrangement with respect to each other and the substrate. Research on zwitterionic molecules assembled alternately has been carried out to achieve these goals although the integrity of the films was not easily controlled.1a The interpenetration of polyelectrolyte chains between adjacent layers has been addressed.2 Therefore, precise sterochemical and positional control of the functional species at the molecular level have not been achieved by the LBL self assembly technique to our knowledge. The Langmuir-Blodgett (LB) deposition technique offers control over the architecture of the films at the molecular level to offer certain desirable properties tailored to suit the application required. However, it is very difficult for the technique and complicated for the instrument and the molecules suitable for this technique are very limited.3 On the other hand, the further application of conjugate polymers or organic molecular semiconductors as the organic based FET or OLED has been limited due to their low electron mobility between the crystal “grains” boundaries. The π stacking is expected to provide more efficient orbital overlap and thereby facilitate the carrier transport.4 In this Chapter we will discuss the 102 fabrication of self-assembled thin films of perylene diimides carrying four positive or negative charges respectively (p-PDI and n-PDI see Figure 3.1) on quartz or silicon substrates using molecular layer-by-layer (MLBL) deposition from aqueous solution. The application of this technique to some other charged aromatic species (pyrenetetrasulfonic acid tetrasodium salt (n-PSA) and terylene diimide(n-TDI)) was also investigated. The reasons for choosing PDIs as the building block are: 1) isolated PDI moieties have a large extinction coefficient in the visible, a high fluorescent quantum yield and very good photostability and are good candidates for n-type semiconductors, photon harvesting arrays (photovoltaic cells) and OLEDs; 2) the perylene structure is very liable to form assemblies because of the strong π-π interaction between them and the π stacking is expected to facilitate energy or charge transport.4,5 The π stacking can also lead to strong quenching of the fluorescence, as we will see in the following. Thus the fluorescence intensity serves as a measure of the extent to which the perylene diimide moieties can be isolated from each other. 4.2 Experimental Section 4.2.1 Materials The p-PDI (CAS[817207-11-7]), n-PDI (CAS[694438-88-5]) and n-TDI, whose chemical structures shown in Figure 3.1, were synthesized by Müllen et al.8 Pyrenetetrasulfonic acid tetrasodium salt (n-PSA) was purchased from Molecular Probes and used as received. Molecular dynamics simulations indicated that p-PDI has the dimensions of 2.13x1.33x1.89 nm (LWH) and n-PDI 2.13x1.15x1.49 nm (LWH) (Figure 3.1). The perylene core itself has a dimension of 0.72x0.53 nm (LW). The peak molar extinction coefficients in water were measured with 3.28x104 and 2.97x104 M-1 cm-1 for 103 p-PDI (λ = 591 nm) and n-PDI (λ = 568 nm) respectively. Beer’s law was verified for these solutions, suggesting that aggregation of the two PDIs is absent over this concentration range. As discussed in Chapter 3, both of these compounds are fluorescent in the aqueous phase, with the n-PDI having a higher quantum yield of fluorescence by a factor of approximately 4 (φfln-PDI = 0.54 and φflp-PDI = 0.14). However if n-PDI and pPDI are mixed in a 1:1 ratio the fluorescence is strongly quenched (by a factor of ca. 13 relative to p-PDI at an equivalent optical density). As will be discussed later, an analogous quenching also occurs in the MLBL assembly. The absorption spectra of nTDI was shown in Figure 3.11 with it chemical structure illustrated in Figure 3.1. The molar extinction coefficients of n-TDI are 4,000(437nm), 29,000(640nm) and 18,000 M1 cm-1 (685nm). Chemical structure of n-PSA is illustrated in Figure 4.5. 4.2.2 Molecular Layer-by-Layer procedures. The MLBL procedure was carried out by dipping the cleaned quartz substrates alternately into the 0.l g/l p-PDI or n-PDI (or n-TDI or n-PSA) solutions for 10 minutes, starting with the p-PDI layer. After each dipping, there were three washes with DI water to remove the physically absorbed PDI dye. After this step, a gentle stream of argon gas was used to dry the films before they were put into the next negatively charged dye solution. All the absorption and fluorescence spectra were taken after each dipping and drying process. These routines were repeated until desired number of double layers was achieved. The films are denoted in the following by (p-PDI/n-PDI or n-PSA or n-TDI)n where n is the number of double layers. 4.2.3 Instruments and measurements 104 UV-Vis and fluorescence spectra were collected on the same instruments described in chapter 2 and chapter 3. For the AFM measurements a commercial microscope (D-3000, Digital Instruments, Santa Barbara, CA) was used. Images were recorded in tapping mode in air employing cantilevers with a nominal radius of less than 10 nm, resonance frequency at 300 kHz and a spring force constant at 40N/m. 4.3 Results and Discussions 4.3.1 Molecular Layer-by-Layer of p-PDI and n-PDI 4.3.1.1 Absorption spectra of (p-PDI/n-PDI)n The growth of the MLBL PDI thin films was examined by using UV-Vis absorption spectroscopy. A typical series of absorption spectra for the formation of (pPDI/n-PDI)n films on the quartz is shown in Figure 4.1a and Figure 4.1b shows the absorbance at 577nm as a function of the number of monolayers of p-PDI and n-PDI. The strong absorbance between 350nm and 650nm is the typical absorption band of the perylene diimides. For the first two layers there is a small (5nm) blue shift in the peak positions (Table 4.1). After the deposition of several p-PDI/n-PDI double layers, the maximum of the absorption peak for perylene diimide core shifted from 567nm to 577nm. The direct exposure of the first two PDI layers to the quartz surface might contribute to the slight blue shift. A very systematic change in the absorption intensity for both perylene diimide cores and phenyl groups can be easily observed with the increasing of PDI layers, which shows the consistency of the amount of absorbed PDI molecules in the film. The absorbance for phenyl groups as a function of number of PDI layers is shown in Figure 4.1c. The absence of any obvious change such as broadening or shift of the absorption spectra implies that no further aggregates were formed upon MLBL 105 process. For PDI compounds without the bay substituents there is a very strong red-shift in the absorption and fluorescence spectrum upon aggregation.6 The good linearity of the absorbance with the number of PDI monolayers indicates a constant increase in the density of PDI molecules deposited into the film upon each dipping cycle, even for the first one or two monolayers. The peak positions for UV-Vis absorption of (p-PDI/n-PDI)n are the same as those for the mixing of p-PDI and n-PD((1:1 ratio) in solution shown in Figure 3.4, which indicates that the incorporated p-PDI and n-PDI is in a 1:1 ratio for each bilayer. Control experiments dipping only into a p-PDI solution failed to increase the absorbance progressively, even after 10 dipping cycles, which implies that the π-π interaction alone was not sufficient to make the PDI layers (see Figure 4.2). The following rough calculation suggests that only a monolayer of p-PDI or nPDI was formed for every deposition. The surface density, Γ, of PDI for every deposition in (p-PDI/n-PDI)n films can be calculated using Γ =[Aλελ-1NA]x10-3, where Aλ is the absorbance of PDI in the film at a given wavelength, ελ is the extinction coefficient of PDI in solution (M-1 cm-1) at λ, and NA is Avogadro’s number.7 Using the average molar extinction coefficient of 3.0x104 M-1 cm-1 at 578nm for p-PDI and n-PDI, an average surface PDI surface density of 3.64x1013 molecules per cm2/layer is obtained for (p-PDI/n-PDI)n films, which corresponds to an average area per PDI of 2.78 nm2. Assuming a single PDI molecule occupies an area of approximately 2.62 (LxW) nm2 (the average from the earlier estimate of molecular dimensions) the average surface coverage is ~94% which reflects close packing of PDI molecules in each monolayer. 4.3.1.2 Characterization of (p-PDI/n-PDI)n 106 The TM-AFM image shown in Figure 4.3b yields a surface roughness of 2.4nm for (p-PDI/n-PDI)10 annealed at 80ºC over night. As we can be seen from the image, some domain structures exist, which can be attributed to aggregates of either p-PDI or nPDI in water. Measuring the depth of a scratch made on this sample indicates a total film thickness of 14 nm, leading to a PDI layer thickness of 0.70nm. This value is smaller than the PDI thickness (dimension H above) obtained from the molecular dynamic simulation, suggesting the collapse of the PDI aggregates upon deposition. Ellipsomerty was also used for the measurements of the film thickness. For this experiment (p-PDI/n-PDI)n was built onto a silicon wafer. For the fitting of the experiment data, a bare silicon wafer was used to build up a SiO2/Si model and then it was used for the fitting of the experiment data over 700nm for the thickness, because there is no absorbance for the PDI films above 700nm. All the MSE values after fitting were less than 10. The thickness curve is shown in Figure 4.3b. A thickness of 0.56nm for each PDI monolayer can be obtained by this approach. The refractive index was fitted to be around 1.57. 4.3.1.3 Emission spectra of (p-PDI/n-PDI)n Figure 4.4 displays the fluorescence spectra of (p-PDI/n-PDI)n films (excitation wavelength at 540nm) and the fluorescence intensity as a function of number of PDI monolayers. For all the fluorescence spectra only one peak can be observed and the shape and wavelength maxima are very similar to the solution phase spectra.6 The peaks are blue shifted for the first two PDI monolayers and then reach the same peak position at 614nm, which is very consistent with the UV-Vis spectra. Note that the fluorescence intensity decreased after the first double layer, indicating the presence of strong fluorescence quenching with the absorption of second PDI double layer. After that, the 107 fluorescence increased linearly with the number of PDI monolayers. The intensity when n-PDI is on the outside layer is higher than for p-PDI as the outside layer due to the higher quantum yield of n-PDI. We also note that there is essentially no shift in the fluorescence maximum compared to the solution phase. The fact that the fluorescence intensity is sensitive to the outermost layer suggests that excitation energy is transferred to the outer layer before fluorescence occurs (e.g. we hypothesize that the outer layer is acting as a energy trap, regardless of which PDI species is present). If there were no energy transfer, then the fluorescence intensity should be just a simple average of the contribution from each species, since our film is optically thin (e.g. all chromophores have an equal probability to be excited). By carefully comparing the fluorescence intensity of the films with an aqueous solution of p-PDI contained in a small path length cell with a similar OD (the thin cell was used to eliminate any optical artifacts in the front-face fluorescence mode) we find that the fluorescence intensity for the (p-PDI/n-PDI)n film is approximately 13 times lower than for the solution species, just as was found for the 1:1 complexes formed in the solution phase(see Chapter 3). 4.3.2 Molecular Layer-by-Layer of p-PDI and n-PSA Instead of using two very similar molecules for MLBL, the smaller dye molecule PSA was also investigated to test its feasibility for MLBL. The photophysics of n-PSA in water is shown in Figure 4.5a along with the chemical structure of n-PSA. A very structured absorption and emission spectra were observed with three absorption peaks at 379nm, 354nm, and 335 nm (attributed to the transition of S0-S1 for 0,0-1,0, 0,0-1,1, 0,0- 108 1,2, respectively). n-PSA is very fluorescent in water with a QY close to unity and a. Stokes shift of about 21nm. 4.3.2.1 Absorption spectra of (p-PDI/n-PSA)n The absorption spectra of (p-PDI/n-PSA)n were shown in Figure 4.5b and the absorbance at 379nm for PSA (corrected for a slight contribution from p-PDI) and 575nm for p-PDI is plotted as a function of number of double layers in Figure 4.5c. The absorption for p-PDI lies primarily in the range of 400nm to 600nm and n-PSA 340nm to 400nm. The n-PSA absorption is considerably less structured than observed in the solution phase. No formation of n-PSA aggregates could be observed as seen from Figure 4.5a. No obvious stripping was found for either p-PDI or n-PSA deposition. The absorbance for p-PDI and n-PSA increases linearly with the number of p-PDI layers. However, the loading efficiency for p-PDI is not as high as for the p-PDI/n-PDI case. The uptake of p-PDI for (p-PDI/n-PSA)n is about 70% of that for (p-PDI/n-PDI)n. 4.3.2.1 Emission spectra of (p-PDI/n-PSA)n The emission spectra of (p-PDI/n-PSA)n films were taken using excitation wavelengths at both 360nm and 540nm. The emission spectra for the 1st layer of p-PDI and (p-PDI/n-PSA)1 are illustrated in Figure 4.6a. As we can see there is fluorescence emitted from the 1st layer of p-PDI due to the weak absorbance of p-PDI at 360nm. The fluorescence centered at 610nm is from S1-S0 and 420nm from S2-S0. The fluorescence for S2-S0 is due to the twist of the perylene diimide as had been reported by Qu et al..8 Upon deposition of n-PSA onto the p-PDI layer, no fluorescence from n-PSA could be observed but there was a fluorescence increase for p-PDI. We attribute this to the energy transfer from n-PSA to p-PDI. The subtraction of the 1st layer fluorescence of p-PDI from 109 the fluorescence of (p-PDI/n-PSA)1 is shown as the black curve in Figure 4.6a, indicating strong energy transfer from n-PSA to p-PDI. If the (p-PDI/n-PSA)n-1/p-PDI films were excited at 540nm, their emission spectra are given in Figure 4.6b. For n=1, 2, and 3, the fluorescence was strongly quenched and then changed little after that. We attribute this quenching effect to the electron transfer mechanism within the p-PDI and n-PSA layer. This scheme is shown in Figure 4.6c. After photoexcitation, the exciton was formed in p-PDI layer. Because n-PSA has larger band gap, one electron from HOMO of n-PSA can be transferred to the hole of HOMO in p-PDI*, leading to the fluorescence quenching of p-PDI. The very close packing of the double layers makes this process possible (the thickness of each layer in (p-PDI/n-PDI)n is 0.6nm). While with increasing of double layers (n), the ratio of number of layers for p-PDI and PSA became closer to 1, the fluorescence became little changed. 4.3.3 Molecular Layer-by-Layer of (p-PDI/n-TDI)n A larger water soluble π-conjugated molecule, n-TDI, was also investigated for its suitability for the MLBL process. Its absorption and fluorescence spectra were shown in Figure 3.11 in chapter 3. It is believed that H-aggregates of n-TDI are formed in water, due to its non-fluorescent behavior in water.9 4.3.3.1 Absorption spectra of (p-PDI/n-TDI)n The absorption spectra for (p-PDI/n-TDI)n were demonstrated in Figure 4.7a. The spectra showed a very systematic increase of both p-PDI and n-TDI. No stripping effect was observed for either p-PDI or n-TDI deposition, indicating the strong interaction between n-TDI and p-PDI layers. The deposition of n-TDI will lead a little increase in the p-PDI absorption region due to the spectral overlap between p-PDI and n-TDI. The 110 absorbance from p-PDI and n-TDI increased linearly with the number of double layers as shown in Figure 4.7b. The absorbance from p-PDI was corrected from n-TDI absorbance at 575nm. It is worth mentioning that the loading efficiency of p-PDI is higher than that for (p-PDI/n-PDI)n. This may be due to the larger aggregates formed by n-TDI in water. A molecule similar to n-TDI was shown to have the aggregates on the order of 100nm in water.9 The absorbance at 284nm for phenyl groups on both p-PDI and n-TDI increased linearly with the number of deposition monolayers, illustrated in Figure 4.7c. 4.3.3.2 Emission spectra of (p-PDI/n-TDI)n Emission spectra from (p-PDI/n-TDI)n were shown in Figure 4.8. The emission spectrum from the 1st p-PDI layer was shown as the red curve in Figure 4.8 and then the fluorescence was totally quenched upon the next deposition of n-TDI layer. No fluorescence emission was observed for the following p-PDI and n-TDI depositions. Due to the large spectra overlap between the p-PDI emission spectra and n-TDI absorption (see later), the energy transfer should be very efficient between the p-PDI and n-TDI layers. 4.3.4 Molecular Layer-by-Layer of (p-PDI/(n-TDI:n-PSA))n A mixture solution (0.1g/l) of n-TDI and n-PSA (1:1 by mol:mol) was employed for the further investigation of molecular dimension on the behavior of MLBL. The absorption spectra for (p-PDI/(n-TDI:n-PSA))n is shown in Figure 4.9a. Very similar spectra to (p-PDI/n-TDI)n were observed, except that there was also an absorbance from n-PSA around 360nm. The p-PDI loading these two cases is about the same (compare Figure 4.9b and 4.7b). However, The TDI uptake for each deposition in the n-TDI: nPSA mixture is about 75% of that in n-TDI solutions alone (comparing Figure 4.9b and 111 4.7b). The smaller uptake of n-TDI was compensated by the incorporation of n-PSA into the films to finish the charge neutralization and over compensation. Again, no n-PSA extraction was observed upon p-PDI deposition. This experiment demonstrated that the larger π-conjugate molecule (n-TDI) is preferentially absorbed onto the p-PDI layer. 4.3.5 Energy Transfer for TDI/(p-PDI/n-PDI)n FRET has been frequently employed as a useful “spectroscopic ruler” for the studying the macromolecular interaction in biological or polymeric systems.10 In this study it is utilized to study the PDI organization and proved the layered PDI structure in the formed films of (p-PDI/n-PDI)n. n-TDI was selected as the acceptor and absorbed onto the quartz as the foundation layer. Films of (p-PDI/n-PDI)n-1/p-PDI were built up on the n-TDI foundation layer. The strong spectral overlap between fluorescence of the (pPDI/n-PDI)10 MLBL and the absorption of n-TDI is shown in Figure 4.10, which can lead to long-range energy transfer from (p-PDI/n-PDI)n(Donor) to the n-TDI(Acceptor) layer. The R0 for (p-PDI/n-PDI)n films with n-TDI can be calculated according to the following equations. 8.75 × 10 −5 κ 2QD J R0 = n4 (1) J = ∫ FD ( λ )ε A (λ )λ4 dλ (2) 0 Where κ2 is the orientation factor between donor and acceptor molecules, QD0 is the quantum yield of donor fluorescence in the absence of acceptor, and n is the index of refraction(here is taken as 1.57 as obtained from the ellipsometry measurements). J is the spectral overlap between the donor emission spectrum and acceptor absorption spectrum. FD is the normalized donor fluorescence spectrum and εA is the wavelength dependent 112 molar extinction coefficient of the acceptor. R0 is in units of angstroms, whereas λ is in nanometers and εA in M-1cm-1. J was obtained at 2.78994×1015, leading to a R0 of 5.74nm if κ2 is assumed to be 4 (PDI and n-TDI dipole moments are parallel). 4.3.5.1 Absorption spectra for n-TDI/(p-PDI/n-PDI)n The absorption spectra of n-TDI/(p-PDI/n-PDI)n were shown in Figure 4.11a, indicating the progressive building up of the p-PDI/n-PDI MLBL films on top of TDI monolayer. Absorption spectra of n-TDI/(p-PDI/n-PDI)n after subtracting the absorption spectrum of n-TDI foundation layer are shown in Figure 4.11b. The smaller peak around 700nm is interpreted as arising from the interaction between the n-TDI and deposited (pPDI/n-PDI)n. This peak was also observed in the case of (p-PDI/n-TDI)n. In Figure 4.11c is plotted the absorbance at 580nm and 700nm as a function of the number of PDI layers. The PDI loading for each layer deposition is about the same as that for (p-PDI/n-PDI)n discussed earlier as illustrated in Figure 4.1. 4.3.5.2 Energy Transfer for n-TDI/(p-PDI/n-PDI)n The dependence of the donor and acceptor separation distance (d) on the donor fluorescence intensity can be written as: 2b Id 1 = (3) I 0 1 + (d 0 / d ) x Where Id is the fluorescence intensity for donor molecules spaced an average distance of d away from the center of acceptor, I0 is the fluorescence intensity of the donor molecules in the absence of acceptor molecules, and d0 is the critical spacing between donor and acceptor corresponding to a 50% probability of energy transfer. Both d0 and x are a function of the geometry assumed. For example, x is equal to 6 in case of a donor molecule transferring energy to an acceptor molecule in 3-D space, x is equal to 4 113 in the case of a plane donor molecules to a plane of acceptor molecules, and x is equal to 2 in case of energy transfer from a J-type aggregates in a planar array. Eq. 4 can be obtained by the transforming of Eq. 3, shown as the following: Log[(I0/Id)-1]=-x Log(d)+ x Log(d0) (4) Therefore, the plotting of Log[(I0/Id)-1] versus Log(d) can yield the two parameters x and d0. Since the molecular layer-by-layer technique is used, we first assumed that these MLBL films are comprised of discrete, non-interpenetrating layers of PDI monolayer. As discussed before, each monolayer of PDI is about 6Aº thick (dPDI). The estimation of the average distance d from (p-PDI/n-PDI)4/p-PDI to n-TDI is illustrated in Figure 4.12a. The thickness of the n-TDI monolayer (dTDI) is assumed to be 12Aº. d for nth p-PDI layer can be derived as follows: d=1/2dTDI+1/2(2n-1)dPDI=6n+3 Aº (5) The fluorescence intensity of n-TDI/(p-PDI/n-PDI)n-1/p-PDI and (p-PDI/n-PDI)n1/p-PDI versus the number of p-PDI layers(n) are shown in Figure 4.12b. For nth p-PDI layer, I0 is the fluorescence from (p-PDI/n-PDI)n-1/p-PDI and Id is from n-TDI/(p- PDI/n-PDI)n-1/p-PDI. A significant fluorescence quenching was observed during the first several p-PDI layers. After 7 p-PDI layers, the fluorescence increased linearly with the number of p-PDI layers, but still with a decrease in fluorescence intensity compared with the MLBL films for p-PDI/n-PDI without a n-TDI foundation layer. The plot of Log[(I0/Id)-1] vs. Log(d) is shown in Figure 4.12c, yielding as the slope x = 2.03 with an intercept of 3.61. It has been reported that x =2 is appropriate for energy transfer from J-aggregates monolayer assemblies to an acceptor monolayer.10 This 114 indicates that the orientation of PDI within the MLBL films is analogous to J-type aggregates. The value of d0 was obtained from the intercept is 60.4Aº. The energy transfer efficiency for n-TDI/(p-PDI/n-PDI)n-1/p-PDI is shown in Figure 4.13. The 50% energy transfer corresponds to 9.5 p-PDI layers, resulting in a d0 of 60Aº from Eq. 5, which is very consistent with the calculated value of 57.4Aº and simulated value of 60.4Aº. Another approach was used to model energy transfer in the p-PDI/n-PDI films. In this model, we will deal with the energy transfer for the nth p-PDI as the average contribution from 1st, 2nd,·····, n-1th, nth layer of p-PDI. Therefore, the donor fluorescence dependence can be formulated in the following equation: In 1 n 1 = ∑ I 0 n 1 1 + (d 0 / d n ) x (6) Where d0 is assumed to be equal to 60Aº(what we got from above discussion) and the calculation of dn is shown in Eq.5. The plot of In/I0 as a function of p-PDI layers for the experimental data and simulated data for x=2, 3 and 4 is shown in Figure 4.13b. The best fit with experimental data is in case that x is equal to 2, which is consistent with the conclusion we obtained above. 4.4 Summary A new layer-by-layer technique that uses a combination of π-π and electrostatic interactions has been shown to be very versatile for making monolayer self-assembled films from the water soluble π-conjugated dye molecules, which we refer to as the as Molecular Layer-by-Layer (MLBL in short). We have demonstrated that it is possible to fabricate monolayer assemblies by the MLBL technique for two very similar dye molecules such as p-PDI and n-PDI. Both the 115 absorption and emission spectra of MLBL films are identical to the complex of p-PDI: nPDI (1:1) in aqueous solutions. The surface coverage of PDI in the monolayer was calculated to be as high as 94%. However, smaller domains were observed from the AFM image with a RMS roughness of around 2.4nm. This domain structure was attributed to the PDI aggregates present in aqueous solutions, as illustrated in Chapter 3. The strongly quenched fluorescence is evidence for π-stacking and the dependence of the fluorescence intensity on the outermost layer suggests efficient energy transfer between the PDI moieties. In addition to using very similar size dye molecules, a smaller dye molecule ( nPSA) and larger molecule (n-TDI) were also investigated for the fabrication of MLBL films with p-PDI. Comparing with (p-PDI/n-PDI)n, a smaller loading efficiency was observed for (p-PDI/n-PSA)n and a higher loading efficiency for (p-PDI/n-TDI)n was observed. Very efficient energy transfer was observed from n-PSA to p-PDI upon the excitation of n-PSA. When p-PDI was excited at 540nm, fluorescence quenching was also observed, which we presume originates from electron transfer from n-PSA to p-PDI* (Figure 4.6c). If a 1:1 mole:mole mixture of n-TDI and n-PSA was utilized as the dipping solution for the negatively charged layer, MLBL films were built up for (p-PDI/(n-TDI:nPSA))n. The p-PDI uptake under this condition is very similar with the case for (p-PDI/nTDI)n. However, this experiment demonstrated that the larger π-conjugate molecule (nTDI) is preferentially absorbed onto the p-PDI layer, almost certainly a reflection of the strong π-stacking interaction. The uptake of n-TDI is about 75% of that for (p-PDI/nTDI)n. No extraction of dye molecules was observed upon depositing of the next dye molecule layer. 116 An energy transfer study for n-TDI/(p-PDI/n-PDI)n indicated that the PDI orientation in the planar films is analogous to J-type aggregation. The critical energy transfer distance was estimated from calculation and experiment to be approximately 60Aº. We think the MLBL technique could be applied for making films for future applications in thin film transistors, OLED or other organic-photonic devices from appropriate charged conjugated dye molecules. 4.5 References 1. (a) Decher, G., Hong, J. Makromol. Chem. Symp. 1991, 46, 321. (b) Decher, G. Science 1997, 277, 1232. 2. (a) Schlenoff, J. B., Dubas, Stephan, T., Farhat, T. Langmuir 2000, 16, 9968. (b) Baur, J. W., Rubner, M. F.; Reynolds, J. R.; Kim, S. Langmuir 1999, 15, 6460. 3. Ulman,A. An Introduction to Ultrathin Organic Films from Langmuir-Blodgett to Self Assembly. Academic Press: New York, 1991, Chapter 3. 4. (a) Moon, H., Zeis, R., Borhent, E.J., Besnard, C., Lovinger, A. J.; Siegrist,T.; Kloc, C.; Bao, Z. N. J. Am. Chem. Soc. 2004, 126, 15322. (b) Curtis, M.D., Cao, J., Kampf, J.W. J. Am. Chem. Soc. 2004, 126, 4318. 5. (a) Malenfant, P.R.L., Dimitrakopouls, C.D., Gelmore, J.D., Kosbar, L.L., Graham, T.O., Curioni, A., Andreoni, W. Appl. Phys. Lett. 2002, 80, 2517. (b) Chesterfield, R.J.; Mckeen, J.C.; Newman, P.R.; Ewbank, P.C.; da Silva Filho, D. A., Brédas, J-L., Miller, L. L.; Mann, K. P., Frisbie, C.D. J. Phys. Chem. B, 2004, 108, 19281. 117 6. (a) Law, K.Y. Chem. Rev. 1993, 93, 449. (b) Liu, S-G., Sui, G., Cormier, R. A., Leblanc, R. M., Gregg, B. A. J. Phys. Chem. B 2002, 106, 1307. (c) Wang, W., Han, J. J., Wang,L-Q., Li, L-S., Shaw; W. J., Li, A. D. Q. Nano Lett. 2003, 3, 455. 7. (a) Caruso, F., Kurth, D. G., Volkmer, D., Koop, M. J., Müller, A. Langmuir 1998, 14, 3462. (b) Li. D., Swanson, B. I., Robison, J. M., Hoffbauer, M. A. J. Am. Chem. Soc. 1993, 115, 6975. 8. Qu. J., Zhang, J., Drimsdale, A. C., Müllen, K., Jaiser, F., Yang, X., Neher, D. Macromolecules 2004, 37, 8197. 9. Jung, C., Müller, B., Lamb, D.C., Nolde, F., Müllen, K., Bräuchle, C. J. Am. Chem. Soc. 2006, 128, 5283. 10. (a) Stryer, L. Annu. Rev. Biochem. 1978, 47, 819. (b) Bokinsky, G.; Zhuang, X.W. Acc. Chem. Res. 2005, 38, 566. 11. Nakahara, H., Fukuda, K., Möbius, D., Kuhn, H. J. Phys. Chem. 1986, 90, 6144. 12. Johnson, G. E. Macromolecules 1980, 13, 145. 118 0.06 Absorbance a 0.04 0.02 0 Absorbance at 580nm 250 0.04 350 450 550 Wavelength(nm) 650 750 850 b 0.03 0.02 0.01 0 0 2 4 6 8 10 12 14 Number of PDI Layers 4 6 8 10 12 Number of PDI Layers 16 18 20 0.06 c Absorbance at 284nm 0.05 0.04 0.03 0.02 0.01 0 0 2 14 16 18 20 Figure 4.1 (a) UV-Vis absorption spectra of the monolayer of p-PDI (Black) and n-PDI (Red). (b) The peak absorbance around 575 nm as a function of monolayer PDI with pPDI as the first layer. (c) The peak absorbance around 284nm as a function of monolayer PDI with p-PDI as the first layer. 119 Absorbance 0.006 0.004 0.002 0 250 350 450 550 Wavelength(nm) 650 750 Figure 4.2 The absorption spectra of p-PDI dipping cycles up to 10. Each dipping is about 10 minutes. 120 30 b Thickness(nm) 25 20 y = 0.5699x 15 10 5 0 0 10 20 30 40 50 Number of PDI Layer Figure 4.3 (a) The TM-AFM image of (p-PDI/n-PDI)10 annealed at 80ºC overnight. (b) Ellipsometric film thickness as a function of PDI monolayers. 121 Fluo. Inten. a 560 610 660 710 Wavelength(nm) 760 Fluo. Inten.(a.u.) b 0 1 2 3 4 5 6 7 8 Number of Double Layer 9 10 11 Figure 4.4 (a) Fluorescence emission spectra from the monolayer of p-PDI/n-PDI with pPDI as the first layer (b) The fluorescence intensity as a function of double layer p-PDI/nPDI (diamonds correspond to n-PDI as the outside layer and the circles p-PDI as the outside layer). The lines between points illustrate the order of deposition (e.g. p-PDI first, followed by n-PDI). 122 a Absorbance 0.04 NaO4S SO4Na NaO4S SO4Na 0.03 0.02 Fluo. Inten. 0.05 0.01 0 300 350 Absorbance 0.016 400 Wavelength(nm) 450 500 b 0.012 0.008 0.004 0 300 400 500 600 Wavelength(nm) 700 800 Absorbance 0.016 c 0.012 0.008 0.004 0 0 1 2 3 4 5 6 7 Number of Double layer 8 9 10 Figure 4.5 (a) Absorption and emission spectra of n-PSA in water with the inset showing n-PSA’s chemical structure. Ex=360nm. (b) Absorption spectra of monolayer of p-PDI and n-PSA starting with p-PDI layer. (c) Peak absorbance as a function of double layer (p-PDI/n-PSA) for p-PDI at 575nm (circles) and n-PSA at 379nm (diamonds) after correcting from the absorbance at 379nm for p-PDI. 123 Fluo. Inten. a 380 420 460 500 540 580 Wavelength(nm) 620 660 700 Norm. Fluo. Inten. 1 Fluo. Inten. b 0.8 0.6 0.4 0.2 1 560 610 c 2 3 4 5 6 7 8 9 Number of p-PDI Layers(n) 660 710 Wavelength(nm) 10 760 LUMO LUMO eT HOMO HOMO n-PSA p-PDI* n-PSA p-PDI Figure 4.6 (a) Emission spectra of 1st p-PDI (Red) monolayer and (p-PDI/n-PSA)1 (Pink). Black curve is the subtraction of 1st p-PDI and (p-PDI/n-PSA)1. Ex=360nm. (b) Emission spectra of (p-PDI/n-PSA)n-1/p-PDI with Ex=540nm. (c) Electron transfer mechanism from the HOMO of PSA to p-PDI exciton. 124 0.06 a Absorbance 0.05 0.04 0.03 0.02 0.01 0 240 300 360 420 480 540 600 660 720 780 840 900 Wavelength(nm) 0.03 Absorbance b 0.02 0.01 0 Abosrbance at 284nm 0 1 0.05 2 3 4 5 6 Number of Double Layers 7 8 9 c 0.04 0.03 0.02 0.01 0 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Number of Depsition Layers Figure 4.7 (a) UV-Vis absorption spectra of the monolayer of p-PDI (Black) and n-TDI (Red). (b) The peak absorbance around 575 nm for p-PDI (black circles) and 700nm for n-TDI (red diamonds) as a function of double layers. (c) The peak absorbance around 284nm as a function of deposition layers with p-PDI as the first layer. 125 Fluo. Inten. (a.u.) 560 600 640 680 Wavelength(nm) 720 760 Figure 4.8 Emission spectra of (p-PDI/n-TDI)n with Ex=540nm. 126 800 0.05 a Absorbance 0.04 0.03 0.02 0.01 0 240 300 360 420 480 540 600 660 720 780 840 900 Wavelength(nm) 0.03 Absorbance b 0.02 0.01 0 0 1 2 3 4 5 6 Number of Double layers 7 8 9 Figure 4.9 (a) UV-Vis absorption spectra of the monolayer of p-PDI (Black) and nTDI:n-PSA (Red). (b) The peak absorbance around 575 nm for p-PDI (black circles) and 700nm for n-TDI (red diamonds) as a function of the number of double layers 127 Fluo. Intensity or ε 560 610 660 710 760 Wavelength(nm) 810 860 Figure 4.10 Emission spectrum of (p-PDI/n-PDI)10(Red) and absorption spectrum of nTDI (Green). 128 Absorbance 0.06 a 0.04 0.02 0 Absorbance 340 420 500 580 660 Wavelength(nm) 740 820 900 b 0.06 0.04 0.02 0 c 0.06 0.04 0.0021 0.02 0 Absorbance at 700nm Absorbance at 579nm 250 300 350 400 450 500 550 600 650 700 750 800 850 Wavelength(nm) 0.0042 0 0 2 4 6 8 10 12 14 16 18 20 22 24 Number of PDI Deposition Layers Figure 4.11 (a) UV-Vis absorption spectra of n-TDI/(p-PDI/n-PDI)n. Green: n-TDI, Black: p-PDI and Red: n-PDI. (b) Absorption spectra of n-TDI/(p-PDI/n-PDI)n upon subtracting the absorption spectrum from n-TDI foundation layer. Black: p-PDI and Red: n-PDI. (c) The peak absorbance around 579 nm as a function of monolayer PDI with pPDI as the first layer. The absorbance was corrected from n-TDI spectrum (black circles). Red diamonds represent the absorbance around 700nm shown in Figure 4.11b as a function of the number of PDI deposition layers. 129 a d Fluo.Inten. b 0 1 Log[(I0/Id)-1] 2.5 2 3 4 5 6 7 8 Number of p-PDI layers 9 10 11 12 c 2 1.5 1 0.5 y = -2.0269x + 3.6097 0 -0.5 0.7 0.9 1.1 1.3 1.5 Log(d) 1.7 1.9 2.1 Figure 4.12 (a) Schematic representation of deriving the average distance from (p-PDI/nPDI)4/p-PDI to the center of n-TDI layer. (b) Fluorescence emission intensity for (pPDI/n-PDI)n and n-TDI/(p-PDI/n-PDI)n as a function of PDI double layers. (c) Plot of Log[(I0/Id)-1] versus Log(d), yielding a slope of 2.02 and intercept 3.61. 130 Energy Transfer Efficiency 1 0.8 a 0.6 0.4 0.2 0 1 0.6 3 4 5 6 7 8 Number of p-PDI Layers 9 10 11 12 b 0.5 I0/Id 2 0.4 0.3 Exp. Data x=2 x=4 x=6 0.2 0.1 0 0 1 2 3 4 5 6 7 8 Number of p-PDI Layers 9 10 11 12 Figure 4.13 (a) Energy transfer efficiency as a function of number of p-PDI layers. (b) pPDI fluorescence dependence as a function of number of p-PDI layers. ● represents the experimental data; ▲: x=2; ♦: x=4; ■: x=6. 131 Chapter 5: Incorporation of Water Soluble Perylene Diimides into Polyelectrolyte Films by the Layer-by-Layer Method 5.1 Introduction Organized perylene diimide thin films have been prepared previously by vapor deposition, spin casting, self-assembly via hydrogen bonding and electrostatic interaction, and Langmuir-Blodgett techniques.1 These films have been shown potential applications for making heterojunction photovoltaics and n-type organic semiconductors. The discovery of layer-by-layer (LBL) technology has inspired researchers to make functional films with desired compositions and controlled thickness during the last 15 years.2 Because of its simplicity, reproducibility and film uniformity over a large area the LBL technique has been widely used in fabricating all kinds of hetero-structural films incorporating dendrimers, quantum dots, polymer micelles, proteins and even carbon nanotubes in support of applications such as chemical sensors, fuel cells, nano- or ultra filtration, OLED etc.3 Recently the introduction of small dye molecules into the LBL film fabrication has drawn considerable interest due to potential applications such as photon harvesting arrays, OLED, controlled drug release and so on. However, a typical problem encountered during the LBL fabrication process is the extraction of small molecules by the next polyelectrolyte layer deposition.4 In this chapter we demonstrate that water soluble PDIs can avoid this problem. The Förster Resonance Energy Transfer study between PDI layers and brilliant green was carried out to elucidate the film structure. In possible future work the incorporation of PDI into films composed of water soluble conducing polymers such as sulfonate polyaniline will be very interesting for the study of 132 OLED and photovoltaics and photon harvesting studies instead of the inert polyelectrolytes such as PSS or PDAC discussed in this Chapter. 5.2 Experimental Section 5.2.1 Materials Same as stated in the experimental sections in Chapter 3.2.1 and Chapter 4.2.1. 5.2.2 Layer-by-Layer Procedures The quartz substrates were dipped in fresh Piranha solution for 10 minutes (H2SO4:H2O2=7:3 v/v, extremely corrosive, be careful!!) followed by RCA solution for 10 minutes (NH4OH:H2O2:H20=6:2:2, v/v/v), both at 60°C, and then baked at 420 ºC for 6 hrs. The advancing contact angle with water was measured to be approximately 5º for all the substrates. Then the quartz was coated with the PEI layer to make it evenly charged with a high density of cations. The LBL dipping time for all solutions was 10 minutes, with both sides of the quartz substrate exposed to the dipping solution. A gentle stream of argon gas was employed to dry the films between each dipping step. For n-PDI LBL film fabrication a double layer of PSS/PDAC was used as a foundation layer before the n-PDI layer was deposited. Films with 1, 3 and 5 separation layers (SLs) between the adjacent n-PDI layers were fabricated, whose film structure can be denoted as PEI/PSS/PDAC/(n-PDI/PDAC/(PSS/PDAC)m)n. 1, 3, and 5 SLs correspond to m = 0, 1 and 2 respectively. If p-PDI was used as the PDI building block, the films can be represented as PEI/PSS/PDAC/PSS/(p-PDI/PSS/(PDAC/PSS)m)n with a 3 layer foundation. It is worth mentioning that the number of PSS layers in p-PDI LBL films is nearly twice that in n-PDI films (for n-PDI films the number of PSS layers is NPSS = 1 + mn and for p-PDI films NPSS = 2 + (m + 1)n). Films with 2SLs between p-PDI and n- 133 PDI layers were also fabricated, whose film structure can be symbolized as PEI/PSS/PDAC/PSS/(p-PDI/PSS/PDAC/n-PDI)n. “Precursor films” of PEI/(PSS/PDAC)n (n=1, 2 and 3) were fabricated in 0.5 M NaCl for the study of the rate of diffusion of nPDI into the LBL films. For the study of the rate of diffusion of p-PDI into LBL films precursor films of PEI/(PSS/PDAC)n/PSS (n = 0, 1, and 2) were fabricated in 0.5 M NaCl. 5.2.3 Spin-Assisted Layer-by-Layer (SA-LBL) Brilliant Green (BG) was used as energy acceptor for the energy transfer study. However, standard dipping experiments showed significant BG penetration into the LBL films, which totally quenched the PDI fluorescence. Therefore, a strategy of spin-casting BG on top of the dried non-swollen solid films was employed so that the short exposure time of the BG to the LBL would reduce the BG penetration. BG was immediately spun cast at a speed of 2000 rpm after being drop cast onto the PDI films and then the film was washed four times by DI water at the same speed. BG was spun-coated onto both sides of the LBL films. 5.2.4 UV-Vis and Fluorescence Spectroscopy Measurements Both UV-Vis and fluorescence spectroscopy were measured to monitor the LBL deposition process. Absorption spectra were obtained with a HP 8453 diode array spectrometer. The fluorimeter used was a SPEX Fluorolog-τ2 equipped with a 450 W xenon light source, Czerny-Turner double grating excitation and emission monochromators. A photomultiplier voltage of 950V was typically used and the excitation and emission slit widths were set at 2/2/4/4mm for LBL samples. The quartz disk was fixed into a homemade sample holder that allowed the fluorescence to be 134 collected in front face mode. In order to average out positional uncertainties all the fluorescence spectra were obtained in triplicate for every PDI layer deposition. 5.2.5 Atomic Force Microscopy (AFM) Measurements For the AFM measurements a commercial microscope (D-3000, Digital Instruments, Santa Barbara, CA) was used. Images were recorded in tapping mode in air employing commercial available cantilevers with a nominal radius of less than 10 nm, resonance frequency at 300 kHz and a spring force constant at 40N/m. The rms surface roughness of n-PDI LBL films were 3.5nm and 2.6 nm, before or after n-PDI deposition. 5.2.6 Förster Resonance Energy Transfer (FRET) Study FRET has been frequently employed as a useful “spectroscopic ruler” for the studying the macromolecular interaction in biological or polymeric systems.5 In this study it is utilized to study the PDI distribution in the fabricated LBL films. R0, which is the characteristic distance at which 50% energy transfer efficiency is achieved, is calculated by the following equations: 6 R0 = 9000(ln 10)κ 2 * QD J 128π 4 Nn 4 ∞ J =∫ 0 FD (ν )ε A (ν ) ν4 dν (1) (2) 3 7.66 × 10−8 C0 = (3) R0 QD is the quantum yield of the donor (p-PDI or n-PDI); FD(ν) is the normalized fluorescence spectrum of the donor; εA(ν) is the molar extinction coefficient of the acceptor in M-1cm-1, J is overlap integral with the units cm4M-1cm-1, which is further expressed in Eq. 2, and ν is the wave number in cm-1.5 <κ2> is the orientational factor 135 for dipoles which is taken to be 2/3 for the R0 calculations presented here. In homogeneous solution the characteristic concentration (C0) required for 76% quenching is given by Eq. 3, given in mM in the following. The absorption and emission spectra for p-PDI or n-PDI overlap, and their R0 values for self-transfer in aqueous solutions were calculated to be 3.58 nm (C0 = 9.79 mM) and 4.50 nm (C0 = 4.93 mM), respectively. R0 values for p-PDI/BG and n-PDI/BG are 4.7 and 5.2 nm, corresponding to C0 at 4.3 and 3.2mM, respectively. The rate of energy transfer is also depends on the orientation of the transition dipoles of the donor and acceptor and is usually expressed by the equation kET = kD0 <κ2> (R0/RDA)6 (4) In this equation <κ2> reflects the appropriate average value of the angular dependence of the dipole-dipole interaction and can vary from 0 to 4. For rapidly rotating molecules the value is 2/3, as we used for the above calculations, but for randomly oriented and static molecules the value of <κ2> is 0.476.5b All J, R0 and C0 are listed in Table 5.1. These R0 values are on the order of the thickness of individual deposited polyelectrolyte layers and suggest that inter- and intra-layer energy transfer could occur (see later discussion). 5.3 Results and Discussions 5.3.1 Dye Penetration into PEI/(PSS/PDAC)n LBL Films A control experiment was carried out to assess the ability of n- or p-PDI to diffuse into “precursor films” PEI/(PSS/PDAC)n (n = 1, 2 and 3 were investigated here) (see Experimental). The absorbance and fluorescence intensity as a function of the dipping time is illustrated in Figure 5.1a and b respectively. The absorption and fluorescence 136 emission spectra for n = 3 are similar to the spectra of the PEI/PSS/PDAC/(nPDI/PDAC/PSS/PDAC)n LBL film (see Figure 5.2-5.3). The time required for the n-PDI concentration to reach equilibrium in the film is about 10 minutes, which sets the disposition time scale for the PDI LBL film fabrication. It can be seen that for thicker PEI/(PSS/PDAC)n films more n-PDI is absorbed. This implies the presence of extensive penetration and migration of the PDI molecules or their solution-phase aggregates within the LBL films, such that the PDI is not necessarily deposited in a well-defined “layer”. Similar experimental results were obtained for p-PDI and (PSS/PDAC)n/PSS (n=0, 1 and 2) LBL films, except that the fluorescence intensity decreased for longer dipping times. This indicates that self-quenching of p-PDI occurred after being incorporated into the polyelectrolyte. The absorbance and fluorescence intensity as a function of dipping time were illustrated in Figure 5.3. 5.3.2 LBL of n-PDI or p-PDI with PSS and PDAC 5.3.2.1 LBL with 0.5M NaCl in Polyelectrolyte Solution A typical series PDI/PDAC/PSS/PDAC)n of absorption spectra of PEI/PSS/PDAC/(n- with 3 SLs and different numbers of n-PDI deposition layers are shown in Figure 5.4a, together with the inset showing the absorbance at the maximum (~580 nm) as a function of number of n-PDI layers. In this case, PSS and PDAC solutions were made in 0.5M NaCl. The absorption spectrum is almost identical to the complex between n-PDI and PDAC in homogeneous solution (Figure 3.13a). The maxima peak position for n-PDI LBL with 3SL is located at 576nm, which is an 8 nm red shift compared to n-PDI in aqueous solution. The linearly increased absorbance of the nPDI is unchanged when the next double layer (PDAC/PSS) is deposited (see the inset of 137 Figure 5.4a), which indicates that no n-PDI was extracted by the subsequent PDAC and PSS depositions. Figure 5.4b shows the same result when p-PDI is used as the building block with PSS and PDAC for making PEI/PSS/PDAC/PSS/(p-PDI/PSS/PDAC/PSS)n. The absorption is identical to the complex between p-PDI and PSS in homogeneous solution. Again, no extraction of p-PDI was observed upon the deposition of PSS/PDAC double layer (see inset in Figure 5.4b. This behavior is unusual as extraction of small dye molecules is often observed by the deposition of subsequent polyelectrolyte layers.4 The emission peak position for the fluorescence in 3SL n-PDI LBL films is at 614 nm (Figure 5.5a), a 3 nm blue shift comparing with the n-PDI solution.6 The fluorescence intensity showed a good linearity with the number of n-PDI layers, although there is a discernable change in the fluorescence intensity when the polyelectrolyte double layer is deposited on top of the PDI. The non-zero intercept of the fluorescence intensity vs. the number of n-PDI layers indicates that the fluorescence quantum yield is diminished for n > 1, presumably because of additional self-quenching and interlayer FRET. A typical double layer of PSS/PDAC has a thickness about 12nm, as verified by the ellipsometry measurements.7 The R0 for n-PDI/n-PDI is 4.5nm. Therefore, no efficient FRET will occur if the n-PDI is sharply confined to a single thin layer. The dipping experiments with precursor films showed the strong tendency of PDI to penetrate into the LBL films. Therefore, if there is migration of n-PDI within the films this may permit FRET. By carefully comparing the fluorescence intensity of the films with an aqueous solution of nPDI contained in a small path length cell with a similar OD (the thin cell was used to eliminate any optical artifacts in the front-face fluorescence mode) we find that the fluorescence intensity for the LBL film is approximately 33 times lower than for the n- 138 PDI solution species. This value is much smaller than the quenching factor (~220) of nPDI by PDAC in homogeneous solution (see Figure 3.13a and 3.14a, Chapter 3). We ascribe this loss of fluorescence to self-quenching as the local concentration of the PDI moieties is estimated to be fairly high (see later discussion). The fluorescence spectra of p-PDI LBL films having 3 SLs are displayed in Figure 5.5b along with the inset showing the fluorescence intensity versus the number of p-PDI layers. A single emission peak was observed at 620nm, with a red shift about 2 nm compared to the p-PDI solution. A quenching factor of 25 was obtained from the thin cell measurements, compared to an enhancement by a factor of ca. 4 for the p-PDI/PSS complexation at higher PSS concentration. We always find that the deposition of PSS onto p-PDI increases the fluorescence intensity and the subsequent deposition of PDAC decreases it. This is illustrated in Figure 5.6a for p-PDI and 5 SLs. We can propose that the first increase with the addition PSS layer is due to the migration of p-PDI molecules into it, thereby decreasing of the p-PDI local concentration and hence diminishing self-quenching (a large excess of PSS can also increase p-PDI’s fluorescence intensity in the solution phase, as shown in Figure 3.14b). Addition of the PDAC layer reverses this result as the PDAC competes with the p-PDI for the anionic PSS groups, once again compressing the p-PDI layer and enhancing self-quenching. If this is the mechanism for the phenomenon presented in Figure 5.6a, then it surprises us that it persists for so many depositions and implies considerable co-mixing of the individual layers in the LBL film.31 In Figure 5.6b is given the n-PDI LBL films with 5SLs. It can be seen that the fluorescence did not change as much as that for the p-PDI case (Figure 5.6a). The deposition of PDAC following the n-PDI layers leads to a smaller fluorescence 139 decrease, but not as much as that for n-PDI quenched by PDAC in homogeneous solution (Figure 3.13a and Figure 3.14a). Analogous to the p-PDI results above, depositing PSS next to PDAC resulted in an appreciable fluorescence increase. 5.3.2.2 LBL with no NaCl in Polyelectrolyte Solution In Figure 5.7a and b are displayed the UV-Vis absorption spectra of n-PDI or pPDI with PSS and PDAC with 3 SLs without the use of NaCl during the LBL procedure. Insets showed the absorbance around 580nm as a function of PDI layers. Similar spectra and peak positions were observed as when 0.5 M NaCl was used. However, two main different features were observed here compared with the LBL when 0.5 M NaCl salt was employed. Firstly, the loading capacity of PDI into the films is about 4x smaller than when NaCl is present. We presume that this phenomenon originates from the fact that thicker deposition layers of PSS and PDAC are achieved when 0.5 M NaCl was used during the LBL process.7 Secondly, some minor extraction of n-PDI by PDAC or p-PDI by PSS was observed under this deposition condition. This is undoubtedly a result of the diminished screening which enhances the electrostatic attraction between the PDI and the oppositely charged polyelectrolyte. The enhanced extraction of n-PDI by PDAC/PSS compared with that of p-PDI by PSS/PDAC is due to the less hydrophobic nature of nPDI. Figure 5.8a and b display the fluorescence emission spectra as a function of the number of n-PDI and p-PDI layers respectively with 3SLs under the condition where no NaCl was used. The insets showed the fluorescence intensity as a function of PDI layers. Peak positions were centered at 614nm and 625nm for n-PDI and p-PDI LBL, which are corresponding to the blue shift of 4nm and 5nm comparing to their aqueous solution 140 phases, respectively. It can see clearly seen that the overall fluorescence intensities for the same PDI layer with no NaCl used during the LBL procedures are higher than that with 0.5 M NaCl utilized (Figure 5.5a and b correspondingly). Thin cell experiments showed quenching factors of about 15 and 18 for n-PDI and p-PDI LBLs relative to their solution phases. This is attributed to the fact that the loading efficiency of PDI is lower when no NaCl was used in the dipping media, leading to less self-quenching. An obvious fluorescence increase was observed after carrying out a double layer polyelectrolyte deposition on top of n-PDI. As has been shown in Figure 5.5b, this might be due to the compressing of n-PDI from PDAC layer to n-PDI layer. Therefore, the quenching from PDAC is diminished. Formation of LBL films of p-PDI and n-PDI with 1 and 5 SLs was also carried out. The very strong interactions between PDIs can make it possible to build up the PDI/PSS or PDAC LBL films with 1SL. The characteristics of p-PDI and n-PDI LBL films such as maxima peak positions for both absorption and fluorescence emission spectra, slopes of absorbance as a function of the number of PDI layers and quenching factor (e.g. comparison with the thin cell measurements) are given in Table 5.2 for 1, 3, and 5 SLs. As we can see in Table 5.2, the amount of PDI deposited (abs slope) was essentially independent of the number of SLs (1, 3 and 5), implying that each PDI layer (which presumably achieved charge overcompensation) acts as a barrier for the next one deposited. When 0.5M NaCl was used during the LBL, the loading efficiency of PDI is about 4 times higher than that when no NaCl was used. The fluorescence quenching is diminished as the number of SLs increases, presumably because there is less selfquenching due to the migration of PDI into the adjacent polyelectrolyte layers. This effect 141 is also illustrated in Figure 5.9, which shows the fluorescence intensity increasing steadily with the number of separation layers but the amount of PDI increases linearly with the number of PDI depositions. Therefore, the separation layers between the PDI layers behave like inert buffer layers that will accommodate the same amount of PDI molecules but with a lower overall local concentration. 5.3.2.3 Characterization of p-PDI or n-PDI in the LBL films Two TM-AFM images are illustrated in Figure 5.10a and b for p-PDI LBL films with 3SLs and 0.5 NaCl used for the polyelectrolyte solutions. The surface became smoother when p-PDI was absorbed on top of the films. The RMSs before and after pPDI deposition were measured to be 3.5 and 2.6 nm, respectively, which was similar to the case of 1, 3, 6, 8-pyrene tetrasulfonic acid salt self assembled with PSS and PAH.4b In the literature, two main species of PDI aggregates are distinguished: Haggregates with the face-to-face or card-pack stacking configuration leading to an absorption blue shift in respect to the solution phase and fluorescence annihilation and Jaggregates with the head-to-tail configuration leading to the red shift of both absorption and fluorescence spectra.8 For our PDI LBL films there is only a slight red shift of the absorption spectra and blue shift of the emission spectra, compared with their solution phases. Thus we conclude that there is no evidence for the formation of H- or Jaggregates in our fabricated LBL films, and therefore the n-PDI or p-PDI moieties are randomly embedded into the adjacent polyelectrolyte films (due to the fact that the loading efficiency for 1SL is the same as that for 3 and 5 SLs). However, as we will see later, there are very strong interactions between the loaded PDIs. 142 Based on the estimated polyelectrolyte layer thickness of 6 nm when 0.5M NaCl is used,7 the local n-PDI and p-PDI concentration in a single layer can be deduced from Beer’s Law as 0.64M and 0.56 M respectively (this estimate ignores diffusion of the PDI into adjacent layers, which as discussed above almost certainly occurs). Therefore, the loading efficiency of PDI in the LBL films is much higher than that for the NaCl-free polyelectrolyte solutions. This concentration corresponds to a volume per molecule of ca. 2.3-2.6 nm3, which is smaller than the approximate molecular volume in water (3.65 nm3) estimated from MD simulations (see 3.9). Thus there are ample opportunities for molecular interactions or the formation of larger aggregates that could lead to quenching. These estimated local concentrations are also much higher than C0 (see eq 3 above) for these molecules, suggesting that facile intralayer Förster energy transfer could occur. If there is no NaCl utilized during the LBL process, the local concentration of n-PDI and pPDI was calculated on the order of 0.25M. As discussed above, the decrease of the quenching factor with the number of SLs is attributed to the n-PDI or p-PDI’s partial migration into the adjacent layers. The diffusion away from such a high local concentration of PDI into the adjacent layers could easily provide a concentration higher than C0 (9.79 and 4.93mM for p-PDI and n-PDI respectively Table 5.1), thus leading to the interlayer FRET. The scheme in Figure 5.11 represents these ideas. 5.3.3 Energy Transfer Study Between PDI films and BG. The following experiments were designed to test the interlayer energy transfer efficiency, since other workers have suggested LBL films containing dyes as photon harvesting devices.9 Our objective in these experiments was to place a layer of an energy acceptor at the outermost layer of a film containing PDI molecules and see if there was 143 extensive energy migration between the PDI layers by measuring the fluorescence quantum yield as a function of the number of films cast. Brilliant Green (BG), which has been widely used as a optical probe or biological dye was chosen as a energy acceptor due to its good spectral overlap with the PDI emission (see earlier R0 and C0 estimates, Table 5.1) As was pointed out above, the double layer thickness of PDAC/PSS is around 12 nm, 7 in which case FRET between the PDI moieties in different layers is not expected to be efficient (see the R0 estimates in Table 5.1). Since we found that BG from bulk water is able to penetrate into the as-formed LBL films, a spin-casting technique was employed in an attempt to confine the BG to the outermost layer of the LBL film (see the Experimental Section), but as we will see in the following, it does not seem that confinement of the BG to the outermost layer was very successful. The FRET efficiency was measured by comparing the fluorescence of a particular film before and after deposition of BG. First we measured the FRET efficiency versus the concentration of spin-casting BG solution for the film PEI/PSS/PDAC/(nPDI/PDAC/PSS/PDAC)3/n-PDI/PDAC/PSS/BG (2SL between outermost n-PDI and BG layers, see Figure 5.12a) and for the film PEI/PSS/PDAC/PSS/(p- PDI/PSS/PDAC/PSS)3/p-PDI/PSS/PDAC/PSS/BG (3SLs between outermost p-PDI and BG layers see Figure 5.12b). Both curves are very similar except that the BG concentration for n-PDI films is much larger than for p-PDI to achieve similar quenching efficiencies. Likewise the plot of FRET vs. the number of layers is very similar for n-PDI and p-PDI except for the very large difference in the BG concentration required (Figure 5.13a and b). Given the similarity in the R0 values for n-PDI and p-PDI for energy transfer to BG the data in Figure 5.13 cannot be due to the long distance energy transfer 144 between PDI molecules but instead indicate extensive BG penetration into the films (at least on the order of 6-7 layers). We suggest that the very large difference between the required BG concentrations to achieve efficient PDI quenching is because of the different local environment of the PDI moiety. A layer of p-PDI is surrounded on both sides by PSS, which would be expected to strongly attract the cationic BG. Thus if there is significant intermingling of the PSS layers and the p-PDI, the BG could be brought into relatively close proximity to the p-PDI.10 By the same argument, the BG would be excluded from the immediate vicinity of the n-PDI (these qualitative ideas are represented in Figure 5.14). Thus it would seem that these experiments designed to test for inter-layer energy transfer have revealed more about the diffusion of a relatively large dye molecule (BG) into the film and the segregation of this dye within the multilayer assembly. 5.4 Summary In this work we have explored the incorporation of charged water soluble perylene diimide dyes into polyelectrolyte multilayers using the layer-by-layer (LBL) dipping technique. An underlying motivation for this study is the potential for such layers to act as photon harvesting arrays given the fairly large R0 values for same-species Förster resonance energy transfer (calculated to be 3.58 nm and 4.50 nm for p-PDI/ p-PDI and nPDI/ n-PDI respectively), which are on the order of the thickness of individual polyelectrolyte layers. These PDI species have relatively high fluorescence quantum yields in water and it was hoped that this property would be retained in polyelectrolyte films, but in fact there was quite considerable fluorescence quenching (by factors from 9 145 to 43 – see Table 5.2), which we have interpreted as self-quenching (we estimate the local concentration in the layers to be on the order of 0.5 M). Both p-PDI and n-PDI could be incorporated into PSS and PDAC polyelectrolyte films by the standard dipping LBL technique. Unlike many other cases of dye incorporation into polyelectrolyte films, the PDI moieties were not stripped by subsequent polyelectrolyte depositions. Each PDI layer (with variable numbers of separation layers) is evidently able to block excess deposition of PDI in subsequent depositions. The evidence for this is presented in Figure 5.9 and Table 5.2. For PSS/PDAC “precursor films’ prepared without PDI the amount of PDI uptake increases with the dipping time to a limiting value that increases with the number of separation layers (Figure 5.1 and Figure 5.3) (that is, the thicker the film the more PDI can be adsorbed). However for LBL films in which PDI is absorbed with variable numbers of separation layers (SL) there is essentially no difference in the PDI uptake (see Table5.2) suggesting that new PDI cannot intrude on the volume of the polyelectrolyte previously saturated by PDI. Increasing the number of SL increases the fluorescence intensity, as seen by the smaller quenching factor in Table 5.2. This effect is also illustrated in Figure 5.9, which shows the fluorescence intensity increasing steadily with the number of separation layers but the amount of PDI increases linearly with the number of PDI depositions. We wished to study interlayer energy transfer by depositing a good energy acceptor dye (brilliant green, BG) on the outer layer of the LBL film assembly, but we found that BG (a dye of moderate size and a net charge of +1) is able to penetrate well past the outer film surface. Furthermore we found a very large difference in the 146 effectiveness of BG in quenching the fluorescence of p-PDI and n-PDI, despite the very similar R0 values (4.7 and 5.2 nm for p-PDI/BG and n-PDI/BG respectively). The cationic BG is expected to reside preferentially in regions of high PSS density and since PSS is the layer that surrounds (and interpenetrates) the p-PDI, we propose that the BG and p-PDI are brought into juxtaposition, leading to very efficient FRET. The n-PDI layer will be surrounded on both sides by PDAC, which is expected to exclude BG with the result that approximately 100x more BG is required to produce approximately the same quenching effect as for p-PDI. These ideas are illustrated in Figure 5.14. Therefore the plot of the efficiency of energy transfer vs. the number of layers (Figure 5.13) does not allow us to differentiate between the penetration of BG into the film and inter-layer energy transfer. Water-soluble perylene diimide dyes have potential applications as biochemical dyes16(a),34 and because of their photostability and good fluorescence quantum yield can be used in single molecule experiments.35 As we have shown here they can be readily incorporated into polyelectrolyte films by the LBL technique, but we assume that improvement of their fluorescence quantum yield in such arrays would require even more steric stabilization and, most likely, a larger number of charged groups per molecular species. 5.5 References 1. (a) Cormier, R. A., Gregg, B. A. Chem. Mater. 1998, 10, 1309. (b) Würthner, F., Thalacker, C., Sautter, A. Adv. Mater. 1999, 11, 754. (c) Antunes, P. A., Constantino, C. J. L., Aroca, R. F., Duff, J. Langmuir 2001, 17, 2985. (d) Klebe, G., Graser, F., Hädicke, E., Berndt, J. Acta Crystallogr. 1989, B45, 69. 147 2. (a) Decher, G., Hong, J. Makromol. Chem. Symp. 1991, 46, 321. (b) Decher, G. Science 1997, 277, 1232. 3. (a) Wang, Y., Tang, Z. Y., Correa-Duarte, M. A., Liz-Marza’n, L. M., Kotov, N. A. J. Am. Chem. Soc. 2003, 125, 2830. (b) Wu, A., Yoo, J.K., Rubner, M.F. J. Am. Chem. Soc. 1999, 121, 4883. (c) Hiller, L., Mendelsohn, J. D., Rubner, M. F. Nat. Mater. 2002, 1, 59. (d) Tang, Z., Wang, Y., Kotov, N. A. Langmuir 2002, 18, 7035. (e) Park, J., Hammond, P.T. Adv. Mater. 2004, 6, 520. (f) Caruso, F., Caruso, R. A., Möhwald, H. Chem. Mater. 2001, 13, 1076. (g) Harris, J. L., Stair, J.L., Bruening, M.L. Chem. Mater. 2000, 12, 1941. 4. (a) Tedeschi, C., Caruso, F., Möhwald, H., Kirstein, S. J. Am. Chem. Soc. 2001, 122, 5841. (b) Caruso, F., Lichtenfeld, H., Donath, E., Möhwald, H. Macromolecules 1999, 32, 2317. 5. (a) Birks, J.B., “Photophysics of Aromatic Molecules”, Wiley-Interscience 1971. (b) Lakowicz, J.R. “principles of Fluorescence Spectroscopy”, 2nd edn., Kluwer Academic/Plenum Publishers, New York, 1999. 6. (a) Würthner, F., Thalacker, C., Diele, S., Tschierske, C. Chem.-Eur. J. 2001, 7, 2245. (b) Syamakumari, A., Schening, A., Meijer, E.W. Chem.-Eur. J. 2002, 8, 3353. 7. Park. J., Hammond, P.T. Adv. Mater.2005, 16, 520. 8. Ford, W. E. J. Photochem. 1987, 37, 189. 9. Dai, Z., Dähne, L., Donath, E., Möhwald, H. J. Phys. Chem. B. 2002, 106, 11501. 10. Kellog, G. J., Mayes, A. M., Stockton, Langmuir 1996, 12, 5190. 148 W.B., Ferreira, M., Rubner, M. F. 0.04 Abs. at 580nm a 0.03 n=1 n=3 0.02 n=2 0.01 0 0 2 4 6 8 10 Dipping Time(Min) 12 14 16 Fluo. Inten.(a.u.) b n=1 0 2 4 6 n=2 8 10 12 Dipping Time(Min) n=3 14 16 Figure 5.1 (a) Absorbance at 580nm and (b) fluorescence intensity of n-PDI as a function of dipping time for “precursor” LBL films of PEI/(PSS/PDAC)n (n=1, 2, and 3). 149 Absorbance 0.04 a 0.03 0.02 0.01 0 340 400 460 520 580 640 Wavelength(nm) 700 760 Fluo. Inten. b 560 600 640 680 Wavelength(nm) 720 760 800 Figure 5.2 (a) and (b) Absorption and fluorescence emission spectra of n-PDI dipping into the preassembled PEI/(PSS/PDAC)6 as a function of immersing time. 150 Abs at 587nm 0.016 a 0.012 0.008 0.004 n=0 n=1 n=2 0 0 1 2 3 4 5 6 7 8 9 Dipping Time(min) 10 11 12 13 14 15 Fluo. Inten. b n=0 0 1 2 3 4 5 n=1 n=2 6 7 8 9 10 11 12 13 14 15 Dipping Time(Min) Figure 5.3 (a) Absorbance at 587nm and (b) fluorescence intensity of p-PDI as a function of dipping time for “precursor” LBL films of PEI/(PSS/PDAC)n-1/PSS (n=1, 2, and 30). 151 a Absorbance 0.06 n-PDI 0.06 0.04 0.02 0.04 0 0 0.02 1 2 3 4 5 0 340 440 540 640 740 Wavelength(nm) 0.06 940 0.06 b Absorbance 840 p-PDI 0.04 0.04 0.02 0 0 0.02 1 2 3 4 5 0 340 440 540 640 740 Wavelength(nm) 840 940 Figure 5.4 (a) and (b) Absorption spectra of n-PDI and p-PDI layer-by-layer assembly with PSS and PDAC solutions made in 0.5 M NaCl and 3 SLs, respectively. Black spectra are for the PDI layer as the outside layers and red when the following double layer of either PSS/PDAC or PDAC/PSS is added with insets showing the absorbance as a function of the number PDI layers. 152 Fluo. Inten. 4.E+05 a 3.E+05 n-PDI 2.E+05 0 1 2 3 4 5 1.E+05 0.E+00 560 600 640 680 720 Wavelength(nm) 760 800 1.E+06 b Fluo. Inten. 8.E+05 p-PDI 6.E+05 4.E+05 0 1 2 3 4 5 2.E+05 0.E+00 560 600 640 680 720 760 800 Wavelength(nm) Figure 5.5 (a) and (b) Fluorescence emission spectra of n-PDI and p-PDI with PSS and PDAC (0.5M NaCl) LBL films. Black spectra indicate the emission spectra when PDI layer was formed as the outside layers and red when the following double layer of either PSS/PDAC or PDAC/PSS formed with insets showing the fluorescence intensity as a function of the number PDI layers. 153 Fluo. Inten. a p-PDI PSS PDAC PSS PDAC PSS Fluo. Inten. b n-PDI PDAC PSS PDAC PSS PDAC Figure 5.6 (a) Fluorescence intensity as a function of sequential deposition of polyelectrolyte layers following the third p-PDI deposition (b) Fluorescence intensity as a function of sequential deposition of polyelectrolyte layers following the third n-PDI deposition. All the depositions were performed with 5SLs and 0.5M NaCl in the polyelectrolyte solutions. 154 0.02 Absorbance 0.02 a 0.016 n-PDI 0.012 0.01 0 0.008 0 1 2 3 4 5 820 880 940 0.004 0 340 400 460 520 580 640 700 760 Wavelength(nm) 0.02 0.02 0.015 Absorbance 0.015 b 0.01 0.005 p-PDI 0.01 0 0 1 2 3 4 5 0.005 0 340 400 460 520 580 640 700 Wavelength(nm) 760 820 880 940 Figure 5.7 (a) and (b) Absorption spectra of n-PDI and p-PDI with PSS and PDAC (3SLs) in the LBL films (with no NaCl in the polyelectrolyte solution). Black spectra indicate the absorption or emission spectra when PDI layer was formed as the outside layer and red when the following double layer of either PSS/PDAC or PDAC/PSS was deposited. The insets show the absorbance at 580nm as a function of the number PDI layers before and after deposition of double layer onto the PDI layer. 155 Fluo. Inten a n-PDI 0 560 600 640 680 1 2 720 760 Wavelength(nm) 3 800 4 840 5 880 Fluo. Inten b p-PDI 0 560 600 640 1 680 720 760 Wavelength(nm) 2 3 800 4 840 5 880 Figure 5.8 (a) and (b) fluorescence spectra for n-PDI and p-PDI. Black spectra indicate the absorption or emission spectra when PDI layer was formed as the outside layer and red when the following double layer of either PSS/PDAC or PDAC/PSS was deposited. The insets show the or fluorescence intensity as a function of the number PDI layers. 156 Abs. at 577nm 1 0.06 0.8 y = 0.0107x 0.04 0.02 3 0.6 0.4 7 5 9 1 0.2 Norm. Fluo. Inten. 1.2 0.08 0 0 0 1 2 3 4 5 Number of n-PDI Layers 6 7 Figure 5.9 Absorption vs. the number of n-PDI layers deposited (●) and the normalized fluorescence intensity (■) for each layer. The number of separation layers are indicated between each n-PDI deposition (SL=1, 3, 5, 7, 9). 157 a b Figure 5.10 TM-AFM angle view images of (a) before p-PDI deposition and (b) after pPDI depositions. 158 Blue: PSS Black: PDAC Red: PDI Intralayer-FRET Interlayer-FRET Intralayer-FRET Figure 5.11 Schematic representation of p-PDI in LBL films with 3SLs along with showing the intralayer and interlayer FRET process. 159 0.8 ET Eff. 0.6 0.4 a n-PDI 0.2 0 0 0.001 0.002 0.003 0.004 0.005 0.006 0.007 a Spin-Coating Concentration of BG(M) 1.2 ET Eff. 1 0.8 0.6 b p-PDI 0.4 0.2 0 0 0.0001 0.0002 0.0003 0.0004 0.0005 0.0006 Spin Coating Concentration of [BG]/M Figure 5.12 (a) and (b) Energy transfer efficiency as a function of spin-casting BG concentration for 4 n-PDI and p-PDI LBL films with 3SLs. 160 1 ET Eff. 0.8 a n-PDI 0.6 0.4 0.2 0 2 3 4 5 6 7 8 9 10 11 12 13 Number of n-PDI layers ET Eff. 0.8 p-PDI b 0.6 0.4 0.2 0 2 3 4 5 6 7 8 9 10 11 12 13 Number of p-PDI Layers Figure 5.13 (a) Energy transfer efficiency as a function of number of n-PDI layers at BG concentration of 1.3x10-3M. (b) Energy transfer efficiency as a function of number of pPDI layers at BG concentration of 8.3x10-6M. 161 (p-PDI/3SL)n/p-PDI/PSS/PDAC/PSS/BG Red: p-PDI or n-PDI Blue: PSS Layer Black: PDAC Layer Green: BG Layer (n-PDI/3SL)n/n-PDI/PDAC/BG Figure 5.14 Schematic representation of BG distribution in p-PDI and n-PDI LBL films with 3SLs. 162 Table 5.1 FRET parameters for D-A pairs of p-PDI, n-PDI and BG D-A p-PDI/p-PDI n-PDI/n-PDI p-PDI/BG n-PDI/BG J 7.99x1014 8.37x1014 4.03x1015 5.22x1015 R0(nm) 3.58 4.50 4.68 5.18 163 C0(mM) 9.79 4.93 4.32 3.25 Table 5.2 Characteristics of UV-Vis and fluorescence spectra of p-PDI and n-PDI LBL films with 1, 3, and 5 SLs with and without using NaCl. p-PDI/NaCla n-PDI/NaCla p-PDI n-PDI SLs Abs.b (x1000) Peakc 1 3 5 1 3 5 1 3 5 1 3 5 9.6 10.9 9.8 2.6 2.8 2.8 11.2 11.5 11.6 3.4 3.1 3.0 587 592 588 588 588 588 575 576 577 575 575 575 Extractiond N N N Y Y Y N N N Y Y Y Fluo. Peakc 621 620 621 619 620 620 612 614 613 613 615 614 Quen. Factor.e 35 25 9 15 19 8 43 33 22 17 14 12 a. 0.5 M NaCl was used for the polyelectrolyte solutions. b. Slopes of absorbance around 580 nm from either p-PDI or n-PDI as outside layer versus the number of PDI layers; c. S0ÆS1 UV-Vis peak position or S1ÆS0 fluorescence emission peak position for either p-PDI or n-PDI as the outmost layers; d. N means no extraction and Y means having extraction; e. Quenching factors obtained by comparing the fluorescence intensity of the p-PDI and n-PDI aqueous solutions in thin cells with p- or nPDI LBL films. 164 Chapter 6 Some Preliminary Results and Suggestions for Future work Two projects were initiated as part of this Ph. D. research which suggests some new and interesting directions for future work with PDI materials. These are briefly described in the following sections. 6.1 Synthesis of n-type and p-type PDI materials 6.1.1 Introduction The drive towards inexpensive electronic technologies involving organic thin-film field-effect transistors (OTFTs) has resulted in part from increasing demand for pervasive computing devices that will require light weight construction and inexpensive components for memory, logic, and displays. Moreover, OTFTs are well known to have many advantages such as large-area coverage, structural flexibility, shock resistance, and low-temperature processing, in comparison with the conventional silicon based TFTs.1 Historically, OTFTs based on p-type materials such as pentacene, poly(3-hexylthiophene) or oligothiophenes in which holes are the majority carriers, have received most of the attention. The highest hole mobility for pentacene TFTs was 1.5 cm2V-1s-1.2 Recently, the hole mobility had been reported to reach 8-20cm2V-1s-1 in rubrene single crystal TFTs.3 High performance TFTs based on n-type materials in which electrons are the majority carriers, are also desired since they will enable the fabrication of complimentary circuits. However, the performance of the best n-type materials is not quite as good as the best ptype materials, although there has been increased attention to n-type materials such as phthalocyanine and fluorinated polythiophene with the corresponding improvements in the field effect mobility, the on-to-off current ratio and the threshold voltage.4 Perylene diimides are good candidates for n-type conductors because (1) they have relatively large 165 electron affinities, (2) they typically assemble in π-stacks in the solid state which enhances the intermolecular π-orbital overlap and facilitates charge transport, and (3) the nature of π-orbital interactions can be tuned with high precision by changing the substituents on the imide N atoms, offering the opportunity to optimize transport properties by systematic structure variations, which in turn influences the crystal packing. Until now, the highest mobility of PDIs can reach to 2 cm2V-1s-1 with on-to-off current ratios of 107.5 Although both p-type and n-type materials with very good properties for the fabrication of TFTs have been independently discovered, there has not be any report on materials which can have the n-type and p-type properties in one molecule. In this research we are trying to combine the perylene diimides and fused benzene ring molecules such as naphthalene and phenanthrene in order to obtain materials which can show both the n-type and p-type field effect. In this work we introduce the naphthalene or phenanthrene groups in the bay area and not on the imide head-tail positions. The double substitutions in the bay area of PDIs are expected to impose less twisting of the perylene core compared with the tetra substituents in the bay region. 6.1.2 Synthesis The synthetic routes for the PDI compounds are shown in Figure 6.1, which involves three steps. 6.1.2.1 Bromination of Perylene Dianhydride(PDA) The bromination of PDA was repeated according to the patent procedure as shown in the first step in Figure 6.1.6 A mixture of 31.3 (80.0mmol) of PDA and 480g of 100% sulfuric acid was stirred for 12hrs at room temperature, and subsequently I2 (0.77g, 3.0mmol) was added. The reaction mixture was heated to 85ºC, and 29g (177mmol) of 166 bromine was added dropwise over a time period of 8hrs. After bromine addition, the reaction mixture was heated for an additional 10 hrs at 85ºC and cooled to room temperature. The excess bromine was removed by a gentle stream of N2 gas and 65 mL of water was added carefully. The resulting precipitate was separated by filtration through a G4 funnel, washed with 300g of 86% sulfuric acid and a large amount of water, and dried in a vacuum to give 37.6g (87%) of red powder. The crude product (written as PDA-Br2) could not be purified since it is insoluble in organic solvents. PDA-Br2 was used for the subsequent imidization with cyclohexylamine. 6.1.2.2 Imidization of crude Bromination Product7 A suspension of PDA-Br2 (0.95g, 1.72mmol) obtained in the above reaction, cyclohexylamine (0.502g, 5.07mmol), and acetic acid (0.50g, 8.33mmol) in 30mL of Nmethyl-2-pyrrolidinone was stirred at 85ºC under Argon gas protection for 6 hrs. After mixture was cooled to room temperature, the solution was poured into 300mL of 1M HCl for precipitation overnight. The precipitate was separated by filtration, washed with 100mL of MeOH, and dried in a vacuum. The crude product was purified by silica gel column chromotography with CH2Cl2 as eluent. This product is denoted as PDI-Br2. This product was characterized by both TLC and Mass Spectroscopy for purification. 6.1.2.2 Nucleophilic Replacement of Bromine atoms8 PDI-Br2 (0.90g, 1.3mmol), 1-Naphthanol (1.5g, 10.4mmol) and K2CO3 (0.3g, 2.2mmol) were heated in 50mL of NMP at 85ºC overnight under argon gas protection. After cooling to room temperature, the reaction mixture was poured into 150mL of 1M HCl for precipitation. The crude product was separated by vacuum filtration. Pure product of PDI-NP1 was obtained by running silicon gel column chromatography with 167 CH2Cl2/MeOH (9:1) as the eluent. Its mass spectroscopy and H-NMR were shown in Figure 6.2a and b respectively. The perylene core chemical shift resides above 8ppm and naphthalene around 6 and 8ppm. They are both multipeaks. CDCl3 was used for H-NMR measurements. Following the same protocol as above, the pure product of PDI-NP2 and PDI-PN9 were synthesized. Similar Mass Spectroscopy and H-NMR with PDI-NP1 were obtained for PDI-NP2, as shown in Figure 6.3. The mass spectroscopy for PDI-PN9 was shown in Figure 6.4 with a very strong molecular ion peak. The chemical structures of these 3 PDIs are shown in Figure 6.5. 6.1.3 Photophysics of the synthesized PDIs The normalized absorption and emission spectra of PDI-NP1, PDI-NP2 and PDIPN9 in DCE are shown in Figure 6.6. The absorption spectra for PDI-NP1 and PDI-NP2 are very similar with three absorption peaks that are typical for a PDI core except there is a 4nm red shift for PDI-NP1. This red shift is attributed to the higher steric hindrance originating from the 1-naphthanol substituent. Comparing to PDI-NP1 and PDI-NP2, there is stronger S0,0-S1,1 absorption peak observed for PDI-PN9, which might be attributed to dimer formation for PDI-PN9 in DCE due to the larger π-π interactions from phenanthrene in the bay region.9 Emission spectra at Ex=505nm are shown in Figure 6.6b. A single emission peak at 567nm was observed for PDI-NP1 and PDI-NP2, which is typical for bay area substituted PDIs. The same position peak for PDI-NP1 and PDINP2 indicates no significant coupling and interaction between naphthalene and PDI core is present. PDI-NP2 has a higher fluorescence quantum yield of 0.65 and PDI-NP1 has a quantum yield about 0.11. The lower quantum yield of PDI-NP1 is due to the larger steric 168 hindrance from 1-naphthanol substituent leading to more twisting of the PDI core. The Stokes shifts are 25 and 29nm for PDI-NP1 and PDI-NP2. Two emission peaks are located a 573nm and 620nm observed for PDI-PN9, leading to a Stokes shift of 32nm. The quantum yield is about 0.01 for PDI-PN9 presumably due to aggregation or the strong coupling between phenanthrene and PDI core. Emission spectra with the excitation wavelength chosen for the bay region substituents are shown in Figure 6.6c. Typical naphthalene emission spectra were observed for PDI-NP1 and PDI-NP2. The excitation for the phenanthrene substituent also resulted in the emission from phenanthrene alone. All these results indicate that no coupling exists between perylene core and bay substituent. In other words, the bay substituent chromophore and perylene core behave independently. Further investigation of the electrochemical properties of these compounds will provide us more information about this point. 6.1.4. Future work The decomposition temperature for PDI-NP1 and PDI-NP2 is around 410ºC. DSC measurements have shown a Tg of 277 ºC of PDI-NP1. The Tg for PDI-NP2 is not very obvious. This indicated the good thermostability of synthesized PDIs. Future research of using these synthesized for the TFT study will be conducted to characterize the electron and hole carrier mobility. 6.2 Self-assembly of n-type semiconductor PDIs 6.2.1 Introduction One-dimensional nanostructures (such as nanowires) of semiconductor materials have attracted increased interest in recent years due to their promising applications in 169 optical and electronic nanodevices.10 However, most nanowires reported to date are based on the inorganic materials. A few one-dimensional nanostructures (e.g. nanofibers) have been fabricated with conducting polymers such as polyaniline and polyacetylene, but the crystalline structure of polymer nanofibers is often difficult to control due to the complicated intermolecular interaction.11 Recently, some works have proven that selfassembly of through strong π-π stacking is an effective approach to one-dimension nanostructures for aromatic molecules, particularly the larger macrocyclic aromatic molecules such as hexabenzocoronene.12 Very recently, Zang Ling et al. used “phase transfer” self-assembly between good and poor solvents and a surface annealing based self-assembly method for making organized nanobelts of PDIs by controlling the π-π stacking and the hydrophobic interactions between the side chains linked at the imide positions.13-15 This implies a potential method to enhance the charge carrier mobility, which is believed be favored along the π-π stacking direction. Utilizing the solution based “phase transfer” self-assembly method, nanobelt or even nanotubes can be obtained with our synthesized PDIs. 6.2.2 Self-assembly of PDIs 6.2.2.1 Chemical structures of PDIs The synthesis of PDIs was described in Chapter 2. These PDIs have branched chains originating from a tertiary nitrogen (their Chemical Structures are also shown in Figure 6.5). These three PDIs studied here are denoted as PDI-DMPA, PDI-DEPA and PDI-DMEA. All three have good solubility in chloroform and very low solubility in hexane. 6.2.2.2 Solution based Self-assemblying 170 The solution based self-assembly of PDIs was performed using a so-called “phase transfer” method.13 Briefly, the nanostructure growth of the PDIs assembly was acheived through slow crystallization at the interface between a “good” and a “poor” solvent, where the slow “phase transfer” between the two solvents decreases the solubility at the interface. The poor solvent (e.g., hexane) is normally quite different (e.g., in term of polarity and density) from the good solvent (e.g. chloroform), thus providing the possibility to keep the two solvents in separate phases for an extended period (longer than one day). Typically, a larger amount (15:1, v/v) of hexane was carefully transferred atop a concentrated chloroform solution of PDI solutions (~0.1mM) in a test tube. Within 20 minutes, red crystals formed at the interface, followed by very slow diffusion into the upper phase of hexane. The nanobelts can be transferred into the hexane for dilution by pipeting. Then the suspension was drop-cast onto the clean silicon wafer. The images were then measured by SEM. Scanning Electron Microscopy (SEM) images were taken on a LEO 1530 scanning electron microscope, which has a GEMINI field emission column with a thermal field emitter. The voltage was set at 10 kV. The samples were prepared by casting a hexane solution of PDI nanoassemblies onto a clean silicon wafer, followed by drying in air. Typical images of SEMs for PDI-DEPA are shown in Figure 6.7. Nanobelts were formed for PDI-DEPA with >10µm in length and 400nm in width, which is around the same size as Ling Zang et al. obtained for their PDIs. Large area nanobelt piles were found over the surfaces for PDI-DMPA shown in Figure 6.8, and they showed quite 171 narrow size (width) distribution. The π-π stacking was found to be along the nanobelt optical axis by Ling Zang et al. 14 SEM images of PDI-DMEA nanotubes are shown in Figure 6.9. Figure 6.11a showed a chunk of broken nanotubes. A wall thickness of 500nm was obtained for the nanotube from the Figure 6.9b and c. Evidently the smaller size of the side chains of PDIDMEA make it possible to form nanotubes. The nanotube structures are very fragile as shown in Figure 6.9a. As far as we know, this is the first report of nanotube synthesis from organic semiconductors. 6.2.3 Future of Self-assembly of PDIs The PDIs we synthesized have a tertiary amine in the side chains, which is different from the PDI molecules that Ling Zang studied. Therefore, the protonation of tertiary amine by dicarboxylic acid molecules such as butanedioic acid or hexanedioic acid will lead to the crosslinking of the PDI side chains in the nanoassemblies. The structural stability upon diacid crosslinking can be tested by dissolving the nanoassemblies into chloroform to see if the nanostructure can be retained or not. In the same way, the crosslinking of diamines by di-bromo agents could also be examined. We have prepared a fairly large number of compounds of this class, with chemically distinct branched structures (see Chapter 2), which may modify the kinds of linear structures that can be formed by the phase-transfer or related methods. 6.3 References 1. (a) Dimitrakopoulos, C. D., Mascaro, D. J. IBM J. Res. Dev. 2001, 45, 1. (b) Dimitrakopoulos, C. D., Malenfant, P. R. L. Adv. Mater. 2002, 14, 99. 172 2. Lin, Y.-Y., Gundlach, D. J., Nelson, S. F., Jackson, T. N. IEEE Electron Device Lett. 1997, 18, 606. 3. (a) Sundar, V. C., Zaumseil, J., Podzorov, V., Menard, E., Willett, R. L., Someya, T., Gershenson, M. E., Rogers, J. A. Science 2004, 303, 1644. (b) Podzorov, V., Menard, E., Borissov, A., Kiryukhin, V., Rogers, J. A., Gershenson, M. E. Phys. Rev. Lett. 2004, 93, 086602. 4. (a) Bao, Z., Lovinger, A. J., Brown, J. J. Am. Chem. Soc. 1998, 120, 207. (b) Bao. Z. Adv. Mater. 2000, 12, 227. (c) Facchetti, A., Mushrush, M., Katz, H. E., Marks, T. J. Adv. Mater. 2003, 15, 33. 5. Chesterfield, R. J., Mckeen, J. C., Newman, C. R., Ewbank, P. C., da Silva Filho, D. A., Brédas, J-L., Miller, L. L., Mann, K. R., Frisbie, C. D. J. Phys. Chem.B 2004, 108, 19281-19292. 6. Böhm, A., Arms, H., Henning, G., Blaschka, P. (BASF AG) German Pat. DE 19547209A1, 1997. Chem. Abstr. 1997, 127, 96569g. 7. Würthner, F., Stepanenka, V., Chen, Z., Saha-Möller, C. R., Kocher, N., Stalke, D. J. Org. Chem. 2004, 69, 7933. 8. (a) Qu, J., Kohl, C., Pottek, M., Müllen, K. Angew. Chem., Int. Ed. 2004, 43, 1528. (b) Kohl, C., Weil, T., Qu, J., Müllen, K. Chem.-Eur. J. 2004, 10, 5297. 9. van der Boom, T., Evmeneko, G., Dutta, P., Wasielewski, M. R. Chem. Mater. 2003, 15, 4068. 10. Law, M., Goldberger, J., Yang, P. Annu. Rev. Mater. Res. 2004, 34, 83. 11. (a) Huang, J. X., Virji, S., Weiller, B. H., Kaner, R. B. J. Am. Chem. Soc. 2003, 125, 314. (b) Lee, H. J., et al. J. Am. Chem. Soc. 2004, 126, 16722. 173 12. Kastler, M., Pisula, W., Wasserfaller, D., Pakula, T., Müllen, K. J. Am. Chem. Soc. 2005, 127, 4286. 13. Balakrishnan, K., Datar, A., Naddo, T., Huang, J., Oikter, R., Yen, M., Zhao, J., Zang, L. J. Am. Chem. Soc. 2006, ASAP. 14. Balakrishnan, K., Datar, A., Oikter, R., Chen, H., Zuo, J., Zang, L. J. Am. Chem. Soc. 2005, 127, 10496. 15. Datar, A., Oikter, R., Zang, L. Chem. Comm. 2006, 1649. 174 Br O O O O i: Br2, 100% H2SO4/I2 O 80oC, 12h, 87% O O O O O O O Br ArOH=1-Naphthalenol PDI-NP1 2-Naphthalenol PDI-NP2 9-Phenanthrol PDI-PN9 ii: Cyclohexylamine, NMP, 85oC, overnight Ar O Br O O C6H11 N N C6H11 O Ar-OH, K2CO3 O O iii:NMP, 85oC, C6H11 N N C6H11 O O O O Br Ar Figure 6.1 Synthetic routes to Compounds PDI-NP1, PDI-NP2, and PDI-PN9 175 a b Figure 6.2 Mass Spectrum of PDI-NP1 by CI+ (a) and H-NMR in CDCl3 (b). 176 a b Figure 6.3 Mass Spectrum of PDI-NP2 by CI- (a) and H-NMR in CDCl3 (b). 177 Figure 6.4 Mass Spectrum of PDI-PN9 by CI+. 178 O O O O O O O O O N N N N N N O O O O O O O PDI-NP1 O PDI-NP2 O H3 C H3 C PDI-PN9 O NH2CH2CH2C N CH3 N CH2CH2CH2N O CH3 O PDI-MAPA O H3CH2C H3CH2C O NH2CH2CH2C N N CH2CH2CH2N O CH2CH3 CH2CH3 O PDI-DEPA O H3C H3 C O NH2CH2C N N CH2CH2N O O PDI-DMEA Figure 6.5 Chemical Structures of Synthesized PDIs 179 O CH3 CH3 a Absorbance 1.2 0.8 0.4 0 220 340 400 460 520 580 Wavelength(nm) 640 700 b Norm. Fluo. Inten. 0.016 0.8 0.012 0.6 0.4 0.008 0.2 0.004 0 0 520 570 1 Norm. Fluo. Inten. 760 0.02 1 Norm. Fluo. Inten. 280 620 670 720 Wavelength(nm) 770 c 0.8 0.6 0.4 0.2 0 270 300 330 360 390 420 Wavelength(nm) 450 480 Figure 6.6 Normalized absorption (a) and emission spectra (b and c) of PDI-NP1 (Red), PDI-NP-2(pink) and PDI-PN9 (Blue) in DCE. Emission spectra in Figure 6.7b were taken at Ex=505nm. Emission spectra in Figure 6.7c were taken at Ex=260nm for PDINP1 and PDI-NP2 and Ex=290nm. 180 Figure 6.7 SEM images of PDI-DEAPA. 181 Figure 6.8 Large area SEM images of PDI-DMPA 182 Figure 3.9 SEMs images of PDI-DMEA. 183 Bibliography 1. Hunter, C. A., Sanderes, J. K. M. J. Am. Chem. Soc. 1990, 112, 5525. 2. Cao, T., Wei, L., Cao, W. Langmuir 2002, 18, 750. 3. Wei, X-L., Wang, Y. Z., Long, S. M., Bobeczko, C., Epstein J. Am. Chem. Soc. 1996, 118, 2545. 4. Tomiki, I. J. Mater.Chem. 2003, 13, 2037. 5. Liu, S., Sui, G., Cormier, R. A., Leblane, R. M., Gregga, B. A. J. Phys. Chem. B. 2002, 106, 1307 6. Horowitz, G. Adv. Mater. 1998, 10, 365. 7. Nueteboom, E. E., Meskers, S. C. J., Meijer, E. W., Janssen, R. A. Macromol. Chem. Phys. 2004, 205, 217. 8. Liu, Y., Wang, N., Li, Y., Liu, H., Li, Y., Xiao, J., Xu, He., Xu, X., Huang, C., Cui, S., Zhu, D. Macromolecules 2005, 38, 4880. 9. Köhler, K., Shchukin, D. G., Sukhorukov., G. B., Möhwald, H. Macromolecules 2004, 37, 9546. 10. Gao, C., Wang, B., Feng, J., Shen, J. Macrmolecules 2004, 37, 8836. 11. Zhao, L., Cebeci, F. C., Cohen, R., Rubner, M. Nano. Lett. 2004, 7, 1349. 12. Liu, N., Dunphy, D., Atanassov, P., Bunge, S. D., Chen, Z., Lopez, G. P., Boyle, T. J., Brinker, C. J. Nano. Lett. 2004, 7, 551. 13. Dante, S., Advincula, R., Frank, C. W., Stroeve, P. Langmuir 1999, 15, 193. 14. Cho, J., Quinn, J. F., Caruso, F. J. Am. Chem. Soc. 2004, 126, 2270. 184 15. Chiu, J. J., Kim, B. J., Kramer, E. J., Pine, D. J. J. Am. Chem. Soc. 2005, 127, 5036. 16. Lee, M., Cho, B. K., Zin, W. C. Chem. Rev. 2001, 101, 3869. 17. Klok, H-A., Lecommandous, S. Adv. Mater. 2001, 13, 1217. 18. Lazzari, M., Lopez-Quintela, M. Adv. Mater. 2003, 15, 1583. 19. Ikkala, O., Brinke, G. T. Science 2002, 295, 29, 2407. 20. Park, C., Yoon, J., Thomas, E. L. Polymer 2003, 44, 6276. 21. Beijonne, D., Pourtois, G., Hennebicq, E., Herz, L. M., Friend, R. H., Scholes, G. D., Setayesh, S., Müllen, K., Brédas, J. L. PNAS 2002, 99, 10982. 185 Vita Tingji Tang was born to Jiaen Tang and Chengfeng Lu on May 11, 1977 in Nanluo village, Penglai City, Shandong Province, P.R. China. After completing his work at 2nd place in Xujiaji Middle School, he went to Penglai No. 1 High School in 1992. Due to his strong performance in the college entrance examination in 1995, he entered Qingdao Institute of Chemical technology (now as Qingdao University of Science and Tehnology) and earned his Bachelor of Engineering degree in Rubber Science and Engineering in 1999. Then he continued to pursue his Master Degree of Science in Polymer Chemistry and Physics from Peking University in 2002. In fall, 2002, he came to the United States and entered the Graduate School of the University of Texas at Austin. He was appointed as Graduate Teaching and Research Assistant in the course of his graduate studies. He married Yanbo Wang In 2002 and had a son, Bryan Benshuo Tang, in 2005. Permanent Addess: Nanoluo Village, Beigou Town, Penglai City, Shandong Province, P. R. China, 265602. This dissertation was typed by the author. 186