315
Protein electrostatics: A review of the equations and methods used
to model electrostatic equations in biomolecules – Applications in
biotechnology
Maria Teresa Neves-Petersen and Steffen B. Petersen*
Department of Physics and Nanotechnology, University of Aalborg, Biostructure and
Protein Engineering Group, Sohngaardsholmsvej 49, DK-9000 Aalborg, Denmark
Abstract. The molecular understanding of the initial interaction between a protein and, e.g., its
substrate, a surface or an inhibitor is essentially an understanding of the role of electrostatics in
intermolecular interactions. When studying biomolecules it is becoming increasingly evident that
electrostatic interactions play a role in folding, conformational stability, enzyme activity and
binding energies as well as in protein–protein interactions. In this chapter we present the key basic
equations of electrostatics necessary to derive the equations used to model electrostatic interactions
in biomolecules. We will also address how to solve such equations. This chapter is divided into two
major sections. In the first part we will review the basic Maxwell equations of electrostatics
equations called the Laws of Electrostatics that combined will result in the Poisson equation. This
equation is the starting point of the Poisson–Boltzmann (PB) equation used to model electrostatic
interactions in biomolecules. Concepts as electric field lines, equipotential surfaces, electrostatic
energy and when can electrostatics be applied to study interactions between charges will be
addressed. In the second part we will arrive at the electrostatic equations for dielectric media such as
a protein. We will address the theory of dielectrics and arrive at the Poisson equation for dielectric
media and at the PB equation, the main equation used to model electrostatic interactions in
biomolecules (e.g., proteins, DNA). It will be shown how to compute forces and potentials in a
dielectric medium. In order to solve the PB equation we will present the continuum electrostatic
models, namely the Tanford–Kirkwood and the modified Tandord–Kirkwood methods. Priority
will be given to finding the protonation state of proteins prior to solving the PB equation. We also
present some methods that can be used to map and study the electrostatic potential distribution on
the molecular surface of proteins. The combination of graphical visualisation of the electrostatic
fields combined with knowledge about the location of key residues on the protein surface allows us
to envision atomic models for enzyme function. Finally, we exemplify the use of some of these
methods on the enzymes of the lipase family.
Keywords: protein electrostatics, laws of electrostatics, Maxwell equations of electrostatics, Poisson
equation, Poisson–Boltzmann equation, Tanford–Kirkwood model, electrostatic potential
distribution, molecular surface, pKa, dielectric constant, titratable residues, Debye–Hückel.
INTRODUCTION
1. Understanding the basic equations of electrostatics in order to model
electrostatic interactions in proteins
‘‘Nature has simplicity and therefore a great beauty’’
Richard P. Feynman
Physics, the old Greek name for Nature, is the starting point of any field in
science allowing us to describe how Nature works, even if we do not understand
*Corresponding author: Tel: þ 45 9635 8469. Fax: þ 45 9635 9129. E-mail: sp@bio.auc.dk
BIOTECHNOLOGY ANNUAL REVIEW
VOLUME 9 ISSN 1387-2656
DOI: 10.1016/S1387-2656(03)09010-0
ß 2003 ELSEVIER SCIENCE BV
ALL RIGHTS RESERVED
316
why Nature works that way. We cannot explain why Nature behaves in this
peculiar way. Most theories will continue to evolve with time, others will not
stand the test of time, and only a few pass this test. Some of the very basic
questions we can ask within the scope of this chapter, dedicated to modelling
electrostatic interactions in proteins, are: how does a charge perturb the space
around it as it does? Why is the space surrounding a charge perturbed by it?
What is the nature of this space? What are the fundamental equations of
electrostatics? How were the equations used to model electrostatic interactions in
proteins derived? In some sense we are very fortunate that some of these
questions do not have immediate and perfectly understandable answers. This
allows us to have the pleasure of speculation throughout our lives.
In order to guide the reader this chapter has been divided in two main sections:
(1) the basics of electrostatics; and (2) modelling protein electrostatics in
proteins. The first section is dedicated to fundamental equations and laws of
electrostatics that will hopefully fulfil the curiosity of the mind interested in the
physics of electrostatics. We will go through the theory needed to derive the two
laws (two of the Maxwell’s equations) of electrostatics. And why? Well, these two
laws combined into a single equation will allow us to arrive at the Poisson
equation of the electrostatic potential that in the second part of this chapter will
be used as the starting point for the study of electrostatic interactions in proteins.
On the other hand, if we know how these equations came about we will know
under which conditions such equations are valid, i.e., the limitations of the laws
of electrostatics. Knowing these limitations we will be better prepared to criticise
the advantages and limitations of the methodologies used to model electrostatic
interactions in proteins. The fulfilment that we feel when we understand how a
particular formula came about and under which circumstances it can be used
brings us the depth and the necessary knowledge needed in order to apply it
properly in, e.g., the applied science domain.
In Part 1 of this chapter we will consider electrostatic interactions between
charges in vacuum, not including the effect that the dielectric constant of the
media might have on such interactions. But much of electrostatic interactions
have to do with charges and fields in media whose respective electric responses
must be taken into account. In order to model electrostatic interactions in
proteins we will consider the solute – protein molecules and the solvent molecules
as dielectric media characterised by a particular dielectric constant. Therefore,
we call them dielectrics. In Part 2 we shall then derive the equations of
electrostatics when there are dielectrics. We will see that the Poisson equation
derived in Part 1 from the fundamental equations of classical electrostatics is the
starting point for modelling electrostatic interactions in proteins since it will
allow us to arrive at the Poisson equation for a dielectric medium. In addition,
it is reasonable to assume that the protein surrounds itself with an atmosphere
of counterions, as described by the Debye–Hückel theory of electrolytes [1]. In
this case the Poisson–Boltzmann (PB) equation, usually in its linear form, is
solved. Solving such equation correctly parameterised will allow us to know the
317
electrostatic potential distribution at the location of each atom belonging to the
protein.
Summarising, in Part 2 we will cover the equations used to model electrostatic
interactions in proteins, the different models that have been used to describe the
electrostatic interactions in such macromolecules, as well as one method used to
find the electrostatic potential distribution in proteins, i.e., methods used to solve
the PB equation.
We will start with simple delightful concepts of physics. Slowly the complexity
will increase and we will see ourselves travelling from the world depicting the
interaction between a pair of point charges to the beginning of the universe of
complexity of an arbitrary distribution of charges. The beginning of this universe
is still simple and understandable through simple concepts of physics. Slowly
we will get closer to the protein and realise that the way of computing the
electrostatic potential is different. The concepts of electrical force, electric field
and electric potential will be introduced since they will many times be mentioned.
In the following sections, a vector will be written in bold throughout the text and
with an arrow above its symbol if in an equation. Both vectors and scalars will be
written in italic.
PART 1 – THE BASICS OF ELECTROSTATICS
2. Electromagnetic forces: electrostatics and electrodynamics
2.1. Introduction to electrical forces
‘‘Let us consider a force like gravitation which varies inversely as the square of the distance, but which
is about a billion–billion–billion–billion times stronger. And with another difference. There are two
kinds of ‘‘matter’’, which we can call positive and negative. Like kinds repel and unlike kinds attract –
unlike gravity where there is presumably only attraction. What would happen? A bunch of positives
would repel with an enormous force and spread out in all directions. A bunch of negatives would do
the same. But an evenly mixture of positives and negatives would do something totally different. The
net result would be that the terrific forces would balance themselves out almost perfectly, by forming
tight, fine mixtures of the positive and the negative, and between two separate gathering of such
mixtures there would be practically no attraction or repulsion at all. The electrical force is such a
force. All the matter is a mixture of positive protons and negative electrons which are attracting and
repelling with this great force. So perfect is the balance, however, that when we stand near someone
else we do not feel any force at all. If there were even a very small unbalance we would know it. If a
person was standing at an arm’s length from someone else and each of them had one percent more
electrons than protons, the repelling force would be incredible. The repulsion would be enough to lift
a ‘‘weight’’ equal to that of the entire earth!
The force that holds the atoms together, and the chemical forces that hold molecules together,
are really electrical forces acting in regions where the balance of charge is not perfect, or where the
distances are very small. We would like to give another example that illustrates the magnitude and
relevance of electrical forces. Let us think about a nucleus. In a nucleus there are several protons, all
of which are positive. Why don’t they push themselves apart? And what would happen to the
nucleus if they did? It turns out that in nuclei there are, in addition to electrical forces, non-electrical
forces, called nuclear forces, which are greater that electrical forces and which are able to hold
318
the protons together in spite of the electrical repulsion. The nuclear forces, however, have a short
range – their force falls off much more rapidly than 1/r2. And this has an important consequence. If
a nucleus has too many protons in it, it gets too large, and it will not stay together. An example is
uranium, with 92 protons. The nuclear forces act mainly between each proton (or neutron) and its
nearest neighbour, while the electrical forces act over larger distances, giving a repulsion between
each proton and all of the others in the nucleus. The more protons in a nucleus, the stronger is the
electrical repulsion, until, as in the case of uranium, the balance is so delicate that the nucleus is
almost ready to fly apart from the repulsive electrical forces. If such a nucleus is just ‘‘tapped’’
lightly (as can be done by sending in a slow neutron), it breaks into two pieces, each with positive
charge, and these pieces fly apart by electrical repulsion. The energy which is liberated is the energy
utilised to create the atomic bomb. This energy is usually called ‘‘nuclear’’ energy, but it is really
‘‘electrical’’ energy released when electrical forces have overcome the attractive nuclear forces.’’
Richard P. Feynman
Like a gravitational force, electrical forces decrease as the square of the
distance between charges. This relationship is called Coulomb’s law, and will be
addressed in this chapter. But this law is not precisely true when charges are
moving – the electrical forces depend also on the motions of the charges in a
complex way. One part of the force between moving charges is called the
magnetic force. It is really one aspect of an electrical effect. This is why the
subject is called ‘‘electromagnetism’’.
The above-mentioned forces all depend on the distance between the bodies,
but other things being equal they can be ranked as follows in order of magnitude:
Strong nuclear > Electromagnetic > Weak nuclear > Gravitational
It is rather peculiar that both gravitational and electrostatic forces follow the
same fundamental equation.
2.2. Electromagnetism
It has been found from experiment that the force that acts on a particular charge –
no matter how many charges there are or how they are moving – depends only
on the position of that particular charge, on the velocity of the charge and on the
amount of charge. We can write the force F on a charge q moving with a velocity
v as (non-relativistic force)
Lorentz force
F~ ¼ q E~ þ v~ B~
ð1Þ
E is the electric field and B the magnetic field at the location of the charge. The
important thing is that the electrical forces from all the other charges in the
universe can be summarised by giving just these two vectors. Their values will
depend on where the charge is, and may change with time. Furthermore, if we
replace that charge with another charge, the force on the new charge will be just
in proportion to the amount of charge so long as all the rest of the charges in the
world do not change their positions or motions. In reality, each charge produces
319
forces on all other charges in the neighbourhood and may cause these other
charges to move, and so in some cases the fields can change if we replace our
particular charge by another.
2.3. Principle of superposition
One of the most important simplifying principles about the way the fields are
produced is this: suppose a number of charges moving in some manner would
produce a field E1, and another set of charges would produce E2. If both sets of
charges are in place at the same time (keeping the same locations and motions
they had when considered separately), then the field produced is the sum
E~ ¼ E~1 þ E~2
ð2Þ
This fact is called the principle of superposition of fields. It holds also for magnetic
fields. This principle means that if we know the law for the electric and magnetic
fields produced by a single charge moving in an arbitrary way, then all the laws
of electrodynamics are complete. If we want to know the force on charge A we
need only to calculate the E and B produced by each of the charges B, C, D, etc.,
and then add the Es and the Bs from all the charges to find the fields, and from
them the forces acting on charge A. However, it is not simple to give a formula
for the force that one charge produces on another. It is true that when charges
are standing still the Coulomb’s force law is simple, but when charges are moving
about the relations are complicated by delays in time and by the effects of
acceleration, among others.
3. Maxwell’s equations
The complete classical theory of the electromagnetic field is contained in the
following four equations, the Maxwell’s equations [2].
Maxwell’s equation:
~ E~ ¼ r
"0
ð3Þ
~
~ E~ ¼ @B
r
@t
ð4Þ
~
~
~ B~ ¼ @E þ j
c2 r
@T "0
ð5Þ
~ B~ ¼ 0
r
ð6Þ
320
where (rho), the ‘‘electric charge density’’, is the amount of charge per unit
volume, and j, the ‘‘electrical current density’’, is the rate at which charge flows
through a unit area per second, and the gradient operator is defined as
@ @ @
~
r¼
, ,
@x @y @z
The situations that are described by these equations can be very complicated.
The easiest circumstance to treat is one in which nothing depends on time –
called the static case, electrostatics or magnetostatics.
4. Electrostatics
Electrostatics is the branch of electromagnetism dealing with static electric fields
and will be further developed in the present chapter. Its application to the study
of the interaction between charged atoms in the proteins and solvent is largely
dependent on the following approximations:
– All charges are permanently fixed in space, or if they move, they move as a
steady flow in a closed circuit. In these circumstances, all of the terms in the
Maxwell’s equations which are time derivatives of the field are zero. This
implies that we assume that the behaviour of a molecule in solution can be
described in terms of a spatial and temporal average static structure. Protein
structures determined by X-ray diffraction or NMR are normally used as
models for the average structure. The electrostatic field and interactions
between charged groups in the average structure can be taken as an average of
the instantaneous charges in the real, or dynamic, structure.
– A charged particle is instantaneously aware of a change in position of any
other charge [3], i.e., relativistic or retardation effects do not play a role.
– The electric field lines can originate or terminate only on electric charges. A
given line of electric field in space is continuous and unbroken from its origin.
– The electric field of a point charge at rest as having an isotropic radial pattern
centred on the charge.
– As long as the principle of superposition is valid (section 2.3 and section 9.1 in
Part 2).
– The coupling between electric and magnetic fields can be neglected. Electricity
and magnetism are distinct phenomena so long as charges and currents are
static, allowing electrostatics to be studied independently of magnetism.
Under these circumstances, all of the terms in the Maxwell equations which
are time derivatives of the field are zero. In this case, the Maxwell equations for
electrostatics become [2]:
~ E~ ¼ r
"0
ð7Þ
~ E~ ¼ 0
r
ð8Þ
321
We are saying that what is true for electrostatics is false for electrodynamics,
because all terms with time derivatives are left off. Thus, the Coulomb’s law is in
general false (true only for statics) whereas Lorentz’ law is always true.
These two equations are the laws of electrostatics and in this chapter we will
work through a number of calculations which will help us to deduct the first
equation also called the Gauss’ law, and the second equation. It will be shown
that Gauss’ law is equivalent to the Coulomb’s law mentioned in the next section.
4.1. Coulomb’s law
The Coulomb’s law states that between two charges at rest there is a force
directly proportional to the product of the charges and inversely proportional to
the square of the distance between them. The force is along the straight line from
one charge to the other.
Coulomb’s law:
1 q1 q2
F~1 ¼
e~12 ¼ F~2
4p"0 r212
ð9Þ
F1 is the force on charge q1, e12 is the unit vector in the direction to q1 from
q2, and r12 is the distance between q1 and q2. The force F2 on q2 is equal and
opposite to F1.
The constant of proportionality, for historical reasons, is written as 1/4p"0.
In the mks unit system it is defined as exactly 107 times the speed of light
squared. Since the speed of light is approximately 3 108 m/s, the constant is
approximately 9 109, and the unit turns out to be newton times meter2 per
coulomb2 or volt times meter per coulomb.
When there are more than two charges present we must supplement
Coulomb’s law with another fact of nature: the force on any charge is the vector
sum of the Coulomb forces from each of the other charges. This is called ‘‘the
principle of superposition’’. However, this principle cannot be applied in dealing
with non-linear phenomena, since higher-order terms have to be included in
order (see Part 2, section 9.1).
4.2. Electric field
When applying Coulomb’s law, it is convenient to introduce the idea of an electric
field. We say that the field E(1) is the force per unit of charge on q1 (due to all
other charges). Dividing Eq. (9) by q1, we have, for one other charge besides q1,
1 q2
E~ð1Þ ¼
e~12
4p"0 r212
ð10Þ
322
E(1) also describes something about point (1) even if q1 was not there – assuming
that all other charges keep their same positions. E(1) is the electric field at point
(1). If there are many charges present, the field E at any point (1) is a sum of the
contributions from each of the other charges. Each term of the sum will look like
Eq. (10). Let qj be the magnitude of the jth charge, and r1j the displacement from
qj to the point (1), we write
E~ð1Þ ¼
X
j
1 qj
e~1j
4p"0 r21j
ð11Þ
Electric fields are vector functions of x, y and z (static conditions). It is
precisely because E can be specified at every point in space that it is called a
‘‘field’’. A ‘‘field’’ is any physical quantity which takes on different values at
different points in space (see Fig. 1).
Often it is convenient to ignore the fact that charges come in packages like
electrons and protons, and think of them as being spread out in a continuous
smear – or in a ‘‘distribution’’, as it is called. This is acceptable as long as we are
not interested in what is happening on too small scale. We describe a charge
distribution by the ‘‘charge density’’, (x, y, z). If the amount of charge in a small
volume V2 located at the point (2) in q2, then is defined by
q2 ¼ ð2ÞV2
ð12Þ
To use Coulomb’s law with such a description, we replace the sums of Eq. (11) by
integrals over all volumes containing charges. Then we have
1
E~ð1Þ ¼
4p"0
Z
all space
ð2Þ~
e12 dV2
r212
ð13Þ
Fig. 1. A vector field can be represented by drawing lines which are tangent to the direction of the
field vector at each point, and by drawing the density of lines proportional to the magnitude of the
field vector. The magnitudes and directions of the arrows indicate the values of the vector field at the
points from which the arrows are drawn.
323
With the integrals we can find the fields produced by a sheet of charge, from a
line of charge, from a spherical shell of charge or from any specified distribution.
We shall go on to discuss the electric potential.
4.3. Electric potential
The idea of electric potential is related to the work done in carrying a charge
from a point to another. There is some distribution of charge, which produces an
electric field. We ask about how much work it would take to carry a small charge
from one place to another. The work done against the electrical forces in
carrying a charge along some path is the negative of the component of the
electrical force in the direction of the motion, integrated along the path. If we
carry a charge from one point a to point b,
Z
b
W ¼
F~ d l~ ¼ Z
a
b
ðFx dx þ Fy dy þ Fz dzÞ
ð14Þ
a
where F is the electrical force vector on the charge at each point, and dl is the
differential vector displacement along the path (see Fig. 2).
It is more interesting for our purposes to consider the work that would be done
in carrying one unit of charge. Then the force on the charge is numerically the
same as the electric field. Calling the work done against electrical forces in this
case Wunit, we write
Z
b
Wunit ¼ E~ d l~ ¼ a
Z
b
ðEx dx þ Ey dy þ Ez dzÞ
ð15Þ
a
We consider first what happens in the field due to a single charge q. Let point a
be at the distance r1 from q, and point b at r2. Now we carry a different charge,
which we call the ‘‘test’’ charge, and whose magnitude we choose to be one unit,
from a to b. Let us start with the easiest possible path to calculate. We carry
our test charge first along the arc of a circle, then along the radius, as shown in
Fig. 3(a). To calculate the work done we think the following way [2]: first, there is
no work done at all on the path from a to a0 . The field is radial (from Coulomb’s
law), so it is at right angles to the direction of motion. Next, on the path from a0
to b, the field is in the direction of motion and varies with 1/r2. Thus, work done
on the test charge in carrying it from a to b would be [2]
Z
b
a
q
E~ d l~ ¼ 4p"0
Z
b
a0
dr
q
1 1
¼
r2
4p"0 ra rb
ð16Þ
324
Fig. 2. The work done in carrying a charge from a to b is the negative of the integral of F dl along
the path taken.
Fig. 3. The work when carrying this charge from a to b is the same along any chosen path.
From Ref. [2].
Let us imagine a second possible path from a to b (Fig. 3b). It goes for a while
along an arc of a circle, then radially for a while, then along an arc again, then
radially, and so on. Every time we go along the circular parts, we do no work.
Every time we go along the radial parts, we must just integrate 1/r2. Along the
first radial stretch, we integrate from ra to ra0 , then along the next radial stretch
from ra0 to ra00 , and so on. The sum of all these integrals is the same as a single
integral directly from ra to rb. We get the same answer for this path that we did
for the first path we tried. It is clear that we would get the same answer for any
path, smooth or not.
Z
W unit ¼ a!b
b
E~ d l~
ð17Þ
a
Since the work done depends only on the endpoints, it can be represented as the
difference between two numbers. Let (a) stand for the work done against the
field in going from a reference point P0 to a, and let (b) be the work done in
325
Fig. 4. The work done in going along any path from a to b is the negative of the work from some
point P0 to a plus the work from P0 to b.
going from P0 to b (Fig. 4). The work in going from a to b can be written as
Z
b
E~ d l~ ¼ ðbÞ ðaÞ
ð18Þ
a
Once we have chosen some arbitrary reference point, a number is determined for any point in space: is then a scalar field. It is a function of x, y, z.
We call this scalar function the electrostatic potential at any point:
Electrostatic potential:
Z
P
ðPÞ ¼ E~ d l~
ð19Þ
P0
For convenience, we will often take the reference point at infinity, where the
potential is considered zero. Then, for a single charge at the origin, the potential
is given for any point (x, y, z) using the following equation:
ðx, y, zÞ ¼
q 1
4p"0 r
ð20Þ
The electric field from several charges can be written as the sum of the electric
field from the first, from the second, from the third, etc. When we integrate the
sum to find the potential we get a sum of integrals. Each integral is the potential
from one of the charges. We conclude that the potential from several charges
is the sum of the potentials from all the individual charges. There is a
superposition principle also for potentials. Using the same kind of arguments by
which we found the electric field from a group of charges and for a distribution
of charges, we can get the complete formulas for the potential at a point
we call (1):
ð1Þ ¼
X
j
1 qj
4p"0 r1j
ð21Þ
326
The potential has physical significance: it is the potential energy which a unit
charge would have if brought to the specified point in space from some reference
point.
ð1Þ ¼
1
4p"0
Z
ð2Þ dV2
r12
ð22Þ
4.4. Electric field and electrostatic potential
The electric field vector E can be obtained easily from the electrostatic potential
by taking its derivative. Consider two points, one at x and one at (x þ dx), but
both at the same y and z, and ask how much work is done in carrying a unit
charge from one point to the other. The path is along the horizontal line from x
to x þ dx. The work done is the difference in the potential at the points:
W ¼ ðx þ x, y, zÞ ðx, y, zÞ ¼
@
x
@x
ð23Þ
But the work done against the field for the same path is
Z
W ¼ E~ d l~ ¼ Ex x
ð24Þ
We see that
Ex ¼ @
@x
ð25Þ
Similarly,
Ey ¼ @
@y
Ez ¼ @
@z
ð26a,26bÞ
or, summarising
~
E~ ¼ r
ð27Þ
This equation is the differential for Eq. (19). Any problem with specified charges
can be solved by computing the potential from Eq. (21) or Eq. (22) and using
Eq. (27) to get the field. Equation (27) also agrees with what was found from
vector calculus, that for any scalar field Z
b
a
~ d l~ ¼ ðbÞ ðaÞ
r
ð28Þ
327
The advantage of computing rather than E is that there is only one integral for
while there are three for E (because E is a vector). It turns out that in many
practical cases it is easier to calculate and then take the gradient to find the
electrical field, than it is to evaluate the three integrals for E.
4.5. First law of electrostatics: Gauss’ law
Let us consider a surface, for example a sphere with radius r, centred on a point
charge q, as shown in Fig. 7. What is the flux of E out of the closed surface that
contains the point charge q? If the radius of the little sphere is r, the value of E
everywhere on its surface is
1 q
4p"0 r2
and is directed always normal to the surface. We find the total flux through S0 if
we multiply this normal component of E by the surface area:
Flux through the surface S0
1 q
q
ð4pr2 Þ ¼
4p"0 r2
"0
ð29Þ
Flux ¼ (average normal component) (surface area)
a number independent of the radius of the sphere. The flux through S is also
q/"0, a value independent of the shape of S so long as the charge q is inside.
Let us consider the volume enclosed between the two surfaces S and S0 ,
that has no charge in it. Let us consider the surface shown in Fig. 5. If the
E field is like a flow, the net flow out of this box should be zero. That is what
we get if by the ‘‘flow’’ from this surface we mean the surface integral of the
normal component of E – that is, the flux of E. On the radial faces, the normal
component En of the electric field is zero. On the spherical faces, the normal
component En is just the magnitude of E – minus for the smaller face and plus for
the larger face. The magnitude of E decreases as 1/r2, but the surface area is
proportional to r2, so the product is independent of r. The flux of E into face a is
just cancelled by the flux out of face b. The total flow out of S1 is zero, which is to
say that for this surface
Z
E~ d a~ ¼
S1
Z
Z
E da cos ¼
S1
En da ¼ 0
ð30Þ
S1
where E is the electric field vector, da is an infinitesimal element of some surface
over which we want to integrate the field, and is the angle between E and da.
If for example the surface were to lie in the x–y plane, then in magnitude
328
da ¼ dx dy. The direction of the vector da is considered perpendicular to the
surface at each point on it. For an integral over a closed surface, the direction of
da is that of the outward normal. In the simple case were E has a constant
magnitude, and makes a constant angle with the surface normal, the integral
becomes ES cos , where S is the surface area.
The volume enclosed by surface S0 and S can be considered made of several
volumes as shown in Fig. 5. Therefore, the flux of E into the volume V through
surface S0 is cancelled by the flux of E out of the volume V from surface S. The
total flow is then zero (Fig. 6).
We can write our conclusions as follows [2]:
Z
En da ¼ 0 if q is outside S
ð31Þ
any surface S
Z
En da ¼
any surface S
q
"0
if q is inside S
ð32Þ
Now let us suppose that there are two charges, a charge q1 at one point and a
charge q2 at another point. The electric field whose normal component we
integrate for the flux is the field due to both charges. That is, if E1 represents the
electric field that would have been produced by q1 alone, and E2 represents the
electric field produced by q2 alone, the total electric field is E ¼ E1 þ E2. The flux
through any closed surface S is
Z
Z
Z
ðE1n þ E2n Þ da ¼
S
E1n da þ
S
E2n da
ð33Þ
S
Fig. 5. The flux of E out of the surface S1 is zero.
Fig. 6. The flux of E through a spherical surface containing a point charge q is q/"0. The total flux
through the volume V between the two surfaces S0 and S is zero.
329
The flux with both charges present is the flux due to a single charge plus the
flux due to the other charge. If both charges are outside S, the flux through S is
zero. If q1 is inside S but q2 is outside, then the first integral gives q1/"0 and the
second integral gives zero. If the surface encloses both charges, each will give its
contribution and we have that the flux is (q1 þ q2)/"0. The general rule is clearly
that the total flux out of a closed surface is equal to the total charge inside,
divided by "0.
This result is an important general law of the electrostatic field, called the
Gauss’ law.
Gauss’ law
Z
En da ¼
any closed surface S
Qint
"0
ð34Þ
where
Qint ¼
X
qi
ð35Þ
inside S
If we describe the location of charges in terms of a charge density , we can
consider that each infinitesimal volume dV contains a ‘‘point’’ charge dV. The
sum over all charges is then the integral
Z
Qint ¼
dV
ð36Þ
volume inside S
From this derivation we see that Gauss’ law follows from the fact that the
exponent in Coulomb’s law is exactly two. A 1/r3 field, or any 1/rn field with
n 6¼ 2, would not give Gauss’ law. So the Gauss’ law is just an expression, in a
different form, of the Coulomb’s law. The two are quite equivalent so long as we
keep in mind the rule that the forces between charges are radial.
4.5.1. Differential form of Gauss’ law
Gauss’ law can be thought of as being an integral formulation of the law of
electrostatics. We can obtain a differential form (i.e., a differential equation) by
using the divergence theorem. The divergence theorem states that for any wellbehaved vector field C(x) defined within a volume V surrounded by the closed
surface S, the relation
Gauss’ Theorem
I
Z
~
~ C~ dV
r
C n~ da ¼
S
V
ð37Þ
330
holds between the volume integral of the divergence of C and the surface integral
of the outwardly directed normal component of C. The equation in fact can be
used as the definition of the divergence. The Gauss’ theorem is demonstrated in
Appendix A by applying Gauss’ law to an infinitesimal cubic surface. Gauss’
theorem tell us that the flux of E out of such cube is r E times the volume dV of
the cube. The charge inside of dV, by the definition of , is equal to dV, so
Gauss’ law gives
~ E~ dV ¼ dV
r
"0
ð38Þ
~ E~ ¼ r
"0
ð39Þ
which is the differential form of the Gauss’ law of electrostatics. The differential
form of the Gauss’ law is the first fundamental equation of electrostatics.
4.6. Second law of electrostatics
The second law of electrostatics states that the circulation of the electrical field is
zero [2].
~ E~ ¼ 0
r
ð40Þ
For any vector field the circulation around any imaginary closed curve is
defined as the average tangential component of the vector multiplied by the
circumference of the loop.
Circulation ¼ ðaverage tangential componentÞ ðdistance aroundÞ
If we have an arbitrary curve in space and measure the circulation of the electric
field around the curve, we will find that it is not, in general, zero. However, it is
zero for the Coulomb field.
In order to derive the second law of electrostatics we need to use and
demonstrate two theorems. The demonstration of these theorems can be found in
Appendix A.
Theorem 1. The line integral of a scalar field
Z
ð2Þ ð1Þ ¼
2
~ d l~
r
1
any curve from 1 to 2
ð41Þ
331
where the function (x,y,z) is a scalar field that assumes the value
(x2, y2, z2) and (1) at point (x1, y1, z1).
(2)
at point
Theorem 2. The Stokes’ Theorem
I
C~ d l~ ¼
Z ~ C~
r
S
n
da
ð42Þ
where C is a vector field and S is any surface bounded by G.
The cross product r C is a vector whose components we can write by the
usual rule for cross product:
~ C~ ¼ ry Cz rz Cy ¼ @Cz @Cy
r
x
@y
@z
ð43Þ
~ C~ ¼ rz Cx rx Cz ¼ @Cx @Cz
r
y
@z
@x
ð44Þ
~ C~ ¼ rx Cy ry Cx ¼ @Cy @Cx
r
z
@x
@y
ð45Þ
The combination r C is called ‘‘the curl of C’’.
After these two theorems have been demonstrated and accepted, it is
straightforward to derive the second law of electrostatics:
~ E~ ¼ 0
r
ð46Þ
Let us imagine a closed line from point 1 to point 2 as shown in Fig. 2, where 1
and 2 were named a and b. Since E ¼ r , Theorem 1 tells us that the integral
of the vector r around any closed loop must be zero:
Z
2
~ d l~ ¼ ð2Þ ð1Þ
r
ð47Þ
~ d l~ ¼ ð1Þ ð2Þ
r
ð48Þ
1
Z
1
2
Therefore,
I
~ d l~ ¼
r
12
Z
2
1
~ d l~þ
r
Z
1
~ d l~ ¼ ð2Þ ð1Þ þ ð1Þ ð2Þ ¼ 0
r
2
ð49Þ
332
Using Stokes’ theorem, we can conclude that
Z
~ r
~ da ¼ 0
r
ð50Þ
over any surface. But if the integral is zero over any surface, the integrand must
be zero.
~ r
~ ¼ 0
r
ð51Þ
The second law of electrostatics follows directly from Coulomb’s law.
4.7. Equations of the electrostatic potential: Poisson and Laplace equations
There are two laws of electrostatics: that the flux of the electric field from a
volume is proportional to the charge inside – Gauss’ law, and that the circulation
of the electrical field is zero – E is a gradient. These two laws are summarised in
the Maxwell equations for electrostatics:
~ E~ ¼ r
"0
ð52Þ
~ E~ ¼ 0
r
ð53Þ
In fact, the two equations can be combined into a single equation. From the
second equation, we know at once that we can describe the field as the gradient
of a scalar
~
E~ ¼ r
ð54Þ
We may completely describe any particular electric field in terms of its potential
. We obtain the differential equation that must obey by substituting Eq. (54)
into Eq. (52), to get
~ r
~ ¼ r
"0
ð55Þ
The divergence of the gradient of is the same as r2 operating :
2
2
2
2
~ r
~ ¼ r
~ ¼@ þ@ þ@ r
@x2 @y2 @z2
ð56Þ
333
so we write Eq. (55) as
Poisson equation
r2 ¼ "0
ð57Þ
The operator r2 is called the Laplacian, and Eq. (57) is called the Poisson
equation. In regions of space that lack a charge density, the scalar potential
satisfies the Laplace equation:
Laplace equation
r2 ¼ 0
ð58Þ
The entire subject of electrostatics, from a mathematical point of view, is merely
a study of the solutions of the single equation 57. Once is obtained by solving
the Poisson equation we can find E immediately from Eq. (54).
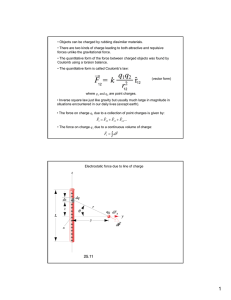
5. Field lines and equipotential surfaces
A geometrical description of the electrostatic field will now be given. The two
laws of electrostatics, one that the flux is proportional to the charge inside and
the other that the electric field is the gradient of a potential, can also be
represented geometrically. We shall illustrate this with two examples.
First, we take the field of a point charge. Lines in the direction of the field can
be drawn – lines which are always tangent to the field. These are called field lines.
The lines show everywhere the direction of the electric vector. We also want to
represent the magnitude of the vector. We can make the rule that the strength of
the electric field will be represented by the ‘‘density’’ of the lines. By the density
of the lines we mean the number of lines per unit area through a surface
perpendicular to the lines. With these two rules we can have a picture of the
electric field. For a point charge, the density of the lines must decrease with 1/r2.
But the area of a spherical surface perpendicular to the lines at any radius r
increases as r2, so if we keep the same number of lines for all distances from the
charge, the density will remain in proportion to the magnitude of the field. We
can guarantee that there are the same number of lines at every distance if we
insist that the lines are continuous – that once a line is started from the charge, it
never stops. In terms of the field lines, Gauss’ law says that lines should start at
plus charges and stop at minus charges. The number which leaves a charge q
must be equal to q/"0.
Now we can find a similar geometrical picture for the potential . The easiest
way to represent the potential is to draw surfaces on which is constant. We call
them equipotential surfaces – surfaces of equal potential. What is the geometrical
334
relationship of the equipotential surfaces to the field lines? The electric field is the
gradient of the potential. The gradient is in the direction of the most rapid
change of the potential, and is therefore perpendicular to an equipotential
surface. If E were not perpendicular to the surface, it would have a component in
the surface. The potential would be changing in the surface, but then it would not
be an equipotential. The equipotential surfaces must then be everywhere at right
angles to the electric field lines. For a point charge all by itself, the equipotential
surfaces are spheres centred at the charge (Fig. 7).
Fig. 7. Field lines and equipotential surfaces for a positive point charge.
The electric field lines are directed away from a positive charge and towards a
negative charge (Fig. 8).
Fig. 8. Field lines and equipotential surfaces for a pair of point charges (positive charge
þ 8, negative charge 8). Image created with the Consortium for Upper-Level Physics Software
(CUPS) [4].
6. Electrostatic energy and electric fields from an arbitrary distribution of charges
The simple case of a pair of point charges is quite rare. We shall now start to
channel this dissertation towards the biological world of proteins, richly
populated by charged residues.
335
6.1. Electrostatic energy of an arbitrary distribution of charges
We wish now to consider the energy of electrostatic systems. The law of the
energy of interaction in electrostatics is very simple. Suppose we have two
charges q1 and q2 separated by the distance r12. There is some energy in the
system, because a certain amount of work was required to bring the charges
together. We have already calculated the work done in bringing two charges
together from a large distance. It is
q1 q2
4p"0 r12
We also know, from the principle of superposition, that if we have many charges
present, the total force on any charge is the sum of the forces from the others. It
follows, therefore, that the total energy of system of a number of charges is
the sum of the terms due to the mutual interaction of each pair of charges. If
qi and qj are any two of the charges and rij is the distance between them, the
energy of that particular pair is
qi qj
4p"0 rij
ð59Þ
The total electrostatic energy U is the sum of the energies of all possible pairs of
charges:
U¼
X
all pairs
qi qj
4p"0 rij
ð60Þ
It is important to notice that the last equation excludes infinite self-energy terms
(i ¼ j), which correspond to the work of assembling a charge into a point.
Usually, these Coulombic self-energy terms are of no interest at all, because the
use of point charges is precisely done to focus on the charge configurations only.
Thus, i ¼ j are usually excluded from the usual definition of electrostatic potential
energy for a set of point charges. It should be understood that in actual
calculations with point charges the Coulombic self-energy terms should be
eliminated from the electrostatic potential energy.
If we have a distribution of charge specified by a charge density , the sum of
Eq. (60) is, of course, to be replaced by an integral. As usual, we consider that
each volume element dV contains the element of the charge dV. Then Eq. (60)
should be written
U¼
1
2
Z
all space
ð1Þð2Þ
dV1 dV2
4p"0 r12
ð61Þ
336
Notice that the factor 1/2 was introduced because in the double integral over dV1
and dV2 we have counted all pairs of charge elements twice. Next we notice that
the integral over dV2 in Eq. (61) is just the potential at (1). That is,
Z
ð2Þ
dV2 ¼ ð1Þ
4p"0 r12
ð62Þ
so that Eq. (61) can be written as
U¼
1
2
Z
ð1Þð1Þ dV1
ð63Þ
Or, since the point (2) no longer appears, we can simply write
1
U¼
2
Z
dV
ð64Þ
This equation can be interpreted as follows. The potential energy of the charge
dV is the product of this charge and the potential energy at the same point.
The total energy is therefore the integral over dV. But there is again the
factor 1/2. It is still required because we are counting energies twice. The mutual
energy of two charges is the charge of one times the potential at it due to the
other. Or, it can be taken as the second charge times the potential at it from the
first. Thus, for two-point charges we could write
U ¼ q1 ð1Þ ¼ q1
q2
4p"0 r12
ð65Þ
or
U ¼ q2 2 ¼ q2
q1
4p"0 r12
ð66Þ
Notice that we could also write
1
U ¼ ½q1 ð1Þ þ q2 ð2Þ
2
ð67Þ
This energy is located in space, where the electric field is. This seems reasonable
because we know that when charges are accelerated they radiate electric fields.
We would like to say that when light or radio waves travel from one point to
another, they carry their energy with them. But there are no charges in waves. So
we would like to locate the energy where the electromagnetic field is and not
337
at the charges from which it came. We thus describe the energy, not in terms of
the charges, but in terms of the fields they produce. We can, in fact, show that
Eq. (64) is numerically equal to (see Appendix A)
"0
U¼
2
Z
E~ E~ dV
ð68Þ
We can then interpret this formula as saying that when an electric field is present,
there is located in space an energy whose density (energy per unit volume) is
u¼
"0 ~ ~ "0 E 2
EE ¼
2
2
ð69Þ
In Appendix A we show that Eq. (68) is consistent with the laws of electrostatics.
Eq. (68) is derived using the Poisson equation.
PART 2 – MODELLING PROTEIN ELECTROSTATICS IN PROTEINS
7. Perspective and overview
Electrostatic interactions in macromolecular systems arise from the following
sources: the presence of local charges, the polarisation stemming from the
non-spherical distribution of electron density around atoms, the redistribution
of electrons caused by local electrical fields (electronic polarisation) and the
reorientation of polar groups in the solute and solvent molecules in response to
the electric field (orientation polarisation) [5]. The treatment of each of these
factors has its own challenges. Can charge distributions be adequately represented
by only partial atomic charges at the atom centres? Can electronic polarisation
within the macromolecule be ignored or, if not, is it best approximated by point
inducible dipoles (PIDs) on the atoms or bonds, or by a continuum dielectric? Can
polar group reorientation be treated in any way other then by some form of
simulation of motion? Can the very large and environment-specific reorientation of
the solvent molecules be represented by a continuum dielectric, or is it necessary to
introduce explicit solvent structure? How can each of these approximations be
tested? Electrostatic calculations attempt to model these complex and often
subtle effects. Electrostatic models should provide insight into the role of
electrostatics in macromolecule structure and function, fit appropriate
experimental data, and allow us to make predictions about macromolecular
structure and function.
Arguably, there is no theoretical difficulty in treating all of the electrostatic
contributions listed above adequately. Molecular dynamic simulations can
provide sufficient sampling of configurational space of both the macromolecules
338
and solvent structures. Charge distributions can be made as elaborate as
necessary, in the form of multipole expansions and, electronic polarisation
can be treated by polarisation tensors. The difficulty lies in the development
of accurate parameterisations of these effects and in the time used by such
calculations. Thus, current work in the field of electrostatic simulations is
largely concerned with investigating the efficacy of the various possible
approximations, both in reproducing experimental observations and in reducing
computation time.
The fundamentals of classical electrostatics, that allows us to arrive at the
Poisson equation, were stated concisely in Part 1 (section 4.7). This equation is
the starting point for modelling electrostatic interactions in proteins. The
apparent simplicity of such equations, however, can hide the substantial
difficulties involved in applying them to complex systems. The problem is
particularly acute in studies of proteins and nucleic acids owing to the vast
amount of structural information about these macromolecules now available. In
contrast to traditional models in which proteins were treated as low-dielectric
media spheres and DNA as a charge cylinder, most current questions of interest
are asked at the atomic level. The question of how best to represent atomic and
molecular properties within the framework of electrostatic theory poses new
conceptual as well as numerical difficulties.
It is common to encounter the opinion that models based on classical
electrostatics have been superseded, or even invalidated, by the advent of
computer simulations of atomic motions. A criticism sometimes expressed is that
classical electrostatics in not valid on a microscopic scale. Thus, the theory must
be applied in a physical meaningful way to the system being studied. Classical
electrostatics remains a rigorous and intuitively appealing approach to a wide
range of microscopic phenomena.
In the following sections we will cover the equations used to model
electrostatic interactions in proteins, the different models that have been used to
describe the electrostatic interactions in such macromolecules, as well as one of
the methods used to find the potential distribution in proteins (see Fig. 9). Such
potential maps are a major source of information when trying to correlate
protein structure and function and stability. One of the first steps needed to find
the potential distribution on proteins is to compute the charge carried by each
titratable residue as a function of pH. DelPhi is a widely used program that offers
the possibility of finding the charge distribution of a protein [6]. However, this
program does not, for example, consider Tyr residues as titratable residues, and
at any pH value (from 1 to 14) it considers that Tyr is a neutral amino acid.
Unaware of this serious mistake, a large number of scientists still use such
program in order to find the charge distribution on a protein as a function of pH.
Also, this and other programs, assume that, for example, all Asp residues in a
protein have a pKa value of 4, all Glu residues a pKa of 4.5, all His display a pKa
of 6.4, all Lys have a pKa of 10.4. As we will see in Appendices A and B, other
charged sites and the local environment in the protein may shift the pKa of a
339
Fig. 9. Electrostatic potential distribution on the molecular surface of the lipase/esterase cutinase at
pH 8.5, the pH optimum of this enzyme. The arrow points into the active site. Blue colour
represents positive potential, white colour neutral potential and red colour negative potential.
given site from its typical value by several pH units. Therefore, it is of crucial
importance to use a methodology that takes into account such effects when
calculating the protonation state of each titratable residue as a function of pH.
We have selected the program TITRA, written by Paulo Martel and Steffen B.
Petersen [7,8] for such computations, since the computed pKa values by TITRA
have been shown to be in good agreement with the experimental values, for a
large number of selective protein [7].
8. Classical continuum electrostatics – the two dielectric media
The approaches to model electrostatic interactions in chemical and biochemical
systems, either from a purely theoretical or a computational point of view, can be
divided in two broad types. The earlier models obviated the atomic level
description by treating solute and solvent as homogeneous dielectric media
where charges were distributed in a discrete or continuous fashion [1,9–11]. In
this way, the treatment of atomic electrostatic interactions was reduced to a
problem of classical continuum electrostatics (CE), based on classical electrostatics. These models were justifiable given the lack of atomic-level information
about biological molecules at the time they were developed, and the limited
computational facilities. With the advent of computers and high-resolution,
molecular structure techniques, new methods were introduced for calculations
based on simulations at the atomic level, namely Monte Carlo [12] and molecular
dynamics (MM) [13,14]. These atomic-level methods became a common practice
in chemical–physical studies and were later extended to a wide range of systems
of chemical and biological interests [15–18]. The atomic detail of these methods
leads to a neglect of CE-based methods, whose less-detailed nature is regarded as
a crude approximation. However, the development of fast numerical and
computational methods made it possible to achieve a quantitative level in CE
340
calculations and caused a revival in the use of CE methods [19,20]. The work
presented in this chapter is primarily based on CE methods.
In the most simplistic approach to model electrostatic interactions in proteins,
one can assume that charges on a protein interact through a medium
characterised by a single dielectric constant, and that all interactions can be
described by Coulomb’s law (Part 1, Eq. (9)). However, this approach fails since
the protein and the solvent have very different dielectric properties. A more
realistic approach is to explicitly consider that the protein and solvent region
have different dielectric constants. This means that the interactions can no longer
be computed using Coulomb’s law. Instead, the Poisson equation of the system
of charges and dielectrics has to be solved [21,22]. In addition, it is reasonable to
assume that the protein surrounds itself with an atmosphere of counterions, as
described by the Debye–Hückel theory of electrolytes [1,21,23,24]. In this case
the PB equation, usually in its linear form, is solved. When the system has some
symmetry, it is often possible to express the solution of either the Poisson or the
Poisson–Boltzmann equations in an analytic form. A simple approximation is to
consider the protein to be a sphere with the charges placed at a small distance
beneath the surface and surrounded by an ionic atmosphere [25]. Although
proteins are never perfectly spherical, this model was shown to give satisfactory
results in many cases [26], especially when the interactions are corrected
according to the solvent accessibility of the residues [27]. Although these simple
spherical models do not include atomic detail to any substantial extent, they have
the advantage of being analytically solvable and computationally accessible with
present-day computers.
The dielectric properties of a system are described by the dielectric constant
which reflects the reorientation of dipoles under the local electric field. These
dipoles are essentially of two types: permanent and induced. Permanent dipoles
occur when the distribution of charge over neighbouring atoms is not symmetric.
Typical examples are the peptide bond and the water molecule. Induced dipoles
arise from electronic polarisation, i.e., from the distortion of electron clouds
immersed in an electric field. In liquid water, the relative freedom of the
molecules allows a high-dipolar rotation and consequently a high-dielectric
constant (78.5 at 298 K). The contribution of electronic polarisation to this
overall value is very small, 4. In contrast, permanent dipoles in the protein
interior are virtually fixed and the orientation of the induced dipoles leads to a
much smaller dielectric constant. Both experiment and theory point to 2–4 for
the protein dielectric constant (see section 10.1), where electronic polarisation is
considered to be the most important contribution [7]. However, previous studies
on the interpretation and modelling of the pKa shifts introduced on particular
titratable residues of subtilisin upon mutation of titratable residues has
shown that the dielectric constant between charges in a protein could range from
45 to 120 [28].
The resulting dielectric regions can be seen as a cavity (see Fig. 10) with
a low-dielectric constant "p (the protein) immersed in a continuous medium
341
Fig. 10. Continuum electrostatic model of a protein in a solution. "p, protein dielectric constant; "s,
solvent dielectric constant; I, solvent ionic strength.
with a high-dielectric constant "s (the solvent). In this model, formal charges
are assigned to all titratable residues, depending on pH and pKa, and bound
ions can be included. In more detailed models, partial charges on all atoms
can be included. The charge sources have been divided into two groups,
the background charges and the titratable charges. While some of the atomic
charges are independent of the protonation state of the molecule (background
charges, e.g., partial charges carried by the peptide bond atoms and partial
charges carried by non-titratable polar groups, such as the hydroxyl group
of serine and threonine, or charges carried by metal ions such as Ca2 þ ), partial
atomic charges in the vicinity of the titratable protons of ionic (titratable)
residues (Asp, Glu, Lys, Arg, His, Tyr, free Cys, N- and C-terminus) are
generally pH dependent, as a consequence of the protonation/deprotonation
reactions. In some cases, the contribution of the background charges is not
included and the formal charges of the titratable residues are taken as the only
electrostatic field sources in the protein. The spatial location of the titratable
moieties on the protein derives from the coordinate information obtained from
X-ray or NMR studies. In the absence of such information one may have to rely
on homology-based modelling. The charges on the solvent molecules, on the
other hand, are assumed to be averaged out in the dielectric-based continuum
description. However, the polarisation of the molecular surface reflects the
orientation of the water molecules throughout the solvent. If there are ions
present in the aqueous phase, their distribution will be affected by the protein
charges, and in the CE model this effect is normally accounted for through the
use of a counterionic charge. The counterions cannot approach the protein more
than allowed by their ionic radii, which defines an ionic exclusion boundary. The
counterion distribution is usually assumed to be determined simply by the
electrostatic potential and the solution ionic strength, I, as in the Debye–Hückel
theory of the solutes [23,24].
We shall now derive the electrostatic equations for dielectrics, namely
the Poisson equation for dielectrics. This equation is the starting point for
the determination of the electrostatic potential in a protein once we know the
distribution of charge in the protein and its dielectric constant has been chosen.
342
We will also gain better insight on why our choice of the uniform dielectric model
is a very reasonable choice to account for the electronic polarisation.
9. Equations of electrostatics for dielectrics
In Part 1 we considered electrostatic interactions between charges in vacuum, but
we did not include the effect that the dielectric constant of the media might have
on such interactions. We therefore made no distinction between microscopic
fields and macroscopic fields. Air is sufficiently tenuous that the neglect of its
dielectric properties causes no great error. Our results so far are applicable there.
But much of electrostatics concerns itself with charges and fields in media whose
respective electric responses must be taken into account.
As mentioned above, we will model the solute protein molecules and the
solvent molecules as dielectric media, characterised by a particular dielectric
constant. Therefore, we call them dielectrics. Before we enter such discussion, we
shall now derive the equations of electrostatics when there are dielectrics. In
order to arrive at these equations we need to discuss another of the peculiar
properties of matter that arises under the influence of the electric field.
9.1. Macroscopic theory of dielectrics – the polarisation vector P
Let us start with a simple but relevant question: Why should a field induce a
dipole moment in an atom? We will here give an example to illustrate a possible
mechanism. An atom has a positive charge on the nucleus, which is surrounded
by negative electrons. In an electric field, the nucleus will be attracted in one
direction and the electrons in the other. The orbits or wave patterns of the
electrons (or whatever picture is used in quantum mechanics) will be distorted to
some extent, as shown in Fig. 11. The centre of gravity of the negative charge will
be displaced and will no longer coincide with the positive charge of the nucleus.
If we look from a distance, such a neutral configuration is equivalent, to a first
approximation, to a little dipole p.
It seems reasonable that if the field is not too strong, the amount of induced
dipole will be proportional to the field. That is, a small field will displace the
charges a little bit and a large field will displace them further – and in proportion
Fig. 11. An atom in an electric field has its distribution of electrons displaced with respect to the
nucleus. The centre of positive and negative charges no longer overlap.
343
to the field – unless the displacement gets too large. For the remainder of this
chapter, it will be supposed that the dipole moment is exactly proportional to the
field. Materials that show a linear response to weak fields eventually show nonlinear behaviour at high-enough field strengths, where the applied electric field
no longer induces an electric polarisation proportional to the magnitude of the
applied field. Under such conditions the electronic or ionic oscillators are driven
to large amplitudes. The linear relation between the polarisation vector and the
electric vector (described in Eq. (75)) is no longer valid, and the magnitude of the
electric polarisation induced in the medium by the electric field can be expressed
in a Taylor series expansion as
P ¼ "0 ðE þ 2 E 2 þ 3 E 3 þ Þ
Non-linear response
P~ ¼ "0 E~
Linear response
where is the linear susceptibility of the material, and 2 and 3 are the secondorder and third-order non-linear optical susceptibilities, respectively. The first
term "0E represents linear effects in which the polarisation of the medium is
simply proportional to E. Unless the E field is very large, the coefficients of the
higher-order terms are too small to allow high-power terms to influence the
polarisation appreciably. Only with the availability of intense, coherent light
have these higher-order terms become significant. Interestingly, the non-linear
term "02E2 in the case of optical electric fields is responsible for the frequency
doubling when light passes through a prism. However, we will not go into detail
into this matter. We will now assume that in each atom there are charges q
separated by a distance , so that q is the dipole moment per atom. If there are N
atoms per unit volume, there will be a dipole moment per unit volume equal to
Nq. This dipole moment per unit volume will be represented by a vector, P.
Needless to say, it is in the direction of the individual dipole moments, i.e., in the
direction of the charge separation [2]:
P~ ¼ Nq~
ð70Þ
In general, P will vary from place to place in the dielectric. However, at any point
in the material, P is proportional to the electric field E. The constant of
proportionality, which depends on the ease with which the electron is displaced,
will depend on the kinds of atoms in the molecule.
9.2. Polarisation charges
9.2.1. Uniform polarisation in the dielectric
Let us consider a material in which there is a certain dipole moment per unit
volume. Will there be on average any charge density produced by this? Not if P is
uniform. If the positive and negative charges being displaced relative to each
344
other have the same average density, the fact that they are displaced does not
produce any net charge inside the volume. So, we need to look only at what
happens at the surfaces. At one surface the negative charges, the electrons, have
effectively moved out a distance . At the other surface they have moved
in, leaving some positive charge effectively moved out a distance , as shown in
Fig. 12. We will have a surface density of charge, which will be called the surface
polarisation charge.
This charge can be calculated as follows. If A is the area of the plate, the
number of electrons that appears at the surface is the product of A and N, the
number per unit volume, and the displacement , which we assume here
perpendicular to the surface. The total charge is obtained by multiplying by the
electronic charge qe. To get the surface density of the polarisation charge induced
on the surface, we divide by A. The magnitude of the surface charge density is
pol ¼ Nqe ð71Þ
But this is just equal to the magnitude P of the polarisation vector P,
pol ¼ P
ð72Þ
The surface density of charge is equal to the polarisation inside the material. The
surface charge is, of course, positive on one surface and negative on the other.
Now let us assume that our dielectric also has surface charges, which we will
call charge. It should be emphasised that pol exists only because of charge. Like
in a parallel-plate capacitor, if charge is removed by discharging the capacitor,
pol will disappear, not by going out on the discharging wire, but by moving back
into the dielectric material – by the relaxation of the polarisation inside the
material [2]. We are trying to establish an analogy with the surface of the protein,
where charge can be seen as the charge carried by the titratable residues.
We can now apply Gauss’ law (see Part 1) to the Gaussian surface S in Fig. 13.
The electric field E in the dielectric is equal to the total surface charge density
divided by "0. It is clear that pol and charge have opposite signs, so
E¼
charge pol
"0
ð73Þ
Fig. 12. A dielectric slab in a uniform field. The positive charges displaced the distance with
respect to the negatives.
345
Fig. 13. A dielectric in-between two charged plates.
The field E0 between the surface of the dielectric and the outmost charged surface
is higher than the field E. It corresponds to charge alone. But we are concerned
about the field inside the dielectric which, if the dielectric nearly fills the gap, is
the field over nearly the whole volume. Using Eq. (72), we can write:
E¼
charge P
"0
ð74Þ
This equation does not tell us what the electric field is unless we know what P is.
Here, however, we are assuming that P depends on E – in fact, that it is
proportional to E. This proportionality is usually written as
P~ ¼ "0 E~
ð75Þ
The constant (Greek ‘‘khi’’) is called the electric susceptibility of the dielectric.
Then Eq. (74) becomes
E¼
charge
1
"0 ð1 þ Þ
ð76Þ
which gives us the factor 1/(1 þ ) by which the field is reduced.
The factor (1 þ ) is a property of the material. It is its dielectric constant.
Dielectric constant
k¼1þ
ð77Þ
Let us consider something a bit more complicated – the situation in which
the polarisation P is not everywhere the same. We shall not get lost! This is our
way to understand the Poisson equation for an inhomogeneous medium, the
starting equation for finding out the electrostatic potential distribution in
proteins. This way we will understand from where this so-spoken equation
came about!
346
9.2.2. Non-uniform polarisation in the dielectric
If the polarisation is not constant, we would expect in general to find a
charge density in the volume, because more charge might come into one side of a
small-volume element than leaves it on the other. How can we find out how
much charge is gained or lost from a small volume?
First, we shall compute how much charge moves across any imaginary
surface when the material is polarised. The amount of charge that goes
across a surface is just P times the surface area if the polarisation is normal to
the surface. Of course, if the polarisation is tangential to the surface, no
charge moves across it. This is the same line of thinking as applied in Part 1.
Nothing new!
Following the same arguments we have already used, it is easy to see that the
charge moved across any surface element is proportional to the component of P
perpendicular to the surface. In general, Eq. (72) should be written as,
pol ¼ P~ n~ ¼ Pn cos ð78Þ
where n is the outward unitary vector normal to the surface, and the angle
between the vectors P and n [2].
If we are thinking of an imagined surface element inside the dielectric, Eq. (78)
gives the charge moved across the surface but does not result in a net surface
charge, because there are equal and opposite contributions from the dielectric on
the two sides of the surface.
The displacements of the charges can, however, result in a volume charge
density. The total charge displaced out of any volume V by the polarisation is the
integral of the outward normal component of P over the surface S that bounds
the volume (see Fig. 14).
An equal excess charge of the opposite sign is left behind. Denoting the net
charge inside V by Qpol we write
Z
Qpol ¼ P~ n~ da
S
Fig. 14. A non-uniform polarisation P can result in a net charge in the body of a dielectric.
ð79Þ
347
We can attribute Qpol to a volume distribution of charge with the density pol,
and so
Z
Qpol ¼
pol dV
ð80Þ
V
Combining the two equations yields
Z
vol dV ¼ Z
V
P~ n~ da
ð81Þ
S
We have a kind of Gauss’ theorem that relates the charge density from
polarised materials to the polarisation vector P. Using Eq. (81) with the
Gaussian surface of Fig. 13, the surface integral gives PA, and the charge inside
is polA, so we get again that ¼ P.
Just as we did for Gauss’ law of electrostatics, we can convert Eq. (81) to a
different form – using Gauss’ mathematical theorem:
Z
P~ n~ da ¼
S
Z
~ P~ dV
r
ð82Þ
V
we get
~ P~
pol ¼ r
ð83Þ
If there is a non-uniform polarisation, its divergence gives the net density of
charge appearing in the material. We emphasise that this is a perfectly real charge
density. We shall call it ‘‘polarisation charge’’ only to remind ourselves how it
got there.
Now we are ready to write the electrostatic equations with the dielectrics, i.e.,
the Poisson equation for an inhomogeneous medium, the starting equation for
finding out the electrostatic potential distribution in proteins (see Fig. 9).
9.3. Poisson equation for a dielectric inhomogeneous medium
Now let us combine the above result with the theory of electrostatics. The
fundamental equation is (see Part 1, section 4.5)
~ E~ ¼ r
"0
ð84Þ
The here is the density of all electric charges. It is convenient to separate into
two parts. Again we call pol the charges due to non-uniform polarisation, and
call charge all the rest, usually the charge at known places in space. In the protein
348
world, it is the charge carried by titratable residues and by background charges,
as we will allude to incoming sections.
Equation (84) then becomes
~ E~ ¼ charge þ pol
r
"0
ð85Þ
Since (Eq. (83))
~ P~
pol ¼ r
we get
~ ~
~ E~ ¼ charge r P
r
"0
ð86Þ
Substituting P by (Eq. (75))
P~ ¼ "0 E~
we get
~
~ E~ þ P
r
"0
!
!
~
"
charge
E
0
~ E~ þ
¼r
¼
"0
"0
ð87Þ
equivalent to
~ ½E~ð1 þ Þ ¼ charge
r
"0
ð88Þ
or
Poisson equation for dielectrics in the SI system of units
~ kE~ ¼ charge
r
"0
ð89Þ
where k ¼ 1 þ .
These are the equations of electrostatics, in the SI system of units. We have not
taken the dielectric ‘‘constant’’, k, out of the divergence. That is because it may
not be the same everywhere. If it has everywhere the same value, it can be
factored out and the equations are just those of electrostatics with the charge
density charge divided by k. In the form we have given, the equations apply to the
general case where different dielectrics may be in different places in the field.
Then the equations may be quite difficult to solve.
As we saw in Part 1 (section 4.4, Eq. (27)),
~ ð~rÞ
E~ ¼ r
ð90Þ
349
So, replacing E by r (r) in Eq. (89) we get
~ ½kr
~ ð~rÞ ¼ charge
r
"0
ð91Þ
~ ½kr
~ ð~rÞ þ charge ¼ 0
r
"0
ð92Þ
or
There is a matter of some historical importance [2] that should be mentioned
here. In the early days of electricity, the atomic mechanism of polarisation was
not known and the existence of pol was not appreciated. The charge charge was
considered to be the entire charge density. In order to write Maxwell’s equations
in a simple form, a new vector D (electric displacement vector) was defined to be
equal to a linear combination if E and P:
D~ ¼ "0 E~ þ P~
ð93Þ
As a result, Eq. (87) was written in an apparently very simple form:
~ D~ ¼ charge
r
ð94Þ
This equation can be solved if another equation is given for the relationship
between D and E. When Eq. (75) holds, this relationship is
D~ ¼ "0 ð1 þ ÞE~ ¼ k"0 E~
ð95Þ
This equation is usually written
D~ ¼ "E~
ð96Þ
where " is still another constant for describing the dielectric property of
materials. It is called the ‘‘permittivity’’. Now we see why we have "0 in our
equations, it is the ‘‘permittivity of empty space’’.
Evidently,
" ¼ k"0 ¼ ð1 þ Þ"0
ð97Þ
One more point should be emphasised. An equation like (96) is an attempt
to describe a property of matter. But matter is extremely complex, and
such an equation is in fact not correct (see section 9.1). For instance, if E gets
too large, then D is no longer proportional to E. For some substances, the
proportionality breaks down even with relatively small fields. Also, the
‘‘constant’’ of proportionality may depend on how fast E changes with time.
350
Therefore, this kind of equation is kind of approximation, like Hook’s law. It
cannot be a deep and fundamental equation. On the other hand, the fundamental
equations for E, Eqs. (7) and (8), represent our deepest and most complete
understanding of electrostatics.
All these equations are valid in the SI system of units. The two systems of
electromagnetic units in most common use today are the SI and the Gaussian
systems. The SI system has the virtue of overall convenience in practical,
large/scale phenomena, especially in engineering applications. The Gaussian
system is more suitable for microscopic problems involving the electrodynamics
of individual charged particles, etc. Usually, in review papers about
electrostatics, Eq. (92) is presented in the Gaussian system of units as
Poisson equation for dielectrics in the Gaussian system of units
~ ½"ð~rÞr
~ ð~rÞ þ 4pð~rÞ ¼ 0
r
ð98Þ
where "(r) is the dielectric constant of a given system with charge density (r) at
each point r in space.
Conversion of equations and amounts between SI units and Gaussian units is
discussed in detail in the appendix on Units and Dimensions by Jackson [29]. We
started deducting the basic electrostatic equations in Part 1 using the SI systems
of units since when they were developed this system of units was most adequate.
From now on, since we will be dealing with microscopic systems, we shall use the
equations in the Gaussian system of units. In the Gaussian system of units, in
practical terms, corresponds to the elimination of most conversion constants. In
particular, the SI conversion factor 1/4p"0 does not occur in the Coulomb
equation and the factor 1/"0 of the Poisson equation is substituted by the factor
4p (Eq. (98)). Also, the difference between (relative) dielectric constant and
electric permittivity (Eq. (98)) disappears.
In Table 1 are listed some of the definition of the Lorentz’ force equation, "0,
permittivity k and conversion factors in the Gaussian and SI systems of
electromagnetic units.
Table 1. Definitions of key equations and amounts in two systems of electromagnetic units.
Gaussian system of units*
Lorentz force equation
Poisson equation in vacuum
Poisson equation for dielectrics
Conversion factors 1/4p"0
Dielectric constant versus permittivity
"0
D
*
From Refs. [2,29].
SI system of units*
E þ v/c B
EþvB
r2 (r) ¼ 4p(r)
r2 ¼ /"0
r ["(r) r (r)] þ 4p(r) ¼ 0 r [k(r) r (r)] þ (r)/"0 ¼ 0
Absent in Coulomb equation Present in Coulomb equation
"(r) ¼ k(r)
"(r) 6¼ k(r)
1
107/4pc2
D ¼ E þ 4pP
D ¼ "0E þ P
D ¼ "(r)E
D ¼ k(r)"0E
351
9.4. Maxwell’s equations in empty space and in dielectric media
In Part 1 we considered electrostatic interactions and fields in the presence of
charges, but no other ponderable media. We will now write and compare the
Maxwell equations of electrostatics in vacuum and in a dielectric medium [2,29].
Maxwell’s equations in vacuum
Gaussian system of units
r E0 ¼ 4p
r E0 ¼ 0
SI system of units
r E0 ¼ /"0
r E0 ¼ 0
Maxwell’s equations in a dielectric medium
Gaussian system of units
r ("E) ¼ 4p
r ("E) ¼ 0
SI system of units
r (kE) ¼ /"0
r (kE) ¼ 0
If the dielectric medium is not only isotropic but also uniform, we have the
following equations:
Maxwell’s equations in a isotropic and uniform dielectric medium
Gaussian system of units
r E ¼ 4p/"
r ("E) ¼ 0
SI system of units
r E ¼ /k"0
r (kE) ¼ 0
10. Does the uniform dielectric media account for electronic polarisability?
One of the questions we asked at the beginning of this chapter was: Can
electronic polarisation within the macromolecule be ignored or, if not, is it best
described by PIDs on the atoms or bonds, or by a continuum dielectric? As we
just saw in section 9.2, one way to account for electronic polarisability is to
incorporate its effects into a dielectric constant – to assume that all charges and
permanent dipoles interact with one another as if they were embedded in a
medium that has a particular dielectric constant.
Until recently, electronic polarisability has usually been neglected in potential
energy force fields used in molecular mechanic simulations because the effects
cannot be easily reduced to a set of two-body interactions. For example, if a charge
on a particular atom polarises the electrons on neighbouring atoms, those electron
clouds will also polarise one another, leading to a complex many body interaction.
We shall see now and compare three ways to account for electronic
polarisability: the already mentioned uniform dielectric model, the induced
dipole model and the local dielectric-constant model. A central question
concerning the use of a dielectric constant over a region of space involving many
atoms asks if using a single, spatially invariant parameter that ignores the atomic
nature of matter is valid. This problem will be considered after three microscopic
352
models are presented. The comparison of such models will give us a better insight
on why our choice of the uniform dielectric model is a very reasonable choice to
amount for the electronic polarisation.
10.1. Uniform dielectric model
This model assumes that all nuclei and dipoles are kept in fixed positions and
therefore, the dielectric response is determined almost entirely by electronic
polarisation. It is assumed that the nuclei will not reorient in the presence of an
electric field. The contribution of the elastic displacement of nuclei or of dipoles to
the dielectric constant is neglected. Clearly the largest contributions will arise from
the electrons with the smallest binding energies, i.e., from valency electrons. The
displacement of the electrons is also considered elastic. For all frequencies which
are less that 0 (a particular resonant frequency of an electron bound in an atom)
by a sufficient amount, the dielectric constant should be independent of frequency.
Thus, for <
< 0 the dielectric constant " should be equal to the static dielectric
constant "stat and should satisfy the Maxwell’s relation " ¼ n2 [2,30]. That is,
" ¼ "stat ¼ n2
ð99Þ
should hold between the static dielectric constant and the refraction index at
frequencies for <
< 0, which is 2 for most polar and non-polar organic liquids.
As mentioned in section 8, both experiment and theory point to section 10 for the
protein dielectric constant, where electronic polarisation is considered to be the
most important contribution.
10.2. Induced dipole model
The most common means of representing electronic polarisability at the
molecular levels assign PIDs to atoms, bonds or groups [31–33]. In the simplest
case, the induced dipole moment (before represented as p) is presumed to be
linearly related to the field by an isotropic polarisability , ¼ E.
For a collection of charges and PIDs, the field depends on the charges and
dipole moments, while each induced dipole moment in turn depends on the field
it experiences from the charges and all other dipoles. This leads to a set of
simultaneous linear implicit equations for the dipole moments:
~ i ¼ i
X
~ Þij ½E~ðqÞij þ E~ð
ð100Þ
j6¼i
where, E(q)ij and E()ij are the electric fields due to the charges and the dipoles,
respectively, and the subscripts i and j run over all the charges/dipoles. This
matrix equation can be solved analytically only for two-body case because, as
353
pointed out above, electronic polarisation involves a many body interaction that
cannot be decomposed into a sum of pairwise interaction. Generally, an iterative
procedure is used in which an initial estimate for the dipole moments is
substituted into the right side of Eq. (100), giving rise to an improved estimate of
the dipole moments. This procedure is repeated until a self-consistent set of fields
and dipole moments results [31,33].
The PID model assumes usually that an atom has a uniform polarisability that
can be represented by an induced dipole placed at the nucleus. Two difficulties
with this model are: (a) atomic polarisabilities taken from experiment or theory
on isolated atoms are not necessarily accurate for atoms in molecules [34]; and
(b) nearby inducible dipoles can mutually increase each other’s polarisation
without limit causing a polarisation catastrophe [35]. The ad-hoc exclusion of
interactions between neighbouring atoms has been used to circumvent this
problem [36–38].
10.3. Local dielectric-constant model
An alternate way of representing the electronic polarisability treats atoms or
groups of atoms as polarisable bodies, each with its own local dielectric constant
(LDC) [35,39,40]. The LDC model effectively distributes the dielectric response
over the van der Waals volume occupied by the atoms’ electrons. This model
make fewer approximations than the other two models, since it assumes neither
that the response is uniform throughout space nor that the response arises from
infinitesimal dipoles. In the simplest form of the LDC model, each atom is
represented as a sphere of constant dielectric, "1. The equivalent point
polarisability in the PID model, i, would be [41]
¼
3Vð"i 1Þ
4pð"i þ 2Þ
ð101Þ
where V is the volume of the sphere.
Figure 15 schematically illustrates the relationship between the uniform
dielectric, PID and LDC representations [35]. The LDC and PID models are
equivalent for two special cases: when the atom is in a homogeneous medium or
when it is exposed to a uniform field [35]. In general, however, the polarisability
response involves higher-order terms then dipoles, and on atomic dimensions the
errors in the PID approximation can be quite large [39,41]. The LDC model may
also be extended to use non-uniform and anisotropic dielectric distributions [40].
The LDC and PID models shown in Fig. 15 appear microscopic, while the
uniform dielectric model appears macroscopic. It can also be seen in this figure
that the uniform dielectric and the LDC model differ in the absence of cavities
between the atoms in the former and the assumption of the same dielectric
constant for each atom.
354
Fig. 15. Schematic diagram illustrating three different models for molecular response to electric
fields: The Uniform Dielectric Constant (UDC), Local Dielectric Constant (LDC) and Point
Inducible Dipoles (PID). For models, UDC and LDC, the mean-induced dipole density per unit
volume at any point P(r) is given by h(r)i/V ¼ ["(r)1]E(r)/4p, where E(r) is the Maxwell field at
the point and "(r).
Since atoms in proteins are closely packed and are neither spherical nor static,
it is not unreasonable to consider them as filling space. Moreover, the highfrequency dielectric constant of organic liquids depends only weakly on the
identity of the solvent molecule. Thus, the use of a single dielectric constant to
account for the electronic polarisation response of an entire macromolecule
appears to be a very reasonable approximation. It should be emphasised that it is
not clear which of the three models is actually most appropriate for applications
to biological systems. The PID and LDC models are truly microscopic, but they
are numerically complex and the PID model in particular entails a number of
questionable assumptions. Moreover, both require knowledge of polarisabilities
for a large number of atoms in different molecules and thus involve a significant
number of parameters. On the other hand, experimental and theoretical evidence
suggest that proteins have an average dielectric response that can be
approximated with a dielectric constant of about 4 [42–45], while water has a
dielectric constant of approximately 80 at room temperature. Thus, empirically,
the protein system can be viewed as uniform and therefore at least two dielectric
constants must be used.
11. Mobile ions – the Poisson–Boltzmann equation
As we saw in Section 9.3, after having characterised a given system by its
dielectric constant "(r) and charge density (r) at each point r in space, the
electrostatic potential (r) can be determined as the solution of Poisson’s
equation for an inhomogeneous medium:
Poisson equation for dielectrics in the Gaussian system of units
~ ½"ð~rÞr
~ ð~rÞ þ 4pð~rÞ ¼ 0
r
ð102Þ
355
The presence of mobile counterions in solution can be represented implicitly. The
chemical potential of each ion is assumed to be uniform throughout the solution.
The entropic and electrostatic contributions to the chemical potential of an ion at
any point r are kT ln C(r) and q (r), respectively, where C(r) is the local
concentration, q its charge and (r) is the mean potential. This leads to a
Boltzmann’ expression for the ion concentration [35]:
Cð~rÞ ¼ Cð1Þ exp½qðrÞ=kT
ð103Þ
where C(1) is the bulk ion concentration. When incorporated into the Poisson
equation, this yields the most general of the widely used CE equations, which,
after linearization gives the PB equation:
Linearized PB equation
~ ½"ð~rÞr
~ ð~rÞ k02 ð~rÞ"ð~rÞð~rÞ þ 4pp ð~rÞ ¼ 0
r
ð104Þ
where the charge density p(r) refers only to the protein charges, and the
counterionic term effect is totally contained in the second term of the equation.
The parameter k0 is the so-called reciprocal Debye length that assumes the value
k0 ¼
8pe2 Na I
"out kB T
1=2
ð105Þ
if a point in space is accessible to other ions, and zero otherwise. We use the
prime notation to distinguish it from the permittivity k(r) defined in section 9.3.
Na is the Avogadro number, e is the proton charge, I the ionic strength, kB the
Boltzmann constant and "s the solvent dielectric constant.
The ionic strength I of the solution is defined as
I¼
1X 2
cj zj
2 j
with the sum over all ionic species in solution with charge zj, and cj their bulk
concentration (ions per volume). The ion exclusion boundary which delimits the
region inaccessible to the ions is usually defined as the closest distance that an
ionic centre can approach the reference ion, i.e., the boundary lies at one ionic
radius from the surface of the reference ion.
One of the advantages of the linear form of the PB equation is the linearity
of CE (classical CE methods), i.e., the superposition of the potential arising
from independent charges. However, when linearity breaks down, higher-order
356
terms have to be introduced (see Eq. (106), non-linear form of the PB equation,
NLPBE).
~ ½"ð~rÞr
~ ð~rÞ k02 ð~rÞ"ð~rÞ sinh½ð~rÞe=kB TkB T=e þ 4pp ð~rÞ ¼ 0
r
sinhðxÞ ¼ x þ
x3 x5 x7
þ þ þ 3! 5! 7!
ðx2 < 1Þ
ð106Þ
There is some controversy about the validity of the NLPBE [23,24,46,47] but this
will not be of concern to us here. We will consider here only the case where
linearity holds (i.e., the Poisson or LPB equation).
The PB equation incorporates electronic and dipole polarisation through " and
ion screening through k0 , and it allows shape effects to be modelled through the
spatial variation of ", k0 and . The linearity of the PB equation (104) implies that
the superposition principle is still valid (see section 9.1) and that a pairwise
decomposition of the interaction of the system also holds.
12. Forces and potentials with dielectrics
Let us ask now what would be the Coulombic force between two charges in a
dielectric. In a medium of dielectric constant k, all forces will be reduced by this
same factor. It means that the Coulombic force equation (9) and the Coulombic
potential equation (20) have to be replaced by:
In the SI system of units
F~ ¼
1 q1 q2
e~12 ¼ F~2
4p"0 k r212
ð107Þ
q 1
4p"0 k r
ð108Þ
ðx, y, zÞ ¼
where k is the dielectric constant of the dielectric material, here assumed to be the
same everywhere in the material, like we will assume for molecules such as
proteins.
or
In the Gaussian system of units
1 q1 q2
F~1 ¼
e~12 ¼ F~2
" r212
ðx, y, zÞ ¼
1q
" r
where " is the dielectric constant of the dielectric material.
ð109Þ
ð110Þ
357
13. Solving the Poisson–Boltzmann equation with continuum electrostatic models
The use of CE methods at the molecular level goes back to the Born model of
ionic solvation [9,23] where the Gibbs free energy of solvation is regarded as the
electrostatic work (i.e., U) of transferring a charged sphere, the ion, from
vacuum to a high-dielectric medium, the solvent. This approach arises from an
analogy between the microscopic system and a familiar macroscopic model, and
seems physically reasonable and intuitive. In fact of the simplicity and relative
success of the Born model, it has been widely used and extended in other
developments, among the most important being the Debye–Hückel theory of
strong electrolyte solution [1,23,24,46,48]. The model by Debye and Hückel is
essentially the Born model plus an hypothesis concerning the distribution of ions
around a reference ion. The theory of electrolytes of Debye and Hückel allowed
the inclusion of the effect of the ion concentration through the formulation of the
LPBE (Eq. (104)).
The solution of the Poisson equation for dielectrics (Eq. (102)) and the
solution of the linearized PB equation (Eq. (104)) can be obtained from
analytical or numerical solutions, depending on the complexity of the problem.
When the system has some symmetry it is usually possible to express the solution
in an analytic form. But symmetry is not a common feature of real proteins. The
first introduction of asymmetry in CE models was done by Kirkwood [11] who
later generalised the Debye–Hückel model to include an arbitrary number of
charges inside the sphere. The model was later extended to ellipsoids [49]. With
the advent of computers, the necessity for analytical solutions became less
relevant, and the PB equation can now be solved numerically for molecules with
arbitrary shape and charge distribution. The resulting general CE method, which
has been widely applied in proteins, is represented in Fig. 10. The solute charges
included in the model may vary, but current applications usually include the
(partial) charges of all atoms. As in the original Born and Debye–Hückel models,
only two dielectric regions are usually considered: the solvent region, with "
equal to the solvent bulk macroscopic constant ( ffi 80 for water at room
temperature), and the solute region, with " in the range 2–4, as mentioned in
sections 8 and 10.1.
The boundary between the two regions can be obtained by using one of the
commonly used definitions of molecular surface [51] such as the Connolly
molecular surface [51] equivalent to the Richards contact and reentrants surfaces
[52]. In most cases, the surface is determined by rolling a spherical probe with the
radius of a solvent molecule, e.g., water, on the surface of a molecule.
The first application of CE models to proteins was done by Linderstrøm–Lang
[53] who modelled a protein molecule as a sphere with its total charge smeared
uniformly over the surface. Thus corresponds to assuming that charged groups
(in particular titratable sites) are equally likely to lie at any position on the
surface, a reasonable assumption considering that at the time the ideas on
protein structure were mostly speculative. The model and the corresponding
358
solution for the electrostatic potential are very similar to the ones in the original
Debye–Hückel theory.
13.1. Tanford–Kirkwood continuum model
The asymmetric Kirkwood model is better suited to a protein native structure
than the Linderstrøm model and can be applied directly to proteins once the
charge positions on the protein are known. This was the model used by Tanford
and Kirkwood in their theory of protein titration, and is shown in Fig. 16.
Tanford and Kirkwood [54] calculated the electrostatic free energy for a set of
discrete point charges (as opposed to the smeared charges of the Debye–Hückel
theory) on a spherical surface of radius b and ion exclusion layer a (closest
possible approach distance of an ion). Each pair of charges is considered to be
placed at the surface of the sphere, which is assumed to form a continuous
medium of low-dielectric region, surrounded by solvent with an external
dielectric constant "s, and mobile counterions whose Coulombic screening is
proportional to square root of ionic strength. The protein is considered as a
sphere of a given radius, such that the volume of the sphere is the same as that of
the protein. The formula used to compute the interaction energy between two
charges is given in Refs. [3,7,8] where the Tanford–Kirkwood (TK) model is
further presented.
13.2. Modified Tanford–Kirkwood model
A solvent static accessibility parameter [50] for each protein charge site was
incorporated by Shire et al. [27] into the TK discrete-charge electrostatic theory.
This modification was introduced to overcome the uncertainty of an adjustable
charge-burial parameter beneath the dielectric interface, which was required in
the original treatment to fit the protein-titration curves [55,56] and to allow for
the irregular protein–solvent interface. The use of this parameter has been
Fig. 16. The Tanford–Kirkwood model. The protein is considered spherical. Usually, charges (e.g.,
qi and qj) are considered to be at the same depth from the surface of the sphere (i.e., the same di) and
separated by the experimental (e.g., crystallographic) rij distances. "p, protein dielectric constant; "s,
solvent dielectric constant; I, solvent ionic strength; b, protein radius; a, protein radius plus ion
exclusion boundary radius.
359
difficult to justify on physical grounds, but it has nevertheless been effective in
improving experimental agreement for protein-titration curves.
In this solvent-accessibility (SA) discrete-charge treatment, the fractional
solvent accessibility for each group was incorporated into the calculation of the
pairwise electrostatic interactive energy Uij [27]. A formalism was adopted that
linearly reduced the TK pairwise interactive energy [57,58] at a dielectric
interface by the charge pairs’ solvent exposure
Uij0 ¼ Uij ð1 SAij Þ
ð111Þ
where SAij is the average accessibility of sites i and j. This approach is usually
referred to as the Modified Tanford–Kirkwood (MTK) method. The method has
additional, implicit, theoretical difficulties [35,43], but it nevertheless represents
an important step in the development of methods that map protein-structural
information onto the parameters of the PB equation. Another version of the
original TK model can be obtained by placing some of the charges in the outer
(solvent) region [59].
Different accessibilities of the various groups reflect the ability of the protein
to restrict both solvent interactions and the effective sequestering of counterions.
When SAij exceeds 0.95, the interaction energy between the two charge sites is
small and neither markedly perturbs the other. For lower values of SAij , the
protein prevents access of solvent and mobile counterions to the high local field
of the charge sites. Hence, the charge sites with low solvent accessibility are
allowed to interact as calculated by the TK formalism. The use of the SAij factor
in reducing electrostatic free energy results in a higher-effective Coulombic
shielding for solvent-exposed sites. This shielding, which is due to higher-effective
LDC, has been interpreted as a local ionic strength [58].
The effects on charge-site interaction mediated by steric constraints on counterion approach are shown in Fig. 17. Figure 17A shows the field calculated for
two univalent ions immersed in a uniform dielectric with no mobile ions. In panel B
an ion is allowed to approach the midpoint between the two cationic sites, controlling this way the field distribution. Other geometrical restrictions are possible.
Fig. 17. Panel A shows the field calculated for two univalent ions immersed in a uniform dielectric
with no mobile ions. In panel B an ion is allowed to approach the midpoint between the two cationic
sites, controlling this way the field distribution.
360
13.2.1. Summary
While it would appear that any attempt to portray molecular-level interactions in
the context of a dielectric formalism should be treated as suspect, it is clear that a
continuum model with dielectric boundaries which tries to incorporate the effects
of a protein–solvent interface as well as the presence and distribution of mobile
ions is preferable to a ‘‘vacuum’’ or uniform CE model.
Despite the inherent assumptions and limitations of this type of formalism, the
electrostatic treatment, which incorporates a static accessibility modification into
the TK discrete-charge dielectric boundary theory, has provided a simple
and efficient computational procedure yielding quantitative and qualitative
predictions that are in agreement with experimental data. The electrostatic
consequences of the peptide dipoles can be included but are usually ignored
because of their weak contribution to long-range electrostatic interactions when
compared to the formal charge effects. The PB equation incorporates electronic
and dipole polarisation through " and ion screening through k0 , and it allows
shape effects to be modelled through the spatial variation of ", k0 and .
A more realistic representation of the protein molecule, corresponding to the
type of model shown in Fig. 16, usually implies a loss of any geometrical
symmetry, meaning that one has to resort to numerical methods to solve the
Poisson or Poisson–Boltzmann equations. The method of finite differences is the
most common one in protein applications and was used in the work presented in
this chapter to solve the linear form of the PB equation. This method is generally
referred to as FDPB (finite-difference Poisson–Boltzmann) method. A
description of this method is given in section 13.5.3.
The choice of a particular CE method is usually the result of a compromise
between the atomic detail of the model and the computation time. With the
current computer power, a finite difference calculation for any fairly large
protein should be feasible. The high detail of some of the methods does not
necessarily imply a high accuracy, because a proper parameterisation is necessary
(like in Molecular Mechanics, MM, methods). For example, the results differ
when partial charge sets from different MM force fields are used, which suggest
that a specific parameterisation for each CE method may be the more correct
procedure [60]. Another criterion for the choice of a particular CE method
should be the electrostatic quantities one is interested in. For example, though
the visualisation of the electrostatic potential around the protein can give
valuable insight on its function [61–64], its calculation is only meaningful when
some molecular detail is included. More simple methods like the TK one can be
used to compute electrostatic energies but their electrostatic potentials are not
particularly useful, since the spherical approximation makes the method
inappropriate to map atomic-level properties.
The major use of CE methods is in computing the free-energy difference of
processes involving charge changes, which, following Born, Debye and Hückel,
is simply taken as the difference of U between the final and the initial states.
The CE potential energy of each state is often called ‘‘electrostatic free energy’’.
361
A particularly important type of interaction which is not included in the CE
model is the apolar interaction with the solvent, which gives rise to the so-called
hydrophobic. The processes usually considered are ionisation changes such as
the ones occurring in redox or titration reactions of protein molecules. The later
is of major interest to us here and is discussed in the following sections. For an
overview of the applications, see Refs. [26,35,65]. Although this type of model
has been mostly pursued for protein applications, it has also been applied to
small molecules [60,66–69].
Many times we would like to know the energy associated of a particular
interaction express in different units. Appendix B provides us the useful tool that
will give us such information.
13.3. Finding the protonation state of proteins prior to solving the PB equation
The visualisation of the electrostatic potential distribution in and around
macromolecules can give valuable insight on its function and stability. In order
to find the potential distribution on the molecular surface of each protein, at
different pH values, we will solve the linearized PB equation, LPBE (Eq. (104)),
with the FDPB method described in section 13.5.3.
Linearized Poisson–Boltzmann equation
~ ½"ð~rÞr
~ r~ r
8pe2 Na I
"ð~rÞð~rÞ þ 4pp ð~rÞ ¼ 0
"out ð~rÞkB T
As mentioned in previous sections, in order to solve this equation, i.e., in order to
find (r), we need to characterise the system, thus we need to know the (Fig. 18)
– Dielectric constant of the solute (protein, "(r) ¼ "p)
– Dielectric constant of the solvent ("out(r) ¼ "s)
– Charge density of the protein at each point r in space (p(r))
– Ionic strength of the solvent (I)
– Temperature (T)
The location of the charges can be given by the experimentally determined 3D
structure of the protein (by X-ray diffraction or NMR). As mentioned earlier in
this chapter, the protein’s dielectric constant is usually set to 4 and the solvent
(water) dielectric constant is approximately 80 at room temperature. The ionic
strength of the solution can be set to any value. In the present work we assume it
to be 0.14 M, the physiological ionic strength.
The key question now is how can we find the charge, (p(r)), that each
titratable residue will carry at a particular pH value? We will see in the following
section that it is not a trivial matter to find the protonation state of each
titratable residue. At a first glance, the theoretical task finding the charged state
of each titratable residue seems straightforward – given the pKa of the titratable
362
Fig. 18. Solving the Poisson–Boltzmann equation.
residue (available in any biochemistry handbook, see Table 2) it would be a
trivial matter to tell whether a given group is charged or not at a particular pH
value. However, the situation is far more complicated because the other charged
sites and the local environment in the protein may shift the pKa of a given site
from its typical value by several pH units (see Tables 3 and 4). In fact, as shown
below, even the usual concept of pKa becomes, to some extent, inappropriate.
The TITRA program, written by Martel and Petersen, will be the tool used for
the calculation of the average protonation state of the titratable sites. The details
will be outlined in the following sections. The TITRA program [7,8] is a protein
titration program implementing the modified TK sphere model for site–site
interactions [7,54] and the Tanford–Roxby iterative mean field approximation
[56] for calculation of the average protonation state of the titratable sites.
In Refs. [7,8] is discussed how to model the effects of pH on proteins in order
to find the charge distribution of a protein at a particular pH value. We will
present and discuss some methods from the point of view of their
implementation and use, introducing the software tools that we use in order to
visualise electrostatic charge and potential distribution in macromolecules.
13.4. Modelling the effects of pH on proteins in order to find the charge distribution
of a protein at a particular pH value
Enzymes require that the catalytic residues have the appropriate protonation
state in the active pH range. Thus, pH is of key importance for enzyme activity.
363
Table 2. pKa’s of titratable groups*.
Group
pKa
model
Amino acid -COOH
Asp (COOH)
Glu (COOH)
His (imidazole)
Amino acid -NH2
Lys ("-NH2)
Arg (guanidine)
Tyr (OH)
Cys (free SH)
Model compounds (pK
3.6
4.0
4.5
6.4
7.8
10.4
12
9.7
9.1
)
Usual range in proteins
2–5.5
5–8
8
10
–
9–12
8–11
*Data from Refs. [71,72].
Table 3. Some highly perturbed pKa’s in proteins*.
Enzyme
Residue
pKa
Lysozyme
Lysozyme–glycolchitin complex
Carbopeptidase A
Acetoacetate decarboxylase
Chymotrypsin
-Lactoalbumin
Rhodanese
Papain
Glu35
Glu35
Glu27
Lys ("-NH2)
IIe-16 (-NH2)
COOH
Cys247
His159
Cys25
Asp32
6.5
8.2
7.0
5.9
10.0
7.5
6.5
3.4
3.3
1.5
Pepsin
*Data from Refs. [71,72].
Usually, proteins become unstable at extreme pH values, not only because of
acid- and base-catalysed reactions but also because of changes in the formal
charge states of the titratable groups. Ever since these principles were recognised,
there has been great interest in underlying the physical basis of the pH-dependent
phenomena in proteins. It is clear that a successful structure-based model for the
prediction such phenomena would contribute significantly to our understanding
of enzyme mechanisms, protein stability and molecular recognition.
The direct result of a pH change is a modification in the equilibrium
concentrations of the protonated and deprotonated forms of the titratable
sites. The most pronounced consequence of this modification is a corresponding
change in the average charge of the titratable sites. Therefore, electrostatic
interactions are widely believed to be the primary forces controlling pHdependent phenomena. As a consequence, the development of the PB method for
computing detailed electrostatic fields in and around macromolecules has led to a
burst of new activity in the theory of pH-dependent phenomena [70].
364
At a first glance, the theoretical task of explaining and predicting these
pH-dependent electrostatic changes may seem straightforward – given the pKa of
the titratable residue (available in any biochemistry handbook, see Table 2) it
would be a trivial matter to tell whether a given group is charged or not at a
particular pH value. However, the situation is far more complicated because the
other charged sites and the local environment in the protein may shift the pKa of
a given site from its typical value by several pH units (see Table 2, Table 3 and
Fig. 19, Fig. 20). In fact, as shown below, even the usual concept of pKa becomes,
to some extent, inappropriate.
Fig. 19. The titration behaviour of a residue is dependent on the local environment.
Fig. 20. The pK value of each carboxylic group in a dicarboxylic acid will reflect the electrostatic
environment.
Following the nomenclature of Bashford and Karplus [73] we will use the
following terms:
pKmodel – is the pKa of a titrating group in a small model compound, supposedly
free from the action of other titrating groups. It can be measured by NMR or
other titration methods.
pKint – is the pKa of a titrating site with all other groups in the protein
neutralised. This quantity depends not only on the residue type but also on its
location in the protein. It is pH independent.
pKeff – is the pKa displayed by a given group at a given pH by the fully charged
protein. This quantity changes with pH throughout the titration due to the
mutual interactions between groups.
pK1/2 – is the pH at which the residue is half protonated.
The protonation equilibrium is fully described by the pKa of the site through
the familiar Henderson–Hasselbach equation of the acid–base equilibrium [21]:
pKa ¼ pH log
f
1f
ð112Þ
365
where f is the degree of protonation, i.e., the fraction of molecules that has the
site protonated. From this equation, it can be predicted that pKa is the pH value
at which the site is half protonated.
The pKa value measured in solution for the model compound (pKmodel),
typically Gly-X-Gly, where X is the residue in question, reflects an aqueous
environment for the residue, considered completely solvent accessible. However,
when the titratable residue is transferred from the model compound into a specific
site in the protein, new terms contribute to the energetics of its titration [7,70]:
– The Born, or desolvation term, represents the free energy change in the
protonation reaction du to burying the residue in the protein low dielectric.
– ‘‘Background’’ term describes the free energy change coming from the
interaction of the residue with the other non-titratable charges in the protein
(e.g., peptide-bond dipoles and polar atoms).
Together, those two terms account for the difference between pKint and pKmodel.
A third energetic term comes from the interaction of the residue with all other
titratable residues in the protein. The magnitude of this term is pH dependent.
The pKa value resulting from the insertion of the amino acid residue into a
neutral protein is usually referred to as the intrinsic pKa, pKint, and may be
written as:
pK int ¼ pK model þ
1
Genv
ð2:3kB TÞ
ð113Þ
where Genv is the free-energy change due to moving the residue from water
into the neutral form of the protein (see Fig. 21).
Genv ¼ Genv ðAÞ Genv ðAHÞ
ð114Þ
If we only had one titratable residue in the protein molecule, the protonation
equilibrium would be given by Eqs. (112) and (113), with pKa ¼ pKint. However,
when other titratable or permanently charged sites exist, the electrostatic
interaction between them needs to be considered as well. Thus, the way in which
the pKint of a given site is affected by a closely positioned one depends on
whether the latter is charged or not. But, conversely, the protonation state of the
second group will also depend on the protonation of the first. Another way of
stating the problem is to say that a protein with s titratable sites has 2s possible
protonation states, and in order to characterise the protonation equilibrium of a
Fig. 21. Thermodynamic cycle to compute the effect of inserting a titratable amino acid (A) in a
protein molecule (P).
366
single titratable site we have to specify the populations of each two forms at each
of the 2s forms of the protein at each pH value. The probability of each
protonation state can be computed [54,73–76] and this task is sometimes referred
to as the multiple-state titration problem. Thus, in order to account for the
additional interactions that an amino acid residue displays with other charged
sites in the protein, an effective pKa is defined:
pK eff ¼ pK int þ
1
Ginter
2:3kB T
ð115Þ
where Ginter is the electrostatic contribution due to the interaction with other
charged residues. Since the interaction term is a pH-dependent quantity, the pKeff
itself becomes pH dependent and it can no longer be equated with the pH
corresponding to half protonation.
13.5. Methods: a practical approach
Overview
After presenting a view of electrostatic interactions from the point of view of CE
and how to model the effect of pH on proteins, it seems appropriate to present
and discuss some methods from the point of view of their implementation and
use, introducing the software tools that used for calculating and displaying the
electrostatic energy and potential in a macromolecule.
13.5.1. Program TITRA – computing the electrostatic interaction energy between
charged sites and their protonation state in order to calculate the pKa of each
titratable residue
The TITRA program, is a protein titration program implementing the TK sphere
model for site–site interactions (see Fig. 16) [7,54] and the Tanford–Roxby
iterative mean field approximation [56] for calculation of the average protonation state of the titratable sites. In Refs. [7,8] is outlined the general workings
of the program. The general flow of the TITRA program is shown in Fig. 22.
First, files containing atomic (AA)- or solvent (SA)-exposed area of individual
atoms, pKint for each of the titratable sites and TK model parameters are read,
and user options and arguments processed. A set of titratable residues and
atomic locations for charge placement are selected according to default internal
rules and/or information specified in user input files.
Values for the site–site coupling function Wij (pairwise electrostatic interaction
energy needed in order to calculate the total interaction energy between charges
in the protein, described in Refs. [7,8]) are then computed, using the TK formula,
for a range of distances specified by the cut-off values, and stored in a table for
later use. The pairwise interaction energies Wij between charges i and j, placed at
a certain depth under the surface of a sphere of radius b and ion exclusion radius
367
Fig. 22. Flowchart describing the steps within the program TITRA.
a, and at a distance rij from each other (see Fig. 16), are calculated assuming
the TK [11,54] model of a protein. As shown in Fig. 16, the positions of m
titratable sites are indicated by points. There are interactions between only those
points which bear a charge. If they bear charges, these will be point charges
embedded in a spherical cavity of dielectric constant "s. The external dielectric
constant is "p.
The fractional charge of each site is computed at the starting pH value, using
the pKint value for that group and Eq. (112). The total electrostatic potential at
each group, generated by the remaining groups, is determined using the
previously calculated partial charges and Wij coupling terms. Because TITRA
uses the MTK model, the interaction terms Wij are further corrected with a
scaling factor [27],
Wij0 ¼ Wij 1 SAij
ð116Þ
The solvent accessibility values will be computed by the ACC_RUN program
described in the next section.
In TITRA there is currently no provision to calculate pKint values from the
pKmodel values. Instead, the former have to be provided beforehand when this is
368
found necessary or when there are experimental data indicating a large shift from
pKmodel for a particular residue which cannot be explained through interaction
with other residues. The user is allowed to edit the pKmodel values for individual
residues in one of the TITRA input files. To set pKint equal to pKmodel
corresponds to assuming that the titratable sites’ environment is not significantly
changed upon inclusion in the protein, which may be a reasonable assumption
for solvent-exposed sites.
A number of user options may change details of the above-sketched
procedure. Energy values may be read from a pre-computed table stored in disk,
or a set of site–site coupling constants Wij may be read from a file. The format of
the pKint input file allows the values of selected residues to be pre-set or fixed at
given pKa or charge values (fixing the charge value of a site creates a background
charge, with a pH-independent value) [7].
13.5.2. Program ACC_RUN
ACC_RUN is a simple program that computes contact solvent accessibilities [52].
Each atom is modelled as a collection of evenly distributed points on the surface
of a sphere. The atom is considered solvent accessible if a water-probe tangent to
one or more of these points does not overlap any other protein atoms (the water
probe is usually modelled as a 1.4 Å sphere). The solvent accessibility is calculated from the fraction of exposed dots on the surface of each atomic sphere.
The program takes as input a PDB file and a water-probe radius value (default
value 1.4 Å), and output solvent-accessibility files for all individual atoms as well
as the side chains. The side-chain file contains accessibilities for all side chains,
normalised with the standard areas for tripeptides Gly-X-Gly in extended
conformation [27], while the atomic accessibility file contains absolute solventexposed atomic areas in Å2. The program is written in C and runs under SGI
IRIX and Linux. The two accessibility files produced by ACC_RUN are
required as input for a TITRA calculation.
13.5.3. Solution of the Poisson–Boltzmann equation using finite-difference
grid method
The PB equation appears to be a good model since it accounts for both the effects
of dielectrics and ionic strength. Unfortunately, this equation can be solved
analytically only for systems with simple dielectric boundary shapes, such as
spheres and planes. In particular, the linear PB solution for a single point charge
qi placed in the origin of the coordinate system has the Debye–Hückel form
i ¼
qi
expðk0 rÞ
"r
ð117Þ
Most molecules of interest have complex shapes, and their conformations may
have a significant effect on the resulting electrostatic properties. The alternative
to analytical solutions is to use numerical techniques to find an approximate
solution.
369
Three principal methods have been developed to the point where they can be used
to attempt to calculate experimental data. Solutions of the PB equation using finitedifference grid methods treat the protein and solvent as two dielectric continuums
but, unlike older TK implementations, allow for the detailed shape of the protein
surface. A semi-solvent continuum approach places induced dipoles on a grid for the
solvent and on atomic centres within the protein and is therefore termed the protein
dipoles/Langevin dipoles (PDLD) method. Free energy perturbation calculations
allow some experimental electrostatic quantities to be derived from Molecular
Dynamics (MD)andMonte-Carlo (MC)simulations usingexplicit solventmolecule
descriptions but usually ignoring electronic polarisation. These methods have been
extensively reviewed during the past couple of years. Harvey [43] has provided a
full, thoughtful and relatively objective survey of methods together with a
description of the background basic theory. Davis and McCammon [77] have
given a useful outline of the theoretical basis of each of the contemporary
methods. Sharp and Honig [47] have given an excellent if somewhat partisan
review, focussing on the FDPB method. Warshel and Åqvist [78] have
championed the PDLD approach and discussed the relationship between the
results of calculations and basic electrostatic concepts. Beveridge and Dicapus
[79] have reviewed the use of free-energy perturbation calculations. Bashford [80]
has outlined the methods and tests that have been made with model systems, as
well as the state of the art for their applications to macromolecules.
13.5.3.1. Finite difference approximation – the program DelPhi
DelPhi is a software package that calculates the electrostatic potential in
and around macromolecules, using a finite-difference solution to the non-linear
PB equation [81]. It was developed by Barry Honig and co-workers at Columbia
University [42,82–84] and marketed by Biosym Technologies, Inc. [85]. Typical
uses for DelPhi include calculating electrostatic potential in and around a protein
and displaying isopotential contour maps to gain qualitative information on
protein–substrate interactions, determining the effects of site-directed mutagenesis on the pKas of important residues, on binding energies and on catalytic rates.
The FDPB method involves mapping the molecule onto a three-dimensional
cubical grid, with spacing between grid points of size h (as shown in Fig. 23
in two dimensions). The interior of the solvent-accessible surface is assigned
one dielectric, and the exterior is assigned another. A molecule such as a protein
has a low-dielectric constant since its dipolar groups are frozen into a hydrogenbonded lattice and cannot reorient in an external electrostatic field. A value near
2 measures its electronic-polarisation response while a value near 4 includes some
additional contributions from dipole reorientation. Water, on the other hand,
has a very high-dielectric constant (78.5 at 298 K) since its dipoles reorient more
freely. Therefore, a protein molecule in aqueous solution yields a system with
two very different dielectric media.
The PB equation must be satisfied everywhere in the grid, and in particular, at
each grid point. If the cube of side h surrounding a grid point is considered, as
370
Fig. 23. Two-dimensional mapping of the molecule on a DelPhi grid. From Ref. [6].
Fig. 24. Cube of side h surrounding the grid point. The black circles are the six surrounding points.
Note that associated with each grid point i is a charge qi, a modified Debye–Hückel reciprocal
length k00 , and a potential 1. The dielectric values, however, are associated with the midpoints of
the lines between the grid points. The modified Debye–Hückel parameter, k0 ¼ (")1/2k, is defined for
convenience of implementation of the finite-difference formulas. From Ref. [6].
shown in Fig. 24, the derivatives in the equation can be replaced by finite
differences over this cube, and the continuous functions , and " can be
replaced by their values at the grid points [83].
Using this strategy, a finite-difference formula can be obtained in which the
potential at any grid point depends on the charge at the grid point, the value 00 at
the grid point, the grid spacing h, and the potential and dielectric values of the six
neighbouring grid points [83],
P
"i i þ 4pq0 =h
0 ¼ P
2
6
0
i¼1 "i þ 0 h
6
i¼1
ð118Þ
where 0 is the potential at each node of the cubic grid with spacing h, i the
potential at each of the six nearest neighbours, "i the dielectric constant at the
midpoint of 0 and i, q0 the charge assigned to the grid node.
371
Fig. 25. Flowchart representing the steps necessary for displaying electrostatic potential maps onto
the protein molecular surface. The files needed for the different programs are: protein.acc (side-chain
static-accessibility file); protein.atom.acc (atomic accessibility file); protein.tcv (titration curve data);
protein.pks (information about each titratable site: Residue_name, Residue_number, pH, pKint,
pKeff, Partial_charge) [8]; protein.crg (information about each titratable site: pH, Residue_name,
Residue_number, Partial_charge); protein.pdb (coordinates of the residues); delphi.param (solvent,
solute and grid parameters); protein.grd (potential map file); protein.frc (optional file: lists the
coordinates, charges, potential and field components for a specified set of atoms).
372
13.5.3.2. Program DelPhi – input and output files
The input files for DelPhi (see Fig. 25) include a coordinate file (in PDB
format), an atomic radii file, an atomic charge file and a parameter file
containing various parameters and options that control the program’s
behaviour. These include the grid step, its extent and placement relative to
the protein molecule, as well as the ionic strength, the dielectric constants for
protein and solvent, as well as the maximum number of iterations and
boundary conditions. Specification of both charged and non-charged atoms is
required because both contribute to the overall protein surface and, in
particular, to the definition of the protein–solvent interface. The program
outputs a grid file containing potentials for every grid point and a file
containing the potential and electrostatic field vectors at the location of each
atom in the system. The grid file can be read by Biosym’s viewer program,
InsightII [86] and colour-coded equipotential surfaces can be displayed at
defined kT/e values. DelPhi charge files can be generated by another application,
e.g., TITRA, to allow the display of equipotential surfaces. DelPhi calculation
times depend primarily on the total number of grid nodes, but also on the chosen
ionic strength and number of point charges of the system, the first two having an
effect upon the rate of convergence of the iteration. Setting up the molecular
surface dielectric boundary takes very little time, due to the use of an efficient
algorithm [87]. The DelPhi computations are not time consuming when
compared with, e.g., protein molecular dynamics calculation.
13.5.4. Program Grasp
The program Grasp [88] was developed as a consequence of the need for
visualising electrostatic potentials at surfaces, in particular, the surface of
biological molecules, where the surface is modelled as a solid surface. The
program DelPhi, which calculates electrostatic potentials from the PB equation,
can be used to obtain quantitative numbers for a variety of biochemical
phenomena but visualisation has been limited to qualitative isopotential
contouring. The limitation of this approach is that the contours do not highlight
local topology or shape. They often extend significant distances away from the
surface of the molecule while one expect most of the interactions to be close to
the molecule, in fact at the surface of molecules. Whereas DelPhi can give
detailed information about the molecular electrostatic signature or shape, it does
not permit concurrent viewing of the electrostatic potential and the molecular
surface. On the other hand, Grasp allows for the production of a solid surface,
colour coded with the local electrostatic potential.
Grasp has proven to be an ideal tool for the study of the electrostatics of many
families of enzymes, where the details of the molecular surface can be viewed
simultaneously with the electrostatic potential features. The flowchart
representing the necessary steps for displaying the electrostatic potential maps
onto the molecular surface of a protein is displayed in Fig. 25.
373
14. Applications
In section 1 we referred to why is it so relevant to model electrostatic interactions
and to obtain the electrostatic potential distribution on each atom of a
macromolecule or displayed on its molecular surface. All long-range intermolecular forces are thought to be essentially electrostatic in origin. Therefore,
the molecular understanding of the initial interaction between a protein and, e.g.,
its substrate or inhibitor is essentially an understanding of the role of
electrostatics in intermolecular interactions, such as molecular recognition.
Electrostatic interactions are widely believed to be the primary factors upon
which the pH-dependent phenomena are dependent. The protonation state of the
catalytic residues and of the residue nearby the active site may influence the
charge and potential distribution in the catalytic/binding region of the protein. If
a substrate and/or the product(s) of the reaction also carry charge, its strong
or weak interaction with the active-site region of the enzyme will depend on
the charge/potential of this same region. Ever since these principles were
recognised, there has been great interest in uncovering the physical basis of the
pH-dependent phenomena in proteins (see Ref. [63] and references therein). The
role of electrostatic interaction on enzyme activity, specificity, stability and ion or
ligand binding has been partially unravelled by several previous studies (see Ref.
[63] and references therein). It is clear that a successful structure-based model for
the prediction of such phenomena would contribute significantly to our
understanding of enzyme mechanisms, protein stability and molecular
recognition.
In the following section we will address several applications of electrostaticsderived knowledge and its use.
14.1. Interpretation of electrostatic potential maps displayed on the molecular
surface of an enzyme
Since the charge carried by a protein will be pH dependent, the electrostatic
interaction between the residues in the protein and the electrostatic potential at
each location of the protein will be modulated by pH. Enzymatic activity is also
pH dependent as well as protein-structural stability. Therefore, it is relevant to
correlate the protein’s activity, with its structural stability and electrostatic
energy/potential as a function of pH.
All over the surface of the protein the effects of titration can be observed.
Regions displaying positive potential at pH 4 have become neutral or even carry
negative potential at later pH values. This is the result of the titration of the
different titratable residues in the protein. As pH goes from acidic to alkaline, the
total charge of the protein goes from positive to negative due to the tiltration of
C-terminus, Glu, Asp, His, N-terminus, Tyr and Cys (when free), Arg and Lys.
That is the reason for the different potential distribution on the molecular surface
of a protein as a function of pH.
374
The electrostatic potential distribution as a function of pH on the molecular
surface of Fusarium solani pisi cutinase is displayed in Fig. 26. We can observe
that when changing pH to very acidic conditions we observe an increasing
polarisation of the active-site pocket, which present a more and more positive
potential, and when changing pH to more basic conditions the active-site
potential becomes more and more negative. The same is observed on the activesite flanking regions. We can also observe that the molecular surface at the
bottom of the active-site cleft (pointed by an arrow in Fig. 26) still displays a
positive potential at pH 6 due to the presence of the fully or partially positively
charged catalytic His residue (blue colour at the edge of the arrow, Fig. 26). It
can also be observed that as early as pH 4, a negatively charged residue located
just above the arrow (Glu44, pKmodel 4.5) is contributing to a region of negative
potential. In the pH ranges where highest activity is reported for F. solani pisi
cutinase (pH around 8.5 against tributyrin), the molecular surface at the activesite entrance is displaying negative potential (see map at pH 8.5, Fig. 26). At this
pH the catalytic His residue has titrated, therefore it has lost its positive. From
pH 8.5 onwards the potential in the active-site environment becomes more and
more negative due to the deprotonation of the Tyr residues that are in or very
close to the active cleft.
Fig. 26. Electrostatic potential maps displayed on the molecular surface of Fusarium solani pisi
cutinase at different pH values: 4, 6, 8.5 and 10. Blue colour represents positive potential and red
colour negative potential. The arrow points to the catalytic cleft. The units of the potential energy
values are kT/e.
375
The units of the potential-energy values reported in any electrostatic potential
maps distribution in the present thesis are kT/e. What is the physical meaning
of the kT/e energy levels, where k is the Boltzmann constant? The significance of
the kT/e energy levels comes from the fact that the average thermal energy
of the particles in a solvent at temperature T is kT. Since the electrostatic
energy W of a particle experiencing an electrostatic potential is given by
W ¼ q , the regions where the potential energy level is, in absolute value, above
kT are those where the electrostatic energy of charged particle is above the
thermal noise, and therefore ready to be electrostatically driven by the action of a
protein field.
14.2. Thermal stability, activity and Coulombic electrostatic energy
The Coulombic electrostatic energy in kT units of the whole molecule as a
function of pH computed by DelPhi as a function of pH for F. solani pisi cutinase
is shown in Fig. 27 (using the charge file predicted by TITRA at a particular pH
value, as described in the paper by Petersen et al. [63]). The dielectric constant of
the protein was set to 4.
The shape of the displayed electrostatic energy versus pH profiles in Fig. 27
resembles the Tm versus pH profiles displayed in Figs. 28 and 29. In both the
figures there is plateau from pH 6.0 to 8.5 for native cutinase. Electrostatic
interactions are thought to have a critical role in defining the thermostability of
the studied enzymes. A decrease in Tm is correlated with a reduction of the electrostatic energy. Figure 27 shows that there is a rapid decrease of the electrostatic
energy after pH 10, and this is correlated with the titration of the nearby six
tyrosine residues present in cutinase. Their deprotonation renders them negatively charged, giving rise to electrostatic repulsion. Also, usually above pH 10
residues like Lys loose their capacity of stabilising the deprotonated/negatively
charged Tyr residues since they start titrating and therefore loosing their positive
charge. Later on the same happens for Arg residues (pKmodel around 12). From
Figs. 27 and 29(b) it can be seen that loss of enzymatic activity above pH 9.5–10
is correlated with loss of structural thermal stability and with an unfavourable
increase of the Coulombic energy.
The Coulombic electrostatic energy in kT units of the native cutinase at a
particular pH value plotted as a function of Tm determined at the same pH value
(determined by differential-scanning calorimetry, DSC, by Petersen et al. [63,89])
is displayed in Fig. 30. It can be observed that a positive, thus destabilising,
electrostatic energy correlates with the lowest Tm observed (at pH 3). On the
other hand, the most negative values of electrostatic energy (thus contributing to
the proteins’ structural stability) is correlated with the highest Tm values
observed for native cutinase (see Fig. 30).
The small variations of the electrostatic energy observed in the pH range from
5.2 to 10.0 correspond to a plateau region of the Tm versus pH plot in the same
pH range.
376
Fig. 27. Coulombic electrostatic energy in kT units of the whole molecule has a function of pH
computed by DelPhi as a function of pH for native Fusarium solani pisi cutinase (using the charge
file predicted by TITRA at a particular pH value as described above). The dielectric constant of the
protein was set to 4.
Fig. 28. Changes in thermal stability for native cutinase investigated by CD spectroscopy at pH 4.0,
6.0, 8.5 and 10.0 at a scan rate of 90 C/h [63].
The authors are fully aware that electrostatic interactions alone cannot
fully explain the thermal stability of the protein as a function of pH. The
hydrogen-bond network as well as the hydrophobic interactions will definitely
play an important role for protein stability. However, the correlation observed in
Fig. 30 is very significant. We believe that it can be used to predict what changes
in Tm the introduction or the removal of salt bridges in native cutinase would
377
Fig. 29. (a) pH-thermal stability profile of Fusarium solani pisi cutinase. Tm determined by
differential scanning calorimetry, DSC. (b) pH-activity profile of F. solani pisi cutinase determined
by the pH-Stat methodology [63].
bring. However, the Coulombic calculation by DelPhi only reflects the energy
necessary to bring the charges present from infinity to their location on the
protein using the protein’s dielectric constant. The solvent effects were included
only during the charge-file calculation by TITRA, as described in section 13.5.1
(implicit solvent effect).
378
Fig. 30. The Coulombic electrostatic energy in kT units of the native cutinase at a particular pH
value plotted as a function of Tm determined at the same pH value determined by differential
scanning calorimetry, DSC.
14.3. Engineering the pH optimum of the triglyceride lipase cutinase from
F. solani pisi
The optimisation of enzymes for particular purposes or conditions remains an
important target in virtually all protein-engineering endeavours. In NevesPetersen et al. [63] we have presented a successful strategy for altering the pH
optimum of the triglyceride lipase cutinase from F. solani pisi. The computed
electrostatic pH-dependent potentials in the active-site environment are
correlated with the experimentally observed enzymatic activities. At pH
optimum a distinct negative potential is present in all lipases and esterases
that we studied so far [62]. This has prompted us to propose the ‘‘The
Electrostatic Catapult Model’’ as a model for product release after cleavage of
the ester bond [62,63]. The origin of the negative potential is associated with the
titration status of specific residues in the vicinity of the active-site cleft. In the
case of cutinase, the role of Glu44 was systematically investigated by mutations
into Ala and Lys. All charge mutants displayed altered titration behaviour of
active-site electrostatic potentials. Typically, the removal of the residue Glu44
pushes the onset of the active-site negative potential towards more alkaline
conditions. We therefore predicted more alkaline pH optima, and this was indeed
the experimentally observed. The experimentally observed pH optimum of E44K
mutant was 10.5 when compared to 8.5 for native cutinase.
In Fig. 31 is displayed the effect of carrying out a charge mutation (Glu into
Ala or Lys) in the active site of the F. solani pisi cutinase on the electrostatic
potential distribution map displayed on the molecular surface of the enzymes. It
can clearly be seen that when the glutamic-acid residue (Glu44) is replaced by an
379
Fig. 31. Electrostatic potential maps displayed on the molecular surface of native, E44A and E44K
mutant cutinases from Fusarium solani pisi at pH value 8.5. The black arrow indicates location of
the active site Ser120O. The green arrow indicates the location of Glu44 on the molecular surface
of native cutinase. Blue colour represents positive potential, white colour neutral potential and red
colour negative potential. The potential scale used ranged from 5kT/e to þ 5kT/e.
Ala or a Lys, the negative potential observed at the bottom of native cutinase at
pH 8.5 is not present at the bottom of the active site of the mutant enzymes.
Acknowledgements
M.T.N.P. acknowledges the support from the Danish Research Agency, Novo
Nordisk A/S, and Novozymes A/S.
Appendix A
The goal of this appendix is to derive three theorems needed to derive the two
laws of electrostatics as presented in Part 1. On the other hand, we will derive
Eq. (68) for the energy in the electrostatic field (from Ref. [2]).
Theorem 1 – The line integral of r
Z
2
ð2Þ ð1Þ ¼
~ Þ d l~
ðr
1
Theorem 2 – Gauss’ Theorem
Z
C~ n~ da ¼
Z
S
~ C~ dV
r
V
Theorem 3 – Stokes’ Theorem
I
C~ d l~ ¼
Z
~ C~Þn da
ðr
S
380
Equation (3.75)
"0
U¼
2
Z
E~ E~ dV
Vector integral calculus
It is relevant to get some understanding of the significance of the derivatives of
fields. This way, we will have a better feeling for what a vector-field equation
means. We will try to find the meanings of the divergence and curl operations.
The interpretation of these quantities is best done in terms of certain vector
integrals and equations relating such integrals. We will derive these integral
formulas. The equations we shall present in here are really mathematical
theorems useful for interpreting the meaning and the content of the divergence
and the curl. These mathematical theorems are, for the theory of fields, what the
theorem of the conservation of energy is to the mechanics of particles. General
theorems like these are important for a deeper understanding of physics. We find
them delightful, enlightening!
The line integral of r
We will take up first an integral involving the gradient. The relation contains a
very simple idea: since the gradient represents the rate of change of a field
quantity, if we integrate that rate of change, we should get the total change.
Suppose we have a scalar field (x, y, z). At any two points (1) and (2), the
function
will have the values (1) and (2), respectively. We shall use
the convenient notation, in which (2) represents the point (x2, y2, z2) and (2)
means the same as (x2, y2, z2).
If G (gamma) is any curve joining (1) and (2), as in Fig. 32, the following
relation is true:
Theorem 1
Z
2
ð2Þ ð1Þ ¼
~ Þ d l~
ðr
ðA1Þ
1
Fig. 32. The terms used in Eq. (A1). The vector r
is evaluated at the line element dl.
381
The integral is a line integral, from (1) to (2) along the curve , of the dot
product of r – a vector – with dl – another vector which is an infinitesimal
element of the curve [directed away from (1) and towards (2)].
First, we should review what we mean by a line integral. Consider a scalar
function f(x, y, z), and the curve joining two points (1) and (2). We shall mark
off the curve at a number of points and join these points by straight-line
segments, as shown in Fig. 33. Each segment has the length si, where i is an
index that runs 1, 2, 3, . . ..
By the line integral
Z
ð2Þ
f dl
ðA2Þ
ð1Þ
along it is understood the limit of the sum
X
fi li
where fi is the value of the function at the ith segment. The limiting value is what
the sum approaches as we add more and more segments.
The integral in Eq. (A1) has the same meaning, although it looks different.
Instead of f, we have another scalar – the component of r in the direction of
l. If we write (r )t for this tangential component, it is clear that
~
~
l ¼ r
l~
r
ðA3Þ
t
The integral in Eq. (A1) means the sum of such terms
The component of r along a small displacement s is the rate of change of
in the direction of l. Consider the line segment l from (1) to point a in Fig. 33.
According to the definition,
1
~
¼ ðaÞ ð1Þ ¼ r
l~1
1
Fig. 33. The line integral is the limit of a sum.
ðA4Þ
382
Also, we have
~ Þ l~2
ðbÞ ðaÞ ¼ ðr
ðA5Þ
Where, of course, (r )1 means the gradient evaluated at the segment l1 and
(r )2 the gradient evaluated at the segment l2. If we add Eqs. (A4) and (A5),
we get
~
~
ðbÞ ð1Þ ¼ r
l~1 þ r
l~2
1
2
ðA6Þ
We can see that if we keep adding such terms, we get the result
ð2Þ ð1Þ ¼
X ~
r
l~i
ðA7Þ
i
The left-hand side does not depend on how we choose our intervals – if (1) and
(2) are kept always the same – so we can take the limit of the right-handed side.
We have therefore proved Eq. (A1). This theorem is correct for any curve from
(1) to (2) and is independent on how the points a, b, c, . . . are chosen.
Gauss’ theorem – the flux from a cube
The flux of a vector field
Before we consider our next integral theorem – a theorem about the divergence –
we would like to generalise a certain idea which has an easily understood physical
significance in the case of, e.g., heat flow, to the case where the vector does not
represent the flux of anything. For instance, it might be the electric field.
Suppose that we have some closed surface S which enclosed the volume V and
we would like to find how much heat is flowing out of this surface S. For this
purpose we have to define the vector h, which represents the heat that flows
through a unit area in a unit time. We shall write da for the area of an element of
the surface. The symbol stands for a two-dimensional differential. If, of course,
the area happened to be in the xy-plane we would have da ¼ dx dy. Later we shall
have integrals over volume and for these it is convenient to consider a differential
volume that is a little cube. So, when we write dV we mean dV ¼ dx dy dz.
The heat flow out through the surface element da is the area times the
component of h perpendicular to da. Defining n as a unit vector pointing
outwards at right angles to the surface, the component of h that we want is
hn ¼ h~ n~
ðA8Þ
The heat flow through da is then
h~ n~ da
ðA9Þ
383
To get the total heat flow through any surface we sum the contributions from all
the elements of the surface. In other words, we integrate Eq. (A9) over the whole
surface:
Total heat flow through S:
Z
h~ n~ da
S
If the vector represents the electric field, E, we can certainly still integrate the
normal component of the electric field over an area if we wish. Although it is not
the flow of anything, we still call it the ‘‘flux’’. We say
Flux of E through the surface S:
Z
E~ n~ da
S
We generalise the word ‘‘flux’’ to mean the ‘‘surface integral of the normal
component’’ of a vector.
The flux from a cube
We now take the special case of a small cube and find an interesting formula
for the flux out of it. Consider a cube whose edges are lined up with the axes as
in Fig. 34.
Let us suppose that the coordinates of the corner nearest the origin are x, y, z.
Let x be the length of the cube in the x direction, y be the length in the y
direction, and z be the length in the z direction. We wish to find the flux of a
vector field C through the surface of the cube.
Z
Cx dy dz
Fig. 34. Computation of the flux of C out of a small cube.
384
We shall do this by making a sum of the fluxes through each of the six faces.
First, consider the face marked 1 in the figure. The flux outward on this face is
the negative of the x component of C, integrated over the area of the face. This
flux is
Since we are considering a small cube, we can approximate this integral by the
value of Cx at the centre of the face – which we call the point (1) – multiplied by
the area of the face, yz:
Flux out of 1:
Cx ð1Þyz
Similarly, for the flux out of face 2, we write
Flux out of 2:
Cx ð2Þ ¼ Cx ð2Þyz
Now Cx(1) and Cx(2) are, in general, slightly different. If x is small enough, we
can write
Cx ð2Þ ¼ Cx ð1Þ þ
@Cx
x
@x
There are, of course, more terms, but they will involve (x)2 and higher powers,
and so will be negligible if we consider only the limit of small x. So, the flux
through face 2 is
Flux out of 2:
@Cx
x yz
Cx ð1Þ þ
@x
Summing the fluxes for faces 1 and 2, we get
Flux out of 1 and 2:
@Cx
xyz
@x
The derivative should really be evaluated at the centre of face 1, that is, at
[x, y þ (y/2), z þ (z/2)]. But in the limit of an infinitesimal cube, we make a
negligible error if we calculate it at the corner (x, y, z).
385
Applying the same reasoning to each of the other pairs of faces, we have
Flux out of 3 and 4:
@Cy
xyz
@y
and
Flux out of 5 and 6:
@Cz
xyz
@z
The total flux through all the faces is the sum of these terms. We find that
Z
cube
@Cx @Cy @Cz
~
þ
þ
xyz
C n~ da ¼
@x
@y
@z
and the sum of the derivatives in just r C. Also, xyz ¼ V, the volume of
the cube. So we can say that for an infinitesimal cube
Z
~ C~ V
C~ n~ da ¼ r
surface
We have shown that the outward flux from the surface of an infinitesimal cube is
equal to the divergence of the vector multiplied by the volume of the cube. We
now see the ‘‘meaning’’ of the divergence of a vector. The divergence of a vector
at the point P is the flux – outgoing ‘‘flow’’ of E – per unit volume, in the
neighbourhood of P.
We have connected the divergence of C to the flux of C out of each
infinitesimal volume. For any finite volume we can use the fact we proved
above – that the total flux from a volume is the sum of the fluxes out of each
part. We can, that is, integrate the divergence over the entire volume. This
gives us the theorem that the integral of the normal component of any
vector over any closed surface can also be written as the integral of the
divergence of the vector over the volume enclosed by the surface. This theorem is
named after Gauss.
Gauss’ Theorem
Z
C~ n~ da ¼
S
Z
~ C~ dV
r
V
386
Stokes’ theorem – the circulation around a square
The circulation of a vector field
We wish now to look at the curl in somewhat the same way we looked at the
divergence. We obtained Gauss’ theorem by considering the integral over a
surface, although it was not obvious at the beginning that we were going to be
dealing with the divergence. It was not at all clear that this would be the result.
And with an equal apparent lack of justification, we shall calculate something
else about a vector and show that it is related to the curl. This time we calculate
what is called the circulation of a vector field. If C is any vector field, we take its
component along a curved line and take the integral of this component all the
way around a complete loop. The integral is called the circulation of the vector
field around the loop. We have already considered a line integral r earlier in
this appendix. Now we do the same kind of thinking for any vector field C.
Let be any closed loop in space. The line integral of the tangential
component of C around the loop is written as
I
I
C~ d l~
Ct dl ¼
We should note that the integral is taken all the way around, not from one point
to another as we did before. This integral is called the circulation of the vector
field around the curve .
Playing with the same kind of rational we did for the flux, we can show that
the circulation around a loop is the sum of the circulations around two partial
loops. Suppose we break up our curve into two loops by joining two points (1)
and (2) on the original curve by some line that cuts across as shown in Fig. 35.
There are now two loops, 1 and 2. 1 is made up of a, which is that part of the
original curve to the left of (1) and (2), plus ab, the ‘‘short cut’’. 2 is made up of
the rest of the original curve plus the short cut.
The circulation around 1 is the sum of an integral along a and along ab.
Similarly, the circulation around 2 is the sum of two parts, one along b and the
other along ab. The integral along ab will have, for the curve 2, the opposite
sign from what it has for 1, because the direction of travel is opposite – we must
Fig. 35. The circulation of C around the curve i is the line integral of Ct, the tangential component
of C. The circulation around the whole loop is the sum of the circulations around the two loops:
1 ¼ a þ ab and 2 ¼ b þ ab.
387
take both our line integrals with the same ‘‘sense’’ of rotation. Following the
same kind of argument we used before, we can see that the sum of the two
circulations will give just the line integral around the original curve . The parts
due to ab cancel.
We can continue the process of cutting the original loop into any number of
smaller loops. When we add the circulations of the smaller loops, there is always
a cancellation of the parts on their adjacent potions, so that the sum is equivalent
to the circulation around the original single loop.
Now let us divide our original loop into a number of small loops that all lie on
the surface we have chosen, as in Fig. 36. No matter what the shape of the
surface, if we choose our small loops small enough, we can assume that each of
the small loops will enclose an area which is essentially flat. Also, we can choose
our small loops so that each is very nearly a square. Now we can calculate
the circulation around the big loop by finding the circulations around all of the
little squares and then taking their sum.
Fig. 36. Some surface bounded by the loop is chosen. The surface is divided into a number of
small areas, each approximately a square. The circulation around is the sum of the circulations
around the little loops.
The circulation around a square; Stokes’ theorem
How shall we find the circulation for each little square? We could easily make the
calculation if it had a special orientation. For example, if it was in one of the
coordinate planes. Since we have not assumed anything yet about the orientation
of the coordinate axes, we can just as well choose the axes so that the one little
square we are concentrating on at the moment lies in the xy-plane, as in Fig. 37.
Fig. 37. Computing the circulation of C around a small square.
388
If our result is expressed in vector notation, we can say that it will be the same no
matter what the particular orientation of the plane.
We want know to find the circulation of the field C around the little square.
It will be easy to do the line integral if we make the square small enough that
the vector C does not change much along any one side of the square.
The assumption is better the smaller the square, so we are really talking
about infinitesimal squares. Starting at point (x, y) – the lower left corner of
the figure – we go around in the direction indicated by the arrows. Along the first
side (1) the tangential component is Cx(1) and the distance is x. The rust part
of the integral is Cx(1)x. Along the leg, we get Cy(2)y. Along the third, we get
Cx(3)x, and along the fourth, Cy4y. The minus signs are required because
we want the tangential component in the direction of travel. The whole line
integral is then
I
C~ d s~ ¼ Cx ð1Þx þ Cy ð2Þy þ Cx ð3Þx þ Cy ð4Þy
Now let us look at the first and third pieces. Together they are
½Cx ð1Þ Cx ð3Þx
If we take into account the rate of change of Cx, we write
Cx ð3Þ ¼ Cx ð1Þ þ
@Cx
y
@x
Since we ultimately think of the limit as y ! 0, the terms in (y)2 are neglected.
Combining the two previous equations, we find that
½Cx ð1Þ Cx ð3Þy ¼ @Cx
xy
@y
The derivative can, to our approximation, be evaluated at (x, y).
Similarly, for the other two terms in the circulation, we may write
Cy ð2Þy Cy ð4Þy ¼
@Cy
xy
@x
The circulation around the square is then
@Cy @Cx
xy
@x
@y
389
which is interesting, because the two terms in the parentheses are just the
z component of the curl. Also we note that xy is the area of the square. So we
can write the circulation as
~ C~ a
r
z
But the z component really means the component normal to the surface element.
We can therefore write the circulation around a differential square in an
invariant vector form:
I
~ C~ da ¼ r
~ C~ n~a
C~ d s~ ¼ r
n
Our result says that the circulation of any vector C around an infinitesimal
square is the component of the curl of C normal to the surface, times the area of
the square.
The circulation around any loop can now be easily related to the curl of the
vector field. We fill the loop with any convenient surface S and add the
circulations around a set of infinitesimal squares in this surface. The sum can be
written as an integral. Our result is a very useful theorem called Stokes’ theorem.
Stokes’ Theorem
I
Z ~
~
~ C~ da
r
C dl ¼
S
n
where S is any surface bounded by .
Energy in the electrostatic field
To show that Eq. (68) is consistent with our laws of electrostatics, we begin by
introducing into Eq. (64)
Z
1
dV
U¼
2
the relation between and that we obtained in Part 1:
Poisson equation
¼ "0 r2 We get
"0
U¼
2
Z
r2 390
Writing out the components of the integrand, we see that
@2 @2 @2 r ¼ þ
þ
@x2 @y2 @z2
2
2
2
@
@
@
@
@
@
@
@
@
þ
þ
¼
@x
@x
@x
@y
@y
@y
@y
@z
@z
~ r
~ r
~ r
~
¼r
since
@
@ @ @
@2 ¼
þ 2
@x @x @x @x
@x
Our energy integral is then
"0
U¼
2
Z Z
"0
~
~
~ r
~ dV
r r dV r
2
We can use Gauss’ theorem to change the second integral into a surface integral:
Z
Z
~ r
~ dV ¼
r
vol
~ n~ da
r
surface
We evaluate the surface integral in the case that the surface goes to infinity (so
the volume integrals become integrals over all space), supposing that all the
charges are located within some finite distance. The simple way to proceed is to
take a spherical surface of enormous radius R whose centre is at the origin of
coordinates. We know that when we are very far away from all charges, varies
as 1/R and r as 1/R2. Since the surface area of the large sphere increases as R2,
we see that the surface integral falls off as (1/R)(1/R2)R2 ¼ (1/R) as the radius of
the sphere increases. So if we include all space in our integration (R ! 1), the
surface integral goes to zero and we have that
"0
U¼
2
Z
all space
Z
"0
~
~
r r dV ¼
E~ E~ dV
2 all space
We see that it is possible for us to represent the energy of any charge distribution
as being the integral over an energy density located in the field.
391
Appendix B
A note on units and dimensions
Many times we would like to know the energy associated of a particular
interaction express in different units. The goal of this appendix is exactly to
provide us the useful tool that will give us such information.
If we take the electrostatic interaction energy between two charges in units of
charge squared per unit length, the Coulomb equation for the electrostatic
potential becomes simply
W¼
q1 q2
d
in atomic units (a.u.), being q1 and q2 in units of protonic charge and d in Å
(angstrom). We then have the relations:
Wðkcal=molÞ ¼ 331:842 Wða:u:Þ
and
Wðkjoule=molÞ ¼ 1388:4269 Wða:u:Þ
It is simpler to do all calculations in atomic units and then convert to the units of
interest using the above relations.
Molecular electrostatic energies and potentials are often expressed respectively
in kBT and kBT/e units, kT being an approximate value for the thermal noise of a
system. These units depend on the absolute temperature of the system, and
therefore are only meaningful when a value for T is given. At T ¼ 298 K we have:
WðkB TÞ ¼ 560:780 Wða:u:Þ
In atomic units the proton charge e is unitary and kBT are therefore numerically
equal to the corresponding kBT/e potential values.
When working with pKa calculations, it is also customary to express the
electrostatic interaction energies in terms of the induced pKa shift. These units are
dimensional and therefore independent of the unit system. At 298 K we have that
WðpKa unitsÞ ¼ Wða:u:Þ=½lnð10Þ kB T ¼ 243:499 Wða:u:Þ
From the above results we can derive other useful conversions:
1 pKa unit ¼ 1:3628 kcal=mol ¼ 2:303kB T
392
and
1 kB T ¼ 0:59175 kcal=mol
both at T ¼ 298 K.
References
1. Debye VP and Hückel E. Zur Theorie der Elektrolyte. Phys Z 1923;9(24):185–206.
2. Feynman RP, Leighton RB and Sands M. The Feynman Lectures on Physics, Vol. I. London:
Addison-Wesley Publishing Company, 1964.
3. Neves-Petersen, MT. The role of electrostatic interactions and light in protein structure and
function. PhD Thesis, University of Aalborg, 2000.
4. Brandt D, Hiller JR and Moloney MJ. In: Modern Physics Simulation. The Consortium for
Upper-Level Physics Software Ehrlich Robert, MacDonald William, Dworzecka Maria (eds.).
USA: John Wiley & Sons, Inc, 1995.
5. Moult J. Electrostatics. Curr Opin Struct Biol 1992;2:223.
6. Biosym/MSI, DelPhi and Solvation, version 95.0, San Diego, CA.
7. Martel, P. PhD Thesis, Universidade Nova de Lisboa (1996).
8. Petersen MTN, Martel P, Petersen EI, Drabløs F and Petersen SB. Surface and electrostatics
of cutinases. Methods Enzymol. 1997;284:130–154.
9. Born M. Volumen und Hydratationswärme der Ionen. Z Phys 1920;1:45–48.
10. Onsager L. Electric moments of molecules in liquids. J Am. Chem. Soc. 1936;58:1486–1493.
11. Kirkwood JG. Theory of solutions of molecules containing widely separated charges with
special application to zwitterions. J Chem Phys 1834;2(7):351–361.
12. Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH and Teller E. Equation of state
calculations by fast computing machines. J Chem Phys 1953;21(6):1087–1092.
13. Alder BJ and Wainwright TE. J Chem Phys 1959;31:459.
14. Rahaman A. Phys Rev 1964;136A:405.
15. Allen MP and Tildesley DJ. Computer Simulation of Liquids. Oxford: Clarendon, 1987.
16. Ciccotti G, Frenkel D and McDonald IR. Simulation of Liquids and Solids. Amsterdam:
North-Holland, 1987.
17. McCammon JA and Harvey SC. Dynamics of Proteins and Nucleic Acids. Cambridge:
Cambridge University Press, 1987.
18. van Gunsteren WF and Weiner PK. Computer Simulation of Biomolecular System. Leiden:
Escom, 1989.
19. Rashin AA and Bukatin, MA. A view of thermodynamics of hydration emerging from
continuum studies. Biophys Chem 1994 Aug;51(2–3):167–190; discussion 190–192.
20. Honig B and Nicholls A. Classical electrostatics in biology and chemistry. Science
1995;268:1144.
21. Atkins PW. Physical Chemistry. Oxford: Oxford University Press, 1994.
22. Jackson JD. Classical Electrodynamics. New York: Wiley, 1975.
23. Bockris JO and Reddy AKN. Modern Electrochemistry. London: MacDonald, 1970.
24. McQuarrie DA. Statistical Mechanics. New York: Harper, 1976.
25. Baptista A, Brautaset T, Drablos F, Martel P, Valla S and Petersen SB. In: Carbohydrate
Bioengineering Petersen SB, Svensson B, Pedersen S (eds.). Amsterdam: Elsevier Science BV,
1995;181–204.
26. Matthew JB. Electrostatic effects in proteins. Annu Rev Biophys B Chem 1985;14:387–417.
27. Shire SJ, Hanania GIH and Gurd FRN. Electrostatic effects in myoglobin. Hydrogen ion
equilibria in sperm whale ferrimyoglobin. Biochemistry 1974;13:2967–2974.
393
28. Fersht AR and Sternberg MJE. Can a simple function for the dielectric response model
electrostatic effects in globular proteins? Protein Engineering 1989;2(7):527–530.
29. Jackson JD. Classical Electrodynamics, 3rd ed. New York: Wiley, 1998.
30. Frohlich H. Theory of Dielectrics – Dielectric Constant and Dielectric Loss, 2nd ed. Oxford:
Oxford University Press, 1958.
31. Alder BJ, Alley WE and Pollock EL. Validity of macroscopic concepts for fluids on a
microscopic scale. Ber Bunsen Phys Chem 1981;85(11):944–952.
32. Allison SA, Northrup SH and McCammon JA. Simulation of biomolecular diffusion and
complex formation. Biophys J 1986;49(1):167–175 Jan.
33. Warshel A and Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic
and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol
1976;103(2):227–249 May 15.
34. Applequist J, Carl JR and Fung K. Atom dipole interaction model for molecular
polarizability. Application to polyatomic molecules and determination of atom polarizabilities. J Am Chem Soc 1972;94:2952.
35. Sharp KA and Honig B. Electrostatic interactions in macromolecules: theory and applications.
Annu Rev Biophys Biophys Chem 1990;19:301–332.
36. Rullman JA, Bellido MN and van Duijnen PT. The active site of papain. All-atom study of
interactions with protein matrix and solvent. J Mol Biol 1989;206:101.
37. VanBelle D, Couplet I, Prevost M and Wodak S. Calculations of electrostatic properties
in proteins. Analysis of contributions from induced protein dipoles. J Mol Biol
1987;198:721–735.
38. Warshel A and Russel S. Calculations of electrostatic interactions in biological systems in
solutions. Q Rev Biophys 1984;17(3):283–422.
39. Orttung WH. Polarizability density of inert-gas atom pairs. 1. J Phys Chem-US
1985;89(14):3011–3016.
40. Orttung WH and Vosooghi D. Polarizability densities within atoms. 1. Simple one-electron
systems. J Phys Chem-US 1983;87(8):1432–1437.
41. Bottcher CJF. In Theory of Electric Polarization. Amsterdam: Elsevier, 1973.
42. Gilson M and Honig B. Calculation of electrostatic potentials in an enzyme active site. Nature
(London) 1987;330:84.
43. Harvey S. Treatment of electrostatic effects in macromolecular modelling. Proteins: Structure,
Function and Genetics 1989;5:78.
44. Nakamura H, Sakamoto T and Wada A. A theoretical study of the dielectric constant of
protein. Protein Eng 1988;2(3):177–183 Sep.
45. Takashima S and Schwan HP. J Phys Chem 1965;69:4176.
46. Hill TL. Statistical Mechanics. New York: Dover, 1987. Originally published in McGraw-Hill,
New York, 1956.
47. Sharp KA and Honig B. Calculating total electrostatic energies with the nonlinear PoissonBoltzmann equation. J Phys Chem 1990;94:7648–7692.
48. Ben-Naim A. Statistical Thermodynamics for Chemist and Biochemists. New York: Plenum
Press, 1992.
49. Lee B and Richards FM. The interpretation of protein structures: estimation of static
accessibility. J Mol Biol 1971;55:379–400.
50. Westheimer FH and Kirkwood JG. J Chem Phys 1938;6:513–517.
51. Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science
1983;221(4612):709–713 Aug 19.
52. Richards FM. Areas, volumes, packing and protein structure. Annu Rev Biophys Bioeng
1977;6:151–176.
53. Linderstrøm-Lang K. On the ionization of proteins. CR Trav Lab Carlsberg 1924;15:1–29.
54. Tanford C and Kirkwood JG. Physical and inorganic chemistry. J Am Chem Soc
1957;79(20):5333–5339.
394
55. Orttung WH. Proton binding and dipole moment of hemoglobin. Refined calculations.
Biochemistry 1970;9(12):2394–2402 Jun 9.
56. Tanford C and Roxby R. Interpretation of protein titration curves. Application to lysozyme.
Biochemistry 1972;11:2192.
57. Matthew JB, Friend SH and Gurd FRN. In: Hemoglobin and Oxygen Binding Ho C New
York: Elsevier/North Holland, 1982;231.
58. Matthew JB and Richards FM. Anion binding and pH-dependent electrostatic effects in
ribonuclease. Biochemistry 1982;21(20):4989–4999 Sep 28.
59. States DJ and Karplus M. A model for electrostatic effects in proteins. J Mol Biol
1987;197:122–130.
60. Sitkoff D, Sharp KA and Honig B. Accurate calculation of hydration free energies using
macroscopic solvent models. J Phys Chem 1994;98:1978–1988.
61. Fojan P, Jonson PH, Petersen MTN and Petersen SB. What distinguishes a lipase from an
esterase: a new practical approach. Biochimie 2000;82:1033–1041.
62. Neves-Petersen MT, Fojan P and Petersen SB. How do lipases and esterases work: the
electrostatic contribution. J Biotechnol. 2001;85:115–147.
63. Neves-Petersen MT, Petersen EI, Fojan P, Noronha M, Madsen RG and Petersen SB.
Engineering the pH-optimum of a triglyceride lipase: from predictions based on electrostatic
computations to experimental results. J Biotechnol. 2001;87(3):225–254 May 18.
64. Petersen SB, Fojan P, Petersen EI and Petersen MTN. The thermal stability of the
Fusarium solani pisi cutinase as a function of pH. Journal of Biomedicine and Biotechnology
2001;1(2):1–8.
65. Martelt PJ, Baptista A and Petersen SB. Lipases and esterases: a review of their sequences,
structure and evolution. Biotechnol. Annu. Rev. 1996;2:315–372.
66. Rashin AA and Namboodiri K. A simple method for the calculation of hydration enthalpies
of polar molecules with arbitrary shapes. J Phys Chem 1987;91:6003–6012.
67. Jean-Charles A, Nicholls A, Sharp K, Honig B and Tempczyk A. Electrostatic contributions
to solvation energies: comparison of free energy perturbation and continuum calculations. J
Am Chem Soc 1991;113:1454–1455.
68. Mohan V, Davis ME, McCammon JA and Pettit BM. Continuum model-calculations of
solvation free-energies – accurate evaluation of electrostatic contributions. J Phys Chem
1992;96(15):6428–6431.
69. Ewing TJA and Lybrand TP. A comparison of perturbation-methods and Poisson-Boltzmann
electrostatics calculations for estimation of relative solvation free-energies. J Phys Chem
1994;98:1748–1752.
70. Antosiewics J, McCammon JA and Gilson MK. Prediction of pH-dependent properties of
proteins. J Mol Biol 1994;238:415–436.
71. Creighton TE. Proteins: Structures and Molecular Properties. USA: W.H. Freeman and
Company, 1993.
72. Fersht A. Enzyme structure and mechanism. New York: W.H. Freeman and Company, 1985.
73. Bashford D and Karplus M. pKa’s of ionizable groups in proteins: atomic detail from a
continuum electrostatic model. Biochemistry 1990;29:10219.
74. Bashford D and Karplus M. Multiple-site titration curves of proteins – an analysis of exact
and approximate methods for their calculation. J Phys Chem-US 1991;95(23):9556–9561.
75. Yang AS, Gunner MR, Sampogna R, Sharp K and Honig B. On the calculation of pKas in
proteins. Proteins 1993;15:252–265.
76. Gilson MK. Multiple-site titration and molecular modeling: two rapid methods for computing
energies and forces for ionizable groups in proteins. Proteins 1993;15(3):266–282 Mar.
77. Davis ME and McCammon JA. Electrostatics in biomolecular structure and dynamics. Chem
Rev 1990;90(3):509–521.
78. Warshel A and Åqvist J. Electrostatic energy and macromolecular function. Annu Rev
Biophys Biophys Chem 1991;20:267–298.
395
79. Beveridge DI and Dicapua FM. Free energy via molecular simulation: applications to
chemical biomolecular systems. Annu Rev Biophys Biophys Chem 1989;18:431–492.
80. Bashford D. Electrostatic effects in biological molecules. Curr Opin Struct Biol
1990;1:175–184.
81. Rose GD, Geselowitz AR, Lesser GJ, Lee RH and Zehfus MH. Hydrophobicity of amino-acid
residues in globular-proteins. Science 1985;229(4716):834–838.
82. Honig B, Sharp K and Yang AS. Macroscopic models of aqueous-solutions – biological and
chemical applications. J Phys Chem-US 1993;97(6):1101–1109.
83. Klapper I, Hagstrom R, Fine R, Sharp K and Honig B. Focusing of electric fields in the active
site of Cu-Zn superoxide dismutase: effects of ionic strength and amino-acid modification.
Proteins 1986;1(1):47–59.
84. Gilson M and Honig B. Energetics of charge-charge interactions in proteins. Proteins:
Structure, Function and Genetics 1988;3:32–52.
85. Gilson M and Honig B. Calculation of the total electrostatic energy of a macromolecular
system: salvation energies, biding energies, and conformational analysis. Proteins: Structure,
Function and Genetics 1988;4:7–18.
86. InsightII, 1995, version 95.0. Biosym/MSI, San Diego, CA.
87. Nicholls A and Honig B. A rapid finite-difference algorithm, utilizing successive overrelaxation to solve the Poisson-Boltzmann equation. J Comput Chem 1991;12(4):435–445.
88. Nicholls A, Sharp K and Honig B. Protein folding and association: insights from the
interfacial and thermodynamic properties of hydrocarbons. Proteins 1991;11(4):281–296.
89. Petersen SB, Jonson PH, Fojan P, Petersen EI, Petersen MTN, Hanssen S, Ishak RJ and
Hough E. Protein Engineering the surfaces of enzymes. J Biotechnol. 1998;66:11–26.