: 100 學年度第 1 學期 班級: 碩研電機二甲 ...
advertisement

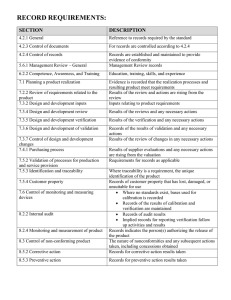
醫儀品質驗證系統 授課老師:陳世中 評分: 100 學年度第 1 學期 班級: 碩研電機二甲 學號: M9920133 學生姓名: 陳玉銘 作業次號 第 7 次 週 繳交日期 週次 第 11 主 講 人 主題 醫療器材優良製造規範 2011 年 11 月 24 日 鄭宗記 上課日期 2011 年 11 月 24 日 報告內容至少一頁(可自行延伸) 字體:中文標楷體 英文 Times New Roman 字體大小:12 列距:單行 內容重點(含圖、文、表) Premarket: Clinical Trials Regulation、Premarket Assessments、GMP Inspections Postmarket: Adverse Event Reporting、Compliance Testing、GMP Inspections、Recalls 質量管理體系-ISO 9000 的過程方法: ISO 13485 的目的: The main objective was to help foster efforts of international harmonization in medical regulatory requirements for Quality Management Systems. ISO 13485:2003 by it self is not sufficient, but must be complemented by specific requirements of local regulatory authorities. ISO 13485 的主要結構: 1.Scope, 2.Normative reference, 3.Terms & definitions, 4.quality management system, 5.management responsibility, 6.resource management, 7.product realization, 8.Measurement, analysis and improvement Clause 5-4 規劃: Quality objectives – are established at relevant functions and levels within the organization. – shall be measurable and consistent with the quality policy Quality management system planning – the planning is carried out in order to meet the requirements given in “General requirements”(4.1), as well as the quality objectives, and – integrity of the quality management system is maintained when changes to the quality management system are planned and implemented. Clause 7 Product realization: Planning of product realization - Customer-related processes - Design and development (D&D) - Purchasing - Production and service provision - Control of monitoring and measuring devices Clause 7-1 Planning of product realization: Determine the following: – quality objectives and requirements for the product; – the need to establish processes, documents, and provide resources specific to the product; – required verification, validation, monitoring, inspection and test activities specific to the product and the criteria for product acceptance; – records needed to provide evidence that the realization processes and resulting product fulfill requirements. Clause 7-3 Design Control: - General - Design and development planning - Organizational and technical interfaces - Design input - Design output --------- Design Transfer ---------- Design review - Design verification - Design validation - Design changes Clause 7-3-6 Design & development validation: • Clinical evaluation may be involved in the validation of the design of medical devices. – The conduct of clinical investigations should conform to applicable regulations/standards. • As part of design validation, the organization shall perform and maintain records of clinical evaluations. • Clinical evaluation may include – a compilation of relevant scientific literature, – historical evidence that similar designs and/or materials are clinically safe, or – a clinical investigation or trial, to ensure that the device performs as intended. • National or regional regulations may require actual clinical investigations or trials. Clause 8-5 Improvement: • General – shall establish and maintain documented procedures for the issue of advisory notices for medical devices. – shall maintain records of all customer complaint investigations. • When the investigation determines that the activities at remote premises contributed to the customer complaint, relevant information shall be communicated between the organization and the remote premises. – If regulations require, the organization shall establish and maintain documented procedures to notify the regulatory authority(ies) of those adverse events which meet the reporting criteria. Clause 8-5-2 Improvement(Corrective action): - Corrective & Preventive Action (CAPA) • define requirements for – reviewing nonconformities (including customer complaints), – determining the causes of nonconformities, – evaluating the need for action to ensure that nonconformities do not recur, – determining and implementing action needed, – records of the results of any investigation and of the actions taken (see 4.2.4), and – reviewing corrective action taken and its effectiveness. 課後心得:及參考文獻 初步認識了醫療器材製造的規範與 ISO 13485 的目的。相信對往後在醫材方面有很大的幫助。 參考資料:演講者的講義(醫療器材優良製造規範.ppt)