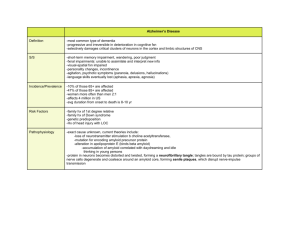
Neurodegenerative disease April 2008
advertisement

Neurodegenerative disease April 2008 QuickTime™ and a TIFF (LZW) decompressor are needed to see this picture. Pt. 4 Major categories of neurologic disease 527 30 31 32 33 34 35 36 37 38 39 40 41 42 43 Disturbances of cerebrospinal fluid and its circulation 529 Intracranial neoplasms and paraneoplastic disorders 546 Infections of the nervous system (bacterial, fungal, spirochetal, parasitic) and sarcoidosis Viral infections of the nervous system, chronic meningitis, and prion diseases 631 Cerebrovascular diseases 660 Craniocerebral trauma 747 Multiple sclerosis and allied demyelinative diseases 771 The inherited metabolic diseases of the nervous system 797 Developmental diseases of the nervous system 850 Degenerative diseases of the nervous system 895 The acquired metabolic disorders of the nervous system 959 Diseases of the nervous system due to nutritional deficiency-983 Alcohol and alcoholism 1004 Disorders of the nervous system due to drugs, toxins, and other chemical agents-1016 592 Characteristics of neurodegenerative disease • Insidious in onset • Progressive course • Selective death/dysfunction of neurons Characteristics of neurodegenerative disease • • • • Insidious in onset Progressive course Selective death/dysfunction of neurons Etiology unclear Examples of neurodegenerative disease: • • • • Alzheimer’s disease Parkinson’s disease Frontotemporal dementia Amyotrophic lateral sclerosis (Lou Gehrig’s disease) • Spinocerebellar ataxia • Huntington’s disease Neurodegenerative disease is common Neurodegenerative disease prevalence: • Alzheimer’s disease: – 1-2% age 65-75; 50% over age 85 • Parkinson’s disease – 13/100,00; 0.5-1% age 60-69; 1-3% over age 80 • Frontotemporal dementia – 1 per 10,000? • ALS – 1-2 per 100,000 per year • Spinocerebellar ataxias – 0.3-3 per 100,000 • Huntington’s disease – 1 in 10,000 Organization of presentation: • • • • • • Clinical presentation and diagnosis Pathology Genetic risk factors Environmental risk factors Pathogenesis (stories we tell) Treatments available (ie, the need for better treatments) Alzheimer’s presentation • Starts with memory loss--repetitive stories, repetitive questions, forgotten events, progressing to the point that ADLs are affected. • By the time of diagnosis, a second “cognitive domain” is affected (language, spatial function, executive dysfunction) Alzheimer’s course: • Progressive loss of cognitive abilities and ADLs, leading ultimately to a vegetative state, and finally death (infection, malnutrition, MI, CVA) • Average time from dx to death = 8-10 years • Rate of progression is variable • Behavior changes (psychosis, depression, apathy, agitation) are especially variable • “If you’ve met one patient with Alzheimer’s……you’ve met one patient with Alzheimer’s” Alzheimer’s pathology • • • • Amyloid plaques Neurofibrillary tangles Neuronal death and brain atrophy Cholinergic projection system withers Senile plaques • Extracellular deposits • Plaques described as “diffuse”, “neuritic”, or “cored” • These may represent different ages of plaque • Neuritic plaques are one of the pathologic criteria for diagnosis of Alzheimer’s disease • Composed chiefly of beta amyloid Beta amyloid • Beta amyloid is a 39-43 amino acid peptide • Derived from 700 amino acid amyloid precursor protein (APP) • APP may be processed to “amyloidogenic” or “non-amyloidogenic” pathways -Amyloid Plaques SIGMA-ALDRICH Neurofibrillary tangles • Intracellular inclusion • Chief component is hyper-phosphorylated tau • Tau is a normal intracellular protein which stabilizes microtubules Alzheimer’s pathology • • • • Amyloid plaques Neurofibrillary tangles Neuronal death and brain atrophy Cholinergic projection system withers Alzheimer’s disease: genetic risk factors (autosomal dominant) • Amyloid precursor protein • Presenilin-1 • Presenilin-2 Alzheimer’s disease: genetic risk factors (sporadic) • Apolipoprotein E – Alleles: E2, E3, E4 – E4 is present in 15% of population – E4 is present in 45-50% of Alzheimer’s Alzheimer’s disease: environmental risk factors • • • • Low educational attainment Head injury Depression Vascular risk factors (HTN, DM, hypercholesterolemia) Alzheimer’s disease: stories re pathogenesis • Braak staging (“tau hypothesis”?) • Amyloid hypothesis • Cholinergic hypothesis Braak staging • Based on the predictable spread of tangle pathology – First entorhinal cortex – Then entorhinal cortex + hippocampus – Then entorhinal cortex + hippocampus + association cortex Braak staging of AD Braak 1-2 Braak 3-4 Tangle histology Braak 5-6 Atrophy on MRI* Entorhinal Entorhinal + Entorhinal + cortex hippocampus hippocampus + cortex Same as Same as Same as above above above Clinical status Healthy aging Isolated AD-type memory loss dementia (“MCI”) *(Kaye et al 1997, Silbert et al 2003) Amyloid hypothesis of AD • Holds that neurotoxicity of beta amyloid drives the neurodegenerative process • But: – beta amyloid is produced under physiologic conditions--how could it be toxic? Merlini and Bellotti, NEJM 349:583-596, 2003 Merlini and Bellotti, NEJM 349:583-596, 2003 Evaluating the amyloid hypothesis-pros and cons • • • • Clinicopathologic correlation Genetics of AD Cell culture studies Animal studies Pathologic correlates of dementia severity (Terry, 1991) • • • • Amyloid plaques: Neurofibrillary tangles: Neuronal loss: Synaptic density: poor better same as tangles best • So…..clin-path studies do not support the amyloid hypothesis Evaluating the amyloid hypothesis-pros and cons • Genetics of AD------------------pro – Autosomal dominant AD associated with mutations in amyloid precursor protein (APP) – Trisomy 21 also associated with overexpression of APP and AD – “presenilin” initially identified in autosomal dominant AD, since shown to be a component of gamma secretase-- enzyme which processes APP to beta amyloid Evaluating the amyloid hypothesis-pros and cons • • • • Clinicopathologic correlation--con Genetics of AD------------------pro Cell culture studies--------------pro Animal studies-------------------+/- Cholinergic hypothesis of AD • Based in part on clin-path observation of correlation between cholinergic markers and dementia severity Other putative mechanisms: • • • • • • • Inflammation Oxidative damage Ubiquitin-proteasome dysfunction Mitochondrial dysfunction Metal dyshomeostasis (copper, iron) Excitotoxicity Axonal transport dysfunction Treatments available for AD • Clinical trials of multiple cholinergic agents have shown enough efficacy to be FDA-approved, but none is dramatically effective • Anti-amyloid therapies are in Phase 3 trials • Anti-tau therapies are in earlier trials • Gene therapy with NGF is also under way targeting the cholinergic system Parkinson’s disease-clinical • Cardinal signs: – – – – Tremor Rigidity bradykinesia Gait impairment Parkinson’s-clinical • Traditionally considered purely a disorder of movement • Now appreciated to include autonomic nervous system dysfunction (before motor impairment) and cognitive dysfunction (after motor impairment) Parkinson’s disease pathology Parkinson’s pathology: Lewy bodies • Intraneuronal inclusions comprised of alpha synuclein and other proteins • Initially thought to be confined to substantia nigra and other projection systems that deteriorate in PD • Subsequently identified throughout the nervous system, from brainstem to cortex • Incidental Lewy bodies seen in as many as 7-10% of asymptomatic individuals over age 60 Parkinson’s genetic risk factors • “familial cases are on record, but the evidence is rather unsubstantial…” Adams and Victor 1985 • “Though there is no evidence to indicate a hereditary factor, a familial evidence is claimed by some.” Merritt’s Textbook of Neurology, 1984 Parkinson’s genetic risk factors But..despite the number of genes implicated in rare sub-types of PD, Most cases of “garden-variety” PD are not explained by genes (as in AD) Parkinson’s: environmental risk factors • • • • Age Male gender Rural living Smoking is protective Parkinson’s pathogenesis: Braak staging suggests a progressive “synuclein-opathy”: • Lewy bodies spread caudal-->rostral • Stage 1-2: Lewy bodies in medulla and olfactory bulb (asymptomatic) • Stage 3-4: Lewy bodies in substantia nigra, locus coeruleus, cholinergic basal forebrain (parkinson’s symptoms appear when >80% of nigral neurons gone)) • Stage 5-6: Lewy bodies in forebrain (dementia) Parkinson’s pathogenesis: other models: • MPTP model – Gives rise to selective neuronal death and parkinsonism, but no Lewy bodies • Rotenone model (Greenamyre) – Chronic intravenous infusion of mitochondrial complex I inhibitor in rats – Produces selective neuronal death, parkinsonism, and Lewy bodies Parkinson’s treatments: • Dopaminergic therapy has dramatic symptomatic effects • Surgical therapies--both ablative and deep brain stimulator therapies have symptomatic effects • Gene therapy with trophic factors is under investigation • No proven neuroprotectant therapy to date Frontotemporal dementia-clinical • Presents as personality change and disinhibition, in the absence of significant memory loss • Also may present as a primary disorder of language • Progresses to a more generalized dementia over time Frontotemporal dementiapathology • Clinical syndrome with a variety of underlying pathologies (Pick’s disease, “DLDH”, others) • Many have neurofibrillary tangles Frontotemporal dementiagenetics • Most cases of FTD are sporadic • A mutation in the tau protein is a cause of FTD in a minority of cases (FTDP=17). • Tau is a normal intracellular protein which stabilizes microtubules. FTD-treatment • Nothing available Amyotrophic lateral sclerosisclinical • Also known as “motor neuron disease” or “Lou Gehrig’s disease” • Presents as slowly progressive weakness and muscle wasting. • Death within 2-5 years in most patients due to respiratory failure • Concomitant FTD in a sub-population of patients (subclinical neuropsych changes may be more common) ALS-pathology ALS-genetics • About 10% of cases are familial • About 2-3% are caused by mutations in Cu/Zn SOD ALS-treatment • Riluzole, a glutamate antagonist, prolongs survival by a few months • Treatment trials with trophic factors have failed • No other symptomatic or neuroprotectant therapy • No SOD-directed therapy Spinocerebellar ataxia-clinical • Slowly progressive gait disorder, slurred speech, and clumsiness • Age of onset widely variable--from early childhood to late life • Patients look like they are intoxicated with alcohol Spinocerebellar ataxia-pathology Spinocerebellar ataxia-genetics (autosomal dominant) How and why does a polyglutamine repeat in several different proteins--give rise to a single phenotype? (there are other phenotypes associated with other polyglutamine repeats) Neurodegeneration-summary signs region histology protein AD dementia HC, ctx Plq, NFT AB, tau PD motor Lewy b. alphasyn FTD dementia ctx NFT tau ALS weakness Motor n. MN loss SOD SCA ataxia HD S. nigra Cb, SC Cerebel’r many atrophy Chorea, Caudate, inclusion Huntingdementia ctx tin Unanswered questions • How does the identification of the deranged protein in each disease explain the selective vulnerability of neurons? • Are the mutated proteins themselves neurotoxic? Or what? • What can we learn from transgenic mouse models? (examples: APP, tau mutants) Questions?