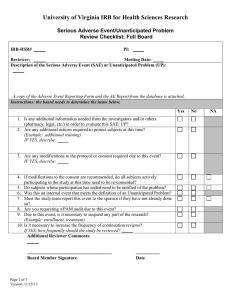
Instructions for completing SAE Report Form
advertisement

IRB Version Date: 2/2012 Saint Louis University Institutional Review Board (IRB) INSTRUCTIONS FOR COMPLETING THE SERIOUS ADVERSE EVENT (SAE) Guidelines require study investigators to report any fatal or life-threatening event within 3 calendar days and any serious or unanticipated event within 7 calendar days. NOTE: The SAE form is to be completed for events related (possible, probable or definite) to a particular human research protocol approved by the SLU IRB. For questions on reporting SAEs to the IRB, please refer to the Requirements for Reporting Events Relating to Subject Safety. Step 1: Download the SAE report form. Handwritten forms are not acceptable. Add the IRB# at the top right corner. Fill in the date you are completing the report. Complete the contact and study title information. Step 2: Question #1 pertains to the type of SAE reporting information that is being submitted. If you answer “yes” to questions 1a, 1b, or 1c, the IRB will require the investigator completing this form to provide the details about the SAE. In addition to completing the table in Appendix A, question #2 will need to be answered for each event being reported. The relevant information used to determine whether an adverse event is related must be provided in a “MedWatch” type of format, discharge summary, ER report, or clinical summary. For an example of the type of information in a MedWatch form, see the following website: http://www.fda.gov/medwatch/getforms.htm Check the box in 1c if the SAE is an Unanticipated Problem. An Unanticipated Problem (UP) is an adverse event that is: Any unexpected serious adverse event or problem reasonably related to a SLU protocol that was not previously identified in the informed consent document, research protocol, or investigator’s brochure, AND a. The event is not consistent with or exceeds the nature, severity or frequency described in the protocol, consent form, or investigator’s brochure, OR b. The event relates to the rights, safety, or welfare of the subjects . Step 3: Question #2 pertains to SAEs that occurred within our institution or where a SLU investigator is the head or coordinator of a multicenter study and is responsible for reporting SAEs or if the event is an Unanticipated Problem. Provide a) the event number from the table and b) a description of each event and a description of medical actions taken and the findings. Please include the timeline of events, whether the event occurred while the subject was actively receiving treatment, relevant medical history, as well as any other contributing information to support the designated relationship (column H on table) of the SAE. Attached documentation does not replace an answer to this question. Step 4: Questions 3 and 4 pertain to informed consent. FDA does not require re-consenting of subjects who have completed their active participation in the study, or from subjects who are still actively participating when the changes occur, if the changes will not affect their participation. According to FDA regulations, 21CFR 50.25(b)(5): “Those subjects who are presently enrolled and actively participating in the study should be informed of the change if it might relate to the subject’s willingness to continue their participation. 1 IRB Version Date: 2/2012 Step 5: Questions 5 and 6 pertain to SAEs that occur at a Tenet or SSM property. While the SLU IRB serves as the IRB of record for all research of SLU personnel at these locations, each individual property provides administrative research oversight to that research. All SAEs that occur at these locations must be reported for administrative oversight. Appendix A The following are instructions for completing the boxes in the table in Appendix A. Note: The text boxes will expand as needed. A) B) C) D) E) F) G) H) Please number the events in consecutive order (e.g., 1, 2, 3). The most recent events should be placed last. When adding events to the table, highlight in yellow so that the IRB reviewer will know which event(s) are new. Follow-Up SAEs: If you are submitting a follow-up to a previously submitted SAE, insert a row into the table directly below the original SAE that is being followed-up. To insert a row, place your curser in the row of the original SAE. Then click on Table in the windows toolbar on the top of your screen. Next, select “Insert” from the drop down menu, then select “Rows Below”. If the original SAE is numbered 1, you should number the follow-up 1.1, 1.2, 1.3, and so on. Enter the original date of occurrence of the SAE. Check the appropriate site of occurrence, SLU or other institution. Check the appropriate option of submission: initial or follow-up Follow-up would only be marked if the original SAE had been previously submitted to the IRB office for the same subject. Fill in the unique subject ID number for each subject submitted. List the code letter for the appropriate item(s). H = Hospitalization PH = Prolonged Hospitalization PD = Resulted in permanent disability DISC = Discontinuation of drug/device for medical reasons DCA = Development of a congenital anomaly BD = Development of a birth defect CA = Cancer A = Autopsy performed LT= Life Threatening D = Death P = Pregnancy SME = Other Important/Significant Medical Event (Examples of such events include allergic bronchospasm requiring intensive treatment in an emergency room or at home, blood dyscrasias or convulsions that do not result in inpatient hospitalization, or the development of drug dependency or drug abuse.) Indicate “Other” and provide description for events that do not fall under one of the above categories. Indicate the “key words” of the SAE (pneumonia, renal failure, rash, etc.) There may be several key words. List all that are applicable. Choose option 1-4 to represent the relationship of causality of the SAE to the study (which may include the study drug or ancillary drugs, study procedures, the use of placebo, or randomization procedure). If sponsor reports do not use these designations of causality (for example, the reports state “yes” or “no”), then state their designation. If (3) “probable” or (4) “definite” is chosen, or the investigator believes that there is a “sufficient number of similar AE’s” of (1) “not likely” or (2) “possible” events to cause concern, then a change-in-protocol form should be submitted within 3 weeks. The risk section of the protocol (#11) and parts of the consent form, including the discussion of risks (item #4), should be modified. The IRB may query the investigator to ascertain further details of the SAE’s that would determine the necessity of protocol and consent modifications. 2 IRB Version Date: 2/2012 Include the number of previously submitted similar SAE’s (information from this cumulative table including the current event being reported). J) Indicate if this SAE is already mentioned as a possible side effect in the consent form. K) Indicate whether the protocol and consent form need to be modified to include the new information. L) Indicate whether the SAE is also an Unanticipated Problem (UP). IMPORTANT: Indicate which events are new by highlighting in yellow or by (lightly) shading the applicable row(s). If the highlighting or shading is too dark, the text will be unreadable on copies made of the report. Since the table is cumulative, only new events should be highlighted each time the SAE report is submitted. Add boxes as needed by putting the cursor in the last box of the table and hitting the ‘tab’ key. This will add a new row to the table. I) Step 6: The principal investigator should sign and date the SAE form. Step 7: Attach any supporting documentation for the SAEs. Please attach them in the order that the new (highlighted) SAEs are listed in the table. Step 8: Attach any revised materials. Be sure to highlight where revisions were made. Step 9: Submit materials to the IRB office. Once reviewed, the form will be returned to the investigator. If the IRB requires additional information, the investigator must respond within 3 weeks. 3