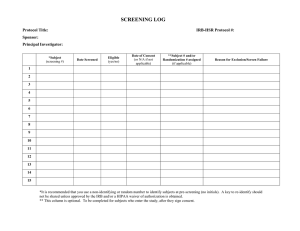
Research Common Application Step 2 Continuation UTHSCSA IRB Application
advertisement

UT Health Science Center San Antonio Common Research Application Step 2 Continuation – UTHSCSA IRB Application The Common Application consists of Step 1 – Intent to Conduct Research, Step 2 – Institutional Application and Step 2 Continuation –UTHSCSA IRB Application (when using UTHSCSA IRB). These three forms may be submitted in one of two combinations: A) Step 1 alone (Step 2 to follow later) –OR- B) All three together. Items marked with the icon indicate fields that the institution and the IRB share. Using this form – To check the checkboxes, click once on the box. To enter text in the text boxes, click once on the gray box and then type your response. If you are a Mac user and are having trouble using this form, try this alternate version of the form. UTHSCSA Tracking Number Item 40 Is your study eligible for Expedited Review? ☐ No continue to Item 41 ☐ Yes submit Form B-1 - Expedited Certification Form Applicable during initial IRB review of a new study Item 41 Special Confidentiality Considerations Select all applicable ☐ Drug or Substance Abuse research - - -[consider obtaining a certificate of confidentiality] ☐ Certificate of Confidentiality - - - - - - - - [include statement in consent and provide the approval] ☐ Genetic Research - - - - - - - - - - - - - [include risks of genetic research in consent & confidentiality plan in Step 2 institutional-Data Security plan] ☐ HIV or Hepatitis Screening - -[Review the IRB Instructions - include state reporting requirements in consent] ☐ Documentation in the Medical Record ----[consider altering the study title to eliminate increased risk of research participation disclosure] ☐ N/A – no special confidentiality considerations - continue to Item 42 Item 42 Data Safety Monitoring ☐ A Data Safety Monitoring Plan (DSMP) is required by either: Select all applicable ☐ A Data Safety Monitoring Plan (DSMP) is required by: The IRB (because this is a greater than minimal risk study) ☐ A Data & Safety Monitoring Board (DSMB) will be used ☐ N/A – none of the situations listed above apply The NIH (i.e. grant) or the FDA (i.e. research with IND or IDE) vMay31-2016 1 Submit Form R - Monitoring Participant Safety and Data Integrity continue to Item 43 Item 43 Categories of subjects Select all applicable Note for chart review research: Answer based on whether subjects will fall into a category at the time you are reviewing the records. This study includes: ☐ Decisionally Impaired Individuals - - - - - - - - - - -- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -☐ Decisionally Impaired and Institutionalized Individuals - - - - - - - - - - - - - - - - - - - - - - - - - - - - ☐ Individuals with diminished decision-making capacity - - - - - - - - -- - - - - - - - - - - - - - - - - - - - - Submit Form T Form T Form T (not including incompetent or impaired decision making capacity) ☐ Pregnant Women *Pregnant Women will not be participants at the VA - - - - - - - - - - - - - - - - - ☐ Pregnant Women (for follow-up) - - -- - - - - - -- - -- - - - - - -- - -- - - - - - -- - -- - - - - - -- - -- - - - - - ☐ Fetal Material *Fetal Material will not be used at the VA - - - - - - - - - - - - - - - - - - - - - - - - ☐ Neonates of uncertain viability or nonviable neonates - - - - - - - - - - - - - - - - - - - - - - - - - - - - Form U Form U-Follow and pregnant person consent Form U Form U *Neonates will not be participants at the VA *Prisoners will not be participants at the VA - - - - - - - - - - - - - - - - - - - - - - - - Form V Form W *Children will not be participants at the VA Form W ☐ Prisoners ☐ Children (17 yrs or less), includes viable neonates - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - ☐ ☐ ☐ ☐ ☐ Wards of the State - - - - - - - - - - - - - - - - - - - - -- - - - - - - - - - - - - - - -- - - - - - - - - - - - - - - - - - - Students under the supervision of any investigator Employees under the supervision of any investigator Embryonic stem cell research Enrollment of non-veterans in VA-approved research, include justification (i.e. insufficient veterans available to complete study, survey of VA employees, study of active duty): ☐ Other vulnerable population: Item 44 Research Consent & HIPAA Authorization Wizard Items 43 – 47 are designed to help you determine the appropriate waivers and documents. In Step 1, Item 9 – which answer did you choose? ☐ No – this study does not involve interaction or intervention with living individuals - continue to Item 45 ☐ Yes – this study involves interaction or intervention with living individuals - continue to Item 46 Item 45 Study does not involve interaction or intervention with living individuals Will you obtain private identifiable information collected for ☐ No continue to next section other purposes? ☐ Yes Submit Form F (complete the Waiver of Consent section) ☐ Yes but a Waiver of Consent is not needed because subjects will provide informed consent or informed consent will be waived as part of their participation in a contributing protocol (i.e. data coordinating center activities). If yes, Describe the source: Describe the source of the information: (medical records of patients seen in PI’s clinic, research records from HSC2010XXXH, etc.) vMay31-2016 2 UT Health Science Center San Antonio Common Research Application Step 2 Continuation –UTHSCSA IRB Application If yes, ☐ No continue to next section Does the information reviewed contain both health ☐ Yes submit Form J – HIPAA Waiver information and identifiers? ☐ Yes but a HIPPA Waiver is not needed because subjects will Will you obtain anonymous tissue specimens to test the safety or effectiveness of an in vitro diagnostic device? provide authorization or authorization will be waived as part of their participation in a contributing protocol (i.e. data coordinating center activities). ☐ No continue to Item 46 ☐ Yes contact IRB Associate Director for applicable form(s) Item 46 Issues related to identifying prospective subjects Do you plan to review existing information (e.g., clinical or research records) to identify individuals who may be eligible to participate? ☐ No continue to Item 47 ☐ Yes submit Form F (complete the Waiver of Consent section) If yes, Describe the source: Describe the source of the existing information: (medical records of patients seen in PI’s clinic, research records from HSC2010XXXH, etc.) If yes, Does the information reviewed contain PHI? Will individuals who are not members of the study team refer subjects to study staff? (i.e., clinical providers) Will participants self-identify? (i.e., response to advertisements, word-of- ☐ No continue to Item 47 ☐ Yes submit Form J – HIPAA Waiver ☐ No ☐ Yes ☐ No ☐ Yes mouth) ☐ No ☐ Yes (attach recruitment materials) If yes, Will recruitment materials be used? Item 47 First Contact - Recruitment select how and where initial contact will be made with potential subjects; list all applicable ☐ N/A – this study does not involve interaction with living individuals (you answered no - Step 1, Item 9) - continue to Item 54 How will contact be made: Telephone call Email Mail Waiting room (public) During scheduled visit (private) Other method: From study staff to participants ☐ ☐ ☐ ☐ ☐ ☐ From participants to study staff ☐ ☐ ☐ ☐ ☐ ☐ Document1 vSept3-2014 3 Briefly describe the plan to contact subjects including differences between groups (if any) --include details regarding relationship with subjects (i.e., members of treatment team will contact…) Indicate how special VA rules will be applied: Refer to the VA Handbook for additional information ☐N/A – VA not a study site ☐Although email and telephone may be used as a first contact method at other study sites above, it will not be used for VA subjects Item 48 Screening procedures Will any screening procedures be completed by subjects prior to obtaining consent? ☐ N/A not obtaining informed consent ☐ No – continue to Item 49 ☐ Yes If yes, Select all applicable What type(s) of screening procedures are planned? ☐ ☐ ☐ ☐ Screening questions Withhold food and/or water overnight Withhold medication overnight: List Other: describe If yes, How do you plan to handle consent for screening procedures? Full (long) informed consent will be provided (either verbally or in writing), however a consent document will not be signed until after screening Abbreviated information about the study will be provided to subjects (not including all required elements of consent) and a consent document will not be signed until after screening If recording the answers to screening questions – is the information health related? Item 49 How will you consent subjects to participate in the research? ☐ Will not obtain consent from: (select one) ☐any participant ☐some participants Select all applicable Will obtain consent without a signature Documentation section) ☐ submit Form F (complete the Alteration of Consent and Waiver of Documentation sections] ☐ No– continue to Item 49 ☐ Yes submit Form J – HIPAA Waiver If some participants selected, which ones? ☐ Submit Form F (complete the Waiver of If selected, ☐ No ☐ Yes submit Form J – HIPAA Waiver ☐ Yes but a HIPPA Waiver is not needed Is the information collected for the study health information? ☐ ☐ submit Form F (complete the Waiver of If selected, select at least one of the following describing how you will obtain consent: ☐ Verbal in person ☐ Telephone ☐ Online ☐ Survey (or other data collection tool) ☐ Other vMay31-2016 4 Consent section) OR ☐ Waiver of Consent is not needed because subjects will provide informed consent or informed consent will be waived as part of their participation in a contributing protocol (i.e. data coordinating center activities). because subjects will provide authorization or a waiver of authorization will be obtained as part of their participation in a contributing protocol (i.e. data coordinating center activities). submit Form F (complete the Waiver of Documentation section) Also submit either: the text of the information that will be provided (i.e., script, consent paragraph) or an Information sheet -Form D-IS If selected, ☐ ☐ ☐ ☐ ☐ Is the information collected for the study health information? Will exclude some of the required elements of consent in the consent form, information sheet, or consent process Will obtain full consent ☐ research at non-VA site using the long consent ☐ research at a VA site form for: ☐ only collecting biological specimens or data for a bank or repository at non-VA site ☐ only collecting biological specimens or data for a bank or repository at a VA site Will use deception during consent – not providing subjects will all information about the study to prevent potential biases in the study Will consent pregnant partners of participants Will obtain consent for a research study and an optional sub-study to bank specimens and/or data Item 50 Initial Consent Procedure Select all applicable Item 51 Waiting period between informing the prospective subject and obtaining consent ☐ No ☐ Yes submit Form J – HIPAA Waiver submit Form F (complete the Alteration of Consent and Waiver of Documentation sections) submit Form D -UTHSCSA Consent submit Form D-1- VA Consent submit Form E- Repository Consent submit Form E-1- VA Repository Consent Review the deception instructions to determine whether to submit Form F (Alteration of Consent section) ☐ sub-study is an external repository/registry -submit Pre-filled Form U for Pregnant Partners, and submit Partner Consent Form submit Form E- Repository Consent or review the Informed Consent Instructions for alternatives ☐ sub-study is an internal repository/registry Use the approved consent for existing Repository Study Insert HSC# for Repository: ☐ ☐ ☐ The IRB Policy on Informed Consent Process will be followed The IRB Policy on Informed Consent Documentation will be followed A different consent or documentation procedure will be used as follows: ☐ ☐ ☐ N/A this study does not involve interaction with living individuals - continue to Item 54 Prospective subjects can take as long as they wish to make a decision Prospective subjects are expected to make a decision immediately or within a specified timeframe described here: Select all applicable Item 52 Informed Consent for Research Involving Non-English Speaking Subjects Select one ☐ ☐ Only individuals who speak English will be enrolled Individuals who do not speak Select one ☐There is no expected direct benefit for those participating. ☐There is an expected direct benefit for those participating. Excluding non-English speaking individuals is acceptable because: Describe: The translated consent will be submitted to the IRB: Select one ☐ Immediately following approval of the English consent. vMay31-2016 5 English will be enrolled ☐ ☐ Only after a potential non-English speaking participant is identified. Since this plan will delay enrollment pending IRB approval of a translated consent, provide justification that prospective non-English speaking subjects will not be excluded from beneficial research. ☐ Select one ☐ ☐ ☐ ☐There is no expected direct benefit for those participating. ☐There is an expected direct benefit for those participating. Any delay in obtaining a translated consent is acceptable because: Describe: N/A ☐ study does not involve interaction with living individuals (answered no - Step 1, Item 9) ☐ study includes a waiver of informed consent for all subjects (see Item 49) Item 53 Privacy Protections Describe the privacy protections that will be used to ensure privacy is protected during recruitment, consent, and the research interventions: Use of drapes or other barriers Activities occur in a private room Activities occur in a semi-private room Other method: Item 54 Nature of the Study Select one Recruitment Consent Research Intervention ☐ ☐ ☐ ☐ ☐ ☐ ☐ ☐ ☐ ☐ ☐ ☐ Interventional Primary Purpose Study Phase Individuals are assigned by an investigator based on a protocol to receive specific interventions. Subjects may receive diagnostic, therapeutic or other types of interventions. The assignment of the intervention may or may not be random. The individuals are then followed and biomedical and/or health outcomes are assessed. Select one ☐Treatment ☐Prevention ☐Diagnostic ☐Supportive Care ☐Screening ☐Health Services Research ☐Basic Science ☐Other: Select one ☐N/A ☐Phase 0 ☐Phase 1 ☐Phase 1/Phase 2 ☐Phase 2 ☐Phase 2/ Phase 3 ☐Phase 3 ☐Phase 4 Observational Time Perspective Biomedical and/or health outcomes are assessed in predefined groups of individuals. Subjects in the study may receive diagnostic, therapeutic, or other interventions, but the investigator does not assign specific interventions to the subjects of the study. Select all that apply ☐Prospective ☐Retrospective ☐Cross-sectional ☐Other: Repository Submit Form C-1- Repository Details A repository or data registry is created to store specimens and/or data (i.e., materials) for use in future research. Also referred to as data or specimen warehouse, or bio-banks. NOTE: Only choose this option if the repository is NOT part of another local protocol (substudy) Coordinating Center Submit Form Y – Coordinating Center Details A coordinating center is created to facilitate collaborative clinical research by addressing the coordination and integration of all essential administrative and research data and activities conducted at multiple individual sites. The coordinating center has a principal investigator who provides scientific leadership to the individual sites. The PI may be a sponsor investigator and holder of an IND or IDE or the awardee of a large multi-site grant. vMay31-2016 6 NOTE: Only choose this option if the coordinating center is NOT part of another local protocol Item 55 Current Practice ☐ N/A – this study does not test or evaluate an intervention (Step 2 Institutional – Item 28) - continue to Describe the current local practice so that the IRB can understand how the research interventions compare. -orDescribe current practice including any differences at your study sites: Item 56 Item 56 Will a placebo be used in place of standard therapy? ☐ No ☐ Yes submit Form O-2 – Use of Placebo in Place of Standard Therapy vMay31-2016 7 Item 57 Safety precautions for greater than minimal risk intervention(s) being tested List the greater then minimal risk study intervention(s) being tested To add rows use copy & paste ☐ N/A - this study does not test or evaluate a greater than minimal risk intervention - - continue to Item 58 Describe the procedures for protecting against or minimizing any potential risks Where appropriate, discuss provisions for ensuring necessary medical or professional intervention or equipment provided in the event of adverse events, or unanticipated problems involving subjects. If the safeguards will be different between/among groups, describe here Item 58 List the potential benefits of the study intervention(s) and/or monitoring procedure(s) likely to contribute to enrolled subjects’ well-being. If there are no benefits to enrolled subjects, state “none”. *All risks and discomforts associated with the intervention(s), study directed drugs, devices, and procedures will be listed in the informed consent document according to probability (likely, less likely or rare) and magnitude (serious or not serious). If you do not have a consent document, you must complete the risk table in Form BC (Protocol Template Form). To add rows use copy & paste Item 59 Describe the potential benefits or importance of this study to others not enrolled in this study (i.e. what is the clinical relevance of this study): Item 60 You must provide a detailed protocol with your IRB application. Indicate which of these documents you are including ☐ Sponsor Study Protocol ☐ Protocol Template Form vMay31-2016 8