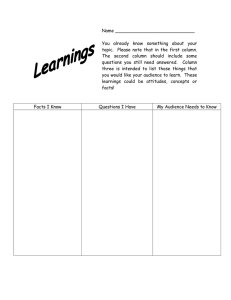
i MIXED-MODE SILICA MONOLITH AS AN EFFICIENT SEPARATION MEDIUM FOR CAPILLARY ELECTROCHROMATOGRAPHY
advertisement

i MIXED-MODE SILICA MONOLITH AS AN EFFICIENT SEPARATION MEDIUM FOR CAPILLARY ELECTROCHROMATOGRAPHY ALIA SOFIE BINTI JEMALE Universiti Teknologi Malaysia v MIXED-MODE SILICA MONOLITH AS AN EFFICIENT SEPARATION MEDIUM FOR CAPILLARY ELECTROCHROMATOGRAPHY ALIA SOFIE BINTI JEMALE A thesis submitted in fulfillment of the requirement for the award of the degree of Master of Science (Chemistry) Faculty of Science Universiti Teknologi Malaysia AUGUST 2010 iii To my beloved mother, Hajjah Jamelah, and father, Haji Jemale. and To dearest husband, Nor Azlan Mohd Aris iv ACKNOWLEDGEMENT First and foremost, I would like to express my countless gratitude to my project supervisor, Assoc. Prof. Dr. Jafariah Jaafar who made a multitude of useful comments and perceptive suggestions for this project. Her encouragement, patience and willingness to motivate me also contribute tremendously in the completion of this thesis This project would not have been accomplished without the unconditional love and support of my family members. My highest appreciation goes to my beloved parents, Hajjah Jamelah and Haji Jemale. Their understanding and prayer make the flow of this thesis a lot easier and enjoyable. An honorable mention also goes to my dearest husband, Nor Azlan for the support and understanding throughout the completion of this project. Special thanks to the Department of Chemistry and Faculty of Science for providing me with a good environment and project facilities. Also, I am thankful to all the lab assistants in the Department of Chemistry for being co-operative and helpful in assisting me on technical matters. Finally, I would like to thank all my friends, especially Nurul Asyikin, Yuhanees and Noor Fairuzah, as well as other individuals who are not listed here for giving me endless support and valuable insight. v ABSTRACT Silica monolith column having mixed-mode i.e., hydrophobic (C18) and anion exchange interaction was successfully synthesized by on-column modification of the hybrid tetramethoxysilanes (TMOS) and methyltrimethoxysilanes (MTMS) using octadecyldimethyl-(N-N-diethylamino)silane (ODS) and ion exchange monomer of N-[3-(dimethylamino)propyl]acrylamide (DMAPAA-Q) methyl chloride-quaternary salt, for the surface modification. The preparation of the ODS/DMAPAA-Q silica monolith column was carried out in a fused silica capillary column of 75 µm internal diameter. At each modification stage, the ODS/DMAPAA-Q silica monolith column was chromatographically characterized with micro liquid chromatography (µLC) and produced separation efficiencies of up to 43000 plates/min at the optimum velocity using the test mixture of alkylbenzenes, and 41800 plates/min for selected nucleotides. The ODS/DMAPAA-Q silica monolith column was also characterized physically with scanning electron microscope (SEM), and showed to have an average through-pore size and skeleton size of 2.2 µm and 2.0 µm, respectively. The ODS/DMAPAA-Q silica monolith column was then characterized in capillary electrochromatography (CEC) for the separation of inorganic and organic ions using a 50 mM phosphate pH 6.9 background electrolyte. The inorganic anions and organic ions were separated within 10 mins with column efficiency up to 114900 plates/min. The results suggest that modifying a silica monolith capillary column through in-situ polymerization of a monomer carrying a functional group can yield high efficiency columns for the ion-exchange-mode separation as well as for reversed-phased separations. The analysis of the porous silica monolith columns with CEC yields higher performance than CZE analysis for the separation of anions. vi ABSTRAK Turus silika monolit yang mempunyai mod saling tindakan campuran iaitu hidrofobik (C18) dan penukar anion telah disintesis dengan jayanya melalui pengubahsuaian langsung ke atas permukaan silika di dalam turus hibrid tetrametoksisilana (TMOS) dan metiltrimetoksisilana (MTMS) menggunakan oktadekilmetil-(N-N-dietilamino)silana (ODS) dan monomer penukar ion iaitu garam metil klorida kuartenar N-3[-(dimetilamino)propil]akrilamida (DMAPAA-Q). Penyediaan turus silika monolit dilakukan di dalam turus rerambut silika terlakur 75 µm diameter dalaman. Di setiap peringkat pengubahsuaian, turus silika monolit ODS/DMAPAA-Q dicirikan secara kromatografi dengan kromatografi cecair mikro, (µLC) dan menghasilkan kecekapan yang tinggi sehingga 43000 plat/min pada kelajuan optimum menggunakan campuran uji alkilbenzena, dan 41800 plat/min untuk nukleotida terpilih. Turus silika monolit ODS/DMAPAA-Q juga dicirikan secara fizikal dengan mikroskop pengimbas elektron (SEM), dan menunjukkan purata saiz lubang terus dan saiz partikel masing-masing adalah 2.2 µm dan 2.0 µm. Turus silika monolit ODS/DMAPAA-Q kemudiannya dicirikan secara kapilari elektrokromatografi, (CEC) untuk pemisahan ion tak organik dan organik menggunakan 50 mM fosfat pH 6.9 sebagai elektrolit latarbelakang. Ion tak organik dan organik telah dipisahkan dalam masa 10 min dengan kecekapan turus sehingga 114900 plat/min. Keputusan ini menunjukkan pengubahsuaian turus silika monolit secara pempolimeran in-situ dengan monomer pembawa kumpulan berfungsi boleh menghasilkan turus berprestasi tinggi bagi pemisahan dalam mod penukar ion dan juga pemisahan fasa terbalik. Analisis dengan teknik CEC juga menghasilkan prestasi yang lebih tinggi berbanding analisis CZE bagi pemisahan anion. vii TABLE OF CONTENTS CHAPTER 1 2 TITLE PAGE DECLARATION ii DEDICATION iii ACKNOWLEDGEMENTS iv ABSTRACT v ABSTRAK vi TABLE OF CONTENTS vii LIST OF TABLES x LIST OF FIGURES xii LIST OF ABBREVIATIONS xiv LIST OF SYMBOLS xvi INTRODUCTION 1 1.1 General Introduction 1 1.2 Problem Statement 4 1.3 Significance of Study 4 1.4 Objectives of Research 5 1.5 Scope of Research 5 LITERATURE REVIEW 7 2.1 Research Background 7 2.2 CEC 8 2.3 Electroosmosis 12 2.4 Separation in CEC 14 2.5 Column Technology in CEC 16 viii 2.6 Monolith Column 18 2.7 Monolith Column in CEC 19 2.7.1 Polymer-Based Monolith in CEC 23 2.7.2 Silica-Based Monolith in CEC 24 Mixed-Mode Silica Monolith as an Electrochromatographic Separation Medium Determination of Corrected Retention Factor 26 2.8 2.9 3 28 EXPERIMENTAL 32 3.1 Chemicals and Reagents 32 3.2 Synthesis of the ODS/DMAPA-Q Silica Monolith Capillary Column Instrumentation 33 35 3.3.1 CEC and CZE 35 3.3.2 µLC 36 3.3.3 SEM 36 3.3 3.4 3.5 Preparation of Chemicals 37 3.4.1 Buffer 37 3.4.2 Stock Solution 37 Experimental Procedures 37 3.5.1 CEC Procedures 37 3.5.2 CZE Procedures 38 3.5.3 µLC Procedures 38 3.6 Qualitative Analysis 39 3.7 Data Analysis 39 3.7.1 Mobility of Analyte 39 3.7.2 Separation Efficiency 40 3.7.3 Corrected Retention Factor 41 ix 4 RESULTS AND DISCUSSION 4.1 4.2 4.3 4.4 4.5 5 Synthesis of the ODS/DMAPAAQ Silica Monolith Capillary Column SEM Physical Characterization of ODS/DMAPAAQ Silica Monolith Capillary Column µLC Chromatographic Characterization of ODS/DMAPAAQ Silica Monolith Capillary Column ODS/DMAPAA-Q Silica Monolith Capillary Column as a Separation Medium in Capillary Electrochromatograph 4.4.1 Anions Separation on ODS/DMAPAA-Q Silica Monolith Capillary Column 4.4.2 Determination of Corrected Retention Factor of the Anions on ODS/DMAPAA-Q Silica Monolith Capillary Column 4.2.3 Stability of the ODS.DMAPAA-Q Silica Monolith Capillary Column Comparison of Anion Separation with CZE 4.5.1 Reproducibility of Anion Separation with CZE 43 43 45 46 54 54 56 59 60 62 CONCLUSION AND SUGGESTION 64 5.1 Conclusion 64 5.2 Suggestion 67 REFERENCES 69 x LIST OF TABLES TABLE TITLE PAGE NO. 2.1 Comparison of electrodriven separation method 9 2.2 Landmarks in CEC 11 2.3 Features, roles and limitation of monolithic column 19 4.1 Pore properties of ODS/DMAPAA-Q silica monolith capillary column 46 Theoretical plates, Neff, plate height, H, retention factor, k and separation factor, of the ODS silica monolith column for alkylbenzenes separation 49 Theoretical plates, Neff, plate height, H, retention factor, k and separation factor, of the ODS/DMAPAA-Q silica monolith capillary column for alkylbenzenes separation 51 Theoretical plates, Neff, plate height, H, retention factor, k and separation factor, of the ODS/DMAPAA-Q silica monolith capillary column for nucleotides separation 53 The applied mobility, µapp, theoretical plate, Neff and plate height, H for the separation of ions and anions on the ODS/DMAPAA-Q silica monolith capillary column Data calculated from CZE experiment 55 57 Data calculated from CEC experiment with the ODS/DMAPAA-Q silica monolith capillary column 57 Retention factor, k, of inorganic anions separated on DMAPAA-Q silica monolith capillary column with µLC and corrected retention factor, kCEC,, on DMAPAA-Q silica monolith capillary column with CEC (Jaafar et al., 2008) 58 Repeatibility of the ODS/DMAPAA-Q silica monolith capillary column for short-term stability 59 4.2 4.3 4.4 4.5 4.6 4.7 4.8 4.9 xi 4.10 Reproducibility of ODS/DMAPAA-Q silica monolith capillary column for long-term stability 60 4.11 Efficiency of anion separation on CZE 62 4.12 Repeatibility of anions separation with CZE for short-term stability 63 Reproducibility of anions separation with CZE for long-term stability 63 4.13 xii LIST OF FIGURES FIGURE NO. 2.1 2.2 2.3 2.4 2.5 2.6 TITLE Flow profiles in (A) Pressure driven; (B) electroosmotic driven (A) Representation of the surface of fused silica tubing. (B) Formation of an electrical double layer near the surface of fused silica tubing Differential of a solute migration in a column under voltage gradient PAGE 10 12 14 CEC of neutral and charged solute. Teof denotes retention of EOF marker 15 Schematic preparation of direct synthesis for CEC monolith capillary 21 Schematic preparation of post modification synthesis for CEC monolith capillary 22 2.7 A generalized reaction scheme for a sol-gel reaction 3.1 Monomers charged onto the MAS bonded column: A) octadecyldimethyl-(N-N-diethylamino)silane, ODS B) N-[3-(dimethylamino)propyl]acrylamide methyl chloride-quaternary salt, DMAPAA-Q 34 3.2 CEC / CZE schematic diagram 35 4.1Proposed st Proposed structure of the ODS/DMAPAA-Q silica monolith packing 44 SEM micrograph of 200 µm I.D. ODS/DMAPAAQ silica monolith capillary column. A: 850x magnification B: 3500x magnification 45 Chromatogram of uracil at MAS modification stage. Mobile phase: 80% aqueous methanol. Column temperature: 23˚C. Pressure: 52 bar. Linear velocity: 1.0 mm/s 47 4.2 4.3 25 xiii 4.4 4.5 4.6 4.7 4.8 Chromatogram for mixtures of uracil and alkylbenzenes, C6H5(CH2)nH, (n=0-6) at ODS modified stage. Mobile phase: 80% aqueous methanol. Column temperature: 23˚C. Pressure: 52 bar. Linear velocity: 1.0 mm/s 48 Chromatogram for mixtures of uracil and alkylbenzenes, C6H5(CH2)nH, (n=0-6) at ODS/DMAPAA-Q modified stage. Mobile phase: 80% aqueous methanol. Column temperature: 23˚C. Pressure: 52 bar. Linear velocity: 1.0 mm/s 50 Chromatogram obtained for mixtures of nucleotides at ODS/DMAPAA-Q modified stage: Mobile phase 50 mM phosphate (pH 2.8). Column temperature: 25˚C. Pressure: 34.3 bar. Linear velocity: 1.0 mm/s 52 Electropherogram obtained for mixtures of four inorganic anions and one organic acid. Mobile phase: 50 mM phosphate pH 6.9. Column temperature 25°C. Applied voltage: -15 kV. Separation voltage: -3 kV for 3 secs. 54 Electropherogram obtained for mixtures of four inorganic anions and one organic acid. Mobile phase: 50 mM phosphate pH 6.9. Column temperature 25°C. Applied voltage: -15 kV. Separation voltage: -3 kV for 3 secs. 61 xiv LIST OF ABBREVIATION AX - Anion exchange APS - Ammonium persulfate CE - Capillary Electrophoresis CZE - Capillary Zone Electrophoresis CEC - Capillary Electrochromatography CX - Cation exchange DMAPAA-Q - N-3[-(dimethylamino)propyl]acrylamidemethyl] EOF - Electroosmotic flow EDMA - Ethylene dimethacrylate HPLC - High Performance Liquid Chromatography MAS - 3-methacrylamidopropyltriethoxysilane MEAMS - 2-(methacryloyloxy)ethyltrimethylammonium methyl sulfate MEKC - Micellar Electrokinetic Chromatography MTMS - Methyltrimethoxysilanes ODS - Octadecyldimethyl-(N-N-diethylamino)silane OT - Open tubular PEG - Poly(ethylene glycol) RP - Reversed phased RP/AX - Reversed phased / anion exchange SAX - Strong anion exchange SCX - Strong cation exchange SEM - Scanning Electron Microscope SEMA - 2-(sulfooxy)ethyl methacrylate SWNT - Single-wall carbon nanotubes TEOS - Tetraethoxysilane TCM - Traditional Chinese medicine xv TMOS - Tetramethoxysilanes VBC - Vinylbenzyl chloride µLC - Micro Liquid Chromatography xvi LIST OF SYMBOLS eo Electroosmotic mobility ζ, Zeta potential r Permittivity of the mobile phase Viscosity of the mobile phase E Electric field strength dp Diameter of the stationary phase particle L Column length v Pressure-driven flow velocity Porosity p Pressure drop kc Chromatographic retention factor kapp Apparent retention factor t0 Hold up time tr Retention time t’0 Corrected hold up time µep Electrophoretic mobility Leff Effective length of capillary Ltot Total length of capillary V Applied voltage tM Migration time w1/2 Width of peak at half height Veo-0 Electroosmotic velocity Vep Electrophoretic velocity Veff Effective velocity Veff-CZE Effective velocity in CZE xvii ECZE Energy in CZE Vep-CZE Electrophoretic velocity in CZE Veo Eletcroosmotic velocity in CZE Neff Theoretical plates H Plate height k Retention factor Separation factor CHAPTER 1 INTRODUCTION 1.1 General Introduction Being the youngest member in the family of separation technique, capillary electrochromatography (CEC), is a micro column separation technique that combines the features of both capillary zone electrophoresis (CZE) and micro liquid chromatography (µLC) (Bartle and Mayers, 2001, Wu et al., 2008). In CEC, the electroosmotic flow (EOF) generated on the surface of the stationary phase, is used to drive the mobile phase instead of hydrodynamic flow. Due to the plug-flow profile of the EOF, CEC exhibits separation of higher efficiency than in the pressuredriven µLC. Separations of ionic solutes in CEC are commonly achieved with ionexchange column packed with particles (Gu et al., 2006; Norton and Shamsi, 2008, Tanaka and Kobayashi, 2003). However, there are several limitations that need to be solved on packed-CEC. One of the limitations of the conventional packed column is the necessity to fabricate frits, which is required for retention of the packed particles within the column. In addition, packed column also have the tendency to form bubbles around the packing materials or at the frits. Such problems often results in an unstable baseline, non-reproducible migration time and even, current breakdown. Moreover, in packed-CEC, the packing procedure is often more difficult compared to the normal High Performance Liquid Chromatography (HPLC) column due to the narrow inner diameter of the capillary (Bartle and Mayers; 2001). It is also observed that the solute band in this type of chromatographic column is broadened due to the 2 presence of multiple flow paths of different length and velocity, and slow equilibration of the solute between the mobile phase and the stationary phase, especially in nm-sized pores of µm-sized particles (Guiochon, 2007, Tanaka and Kobayashi, 2003). As to obtain separation with high efficiency, the use of small sized particles helps to reduce the contribution of the above mentioned factors, therefore enables faster equilibration and narrower bands to be achievable (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008, Klampfl et al., 2000, Lin et al.,2006; Minakuchi et al., 1996; Motokawa et al., 2002; Tanaka and Kobayashi, 2003; Tang et al., 2000). The introduction of monolith stationary phases in the field of column technology is due to the problematic issues associated with packed column. The major chromatographic features of these monolith columns are providing high permeability based on its large through-pores, as well as high efficient separation with large number of theoretical plates per unit pressure drop based on small-sized skeleton (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008, Klampfl et al., 2000, Lin et al.,2006; Minakuchi et al., 1996; Motokawa et al., 2002; Pesek et al., 2008; Roux et al., 2008; Tanaka and Kobayashi, 2003; Tang et al., 2000). Guichon (2007) defined monolith column as made up of one piece of continuous and porous material that is sealed against the wall of the tube. Due to this characteristic, the mobile phase will tends to percolate through it, avoiding bypassing any significant length of the bed. Other chromatographic advantages offer by this monolith material also include low backpressure and fast analytes mass transfer. In the aspect of monolith column preparation, the simplified preparation procedures which can be directly performed in-situ within capillaries has brought the monolith material to be gradually fabricated for application in microscale chromatographic separation such as in the pressure-driven µLC (Jaafar et al., 2008; Pesek et al., 2008; Roux et al., 2008; Wu et al., 2008) and the electro-driven CEC (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008; Klampfl et al., 2000; Lin et al.,2006; Wu et al., 2008). Technically, the preparation of monolith columns in electro-driven microscale capillaries is similar to that in pressure-driven µLC, except for the consideration of the generation of EOF for driving the mobile phase, which requires the incorporation of charged moieties onto the surface of the monolith. 3 Monolith stationary phase can be divided into two main groups which are silica-based monolith (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008, Klampfl et al., 2000; Lin et al.,2006; Minakuchi et al., 1996; Motokawa et al., 2002) and organic polymer-based monolith (Uysal et al., 2009, Wang et al., 2009). Organic polymer-based monoliths are often prepared via a onestep polymerization of an organic monomer in the presence of a cross-linker, initiator and porogen. The results can be classified as either rigid or soft monoliths. As for silica-based monolith, it can be prepared by a sol-gel process whereby, alkoxysilanes will undergo a hydrolysis and polycondensation reaction catalyzed by acetic acid in the presence of porogens. The macroporous structure of the silica monolith prepared by sol-gel process is controlled by the composition of the starting materials. Therefore the size of silica skeletons and through-pore can be varied independently. Silica-based monolith column usually provides greater mechanical stability compared to the organic polymer-based monolith as the material show less swelling in solvents. One drawback of silica is its degradation above pH 7.5, whereas polymer-based stationary phase can withstand higher pH values. However, to circumvent this problem, weak acid eluents can be used instead of highly-alkaline eluents. The improvement of selectivity in CEC is powered by the utilization of various packing materials as its stationary phase. Besides commercially available capillaries, novel stationary phase are also synthesized to provide sites of required interaction. Not only to widen the range of selectivity, novel aspect of the material is their ability to promote strong and constant EOF. Since stationary phase is the heart of separation technique, improvement of column technology is foremost important for the continued growth in CEC. Here, we prepared a mixed-mode silica monolith column synthesized by modifying the hybrid silica monolith with C18 (ODS) and quaternary ammonium functional group (DMAPAA-Q), producing a mixed-mode of reversed-phase (RP) and a strong anion exchange stationary (SAX) phase for CEC. As a mixed-mode type of column, two different interactions of analytes towards the stationary phase is offered, thus enabling hydrophobic and positively charge compounds to be 4 separated. Physical characterization of the column exhibits skeleton and throughpore size within the range of that reported by Nakanishi et al. (1996), and it is expected that this column will produce good or similar performance in terms of its separation efficiency compared to other silica monolith column reported previously. A comparison study of the anions separation was also carried out on the CZE mode with a fused silica capillary. 1.2 Problem Statement Reversed phase column with ODS functionality is widely applied as the stationary phase in chromatography due to its proven capability of separating neutral and hydrophobic analytes. Recently, strong anion exchange (SAX) column of DMAPAA-Q moiety has been proven successful in separating nucleotides and inorganic ions (Jaafar et al., 2008). Incorporating the SAX functionality to an ODS column could afford a dual separation mode which can be applied to a wider range of analytes of organic species having hydrophobic properties; anion species, organic and inorganic; and hydrophilic substances, particularly amines. Better column performance could be expected from this mixed-mode column due to the additional interaction yield from both the moieties. 1.3 Significance of Study In obtaining high efficient separation, silica monolith material is an ideal chromatographic separation medium to be applied in CEC, as the use of monolith helps to reduce the contribution of the band-broadening factors, therefore enables faster equilibration and narrower bands to be achievable. It is an added value in this research as not only physical characterization is carried out at the final stage, chromatographic characterization of the ODS/DMAPAA-Q silica monolith column at each modification stage is also conducted using µLC at certain chromatographic conditions. Analytes separation on normal CZE is also conducted as to distinguish 5 the separation influences on CEC that generate higher efficiency compared to the conventional CZE method. 1.4 Objectives of Research The objectives of this research are to: (i) synthesize a silica monolith column having mixed-mode interaction of reversed-phased and ion-exchange using ODS and DMAPAA-Q monomer. (ii) conduct a chromatographic evaluation using µLC at each modification stage of silica monolith column. Those stages consist of (i) MAS-modified stage, (ii) ODS modified stage and (iii) ODS/DMAPAA-Q modified stage. (iii) conduct a physical characterization on the ODS/DMAPAA-Q silica monolith capillary using Scanning Electron Microscope (SEM) with the aid of Image Pro-Plus Analyzer. (iv) evaluate the performance of the ODS/DMAPAA-Q silica monolith column as an electrochromatographic separation medium in CEC using selected anionic compounds. (v) compare the efficiency and separation mode between the normal CZE and CEC for the separation of selected inorganic anions and ionic compounds. 1.5 Scope of Research This research is carried out to explore the capability of mixed-mode monolithic silica as an electrochromatographic separation medium on selected anionic compounds (bromide, bromated, iodate, nitrate and benzoic acid) with CEC. 6 The ODS/DMAPAA-Q silica monolith is synthesized with the collaboration of Tanaka’s Laboratory, Kyoto Institute of Technology, Japan. The ODS/DMAPAA-Q silica monolith prepared in the fused-silica capillary of 75 m I.D. from the mixture of TMOS and MTMS first evaluated under µLC mode with specific chromatographic condition proposed by Motokawa et al. (2002). Evaluation based on column separation efficiency, are at the unmodified stage, modified ODS stage and lastly at the modification stage with ODS/DMAPAA-Q. The synthesized capillary is then tested for the separation of some common nucleotides. Since the mixed-mode monolithic silica is obtained in the form of capillary rod, physical characterization is limited to using SEM. The morphology study is conducted with the aid of Image Pro-Plus analyzer under cross-sectional view obtained from SEM. The ODS/DMAPAA-Q silica monolith capillary with the mixed-mode functionalities is then applied as an electrochromatographic separation medium in CEC. The column is tested for the separation of selected anionic compounds and its performance is evaluated based on the calculation of theoretical plates, N and plate height, H. In this part of research, an optimum separation conditions are applied, adopted from the previous study (Jaafar et al., 2008). Lastly, a comparison study of the ion analysis with the normal CZE mode is conducted and results obtained are discussed based on its separation elution and efficiency. 7 CHAPTER 2 LITERATURE REVIEW 2.1 Research Background Taking the advantage offered by CEC, this study is focused on the ability of silica monolith material with dual functionality for the separation of anionic compounds. Monolithic column when coupled with CEC exhibit sharp, narrow and compact band of analyte due to the plug flow profile generated across the column (Bartle and Mayers, 2001; Wu et al., 2008). The characteristic of the monolith material itself that consist of small skeleton and large through-pore size also contributed to the high efficiency separation, producing the lowest plate height (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008, Klampfl et al., 2000, Lin et al.,2006; Minakuchi et al., 1996; Motokawa et al., 2002; Pesek et al., 2008; Roux et al., 2008; Tanaka and Kobayashi, 2003; Tang et al., 2000). The chromatographic capillary investigated in this work is a silica-based monolith created with sol-gel method patented by Nakanishi and Soga (1997). This method is developed over the course of five years documented in a series of papers published by two groups at Kyoto University and Kyoto Institute of Technology, Japan. A key feature of this synthesis is product riddled with pores in two size ranges: tiny ones that provide high surface area and access to the silica surfaces on which separations occur and the large ones that facilitate easy flow of material through the monolith. 8 The simplicity of derivatization process of the functional group on monolithic silica has captivated researchers’ attention to synthesize column of mixed-mode characteristic which is normally a combination of two different functionalities. The mixed-mode column of reversed-phased/anion exchange (RP/AX) (Ding et al., 2006; Fu et al., 2004; Klampfl et al., 2000) were synthesized and its column performance was tested for numerous anionic compounds, as well as inorganic anions. This type of mixed-mode monolithic silica column, besides widening the range of analytes selectivity, also promotes strong and constant EOF. In CEC of the mixed-mode type of column, the elution trend of the analytes could be under the influence of either electrophoresis or chromatographic mechanisms. This phenomenon is clarified through the migration of EOF marker (Bartle and Mayers, 2001; Ding et al., 2006; Fu et al., 2004; Klampfl et al., 2000). In this research, a hybrid-typed monolithic silica column is prepared from the mixture of TMOS and MTMS before surface modification with ODS and DMAPAA-Q monomer to exhibit mixed-mode characteristic of reversed-phased and strong anion exchange functionalities. The synthesized ODS/DMAPAA-Q monolithic silica column is then applied as the electrochromatographic separation medium on CEC, an intermediate technique bridging the gap between µLC and CZE, for the separation of anionic analytes. 2.2 CEC Separation techniques such as CE and HPLC are particularly useful in the analysis of complex mixtures because of their high resolving power. Continued refinement of these techniques has led to a novel separation method known as CEC, also referred to as capillary electrokinetic chromatography, which is a combination of HPLC and conventional CZE (Bartle and Mayers, 2001). As in CZE and micellar electrokinetic chromatography (MEKC), small diameter of typically 50-100 µm capillary column with favourable surface area-to- 9 volume ratio are employed to minimize thermal gradients from ohmic heating, which can have an adverse effect on band widths. CEC differs crucially from CE and MEKC. However, in terms of separation principle, CEC, CZE and MEKC are identical as these separation techniques are based on partition between the liquid and solid phases. In summary, the comparison of these three electro-driven separation methods are tabulated in Table 2.1 (Bartle and Mayers, 2001). Table 2.1: Comparison of electrodriven separation method CE MEKC CEC Separation principle Different mobilities of ions in electric field Partition between bulk solution and micelle moving in opposite direction of analyte Partition between solid stationary phase and mobile phase Typical column diameter (µm) 50-100 50-100 50-100 Stationary phase None None Particle packed, open-tubular, or monolithic Mobile phase Sample type Electrolyte solution Electrolyte solution Electrolyte solution Charged species Neutral and charged species Neutral and charged species In CEC, the flow of mobile phase is driven through the column by an electric field, a phenomenon known as electroosmosis, rather than by applied pressure in normal HPLC. This electroosmotic flow (EOF) is generated by applying a large voltage across the column. The positive ions of the added electrolyte will accumulate in the electrical double layer of the column packing, move towards the cathode, and drag the liquid mobile phase with them (Bartle and Mayers, 2001). This resulted in a plug flow profile characteristic of the EOF, in comparison to the parabolic flow profile in HPLC. Figure 2.1 illustrates the differences in flow profile of CEC and HPLC. 10 Figure 2.1: Flow profiles in (A) Pressure driven; (B) Electroosmotic driven Avoiding the use of pressure results in a number of important advantages for CEC over HPLC (Bartle and Mayers, 2001). Firstly, in the pressure-driven separation mode, the flow rate through the packing inside the column depends directly on the particle diameter and inversely on the column length. For such case, the particle diameters of the column packing are seldom less than 3 µm, with column length restricted to approximately 25 cm long. By contrast, in CEC, the electrically driven flow rate is independent of particle diameter and column length, therefore in principle, smaller particles and longer columns can be used and thus, higher efficiency is generated in CEC than that offered in HPLC. A second consequence of employing the electrodriven separation mode is that increase in column efficiency is expected since the plug-like flow velocity profile in the EOF reduces dispersion of the solute band as it passes through the column. Thus, these combined effects of reduced particle diameter, increased in column length and the plug flow characteristic leads to CEC separation of higher efficiency and substantially improved resolution. Strain (1939) first reported the use of EOF in chromatography. He recognized the difference between electrophoresis and electrochromatography on the one hand, 11 and partitioning of analytes between the mobile and stationary phase on the other. The importance of the EOF in electrochromatography was then recognized, since the aid of electrophoretic mobility and electroosmosis in this electrodriven separation mode enabled the movement of analytes through the separation medium. The pioneer of CEC was however by the Pretorius group (1974). They reported that the EOF used to drive the mobile phase, that generated a plug flow profile in contrast to the hydrodynamic flow in HPLC, yield separation with the lowest plate height. They also pointed out the absence of pressure drop across the column if the EOF was used. Significant progress in CEC began in 1980 when Joergenson and Lukacs (1981) demonstrated the use of electroosmosis in capillaries and showed the possibilities for low reduced plate heights. Tsuda (1987) then showed that CEC was possible in coated open tubular (OT-CEC) and recognized the factors that control the EOF as well as the importance of practical effects, such as bubble formation in packed columns. The detail theoretical analysis of CEC and its technique development was published by Knox and Grant (1987), followed by practical demonstrations by the same group in 1991. The important observation made was that column driven electrically showed higher efficiencies than the same column with pressure drive. Some landmarks in the history of CEC are listed in Table 2.2. Table 2.2: Landmarks in CEC Landmarks in CEC Year Reference First report of use of EOF in chromatography 1939 Strain, 1939 Use of EOF in column chromatography 1974 Pretorius et al., 1974 Electroosmosis in capillaries 1981 Jorgenson and Lukacs, 1981 CEC in open tubular column 1987 Tsuda, 1987 Theory of CEC and technique development 1987, 1991 Knox and Grant, 1987; Knox and Grant, 1991 12 Since the re-emergence of CEC, there has been a very rapid increase in the number of publications and reviews relating to CEC. To this date, due to the attractive potential of CEC, it is widely applied as the separations technique for the analysis of drugs (Aturki et al., 2009), food (Uysal et al., 2009) and pharmaceutical analysis (Adu et al., 2008), enantiomeric separation (Fanali et al., 2008), as well as for the determination of organic acids (Klampfl, 2007) and inorganic metals (Breadmore et al., 2001; Kuban et al., 2008; Wang et al., 2009). 2.3 Electroosmosis Electroosmosis is described as the movement of liquid relative to a stationary charged surface under an applied field (Bartle and Mayers, 2001). Substances tend to acquire a surface charge as a result of ionization of the surface and/or by the interaction of ionic species. In a fused silica capillary, the ionization of silanol groups give rise to a negatively charged surface, which affects the distribution of nearby ions in solution. Ions of opposite charge (counter-ions) are attracted to the surface to maintain the charged balance whilst ions of like charge (co-ions) are repelled. The resultant is the formation of electrical double layer, illustrated in Figure 2.2. Figure 2.2: (A) Representation of the surface of fused silica tubing. (B) Formation of an electrical double layer near the surface of fused silica tubing. 13 The counter-ions are arranged in two layers, which are the fixed and diffuse layer, with a surface of shear beyond the interface. The voltage drop between the wall and the surface of shear is known as the zeta potential, ζ. In the diffuse layer, the potential falls exponentially to zero, and the distance over which it falls is known as the double layer thickness, . When voltage is applied, the solvated cations in the diffuse layer migrate towards the cathode, dragging the solvent molecules along with them. The linear velocity of the EOF, eo , is described by Smoluchowski (Knox, 1988) in Equation 2.1. This shows how the EOF is governed by ζ, the permittivity and viscosity of the mobile phase, r and respectively, and the electric field strength, E. eo 0 r E (2.1) The flow profile in CEC is assumed to be near-plug-like since it originates from the capillary wall, but in reality it depends on the capillary internal diameter, d and . The use of microscope optic to image flow profiles in narrow capillaries has produced conflicting results. While the plug flow profile predicted from theory has been observed by Taylor and Yeung (1993), Tsuda et al. (1993) found a higher EOF at the capillary wall than at the centre. Therefore, the importance of the EOF profile in CEC will necessitates further research. From Equation 2.1, it can be seen that neither the diameter of the stationary phase particle (dp) nor the column length (L) affect the mobile phase velocity. This is in contrast with pressure-driven flow velocity, v , which was described by KonzenyCarman in Equation 2.2, where is the porosity and p is the pressure drop. 2 d 2 p p v 180(1 ) 2 L _ (2.2) 14 In CEC, both the capillary wall and the column packing carry surface charges that are capable of supporting the EOF. To date, most of the separation analysis carried out on CEC (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008; Klampfl et al., 2000; Lin et al.,2006; Motokawa et al., 2002; Pesek et al., 2008; Roux et al., 2008; Uysal et al., 2009; Wang et al., 2009) suggests that the column packing is responsible for the generation of EOF. This is because, there is a greater number of free silanol groups presents since the solid packing has a far larger surface area compared to that provide by the internal silica wall. 2.4 Separation in CEC Separation in CZE is a consequence of differential migration of charged and neutral species, as depicted in Figure 2.3. When a negative separation voltage is applied, depending on the charge of analytes, cations will be attracted to the cathode, followed by neutral solute and lastly the anions. Figure 2.3: Differential of a solute migration in a column under voltage gradient CEC may be compared with CZE and classified in the hierarchy of separation methods employing liquids by unified description proposed by Rathore and Horvath (1996). The differential migration of solute bands can be divided into components that are separative (selective interaction with the stationary phase, or differences in electrophoretic migration velocities), and components that are non-separative (migration not contributing to the separation. 15 In CEC of neutral solute (Bartle and Mayers, 2001), depicted as in Figure 2.4 (A), the solute and the mobile phase move in the same direction, and the sample components emerge in the order of retardation by the stationary phase. However, if the solute is charged, there are three operational modes depending on the direction and magnitude of electrophoretic migration with respect to the direction of EOF (Figure 2.4) (Bartle and Mayers, 2001). Figure 2.4: CEC of neutral and charged solute. T eof denotes retention of EOF marker 16 Referring to Figure 2.4 (B), a negative voltage is applied for the separation. The positive charged species emerged before the EOF marker due to the strong attraction at the cathodic end. Therefore, the EOF is of co-directional, whereby the migration velocities of charged species are always greater than that of the EOF marker. Similarly, a negative separation voltage was also applied as in the case of 2.4 (C). However, since the solute exhibit negative charged, it is strongly attracted to the anodic end, resulting it to emerge after the EOF marker. A counter-directional EOF is generated because the EOF velocity is greater than the electrophoretic velocity of a charged component. Unlike in the three other cases, in 2.4 (D) a positive separation voltage was applied. In this case, the EOF marker is not detected. A counterdirectional EOF is generated where the EOF velocity is less than the electrophoretic velocity. The detection of a charged component is only possible with instrument of reversed polarity. 2.5 Column Technology in CEC In the development of CEC, three types of column configuration had been reported (Gu et al., 2007; Tang and Lee, 2000; Yuan et al., 2006). These include packed (Norton and Shamsi, 2008), open-tubular (Zaidi et al., 2009) and monolith stationary phase (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Guiochon, 2007; Jaafar et al., 2008; Klampfl et al., 2000; Lin et al., 2006; Minakuchi et al., Motokawa et al., 2002; 1996; Tanaka and Kobayashi, 2003; Tang et al., 2000). Generally, a packed column is a capillary packed with packing materials that are confined between two end-frits, while an open-tubular column is a capillary bonded with a wall-supported stationary phase that can be a coated polymer, bonded molecule monolayer, or a synthesized porous layer network. As for the monolith stationary phase, it is usually made up of a porous continuous bed that is formed in situ inside the capillary. Normally in CEC, it uses fused silica capillary with inner diameter measured from 50 m to 100 m is used (Bartle and Mayers, 2001; Tang and Lee, 2000). Small diameter capillaries are employed as to minimize thermal gradient from ohmic 17 heating, which will cause an adverse effect on band widths. Fused silica capillary is the preferred choice for CEC due to its superior properties such as high electrical and pressure resistance, good UV transparency, high thermal conductivity, good chemical inertness, good flexibility as well as high mechanical strength. In the early beginning of CEC, the capillary is packed with typical HPLC stationary phase such as octadecyl silica (ODS) (Gu et al., 2007; Norton and Shamsi, 2008; Tang and Lee, 2000; Yuan et al., 2006). However, there are several problems that need to be solved on packed-CEC. One of the limitations of the conventional CEC is the necessity to fabricate frits, which are required for retention of the packed particles within the column. In addition, packed capillaries also have the tendency to form bubbles around the packing materials or at the frits. Such problems often results in an unstable baseline, non-reproducible migration time and even, current breakdown. In packed-CEC, the packing procedures are more difficult compared to HPLC due to the narrow inner diameter of the capillary. Since problems encountered using packed-CEC, as an alternative approach, is the introduction of open-tubular capillary electrochromatography (OT-CEC) (Gu et al., 2007; Tang and Lee, 2000; Yuan et al., 2006; Zaidi et al., 2009). In OT-CEC, the stationary phase is coated to the capillary wall. This helps to increase the separation efficiency than in packed-CEC as the eddy diffusion contribution is eliminated. In OT-CEC, inner diameter of capillary smaller than 25 m is normally being used. Still, there are some drawbacks of such alternative as the loadability is very low, and lower retention factors are obtained than on packed-CEC. Interest regarding monolith porous materials as chromatographic stationary phase in capillary electrochromatography (CEC) is the result of many advantages over packed-CEC and OT-CEC (Gu et al., 2007; Norton and Shamsi, 2008; Tang and Lee, 2000; Yuan et al., 2006; Zaidi et al., 2009). In the past few years, the monolith types of stationary phase, with wall supported porous continuous bed columns have been developed for CEC. The monolith technology is widely made into application as to create separation media that fulfill the requirement needed for specific application. 18 2.6 Monolith Column Separation of solute in liquid chromatography is commonly achieved by using a column packed with particles (Gu et al., 2006; Norton and Shamsi, 2008; Tanaka and Kobayashi, 2003). The particles are to provide large surface areas to adsorb solutes onto the stationary phase from the mobile phase. Chromatography was first carried out in this format at the beginning of 20 th century. Although most analytical columns still consist of a packed bed, monolith materials has been recently proposed as a chromatographic separation medium of higher performance (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Guiochon, 2007; Jaafar et al., 2008; Klampfl et al., 2000; Lin et al., 2006; Minakuchi et al., Motokawa et al., 2002; 1996; Tanaka and Kobayashi, 2003; Tang et al., 2000). Previously, with particles packed stationary phase, the solute band is broadened, either in a pressurized flow of electrodriven flow of solvent by the presence of multiple flow paths of different length and velocity, slow equilibrium of a solute between mobile phase and stationary phase and the diffusion of solute in the mobile phase (Guiochon, 2007; Minakuchi et al., 1996; Tanaka and Kobayashi, 2003). The higher efficiency separation is obtainable with the use of smaller particles sizes, as it reduces the above-mentioned factors. Monolith column can be defined as a column consisting of one piece of solid that possesses interconnected skeletons and interconnected flow paths (throughpores) (Guiochon, 2007; Tanaka and Kobayashi, 2003). A monolith column with small-sized skeletons and large through-pores, having a large ratio (throughpore/skeleton size), commonly 1-4 can simultaneously provide high permeability and high column efficiency. In contrast to the particle-packed column, monolith column provide higher porosities, which is 65-70% for monolith prepared in a mold and higher than 80% for those prepared in a capillary, compared to 40% for a particlepacked column. The features, roles and limitation of monolithic column are summarized as in Table 2.3 (Tanaka and Kobayashi, 2003). 19 Table 2.3: Features, roles and limitation of monolithic column Monolith Column Features 1) Continuous one-piece structure, fritless (mechanical stability) 2) High porosity (high permeability) 3) Small skeleton, large through-pores (high efficiency at high speed) 4) Facile design and surfaces (selectivity) Roles 1) High-efficiency and high speed separations beyond the limit of particle-packed column 2) Stable and high-performance miniaturized columns for LC and chip channels 3) Column for hyphenation with MS 4) Material for sample preparation by SPME and SPE Limitation 1) Limited availability in size (50-100 µm, ca. 4-8 mm diameter) and in chemical structure at present 2) Limited availability of skeleton size and through-pore size 3) Relatively low sample loading capacity (1/2-1/10 of a particlepacked column Monolithic columns offer faster analysis by bypassing the limitations imposed by pressure via through-pores, which allow higher flow rates than particulate columns at reasonable column backpressures just like other continuous media. Monolithic columns consist of a single, rigid or semi-rigid, porous rod. Within the monolithic structure, analyte capacity is usually provided by smaller mesopores. 2.7 Monolith Column in CEC Monolith stationary phases have been widely used in fast efficiency separation system. The preparation and application of monolith capillary in CEC have gained increasing interest due to the distinct features of the porous monolith material, such as the facile preparation, versatile surface chemistry, fritless design, good permeability and fast mass transfer. The preparation of porous monolith capillary is actually similar to that in HPLC. However, charged groups should be 20 incorporated to the surface of the monolith to generate EOF since EOF is the driving force for the mobile phase. In general, the derivatization of the functional groups onto the polymer or silica monolith is fabricated using single step (direct synthesis) or multiple steps (post-modification) procedures (Wu et al., 2008). Technically, the preparation of CEC monolith capillaries is similar to that in µLC. This is demonstrated by Ishizuka and coworkers (1998) for the preparation of ODS modified column, containing a layer of octadecyl ligand on a silica monolith. Although their earlier work was focused on µLC, their preparation process was then adapted by most researchers to synthesize column for the utilization as stationary phase in CEC. Direct synthesis of polymer based and silica based monolith column was practiced by some researchers as this technique offers advantages such as timesaving, simplicity and also produces good reproducibility of column (Ding et al., 2006; Lin et al., 2006). Nevertheless, obtaining the reliable silica monolith support with desired chromatographic properties is not an easy task, due to the complexity of the reaction that consist of a mixture containing monomers, cross-linker and initiator in the presence of porogenic solvent. While this kind of silica monolith still needs to go further investigation as to obtain support with the expected chromatographic properties, based on the original method introduced by Minakuchi et al. (1996), the study on the post-modification of the silica monolith column has been developing to fit the various needs for the wide separation of compounds in µLC and CEC (Fu et al., 2004; Jaafar et al., 2008). Through the post-modification step, monolithic columns allow completely independent control of the porous and the chemical properties. The schematic preparation steps for the monolith in CEC using direct and post-modification method are depicted in Figure 2.5 and 2.6 respectively. Monolith stationary phases applied in CEC are of two types, which are the polymer-based monolith and the silica-based monolith. Examples of these types of monolith will be discusses briefly in the following sub-topics. 21 Bare capillary Polymerization mixture a) Silanization b) Filling Partially filled capillary c) Polymerization Thermal initiation UV initiation d) Washing Mechanical pump Electroosmotic flow Monolithic column ready for use Figure 2.5: Schematic preparation of direct synthesis for CEC monolith capillary 22 Bare capillary a) Silanization Silanized capillary Polymerization mixture b) Silanization Partially filled capillary c) Polymerization UV initiation Thermal initiation d) Washing Mechanical pump Electroosmotic flow Capillary filled with monolithic matrix e) Coupling of chromatographic ligand f) Coupling of charged group Functionalized monolithic CEC column g) Creation of on-column detection window Monolithic column ready for use Figure 2.6: Schematic preparation of post modification synthesis for CEC monolith capillary 23 2.7.1 Polymer-Based Monoliths in CEC Reversed-phase (RP) stationary phases is most widely applied in CEC. As the most common polymer monolith column in capillary, poly(methacrylate), poly(acrylamide), poly(PS-DVB)-based monolith column were the good candidate to RP stationary phases in CEC. Li et al. (2005) incorporated single-wall carbon nanotubes (SWNTs) into the preparation of polymer monolith column via in situ copolymerization of vinylbenzyl chloride (VBC) and EDMA for CEC. The prepared poly(VBC-EDMA-SWNT) monolith column was evaluated by separating a mixture of small organic molecules. It was observed that the incorporation of SWNTs indeed enhanced the retention of small neutral molecules on the monolith column. Another type of RP polymer monolith was prepared by Ding et al. (2006), of a lysine immobilized poly(GMA-co-EDMA). On this post-modified monolith column, the hydrophobic interaction was responsible for separation of neutral analytes, and the separation of ionic or ionizable analytes in CEC involved electrostatic interaction and electrophoretic migration, in addition to the hydrophobic interaction. Ion-exchange is also an important separation mode in CEC. Thus, ionexchange stationary phase with high degree of ionic interaction for CEC is always desirable. Wu et al., (2002) developed SCX polymer monolith stationary phase by in situ copolymerization of 2-(sulfooxy)ethyl methacrylate (SEMA) and ethylene dimethacrylate (EDMA). The sulfate group provided by the monomer SEMA on the monolith bed was used for the generation of electroosmotic flow from anode to cathode, and these negatively charged groups also served as a SCX stationary phase. But, because of the hydrophobicity of the polymeric skeleton, the CEC separation of peptides was explained under both electrostatic interaction of SCX and hydrophobic interaction of RP. An anion-exchange stationary phase was prepared by Bisjak et al. (2005) by grafting the ionizable amino groups onto the poly(GMA-co-DVB) monolith by the post modification with deithylamine via the ring opening of the epoxy groups. Wieder et al. (2006) further extended this approach to prepare the SAX stationary phase by a two-step derivatization of poly(GMA-co-DVB) monolith first with 24 diethylamine and then with diethyl sulfate to form the designated quaternary ammonium group via the ring opening epoxy groups from GMA. The coating of latex-nanoparticles onto the polymer-based monoliths has also been applied for the functionalization of monolith columns. Hilder et al. (2004) prepared an anion-exchange polymer monolith column for the separation of polysaccharides by coating 60 nm quaternary ammonium latex-nanoparticles on a poly(BMA-co-EDMA-co-AMPS) monolith matrix via the electrostatic interaction. The SEM images before and after coating of latex particles on the monolith demonstrated that the macroporous structure of the monolith did not change but the microglobules of coated nanoparticles played as the anion-exchange stationary phase were found on the surface of the monolith. By applying this method, Hutchinson et al. (2005) and Zakaria et al. (2005) realized the separation of inorganic anions over a short separation period, and the elution order indicated the contribution of both ionexchange and electrophoresis mechanisms on the latex-nanoparticles coated polymer-based monolith column. However, because the electrostatic adsorption of quaternary ammonium latex onto the monolithic matrix was restricted by the amount of negatively charged groups on monolith, the increase of the density of surface active groups will accordingly improve the adsorption of latex-nanoparticles and consequently increase the ion-exchange capacity of the latex-coated monolith stationary phase. 2.7.2 Silica-Based Monoliths in CEC Silica-based monolith columns provide good mechanical stability and specific mesoporous and macroporous structure as well as the variety of chemical modification for CEC. The aspirations of exploring the performance of silica monolith column in CEC have driven researchers to develop a variety of new methods for the preparation of silica-based monolith column. Silica-based monolith columns can be prepared by hydrolysis and polycondensation of alkoxysilanes catalyzed by acetic acid in the presence of porogenic agent using the sol-gel method. 25 Figure 2.7 shows a generalized reaction scheme for a sol-gel reaction. After drying and heating, the sol-gel network is derivatized by on-column silylation reaction. Figure 2.7: A generalized reaction scheme for a sol-gel reaction For modifying the surface feature for chromatographic separation, the functional groups can be introduced onto the silica monolith matrix either by post-modification or direct incorporation of desired functional monomers during the fabrication process. Hayes and Malik (2000) prepared porous silica monolith inside the fused silica capillaries for the use as a separation bed in CEC. In this method, a sol-gel precursor, N-octadecyldimethyl [3-(trimethoxysilyl)propyl] ammonium chloride was incorporated into the sol solution for the preparation of the silica-based monolith column. The prepared silica-based monolith with C18 moiety could be directly applied in CEC, and therefore, the post-modification of the silica-based monolith is avoided. Constantin and Fretag (2003) developed a similar one step sol-gel process for the preparation of a C8 silica monolith column inside the fused silica capillary, which also does not require the additional derivatization. Different from the previous 26 method, Dulay et al. (2002) reported the chemical modification approach of sol-gel monoliths by silanizing the sol-gel surface with organochlorosilane or organoalkoxysilane coupling reagents to form the stationary phases with the moieties of pentafluorophenylpropyldimethyl, pentafluorophenyl, 3, 3, 3-trifluoropropyl, noctadimethyl, perfluorohexyl, and aminopropyl, respectively. After the derivatization, the modified monoliths had higher stability at pH values below 4 compared to the underivatized monolith matrix. The separations of different mixtures containing nucleosides, positively charged peptides and taxol derivatives were observed on these phase-bonded PSG columns in CEC. Using mercapto silane reagent, Xie et al. (2005) prepared the SCX silicabased monolith column for SCX separation in CEC. In this method, the silica monolith matrix from a sol-gel process was first chemically modified by (3mercaptopropyl)-trimethoxysilane), and then followed by in situ oxidation of thiol groups to sulfonic groups to produce the SCX stationary phase. The SCX stationary phase was characterized by its substantial and stable electroosmotic flow (EOF), and it was observed that the EOF value of the prepared column remained unchanged at different buffer pH values and slowly decreased with increasing phosphate concentration in the mobile phase. The silica monolith column with SCX stationary phase was then employed in CEC for the separation of β-blockers and alkaloids, which were extracted from the traditional Chinese medicines (TCMs). 2.8 Mixed-Mode Silica Monolith as Electrochromatographic Separation Medium Initially, the stationary phases that had been applied in CEC are the cation exchange (CX) (Xie et al., 2005), RP (Motokawa et al., 2002; Pesek et al., 2008; Roux et al., 2008) and anion exchange (AX) (Jaafar et al., 2008; Lin et al.,2006) functionalities modified on silica monolith. Minakuchi and co-workers (1996) prepared an ODS modified column, containing a layer of octadecyl ligand on a silica monolith. Although their earlier work was focused on µLC, their preparation process was then adapted by most researchers to synthesize column for the utilization as 27 stationary phase in CEC. An AX column was synthesized and evaluated by Jaafar et al. (2008) for the separation of some common nucleotides and inorganic anions by CEC. Polymerization of the DMAPAA-Q moities on the column exhibit a strong anion exchange functionality, produced up to 90,000 theoretical plates with a BGS of 50 mM phosphate pH 6.9. Column short term stability was reported to be moderate with RSD's values ranging from 4.7-8.8%. Another CEC AX column containing a propyl-N,N,N-trimethylammonium group was also reported by Lin et al. (2006), and its performance was examined using mixtures of inorganic anions. A plate height of 7.5 µm was obtained from HCrO4 - ion with 40 mM phosphate buffer pH 3.5, while its short term and long term stability were reasonably good with RSD's values of 1.73.9% and 5.5-8.2% respectively. In the separation of anions, mixed-mode phases offer more optimization potential that RP or AX chromatography. Mixed-mode retention mechanism results in which both electrophoretic mobility and hydrophobic interactions with the stationary phase contribute to the overall retention of the analytes. Scherer and Steiner (2001) synthesize a porous encapsulated silica monolith with a mixed-mode of SAX and RP. The column exhibit high efficiency, H=7 µm for neutral solutes in 5 mmol/L phosphate, pH 4.0-water-acetonitrile eluent, while slightly higher H values were obtained for some carboxylate ions. A mixed-mode column of RP/SAX with the introduction of C6-alkyl chain and quarternary ammonium group modified on the surface of the silica monolith was reported by Klampfl et al. (2000). A BGE of 5 mM Bis-Tris (pH 6.5 with HCl)-45% acetonitrile and addition of 2.5 mM chloride was used to separate the inorganic anions, organic acid, organic bases and neutral compounds. This column exhibited a high efficiency, N=197,000 for phenol but slightly lower theoretical plates of 7000 for salicylic acid. Fu et al. (2004), prepared a mixed-mode of RP/SAX type of column on a silica monolith modified with charged monomer of 2- (methacryloyloxy)ethyltrimethylammonium methyl sulfate (MEAMS) and ethylene dimethacrylate (EDMA) for the separation of neutral and ionic compounds. The column yields plate height of 5.2 µm and 5.5 µm for thiourea and benzyl alcohol respectively, using a 10 mM phosphate buffer containing 40% (v/v) acetonitrile at 28 pH 2.0. The separation of analytes in the column is predominantly under the influence of electrophoresis, due to the late elution of thiourea; an EOF marker. A RP/WAX silica monolith column synthesized by Ding et al. (2006) used a mixture of three precursors, tetraethoxysilane (TEOS), aminopropyltriethoxysilane (APTES) and octytriethoxysislane (C8-TEOS). Fast and efficient separation of six aromatic acid was obtained using a 10 mM phosphate buffer (pH 4.7) and 20% acetonitrile with column efficiency up to 160,000 theoretical plates by CEC. The early elution of its EOF marker, thiourea, at approximately 3 mins with a plate height of 5.7 µm, suggested that separation is determined under chromatographic mechanisms. For both of the above-mentioned cases, separations of the analytes were carried out with a electrokinetic injection and a positive applied voltage. 2.9 Determination of Corrected Retention Factor In column chromatography, retention factor, k, is a measure of the time the sample component resides in the stationary phase relative to the time it resides in the mobile phase (Kucera, 1985; Swadesh, 2001; Dong, 2006). k also expresses how much longer a sample component is retarded by the stationary phase than it would take to travel through the column with the velocity of the mobile phase. Retention in CEC of charged solute depends on both chromatographic retention and electrophoresis. Electrophoresis drives the charged solute through the capillary at a rate which depends on it mass-to-charge ratio while the chromatographic interaction is based on its chemical constituents towards the stationary phase. Several researchers (Al-Rimawi and Pyell; 2007; Knox and Grant, 1996; Rathore and Horvath; 1999; Tsuda, 1988) have proposed different CEC retention models. Tsuda (1988) introduced the following relationship as in Equation 2.3. 29 L tR u pre (2.3) (1 k ' LC ) u ep In Equation 2.3, upre and uep are corresponding migration velocities under pressure and electric field mode. The variables, tR and L are solute retention and capillary length, while k’LC is the chromatographic retention factor. Here CEC retention is separated into two components, one being the chromatographic factor and another, the electrophoretic factor. According to Equation 2.3, when upre and uep have the same mathematical sign, the CEC retention will decrease; otherwise, it will increase. However, the CEC retention factor, k’ is not included in Equation 2.3, and therefore cannot be further evaluated. Knox and Grant (1996) proposed that the overall migration velocity (ux) of any charged solute in CEC could be expressed as in Equation 2.4, where k’LC is the chromatographic retention factor of a solute and ueo is the electroosmotic flow velocity. ux u eo u ep 1 k ' LC (2.4) Knox and Grant (1996) equation also describes the CEC retention in terms of migration velocity. According to Equation 2.2, the CEC retention consists of two independent factors, which are chromatographic and electrophoretic. However, similarly as in Equation 2.3, CEC retention factor, k’ is also not expressed in the Equation 2.4. The omission of the k’ thus restricted the physical understanding of the CEC retention factor. Rathore and Horvath (1999) introduced the concept of virtual migration distances into CEC. Based on this theory, the migration process in CEC can be divided into separative and non-separative components. The separative components describe selective interaction with the stationary phase and differences in the 30 electroporetic migration velocity while the non-separative component describes migration by convection that does not contribute directly to separation. Rathore and Hovath (1999) defined CEC retention factor, k’ as in Equations 2.5 and 2.6, where k’e is the electrophoretic velocity factor. k ' k ' LC k ' e k ' e k 'e u ep u eo (2.5) (2.6) All features of CZE and liquid chromatography (LC) are combined in Equation 2.5. In principle, this model should be applicable to evaluate the CEC retention factor. Recently, Al-Rimawi and Pyell (2007) proposed the corrected retention factor, kc in CEC. The determination of corrected retention factor for charged analyte in CEC is different from the normal apparent value, kapp, as the kc reflect both chromatographic interaction and electrophoretic migration. Apparent retention factor, kapp can be calculated as in Equation 2.7, where tr is the retention time of analyte and t0 is the hold-up time. k app tr t0 t0 (2.7) For the determination of corrected retention factor, kc, Al-Rimawi and Pyell (2007) modified Equation 2.5 (Rathore and Horvath, 1999) into Equation 2.8, where tr is the migration time of analyte ant t’0 is the corrected hold up time. kc t r t '0 t '0 (2.8) 31 In calculating the kc, it is necessary to take into consideration the electrophoretic mobility, µep of the analyte. The electrophoretic mobility, µep can be determined from CZE experiment with an empty fused silica capillary. The electrophoretic mobility, µep is expressed as Equation 2.9, where Leff and Ltot is the effective length and the total length of capillary, V is the applied voltage, tM is the migration time of analyte and t0 is the hold up time. ep Leff Ltot 1 1 V t M t 0 (2.9) 32 CHAPTER 3 EXPERIMENTAL 3.1 Chemicals and Reagents All aqueous solutions were prepared with deionized water of 18 M.cm purified by a Mili-Q A10 system from Millipore (Massachusetts, USA). All chemicals were used without further purification, unless otherwise stated. Toluene purchased from Nacalai Tesque (Kyoto, Japan) was distilled from calcium hydride after refluxing the mixture for 2 h. Salts for dihydrogenphosphate; background electrolyte NaH2PO4.2H2O and (BGE) consist disodium of sodium hydrogenphosphate; Na2HPO4.12H2O were supplied by GCE Laboratory Chemicals (Eppelheim, Germany), while analysis grade phosphoric acid; H3PO4 was from QRёC (Auckland, New Zealand). 1 N of sodium hydroxide; (NaOH) and methanol; MeOH of HPLC grade were both purchased from J. T. Baker (New Jersey, USA). Salts for sample solutions were of potassium bromide; KBr, potassium iodate; KBrO3 and potassium iodide; KI, purchased from GCE Laboratory Chemicals (Eppelheim, Germany). Potassium nitrate; KNO3, and salt for EOF marker, thiourea; (NH2)2CS were supplied by QRёC (Auckland, New Zealand). Standards of adenosine-5’-monophosphate; (5’-AMP), cytidine-5’-monophosphate; (5’-CMP), guanosine-5’-monophosphate; (5’-GMP) and uridine-5’-monophosphate; (5’-UMP) 33 were purchased from Wako (Osaka, Japan), while the sample solution of alkylbenzenes were from Sigma-Aldrich (Wisconsin, USA). Tetramethoxysilane, TMOS and methyltrimethoxysilane, MTMS were purchased from Shin-Etsu Chemical (Tokyo, Japan). Solution of poly(ethylene glycol), PEG, urea, acetic acid; CH3COOH were from Nacalai, Tesque (Kyoto, Japan). N,N-dimethylaminopropylacrylamide methyl chloride quaternary salt, (DMAPAA-Q) was from Kohjin (Tokyo, Japan), and ammonium persulfate (APS) was from Wako (Osaka, Japan). 3.2 Synthesis of the ODS/DMAPAA-Q Silica Monolith Capillary Column The preparation conditions of the ODS/DMAPAA-Q silica monolith capillary column were similar to the procedures reported earlier (Motokawa et al., 2002). Typical conditions were as follows: Mixture of TMOS and MTMOS (9 mL) was added to a solution of PEG (1.05 g) and urea (2.03 g) in 0.01 M acetic acid (20 mL) and stirred at 40°C for 45 mins. The resultant homogeneous solution was charged into a fused-silica capillary tube from Polymicro Technologies (Arizona, USA), which had been treated in advanced with 1 N NaOH solution at 40°C for 3 h, and allowed to react at 40°C. Gelation occurred within 2 h and the gel was subsequently aged in the capillary overnight at the same temperature. Then, the temperature was raised, and the monolithic silica was treated for 3 h at 120°C to complete the formation of the mesopores with ammonia generated by the hydrolysis of urea, followed by water and methanol washes. After drying, heat-treatment was carried out at 330°C for 25 h, resulting in the decomposition of organic moieties in the capillary. Surface modification of the silica monolith was carried out on-column with an anchor group, 3-methacrylamidopropyltriethoxysilane (MAS). Then each monomer solution (Figure 3.1) was charged into the MAS bonded column. Firstly, solution of octadecyldimethyl-N,N-diethylaminosilane monomer, ODS (2 mL) in 8 mL of toluene was continuously fed under a pressure of 50 mbar at 60°C for 3 h. 34 This was followed by continuous feeding of DMAPAA-Q monomer (0.2 mL) under similar conditions as mentioned above. CH A) 3 N + N Si(CH2CH3) H3) CH 3 B) CH3 Figure 3.1: C N O H (CH2)3 N+ CH3 Cl- CH3 Monomers charged onto the MAS bonded column: A) octadecyldimethyl-(N-N-diethylamino)silane, ODS B) N-[3(dimethylamino)propyl]acrylamide methyl chloride-quaternary salt, DMAPAA-Q Then both ends of the column were dipped in the reaction mixture in a 1 mL vial and allowed to react at 68°C in a water bath for 2 h. After the reaction, the column was washed quickly using water with a HPLC pump at 15 MPa for 6 h. The washing procedure was repeated twice to obtain the mixed-mode ODS/DMAPAA-Q silica monolith capillary column. 35 3.3 Instrumentation 3.3.1 CEC and CZE CEC and CZE experiments were carried out on a HP3D CE Instrument from Agilent Technologies (Waldron, Germany), equipped with a diode array detector and ChemStation software. The schematic diagram of both the CEC and CZE mode is depicted as in Figure 3.2. Figure 3.2: CEC / CZE schematic diagram Separations in CEC were carried out on a packed column, 34 cm x 75 μm I.D. x 360 μm O.D. capillary. Off-column detection was performed for the CEC measurement by connecting the ODS/DMAPAA-Q silica monolith capillary column to an empty fused-silica capillary column from Polymicro Technologies (Arizona, USA), 10 cm x 75 μm I.D. x 360 μm O.D. using a 1 cm union, Teflon tubing of 33 mm I.D., part number 6010 from GL Science (California, USA). A detection window about 2 mm in length, 1.5 cm from the union joint was made by burning out the polyamide coating by a capillary burner. The capillary was then inserted into the alignment of the CE cassette holder. UV detection wavelength was measured at 200 nm. 36 Separations on CZE were carried on a fused silica column from Polymicro Technologies (Arizona, USA), 50 cm x 75μm I.D. x 360 μm O.D.. A detection window about 2 mm in length, 8.5 cm from the end was made by burning out the polyamide coating by a capillary burner. The capillary was then inserted into the alignment of the CE cassette holder. UV detection wavelength was also measured at 200 nm. 3.3.2 µLC The µLC evaluation of the ODS/DMAPAA-Q silica monolith capillary column was carried out using a LC-10AD VP pump from Shimadzu (Kyoto, Japan) and a UV detector, model CE1575 from JASCO (Tokyo, Japan) operated at 254 nm, a data processor model C-R6A from Shimadzu (Kyoto, Japan) and a injector valve model 7125 from Rheodyne (California, USA) fitted with a T-union which serves as a splitter, with one end connected to the ODS/DMAPAA-Q silica monolith capillary column, and the other end to a flow restrictor of an empty fused silica capillary tubing. 3.3.3 SEM Scanning Electron Microscopy from JEOL (Montana, USA), was used for the assessment of the structural parameters in the monolithic column. The size of skeleton and through-pore were calculated manually using the Image Pro-Plus software from MediaCybernatic Inc (Montana, USA). The ODS/DMAPAA-Q column was cut into duplicate fragments of 0.50 cm respectively before mounted perpendicularly onto a 12 mm pin-type aluminum stub using carbon paste. High resolution images were obtained by coating the capillary with platinum (~40 nm thick), operated at high vacuum mode using an accelerating low voltage of 2 kV. 37 3.4 Preparation of Chemicals 3.4.1 Buffer The BGE of 50 mM phosphate buffer at pH 6.9 was prepared using the following procedures. 0.1950 g of NaH2PO4.2H2O was mixed with 0.4477 g of Na2HPO4.12H2O in a 50 mL volumetric flask and top-up with deionized water. Before being used, the pH was measured using pH/Ion 5.5 Eutech Instrument pHmeter (Singapore). Prior to use, buffer solutions were filtered through a 0.22 μm Millipore syringe (Massachusetts, USA), sonicated for 10 min, and 30 min degassing using a Branson Ultrasonic cleaner (Connecticut, USA). 3.4.2 Stock Solution A stock solution of 1000 ppm for each studied ion was prepared by weighing appropriate amount of salt before made up to mark with deionized water. Each of the stock solution was stored at 4˚C in a refrigerator until needed. Prior to analysis, the standard solution mixture was prepared by diluting certain amount of stock solution with deionized water. 3.5 Experimental Procedures 3.5.1 CEC Procedures Prior to CEC measurement, the ODS/DMAPAA-Q silica monolith capillary column was preconditioned by a syringe pump from KD Scientific (Massachusetts, USA), with a pump flow of 10 μL/h for 3 h using methanol. The capillary was then further conditioned via the CE instrument with water under 8 bar of nitrogen pressure, applied to the inlet vial for 1 hr and followed by a BGE for another 1 h. The 38 column was further conditioned electrokinetically at -5 kV applied voltage with the background electrolyte until a stable baseline was achieved. Standard solutions mixture was introduced into the ODS/DMAPAA-Q column through electrokinetic injection mode, -3 kV for 3 s from the cathodic end (Jaafar et al. 2008). The EOF was determined by using thiourea as a marker. Between each injection, the capillary was flushed with running electrolyte for 2 min to ensure repeatibility between runs. 3.5.2 CZE Procedures The fused silica capillary was preconditioned with 1 N NaOH (30 min), methanol (30 min), deionized water (30 min) and finally BGE (30 min). Standard solutions mixture was introduced into the fused-silica capillary through electrokinetic injection mode, at -3 kV for 3 s from the cathodic end. The EOF was determined by using thiourea as a marker. Between each injection, the capillary was flushed with 0.1 N sodium hydroxide (3 min), methanol (3 min), deionized water (3 min) and BGE (3 min) to ensure repeatibility between runs. 3.5.3 µLC Procedures µLC experiments were performed on a LC-10AD VP pump and a UV detector, operated at 210 nm for the separations of the alkylbenzenes (n=0-6). Separations were carried out on ODS/DMAPAA-Q monolithic silica column, and samples were injected in a split-flow injection mode at a linear velocity, u = 1.00 mm/s. Prior to being used, the ODS/DMAPAA-Q monolithic silica column was preconditioned with the mobile phase consisting of 80% methanol until a stable baseline was achieved. Temperature of the column was kept constant at 23°C with pressure, P of 52.0 bar. 39 µLC experiments for the separations of nucleotides (5’-CMP, 5’-AMP, 5’UMP and 5’-GMP) were also carried on the same instruments under similar injection mode at u=1.00 mm/s. Detection was determined at 254 nm. Prior to being used, the ODS/DMAPAA-Q monolithic silica column was pre-conditioned with methanol (1 h), degassed deionized water (1 h) and finally with the mobile phase of 50 mM phosphate buffer (pH 2.8) until a stable baseline was achieved. Temperature of the column was kept constant at 24°C with P of 34.3 bar. 3.6 Qualitative Analysis In qualitative analysis, each of the analyte (100 ppm) was injected into CEC or CZE individually. The migration time of each analyte was noted from the electropherogram. A mixture of standard solution was also prepared prior to the determination via CEC or CZE. Each peak in the standard solution mixture found in the chromatogram was analyzed by comparing their retention time with the retention time of the individual standard injected under identical CEC or CZE condition. 3.7 Data Analysis 3.7.1 Mobility of Analyte In CEC and CZE, the mobility of analytes, defined as applied mobility, µapp and the electrophoretic mobility, µ eo of EOF marker were calculated using Equations 3.1 and 3.2 respectively (Camilleri, 1998; Henry, 2006). The effective mobility was calculated using Equation 3.3(Camilleri, 1998; Henry, 2006). µapp = Leff Ltot kV t m (3.1) 40 µeo = Leff Ltot (3.2) kV t 0 µeff = µapp - µeo Where (3.3) Leff : Effective length of capillary (m) Ltot : Total length of capillary (m) kV : Applied voltage (V) tm : Migration time of analyte (s) t0 : Migration time of EOF marker 3.7.2 Separation Efficiency Separation efficiency is evaluated with regard to the value of theoretical plates, N and plate heights, H. For both parameters, the calculations were as in Equations 3.4 and 3.5 respectively. 2 t N = 5.54 R w1 / 2 Where w1/2 : Width of peak at half height H= Leff N (3.4) (3.5) 41 3.7.3 Corrected Retention Factor The CEC retention factor, kCEC can be calculated using Equation 3.6. Veo 0 Veff Vep kCEC = Vep Where Veo-0 : Electroosmotic velocity Vep : Electrophoretic velocity Veff : Effective velocity (3.6) The Veo-0, Vep and Veff of the EOF marker and charged species in CEC is given by Equation 3.7, 3.8 and 3.9 respectively. Veo-0 = Vep = Veff = Leff t0 Leff (3.7) (3.8) tm eff CZE kV Leff (3.9) To calculate the effective mobility in CZE, µeff-CZE, a CZE experiment using fused silica capillary was carried out under identical chromatographic conditions as applied in CEC. µeff-CZE is calculated using Equation 3.10. 42 µeff-CZE = Veff CZE (3.10) ECZE Where Veff-CZE : Effective velocity in CZE ECZE : Energy in CZE The ECZE and Veff-CZE value are also obtained from CZE experiment. The equations for both ECZE and Veff-CZE are shown in Equations 3.11 and 3.12 respectively. ECZE = kV Leff (3.11) Veff-CZE = Vep-CZE + Veo-CZE Where (3.12) Vep-CZE : Electrophoretic velocity in CZE Veo : Eletcroosmotic velocity in CZE The VCZE and Veo-CZE value are from the CZE experiment. The equations for Vep-CZE and Veo-CZE are as in Equations 3.13 and 3.14 respectively. Vep-CZE = Veo-CZE = Leff tm Leff t0 (3.13) (3.14) 43 CHAPTER 4 RESULTS AND DISCUSSION 4.1 Synthesis of ODS/DMAPAA-Q Silica Monolith Capillary Column The ODS/DMAPAA-Q silica monolith capillary column was synthesized according to previous report (Motokawa et al., 2002), and consisted of two-steps process. It involved the preparation of the hybrid silica monolith as the backbone for the attachment of monomers, using the sol-gel method, and followed by surface modification through co-polymerization process to obtain the mixed-mode ODS/DMAPAA-Q functionalities. As a hybrid type of silica monolith, the preparation involved the addition of TMOS and MTMS with the presence of PEG and urea to firstly form the silica network. The role of PEG and urea was to set the monolithic structure during the gelation and ageing process. An advantage offered by hybrid silica monolith was that it could effectively avoid shrinkage and cracking of the column packing (Ding et al., 2006; Motokawa et al., 2002). In this experiment, a sol-gel reaction was employed for the formation of silica monolith backbone. The network was achieved via hydrolysis of an alkoxy silicate, followed by condensation and polymerization reaction. The silanol groups at the inner surface of the capillary took part in this process to produce the monolithic bed bonded to the capillary wall. The ODS/DMAPAA-Q silica monolith capillary column was prepared in a 100 cm fused-silica capillary with 75 µm I. D., since reported work (Motokawa et al., 2002) and review regarding the column technology for liquid chromatography (Gu et al., 2007; Tang et al., 2000; Wu et al., 2008; Yuan 44 et al., 2006), showed some difficulty when prepared in a 50 µm I.D. capillary column. Although it is easier to prepare the monolith in capillaries of 100 µm and 200 µm I.D., it was reported that larger I.D. of capillary will create the effect of ohmic heating which leads to the current leaking during analysis (Bartle and Mayers, 2001). After synthesizing the silica network, next is the process of surface modification. The derivatization of the mesoporous monolithic silica to C 18 with ODS before the attachment of DMAPAA-Q, both by on-column polymerization results in a reversed phase and strong anion exchange silica monolith capillary column. Figure 4.1 shows the proposed structure of the polymer inside the capillary. The C18 group which is a hydrophobic compound enables separation to be carried out under the RP-mode while the ammonium groups from the DMAPAA-Q moieties served to yield negatively charged surface, thus generating anodic EOF. The presence of dual functionalities inside the capillary, either with hydrophobic, and anion (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Klampfl et al., 2000) or cation moieties create separation medium that has the ability to generate strong and constant EOF. H O Si N H O Hybrid silica monolith Figure 4.1: Si O O N N-(3-triethoxysilylpropyl) methacrylamide, MAS ODS-DEA MONOMER DMAPAA-Q MONOMER Functional group Proposed structure of the ODS/DMAPAA-Q silica monolith packing 45 4.2 SEM Physical Characterization of ODS/DMAPAA-Q Silica Monolith Capillary Column The ODS/DMAPAA-Q silica monolith was characterized using SEM to obtain an estimate of the skeleton and through-pore sizes. The SEM micrographs of the cross-section of the monolith at 850x and 3500x magnification are shown in Figure 4.2. ODS/DMAPAA-Q monolithic silica column showed a continuous porous network and was successfully prepared without any shrinkage as shown in the micrograph. A B Figure 4.2 SEM micrograph of 200 µm I. D. ODS/DMAPAA-Q silica monolith capillary column. A: 850x magnification B: 3500x magnification. 46 The pore properties of the ODS/DMAPAA-Q silica monolith capillary column are listed in Table 4.1. The ODS/DMPAA-Q silica monolith capillary column prepared from the mixture of TMOS/MTMS gave network structure with larger through-pore and skeleton size, as well as higher domain and smaller ratio value compared to the reported columns of MS-H(50)-II (Motokawa et al., 2002). Minakuchi et al. (1996) reported that silica monolith capillary columns showed pore properties of small-sized skeletons (1-3 µm), large through-pores size (1.5-5 µm) and large through-pores size/skeleton size ratios (~1.2-1.5), as such properties could create a column with performance beyond the limit of particle-packed column especially when operated at high flow velocities. A good attachment of the silica monolith to the capillary wall was attributed to the small domain size (Ding et al., 2006; Fu et al., 2004; Jaafar et al., 2008, Lin et al., 2006; Motokawa et al., 2002) showed by the ODS/DMAPAA-Q silica monolith capillary column, thus reducing the void volume of the column. Table 4.1: a Pore properties of the ODS/DMAPAA-Q silica monolith capillary column Through-pore size Ratioa Domain sizeb (μm) (μm) Capillary Skeleton size (μm) ODS/DMAPAA-Q 2.0 2.2 1.1 4.2 MS(50)-D (Motokawa et al., 2002) MS-H(50) II (Motokawa et al., 2002) 1.0 2.0 1.0 3.0 1.5 2.0 1.3 3.5 : through-pore size to skeleton size ratio : skeleton size + through-pore size b 4.3 µLC Chromatographic Characterization of ODS/DMAPAA-Q Silica Monolith Capillary Column The ODS/DMAPAA-Q silica monolith capillary column was characterized under chromatographic mode utilizing µLC after each modification step. These steps consist of modification stage with MAS as the anchoring group, modification stage 47 with the ODS-DEA monomer, and lastly modification stage with the introduction of DMAPAA-Q monomer to yield silica monolith capillary column with mixed-mode functionalities of ODS/DMAPAA-Q. The purpose of conducting chromatographic characterization at each modification stage was to ensure that the monomers were successfully polymerized inside the capillary column, through the evaluation of the chromatograms obtained. All chromatographic conditions and the choice of test compounds applied in this part of experiment were based on method reported by Motokawa et al. (2002). The modified silica monolith capillary column with the MAS-anchoring group was applied for the separation of uracil, an unretain marker, and alkylbenzenes (n=0-6). Alkylbenzenes were selected as the test compounds due to the existence of methyl group that provide hydrophobic characteristic. At this stage, as shown in Figure 4.3, the uracil peak was observed at ca. 6.7 min, showing that the MAS group was successfully anchored onto the silica monolith backbone. Figure 4.3: Chromatogram of uracil at MAS modification stage. Mobile phase: 80% aqueous methanol. Column temperature: 23˚C. Pressure: 52 bar. Linear velocity: 1.0 mm/s. The MAS-silica monolith capillary column was then co-polymerized with ODS-DEA monomer to yield the reversed-phase functionality. The chromatogram, shown in Figure 4.4 indicated that ODS-silica monolith capillary column yield 48 excellent peak symmetry within 12 min of analysis for the separation of alkylbenzenes. From the chromatogram, it was observed that the ODS-silica monolith capillary column yielded excellent peaks symmetry within 12 min of analysis for the separation of alkylbenzenes. Peak Identifications: (1) Uracil (2) C6H5(CH2)nH; n = 0 (3) C6H5(CH2)nH; n = 1 (4) C6H5(CH2)nH; n = 2 (5) C6H5(CH2)nH; n = 3 (6) C6H5(CH2)nH; n = 4 (7) C6H5(CH2)nH; n = 5 (8) C6H5(CH2)nH; n = 6 Figure 4.4: (2) (3) (4) (5) (6) (1) (7) (8) Chromatogram for mixtures of uracil and alkylbenzenes, C6H5(CH2)nH, (n=0-6) at ODS modified stage. Mobile phase: 80% aqueous methanol. Column temperature: 23˚C. Pressure: 52 bar. Linear velocity: 1.0 mm/s. Results showed that the ODS-silica monolith capillary column provide similar selectivity in terms of the alkylbenzenes separation orders as reported previously (Motokawa et al., 2002) on a silica monolith capillary column. According to Motokawa et al. (2002), the selectivity of the column was not significantly affected by the type of chromatographic support such as in monolith or particle packed column. This was because, as long as a sufficient dense layer of chromatographic ligand can be grafted to the silica surface, the selectivity of a column will not be much affected. It was also found out that the compounds were eluted in the order of increasing hydrophobicity. The column performance evaluation in terms of theoretical plate, Neff, plate height, H, retention factor, k and separation factor, α of the ODS-silica monolith column for alkylbenzenes are tabulated in Table 4.2. 49 Table 4.2: Theoretical plates, Neff, plate height, H, retention factor, k and separation factor, of the ODS silica monolith capillary column for alkylbenzenes separation Alkylbenzenes, C6H5(CH2)nH Neff H (m) k n=0 30 900 12.9 0.12 - n=1 36 000 11.1 0.22 1.80 n=2 39 100 10.2 0.26 1.18 n=3 25 700 15.6 0.33 1.27 n=4 29 300 13.7 0.42 1.27 n=5 34 500 11.6 0.54 1.29 n=6 43 000 9.3 0.72 1.33 At the optimum linear velocity, the ODS-silica monolith capillary column produced 25700–43000 theoretical plates/min with a minimum plate height of 9.3 m. These results were very similar to or greater than the efficiency of a ODS-DEA MS-H(100)-II hybrid silica monolith capillary column prepared from the mixture of TMOS/MTMS using similar procedures (Motokawa et al., 2002). This was presumably due to the network of silica monolith of the MS-H(100)-II capillary column when being prepared in a larger I. D. capillary, although was easier in term of preparation, it however tends to easily shrink and therefore could cause adverse affect on the column performance. Another reason that contributed to this scenario was because of the different skeleton and through-pore sizes yield from both capillaries. Although both types of capillary column were prepared from the same procedure and feed, the effect of I. D. will somehow cause differences in the column performance. The retention time of neutral solutes like alkylbenzenes on the modified ODS-silica monolith capillary column depends on the carbon content and the density of ligands of the stationary phase (Motokawa et al., 2002; Roux et al., 2008). This retention behavior can therefore be used to evaluate the hydrophobicity of the stationary phase. The hydrophobicity was usually determined by the ratio of the retention factors of two neutral solutes differing in a single CH2 groups such as ethylbenzene and toluene. The retention factors of the alkylbenzenes on the ODS- 50 silica monolith capillary column were found to be slightly larger than those obtained from the column prepared only from TMOS (Motokawa et al., 2002). This was due to the more hydrophobic nature of the hybrid capillary column that possessed abundant methyl groups as well as C18 alkyl chains after chemical modification. The separation factor for hexylbenzene and amybenzene, αCH2, as shown in Table 4.2 was 1.33 for the hybrid ODS modified column. Compared to the value from previous ODS MS-H(100)II silica monolith capillary column, the value obtained was 1.51 (Motokawa et al., 2002). Therefore, it can be concluded that the new synthesized column was much better in term of its performance due to the existence of small skeleton and large through-pore sizes. Since the column exhibit excellent peak symmetry and separation, further modification with the co-polymerization of strong anion monomer, DMAPAA-Q was carried out. The capillary column was again, evaluated under the same chromatographic condition as in previous step. The chromatogram obtained for the mixed-mode ODS/DMAPAA-Q silica monolith capillary column is shown in Figure 4.5. Peak Identifications: (1) Uracil (2) C6H5(CH2)nH; n = 0 (3) C6H5(CH2)nH; n = 1 (4) C6H5(CH2)nH; n = 2 (5) C6H5(CH2)nH; n = 3 (6) C6H5(CH2)nH; n = 4 (7) C6H5(CH2)nH; n = 5 (8) C6H5(CH2)nH; n = 6 Figure 4.5: (2) (3) (4) (5) (6) (7) (8) (1) Chromatogram for mixtures of uracil and alkylbenzenes, C6H5(CH2)nH, (n=0-6) at ODS/DMAPAA-Q modified stage. Mobile phase: 80% aqueous methanol. Column temperature: 23˚C. Pressure: 52 bar. Linear velocity: 1.0 mm/s. 51 At the optimum linear velocity of ~1.0 mm/s, rapid analysis was obtained for the separation of the alkylbenzenes on the ODS/DMAPAA-Q silica monolith capillary column, compared to the ODS-silica monolith capillary column modified at the second stage. However, in this stage, the drawback of this capillary column was that, a base line separation for the first three alkylbenzenes was not achieved probably due to weak interaction of the analytes towards the C 18 sites as the surface was more shielded with the introduction of the anion exchange functional group. The performance in terms of theoretical plates, Neff and plate height, H, for the base line separated peaks at the ODS/DMAPAA-Q column are listed in Table 4.3. Table 4.3: Theoretical plates, Neff, plate height, H, retention factor, k and separation factor, of the ODS/DMAPAA-Q silica monolith capillary column for alkylbenzenes separation Alkylbenzenes, C6H5(CH2)nH n=0 n=1 n=2 n=3 Neff 39 800 H (m) 10.1 K 0.06 0.08 0.11 0.14 1.33 1.38 1.27 n=4 n=5 n=6 24 300 26 800 30 300 16.5 14.9 13.2 0.18 0.27 0.35 1.29 1.50 1.30 The separation of alkylbenzenes was achieved within 9 min of analysis time with the ODS/DMAPAA-Q silica monolith capillary column. At this stage, the ODS/DMAPAA-Q silica monolith capillary column was compared to the ODS-silica monolith capillary column that was synthesized in the previous stage, and showed to have a poorer column performance as it produced an average of 30300 theoretical plate and H=13.7 µm for propylbenzene, butylbenzene, pentylbenzene and hexylbenzene. Although the separation time of the alkylbenzenes were faster in the ODS/DMAPAA-Q silica monolith capillary column compared with the ODS-silica monolith capillary column synthesized in the second stage, the average retention factor and the separation factor for the alkylbenzenes were of smaller value than those obtained using ODS silica monolith column. This was due to the relatively weaker hydrophobic surface of the ODS/DMAPAA-Q silica monolith capillary 52 column, probably due to the existence of the quaternary ammonium functional group. The anion exchange property of the ODS/DMAPAA-Q silica monolith capillary column was examined using 50 mM phosphate buffer (pH 2.8) and four nucleotides, 5’-AMP, 5’-CMP, 5’-GMP and 5’-UMP. Figure 4.6 shows the separation of the nucleotides on the ODS/DMAPAA-Q silica monolith capillary column. Peak Identifications: (1) 5’-CMP (2) 5’-UMP (3) 5’-AMP (4) 5’-GMP *Unknown peak * Figure 4.6: * Chromatogram obtained for mixtures of nucleotides at ODS/DMAPAA-Q modified stage: Mobile phase 50 mM phosphate (pH 2.8). Column temperature: 25˚C. Pressure: 34.3 bar. Linear velocity: 1.0 mm/s. Using the 50 mM phosphate buffer at pH 2.8, 5’-CMP and 5’-AMP were protonated, while 5’-UMP and 5’-GMP were in their neutral state, therefore it was expected that the former pair is more retained that the latter pair (Watanabe et al., 2009). However, it was observed that among the protonated and neutral pairs, nucleotides with purine base (5’-AMP and 5’-GMP) possess greater retention than those with pyrimidine (5’-CMP and 5’-UMP) bases. This result was explained by the differences in the hydrophobic nature of these nucleotides. A more hydrophobic pair was retained longer inside the ODS/DMAPAA-Q silica monolith capillary column while the less hydrophobic pair was eluted earlier. All the four nucleotides were base-line separated within 11 min of analysis time. From the chromatogram in Figure 4.6, it was observed that the order of 53 nucleotides elution were different on a strong anion exchange column, DMAPAA-Q silica monolith capillary column reported previously (Jaafar et al., 2008) for the retention time order of 5’-AMP and 5’-GMP. The nucleotides separation on the DMAPAA-Q silica monolith capillary column was much faster, within 6 min of analysis time. The ODS/DMAPAA-Q silica monolith capillary column performance based on calculated theoretical plates, Neff, plate height, H, retention factor, k and separation factor, for the nucleotides separation are listed in Table 4.4. As tabulated in Table 4.4, the values of the retention factor were less than 1, resulted in a moderate retention time for all of the analytes. Table 4.4: Theoretical plates, Neff, plate height, H, retention factor, k and separation factor, of the ODS/DMAPAA-Q silica monolith capillary column for nucleotides separation Nucleotides Neff H (m) k 5’-CMP 5’-UMP 5’-AMP 41800 38000 21900 8.1 8.9 15.5 0.06 0.52 0.53 8.67 1.02 5’-GMP 18300 18.6 0.56 1.06 The separation factor of α5’-UMP/5’-CMP=8.76 was high presumably due to the strong interaction of 5’-UMP towards the stationary phase, which results in longer retention time compared to 5’-CMP. Nevertheless, at the optimum linear velocity, the column was efficient, producing an average 30000 plates/min and H=12.8 µm for the four nucleotides. These values were lower in comparison with the DMAPAA-Q silica monolith capillary column reported by Jaafar et al. (2008), which exhibit higher column efficiency with an average theoretical plates and plate height of 35000 plates/min and 9.5 µm respectively. 54 4.4 ODS/DMAPAAQ Silica Monolith Capillary Column as Separation Medium in Capillary Electrochromatography 4.4.1 Anions Separation on ODS/DMAPAA-Q Silica Monolith Capillary Column The ODS/DMAPAA-Q silica monolith capillary column was tested for the separation of inorganic and organic ions on CEC. The pH and concentration of BGE, and separation pressure and temperature of capillary was adopted from previous study (Jaafar et al., 2008). Phosphate buffer employed in this study as the BGE has a low UV absorbance, providing a stable baseline compared to either acetate or ammonium buffers. Since solutes exhibit a negative charge, an electrokinetic injection from the cathodic end was applied so that solutes can migrate towards the anodic end. As compared to the hydrodynamic injection, the electrokinetic injection was applied as it produced good repeatability and reproducibility between runs. Figure 4.7 shows the electropherogram of results on the separation of a standard mixture of four inorganic anions and one organic ion. It was observed that the anions were base-line separated within 10 min in the order of bromate, nitrate, bromide, iodate and benzoic acid. No obvious tailing peaks were observed for the inorganic anions except for benzoic acid. mAU 2 Peak Identifications: 3535 (1) Bromate, 20 ppb (2) Nitrate, 30 ppb (3) Bromide, 20 ppb (4) Iodate, 20 ppb (5) Benzoic acid, 20 ppb 3030 3 2525 2020 1 1515 4 1010 5 5 5 0 0 2 Figure 4.7: 4 6 8 10 min Electropherogram obtained for mixtures of four inorganic anions and one organic acid. Mobile phase: 50 mM phosphate pH 6.9. Column temperature 25°C. Applied voltage: -15 kV. Separation voltage: -3 kV for 3 secs. 55 Thiourea, a neutral solute, was used to determine the t 0. Functioning as a neutral marker, the late migration of thiourea at 15 min suggests that the separation of solute was predominantly under the influence of electrophoresis. The applied electrochromatographic condition generated a co-directional EOF where the migration velocities of charged species were always greater than the EOF marker, thus enabling the anions to emerge before the EOF marker (Bartle and Mayers, 2001). The migration of EOF marker, after the migration of anions was also reported in several previous studies (Jaafar et al., 2008; Klampfl et al., 2001). An advantage of a mixed-mode capillary column was its ability to provide two different mode of interaction of solute towards the stationary phase (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Klampfl et al., 2000). In the case of the ODS/DMAPAA-Q silica monolith capillary column, the separation of the inorganic anions was due to the interaction with the quaternary ammonium group that exhibit positive charge surface. Benzoic acid was retained longest by the column and resulted in a tailing peak. The retention of benzoic acid was probably governed by several different retention mechanisms, namely electrophoretic, ionexchange, as well as hydrophobic interaction with the alkyl-chains on the stationary phase (Klampfl et al., 2001). The applied mobility, µapp, number of theoretical plates, Neff, and plate heights, H, of each analytes are listed in Table 4.5. Table 4.5: The applied mobility, µapp, theoretical plate, Neff and plate height, H for the separation of ions and anions on the ODS/DMAPAA-Q silica monolith capillary column Analyte µapp (m2Vs-1) Neff H (µm) Bromate -3.37 x 10-8 98 100 3.5 Nitrate -3.09 x 10-8 114 900 3.0 Bromide -2.59 x 10-8 82 600 4.1 Iodate -2.24 x 10 -8 73 100 4.7 Benzoic acid -1.37 x 10-8 16 900 20.1 56 In CEC, the movement of solutes through the column was a result of the EOF generated by the applied field. The plug-flow profile produced an improvement of separation efficiency in CEC. Good efficiency with low plate height of H=3.0-4.7 µm was obtained from the ODS/DMAPAA-Q silica monolith column, except for benzoic acid. Overall, for the separation of inorganic anions, the efficiency of the ODS/DMAPAA-Q silica monolith capillary column was slightly higher but with lower ion mobility compared to the DMAPAA-Q strong anion exchange capillary column (Jaafar et al., 2008). The ODS/DMAPAA-Q silica monolith capillary column also provided lower plate height values than other mixed-mode (Klampfl et al., 2001) and ion exchange (Lin et al., 2006) monolithic columns reported. However, compared to other mixed-mode capillary columns (Breadmore et al., 2001; Ding et al., 2006; Fu et al., 2004; Klampfl et al., 2001), limited selectivity of analytes for ODS/DMAPAA-Q silica monolith capillary column was obtained. 4.4.2 Determination of Corrected Retention Factor of the Anions on ODS/DMAPAA-Q Silica Monolith Capillary Column All charged analytes separated on CEC have an electrophoretic mobility, µ ep, in addition to the retention due to chromatographic interaction (Klampfl et al., 2000). The studied inorganic anions and organic ions produced an effective electric charge depending on the pH of the mobile phase and therefore have an µ ep which will affect the elution time. In an attempt to describe the retention of charged species, Al-Rimawi and Pyell (2007) defined the corrected CEC retention factor, kCEC as in the Equation 3.6, which includes the interaction of analyte towards the ODS/DMAPAA-Q silica monolith column, and the contribution of the applied voltage generated towards the mobility of the analytes. The proposed equation was modified from the original equation defined by Rathore and Horvath (2002). In order to calculate the kCEC, it was necessary to take into consideration the effective mobility, µ eff of the analyte. 57 The µeff was calculated based on experiment performed on CZE using fused-silica capillary, with identical background electrolyte and separation conditions as in CEC. The energy generated from the CZE experiment is -58823 Vm-1, calculated using Equation 3.11. Other data related to the determination of the µ eff are tabulated in Table 4.6. Table 4.6: Data calculated from CZE experiment Bromate tm (s) 315.6 t0 (s) - Vep-CZE (ms-1) 8.08 x 10-4 Veff-CZE (ms-1) 2.18 x 10-3 eff (m2Vs-1) -3.71 x 10-8 Nitrate 325.8 - 7.83 x 10-4 2.15 x 10-3 -3.66 x 10-8 Bromide 435.0 - 5.86 x 10-4 1.97 x 10-3 -3.35 x1 0-8 Iodate 531.06 - 4.80 x 10-4 1.85 x 10-3 -3.15 x 10-8 Thiourea - 185.04 1.37 x 10-3 - - Analyte Referring to the calculated µeff from the CZE experiment, it is possible to calculate the kCEC. To further calculate the kCEC, data were based on CEC experiment, and the values were calculated using Equation 3.6-3.10. All data calculated are listed in Table 4.7. The values for kCEC for the other four inorganic anions were between 0.92-1.49, indicating that all analytes have sufficient interaction with the stationary phase. Table 4.7: Analyte Data calculated from CEC experiment with ODS/DMAPAA-Q silica monolith capillary column Bromate tm (s) 230.76 t0 (s) - Vep-CZE (ms-1) 1.30 x 10-3 Veo-0 (ms-1) - Veff (Vm-1) 2.18 x 10-3 kcec 0.92 Nitrate 249.48 - 1.20 x 10-3 - 2.15 x 10-3 1.06 Bromide 297.72 - 1.01 x 10-3 - 1.97 x 10-3 1.27 Iodate 343.86 - 0.87 x 10-3 - 1.85 x 10-3 1.49 Thiourea - 932.22 - 0.32 x 10-3 - - 58 In comparing the retention factor, k, between CEC and the pressure-driven separation, µLC, the normal chromatographic k values of bromate, nitrate, bromide and iodate from DMAPAA-Q silica monolith capillary column (Jaafar et al., 2008) were calculated manually using the following Equation 5.1. The k values calculated are listed in Table 4.8 k= tr t0 t0 Table 4.8: (5.1) Retention factor, k, of inorganic anions separated on DMAPAA-Q silica monolith capillary column with µLC and corrected retention factor, kCEC, on DMAPAA-Q silica monolith capillary column with CEC (Jaafar et al., 2008) Analyte K kCEC Iodate 0.05 0.87 Bromate 0.24 0.95 Nitrate 0.48 1.18 Bromide 0.52 1.24 Although the DMAPAA-Q silica monolith capillary column (Jaafar et al., 2008) only possesses anion exchange functionality compared to the dual functionalities of C18 and anion exchange on the ODS/DMAPAA-Q silica monolith column, the k values from DMAPAA-Q silica monolith column on µLC are still comparable to the kCEC of the ODS/DMAPAA-Q silica monolith column on CEC. In comparison with the kCEC value on CEC, it was observed that the value of k was found to be lower on µLC. Jaafar et al. (2008), observed that the kCEC values generated from CEC were higher than the k value produced from µLC, due to the additional separation based on electrophoretic mobility, µ ep, of the charged analytes. In comparison, the kCEC values were higher on the ODS/DMAPAA-Q silica monolith as analytes are retained slightly longer inside the column, allowing the analytes to have sufficient time for interaction and therefore producing separation of high efficiency. Also, according to Rathore and Horvath (1999), the value of retention 59 time in CEC was always larger than in µLC when the electroporetic and electroosmotic velocity are in the same direction. 4.4.3 Stability of the ODS/DMAPAA-Q Silica Monolith Capillary Column The stability of the ODS/DMAPAA-Q silica monolith capillary column was examined based on the measurement of relative standard deviation (RSD) of the migration time of the inorganic anions, organic ions and EOF marker. Repeatability measurements of solutes migration time on the same day were used to determine the short-term stability of the column. As shown in Table 4.9, the short-term stability of the column was excellent with RSDs ranging from 0.6–1.8%. The repeatability was found to be better than the DMAPAA-Q silica monolith capillary column reported by Jaafar et al. (2008). Table 4.9: Repeatability of the ODS/DMAPAA-Q silica monolith capillary column for short-term stability Analyte Migration time (min) ± SD RSD; n=2 (%) Bromate 3.85 ± 0.05 1.3 Nitrate 4.16 ± 0.05 1.2 Bromide 4.96 ± 0.07 1.4 Iodate 5.73 ± 0.08 1.4 Benzoic Acid 9.35 ± 0.09 1.0 Thiourea 15.54 ± 0.09 0.6 Similarly, in evaluating the performance of the ODS/DMAPAA-Q silica monolith capillary column for its long term stability, migration time data for the solute for three successive days were examined. Data as tabulated in Table 4.10 indicated that the column showed good long-term stability (RSDs of 3.0-6.7%). 60 Table 4.10: Reproducibility of the ODS/DMAPAA-Q silica monolith capillary column for long-term stability Analyte Migration time (min) ± SD RSD; n=2 (%) Bromate 3.86 ± 0.26 6.7 Nitrate 4.10 ± 0.21 5.1 Bromide 4.82 ± 0.29 6.0 Iodate 5.66 ± 0.25 4.4 Benzoic Acid 9.30 ± 0.28 3.0 Thiourea 15.5 ± 0.21 1.4 Although higher values of reproducibility were obtained for the ODS/DMAPAA-Q silica monolith capillary column compared to an anion exchange capillary column containing a propyl-N,N,N-trimethylammonium group on silicon alkoxide monolith, as reported by Lin et al. (2006), these values were still acceptable for a CE method (Jaafar et al., 2008). 4.5 Comparison of Anions Separation with CZE Separation of the anions was also performed on CZE under identical condition as in CEC. The electropherogam generated is illustrated in Figure 4.8. CZE with fused-silica capillary and BGS of 50 mM phosphate pH 6.9 was only capable of separating four inorganic anions. Benzoic acid was unable to be detected within 20 min of analysis time. A baseline separation was achieved for the inorganic anions within 10 min in the order of bromide, nitrate, bromate and iodate. 61 mAU Peak Identifications: 20 17.5 (1) Bromate, 20 ppb (2) Nitrate, 30 ppb (3) Bromide, 20 ppb (4) Iodate, 20 ppb (5) Benzoic acid, 20 ppb (not detected) 15 12.5 10 4 7.5 3 1 5 2 2.5 0 -2.5 2 Figure 4.8: 4 6 8 10 min Separation of the test mixture of four inorganic anions and one organic acid. Mobile phase: 50 mM phosphate pH 6.9. Applied voltage: -15 kV. Separation voltage: -3 kV for 3 s. Capillary column temperature: 25°C. In conventional CZE with utilization of acidic buffer, a negatively charged species was normally separated under the influence of counter-EOF when no EOF modifier was added into the BGS (Breadmore et al., 2001; Klampfl 2007). As a result, the determination of EOF marker requires a relatively longer time, and in some reports it was unable to be determined at all (Breadmore et al., 2001; Bartle and Mayers, 2001; Klampfl 2007). In this experiment, thiourea gave no signal within 20 min of analysis time. However, when a positive mode of separation potential was applied, thiourea showed a peak at 3.01 min. This phenomenon supports the evident that under the previous approach, anions were separated under the influence of counter-EOF. The separation of anions under counter-EOF lengthens the analysis time as the anions tend to migrate in the opposite direction to the EOF. This was observed through the values of µapp as tabulated in Table 4.11. Evaluation of separation efficiency in terms of Neff and H yielded lower performance than in CEC which might be due to the effect of counter-EOF. 62 Table 4.11: Efficiency of anion separation on CZE Analyte µapp (m2Vs-1) Neff H (µm) Bromide -3.30 x 10-8 46 300 7.7 Nitrate -3.20 x 10-8 39 600 9.0 Bromate -2.39 x 10-8 46 500 7.6 -8 43 400 8.2 Iodate -1.96 x 10 Although baseline separation of the inorganic anions was achievable in CZE with the same condition as applied in CEC, results clearly highlighted that CEC with the utilization of the mixed-mode monolithic silica column provided different selectivity from CZE for bromide and bromate. Separation in CZE was determined by the electrophoretic mobilities of the solute (Breadmore et al., 2001; Klampfl 2007) while in CEC, chromatographic retention was available to help improve the separation selectivity (Breadmore et al., 2001; Ding et al., 2006; Fu et al., Jaafar et al., 2008; 2004; Klampfl et al., 2000; Lin et al., 2006). The characteristic of the stationary phase which is attached to the quaternary ammonium functional group provides positive charge surface, thus enabling the separation of solutes under the direction of EOF. Unlike in CEC, separation in CZE under co-EOF was impossible without the addition of EOF modifier. 4.5.1 Stability of Anions Separation with CZE The repeatability and reproducibility of the anions separation with CZE was examined based on the measurement of relative standard deviation (RSD) of the migration time of the inorganic anions, organic ions and EOF marker. Repeatability measurements of solutes migration time on the same day were used to determine the short-term stability. As shown in Table 4.12, the short-term stability for the anions separation is excellent with RSDs ranging from 0.2–0.6%. 63 Table 4.12: Repeatability of anions separation with CZE for short-term stability Analyte Migration time (min) ± SD RSD; n=2 (%) Bromide 5.26 ± 0.03 0.4 Nitrate 5.43 ± 0.03 0.6 Bromate 7.25 ± 0.02 0.4 Iodate 8.85 ± 0.02 0.2 Thiourea 3.08 ± 0.01 0.3 Similarly, in evaluating the performance of the analytes separation for its long term stability, migration time data for the solute for three successive days were examined to determine its reproducibility. Data as tabulated in Table 4.13 indicated that the CZE separation also showed excellent long-term stability (RSDs of 0.31.1%). Table 4.13: Reproducibility of anions separation with CZE for long-term stability Analyte Migration time (min) ± SD RSD; n=2(%) Bromide 5.25 ± 0.05 0.9 Nitrate 5.42 ± 0.06 1.1 Bromate 7.24 ± 0.04 0.6 Iodate 8.84 ± 0.03 0.3 Thiourea 3.08 ± 0.02 0.6 64 CHAPTER 5 CONCLUSION AND SUGGESTION 5.1 Conclusion The silica monolith capillary column having mixed-mode hydrophobic (C18) and anion exchange functionalities was successfully synthesized by on-column polymerization onto the hybrid TMOS/MTMS silica backbone using ODS and ionexchange monomer, DMAPAA-Q in capillary of 75 µm I. D.. The ODS/DMAPAAQ silica monolith capillary column showed properties of continuous porous silica with average through-pore size of 2.2 µm and skeleton size of 2.0 µm, while the domain size of this column was calculated to be 4.2 µm. It was observed that there is no shrinkage or cracking of the column packing and also, no polymer agglomerate was formed through the co-polymerization of monomers and the anchor group. A multiple step synthesis of the ODS/DMAPAA-Q column was adopted as the technique offers flexibility and allows completely independent control of the porous and the chemical properties. A hybrid silica monolith backbone was first synthesized with mixture of TMOS/MTMS using the sol-gel method. The hybrid silica monolith provides more hydrophobicity of the column nature due to the abundant methyl group present from the mixture of TMOS/MTMS as well as the C 18 carbon alkyl chains after undergoing the chemical bonding. Therefore, DMAPAAQ, besides serving as the anion exchange monomer, it is also responsible for the generation of the EOF across the column. 65 The ODS/DMAPAA-Q silica monolith was characterized chromatographically using µLC. The silica monolith was first modified with MASanchoring group and tested for the separation of uracil and alkylbenzene. The uracil peak was observed at ca. 6.7 min while alkylbenzene peaks was not retained, showing that there is no interaction of the solutes toward the stationary phase. Copolymerization on the MAS-silica monolith capillary column with ODS-DEA monomer yield a reversed-phase column with excellent column efficiency of up to 43000 plates/min and H=9.3 µm for alkylbenzenes (n=0-6) separations. The modified ODS-silica monolith capillary column was then subjected with the co-polymerization of DMAPAA-Q monomer to afford a mixed-mode capillary column, ODS/DMAPAA-Q silica monolith. However, the performance between the ODS/DMAPAA-Q silica monolith and the ODS-silica monolith was poorer as not all alkylbenzenes were baseline separated, and the column efficiency too decreased with theoretical plates and plate height of 24300-39800 plates/min and H=10.1-16.5 µm respectively. Nevertheless, the evaluation on the ODS/DMAPAA-Q silica monolith capillary column towards the separation of anionic mixture of selected nucleotides were again excellent with the average value of N=41800 and H=9.6 m. The results gathered from this part of experiment, showed that the ODS-DEA and DMAPAA-Q monomer were successfully co-polymerized onto the hybrid silica monolith, while the last evaluation for the separation of nucleotides suggested that the ODS/DMAPAA-Q column has the capability of fast separation of anions. Application of the ODS/DMAPAA-Q column on CEC exhibited good performance for the inorganic anions; bromate, nitrate, bromide, iodate, with large number of theoretical plate, although tailing peak was observed for late eluting anion; benzoic acid. The ODS/DMAPAA-Q column gave an enhanced separation efficiency of up to N=114900 and H=3.0 µm due to the production of sharp, narrow and compact band. In the ODS/DMAPAA-Q silica monolith capillary column, a negative separation mode was applied to avoid the counter-EOF due to the positive charged surface of the column. 66 The corrected retention factor, kCEC for the separation of charged ions on the ODS/DMAPAA-Q column was calculated using modified equation proposed by AlRimawi and Pyell (2007). kCEC has to be determine since in CEC, two types of separation mode are combine, electrophoretic from CZE and chromatographic from µLC. The calculated kCEC were within 2.02-2.89, were slightly of higher value than the DMAPAA-Q silica monolith capillary column. The kCEC values yield from CEC was also found to be higher than in µLC, suggesting that the electrophoretic and electroosmotic velocities generated across the column are in the same direction. The kCEC values also showed that the analytes are provided with enough time for interaction towards the stationary phase, and also interaction based on analyte electrophoretic mobility, µep. Overall, the separation of ions and anions of this column was predominantly under the electrophoresis basis due to the late elution of thiourea, a EOF marker. However, the interaction of solute towards the stationary phase was a combination of eletcrophoretic, ion-exchange as well as the hydrophobic interaction with the alkylchains. The short term stability of the ODS/DMAPAA-Q silica monolith capillary column was excellent with RSDs ranging from 0.2–0.6% while its long term stability was good (RSDs of 0.3-1.1%) for a period of three successive days. Although the plug-flow profile was also generated on conventional CZE, the comparison of separation efficiency for anions obtained from CZE was lower. The values of theoretical plates and plate height were between 39600-46500 plates/min and 9.0-7.6 µm respectively. In another aspect, the mobilities of the ions were higher than in the ODS/DMAPAA-Q column, indicating that the ions were slightly faster eluted with the CZE. Lower efficiency and ions mobilities observed with the CZE was due to the counter-EOF generated across the column. Unlike in CEC, separation in CZE under co-EOF is impossible without the addition of EOF modifier. The short-term repeatability and long-term reproducibility were excellent with RSDs values of 0.1-0.4% and 0.8-1.0% respectively. As a conclusion, the ODS/DMAPAA-Q silica monolith capillary column besides being used as a stationary phase in µLC, it also showed potential as a 67 electrochromatographic separation medium on CEC. As a monolith separation medium, the analytes were separated within the shortest analysis of time, both on µLC and on CEC. An enhanced in the separation efficiency of the column was observed on CEC than on µLC and CZE. Highest theoretical plate, along with the lowest plate height was due to the separation medium itself, and the plug-flow profile generated across the column that produces sharp, narrow and compact band of analyte. The evaluation of ODS/DMAPAA-Q silica monolith capillary column with CEC demonstrated it to be useful separation technique that could offers separation of high efficient for the inorganic anions as compared to the conventional CZE. 5.2 Suggestion Although results obtained for the inorganic anions separation on the ODS/DMAPAA-Q silica monolith capillary column was good, the column should be further evaluated for its performance for the separation of organic species having hydrophobic properties, organic and inorganic ions as well as hydrophilic substance such as amines. Good examples of separation of these types of analytes, separately and in mixtures would make this work more significant. The behavior of amines on the ODS/DMAPAA-Q silica monolith capillary column will be of interest as these compounds provide problems with the reversed-phase mode such as tailing peak and longer analysis time. The advantage of the ODS/DMAPAA-Q silica monolith capillary column shown by the separation impedance, E, is also useful to describe the total performance of column based on the required time and column backpressure to produce one theoretical plate. Study on the effect of domain size (through-pore size + silica skeleton size) and the ratio between through-pore size and skeleton size on the performance of the column is important for the complete characterization of the column. Study on the factors determining band-broadening should also be carried out for the 68 ODS/DMAPAA-Q silica monolith capillary column. In the silica monolith, the band-broadening is dominated by A-term, or the contribution of slow mass transfer in the mobile phase, especially when the silica monolith posses large through-pore size. A slower mass transfer (C-term of the van Deemter equation) is predicted for the ODS/DMAPAA-Q silica monolith capillary column when evaluated under pressure driven compared to electro-driven conditions. The theoretical and experimental study on band-broadening will give some fundamental knowledge in optimizing the synthesis of mixed-mode silica monolith capillary column. Further work on the fundamental characterization of the ODS/DMAPAA-Q silica monolith capillary column must be conducted to study the band broadening effect. The van Deemter plot is necessary to study the contribution of A-, B- and Cterms to band broadening. A detail study on band broadening in the ODS/DMAPAA-Q silica monolith capillary column should be compared with column of ODS and DMAPAA-Q functionality respectively. Further study on the ODS/DMAPAA-Q silica monolith stationary phase is also necessary in order to reduce A- and C- terms contributions by increasing homogeneity of silica structure as well as optimizing bonded polymer structures. 69 REFERENCES Adu, J. K., Eueby, M. R., Tettey, J. N. A. and Shellern, G. G. (2008). “Capillary Electrochromatography of Pharmaceuticals”. Sep. Sci. and Tech. 9. 439-476. Al-Rimawi, F. and Pyell, U. (2007). “Investigation of the Ion-Exchange Properties of Methacrylate-Based Mixed-Mode Monolithic Stationary Phases Employed as Stationary Phases in Capillary Electrochromatography”. J. Chromatogr. A. 1160. 326-335. Aturki, Z., O’razio, G., Fanali, S., Rocco, A., Bortolotti, F., Gottardo, R. and Tagliara, F. (2009). “Capillary Electrochromatographic Separation of Illicit Drugs Employing a Cyano Stationary Phase”. J. Chromatogr. A. 1216. 36523659. Bartle, K. D. and Mayers, P. Capillary Electrochromatograpy. 2nd. ed. U.K.: The Royal Science of Chemistry. 2001. Bisjak, C. P., Bakry, R., Huck, C. W. and Bonn, G. K. (2005). “AminoFunctionalized Monolithic Poly(glycidyl methacrylate-co-divinylbenzene) Ion-Exchange Stationary Phases for the Separation of Oligonucleotides”. Chromatographia. 62. S31-S36. Breadmore, M. C., Hilder, E. F., Mcka, M. and Haddad, P. R. (2001). “Determination of inorganic anions by Capillary Electrochromatography”. Trends in Anal. Chem. 20. 355-364. Camilleri, P. Microchip Capillary Electrophoresis: Theory and Practise. 2nd. ed. U.S.A.: CRC Press LLC. 1998. Constantin, S. and Freitag, R. (2003). “One-Step Synthesis of Monolithic Silica Nanocomposites in Fused Silica Capillaries”. J. Sol-Gel Sci. Technol. 28. 7180. 70 Ding, G., Da, Z., Yuan, R. and Bao, J. J. (2006). “Reversed-Phased and Weak Anion-Exchange Mixed-Mode Silica-Based Monolithics Column for Capillary Electrochromatography”. Electrophoresis. 27. 3363-3372. Dong, M. W. Modern HPLC for Practicing Scientist. N. J.: John Wiley and Sons, Inc.. 2006. Dong, X., Dong J., Ou, J., Zhu, Y. and Zou, H. (2006). “Capillary Electrochromatography with Zwitterionic Stationary Phase on the LysineBonded Poly(glycidyl methacrylate-co-ethylene dimethacrylate) Monolithic Capillary Column”. Electrophoresis. 27. 2518-2525. Dulay, M. T., Quirino, J. P., Bennett, B. D. and Zare, R. N. (2002). “Bonded-Phase Photopolymerized Sol-Gel Monoliths for Reversed Phase Capillary Electrochromatography”. J. Sep. Sci. 25. 3-9. Fanali, S., D’Orazio, G.., Lomsadze, K. and Chankvetadze, B. (2008). “Enantioseparation with Cellulose Tris-(3-chloro-4-methylphenylcarbamate) in Nano-Liquid Chromatography and Capillary Electrochromatograhy”. J. Chromatogr. B. 875. 396-303. Fields, S. M. (1996). “Silica Xerogel as a Continuous Column Support for HighPerformance Liquid Chromatography”. Anal. Chem. 68. 2709-2712. Fu, H., Xie, C., Xiao, H., Dong, J., Hu, J. and Zuo, H. (2004). “Monolithic Columns with Mixed-Modes of Reversed-Phase and Anion-Exchange Stationary Phase for Capillary Electrochromatography”. J. Chromatogr. A. 1044. 237-244. Giddings, J. C. Unified Separation Science. N. Y.: Wiley. 1991. Gu, X., Qu, Q. and Yan, C. (2007). “Advances in Column and Stationary Phase of Capillary Electrochromatpgraphy”. Chinese J. Chromatogr. 25. 157-162. Guiochon, G. (2007). “Monolithic Columns in High-Performance Liquid Chromatography”. J. Chromatogr. A. 1168. 101-168. Hayes, J. D. and Malik, A. (2000). “Sol-Gel Monolithic Columns with Reversed Electroosmotic Flow for Capillary Electrochromatogrphy”. Anal. Chem. 72. 4090-4099. Henry, C. S. Microchip Capillary Electrophoresis. N. J.: Human Press Inc.. 2006. Hilder, E. F., Svec, F. and Frechet, J. M. (2004). “Shielded Stationary Phases Based on Porous Polymer Monolith for the Capillary Electrochromatography of Hughly Basic Biomolecules”. Anal. Chem. 76. 3887-3892. 71 Hutchinson, J. P., Zakaria. P., Bowie, A. R., Macka, M., Avdalovic, N. and Haddad, P. R. (2005). “Latex-Coated Polymeric Monolith Ion-Exchange Stationary Phases. 1. Anion-Exchange Capillary Electrochromatography and In-Line Sample Preconcentration in Capillary Electrophoresis”. Anal. Chem. 77. 407416. Ishizuka, N., Kobayashi, H., Minakuchi, H., Nakanishi, K., Hirao, K., Hosoya, K., Ikegami, T. and Tanaka, N. (2002). “Monolithic Silica Columns for High Efficiency Separations by High-Performance Liquid Chromatography”. J. Chromatogr. A. 960. 85-96. Ishizuka, N., Minakuchi, H., Nakanishi, K., Soga, N., Hosoya, K. and Tanaka, N. (1998). “Chromatographic Properties of Miniaturized Silica Rod Columns”. J. High Resolut. Chromatogr. 21. 477-479. Jaafar, J., Watanabe, Y., Ikegami, T., Miyamoto, K. and Tanaka, N. (2008). “Anion Exchange Silica Monolith for Capillary Liquid Chromatography”. Anal. Bioanal. Chem. 391. 2551-2556. Jorgenson, J. W. and Lukacs, K. D. (1981). “High Resolution Separation Based on Electrophoresis and Electroosmosis”. J. Chromatogr. 218. 209-212. Klampfl, C. W. (2007). “Determination of Organic Acids by CE and CEC Methods”. Electrophoresis. 19. 5479-5482. Klampfl, C. W., Hidler, E. F. and Haddad, P. R. (2000). “Investigation on the Behaviour of Acidic, Basic and Neutral Compounds in Capillary Electrochromatography on a Mixed-Mode Stationary Phase”. J. Chromatogr. A. 888. 267-274. Knox, J. H. (1988). “The Effects and Band Spreading in Capillary ElectroSeparation”. Chromatographia. 26. 329-337. Knox, J. H. and Grant, I. H. (1987). “Miniaturization in Pressure and Electroendosmotically Driven Liquid Chromatography: Some Threotical Considerations”. Chromatographia. 24. 135-143. Knox, J. H. and Grant, I. H. (1991). “Electrochromatography in Packed Tubes Using 1.5 to 50 µm Silica Gels and ODS Bonded Silica Gels”. Chromatographia. 32. 317-328. 72 Kuban, P., Kuban, P., Kuban, V., Hauser, P. C. and Bocek, P. (2008). “Capillary Electrochromatography of Inoganic Cations in Open Tubular Columns with Controllable Capacity Multilayered Stationary Phase Architecture”. J. Chromatogr. A. 1190. 377-382. Kucera, P. Microcolumn High-Performance Liquid Chromatography. 2nd. ed. U.S.A.: Elsevier Science Publishers B. V. 1985. Li, Y., Chen, Y., Xiang, R., Ciupru, D., Pfefferle, L. D., Horvath C. and Wilkins J. A. (2005). “Incorporation of Single-Wall Carbon Nanotubes into an Organic Polymer Monolithic Stationary Phase for µ-HPLC and Capillary Electrochromatography”. Anal Chem. 77. 1398-1406. Lin, J., Lin, J, Lin, X. and Xie, Z. (2009). “Preparation of a Mixed-Mode Hydrophillic Interaction/Anion-Exchange Polymeric Monolith Stationary Phase for Capillary Liquid Chromatography of Polar Analytes”. J. Chromatogr. A. 1216. 801-806. Lin, T.-A., Li, G.-Y. and Chaw, L.-K. (2006). “Sol-Gel Monolithic Anion-Exchange Column for Capillary Electrochromatography”. Anal. Chim. Acta. 576. 117123. Motokawa, M., Kobayashi, H., Ishizuka, N., Minakuchi, H., Nakanishi, K., Jinnai, H., Hosoya, K., Ikegami, T. and Tanaka, N. (2002). “Monolithic Silica Columns with Various Skeletons Sizes and Through-Pore Sizes for Capillary Chromatography”. J. Chromatogr. A. 961. 53-63. Nakanishi, K. and Soga, N. (1997). U. S. Patent No. 5624875. Retrieve on November 5, 2009, from http://www.wikipatents.com/. Norton, D. and Shamsi, Electrochromatography S. and A. (2008). Capillary “Packed-Column Capillary Electrochromatography-Mass Spectrometry Using a Lithocholic Acid Stationary Phase”. Electrophoresis. 29. 2004-2015. Pesek, J. J., Matyska, M. T. and Sukul, D. (2008). “Capillary Liquid Chromatography and Capillary Electrochromatography Using Silica Hydride Stationary Phases”. J. Chromatogr. A. 1191. 136-140. Pesek, J. J., Matyska, M. T. and Velpula, S. (2006). “Open Tubular Capillary Electrochromatography Migration Behavior of Enkephalins in Etched Chemically Modified Fused Silica Capillaries”. J. Chromatogr. A. 1126. 298303. 73 Preinerstorfer, B., Linder, W. and Lammerhofer, M. (2005). “Polymethacrylate Type Monoliths Functionalized with Chiral Amino Phosphoric Acid-Derived Strong Cation Exchange Moities for Enantioselective Non-aqueous Capillary Electrochromatography and Investigation of the Chemical Composition of the Monolithic Polymer”. Electrophoresis. 26. 2005-2018. Pretorius, V., Hopkins, B. J. and Schieke, J. D. (1974). “A New Concept of High Speed Liquid Chromatography”. J. Chromatogr. A. 99. 23-30. Rathore. A. S. and Hovath, Cs. (1996). “Separation Parameters Via Virtual Migration Distances in High-Performance Liquid Chromatography, Capillary Zone Electrophoresis and Electrokinetic Chromatography”. J. Chromatogr. 743. 231-246. Rathore, A. S. and Horvath, Cs. (2002). “Chromatographic and Electrophoretic Migration Parameters in Capillary Electrochromatography”. Electrophoresis. 23. 1211-1216. Roux, R., Abi Jaoude, M. and Demesmay, C. (2009). “Improvement of Chromatographic Performances of In-Situ Synthesized Hybrid C8 Silica Monoliths by Reduction of Structural Radial Heterogeneties”. J. Chromatogr. A. 1216. 3857-3863. Roux, R., Jaoude, M. A., Demesmay, C. and Rocca, J-L. (2008). “Optimization fo the Single Step Synthesis of Hybrid C8 Silica Monoliths Dedicated to NanoLiquid Chromatography and Capillary Electrochromatography”. J. Chromatogr. A. 1209. 120-127. Scherer, B. and Steiner, F. (2001). “Application of Hydrophobic Anion Exchange Phases in Capillary Electrochromatography”. J. Chromatogr. A. 924. 197209. Strain, H. H. (1939). “On the Combination of Electrophoretic and Chromatographic Adsorption Methods”. J. Am. Chem. Soc. 61. 1292-1293. Swadesh, J. K. HPLC: Practical and Industrial Applications. 2nd. ed. U.S.A.: CRC Press LLC. 2001. Tanaka, N. and Kobayashi, H. (2003). “Monolithic Columns for Liquid Chromatography”. Anal. Bioanal. Chem. 376. 298-301. Tanaka, N., Kobayashi, H., Ishizuka, N., Minakuchi, H., Nakanishi, K., Hosoya, K. and Ikegami, T. (2002). “Monolithic Silica Columns for High-Efficiency Chromatography Separation”. J. Chromatogr. A. 965. 35-49. 74 Tang, Q. and Lee, M. L. (2000). “Column Technology for Capillary Electrochromatography”. Trends in Anal. Chem. 19. 648-663. Taylor, J. A. and Yeung, E. S. (1993). “Imaging of Hydrodynamic and Electrokinatic Flow Profiles in Capillaries”. Anal. Chem. 65. 2928-2932. Tsuda, T. (1987). “Electrochromatography Using High Applied Voltage”. Anal. Chem. 59. 521-523. Tsuda, T., Ikedo, M, Jones, G., Dadoo, R. and Zare, R. N. (1993). “Flow Profile of Electroosmosis”. J. Chromatogr. 632. 201-207. Uysal, U. D., Aturk, Z., Raqqi, M. A. and Fanall, S. (2009). “Separation of Catechins and Methylxanthines in Tea Samples by Capillary Electrochromatography”. J. Sep. Sci. 32. 1002-1010. Wang, G.-R., Huang, K.-P., Huang, B.-Y. and Liu, C.-Y. (2009). “Preparation and Characterization of Monoliths Covalently Bonded Chelating Groups for Capillary Electrochromatographic Separation of Metals Ions”. J. Chromatogr. A. 34. 6245-6251. Watanabe, Y., Ikegami, T., Horie, K., Hara, T., Jaafar, J. and Tanaka, N. (2009). “Improvement of Separation Efficiencies of Anion-Exchange Chromatography Using Monolithic Silica Capillary Columns Modified with Polyacrylates and Polymethacrylates Containing Tertiary Amino or Quaternary Ammonium Groups”. J. Chromatogr. A. 1216. 7394-7401. Wieder, W., Bisjak, C. P., Huck, C. W., Bakry, R. and Boon, G. K. (2006). “Monolithic Poly(glycidyl methacrylate-co-divinylbenzene) Capillary Columns Functionalized to Strong Anion Exchangers for Nucleotide and Oligonucleotides Separation”. J. Sep. Sci. 29. 2478-2484. Wu, R., Hu, L., Wang, F., Ye, M. and Zou, H. (2008).”Recent Developments of Monolithic Stationary Phases with Emphasis on Microscale Chromatographic Separation”. J. Chromatogr. A. 1184. 369-392. Wu, R., Zou, H., Fu H., Jin W. and Ye, M. (2002). “Separation of Peptides on Mixed-Mode of Reversed-Phased and Ion-Exchange Capillary Electrochromatography with a Monolithic Column”. Electrophoresis. 23. 1239-1245. Xie, C., Hu, J., Xiao, H., Su, X., Dong, J., Tian, R., He, Z. and Zou, H. (2005). “Preparation of Monolithic Silica Column with Strong Cation Exchange Phase for Capillary Electrochromatography”. J. Sep. Sci. 28. 751-756. 75 Yuan, R., Wang, H., Lui, D., Guo, Y., and Fu, H. (2006). “Recent Advances in Capillary Electrochromatography”. Progress in Chem. 21. 1181-1187. Zaidi, S. A., Han, K. M., Kim, S. S., Hwang, D. G. and Cheong, W. J. (2009). “Open Tubular Layer of S-Ofloxacin Imprinted Polymer Fabricated in Silica Capillary for Chiral CEC Separation”. J. Sep. Sci. 32. 996-1001. Zakaria, P., Hutchinson, J. P., Avdalovic, N., Liu, Y. and Haddad, P. R. (2005). “Latec-Coated Polymeric Monolith Ion-Exchange Stationary Phases. 2. Micro-Ion Chromatography”. Anal. Chem. 77. 417-423.