CLASSIFICATION OF BREAST CANCER MICROARRAY DATA USING RADIAL BASIS FUNCTION NETWORK
advertisement

CLASSIFICATION OF BREAST CANCER MICROARRAY DATA USING
RADIAL BASIS FUNCTION NETWORK
UMI HANIM BINTI MAZLAN
UNIVERSITI TEKNOLOGI MALAYSIA
CLASSIFICATION OF BREAST CANCER MICROARRAY DATA USING
RADIAL BASIS FUNCTION NETWORK
UMI HANIM BINTI MAZLAN
A project report submitted in partial fulfillment of the
requirements for the award of the degree of
Master of Science (Computer Science)
Faculty of Computer Science and Information Systems
Universiti Teknologi Malaysia
October 2009
iii
To my beloved dad, my lovely mum and my precious siblings
iv
ACKNOWLEDGMENT
In the name of Allah, Most Gracious, Most Merciful
Praise to The Almighty, Allah S.W.T for giving me the strength and passion
towards completing this research throughout the semester. By His willing and bless, I
had managed to complete my work within the period given.
First of all, my thanks go to Dr. Puteh Bt Saad, who responsible as my Master
Project’s supervisor for her guidance, counsels, motivation, and most importantly for
sharing her thoughts with me.
My deepest thanks go to my lovely family for their endless prayers, love and
support. Their moral supports that always right by my side had been the inspiration
along the journey and highly appreciated.
I would also like to extend my heartfelt appreciation to all my friends for
their valuable assistance and friendship.
May Allah S.W.T repay all their sacrifices and deeds with His Mercy and Bless.
v
ABSTRACT
Breast cancer is the number one killer disease among women worldwide.
Although this disease may affect women and men but the rate of incidence and the
number of death is high among women compared to men. Early detection of breast
cancer will help to increase the chance of survival since the early treatment can be
decided for the patients who suffer this disease. The advent of the microarray
technology has been applied to the medical area in term of classification of cancer
and diseases. By using the microarray, thousands of genes expression can be
determined simultaneously. However, this microarray suffers several drawbacks such
as high dimensionality and contains irrelevant genes. Therefore, various techniques
of feature selection have been developed in order to reduce the dimensionality of the
microarray and also to select only the appropriate genes. For this study, the
microarray breast cancer data, which is obtained from the Centre for Computational
Intelligence will be used in the experiment. The Relief-F algorithm has been chosen
as the method of the feature selection. As the comparison, another two methods of
feature selection which are Information Gain and Chi-Square will also be used in the
experiment. The Radial Basis Function, RBF network will be used as the classifier to
distinguish between the cancerous and non-cancerous cells. The accuracy of the
classification will be evaluated by using the chosen metric namely Receiver
Operating Characteristic, ROC.
vi
ABSTRAK
Barah payudara merupkan pembunuh nombor satu di kalangan wanita di
seluruh dunia. Walaupun penyakit ini boleh menyerang wanita dan lelaki namun
kadar kejadian dan kematian adalah lebih tinggi di kalangan wanita berbanding
lelaki. Pengesanan awal barah payudara boleh membantu meningkatkan peluang
untuk hidup memandangkan rawatan awal boleh disarankan kepada pesakit yang
menghidapi penyakit ini. Kemunculan teknologi mikroarray telah diaplikasikan
dalam bidang perubatan untuk pengkelasan bagi penyakit barah. Dengan
menggunakan mikroarray, berjuta-juta pengekspresan gen boleh ditentukan secara
serentak. Walaubagaimanapun, mikroarray ini berhadapan dengan beberapa
kelemahan seperti mempunyai dimensi yang tinggi dan juga mengandungi gen-gen
yang tidak relevan. Oleh sebab itu, pelbagai teknik pemilihan fitur telah dibangunkan
bertujuan mengurangkan dimensi mikroarray dan hanya memilih gen-gen yang
bersesuaian sahaja. Bagi kajian ini, data barah payudara mikroarray yang digunakan
dalam eksperimen diperoleh dari Centre for Computational Intelligence. Algoritma
ReliefF telah dipilih sebagai teknik untuk pemilihan fitur. Sebagai perbandingan, dua
lagi teknik pemilihan fitur iaitu Information Gain dan Chi-square juga akan
digunakan dalam eksperimen. Bagi membezakan di antara sel barah dan bukan
barah, rangkaian Radial Basis Function, RBF, akan digunakan sebagai pengkelas.
Ketepatan pengkelasan akan dinilai oleh metrik yang telah dipilih iaitu Receiver
Operating Characteristic, ROC.
vii
TABLE OF CONTENTS
CHAPTER
1
TITLE
PAGE
DECLARATION
ii
DEDICATION
iii
ACKNOWLEDGEMENTS
iv
ABSTRACT
v
ABSTRAK
vi
TABLE OF CONTENTS
vii
LIST OF TABLES
xi
LIST OF FIGURES
xii
LIST OF ABBREVIATIONS
xiv
LIST OF APPENDICES
xv
INTRODUCTION
1.1
Introduction
1
1.2
Problem Background
3
1.3
Problem Statement
4
1.4
Objectives of the Projects
5
1.5
Scopes of the Projects
5
1.6
Importance of the Study
6
1.7
Summary
7
viii
2
LITERATURE REVIEW
2.1
Introduction
8
2.2
Breast cancer
9
2.2.1 What is Breast Cancer
9
2.2.2 Types of Breast Cancer
9
2.2.2.1 Non-invasive Breast Cancer
10
2.2.2.2 Invasive Breast Cancer
11
2.2.3 Screening for Breast Cancer
12
2.2.4 Diagnosis of Breast Cancer
13
2.2.4.1 Biopsy
2.3
2.4
Microarray Technology
15
2.3.1 Definition
15
2.3.2 Applications of Microarray
15
2.3.2.1 Gene Expression Profiling
17
2.3.2.2 Data Analysis
19
Feature Selection
20
2.4.1 Relief
24
2.4.2 Information Gain
25
2.4.3 Chi-Square
26
Radial Basis Function
27
2.5.1 Types of Radial Basis Function, RBF
27
2.5.2 Radial Basis Function, RBF, network
30
Classification
34
2.6.1 Comparison with others Classifiers
35
2.7
Receiver Operating Characteristic, ROC
37
2.8
Summary
38
2.5
2.6
3
14
METHODOLOGY
3.1
Introduction
39
3.2
Research Framework
40
3.3
Software Requirement
41
3.4
Data Source
41
3.5
HykGene
41
ix
3.5.1 Gene Ranking
3.5.1.1 ReliefF Algorithm
44
3.5.1.2 Information Gain
47
2
3.5.1.3 χ -statistic
3.5.2 Classification
3.6
4
47
50
3.5.2.1 k-nearest neigbor
50
3.5.2.2 Support vector machine
51
3.5.2.3 C4.5 decision tree
51
3.5.2.4 Naive Bayes
52
Classification using Radial Basis Function
Network
52
3.7
Evaluation of the Classifier
54
3.8
Summary
56
DESIGN AND IMPLEMENTATION
4.1
Introduction
57
4.2
Data Acquisition
58
4.2.1 Data format
61
Experiments
63
4.3.1 Genes Ranked
63
4.3
4.3.1.1 Experimental Settings
4.3.2 Classifications of top-ranked genes
4.3.2.1 Experimental Settings
4.3.3 Evaluation of Classifiers Performances
4.3.3.1 Experimental Settings
4.4
5
43
Summary
63
64
64
65
65
67
EXPERIMENTAL RESULTS ANALYSIS
5.1
Introduction
68
5.2
Results Discussion
69
5.2.1 Analysis Results of Features Selection
Techniques
5.2.2 Analysis Results of Classifications
69
72
x
5.2.3 Analysis Results of Classifier Performances 74
5.3
6
Summary
77
DISCUSSIONS AND CONCLUSION
6.1
Introduction
79
6.2
Overview
79
6.3
Achievements and Contributions
81
6.4
Research Problem
82
6.5
Suggestion to Enhance the Research
82
6.6
Conclusion
83
REFERENCES
85
APPENDIX A
95
APPENDIX B
99
xi
LIST OF TABLES
TABLE NO
2.1
TITLE
Previous researches using microarray
PAGE
16
technology
2.2
A taxonomy of feature selection techniques
22
2.3
Previous researches on classification using RBF
35
network
4.1
EST-contigs, GenBank accession number and
59
oligonucleotide probe sequence
5.1
Results on breast cancer dataset using Relief-F
69
5.2
Results on breast cancer using Information Gain
69
5.3
Results on breast cancer using χ2-statistic
70
5.4
Results on classification using different
72
classifiers
5.5
TPR and FPR of classifiers
75
5.6
ROC Area of Classifiers
77
xii
LIST OF FIGURES
FIGURE NO
1.1
TITLE
PAGE
Ten most frequent cancer in females, Peninsular
3
Malaysia 2003-2005
2.1
Ductal Carcinoma In-situ, DCIS
10
2.2
The cancer cells spread outside the duct and
11
invade nearby breast tissue
2.3
Gene Expression Profiling
18
2.4
Feature selection process
21
2.5
Gaussian RBF
28
2.6
Multiquadric RBF
29
2.7
Gaussian(green), Multiwuadric (magenta),
30
Inverse-multiquadric (red) and Cauchy (cyan)
RBF
2.8
The traditional radial basis function network
32
2.9
The ROC curve
38
3.1
Research Framework
40
3.2
HykGene: a hybrid system for marker gene
42
selection
3.3
The expanded framework of HykGene in Step1
44
3.4
Original Relief Algorithm
45
3.5
Relief-F algorithm
46
xiii
3.6
χ²-statistic algorithm
49
3.7
The expanded framework of HykGene in Step 4
50
3.8
Radial Basis Function Network architecture
53
3.9
AUC algorithm
55
4.1
The data matrix
58
4.2
Fifteenth of the instances of breast cancer
60
microarray dataset
4.3
Microarray Image
61
4.4
Header of the ARFF file
62
4.5
Data of the ARFF file
62
4.6
ROC Space
66
4.7
Framework to plotting multiple ROC curve
67
using Weka
5.1
Graph on cross validation classification
70
accuracy using 50-top ranked genes
5.2
Graph on cross validation classification
71
accuracy using 100-top ranked genes
5.3
Percentage of Correctly Classified Instances
73
using 50 top-ranked genes
5.4
Percentage of Correctly Classified Instances
73
using 100 top-ranked genes
5.5
ROC Space of 50 top-ranked genes
75
5.6
ROC Space of 100 top-ranked genes
75
5.7
ROC Curve of 50 top-ranked genes
76
5.8
ROC Curve of 100 top-ranked genes
76
xiv
LIST OF ABBREVIATIONS
AUC
-
Area Under Curve
ARFF
-
Artificial Relation File Format
cDNA
-
Complementary Deoxyribonucleic Acid
cRNA
-
Complementary Ribonucleic Acid
DNA
-
Deoxyribonucleic Acid
EST
-
Expressed Sequence Tag
FPR
-
False Positive Rate
k-NN
-
k-nearest neighbor
MLP
-
Multilayer Perceptron
mRNA
-
Messenger Ribonucleic Acid
NB
-
Naive Bayes
RBF
-
Radial Basis Function
RNA
-
Ribonucleic Acid
ROC
-
Receiver Operating Characteristics
SOM
-
Self Organising Maps
SVM
-
Support Vector Machine
TPR
-
True Positive Rate
xv
LIST OF APPENDICES
APPENDIX
TITLE
PAGE
A
Project 1 Gantt Chart
95
B
Project 2 Gantt Chart
99
CHAPTER 1
INTRODUCTION
1.1
Introduction
Cancer or known as malignant neoplasm in medical term is the number one
killer diseases in most of the countries worldwide. The number of deaths and people
suffering with cancer are increasing year by year. This number will continue to
increase with an approximate 12 million deaths in 2030 (Farzana Kabir Ahmad,
2008). This killer disease may affect people at all ages even fetuses and also the
animals but the risk increases with age. According to the National Cancer Institute,
cancer is defined as a disease causes by uncontrolled division of abnormal cells.
Cancer cells are capable to invade other tissues and can spread through the blood and
lymph systems to other parts of the body.
There are various factors that can cause cancer namely mutation, viral or
bacterial infection, hormonal imbalances and others. Mutation can be classified into
two categories which are chemical carcinogens and ionizing radiation. Carcinogens
are the mutagen, substances that cause DNA mutations, which cause cancer. Tobacco
2
smoking which is accounted for 90 percent of lung cancer (Biesalski HK, Bueno de
Mesquita B, Chesson A, et al., 1998) and prolonged exposure asbestos are the
examples of the substances (O'Reilly KM, Mclaughlin AM, Beckett WS, Sime PJ,
2007).
Radon gas which can cause cancer while prolonged exposure to ultraviolet
radiation from the sun that can lead to melanoma and other skin malignancies
(English DR, Armstrong BK, Kricker A, Fleming C,1997) are the sources of the
ionizing radiation. Based on the experiment and epidemiological data, it shows that
viruses become the second most important risk factor of the cancer development in
human (zur Hausen H, 1991). Hormonal Imbalances can lead to cancer because
some hormones are able to behave similarly like the non-mutagenic carcinogens.
This condition may stimulate the uncontrolled cell growth.
Cancer can be classified into various types with various names which are
mostly named based on the part of the body where the cancer originates (American
Cancer Society). Lung cancer, breast cancer, ovary cancer, colon cancer are several
examples of the common cancer types. Breast cancer is the disease which fears every
woman since it is the number one killer cancer for woman. Breast cancer remains a
major health burden since it is a major cause of cancer-related morbidity and
mortality among female worldwide (Bray, D., and Parkin, 2004).
It has been reported that, each year, more than one million new cases of
female breast cancer are diagnosed, worldwide (Ferlay J, Bray F, Pisani P, Parkin
DM, 2001). In United States of America, an estimated 192,370 new cases of invasive
breast cancer and an estimated 40,610 breast cancer death are expected in 2009
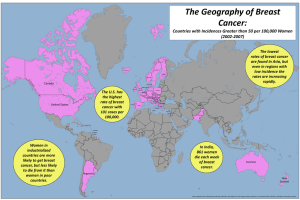
(American Cancer Society, 2009). The following figure shows the percentage of ten
most cancers in female in Malaysia from 2003 to 2005. From the figure, significant
shows that breast cancer hold the highest percentage among all the cancers in female.
3
As discussed in this part, it is proven that breast cancer is one of the main
health problems. A lot of researches done to study the best way to prevent, detect and
treat the breast cancer in order to reduce the number of deaths. This research also
study about detection of breast cancer using the microarray data. The microarray data
of the breast cancer will be classified to determine whether it is benign (noncancerous) or malignant (cancerous) using Radial Basis Function, RBF, after doing
the feature selection.
Figure 1.1: Ten most frequent cancer in females, Peninsular Malaysia 2003-2005
(Lim, G.C.C., Rampal, S., and Yahaya, H., 2008)
1.2
Problem Background
This project will use the microarray data of breast cancer. According to
Tinker et al., 2006, microarray has become a standard tool in many genomic research
laboratories because it has revolutionized the approach of biology research. Now,
scientist can study thousand of genes at once instead of working on a single gene
4
basis. However, microarray data are often overwhelmed, over fitting and confused by
the complexity of data analysis. Using this overwhelmed data during classification
will lead to the increase of the dimensionality of the classification problem presents
computational difficulties and introduces unnecessary noise to the process.
Other than that, due to improper scanning, it also contains multiple missing
gene expression values. In addition, mislabeled data or questioned tissues result by
experts also the drawback of microarray that decreases the accuracy of the results
(Furey et al., 2000). However, feature selection can be utilized to filter the
unnecessary genes before the classification stage is implemented. Based on Farzana
Kabir Ahmad, 2008, (cited from Ben-Dor et al., 2000; Guyon et al., 2002; Li, Y. et
al., 2002; Tago & Hanai, 2003 ) in many large scale of gene expression data analysis,
feature selection has become a prior step and a pre-requisite. The high reliance of
this gene selection technique is due to its important purpose that requires small sets
of genes in order to informatively sufficient to differentiate between cancerous and
non-cancerous cells.
1.3
Problem Statement
The main issue in the classification of microarray data is the presence of
noise and irrelevant genes. According to Liu and Iba, 2002, many genes are not
relevant to differentiate between classes and caused noise in the process of
classification. Hence, this will lead to the drowning out of the relevant ones Shen et
al., 2007. Therefore, the prior step before doing the classification which is feature
selection is important in order to reduce the huge dimension of microarray data.
5
1.4
Objectives of the Projects
The aim of the project is to classify a selected significant genes from
microarray breast cancer data. In order to achieve the aim, the objectives are as
follows:
i. To select significant features from microarray data using a suitable
feature selection technique.
ii. To classify the selected features into non-cancerous and cancerous using
Radial Basis Function network.
iii. To evaluate the performance of the classifier using Receiver Operating
Characteristic, ROC.
1.5
Scopes of the Project
This project will use a breast cancer microarray data which is obtained from
Centre for Computational Intelligence. This data originates from the Zhu, Z., Ong,
Y.S., and Dash, M., (2007). The dataset contains of 97 instances with 24481 genes
and 2 classes.
6
1.6
Importance of the Study
Early detection of breast cancer can help to avoid the cancer from getting
worse and eventually leads to the decreasing of the rate of death. Mammogram is one
of the tools that can detect early stage of breast cancer. However, biopsy is needed in
order to confirm the present of the malignant (cancerous) tissue that cannot be
performed by the mammogram. From this biopsy process the microarray data will be
obtained.
As discussed in the problem background there are several drawbacks of
microarray data. However, it can be solved by implementing the feature selection
before it can proceed to the classification. In this study, several feature selection will
be studied before choosing the most suitable method. Feature selection is very
important since it will determine the accuracy of the result after the classification.
The accuracy of the result is also important because it will classify between
the cancerous and non-cancerous. Since the purpose of biopsy is to confirm the
presence of the cancer cells after the early detection, the confirmation must not take a
long time. The best feature selection method must be used before classifying the
microarray data in order to reduce the time and produce the accurate result.
Through this project, the best feature selection method will be selected and
implemented to microarray data before it will be classified. The performance of the
classifiers will be evaluated based on the suitable metric. The result of this study will
be discussed in order to give the information for the future research in breast cancer
area.
7
1.7
Summary
As a conclusion, this chapter discusses the overview of the project. This
project will classify the breast cancer microarray data using the Radial Basis
Function to two classes namely benign (non-cancerous) and malignant (cancerous).
Feature selections will be studied and the best method will be chosen to implement
the microarray data before the classification.
CHAPTER 2
LITERATURE REVIEW
2.1
Introduction
Breast cancer can affect both genders but women have higher risk compares
to men. Early detection of breast cancer can ensure a better chance to cure the
disease at the early stage. There are three types of screening tools for early detection
of breast cancer. The best tool is the screening mammogram which produces the Xrays pictures. However, the cancer cells cannot be confirmed via the mammogram
alone, so the diagnosis is required to detect the existence of cancer cells. The
microarray data obtained from the biopsy can be used to distinguish the benign
(non-cancerous) and malignant (cancerous) tissue. Because the microarray data also
contains unnecessary genes, feature selection needs to be implemented in order to
filter it before the classification can be made using Radial Basis Function (RBF)
technique.
In this chapter, five main sub topics will be discussed; Breast Cancer,
Microarray Technology, Feature Selection, Radial Basis Function technique and
9
Receiver Operating Characteristic, ROC. The first subtopic will explain about the
types, screening tools and diagnosis of breast cancer. Microarray data produces by
the biopsy will be discussed in the following subtopic. Next, feature selection
method will be discussed to determine the best method that can be implemented in
this study. Finally the classification technique followed by the evaluation of the
classifier using chosen metric will be discussed. Furthermore, several previous
researches will be briefly explained for comparison.
2.2
Breast Cancer
2.2.1
What is Breast Cancer
According to the National Breast Cancer Foundation and American Cancer
Society, breast cancer is a disease in which malignant tumor which is a group of
cancer cells, forms in the tissue or cells of the breast. The malignant tumor is able to
invade surrounding tissues or spread to further area of the body, also called as
metastasize. Woman has the higher risk of developing the cancer compares to man.
2.2.2
Types of Breast Cancer
Breast cancer can be categorized by the areas where the cancer cells begin to
develop; ducts or lobules in which the milk produced. The different types of breast
cancer can be classified into two most common groups which are non-invasive and
10
invasive breast cancer. Besides the invasive and non-invasive, there are many other
common types of breast cancer.
2.2.2.1 Non-invasive Breast Cancer
The term non-invasive or also known as in-situ in breast cancer means that
the cancer cells remain confined to ducts or lubules. This category of breast cancer
did not invade surrounding tissue of the breast and spread to others part of the body.
There are two types of breast cancers that belong to this group which are:
i)
Ductal Carcinoma In-situ, DCIS
This type of breast cancer is the early stage of breast cancer in which
the cancer cells stay in the milk duct of the breast where it started. Although
it can grow, it does not affect the surrounding breast tissue or spread to lymph
nodes and other organs.
Figure 2.1: Ductal Carcinoma In-situ, DCIS
11
ii)
Lobular Carcinoma In-situ, LCIS
LCIS tends to develop into invasive breast cancer eventhough it has
been classified as non-invasive, in which the abnormal cells begins in the
milk-producing glands at the end of breast ducts.
2.2.2.2 Invasive Breast Cancer
Invasive or infliltrating breast cancer is a contrary to the non-invasive or insitu breast cancer. Like non-invasive, this category also have two types of breast
cancer:
Figure 2.2: The cancer cells spread outside the duct and invade nearby breast
tissue
i)
Invasive Ductal Carcinoma, IDC
Invasive ductal carcinoma is the most common type of breast
cancer where it begins in the duct of the breast. The cancer cells then
12
spread through the wall of the ducts and grows into the fatty tissue of
the breast. It can spread to the lymph nodes and to other parts of the
body later on.
ii)
Invasive Lobular Carcinoma, ILC
Like the non-invasive lobular carcinoma, invasive lobular
carcinoma also begins in the milk-producing glands. However, ILC
can spread beyond the lobules (milk-producing glands) to the other
parts of the body.
2.2.3
Screening for Breast Cancer
There are three types of screening test for breast cancer that can detect the
cancer at the early stage. The three screening test are as follows (National Cancer
Institute ):
i)
Screening Mammogram
Mammogram is the screening tool that produces the breast
picture from the X-rays which can help early detection of breast
cancer. This screening tool can often shows a breast lump before it
can be felt and also a cluster of tiny specks of calcium known as
microcalcifications. Cancer, precancerous or other condition can be
formed from that lump or specks.
Although mammograms can help early detection of breast
cancer, it also has several drawbacks which are:
13
a)
False negative because the mammogram may miss some
cancers.
b)
False positive causes by the mammogram may show the result
that is not the cancer.
ii)
Clinical Breast Exam
The clinical breast exam will be done by the doctor and during
the process the differences in size or shape of patient’s breasts will be
inspected. The doctor will examine the breast skin for signs of rash,
dimpling or other abnormal signs. Other examination method is by
squeezing the nipple to check for fluid or by using the pads of the
fingers. The entire breast including underarm and collarbone area will
be checked for the existence of any lumps. Lymph nodes will also be
inspected to ensure that they are in the normal size.
iii)
Breast-self Exam
Screening test for breast cancer can be done by any individual
once a month. This breast-self exam can be carried out even while in
the shower with several simple steps to detect any changes of the
breast. However, studies have shown that the reducing number of
deaths from breast cancer is not solely caused by breast-self exam.
2.2.4
Diognosis of Breast Cancer
There are three procedures in early detection of breast cancer. If there is any
differences discovered during the screening process, diagnosis stage will be required.
Mammograms is the most effective tool to detect breast cancer at the early stage
14
(American Cancer Society) although it has a few disadvantages. Because of the
disadvantages, the follow-up test is required to confirm the present of the cancer. The
biopsy is the follow-up test that can help to diagnose the existence of the malignant
tissue (cancerous).
2.2.4.1 Biopsy
Biopsy is a minor surgery to remove fluid or tissue from the breast to confirm
the presence of the cancer. The biopsy can be done in using three techniques
(National Cancer Institute ):
i)
Fine-needle aspiration
A thin needle will be used to remove the fluid from a breast
lump. The lab test will be run if the fluid consists of cancer cells.
ii)
Core biopsy/ Needle biopsy
To remove the breast tissue, a thick needle will be used and
the cancer cells will be examined afterwards.
iii)
Surgical biopsy
A sample of tissues will be checked for cancer cells after it has
been removed through the surgery. There are two types of surgical
biopsy which are incisional biopsy where a sample of a lump or
abnormal area will be taken and excisional biopsy which takes the
entire lump or area.
15
2.3
Microarray Technology
2.3.1
Definition
Microarray technology is defined as a method to study the activity of a huge
number of genes simultaneously. A high speed robotics is used in this method to
apply tiny droplets consisting of biologically active materials such as DNA, proteins
and chemicals to glass slides precisely. Microarrays are used in Functional Genomics
DNA to monitor gene expression at RNA level. Furthermore, it is also used to
identify genetic mutations and to discover the novel regulatory area in genomics
sequences. In the protein or chemicals materials, microarrays are used to monitor
protein expression or to identify the interactions of protein-protein or drug-protein.
Normally, in a single experiment, there are ten of thousands of genes interactions can
be monitored (Compton, R., et al., 2008).
2.3.2
Applications of Microarray
The applications of microarray were first applied in the mid 90s (Schena, M.,
et al., 1995). Since that, microarray technology has been successfully applied to
almost every field of biomedical research (Alizadeh AA et al., 2000; Miki R., et al.,
2001; Chu S., et al., 1998; Whitfield M.L., et al., 2002; van de vijver MJ et al., 2002;
Boldrick JC et al., 2002). Gene expression profiling is the most common application
of microarray technology (Russell Compton et al., 2008). Others application of
microarray technology are Genotyping of Human Single nucleotide polymorphism
(SNPs), Microarrays analysis of Chromatin Immunoprecipitation (Chip-on-chip) and
Protein Arrays.
16
Microarrays opened up an entire new world to researches (Fodor, S.P. et al.,
1991; Fodor, S.P. et al., 1993; Pease, A.C. et al., 1994). To study a unique single
aspect of the host or pathogen relationship is no longer a restriction but one can
explore a genome-wide view of the complex interaction. This helps scientist to
discover and understand disease pathways to ultimately develop better methods of
detection, treatment and prevention. Microarray technology has provided an
understanding of the tumor subtypes and their attendant clinical outcomes from the
molecular phenotyping of human tumors (Abu Khalaf, M.M. et al., 2007). In the
breast cancer study, it has been reported that the invasive breast cancer can be
divided into four main subtypes through unsupervised analysis by using microarray
technology (Sorlie, T. et al., 2001). In recent studies, the subset of genes that is
identified by microarray profiling has been suggested to be a prediction of a response
to chemotherapy in breast cancer, the taxanes (Abu Khalaf, M.M. et al., 2007).
The following table shows the previous researches using microarray
technology to detect, cure, or prevent various diseases including breast cancer.
Table 2.1: Previous researches using microarray technology
Author
Disease
Keltouma Driouch et al., 2007
Breast cancer
Lance D Miller and Edison T Liu, 2007
Breast cancer
Sofia K Gruvbeger-Saal et al., 2006
Breast cancer
Argryis Tzouvelekesis et al., 2004
Pulmonary
Gustavo Palacios et al., 2007
Infectious
Christian Harwanegg and Reinhard Hiller, 2005
Allergic
17
2.3.2.1 Gene Expression Profiling
In gene expression profiling or also known as mRNA experiment, the
thousand of genes levels are monitored simultaneously to study the effects of certain
treatments, diseases and developmental stages on gene expression (Adomas A. et al.,
2008). Typically, microarrays consist of several thousand single-stranded DNA
sequences, which are “arrayed” at specific locations on a synthetic “chip” via
covalent linkage. These DNA fragments provide a matrix of probes to label the
complementary RNA (cRNA) fluorescently which is derived from the sample of
interest. Each of gene expression is quantified by the intensity of the fluorescence
that emits from the specific location on the array matrix which proportional to the
amount of the gene product (Sauter, G. and Simon, R., 2002). There are two main
types of microarrays used to evaluate the clinical samples (Ramaswamy, S. and
Golub TR, 2002):
i)
cDNA array
This type of array also known as spotted arrays is build using
annotated cDNA probes or by reverse transcription of mRNA from a known
source. The spotted arrays used to generate cDNA probes for specific gene
product that will spot on glass slide systematically (Ramaswamy, S. and
Golub TR, 2002). By using human Universal RNA Standards, the sample
testing is extracted and reverse transcribed in parallel along with test samples.
It will be labeled using fluorescent dye, Cy3, while the cRNA from test
sample will be labeled with fluorescent dye, Cy5. The hybridization of each
Cy5-labeled test sample cRNA with the Cy3-labeled reference probe to the
spotted microarrays is done simultaneously. From the hybridization, the
fluorescence intensities of the scanned images are quantified, normalized, and
corrected to produce the transcript abundance of a gene as an intensity ratio
with respect to the reference sample (Cheung VG et al., 1999).
18
ii)
Oligonucleotides microarrays
25 pair base pair DNA oligomers are used in oligonucleotides
microarrays to be printed onto the matrix of array (Lockhart DJ et al., 1996).
This array that was designed and patented by Affymetric uses combination of
photolithography and combinatorial chemistry. This allows the thousands of
probes to generate simultaneously. In this array, the Perfect Match/Mismatch
(PM/MM) probe strategy is used to avoid nonspecific hybridization. By using
that strategy, a second probe which is identical to the first except for the
mismatched probe is laid adjoin to the PM probe. A signal from the MM
probed is subtracted from the PM probe signal to indicate the nonspecific
binding.
Figure 2.3: Gene Expression Profiling
Reprinted from Sauter, G., and Simon, R. (2002)
19
2.3.2.2 Data Analysis
In gene expression profiling, there are four levels of analysis that have been
applied. The levels of analysis are as follow (Compton, R., et al., 2008):
i)
Using the statistical test to identify the differences of genes expression
between two or more samples.
ii)
By using data reduction technique to simplify the complexity of the
dataset. The cluster of similar expression profile genes is identified
across different experimental.
iii)
A statistical model such as classifier used to identify genes predictive
of samples classes (cancerous and non-cancerous) or to the expression
of interesting biomaker. The evaluation of the accuracy of the model
is really important in this statistical modeling. Holdout method is the
classic approach in which the correct class labels are known and the
data is divided into a set of training and test. The training set builds
the model while the test set provides a group of samples that the
model unseen previously. Then, it can be used to test if the model
predicts their class membership on an independent population
correctly. Gene selection is required using this approach. The gene
may be tested individually (univariate gene selection) or in association
(multivariate selection strategies).
iv)
By using the reverse engineering algorithm to infer the transcriptional
pathway structures within the cells.
20
2.4
Feature Selection
Feature selection is the process of choosing the most suitable features when
building process model (Kasabov, 2002). The main purpose of feature selection in
pattern recognition, statitistic and data mining communities (text mining,
bionformatics, sensor networks) is to select a subset of the input variables by
eliminating features that have little or no predictive information. The
comprehensibilty of the resulting classifier model can significantly be improved by
the feature selection. Feature selection often builds a model of better generalization
to unseen point (YongSeong Kim, et al., 2003). The potential benefits of feature
selection as stated by (Gianluca Bontempi and Benjamin Haiben-Kains,2008) are as
follow:
i)
Facilitate the visualization and comprehensibilty of the data.
ii)
The measurement and storage requirement can be reduced.
iii)
Reduce the times of training and utilization.
iv)
Improve the performance of prediction by defying the curve of
dimensionality.
Feature selection is important for various reasons in many supervised
learning problems namely generalization performance, running time requirements,
and constraints and interpretational issues imposed by the problem itself (J.Weston et
al., 2000). The main goal of feature selection in supervised learning is to search for a
feature subset with a higher classification accuracy (YongSeong Kim, et al., 2003).
21
Evaluation function
Feature set
Selection of
starting feature set
Goodness
Generation of next feature set
Stopping criteria
Figure 2.4: Feature selection process
There are three main approaches of feature selection which are (YongSeong
Kim, et al., 2003) :
i)
Filter methods
The filter methods are the preprocessing methods that attempt to
evaluate the merits of features from the data. The performance of the learning
algorithm which is affected by the chosen feature subset will be ignored.
ii)
Wrapper methods
The subsets of variables is evaluated based on their usefulness to a
given predictor using these methods. In order to select the best subset, its own
learning algorithm will be used as a part of the evaluation function. A
problem of stochastic state space search is identified as the problem in these
methods.
iii)
Embedded methods
These methods implement variable selection as part of the learning
procedure and often specific to given learning machines.
22
Table 2.2: A taxonomy of feature selection techniques
Search Method
Filter
Univariate
Advantages
• Fast
• Scalable
• Independent of the classifier
Disadvantages
• Ignores features
dependencies
• Ignores interaction with
the classifier
Example
• χ2
• Euclidean distance
• Information gain, Gain ratio (BenBassat,
1982)
• t-test
Mutivariate
• Model features dependecies
• Independent of the classifier
• Better than wrapper method
in complexity computation
• Slower compares to
univariate techniques
• Less scalable compares to
univariate techniques
• Ignores interaction with
the classifier
• Correlation-based feature selection, CFS
(Hall, 1999)
• Markov blanket filter, MBF (Koller and
Sahami, 1996)
• Fast correlation-based feature selection,
FCBF (Yu and Liu, 2004)
• Relief ( Kenji Kira and Larry A. Rendell,
1992)
Wrapper
Deterministic
• Simple
• Risk of overfitting
• Interacts with the classifiers
• Prones to trap in a local
• Model feature dependencies
optimum compares to
randomized algorithm
• Sequential forward selestion, SFS
(Kittler, 1978)
• Sequential backward elimination, SBE
(Kittler, 1978)
• Plus q take -away r (Ferri et al., 1994)
23
• Less computationally
intensive compared to
• Classifier dependent
selection
• Beam search (Siedelecky and Sklansky,
1988)
randomized methods
Randomized
• Less prone to local optima
• Computationally intensive
• Simulated annealing
• Interacts with the classifier
• Classifier dependent
• Randomized hill climbing (Skalak, 1994)
• Model features dependencies
selection
• Higher risk of overfitting
compared to deterministic
Embedded
• Interacts with the classifier
• Computational complexity is
• Classifier dependent
selection
• Genetic Algorithms (Holland, 1975)
Estimation of distribution algorithm (Inza
et al.,2000 )
• Decision trees
Weighted naive Bayes (Duda et al., 2001)
better than wrapper method
• Feature selection using the weight vector
• Model features dependencies
of SVM (Guyon et al., 2002; Weston et
al., 2003)
24
Among the three main approaches, the filter approach are the computational
efficient and robust against overfitting. Although it may introduce bias but it may
have considerably less variance (Gianluca Bontempi and Benjamin HaibenKains,2008). Hence, a larger size of dataset can be handled by the filter method
compares to wrapper method (Guyon, I and Elisseeff A, 2003; Kohavi, R and John
G.H.,1997; Liu, H. and Motoda, H., 1997; Ahmad, A. and Dey, L., 2005; Dash, M.
And Liu, H.,1997). The common filter method drawback is the interaction with the
classifier will be ignored which means the search in the feature subset space is
separated from the search in the hypothesis space. The most suggested technique to
solve this problem is univariate. However, univariate technique may lead to worse
performance of classification due to the ignoring feature dependencies compares to
other techniques. Thus, various multivariate filter techniques are proposed to
overcome the problem which aims at the incorporation of feature dependencies to
some degrees (Saeys, Y. et al., 2007).
Relief is one type of the multivariate filter technique which leads to better
performance improvements and proves that the filter methods are capable to handle
the non-linear and multi-modal environments (Reisert, M., and Burkhardt, 2006).
This method is the most well known and deeply studied which estimation is better
than the usual stastical attribute such as correlation or covariance. This is because it
takes into account the attribute interrelationship (Arauzo-Azofra, A., et al., 2004). It
has been reported that this method has shown a good performance in various domain
(Sikonja, M.R. and Kononenko, I., 2003).
2.4.1
Relief
Relief is a feature selection method based on attribute information that was
introduced by Kira and Rendell (Kira, K., and Rendell, L.A., 1992). The original
25
Relief only handled boolean concepts problem but the extend of it has been
constructed to be used in classifications problems (Relief-F) (Kononenko, I., 1994)
and also in regression (RRelief-F) (Sikonja, M.R and Kononenko, I, 1997). In ReliefF, one nearest neighbor of E1 will be search from every class. With these neighbor,
the relevance of every feature f ∈ F will be evaluated by the Relief and accumulated
into the W[ f ] based on the equation (2.1).
W[ f ] = W[ f ] – diff ( f, E1, H ) + Σ
P ( C ) x diff (f, E1, M(C))
C ≠ class(E1)
(2.1)
H is a hit of the nearest neighbor which is from the same class while M(C) is the miss
from the different class of class C. W [ f ] is divided by m to get the average
evaluation in range of [-1,1]. The diff ( f, E1, H ) function computes the grade in
which the values of features f is different in examples E1 and E2 as in equation (2.2).
(2.2)
f is dicrete
f is continuous
diff ( f, E1,E2 ) = 0 if value ( f, E1 ) = value ( f, E2 ) |value ( f, E1 )- value ( f, E2 )|
1 otherwise
max (f) – min (f)
The value ( f, E1 ) indicate the value of feature f on example E1 while the max (f) is
the maximum value f gets. The distance used is the sum of differences considering
nearest neighbor given by diff function, of all features.
2.4.2
Information Gain
Information gain is commonly used as a surrogate for approximating a
conditional distribution in the classification setting (Cover, T., and Thomas, J).
26
According to Mitchell (1997), by knowing the value of a feature, information gain
measure the number of bits of information obtained for class prediction. The
following is the equation of information gain :
InfoGain = H(Y) – H(Y|X)
(2.3)
where
X and Y is the feature and
H(Y) = - Σ p (y) log2 (p(y))
(2.4)
yεY
H(Y|X) = - Σ p (x) Σ p (y|x) log2 (p(y|x))
xεX
2.4.3
(2.5)
yεY
Chi-Square
The Chi-square algorithm will evaluate genes individually with respect to the
classes. This algorithm is based on comparing the obtained values of the frequency
of a class because of the split to the expected frequency of the class. From the N
examples, let Nij be the number of samples of the Ci class within the jth interval
while MIj is the number of samples in the jth interval. The expected frequency of Nij
is Eij = MIj | Ci | lN . Therefore, the Chi-squared statistic of a gene is then defined as
follow (Jin, X., et al., 2006):
C
l
χ = Σ Σ (Nij - Eij)2
2
i=1 j=1
Eij
where I is the number of intervals.
The larger the the χ2 value, the more informative the corresponding gene is.
(2.6)
27
2.5
Radial Basis Function
A Radial Basis Function, RBF, was introduced by Powell to solve the
problem of the real multivariate interpolation (Powell M.J.D., 1997). In a neural
network, radial basis function is a set of functions formed by a hidden unit which
composes a random basis for the input vectors (Haykin, S., 1994). This function is a
real-valued in which the value merely depends on the distance from the origins. The
most general formula is:
h (x) = φ (x - c)2
(2.7)
2
r
where
φ is the function used (Gaussian, Quadratic, Inverse Multiquadric and
Cauchy)
c is the centre
r = R is the metric, Euclidean
2.5.1
Types of Radial Basis Function, RBF
Radial functions are class of function that has the characteristic feature where
response decreases or increases monotonically with distance from the origin. If the
radial functions is linear, all the center, the distance scale and the precise shape will
be fixed as a parameters of the model (Mark J.L.Orr, 1996). There are two common
types of Radial Basis Function based on the types of radial functions used. The types
of Radial Basis Function are as listed :
28
i) Gaussian Radial Basis Function
φ (z) = e –z
(2.8)
Figure 2.5: Gaussian RBF
ii) Multiquadric Radial Basis Function
φ (z) = (1 + z) ½
(2.9)
29
Figure 2.6: Multiquadric RBF
Among all of the types, Gaussian is a typical radial function and the most
commonly used. This is because Gaussian RBF monotonically decreases with the
origin and has a local response which gives a significant response only in a
neighborhood near the origin. Other than that, Gaussian RBF is biologically plausible
caused by its finite response. Multiquadric RBF is contrary to Gaussian RBF since it
monotonically increases with the distance from the origin and have a global
response. This is why the Gaussian RBF is commonly used compares to the
Multiquadric RBF. The following figure illustrates the different result of RBF types
with c = 0 and r = 1.
30
Figure 2.7: Gaussian (green), Multiquadric (magenta), Inverse-multiquadric (red)
and Cauchy (cyan) RBF
2.5.2
Radial Basis Function, RBF, Network
A neural network is designed as a machine to model the way of the brain
performs a particular tasks or function of interest (Haykin S., 1994). Neural network
has been successfully applied in various area including financial, engineering and
mathematics because of its useful properties and capabilities such as nonlinearity,
input-output mapping and neurobiological analogy (Mat Isa, N.A., et al., 2004). The
application of neural network has been expanded in the medical area with two
common applications which are image processing (Sankupellay, M., and
Selvanathan, N., 2002; Chang, C.Y., and Chung, P.C., 1999; Kovacevic, D., and
Loncaric, S., 1997) and diagnostic system (Kuan, M.M., et al., 2002; Ng, E.Y.K., et
al., 2002; Mat Isa, N.A., et al., 2002; Mashor, M. Y., et al., 2002). Neural network
has been proven to perform very well in both applications. A high capability can be
31
reached either to increase the medical image contrast or to detect certain region in the
medical image processing while in disease diagnostic system, it performs as good as
human experts or sometimes better than them (Sankupellay, M., and Selvanathan, N.,
2002; Chang, C.Y., and Chung, P.C., 1999; Kovacevic, D., and Loncaric, S., 1997;
Kuan, M.M., et al., 2002; Ng, E.Y.K., et al., 2002; Mat Isa, N.A., et al., 2002;
Mashor, M. Y., et al., 2002).
Radial Basis Function, RBF, networks is the artificial neural network type for
application of supervised learning problem (Orr, M.J.L., 1996). By using RBF
networks, the training of networks is relatively fast due to the simple structure of
RBF networks. Other than that, RBF networks is also capable of universal
approximation with non-restrictive assumptions (Park. J. et al., 1993). The RBF
networks can be implemented in any types of model whether linear on non-linear and
in any kind of network whether single or multilayer (Orr, M.J.L. 1996). The most
basic design of RBF networks as first proposed by Broomhead and Lowe in 1988 is
traditionally associated with radial functions in a single layer network. The following
figure shows the traditional RBF network with each of the n components of input
vector x feeds forward to m basis function. This yield a output that linearly combined
with weights {wj}jm=1 into the network output f(x).
32
Figure 2.8: The traditional radial basis function network
The first layers indicate the input layer which is the source nodes or sensory
units while the second layer is a hidden layer of high dimension. The last layer which
is the output layer will give the network response to the activation pattern that is
applied to the input layer. From the input space to the hidden unit space, the
transformation is non-linear but the transformation is linear from the hidden space to
the output space (Haykin, S., 1994).
To implement RBF network, we need to identify the hidden unit activation
function, the number of the processing units, a criterion for modeling the given task
and a training algorithm for finding the parameters of the network. The network will
be trained to find the RBF weight while the training set, a set of input-output pairs
will optimize the network parameters so that the network output will fit with the
given inputs. To evaluate the fits, the means of cost function or assumed as mean
square error will be used. The network can be used after the training with the data in
which the underlying statistics is similar to that of the training set. The adaptation of
the network parameters to the changing data statistics is caused by the on-line
training algorithms (Moody, J., 1989).
33
There are various functions that have been tested as activation function for
RBF networks (Matej, S., Lewitt, R.M, 1996) but Gaussian is preferred in the
application of pattern classification (Poggio, T., Girosi, F., 1990; Moody, J., 1989;
Bors, A.G., Pitas, I., 1996; Sanner, R.M., Slotine, J.J.E., 1994; Bors, A.G., Gabbouj,
G., 1994). The following is the Gaussian activation function for RBF network:
φj (X) = exp [(X - μj)T Σj-1 ( X - μj)]
(2.10)
for j = 1,..,L where
X is the input feature vector.
L is the number of hidden units.
μj and Σj are the mean and the covariance matrix of the jth Gaussian function.
The mean of the vector μj is representing the location while Σj models the shape of
activation function. The output layer applies a sum of weight of hidden unit output
according the following formula:
ψk(X) = Σ λjk φ j (X)
(2.11)
for k = 1,..,M where
λjk are the output weight, each corresponding to the connection between a
hidden unit and an output unit.
M represent the number of output units.
For classification problem, if the weight λjk > 0, the activation field of the hidden unit
j contained in the activation field of the output unit k.
34
2.6
Classification
Classification can also be termed as supervised learning (Tou, J.T. and
Gonzalez, R.C., 1974; Lin, C.T., and Lee, C. S. G., 1996). In the classification
problems, the previous unseen patterns will be assigned to their respective classes
according to previous examples from each class. This will cause the output of the
learning algorithm as one of a discrete set of possible classes (Mark J.L.Orr, 1996).
The feature entries will be represented by the input, the hidden layer unit correspond
to the subclasses while the class corresponded by each output (Tou, J.T. and
Gonzalez, R.C., 1974). There are several methods of classification (Bashirahamad
S.Momin et al., 2006):
i)
Decision Tree
A decision space divided into piecewise constant region (Pedrycz, W.,
1998)
ii)
Probabilistic or generative models
Compute the probabilities based on Bayes’ Theorem (Tou, J.T. and
Gonzalez, R.C., 1974).
iii)
Nearest-Neighbor classifiers
Calculate the minimum distance from instances or prototype (Tou,
J.T. and Gonzalez, R.C., 1974).
iv)
Artificial Neural Networks (ANNs)
The division is by nonlinear boundaries. The learning incorporated, in
the environment of data-rich by encoding information in the
conversion weights (Haykin, S., 1994).
35
2.6.1
Comparison with others Classifiers
The following table shows the previous researches that have be done on
classification with various type of data using the RBF network.
Table 2.3: Previous researches on classification using RBF network
Author
Classification
Wang, D.H., et al., 2002
Protein sequences
Li, X., Zhou, G., and Zhou, L., 2006
Novel data
Leonard, J.A., and Kramer, M.A., 1991
Process Fault
Horng, M-H., 2008
Texture of the Ultrasonic Image
of Rotator Cuff Diseases
Kégl, B., Krzyzak, A., and Niemann, H.
Nonparametric (Pattern
(1998)
Recognition)
Based on (Wang, D.H., et al.,2002) Self Organizing Mapping (SOM) networks
(Wang, H.C et al.,1998; Ferran EA., 1994) and Multilayer Perceptrons (Wu C.H. et
al., 1995; Wu C.H., 1997) are the two types of neural models applicable for protein
sequences classification task. By using the SOM networks, the relationship within a
set of proteins can be discovered by clustering them into different groups. However
SOM networks have two main limitation when applied in protein sequences
classification. The limitations are:
i) Time consuming and the difficulty of the results interpretation.
ii) The size selection of Kohonen layer is subjective and often involves the trial
and error procedure.
There are several undesired features arising from system designed of MLP based
classification system which are:
36
i)
The determination of the neural metwork architecture.
ii)
Lack of intepretability of the ”black box”.
iii)
The training time needed as the number of inputs is over 100.
In (Li, X., Zhou, G., and Zhou, L., 2006), RBF network has been compared to
the Support Vector Machine (SVM). Although data classification is one of the main
applications that RBF networks have applied (Oyang, Y.J et al., 2005; Sun, J. et al.,
2003), recent research has focused more on SVM. This is because it has been
reported by several latest research that SVM generally capable to deliver higher
classification accuracy compared to other existing data classification algorithms.
However, SVM suffer with one serious disadvantage namely the time taken to carry
out the model selection which could be unacceptably long for some contemporary
applications. All the proposed approaches for expediting the model selection of SVM
lead to lower prediction accuracy.
Backpropagation network and Nearest Neighbor classifiers have been
compared in (Leonard, J.A., and Kramer, M.A., 1991). The comparison have shown
that the backpropagation classifier can produce nonintuitive and nonrobust decision
surfaces due to the inherent nature of the sigmoid function, the definition of the
training set and the error function used for training. Other than that, there is no
standard training scheme mechanism for identifying unknown classes of regions in
backpropagation network. For the Nearest Neighbor classifier, it becomes inefficient
when the number of the training point is high. This is because it needs a complete
search via the set of training examples to classify each test point.
37
2.7
Receiver Operating Characteristic, ROC
The Reciever Operating Characteristic, ROC, analysis emerges from the
statistical decision theory (Green and Swets, 1996). In the medical area, this theory
was first applied during the late 1960s. This theory analysis has been increasingly
used as evaluation tool in computer science to assess distinction effects among
different methods (Westin, L. K., 2001 ). In neural classifier, ROC analysis is easy to
implement, yet rarely used to evaluate the performance of neural classifier outside of
the medical and signal detection field. The advantages of ROC analysis are the
robust description of the network’s predictive ability and an easy way to change the
existence network based on differential cost of misclassification and varying prior
probabilities of class occurences.
Reciever Operating Characteristic, ROC, curve is the ultimate metric to
detect algorithm performance (John Kerekes, 2008). The ROC curve is defined as
sensitivity and specificity which are the number of true positive decision over the
number of actually positive cases and the number of true negative decisons over the
number of actually negative cases, respectively. This method leads to the
measurement of diagnostic test performance (Park, S.H., et al., 2004) . Based on the
ROC curve, area under the curve, AUC, is the most commonly used and
comprehensive of the performance measures (Downey, T.J., Jr., et al., 1999). The
following equation is the formula of the AUC. The TPR is referred to True Positive
Rate while the FPR is referred to False Positive Rate.
1
ROC = (TPR)( FPR)
2
(2.8)
3
38
Figure 2.9: The RO
OC curve
2
2.8
Sum
mmary
pter discussees about the breast canceer, microarraay
In brrief, this chap
t
technology,
feature selecction, radial basis function, RBF nettwork, and reeceiver
o
operating
ch
haracteristic, ROC. The bbiopsy is thee way to confirm the occcurrence of
c
cancer
cells in the breastt after the m
mammograph
hy. By doing biopsy, the DNA
m
microrrays
w be obtainned. In order to select thhe useful genne, feature seelection needds
will
t be appliedd first. Theree are variouss techniques of feature seelection whiich are
to
c
categorized
into three methods.
m
The filter methood with the m
multivariate techniques
t
n
namely
ReliefF has beenn chosen to bbe applied inn this study. Based on th
he previous
r
researches,
t Radial baasis functionn network haas been show
the
wn the best network
n
in
c
classification
n problem. Therefore,
T
it has been ch
hosen as the classifier to distinguish
t cancerouus and non-ccancerous. Thhe receiver operating
the
o
chharacteristic, ROC curve
m
metric
is useed to evaluatte the perform
mance of thee classifier.
CHAPTER 3
METHODOLOGY
3.1
Introduction
This chapter elaborates the processes or steps which are involved during the
design and implementation phase. The process flows will be illustrated in the
research framework which contains four main steps. Begin with the data acquisition,
the format of the data will be explained briefly. Other than that, the HykGene
software that will be used in the experiment will be discussed according to its
framework.
40
3.2
Research Framework
The following figures describe the research framework for this study. There
are four main steps or processes involved which are data acquisition, feature
selection, classification and evaluation. In the data acquisition steps, the data set of
breast cancer microarray data is obtained. Then, we can proceed to the second step
which involved feature selection. This step is very important because it leads to the
accuracy of the classification. The classification of the microarray data will be
implemented to classify the cancer cells and the normal cells. The final step in this
framework is to evaluate the performance of the classifier using ROC.
Start
Data Acquisition
Feature Selection
Classification
Evaluation
End
Figure 3.1: Research Framework
41
3.3
Software Requirement
In this study, the software requirements include the Matlab version 7.0 Neural
Network toolbox to develop the feature selections function and the classifier namely
radial basis function, RBF, network. Other than that, another two software will be
used which are WEKA and HykGene to support the feature selection function.
3.4
Data Source
The breast cancer microarray data will be obtained from the Centre for
Computational Intelligence (http://www.c2i.ntu.edu.sg/) for this study. The dataset
which contains 97 instances with 24461 genes and 2 classes are in the ArtificialRelation File Format, ARFF. An ARFF format will be explain in detail in the next
chapter.
3.5
HykGene
HykGene is a hybrid approach for selecting marker genes for phenotype
classification using microarray gene expression data and have been developed by
Wang, Y. et al., (2005). The following figure indicates the framework of the
HykGene.
42
Figure 3.2: HykGene: a hybrid system for marker gene selection
(Wang, Y. et al., 2005)
In the fist step, feature filtering algorithms is applied on the training data to
select a set of top-ranked informative genes. Next, to generate a dendogram,
hierarchical clustering is applied on the set of top-ranked genes. Then, in the third
step a sweep- line algorithm is used to extract clusters from the dendogram and the
marker gene will be selected by collapsing clusters. The LOOCV will determine the
best number of marker genes. In the last step, only the marker genes will be used as
the features for classification of test data.
For the first step which is gene ranking, feature filtering algorithms are
applied to rank the genes. Features or genes are ranked based on the statistic over the
features that indicate which features are better than others. In this HykGene
software, three popular feature filtering methods have been used which are ReliefF,
Information Gain and Chi-Square or χ2-statistic.
After done with the feature filtering on the whole set of genes using training
data, the top-ranked genes will be pre-selected from the whole set. Normally, there
are 50 to 100 genes are pre-selected. Next, the hierarchical clustering (HC) will be
43
applied on the set of pre-selected genes. These will resulting a dendogram, or
hierarchical tree where heights represent degrees of dissimilarity. Due to the high of
correlation of the some genes in the pre-selected set of genes, the cluster will be
extracted by analyzing the dendogram output of the HC algorithm. Then, each cluster
will be collapsed into one representative gene from each cluster. These extracted
representative genes were used as marker genes in the classification step.
Finally, the test data will be classified using one of the four classifiers which
are k-nearest neighbor (k-NN), linear support vector machine (SVM), C4.5 decision
tree and Naive Bayes (NB).
3.5.1
Gene Ranking
The estimation accuracy of the features or attributes quality is important in
the preprocessing step of classification. Therefore, to produce an ideal estimation of
attributes in feature selection, the noisy and irrelevant features need to be eliminated.
Other than that, the features which are having the specific characteristics of a class
need to clearly select. As illustrated before in the HykGene framework, feature
selection or gene ranking is the first step which involved three feature filtering
method. Thus, the original framework can be expanded into the following figure in
order to show the methods that involved as mentioned previously more clearly.
44
ReliefF
Training
data
Step1: Ranking
Genes
Information
Gain
Topranked
Genes
χ2-statistic
Figure 3.3: The expanded framework of HykGene in Step1
3.5.1.1 ReliefF Algorithm
The main idea of the Relief algorithm that was proposed by (Kira and
Rendell, 1992) is to estimate the quality of attributes that have weights greater than
the threshold using the distinction of an attribute value between a given instance and
the two nearest instances namely Hit and Miss. The following is the original
algorithm of Relief.
45
Input : a vector space for training instances with the value of
attributes and class values
Output : a vector space for training instances with the weight W of
each attribute
Set all weights W[A]=0.0
for i=1 to m do begin
randomly select instance Ri
find nearest hit H and nearest miss M
for A=1 to all attribute do
W(A) = W(A) – diff (A, Ri, H)/m + diff (A, Ri, M)/m
end
Figure 3.4: Original Relief Algorithm
(Park, H., and Kwon, H-C., 2007)
The drawback of the original Relief algorithm is that it cannot deal with incomplete
data and multiclass datasets. To overcome the disadvantages of original algorithm,
the extension of Relief-F algorithm was suggested by Kononenko (1994). This
extension algorithm is more robust, meaning it can handle incomplete and noisy data
as well as multi-class dataset. The updated Relief-F algorithm is as follow.
46
Input : a vector space for training instances with the value of
attributes and class values
Output : a vector space for training instances with the weight W of
each attribute
Set all weights W[A]=0.0
for i=1 to m do begin
randomly select instance Ri
find k nearest hit Hj
for each class C≠ class(Ri) do
find k nearest miss Mj(C) from class C
for A=1 to all attribute do
k
k
W(A) = W(A) - Σ diff (A, Ri, Hj)/(m, k) + Σ
j=1
k
[P(C)/
C≠ class(Ri)
1-P(class(Ri)) Σ diff(A, Ri, Mj(C))]/(mk)
j=1
end
Figure 3.5: ReliefF algorithm
(Park, H., and Kwon, H-C., 2007)
According to the algorithm, the nearest hits Hj is finds k the nearest neighbors which
is from the same class while the nearest misses Mj(C) is k the nearest neighbors
which is from a different class. W (A) which is the quality estimation for all attributes
A is updated based on their values for Ri, hits Hj and M(C). Each probability weight
must be divided by 1-P (class (Ri)) due to the missing of the class of hits in the sum
of diff (A, R, Mj(C)).
47
3.5.1.2 Information Gain
The second method of feature selection that will be used in the experiment is
Information Gain. In the Hykgene software, the equation of the information gain
method applied is as following.
Let {ci}mi=1 denote the of classes. Let V be the set of possible values for feature f.
Thus, the information gain of a feature f is defined as:
m
m
G(f) = - Σ P(ci)log P(ci) + Σ Σ P (f = v) P(ci | f = v) log P (ci | f = v)
i=1
(3.1)
vεV i=1
In the information gain, the numeric features need to be discretized. Therefore,
HykGene software has used entropy-based discretization method (Fayyad and Urani,
1993) that has been implemented in WEKA software (Witten and Frank, 1999).
3.5.1.3 χ²-statistic
Chi- Square or χ2 –statistic is one more feature selection method that will be
used. The equation of this method that has been used by HykGene software is as
follow:
m
χ (f) = Σ Σ [Ai ( f = v) – Ei( f = v)]2
2
vεV i=1
Ei( f = v)
where
V is the set of possible values for feature f,
(3.2)
48
Ai ( f = v) is the number of instances in class ci with f = v
Ei( f = v) is the expected value of Ai ( f = v)
Ei( f = v) is computed with:
Ei( f = v) = P ( f = v)P(ci)N
(3.3)
where
N is the total number of instances
Like information gain, this method also requires numeric features to be discretized,
so the same discretization method is used. The following is the example of the chisquare algorithm which is consists of two phases. For discretization, in the first
phase, it begins with a high significance level (sigLevel) for all numeric attributes.
The process involved in phase 1 will be iterated with a decreased sigLevel until an
inconsistency rate, δ is exceeded in the discretized data. In the Phase 2, begin with
sigLevel0 determined in Phase 1, each attribute i is associated with a sigLevel [i] and
takes turns for merging until no attribute’s value can be merged. If an attribute is
merged to only one value at the end of Phase 2, it means that this attribute is not
relevant in representing the original dataset. Feature selection is accomplished when
discretization ends.
49
Phase 1:
set sigLevel = .5;
do while (InConsistency (data) < δ) {
for each numeric attribute {
Sort(attribute, data);
chi-sq-calculation(attribute, data)
do {
chi-sq-calculation(attribute, data)
} while (Merge (data))
}
sigLevel0 = sigLevel;
sigLevel = decreSigLevel(sigLevel);
}
Phase 2:
set all sigLvl [i] = sigLevel0
do until no-attribute-can be-merged {
for each attribute i that can be merged {
Sort(attribute, data);
chi-sq-initialization(attribute, data);
do {
chi-sq-calculation(attribute, data)
} while (Merge(data))
if (InConsistency (data) < δ)
sigLvl [i] = decreSigLevel(sigLvl [i]);
else
attribute i cannot be merged
}
}
Figure 3.6: χ²-statistic algorithm
(Liu, H., and Setiono, R., 1995)
50
3.5.2
Classification
Classification is a system capability to learn from a set of input or output
pairs which are composed of two set namely training and test. The input of the
training set is the features vector and class label while the output is the classification
model. For the test set, the inputs are the features vector and also the classification
model while the output is the predicted class. The classification is the last step
applied in the HykGene software after selecting the marker genes. There are four
classifiers that can be chosen as indicate in the following expanded framework of
HykGene.
k- nearest
neighbor
Test
data
Step 4:
Classification
using Marker
Genes Only
Support vector
machine
C4.5 decision
tree
Naïve Bayes
Figure 3.7: The expanded framework of HykGene in Step 4
3.5.2.1 k- nearest neighbor
The k- nearest neighbor or k-NN classifier (Dasarathy, B., 1991) is a wellknown non-parametric classifier. For the classification of a new input x, the k-NNs
are retrieved from the training data. Then, the input x is labeled with the majority
class label corresponding to the k-NNs.
51
3.5.2.2 Support vector machine
Support vector machine, SVM, is a type of the new generation of learning
system based on recent advances in statistical learning theory (Vapnik, V.N., 1998).
In the HykGene software, a linear SVM is used in order to find the separating
hyperplane with the largest margin. This is defined as the sum of distances from a
hyperplane (implied by a linear classifier) to the closest positive and negative
examplars with an expectation that the larger the margin, the better the generalization
of the classifier.
3.5.2.3 C4.5 decision tree
The C4.5 decision tree classifier is based on a tree structure where non-leaf
nodes represent test on one or more features and leaf nodes reflect classification
results. The classification of an instance is starting at the root node of the decision
tree, testing the attributes specified by this node then moving down the tree branch
corresponding to the value of the attribute. The process is iterated at the nodes on
that branch until a leaf node is reached. The J4.8 algorithm in Weka is used in the
HykGene software which is a Java implementation of C4.5 Revision 8.
52
3.5.2.4 Naïve Bayes
The fourth classifier used in the HykGene software is the Naïve Bayes
classifier which is a probabilistic algorithm based on Bayes’ rule. This classifier
approximates the conditional probabilities of classes using the joint probabilities of
training sample observation and classes.
3.6
Classification using Radial Basis Function Network
There are various approaches to solve the classification problem but as
discussed in the previous chapter, radial basis function network have been chosen as
the classifier. This is because others than previous discussion of the advantages of
RBF network, data classification is one of the main applications in RBF network.
To construct or build the classifier, a neural network will be embedded with
the radial basis function network in which each of the hidden unit implements a
radial activation function. The weighed sum of hidden unit outputs will be produce
by the output units. The RBF network is nonlinear from the input but the output is
linear. Typically, the RBF network is a three-layer, feed forward network which is
composed of input layer, hidden layer and output layer. As discussed in the literature
review, Gaussian has been chosen as the activation function. The following figure is
the architecture of radial basis function, RBF, network.
53
Figure 3.8: Radial Basis Function Network architecture
The Gaussian activation function that has been chosen as activation function
in the hidden layer has the following formula:
φj (X) = exp[-║ X-μj║2]
2σ2j
(3.1)
For j=1,…,L, where
X is the input selected l-dimensional feature vector
L is the number of hidden units
μj is the mean
σj is the standard deviation of the jth Gaussian activation function
the norm is the Euclidean
The following equation (3.2) is the formula of the output layer which is implements a
weighted sum of hidden-unit outputs.
L
ψk (X) = Σ λjk φj(X)
j=1
for k = 1,…,M where
(3.2)
54
λjk are the output weights, each corresponding to the connection between a
hidden unit an output unit
M refer to the number of output units
The weights λjk show the contribution of a hidden unit to respective output
unit.
The output of the radial basis function is often limited to the range (0, 1) by using the
sigmodial function given as follow:
Yk (X) =
1
, k =1,…,M
1+ exp [-ψk(X)]
(3.3)
A training mechanism is to minimize the sum of square error between the desired
output and the actual output Yk (X).
L
MSE = ½ Σ Σ (tpi - ypi) = ½Σ E (p)
p j=1
(3.4)
L
where
tp and yp are the i-th target and the network output for the p-th input respectively
3.7
Evaluation of the Classifier
Receiver operating characteristic, ROC, curves are widely used in the
medical area to evaluate the performance of a diagnostic test. This method consists a
lot of information for comprehensibility and improving classifiers performance.
However, it requires visual inspection because the best classifiers are hard to
recognize when the curves are mixed. Therefore the area under the ROC curve will
be used to help in deciding the suitable one for the problem. The Area Under Curve,
55
AUC, will summarized the statistic of diagnostic performance and evaluate the
ranking in terms to split of the classes. The following figure shows the algorithm of
AUC.
Algorithm. Calculating the area under an ROC curve
Inputs: L, the set of test examples: f(i), the probabilistic classifier’s
estimate that examples i is positive: P and N, the number of positive and
negative examples.
Outputs: A, the area under the ROC curve.
Require: P > 0 and N > 0
1. Lsorted
L sorted decreasing by f scores
2. FP
TP
0
3. FPprev
TPprev
0
4. A
0
5. f prev
-∞
6. i
1
7. while i ≤ | Lsorted | do
8.
if f(i) ≠ f prev then
9.
A
A+ TRAPEZOID_AREA (FP, FPprev,TP, TPprev)
10.
f prev
f(i)
11.
FPprev
FP
12.
TPprev
TP
13. end if
14. if i is positive example then
15.
TP
TP + 1
16. else /* i is negative example */
17.
FP
FP + 1
18. end if
19. i
i+1
20. end while
21. A
A+ TRAPEZOID_AREA (N, FPprev, N, TPprev)
22. A
A / (P x N) /* scale from P x N onto the unit square */
23. end
1. function TRAPEZOID_AREA(X1, X2, Y1, Y2)
2. Base
| X1 – X2|
3. Heightavg
(Y1 + Y2) / 2
4. return Base x Heightavg
5. end function
Figure 3.9: AUC algorithm
(Fawcett, T., 2005)
56
Based on the algorithm, the trapezoids are used rather than rectangles and thus, the
successive region of trapezoids added to A in order to average the effect between
points. The algorithm divides A by the total possible area to scale the value to the
unit square.
3.8
Summary
In this chapter, the processes or steps that involve in the study were briefly
discussed. The flow of the process has been described by the research framework
which consists of four main steps. In the HykGene software, three filtering methods
and four classifiers can be chosen to carry out the experiment. This chapter will be
guidelines in the design and implementation phase for the next chapter.
CHAPTER 4
DESIGN AND IMPLEMENTATION
4.1
Introduction
In this chapter, all experiments that were carried out on breast cancer
microarray data will be discussed in detail. These experiments involve features
selection, classification and evaluation of the classifiers. Breasts cancer microarray
data is needed to proceed with the experiments. The data sets will be explained
thoroughly in the first section of this chapter and followed by the elaboration of the
features selection techniques. The following section explains about the classifications
of selected genes by several classifiers. Finally, the last section will discuss the
classifier evaluation done by ROC.
58
4.2
Data Acquisition
Figure 4.1: The data matrix
(Wang, Y. et al., 2005)
The microarray data can be stored in an N x (M + l) matrix as shown in
Figure 4.4. Each of the first M columns represents a gene while each row represents a
tissue sample. gi,j is a numeric value indicate the gene expression level of gene i in
sample j. lj in the last column is the class label for sample j. Each gene is labeled by
Expressed Sequence Tag (EST). EST is used to identify gene transcripts and also the
tools in discovered and determined genes (Adams, M.D. et al., 1991). Several groups
such as Phil Green’s group had assembled EST into EST – contigs in order to reduce
the number of EST for downstream gene discovery analysis.
The EST-contigs are oriented by a procedure which involves the Basic Local
Alignment Search Tool (BLAST) similarity searching contradict a database of
mRNAs and putatively oriented ESTs, a procedure estimated to be 90 percent
accurate (Burchard, J., unpublished ). EST-contigs with significant BLAST hits were
correspond with the accession number of the sequence to which they had the best hit.
The accession numbers of these sequences were altered by addition of “_RC” to the
end presenting the distinct in orientation from the original sequence.
59
Oligonucleotides are often used as probes for detecting DNA or RNA
samples because they readily bind to their respective complementary nucleotide
sequences. The following table mapping the EST-contigs with GenBank accession
number and oligonucleotide probe sequence.
Table 4.1: EST-contigs, GenBank accession number and oligonucleotide probe
sequence
Phil Green Contig
GenBank
Oligonucleotide Probe Sequence
Accession
Number
Contig45645_RC
AI151500
CACACTCTCTTCGAACCCAATTGTGGGTGTA
GCAATGAAAGCAATATGATATGCTGCAGT
Contig44916_RC
AI279346
TTGTCCCTTTTGTCTCAAAATTTTCAAAACAG
CAACACCATACATGTGTCATTATAGGGG
Contig29982_RC
AI277612
TAAATGTCCTAATGATGAGAAACTCTCTTTC
TGACCAATTGCTATGTTTACATAACACGC
As mentioned in Chapter 3, the breast cancer microarray data used in this study is
obtained from the Centre for Computational Intelligence. The data contains 78
training data and 19 testing data set. For the training data, 34 samples are from the
patients who had developed distances metastases within 5 years which is labeled as
relapse. The rest of the 44 samples are from patients who remained healthy from the
disease after their initial diagnosis for at least 5 years which is labeled as non-relapse.
In the training data, there are 12 relapse and 7 non-relapse samples correspondingly.
The following figure shows fifteenth of the instances of the microarray breast cancer
dataset with certain of its attributes.
60
Figure 4.2: Fifteenth of the instances of breast cancer microarray dataset
The values of Ratio have been extracted from the original microarray data
with the replacement of all Not a Number or NaN to 100.0. If the ratio is greater than
one, it indicates that the binding of the test sample is greater than the control sample
which is known as up-regulation. If the ratio is smaller than one, it shows that the
binding of the test sample is less than the control sample and known as downregulation. Up-regulation and down-regulation are the regulations of gene
expression. Down-regulation is the process which the quantity of cellular component
such as RNA protein is decreased by the cells in response to an external variable
whereas up-regulation is an increase of cellular component.
In the microarray image, the green spot represents down-regulation while the
red spot indicates up-regulation. If the spot shows incandescent yellow, it indicates
that the binding between samples are equal or constant regulation whereas if the spot
appears in black, it signifies that there is no detectable signal.
61
Figure 4.3: Microarray Image
4.2.1
Data Format
The breast cancer microarray data obtained from the Centre for
Computational Intelligence is in Attribute-Relation File Format, ARFF. This file
format is an ASCII text file that illustrates a list of instances sharing a set of
attributes. An ARFF file is used as the format of the data because this file has been
developed to be implemented with WEKA machine learning software. The ARFF
file contains two different sections which are Header information and Data
Information. The Header of the ARFF file consists of relation declaration and
attributes declarations while in the Data section, it contains data declaration line and
actual instances lines. The following figures show the Header and Data information
of the ARFF file.
62
@relation Breast
@attribute Contig45645_RC numeric
@attribute Contig44916_RC numeric
@attribute D25272 numeric
@attribute J00129 numeric
@attribute Contig29982_RC numeric
@attribute Contig26811 numeric
Figure 4.4: Header of the ARFF file
@data
-0.299,0.093,-0.215,-0.566,-0.596,-0.195,0.039,-0.409,-0.352,0.066,0.136,
-0.336,-0.001,-0.272,-0.012,-0.141,0.037,-0.055,-0.141,0.146,-0.087,0.028,
-0.172,-0.005,-0.064,-0.147,0.039,-0.148,-0.081,0.061,0.037,-0.112,0.106,
-0.117,-0.069,0.064,0.086,0.062,-0.054,- 0.007,0.259,0.383, 0.049,0.383,0.094,
0.152,-0.161,0.121,0.021,0.246,-0.042,0.08,0.006,-1.082,- 0.085,0.213,100,
0.009,-0.024,0.049,0.145,0.071,-0.097,0.035,0.083,-0.119,-0.087,-0.087,-0.188,
-0.447,0.058,0.283,0.093,0.098,0.08,-0.12,0.039,-0.252,0.389,-0.257,-0.088,0.356,-0.43,0.148,0.063,-0.035,-0.069,0.27,0.363,-0.073,-0.018,-0.005,- 0.054,
0.006,-0.052,-0.129,0.008,-0.169,-0.063,0.029,0.054,-0.137,-0.107,-0.185,.035,0.109,0.289,-0.08,-0.15,-0.006,0.258,-0.263,0.15,0.324,-0.128,0.071,0.03,0.055,-0.238,-0.207,0.525,100,-0.149,-0.064,0.369,0.036,-0.041,-0.211,0.073,0.063,-0.229,0.146,-0.23,-0.112,-0.076,0.06,0.264,0.077,0.135,0.033,0.222,0.105,0.193,0.024,0.115,-0.235,-0.282,-0.063,- 0.133,0.074,
0.155,0.003,0.069,0.083,0.077,-0.125,0.164,0.381,0.074,0.096
Figure 4.5: Data of the ARFF file
The @relation, @attributes and @data declaration are case insensitive. The
sequence of the attribute declarations of the @attribute statements is ordered. In the
dataset, each attribute has its own @attribute statement that defines the name of its
attribute and its data type uniquely. The sequence order of the attribute indicates the
position of the column in the data section of the file. There are four types of the data
type which supported by WEKA namely numeric attributes, nominal attributes,
string attributes and date attributes.
63
4.3
Experiments
4.3.1 Genes Ranked
4.3.1.1 Experimental Settings
The process of selecting informative genes was implemented in Java and
Matlab using Weka 3.6.0 and PRTools 3.1.7. All three features selection techniques
in HykGene software were used in the experiments on breast cancer microarray data.
For comparison and demonstrating the effectiveness of each feature selection
algorithm, classification using four classifiers in the HykGene software was
conducted. The classifications had been done on 50 and 100 top-ranked genes which
were produced by each filtering methods.
Based on the cross validation classification accuracy, the best features
selection techniques can be chosen for this study. This is because the performance of
the classifiers is also depends on the selected genes during classification. If the
accuracy is high it means that the classifier had classified selected genes with high
number of informative genes and vice versa.
64
4.3.2 Classifications of top-ranked genes
4.3.2.1 Experimental Settings
One of the binary classification tasks is medical testing to determine whether
a patient has certain disease or not. Binary classification is a task to classify the
members of the given set of objects into two groups on the basis whether they have
some property or not. The classification property of the medical testing is the disease.
In order to classify the selected genes of breast cancer microarray data into two
classes which are relapse and non-relapse, the experiments were done using Weka
3.6.0.
Before classifying the selected genes, they need to be split into training and
testing set. This is because supervised learning or classification is the machine
learning method for deducing a function from training data. A training set is a set of
data used to discover potentially predictive relationships while a testing set is a set of
data used to evaluate the strength and utility of a predictive relationship. Therefore,
the 50 and 100 top ranked genes of breast cancer microarray data were split into both
set of 66 percent.
There are three others classifiers beside RBNF were used in the experiment
such as Multilayer Perceptron (MLP), Naïve Bayes (NB) and C4.5. MLP is the
feedforward neural network model while NB is a classifier which applies Bayes’s
Theorem. A C4.5 is an algorithm used to generate decision tree which can be applied
in classification and known as statistical classifier. These three classifiers are also
used in the experiments in order to asses the effectiveness of chosen classifier
(RBNF) based on the accuracy of correctly classified instances.
65
4.3.3 Evaluation of Classifiers Performances
4.3.3.1 Experimental Settings
Mostly, the prognostic performance will be defined by the percentage of the
prognostic decisions that turned out to be correct. However, this would be wrong to
say before looking at the ROC analysis results (Tokan, F., Turker, N., and Tulay, Y.,
2006). With performing ROC analysis, the real performances of the networks can be
found. This can help to decide which classifier has the best performances for breast
cancer microarray data.
ROC can be represented by plotting the fraction of the true positive versus
false positive. A ROC space which depicts relative trade-offs between true positive
and false positive is defined by FPR and TPR as x and y axes respectively. TPR or
also known as sensitivity determine the performance of the classifier on classifying
instances correctly among all positive samples. FPR or also called as 1-specificity
define the number of incorrect positive results among negative samples. The
following figure illustrates the ROC space.
66
Figure 4.6: ROC Space
Based on the figure, we can see that ROC space is divided by the diagonal
line which is referred to as line of no discrimination (random guess). This line splits
the ROC space in area of good or bad classification. A good classification is shown
by the point above the diagonal line while the points below the line show the wrong
results. The perfect classification yield when a point in upper left corner or its
coordinate is (0,1) of the ROC space. This result represents 100 percent of sensitivity
which means no false negatives and specificity which means no false positives.
With the values of FPR and TPR, a ROC curve can be drawn. However, the
curves of the networks are hard to be investigated by visual inspection in ROC curve.
Therefore, the area under ROC curve, AUC, would be helpful to compare the
performance of the networks. The perfect classifier has an AUC of 1 while
completely random classifier has an AUC of 0.5. In order to evaluate the
performances of the classifiers in this study, the experiments were also carried out
using Weka 3.6.0. The following figure depicts the framework to plotting multiple
ROC curves using Weka.
67
Figure 4.7: Framework to plotting multiple ROC curve using Weka.
4.5
Summary
Overall, this chapter discussed the experiments that have been carried out
throughout the study. Features selection was the first experiments performed
followed by the classifications. There are three methods used which are Relief-F,
Information Gain and Chi Square in the features selection experiments. The selected
genes yield by the chosen features selection technique will be further used in the
classifications experiments. Four classifiers were involved in the classification
experiments where the performance of each of them is evaluated by ROC. All the
experimental results will be further analyzed in the next chapter.
CHAPTER 5
EXPERIMENTAL RESULTS ANALYSIS
5.1
Introduction
The experimental results will be analyzed and discussed in this chapter.
Comparisons of the feature selection results yielded by three types of techniques will
determine the best method for genes selection. The classification results of each
classifier will be analyzed based on the accuracy percentage and time taken to build
the network. These classifiers will be evaluated to show the performance of the best
classifier according to the area under ROC.
69
5.2
Results Discussion
5.2.1
Analysis Results of Features Selection Techniques
The experimental results of features selection are tabulated in the tables 4.1
until 4.3. Figure 4.4 and 4.5 indicate the graph of the cross validation classification
accuracy based on SVM in order to show the comparison clearly.
Table 5.1: Results on breast cancer dataset using Relief-F
50-top ranked genes
100-top ranked genes
Cross Validation
Cross Validation Classification
Classification Accuracy (%)
Accuracy (%)
k-NN
86.60
87.63
SVM
86.60
83.51
C4.5
68.04
65.98
Naive Bayes
80.41
78.35
Classifier
Table 5.2: Results on breast cancer using Information Gain
50-top ranked genes
100-top ranked genes
Cross Validation
Cross Validation Classification
Classification Accuracy (%)
Accuracy (%)
k-NN
79.38
77.32
SVM
73.20
72.16
C4.5
76.29
70.13
Naive Bayes
58.76
57.73
Classifier
70
Table 5.3: Results on breast cancer using χ2-statistic
50-top ranked genes
100-top ranked genes
Cross Validation
Cross Validation Classification
Classification Accuracy (%)
Accuracy (%)
k-NN
79.38
74.23
SVM
80.41
74.23
C4.5
74.23
70.10
Naive Bayes
55.67
59.79
Classifier
50 Top‐ranked genes
90
Percentage
85
80
75
Cross Validation Classification Accuracy
70
65
ReliefF
Information Gain
Chi‐Square
Method
Figure 5.1: Graph on cross validation classification accuracy using 50-top ranked
genes
71
Percentage
100 Top‐ranked genes
86
84
82
80
78
76
74
72
70
68
66
Cross Validation Classification Accuracy
ReliefF
Information Gain
Chi‐Square
Method
Figure 5.2: Graph on cross validation classification accuracy using 100-top ranked
genes
From the results, we can observe that all four classifiers yielded high cross
validation classification accuracy using the selected genes of ReliefF compared to
Information Gain and Chi-Square. This proves that ReliefF algorithm is a good
method in selecting informative genes on breast cancer dataset. The efficiency of
ReliefF algorithm to estimate attributes quality in problems with strong dependencies
between attributes (Marko, R.S., and Igor, K., 2003) caused this result. Unlike
ReliefF, Information Gain and Chi-Square methods are ignoring the features
dependencies which lead to their bad performances. Therefore, ReliefF algorithm
was chosen as the features selection technique in this study.
72
5.2.2
Analysis Results of Classifications
As discussed in previous section, ReliefF algorithm obtained a better result
during feature selection. Thus, the classifications were carried out on the 50 and 100
top-ranked genes which were selected by ReliefF algorithm. These classifications
process were done using four types of classifiers which are Radial Basis Functions
Network, RBNF, Multilayer Perceptron, MLP, Naïve Bayes, NB and C4.5. The
following table shows the comparison of the classification results yielded by four
different classifiers.
Table 5.4: Results on classification using different classifiers
Classifier
Top-ranked genes
Time taken (s)
Correctly
Classified
Instances (%)
Radial Basis
50
0.08
90.91
100
0.08
84.85
Multilayer
50
6.5
84.85
Perceptron, MLP
100
23.72
81.82
Naïve Bayes
50
0.02
81.81
100
0.03
78.79
50
0.03
57.58
100
0.08
57.58
Function Network,
RBFN
C4.5
The classifiers performance is compared based on the percentage of correctly
classified instances and time taken to build the model as shown in the table 4.5.
Referring to the table, it is clear that Radial Basis Function Network produced the
highest result compared to other classifiers although the time taken by RBNF to carry
out the model was not the smallest among all classifiers. From the table, we can also
observe that the correctly classified instances percentage of all classifiers decrease
7
73
w
when
classiffications werre carried ouut on 100 top
p-ranked gennes. This pro
oves that the
h
highest
classsification acccuracy can bbe achieved with
w the smaallest numbeer of genes.
F
Figure
4.6 illlustrate the classificationn results in graph.
g
Claassificattion usin
ng 50 top
p‐ranked
d genes
Percentage/Time
100
9
90.91
84.8
85
81.81
80
57.58
60
Correcttly Classified Instancce %
40
20
0.08
6.5
0.02
0.03
Time Taken (s)
0
RBNF
MLLP
NB
C4.5
Classifier
F
Figure
5.3: Percentage of
o Correctlyy Classified Instances
I
usiing 50 top-raanked genes.
Claassification usingg 100 top‐ranked genes
Percentage/Time
100
8
84.85
82
81.8
78.79
80
57.58
60
40
20
Correcttly Classified Instancce %
23.72
0.08
0.03
0.08
Time Taken (s)
0
RBNF
MLLP
NB
C4.5
Classifier
F
Figure
5.4: Percentage of
o Correctlyy Classified Instances
I
usiing 100 top-rranked genees.
74
As stated by Dian Hui Wang et al., (2002), one of the MLP drawbacks is the
time needed to train the data is more longer as the number of input is over 100.
According to the obtainable results, it is clearly shows that the time taken by MLP to
carry out the model especially when 100 top-ranked genes used was very long
compared to other classifiers. The differences of the time taken by MLP can be said
quite huge compared to RBNF, NB and C4.5. Time taken by MLP when 150 topranked genes were used is 52.24 seconds, which strengthen citation. Unlike MLP,
RBNF took less time to train the data due to its simple structure.
The extracted features of genes are distributed in a high dimensional space
with complex characteristics. This caused it difficult to satisfy a model that using
some parameterized approaches like Naïve Bayes and yield a bad result. The bad
performance of C4.5 is due to the decision tree techniques that generate complex
rules and difficult to understand. In brief, RBNF is the best classifier to classify
breast cancer microarray data.
5.2.3
Analysis Results of Classifiers Performance
As the first step of ROC analysis, the true positive rate or sensitivity and true
positive rate or 1-specificity values of RBNF, MLP, NB and C4.5 are given in the
following table. Plots of the results in the ROC space are shows in the Figure 5.6.
75
Table 5.5: TPR and FPR of classifiers
Classifier
Top-ranked
True Positive
False Positive,
genes
Rate, TPR
FPR
50
0.909
0.067
100
0.848
0.149
50
0.848
0.149
100
0.818
0.19
50
0.818
0.19
100
0.788
0.231
50
0.576
0.463
100
0.576
0.463
Radial Basis Function
Network, RBFN
Multilayer Perceptron, MLP
Naïve Bayes, NB
C4.5
1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
ROC Space RBNF
ML
NB
C4.5
TPR or Sensitivity
TPR or Sensitivity
ROC Space 1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
RBNF
MLP
NB
C4.5
0 0.10.20.30.40.50.60.70.80.9 1
0 0.10.20.30.40.50.60.70.80.9 1
FPR or (1 ‐ Specificity)
FPR or (1 ‐ Specificity)
Figure 5.5: ROC Space of 50 top-ranked
Figure 5.6: ROC Space of 100 top-
ranked genes
genes
Based on the graphs, the result of RBNF clearly is the best among other classifiers.
The results of MLP and NB are slightly differs with RBNF while the result of C4.5
almost lies on the random guess line or the diagonal line. The second step of ROC
analysis is the ROC curves. Therefore, the ROC curves of the networks which
76
contain information for understanding the performances of the networks are obtained
as shown in the following figures. However, the curves of the networks are hard to
be investigated based on the same illustration.
Figure 5.7: ROC Curve of 50 top-ranked genes
Figure 5.8: ROC Curve of 100 top-ranked genes
77
Therefore, to compare the performances of the classifiers, results of the area under
the ROC curves as given in Table 5.6 are used.
Table 5.6: ROC Area of Classifiers
Classifier
Top-ranked genes
ROC Area (Area
Under Curve)
Radial Basis Function
50
0.94
100
0.882
Multilayer
50
0.94
Perceptron, MLP
100
0.932
Naïve Bayes
50
0.887
100
0.838
50
0.556
100
0.556
Network, RBFN
C4.5
Based on the table, we can observe that RBNF with area of 0.94 for 50 top-ranked
genes can be chosen as the best classifiers for breast cancer microarray data.
Although MLP also have the same area with RBNF, it has low FPR-TPR compares
to RBNF. Naïve Bayes with area of 0.887 and 0.838 comes after MLP while C4.5
yields the lowest area for both top-ranked genes.
5.3
Summary
In this chapter, the experimental results of features selection, classifications
and evaluation of the classifiers had been analyzed. From the results analysis of
78
features selection techniques, ReliefF algorithm was chosen as the best technique in
selecting informative genes. Radial Basis Function Network was proven as the best
classifier for classify microarray breast cancer data based on its classification
accuracy. Furthermore, the evaluation of its performance according to area under
ROC curve was strongly supportive as well.
CHAPTER 6
DISCUSSIONS AND CONCLUSION
6.1
Introduction
This final chapter discusses the overall of the research. The achievements that
have been obtained will be stated. The problems arise during the study will be
identified. Appropriate suggestions will be proposed to overcome the problems
followed by the conclusion to wrap up the study.
6.2
Overview
As stated in chapter 1, the goal of this study is to classify the breast cancer
microarray data. Several objectives and scopes were determined as the guides to
achieve the goal. The objectives are as follow:
80
i)
To select significant features from microarray data using a suitable
feature selection technique.
ii)
To classify selected features into non-cancerous and cancerous using
Radial Basis Function, RBF, network.
iii)
To evaluate the performance of the classifiers using Receiver
Operating Characteristic, ROC.
Related topics have been discussed in the literature sections which are breast
cancer, microarray technology, features selection, classifiers and the metric to
evaluate classifiers performance. These topics gave the views and idea to understand
the overall study before its implementation. This literature study is vital in the
research because it is the second step of the study in order to achieve all the
objectives.
After collecting the required information in the literature study, the next step
to achieve the objectives is the methodology. The methodology is the guidelines to
design appropriate implementations for the research so that the objectives can be
achieved. After the methodology has been determined, experiments can be carried
out to obtain the results.
Finally the experimental results were analyzed before the objectives can be
achieved. Therefore, the following section discussed the achievements and
contributions of this research. Moreover, the problem encountered during the
implementation will be discussed as well as to propose the best solution for it.
81
6.3
Achievements and Contributions
The achievements obtained from this research can also be considered as the
contributions. The following are the achievements that have been found in this
research:
i)
The best feature selections technique in selecting informative genes
for microarray breast cancer data has been identified. Relief F
algorithm is chosen as the best technique among Information Gain and
Chi-Square in selecting informative genes for breast cancer
microarray data based on the accuracy of the classifiers during
classification.
ii)
The selected features selection classified using four types of classifier
and their classification accuracy were compared. Radial Basis
Function Network has been proven as the efficient classifier in
classifying breast cancer microarray data since it shows the highest
percentage of accuracy (90.91% for 50-top ranked genes) among other
classifiers.
iii)
The performances of the classifiers were evaluated using Receiver
Operating Characteristic and compared based on the area under curve.
Among the four classifiers, Radial Basis Function Network also shows
the best performance with area of 0.94 and TPR-FPR of 0.909-0.067
for 50 top-ranked genes.
iv)
The classification of the accuracy decrease when the classifier
classified 100 top-ranked genes compare to 50 top-ranked genes. This
proved that the efficiency of the classification depends on the number
of the genes.
82
Based on the stated achievements, it can be concluded that all the objectives for this
research have been achieved.
6.4
Research Problem
Although several achievements have obtained from this study, there is still a
problem arises during the implementations of this research. The problem is as
follows:
i)
The time taken by the chosen features selection technique, ReliefF,
was too long compared to other two techniques even though it was an
effective method during the selection of informative genes.
6.5
Suggestion to Enhance the Research
It is necessary to overcome the problem arises in the study in order to
improve this research. Since the problem is due to the time taken by ReliefF, another
type of feature selection techniques is suggested to compare ReliefF algorithm. The
suggested techniques are other types of novel extended Relief method such as
orthogonal Relief (O-Relief), extended ReliefF (E-ReliefF) or the hybrid of ReliefFmRMR (minimum redundancy maximum relevance).
83
6.6
Conclusion
Breast cancer is the number one killer disease among woman compares to
other diseases. Early detection of breast cancer will help to secure a better chance of
survival. There is a various ways to detect, cure and treat breast cancer nowadays. A
lot of researches also have been done in this area in order to reduce the number of
deaths caused by breast cancer. The advent of microarray technology will increase
the number of studies of breast cancer due to its advantages.
The microarray technology allows thousands of the genes expressions to be
determined simultaneously. This advantage of microarray has lead to the application
in medical area namely management and classification of cancer and infectious
diseases. However, microarray technology also suffers several drawbacks. The
disadvantages of the microarray are the high dimensionality of data and also consist
of irrelevant information to classify disease accurately.
However, there are various techniques of feature selection that can be used to
solve the arising problem in microarray in order to improve the accuracy of the
classification. By using feature selection, only the appropriate subset of genes will be
selected among the microarray. There are three approaches of feature selection which
are filter, wrapper and embedded method. Based on the previous researches, filter
method with multivariate techniques is the best and commonly used among other
methods.
The goal of the classification is to distinguish between the cancerous
(malignant) and non-cancerous (benign). There are various classifier whether
traditionally or currently develop that have been studied to classify the microarray.
However, not all the classifier shows the high performance of accuracy. The suitable
metric can be used so as to evaluate the performance of the classifier.
84
In a nutshell, it is hopeful that this study can give valuable information in the
breast cancer researches using microarray technology in order to reduce the number
of deaths caused by breast cancer.
85
REFERENCES
Abu-Khalaf, M.M., Harris, L.N., and Chung, G.C. (2007). Chapter 10 DNA and
Tissue Microarrays. In Patel, H.R.H., Arya, M., and Shergill, I.S. Basic
Science Techniques in Clinical Practice. London: Springer
Adomas, A., Heller, G., Olson, A., Osborne, J., et al. (2008). Comparative analysis
of transcript abundance in Pinus sylverstric after challenge with a
saprotrophic, pathogenic or mutualistic fungus. Tree Physiology. 28, 885-897
Ahmad, A., and Dey, L. (2005). A feature selection technique for classificatory
analysis. Pattern Recognition Letters. 26(1), 43-56
Alizadeh A.A., Eisen M.M., Davis, R.E., Ma, C., Lossos, I.S., et al., (2000). Diffuse
types of diffuse large B-cell lymphoma identified by gene expression
profiling. Nature. 403, 503-511
Arauzo-Azofra, A., Benitez, J.M., Castro, J.L. (2004). A feature set measure based
on Relief. The 5th International Conference on Recent Advances in Soft
Computing (RASC2004). Nottinhham Trant Univ. 104-109
Ben-Bassat, M. (1982) Pattern Recognition and reduction of dimensionality. In
Krishnaiah, P. and Kanal, L., (eds) Handbook of Statistics II, Vol I. NorthHolland, Amsterdam; 773-791
Ben-Dor, A., Bruhn, L., Friedman, N., Nachman, I., Schummer, M. E., and Yakhini,
Z. (2000). Tissue classification with gene expression profiles. Journal of
Computational Biology. 7; 559-584
Biesalski HK, Bueno de Musqeito B, Chesson A, et al., (1998). European Consensus
Statement on Lung Cancer: Risk Factors and Prevention. CA : a cancer
journal for clinicians. 48(3),167-176
86
Boldrick, J.C., Alizadeh, A.A., Diehn, M., Dudoit, S., Liu, C.L., et al. (2002).
Stereotyped and specific gene expression programs in human innate immune
responses to bacteria. Proceedings of the National Academy of Sciences of
the USA. 99, 972-977
Bors, A. G., Gabbouj, G., (1994). Minimal topology for radial basis function neural
network for pattern classification. Digital Signal Processing: a review
journal. 3, 302-309
Bors, A.G., Pitas, I., (1996). Median radial basis functions neural network. IEEE
Trans. On Neural Networks. 7(6), 1351-1364
Bray, F., D., P. M., and Parkin, M (2004). The changing global patterns of female
breast cancer incidence and mortality. Breast Cancer Research. 6(6)
Burchard, J., unpublished.
Chang, C. Y., and Chung, P. C. (1999). A Contextual-Constraint Based Hopfield
Neural Cube for Medical Image Segmentation. Proceedings of The IEEE
Region 10 Conference on TENCON. 2, 1170-1173
Cheung, VG., Morley, M., Aguilar, F., Massimi, A., Kucherlapati, R., and Childs, G.
(1999). Making and reading microarrays. Nature Genetics. 21, 15-19
Christian Harwanegg and Reinhard Hiller. (2005). Protein microarrays for the
diagnosis of allergic diseases: State-of-the-art and future development.
Clinical Chemistry and Laboratory Medicine. 29 (4), 272-277. Walter de
Gruyter.
Chu, S., DeRisi, J., Eisen, M., Mulholland, J., et al. (1998). The transcriptional
program of sporulation in budding yeast. Science. 282, 699-705
Compton, R., Lioumi, M., and Falciani, F. (2008). Microarray Technology.
Encyclopedia of Molecular Pharmacology. Berlin Heidelberg, New York:
Springer-Verlag
Cover, T., and Thomas, J. (1991). Elements of Information Theory. New York :John
Wiley and Sons
Dasarathy, B.(1991). Nearest Neighbor Norms: NN Pattern Classification tehniques.
IEEE Computer Society Press, Los Alamitos, CA,USA
Dash, M., and Liu, H. (1997). Feature selection for classification. Intelligent Data
Analysis. 1, 131-156
87
Downey, T.J., Jr., Meyer, D. J., Price, R.K., and Spitznagel, E.L. (1999). Using the
receiver operating characteristic to asses the performance of neural classifier.
International Joint Conference on Neural Networks (IJCNN’99). 10-16 July.
Washigton, DC, USA. 5, 3642-3646
Driouch, K., Landemaine, T., Sin, S., Wang, SX., and Lidereau, R. (2007). Gene
arrays for diagnosis, prognosis and treatment of breast cancer metastasis.
Clinical & Experimental Metastasis. 24(8), 575-585
Duda, P., et al. (2001). Pattern Classification. Wiley, New York.
English, DR., Armstrong, BK., Kricker, A., Fleming, C. (1997). Sunlight and cancer.
Cancer Causes and Contol. 8(3); 271-83, 283
Farzana Kabir Ahmad (2008). An Integrated Clinical and Gene Expression Profiles
for Breast Cancer Prognosis Model. Master, Universiti Utara Malaysia
Fawcett, T. (2005). An introduction to ROC analysis. Pattern Recognition Letters.
Palo Alto, USA. 27(2006), 861-874
Fayyad, U., and Irani, K.(1993). Multi-interval discretization of continuous-values
attributes for classification learning. Proceedings of the 13th International
Joint Conference on Artificial Intelligence. Portland, OR. 1022-1027
Ferlay J., Bray F., Pisani P., and Parkin D. (2001). GLOBOCAN 2000: Cancer
incidence, mortality and prevalence worldwide
Ferran, E.A., Pflugfedder, B., and Ferrarap. (1994). Self Organized neural maps of
human protein sequences. Protein Science. 3,507-521
Ferri, F., et al. (1994). Pattern Recognition in Practice IV, Multiple Paradigms,
Comparative Studies and Hybrid Systems. Elsevier, Amsterdam. 403-413
Fodor, S.P., et al. (1991). Ligth-directed, spatially addressable parallel chemical
synthesis. Science. 251, 767-773
Fodor, S.P., et al. (1993). Multiplexed biochemical assays with biological chips.
Nature. 364, 555-556
Furey, T.S., Cristianni, N., Duffy N., Bednarski, D. W., Schummer, M. and Haussler,
D. (2000). Support vector machine classification and validation of cancer
tissue samples using microarray expression data. Bioinformatics. 16(10), 906914
Gianluca Bontempi and Benjamin Haibe-Kains. 2008. Feature selection methods for
mining bioinformatics data. Bruxelles, Belgium: ULB Machine Learning
Group
88
Green, D. M., and Swets, J. A. (1996). Signal detection theory and psychophysics.
New York: John Wiley and Sons, Inc
Gruvberger-Saal, S.K., Cunliffe, H.E., and Carr, M.K. 2006. Microarrays in breast
cancer research and clinical practice- the future lies ahead. Endocrine-Related
Cancer. 13, 1017-1031
Guyon, I., Weston, J., Stephen Barnhill, M. D., and Vapnik, V. (2002). Gene
Selection for Cancer Classification using Support Vector Machines. Machine
Learning. 46; 389-422
Guyon , I., and Elisseeff, A. (2003). An introduction to variable and feature
selection. Journal of Machine Learning Research. 3, 1157-1182
Hall, M. (1999). Correlation-based feature selection for machine learning. Doctoral
Philosophy, Waikato University, New Zealand.
Haykin, S. (1994). Neural Networks: A Comprehensive Foundation. New York:
Macmillan College Publishing Co. Inc
Holland, J. (1975). Adaptation in Natural and Artificial Systems. University if
Michigan Press, Ann Arbor.
Horng, M-H. (2008). Texture Classification of the Ultrasonic Images of Rotator Cuff
Disease based on Radial Basis Function Network. International Joint
Conference on Neural Networks (IJCNN 2008). Hong Kong. 91-97
Inza, I., et al. (2000). Feature subset selection by Bayesian networks based
optimization. Artificial Intelligence. 123, 157-184
Jin, X., Xu, A., Bie, R., and Guo, P. (2006). Machine Learning and Chi-Square
Feature Selection for Cancer Classification Using SAGE Gene Expression
Profiles. In Li, J. et al. (Eds) Data Mining for Biomedical Applications. (pp:
106-115). Berlin Heidelberg: Springer-Verlag
Kasabov, N.K. (2002). Evolving Connectionist Systems, Methods and Applications in
Bioinformatics, Brain Study and Intelligent Machines. Verlag: Springer
Kerekes, J. (2008). Receiver Operating Characteristic Curve Confidence Intervals
and Regions. IEEE Geoscience and Remote Sensing Letters. 5(2), 251-255
Kim, YS., Street W.N., Menczer, F. (2003). Feature selection in data mining. In John
Wang (Ed.) Data Mining: opportunities and challenges. (80-105) Hershey,
PA, USA : IGI Publishing
89
Kira, K., and Rendell, L.A. (1992). A practical approach to feature selection.
Proceedings of the ninth international workshop on Machine learning.
Morgan Kaufmann Publishers Inc. 249-256
Kittler, J. (1978). Pattern Recognition and Signal Processing, Chapter Feature Set
Search Algorithms Sijthoff and Noordhoff, Alphen aan den Rijn, Netherlands;
41-60
Kohavi, R., and John, G.H. (1997). Wrappers for feature subset selection. Artificial
Intelligence. 97, 273-324
Koller, D. and Sahami, M. (1996). Toward optimal feature selection. Proceedings of
the Thirteenth International Conference on Machine Learning, Bari, Italy,
284-292
Kononenko, I. (1994). Estimating attributes: Analysis and extensions of RELIEF.
European Conference on Machine Learning. 171-182
Konvacevic, D., and Loncaric, S. (1997). Radial Basis Function Based Image
Segmentation Using A Receptive Field. Proceedings of Tenth IEEE
Symposium on Computer-Based Medical Systems. 126-130
Kuan, M. M., Lim, C. P., Ismail, O., Yuvaraj, R. M., and Singh, I. (2002).
Application of Artificial Neural Networks to The Diagnosis of Acute
Coronary Syndrome. Proceedings of International Conference on Artificial
Intelligence in Engineering and Technology. Kota Kinabalu, Malaysia. 470475
Kégl, B., Krzyzak, A., and Niemann, H. (1998). Radial Basis Function Network in
Nonparametric Classification and Function Learning. Proceedings of the
Fourteenth International Conference on Pattern Recognition. 16-20 August.
1, 565-570
Leonard, J.A., and Kramer, M.A. (1991). Radial basis function network for
classifying process faults. Control Systems Magazine, IEEE. 11(3), 31-38
Li, Y., Campbell, C., and Tipping, M. (2002). Bayesian automatic relevance
determination algorithms for classifying gene expression data.
Bioinformatics. 18(10); 1332-1339
Li, X., Zhou, G., and Zhou, L. (2006). Novel Data Classification Method on Radial
Basis Function Networks. Proceedings of the Sixth International Conference
on Intelligent Systems Design and Applications (ISDA’06)
90
Lim, G.C.C., Rampal, S., and Yahaya, H. (Eds.) (2008). Cancer Incidence in
Peninsular Malaysia, 2003-2005. Kuala Lumpur: National Cancer Registry
Lin, C.T., and Lee, C. S. G. (1996). Neural Fuzzy Systems - A Neuro-Fuzzy
Synergism to Intelligent Systems. New Jersey: Prentice Hall.
Liu, H., and Setiono, R. (1995). Chi2: feature selection and discretization of numeric
attributes. Proceedings of Seventh International Conference on Tools with
Artificial Intelligence. 11 May-11 August 1995. 388-391
Liu, H., and Motoda, H. (1998). Feature Selection for Knowledge Discovery and
Data Mining. Boston: Kluwer Academic Publishers.
Liu. J. and Iba, H. (2002). Selecting Informative Genes Using a Multiobjectives
Evolutionary Algorithm. Proceedings of the 2002 congress. 297-302
Lockhart, DJ., Dong, H., Byrne, MC., et al. (1996). Expression monitoring by
hybridization to high-density oligonucleotide arrays. Nature Biotechnology.
14, 1675-1680
Markov, R.S., and Igor, K. (2003). Theoretical and empirical analysis of relief and
rreliefF. Machine Learning Journal. 53,23-69
Mashor, M. Y., Mat Isa, N. A., and Othman, N. H. (2002). Automatic Pap Smear
Screening Using HMLP Network. Proceedings of International Conference
on Artificial Intelligence in Engineering and Technology. Kuala Lumpur,
Malaysia. 453-457
Mat Isa, N.A., Mashor, M. Y., and Othman, N.H. (2002). Diagnosis of Cervical
Cancer Using Hierachical Radial Basis Function (HRBF) Network.
Proceedings of International Conference on Artificial Intelligence in
Engineering and Technology. Kota Kinabalu, Malaysia. 458-463
Matej, S., Lewitt, R.M., (1996). Practical considerations for 3-D image
reconstruction using spherically symmetric volume elements. IEEE Trans. On
Medical Imaging.15(1), 68-78
Mat Isa, N.A., Mat Sakim, H.A., Zamli, K.Z., Haji A Hamid, N., and Mashor, M.Y.
(2004). Intelligent Classification System for Cancer Data Based on Artificial
Neural Network. Proceedings of the 2004 IEEE Conference on Cybernatics
and Intelligent Systems. 1-3 December. Singapore.
91
Miki, R., Kadota, K., Bono, H., Mizuno, Y., Tomaru, Y., et al. (2001). Delineating
developmental and metabolic pathways in vivo by expression profiling using
the RIKEN set of 18,816 full-length enriched mouse cDNA arrays. Proc Natl
Acad Sci USA. 98(2), 199-204
Miller, L.D and Liu, E.T. 2007. Expression genomics in breast cancer research:
microarrays at the crossroads of biology and medicine. Breast Cancer
Research. 9, 206
Mitchell, T.M. (1997). Machine Learning. New York : McGraw-Hill
Momin, B. A., Mitra, S., and Gupta, R. N. (2006). Reduct Generation and
Classification of Gene Expression Data. International Conference on Hybrid
Information Technology (ICHIT’06). 6-11 November. 1, 699-708
Moody, J., (1989). Fast learning in networks of locally-tuned processing units.
Neural Computation. 1, 281-294
National Cancer Institute. (2005). What You Need To Know About Breast Cancer.
National Institutes of Health, U.S Department of Health and Human
Services
Ng, E. Y. K., Peh, Y. C., Fok, S. C., Ng, F. C., and Sim, L. S. J. (2002). Early
Diagnosis of Breast Cancer Using Artificial Neural Network with Thermal
Images. Proceedings of Kuala Lumpur International Conference on
Biomedical Engineering. Kuala Lumpur , Malaysia. 458-463
Orr, M. J. L. (1996). Radial Basis Function Networks. Edinburgh, Scotland
Oyang, Y.J., Hwang S.C., Ou, Y.Y., et al. (2005). Data Classification with radial
basis function networks based on a novel kernel density estimation algorithm.
IEEE Transaction on Neural networks. 16(1), 225-236
O’reilly KM, MClaughin AM, Backett WS, Sime PJ (2007). Asbestos-Related Lung
Disease. American Academy of Family Physician. 75(5); 683-8,690
Palacios, G., Quan, P-L., Jabado, O.J., Conlan, S., Hirschberg, D.L., et al. (2007).
Panmicrobial Oligonucleotide Array for Diagnosis of Infectious Diseases.
Emerging Infectious Diseases. 13, 73-81
Park, H., and Kwon, H-C. (2007). Extended Relief algorithms in instance-based
Feature Filtering. Sixth International conference on Advanced Language
Processing and Web Information Technology. 123-128
Park, J., and Sandberg, I.W. (1993). Approximation and Radial-Basis-Function
Networks. Neural Computation. 5, 305-316
92
Park, S. H., Goo, J. G., and Jo, C-H. (2004). Receiver Operating Characteristic
(ROC) Curve: Practical Review for Radiologists. Korean Journal of
Radiology. 5, 11-18
Pease, A.C. et al. (1994). Light-generated oligonucleotide arrays for rapid DNA
sequence analysis. Proceedings of National Academy Science of USA. 91,
5022-5026
Pedrycz, W., editor. (1998). Computational Intelligence : An Introduction. NY: CRC
Press, Boca Raton
Poggio, T., Girosi, F., (1990). Networks for approximation and learning.
Proceedings IEEE. 78(9), 1481-1497
Powell, M.J.D. (1977). Restart Procedures for the Conjugate Gradient Method.
Mathematical Programming. 12, 241-254
Ramaswamy, S. and Golub, TR. (2002). DNA microarrays in clinical oncology.
Journal Clinical Oncology. 20, 1932-1941
Reisert, M., and Burkhardt, H. (2006). Feature Selection for Retrieval Purposes. In
Lectures Notes in Computer Science. Image Analysis and Recognition. 4141,
661-672. Heidelberg: Springer-Verlag
Sankupellay, M., and Selvanathan, N. (2002). Fuzzy Neural and Neural Fuzzy
Segmentation of Magnetic Resonance Images. Proceedings of Kuala Lumpur
International Conference on Biomedical Engineering. Kuala Lumpur,
Malaysia. 47-51
Sauter, G., and Simon, R. (2002). Predictive molecular pathology. New England
Journal Medicine. 347, 1995-1996
Sanner, R.M., Slotine, J.-J. E., (1994). Gaussian networks for direct adaptive control.
IEEE Trans. On Neural Networks. 3(6), 837-863
Saeys, Y. et al. (2007). Gene Selection, A review of feature selection techniques in
bioinformatics. Bioinformatics. 23(19), 2507-2517. Oxford University Press.
Schena, M., Shalon, D., Davis, R., and Brown, P.O. (1995). Quantitative monitoring
of gene expression patterns with a complementary DNA microarray. Science.
270, 467-470
Siedelecky, W. and Sklansky, J. (1998). On automatic feature selection.
International Journal Pattern Recognition. 2; 197-220
93
Sikonja, M.R., and Kononenko, I. (1997). An adaptation of relief for attribute
estimation in regression. In Morgan Kaufmann, editor. Machine Learning:
Proceedings of the Fourteenth International Conference. 296-304
Sikonja, M.R., and Kononenko, I. (2003). Theroretical and Empirical Analysis of
ReliefF and RReliefF. Journal of Machine Learning. 53(1-2), 23-69
Skalak, D. (1994). Prototype and feature selection by sampling and random mutation
hill climbing algorithms. Proceedings of the Eleventh International
Conference on Machine Learning, 293-301
Sorlie, T., Perou, C.M., Tibshirani, R., et al. (2001). Gene expressiom patterns of
breast carcinomas distinguish tumor subclasses with clinical implications.
Proceedings of National Academy Science of USA. 98, 10869-10874
Sun, J., Shen, R.M., Han, P. (2003). An original RBF network learning algorithm.
Chinese journal of computers. 26(11), 1562-1567
Tago, C., and Hanai, T., (2003). Prognosis prediction by microarray gene expression
using support vector machine. Genome Informatics. 14; 324-325
Tinker, A.V., Boussioutas, A., and Bowtell, D.D.L. (2006). The Challenges of gene
expression microarrays for the study of human cancer. Cancer Cell. 9(5),
333-339
Tokan, F., Turker, N., and Tulay, Y., (2006). ROC Analysis as a Useful Tool for
Performance Evaluation of Artificial Neural Networks. In Kollias, S., et al.
(Eds). ICANN 2006, Part II, LNCS 4132. 923-931. Springer-Verlag.
Tou, J.T., and Gonzalez, R.C. (1974). Pattern Recognition Principles. London:
Addison-Wesley.
Tzouvelekis, A., Patlakas, G., and Bouros, D. 2004. Application of microarray
technology in pulmonary diseases. Respiratory Research. 5(26)
van de Vijver, M.J., He, Y.D., van’t Veer L.J., Dai, H., Hart, A.A., et al. A geneexpression signatures as a predictor of survival in breast cancer. New England
Journal of Medicine. 98, 2199-2204
Vapnik, V.N. (1998). Statistical Learning Theory. Wiley-Interscience, New York,
USA
Wang, D.H., Lee, N.K., Dillon, S.T., and Hoogenraad, N.J. (2002) Protein Sequences
Classification using Radial Basis Function (RBF) Neural Networks.
Proceedings of the 9th International Conference on Neural information
Processing (ICONIP’02). 2,
94
Wang, H.C., Daparzo J., De La Fraga, L.G., Zhu, Y.P., Carazo, J.M.(1998). SelfOrganizing tree-growing network for the classification of protein sequences.
Protein Science, 2613-2622
Wang, Y., Makedon, F.S., Ford, J.C., and Pearlman, J. (2005). HykGene: a hybrid
approach for selecting marker genes for phenotype classification using
microarray gene expression data. Bioinformatics. 21(5), 1530-1537
Westin, L. K. (2001). Receiver operating characteristic (ROC) analysis: Evaluating
discriminance effects among decision support systems. Umea, Sweden
Weston, J., Mukherjee, S., Chapelle, O., Pontil, M., Poggio, T., and Vapnik, V.
(2000). Feature selection for SVMs. Advances in Neural Information
Processing Systems. 13
Whitfield, M.L., Sherlock, G., Saldanha, A.J., et al. (2002). Identification of genes
periodically expressed in the human cell cycle and their expression in tumors.
Mol Biol Cell. 13, 1997-2000
Witten, I.H., and Frank, E. (1999). Data mining: practical machine learning tools
and techniques with Java implementations. Morgan Kaufmann, San
Francisco, CA
Wu, C.H., Berry, M., Shivakumar, S., and McLarty, J.(1995). Neural networks for
full-scale protein sequence classification: sequence encoding with singular
value decomposition. Machine Learning. 21, 177-193
Wu, C.H. Artificial neural networks for molecular sequence analysis. (1997).
Computers Chemistry. 21, 237-256
Yu, L. and Liu, H. (2004). Efficient feature selection via analysis of relevance and
redundancy. Journal of Machine Learning Respiratory. 5; 1205-1224
zur Hausen H (1991). Viruses in human cancers. Science. 254(5035); 1167-1173
Zhu, Z., Ong, Y-S., and Dash, M. (2007). Markov blanket-embedded genetic
algorithm for gene selection. Pattern Recognition. 40, 3236-3248
APPENDIX A
PROJECT 1 GANTT CHART
96
97
98
APPENDIX B
PROJECT 2 GANTT CHART
100
101
102
103