WASHINGTON UNIVERSITY Program in Molecular Biophysics Dissertation Committee: Anders E. Carlsson
advertisement

WASHINGTON UNIVERSITY
Program in Molecular Biophysics
Dissertation Committee:
Anders E. Carlsson
Enrico Di Cera
Garland R. Marshall
Jay W. Ponder, Chairperson
David J. States
Michael Zuker
POTENTIAL FUNCTION SMOOTHING
WITH APPLICATIONS TO MOLECULAR DOCKING
by
Reece Kimball Hart
A dissertation presented to the
Graduate School of Arts and Sciences
of Washington University in
partial fulfillment of the requirements for the degree
of Doctor of Philosophy
1998 December
Saint Louis, Missouri
Copyright by
Reece Kimball Hart
1999
Permission to use any portion
of this thesis is hereby granted
provided that the source is
clearly cited.
This thesis is available online at
http://www.in-machina.com/~reece/
ABSTRACT OF THE DISSERTATION
POTENTIAL FUNCTION SMOOTHING
WITH APPLICATIONS TO MOLECULAR DOCKING
by Reece Kimball Hart
Doctor of Philosophy in Molecular Biophysics
Washington University in St. Louis
1998 December
Professor Jay W. Ponder, Advisor
Structure prediction is often modelled as an optimization problem in which a computer must minimize the potential energy of a molecular system in terms of its atomic coordinates. Potential energy surfaces for chemical systems present a large number of local
minima which frustrate the search for global minima. This roughness of conformational
space is the primary impediment to conformational search methods. Most present methods for optimization and conformational search do not address the roughness issue directly, but instead rely on stochastic or heuristic mechanisms to traverse surface barriers.
The method investigated herein mathematically and analytically transforms an
original potential energy surface into a continuous series of progressively smoother surfaces. Among the achievements of this research are a deformable variant of the
AMBER/OPLS potential function and its application to conformational search problems.
A thorough analysis of potential smoothing applied to capped dialanine peptide reveals
that the deformed surfaces retain the most dominant features of the original surface but
have many fewer minima. Consequently, deformed surfaces are easier to search. Strong
ii
qualitative and quantitative correlations are identified between potential smoothing and
simulated annealing, the present state-of-the-art technique for conformational optimization. These observations lead to the conclusion that potential smoothing is a deterministic
analog of simulated annealing.
The analysis of potential smoothing applied to capped dialanine peptide identifies
three characteristics of potential smoothing: merging, shifting, and crossing. Each of these
effects has a direct correlate in simulated annealing. Crossing is shown to account for the
reduced efficacy of potential smoothing and simulated annealing for the global optimization of chemical systems. Analysis of this feature of potential smoothing led to the coupling of potential smoothing to a local search procedure which attempts to correct for
crossing events. The results of the hybrid Potential Smoothing and Search (PSS) method
applied cycloheptadecane and molecular docking of trypsin-benzamidine and HIV
protease-XK263 are presented. Refinements to the concepts presented herein will enable
investigations of greater computational complexity than currently possible.
iii
DEDICATED TO MOM AND DAD
FOR EVERYTHING
iv
Acknowledgements
The longer the journey, the more important the company. Graduate school is indeed
a long adventure and I have been extremely fortunate to have been joined by a supportive
entourage. I am grateful for the encouragement of my wonderful family and close friends:
my parents, Larry and Roianne, my sister, Brooke, and Beth Landers. They made the
good times worth celebrating and the hard times surmountable. They are my pillars.
All graduate students experience potholes on their journey and I was no exception. I
am indebted to Enrico Di Cera and Michael Hodsdon for their belief in me and my abilities. It is not an overestimation of their support to write that I could not have completed
graduate school without them. I will always remember their succor in my times of need.
Of course, this research would not have been possible without the mentorship of Jay
Ponder. I remain in awe of his knowledge and grasp of diverse topics. I especially appreciated his accessibility for discussion. Thank you to the remainder of my committee −
Anders Carlsson, Enrico Di Cera, Garland Marshall, David States, and Michael Zuker −
for their advice and generous efforts in improving the quality of this research and dissertation.
I am indebted to Rohit Pappu, a past post-doctoral fellow in the lab, for the close
collaboration on potential smoothing and the profound physical and mathematical insights
he forged. His efforts greatly improved the quality of this thesis. I am also grateful to
Enoch Huang, a present post-doctoral fellow, and Yong Kong, a past graduate student, for
v
their encouragement and friendship. The biophysics, biochemistry, and bioorganic graduate students − known as the grubbbs − foster a wonderful academic environment. I appreciate their comaraderie and companionship during graduate school.
vi
Vita
Reece Kimball Hart
Date of birth:
1968 November 22
Place of birth:
Long Beach, California
Undergraduate study:
University of California, San Diego
B.A. Molecular Biology, 1990
Graduate study:
Washington University, St. Louis
M.S. Computer Scinece 1994
Washington University, St. Louis
Ph.D. Molecular Biophysics, expected 1998
Contact information:
reece@in-machina.com
http://www.in-machina.com/~reece/
vii
Table of Contents
Abstract of the Dissertation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ii
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . v
Vita . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . vii
Table of Contents. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . viii
List of Figures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xi
List of Tables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xv
Chapter 1:
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
Molecular Structure Prediction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
Potential Energy Functions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Searching Conformational Space . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
Synopsis of the Dissertation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
Chapter 2:
Potential Energy Smoothing: A Deterministic Analog of Simulated Annealing . . . 31
Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
Results. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
CDAP Conformational Network . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
Conformational Clustering Based on Energetic Barrier and Potential Smoothing 52
Energetic Clustering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
Smoothing Clustering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
Potential Smoothing and Simulated Annealing as Global Optimization Methods 67
Comparison of Potential Smoothing and Simulated Annealing . . . . . . . . . 67
Potential Smoothing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
Evidence for the Similarity of Potential Smoothing and Simulated Annealing 77
Molecular Dynamics Simulated Annealing (MDSA) for CDAP. . . . . . . . . 77
Shifting in Potential Smoothing and Simulated Annealing . . . . . . . . . . . . . 80
Crossings in Potential Smoothing and Simulated Annealing . . . . . . . . . . . 85
viii
Merging in Potential Smoothing and Simulated Annealing . . . . . . . . . . . . 90
Discussion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
Appendix A. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
Chapter 3:
Analysis and Application of Potential Energy Smoothing and Search Methods for
Global Optimization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
Potential Function and Parameterization . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
Diffusion Equation Method for Smoothing of Potential Functions . . . . . . . . 112
Coordinate Representations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
Diffusion Coefficients to Modulate Smoothing of Potential Function Terms. 116
Potential Smoothing Protocol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
Smoothing Schedule. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
Compactness Restraints In Smoothing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
PSS Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
"Normal Mode" Local Search (NMLS) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124
Transition State Based Search (TSBS). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
Results. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127
Clusters of Argon Atoms in Γvc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127
Oligopeptides in Γvc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
CH3CO-(L-Ala)n-NHCH3 in Γvt and Γvc . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
Γvc Calculations. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
Cycloheptadecane in Γvc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 146
Optimizations in Γvr . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
Discussion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
Features of Potential Smoothing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
Other variants of search enhanced potential smoothing . . . . . . . . . . . . . . . . . 163
Computational efficiency: PS-NMLS versus DEM . . . . . . . . . . . . . . . . . . . . 164
Selecting Smoothing Windows for Local Searches . . . . . . . . . . . . . . . . . . . . 165
Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
Acknowledgments. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
Appendix A. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
Appendix B . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
Chapter 4:
Flexible Molecular Docking with Potential Energy Smoothing. . . . . . . . . . . . . . . . 178
Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182
ix
Results. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Undirected Docking of Trypsin - Benzamidine. . . . . . . . . . . . . . . . . . . . . . .
Undirected Docking of HIV-1 Protease and XK263 . . . . . . . . . . . . . . . . . . .
Directed Docking of HIV-1 Protease and XK263 . . . . . . . . . . . . . . . . . . . . .
Discussion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
188
188
192
198
203
207
Chapter 5:
Summary of the Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
Glossary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214
x
List of Figures
Chapter 1:
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
Figure 1. A hypothetical molecule illustrating some common terms in potential energy
functions. The terms are bond stretching, in-plane angle bending, torsion rotation, van
der Waals interaction, and electrostatic interaction. Only one example of each term
type is shown, but the total energy is a sum of all terms. . . . . . . . . . . . . . . . . . . . . . . 22
Chapter 2:
Potential Energy Smoothing: A Deterministic Analog of Simulated Annealing . . . 31
Figure 1. Capped Dialanine Peptide (N-Acetyl−Ala−Ala−N-Methylamide; CDAP). Methyl groups are represented as united atoms. The system has 18 atom centers, 48 dimensional Cartesian conformational space, and 7 rotatable bonds. . . . . . . . . . . . . . . 43
Figure 2. Sixteen conformational regions of a Ramachandran map identified by Zimmerman, et al. (cf. Table II). The nine <φ,ψ> regions used for grid search are denoted by
filled circles. The two <φ,ψ> descriptors of the conformational code were determined
using this map. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
Figure 3. The distribution of energies of 142 unique minima (filled) and 1038 unique transition state (hollow) conformations on the undeformed DOPLS PES. The bin size is 5
kJ/mol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
Figure 4. Ramachandran plots of <φ1,ψ1> (left) and <φ2,ψ2> (right) for the 142 minima
discovered by grid search.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
Figure 5. The four lowest energy structures. See also Table III. . . . . . . . . . . . . . . . . . . . 50
Figure 6. Energy barrier clustering of minima. All minima within a branch are connected
by barriers not greater than the value on the ordinate. The two most energetically distinct basins are 1 and 2. There is no pair of minima, one from basin 1 and one from basin 2, connected by a barrier less that 80.455 kJ/mol. The eight structures at E*=38
kJ/mol are used for comparison with potential smoothing.. . . . . . . . . . . . . . . . . . . . . 54
Figure 7. Illustration of topographical changes during potential smoothing. The original
PES (t0) appears at the bottom. PESs at increasing deformations are offset vertically
for clarity. Potential smoothing is characterized by three processes: shifting, merging,
and crossing. Dotted arrows depict an adiabatic change of deformation for a given
xi
conformation. Solid black arrows denote minimizations. Minima are indicated by filled
circles.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
Figure 8. Ramachandran plots of <φ1,ψ1> (top) and <φ2,ψ2> (bottom) for four levels of
surface deformation: t=0, 0.61, 1.08, 1.69. The deformations shown were chosen to exemplify features of the clustering or differences between to the two sets of dihedrals
and are typical for other deformations. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Figure 9. Smoothing clustering of minima. The 142 minima enumerated by grid search
appear as leaves of the tree at the bottom of the figure. The ordinate is the extent of deformation and is discontinuous for readability purposes. As deformation increases
(from bottom to top), minima merge into basins. The eight structures at t=1.69 are the
basis for much of the analysis herein. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
Figure 10. The number of minima during potential smoothing. The number of minima decreases monotonically as minima merge to form conformational basins.. . . . . . . . . . 60
Figure 11. Network of conformations remaining at t=1.69. The eight remaining structures
represent all combinations of cis-trans interconversions of the three peptide bonds and
are denoted by the vertices of the cube. Each vertex shows the rank of the energy on
this surface and the structure identifier in parentheses for comparison with Figure 9.
Paths are denoted by the edges. Transition states were found for every edge of the cube
and nowhere else. The energy rank of transition states is indicated by italic text adjacent to an edge. Edge arrows point from higher to lower energy on the t=1.69 surface.
Note that minimum 4 from the undeformed surface is the global minimum on the deformed surface. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
Figure 12. Trajectories for P1(τmes) and P2(τmes), the occupation probabilities of
minimum1 and minimum 2 as a function of temperature. Past T = 450K, the barrier
separating minimum 1 from the rest of the PES is difficult to traverse. The dynamics
are dominated by a flux into minimum 2 and no flux into or out of minimum 1. The result is that most simulated annealing calculations converge to minimum 2 or higher lying minima. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
Figure 13. Second order curves for shifting of <φ1,ψ1>, <φ2,ψ2> for minimum 2 of
CDAP as a function of deformation and temperature. The curves were generated by interpolating the observed temperature values to a second order fit to the original data of
the variation of torsional angles as a function of deformation. . . . . . . . . . . . . . . . . . . 85
Figure 14. Equilibrium probabilities at various temperatures of the 10 lowest energy
minima on the undeformed surface. Broad basins become favored entropically as temperature increases. In particular, as temperature increases the very narrow global minimum 1 becomes much less favored and the broader basin 4 which represents the DEMbacktrack minimum becomes the dominant state. . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
Figure 15. Correlation of crossing temperature (T) and time (t). The line is a least squares
fit to the data and has a correlation coefficient r2 = 0.95. . . . . . . . . . . . . . . . . . . . . . . 90
xii
Chapter 3:
Analysis and Application of Potential Energy Smoothing and Search Methods for
Global Optimization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
Figure 1. (a) Lowest energy α-helical conformations of CH3CO-(L-Ala)8-NHCH3 in Γvc.
(b) The β-hairpin structure which is 7.4179 kcal/mol lower in energy than the canonical α-helix shown in (a) and is the structure found using PS-NMLS. . . . . . . . . . . . 139
Figure 2. Conformational energies of CH3CO-(L-Ala)10-NHCH3 in Γvc as a function of
increasing deformation t for a canonical α-helix (−), for an α´-helix (- -), and for the
β-hairpin found from a PS-NMLS protocol (...). On the t = 0.0051 surface the βhairpin is lower in energy than the α-helix. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
Figure 3. (a) Energy distribution of the 20,469 unique minima for cycloheptadecane located using an self-consistent NMLS-based search to scan the complete potential energy surface. The number of minima found in each 0.1 kcal/mol energy bin is plotted
as a function of increasing MM2 energy value. (b) Low energy tail of (a) showing the
distribution of minima with MM2 energy values less than 3 kcal/mol above the global
minimum. The search procedure used to generate both panels (a) and (b) found 11
minima within 1 kcal/mol of the global minimum, 68 minima within 2 kcal/mol, and
261 minima within 3 kcal/mol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 148
Figure 4. (left) Global minimum structure for cycloheptadecane with MM2 energy of
19.0680 kcal/mol. (right) Second lowest energy minimum and the structure found by
PS-NMLS algorithm with MM2 energy of 19.0774 kcal/mol. . . . . . . . . . . . . . . . . . 151
Figure 5. Distribution of inter-helical conformational energies for the 1093 unique
minima found from an extensive grid search over 18,000 unique starting positions for
two rigid capped CH3CO-(L-Ala)10-NHCH3 α-helices. The global minimum has an
energy of -16.4124 kcal/mol. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
Figure 6. Conformation of the global energy minimum for the packing of two capped,
right-handed α-helices of sequence CH3CO-(L-Ala)10-NHCH3.. . . . . . . . . . . . . . . 156
Figure 7. Reduction in the number of minima for cycloheptadecane as a function of increasing PES deformation. On the undeformed surface there are 20,469 unique
minima. These minima merge into a single minimum on the t = 25 PES. The figure
shows a plot of the log10 of the number of unique minima remaining on the PES as a
function of increasing smoothing, t. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
Figure 8. One-dimensional schematic of the effect of a smoothing protocol on a potential
energy surface. The original PES is transformed by successive application of a
smoothing operator, where the extent of smoothing is dictated by a control parameter t.
The unsmoothed original surface (t = 0), the surface at an intermediate level of
smoothing (t = t1) and a highly smoothed surface (t = tlarge) are shown. As the surface
xiii
is transformed higher lying minima merge into catchment regions of low lying minima
and barriers between minima are progressively lowered. Open circles are starting or
intermediate points on each surface. Solid circles are local minima. Dashed arrows
show the result of local optimization ending at a local minimum. Solid arrows represent adiabatic movement from a local minimum on one surface to the corresponding
starting point on a rougher surface. A simple smoothing protocol consists of repeated
cycles of local optimization followed by adiabatic transfer to the next surface.. . . . 160
Figure 9. Schematic of a more realistic potential smoothing protocol for molecular search
problems. This figure shows a crossing between the two surviving minima on the t = t2
surface. A reversing schedule encounters the first bifurcation at t = t2. At this level of
smoothing the protocol favors basin B over basin A due to a crossing of relative energies which is an artifact of the averaging process. If bifurcations are sampled where the
relative energies of the alternative basins are inverted from the t = 0 surface, then the
simple method will not converge to the global minimum. Between t = t2 and t = 0 there
exist values of t for which the energy ordering resembles that of the original PES. A
local search process coupled to the smoothing schedule can potentially recognize errors due to earlier energy crossings. For example, a local search represented by the dotted arrow on the t = t1 surface would correctly decide that basin A should be favored
over basin B. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
Figure 10. First-order least squares fit to the smoothing paramater tdetour as a function of
n for application of PS-NMLS to CH3CO-(L-Ala)n-NHCH3 sequences in Γvt for (n =
5−12). The fit could be used to implement windowing schemes to estimate smoothing
values for which an NMLS search protocol is to be used in Γvt. Restricting local search
to a limited window of t values allows a reduction in computational overhead by eliminating unnecessary and redundant local searches. . . . . . . . . . . . . . . . . . . . . . . . . . . 166
Chapter 4:
Flexible Molecular Docking with Potential Energy Smoothing. . . . . . . . . . . . . . . . 178
Figure 1. Benzamidine. Atom numbering for C and N atoms is as in reference 26. . . . 185
Figure 2. XK263 inhibitor. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
Figure 3a-h. Snapshots of 50 Trypsin-Benzamidine docking.. . . . . . . . . . . . . . . . . . . . 190
Figure 4. XK263 flexibility. The 10 regions of torsional flexibility are denoted by arcs
across the arms of the napthalene, benzene, and hydroxyl groups.. . . . . . . . . . . . . . 192
Figure 5a-h. HIV-1 protease - XK263 docking. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 194
Figure 6a-f. Directed docking of XK263 into the active site of HIV-1 protease. . . . . . 200
Chapter 5:
Summary of the Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
Glossary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214
xiv
List of Tables
Chapter 1:
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
Chapter 2:
Potential Energy Smoothing: A Deterministic Analog of Simulated Annealing . . . 31
Table I. Harmonic improper torsion parameters. These values were determined empirically by fitting OPLS-style trigonometric improper torsion to a CHARMM-style harmonic improper torsion with emphasis on small displacement from the ideal value. 40
Table II. Nine low energy regions identified by Zimmerman, et al.31. These values were
used for grid search on the undeformed PES. The <φ,ψ> points are depicted in Figure
2. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
Table III. Ten minima within 15 kJ/mol of the global minimum. The columns are structural identifier (energetic rank on the undeformed DOPLS PES), energy, dihedral
angles (cf. Figure 1), and conformational code. Structures of minima 1-4 are depicted
in Figure 5. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
Table IV. Summary of 1038 paths in the CDAP network. There are 1038 paths between
688 unique pairs of minima. The average Euclidean distance between minima in
<φ,ψ> space (see equation 9) and the average minimum energy barrier. . . . . . . . . . . 52
Table V. Comparison of clustering by the energy barrier and potential smoothing methods
at levels where 8 basins remain (t<=1.69, E*<=38 kJ/mol). Each basin represents a set
of structures on the original PES clustered by either energy barrier (EB) or potential
smoothing (PS) merge time. The number of structures in the intersection of each potential smoothing basin with each energy barrier clustering basin suggests the extent to
which the two techniques cluster similarly. We identified each basin by the member
with the lowest energy. It is important to note that the names are irrelevant; only the
nature of the overlap between basins clustered by the different techniques is meaningful.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
Table VI. Summary of Master equation calculations to "simulated simulated annealing".
tmes is the Master equation simulation time length for each temperature level along the
cooling schedule. The final temperature is Tlow = 100 K. For T = Tlow the equilibrium
probabilities for the global minimum and minimum 2 are: Peq(1) = 0.822 and Peq(2)
= 0.178. However, in order to achieve the very low temperature thermodynamic equilibrium requires inordinately slow cooling schedules or very large values of tmes. In a
molecular dynamics simulated annealing calculation large values for tmes translate to
xv
extremely long molecular dynamics trajectories. Minimum 2 is favored over minimum
1 for T > 450 K. When minimum 1 is favored compared to minimum 2, the activation
barrier is too big and the system is frozen into minimum 2. . . . . . . . . . . . . . . . . . . . . 75
Table VII. Summary of 100 independent 500 ps molecular dynamics simulated annealing
trajectories applied to CDAP. The initial conformer for each run is that of minimum
142, the highest lying minimum on the PES. The initial temperature was set to 5000K.
Results are insensitive to the choice of the starting temperature provided it is sufficiently high. Shorter trajectories (50ps) yield similar results. The table shows the energies of minima found and the frequencies with which they were found in the MDSA
calculations. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
Table VIII. Summary of multiple simulated annealing runs for CDAP from minima 1, 2,
4, 11, 16, 18, 28 and 40. These 8 minima were the ones that remained for large values
of Eb in energy clustering. The starting positions are shown in the gray boxes. Columns under the gray boxes show the number of times a given minimum is found in
100 independent simulated annealing runs from the minimum in the gray box. Column
1 is the identity of the minimum on the PES found from the annealing runs. The global
minimum is found with very low probability compared to minimum 2. Success ratios
for finding minimum 4 are comparable to that of finding minimum 1. . . . . . . . . . . . 80
Table IX. Shifting of minima as a function of deformation, t. Shifting is measured by the
deviations of <φ1,ψ1> and <φ2,ψ2> for minimum 2 of CDAP from their t = 0 values.
For t = 0, <φ1(o),ψ1(o)> = <−83.89°,67.73°> and <φ2(o),ψ2(o)> = <−82.79°,65.8°>.
The deviations increase as the PES becomes smoother and is a measure of the anisotropy within local minima. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
Table X. Shifting of minima as a function of temperature T. Shifting is estimated by the
deviation of (<φ1,ψ1>) and (<φ2,ψ2>) which are time averaged values of torsional
angles for minimum 2 of CDAP generated from 11.5 ns long molecular dynamics
simulations at different temperatures. For T = 0 K, <φ1(o),ψ1(o)> = <−83.89°,67.73°>
and <φ2(o),ψ2(o)> = <−82.79°,65.8°>. The deviations increase as temperature increases. Shifting is in the direction of the smallest energy barrier that connects minimum 2 and minimum 5 on the PES network for CDAP. . . . . . . . . . . . . . . . . . . . . . . 84
Table XI. Comparison of the some of the crossings between pairs of minima as a function
of deformation time t and canonical temperature T for CDAP. . . . . . . . . . . . . . . . . . 89
Table XII. Comparison of simulated annealing and potential smoothing.. . . . . . . . . . . . 95
Chapter 3:
Analysis and Application of Potential Energy Smoothing and Search Methods for
Global Optimization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
Table I. Characterization of the diffusion spaces and diffusion coefficients for the different energy terms of a DOPLS molecular mechanics potential. In all calculations t is set
to a dimensionless parameter that controls the level of smoothing. . . . . . . . . . . . . . 117
xvi
Table II. Results for PS-NMLS and DEM and energy minimizations applied to clusters of
argon atoms. In the table n denotes the size of the cluster. If the number of search directions for PS-NMLS is zero, then a straight DEM protocol finds the global minimum. All energies are in Lennard-Jones (LJ) units. . . . . . . . . . . . . . . . . . . . . . . . . . 131
Table III. Ideal geometries used for constructing capped polyalanine chains using DOPLS
definitions of atom types for varying lengths in Γvt. On this manifold the values of ω
are kept fixed at 180°. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135
Table IV. Summary of results from application of PS-NMLS to varying lengths of
CH3CO-(L-Ala)n-NHCH3 Sequences in Γvt. In Γvt the bonds and angles remain fixed
and the energy minimizations find the lowest or global minimum on the manifold of
fixed bond and angles. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136
Table V. Results of DEM and PS-NMLS applied to capped sequences of polyalanine,
CH3CO-(L-Ala)n-NHCH3 in Γvc. A schedule of s = 3, nd = 100 and td = 10 was used
in this study. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Table VI. Hydrogen-bonding distances for the 23 unique local minimum energy structures
sampled in a local search out of the α-helical local minimum on the t = 0.0045 Surface.
The shaded region of the table corresponds to the type of hydrogen bonds that classify
α-helical structures. All the very low energy structures show typical α-helical hydrogen bonding pattern. The number of α-helical hydrogen bonds decreases with increase
in conformational energy and the higher energy structures sampled in this calculation
are random coil conformations. The table reflects two important features of the local
search sampling. The α-helical basin and the β-hairpin basin are disjoint sets and the
α-helical basin is a very narrow deep well reflected in the fewer number of structures
sampled, compared to the β-hairpin.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 143
Table VII. Hydrogen-bonding distances for 32 unique local minima sampled in a local
search out of the β-hairpin local minimum on the t = 0.0045 surface. The shaded region corresponds to hydrogen bonds for a β-hairpin. All low energy structures sampled
from the β-hairpin local minimum show hydrogen bonding patterns typical of βhairpins. Higher energy structures have fewer β-hairpin hydrogen bonds and some
show a few α-helical hydrogen bonds. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 144
Table VIII. Evolution of the lowest fifteen minima of cycloheptadecane as a function of
increased smoothing. Column 2 shows the MM2 energies for the lowest energy conformers found using an extensive distance geometry search technique. We found 257
unique minima within 3 kcal/mol of the global minimum. A smoothable variant of the
MM2 PES which replaces the Buckingham potential with a 2 Gaussian approximation
has conformational energies on the t = 0 surface as shown in column 3. The spacing
between and ordering of conformational energies is similar to the original MM2 surface. Columns 4−6 show the change in conformational energies as a function of
xvii
smoothing. Increase in smoothing is characterized by a reduction in the conformational
energy spacing between minima and a rearrangement of the rank ordering of minima,
i.e., for 0.001<t<0.01 minimum 2 is the lowest in energy. . . . . . . . . . . . . . . . . . . . . 150
Table IX. Fifteen lowest energy conformers for docked polyalanine helices in descending
order of interhelical energy. The table shows the values for the interhelical energies,
the packing angles Ω and the distance of closest approach d.. . . . . . . . . . . . . . . . . . 155
Table X. Smoothing parameters t = tdetour at which alternate minima were obtained using
a NMLS protocol in Γvt for varying lengths of capped polyalanine CH3CO-(L-Ala)nNHCH3 chains. td = 10.0 and s = 5 in eq 8 of the text . . . . . . . . . . . . . . . . . . . . . . . 165
Chapter 4:
Flexible Molecular Docking with Potential Energy Smoothing. . . . . . . . . . . . . . . . 178
Table I. Van der Waals parameters for benzamidine. . . . . . . . . . . . . . . . . . . . . . . . . . . 185
Table II. Van der Waals parameters for XK263. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
Table III. Success rate of benzamidine docking to trypsin. . . . . . . . . . . . . . . . . . . . . . . 189
Chapter 5:
Summary of the Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
Glossary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214
xviii
Chapter 1:
Introduction
19
Molecular Structure Prediction
A molecular species is specified by a set of atoms which are interconnected by
bonds. However, it is a molecule’s structure − the coordinates of each of its atoms −
which determines its behavior. The prediction of molecular structure from the basic topological information remains an elusive goal. Nonetheless, the dogma that "structure determines function" pervades biology and provides the impetus for tackling this difficult
problem.
The prediction of molecular structure from atomic topology is difficult because an
atom’s coordinates depend on a large number of attractive and repulsive interactions with
its neighbors. The most favorable conformations typically represent compromises of very
many cooperative or competitive interactions. While it is common and convenient to refer
to a single molecular structure, it is more correct to think of molecules as having a small
number of preferred conformations among a vast set of possibilities. A common objective is to determine the best conformations from among the large number of possible conformations, where "best" implies the existence of a metric by which conformations may
be ordered.
Molecular conformations are often compared using potential energy, a quantitative
measure of the net effect of the atomic interactions. Given a particular potential energy
function, one might expect that optimal structures could be identified simply by computing the energy of every conformation within conformational space. However, conformational space is vast and iterating over all conformations would take prohibitively long.
20
Indeed, the number of possible states is so large that Cyrus Leventhal posed the wellknown Leventhal Paradox: Nature could not possibly sample all conformations because
doing so even at unrealistic speeds for moderately sized proteins would take longer than
the age of the Universe1.
A primary objective of modern computational biology is finding a needle in a haystack. The explosive growth in the number of cyrstallographic and NMR structures and
the advent of high-throughput DNA sequencing, in conjunction with very fast modern
computers, has fueled hopes that macro-molecular structures may be predicted by molecular modelling. Such efforts pave the way to the primary challenges of human biology,
including the design of novel therapeutic agents. This dissertation investigates potential
function smoothing, a technique by which the structure prediction task is solved initially
at low resolution where the broad features of the problem are readily apparent and the
multiple minimum problem is lessened, and then at increasing resolution until the full
atomic detail is regained.
Potential Energy Functions
The energy of a molecular system may be computed by many methods, but the
choice of method involves a trade-off between accuracy and speed. In any case, the energy is a state function which is dependent upon the atomic coordinates and must be computed anew for distinct conformations. At one end of the spectrum, quantum mechanical
calculations use few approximations and are capable of extremely accurate energies, but
may take many days to compute the energy of a single small molecular structure. At the
21
other end are simple statistical potentials which make extensive approximations for the
sake of computational feasability. Between these extremes lays empirical potential energy
functions. This class of energy functions provides a reasonably accurate potential energy
for most moderately-sized proteins in a second or so on present-day commodity computers. Potential energy functions are composed of a number of term types, each of which
represents a particular type of molecular interaction. An example is shown in Figure 1.
For a more complete description of empirical potential functions, see Chapter 2.
Figure 1. A hypothetical molecule illustrating some common terms
in potential energy functions. The terms are bond stretching, inplane angle bending, torsion rotation, van der Waals interaction,
and electrostatic interaction. Only one example of each term type is
shown, but the total energy is a sum of all terms.
angle
bending
R1
vdW
interaction
R2
O
R3
torsion
rotation
electrostatic
interaction
H
N
Σ Σ Σ Σ Σ 22
bond
stretching
It must be emphasized that a potential energy function is dependent on the x, y, and
z coordinates of every atom. For a system with N atoms, there are on the order of 3N independent variables over which the system must be optimized. A molecular conformation is
defined by a particular set of values for the variables. The set of possible conformations
accessible to a molecule is called its conformational space. Just as a three-dimensional
system defines a surface in our real world, a n-dimensional system of variables defines an
imaginary hypersurface in conformational space. Because large-dimensional spaces are
not easily visualized, it is helpful to construct analogies between real-world surfaces and
hypersurfaces.
Let us pursue such an analogy now. Imagine standing on rough terrain and being
commissioned to find the lowest points on the landscape. You consider how to proceed.
You might move randomly, but you realize that you’ll never know when to stop looking.
If you stop after some predetermined condition, you will still be left to wonder whether
the lowest point lies just over the next ridge. What’s worse, you would probably give a
different answer next time you were asked. Or you might move in a grid, making small
methodical steps along each dimension. With only two dimensions to search on a physical
terrain, that is conceivable. However, conformational space in the molecular world typically consists of thousands of dimensions, and as was discussed earlier, Leventhal proposed that not even Nature is fast enough to use this brute force approach. Or, perhaps
you simply move downhill as long as you can. Unfortunately, when you get to a low spot,
you will only be guaranteed to have found a local minimum, and over the next hill may be
23
yet a deeper region. Each of these methods represents a particular strategy for conformational searching, which is discussed next.
Searching Conformational Space
Conformational search is typically used to locate conformations with particular
structural features, sample ensembles of conformations, or to optimize a given conformation. Conformational searching methods can be classified into three broad categories: heuristic methods, stochastic methods, and deterministic methods. The primary goal of each
method is to overcome the barriers on the potential surface in order to facilitate the exploration of conformational space.
There are several important traits of conformational search methods. Repeatable behavior is impossible to guarantee in methods which incorporate randomness. As a result,
most investigators apply heuristic and stochastic methods multiple times and assume that
the most frequently occurring result is the correct one. Second, certain algorithms (of any
class) use energy functions which represent biophysical principles better than others. As a
result, these algorithms are better able to provide insights into molecular processes. Another important trait is scalability − that is, the dependence on required computer resources (CPU time or memory). For example, an algorithm with linear time dependence
requires CPU time that scales directly with the size of the problem, whereas one with cubic dependence requires time that scales with the cube of the size of the problem. The latter algorithm might be said to scale poorly, although for practical purposes it might be
useful nonetheless.
24
Scalability is particularly important because it dictates the feasability of widespread
use of an algorithm on mainstream computer equipment. Theoretically, structure prediction is NP-hard2, which means that no solution is possible by an algorithm which has
polynomial time dependence on the problem size. That is, the required time increases exponentially with input size. The NP-hard argument holds for conformational spaces which
have no broad features and no overall shape. Although many NP-hard problems are practically soluble in polynomial time using heuristics, it is unclear that any known heuristics
are are reliable enough for the case of macromolecular structure prediction. Fortunately,
the energy landscapes of chemical systems do have underlying shape which drives the
physical behavior of such systems. Potential smoothing identifies the underlying structure
embedded in the energy function and uses it to focus conformational search efforts into
low energy regions.
Heuristic methods typically rely on the application of intuitive beliefs regarding molecular conformations, but which may not be reliable in all cases. For instance, genetic algorithms essentially divide a molecule into pieces and optimize the fragments independently. This is intuitively appealing and correct in many instances, but it is also possible
for optimal fragments to have non-optimal total energy and, conversely, for non-optimal
fragments to have optimal total energy. Other examples of heuristic methods are evolutionary programming, neural networks, and tabu search3.
Stochastic methods use random perturbations to overcome barriers on the potential
surface. Simulated annealing is an oft-used conformational search tool. The general algorithm is to use a simulation temperature to control the amount of thermal energy in the
25
system. The system is started in a particular conformation and at a high temperature over
which barriers on the surface are easily crossed, and is then slowly cooled. At each temperature, the system is equilibrated by molecular dynamics or Monte Carlo. As T decreases, so does the likelihood of occupying high-energy minima. Simulated annealing is
discussed extensively in Chapter 2.
Deterministic methods are characterized by algorithms which do not use randomness. As a result, they have the appealing trait of repeatable behavior given the same initial conditions and parameters. In practice, many deterministic methods are poor at traversing large barriers in the system, and consequently large conformational changes are
inaccessible. Local optimization techniques such as steepest descent, conjugent gradient,
or Newton’s method are examples of deterministic procedures. Potential smoothing is a
recent class of deterministic methods which gradually lessen the barriers on potential surfaces. The deformed surfaces are smoother and therefore easier to search, but retain the
broadest features of the original surface. In essence, they eliminate the distraction of the
multiple minimum problem.
Whereas most conformational search methods seek methods which traverse barriers
on the potential surface, potential smoothing alleviates the barriers themselves. There are
three primary implementations of potential smoothing: Gaussian Density Annealing4,
Gaussian Packet Annealing5, and the Diffusion Equation Method6. The research in this
dissertation is concerned specifically with the Diffusion Equation Method, although many
of the results are generalizable to the other methods.
26
Synopsis of the Dissertation
This dissertation pertains to the use of potential smoothing as a conformational
search and optimization tool for chemical systems. When research was begun, a detailed
characterization of the effects of smoothing and comparisons with simulated annealing
had not been conducted. Additionally, applications of smoothing had been limited to
pedagogical optimization challenges and small chemical systems such as sodium chloride
crystals and argon clusters.
The goals of this research were to:
1) Recast an existing potential function into a deformable variant
suitable for the study of biochemical systems. This function has
been named the Deformable OPLS (DOPLS) potential function, after the OPLS7 function from which it is derived.
2) Use DOPLS to carefully analyze the nature of the smoothing
process in the Capped DiAlanine Peptide (CDAP) system. In
particular, we sought to relate basins on deformed surfaces
with the clustering of conformational features from the undeformed surface.
3) Compare the effectiveness of potential smoothing and simulated annealing in the CDAP system.
4) Apply potential smoothing to molecular docking, a challenging
problem with pharmaceutical relevance.
27
Chapter 2 addresses the first three goals. Potential smoothing, as implemented in the
diffusion equation method, is used to derive DOPLS, a deformable version of the OPLS
force fields for peptides. A complete enumeration of the local minima and transition
states of N-Acetyl-Ala-Ala-N-Methylamide is the basis for a detailed investigation of
structural effects of potential smoothing and a comparison of the partitioning of conformational space by potential smoothing and simulated annealing. It is shown that the
smoothing process partitions conformational space in a manner consistent with the inherent structure of the undeformed PES and with the partitioning expected by simulated annealing. Three features of potential smoothing − conformational shifting, basin merging,
and energetic crossing − which change the ensemble during potential smoothing are
shown to have direct analogs in simulated annealing by Master equation and molecular
dynamics calculations. Strong qualitative and quantitative correlations between simulated
annealing and potential smoothing are established. This conceptual analysis is used to
quantify the possibility of using either simulated annealing or potential smoothing as tools
for global optimization. It is concluded that potential energy smoothing is essentially the
deterministic analog of simulated annealing. A discussion of possible generalized algorithms which use correlations between the two methods to achieve a coupled conformational search protocol is included.
The analysis in Chapter 2 revealed the need for a local search mechanism to correct
for a characteristic of potential smoothing called "energy crossing". That algorithm, Normal Mode Local Search (NMLS), is described in Chapter 3. Potential smoothing as
implemented in DEM is intuitively appealing and has certain appropriate statistical
28
mechanical properties, but often fails to identify the global minimum even for relatively
small problems. Extensions to DEM capable of correcting its empirical behavior are systematically investigated. Two types of local search (LS) procedures are applied during the
reversing schedule from the smooth deformed PES to the undeformed surface. Changes
needed to generate smoothable versions of standard molecular mechanics force fields
such as AMBER/OPLS and MM2 are also described. The resulting methods are applied
in an attempt to determine the global energy minimum for a variety of systems in different coordinate representations. The problems studied include argon clusters, cycloheptadecane, capped polyalanine, and the docking of α-helices. Depending on the specific
problem, Potential Smoothing and Search (PSS) is performed in Cartesian, torsional or
rigid body space. For example, PSS finds a very low energy structure for cycloheptadecane with much greater efficiency than a search restricted to the undeformed potential
surface. It is shown that potential smoothing is characterized by three salient features. As
the level of smoothing is increased unique minima merge into a common basin, crossings
can occur in the relative energies of a pair of minima, and the spatial locations of minima
are shifted due to the averaging effects of smoothing. Local search procedures improve
the ability of smoothing methods to locate global minima because they facilitate the post
facto correction of errors due to energy crossings that may have occurred at higher levels
of smoothing. PSS methods should serve as useful tools for global energy optimization
on a variety of difficult problems of practical interest.
Results of the Potential Smoothing and Search (PSS) method applied to molecular
docking are presented in Chapter 4. Molecular docking requires efficient exploration of
29
possible orientations for two molecules. The multiple minima problem is frequently assuaged by the use of simulated annealing, genetic algorithms, or multiple-copy searching.
A primary advantage of PSS is the ease with which ligand and/or receptor flexibility is
accommodated. Results for the undirected rigid-body docking of benzamidine to trypsin
and flexible docking of XK263 to HIV-1 protease are presented. Preliminary results for
the prediction for directed flexible docking of HIV-1 protease and XK263 are also reported.
References
1
C. Leventhal, J. Chim. Phys., 65:44-5 (1968).
2
R. Unger and J. Moult, Bull. of Math. Biol., 55(6):1183-98 (1993).
3
D. R. Westhead, D. E. Clark, and C. W. Murray, J. Comput. Aided Mol. Des.,
11:209-28 (1997).
4
P. Amara and J. E. Straub, Phys. Rev. B, 53(20), 13857-63 (1996).
5
D. Shalloway, In Recent Advances in Global Optimization, C.A. Floudas and
P.M. Pardalos, Eds., Princeton University Press, Princeton, 1992, pp. 433-477.
6
J. Kostrowicki, L. Piela, B. J. Cherayil, and Harold A. Scheraga, J. Phys. Chem.,
95, 4113-9 (1991).
7
W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 110.6, 1657-66 (1988).
30
Chapter 2:
Potential Energy Smoothing: A
Deterministic Analog of
Simulated Annealing
31
Introduction
Molecular conformations are often compared via a potential energy function which
defines a potential energy surface (PES) over conformational space. Two separable issues
arise in such studies: the accuracy of the potential energy function itself, and the tractability of sampling the PES. A typical PES is a multidimensional hypersurface characterized
by topographical features of widely varying spatial scale. The broad features of the surface correspond to large scale conformational rearrangements while smaller features represent more localized changes. For this reason, the surface is said to be "rough".
The roughness of a surface impedes efficient sampling and precludes the possibility
that local optimization techniques will converge reliably to the global energy minimum.
Nonetheless, efficient sampling methods must avoid becoming trapped in one of the overwhelming number of local minima. At the same time they must not spend time exploring
regions of conformational space which may have little bearing on the most significant
conformational states and transitions. In the modeling of dynamic properties, extensive
sampling of conformational space is necessary in order to satisfy ergodicity within a
Monte Carlo or molecular dynamics simulation1. Another class of problem, conformational search, requires sampling directed toward finding a structure with specific desired
properties, often a structure of low or global minimum potential energy.
Hierarchical sampling is an important precept for locating low energy regions on a
PES. The general idea of hierarchical sampling is common to stochastic methods such as
simulated annealing2 and hypersurface deformation methods such as potential
32
smoothing3,4. In both methods, a control parameter − temperature or extent of deformation − determines the breadth of sampling. With an appropriate schedule for the control
parameter, a search procedure is gradually focussed on low lying regions on a PES.
Simulated annealing is a widely used stochastic method for molecular structure optimization and refinement. A molecular system is coupled to a heat bath which is initially
equilibrated at a very high temperature. The temperature is then iteratively reduced according to a cooling schedule and conformational space is sampled at each temperature. A
key to the annealing process is the equilibration at each iteration and the gradual lowering
of temperature5. A practical advantage of simulated annealing is that a cooling schedule
can be coupled to either a Monte Carlo (MC)6 or a molecular dynamics (MD)7 protocol
without significant modification.
Aarts and Korst have discussed the use of equilibrium Boltzmann statistics for analyzing simulated annealing8. Analysis in terms of Boltzmann statistics leads to the computation of equilibrium quantities including the average energy, the distribution of energies and the entropy. They elaborate on specific features of simulated annealing including
the conditions for asymptotic convergence to the global minimum, requirements of a
cooling schedule and different sampling regimes that ensue as a function of temperature.
It can be shown that simulated annealing ensures asymptotic convergence to the global
minimum given the caveat that the equilibrium Boltzmann distribution is attained at each
value of the temperature, T. Also the expected value for the energy and the entropy of the
system decrease monotonically with decrease in T provided the equilibrium distribution is
realized at each level. Acceptance ratios for proposed transitions in a MC protocol or the
33
number of transitions between local basins in a MD protocol for simulated annealing goes
through a very sharp transition region between the high and low temperature regions8,9,10.
For large values of T, the average value for the energy and width of the distribution of energies approach constant values. In a MC protocol the high temperature regime is characterized by acceptance ratio values tending to unity. A threshold region for the control temperature can be delineated based on the value the acceptance ratio, χ. The temperature region where χ ≈ 1/2 separates the high temperature region where χ → 1 from the low temperature region for which χ → 0.
In practice, the application of simulated annealing to molecular systems is fraught
with the several problems8,9,11,12. First, obtaining an equilibrium distribution at each temperature level requires extensive sampling or lengthy trajectories that grow exponentially
with the size of the system. The condition for obtaining equilibrium can be relaxed somewhat by stipulating sampling lengths to ensure quasi-equilibrium, i.e., a system tending to
equilibrium. Second, different ensembles result in the high and low temperature regions
respectively. This is characterized by a drastic alteration of the equilibrium occupation
probabilities of the low lying regions with respect to the rest of the PES. For high temperatures all states become equally accessible in equilibrium leading to rapid transitions
between states. This entails that transitions over higher energy barriers are equally likely
as transitions over lower energy barriers. As the temperature is lowered the protocol confronts a very sharp transition region to a regime where the equilibrium ensemble is radically different from the high temperature regime. Crossing the transition region leads to a
drastic reduction in excursions across states. For simulated annealing to work, either the
34
low lying regions need to be significantly populated for values of T higher than the
threshold temperature or the extent of sampling through the transition region has to be
significantly enhanced so the system can populate low lying regions. All of these considerations lead to the need for logarithmically slow cooling schedules. Straub9 has developed metrics to quantify the length of a required trajectory between Thigh and Tlow. He
shows that the number of steps along the cooling schedule is determined to a first approximation by the ratio of the highest energy barrier connecting the global minimum to
the rest of the PES, and the difference in energy between the global minimum and the second lowest minimum on the PES. For molecular systems such as proteins and peptides the
combination of different energy terms leads to a diverse distribution of minima and energy barriers making simulated annealing to the global minimum a difficult challenge.
Whereas simulated annealing facilitates barrier crossing by providing sufficent thermal energy, potential smoothing methods diminish or eliminate the barriers themselves.
As a result, low lying regions are "projected out" as broad conformational basins on deformed surfaces. The basic idea in potential smoothing is to mathematically transform a
multidimensional potential energy function into one which may be variably deformed.
The corresponding PES is dependent on a new parameter which controls the extent of surface deformation, i.e. the extent to which distinct minima cluster into conformational basins. A deformed PES is easier to search, and reversal of deformation combined with conjugate gradient minimization can potentially lead back to the global minimum on the
original PES. Smoothing methods include Scheraga’s Diffusion Equation Method
35
(DEM)3, Straub’s Gaussian Density Annealing4, and Shalloway’s Gaussian Packet
Annealing5 in probability space.
The motivation behind potential smoothing is to use information about PES curvature to transform a rough potential energy function f(x) into one which may be variably
deformed. The resulting deformable function, F(x,t), is dependent on the variables of the
original function, x, and an additional parameter, t, which controls the extent of deformation. In DEM, a deformable function is defined by
( )
∂
2
F(x,t) = lim 1 + β
N→∞
∂x
2
N
f(x) , where β = t N.
1
In the cases where this series converges, the solution F(x,t) satisfies the diffusion equation
∂F
∂F
=D 2
∂t
∂x
for which f(x) is the initial condition, i.e., F(x,t=0) = f(x).
2
2
In recent work15, we analyzed three major features of a typical potential smoothing
algorithm for global optimization. We discussed tuning of smoothing algorithms to for
productive global optimization. The main features of potential smoothing or Gaussian
spatial averaging are mergers, energy crossings and conformational shifting. As the level
of smoothing is increased unique minima merge into a common basin, crossings can occur in the relative energies of a pair of minima, and the spatial locations of minima are
shifted due to the averaging effects. Here we show that these features are exact analogs of
corresponding measurable features that accompany increased temperature in simulated
annealing. An interpretation that emerges from our analysis is that potential smoothing
can be viewed as a deterministic analog of simulated annealing.
36
In developing metrics to compare and contrast features of potential smoothing and
simulated annealing we note that previous work has been done to anneal approximations
to classical distribution functions which lead to smooth energy surfaces3,5,16. These methods rely on an implicit relationship between the extent of PES deformation and the system
temperature. In this work we uncouple the two control parameters, i.e., deformation and
temperature to reflect upon algorithms where the deformation parameter is set
adiabatically3,17 and compare the effects of spatial averaging explicitly to the effect of increased sampling temperature.
The deterministic nature of smoothing can also be used to hierarchically partition a
potential energy surface. Increased deformation leads to a many-to-few mapping of multiple minima into a select few basins. It can be shown that the clustering of conformers
into reduced numbers of basins as a function of deformation reflects the underlying structure to the PES. Particularly it emphasizes the clustering of structurally related conformers separated by lower energy barriers.
Full analysis of the partitioning of a potential surface requires complete enumeration of all the relevant features of the surface. A map of all the local minima and transition states on a PES allows a direct evaluation of the equilibrium thermodynamics of the
system and an accurate measure of thermodynamic averages and dynamical events. A
complete enumeration of minima and transition states using traditional search methods is
possible for systems with up to ~20 degrees of freedom18. For larger systems the number
of minima and transition states to be enumerated increases exponentially with the size of
the system. However, small systems for which the PES can be exhaustively enumerated
37
can serve as models for more complicated multidimensional systems, especially if the
PES for the smaller problem is described by individual potential energy terms that have
varying energy scales and spatial extents. PES topographies of model systems and small
molecules such as clusters and peptides have been used to quantify relaxation to the global minimum for global optimization19,20, characterize the nature of transition states that
determine rate limiting dynamical events in peptides21, study the relationship between
PES topography and dynamics18, and map the partitioning of conformational space into
basins of attraction22,23,24.
The current study uses the deformable OPLS (DOPLS) potential function to analyze
the conformational space of a capped dialanine peptide, N-Acetyl−Ala−Ala−NMethylamide (CDAP). We have thoroughly explored all minima and transition states on
the CDAP PES and provide a detailed characterization of conformational changes during
potential smoothing. Knowledge of the complete surface enables us to quantitatively demonstrate the increased conformational sampling provided by potential smoothing and
simulated annealing. This is particularly relevant in light of the recent results of Huber, et
al.25 who have shown increased sampling of conformational space in a protocol that
couples molecular dynamics to potential smoothing for the refinement of an undecapeptide, Cyclosporin A.
In the next section we describe aspects of the molecular mechanics potential, methods used to generate a fully characterized PES and the potential smoothing protocol. In
the Results section we discuss the partitioning of the undeformed PES, conformational
clustering on smooth surfaces, metrics to quantify smooth surfaces and a comparison of
38
important features of potential smoothing and simulated annealing. We conclude with a
comparison of the CDAP conformational network with a similar study by Czerminski and
Elber21, followed by a discussion of the implications the analogy between potential
smoothing and simulated annealing.
Methods
All calculations and structural manipulations were performed in Cartesian coordinates in vacuo using the TINKER molecular modeling package26.
Deformable OPLS (DOPLS) Potential Function and Parameterization. The
DOPLS potential function is a version of the OPLS potential function7 which is modified
to enable potential smoothing. DOPLS is dependent on the same Cartesian coordinates (r)
as OPLS, as well as a continuous parameter, t, which controls the extent of deformation.
Equation 3 shows the individual energy terms of the DOPLS potential.
( )
Etotal r,t =
∑E
bond +
bonds
+
∑E
dihedrals
∑E
angle +
angles
torsion +
∑∑E
atom pairs
∑E
improper
chiral
charge +
∑∑E
vdW
3
atom pairs
Bond and angle terms are implemented as in the standard OPLS7. Chirality and planarity are enforced using a CHARMM28 style improper torsional energy term as shown
in equation 4. Harmonic improper torsions are required to impose planarity at sp2 atoms
and chirality of sp3 α-carbon atoms. Parameters for the harmonic improper restraint term
were derived by fitting to the minima of a standard OPLS trigonometric improper
39
torsional energy term. The values used for the parameters in equation 4 are shown in
Table I.
(
1
Eimproper = KΘ Θ − Θ0
2
)
2
4
Table I. Harmonic improper torsion parameters. These values were
determined empirically by fitting OPLS-style trigonometric improper torsion to a CHARMM-style harmonic improper torsion
with emphasis on small displacement from the ideal value.
atom types
C-CH3-N-O
N-C-Cα-H
Cα-N-C-CH3
Θ°
(deg)
0.00
0.00
36.5
k
(kJ/mol/deg2)
251.0
23.0
732.0
Because it is easy to compute analytical solutions for a diffusion equation with
Gaussian initial conditions, we use a Gaussian approximation6 to the OPLS 12-6
Lennard-Jones potential as shown in equation 5.
ngauss
EvdW =
∑a e
i
i=1
( )
2
− bir2
, with ai =
a°i ε0,
bi =
b°i
21/6
, ε0 =
r0
εa + εb , r0 = 2 ra + rb
5
where εx and rx are the Lennard-Jones parameters for atom x, < a°i ,b°i > are reference parameters chosen to fit a canonical Lennard-Jones function with well depth ε = 4.184
kJ/mol and a hard sphere radius σ=1Å; ngauss is the number of Gaussians used in the approximation. We set ngauss=2, < a°1,b°1 > =<60614.0 kJ/mol, 905148 Å> and < a°2,b°2 > =<23.2353 kJ/mol, 1.22536 Å> which generates a very good fit over a wide range of interatomic distances6. A small van der Waals envelope of radius of 1 Å and Lennard-Jones
40
well depth ε=0.01 kcal/mol (0.042 kJ/mol) is added to polar hydrogen atoms to prevent
fusion with hydrogen bond acceptor atoms during large scale conformational changes in
potential smoothing.
The bond, angle, and improper torsions are not altered as a function of t. For values
of t > 0, the DOPLS potential function uses the deformable functional form for the torsion, electrostatic and van der Waals terms as shown in equations 6, 7, and 8. These are
obtained using the corresponding t=0 functional forms as initial conditions of a diffusion
equation.
The deformed DOPLS torsional energy is computed as shown in equation 6,
Etorsion(ω,t) =
1
2
∑V (1 + cos(ωj + φ))e
j
− j2Dtorsiont
6
j
where j is the periodicity, Vj is the half-amplitude, φ is the phase (0° or 180° for cis or
trans peptide bonds), and ω is the dihedral angle value. The electrostatic energy is computed as shown in equation 74,
Echarge(rij,t) =
erf
4πε0rij 2
qiqj
Dcharget
rij
7
where qi and qj are the partial charges for atoms i and j respectively and rij is the distances between these atoms. The deformable Gaussian approximation to the OPLS van
der Waals energy is computed as shown in equation 8,
ai
bir2ij
EvdW(rij,t) =
exp −
3/2
1 + 4biDvdWt
i = 1 1 + 4biDvdWt
where ai and bi are as in equation 5.
ngauss
∑
(
)
41
(
)
8
The parameter t controls the extent of potential surface smoothing. Dtorsion, Dcharge,
and DvdW are tunable diffusion coefficients which control the relative rates at which individual terms are smoothed. In the current work, we use Dtorsion= 0.0225 (radian)2,
Dcharge= 1Å2, and DvdW = 1Å2. These values were chosen based on an estimate of the
range and analysis of the type of diffusion space − Cartesian vs. torsional − accessible to
each term15.
The t = 0 DOPLS potential closely approximates the original OPLS potential function for reasonable low energy conformations. The average deviation of the original and
DOPLS potentials is 0.07 kJ/mol at the 142 minima and 0.15 kJ/mol at the 1038 transition
states discovered by grid search as described below. Analytical first and second derivatives of all terms in DOPLS are used in energy minimizations.
Minimization and Conformational Redundancy. All minimization were performed using a truncated newton conjugate gradient method with finite difference matrixgradient vector product30 to a GRMS of 10-4 kJ/mol/Å per atom. Two minimum energy
conformations are considered to be identical if the root mean squared deviation (RMSD)
from the superposition of all atoms is less than 0.001Å. When determining which members in a set of conformations are identical, we obviated a full pairwise O(N2) comparison
by superposing only those pairs within an energy window of 0.01 kJ/mol. The use of a
strict convergence criterion during minimization facilitates the use of energies to discriminate distinct structures. All conformational pairs determined to be identical by superposition had equivalent energies to 7 significant digits.
42
Generation of Capped Dialanine Peptide (CDAP) Conformations. The structure
of N-Acetyl−Ala−Ala−N-Methylamide (CDAP) is shown in Figure 1. CDAP conformations were generated by iterating over cis or trans peptide bonds and nine <φ,ψ> pairs
corresponding to canonical low energy regions shown in Table II31. This enumeration resulted in a set of 2 × 9 × 2 × 9 × 2 = 648 conformations. After minimization from each
starting conformation, 136 unique minima remained. Each minimum was assigned a
unique identifier equal to its energy rank (global minimum is structure 1, etc.) and a conformational code. The conformational code consists of five conformational descriptors,
one for each of the three peptide bonds and the two <φ,ψ> pairs in CDAP. Peptide bond
descriptors are classified as cis (c) or trans (t). A <φ,ψ> descriptor is a letter corresponding to one of 16 regions shown in Figure 2. As described in Results, the set of 136
minima was subsequently expanded to a connected network of 142 minima.
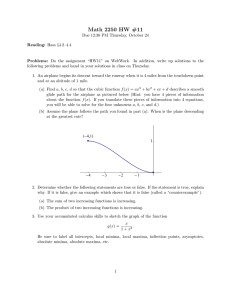
Figure 1. Capped Dialanine Peptide (N-Acetyl−Ala−Ala−NMethylamide; CDAP). Methyl groups are represented as united atoms. The system has 18 atom centers, 48 dimensional Cartesian
conformational space, and 7 rotatable bonds.
O H
O H
CH3 C N φ C
ω
0
1
ψ1
O H
C Nφ C
ω1
CH3
2
ψ2
C N CH3
CH3
43
ω2
Table II. Nine low energy regions identified by Zimmerman, et
al.31. These values were used for grid search on the undeformed
PES. The <φ,ψ> points are depicted in Figure 2.
angle
ωi ∈
<φ,ψ> ∈
values
{
0,
180}
{< -75, -45>,
< -85, 80>,
<-150, 70>,
<-155, 155>,
< -85, 155>,
<-160, -60>,
< 55, 60>,
< 80, -65>,
< 65, 180>}
conformer
(region)
cis
trans
αR (A)
β (C)
β (D)
β (E)
β (F)
αR (G)
αL (a)
(c)
(f)
Figure 2. Sixteen conformational regions of a Ramachandran map
identified by Zimmerman, et al.31 (cf. Table II). The nine <φ,ψ> regions used for grid search are denoted by filled circles. The two
<φ,ψ> descriptors of the conformational code were determined using this map.
180 E
180
F
f
130
110 D
C
ψ (°)
-10
90
g
a
b
B
A
-20
-50
-90
d
H
-180
-180
40
10
-40 G
-140 E
140
h
50
20
e
c
-110
-130
F
f
-110
-40
0
φ (°)
44
40
110
e
-180
180
Paths and Transition States. In order to fully characterize the surface, all transition states were located. Conformational paths connecting pairs of minima were calculated using the method of Czerminski and Elber21,32. The local energetic maxima along
each path were minimized to the nearest stationary point via a truncated Newton method.
Transition states were the subset of unique stationary points whose Hessians had exactly
one negative eigenvalue. Because the Czerminski and Elber method assumes a directionality of the path, we computed paths in both directions. In a few cases, a transition state
found in one direction differed from that found in the opposite direction.
Each transition state connects a pair of minima on the potential surface. In some
cases, more than one transition state connects the same pair of minima. The minima directly connected by a transition state were identified using the following scheme: 1) Two
conformations were generated by perturbing away from the transition state by a small displacement in each direction along the eigenvector corresponding to the negative eigenvalue; 2) the structures were minimized; 3) the identity of each minimized conformation
was determined by comparison with known minima using the conformational redundancy
criterion.
Potential Smoothing Protocol. The potential smoothing protocol consists of a series of minimizations on increasingly deformed potential surfaces corresponding to increasing values of t. We refer to the PES smoothed using t=ti as the ti surface. The undeformed potential surface is defined by t=t0=0. In the first stage of the protocol (i=1), each
minimum on the t0 surface is minimized on the smoother t1=t0+∆t surface. More generally, for iteration i of the protocol (1≤i≤n), each conformation on the ti-1 surface is
45
minimized on the ti surface. As surfaces become smoother, minima which are distinct on
the ti-1 surface may merge into a single minimum on the smoother ti surface. We detect
this event using the redundancy criterion. At the end of every iteration, we record the
identities of the minima which merge and the number of unique minima which remain.
For reasons presented in the Discussion, we use the convention that minima of higher energy merge into those of lower energy based on the energies on the ti-1 surface.
The rate of deformation is dictated by a schedule of n steps in the range [0,tmax]
given by
()
q
ti = tmax
i
, 1 ≤ i ≤ n.
n
For the present investigation, we used q=2, n=120, and tmax=60. A single minimum
remains on the CDAP surface for all t ≥ 51.27. The chosen smoothing schedule results in
a gradual clustering of similar conformations and provides sufficient detail to analyze
small changes in the PES. Similar results are obtained for slower (n=2000) and faster
(n=20) rates of smoothing, and for linear (q=1), cubic (q=3), or quartic (q=4) schedules.
Results
CDAP Conformational Network
CDAP was chosen because we could thoroughly search for minima and transitions
states on the undeformed surface. This set of conformations provided the means by which
we could carefully monitor the evolution of minima during the potential smoothing
46
protocol. These observations enabled the present detailed 1) analysis of changes to the
surface features during potential smoothing; 2) comparison of conformational clustering
by potential energy barriers and potential smoothing; and 3) comparison of global optimization by potential smoothing and simulated annealing.
We initially computed 4262 reaction paths between the pairs of the 136 minima
which differed by zero (2), one (864), or two (3396) conformational descriptors. This initial calculation of reaction paths resulted in 667 unique transition states of which 75 represented paths connecting minima which differed by three descriptors. In light of previous
results for isobuturyl-(ala)3-NH-methyl (IAN) on the CHARMM PES21, we were intrigued that single transitions spanned three degrees of freedom. We computed the 7128
reaction paths between minima which differed by three descriptors and found 312 additional unique transition states. By this point, 10806 of 136 x 135 = 18360 possible reaction paths where each path was generated in both directions. The remaining reaction paths
could be computed quickly and we did so. We found 59 additional transition states. No
paths connected minima which differed by more than three descriptors.
Minimization from 667 + 312 + 59 = 1038 unique transition states led to the discovery of 6 new high-lying minima. These were 130-170 kJ/mol above the global minimum
and connected to existing minima by energetic barriers less than 0.6 kJ/mol. The six new
minima were added to the pool of unique minima and the remaining pairwise paths were
computed; no new minima or transition states were found. Because all minimizations converged to known minima, we believe that the set of local minima and transition states
found from the grid search represents an exhaustive enumeration of the topographical
47
features of the PES. The resulting network on the undeformed DOPLS PES consists of
142 unique minima and 1038 unique transition states which form a connected network of
conformations, i.e., there exists at least one sequence of paths from every minimum to every other minimum.
The distributions of minima and transition state energies are shown in Figure 3. The
minima conformations are distributed widely over regions of the Ramachandran map as
shown in Figure 4, include all cis and trans peptide bond combinations, and cover 140
unique conformational codes. The conformations of the 10 minima within 15 kJ/mol of
the global minimum are presented in Table III and Figure 5.
Figure 3. The distribution of energies of 142 unique minima (filled)
and 1038 unique transition state (hollow) conformations on the undeformed DOPLS PES. The bin size is 5 kJ/mol.
70
Number of Conformations
60
50
40
30
20
10
0
−355
−320
−285
−250
−215
Energy (KJ/mol)
48
−180
−145
Figure 4. Ramachandran plots of <φ1,ψ1> (left) and <φ2,ψ2> (right)
for the 142 minima discovered by grid search.
180
180
90
90
ψ1
ψ2
0
−90
0
−90
−180
−180
−90
0
φ1
90
−180
−180 −90
180
0
φ2
90
180
Table III. Ten minima within 15 kJ/mol of the global minimum.
The columns are structural identifier (energetic rank on the undeformed DOPLS PES), energy, dihedral angles (cf. Figure 1), and
conformational code. Structures of minima 1-4 are depicted in Figure 5.
Id
1
2
3
4
5
6
7
8
9
10
Energy
(kJ/mol)
-352.2535
-351.1075
-343.0721
-342.8826
-342.8717
-342.0407
-338.9203
-337.9781
-337.9781
-337.1430
ω0
(°)
+174
-178
-178
+173
+174
+179
-179
-179
-169
+177
φ1
(°)
-99
-83
-84
-153
-149
+64
-77
+59
-79
-88
ψ1
(°)
+118
+67
+68
+167
+162
-55
+77
-74
-6
+77
49
ω1
(°)
-5
-176
-178
+174
-179
+178
-174
+177
+173
+176
φ2
(°)
-119
-82
+65
-152
-84
-82
-174
-101
-124
-142
ψ2
(°)
+100
+65
-54
+166
+72
+67
-39
-15
+29
+157
ω2
conf.
code
(°)
-177 tCcDt
-179 tCtCt
+179 tCtct
-179 tEtEt
-178 tEtCt
-179 tctCt
+178 tCtBt
-179 tctAt
-179 tBtDt
-178 tCtEt
Figure 5. The four lowest energy structures. See also Table III.
00
000
000
000
CH3
H
C
00
00
N
C
00
00
C
H
O
00
00
C
N
O
O
N
H
C
C
C
00
00
O
N
H
N
C
O
C
C
H
CH3
CH3
C
H
N
CH3
C
C
00
00
C
O
minimum 1, E=-352.2535 kJ/mol
minimum 2, E=-351.1075 kJ/mol
0
0
O
00
000
00
C
CH3
C
O
H
C
C
H
C
CH3
0
00
N
N
N
00
00
0
O
H
H
0
0
N
O
C
CH3
C
O
C
C
C
N
H
C
C
000
000
000
C
C
O
N
CH3
H
minimum 3, E=-343.0721 kJ/mol
minimum 4, E=-342.8826 kJ/mol
A number of important features of the PES topography were obtained by an analysis of the transitions directly accessible to each minimum. For every transition state, ts,
which connects minima m1 and m2, with energies Ets, Em1, and Em2 and such that
Em1<Em2, we computed the high-to-low energy barrier (Eb=Ets12- Em2). When multiple
transition states connect the same pair of minima, we used the transition state of lowest
50
energy. The smallest barriers are typically between high lying minima. There does not appear to be any obvious correlation between Eb and the energies of the minima. There are
on average 15 paths connecting a given minimum and the number of paths connecting
each minimum ranges between 1 and 67. Higher energy minima tend to be connected by
fewer paths than those at lower energy. The lowest energy structure, minimum 1, is a
deep minimum which is connected to 67 transition states. This apparent density of reaction paths belies the fact that all transitions from minimum 1 to other minima are via barriers of 48 kJ/mol or more. Other minima typically have several pathways over much
smaller barriers. For instance, minimum 2 has 5 reaction paths over barriers below 20
kJ/mol, and 9 below 48 kJ/mol.
Paths between minima were classified by the number of conformational descriptors
which differ between the incident minima. We observed paths between minima which differed by zero, one, two, and three descriptors. Of 17 paths which connect minima differing by zero descriptors, 14 are self-connecting or "loopback" paths. We noted that a large
number of paths connected minima which differed by three descriptors and we were concerned that classification by conformational descriptors overestimated the significance of
conformational changes. We investigated the structural differences for these more thoroughly by computing the Euclidean distance spanned by a path for both <φ1,ψ1> and
<φ2,ψ2> as shown in equation 9.
di =
(∆φ ) + (∆ψ )
2
i
2
i
(|
|
|
∆ωi = min ωi,a − ωi,b , 360 − ωi,a − ωi,b
51
|)
9
where di is the <φi,ψi> Euclidean distance between structures a and b. The results are
summarized in Table IV and clearly show that a significant number of paths spanned
large distances of conformational space.
Table IV. Summary of 1038 paths in the CDAP network. There are
1038 paths between 688 unique pairs of minima. The average Euclidean distance between minima in <φ,ψ> space (see equation 9)
and the average minimum energy barrier.
unique
total
<d1> ± s.d.(°)
<d2> ± s.d. (°)
<Eb> ± s.d. (kJ/mol)
loopback
7
14
77 ± 58
number of changed descriptors
0
1
2
2
306
243
3
489
345
16 ± 10
66 ± 77
86 ± 69
111 ± 74
72 ± 76
98 ± 67
6±7
32 ± 26
44 ± 27
3
130
187
113 ± 58
111 ± 60
64 ± 23
Conformational Clustering Based on Energetic Barrier and Potential
Smoothing
We use the term "clustering" to refer to the partitioning of a set of conformations
into mutually exclusive subsets or "hard" clustering. Clustering imposes a partial ordering
of the system in which all but one conformer has a parent basin. We employ an agglomerative clustering algorithm equivalent to that used by Shenkin and McDonald33 for
geometric clustering. For a system with N conformations, there are N(N-1)/2 possible
generalized pairwise distances. Agglomerative clustering identifies the N-1 shortest edges
which join the conformations in a connected network. We associate a directionality with
each edge based upon the energies of the connected conformations, e.g., minimum m1
merges into m2. The result is a set of N-1 ordering relations which are easily and meaningfully represented by tree diagrams in which branches of the tree represent families of
52
structures or conformational basins. Trees neatly depict two important pieces of information: 1) the greatest separation of structures within a single basin, and 2) the minimum
separation between structures in different basins. For example, a branchpoint which joins
branches b1 and b2 at 5 units indicates: 1) that every minimum in basin b1 is separated
from every other minimum in b1 by no more than 5 units, and 2) that every minimum in
basin b1 is separated from every minimum in b2 by at least 5 units. For examples, see
Figure 6 and Figure 9.
Energetic Clustering
We characterized the topography of the undeformed PES by clustering conformations according to energetic barriers between states. In energy barrier clustering, we use
the energetic barrier from the minimum of higher energy to that of lower energy as a measure of the distance between a pair of minima. Specifically, for each path between minima
A and B connected by transition state AB with energies EA, EB and EAB respectively, we
associate the minimum energy barrier Eb=min(EAB-EA, EAB-EB). In cases where minima
A and B are connected by multiple paths, we used the minimum Eb value for the connection. A similar partitioning scheme has been used by Becker and Karplus to generate a canonical disconnectivity graph for a IAN, where Eb can be associated with a thermal energy barrier kT24. We chose to associate with each transition the energetic barrier traversed in crossing from the higher- to lower- energy minimum. We use this convention by
analogy with simulated annealing in which a system becomes gradually confined to low
energy regions because the transitions out of these regions become less probable with decreasing temperature. Thus, barriers act as a sort of one-way trap whose effect is to
53
localize a system in regions of low energy. A minimum subsumes all higher energy
minima which are connected it via barriers less than or equal to some threshold energy,
E*. We use the identifier of the structure of lowest energy conformation within a basin as
a representative of the basin and the conformations therein. The result of this clustering is
shown in Figure 6. E* is equivalent to a threshold thermal barrier kT where T may be the
temperature of a heat bath coupled to the system.
Figure 6. Energy barrier clustering of minima. All minima within a
branch are connected by barriers not greater than the value on the
ordinate. The two most energetically distinct basins are 1 and 2.
There is no pair of minima, one from basin 1 and one from basin 2,
connected by a barrier less that 80.455 kJ/mol. The eight structures
at E*=38 kJ/mol are used for comparison with potential smoothing.
001
80.455
001
70.836
002
001
011
001
58.604
004
001
018
011
011
50.529
028
002
018
46.115
018
37.279
002
040
002
0 1
31.415
001
001
011
17.292
11.290
045
002
011 011 025
20.734
14.238
002
033
27.125
24.419
001 001 054
001 001 037054
054
101
033
033
076
033 033 070
033 107 070
070 121
03303 065
002
006
003
006
006019
011 0 1 068
028
028
040
040040118114
040 40090
04004007090
090
26
04040
08
018 018 072
073
07
06
072
016
062
016
016
016 0 6 046
029
029063
062062116
0 1
011
110
011 0 1 042
028
028
064
8.455
5.828
3.219
001
080
098
038
053
055
060
066
077
059
099
117
037
123
054
100
130
101
112
033
034
036
069
065
071
124
107
070
089
139
128
121
076
084
140
127
018
105
082
056
057
096
132
138
097
072
122
108
141
040
074
083
135
073
106
090
126
118
142
114
004
011
014
020
043
047
094
027
042
081
095
048
049
120
093
110
068
078
092
025
052
045
051
102
028
075
085
030
104
113
064
103
111
125
115
087
088
062
119
131
134
079
116
129
136
133
002
013
010
009
012
005
007
031
015
026
003
017
035
021
006
137
008
019
041
016
039
022
061
024
023
044
046
050
086
058
067
109
029
032
063
091
0.479
54
Smoothing Clustering
The topographical changes which occur during potential smoothing are characterized by three processes: minimum location shifting, energy rank inversion, and merging.
These effects and their influence on the smoothing protocol are depicted in Figure 7 and
discussed below.
We refer to the migration of basins in conformational space during potential
smoothing as shifting. Shifting in the CDAP system is shown in Figure 8. The displacement of <φ1,ψ1> and <φ2,ψ2> on Ramachandran maps depends on the proximity, relative
depth, and relative breadth as measured by the number of minima which constitute the basin. This effect is a consequence of the changing curvature of minima and lowering barriers and results in minima which become more shallow and the basins which become
broader with increasing deformation. Minima tend to shift in the direction of the broadest
features on the surface. In Figure 7, minimum D0 on the t = 0 surface is minimized on the
t = t1 surface. This causes the conformation to shift to a slightly changed conformation,
D1. For CDAP, we found that structures shift very little, except when they merge with
other structures.
55
Figure 7. Illustration of topographical changes during potential
smoothing. The original PES (t0) appears at the bottom. PESs at increasing deformations are offset vertically for clarity. Potential
smoothing is characterized by three processes: shifting, merging,
and crossing. Dotted arrows depict an adiabatic change of deformation for a given conformation. Solid black arrows denote minimizations. Minima are indicated by filled circles.
t4
t3
B4
B5
t2
C3
Merging
t1
B3
Crossing
C2
t0
A2
D1
Shifting
B2
C1
B1
D0
A1
C0
B0
A0
56
Figure 8. Ramachandran plots of <φ1,ψ1> (top) and <φ2,ψ2> (bottom) for four levels of surface deformation: t=0, 0.61, 1.08, 1.69. The
deformations shown were chosen to exemplify features of the clustering or differences between to the two sets of dihedrals and are
typical for other deformations.
t=0.00
t=0.61
t=1.08
t=1.69
φ1
φ1
φ1
φ1
180
90
ψ1
0
90
180
180
90
ψ2
0
90
180
180
90
0
φ2
90
180
180
90
0
φ2
90
180
180
90
0
φ2
90
180
180
90
0
φ2
90
180
The rank energy order of a pair of minima may invert, which we refer to as a crossing. Crossing is depicted in Figure 7 with minima A1, B1, A2, and B2. Notice that E(A1) <
E(B1) on the t = t1 surface but that E(A2) > E(B2) on the t = t2 surface. From the example
above, a reversing procedure3 may follow B2 at t2 > t1, oblivious to the inversion of energies at a lesser deformation. Crossings have serious consequences when potential smoothing is applied to global optimization and we and others have developed techniques to circumvent this effect15,34.
Two minima mi and mj which are unique at some deformation will eventually
merge into the same basin conformation b at some deformation tm. To be faithful to the
57
crossing phenomenon identified above, we chose the convention of using the energies at
the previously deformed surface rather than the undeformed PES to determine merge direction. Thus, minimum 4 is the root of the tree in Figure 9 because it crossed with minimum 1 at t = 1.22. If the deformation process is characterized by crossing-free mergers
then the minimum that survives on the highly deformed surface is related to the global
minimum. Merging is depicted in Figure 7 using minima C1, D1, and C2. When conformations C1 and D1 on the t=t1 surface are minimized on the t=t2 surface, they converge
to the same conformation C2. Merging is the basis for conformational clustering on deformed potential surfaces and results in the dramatic reduction of the number of minima
shown in Figure 10.
58
Figure 9. Smoothing clustering of minima. The 142 minima enumerated by grid search appear as leaves of the tree at the bottom of
the figure. The ordinate is the extent of deformation and is discontinuous for readability purposes. As deformation increases (from
bottom to top), minima merge into basins. The eight structures at
t=1.69 are the basis for much of the analysis herein.
60.000
55.060
50.340
44.950
39.870
35.090
29.900
25.120
20.170
15.250
10.590
5.800
004
004
004
004
004
001
004
004
023
011
011
023
023
011
018
023
023
036
030
030
030
040
1.870
1.690
1.530
1.370
1.220
1.080
0.950
0.830
0.720
0.610
0.510
001
080
098
038
054
100
059
099
117
066
077
101
130
037
123
053
055
060
112
002
004
010
005
009
012
013
021
041
006
015
026
003
017
007
031
008
019
035
011
014
020
094
043
047
025
052
027
110
141
042
048
049
093
081
095
045
051
102
120
122
018
082
105
072
108
068
078
092
056
057
097
096
132
138
016
022
137
039
061
058
067
023
024
046
050
032
063
044
086
029
091
109
128
033
034
036
076
084
070
121
069
107
065
071
124
140
089
139
127
028
079
131
134
030
075
104
113
085
062
119
064
103
129
111
125
136
087
088
116
133
115
135
040
090
083
126
074
114
073
118
106
142
0.420
0.340
0.270
0.210
0.150
0.110
0.070
0.020
59
Figure 10. The number of minima during potential smoothing. The
number of minima decreases monotonically as minima merge to
form conformational basins.
150
Number of Minima
100
50
0
0.0
5.0
10.0
Time
15.0
20.0
Whereas Shenkin and McDonald33 cluster structures which are geometrically similar, we cluster based upon the smoothing parameter t and energy barrier Eb. In smoothing
clustering, the distance between structures is the deformation, t, at which they merge. In
energetic clustering, the distance is lowest high-to-low barrier, Eb. We use the clustering
results provided by these methods for drawing comparisons between potential smoothing
and temperature controlled algorithms for conformational space searching.
Distant regions of conformational space are typically clustered as a single merging
of one basin into another. The <φ,ψ> centers of basins are essentially stationary until they
60
merge. In other words, basins merge not because their fiducial centers are gradually
drawn together, but because the barrier between them is eliminated. The surviving minimum which represents a basin of conformations is usually located near the larger of the
merging minima. This observation is important because it suggests that a basin of structures is located near a conformation which represents the most prevalent members of the
basin, not a meaningless average structure.
Snapshots for the evolution of minima on the t = 0, 0.61, 1.08, 1.69 surfaces are
shown in Figure 8. The 142 minima on the undeformed PES are scattered over all quadrants of the <φ,ψ> map and aggregate into several groups within each quadrant, nominally occupying the canonical αR, αL, and β regions, and a low energy region near <+50,50> at t=0.610. For t=1.08, only the αR and β regions remain. Similar patterns of clustering are seen for both <φ1,ψ1> and <φ2,ψ2>.
Figure 8 illustrates several important trends in smoothing clustering. Basins shift in
a manner determined by the relative breadth and depth under the influence of the other
basins on the surface. Basins become broader during smoothing and particularly as a result of a merging. As a result, the "weight" of a basin reflects the number, energies, and
heterogeneity of the minima from the undeformed surface which constitute the basin. We
observe that those with greater weight survive the merging process. We are presently investigating the extent to which basin weights correspond to conformational space volumes
and are consistent with statistical mechanical expectations.
Figure 8 shows that basin conformations which remain on moderately deformed
surfaces are reasonable chemical structures. This occurs because basins are approximately
61
stationary in conformational space, except when merging. We emphasize that merging occurs because the barrier between two minima is eliminated, and not because two minima
shift into each other. Evidence of this effect may be seen in Figure 8 by noting that the
<φ1,ψ1> and <φ2,ψ2> coordinates of the β cluster is relatively stationary for t=0.61, 1.08,
and 1.69. Because basins do not migrate significantly except when merging, conformational search on deformed surfaces provides a meaningful sampling of the conformations
represented by a basin.
An essential feature of potential smoothing is its ability to deterministically simplify
a potential surface in a way which preserves the large scale features of the PES. Merging
is a consequence of the shape of a PES. At low t, high frequency features of the PES are
subsumed into nearby basins. At high t, only the lowest frequency features of the PES remain. Because the minima which remain on a deformed PES represent those dominant
features, we refer to potential smoothing as a projection method, i.e., the catchment regions are deterministically projected from the original PES.
As deformation of a PES is increased, barriers between minima are gradually eliminated. When this happens, two minima merge into a common basin. At every level of the
smoothing protocol, the merging of minima is recorded. We interpret the value of t at
which such a merging occurs as a "distance" between the conformations which merge.
Because redundant structures are eliminated throughout the smoothing protocol, we obtain exactly N-1=141 merger relations. That is, the smoothing protocol implicitly clusters
minima based upon conformational similarity on deformed surfaces. From these
62
distances, a tree of the potential smoothing clustering process is generated as shown in
Figure 9.
We establish the connectivity of the minima on the t=1.69 surface as was done for
all minima on the undeformed DOPLS surface. At this level of smoothing, the system exhibits all eight permutations of the three cis or trans peptide bond conformations. A diagram of this network is shown in Figure 11. Further collapse of this cube occurs by nearly
simultaneous merging of parallel transitions: ω1 (minimum 1→4, 36→23, 18→11,
40→30) for t≈7, ω2 (11→4, 30→23) for t≈39, and ω0 (23→4) for the t = 51.27 surface. It
is important to emphasize the structural significance of the clustering represented in Figure 11. The barrier to rotation of a single peptide bond is ~84 kJ/mol and is expected to be
the origin of the most prominent features of the undeformed surface. Each of the 8
minima remaining on the t = 1.69 surface represent a combination of these three structural
characteristics. This suggests that the smoothing process simplifies the original PES by
clustering conformations into broad basins according to the distinguishing PES features,
i.e., cis-trans isomers. Conformational search on this smooth surface will correspond to
cis-trans isomerization and will not be inhibited by the large number of minima within
each of the basins.
63
Figure 11. Network of conformations remaining at t=1.69. The eight
remaining structures represent all combinations of cis-trans interconversions of the three peptide bonds and are denoted by the vertices of the cube. Each vertex shows the rank of the energy on this
surface and the structure identifier in parentheses for comparison
with Figure 9. Paths are denoted by the edges. Transition states
were found for every edge of the cube and nowhere else. The energy
rank of transition states is indicated by italic text adjacent to an
edge. Edge arrows point from higher to lower energy on the t=1.69
surface. Note that minimum 4 from the undeformed surface is the
global minimum on the deformed surface.
16
6
8
(30)
(40)
7,8
15
6
2
13
(23)
(36)
14
5
(11)
1,2
9,10
4,5
1
(4)
18
4
7
(18)
11,12
3
3
(1)
ω0
cis-
trans-
ω2
cisω
cis- 1
Computational Time. All calculations were performed on a Digital 2100 Server
with four 250 MHz alpha CPUs running Digital Unix 4.0 utilizing coarse parallelization
of each procedure. Total time for grid generation and network computation (~40000 minimizations, ~700 superpositions) was about 4 hours. The potential smoothing protocol required about 20 minutes. Energetic clustering required negligible CPU time.
64
Comparison of Smoothing Clustering and Energetic Clustering. We compare
clustering using energy barriers and smoothing levels when these methods reduce the
original set of 142 minima to 8 important basins. This corresponds to a smoothing level of
t = 1.69 (Figure 9) and an energy barrier of E* = 38 kJ/mol (Figure 6). In each clustering
technique, every minimum on the undeformed PES is a member of exactly one basin. In
Figure 9 and Figure 6, the minima which constitute a basin are the leaves of the tree
which appear beneath a branch.
We compare the clustering techniques by examining which minima are clustered together and the structural similarities and differences in each basin. Similarities between
the two types of clustering for t=1.69 and E*=38 kJ/mol are shown in Table V. If the two
methods are exactly equivalent, the overlap (set intersection) of members for each basin
would be identical and Table V would contain no off-diagonal elements. This is true for
all but the intersection of the E1 and S36 basins. E1 consists of a total of 36 conformers of
which 19 are trans-cis-trans (ω0-ω1-ω2) and 17 cis-cis-trans conformers. The total population of S1 is 19 trans-cis-trans conformers. The overlap of E1 and S1 is the 19 transcis-trans conformers. The remaining 17 cis-cis-trans conformers in E1 are split between
S36 and S23. This suggests that clustering in terms of potential smoothing shows more
uniformity in conformational types compared to clustering in terms of energy barriers. It
also shows that the partitioning of conformational space is similar and, therefore, that
smoothing clustering and barrier clustering are qualitatively similar.
65
Table V. Comparison of clustering by the energy barrier and potential smoothing methods at levels where 8 basins remain (t<=1.69,
E*<=38 kJ/mol). Each basin represents a set of structures on the
original PES clustered by either energy barrier (EB) or potential
smoothing (PS) merge time. The number of structures in the intersection of each potential smoothing basin with each energy barrier
clustering basin suggests the extent to which the two techniques
cluster similarly. We identified each basin by the member with the
lowest energy. It is important to note that the names are irrelevant;
only the nature of the overlap between basins clustered by the different techniques is meaningful.
PS
1
4
11
18
23
30
36
40
EB
pop.
19
19
22
14
19
23
16
10
1
36
19
2
19
11
17
18
22
16
13
28
11
40
3
1
18
2
11
20
3
1
4
1
1
17
22
16
1
10
66
Potential Smoothing and Simulated Annealing as Global Optimization
Methods
Comparison of Potential Smoothing and Simulated Annealing
We have analyzed the clustering of CDAP conformers in terms of potential smoothing (t) and energetic barrier height (Eb). The latter is similar to the mapping procedure in
a canonical ensemble formulated by Becker and Karplus24. Energy barriers can be ascribed a temperature, i.e., Eb is equivalent to a particular value for kT. Therefore the two
clustering methods reflect a comparison between deformation and temperature. Similarities in PES partitions derived from the two clustering mechanisms lead to a quantitative
comparison of potential smoothing and simulated annealing.
In simulated annealing2 the system is coupled to a heat bath at some high initial
temperature Thigh. Thigh is typically set to be commensurate with the highest energy barrier on the PES. At this temperature, the system is characterized by rapid transitions between high and low lying minima because all energy barriers can be negotiated easily. After an equilibration period, the temperature is lowered down to Tlow according to a prescribed cooling schedule. Very slow cooling schedules are required as the system goes
through the sharp transition region between the non-native high temperature ensemble
and the native ensemble. A decrease in temperature is associated with an increased likelihood of occupying low energy states and reduced likelihood of transitions between
minima. A single trajectory between Thigh and Tlow is generated by coupling the cooling
schedule to either a Monte Carlo35 or molecular dynamics36 protocol. The relative occupation probabilities for the different minima change as a function of temperature and
may eventually lead to a reordering in relative equilibrium occupation probabilities of the
67
minima. This feature is analogous to crossings between pairs of minima in potential
smoothing.
Potential smoothing is a deterministic analog of simulated annealing. In the following sections we prove this assertion by comparing specific features of the two methods.
These features include shifting of minima, crossings between pairs of minima, mergers of
minima and efficiency of the two methods in converging to the global minimum. We use
the fully enumerated PES of CDAP to quantify these comparisons.
Potential Smoothing
In smoothing, conformational shifts, crossings and mergers determine the new ensemble obtained as a function of PES deformation. Changes in ensemble as a function of
temperature are due to shifting of the average location of the minima, crossings in relative
occupation probabilities and the reduced likelihood of populating low lying regions.
These three features play an important role in using either method as a tool for global optimization. In the following section we quantify the efficiency of potential smoothing and
simulated annealing algorithms as global optimization tools for CDAP. The global minimum for CDAP has an energy of −352.2535 kJ/mol and the second lowest minimum has
an energy of −351.1075 kJ/mol (see Table III).
We used the diffusion equation method of Scheraga and coworkers for global optimization based on potential smoothing. A smoothing algorithm for global optimization
proceeds as follows:
• The original potential function V(r) is replaced by a transformed function U(r,t), where t is a control parameter that determines the extent of
68
smoothing. The parameter t is initially set to a large value at which level
minimizations from random starting conformations converge to a
unique minimum on the deformed convex PES.
• The smoothing parameter is slowly reduced according to a chosen reversal schedule3 followed by minimizations until t = 0 at which point
the original PES is reached.
A crossing between minimum 4 and the global minimum occurs for t = 0.1372, i.e.,
V(4) < V(1) for all values of t ≥ 0.1372. For t > 51, only a single minimum remains on the
deformed PES. The smoothing process projects a basin related to minimum 4 for large t
(see Figure 9). For the DEM reversal schedule, we choose the deformation ti according to
( )
n−i
t =t
i
max n
q
, tmax = 60, n = 100, and q=3. The level of smoothing is adiabatically
lowered from followed by a minimization. Our results are insensitive to tuning the
smoothing schedule for reversal. A quadratic or quartic schedule yields identical results
for the DEM calculation. This reversal procedure converges to minimum 4 on the PES
for t = 0. As shown in Figure 10, the number of minima on the PES is reduced through a
series of mergers. However, the mergers of minimum 1, 2 and 3 with minimum 4 are preceded by crossings, i.e., there is a level of smoothing for which V(i) < V(4), i ∈ {1,2,3}.
These crossings preclude the possibility that a procedure like DEM will converge to the
global minimum.
The main advantage of potential smoothing lies in projecting a low energy basin
from the PES. This is always true if the underlying PES has sufficient underlying
69
structure. For the case of CDAP, DEM can be modified to converge to the global minimum by coupling a secondary local search procedure to the reversal schedule. At chosen
levels of smoothing along the backtrack a secondary procedure is used to search the vicinity of the current minimum. If an alternate minimum can be found, the system is
moved to the new location. For CDAP we applied two local search procedures that successfully found the global minimum by locating the errors due to crossings for values of t
lower than 0.13. Details of these secondary search methods are published elsewhere15. In
summary, potential smoothing algorithms such as DEM will not succeed as global optimization tools if the projected basin on largely deformed surfaces is not related to the global
minimum, which is true the case if any of the crossings involve the global minimum.
Simulated Annealing
We propose that potential smoothing is analogous to simulated annealing. If this hypothesis is correct, then we ought to expect that simulated annealing should not be very
successful in finding the global minimum for CDAP. Because simulated annealing is a
stochastic process, we repeat the simulated annealing protocol multiple times and compute the likelihood of finding the global minimum. We use two approaches to study relaxation to the global minimum using simulated annealing. The fully enumerated PES of all
the minima and the network of transition states can be used in a Master equation
formalism37,19 to quantify ensemble kinetics. The Master equation describes the time
evolution of the probability Pi(τ) of finding the system in minimum i at time τ. In this approach we populate the highest lying minimum, minimum 142, and compute the time dependent occupation probabilities for different temperatures along a prescribed cooling
70
schedule. Success of simulated annealing is based on the occupation probability for the
global minimum at the end of the Master equation calculation. A second approach is
based on molecular dynamics simulated annealing36 calculations using a very slow cooling schedule.
Simulation Duration in Simulated Annealing. The ability of simulated annealing
to accurately converge to regions of low energy depends critically on 1) rate of cooling
and 2) the completeness of equilibration for each temperature level 2,19. Straub 9 has developed a metric to show that these two parameters are equivalent to the number of steps
required for slow cooling. The length of the required simulation to isolate the global minimum in simulated annealing grows exponentially with important size scales on the PES.
We provide a brief review of Straub’s metric to quantify simulated annealing and apply it
to the PES of CDAP.
Efficient sampling of the PES at a given temperature T requires a simulation of
Emax
b
) 9. Emax
= Γ exp(
length τ
is the highest energy barrier connecting the global
b
sim
r
kT
minimum to the rest of the PES and Γr is the characteristic time for vibrational motion.
The lower bound for the total simulation time τsim can be written in terms of the number
of steps required along the cooling schedule, Nstep:
max
P (1)
τ
Eb
sim
eq
exp
≡ Xexp(E )
N
=
≈
step
gap
1-P (1)
Γ
∆E
eq
r
71
10
Here ∆E is the difference in energy between the global minimum and the second
lowest minium, and Peq(1) is the equilibrium probability for the global minimum at low
temperature.
For the PES of CDAP the appropriate parameters are Γr ≈ 10-13 s and the highest
energy barrier connecting minimum 1 is 138.69 kJ/mol. The equilibrium probability at
low temperatures is about 0.9, i.e., X ≈ 9, ∆E is 1.15 kJ/mol and Egap = 121.0 kJ/mol. According to equation 10, Nstep has to be ~1053 in order to successfully converge to the global minimum. This is an extreme estimate for the length of a proposed simulated annealing run to converge to the global minimum. Clearly at least one or more of one hundred
independent runs will find their way to the global minimum for a problem as small as
CDAP. Equation [9] really stresses the role that variation in size scales on a PES, as measured by Egap, plays in determining the likelihood of success for a single annealing trajectory.
Master Equation Calculations to Simulate Simulated Annealing. In order to
simulate simulated annealing, we use the Master equation formalism to study relaxation
of CDAP to the global minimum. Details of the Master equation formalism are given in
Appendix A. The algorithm used for this purpose is as follows: We set up a cooling
schedule between Thigh = 600K and Tlow = 100K. The upper limit for Thigh is a conservative estimate for the validity of the Master equation formalism. We start the simulation
under by setting the temperature to Thigh, P(142) = 1 and P(i) = 0 for i≠142; Pi(τ) is computed for each of the states by setting τ = τmes and using the solution given in appendix
equation A4. τmes ≥ 1ns because the Master equation formalism is not valid for smaller
72
time intervals. The distribution of probabilities Pi(τmes) for T = Thigh is used as the initial
condition for T = Thigh − ∆T, the next temperature value from the cooling schedule. This
procedure is repeated until T = Tlow is reached.
The two parameters that determine results of the Master equation simulation are the
cooling schedule and the value for τmes at each value of T. We found that the results of
our calculations were very sensitive to the values chosen for τmes and were relatively insensitive to details of cooling schedule between Thigh and Tlow. This is because the Master equation formalism does not simulate real time dynamics. τmes is typically a few orders of magnitude longer than the characteristic relaxation times for vibrational motions
which are directly influenced by temperature fluctuations. Within a Master equation formalism, τmes is the equivalent of the Γsim or Nstep parameters for the cooling schedule
discussed above.
Results of Master equation simulations of simulated annealing for different values
of τmes are shown in Table VI. For small values of τmes the probability, at the end of the
simulation, of populating the global minimum or the second lowest minimum is very
small. This behavior changes with increasing τmes which is equivalent to a more elaborate
cooling schedule. For τmes ≥ 1ns, the final values for the occupation probabilities of minimum 1 and minimum 2 are approximately 0.14 and 0.7 respectively. This is to be contrasted with the thermodynamic equilibrium for T = 100K which is the value of Tlow in all
of our simulations. For T = 100K, Peq(1) = 0.82, and Peq(2) = 0.17. The results of our
simulation do not reflect the equilibrium situation. A sample trajectory for P1 and P2 as a
function of temperatures along the cooling schedule is shown in Figure 12 for τmes = 1 ns.
73
Minimum 1 shows an increased occupation probability for T > 450K. At this level the
system is characterized by frequent basin hopping. Through this temperature range the
equilibrium probability for minimum 2 is nearly the same as that of minimum 1. The
changed ensemble leads to a higher population of minimum 2 during the simulation, i.e.
Peq(2) > Peq(1) for 400K ≤ T ≤ 600K. For temperatures lower than 400K, all barriers
connecting minimum 1 to the rest of the PES become more pronounced leading to a flux
into minimum 2. Minimum 2 has considerably lower barriers connecting the rest of the
PES. Low temperature dynamics are dominated by flow into minimum 2 and negligible
transition into or out of minimum 1. The transition between the simulated scenario and
thermodynamic equilibrium for T = 100K is very sharp and requires values of τmes on the
order of 1030 s. This is consistent with the estimates for τsim obtained above.
74
Table VI. Summary of Master equation calculations to "simulated
simulated annealing". τmes is the Master equation simulation time
length for each temperature level along the cooling schedule. The
final temperature is Tlow = 100 K. For T = Tlow the equilibrium
probabilities for the global minimum and minimum 2 are: Peq(1) =
0.822 and Peq(2) = 0.178. However, in order to achieve the very low
temperature thermodynamic equilibrium requires inordinately
slow cooling schedules or very large values of τmes. In a molecular
dynamics simulated annealing calculation large values for τmes
translate to extremely long molecular dynamics trajectories. Minimum 2 is favored over minimum 1 for T > 450 K. When minimum 1
is favored compared to minimum 2, the activation barrier is too big
and the system is frozen into minimum 2.
τmes
P1: Probability of
populating the
ground state
1.0 ns
6.878e-04
P2: Probability of
populating the second lowest minimum
0.0213
10 ns
6.849e-03
0.1776
0.1 µs
0.0568
0.586
1.0 µs
0.1456
0.702
10 µs
0.1462
0.703
0.1 ms
0.1462
0.703
1.0 ms
0.1462
0.703
10.0 ms
0.1462
0.705
75
Figure 12. Trajectories for P1(τmes) and P2(τmes), the occupation
probabilities of minimum1 and minimum 2 as a function of temperature. Past T = 450K, the barrier separating minimum 1 from
the rest of the PES is difficult to traverse. The dynamics are dominated by a flux into minimum 2 and no flux into or out of minimum
1. The result is that most simulated annealing calculations converge
to minimum 2 or higher lying minima.
1
0.8
Probability
0.6
0.4
0.2
0
100
150
200
250
300
350
400
450
500
550
600
Temperature (K)
In summary, the Master equation simulations set an upper limit on the extent of
simulation required if a simulated annealing protocol is to converge to the global minimum. Enhanced sampling at higher temperatures does not facilitate convergence to the
global minimum because the high temperature ensemble is non−native. This is a consequence of various crossings in the relative occupation probabilities for pairs of minima.
The situation is analogous to a potential smoothing algorithm for global optimization.
The advantage of simulated annealing over potential smoothing lies in the Boltzmann machinery that provides at least a slim chance for excitation out of a higher lying minimum
when the native ensemble prevails. However, it is easier to tune potential smoothing
76
because it is a deterministic method. If the original projection due to smoothing is in the
vicinity of the global minimum, which will be true if there is inherent structure to the
PES, then a secondary search procedure will be useful in correcting the errors introduced
due to crossings 15. A tunable potential smoothing will be considerably more efficient
than a tunable simulated annealing protocol for global optimization. Reasons for this assertion are discussed in the following sections.
Evidence for the Similarity of Potential Smoothing and Simulated Annealing
Molecular Dynamics Simulated Annealing (MDSA) for CDAP
We carried out multiple molecular dynamics simulated annealing (MDSA) runs for
CDAP. Our estimates above suggest a very low likelihood of converging to the global
minimum using simulated annealing. The initial conformation in all calculations is minimum 142. We generated 100 independent 500 ps trajectories of MDSA using the following parameters for each trajectory: a 1.0 fs timestep, 5x105 steps of MD and a sigmoidal
cooling schedule between 5000 K and 0 K.
Table VII shows a summary of the 100 independent MDSA runs for CDAP. Starting from minimum 142 only 8 of the 100 trajectories converge to the global minimum.
Forty seven of the trajectories converge to minium 2, 6 to minimum 4 and 15 to minimum
8. This result is consistent with the estimates from our Master equation simulations of
simulated annealing. According to these estimates, we will require trajectories many orders of magnitude longer than 500 ps in order to ensure that a significant number of the
MDSA trajectories converge to the global minimum. Convergence to minimum 2 is a
77
consequence of very slow transitions between minimum 2 and the global minimum due to
the high energy barrier separating these two minima. The smallest barrier for a transition
from minimum 2 to minimum 1 is 80.45 kJ/mol. At high enough temperatures where this
transition is rapid the system is in fast equilibrium between all the accessible minima.
Barriers between higher lying minima and minimum 2 are considerably smaller than
80.45 kJ/mol. The thermodynamic ensemble changes at temperatures higher than 450 K
vis-à-vis the relative occupation probabilities of minimum 1 and minimum 2. A consequence of this crossing is that for temperatures when the global minimum becomes
largely favored the barrier between minimum 2 and minimum is too high to negotiate via
thermal activation. Barriers connecting minimum 2 to other low lying minima are considerably smaller and lead to an increased population of minimum 2.
78
Table VII. Summary of 100 independent 500 ps molecular dynamics simulated annealing trajectories applied to CDAP. The initial
conformer for each run is that of minimum 142, the highest lying
minimum on the PES. The initial temperature was set to 5000K.
Results are insensitive to the choice of the starting temperature provided it is sufficiently high. Shorter trajectories (50ps) yield similar
results. The table shows the energies of minima found and the frequencies with which they were found in the MDSA calculations.
CDAP Minimum
found in MDSA
calculations
Energy
(kJ / mol)
1
-352.2535
Number of occurrences of
a minimum from the
CDAP PES in 100 independent MDSA runs
8
2
-351.1075
47
3
-343.0721
4
4
-342.8826
6
5
-342.8717
1
6
-342.0407
3
8
-338.3676
15
11
-335.9982
1
14
-333.7560
2
16
-331.2054
6
18
-330.6301
1
22
-327.4670
2
23
-327.2436
1
25
-325.9273
1
28
-320.8990
2
79
Table VIII. Summary of multiple simulated annealing runs for
CDAP from minima 1, 2, 4, 11, 16, 18, 28 and 40. These 8 minima
were the ones that remained for large values of Eb in energy clustering. The starting positions are shown in the gray boxes. Columns
under the gray boxes show the number of times a given minimum is
found in 100 independent simulated annealing runs from the minimum in the gray box. Column 1 is the identity of the minimum on
the PES found from the annealing runs. The global minimum is
found with very low probability compared to minimum 2. Success
ratios for finding minimum 4 are comparable to that of finding
minimum 1.
Minimum Start
found from Min.
annealing
1
run
1
19
2
18
3
3
4
7
5
5
6
6
11
1
14
1
Start
Min.
2
Start
Min.
4
Start
Min.
11
Start
Min.
16
Start
Min.
18
Start
Min.
28
Start
Min.
40
12
19
3
7
4
0
4
8
9
16
4
12
4
3
5
4
5
10
1
13
4
1
9
6
7
14
2
10
3
3
3
5
14
15
4
10
2
1
2
6
5
17
4
7
3
3
5
5
11
12
2
3
5
2
8
9
Shifting in Potential Smoothing and Simulated Annealing
In potential smoothing, a potential function V(r) in d−dimensions is replaced by a
smoothed version, U(r,t), where the parameter t controls the extent of smoothing. The
smoothed version U(r,t) is derived from an infinite integral of K(r,t) = ρG(r,t)V(r) over
r. The integral of K(r,t) represents a spatial average of the potential function V(r). The
kernel ρG(r,t) is a spherical isotropic Gaussian of width 2dt centered about ro which is a
local minimum of V(r).
If V(r) were perfectly isotropic, the averaging procedure would yield a smoothed
function U(r,t) for which the location of the minimum ro remains unchanged. However,
most molecular mechanics functions are inherently anisotropic leading to a shifting of ro
80
as a function of t. The shifting of ro for an anisotropic potential will be in the direction of
the smallest barrier that separates the local minimum from the rest of the PES. For instance if V(r) represents a three-dimensional DOPLS Gaussian van der Waals interaction
potential given by V(r) = a1exp( − b1r2) + a2exp( − b2r2), then ro can be written as
r =
o
b a
1 1
ln −
b a
2 2
.
a
i
b
i
and b →
. As a
For non-zero values of t, a →
i
i
3/2
4b t)
(1
+
(1 + 4b t)
i
i
consequence, ro shifts as a function of t.
We use minimum 2 for a comparison of shifting in potential smoothing and molecular dynamics. Table IX shows values for shifting in terms of the torsional angles <φ1,ψ1>
and <φ2,ψ2> for minimum 2. The original values for these angles are <φ1(o),ψ1(o)> =
<−83.89°, 67.73°> and <φ2(o),ψ2(o)> = <−82.79°, 65.8°>. Only small values of t are
shown because the shifting is influenced by the basin that minimum 2 merges into for
larger values of t.
81
Table IX. Shifting of minima as a function of deformation, t. Shifting is measured by the deviations of <φ1,ψ1> and <φ2,ψ2> for minimum 2 of CDAP from their t = 0 values. For t = 0, <φ1(o),ψ1(o)> =
<−83.89°,67.73°> and <φ2(o),ψ2(o)> = <−82.79°,65.8°>. The deviations increase as the PES becomes smoother and is a measure of the
anisotropy within local minima.
Angular Deviations (°)
t
φ1
ψ1
φ2
ψ2
0.00000
-83.89
67.73
-82.79
65.80
0.00005
-83.91
67.74
-82.80
65.81
0.00040
-83.98
67.82
-82.88
65.89
0.00135
-84.19
68.04
-83.11
66.10
0.00320
-84.61
68.47
-83.55
66.52
0.00625
-85.30
69.20
-84.27
67.21
0.01080
-86.31
70.32
-85.33
68.28
0.01715
-87.72
72.00
-86.79
69.84
0.02560
-89.57
74.37
-88.67
72.06
0.03645
-91.93
77.84
-91.00
75.25
0.05000
-94.91
83.18
-93.79
79.96
Shifting of minima can also be observed as a function of temperature. If the PES of
CDAP were perfectly isotropic the time averaged values of <φ1,ψ1> and <φ2,ψ2>, for
minimum 2, from long molecular dynamics trajectories at different temperatures would
show very small deviations about the original values of <φ1(o),ψ1(o)> = <−83.89°,
67.73°> and <φ2(o),ψ2(o)> = <−82.79°, 65.8°>. However, for minimum 2 of CDAP, the
time averaged values of <φ1,ψ1> and <φ2,ψ2> change as a function of temperature in
keeping with the anisotropy of the PES. We generated 11.5 ns dynamics trajectories for
different values of the system temperature T. The first 500 ps were set aside as
82
equilibration steps. Snapshots were generated for every picosecond of simulation. These
snapshots were used to compute the values of <φ1,ψ1> and <φ2,ψ2> along the trajectory.
Time averaged values for torsional angles from a single trajectory at temperature T were
N
N
∑
∑
s
1
computed as: < φ >
≡ (
i i = 1,2 N
s
1
φ ) and < ψ >
≡ (
ψ ) where Ns repi
i i = 1,2 N
i
s
s
i=1
i=1
resents the number of snapshots from a trajectory. Shifting is measured by the deviation
of <φ1,ψ1> and <φ2,ψ2> from <φ1(o),ψ1(o)> and <φ2(o),ψ2(o)>, which are the T = 0K values. Only trajectories for lower values of T are used because these trajectories do not
show any basin hopping and can be interpreted accurately as shifting of the local minimum not influenced by non local basins on the PES. Results for the time averaged values
of <φ1,ψ1> and <φ2,ψ2> of minimum 2 computed from trajectories at different temperatures are summarized in Table X. Shifting of the torsional angles is in the direction of the
lowest barrier that connects minimum 2 to minimum 5.
83
Table X. Shifting of minima as a function of temperature T. Shifting is estimated by the deviation of (<φ1,ψ1>) and (<φ2,ψ2>) which
are time averaged values of torsional angles for minimum 2 of
CDAP generated from 11.5 ns long molecular dynamics simulations
at different temperatures. For T = 0 K, <φ1(o),ψ1(o)> =
<−83.89°,67.73°> and <φ2(o),ψ2(o)> = <−82.79°,65.8°>. The deviations increase as temperature increases. Shifting is in the direction
of the smallest energy barrier that connects minimum 2 and minimum 5 on the PES network for CDAP.
Angular Deviations (°)
T
φ1
-84.42
ψ1
68.32
50
φ2
-83.46
ψ2
66.43
75
-84.82
68.66
-83.58
66.68
100
-84.93
68.99
-84.00
66.98
125
-85.05
69.37
-84.53
67.16
150
-85.56
69.83
-84.89
67.61
175
-85.82
70.07
-85.23
68.00
200
-86.80
71.40
-86.03
68.15
225
-88.32
72.34
-87.06
68.92
250
-88.94
72.84
-88.72
69.12
The data in Table IX and Table X show correlations between shifting of minimum 2
measured as a function of smoothing and temperature. The data in Table IX and Table X
is used to correlate values for t and T for shifting of each of the four torsional angles.
These curves are shown in Figure 13 and reinforce the strong analogy between smoothing
a PES and increasing the simulation temperature.
84
Figure 13. Second order curves for shifting of <φ1,ψ1>, <φ2,ψ2> for
minimum 2 of CDAP as a function of deformation and temperature. The curves were generated by interpolating the observed temperature values to a second order fit to the original data of the
variation of torsional angles as a function of deformation.
φ1
250
200
T Kelvin
T Kelvin
200
150
100
150
100
50
50
0
0.005
0.01
t
0.015
0.02
0
φ2
250
0.005
0.01
t
0.015
0.02
ψ2
250
200
T Kelvin
200
T Kelvin
ψ1
250
150
100
150
100
50
50
0
0.005
0.01
t
0.015
0.02
0
0.005
0.01
0.015
t
Crossings in Potential Smoothing and Simulated Annealing
Consider a system characterized by n-minima coupled to a heat bath at some temperature T. The partition function for the n minima of the canonical ensemble is written in
terms of the Helmholtz free energy Fm = Em − TSm as
Zmin =
∑
n
m=1
(
)
exp − βFm
11
where β = (kT)−1; Em and Sm are the potential energy and configurational entropy of
minimum m at temperature T. The canonical partition function of the minima can be rewritten in the harmonic approximation as 38
85
Zmin =
∑
(
)
exp − βEm
n
nv
m=1
(βh)
12
∏Λ
nv
m, j
j=1
where nν is the number of vibrational degrees of freedom, h is Planck’s constant, and Λm,j
is the vibrational frequency of the jth normal mode at minimum m. Configurational entropy in the harmonic approximation is related to the product of the vibrational normal
mode frequencies Λm,j at minimum m38. The equilibrium occupation probability for minimum m is written in terms of the canonical partition function Zmin as:
exp( − βEm)
1
Peq
m =
n
Zmin
n
(βh) ∏Λm, j
j=1
k
k
13
The equilibrium probabilities for the 10 lowest minima of CDAP as a function of
temperature are shown in Figure 14. For very low temperatures the global minimum has
an equilibrium probability greater than 0.9. The figure shows that as temperature increases, equilibrium probability of the global minimum decreases while the equilibrium
probabilities of higher lying minima increase, and that the relative free energies of pairs
of minima also change.
86
Figure 14. Equilibrium probabilities at various temperatures of the
10 lowest energy minima on the undeformed surface. Broad basins
become favored entropically as temperature increases. In particular, as temperature increases the very narrow global minimum 1
becomes much less favored and the broader basin 4 which represents the DEM-backtrack minimum becomes the dominant state.
1
0.9
0.8
0.7
Peq
0.6
0.5
0.4
002
0.3
001
004
0.2
005
0.1
010
0
100
150
200
250
300
T
350
400
450
500
550
Figure 14 also shows some reordering of equilibrium probabilities as a function of
temperature. For a pair of minima mi and mj, Peq(mi) may be less than Peq(mj) at some
temperature T=T1. For a temperature T=T2 > T1 there can be a reordering of minima, i.e.,
Peq(mj) is now less than Peq(mi) indicating an crossing in relative free energies for pairs
of minima. This reflects a changing ensemble as a function of temperature. We correlate
temperature and smoothing values by comparing crossings of a pair of minima mi and mj
in terms of t and T.
87
Consider two minima mi and mj that cross at some smoothing level t1 and two other
unique minima mk and ml which cross at some smoothing level t2 > t1. If the relative occupation probabilities Peq(mi) and Peq(mj) cross at T = T1 and similarly Peq(mk) and
Peq(ml) cross at T = T2, a correlation between smoothing and temperature controlled
crossings exist if T2 > T1.
In some cases, the same pair of CDAP minima cross in terms of relative energy during potential smoothing and in terms of equilibrium probability as a function of temperature. These pairs are listed in Table XI and are the basis for the correlation between t and
temperature T shown in Figure 15. At higher temperatures the harmonic approximation is
inaccurate and similarly for higher values of smoothing a reduction in the number of
minima occurs due a series of mergers and the crossings may not pertain to the original
PES. Figure 14 shows only the low T and low t region, i.e., T < 300K and t < 1.6. The linear correlation coefficient for the plot of t and T in Figure 15 is r2 = 0.95.
88
Table XI. Comparison of the some of the crossings between pairs of
minima as a function of deformation time t and canonical temperature T for CDAP.
crossing
t
T (K)
4⊗3
5⊗3
9⊗8
10 ⊗ 8
10 ⊗ 9
12 ⊗ 8
10 ⊗ 7
9⊗7
13 ⊗ 8
10 ⊗ 3
9⊗6
12 ⊗ 6
9⊗3
12 ⊗ 3
13 ⊗ 6
13 ⊗ 3
13 ⊗ 9
7⊗3
4⊗2
4⊗1
0.00125
0.00125
0.00288
0.0035
0.0048
0.0086
0.0097
0.0124
0.0130
0.0137
0.0144
0.0159
0.0184
0.0190
0.0202
0.0243
0.0439
0.02304
0.05895
0.15875
6.0
6.0
26.0
42.0
59.0
88.0
67.0
76.0
125.0
175.0
215.0
196.0
259.0
223.0
230.0
257.0
253.0
583.0
432.0
527.0
89
Figure 15. Correlation of crossing temperature (T) and time (t). The
line is a least squares fit to the data and has a correlation coefficient
r2 = 0.95.
300
250
T
200
150
100
50
0
0
0.005
0.01
0.015
0.02
0.025
t
Merging in Potential Smoothing and Simulated Annealing
Two unique minima on the undeformed surface will eventually merge into a common basin at some level of smoothing. The multiple minimum problem is circumvented
by reducing the number of minima through a series of such mergers with increasing t.
Mergers of minima can also be obtained with increasing temperature. An increased
temperature is accompanied by an increased probability of populating higher lying
minima. Consequently the probability of populating lower lying minima decreases as temperature increases. If the temperature T is sufficiently high, all minima on the PES become equally accessible. For the purposes of comparing simulated annealing and potential
90
smoothing, we define a merging temperature, Tm, as the lowest temperature such that
|Peq(mi) - Peq(mj)| < ε. Simulated annealing mergers are qualitatively different from potential smoothing mergers because they refer to relative probability of two minima. Figure
14 shows the changing equilibrium probabilities for some of the low lying minima of
CDAP vis a vis the global minimum. The values for the equilibrium probabilities will asymptote to a common value at high temperatures. The prescription outlined in the previous section cannot be used to compute equilibrium probabilities in the high temperature
limit because the harmonic approximation used to estimate basin volumes for conformational entropy of each minimum is inaccurate at high temperatures.
Discussion
Comparison of CDAP and IAN. Czerminski and Elber described21 a network of
138 minima and 490 transitions states of isobuturyl-(ala)3-NH-methyl (IAN) on the
CHARMM PES. IAN consists of 26 united-atom centers; CDAP consists of 18. Thus, one
might intuitively expect more minima and transition states in IAN than in capped dialanine peptide, but this is not observed. These differences are probably attributable to the
diversity of initial conformations used to discover minima and transition states on the
PES, and differences between the DOPLS and CHARMM PESs. In both systems, reaction path predictions led to the discovery of transition states, and minimization from those
states was used to connect minima on a path. Czerminski and Elber 32,21 started from canonical α-helix (<φ,ψ>=<-60°,-60°>) and β-sheet (<φ,ψ>=<-120°,120°>) conformations.
Their reaction path predictions frequently discovered transition states which connected
91
"unknown" minima and they accumulated 136 additional minima by this means. In contrast, we began with 136 conformations generated by grid search in a much larger volume
of conformational space and accumulated only 6 more by path calculations. We conclude
that the reaction path method has a strong tendency to traverse the volume of conformational space bounded by the initial structures used to compute reaction paths.
We found many more transition states for CDAP than were reported for IAN. Czerminski and Elber 32,21 found 502 transition states of 139 x 138 / 2 = 9591 possible unique
pairwise paths (5.2%). In contrast, the present search identified transitions states to occur
at much higher density: we found 1038 transition states for a possible 10011 paths
(10.5%). These comparisons include loopback and redundant paths.
The 1038 transition states represent only 688 unique connections between minima,
i.e., many transition states represent redundant paths between the same pairs of minima.
On average and including the loopback minima found in both systems, each IAN minimum is connected to 2 x 393 / 139 ≈ 5.6 others, whereas the minima for CDAP are connected to 2 x 688 / 142 ≈ 9.7 others. This implies that peptide conformations are much
more extensively interconnected than we initially expected.
The distinction between our work and that of Czerminski and Elber is our observation that paths exist between very distant regions of conformational space. They note that
most transitions involve 1 or 2 dihedral angle changes, but we find that 18% of the paths
connect structures which differ in 3 conformational descriptors. We found no paths between minima which differ by more than 3 descriptors. The data are summarized in Table
IV. To the extent that the DOPLS potential function may be used to predict system
92
kinetics, this observation suggests that conformational transitions may involve simultaneous changes of several descriptors. It is likely that transitions in larger molecular systems involve even more concerted motions.
Finally, Czerminski and Elber estimate 4(n/2) minima for a system with n soft torsional modes, i.e., non-peptide dihedrals in IAN and CDAP. For CDAP, n = 4 and their
estimate significantly understates the number of minima we discovered.
Our results suggest that potential energy surfaces are significantly rougher than
would have been expected based on results in the IAN system. The roughness of a potential surface impinges critically on the difficulty of global optimization. As a surface becomes rougher, the performance of global optimization methods diminishes in terms of
the rate of convergence, radius of convergence, and likelihood of success. Thus, complex
potential surfaces increases the incentive to develop sophisticated conformational search
procedures. We believe that potential smoothing offers significant advantages over simulated annealing as measured by rate of convergence, radius of convergence, and likelihood of success.
Potential Smoothing and Simulated Annealing. We have presented a detailed
analysis of potential smoothing applied to a small peptide. This work integrates potential
smoothing concepts proposed by Scheraga and coworkers 3, topography issues framed by
Czerminski and Elber 32, and ensemble kinetic simulations of Becker and Karplus24. We
have used the fully enumerated PES of CDAP to demonstrate the analogies between potential smoothing and simulated annealing. In both cases the ensemble depends on the
control parameter, t or T. The partitioning of the PES as studied by an energetic clustering
93
scheme is similar for both methods. Potential smoothing is an averaging scheme that reflects inherent structure on the PES and is in the spirit of the partitioning of phase space
using thermal barriers as metrics.
We refer to potential smoothing as a projection method because an important basin
is projected out by eliminating barriers between minima. The projection of basins for different levels of smoothing is deterministic. In contrast, simulated annealing is a trajectory
method. Conformational space is sampled by generating a trajectory via thermal activation over barriers. The efficiency of the method in isolating low energy conformers is determined by the extent of the PES covered. The number of barriers to be negotiated by
simulated annealing grow exponentially with the size of the system. Therefore, projection
methods will show increased efficiency for larger problems.
We have highlighted three important features of potential smoothing and described
analogous features in simulated annealing. In addition, we have compared the likelihood
of finding the global minimum for CDAP using either a potential smoothing protocol or
molecular dynamics simulated annealing. In both cases failure to consistently find the
global minimum is associated with changes to the native ensemble. In previous work15,
we have shown that potential smoothing can be modified to be a useful protocol for global optimization by coupling secondary search procedures such as normal mode searches
and hopping over transition states to the reversal schedule of minimizations in the diffusion equation method for potential smoothing. The salient features of potential smoothing
and simulated annealing are compared in Table XII.
94
Table XII. Comparison of simulated annealing and potential
smoothing.
SIMULATED ANNEALING
POTENTIAL SMOOTHING
Control Parameter
Control Parameter
Simulation temperature, T, determines the Dimensionless parameter, t, determines
size of the largest energy barrier to be ne- the extent of hypersurface deformation.
gotiated.
Conceptual Paradigm
Simulated annealing uses the analogy between minimization and slow cooling of
solids. A cooling schedule between high
and low temperatures preferentially increases the population of low energy
states. The system to be minimized is frozen into lower energy (ordered) states as
temperature decreases.
Conceptual Paradigm
Potential smoothing analytically transforms the PES by reducing the number of
minima and the heights of barriers between minima. The analytical transformation that yields the smooth surface can be
reversibly applied to follow the minimum
energy region on a smooth surface back to
the original PES.
Sampling Method
Sampling Method
Simulated annealing generates a trajectory Potential smoothing projects out an imporby thermal activation over energy barriers. tant basin by eliminating barriers between
minima.
Stochastic Conditioning
Deterministic Conditioning
The likelihood of transitions between Once the catchment region is isolated, an
minima is determined by the Boltzmann adiabatic reversal schedule of energy
condition.
minimizations always finds the same minimum.
Mergers on the PES
Mergers on the PES
Two minima become equally accessible At some deformation, two unique minima
when the energy barrier Eb between them merge into a common basin with the same
is commensurate with kT.
structure and energy.
Relative Probabilities of Minima
Relative Probabilities of Minima
The energy order of minima may change The energy order of minima may change
with T.
with t. (an energy "crossing")
95
SIMULATED ANNEALING
POTENTIAL SMOOTHING
Nature of Ensembles at High T
Nature of Ensembles at High t
Shifting, crossings and merging lead to Shifting, crossings and merging lead to
non native ensembles at high tempera- non native ensembles on highly deformed
tures. The transition region that divides the
hypersurfaces.
native and non native ensembles is very
sharp.
Conditions for Failure of Simulated AnConditions for Failure of Potential
nealing as a Global Optimization Tool
Smoothing as a Global Optimization
Simulated annealing may fail for global
Tool
optimization if:
Potential smoothing may fail for global
optimization if:
•The cooling schedule is not logarithmic;
•Crossings occur at low temperatures •Projection is unrelated to the global minimum;
where transitions are slow and unlikely.
•Smoothing is not characterized by pure
mergers;
•Crossings involve the global minimum.
Tuning Simulated Annealing to Succeed Tuning Potential Smoothing to Succeed
for Global Optimization and Conforma- for Global Optimization and Conformational Search
tional Search
Simulated annealing can be tuned by:
Potential smoothing can be tuned by:
•Very slow cooling schedules to mimic
Boltzmann machines;
•Adaptive simulated annealing for biased
sampling of conformational space;
•Mean field methods to generate multiple
trajectories for increased sampling5,4.
•Coupling local or other search methods to
a reversal schedule to correct for crossings;
•Space covered maps can be generated on
smooth surfaces because the details of the
PES are reduced through the many-to-few
mapping due to smoothing. The PES map
can be used to generate the important
families of low energy conformations39.
96
In simulated annealing there is a finite probability of correcting for crossings along
the cooling schedule. The system may find itself in a high lying minimum at some intermediate temperature TI due to a crossing at some higher temperature. If kTI is commensurate with the size of the barrier separating the true global minimum from the high
lying minimum, then there is a finite probability that the system will make a transition
from its current location into a low lying minimum during the length of the simulation.
The likelihood of such transitions becomes smaller as temperature decreases. Simulated
annealing will fail if the transition temperature Tf at which there are no artifacts due to
crossings is close to the temperature Tg at which the dynamics of the system is extremely
slow due to large barriers for transitions between minima. Doye and Wales 19 have argued that relaxation to the global minimum is facilitated by a large value for the ratio of
Tf / Tg.
Both simulated annealing and potential smoothing can be generalized from being
single trajectory and single projection methods to multiple trajectory and multiple projection methods4,39 respectively. Furthermore the correlation between the control parameters
for the two methods can be used to develop an algorithm to couple the effects of the two
parameters as suggested in the Gaussian Density Annealing method of Straub and coworkers 4 and the Packet Annealing Method of Shalloway and coworkers5.
97
Appendix A
Master Equation Formalism
The fully enumerated PES for CDAP can be used to quantify near equilibrium dynamics of the system at different temperatures using classical transition state theory 38
and the Master equation formalism for reaction rate theory 37. This formalism is valid on
time scales that are a few order of magnitudes slower than the fastest relaxation times for
the system which are on the order of a few picoseconds.
Equation A1 describes the time evolution of the probability Pi(τ) of finding the system in minimum i, and is written as:
dP
i
=
dτ
∑[RijPj(τ) − RjiPi(τ)]
A1
j
where Rij are elements of the rate matrix that determine the transition rates from state j to
state i. The rates are in units of (1/s). Equation A1 can be written in matrix notation as:
dP
= RP(τ)
dτ
A2
Properties of the Rate Matrix R
The elements of R, are greater than or equal to zero but never negative. The sum
over each column of the matrix is exactly zero to ensure conservation of probability. The
elements are not necessarily symmetric, i.e., Rij is not generally equal to Rji. The rate matrix R can be symmeterized using equilibrium probabilities and detailed balance40. The
98
rates are prescribed according to transition state theory 38 and Eyring’s formula for a transition from state j to state i in terms of the energy barrier Eij between states i and j, the
b
temperature parameter RT, and the partition functions Zij≠ and Zj for the transition state
and minimum j respectively. Rij is given by the Arrhenius rate formula of the form 37:
-E
Z≠
ij
RT ij
R =
exp
ij
KT
h Z
j
[ ]
A3
The solution to equation A3 can be written as P(τ) = exp(τR)P(0) and is obtained
by diagonalizing the rate matrix R. The eigenvalues and eigenvectors are used to compute
the time dependent solutions P(τ) given an initial distribution of P(0) according to equation A4 shown below:
P(τ) = P
eq
+
∑
C V exp(λ τ)
i i
i
i|λ < 0
i
A4
Here Ci is a normalization factor determined using the initial conditions, Vi are the
eigenvectors and λi are the eigenvalues. The eigenvalues of R are characteristic relaxation
rates for the different eigenstates of the system at temperature T.
For the system of 142 minima in CDAP we obtain 142 eigenvalues one of which is
zero corresponding to the equilibrium of the system. All non-zero eigenvalues are negative since the Master equation is a gain-loss equation in terms of the state occupation
99
probabilities. The set of relaxation times for the system {Γri} can be written as
1
|λ |
i
the λ’s denote the non-zero eigenvalues of the rate matrix.
100
where
References
1
M.P. Allen and D.J. Tildesley, Computer Simulation of Liquids, Oxford University Press, New York, 1987.
2
S. Kirkpatrick, C.D. Gelatt, and M.P. Vecchi, Science, 220, 671 (1983).
3
L. Piela, J. Kostrowicki, and Harold A. Scheraga, J. Phys. Chem., 93, 3339-46
(1989).
4
J. Ma and J.E. Straub, J. Chem. Phys., 101, 533 (1994).
5
K. Barker and D. Henderson, Rev. Mod. Phys., 48, 587-671 (1976).
6
K. Binder and D. W. Heerman, Monte Carlo Simulation in Statistical Physics,
Introduction, 3rd Ed., Springer-Verlag, New York, 1997.
7
A. T. Brünger, J. Kuriyan and M. Karplus, Science, 235, 458-460 (1987).
8
E. Aarts and J. Korst, Simulated Annealing and Boltzmann Machines, Wiley &
Sons, New York, 1990.
9
J.E. Straub, In Recent Developments in Theoretical Studies of Proteins, R.E. Elber, Ed., World Scientific, Singapore, 1996, p. 135-196.
10
B.J. Berne and J.E. Straub, Curr. Opn. Struc. Biol., 7, 181 (1997).
11
P. J. M. van Laarhoven and E. H. L. Aarts, Simulated Annealing: Theory and Applications, D. Reidel, Dordrecht, 1987.
12
J. P. K. Doye and D. J. Wales, Phys. Rev. Lett., 80, 1357 (1998).
13
P. Amara and J. E. Straub, Phys. Rev. B, 53(20), 13857-63 (1996).
101
14
D. Shalloway, In Recent Advances in Global Optimization, C.A. Floudas and
P.M. Pardalos, Eds., Princeton University Press, Princeton, 1992, pp. 433-477.
15
R.V. Pappu, R.K. Hart and J.W. Ponder, J. Phys. Chem. B, 102, 9725-42 (1998).
16
C. Tsoo and C. L. Brooks III, J. Chem. Phys., 101, 6405-6411 (1994).
17
I. Andricioaei and J. E. Straub, J. Comput. Chem., 19, 1445-1455 (1998).
18
K.D. Ball, R.S. Berry, R.E. Kunz, F-Y. Li, A. Proykova, and D.J. Wales, Science, 271, 963 (1996).
19
J.P.K. Doye and D.J. Wales, J.Chem.Phys., 105, 8428-8445 (1996).
20
J.P.K. Doye and D.J. Wales, Cond-Mat/979019, September (1997).
21
R. Czerminski and R. Elber, J. Chem. Phys., 92, 5580-601 (1990).
22
M. Oresic and D. Shalloway, J. Chem. Phys., 101(11), 9844-57 (1994).
23
F.H. Stillinger and T.A. Weber, J. Phys. Chem, 87, 2833 (1983).
24
O. M. Becker and M. Karplus, J. Chem. Phys., 106.4, 1495-1517 (1997).
25
T. Huber, A.E. Torda, and W.F. van Gunsteren, J. Phys. Chem. A, 101, 5926-30
(1997).
26
J.W. Ponder, TINKER: Software Tools for Molecular Design, version 3.7,
http://dasher.wustl.edu.
27
W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 110.6, 1657-66 (1988).
28
B. Brooks, R. Bruccoleri, B. Olafson, D. States, S. Swaminathan, and M. Karplus, J. Comp. Chem., 8, 132 (1983).
29
J. Kostrowicki, L. Piela, B. J. Cherayil, and Harold A. Scheraga, J. Phys. Chem.,
95, 4113-9 (1991).
102
30
J. W, Ponder and F. M. Richards, J. Comput. Chem., 8, 1016-24 (1987).
31
S. S. Zimmerman, M. S. Pottle, G. Némethy, and H. A. Scheraga, Macromolecules, 10.1, 1-9, Jan-Feb 1977.
32
R. Czerminski and R. Elber, Proc. Nat. Acad. Sci., 86, 6963-7 (1989).
33
P. S. Shenkin and D. Q. McDonald, J. Comput. Chem., 15.8, 899-916 (1994).
34
S. Nakamura, H. Hirose, M. Ikeguchi and J. Doi, J. Phys. Chem., 99, 8374-8378
(1995).
35
N. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller and E.J. Teller,
J. Chem. Phys., 21, 1087 (1953).
36
J. Kuriyan, A. T. Brünger, M. Karplus and W. Hendrickson, Acta Cryst. A, 45,
396 (1989).
37
W. Frost, In Theory of Unimolecular Reactions, Academic Press, New York,
1973.
38
P. Hanggi, P. H. Talkner, M. Borkovec, Rev. Mod. Phys., 62(2), 251 (1990).
39
R. V. Pappu, R. K. Hart, and J. W. Ponder, J. Am. Chem. Soc. 120, xxxx (1998).
(submitted)
40
J. Wei, and C. D. Prater, Adv. in Catalysis, 364, 203 (1962).
103
Chapter 3:
Analysis and Application of
Potential Energy Smoothing and
Search Methods for Global
Optimization
104
Introduction
Global optimization is an important issue in the characterization of complex systems such as glasses, clusters and large biomolecules. Techniques have emerged over the
past few years, all of which exhibit varying degrees of success in application to well established global optimization test problems. The current algorithms can be classified into
four overlapping categories: (1) deterministic methods, (2) stochastic methods, (3) heuristic methods and (4) smoothing methods. Selected methods from each of these four categories have been extensively reviewed1,2.
Deterministic methods include space covering techniques such as branch-and-bound
search3, systematic search methods4, and generalized descent methods5. These methods
are useful for small problems, or for larger systems with well-established constraints, but
will in general fail for large problems due to the exponential increase in the size of the
space to be searched.
Stochastic methods include Bayesian statistical models6 and simulated annealing7.
An underlying theme in many stochastic search procedures is the use of Monte Carlo
sampling enhanced by the Metropolis criterion8. Some notable Monte Carlo techniques
include reweighting methods such as multicanonical sampling9, Monte Carlo with minimization (MCM)10, a revision of MCM referred to as "basin hopping"11, a Molecular
Dynamics−Minimization
procedure
also
related
to
MCM12,
Carlo/Stochastic dynamics methods13, and the random kick method14.
105
mixed
Monte
A widely used stochastic method is simulated annealing7 which is an important tool
for global optimization on rugged energy landscapes. In simulated annealing, the system
is coupled to a heat bath which is initially at some high temperature. At high temperature,
the system is characterized by rapid transitions between high and low lying minima. The
temperature is then slowly lowered according to a prescribed cooling schedule and the
system is allowed to equilibrate at each level using either a Monte Carlo Metropolis
criterion8 or molecular dynamics in dynamical simulated annealing15. A decrease in temperature is associated with an increased likelihood of occupying low-energy states and reduced likelihood of jumping out of minima. The approach is analogous to the slow cooling or annealing of a system through the transition region between the liquid and solid
phases. The success of simulated annealing is largely determined by the cooling schedule,
the size of the largest barrier on the potential energy surface (PES), and the separation between the global minimum and other low lying conformations 16,17. While simulated annealing has been of limited use in the global optimization of proteins and other biopolymer systems18, it is the de facto standard for refinement of x-ray and NMR-determined
structures of biomolecules19. Adaptive simulated annealing20 couples a refined choice of
parameters to the Metropolis criterion for increased sampling of important regions of conformational spaceand has been applied to predict the conformations of Met-enkephalin
and a 14-residue poly-lysine α-helix21. A recent variant of simulated annealing is derived
from generalized Tsallis statistics22. The classical Boltzmann machinery can be recovered
as a special case of this generalized formalism. Generalized simulated annealing23 has
been revised to satisfy detailed balance and has been shown to reduce to a steepest
106
descent algorithm at low temperatures24. It is still unclear if the use of generalized statistics will yield significant improvements over the traditional Boltzmann formulation for
larger biomolecules.
Many heuristic algorithms are based on a reduction of the global problem into
smaller subsets for optimization. The assembly of the smaller optimized parts leads to the
final answer. Algorithms of this type include the build-up procedure25,26, constrained systematic search algorithms27, scanning methods which replace exhaustive enumeration
with Monte Carlo methods28, and genetic algorithms29. These methods lead to the iterative generation of sets of conformations from which the lowest energy conformer is selected.
An emerging concept in global optimization is potential smoothing. The basic idea
is to analytically transform a multidimensional PES by reducing the number of unique
minima and the heights of barriers. Such a transformation can project out a catchment region that may be related to the global minimum. The spirit of smoothing methods is contained in previous work which showed that short range potentials generate large numbers
of local minima, and softening these potentials leads to a reduction in the number of local
minima30,31. This conceptual framework has been used in the Diffusion Equation Method
(DEM) for potential smoothing developed by Scheraga and coworkers32. Important generalizations to DEM include Gaussian Density Annealing (GDA and AGDA), and other
Gaussian Phase Packet dynamics methods developed by Straub and coworkers 33,34,35.
Methods such as the Monte Carlo Minimization (MCM) of Li and Scheraga10 and "basin
hopping" of Doye and Wales11 fall into the category of PES smoothing, though the
107
mechanism of smoothing is different from the DEM32 and AGDA34 class of methods discussed in this work.
Smoothing of a PES can also be extended to transform the Gibbs distribution function and is the basis of the packet annealing methods of Shalloway36. The success of a
smoothing algorithm for global optimization is contingent upon a connection between the
deformed and the undeformed surfaces and sufficient structure underlying the original
rough PES. Straub17 and Church, et al.37 have reviewed methods for potential smoothing
and compared the efficiency of smoothing algorithms with different types of simulated
annealing protocols.
We are initiating a series of systematic studies to establish concepts and quantify
metrics to compare and contrast the efficiency, extent of sampling, CPU intensity and accuracy of specific candidates from the four major classes of global optimization outlined
above. In this work, we detail methods to modulate diffusional smoothing as a tool for
global optimization and use recently developed strategies to generalize its scope for global optimization38. These generalizations come under the category of potential smoothing
and search methods which will be referred to as PSS. The basic ideas presented in this
work can be adapted to generalized smoothing algorithms such as GDA33 and AGDA34.
Here we concentrate on tuning the simplest version of potential smoothing, i.e., DEM.
The focus of this work is to develop an understanding of the smoothing paradigm by applying it to a cross section of molecular conformation problems. Examples are used to
quantitatively illustrate the features of smoothing a rough PES, the limitations of the
original smoothing protocol, and the efficiency of search enhanced generalizations on
108
large conformational problems. Applications studied include (i) varying sizes of argon
atom clusters, (ii) cycloheptadecane, and (iii) regular conformations of polypeptides.
In the next section, we describe in detail the different methods for implementing potential smoothing. This is followed by a description of results from the application of
PSS. We conclude with a discussion of the features of potential smoothing, limitations of
the methods used in this work, and possible future extensions.
Methods
All calculations were performed using the TINKER modeling package, which
implements a self-contained force field engine providing access to several molecular mechanics force fields39. We use a modified version of the united−atom AMBER/OPLS40,41
force field in applications of potential smoothing to conformational energy surfaces of
polypeptides and clusters of argon atoms. A similarly modified MM242 parameter set was
used in calculations on conformations of cycloheptadecane.
Potential Function and Parameterization
The AMBER/OPLS and MM2 force fields were modified to obtain analytical solutions of a diffusion equation corresponding to each energy term. Modified AMBER/OPLS
or MM2 force fields can be deformed to reduce the number of minima on the potential
energy hypersurface. This is done by controlling the level of deformation denoted by an
independent parameter, t.
109
The modified AMBER/OPLS force field, which will be referred to as DOPLS for
deformable OPLS, is a function of the atomic coordinates and t. The DOPLS force field is
of the form
V
total
=V
bond
+V
angle
+V
torsion
+V
improper
+V
vdw
+V
charge
1
The first two terms are bond stretching and valence angle bending energies. Torsional
terms characterize barriers for internal bond rotation. A CHARMM-style harmonic improper dihedral term is included in DOPLS to impose planarity at sp2 atoms and chirality
of sp3 united atoms. In AMBER/OPLS nonbonded terms include van der Waals energies
modeled using a 12-6 Lennard-Jones function and electrostatic energies modeled using a
Coulomb potential for charge−charge interactions. Their modification in DOPLS is discussed below.
Since it is easy to compute solutions to a diffusion equation for Gaussian−like initial
conditions, energy functions for van der Waals interactions are approximated as a sum of
either 2 or 4 Gaussians. The functional form of the Gaussian approximation for van der
Waals interactions is shown in equation (2).
n
gauss
V
vdw
(r ) =
ij
∑
a
k(ij)
exp( −b
2
r )
k(ij) ij
2
k =1
We use a two-Gaussian approximation in this work for each pairwise van der Waals interaction. The Gaussians are centered about the origin and are of opposite sign. Typical van
der Waals interactions present large repulsive barriers for values of rij less than the radius
of the excluded volume shell. The rij = 0 region of a Lennard-Jones 12-6 function is
110
rendered inaccessible in the Gaussian approximation of equation (2) by modulating the
height of the repulsive Gaussian, a1(ij). Parameters for the two Gaussian approximation
are chosen to fit a canonical Lennard-Jones function with an atomic radius (σ) and a well
depth (ε) of one. We use values of (a1,b1) = (14487.1 kcal/mol, 9.05148Å) and (a2,b2) =
(−5.55338 kcal/mol, 1.22536 Å) for σ = 1Å and ε = 1.0 kcal/mol. The pairwise Gaussian
parameters are scaled according to the σ and ε values for each pairwise interaction using
the values prescribed by the force field. DOPLS parameters also include a small non−zero
van der Waals radius on polar hydrogen atoms to avoid fusion of atoms due to favorable
electrostatic interactions. Values of σ = 0.5Å and ε = 0.010 kcal/mol were used for all polar hydrogen atoms. This choice has very little effect on the final energy of reasonable,
low energy structures.
The final component of DOPLS is the inclusion of a CHARMM43 improper dihedral term of the form:
V
improper
=
1
2
K (Θ − Θ )
2 Θ
o
3
This term imposes planarity at sp2 atoms and correct chirality at sp3 α-carbon and other
tetrahedral atoms. Values of KΘ were chosen to best reproduce the low energy regions of
the standard AMBER/OPLS trigonometric improper torsional term. The change in functional form was necessary to maintain the desired planarity and to avoid chirality changes
on deformed energy surfaces.
These modifications to AMBER/OPLS result in only very small structural and energetic deviations from the original force field for low energy minima on the undeformed, t
= 0, DOPLS potential surface. However, each modification is required either for efficient
111
evaluation or desired limiting behavior as the amount of deformation of the surface increases with increasing t.
MM2 substitutes out-of-plane bending for the improper dihedral term and also includes a bond stretching-angle bending cross term. In addition, MM2 replaces the
Lennard-Jones function with a Buckingham exp-6 function for van der Waals interactions. In smoothing applications, the exp-6 potential is replaced by a Gaussian approximation similar to the form shown in Equation (2). Parameters for the two Gaussian approximation are chosen to fit the original MM2 function, 290000 exp(-12.5r*/r) - 2.25
(r*/r)6. We use values of (a1,b1) = (3423.562 kcal/mol, 9.692Å) and (a2,b2) = (−6.503
kcal/mol, 1.585 Å) for r* = 1Å and ε = 1.0 kcal/mol. The pairwise Gaussian parameters
are scaled according to the r* and ε values for each pairwise interaction using the values
prescribed by the force field.
Diffusion Equation Method for Smoothing of Potential Functions
In DEM32, a molecular mechanics potential function is deformed by iterative application of a smoothing operator. A multidimensional potential function with its characteristic roughness is transformed to ϒ(r1,r2,...rn;t) using an operator of the form
∂ϒ
∂t
= ℑ{r ,r ,...r }ϒ, where t is a dimensionless parameter that controls the level of
1 2
N
smoothing. The operator on the right side is a multidimensional diffusion operator. Here
ϒ(r1,r2,...rn;0) = V(r1,r2,...rn) is the original undeformed potential function. The diffusion
operator is applied to each of the pairwise molecular mechanics terms individually. We
assume that the sum of individually deformed parts is equivalent to the deformed
112
potential energy function. This assumption is accurate for distance-dependent potential
functions. If a potential function is written as a sum of pairwise distance-dependent terms,
i.e., V(x) =
∑∑V (|x − x |)
ij
i
i
j
, where |x − x | denotes the distance between atoms i
i
j
j<i
and j, it can be shown that computing a smoothed form for V(x) requires only the
smoothed form for the pair potential Vij44. This principle is strictly valid only for pair potentials that depend on the scalar distance between atoms. It can be extended to a sum of
distance-dependent molecular mechanics terms provided the distance ranges are the same
for each term.
In applying the diffusion equation to molecular mechanics functions as initial conditions for diffusion, the pairwise distance can either be a Cartesian or angular distance. For
example, torsional potentials are smoothed in terms of torsional angles instead of the 1-4
distance. Smoothing torsional potentials in terms of torsional angles is qualitatively similar to smoothing an equivalent potential over the 1-4 distance especially if the 1-2 and 1-3
distances are kept fixed. However, the choice of smoothing the torsional potential in
terms of angular coordinates is the only tractable way to smooth torsional potentials in
Cartesian space when the 1-2 and 1-3 distances are not fixed. Details regarding the
smoothing of torsional potentials are presented in Appendix A.
The diffusion equation in one dimension with no sources or sinks is of the form
2
∂ρ
∂ ρ
=D
.
2
∂t
∂x
113
4a
Here ρ is similar to either temperature in heat conduction or concentration of particles in a
diffusion controlled process and D is a diffusion coefficient in units of Å2. Solutions to a
diffusion equation can be obtained analytically provided we know the initial value of ρ
and the boundary conditions. The general solution to the semi-infinite one-dimensional
diffusion equation shown in equation 4a is
1
ρ(x,t) =
(
exp
4πDt
−(x − xo)2
4Dt
)
4b
for x = xo at t = 0. If ρ(x,t) denotes a one-dimensional distribution of Brownian particles,
then the rms displacement of the Brownian particles as a function of time is
2
x
rms
2
= x + 2Dt
o
4c
implying that the mean squared displacement between Brownian particles increases linearly with time. This is an important feature in the use of a diffusion equation formalism
for deforming potential functions.
For instance, during diffusion of a DOPLS Gaussian van der Waals interaction
term, the location of the potential minimum is pushed out to larger values of r as Vvdw(r)
is deformed. For a two-Gaussian approximation to the van der Waals potential, the location of the potential energy minimum may be written in terms of the Gaussian parameters
as rmin = ln(
− b1a1
). For non-zero values of the smoothing parameter t, the location of the
b2a2
potential energy minimum is shifted and can be written as rmin = ln(
114
− bt1at1
) where
bt2at2
atk =
ak
(1 + 4t)
3/2
and btk =
bk
(1 + 4t)
. The ratio
− b1a1
increases with increasing t, shifting the
b2a2
location of rmin. A qualitative description of potential function smoothing is that pairwise
interactions between localized atoms are altered to be interactions between diffuse atoms.
On smooth surfaces atoms are delocalized and the atomic positions are described by probability distributions. Interactions between the average location of atoms can be computed
using the location and width of the Gaussian distribution for each atom. Interaction between diffuse as opposed to localized atoms leads to a reduction in the combinatorial
problem and hence a reduction in the number of accessible minima.
Coordinate Representations
We study applications of potential smoothing in three types of spaces: Cartesian
(Γvc), torsional (Γvt) and rigid body (Γvr) space. In Γvt, bonds and angles are kept fixed at
their starting positions and conformational changes come about due to changes in torsional angles. The DOPLS potential energy function in Γvt is written as
V(ϖ ,ϖ ,...,ϖ ) = V
1
2
n
torsion
+V
improper
+V
vdw
+V
charge
5
In Γvr, different conformations are distinguished by six rigid body degrees of freedom for each molecule, corresponding to three translations and three rotations. The potential function is of the general form
m
V (rbc ,rbc ,...,rbcm) =
1
2
n
i
n
j
∑ ∑∑ ∑ V(i,k),(j,l)
6
i =1 j < i k =1 l =1
In this work, rigid bodies always correspond to distinct molecules, so the summand denotes nonbonded interactions between atoms on different molecules. m is the number of
115
molecules and ni and nj are the number of atoms in each of the molecules i and j respectively.
Diffusion Coefficients to Modulate Smoothing of Potential Function
Terms.
In analogy with a classical diffusion equation, each of the molecular mechanics
terms represents a different initial condition for diffusion. Individual pairwise energy
functions differ in their distance range, energy scales and pairs of atoms involved. Scaling
the disparate potential energy terms may be accomplished by choosing a set of empirical
diffusion coefficients to control the rate of diffusion of the different terms. Diffusion coefficients can be estimated based on a solution to the appropriate finite one-dimensional
diffusion equations in distance space with the upper and lower bounds stipulated as shown
in Table I. In all of the applications reported here, the bond stretching, angle bending, improper dihedral, out-of-plane bending and bond-angle cross terms are not smoothed. Diffusion coefficients for the Lennard-Jones and Coulomb terms are set to one since these
are nonbonded interactions and the range of these potentials is large. Effective diffusion
coefficients for local geometric interactions are scaled relative to values for the nonbonded terms.
116
Table I. Characterization of the diffusion spaces and diffusion coefficients for the different energy terms of a DOPLS molecular mechanics potential. In all calculations t is set to a dimensionless parameter that controls the level of smoothing.
Energy
Term
Diffusion
space
Distance Interval
Effective
Diffusion Coefficient
Vbond
finite
(r12o,r12max)
Dbond = 0.000156 Å2
Vangle
finite
(r13o,r13max)
Dangle = 0.0014 radian2
Vtorsion
finite
(r14(0),r14(π))
Dtorsion = 0.022 radian2
Vvdw
semi-inifinte
(r14(0),∞)
Dvdw = 1.0Å2
Velectrostatic
semi-inifinite
(r14(0),∞)
Dcharge = 1.0Å2
The DOPLS force field contains terms that are naturally represented in either distance or angular space. Bond stretching, van der Waals and electrostatic terms describe
pairwise interactions in distance space. Angle bending, improper dihedral and torsional
terms describe interactions in angular space. In order to correctly scale the smoothing of
these terms, a mapping of variations in angular space onto variations in distance space is
needed. For example if distances are measured in Å and angles in radians, then in order to
correctly scale the smoothing of different terms an estimate of the change in radians in angular space in terms of Å in distance space is needed. A second consideration is the different ranges of potential functions within the same diffusion space. In distance space,
bond stretching terms involve only nearest neighbor distances, while nonbonded functions
involve larger distances.
117
Our analysis of this problem leads to a choice of very small diffusion coefficients
for bond stretching and angle bending terms. The limit of diffusion coefficients tending to
zero is equivalent to not smoothing the bond and angle terms. Small values for diffusion
coefficients reflect two important considerations (i) the limited range of covalent restraint
terms and (ii) the intrinsic nature of restraint terms to impose severe penalties for all deviations from ideal geometry. This is reasonable since the objective is to explore conformational space without any significant rearrangements of covalent geometry, i.e., making
or breaking of covalent bonds. A similar reasoning is used to justify the use of undeformed bond-angle cross terms and improper dihedral terms.
Torsional potentials are typically a sum of trigonometric terms that impose multifold barriers. Unlike bond stretching and angle bending terms, torsional terms cannot be
ignored since these barriers distinguish between conformations and are considerably
smaller than the barriers imposed by violation of covalent bond and angle restraints. Setting Dtorsion =1 leads to the problem that at fairly small non-zero values for the deformation parameter t, torsional barriers vanish leading to a non-physical exploration of conformational space. Since barriers vanish for small values of t, typical reversal protocols used
in potential smoothing45,46 will not feel these barriers until very small values of t at which
point the method may have already committed to a conformation with high torsional energy. One solution would be to recast the torsional potential in terms of a 1-4 distance 46.
However, a 1-4 distance restraint in Γvc can become severely non-physical because covalent bonds and angles are merely undeformed and not rigidly fixed. A simpler method is
118
to compute the rate of diffusion in torsional space which is then used to estimate an effective torsional diffusion coefficient in distance space.
The method of choosing empirical diffusion coefficients for covalent restraint and
geometry terms has the desired effect of controlling their smoothing relative to nonbonded terms. The current formalism is a generalization of this technique and allows for
increased sampling of conformational alternatives on highly deformed surfaces since torsional terms do get smoothed, albeit slowly. A discussion of the methods used to estimate
empirical diffusion coefficients is provided in Appendix B.
Potential Smoothing Protocol
A typical potential smoothing protocol involves the following steps45,46,47:
1. The conformational energy of a starting structure in Γvc, Γvt or
Γvr is minimized using a local conjugate-gradient or second
derivative minimization method on the t = 0 undeformed surface. In Γvt and Γvr, we use an optimally conditioned quasiNewton method without line searches 48, and in Γvc, we use a
truncated Newton optimization algorithm 49 with a preconditioned linear conjugate-gradient solution of Newton’s equations.
2. The value of the control parameter t is slowly increased according to a prescribed smoothing schedule, and conformational energy is minimized at each step using the methods discussed in
step (1).
119
3. Smoothing of the conformational energy function is carried out
until t = td, where td is the level of smoothing for which all
starting structures from step (1) converge to the same structure
with the same energy. In previous work 45,46, td was chosen to
be the level at which only a single minimum remains on the deformed surface. This is a special case of our condition to
choose td.
4. Starting at t = td, the deformation is reduced followed by conformational energy minimization as discussed in step (1). On
highly deformed surfaces only a few minima remain, so the local optimizer finds a minimum in the same basin as the starting
structure by following the downhill gradient closely. Values of
t are reduced in small intervals ∆t chosen according to a prescribed schedule until t = 0. The reversal process is a fully deterministic procedure and the final value for the conformational
energy at t = 0 is the DEM estimate of the global energy minimum of the molecule.
This protocol may be repeated for different starting structures at step (1) to show
that the chosen td is sufficiently large to ensure convergence to the same minimum at t =
td. Starting at t = td implies the reversal protocol has forgotten the initial conformation and
will follow an invariant deterministic path down to the undeformed surface for a given reversal schedule.
120
Smoothing Schedule
If td is a chosen large value of the deformation parameter for forward smoothing
and nd is the number of points between t = 0 and t = td, the smoothing schedule changes
the deformation parameter according to the formula:
i s
)
d n
t =t (
i
7
d
for forward smoothing and
2n -i
t =t (
i
d
d
n
s
)
8
d
for the reversal, where i = 1,2,3,....,nd and 2 ≤ s ≤ 6 for different applications. For a given
value of s, increasing nd increases the number of points sampled at smaller values of t. As
will be discussed in Results, the optimal choice of values for nd and s varies from problem to problem.
Compactness Restraints In Smoothing
Most applications of smoothing algorithms have used basin functions or confining
potentials to impose compactness conditions during the smoothing process. There are two
classes of problems where basin functions have been used in the past. One example is the
problem of finding the minimum energy configuration of clusters of Lennard-Jones atoms. In DEM smoothing applications45, a basin function of the form aexp(-br2) has been
used to keep atoms from drifting out to infinity as t becomes large. The choice of values
for a and b result in the application of extremely shallow pairwise Gaussians that keep the
system bounded at large values of t. Similarly, Ma and Straub33 have used a harmonic
pair potential to confine the sampling of conformations to the manifold of compact
121
clusters. Basin functions or confining potentials are necessary for noncovalent clusters
since they impose boundedness on the problem, and are required to keep the calculations
numerically stable.
The use of basin functions in application of smoothing to connected systems such as
peptides or hydrocarbons is questionable. At large values of deformation, if bonds and
angles are undeformed in Γvc or kept fixed as in Γvt, flexible molecules sample maximally extended structures. For example, at large t the values of the φ and ψ torsional
angles in peptides are typically near 180°. Unlike an argon cluster problem, there is no
boundary condition to be imposed. However, basin functions restrict sampling to a set of
compact conformations and may bias the results of a smoothing protocol due to limited
sampling of conformations and perturbation of the unconstrained smoothed surface. In
our applications of potential function smoothing, we use basin function restraints for
Lennard-Jones clusters and not for peptides and isolated organic molecules.
PSS Methods
An extension of potential smoothing is to include a local search protocol during the
reversal schedule to search for alternate low lying minima38. Local searches allow for
corrections to be made to estimates of the global minimum at different levels of smoothing. We consider two types of local searches. One alternative is to perform a search in the
vicinity of the local minimum along a randomly chosen direction or along normal modes
out of the local minimum. A second method would be to move the system over transitions
states into adjacent low lying minima. A general local search algorithm adapted for either
of these two methods is as follows:
122
1. At some chosen value of t = tl during the reversal, we reduce
the level of smoothing by ∆t and find a minimum energy conformation using a local optimization protocol. The energy at
the local minimum is stored as Vlocal and its coordinates are
denoted by a vector Rlocal.
2. The system is moved out of the local minimum either along a
set of search directions or to a nearby transition state. The energy of the system at the new location is Vexcite and the system coordinates are denoted by a new Rexcite.
3. From Rexcite, the system is moved to an alternate location
Rnew by an energy minimization. If Vnew < Vlocal, then this
is the new energy and the system is retained at the new location
Rnew.
4. The system is moved from Rnew to explore the vicinity of the
new local minimum by repeating steps 2 and 3 until a new
lower minimum cannot be found.
5. If a new lower minimum cannot be found, we return to step 1,
reduce the value of t and continue steps 2, 3 and 4 until t = 0.
The final t = 0 estimate for the "global" minimum is lower than, or equal to, the minimum
obtained from the DEM protocol without local search.
123
"Normal Mode" Local Search (NMLS)
The algorithm we use for searches in the vicinity of a local minimum is very similar
to the "two-stage" method proposed by Nakamura, et al. 38 In their work, eigenmodes
corresponding to the largest eigenvalues of the Hessian computed at a local minimum are
followed in order to ensure an uphill climb out of a local minimum. We use a generalization of their protocol to different coordinate representations. In Γvc, the true vibrational
normal modes are the appropriate mass weighted eigenvectors of the Hessian, though this
is not strictly true in Γvt or Γvr. However, we will refer to search along the eigenvectors of
the Hessian matrix as "normal mode" local search (NMLS) regardless of the coordinate
representation.
A point k along a search direction i that satisfies the condition Vi,k-1 > Vi,k and
Vi,k-1 > Vi,k+1 where the V’s are the conformational energy values, is chosen to be a new
point Rexcite from which to start a minimization to an alternate minimum. The condition
suggests apparent downhill progress indicating a possible turning point into a new energy
basin. In practice, the minimization can occasionally drop the system back into Rlocal
where it originated.
In Γvc, for connected systems, we use a hybrid scheme for PSS. All minimizations
are done in Cartesian space but the search directions for a guided climb out of the local
minimum are torsional space eigenvectors. The main objective of a local search is to explore conformational space in the vicinity of the local minimum. It is sufficient to explore
structures on the manifold of bonds and angles corresponding to Rlocal , i.e., conformational rearrangements come about from varying torsional angles alone. In Γvr, the Hessian
124
is computed in terms of the 6n rigid body coordinates where n is the number of rigid bodies and the eigenvectors of the Γvr Hessian are the search directions for NMLS.
Transition State Based Search (TSBS)
Consider a system in a local minimum with coordinates Rlocal at some level of
smoothing t. The system can be activated from Rlocal to a nearby transition state region
and quenched to an alternate local minimum Rnew. A subsequent comparison will choose
between Rlocal and Rnew depending on which of the two conformational energy values is
lower, i.e., the system reverts to Rlocal if Vlocal < Vnew or remains in the new metastable
state Rnew if Vnew < Vlocal. In order to implement such a method, we need to be able to
locate saddle points starting from a local minimum.
Methods that rely on the use of two adjacent minima to locate intervening saddle
points by minimization along an orthogonal direction, by propagating a reaction path50 or
evolving a contour of tangency51 are not useful here since we know only one minimum.
Techniques have been developed to locate transition states using the curvature information at a local minimum52. We use the Activation and Relaxation Technique (ART) of
Barkema and Mousseau53 to locate saddle points from a minimum in disordered systems.
This method is briefly outlined below.
Let R1 be the system coordinates at a local minimum. The system is initially perturbed from this local minimum by generating a small random displacement away from
the minimum, i.e., R* = R1 + δ. A saddle point is then reached by iteratively following a
new force vector of the form
G = −∇V − (1 + α) { −∇V • ∆R} ∆R
125
9
where ∆R = R* − R1 is a unit vector parallel to the vector from the original minimum to
the new location of the system. α is a positive non-zero parameter chosen so that 0 < α <
1 and used to ensure that the system does not remain trapped in the local minimum. The
G-vector with α = 0 points in an uphill direction perpendicular to and away from the local
minimum. G is non-zero away from a local minimum or a saddle point, so the simplest
strategy is to start at a local minimum and evolve a trajectory along G from the local
minimum until G becomes very small. A trajectory-based method can be computationally
inefficient. One of the problems in using conjugate-gradient minimizers to optimize along
the uphill direction parallel to G, is there is no object function associated with the force
G. Barkema and Mousseau45 suggest the use of a Levenberg-Marquardt nonlinear leastsquares optimization method designed for following G.
In our experience, the simple method of iteratively following G until it becomes
small leads to saddle point regions on an undeformed energy surface for small peptides in
Γvt. The same method does not work as well in Γvc for peptides and hydrocarbons since
uphill directions often correspond to an unreasonable disruption of covalent geometries. A
potential drawback of the G-vector formalism for our purposes is that the saddle point
generated is not necessarily a saddle point adjacent to the local minimum of interest. In
using this method in a smoothing algorithm, we check for true saddle point convergence
by using a truncated Newton method49 to refine the location of the ART saddle point.
126
Results
Original DEM as well as PS-NMLS and PS-TSBS were applied to conformational
energy optimization problems of argon atom clusters, capped polyalanine peptides, cycloheptadecane and rigid polyalanine helices. Results from these applications are discussed
in detail below.
Clusters of Argon Atoms in Γvc
A DEM smoothing protocol has previously been applied45 to find the global energy
minimum of varying cluster sizes of argon atoms. For these systems, the interatomic interactions are purely van der Waals interactions. Kostrowicki, et al. 45 have used a threeGaussian fit to model the interaction between argon atoms for a DEM smoothing study.
Two Gaussians represent the Lennard-Jones interactions between argon atoms. A third
Gaussian is used as a shallow basin function to keep the clusters bounded during smoothing. In analogous work on potential function smoothing based on annealing an approximate classical distribution, GDA, Ma and Straub34 have used a four Gaussian fit for
the Lennard-Jones term and a harmonic restraint potential to restrict the GDA sampling to
the manifold of compact clusters.
Considerable work has been done toward enumerating the various local and global
minima for argon atom clusters of different sizes11,54. For cluster sizes larger than n = 13,
the number of minima has been estimated using a relation of the form g(n) ≈
exp(a+bn+cn2) where a = −2.5167, b = 0.3572 and c = 0.0286 are parameters derived
127
from a fit to results from a full enumeration of all the minima for n < 13. DEM applied to
clusters of size n = 5−19, 33 and 55 yields the global minimum for n = 5-7, 11, 13-15, 33
and 5545. The lowest energy structures of most argon atom clusters are related to Mackay
icosahedra that show a five fold symmetry. However, there are clusters for which the global minimum is derived from either an fcc symmetry as in n = 38 or Mark’s decahedron
as in, n = 75, 76, 77, 102, 103 and 10413.
For n = 8, Kostrowicki, et al. have shown that DEM yields a dodecahedron of triangle faces which is the global minimum on the undeformed surface of the three Gaussian
approximation. On a true Lennard-Jones surface, the global minimum is a pentagonal bipyramid with two atoms on the outside. It should be noted that the GDA algorithm of Ma
and Straub33 and a refined AGDA protocol55 also find global minima for different cluster
sizes with varying degrees of success. We applied a PS-NMLS to argon atom clusters using a three-Gaussian approximation and parameters ε = 0.2824 kcal/mol and σ =
3.3610Å. The third attractive Gaussian corresponds to a well depth of −1.7 kcal/mol and
width of 0.00001Å, which restricts sampling to compact clusters. We studied all clusters
of size n = 5−39 and n = 55. This list includes clusters not reported in the work of Kostrowicki, et al., clusters for which the original DEM does not recover the global minium45
and certain benchmark "hard" problems such as n = 3813,56.
In our application of DEM, we use td = 300.0 and nd = 100 and s = 3 in equations 7
and 8 and find the global minimum for the n = 5-7, 10-16, and 18-19 clusters starting
from arbitrary structures not near the global minimum. All minimizations were performed
using a truncated Newton algorithm49 with an rms gradient convergence criterion of
128
0.0001 kcal/mol/Å. We note that our implementation of the DEM protocol succeeds in
finding the global minimum for the n = 12 and 19 clusters in contrast to the work of Kostrowicki, et al.
A PS-NMLS method was used in an attempt to find the global minimum for the n =
5-39 and n = 55 clusters. For the n = 8 cluster, the global minimum energy in LennardJones units is -19.8222 and the first excited state, a dodecahedron of triangle faces, has an
energy of -19.7649 LJ units. Both the DEM and PS-NMLS methods recover the first excited state instead of the global minimum. For n = 9 and n = 17, we found the global minimum using a PS-NMLS method with the five largest eigenmodes in Γvc as search directions. Local searches in Γvc were done for all t < 5.0 during the reversal.
Clusters of size n = 38, 75-77 and 102-104 are particularly challenging problems for
global optimization due to the "multiple funnel" structure of the underlying PES56. Multiple funnels refer to potential energy surfaces with multiple similar low energy basins
and very different conformations. For the n = 38 case, the fcc truncated octahedron is the
global minimum with an energy −173.9284 Lennard-Jones (LJ) units57. PES deformation
schemes such as the Distance Scaling Method of Pillardy and Piela57 and the "basin hopping" algorithm of Doye and Wales11 succeed in finding the global minimum for n = 38.
The distance scaling and basin hopping methods generate smoother potential energy surfaces for improved conformational searching. The n = 38 problem is easy for our version
of potential smoothing without local search. The global minimum structure derived from
fcc symmetry can be found from completely random starting structures. For some other
129
problems, particularly n = 31, 34 and 37, we were unable to find the global minimum using PS-NMLS.
For the n = 55, case DEM does not find the global minimum from arbitrary starting
structures. This is because at t = td it is not possible to obtain a unique structure irrespective of the gradient convergence criterion chosen for the truncated Newton optimization.
It is possible that non-unique conformations at large deformations are a consequence of
errors in numerical precision. We applied a PS-NMLS protocol to several different starting structures and a small set of search directions along the Cartesian eigenvectors and always succeeded in finding the global minimum. The n = 13, 55 clusters possess a high degree of symmetry because of their perfect Mackay icosahedral structures. For all clusters,
the results reported above can be recovered from completely random starting conformations. Results from our application of DEM and PS-NMLS protocols are summarized in
Table II.
These results imply that the difficulty of global optimzation problems subtly depends upon symmetry and other nuances of the system, in addition to the system size. Although the local search method presented herein improves the reliability of finding global
minima during the backtracking procedure, the failures identified above suggests that
similar difficulties might be encountered with other strucutre prediction challenges which
exhibit high degrees of symmetry.
130
Table II. Results for PS-NMLS and DEM and energy minimizations
applied to clusters of argon atoms. In the table n denotes the size of
the cluster. If the number of search directions for PS-NMLS is zero,
then a straight DEM protocol finds the global minimum. All energies are in Lennard-Jones (LJ) units.
Cluster Size
n
Global Minimum
PS-NMLS
Minimum
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
55
-9.1039
-12.7121
-16.5054
-19.8222
-24.1134
-28.4225
-32.7660
-37.9676
-44.3268
-47.8452
-52.3226
-56.8157
-61.3180
-66.5309
-72.6598
-77.1770
-81.6846
-86.8098
-92.8445
-97.3488
-102.3727
-108.3156
-112.8736
-117.8224
-123.5874
-128.2866
-133.5864
-139.6355
-144.8427
-150.0445
-155.7566
-161.8254
-167.0338
-173.9284
-180.0332
-279.2485
-9.1039
-12.7121
-16.5054
-19.7649
-23.2698
-28.4225
-32.7660
-37.9676
-44.3268
-47.8452
-52.3226
-56.8157
-61.3180
-66.5309
-72.6598
-77.1770
-81.6846
-81.8098
-92.8445
-97.3488
-102.3727
-108.3156
-112.8736
-117.8224
-123.5874
-128.2866
-133.1836
-139.6355
-144.8427
-149.6721
-155.7566
-161.8254
-166.6315
-173.9284
-180.0332
-279.2485
131
Number of Γvc
Search Directions Used in
PS-NMLS
0
0
0
24
5
0
0
0
0
0
0
0
5
0
0
10
10
15
23
15
15
15
15
25
29
55
87
21
20
96
44
23
105
0
23
10
Oligopeptides in Γvc
DEM and PS-NMLS were applied to N-Acetyl-Ala-Ala-N´-Methylamide in Γvc.
We first exhaustively enumerated all the minima on the undeformed DOPLS surface. This
was done by combining an extensive grid-search around minimum energy regions to a series of truncated Newton conjugate-gradient minimizations. We used a gradient convergence criterion of 0.0001 kcal/mol/Å. Minimum energy regions for the grid search were
chosen based on the work of Zimmerman, et al.58 The combined grid search procedure
finds a total of 142 unique minima. The four lowest minima including the global minimum have energies of Vglobal = −84.1906 kcal/mol , V2 = −83.9167 kcal/mol , V3 =
−81.9962 kcal/mol and V4 = −81.9509 kcal/mol.
We characterized the smoothing of the PES by following each of the 142 minima as
a function of increasing t. We used values of s = 3, td = 55.0 and nd = 400 in Equation (7).
On the t = 55.0 surface only a single minimum remains. We observe that the number of
minima reduces from 142 at t = 0 to 1 for t = 55 through a series of mergers that are a
consequence of diminishing barriers between minima. Exhaustive enumeration allows
complete characterization of the smoothing process and evaluation of the requirements for
finding the global minimum. An analysis of smoothing applied to the fully characterized
PES of N-Acetyl-Ala-Ala-N´-Methylamide will be reported elsewhere59.
An energy crossing between minimum 4 and the global minimum occurs at t =
0.1372, i.e., V4 < Vglobal for all values of t ≥ 0.1372. The smoothing process projects out a
catchment region related to minimum 4 for large t, and a DEM reversing schedule
132
converges to minimum 004 at t = 0, i.e., V4 = −81.9509 kcal/mol. A NMLS protocol using a search along 5 large torsional eigenmodes corrects post facto the consequence of the
crossing at t = 0.1302 and converges to the global minimum. This example illustrates how
a local search method can correct errors due to crossings during the smoothing process.
We tested the ability of a PS-TSBS protocol to find the global minimum for NAcetyl-Ala-Ala-N´-Methylamide. In the TSBS method, the first stage is the location of a
nearby saddle point via the G-vector method described earlier. This is followed by perturbation away from the saddle point along the eigenvectors corresponding to the negative
eigenvalue in order to reach the adjacent minimum.
We tuned the TSBS searches to find the global minimum by correcting for some of
the problems inherent to locating transition states. A point for energy minimization is
found after an NMLS type walk away from the saddle point region indicates a turn toward
a minimum. We found the global minimum using this method in multiple independent
runs starting from random locations on the network of 142 minima. Because transition
states were located using random perturbations away from local minima, deviations from
a DEM reversal were found at found at different values of t for each run.
Despite the success in reproducibly finding the global minimum, the current TSBS
method is unlikely to work for larger systems. The main disadvantage of the current
implementation of PS-TSBS is that the search process becomes non-local, i.e., the system
is moved farther away from the catchment region of interest than is necessary. However,
the concept of moving the system over transition states remains attractive and alternate
methods to restrict the climb along the G-vector modulated by information about the
133
appropriate contour of tangency may be a useful way of refining TSBS methods. For the
rest of this work, we use only the NMLS class of local search enhancements to DEM.
CH3CO-(L-Ala)n-NHCH3 in Γvt and Γvc
Nakamura, et al. 38 have studied DEM conformational energy minimization of a
capped 9-residue polyalanine chain using a modification of an AMBER 4.0 force field.
They showed that a DEM protocol fails to find the global minimum, believed to be an αhelix, while a "two-stage" method recovers a structure slightly lower in energy than the
α-helix that is very similar to the canonical α-helix, differing only in a bifurcated "capping" hydrogen bond at the C-terminus.
We analyzed varying lengths of capped polyalanine sequences CH3CO-(L-Ala)nNHCH3 in Γvt. The bonds and angles are invariant in all of these calculations and were
based on idealized peptide values shown in Table III. Because the torsional barrier for deviation away from ω = 180° is large, the ω angle was kept fixed at 180°, the trans conformation of the peptide bond. It can be shown that t = td = 10 is a sufficiently large extent of
smoothing for random starting structures converge to the same energy and conformation.
Through systematic trials, we found s = 5 to be appropriate for smoothing in Γvt. Values
of nd were varied based on the size of the problem. As the number of residues, n, is increased beyond 7 for CH3CO-(L-Ala)n-NHCH3 in Γvt, the number of points nd between t
= td and t = 0 is increased. If the decrease in smoothing level ∆t is sufficiently small during the reversing schedule, an NMLS protocol is reduced to making a binary decisions in
multidimensional conformational space. If ∆t is not small enough, then a NMLS protocol
may require multiple iterations to converge to the best alternate minimum. For larger
134
values of ∆t, an alternate minimum may be too far from the local minimum to be found
by a NMLS protocol at the current level of smoothing.
Table III. Ideal geometries used for constructing capped polyalanine chains using DOPLS definitions of atom types for varying
lengths in Γvt. On this manifold the values of ω are kept fixed at
180°.
Bonds
Bond Lengths
Angles
Bond Angles
C-CH3
C´=O
N - C´
CαH-N
C´-CαH
N-HN
C-´CαH
CαH-CH3
1.51Å
1.22Å
1.34Å
1.46Å
1.51Å
1.02Å
1.54Å
1.54Å
CH3 -C´- O
CH3 -C´- N
C´- N - CαH
N -CαH -C´
CαH -C´ - O
C´ - N - H
N - CαH - CH3
CαH -C´ - N
C´ - N - CH3
122.5°
114.4°
121.0°
111.0°
122.5°
118.0°
109.5°
112.7°
121.0°
Results from application of PS-NMLS to chains of CH3CO-(L-Ala)n-NHCH3 in Γvt
for n = 5 −12 are described in Table IV. For all of the chains, DEM obtains a γ-turn structure. For n = 5 the γ-turn is the DOPLS global energy minimum. For n = 6 and 8, we find
β-hairpin structures with non-classical reverse turns to be the lowest energy structures.
For n = 7 and n = 9−12, the PS-NMLS method recovers structures very similar to, and
slightly lower in energy than, canonical α-helices. Differences between the two types of
structures are in the C-termini where the N-methyl amide hydrogen forms a bifurcated hydrogen bond with carbonyl oxygens from the residues n−1 and n−2. We refer to such
structures as α´-helices. Values for the torsional φ and ψ are angles are similar to those of
canonical α-helices.
135
Table IV. Summary of results from application of PS-NMLS to
varying lengths of CH3CO-(L-Ala)n-NHCH3 Sequences in Γvt. In
Γvt the bonds and angles remain fixed and the energy minimizations
find the lowest or global minimum on the manifold of fixed bond
and angles.
n
5
6
7
8
9
10
11
12
Number of
Flexible
Torsions
(φ,ψ)
DEM
Energy
(kcal /mol)
PS-NMLS
Energy
(kcal/mol)
PS-NMLS
Structure
Type
Energy of
Canonical
α-helix in
Γvt
10
12
14
16
18
20
22
24
-179.4897
-212.5233
-245.5841
-278.6389
-311.7031
-344.7672
-377.8342
-410.9029
-179.4897
-217.4073
-253.8166
-293.4585
-330.0448
-368.4315
-406.9659
-445.5773
γ-turn
β-hairpin
α´-helix
β-hairpin
α´-helix
α´-helix
α´-helix
α´-helix
-178.2146
-215.5294
-253.3204
-291.4040
-329.7243
-368.1972
-406.7670
-445.4150
Each of the PS−NMLS calculations in Γvt for CH3CO-(L-Ala)n-NHCH3 chains requires approximately a thousand independent minimizations. It is instructive to compare
results from our PS−NMLS calculations to a random search that uses the same number of
local minimizations. For each of the CH3CO-(L-Ala)n-NHCH3 chains, we set the angle ω
to be trans and generated a thousand independent conformations using random values of φ
and ψ between −180° and 180°. Each of the 1000 starting conformations were minimized
using an optimally conditioned quasi−Newton method without line searches48 over φ−ψ
space to an rms gradient convergence of 0.0001 kcal/mol/radian. We were unable to find
the global minimum for any of the polyalanine chains studied using this random search
procedure. The same 1000 randomized starting conformations were used as starting
136
positions for a NMLS local search optimization on the undeformed surface. For n = 5−8,
we found the global minimum approximately 35% of the time, and for n = 9−12, the success in finding the global minimum ranges from 22−30%. The improved success of
NMLS optimizations on the undeformed surface over a random search is an indication
that the iterative scheme in the search procedure facilitates improved sampling of conformational space. On average, we required 6 or more iterations of local search to converge
to the global minimum and this explains the modest dependence of success on chain
length. Larger chains were have not yet been studied. The important virtue of the
PS−NMLS over NMLS on the undeformed surface is that for n = 5−12 all of the 1000
random conformations merge into a single conformation with the same energy for t = td =
10. This means that the smoothing procedure is completely deterministic and does not require a large number of independent minimizations. Also, as the size of the problem increases, the efficiency of using NMLS on the undeformed surface in finding the global
minimum or lower lying minima is greatly reduced.
Γvc Calculations
We studied DEM and PS-NMLS conformational energy minimizations in Γvc for
different lengths of CH3CO-(L-Ala)n-NHCH3 chains. Kostrowicki and Scheraga46 have
discussed implications of deforming penalty function terms such as harmonic bond length
and bond angle restraint terms for DEM smoothing. Because we are not interested in significant rearrangements of covalent geometry, we do not smooth the bond stretching,
angle bending and improper dihedral terms for calculations in Γvc. This is different from
Γvt calculations where bond lengths and bond angles are kept fixed. In Γvc, bonds and
137
angles are allowed to stretch and bend as they would on the undeformed surface. The
bonds and angles deform at different levels of smoothing to compensate for long range
effects of nonbonded interactions.
As in Γvt, we set td = 10.0 in all our calculations. We use a fixed schedule of nd =
100 and s = 3 in equations 7 and 8 for forward smoothing and reversing and study capped
polyalanine chains of length n = 5−11 in vacuo. For all values of n, DEM again finds γturn structures. PS-NMLS finds a set of β-hairpin structures for each of the sequences
studied. The β-hairpin structures found are lower in energy than the α-helices in Γvc for n
= 5−9.
For instance with n = 8, the t = 0 DOPLS energy for a local minimum corresponding to the canonical α-helix is -306.8196 kcal/mol. This structure was located using a
model-built α-helix as the starting conformation and then minimizing to a rms gradient
convergence of 0.0001 kcal/mol/Å. A PS-NMLS protocol yields a β-hairpin structure that
is 7.4179 kcal/mol lower in energy than this α-helix. Results of all Γvc calculations for n
= 5-11 chains of CH3CO-(L-Ala)n-NHCH3 are summarized in Table V. Figure 1 shows
the structures for a n = 8 canonical α-helix and the lower energy β-hairpin obtained from
a PS-NMLS in Γvc. Because 310 helices are higher in energy than alpha helices in vacuo
on the original OPLS surface, 310 helices are not expected to be retained by the PSNMLS algorithm.
138
Table V. Results of DEM and PS-NMLS applied to capped sequences of polyalanine, CH3CO-(L-Ala)n-NHCH3 in Γvc. A schedule of s = 3, nd = 100 and td = 10 was used in this study.
n
DEM Energy
(kcal/mol)
PS-NMLS Energy
(kcal/mol)
Canonical α-helix
Energy (kcal/mol)
5
6
7
8
9
10
11
-183.1443
-216.9159
-250.7094
-285.8123
-318.3087
-352.1148
-385.9248
-197.9965
-238.3538
-272.9906
-314.2375
-350.0894
-387.0713
-423.6528
-188.3236
-227.1757
-266.8064
-306.8196
-346.9622
-387.2196
-427.5763
Figure 1. (a) Lowest energy α-helical conformations of CH3CO-(LAla)8-NHCH3 in Γvc. (b) The β-hairpin structure which is 7.4179
kcal/mol lower in energy than the canonical α-helix shown in (a)
and is the structure found using PS-NMLS.
139
In direct analogy to the Γvt calculation, we expected to see α-helices as the result of
PS-NMLS calculations for longer sequences. For n = 10 and 11, the PS-NMLS method
does not find structures lower in energy than the α-helix, but instead finds β-hairpins that
are higher in energy than the α-helix. For longer chains, we performed exhaustive local
searches along all the torsional eigenmodes and found the higher energy β-hairpins. We
analyzed the reasons for this result by enumerating structures sampled during local
searches. For n = 10, there are 3 unique minima near the global minimum on the DOPLS
surface in Γvc, a β-hairpin (Vβ = −387.0713 kcal/mol), the canonical α-helix (Vα =
−387.2196 kcal/mol) and an α´-helix (Vα´ = −387.5620 kcal/mol) which is the global
minimum. We studied the forward smoothing of these three low energy structures. For
values of t between 0.0051 and 0.1826, the global minimum is in the catchment region of
the β-hairpin. A comparison of conformational energies as a function of t for forward
smoothing is shown in Figure 2. The α-helix becomes higher in energy than the β-hairpin
for t of 0.0051 and greater. For all t > 0.1826 the global minimum is the γ-turn basin. The
PS-NMLS converges to the global minimum up to the t = 0.0051 surface which is a βhairpin. At the next smaller t value in this particular protocol, t = 0.0045, the α-helix is
lower in energy than the β-hairpin. PS-NMLS fails to recognize the crossing of energies
for 0.0045 < t < 0.0051 when the α-helix becomes lower in energy than the β-hairpin.
140
Figure 2. Conformational energies of CH3CO-(L-Ala)10-NHCH3 in
Γvc as a function of increasing deformation t for a canonical α-helix
(−), for an α´-helix (- -), and for the β-hairpin found from a PSNMLS protocol (...). On the t = 0.0051 surface the β-hairpin is lower
in energy than the α-helix.
-377
alpha
alpha’
beta
-378
conformational energy (kcal/mol)
-379
-380
-381
-382
-383
-384
-385
-386
-387
-388
0
0.002
0.004
0.006
smoothing parameter (t)
0.008
0.01
We used the NMLS search strategy to enumerate all local minima sampled from the
β-hairpin and α-helix minima along all of the search directions corresponding to the 20
pairs of φ−ψ angles at t = 0.0045 and t = 0.0051. On the t = 0.0045 surface, the α-helix is
lower in energy than the β-hairpin, Vα = −383.2723 kcal/mol and Vβ = −383.2569
kcal/mol. Structures found from a local search were characterized as α-helical if the distances ri,i+4 between the carbonyl oxygen atoms of residue i and the amide nitrogen atom
of residue i + 4 were between 2.7Å and 3.1Å, implying α-helical hydrogen bonds. If the
structures were such that the distances between the atom pairs (O9 - N2), (O7 - N4), (O4N7) and (O2 - N9) were between 2.7Å and 3.1Å, the structure was deemed a β-hairpin
141
containing inter-strand hydrogen bonds. Bifurcated H-bonds were treated as two independent H-bonds and evaluated as above. Of the 23 unique low energy structures found from
a local search out of the α-helix local minimum, almost all show the hydrogen bonding
pattern of an α-helix. Results of this search are shown in Table VI. Similarly, a local
search out of the β-hairpin minimum at t = 0.0051 finds 32 unique local minima including
the original β-hairpin. More than 90% of the structures show hydrogen bonding patterns
typical of a β-hairpins; see Table VII. The lack of overlap in the conformational energies
listed in these two tables show that local searches out of the α-helical and β-hairpin basins
sample conformationally disjoint regions. Similar results were obtained for calculations
on the t = 0.0051 surface for which the β-hairpin is lower in energy than the α-helix, Vβ =
−382.7399 kcal/mol versus Vα = −382.6904 kcal/mol.
142
Table VI. Hydrogen-bonding distances for the 23 unique local minimum energy structures sampled in a local search out of the α−
helical local minimum on the t = 0.0045 Surface. The shaded region
of the table corresponds to the type of hydrogen bonds that classify
α−helical structures. All the very low energy structures show typical α−helical hydrogen bonding pattern. The number of α−helical
hydrogen bonds decreases with increase in conformational energy
and the higher energy structures sampled in this calculation are
random coil conformations. The table reflects two important features of the local search sampling. The α−helical basin and the βhairpin basin are disjoint sets and the α−helical basin is a very narrow deep well reflected in the fewer number of structures sampled,
compared to the β-haripin.
Rank
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
Energy
-383.2723
-382.9076
-381.8493
-380.2025
-379.6051
-379.4903
-377.3410
-375.4960
-373.6687
-373.0820
-372.6473
-371.9262
-371.5315
-370.9817
-369.4312
-368.7190
-365.4600
-362.7182
-362.7182
-358.1491
-357.0376
-353.3007
-347.6971
i - i+4, distances between carbonyl oxy- i - j distances between cargen (i) and amide nitrogen (i +4) atoms bonyl oxygen (i) and
if the distances are > 2.7Å and < 3.1Å
amide nitrogen (j) atoms if
the distances are > 2.7Å
and < 3.1Å
1-5 2-6 3-7 4-8 5-9 6-10 9-2 7-4 2-9 4-7
2.87 2.96 2.87 2.91 2.91
2.87 2.95 2.88 2.90 2.92 2.95
2.92 2.90 2.94 2.90 2.91
2.87 2.95 2.89 2.93 2.80
2.87 2.98 2.89
2.87 2.95 2.90
2.81
2.92
2.96 2.89 2.92 2.88
3.09
2.87 2.95
2.82
2.89
2.98 2.79
2.88 3.02 2.92
2.92 2.83 2.96
2.93 2.90 3.02
2.86
2.96 2.90
3.01
2.98 2.89 2.83
2.98 2.89 2.83
143
Table VII. Hydrogen-bonding distances for 32 unique local minima
sampled in a local search out of the β-hairpin local minimum on the
t = 0.0045 surface. The shaded region corresponds to hydrogen
bonds for a β-hairpin. All low energy structures sampled from the
β-hairpin local minimum show hydrogen bonding patterns typical
of β-hairpins. Higher energy structures have fewer β-hairpin hydrogen bonds and some show a few α-helical hydrogen bonds.
Rank
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
Energy
-383.2569
-381.8750
-380.7871
-378.7298
-377.0667
-376.5693
-376.3553
-376.2071
-375.9423
-375.5653
-375.0034
-374.3673
-373.6144
-372.4078
-372.2050
-372.1612
-370.1621
-370.0502
-369.6184
-368.6573
-367.5699
-367.1853
-366.6323
-365.6136
-365.5884
-365.0771
-365.0771
-361.5149
-360.0044
-359.5009
-359.0971
-352.6797
i - i+4, distances between carbonyl oxy- i −j distances between cargen (i) and amide nitrogen (i +4) atoms bonyl oxygen (i) and
if the distances are > 2.7Å and < 3.1Å
amide nitrogen (j) atoms if
the distances are > 2.7Å
and < 3.1Å
1-5 2-6 3-7 4-8 5-9 6-10 9-2 7-4 2-9 4-7
2.82 2.86 2.88 2.93
2.76 2.82 2.87 2.93
2.88 2.98 2.90
2.86 2.97 2.91
3.00 2.90 2.80 2.92
2.79 2.81 2.94
2.85
2.94
2.85 2.85 2.86 2.93
2.78 2.84 2.95
2.80 2.82 2.93
2.84 2.90 2.88 2.92
2.83 2.85 2.92
2.83
2.86
2.77 2.95 2.86 3.02
2.84
2.90
2.89
2.85 3.02 2.91
3.02
2.82
2.88
2.95
2.83
2.88
2.81
2.85
2.87
2.79
2.92
2.82
2.82
2.77
2.77
2.92
2.92
2.86
2.99
2.81
2.82
2.89
2.93
2.95
144
2.87
2.99
Three important observations from these local search calculations are: (1) we systematically find more unique local minima out of the β-hairpin minimum than the α´helical minimum, suggesting that the β-hairpin basin may be broader than the α-helical
basin; (2) the hydrogen bonding pattern suggests that the two sets of conformations
sampled from a local search out of an α-helical minimum and β-hairpin minimum are disjoint and are separated by a large barrier; (3) local search cannot surmount the barrier between the narrow α-helix and broad β-hairpin regions for t < 0.0051 and stays within the
catchment region of the β-hairpin all the way back to the t = 0 DOPLS surface.
To test the effect of force field parameterization, we repeated calculations for the n
= 5 and 8 capped polyalanine sequences in Γvc using a deformable version of the
CHARMM22 force field in place of DOPLS . The only modification to the CHARMM22
energy functions was made in the substitution of a Gaussian approximation to the 12-6
Lennard-Jones function for van der Waals interactions. An energy minimized canonical
α-helix for n = 5 using modified CHARMM22 has an undeformed surface energy of
85.4704 kcal/mol. A PS-NMLS protocol finds a structure that is not an α-helix, but which
is lower in energy. The undeformed surface energy for this structure is 73.7718 kcal/mol.
The DOPLS surface minimum energy structure shows an rms α-carbon deviation of
1.8268Å from the undeformed CHARMM surface minimum energy structure. The (φ,ψ)
angles for the DOPLS and CHARMM22 structures are quite similar. For n = 8, the
CHARMM22 α-helix energy is 141.8306 kcal/mol. PS-NMLS finds a β-hairpin structure
with a lower energy of 133.5861 kcal/mol. The DOPLS surface minimum energy
145
β-hairpin for n = 8, shows a deviation of 1.2175Å in α-carbon atom positions from the
undeformed CHARMM surface minimum energy β-hairpin. This calculation demonstrates that PS-NMLS protocols using both DOPLS and CHARMM22 force fields give
qualitatively similar results for Γvc calculations on small polyalanine sequences. We also
performed a Γvt calculation using modified CHARMM22 functions for an n = 12 capped
polyalanine sequence. The PS-NMLS method recovers the α´-helix as the global minimum on the manifold of fixed idealized CHARMM22 bonds lengths and bond angles.
The efficiency of the PS−NMLS method was compared to a random search using
approximately the same number of local minimizations, and to NMLS on the undeformed
surface from random starting conformations. Neither of these methods succeed in finding
the global minimum for n = 5−9. For n = 10 the lowest energy conformer found using the
random search method is 17.2 kcal/mol higher in energy than the PS−NMLS β−hairpin,
and the lowest energy conformer found using the t = 0 surface NMLS is 6.34 kcal/mol
higher in energy than the PS−NMLS structure.
Cycloheptadecane in Γvc
One of the better known benchmark problems for conformational search is the determination of low energy conformations of the highly flexible cycloheptadecane60. This
system continues to attract attention and serve as a test for newly developed search
methods61. While not a particularly large molecule, cycloheptadecane presents a difficult
challenge due to its great flexibility and the close energy spacing of the lower lying
minima. Extensive analysis via a variety of search methods has located exactly 263
minima on the MM2 energy surface within 3.0 kcal/mol of the purported global
146
minimum. Since the full spectrum of energy minima for this molecule has not been described in the literature, we undertook its generation. We used an iterative NMLS protocol with the MM2 energy function to sample the PES of cycloheptadecane. A local search
was carried out from every unique minimum found on the PES. All symmetry-distinct
minima found in this manner were added to the existing map and the procedure was repeated until self-consistent convergence. The structures found are unique minima to an
rms gradient per atom of 0.00001 kcal/mol on the MM2 potential energy surface42 traditionally used in studies of this system. Using the iterative NMLS scheme to hop between
minima, we found 20,469 unique minimum energy structures with an MM2 energy distribution as shown in Figure 3a. Even with the use of an efficient truncated Newton minimization method49, generation of the full distribution required about 13 days of CPU time
on a 250MHz Digital Alpha workstation. The global minimum has an MM2 energy of
19.0680 kcal/mol. A second minimum lies only 0.01 kcal/mol above the global minimum
and has an MM2 energy of 19.0774 kcal/mol. These two structures are separated by about
0.4 kcal/mol from the third best and subsequent structures. The low energy tail of the full
distribution is presented in Figure 3b.
147
Figure 3. (a) Energy distribution of the 20,469 unique minima for
cycloheptadecane located using an self-consistent NMLS-based
search to scan the complete potential energy surface. The number
of minima found in each 0.1 kcal/mol energy bin is plotted as a
function of increasing MM2 energy value. (b) Low energy tail of (a)
showing the distribution of minima with MM2 energy values less
than 3 kcal/mol above the global minimum. The search procedure
used to generate both panels (a) and (b) found 11 minima within 1
kcal/mol of the global minimum, 68 minima within 2 kcal/mol, and
261 minima within 3 kcal/mol.
300
number of minima (0.1 kcal/mol bins)
250
200
150
100
50
0
20
25
30
energy (kcal/mol)
35
40
30
number of minima (0.1 kcal/mol bins)
25
20
15
10
5
0
19
19.5
20
20.5
energy (kcal/mol)
148
21
21.5
22
Application of the PS-NMLS algorithm to cycloheptadecane in Γvc succeeds in
finding the second lowest minimum. We used a maximum deformation of td = 25.0 at
which point only one minimum remains on the smoothed surface. Variations in the reversal protocol (nd = 100 to 150, and s = 2 or 3) coupled with variation in the number of
modes searched during NMLS (values from 3 to 16) also result in the procedure finding
the second lowest minimum.
The PS-NMLS protocol fails to find the global minimum for the same reasons that
it fails for longer polyalanine chains in Γvc. In Table VIII, we summarize the evolution of
minima on the PES as a function of smoothing. There is a crossing of the relative energies
of the global minimum and the second lowest minimum for t ≈ 0.0061. If the PS-NMLS
is to successfully converge to the global minimum, the protocol would have to be able to
sample the global minimum for values of t < 0.0061. Smoothed surfaces at very small t
where the global minimum is favored closely resemble the original undeformed surface.
If the second lowest minimum and the global minimum are in widely separated regions of
conformational space, a local search will not be able to sample the global minimum. Further extensions of PS-NMLS that should be able to find several of the lowest energy
structures of cycloheptadecane, including the global minimum, are discussed below. Figure 4 shows the global minimum for cycloheptadecane and the second lowest energy
structure located by the current PS-NMLS.
149
Table VIII. Evolution of the lowest fifteen minima of cycloheptadecane as a function of increased smoothing. Column 2 shows the
MM2 energies for the lowest energy conformers found using an extensive distance geometry search technique. We found 257 unique
minima within 3 kcal/mol of the global minimum. A smoothable
variant of the MM2 PES which replaces the Buckingham potential
with a 2 Gaussian approximation has conformational energieson
the t = 0 surface as shown in Column 3. The spacing between and
ordering of conformational energies is similar to the original MM2
surface. Columns 4−6 show the change in conformational energies
as a function of smoothing. Increase in smoothing is characterized
by a reduction in the conformational energy spacing between
minima and a rearrangement of the rank ordering of minima, i.e.,
for 0.001<t<0.01 minimum 2 is the lowest in energy.
Rank
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
MM2
Energy
(kcal/mol)
19.0680
19.0774
19.4372
19.4509
19.6571
19.7292
19.7328
19.8400
19.8789
19.9579
19.9869
20.0954
20.1214
20.1470
20.2011
t=0
Energy
(kcal/mol)
19.6627
19.7145
20.0396
20.0627
20.2546
20.3228
20.3453
20.4441
20.4998
20.5813
20.6134
20.7091
20.7466
20.7629
20.8072
t = 0.001 Energy
(kcal/mol)
20.3561
20.3994
20.7415
20.7548
20.9480
21.0136
21.0324
21.1275
21.1875
21.2675
21.3122
21.4031
21.4321
21.4537
21.5034
150
t = 0.01 Energy
(kcal/mol)
26.8127
26.7791
27.2708
27.1992
27.4032
27.4476
27.4308
27.4956
27.5913
27.6577
27.8120
27.8627
27.8163
27.8836
27.9811
t = 0.1 Energy
(kcal/mol)
107.1554
106.3437
107.9972
107.4716
107.7382
107.7838
107.2037
107.1938
107.4179
107.3900
108.2206
108.1181
107.4686
107.8315
108.3395
Figure 4. (left) Global minimum structure for cycloheptadecane
with MM2 energy of 19.0680 kcal/mol. (right) Second lowest energy
minimum and the structure found by PS-NMLS algorithm with
MM2 energy of 19.0774 kcal/mol.
The cycloheptadecane problem illustrates some of the advantages of the PS-NMLS
method for global optimization. It has been estimated that an extensive search procedure
requires approximately 10,000 independent minimizations before converging to the global
minimum and locating the set of low lying minima61. A typical PS-NMLS run for cycloheptadecane with nd = 100, s = 3, and local search along the three highest torsional modes
finds the second lowest minimum in less than 1 hour of CPU time on the computer cited
above and requires only 440 energy minimizations. This number may be reduced further
151
by a coarse reversing schedule (reducing nd with s = 2) while still yielding the same result
as more exhaustive searching. The large reduction in the number of independent minimizations compared with other search techniques derives from the improved sampling on
smooth surfaces.
We calibrated the NMLS procedure on the undeformed surface for cycloheptadecane using an iterative NMLS optimization scheme starting from 500 independent randomized conformations. Each self-consistent run of NMLS on the undeformed surface requires approximately 200 minimizations. Only one of the 500 NMLS runs finds the second lowest minimum, which is the structure found using PS−NMLS. This result is to be
contrasted with the deterministic nature of the PS−NMLS protocol that converges to the
second lowest minimum without regard to the starting structure.
Optimizations in Γvr
We applied PS-NMLS in Γvr for docking two rigid canonical α-helices of CH3CO(L-Ala)9-NHCH3. The variables are the six rigid body degrees of freedom for each helix.
Since formation of complexes involves rigid molecules, we optimize only the intermolecular interactions in Γvr. Considerable work has been done on energetic approaches to
determine the packing of polyalanine α-helices62. It was shown that lower energy packing
orientations are minor variations of antiparallel arrangements. These studies also show
that the most important contributions to packing of polyalanine helices are from van der
Waals interactions with minor contributions from electrostatic interactions. Helix packing
has also been studied based on the packing preferences of sidechains attached to model
152
helical backbones 64-66. These analyses are based on either a knobs-in-holes 63,65 or
ridges-in-grooves64 picture of helix packing.
The orientation of two packed helices with respect to each other can be computed in
terms of two parameters, the distance of closest approach d and the packing angle Ω. The
distance of closest approach is the shortest distance between two points on the two helix
axes. Ω is the angle between the two helix axes when projected onto a plane normal to the
line of closest contact. It is computed as the dihedral angle defined by the points Nt1, cp1,
Nt2 and cp2, where Nti is the N-terminus of helix i and cpi is the point on helix i which is
along the line of contact. Ω varies from −180° to 180°, i.e., the helices are exactly parallel
if Ω = 0 and antiparallel if Ω = ±180°.
For rigid body optimizations, the translational coordinates are defined in terms of
the center of mass of each body and the rotations are the Euler angles that describe independent rotations. We use an "xyz-convention"66 to define the Euler angles. All calculations were done in vacuo and electrostatic terms were excluded.
Γvr was extensively searched to locate minimum energy conformations for the two
polyalanine helices. A grid of 18,000 starting conformations was generated by varying the
distance between the centers of mass, the packing angle between −180° and 180° in 20°
increments, and the rotation angles about each helix axis from 0° to 360° in 40° increments. The network of 18,000 structures includes an even sampling about the parallel, antiparallel and perpendicular orientations. The conformational energy for each of these
starting structures was minimized and duplicate minima removed to yield a set of 1093
unique minima. We used a quasi-Newton method for all minimizations with an rms
153
gradient convergence criterion of 0.0001 kcal/mol/degree-of-freedom. The distribution of
energies for the 1093 conformations is shown in Figure 5. The global minimum from this
grid search has an interhelical van der Waals energy of V = −16.4124 kcal/mol, d = 7.16Å
and Ω = 153.33°. These values correspond to an antiparallel arrangement of the helices
with an approximately 30° twist and denotes a "class a" type of packing of helices described by Walther, et al.65 which is typical for helices from globular proteins. Table IX
show values of d, Ω and V12 for the fifteen lowest minima found from the grid search.
Figure 5. Distribution of inter-helical conformational energies for
the 1093 unique minima found from an extensive grid search over
18,000 unique starting positions for two rigid capped CH3CO-(LAla)10-NHCH3 α-helices. The global minimum has an energy of 16.4124 kcal/mol.
45
number of minima (0.1 kcal/mol bins)
40
35
30
25
20
15
10
5
0
-18
-16
-14
-12
-10
-8
interhelical energy (kcal/mol)
154
-6
-4
Table IX. Fifteen lowest energy conformers for docked polyalanine
helices in descending order of interhelical energy. The table shows
the values for the interhelical energies, the packing angles Ω and
the distance of closest approach d.
Minimum
Inter-Helical
Energy
(kcal/mol)
Ω (deg.)
d (Å)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
-16.4124
-16.2582
-16.0996
-16.0570
-16.0258
-15.8974
-15.7974
-15.5851
-15.2452
-15.5117
-14.9027
-14.8666
-14.7943
-14.5419
-14.4517
153.33
149.82
159.79
146.58
-32.93
-28.92
-25.24
155.42
157.45
153.96
161.98
151.80
158.09
150.36
-26.56
7.1624
6.9569
7.1126
6.9445
6.9680
7.1235
7.0791
7.2866
7.3249
7.1943
7.0153
7.4602
7.1202
7.1636
6.7842
PS-NMLS was tested to see if it could find the global minimum energy conformation for docking the two polyalanine helices. We used a maximum deformation of td = 4.0
beyond which the calculation is numerically unstable without use of a constraining basin
function. We set nd = 150 and s = 3 for the smoothing schedule. Local searches are performed for all values of t < td during the reversing schedule along the twelve eigenvectors
corresponding to the twelve degrees of freedom in Γvr. The global minimum on the t =
4.0 surface has orientation parameters of D = 20.90Å and Ω = 125.86°. Upon returning to
the undeformed surface, the PS-NMLS method finds the global minimum obtained from
the grid search. This result is obtained reproducibly from any arbitrary starting structure.
155
The global minimum conformation of the docked polyalanine helices is shown in Figure
6.
Figure 6. Conformation of the global energy minimum for the packing of two capped, right-handed α-helices of sequence CH3CO-(LAla)10-NHCH3.
N-Terminus Helix A
N-Terminus Helix B
156
Discussion
Features of Potential Smoothing
We illustrate some typical features of the potential smoothing paradigm using onedimensional slices of a rugged PES shown in Figures 7 and 8. The process of transforming a potential surface via smoothing is characterized by three kinds of events: mergers,
crossings and shifting of minima.
Two unique minima on the undeformed surface can merge into a common basin at
some level of smoothing. The multiple minimum problem is circumvented by reducing
the number of minima through a series of such mergers with increasing t. Consider a
smoothing level t = t1 where two formerly distinct minima Mi and Mj merge into the same
basin identified by a common structure and conformational energy. If at t = t1 − ∆t during
a smoothing reversal protocol the two minima reappear and a minimization leads to Mi,
then we label the basin at t = t1 as Mi, i.e., Mj merges into Mi at t = t1. If Mi, and Mj are
equal in depth and width on the undeformed surface then a merger of these two minima
results from eliminating the barrier and an equal translation of the two minima into the
new basin. If the energy gap between the two minima is pronounced, then the higher lying
minimum slides into the broader basin of the lower lying minimum, i.e., the position of
the new single minimum is close to the location of the lower minimum from the undeformed surface. Figure 7 shows the rate at which the number of minima are reduced for
cycloheptadecane as function of increasing deformation. For a smoothing process fully
characterized by mergers, as shown in Figure 8, the minimum that is projected out on a
157
highly smoothed surface is related to the global minimum and a reversal protocol will find
the global minimum.
Figure 7. Reduction in the number of minima for cycloheptadecane
as a function of increasing PES deformation. On the undeformed
surface there are 20,469 unique minima. These minima merge into
a single minimum on the t = 25 PES. The figure shows a plot of the
log10 of the number of unique minima remaining on the PES as a
function of increasing smoothing, t.
4.5
log10(number of minima remaining)
4
3.5
3
2.5
2
1.5
1
0.5
0
0
5
10
15
20
25
deformation (t)
A second feature of potential smoothing is the crossing of relative energies of two
minima. Two unique minima A and B on the undeformed surface can have conformational energies VA and VB such that VA < VB for some t = t1. For t2 = t1 + ∆t, these two
minima persist as unique conformations but their relative energies can be reversed so that
VA > VB. The effect of crossings on a reversal schedule is shown in Figure 9. As the deformation is slowly reduced, minima that merged at a higher level of smoothing reappear
at distinct locations and separated by a small barrier, i.e., the reversal protocol sees a bifurcation. In Figure 9, the minimum in basin B is lower in energy than minimum A on the
158
t = t2 surface. Because the lower minimum at t = t2 is an artifact of a previous energy
crossing at some t1 < t < t2, the DEM reversal procedure will remain trapped in basin B
and not converge to the global minimum. There has been some speculation that crossings
are related to the free energy characteristics of the undeformed surface46, i.e., the minimum energy basin that becomes energetically favored at large t is related to the free energy minimum on the PES. This proposal is reasonable because narrow deep wells merge
into minima defined by broader shallow wells. Generally, the minima that survive on
largely deformed surfaces tend to be entropically favored.
159
Figure 8. One-dimensional schematic of the effect of a smoothing
protocol on a potential energy surface. The original PES is transformed by successive application of a smoothing operator, where
the extent of smoothing is dictated by a control parameter t. The
unsmoothed original surface (t = 0), the surface at an intermediate
level of smoothing (t = t1) and a highly smoothed surface (t = tlarge)
are shown. As the surface is transformed higher lying minima
merge into catchment regions of low lying minima and barriers between minima are progressively lowered. Open circles are starting
or intermediate points on each surface. Solid circles are local
minima. Dashed arrows show the result of local optimization ending
at a local minimum. Solid arrows represent adiabatic movement
from a local minimum on one surface to the corresponding starting
point on a rougher surface. A simple smoothing protocol consists of
repeated cycles of local optimization followed by adiabatic transfer
to the next surface.
t = tlarge
t = t1
t=0
Global Minimum
160
Figure 9. Schematic of a more realistic potential smoothing protocol
for molecular search problems. This figure shows a crossing between the two surviving minima on the t = t2 surface. A reversing
schedule encounters the first bifurcation at t = t2. At this level of
smoothing the protocol favors basin B over basin A due to a crossing of relative energies which is an artifact of the averaging process.
If bifurcations are sampled where the relative energies of the alternative basins are inverted from the t = 0 surface, then the simple
method will not converge to the global minimum. Between t = t2 and
t = 0 there exist values of t for which the energy ordering resembles
that of the original PES. A local search process coupled to the
smoothing schedule can potentially recognize errors due to earlier
energy crossings. For example, a local search represented by the
dotted arrow on the t = t1 surface would correctly decide that basin
A should be favored over basin B.
t = tlarge
t = t2
t = t1
Basin B
Basin A
earch
Local S
t=0
Minimum
B
Minimum
A
161
A third feature of smoothing is shifting in the location of minima and an initial increase in conformational energies associated with minima as the level of smoothing increases. This is a direct consequence of changing the local curvature of minima and lowering barriers, so minima become more shallow and the basins become broader with an
increase in deformation. Figures 7 and 8 show this shifting property as a function of an
increase in smoothing.
Figure 9 also shows how a PSS protocol works in correcting certain kinds of energy
crossings. At t = t1, a reversal protocol will first locate minimum B. Local search starting
from this minimum will explore neighboring basins. If basin A is adjacent to basin B on
the smoothed surface, then a normal mode or transition state directed activation followed
by minimization can locate the lower energy minimum A. Subsequent return to the undeformed surface will then correctly locate the global minimum in basin A. If the crossing
between a pair of minimum energy basins is introduced for a large value of t, then a local
search protocol will most likely find the lower alternate minimum during the reversal. If
there are other crossings on surfaces of very small t, a local search may not be able to correct the energy crossing because the barrier heights and surface roughness are already
comparable to those of the undeformed PES. In essence, "local" search has a greater
range of convergence and can correct crossing errors over larger portions of conformational space on smoother surfaces. This reasoning explains why of PS-NMLS in Γvc finds
very low energy minima, but not the absolute global minima, when applied to longer
chains of capped polyalanine and cycloheptadecane. For crossings built in at very small
162
values of t, a reasonable degree of local search may not be able to sample the global
minimum region.
Potential smoothing can be thought of as a projection method, i.e., an important
catchment region is projected out by reducing barriers between minima. In direct contrast,
barriers are present throughout a simulated annealing protocol and the global minimum is
located by generating a trajectory that uses thermal activation to move over barriers. Since
the number of barriers to be negotiated by simulated annealing grows exponentially with
the size of the system, projection methods are in principle more efficient for larger problems.
Other variants of search enhanced potential smoothing
We have shown in this work that two types of secondary search schemes can be
coupled to DEM for improved sampling of conformational space. NMLS or TSBS methods are not the only search methods that can be coupled to potential smoothing for improved searching. Increased sampling on a smooth PES could also be facilitated by a trajectory- or quench-based search mechanism. These include Molecular Dynamics
(MD)67, methods to promote conformational variation such as poling68, Local Enhanced
Sampling (LES) 69 and Monte Carlo Minimization (MCM) 10 or basin-hopping11.
Wawak, et al.70,71, have coupled an MCM method to two types of potential
smoothing algorithms, DEM and the Distance Scaling Method (DSM) of Pillardy and
Piela57,72. MCM enhanced potential smoothing algorithms were used successfully for ab
initio prediction of crystal packing of hexasulfur and benzene molecules71. Smooth surfaces generated by either DEM or DSM were searched using the basin hopping MCM
163
algorithm. Minima generated using MCM on a smooth surface can be followed back to
the undeformed surface using a DEM or DSM reversal protocol.
Computational efficiency: PS-NMLS versus DEM
In a system with no distance cutoffs on the range of nonbonded interactions, the
CPU time for a single energy/gradient calculation scales as O(N 2) where N is the size of
system. An efficient local optimization procedure will typically require CPU time that
scales as roughly O(N3). The CPU time for DEM scales approximately as 2nd[O(N 3)]
where nd is the number of points along the smoothing schedule. As described above, local
search algorithms are essential for correcting errors due to energy crossings between
minima. The CPU time in PS-NMLS increases relative to DEM due to the increase in the
number of extra O(N3) minimization calculations. The increase is determined by the number of search directions chosen and the number of points along the reversal schedule for
which local searches are performed. In the most extreme case, all possible normal modes,
O(N) of them in general, could be searched. The CPU time for this kind of extensive local
search PS-NMLS scales as O(N 4). For many problems, we have had success in locating
very low or global minimum energy structures using as few as 3-5 search modes, reducing the computational complexity to a small multiple of that for DEM. We are currently
developing various time saving strategies for implementing PS-NMLS protocols, one of
which is outlined below. Other improvements to PS-NMLS, including coupling local
search methods to adiabatic Gaussian density annealing (AGDA)35,55 and generating
families of low energy structures instead of a single estimate of the global minimum, are
also in progress.
164
Selecting Smoothing Windows for Local Searches
An obvious method for increasing computational efficiency is based on shortening
the smoothing window over which NMLS is performed. Table X shows the largest value
of deformation, termed tdetour, at which NMLS found alternate lower minima for varying
lengths of polyalanine chains in Γvt. Values for tdetour increase as a function of the length
of the chain, which is appropriate since the conformational energy surfaces for the larger
peptides become increasingly rough and crossings can occur on highly deformed surfaces.
Figure 10 shows a plot of tdetour as a function of peptide chain length, n. The linear least
squares fit to this data gives tdetour = 0.016 n − 0.0716 with a correlation coefficient of
0.995. An accurate estimate of tdetour provides an upper bound on the window of t values
for which NMLS will be efficacious. For example, polyalanine of length n = 8 has a tdetour
value of 0.0577. Then a PS-NMLS protocol with nd = 300, s = 5 and td = 10.0 will re-
quire about a factor of 3 fewer O[N 3] minimizations using the above tdetour value instead
of 5.0 as an upper bound on the range of t values where NMLS will be used. This will
translate directly into a factor of 3 savings in overall CPU time required.
Table X. Smoothing parameters t = tdetour at which alternate
minima were obtained using a NMLS protocol in Γvt for varying
lengths of capped polyalanine CH3CO-(L-Ala)n-NHCH3 chains. td =
10.0 and s = 5 in eq 8 of the text
n
tdetour
nd
6
7
8
9
10
11
12
0.0223
0.0391
0.0577
0.0792
0.0854
0.1061
0.1179
200
200
300
300
350
350
350
165
Figure 10. First-order least squares fit to the smoothing paramater
tdetour as a function of n for application of PS-NMLS to CH3CO(L-Ala)n-NHCH3 sequences in Γvt for (n = 5−12). The fit could be
used to implement windowing schemes to estimate smoothing values
for which an NMLS search protocol is to be used in Γvt. Restricting
local search to a limited window of t values allows a reduction in
computational overhead by eliminating unnecessary and redundant
local searches.
0.14
largest successful NMLS (tdetour)
0.12
0.10
0.08
0.06
0.04
0.02
0
5
6
7
8
9
10
polyalanine peptide length (n)
11
12
13
Summary
Our results demonstrate that potential smoothing coupled to appropriate modulation
and control mechanisms can be a useful tool for global optimization problems. This assertion is substantiated through the successful application of a set of local search enhanced
potential smoothing protocols to a cross section of conformational problems that vary in
size and complexity of the underlying PES. We have also analyzed limitations of a class
of local search methods used to correct errors inherent in smoothing protocols, and demonstrated in simple terms the effect of smoothing on a typical rough PES.
166
Acknowledgments
This work was supported by a grant from the DOE Environmental Science Management Program. All programs used for calculations reported in this work are part of the
TINKER molecular modeling package and are available via anonymous ftp from
dasher.wustl.edu or WWW from the site http://dasher.wustl.edu/tinker/. RVP would like
to thank Dr. Gerard T. Barkema for helpful discussions and Prof. Garland R. Marshall for
support.
167
Appendix A
In this section we list formulas for diffusional smoothing of DOPLS and MM2 potential function terms. The original undeformed functional forms are the initial conditions
for diffusion.
1. Bond Length and Bond Angle Terms.
For a one-dimensional harmonic potential of the form Kb(x - xo)2 the diffusion equa-
(
)
2
tion solution is of the form Kb x − x0 + 2KbDbondt.
2. Torsional Energy Terms.
Torsional energies are computed as a sum of one, two and three fold sinusoidal bar1
riers. The functional form is 2
∑V (1 + cos ( jω + ϕ)) , where Vj is the amplitude (onej
j
half the barrier height), φ is the phase factor which determines the location of the minima
and ω is the torsional angle. The diffusion equation solution for torsional potentials is of
1
the form 2
∑V (1 + cos ( jω + ϕ) exp( −D
j
torsion
j 2t )) . This is a solution to a diffusion
j
equation in torsional space in terms of the angular coordinates, ω.
In Γvt the torsional potential can be rewritten in terms of a 1-4 distance, r14, using the relation
2
2r − (A + B)
cos(ω) =
14
(A − B)
168
,
where A is the minimum value of the r14 distance at ω = 0 and B is the maximal distance
for ω = 180. This form is valid only when the 1-2 and 1-3 distances remain fixed. Substi1
tution into the original torsional potential, 2
∑V (1 + cos ( jω + ϕ)) , gives an expression
j
j
for the energy as a higher order polynomial in r14. The smoothing of polynomial functions results in smoothed functions that are polynomials of the same degree as the undeformed function, as is illustrated by the covalent terms discussed above. Similarly, the
smoothed forms for trigonometric functions are trigonometric functions scaled by a function of the smoothing parameter. Therefore, the qualitative nature of a smoothed 1-4 distance potential for the torsions is similar to smoothed form in terms of the torsions, provided the 1-4 distance potential is a good approximation to the Fourier series torsional potential.
In Γvc, however, the 1-4 distance potential becomes very complicated because the
change in 1-4 distance is coupled to changes in the 1-2 and 1-3 distances. It is not possible
to obtain a closed form for the undeformed potential in terms of the 1-4 distance. Therefore, we smooth the torsional potential in torsional space and scale the smoothed potential
relative to its distance space counterparts, i.e., the nonbonded electrostatic and van der
Waals terms, through a choice of diffusion coefficients.
3. van der Waals Potential.
A two Gaussian approximation to a 12-6 Lennard-Jones or exp-6 Buckingham function is written as a sum of two Gaussians centered about the origin. The form of these
169
Gaussians is Fvdw(rij,t=0) = aijexp(−bijrij2). The 3-dimensional distance space diffusion
equation in rij ∈ (0,∞) is of the form:
2
∂F
vdw(rij,t)
∂t
∂F
vdw(rij,t )
=D
{
vdw
∂r
2
2
+
rij
∂F
vdw(rij,t )
∂r
ij
}
ij
The solution is of the form:
n
gauss
Fvdw (r ,t) =
ij
4. Coulomb Potential.
a
∑ (1 + 4D
b r
ij
vdw
k= 1
For a Coulomb potential V(rij) =
t)
3/2
exp( −
2
ij ij
1 + 4D
vdw
t
)
1
as "initial condition" in 3-dimensions, the diffurij
sion equation solution is of the form:
1
F
erf (
(r ,t) =
charge ij
r
2D
ij
r
ij
charge
t
)
This formula, adapted from the work of Amara and Straub34 and Moré and Wu73,
satisfies the 3-dimensional distance space diffusion equation of the form:
2
∂F
charge(rij,t)
∂t
∂F
=D
{
charge
charge(rij,t )
∂r
2
ij
170
2
+
rij
∂F
charge (rij,t )
∂r
ij
}.
Appendix B
Many of the standard molecular mechanics potential energy functions such as
CHARMM, AMBER, or OPLS can be written in the general form:
Vtotal = V
bond
+V
angle
+V
torsion
+V
improper
+V
vdw
+V
charge
B1
In potential smoothing, each of these terms is transformed according to a diffusion
equation of the form
∂F
α
∂t
2
=
∂F
α
2
∂x
B2
which is the one-dimensional analog of the multi-dimensional diffusion operator. Here
Fα(x,0) = Vα(x) and α refers to one of the six terms in equation (B1). Using an equation
of the form (B2) with identical diffusion coefficients D for each of the energy functions is
equivalent to claiming that each of these functions have similar distance ranges and energy scales. Values for effective diffusion coefficients can be chosen to scale the relative
smoothing of each of the energy functions.
The bond term and the nonbonded van der Waals and Coulomb interactions are
written in terms of Cartesian distances between the atoms that are part of these interactions. The angle term and torsional terms can be written in terms of the corresponding angular coordinates. From the standpoint of a classical diffusion equation, each of the terms
represent different initial conditions for diffusion. The range of the potentials determine
the extent of diffusion space within which the solutions of equation (B2) need to be obtained. One way to impose different diffusion spaces would be to set appropriate inner
171
and outer boundary conditions for the different initial conditions in equation (B1). An alternative method is to compute effective diffusion coefficients for each of the terms in
(B1) that are determined by the range of the interaction potential. The effective coefficients may be scaled relative to the values for the van der Waals and Coulomb terms
which are set to unity since these nonbonded interactions are of longer range than the local geometry terms.
Effective diffusion coefficients can be computed by a simple comparison of the extents of diffusion spaces. We set D = 1 for the nonbonded terms which have the largest
diffusion extents in diffusion space. In descending order, the extent in distance space for
the torsions, angles and bonds follow the order of rnb > rtorsion > rangle > rbond. Accordingly, the diffusion of the geometry terms scaled relative to the nonbonded terms follows
the relation
Dgeom
Dnb
=
r2geom
r2nb
B3
We set Dnb = 1 and choose rnb and rgeom to correspond to typical upper limits in the
DOPLS forcefield for the nonbonded distance and individual geometry distances. One set
of choices for these values leads to the diffusion coefficients shown in Table I. It should
be stressed that this is purely an empirical choice, but a necessary one because it draws an
exact analogy with the mechanism of diffusion and provides a simple method for modulating the smoothing of diverse potential function terms within a molecular mechanics
formulation.
172
References
1.
Leach, A. R. Rev. Comput. Chem. 1991, 2, 1.
2.
Scheraga, H. A. Rev. Comput. Chem. 1992, 3, 73.
3.
Horst, R.; Tuy, H. J. Opt. Theor. Appl. 1987, 54, 253.
4.
Némethy, G.; Scheraga, H. A. Biopolymers 1965, 3, 155.
5.
Griewank, A. O. J. Opt. Theor. Appl. 1981, 34, 11.
6.
Törn, A.; Zilinskas, A. Global Optimization (Lecture Notes in Computer Science, Vol. 350), Springer-Verlag, Berlin, pp. 117-151, 1989.
7.
Kirkpatrick, S.; Gelatt, C. D.; Vecchi, M. P. Science 1983, 220, 671.
8.
Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E. J.
J. Chem. Phys. 1953, 21, 1087.
9.
Berg, B. A.; Neuhaus, T. Phys. Lett. B 1991, 267, 249.
10.
Li, Z.; Scheraga, H. A. Proc. Natl. Acad. Sci. USA 1987, 84, 6611.
11.
Wales, D. J.; Doye, J. P. K. J. Phys. Chem. A 1997, 101, 5111.
12.
Li, Z.; Laidig, K. E.; Daggett, V. J. Comput. Chem. 1998, 19, 60.
13.
Guarnieri, F.; Still, W. C., J. Comput. Chem. 1994, 15, 1302.
14.
Saunders, M. J. Comput. Chem. 1989, 10, 203.
15.
Brünger, A. T.; Kuriyan, J.; Karplus, M. Science 1987, 235, 458.
16.
van Laarhoven, P. J. M. and Aarts, E. H. L. Simulated Annealing: Theory and
Applications.Kluwer Academic Publishers, 1987.
173
17.
Straub, J. E., In Recent Developments in Theoretical Studies of Proteins, Ed.,
R. E. Elber, World Scientific, Singapore, 1996, 137.
18.
Wilson, S. R.; Cui, W. Biopolymers 1990, 29, 225.
19.
Brünger, A. T.; Rice, L. M. Methd. Enzymol. 1997, 277, 243.
20.
Ingber, L. Math. Comp. Model. 1989, 12, 967.
21.
Wang, Z.; Pachter, R. J. Comput. Chem. 1997, 18, 323.
22.
Tsallis, C. J. Stat. Phys. 1988, 52, 479.
23.
Tsallis, C.; Stariolo, D. A. Physica A 1996, 233, 395.
24.
Andricioaei, I.; Straub, J. E. Phys. Rev. E 1996, 53, R3055.
25.
Simon, I.; Némethy, G.; Scheraga, H. A. Macromolecules 1978, 11, 797.
26.
Pincus, M. R.; Klausner, R. D.; Scheraga, H. A. Proc. Natl. Acad. Sci. USA
1982, 79, 5107.
27.
Dammkoehler, R. A.; Karasek, S. F.; Shands, E. F. B.; Marshall, G. R. J.
Comput.-Aid. Mol. Design 1989, 3, 3.
28.
Merovitch, H.; Vasquez, M.; Scheraga, H. A. J. Chem. Phys. 1990, 92, 1248.
29.
Goldberg, D. E. Genetic Algorithms in Search, Optimization, and Machine
Learning, Addison-Wesley, Reading, MA, 1989.
30.
Hoare, M. R.; McInnes, J. A. Adv. Phys. 1983, 32, 791.
31.
Stillinger, F. H.; Weber, T. J. Stat. Phys. 1988, 52, 1429.
32.
Piela, L.; Kostrowicki, J.; Scheraga, H. A. J. Phys. Chem. 1989, 93, 3339.
33.
Ma, J.; Straub, J. E. J. Chem. Phys. 1993, 101, 533.
34.
Amara, P. and Straub, J. E., Phys. Rev., B. 1996, 53, 13857.
174
35.
Ma, J.; Hsu, D.; and Straub, J. E. J. Chem. Phys., 1993, 99, 4024.
36.
Shalloway, D. in Recent Advances in Global Optimization, Floudas, C. A., Pardalos, P. M., Eds., Princeton University Press, Princeton NJ, pp. 433, 1992.
37.
Church, B.; Oresic, M. and Shalloway, D. In Global Minimization of Nonconvex
Energy Functions: Molecular Conformation and Protein Folding (DIMACS Vol.
23), Ed., P. M. Pardalos, D. Shalloway and G. Xue, Am. Math. Soc., Providence,
1996, 41.
38.
Nakamura, S.; Hirose, H.; Ikeguchi, M.; Doi, J. J. Phys. Chem. 1995, 99, 8374.
39.
Ponder, J. W. TINKER: Software Tools for Molecular Design, Version 3.6,
Washington University School of Medicine, 1998.
40.
Weiner, S. J.; Kollman, P. A.; Case, D. A.; Singh, U. C.; Ghio, C.; Alagona, G.;
Profeta, S., Jr.; Weiner, P. J. Amer. Chem. Soc. 1984, 106, 765.
41.
Jorgensen, W. L.; Tirado-Rives, J. J. Amer. Chem. Soc. 1988, 110, 1657.
42.
Allinger, N. L. J. Amer. Chem. Soc. 1977, 99, 8127.
43.
Brooks, B. R.; Bruccoleri, R. E.; Olafson, B. D.; States, D. J.; Swaminathan, S.;
Karplus, M. J. Comput. Chem. 1983, 4, 187.
44.
Moré, J.; Wu, Z. In Global Minimization of Nonconvex Energy Functions: Molecular Conformation and Protein Folding (DIMACS Vol. 23), Ed., P. M. Pardalos, D. Shalloway and G. Xue, Am. Math. Soc., Providence, 1996, 151.
45.
Kostrowicki, J.; Piela, L.; Cherayil, B. J.; Scheraga, H. A. J. Phys. Chem. 1991,
95, 4113.
46.
Kostrowicki, J.; Scheraga, H. A. J. Phys. Chem. 1992, 96, 7442.
175
47.
Amara, P.; Straub, J. E. J. Phys. Chem. 1995, 99, 14840.
48.
Davidon, W. C. Math. Programming 1975, 9, 1.
49.
Ponder, J. W.; Richards, F. M. J. Comput. Chem. 1987, 8, 1016.
50.
Czerminski, R.; Elber, R. J. Chem. Phys. 1990, 92, 5580.
51.
Ulitsky, A.; Shalloway, D. J. Chem. Phys. 1997, 106, 10099.
52.
Cerjan, C. J.; Miller, W. H. J. Chem. Phys. 1981, 75, 2800.
53.
Barkema, G. T.; Mousseau, N. Phys. Rev. Lett. 1996, 77, 4358.
54.
Northby, J. A. J. Chem. Phys. 1987, 6, 599.
55.
Tsoo, C.; Brooks III, C. L. J. Chem. Phys. 1994, 101, 6405.
56.
Doye, J. P. K. and Wales, D. J.; Phys. Rev. Lett. 1998, 80, 1357.
57.
Pillardy, J. and Piela, L. J. Phys. Chem., 1995, 99, 11805.
58.
Zimmerman, S. S.; Pottle, M. S.; Némethy, G.; Scheraga, H. A. Macromolecules 1977, 10, 1.
59.
Hart, R. K.; Pappu, R. V.; Ponder, J. W. 1998, to be submitted for publication.
60.
Saunders, M.; Houk, K. N.; Wu, Y.-D.; Still, W. C.; Lipton, M.; Chang, G.;
Guida, W. C. J. Am. Chem. Soc. 1990, 112, 1419.
61.
Ngo, J. T.; Karplus, M. J. Am. Chem. Soc. 1997, 119, 5657.
62.
Chou, K. C.; Némethy, G.; Scheraga, H. A. J. Am. Chem. Soc. 1984, 106, 3161.
63.
Crick, F., Acta Cryst. 1953, 6, 689.
64.
Chothia, C.; Levitt, M.; Richardson, D. J. Mol. Biol. 1981, 145, 215.
65.
Walther, D.; Eisenhaber, F.; Argos, P. J. Mol. Biol. 1996, 255, 536.
66.
Goldstein, H. Classical Mechanics, Addison Wesley, Reading, MA,
176
Chapter 4 and Appendix B, 1980.
67.
Huber, T.; Torda, A. E.; van Gunsteren, W. F. J. Phys. Chem. A 1997, 101,
5926.
68.
Smellie, A.; Teig, S. L. and Towbin, P. J. Comput. Chem., 1995, 16, 17.
69.
Roitberg, A. and Elber, R. J. Chem. Phys., 1991, 95, 9277.
70.
Wawak, R. J.; Gibson, K. D.; Liwo, A. and Scheraga, H. A. Proc. Natl. Acad.
Sci., 1996, 93, 1743.
71.
Wawak, R. J.; Pillardy, J; Liwo, A.; Gibson, K. D. and Scheraga, H. A. J. Phys.
Chem. A 1998, 102, 2904.
72.
Pillardy, J. and Piela, L. J. Comput. Chem., 1997, 18, 2040.
73.
Moré, J.; Wu, Z. In Large Scale Optimization with Applications: Molecular
Structure and Optimization, Ed., L. T. Biegler, T. F. Coleman, A. R. Conn and F.
N. Santosa, Springer-Verlag, New York, 1997, 99.
177
Chapter 4:
Flexible Molecular Docking
with Potential Energy
Smoothing
178
Introduction
Virtually all cellular events are consequences of the mutual recognition of one molecule by another. The rapidly growing number of crystal, NMR, and homology-modeled
structures of potential therapeutic targets presents expanding opportunities for the development of engineered drugs which modify molecular interactions. The goal of molecular
docking is to identify the binding location and mode of a hypothetical or known ligand to
a target protein. In practice, molecular docking is used to bootstrap an iterative procedure
of lead compound generation, experimental testing, and refinement.
Some molecular interactions involve species which undergo only small conformational changes upon binding. In light of this observation and the computational intractability of widespread flexibility, it suffices for docking algorithims to treat the docking species as rigid bodies. Therefore, the conformational space available for molecular docking
consists of three translational and three rotational degrees of freedom between the rigid
bodies. However, in many cases there are large conformational changes which occur concommittantly with binding; predictions of associature in these systems will be difficult or
impossible for docking methods which ignore flexibility. Flexible docking may require
many additional degrees of freedom In either rigid or flexible docking cases, conformational space may be explored by any of a number of heuristic, stochastic, and deterministic methods [for reviews, see references 1, 2, 3, 4, 5, 6, and 3].
There are two predominant objectives of docking. In directed docking, a database of
potential ligands, or a ligand with known binding but unknown conformation, is
179
constrained to dock within a defined region. Target regions may be identified by biochemical data, chemical features, cavity identification algorithms8,9, or molecular graphics. In undirected (i.e., fully automated) docking, the entire inter-body conformational
space is accessible. This places greater demands on a docking algorithm to adequately
survey conformational space and on the scoring function to provide rapid and reliable affinity estimates.
Molecular docking methods use either geometric descriptors or energetic scoring
methods to search conformational space3. Geometry-based methods and conformational
hashing10,11,12 search for optimal matchings of shape descriptors, e.g., clefts and ridges.
Candidate matches are subsequently scored; see Oprea and Marshall13 for a recent review
of scoring functions for molecular docking. Geometric docking methods assume static
structures when generating molecular surface descriptors and, consequently, flexibility
concomitant with binding is not easily incorporated into these methods. However, success
has been reported by incorporating minimization14 or explicit torsional searching15 during refinement. In these cases, the scoring function must be soft enough to accept ligand
conformations for which flexible refinement will generate plausible structures, and not so
soft that unreasonable structures are retained. Energetic docking methods have the advantage that receptor or ligand flexibility may be readily accommodated, albeit with increased computational load16. However, the roughness of molecular potential energy
surfaces17 places exquisite demands on a search algorithm and energy function to sample
the vast space of conformations thoroughly and quickly2.
180
Despite continued successes of molecular docking, two important shortcomings exist. First, many geometric docking algorithms provide ligand flexibility as an auxiliary
refinement step after certain rigid-body criteria are accommodated. This is insufficient
for induced fit18 conformational changes, or for systems in which transient structural motions are required for binding. A second obstacle faced by all methods is the vastness of
conformational space and the attendant difficulty in achieving adequate sampling.
Potential smoothing methods transform an original energetic landscape into one
which may be progressively smoothed. As the extent of deformation is increased, the
shallower and narrower minima cluster into conformational basins while preserving the
dominant features of the surface. Potential smoothing techniques include Scheraga’s Diffusion Equation Method (DEM)3, Straub’s Gaussian Density Annealing13, and
Shalloway’s Gaussian Packet Annealing14 in probability space. We have recently shown
that potential smoothing methods are a deterministic analog of simulated annealing22.
In this report, we apply the Deformable OPLS (DOPLS) potential function22,15 to
molecular docking. We examine the performance of undirected docking of trypsinbenzamidine, and of directed and undirected flexible ligand docking of XK263 to HIV
protease. The methods used are generally applicable to systems which undergo conformational changes upon binding.
181
Methods
All calculations were performed using the TINKER modeling package version
3.726. The computation of non-bonded intergroup calculations was made more efficient
by binning atoms by group, thereby obviating checks for group membership for every
non-bonded atom pair. These modifications are provided on the TINKER web site.
Force Field and Parameterization. This investigation uses only van der Waals energy for docking. No restraining potential were employed. We use a Gaussian
approximation29 to the OPLS 12-6 Lennard-Jones potential as shown in equation 5.
ngauss
EvdW =
∑
i=1
(
( )
2
)
21/6
aiexp − bir2 , with ai = a°i ε0, bi = b°i
, ε0 =
r0
εa + εb , r0 = 2 ra + rb
1
where εx and rx are the Lennard-Jones parameters for atom x, < a°i ,b°i > are reference parameters chosen to fit a canonical Lennard-Jones function with well depth ε = 4.184
kJ/mol and a hard sphere radius σ=1Å; ngauss is the number of Gaussians used in the approximation. We set ngauss = 2, < a°1,b°1 > =<60614.0 kJ/mol, 905148 Å> and < a°2,b°2 > =<23.2353 kJ/mol, 1.22536 Å> which generates a very good fit over a wide range of interatomic distances29. Pairwise Gaussian parameters are determined by scaling the reference
parameters as shown in Equation 1.
The deformable Guassian van der Waals function used in this work is an analytical
solution to the diffusion equation for which equation 1 is the initial condition15,22
2
Evdw(r,t) =
∑
i =1
(
ai
−bir 2
exp
1 + 4Dvdwt
(1 + 4Dvdwt)3/2
182
)
2
where t is the deformation parameter that controls the extent of potential smoothing. For
consistency with previous work15,22, Dvdw is set to 1.
Potential Smoothing and Search (PSS) Algorithm. We use Potential Smoothing
and Search (PSS) described previously15. The salient features of this method and modifications for docking are presented below.
0. Select an initial conformation.
For iterations i=0..n of the PSS algorithm, do steps 1-4:
1. Set the extent of deformation, t = t(i) = tmax [(n-i)/n]q.
2. Minimize the system with respect to rigid-body and flexible
torsion coordinates.
3. Perform a Normal Mode Local Search15 (NMLS) in rigid body
coordinates.
4. Perform NMLS of the flexible torsions. This step was performed for HIV protease - XK263 only.
The choice of initial conformations varies for each docking study and is discussed
below. We use a cubic (q=3.0) backtracking schedule. A previous study22 indicates that
the rate of smoothing does not dramatically influence the results. The number of steps, n,
and maximum deformation, tmax, were varied for the problems discussed herein and are
reported in Results. Minimizations were performed using a variable metric minimization
method to an rms gradient of less than or equal to 10-2 kJ/mol/Å.
The NMLS procedure used in steps 3 and 4 is discussed in Chapter 3, but is outlined
here. At a minimum obtained in step 2 or from a previous iteration of NMLS, the hessian
183
of rigid body or torsional degrees of freedom is constructed. Diagonalization of this matrix produces a set of eigenvalue and eigenvector pairs. For the 12 rigid body coordinates,
there are 12 rigid-body eigenvectors, each of which is a mixture of rotation and translation of the rigid bodies. For the 10 rotatable torsions of XK263, there are 10 eigenvectors,
each of which is a mixture of torsional rotations of the napthalene, benzene, and hydroxyl
groups. A number of eigenvectors are chosen as search directions based on increasing
eigenvalue. Torsional NMLS was performed for 6 of 10 torsion eigenvectors for directed
and undirected docking with HIV protease-XK263. Rigid-body NMLS was performed in
all 12 rigid body directions for undirected docking experiments, and in 6 directions as described below for directed docking experiments.
For binding mode predictions of HIV-1 protease-XK263, the six diagonal translational elements of the rigid-body hessian in step 3 are set to a large value, 1016, which is
ten orders of magnitude larger than typical hessian elements. Upon diagonalization, six of
the eigenvectors are predominantly rigid body rotations. The effect of this modification is
to freeze the translations and permit rotations of each body about its center of mass. Note
that translations are still possible during minimization in step 2.
Preparation of Trypsin and Benzamidine Coordinates. Coordinates for trypsin
and benzamidine were extracted from 3PTB26 as deposited in the Protein Data Bank27.
The numbering scheme for benzamidine atoms follows that of Marquart et al.26 and is
shown in Figure 1. The all-atom OPLS27 van der Waals parameters are given in Table I.
The protonated state of benzamidine was used and charges were taken from the 6-31G**
ab initio calculations of Resat et al29.
184
Figure 1. Benzamidine. Atom numbering for C and N atoms is as in
reference 26.
14
13
5
12
18
9
6
1
4
3
7
2
11
17
8
15
16
10
Table I. Van der Waals parameters for benzamidine.
Atoms
σ (Å)
Partial Charge
ε (kJ/mol)
C1
+0.025
C2, C6
-0.162
C3, C5
-0.097
3.550
0.2920
C4
-0.048
C7
+0.737
N8, N9
-0.909
3.250
0.7110
H10, H14
+0.146
H11, H13
+0.160
2.420
0.1250
H12
+0.164
H15..H18
+0.462
1.000
0.0420
For preliminary trypsin-benzamidine docking studies, a sphere of 366 atoms within
10 Å of the benzamidine C7 atom constitute the binding target was used. Subsequent
studies used trypsin in its entirety. Initial conformations for PSS are generated by randomly rotating and translating benzamidine molecules by 10-20 Å relative to the bound
position in 3PTB.
185
Preparation of HIV Protease and XK263 Coordinates. HIV-1 protease and
XK263 coordinates were obtained from 1HVR30. The atom numbering for XK263 is
shown in Figure 2 and all atom OPLS van der Waals parameters are given in Table II.
Figure 2. XK263 inhibitor.
76
60
61
59
17
18
19
62
20
21
58
15
16
22
63
48
57
14
47
2
4
69
50
66
27
49
25
28
24
23
29
70
64
30
6
65
51
54 11
10
33
31
5
72
1
3
68
34
36
13
67
26
75
35
77
56
12
73
52
53
9
84
78
79
38
37
8
74
32
40
39
46
80
41
45
44
43
83
42
81
82
Table II. Van der Waals parameters for XK263.
Atoms
C1
O2
N3, N11
C4, C5, C6, C8, C10, C12, C23, C30
O7, O9
C13, C14, C15, C16, C17, C18, C19, C20,
C21, C22, C24, C25, C26, C27, C28, C29,
C31, C32, C33, C34, C35, C36, C37, C38,
C39, C40, C41, C42, C43, C44, C45, C46
H47, H48, H49, H50, H52, H54, H55, H56,
H64, H65, H71, H72
H51, H53
H57, H58, H59, H60, H61, H62, H63, H66,
H67, H68, H69, H70, H73, H74, H75, H76,
H77, H78, H79, H80, H81, H82, H83, H84
186
σ (Å)
2.1046
1.6612
1.8240
1.9643
1.7510
ε (kJ/mol)
0.4393
0.8786
0.7113
0.2761
0.7113
1.9924
0.2929
1.4031
0.1255
0.5612
0.0418
1.3582
0.1255
For undirected docking, the flaps of HIV protease (residues 46-54) of each monomer were excised. For directed docking, a protease cavity consisting of 1104 atoms within
10 Å of any XK263 atom was used.
187
Results
Undirected Docking of Trypsin - Benzamidine.
In order to establish the feasibility of potential smoothing for trypsin-benzamidine
docking, preliminary results were obtained by using a sphere of atoms around the binding
cavity. The sphere was defined as the 366 trypsin atoms within 10 Å of the benzamidine
C7 atom, as positioned in the crystal structure. The C7 atom is 3.88 Å from the Cγ atom
of D189 which lies at the end of the binding cavity.
Fifty initial conformations were generated by randomly rotating and translating benzamidine from its crystal structure position. Translations were limited to 10-20 Å because
the van der Waals potential is insufficient to attract benzamidine displaced more than 20
Å. Docking results for several values of PSS parameters are in Table III. Figure 3 shows
snapshots of structures during the reversal procedure and the crystal structure conformation. Because the effect of deforming the van der Waals potential is to increase rmin, the
first step of PSS causes the inhibitors to move away from trypsin. Figure 3b depicts the
inhibitors distributed roughly on the surface of a sphere approximately 30 Å from the surface of the cavity atoms.
The structure found by PSS is the same as that of the crystal structure after minimization with electrostatics. These structures appear in Figure 3g & h, The computed structure differs from the crystal structure by a 10° rotation of the benzamidine about the C1C7 carbon bond and a 0.10 Å increase in the C7-D189Cγ distance. In the most successful
docking experiment (n=10, 6 search directions), one of 50 benzamidine ligands docks
188
with the ammidinium group pointing out of the cavity, and minimization does not correct
this error. For clarity, that structure does not appear in Figure 3.
Table III. Success rate of benzamidine docking to trypsin.
10 Å Sphere (N=50)
number of search directions
tmax
number of smoothing steps
successes
average CPU time (min)*
* AlphaServer 2100 5/250
4
2.5
5
40
3.5
6
2.5
5
48
7.0
6
2.5
10
49
10.1
Entire
Trypsin
(N=15)
6
2.5
10
14
363
Results from docking to the 10 Å shell suggested that at least 6 search direction
would be required to achieve reliable undirected docking to intact trypsin. A 10-step
smoothing protocol was chosen to provide a more continuous reversal to the undeformed
surface. The results for intact trypsin docking are presented in Table III. In 14 of the 15
runs, structures identical to those for the 10 Å sphere were obtained. The structure which
failed to dock successfully minimized to a shallow pocket distant from the observed binding cavity and from which the NMLS method was unable to escape. This is an artifact of
the porous artificial molecular surface of the 10 Å shell. Using a slightly larger tmax is expected to cause all structures to converge to a single minimum, as opposed to the two seen
in these results, and all 50 structures should therefore backtrack to the same structure.
189
Figure 3a-h. Snapshots of 50 Trypsin-Benzamidine docking.
a) Starting conformations.
b) t=2.500. All inhibitors lie on a sphere approximately 30 Å from
the nearest cavity atoms.
190
c) t=1.024
d) t=0.600
e) t=0.156
f) t=0
g) after minimization with electrostatics
h) crystal structure
191
Undirected Docking of HIV-1 Protease and XK263
The HIV-1 protease - XK263 system was used to study undirected and directed
docking. In both cases, 10 rotational degrees of freedom in the ligand were permitted as
shown in Figure 4.
Figure 4. XK263 flexibility. The 10 regions of torsional flexibility
are denoted by arcs across the arms of the napthalene, benzene, and
hydroxyl groups.
O
N
N
HO
OH
The starting conformation for undirected molecular docking with HIV protease was
generated by manually translating XK263 20 Å from the bound conformation and minimizing on the t=2.0 surface. As with the benzamidine inhibitors, the increased rmin on deformed surfaces resulted in XK263 minimizing to a position 37 Å from the protease surface. This starting conformation is shown in Figure 5a.
The PSS algorithm was begun using tmax=2.0, n=100. At each level of the protocol,
all 12 rigid-body eigenvectors and 6 of 10 torsional eigenvectors were searched. Snapshots at several levels of the PSS procedure are shown in Figure 5. During PSS, the inhibitor sampled many orientations and locations, but always remained on the side of the
protein surface which is visible in Figure 5. For most of the precedure, XK263 was relatively rigid as a result of the linearization tendency of the van der Waals potential at large
192
deformations. Around t ≈ 0.2, XK263 began moving generally in the direction of the
binding cavity and the napthalene and benzene moieties began rotating freely.
The t=0 structure differs from the crystal structure by 12° and 15° at the proximal
and distal torsions of the napthalene groups respectively, and by 4° at the distal torsion of
the benzene groups. The differences between the PSS and crystal structures is symmetric,
as anticipated from the symmetry of the binding pocket. Furthermore, the t=0 structure is
identical to the minimized crystal structure, which indicates a minor discrepancy between
the crystal structure and AMBER/OPLS minimum. This exhaustive computation required
6 days of CPU time on a Digital Alpha Server 2100.
193
Figure 5a-h. HIV-1 protease - XK263 docking.
a) t=2.000
b) t=0.370
194
c) t=0.054
d) t=0.039
195
e) t=0.021
f) t=0.019
196
g) t=0
h) crystal structure
197
In an effort to reduce the computational load, a subsequent run with 20 smoothing
steps readily identified the binding pocket and correctly predicted the binding mode of the
cyclic urea body and benzene groups, but the napthalene rings were directed into the region which would have been occupied by the protease flaps. This run required 48 hours
of CPU time.
Directed Docking of HIV-1 Protease and XK263
For the directed docking procedure, XK263 was oriented "upside down" within the
binding cavity and manually positioned to minimize steric clashing with the protease.
This initial configuration is shown in Figure 6a. The binding cavity consisted of all protease atoms within 15 Å of any ligand atom in the 1HVR crystal structure. Because the
effect of deforming the van der Waals potential is to increase rmin, it was imperative to
use a small value of t in order to prevent the ligand from being ejected from the cavity
during minimization. PSS was initiated with tmax=0.03 and n=10. NMLS typically identifies rigid body eigenvectors which involve a combination of translation and rotation. In
order to keep NMLS from translating outside of the binding cavity, the diagonal elements
of the rigid body hessian which correspond to translation were set to a large value. Thus,
NMLS pursued 6 rigid-body rotation eigenvectors and 6 torsional eigenvectors in each direction. Some translation occurs during minimization.
As shown in Figure 6, PSS rapidly rights the cyclic urea body of XK263 within the
cavity. Subsequent steps are required to establish the correct orientation of the napthalene
and benzene moieties. The first iteration (t=0.03) of PSS centers XK263 within the cavity,
and the second iteration (t=0.022) inverts the inhibitor. The orientation and conformation
198
of the inhibitor in the final PSS and crystal structures differ insignificantly. The RMSD of
XK263 atoms within the aligned cavities of the final PSS and crystal structures is 1.27 Å.
Total computational time for the directed docking procedure was 10 hours.
In order to verify that this directed docking was sufficiently challenging, we minimized the starting conformation on the undeformed surface. The resulting structure remained inverted but was translated slightly toward the center of the cavity. This structure
closely resembles that from the first step of the PSS procedure in Figure 6a.
It was also possible that significant searching on the undeformed surface would result in the prediction of the correct binding mode. To study this, we initiated a one-step
PSS run on the undeformed surface. The first iteration of the NMLS procedure did not
find any alternate minima of lower energy and terminated.
More challenging directed docking experiments which used randomized initial configurations were unsuccessful. The primary reason for failure is that conformations of
XK263 which are significantly different than those seen in the crystal structure incur large
steric clashes. These clashes are resolved by ejecting the inhibitor from the binding cavity
during minimization. Once XK263 is outside the binding cavity, we have not observed
that NMLS is capable of threading the inhibitor through the small portals on either side of
the binding cavity. We are currently investigating methods to address this issue.
199
Figure 6a-f . Directed docking of XK263 into the active site of HIV1 protease.
a) starting conformation
b) t=0.030
200
c) t=0.022
d) t=0.015
201
e) t=0
f) crystal structure
202
Discussion
We have presented applications of potential smoothing to directed and undirected
molecular docking. To our knowledge, this is the first attempt to use potential smoothing
methods to this class of problems.
In this report, we have used only the van der Waals potential to guide docking procedures. The successes of geometric methods such as DOCK31 evince the importance of
good shape complementarity in molecular recognition. This is understandable in light of
the nature of van der Waals interactions: the energetic penalty for steric overlap is enormous and precludes all such structures, while close packing is modestly favorable.
The location, rmin, of the van der Waals energy minimum increases during smoothing and can be written as rmin = ln(
ak
bk
− bt1at1
. Con) where atk =
and btk =
3/2
bt2at2
(1 + 4t)
(1 + 4t)
sequently, the first iteration of the PSS procedure in the undirected docking experiments
caused the inhibitors to move significantly away from the protein. As the deformation was
reduced, the inhibitors were drawn back to the protein entirely by the shallow attractive
wall of the van der Waals functional form. No restraining terms were used in this study.
While molecular recognition is certainly not driven entirely by shape complementarity, the impressive results of geometric docking methods serves to emphasize the importance of van der Waals fit. This investigation clearly shows the power of potential
smoothing to analytically identify shape complementarity from the information which is
intrinsic to the van der Waals potential.
203
To illustrate this point, benzoic acid was created by manually editing the benzamidine coordinates and minimizing using MM332. Several randomly selected translations
and rotations of benzoic acid were used in PSS runs as above. In all cases, benzoic acid
was predicted to bind in the same orientation as benzamidine. The predicted binding
mode positions the electronegative carboxyl groups of benzoic acid and Asp 189 only 4Å
apart. It must be noted that benzoic acid is expected to be a very poor inhibitor and that
the docking prediction occurs because we have neglected electrostatic effects.
At the heart of molecular docking is the need for an efficient conformational search
method to find the best structures among the many possibilities. Many search procedures
are preoccupied with details of the surface in a small neighborhood near local minima and
do not recognize the larger scale features of the surface which are embedded in the functional form. The goal of search procedures is to rapidly survey conformations in a way
which tends away from unfavorable configurations and toward more promising regions of
conformational space.
Potential smoothing lessens the high frequency details of such landscapes while preserving the broadest and deepest features22. As a surface is smoothed, minima cluster into
basins. On moderately deformed surfaces, a single basin, may represent hundreds of
minima on the undeformed surface. Structurally, these basins represent the docking of
molecular surfaces which have been deformed by the increased van der Waals radii. In
other words, the details of the surface are blurred. Vakser33 has investigated the use of explicit feature blurring in an effort to reduced the effect of structural inaccuracies in molecular docking problems.
204
The protease-XK263 docking studies exemplify the ability of potential smoothing to
accommodate flexibility. A primary advantage of the method presented herein is the ease
with which conformational flexibility is permitted. We chose to excise the flap residues
46-54 in order to simplify the docking process. The present algorithm makes no distinction between the receptor and the ligand: the flaps could have been made flexible with additional computational expense. However, simulations with and without explicit water by
Harte et al.34 suggest that solvation plays an important role in flap motion. This observation has been corroborated by NMR35, which identifies strong correlations in the hydrogen exchange peaks between residues I50 and G51 of opposing flap strands, which led to
a proposal that these residues modulate flap dynamics.
The present implementation of PSS is slow for complicated systems and improvements in this regard are foreseeable. The effect of crossings22 in potential smoothing necessitates searching of some form, and the NMLS search procedure used herein is the
most time-consuming phase of PSS. At the start of every PSS iteration, a minimization is
performed. After diagonalizing the rigid body and flexible torsion hessian matrices, 16 or
22 eigenvectors are used as step vectors for NMLS (the number depends on the problem).
Each direction is search in positive and negative directions. If certain NMLS criteria are
achieved15, a minimization from a point along the search direction is begun. If any of the
minimized structures are lower in energy than the current minimum, the process is repeated. Thus, every level of the PSS procedure entails multiples of 20 or so minimizations.
205
Morris et al.36 recently reported good success in a range of docking problems by
combining the efficient search properties of Lamarckian genetic algorithms (LGAs) with
the simulated annealing. Among the docking cases examined was HIV-1 protease and
XK263, which they were able to repeatedly predict. Their prediction retained the flaps
which were excised in the present investigation. The speed of their method is attributed to
the efficiency of conformation search using a LGA and the use of grid-based energy computations. Gridded energy representations precompute van der Waals, electrostatic, and
other terms for each point in a mesh around the target protein. Such computations greatly
improve the performance of energy calculations but are only valid when the target molecule is rigid. Grid-based energy calculations could be applied to both of the docking systems in this report.
While simulated annealing lessens the barriers between minima, they nonetheless
exist at high temperatures and therefore impede conformational search. In contrast, potential smoothing analytically eliminates barriers, resulting in surfaces with many fewer
minima. Such surfaces are expected to be much easier to search, yet retain the broad scale
features of the original surface22. In principle, any conformational search method will be
more effective on a smooth surface than on an undeformed surface. For that reason, it is
expected that the best performance will result from combining potential smoothing with a
search method which is more efficient than NMLS, even if that method is heuristic or stochastic.
206
References
1
J. M. Blaney and J. S. Dixon, Persp. Drug Disc. Des., 1:301-19 (1993).
2
D. A. Gschwend, A. C. Good, and I. D. Kuntz, J. Mol. Recog., 9:175-86 (1996).
3
I. D. Kuntz, E. C. Meng, and B. K. Shoichet, Acc. Chem. Res., 27(5):117-23
(1994).
4
T. Lengauer and M. Rarey, Curr. Opin. Struct. Biol., 6:402-6 (1996).
5
T. P. Lybrand, Curr. Opin. Struct. Bio., 5:224-8 (1995).
6
R. Rosenfeld, S. Vajda, and C. DeLisi, Annu. Rev. Biophys. Biomol. Struct.,
24:677-700 (1995).
7
D. R. Westhead, D. E. Clark, and C. W. Murray, J. Comput. Aided Mol. Des.,
11:209-28 (1997).
8
C. M. W. Ho and G. R. Marshall, J. Comput. Aided Mol. Des., 4:337-54 (1990).
9
K. P. Peters, J. Fauck, and C. Frömmel, J. Mol. Biol., 256:201-12 (1996).
10
D. Fischer, S. L. Lin, H. L. Wolfson, and R. Nussinov, J. Mol. Biol., 248:459-77
(1995).
11
M. Rosen, S. L. Lin, H. L. Wolfson, and R. Nussinov, Prot. Eng., 11(4):263-77
(1998).
12
A. C. Wallace, N. Borkakoti, and J. M. Thornton, Prot. Sci., 6:2308-23 (1997).
13
T. I. Oprea and G. R. Marshall, Persp. in Drug Disc. and Des., 9/10/11:35-61
(1998).
14
S. Makino and I. D. Kuntz, J. Comput. Chem., 18(14):1812-25 (1997).
207
15
A. R. Leach and I. D. Kuntz, J. Comput. Chem., 13(6):730-48 (1992).
16
A Caflisch, S. Fischer, and M. Karplus, J. Comput. Chem., 18(6):723-43 (1997).
17
H. Frauenfelder and D. T. Leeson, Nature Struct. Bio., 5(9):757-60 (1998).
18
D. E. Koshland, Proc. Nat. Acad. Sci. USA, 44:98-104 (1958).
19
L. Piela, J. Kostrowicki, and Harold A. Scheraga, J. Phys. Chem., 93, 3339-46
(1989).
20
P. Amara and J. E. Straub, Phys. Rev. B, 53(20), 13857-63 (1996).
21
D. Shalloway, In Recent Advances in Global Optimization, C.A. Floudas and
P.M. Pardalos, Eds., Princeton University Press, Princeton, 1992, pp. 433-477.
22
R. K. Hart, R. V. Pappu, and J. W. Ponder, J. Comput. Chem., (to be submitted).
23
R.V. Pappu, R.K. Hart and J.W. Ponder, J. Phys. Chem. B, 102, 9725-42 (1998).
24
J.W. Ponder, TINKER: Software Tools for Molecular Design, version 3.7,
http://dasher.wustl.edu.
25
J. Kostrowicki, L. Piela, B. J. Cherayil, and Harold A. Scheraga, J. Phys. Chem.,
95, 4113-9 (1991).
26
M. Marquart, J. Walter, J. Deisenhofer, W. Bode, R. Huber, Acta Cryst.,
B39:480-90 (1983).
27
F. C. Bernstein, T. F. Koetzle, G. J. B. Williams, E. F. Meyer Jr., M. D. Brice, J.
R. Rodgers, O. Kennard, T. Shimanouchi, M. Tasumi, J. Mol. Biol., 112:535-42
(1977).
28
W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 110.6, 1657-66 (1988).
29
H. Resat, T. J. Marrone, and J. A. McCammon, Biophys. J., 72:522-32 (1997).
208
30
P. Lam, P. Jadhav, C. Eyermann, N. Hodge, Y. Ru, L. Bacheler, J. Meek, M.
Otto, M. Rayner, Y. Wong, C.-H. Chang, P. Weber, Science, 263:380 (1994).
31
I. D. Kuntz, J. M. Blaney, S. J. Oatley, R. Langridge, and T. E. Ferrin, J. Mol.
Biol., 161:269-88 (1982).
32
N. L. Allinger and Y. H. Yuh and J.-H. Lii, J. Am. Chem. Soc., 111:8551 (1989).
33
I. Vakser, Prot. Eng., 8(4):371-7 (1995).
34
W. E. Harte, S. Swaminathan, and D. L. Beveridge, Proteins, 13:175-94 (1992).
35
L. K. Nicholson, T. Yamazaki, D. A. Torchia, S. Grzesiek, A. Bax, S. J. Stahl, J.
D. Kaufman, P. T. Wingfield., P. Y. S. Lam, P. K. Jadhav, C. B. Hodge, P. J.
Domaille, and C-H. Chang, Nature Struct.. Biol., 2(4):274-80 (1995).
36
G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew,
and A. J. Olson, J. Comput. Chem., 19(14):1639-62 (1998).
209
Chapter 5:
Summary of the Results
210
The major contributions of this research are:
1. Development of the Deformable OPLS (DOPLS) potential
function;
2. Detailed characterization of potential smoothing for a peptide
system;
3. Correlation of potential smoothing and simulated annealing;
4. Applications of potential smoothing to molecular docking.
The DOPLS potential function is central to the investigations presented in this dissertation. A primary advantage of DOPLS is the ability to vary the level of detail on the
potential surface via a continuous parameter. The goal of potential smoothing is to focus
conformational search efforts on low resolution potential surfaces before proceeding to
increasingly detailed surfaces. Approximate solutions to a conformational search problem
are found on low resolution surfaces where the multiple minima problem has been significantly ameliorated. Because detail is continuously variable, these approximate solutions
may be refined by iterating the procedure on lesser deformed potential surfaces.
The analysis of potential smoothing applied to Capped DiAlanine Peptide (CDAP)
identified three processes which occur during deformation. These effects − shifting, merging, and crossing − depend primarily on features of the surface such as curvature. Crossings are particularly important because they explain the reduced efficacy of the Diffusion
Equation Method (DEM) for potential smoothing as a global optimization tool. As DEM
was originally conceived, minima on deformed surfaces would be related to those on undeformed surfaces by a straightforward "backtracking protocol". Crossings derail the
211
backtracking protocol. The Normal Mode Local Search (NMLS) was developed in order
to recover reliable global optimization. The intent was for NMLS to search for alternate
minima by following a few select directions from a minimum; in practice, choosing which
vectors to follow is difficult and, as a result, all directions are required for reliable performance. Although NMLS is exhaustive and time intensive, it has been successful in all
cases studied thus far. The combination of potential smoothing and local search is expected to scale better than trajectory-based search methods.
Perhaps the most striking result is the strong correlation between potential smoothing, a deterministic method, and simulated annealing, a stochastic method. Initially, this
was shown qualitatively by comparing the partitioning of CDAP minima by potential
smoothing on one hand and by energetic barriers on the other. In simulated annealing, the
likelihood of traversal between minima is determined by the height of the intervening barrier and the simulation temperature. Consequently, the qualitative similarity of partitioning suggested a correspondence of the extent of deformation and simulation temperature.
Subsequently, analogies to shifting, merging, and crossing effects were identified in simulated annealing and correlated quantitatively with potential smoothing. The implication of
this analysis is that potential smoothing is a deterministic analog of simulated annealing.
The application of potential smoothing to molecular docking is an important proof
of concept. The preliminary results presented herein demonstrate that flexibility is seamlessly accommodated by potential smoothing methods. It is important to recognize that
flexibility in potential smoothing methods is intrinsic to the method; it is not implemented
as a secondary search procedure after rigid body docking. While the performance of PSS
212
docking is uncompetitive when compared to other methods, the causes for the languid results have been identified. In principle, potential smoothing combined with other search
methods ought to be able to deliver outstanding efficiency.
It is stressed that potential smoothing and the method used to search conformational
space are distinct considerations. Deformed surfaces contain fewer minima and should be
conducive to conformational searching. Some hybrid search strategies couple efficient
heuristic or stochastic search methods such as Monte Carlo or genetic algorithms to simulated annealing. Because simulated annealing lessens the barriers between minima but
does not eliminate them, the multiple minima problem remains. It expected that potential
smoothing coupled with these search methods will provide significant benefits to the use
of potential smoothing as an optimization tool.
213
Glossary
214
AGDA − Adiabatic Gaussian Density Annealing. See GDA.
AMBER − Assisted Model Building with Energy Refinement; a common potential energy function for molecular modelling. See "A second generation force field for the
simulation of proteins and nucleic acids", Cornell, WD, Cieplak P, Bayly CI, Gould
IR, Merz KM Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW and Kollman
PA, J. Am. Chem. Soc. 117:5179-5197 (1995).
CDAP − Capped DiAlanine Peptide; chemical formula N-Acetyl-Ala-Ala-N-Methyl; a
small peptide used to investigate detailed changes on potential energy surfaces and
to make strong correlations between potential smoothing and simulated annealing.
See Chapter 2.
CHARMM − A common potential energy function for molecular modelling. See
"CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics
Calculations", B. R. Brooks, R. E.Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comp. Chem. 4:187-217 (1983).
CPU − Central Processing Unit
DEM − Diffusion Equation Method; one incarnation of function smoothing.
DOPLS − Deformable OPLS, a deformable version of OPLS obtained as discussed in
Chapter 2.
GDA − Gaussian Density Annealing, a potential smoothing method which treats each
particle as a point probability distribution.
GRMS − Gradient Root Mean Squared.
HIV − Human Immunodeficiency Virus.
215
IAN − Isobuturyl-(ala)3-NH-methyl, a small peptide used to invesitgate transitions between alpha helixand beta sheet conformations. See "Reaction Path Study of Conformational Transitions and Helix Formation in a Tretrapeptide", R. Czerminski and
R. Elber, Proc. Nat. Acad. Sci. USA, 86:6964-7 (1989).
LES − Local Enhanced Sampling, a sampling method which isolates small molecular
fragments for extensive conformational flexibility while freezing the remainder.
MCM − Monte Carlo with Minimization, a method which periodically minimizes structures derived from Monte Carlo sampling in order to map conformations to structural basins.
MDSA − Molecular Dynamics combined with Simulated Annealing.
NMLS − Normal Mode Local Search. See Chapter 3.
NMR − Nuclear Magnetic Resonance.
OPLS − Optimized Potentials for Liquid Simulations; a common potential energy function for molecular modelling. See "The OPLS Potential Functions for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin.", W. L. Jorgensen
and J. Tirado-Rives, J. Am. Chem. Soc. 110:1657-66 (1988).
PES − Potential Energy Surface.
PSS − Potential Smoothing with Search. See Chapter 3.
RMSD − Root Mean Squared Deviation.
SSA − Simulated Simulated Annealing. See Chapter 2.
TINKER − A package for molecular mechanics. See http://dasher.wustl.edu/tinker/.
TSBS − Transition State Based Search. See Chapter 3.
216