Atherosclerosis Supplements 12 (2011) 221–263
Familial hypercholesterolaemia: A model of care for Australasia
Gerald F. Watts a,∗ , David R Sullivan b , Nicola Poplawski c , Frank van Bockxmeer d ,
Ian Hamilton-Craig e , Peter M. Clifton f , Richard O’Brien g , Warrick Bishop h , Peter George i ,
Phillip J. Barter j , Timothy Bates a , John R. Burnett k , John Coakley l , Patricia Davidson m ,
Jon Emery n , Andrew Martin o , Waleed Farid p , Lucinda Freeman q , Elizabeth Geelhoed r ,
Amanda Juniper a,s , Alexa Kidd t , Karam Kostner u , Ines Krass v , Michael Livingston w ,
Suzy Maxwell s , Peter O’Leary s , Amal Owaimrin x , Trevor G. Redgrave a , Nicola Reid y ,
Lynda Southwell a , Graeme Suthers c , Andrew Tonkin z , Simon Towler aa , Ronald Trent q ,
Familial Hypercholesterolaemia Australasia Network Consensus Group
(Australian Atherosclerosis Society)1
a
Lipid Disorders Clinic, Metabolic Research Centre and Department of Internal Medicine, Royal Perth Hospital, School of Medicine and Pharmacology,
University of Western Australia, Perth, Western Australia, Australia
b Department of Biochemistry and Lipid Clinic, Royal Prince Alfred Hospital, University of Sydney, New South Wales, Australia
c South Australia Clinical Genetics Service, Genetics & Molecular Pathology Directorate, Women’s & Children’s Hospital, Adelaide,
South Australia, Australia
d Cardiovascular Genetics Laboratory, Royal Perth Hospital, University of Western Australia, Western Australia, Australia
e Preventive Cardiology and Lipid Clinic, Gold Coast Hospital, Griffith University, Queensland, Australia
f Baker IDI Heart and Diabetes Institute, Adelaide, South Australia, Australia
g Department of Medicine, Diabetes and Endocrinology, Austin Hospital, University of Melbourne, Victoria, Australia
h Department of Cardiology, Calvary Cardiac Centre, Calvary Health Care, Tasmania, Australia
i Biochemistry and Pathology, Canterbury Health Laboratories, Lipid Clinic, Christchurch Hospital, University of Otago, Christchurch, New Zealand
j Heart Research Institute, University of Sydney, Sydney, New South Wales, Australia
k Core Clinical Pathology & Biochemistry, PathWest Laboratory Medicine WA, Lipid Disorders Clinic, Royal Perth Hospital,
University of Western Australia, Western Australia, Australia
l Department of Paediatrics and Clinical Biochemistry, The Children’s Hospital Westmead, Sydney, New South Wales, Australia
m Cardiovascular and Chronic Care, Curtin University, and Nursing Research, St Vincent’s Hospital, Sydney, New South Wales, Australia
n School of Primary, Aboriginal and Rural Health Care, University of Western Australia, Western Australia, Australia
o Department of Paediatric and Adolescent Medicine, Princess Margaret Hospital, Perth, Western Australia, Australia
p FH Family Support Group of Western Australia, Perth, Western Australia, Australia
q Department of Molecular and Clinical Genetics, Royal Prince Alfred Hospital, University of Sydney, Australia
r School of Population Health, University of Western Australia, Western Australia, Australia
s Office of Population Health Genomics, Department of Health, Government of Western Australia, Australia
t Clinical Genetics, Canterbury Health Laboratories, Christchurch Hospital, New Zealand
u Cardiac Imaging Group, Department of Cardiology, Mater Hospital, University of Queensland, Australia
v Department of Pharmacy Practice, Faculty of Pharmacy, University of Sydney, New South Wales, Australia
w International Cholesterol Foundation, Sutton-Courtney, Oxfordshire, United Kingdom
x Department of Dietetics, Familial Hypercholesterolaemia Clinical Support Service, Auburn Hospital, Sydney, New South Wales, Australia
y Cardiovascular Prevention and Lipid Disorders Clinic, Christchurch Hospital, New Zealand
z Cardiovascular Research Unit, Monash University, Melbourne, Victoria, Australia
aa Health Networks, Department of Health, Government of Western Australia, Australia
∗ Corresponding author at: School of Medicine and Pharmacology, Royal Perth Hospital, University of Western Australia, GPO Box X2213, Perth,
Western Australia 6847, Australia. Tel.: +61 8 9224 0245.
E-mail address: gerald.watts@uwa.edu.au (G.F. Watts).
1 See Appendix 1.
1567-5688/$ – see front matter © 2011 Elsevier Ireland Ltd. All rights reserved.
doi:10.1016/j.atherosclerosissup.2011.06.001
222
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Abstract
Familial hypercholesterolaemia (FH) is a dominantly inherited disorder present from birth that causes marked elevation in plasma cholesterol
and premature coronary heart disease. There are at least 45,000 people with FH in Australia and New Zealand, but the vast majority remains
undetected and those diagnosed with the condition are inadequately treated.
To bridge this major gap in coronary prevention the FH Australasia Network (Australian Atherosclerosis Society) has developed a consensus
model of care (MoC) for FH. The MoC is based on clinical experience, expert opinion, published evidence and consultations with a wide
spectrum of stakeholders, and has been developed for use primarily by specialist centres intending starting a clinical service for FH. This MoC
aims to provide a standardised, high-quality and cost-effective system of care that is likely to have the highest impact on patient outcomes.
The MoC for FH is presented as a series of recommendations and algorithms focusing on the standards required for the detection, diagnosis,
assessment and management of FH in adults and children. The process involved in cascade screening and risk notification, the backbone
for detecting new cases of FH, is detailed. Guidance on treatment is based on risk stratifying patients, management of non-cholesterol risk
factors, safe and effective use of statins, and a rational approach to follow-up of patients. Clinical and laboratory recommendations are given
for genetic testing. An integrative system for providing best clinical care is described.
This MoC for FH is not prescriptive and needs to be complemented by good clinical judgment and adjusted for local needs and resources.
After initial implementation, the MoC will require critical evaluation, development and appropriate modification.
© 2011 Elsevier Ireland Ltd. All rights reserved.
Keywords: Familial hypercholesterolaemia; model of care; adults; children; adolescents; diagnosis; genetic testing; cascade screening; assessment; treatment
Contents
1.
2.
3.
4.
5.
6.
7.
8.
9.
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Summary of recommendations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Overview of algorithms for the model of care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Detection of index cases and diagnosis of FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Assessment of adult patients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Diagnosis and assessment of children and adolescents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Management of adults . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.1. LDL-cholesterol and apoB targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.2. Diet and lifestyle modifications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.3. Pharmacotherapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.3.1. Safety monitoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.3.2. Medication adherence and tolerance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.4. Review intervals and shared care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.5. Assessing atherosclerosis and CHD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
7.6. FH in women . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Management of children and adolescents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8.1. General and lifestyle considerations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8.2. LDL-cholesterol targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8.3. Pharmacotherapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8.4. Assessing atherosclerosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
8.5. Clinical monitoring and continuity of care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
LDL-apheresis and radical therapy for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
223
225
227
227
228
230
232
232
232
232
234
234
234
234
235
235
235
236
237
237
237
238
Abbreviations: ABI, ankle brachial index; ACE, angiotensin converting enzyme; ALT, alanine aminotransferase; ApoA-I, apolipoprotein A-I; apoB, apolipoprotein B; ARMS, amplification refractory mutation system; AST, aspartate aminotransferase; BMI, body mass index; CACS, Coronary Artery Calcium Score;
CCS, coronary calcium score; CHD, coronary heart disease; CIMT, carotid intima-medial thickness; CK, creatine kinase; CRP, C-reactive protein; CTCA,
computerised tomography coronary angiography; CUS, carotid ultrasonography; CVD, cardiovascular disease; DLCNS, Dutch Lipid Clinic Network Score;
EBESA, exon by exon sequence analysis; ECG, electrocardiography; EST, exercise stress test; FH, familial hypercholesterolaemia; FMD, flow-mediated dilatation; FDA, Food and Drug Administration; GP, general practitioner; HDL, high density lipoprotein; InterChol, International Cholesterol Foundation; LDL,
low density lipoprotein; LFT, liver function test; Lp(a), lipoprotein(a); MBS, Medicare Benefits Schedule; MEDPED, Make Early Diagnosis to Prevent Early
Deaths; MLPA, Multiplex Ligation Probe Amplification; MoC, model of care; NATA, National Association of Testing Authorities; NHMRC, National Health
and Medical Research Council; NPAAC, National Pathology Accreditation Advisory Council; PBS, Pharmaceutical Benefits Scheme; PCSK9, proprotein
convertase subtilisin/kexin type 9; PGD, pre-implantation genetic diagnosis; PND, prenatal diagnosis; RCPA, Royal College of Pathologists of Australasia;
SHAPE, Screening for Heart Attack Prevention and Education Task Force; TGA, Therapeutic Goods Administration; TSH, thyroid-stimulating hormone.
10.
11.
12.
13.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
223
9.1. Indications, patient selection and targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9.2. Methods for apheresis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9.3. Monitoring therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9.4. Cost considerations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9.5. Other therapeutic options . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Cascade screening: testing and risk notification of families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10.1. Risk notification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10.1.1. Contacting and informing families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10.2. Co-ordination of cascade screening . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10.3. Risk notification without consent . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10.4. Insurance cover and genetic testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Genetic testing of families . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Laboratory approach to genetic testing for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12.1. Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12.2. A protocol for genetic testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12.3. Assessing the significance of gene variants detected in index cases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
The web of care for FH: the optimal service model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13.1. Focus, aims and objectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13.2. Co-ordination and integration of care . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Inter-specialty links . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13.3. Administrative and information technology support . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13.4. Clinical governance: audit, education, training . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13.5. Patient and family support groups . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13.6. Into the future: chronic care model, commissioning, evaluation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 1. FH Australasia Network Consensus Group and Process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Chair . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Writing committee . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Steering committee . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Contributors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Consensus process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Funding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Disclosures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 2. Dutch Lipid Clinic Network Criteria for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 3. Simon Broome Criteria for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 4. MEDPED Criteria for FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 5. Typical examination features of FH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 6. Hypothetical Pedigree . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 7. ‘Real case’ Pedigree . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Appendix 8. Selected websites for clinical services . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
238
238
238
240
240
240
240
240
242
242
242
243
243
243
245
245
245
247
247
248
249
249
249
250
250
250
250
250
250
251
251
251
251
251
252
252
253
254
254
255
255
1. Introduction
Familial hypercholesterolaemia (FH) is the most common
and serious form of inherited hyperlipidaemia [1]. FH is due
to dominant mutations of genes predominantly affecting the
function of the low-density lipoprotein (LDL) receptor that
clears LDL particles from plasma [2,3], and hence results
in marked elevation in plasma LDL-cholesterol concentration. FH is present from birth and accelerates the onset of all
forms of atherosclerotic cardiovascular disease (CVD), especially coronary heart disease (CHD), by one to four decades
[1,4,5]. Opportunistic diagnosis of FH followed by screening of family members, the so-called cascade screening, can
detect individuals at an early stage of FH [4,6–11]. This
is critically important because it enables early intervention
including lifestyle measures, cholesterol-lowering medica-
tions (particularly statins), and management of other major
cardiovascular risk factors [12–17b]. Alarmingly, less than
10% of cases with FH have been diagnosed in most Western
communities, and only 5% are adequately treated [5,18–20];
Australia and New Zealand being no exception [21–23].
To address this demand in coronary prevention, the FH
Australasia Network established a Consensus Group to devise
a model of care (MoC) for FH from an Australian and New
Zealand (Australasian) perspective. In the present context, the
MoC was conceptualised as an overarching system, based on
theoretical, experiential and evidence-based standards, for
the provision of highest quality health care services for all
patients with FH [24].
This MoC aims to establish a standard of care for FH
patients in a framework within which future evidence and
consensus may be included and developed, thereby extending
224
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Table 1
Grades for recommendations employed for consensus statements.
Grade of recommendation
Description
A
Recommendation can be trusted to
guide practice
Recommendation can be trusted to
guide practice in most situations
Recommendations may be used to
guide practice, but care should be
taken in its application
B
C
These grades were applied to each of the recommendations in Section 2.
Individual members of the Steering Committee were asked to grade the recommendations based on their knowledge of the literature and what they
considered best practice in caring for patients with FH. Gradings were discussed and after full consensus of the committee was reached a final grade
was ascribed to each recommendation. All members of the FH Australasia
Network Consensus Group approved the final gradings.
other proposed clinical care programs from Europe and North
America [12,13,25–29]. The MoC is intended primarily for
lipid disorder clinics in tertiary centres intending to initiate or
develop a clinical service for FH. The MoC has been informed
by published research, clinical experience, expert opinion and
international guidelines for managing FH [5,12–16,30–34].
The major premises for these recommendations were published data on clinical efficacy and outcomes, but information
on cost-effectiveness was also employed where available.
Expert opinion was sourced from diverse stakeholders from
the disciplines of adult medicine, paediatric and adolescent
medicine, clinical genetics, clinical biochemistry, nursing,
pharmacy, general practice, population health, and health
economics; a patient support group was also consulted. The
MoC significantly extends and consolidates other Australian
recommendations on the detection and management of FH
[10,15,32,35–38].
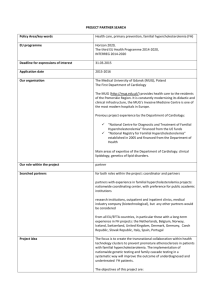
The MoC is presented as a series of recommendations
(Table 1, Section 2) and algorithms (Fig. 1) that if followed could provide a cost-effective, standardised system of
care likely to have the highest impact on patient outcomes.
The recommendations were graded according to a modified
NHMRC classification [39], and reflected the full consensus of an expert committee that was based on knowledge of
the relevant literature and best clinical practice. Algorithms
were chosen to provide easy visualisation of the concepts
underpinning the MoC. The algorithms are accompanied by
background information and explanatory text and reflect the
recommendations of the Consensus Group. This MoC should
not be perceived as prescriptive, but as a tool for guidance.
It should therefore be complemented by good clinical judgment and adjustments made to the model according to local
requirements, protocols and resources. Acknowledging the
lack of objective evidence supporting its clinical-efficacy and
cost-effectiveness, this MoC for FH should be viewed as an
evolving set of diagnostic and care pathways that will need
periodic review, clinical appraisal and modification.
Diagnostic criteria
Appendices 2, 3 & 4
Process
Figure 2
Optimal components
Figure 12
Adults
Figure 2 & 3
Laboratory protocols
Figures 10 & 11
Model of
Care for FH
Clinical protocol
Figure 9
Children/Adolescents
Figure 4
Adults
Figure 5
Children/Adolescents
Figure 6
Process
Figure 8
Fig. 1. Overview of algorithms for model of care for FH.
LDL-Apheresis
Figure 7
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
225
2. Summary of recommendations
Recommendation I
Models of care and components of service
Grade
Figs. 1 and 12
a. Models of care for familial hypercholesterolaemia (FH) should focus on detecting, diagnosing,
assessing and managing index cases, as well as on risk notification and cascade screening of family
members.
b. Adults and children/adolescents will require different models of care.
c. All services need to be integrated across several specialties and incorporated into primary care.
d. Good clinical governance, teaching and training programs, and family support groups are integral to all
models of care.
A
Recommendation II
Identifying index cases
Grade
Fig. 2
a. Index cases of FH should be sought amongst adults with premature cardiovascular disease in primary
and secondary care settings.
b. In adults a simple clinical tool based on the Dutch Lipid Clinic Network Score should be used.
c. All patients with possible-to-definite FH should be referred to a lipid disorders clinic for more detailed
assessment and institution of cascade screening.
A
A
A
A
A
A
Recommendation III
Clinical assessment and management allocation of adults
Grade
Fig. 3
a. Secondary causes of hypercholesterolaemia should first be excluded.
b. The diagnosis of FH should be made using both phenotypic and genetic testing.
c. Patients should be stratified into risk categories according to presence of cardiovascular risk factors
and personal history of cardiovascular disease.
d. Risk stratification should guide the intensity of medical management.
A
A
A
A
Recommendation IV
Clinical assessment and management allocation of children and adolescents
Grade
Fig. 4
a. Children (≥ 5 yr) and adolescents should be tested for FH after the diagnosis of FH has been made in a
parent.
b. Secondary causes of hypercholesterolaemia should first be excluded.
c. With rare exceptions, children and adolescents should only be genetically tested for FH after a
pathogenic variant (mutation) has been identified in a parent or first degree relative.
d. Age- and gender-specific plasma LDL-cholesterol concentration thresholds should be used to make the
phenotypic diagnosis of FH, an LDL-cholesterol ≥ 5.0 mmol/L indicating highly probable/ definite FH;
two fasting lipid profiles are recommended.
e. Patients should be stratified into risk categories according to age, presence of other cardiovascular risk
factors, prematurity of family history of cardiovascular disease and the level of hypercholesterolaemia at
diagnosis.
f. Risk stratification should guide the intensity of medical management.
A
Recommendation V
Management of FH in adults
Grade
Fig. 5
a. All adult patients with FH must receive advice on lifestyle modifications and all non-lipid risk factors
must be addressed.
b. Plasma LDL-cholesterol targets for routine, enhanced and intensive management should be
<4 mmol/L, <3 mmol/L, <2 mmol/L, respectively.
c. Achieving these targets will require a fat-modified diet, plant sterols (or stanols) and a statin with or
without ezetimibe.
d. Niacin, resins and a fibrate may be required with more intensive strategies.
e. Plasma levels of hepatic aminotransferases, creatine kinase and creatinine should be measured before
starting pharmacotherapy. All patients on pharmacotherapy, particularly statins, should have hepatic
aminotransferases monitored; creatine kinase should only be measured when musculoskeletal symptoms
are reported; creatinine should be monitored in those with kidney disease.
f. All women with FH of child-bearing age should have pre-pregnancy counselling
g. Statins and other systemically absorbed lipid regulating agents should be discontinued 3 months before
conception and during pregnancy and breast feeding in women with FH.
h. Non-invasive testing for coronary heart disease and atherosclerosis should be considered in patients
undergoing standard and enhanced treatment, with a step-up in treatment considered if there is evidence
of progression of disease. Non-invasive testing for atherosclerosis need not be carried out more
frequently than every two years.
i. Patients receiving standard or enhanced management should be reviewed every 6–12 months, and those
receiving intensive management should be reviewed according to clinical context, with appropriate
interval assessment of cardiac function and referral to cardiology.
A
A
A
B
A
A
C
A
A
A
A
A
C
B
226
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
(Continued)
Recommendation VI
Management of FH in children and adolescents
Grade
Fig. 6
a. Patients must receive advice on lifestyle modifications and non-lipid risk factors must be addressed.
Effective anti-smoking advice is mandatory.
b. Lowest risk patients should be treated expectantly with a fat-modified diet with or without plant sterols
(or stanols), with statins considered after the age of 10 years in boys and after the menarche in girls.
c. Plasma LDL-cholesterol targets for intermediate and high risk patients should be <4 mmol/L and
<3 mmol/L, respectively.
d. Reaching these targets requires a fat-modified diet, plant sterols (or stanols) and a statin with or
without ezetimibe or a bile acid sequestrant.
e. The preferred statins for initiating therapy are those that are licensed for clinical use in this age group;
in Australia these are pravastatin, fluvastatin or simvastatin, but other statins may be prescribed according
to clinical indications.
f. Weight, growth, physical and sexual development, and well-being should be reviewed regularly in all
patients.
g. Plasma levels of hepatic aminotransferases, creatine kinase and creatinine should be measured before
starting pharmacotherapy. All patients receiving statins should have hepatic aminotransferases
monitored; creatine kinase should be measured when musculoskeletal symptoms are reported; creatinine
should be monitored in those with kidney disease.
h. Carotid artery ultrasonography should be considered for assessing intima-medial thickness and
presence and progression of plaques; this may guide the intensity of medical management.
Ultrasonography need not be carried out more frequently than every 2 years.
i. Consideration should be given to managing children and adults with FH from the same family in a
family centred clinic
A
Recommendation VII
LDL-apheresis for FH
Grade
Fig. 7
a. LDL-apheresis should be considered in patients with homozygous or compound heterozygous FH.
b. LDL-apheresis should be considered in patients with heterozygous FH with documented coronary
heart disease who are refractory to or cannot tolerate cholesterol lowering medication.
c. LDL-apheresis should be considered in children with homozygous or compound heterozygous FH by
the age of 5 years, particularly if the plasma cholesterol concentration remains at 9 mmol/L or above on
medication.
d. LDL-apheresis should be carried out in close collaboration with a centre experienced in apheresis,
such as a transfusion medicine service.
e. The efficacy, tolerability and safety of LDL-apheresis must be reviewed after each treatment
f. The effect of LDL-apheresis on progression of atherosclerosis should be monitored with
echocardiography (aortic valve and root), carotid ultrasonography and/or exercise stress testing.
A
A
B
C
A
C
B
A
C
B
A
A
A
B
Recommendation VIII
Cascade screening: risk notification and genetic/phenotypic testing of families
Grade
Figs. 8 and 9
a. Notification of relatives at risk of FH should not be instituted without the consent of the index case,
with the exception noted below in recommendation c).
b. If no consent is given by the index case, rapport should continue to be built and consideration given to
referral for counselling.
c. Relatives should only be directly notified of their risk without consent of the index case if there is
specific legislative provision for this breach of confidentiality in the relevant jurisdiction.
d. Commonwealth legislation, local state legislation, NHMRC guidelines and local health service
protocols about disclosure of medical information without consent should be consulted.
e. A proactive approach that respects the principles of privacy and autonomy is required.
f. All material sent to relatives and the telephone approach should be clear, comprehensible and not cause
alarm. General and specific modes of communication should be used.
g. Cascade screening should ideally be carried out as a formal collaborative process between lipid
disorders and clinical genetics services. It should also involve close communication and liaison with
primary care physicians and employ a user-friendly family based data management system.
h. Pre-testing counselling should be offered to at risk family members of an index case prior to
phenotypic or genetic testing.
i. If no consent/assent for genetic testing is obtained phenotypic testing for FH should be offered.
j. If genetic testing detects the family mutation, a definitive diagnosis of FH can be made in the tested
individual particularly when the phenotype also suggests FH.
k. If genetic testing does not detect the family mutation, the diagnosis of FH can be excluded, except
when the clinical phenotype is highly suggestive of FH.
A
B
C
A
A
A
A
A
A
A
A
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
227
(Continued)
Recommendation IX
Genetic testing for FH: laboratory approach
Grade
Figs. 10 and 11
a. Genetic testing for FH should be offered to all ‘index cases’ who have a phenotypic diagnosis of FH
(e.g. by Dutch Lipid Clinic Network Score)
b. When the phenotypic diagnosis of FH is unlikely (e.g. by Dutch Lipid Clinic Network Score), genetic
testing of the ‘index case’ need not be carried out.
c. Genetic testing for FH must be carried out in an accredited laboratory.
d. When searching for a mutation in an ‘index case’, genetic testing may be carried out initially using
commercial chip or kit technology.
e. All abnormal genetic test results by chip or kit technology should be confirmed using a different
validated method.
f. When the phenotypic diagnosis of FH is definite or probable (e.g. by Dutch Lipid Clinic Network
Score), but no genetic variant is detected by methods that target specific mutations, comprehensive exon
by exon sequencing is recommended.
g. If genetic testing detects a variant the laboratory report should include an assessment of its significance,
and the report should clearly indicate if the variant is a pathogenic mutation, a previously reported variant
of uncertain significance, a novel variant of uncertain significance or a benign (normal) variant.
h. If the genetic testing protocol does not detect a mutation, the laboratory report should include a caveat
that the result does not exclude FH due to undetected mutations or mutations in untested genes,
particularly if the clinical phenotype is strongly suggestive of FH
A
C
A
B
A
A
A
A
The definitions of the grade for the recommendations are given in Table 1.
3. Overview of algorithms for the model of care
Fig. 1 provides an overview for the MoC for FH. The
sequence of presentation is as follows: detection of index
cases and clinical diagnostic criteria (Fig. 2; Appendices
2–4); diagnosis and assessment of adults, children and adolescents (Figs. 2–4); management of FH in adults, children
and adolescents (Figs. 5 and 6); LDL-apheresis and radical therapy for FH (Fig. 7); cascade screening, including
risk notification and predictive testing (Fig. 8); clinical and
laboratory protocols for genetic testing (Figs. 9–11); optimal components of a clinical service for FH (Fig. 12). The
MoC has initially been devised for use within a specialised
setting, such as a lipid clinic, run out of departments of internal medicine, cardiology or endocrinology in secondary or
tertiary referral centres. The MoC will need to be further
developed to consider current and future potential roles for
primary care providers.
4. Detection of index cases and diagnosis of FH
A key challenge facing the care of FH is the systematic detection of index cases [37,38,40]. The term ‘index
case’ refers to the first individual diagnosed with FH in the
family. Identifying index cases is important because it represents the starting point for family tracing, referred to as
‘cascade screening’, by which the majority of FH cases can
be efficiently detected [4,7–9,11]. There are several diagnostic tools for diagnosing FH clinically, including those from
the Dutch Lipid Clinic Network [34], Simon Broome Registry [12,41] and the US MEDPED Program [42] (see tables
in Appendices 2–4, respectively). There are, however, no
internationally agreed criteria for the phenotypic diagnosis
of FH [8,37,43]. We favour the Dutch Lipid Clinic Network Score (DLCNS) because we consider it simpler for
clinical use and the numerically integrated scoring system
[34,37], which does not fully rely on the plasma level of
LDL-cholesterol, can provide a more sensitive method for
detecting index cases with FH [8,44]. The Simon Broome
system is an alternative tool favoured in the UK [12] that
is comparable to the DLCNS in predicting an FH mutation,
[8] but could overlook patients with true FH who may not
be overtly hypercholesterolaemic. The MEDPED System,
which is based solely on plasma total and LDL-cholesterol
and has some practical appeal, is less sensitive than the other
criteria in predicting an FH mutation and accurate implementation requires that cholesterol measurements be widely
known in family members [42,45]. Illustrative examples of
the typical examination features (arcus cornealis, tendon
xanthomata) of FH are given in Appendix 5. Gender- and
age-specific thresholds for plasma LDL-cholesterol for diagnosing first-degree relatives with FH were recently published
[46], but their value in clinical practice has not yet been
reported. The phenotypic diagnosis of FH should be based
on at least two fasting measures of plasma LDL-cholesterol
[14,47–49]. The value of taking a family history of CHD is
well established [5,12,14–16,33,34,50], but its use is often
neglected in clinical practice [22,51] and this needs rectification [52–54]. Secondary causes of hypercholesterolaemia
(e.g. primary hypothyroidism, proteinuria, cholestasis, and
medications such as corticosteroids) must also be excluded
[14,15], but it is important to note that FH may co-exist with
other cardiovascular risk factors [55–57], most importantly
metabolic syndrome and diabetes [17a]. LDL-cholesterol
is underestimated by the Friedewald equation at plasma
triglycerides >4.5 mmol/L [47], above which a well validated,
direct assay should be employed to measure LDL-cholesterol
228
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[14,47]; measurement of apoB is an alternative [58,59], but
the diagnostic cut-off for diagnosing FH has not yet been
defined. Patients with FH do not, however, classically exhibit
significantly high plasma triglycerides concentrations.
Fig. 2 indicates that potential index cases of FH should
be sought amongst patients aged less than 60 years with
CVD presenting to coronary care, stroke, cardiothoracic
and vascular units [43], as well as amongst similar patients
attending cardiac rehabilitation programs. The greatest yield
will be from screening younger adult patients with CHD
[22,38,41,60]. Where the suspicion of FH is high, preliminary assessment of patients using either a full or modified
DLCNS should be employed [22,34]. A simplified modification of the DLCNS, suitable for use by non-specialists,
involves estimating a score for family and personal clinical history and LDL-cholesterol alone [22,37]; for patients
on statins an upwards adjustment of approximately 30% in
LDL-cholesterol could be employed [61] if the pre-treatment
LDL-cholesterol is unknown. The role of this simple tool
requires evaluation. A persuasive case for universal, as
opposed to selective, screening of children for hypercholesterolaemia has been made, since deficiency in obtaining a
family history of CHD and/or hypercholesterolaemia will
result in children not being tested and treated for FH [62].
It has been proposed that this could be done at immunization
using plasma cholesterol alone and subsequent child–parent
testing where indicated [63] or by universally screening children aged 9–11 years with a standard lipid profile [64a].
However, the acceptability, specificity and cost-effectiveness
of these universal screening strategies for FH are questionable
[4,65,66].
Opportunistic application of the DLCNS should also be
employed in specialist clinics and in primary care. General
practitioners (GPs) are usually the first to encounter individuals who may have unsuspected FH in the community,
and are therefore critically placed for detecting index cases
[12,67,68]. Planned health assessment in middle age, such
as the 45–49 year old health check assessment (Medicare
Benefits Schedule (MBS) Item A27) [69] would be an opportune time for GPs to routinely test for FH, but its uptake
and yield needs evaluation. Children with a positive family
history of hypercholesterolaemia or premature CHD should
be screened for FH [64a], but this is best done as part of a
co-ordinated cascade testing process [9,12]. In primary care
a retrospective search of clinical databases affords another
opportunity for generating new cases of FH [67]. Flagging
of laboratory reports on patients with plasma cholesterol
concentrations > 7 mmol/L is another method for alerting
practitioners about the possibility of FH, but its yield and
cost-effectiveness remain to be reported. There is also a role
for opportunistic application of the DLCNS by community
pharmacists to patients attending to collect a prescription for
a statin [70], with the possibility of on-site testing of relatives with a finger prick cholesterol measurement. Another
opportunity for detecting FH is amongst patients referred
to rheumatologists with tenosynovitis or to plastic surgeons
for the excision of tendon masses. Certain ethnic groups in
the community, such as Christian Lebanese, Afrikaaners and
Lithuanian Jews, in whom the prevalence of FH is particularly
high due to a gene founder effect [1], should also be targeted for screening for the condition; culturally appropriate
processes should be followed.
When the DLCNS is ≥3, we recommend referral to a specialised FH service or lipid clinic for confirmation of the
diagnosis and advice on management and cascade screening. Health providers working in these clinics should have
competence and training in both clinical lipidology and
prevention of CVD [26,71,72]. FH is unlikely when the
DLCNS is <3 [8,44]. This needs to be appropriately communicated to the patient and their GP. For patients on statins,
or other cholesterol lowering medications, a contemporary
plasma LDL-cholesterol concentration will give a falsely
low DLCNS and pre-treatment values must be obtained to
accurately assess the chance of having FH. Hence, we recommend that index cases of FH should be identified in a
two-stage process starting with initial phenotypic assessment
using the DLCNS followed by referral to a specialist FH
service. Employing a DLCNS as low as 3 as a criterion
for referring patients to a lipid clinic may be too sensitive
[8,38], in which case the score may be increased according
to workloads and resources. If resources allow, an alternative
approach would be to genetically screen for FH in all patients
with premature CHD (e.g. age < 50 years) presenting to the
cardiac services referred to in Fig. 2. All patients identified
as having a likelihood of FH should be provided with simple
and clearly written information explaining the importance
of FH and the steps involved in diagnosing the condition in
them and their relatives [8,12,37]. The recommended protocols for risk notification and cascade screening of relatives
and for genetic testing are discussed later and given in the
algorithms in Figs. 8–10.
5. Assessment of adult patients
Whether classified as having possible, probable or definite FH [34], all patients should have a detailed clinical
assessment to investigate other cardiovascular risk factors,
presence of symptomatic or subclinical atherosclerosis and
secondary causes of hypercholesterolaemia. Clinical assessment should ideally be undertaken by a specialist trained in
clinical lipidology [26,71], with skills in preventative cardiovascular medicine [72]. The risk of CVD amongst patients
with FH can vary widely [43,73]. This may relate to the
pre-treatment plasma level of cholesterol, genetic causes
affecting lipid metabolism or arterial biology, and the presence of other major cardiovascular risk factors, in particular
smoking, obesity, hypertension and diabetes [43,55–57,74].
Pathogenic genetic variants (mutations) that lead to very elevated plasma cholesterol and premature CHD may also be
considered a major risk factor [66], as should an elevation
in plasma lipoprotein(a) (Lp(a)) concentration > 0.5 g/L [75].
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
229
Potential sources for identifying index cases with FH:
Coronary Care, Cardiac Rehabilitation, Cardiothoracic Surgery, Stroke Unit, Vascular
Surgery, Ward nursing staff, Specialists, GPs, Laboratory report alerts, Pharmacists,
Clinical Genetics Services, Opportunistically in clinics/hospitals
High index of suspicion for new cases, Retrospective audit for previous cases, Use
simplified modification of phenotypic criteria for FH (Appendix 2 & 3)
Preliminary clinical assessment
Appendix 2: Dutch Lipid Clinic Network Score (DLCNS)
FH likely: DLCNS >3
FH unlikely: DLCNS <3
Review by specialist FH clinic
Inform patient and GP
DLCNS
≥3
<3
Follow-up in clinic
See Figure 3
Fig. 2. Detection of index cases and diagnosis of FH.
Box 1, linked to Fig. 3, gives mandatory, and recommended
(but optional) clinical indices that may be useful in assessing patients with FH. Non-invasive tests for atherosclerosis
and CHD should be considered optional and individualised
to specific clinical situations [76–78], being particularly useful in FH when the family history of CVD is unclear [79].
Carotid ultrasonography (CUS) can be a particularly useful test that recognizes that in FH atherosclerosis is also
accelerated in extra-cranial cerebral arteries and its detection serves as a surrogate for the involvement of coronary
arteries [80–82]. Clinical assessment must take account of
the psychological, intellectual, social and ethnic status of the
patient [8,83–86], and potential need for special methods of
counselling. Inadequate health literacy is not uncommon and
must be addressed [87,88]. Detailed exploration and clear
communication and discussion of the individual’s family history of CVD are essential for effective management of FH
[89,90]. After clinical assessment, patients should be divided
into those at lowest, intermediate and highest risk of CVD;
categories should be modified according to regular clinical
review (see Fig. 5).
This stratification of patients is consistent with other
approaches to cardiovascular risk assessment [14–17a]. At
lowest risk are patients with no other cardiovascular risk
factors (smoking, obesity, diabetes, and hypertension) and
negative tests for subclinical atherosclerosis. At intermediate risk are patients with at least one other cardiovascular
risk factor or subclinical evidence of early atherosclerosis. At
highest risk are patients with a history of symptomatic CVD
(coronary, cerebral or peripheral vascular disease) and/or
a revascularization procedure, or with subclinical evidence
of more advanced atherosclerosis. Subclinical atherosclerosis may be defined according to published guidelines
[77,78] – no evidence: a carotid intima-medial thickness
(CIMT) < 75th percentile (for age and sex) with no carotid
plaques or a Coronary Artery Calcium Score (CACS) of
zero; early evidence: a CIMT > 75th percentile with no
plaques (or stenosis) or a CACS ≥ 1 but <100; more advanced
evidence: presence of carotid plaques (or stenosis) or a
CACS > 100. If electing to do ankle brachial index (ABI),
exercise electrocardiography (ECG) testing and/or computerised tomography coronary angiography (CTCA) in
asymptomatic patients on clinical grounds, abnormal results
by recognised criteria [13,77,78,91] may also be employed
in allocating patients to the higher risk categories shown
in Fig. 3.
230
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
FH diagnosed
Clinical assessment
Lowest risk
No other major CVD risk
factors, no symptomatic or
subclinical CVD
Intermediate risk
>1 other major CVD risk
factors, early subclinical
CVD
Highest risk
Symptomatic CVD,
revascularization, advanced
subclinical CVD
Standard
management
Enhanced
management
Intensive
management
Box 1: Information used for clinical assessment of adults with FH
•
Mandatory: Age, gender, history: CVD (coronary, cerebral and peripheral arterial disease)/revascularisation, history of major
cardiovascular risk factors, psychological and socioeconomic status, family history of hypercholesterolaemia and CVD, BMI, waist
circumference, blood pressure, bruits, arcus cornealis, xanthelasma, tendon xanthomata, triglycerides, total cholesterol, HDL-C,
Lp(a), apoB, smoking status, menopausal status, reproductive status, drug history
•
Recommended/optional: glucose, insulin, CRP, creatinine, TSH, albuminuria, ApoA-I, ABI, EST, carotid and Achilles tendon
ultrasound, CTCA/calcium score
•
Major CVD Risk factors include very elevated LDL-C (>7 mmol/l), low HDL-C, diabetes, smoking, obesity, hypertension, high Lp(a),
family history of very premature CHD.
Fig. 3. Assessment of adult patients.
This subdivision into lowest, intermediate and highest risk
allows management to be classified as standard, enhanced
and intensive, respectively. This risk stratification procedure
will allow the best use of clinical resources, which can in
turn increase cost-effectiveness. Framingham risk scores, or
scores derived from other cardiovascular risk engines, are not
sufficiently reliable to guide management in FH [14,15,38],
particularly in younger patients, in whom a measure of longterm risk based on imaging of subclinical atherosclerosis
may be more appropriate [92]. Another option would be
to offer intensive management to intermediate and highest
risk cases of probable/definite FH. Similarly, enhanced management could be considered for the lowest risk cases with
probable/definite FH and for the possible cases of FH who
are at intermediate or highest risk of CVD. The yield and
cost-effectiveness of all these different therapeutic strategies
requires further evaluation. The importance of assessing other
cardiovascular risk factors beyond hypercholesterolaemia is
particularly underscored by the rising tide of obesity, type
2 diabetes and hypertension in our community [17a,93,94].
Patients considered to have a homozygous FH phenotype
should evidently be classified at exceptionally high risk
[43,95] and be referred for consideration for radical therapy
using LDL-apheresis (see Fig. 7).
6. Diagnosis and assessment of children and
adolescents
One of the most potentially contentious issues in the
care of FH is the identification of the condition in children
and adolescents [48,96–100]. Efforts are well justified by
clear indications that FH remains underdiagnosed particularly in the young [18,21,96] and that untreated patients suffer
cardiovascular damage from an early age [81,96,101,102].
Furthermore, there are reassuring data concerning the safety
and efficacy of dietary and pharmacological treatments
[99,103–108], including confirmation that children receiving
statins achieve normal growth and developmental milestones
and do not exhibit alterations in plasma adrenocortical hormone levels. While there have been concerns about the
psychological impact of ‘disease labelling’ from an early age
[8,84,85], formal studies provide reassurance that this is not
a significant issue [7,86,109]. Despite advances in the overall
care of patients with FH, medical services for children with
the condition remain particularly underdeveloped [110].
Fig. 4 summarizes the recommended approaches to
the diagnosis, assessment (Box 2), and risk stratification
allocation of FH in children and adolescents. We recommend
that FH screening be offered to all children (aged ≥ 5 years)
and adolescents at risk of having FH, particularly in the
context of a co-ordinated cascade testing process. Other
recommendations suggest screening at risk children earlier at
≥2 years of age [64a,b] and universally screening all children
for dyslipidaemias aged 9–11 years [64a]. Under the age of 5
years screening may be justified where there is a strong desire
from the parents or severe FH (homozygous or compound
heterozygous) is suspected, consistent with other guidelines
[12,98]. Because the DLCNS is not applicable to children,
the diagnosis of FH must rely either on serial measurements
of a fasting plasma level of LDL-cholesterol [46,48,97], or
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
231
Parent(s) diagnosed with FH
Pathogenic gene variant (mutation)
identified in parent(s)
LDL-C < 75 percentile
Boys <3.0 mmol/L
Girls <3.3 mmol/L
No
Consent / assent to genetic testing
LDL-C testing of child
Genetic + LDL-C testing of child
LDL-C >75 percentile
Boys >3.0 mmol/L
Girls >3.3 mmol/L
LDL-C <5.0 mmol/L
Pathogenic
mutation detected
No pathogenic
mutation detected
LDL-C >5.0 mmol/L
Reassure patient and
family and inform GP
Clinical assessment
Boys <10yr,
Girls before menarche
Boys >10yr,
Girls after menarche
1 or more of the following:
Early (M <50yr, F <40yr) CVD in family,
>2 CVD risk factors, diabetes, LDL-C >6.0 mmol/L
Lowest risk FH
Expectant
management
Intermediate risk FH
Enhanced
management
Highest risk FH
Intensive management
Box 2: Information used for clinical assessment of children and adolescents with FH
Mandatory: Age, gender, history including cardiovascular risk factors, psychological status, family history of hypercholesterolaemia and CVD, BMI, waist circumference, blood
pressure, bruits, arcus cornealis, xanthelasma, tendon xanthomata, triglycerides, total cholesterol, LDL-C, HDL-C, Lp(a), apoB, smoking status, reproductive status, drug history
Recommended/optional: glucose, insulin, CRP, creatinine, TSH, albuminuria, ApoA-I, CUS for early atherosclerosis, FMD of brachial artery for endothelial function.
Fig. 4. Diagnosis and assessment of children and adolescents. * See text for caveats.
preferably on genetic testing where a recognised mutation
for FH has been detected in a parent [7,8,66]. Secondary
causes of hypercholesterolaemia must be excluded [64a,b].
Irrespective of whether the diagnosis of FH will be made
genetically, plasma LDL-cholesterol must be measured in
all patients since the result is essential to guide therapy
[96–98]. Predictive genetic testing of children for FH is also
well justified as preventive treatment can be instituted before
adulthood with lifestyle measures and pharmacotherapy
[96–98,103,108] (see Fig. 6). Genetic counselling including
discussion of the implications of DNA testing in children
should be provided at the time the parent receives the genetic
results confirming the diagnosis of FH [8,85,111–115].
Because of ethical issues involved in genetically testing
minors [114,115], it is usual and best practice to first genetically test a phenotypically affected parent [64b]. This can
also circumvent issues related to a non-paternity event. In rare
circumstances, such as refusal of a parent to be tested first or
when autosomal recessive FH is suspected [3], genetic testing
may first be carried out in the child. Another special situation
may arise when the child has significant hypercholesterolaemia without the detection of the family’s pathogenic
mutation. In this case, after exclusion of secondary causes of
hypercholesterolaemia and appropriate genetic counselling,
other causative mutations for FH should be sought from the
extended pedigree. Detecting an FH causing mutation would
make the child or adolescent eligible for Pharmaceutical
Benefits Scheme (PBS) government subsidy for a statin at
an LDL-cholesterol > 4.0 mmol/L when the family history of
premature CVD or tendon xanthomata is unclear or unobtainable [116]. When an FH causing mutation is unknown in the
parent, the diagnosis of FH in children should be based on age
and sex adjusted LDL-cholesterol levels, the 95th percentile
being 3.5 mmol/L for boys and 3.8 mmol/L for girls [37,48]
(see Fig. 4). If employing the modified Simon Broome
Criteria, the universal cut-off for probable FH is LDLcholesterol > 4.0 mmol/L [12,41]. At least two consecutive
measurements of LDL-cholesterol over 6 months in fasting
samples should be used to make the phenotypic diagnosis of
FH [49,98]; a non-fasting lipid profile may be employed as
an initial screening test, however. Knowledge of the child’s
plasma LDL-cholesterol and whether a parent is being treated
with a statin may provide a simple clinical tool for diagnosing
FH [117]. Most children with plasma LDL-cholesterol >95th
percentile for age and sex and an autosomal dominant pattern
for inherited hypercholesterolaemia, in whom secondary
causes of dyslipidaemia have been excluded, will have an
FH causing mutation [118]. The thresholds for plasma LDLcholesterol concentration recommended above for making
a phenotypic diagnosis of FH are compatible with those
recently reported to accurately predict an FH causing mutation [46]. Whether using phenotypic or genetic approaches,
screening children for FH requires expertise in working
with and giving advice to families [8,84,98,119,120] and is
best undertaken in close liaison with paediatric services and
where indicated with genetic counsellors [115,121].
232
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
All individuals with at least a possible diagnosis of FH
should be clinically assessed according to mandatory and recommended (but optional) requirements shown in Fig. 4. With
few exceptions, these are generally similar to those recommended previously for adults. Of the available non-invasive
tests for subclinical atherosclerosis in children and adolescents, measurement of CIMT with ultrasonography is the
most promising at present, but it requires special expertise
and if used in risk stratification should be carried out according to recommended protocols [82,122]. With the exception
of homozygous or compound heterozygous FH, boys aged
less than 10 years and girls who have not reached the menarche should generally be considered to have low risk FH and
receive expectant treatment. Boys over the age of 10 years
and girls who have reached the menarche without cardiovascular risk factors or objective evidence of increased CIMT
should be considered to have moderate risk FH and receive
enhanced treatment. Boys over 10 years and girls who have
reached the menarche, with a family history of very premature CVD, two or more major cardiovascular risk factors, or
LDL-cholesterol > 6 mmol/L, should be considered to have
high risk FH and receive intensive treatment [12,96–98]. Certain high risk children and adolescents with FH should ideally
be managed in a joint adult-paediatric FH clinic [123]. Factors that may be used to triage patients for this clinic include
family dynamics, current lifestyle of the family and child,
adherence to treatment, severity of family history of CHD,
and presence of other major cardiovascular risk factors (obesity, hypertension, diabetes and smoking) [123,124]. While
all children with FH should ideally be referred to a paediatric
service, we consider it feasible for affected parents and children to be reviewed together by an adult service in a ‘family
clinic’ [123], provided staff have the required competencies and the environment of the clinic is appropriate. Young
FH patients being reviewed in a paediatric clinic should be
referred to an adult clinic around the age of 16 years, with
appropriate arrangements made for transitional care and with
close involvement of the GP.
7. Management of adults
Fig. 5 shows the protocols for adult patients with FH allocated to standard, enhanced and intensive management. In
parallel with lowering elevated plasma cholesterol, appropriate lifestyle modifications should be emphasised and all
major non-lipid cardiovascular risk factors must be treated
according to expert guidelines [14,16,17a,32,33,125–129];
offering advice and support on smoking cessation is mandatory. Low-dose aspirin should be used in highest risk FH and
considered in intermediate risk FH [128].
(apoB) are given in Fig. 5. These targets have been chosen
to be compatible with other therapeutic guidelines for the
management of hypercholesterolaemia [13–16,32,59]; therapeutic targets should evidently be lower with increasing
CVD risk. Measuring apoB may not, however, be necessary
in leaner FH patients with plasma triglyceride concentrations
<2.0 mmol/L. ApoB accurately reflects the plasma concentration of atherogenic LDL particles. ApoB is particularly
useful, and preferable to LDL-cholesterol, when LDL particle number increases and both size and density fall as a
consequence of hypertriglyceridaemia [58,59].
Hypertriglyceridaemia is a feature of obesity, metabolic
syndrome and type 2 diabetes [17a], all of which are increasing in prevalence in the background population and hence
amongst patients with FH [93,94]. Use of an apoB target
may be restricted to FH patients who exhibit an elevated
triglyceride level > 2.0 mmol/L [17a,58]. Even with contemporary treatments, achieving the absolute targets for
LDL-cholesterol and apoB shown in Fig. 5 may not be
attainable by some patients, particularly those with a higher
baseline plasma cholesterol [61], in which case a more realistic general target of a 40–50% reduction from pre-treatment
levels could be used [12,128].
7.2. Diet and lifestyle modifications
Diet and lifestyle modifications are cornerstones
of the management of all types of dyslipidaemias
[14–17a,32,33,59,130], including FH [13,131]. Diets should
be low in saturated fat and energy and adjusted to achieve
desirable body weight [15,132]. Dietary counselling by a
registered dietician is recommended for all affected individuals and families [133]. Dietary supplementation with
plant sterols or stanols should be considered [134]. Moderate intensity aerobic exercise for at least 30 min on 5 days
of the week should be considered to prevent obesity and
diabetes, with advice appropriately adjusted in those with
established CHD [32]. Several strategies may be employed,
where indicated and feasible, for improving long-term adherence to dietary and life-style changes, including involving
the patient, setting goals, encouraging self-monitoring, frequent and prolonged contact, and motivational interviewing
[135]. However, almost all patients will require medication
to lower the elevation in LDL-cholesterol. Offering effective
treatments and advice on smoking cessation is mandatory
and appropriate management guidelines should be followed
[32]. Alcohol consumption should be limited to no more than
2 standard drinks per day. Stress, anxiety and depression
must be considered in all patients and managed accordingly
[32,136].
7.3. Pharmacotherapy
7.1. LDL-cholesterol and apoB targets
The recommended therapeutic targets for absolute plasma
concentrations of LDL-cholesterol and apolipoprotein B
In Australia, the PBS eligibility criteria for government
subsidy for lipid modifying agents cover almost all adult
patients with FH, particularly if the molecular diagnosis of
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
233
Provide and reinforce information on:
Healthy diet, tobacco avoidance, exercise, family and psychological support and medication adherence.
Treat non-lipid risk factors:
Diabetes, hypertension, obesity, smoking, consider aspirin
Adherence,
tolerability +
safety checks
Standard
Enhanced
Intensive
Review interval: 12 months
Review interval: 3-6 months
Review interval: as per
clinical context
Diet + Plant
Sterols + Statin
± Ezetimibe
Adherence,
tolerability +
safety checks
Consider interval noninvasive testing for
coronary artery disease
/ atherosclerosis
Negative result
Positive result
Diet + Plant
Sterols + Statin
± Ezetimibe
± Niacin
Adherence,
tolerability +
safety checks
Consider interval test
of cardiac function:
exercise ECG / Stress
echocardiogram /
myocardial perfusion
scan
Consider interval noninvasive testing for
coronary artery disease
/ atherosclerosis
Negative or
stable result
Positive result
or progression
of disease
Diet + Plant
Sterols + Statin
± Ezetimibe
± Niacin ± Resin
± Fenofibrate
Negative or
stable result
Clinically
significant
positive result
Fig. 5. Management of Adults (see text for more information on action following initiation of therapy, treatment of special groups, shared care with GP and
radical therapy).
FH has been confirmed with a DNA test [116]. In New
Zealand government subsidies are similar, but the spectrum of
drugs is slightly more restricted [137]. HMG CoA reductase
inhibitors (or statins) are by far the most common and effective drugs to treat FH [104,107,128,129,138–146]. Statins
decrease the incidence of CHD [143–145] and are estimated
to be cost-effective in treating FH [147,148]. Reduction in
the costs of statins will, however, make the medical care
for FH even more cost-effective in the future. All the major
statins are government subsidised in Australia, but only
atorvastatin, simvastatin and pravastatin are subsidised in
New Zealand. After initiation of statin therapy, all patients
should be reviewed at 6–8 weeks to monitor LDL-cholesterol
response, adherence, safety parameters and tolerability [61],
and 6–12 monthly thereafter if targets are achieved and no
problems documented. The statin should be up-titrated to
the maximally recommended tolerable dose that achieves
the therapeutic targets shown in Fig. 5; patients may require
switching to more potent statins [61], such as atorvastatin or
rosuvastatin. Higher risk patients who require greater lowering of plasma LDL-cholesterol and apoB will require other
drugs, especially ezetimibe [149,150], but also niacin, fenofibrate and bile acid binding resins [151–154]. Patients with
higher plasma LDL-cholesterol levels will require a combi-
nation of drugs to achieve therapeutic targets. Combination
drug regimens that target LDL-cholesterol can decrease progression of CHD in patients with FH [155]. Niacin may be
particularly indicated for lowering high plasma Lp(a) concentration [75,151,152]; in FH we recommend an Lp(a) < 0.5 g/L
[75]. Fibrates would be relatively contraindicated with a
history of untreated choleliathisis [156]. For patients not
achieving treatment targets, additional agents should generally only be introduced after at least 12 months of testing
and adjusting the statin regimen and confirming adherence to
medication. In higher risk patients, additional agents should
be considered after an earlier interval (e.g. 4–6 months) of
testing the statin regimen. Residual hypertriglyceridaemia
and low HDL-cholesterol (the so-called ‘atherogenic lipid
profile’) while on a statin is an indication for considering treatment with niacin, a fibrate or higher doses of
supplemental omega-3 fatty acid ethyl esters [152,157].
Hypertriglyceridaemia in FH patients should be managed
according to recently published guidelines [158,159]. There
is outcome evidence supporting use of lower doses of omega3 fatty acids in patients who have sustained a myocardial
infarction [160]. Niacin, fibrates and omega-3 fatty acid
ethyl esters, together with a very low fat diet, will also be
indicated in rare instances of severe hypertriglyceridaemia
234
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
to prevent acute pancreatitis [14,157,161]. There has been
renewed interest from Japan in the therapeutic role of probucol [162], a drug formerly used to treat FH and withdrawn
from the European and US markets because of safety concerns; use of the present formulation of this drug cannot be
recommended.
7.3.1. Safety monitoring
Plasma hepatic aminotransferases (ALT, AST), creatine
kinase (CK) and creatinine levels should be measured routinely as baseline safety checks prior to starting medications
[61,163]. Hepatic aminotransferases should be monitored
according to the approved product information for the drugs,
and checked at least every 3 months if there is a history of
liver disease (e.g. chronic hepatitis or cirrhosis) or more frequently if plasma levels rise to three-fold greater than the
upper reference limit; measurement of serum bilirubin may
also be used to indicate the severity of liver toxicity. Plasma
CK should be measured when musculoskeletal symptoms are
reported. Particular vigilance is required in patients receiving higher doses of a statin, and patients predisposed to statin
side-effects, specifically the elderly and those taking multiple medications including the combination of a statin with
a fibrate [163,164]. Patients on niacin also require monitoring of plasma glucose and uric acid because of a small, but
significantly increased risk of hyperglycaemia and hyperuricaemia [165]. Plasma ALT and AST should be measured
about every 6 months in patients receiving statin–niacin and
statin–fibrate combinations. Evidence of chronic kidney disease, as estimated by an elevation in plasma creatinine and
fall in estimated glomerular filtration rate, would be a precautionary indication to initiate treatment with, or switch to, a
statin that is not eliminated by the kidney [61]. Statins should
not be initiated if the baseline plasma, ALT, AST or CK levels are >3 times the upper reference limit. Discontinuation of
the statin or revision of the dose or regimen is required when
the plasma aminotransferase or CK levels rise to >3 times
the upper reference limit on treatment. Alternative agents
such as a ezetimibe or bile acid sequestrants may need to be
substituted for a statin.
7.3.2. Medication adherence and tolerance
Pharmacists could play a key role by monitoring patients’
use of therapy, flagging non-adherent patients to GPs
and clinics, reducing therapeutic complexity and by more
direct involvement in improving adherence to medication
[166–169]. FH patients who are non-adherent to therapy are
best reviewed in a dedicated clinic that could involve input
from pharmacy, clinical pharmacology, psychology and nursing [170]. Action plan interventions may be more effective
in FH than interventions aimed at altering perceptions about
taking statins [171]. Detailed communication and discussion of an individual’s family history of CVD may improve
adherence to treatment [89,90]. Health literacy must also
be considered [172,173]. The underlying causes of hypercholesterolaemia and adherence to statin therapy remains a
significant issue amongst many at risk patients who have not
specifically been diagnosed with FH [174,175]. This also
needs addressing at a primary care level in the community
[176–178]. Poor control of hypercholesterolaemia and other
risk factors for atherosclerosis is a particular on-going concern in all patients with CHD [179,180].
Patients who are intolerant of medications require special
support and follow-up [170]. Musculoskeletal side-effects
can be frequently reported with statins and require specialist care [170,181]. Potential drug interactions with statins
should be closely monitored, [163] noting the increased risk
with drugs that are metabolised by CYP3A4 with simvastatin and atorvastatin and by CYP2C9 with rosuvastatin and
fluvastatin [61,163]. Ezetimibe is well tolerated and has a
statin dose sparing effect [149,150]. Bile acid binding resins
have frequent gastrointestinal side-effects (e.g. constipation
and abdominal discomfort) and can affect the absorption of
other drugs and fat soluble vitamins [153]; tolerability is
greatest with colesevelam [182]. Resins should be taken with
meals and gastrointestinal side-effects minimised by increasing fluid and fibre intake and use of stool softeners [153].
Flushing is a problem with niacin, but this is diminished with
the newer formulations (Niacin-ER, Tredaptive) and with coadministration of aspirin [151,152]; hyperglycaemia can be
a particular problem in FH patients with impaired glucose
tolerance or diabetes, and hyperuricaemia in those with a
history of gout [165]. Co-administration of fibrates and a
statin can increase the risk of myopathy, but this is diminished significantly when a statin is combined with fenofibrate
[156,163].
7.4. Review intervals and shared care
Uncomplicated patients on routine therapy may be
referred back to the GP for long-term follow-up, but should
also be reviewed annually in a lipid disorders clinic. Those
receiving enhanced care should also be monitored in the lipid
clinic at 3–6 monthly intervals until plasma LDL-cholesterol
targets are achieved and if stable should be reviewed annually
in this clinic with 6 monthly follow-up by the GP. Patients
receiving intensive management should be retained in the
lipid disorders clinic and reviewed at intervals determined by
clinical context and requirements. When changing medication and increasing doses, patients may need more frequent
review. Systems for the shared care of FH patients between
specialties and primary care need to be further investigated
in order to appropriately inform future MoCs for FH [68].
7.5. Assessing atherosclerosis and CHD
CTCA, CUS, ankle-brachial index measurement, stress
echocardiography, treadmill ECG and myocardial nuclear
perfusion scanning are all potential options for monitoring subclinical atherosclerosis and/or symptomatic coronary
disease [76–78] (see Fig. 5). Consistent with expert recommendations [76–78,91], we consider that there is potential
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
value in non-invasive testing in asymptomatic FH patients for
atherosclerosis and coronary disease. However, non-invasive
testing has been better validated for assessing target organ
damage and stratifying risk than for monitoring progression
of disease [13,79]. Nevertheless, clear evidence of progression of atherosclerosis in coronary, carotid or femoral arteries
should be a clinical indication for intensifying treatment and
attaining the recommended targets for LDL-cholesterol and
apoB [13,77,183]. The intervals for repeat testing shown
should be determined by clinical judgment and available
resources. With CUS, the choice of equipment, technical
aspects and operator training must be followed according
to expert recommendations [78,82,122]; imaging protocols
must be comprehensive and standardised, and age- and sexspecific reference values for CIMT and a detailed assessment
of plaques should be employed. CUS is better suited for children and younger patients owing to absence of radiation risk,
while CTCA may be useful to assess plaque burden and
obstructive stenosis in asymptomatic adult patients. Future
developments and refinements in cardiac CT imaging may
allow this imaging modality to be fully incorporated into
clinical algorithms for assessing atherosclerosis and CHD in
FH. All asymptomatic patients should proceed to a functional
test, ideally a stress echocardiogram. All ‘positive’ treadmill
tests, myocardial perfusion scans and stress echocardiograms
should be followed by referral to a cardiologist for the
consideration of invasive coronary angiography and appropriate further care [13,78,110]. Patients with more than 70%
stenosis of the internal carotid artery should be referred for
consideration for revascularization [184,185].
235
in offspring who inherit the condition [189,191]. Bile acid
binding resins are the only safe agents to control hypercholesterolaemia in pregnancy, but only modestly lower plasma
LDL-cholesterol levels and poor tolerability related to gastrointestinal side-effects remains a major problem [129,153].
A newer formulation, colesevelam, is more tolerable than
the older resins [182]. Pregnant women with heterozygous
FH and established CHD, or with homozygous FH, should
be considered for LDL-apheresis [95,129]. During breast
feeding, resins could be employed to lower LDL-cholesterol
where indicated. More data are required on the outcomes of
pregnancy in women with FH and on the effect of statins
on the foetus in the first trimester; an appropriate registry of
patients is recommended.
Particular considerations are also required concerning
future pregnancy when one or both members of a couple
have FH. In these circumstances, there are a number of
options available which enable the couple to avoid having
a child affected by FH. These options include not conceiving, adoption (local and overseas), using donor gametes,
prenatal diagnosis (PND) using chorionic villus sampling or
amniocentesis and pre-implantation genetic diagnosis (PGD)
[192,193]. There are a number of issues associated which
each of these choices and couples may find it useful to meet
with a counsellor who has expertise in reproductive counselling. Couples considering PND or PGD should be referred
to a clinical genetics service for counselling and pre-test work
up, ideally prior to conceiving.
8. Management of children and adolescents
7.6. FH in women
Special considerations apply to the management of FH
in women [43,186]. CHD risk is lower in women than men
with FH [43,73,187]. Women may therefore be less likely
to be treated with a statin at a younger age, but this needs
revision according to a detailed family history of premature
CVD, the presence of other cardiovascular risk factors and
the rate of progression of CIMT [43,55–57,74,75,188]. Low
estrogen-containing oral agents, intra-uterine devices and
barrier methods are the preferred approaches to contraception in women with FH [186]. The latter two options should
be particularly recommended to women older than 35 years
who are of childbearing potential. Pre-pregnancy counselling
is recommended for all women [66,129,186,187]. Statins
and other systemically absorbed lipid regulating medications
should be discontinued 3 months prior to planned conception
and during pregnancy and lactation [186]. However, women
who fall pregnant accidentally while taking a statin could be
re-assured that the likelihood of any foetal complications is
small [189,190]. The risks for future pregnancy and the foetus should be discussed at least annually with all women and
girls of childbearing age. Controlling hypercholesterolaemia
during pregnancy is particularly important in women with
established CHD, and it may also decrease the severity of FH
Fig. 6 summarizes the management of FH in children and
adolescents.
8.1. General and lifestyle considerations
All patients, and ideally all immediate family members, should receive expert advice on lifestyle modifications
including healthy eating, regular exercise and avoidance
of cigarette smoking [12,97,103]. Prevention of obesity,
metabolic syndrome and diabetes is paramount in FH
[97]. Psychological counselling and social support may be
required in special circumstances in certain families with
FH [8,84,124]. Parents of affected children must not smoke.
Non-cholesterol cardiovascular risk factors such as diabetes,
hypertension and obesity should be treated according to relevant expert guidelines [97]. There is clear value in reviewing
patients in a paediatric clinic, but we recommend that the
paediatrician has some expertise in clinical lipidology and
in the prevention of CVD [72]. A good, practical alternative
is a family based clinic in which affected children, adolescents and their parents can be reviewed collectively by a
multidisciplinary team [123].
Lowest risk FH should be treated with a fat modified
diet and plant sterol supplementation [97,103,134,194],
236
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Provide and reinforce information on:
Healthy diet, tobacco avoidance, exercise, family and psychological support and medication adherence.
Treat non-lipid risk factors:
Diabetes, hypertension, obesity, smoking
Expectant management
Enhanced management
Intensive management
Target LDL-C
<4.0 mmol/L
Target LDL-C
<3.0 mmol/L
Treatment
Diet + plant sterols
Treatment
Diet + plant sterols + statin ± ezetimibe ± bile-acid sequestrant
Statins contraindicated in pregnancy: contraceptive advice
in adolescents
Monitoring: 1 – 2 yearly
Weight, growth,
development/well-being,
compliance (lifestyle)
Monitoring
Weight, growth, well-being, physical & sexual development,
hepatic aminotransferases, creatine kinase (if musculoskeletal
symptoms present), adherence (lifestyle, pharmacotherapy)
Boys <10 yr,
Girls before
menarche
Boys >10 yr,
Girls after
menarche
Restratify
as per
assessment
protocol
No
Consider carotid ultrasound
2 – 5 yearly
Consider carotid ultrasound
1 – 3 yearly
Progression of carotid atherosclerosis
(increase in CIMT and/or detection of plaques)
Treat as high risk
childhood FH
Consider treating as high
risk adult FH
Fig. 6. Management of children and adolescents.
with annual or bi-annual monitoring of weight, growth and
developmental milestones. Increased intake of fruit and vegetables may compensate for reduction in plasma carotenoid
concentrations in children consuming a plant sterol-ester
enriched diet [195]. The value and safety of prescribing
statins for children and adolescents with FH is universally
recognised [64a,96–98,105,106,108]. As a general guide,
boys older than 10 years and girls who have reached menarche should be considered for statin therapy. The specific age
at which to introduce a statin is not evidence-based and in
practice should be determined by good clinical judgment
and assessment of several factors. These include the family
history of premature CVD, the prevailing plasma level of
LDL-cholesterol, the type of FH mutation identified and the
presence of other cardiovascular risk factors (e.g. diabetes
mellitus) [1,12,43,55–57,66,73–75,188], as well as the
parents’ perceptions and concerns on use of medication
[123,196]. The LDL-cholesterol treatment targets in young
patients with heterozygous FH will also depend on similar
considerations (Fig. 6). The health literacy of parents must
be addressed when managing children and adolescents with
FH [88,172,173]. Homozygous patients with FH need to be
started on pharmacotherapy as soon as practicable after birth
and definitely by the age of 10 years [95]. In Australia, to
be eligible for PBS subsidy any FH patient aged <18 years
must have a plasma LDL-cholesterol >4.0 mmol/L with a
positive family history of premature, symptomatic CVD
or tendon xanthomata, or known to be positive for an FH
causing mutation [116].
8.2. LDL-cholesterol targets
The recommended targets for plasma LDL-cholesterol
concentration for enhanced and intensive management are
<4 mmol/L and <3 mmol/L, respectively, consistent with
other lipid treatment guidelines [14,15,97,98]. An alternative
global therapeutic target is a 40% reduction in LDLcholesterol [12,37,64b]. For highest risk FH with diabetes,
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
obesity or metabolic syndrome and elevated triglycerides
(>2.0 mmol/L), an apoB target of <1.1 g/L may be used
[17a,58], noting that there are no published guidelines on
the use of apoB or non-HDL cholesterol in children [97,98].
8.3. Pharmacotherapy
The therapeutic targets of LDL-cholesterol in this age
group may be attained with a fat modified diet and a statin
[97,98], but the addition of ezetimibe [98,197,198] or a bileacid sequestrant [98,153,199] may be required. In Australia
only fluvastatin, pravastatin and simvastatin are registered
with the Therapeutic Goods Administration (TGA) for use
in adolescents and children older than 10 years [200]; these
statins should be initiated at the lowest recommended dose,
which should be up-titrated according to cholesterol response
and tolerability. Simvastatin and pravastatin have been shown
to improve arterial function and carotid atherosclerosis,
respectively, in children with FH [104,201]. The efficacy in
lowering cholesterol and the short-term safety of more potent
statins, such as atorvastatin and rosuvastatin, has been confirmed in children with FH [202,203]. Both of these statins
have been approved by the US Food and Drug Administration (FDA) for use in children with FH aged 10 years and
above. Ezetimibe is TGA registered for use in adolescents
and children older than 10 years [200], with no requirement for dose adjustment. Bile-acid sequestrants can affect
the absorption of folate and fat soluble vitamins [153], and
appropriate supplementation will be required with longer
term use of these agents in children. Colesevelam is the
most tolerable form of these agents [182,199], but is not yet
registered for use in Australasia or New Zealand. Bile-acid
sequestrants should be taken with meals and gastrointestinal
side-effects minimised by increasing fluid and fibre intake
[153]. Niacin may be indicated for selected patients with
homozygous or compound heterozygous FH [98], as well
as for heterozygous FH patients with very high Lp(a) and a
very early family history of CHD [75]. Niacin should otherwise rarely be used to treat paediatric FH owing to poor
tolerability and concerns about increased risk of glucose
intolerance, myopathy, hyperuricaemia and hepatitis [98].
By contrast, fibrates are relatively safe and well tolerated
in children, but are rarely indicated except in the treatment of FH patients with significant hypertriglyceridaemia
(>4 mmol/L) or with a homozygous phenotype [14,97,98].
As with fibrates, higher doses of supplemental omega-3 fatty
acid ethyl ester may also be very rarely indicated for patients
with severe hypertriglyceridaemia to prevent acute pancreatitis [14,161]. Omega-3 fatty acid ethyl ester may also improve
arterial function in children with FH [204]. The recommendations for monitoring drug safety and tolerability in paediatric
patients are generally similar to adult FH, but special requirements are noted below and in Fig. 6. More systematically
collected long-tem safety data are required on the use of
statins and other lipid-regulating agents in paediatric FH
[205].
237
8.4. Assessing atherosclerosis
CUS, with specific measurement of CIMT and the detection of early plaques, may be useful in assessing the
progression of early carotid atherosclerosis and hence in risk
stratification of children [78,122,206]. Longitudinal data in
diverse populations support lowering hypercholesterolaemia
in children with FH from the age of at least 10 years [206].
CUS could be carried out every two years in moderate risk
FH and annually in high risk FH. These intervals are only
a guide and should be revised according to clinical context
and available resources. Evidence of an increase in CIMT,
or development of early atherosclerotic plaques, would be an
indication for intensifying treatment in moderate and high
risk cases of FH [183] (see Fig. 5). CUS should be carried
out by a fully credentialled vascular imaging service and
CIMT estimated employing well validated edge-detection
software [82,122]; this facility may not be routinely available to many clinics and consequentially other paediatric
guidelines do not specifically recommend use of CUS in managing FH [64a,b]. Imaging protocols need to be standardised
and age- and sex-specific reference data for CIMT employed
[82,122]. Contemporary CTCA and calcium scoring should
not at present be undertaken in children owing to the radiation doses involved. Uncontrolled dyslipidaemias in young
adulthood predicts coronary calcium in middle-aged [207].
We do not consider that serial measurement of the anklebrachial index plays a role in the management of childhood
or adolescent FH, except in very high risk cases including
homozygous patients. Measurement of endothelial function,
with flow mediated dilatation of the brachial artery, is at
present considered a research tool that is not ready for routine clinical practice [122]. Children with a family history
of CHD in early adulthood will require non-invasive testing
for CHD [13,97] and referral to a paediatric cardiologist is
advised [110]. For patients with homozygous, or compound
heterozygous FH on LDL-apheresis, regular echocardiography of the aortic valve and aortic root, with estimation of the
aortic valve gradient, is recommended [95,97]. These special
cases of very high risk FH should be managed jointly with a
paediatric cardiologist.
8.5. Clinical monitoring and continuity of care
Recommendations for the follow-up of children and adolescents with FH are broadly similar to adults [14,15,97,98].
However, certain issues require specific attention and review
intervals may not need to be as frequent as in adults. Drug
tolerability, interactions and safety need careful surveillance
[97,98,153,156,163,165]. All children and adolescents on
statins require monitoring of physical growth and pubertal development, as well as when indicated plasma levels
of hepatic aminotransferases, creatine kinase and creatinine
[97,98]. Before commencing statin therapy in adolescent
females, specific advice should be given regarding contraceptive choices and the potential risk to the foetus should
238
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
they fall pregnant while on medication [186]. These issues
should be considered at each review. Adolescence is an opportune time to evaluate each patient’s understanding of FH
and provide age-appropriate education as required. Important issues that should be addressed include: the significance
of the autosomal inheritance pattern of FH, with potential
offspring having a 50% chance of inheriting the condition;
the value of maintaining a healthy lifestyle; the importance of
adhering to the medication prescribed and attending appointments for clinical review. Adherence to medical management
is a challenge for many individuals with chronic conditions
and can be a particular issue in adolescence [170,208]. Family counselling and medication support systems, including
nurse or pharmacist co-ordinated programs, to ensure adherence with therapy may be useful, particularly in higher risk
patients [166,167,170]. Enrolling the assistance of personnel skilled in managing adolescent problems may be useful
[209]. Pharmacists may be utilised for monitoring adherence
and flagging non-adherence to medication in the intervals
between clinical reviews, as well as for directly improving
adherence to medication [166,168]. All children and adolescents with FH should be reviewed at least yearly in a
specialist clinic. Highest risk patients should be reviewed
at least every 6 months and homozygotes on LDL-apheresis
more frequently [95,97,210]. Patients who are well controlled
on stable treatment could be managed in primary or shared
care. Appropriate clinical pathways should be developed for
transitioning and transferring adolescent patients with FH
from paediatric to adult clinical care services [211].
9. LDL-apheresis and radical therapy for FH
LDL-apheresis is a radical form of treatment for FH
that entails the extracorporeal removal of apoB-containing
lipoproteins from the circulation [212,213]. Its use in treating
severe FH is supported by published international guidelines
[13,95,214–217] and evidence that LDL-apheresis improves
CHD outcomes, progression of atherosclerosis and aortic
fibrosis, endothelial function and coagulation [212,218,219].
9.1. Indications, patient selection and targets
LDL-apheresis is indicated for patients with homozygous or compound heterozygous FH, as well as for patients
with heterozygous FH with documented CHD who are
refractory to pharmacotherapy [95,216]. FH patients with
very high plasma Lp(a) levels may also benefit from
LDL-apheresis [75,217]. Untreated patients with a homozygous phenotype typically have plasma LDL-cholesterol
>12 mmol/L and should be treated with maximally tolerated
pharmacotherapy for at least 6 months before considering
LDL-apheresis [95,220]. Untreated heterozygous patients
typically have plasma LDL-cholesterol from 5 to 12 mmol/L
[95] and may have true non-responsiveness or intolerance
to cholesterol lowering pharmacotherapy. The recommended
LDL-cholesterol thresholds for selecting patients for apheresis in Fig. 7 are only an approximate guide and should be
modified according to clinical context; an alternative selection criterion could be a <50% reduction in LDL-cholesterol
on maximal pharmacotherapy for both homozygous and heterozygous patients.
To be suitable for LDL-apheresis patients must be psychologically and clinically stable and be committed to the
treatment. Haemorrhagic diatheses and hypersensitivity to
heparin are contra-indications. Clinical assessment should
include baseline echocardiogram for aortic stenosis [215].
Patients unsuitable for LDL-apheresis must continue diet and
medication. LDL-apheresis is an efficacious, well tolerated
and safe treatment for children with severe FH and may be
commenced after the age of 5 years in affected individuals
[95,97,210]. It should also be considered for pregnant women
with uncontrolled FH and stable CHD [221,222].
9.2. Methods for apheresis
There are five apheresis methods that are selective for
LDL: membrane differential filtration, immunoadsorption,
heparin-induced LDL precipitation, dextran sulphate LDL
adsorption and haemoperfusion with direct LDL adsorption
[95,212,216,217]. All are comparable at acutely lowering
LDL-cholesterol by 50–70% following a single treatment.
Plasma exchange (or centrifugal plasma apheresis) may also
be used, but its major disadvantage is that it is not selective for
LDL. The choice of method will depend on local expertise
and resources. Treatment should be carried out in collaboration with a specialty experienced in apheresis, such as a
transfusion medicine service, with whom close communication is mandatory. Plasmapheresis and probably cascade
filtration should be available via all transfusion medicine
services. A dedicated, fully resourced LDL-apheresis unit
should also have a variety of methods of apheresis that can
be tailored to maximise efficacy and tolerability in individual patients [223]. Vascular access is achieved using bilateral
antecubital vein cannulation, but a long-term arteriovenous
shunt or central venous catheter may be required. Anticoagulation with heparin and citrate is required. Typically,
4–6 L exchanges of plasma are carried out for 2–4 h weekly,
biweekly or longer according to therapeutic response.
9.3. Monitoring therapy
The efficacy, safety and tolerability of LDL-apheresis
must be monitored at each treatment and the regimen regulated accordingly [215]. The frequency of LDL-apheresis
should be adjusted to achieve a time-average plasma
LDL-cholesterol concentration between LDL-apheresis
therapy of 6.5 mmol/L and 2.5 mmol/L for homozygotes
and heterozygotes, respectively (Fig. 7); a mean reduction
of >65% in plasma LDL-cholesterol concentration between
LDL-apheresis relative to no form of treatment is another
possible target. To achieve these targets will usually require
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
No
Homozygous or compound
heterozygous FH
Heterozygous FH with
documented CHD
Diet and maximal
drug therapy
Diet and maximal
drug therapy
Review adherence
and tolerability
Review adherence
and tolerability
LDL-C >7 mmol/L
Consider need and suitability
for liver transplantation,
enrolment in clinical trial of
new treatments, or applying
via special access scheme for
drugs in development for FH
No
LDL-C >5 mmol/L
Yes
Not Suitable
Homozygous or compound
heterozygous FH
239
Assess suitability
for LDL-apheresis
Not Suitable
Heterozygous FH
Consider enrolment in clinical
trial of new treatments or applying
via special access scheme for
drugs in development for FH
Suitable
Liaise with transfusion service
to select LDL-apheresis method,
regimen and set efficacy and
safety parameters
Homozygous or compound
heterozygous FH
Apherese 1.5 – 2 plasma
volumes for 2 – 4 hours every 2 –
3 weeks, continue statins and
other drugs, avoid ACE inhibitors
Therapeutic target
Time average
LDL-C <6.5 mmol/L
Heterozygous FH with
documented CHD
Therapeutic target
Time average
LDL-C <2.5 mmol/L
Review efficacy, tolerability and
safety of apheresis at each
treatment
Revise regimen and method of
apheresis; consider need for liver
transplantation
Fig. 7. LDL-apheresis and radical therapy for FH.
an acute reduction of ≥70% in LDL-cholesterol during
each procedure. Statins should be continued since they
slow the post-exchange rebound in LDL-cholesterol, but
use of angiotensin converting enzyme (ACE) inhibitors are
contra-indicated with most systems because of a potential bradykinin reaction; [95,217] angiotensin II receptor
antagonists are suitable alternatives. The overall incidence
of clinical side effects (including hypotension, vasovagal
episodes, symptomatic hypocalcaemia and anaemia) during
apheresis is less than 5%; if present, these may be corrected
using standard medical therapy. Non-specific symptoms
such as nausea, fatigue, dyspnoea and abdominal pain
respond to temporary discontinuation of therapy. Patients
who are persistently intolerant of a particular method of
LDL-apheresis should be trialled on an alternative method,
including plasma exchange if required [212]. Because of the
demands of treatment, psychological status and quality of
life should be monitored in all patients. Accordingly, patients
and parents of affected children undergoing LDL-apheresis
should be offered psychological counselling when indicated.
The long-term efficacy of treatment in homozygous
patients should be assessed about every 2 years using CUS for
carotid atherosclerosis load, and echocardiography for aortic valve and root involvement, proceeding to stress ECG and
possibly coronary angiography, if there is evidence of significant progression of disease [13,78,215,224]. In heterozygous
240
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
patients on LDL-apheresis CUS and exercise testing should
also be employed at suitable intervals to monitor progression
of atherosclerosis [13,183,215]. Many patients undergoing
LDL-apheresis will need to be jointly managed with adult
or paediatric cardiologists [215,224] and appropriate lines of
communication should be established.
9.4. Cost considerations
Besides the complexities of the treatment, the main barrier
to using LDL-apheresis is the financial cost. It is estimated
that on average each procedure costs AU$2200–2600, so the
annual cost of a bi-weekly cycle is approximately AU$53,000
[95]. This is comparable to haemodialysis, but in absolute
terms would be less than 1% of the national expenditure on
haemodialysis. LDL-apheresis is underutilised in Australia.
Health insurance providers, Medicare and tertiary hospitals
should support it as a routine service for patients with severe
forms of FH.
9.5. Other therapeutic options
As shown in Fig. 7, homozygous patients who are not
suitable for LDL-apheresis should be considered for liver
transplantation [225,226] or enrollment in clinical trials of
novel cholesterol lowering treatments [227], such as apoBantisense oligonucleotide therapy and proprotein convertase
subtilisin/kexin type 9 (PCSK9) inhibitors [228,229]. Alternatively, these new treatments could be made available to
patients on a compassionate basis via a special access scheme.
Coronary artery bypass surgery and/or aortic valve replacement should be considered prior to liver transplantation
according to pre-operative cardiac investigations [215,224].
Patients with heterozygous FH who are not suitable for LDLapheresis should also be considered for enrollment in clinical
trials of new cholesterol lowering treatments [228,229], or an
application should be made via a special access scheme for
drugs in development for FH. Partial ileal bypass should be
considered in heterozygous FH patients who are intolerant
of or refractory to pharmacotherapy, noting both the benefits and risks of this surgical procedure [230,231]. There is
very limited experience in Australasia with liver transplantation for severe FH. Portocaval shunting carries a high risk
of encephalopathy and has now been superseded by LDLapheresis and liver transplantation; its use in homozygous FH
may, however, be considered in countries where newer and
more expensive treatments are not available [220]. Finally,
there may be a role in the future for gene therapy in treating
severe FH [232].
10. Cascade screening: testing and risk notification
of families
It is well accepted that the most cost-effective approach
to detecting new cases of FH is family cascade screening
of close relatives using a genotypic or phenotypic strategy
[4,10,31,65,233–236]. Genetic testing of an at-risk individual within a family is often referred to as ‘predictive testing’
[237]; predictive testing on the basis of a known pathogenic
genetic variant (i.e. a mutation) is more accurate than predictive testing based on an individual’s clinical phenotype alone,
and this also applies to FH [7,8,66]. In a condition like FH,
in which the phenotype almost always expresses as hypercholesterolaemia, the descriptor ‘predictive’ could be replaced
by ‘presymptomatic’ or ‘diagnostic’ to describe genetic testing [237]. However, because ‘predictive’ describes a more
general context and is more widely accepted in clinical genetics it is also used in this document when referring to genetic
testing of relatives for FH. Screening for FH using the clinical
phenotype alone is a form of ‘diagnostic’ testing, particularly when assessment provides a definite outcome, as in an
individual with, for example, a DLCNS > 8 [8,34,44].
10.1. Risk notification
Integral to effective and ethical cascade screening is risk
notification [7,8,119,120]. Risk notification is the process of
informing relatives that (1) they are at risk of FH, (2) that
this may have implications for their health, and (3) that clinical and genetic testing (if applicable) is available to clarify
if they do or do not have FH. Fig. 8 provides recommendations, based on expert guidelines and position papers, for
undertaking the process of cascade screening whether using a
genotypic or phenotypic approach [8,113,119,120,238–240].
Fundamental ethical principles of autonomy, beneficence,
non-maleficence and justice are central to this process. Health
illiteracy is not uncommon in individuals in the community
and must be carefully considered and addressed [87,88,173].
10.1.1. Contacting and informing families
The general protocol for cascade screening should begin
by contacting first and then second degree relatives, noting
that cases so detected will then become probands for risk notification of their own first and second degree relatives [6–9].
A typical pedigree tree showing the autosomal dominant pattern of inheritance of FH (clinical phenotype expressed) and
the yield from cascade screening is shown in Appendix 6.
Culturally sensitive strategies may be required when dealing
with some ethnic groups [1,241]. Approaching and liaising
with community leaders early on is advised to facilitate risk
notification and predictive testing occurring in a culturally
appropriate way.
The index case should be invited to discuss family risk
notification with the clinician or nurse, who will construct a
family pedigree and identify first and second degree relatives
who should initially be offered testing for FH. Use of a pedigree drawing tool [242] and a fully integrated information
management system [243] is strongly recommended to facilitate family assessment, work planning, multidisciplinary care
and communication of results to individuals tested and their
doctors [9,12,25]. Appendix 7 shows a pedigree tree for
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Index case consents to risk
notification of relatives
No
Risk notification
Send letter and information sheet to
family member and / or give to index
case to distribute to relatives
No response
Response
Received by
clinical service
Send 2nd letter
/ Telephone
family member
Family member consents to screening
Arrange appointment with FH service
As per local protocol
241
Is there a particularly
strong clinical
indication for risk
notifying relatives
without consent?
No
Yes
No
response
Family member does not
consent to screening
Accept decision not to
be tested
But encourage risk
notification of their 1st and
2nd degree relatives
Fig. 8. Cascade screening: risk notification and approaching families.
an actual family with FH, illustrating the autosomal dominant inheritance of hypercholesterolaemia, premature CVD
and a missense mutation in the LDL-receptor gene. Drawing and detailing such pedigrees is essential for effective
implementation of cascade screening, including appropriate risk notification. Clinical genetics services often have
well-established protocols for managing families with familial disorders and, where possible, cascade screening should
be implemented as a joint collaborative effort of the lipid
disorders and clinical genetics services [12,26,115,121,237].
Comprehensive and well validated resources should be
employed and developed to accurately convey information
to relatives [12,25,37]. The method for risk notification
should be tailored to each family, so that relatives may be
approached either by the index case, the clinical service, or
by both [6,8,119,120,244]. Understanding the reasons why
an index case may not consent to cascade screening of other
family members is important to provide re-assurance and
encourage rapport and disclosure [37,245,246]. Dual risk
notification may be the best option: notification by the clinical service alone may lead to relatives feeling aggrieved that
they were not informed by their related index case; notification by the index case alone may be taken less seriously
than notification by a medical service. Genetic counsellors
must be involved when sensitive family issues are identified
[8,25,110,121,238].
Index cases who contact relatives should be provided with
written information which explains the predictive/diagnostic
testing process and implications, and be encouraged to give
this to their relatives [12,25,37]. Information and letters sent
to relatives should be written in general language to avoid
alarm and concern, while emphasizing the voluntary nature of
testing [119,120], and the health consequences of a diagnosis
of FH being missed when a person decides not to be tested.
Communications should also emphasise the health gains of
diagnosis and treatment [12,25], as well as the benefits for
offspring who may have FH.
In the absence of a response to the first letter, it may be
appropriate to make a second approach by letter or telephone call to the family member [37,244]. However, this
policy should be discussed with relevant local experts on ethical and risk management matters. Multiple contact attempts
could constitute a violation of privacy for some individuals
[119,120] and may be counter-productive to the process of
cascade screening. Reasons why a relative does not wish to
be tested for FH should be explicitly documented [84,86] to
facilitate, where indicated, re-approaching them in the future.
Electronic methods of communication (e.g. e-mail) could be
investigated for their value in enhancing risk notification and
cascade screening for FH.
Prior to testing, family members should be given detailed
verbal and written information about FH. Written information should again be in clear, non-technical language and
preferably accompanied by simplified but explicit diagrams
[25,37,119]. Health illiteracy should always be addressed
[87,88,173]. Family members who consent to being tested
for FH should be offered a standard plasma lipid profile and
a genetic test if the mutation is known in the index case.
Patients without GPs should be encouraged to visit one as
part of the FH counselling process. With consent from the
242
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
individual, the outcome of phenotypic or genetic testing for
FH should be communicated to their GP and other relevant
medical services.
10.2. Co-ordination of cascade screening
Different care pathways may be employed for screening individuals for FH, depending on circumstances and
resources [7,25,26,37]. Family members may be tested via the
lipid disorders clinic, genetic services or their GP. Depending
on knowledge and experience, pre-test counselling may be
best undertaken by a specialist medical practitioner, genetic
counsellor or specialist nurse. FH specialist nurses should
have some competency in genetic counselling [121,247], this
being particularly relevant for efficiently co-ordinating an
outreach screening service. Phenotypic screening for FH may
be carried out by GPs, noting that use of clinical criteria
alone may result in the underdiagnosis of FH in a primary
care setting [248]. However, GPs should not directly request
genetic testing without appropriate specialist training. The
future should see more education of GPs in medical genetics
and this will increase their role in predictive testing of families for FH [249]. This is particularly important in primary
care, given the greater accuracy of genetic testing over clinical criteria in diagnosing FH. FH services that do not employ
health practitioners with formal counselling training could
consider referral to a clinical genetics service for counselling
[26,121,237].
Referral to genetic services should also be considered
when contemplating genetic testing in children or adolescents
[115,238], and when sensitive family issues are identified.
Enquiries concerning PND and PGD for FH are a special
indication for referral to a clinical genetics service [192,193].
Genetic counsellors should be involved in training staff caring
for FH patients and in supporting GPs and outreach services
[7,8,12,26,121].
Cascade screening for FH is best carried out and
co-ordinated centrally by a specialist service ideally run collaboratively by lipid disorders and clinical genetics services
[12,26,71,115,121,237]. However, relatives may choose to
have genetic or phenotypic testing in a primary care setting.
In this case, close liaison and communication will be required
with their GP. If genetic testing is required, support in pretest counselling should be given by a clinical geneticist or a
genetic counsellor [115,121,237]. If the diagnosis of FH is
confirmed genetically, or is highly suspected on phenotypic
criteria, GPs should refer the individual to a specialised FH
service for advice on treatment and follow-up [12,25,26,38].
10.3. Risk notification without consent
If the index case does not consent to risk notification of
family members it is important to explore and understand
the rationale behind this [83,245], and in particular the prevailing family dynamics [83,119,120,246]. By maintaining
professionalism and continuing to patiently and judiciously
explore and resolve key issues [120,246], an effective rapport
can often be built with the index case sufficient to revisit the
option of risk notification after 6–12 months, or sometimes
longer. Referral to a genetic counsellor [121] and involvement
of a patient support group for appropriate lay counselling
[250–252] may all assist in achieving acceptance of risk
notification.
Recent amendments to the Commonwealth’s Privacy Act
1988 in Australia allow private health practitioners “to use or
disclose patients’ genetic information, whether or not they
give consent, in circumstances where there is reasonable
belief that doing so is necessary to lessen or prevent a serious
threat to the life, health or safety of their genetic relatives.”
(Privacy Legislation Amendment Act 2006 (Cth) amendment
to the Privacy Act 1988 (Cth)) [253]. This amendment of the
Privacy Act only applies to the private sector [253]. Practitioners employed in the public sector need to refer to the
privacy legislation in their own state or jurisdiction concerning the release of information without the consent of the
patient [246]. The difference in the privacy laws for public and private patients in Australia is an issue that requires
resolution.
A decision to risk notify without the consent of the
index case should therefore be very carefully and judiciously
taken with attention to privacy legislation in different states,
local health service protocols and the NHMRC guidelines
[239,246]. Health professionals must understand the genetic
basis for FH, take reasonable steps to obtain patient consent
and document the process fully before considering risk notification of relatives without consent. The recent NHMRC
guidelines [239] are not prescriptive and we urge caution
when independently approaching relatives. Nevertheless, FH
is a potentially lethal condition and if there are high risk features in the family, such as a strong history of premature
CVD, contacting of relatives without consent is justifiable,
as indicated in Fig. 8. Refusal of an index patient with FH
to share genetic information with relatives is unusual, but
when present poses a significant challenge to risk notification
and cascade screening [119,120,246]. Best clinical practice
implies that disclosure of information without consent during
cascade screening for FH should be viewed as a last resort
[246]. However, when information is disclosed, it should be
the minimum required, avoid identifying the patient or their
lack of consent, and should only be made available to family
members no further removed than third-degree relatives.
10.4. Insurance cover and genetic testing
All individuals with potential FH should be made aware
and understand the implications of genetic testing for certain types of insurance cover [254–256]. Family history of
CVD, a clinical diagnosis of FH, plasma level of cholesterol and predictive genetic information could all potentially
be employed by the insurance industry to make decisions
about exclusion of specific conditions from insurance coverage and setting an insurance policy premium [256–258].
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
In Australia premiums for insurance products which include
cover for life (term), disability/income protection, trauma,
business and bank loans (but not private health insurance)
are calculated according to the present and past health of the
applicant, their family history and any known genetic test
result(s) in the applicant, their siblings or their parents [256].
However, insurers who are members of the Financial Services
Council have agreed that they will not ask an individual who
is applying for insurance coverage to have a genetic test if
it has not been done already [259]. Similar conditions apply
at present in New Zealand. Better control of hypercholesterolaemia with statins in FH is likely to lower insurance
premiums [258].
11. Genetic testing of families
FH is an autosomal dominantly inherited disorder, affected
individuals having a 50% chance of passing the causative
mutation to each offspring [1–3]. If a pathogenic mutation
is identified in an affected index case, predictive testing of
relatives using genetic testing is a cost-effective, accurate and
acceptable approach for detecting new cases [66,234,260].
However, genetic mutations may not be detected in at least
20% of patients in whom a clinical diagnosis of FH can be
confidently made [3,23,45,66,261,262]. Thus, in a smaller but
significant proportion of families diagnostic testing should be
carried out phenotypically [12,38,68].
One of the main advantages of predictive genetic testing
is its ability to definitively confirm or exclude a diagnosis of
FH in the relatives of an index case [66]. Fig. 9 outlines the
process to be followed when employing predictive genetic
testing in cascade screening. The majority of cases of FH are
due to mutations in the LDLR, APOB and PCSK9 genes [1–3].
Given that a mutation is known to be present in the index case,
genetic testing should be offered to all at-risk family members
who present for predictive testing [8,66]. Because of ethical
issues involved in genetically testing minors [114,115], it is
best and usual practice to first genetically test a phenotypically affected parent. When no consent, or assent, is obtained
for genetic testing, the diagnosis of FH can be made phenotypically according to the DLCNS in an adult [8,34,38,44]
or to the plasma LDL-cholesterol concentration in a child
[46,48,97] (Fig. 9).
Genetic testing of families for FH should be co-ordinated
by a specialist ideally run collaboratively by a lipid disorder clinic and clinical genetics [12,26,71,115,121,237].
The psychological consequences of genetic testing must be
carefully considered and appropriate education and counselling offered in all individuals [8,66,84,86,115,121,237].
Obtaining informed consent, or parental consent plus child
assent in the case of testing children, requires pre-test counselling concerning the implications of the result [115,238].
This is particularly important in children because the future
implications are more difficult to anticipate and the consent
process via parents or guardians is less direct [115]. Age
243
and developmentally appropriate assent/consent should be
obtained from children and adolescents prior to testing and
information should be tailored to the child’s level of comprehension [115,121] and the parent’s level of literacy [87,88].
Genetic testing in children can be carried out without invasive
venepuncture, for example by using a buccal swab specimen
[263].
If the family mutation is not identified by a predictive
genetic test, the diagnosis of FH can be excluded in that family member. However, there may be some situations when the
family member has significant hypercholesterolaemia (and
a phenotype strongly suggestive of FH) but does not carry
the pathogenic mutation identified in other family members.
Having excluded secondary causes of hypercholesterolaemia
[14,15], it may then be appropriate to do a full ‘FH mutation search’ in that individual, including investigation of the
extended pedigree. This mutation search should be preceded
by appropriate pre-test counselling [115,121,237].
As indicated earlier, when one or both members of a couple has FH and there are concerns about having an affected
child, referral for specialist genetic counselling regarding
PND and PGD should be considered [192,193]. Couples opting for pre-natal testing should be seen prior to conception.
PGD will require detailed assessment by an in vitro fertilization service and a specialised DNA laboratory [264].
Services providing PGD must comply with established laboratory requirements and relevant State and Commonwealth
legislation [265].
12. Laboratory approach to genetic testing for FH
12.1. Background
Laboratories providing medical genetic testing in
Australia must comply with requirements developed by
the National Pathology Accreditation Advisory Council
(NPAAC) [264]. Compliance with these standards is regularly assessed in a joint program managed by the Royal
College of Pathologists of Australasia (RCPA) and the
National Association of Testing Authorities (NATA) [266].
NPAAC requirements address the need for appropriate laboratory resources, governance and supervision, training and
continuing accreditation of staff, and appropriate clinical
liaison. Medical testing that may be the basis for clinical
decision-making must be performed in an accredited laboratory.
Accordingly, genetic testing for FH should be carried out
in a NATA/RCPA accredited laboratory [266] that will issue
results to the requesting doctor. The process of screening
for genetic mutations, confirming identified genetic variants,
assessing pathogenicity and issuing a formal report should
ideally not take longer than three months. Supported by further evaluation of its cost-effectiveness, we recommend that
an application is made for genetic testing for FH to be listed
as an MBS item.
244
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Proband or relative carrying gene variant of known
pathogenicity (i.e. mutation)
Counseling of family members
Psychological / genetic / lay counseling. Clinical
genetics and paediatric review if required. Informed
consent / assent sought
No assent/consent
to genetic testing
Assent/consent
to genetic testing
Phenotypic testing
Genetic testing
FH unlikely
If still concerned
about possibility of
FH, continue to build
rapport and
reconsider genetic
testing
Probable to Definite
FH confirmed
Family mutation
present
Discuss risk
notification of close
relatives
Figure 8
Family mutation
not present
FH excluded*
Offer phenotypic or
genetic testing to
relatives
Fig. 9. Genetic testing of families. * See text for caveats.
In the present context, a gene variant is defined as an alteration in the normal sequence of a gene. Genetic testing of an
index case may fail to identify a variant in the genes tested
(variant absent) or it may identify a variant (variant present).
Identified variants may have been previously reported or be
novel. NATA/RCPA accredited laboratories have processes
for assessing the likely significance of an identified gene variant and for classifying the variant as clearly pathogenic (a
mutation), clearly non-pathogenic (a benign variant) or of
uncertain significance (a variant of uncertain significance)
[264]. The assessment of significance is discussed in more
detail in a later section.
FH has significant locus and allelic heterogeneity, i.e.
FH is caused by mutations in a number of different genes
and many different pathogenic mutations have been identified in each implicated gene [112,261,267–270]. Thus, the
main challenges for genetic testing for FH remain the costs
and feasibility of testing the large numbers of patients clinically diagnosed with or suspected to have the condition.
As noted earlier, the likelihood of detecting a pathogenic
gene variant in an individual suspected of being an index
case is directly proportional to the clinical probability of that
individual having FH [1,3,66,112,118], as assessed phenotypically by, for example, the DLCNS [34,38,44]. Hence,
genetic testing of adults with a phenotypic DLCNS < 3 may
not be cost-effective [38,44].
Currently, mutations in three different genes are known to
cause FH [1,3,66]. A pathogenic mutation in one of these
genes is identified in about 70% of phenotypically definite and 20% of phenotypically probable/possible FH [8,66].
About 95% of the identified mutations are in the LDLR gene
and 4–5% in the APOB gene. Mutations are seldom identified in the PCSK9 gene. Detection of a mutation in a family
member allows the definite diagnosis of FH to be made, as
outlined in Fig. 9. However, failure to detect a mutation does
not exclude a diagnosis of FH, particularly if the clinical
phenotype is highly suggestive of FH [12,37,66].
There are several reasons why mutations might not
be detected in patients with a high clinical suspicion of
FH [1,3,66,262]. First, the present laboratory technology
is not sufficiently sensitive nor specific for detecting all
pathogenic mutations. Second, FH is genetically heterogeneous (caused by mutations in a number of different
genes) and not all causative genes have been identified.
Clearly, genetic testing is not helpful in families with FH
due to mutations in unknown genes as these genes cannot
be included in genetic testing. Third, the index case may
not have ‘true’ FH, since a family history of hypercholesterolaemia and early CVD is not sufficiently specific for
making the diagnosis of FH; some of these patients may have
other genetic disorders [66], particularly familial combined
hyperlipidaemia [271,272].
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Most diagnostic laboratories undertaking FH genetic testing will utilize a number of mutation detection methods
[66]. These may include: (1) nucleotide sequence analysis
of each of the exons and flanking splice regions (exon by
exon sequence analysis, EBESA) of the LDLR, APOB and
PCSK9 genes [3,112,261,270,273]; (2) methods that detect
large duplications and deletions in the LDLR gene based on
Multiplex Ligation Probe Amplification (MLPA) [274,275];
(3) methods that screen for specific mutations in the LDLR,
APOB and PCSK9 genes [260,276–280].
12.2. A protocol for genetic testing
Complete screening for all known FH-causing genes is
expensive and time consuming [66]. A laboratory protocol
that is likely to be cost-effective for genetic testing (searching
for a mutation) an ‘index case’ considered to have phenotypic
FH is proposed in Fig. 10. This protocol could form the basis
of a standardised, national protocol for genetic testing for
FH. We emphasize that laboratories may choose to vary the
approach to genetic testing for FH shown in Fig. 10 according
to the available resources: for example, laboratories may opt
to proceed directly to DNA sequencing for an LDL-receptor
mutation of all test samples received [3,66,112,261,270,273].
We recognize that technological advances in genetic testing
and the discovery of new genes causing FH will necessitate
appropriate modification to the protocol [66,110] in Fig. 10.
The steps proposed in Fig. 10 are as follows: (1) individuals with a DLCNS < 3 (unlikely FH) are not offered genetic
testing, because the likelihood of identifying a pathogenic
gene variant is low and with currently available technology
not cost-effective; (2) genetic testing for FH is offered to
all individuals with a DLCNS ≥ 3, which includes all possible, probable and definite phenotypic cases of FH; (3) for
individuals with a score of 3–5 (possible FH), to maximize
cost-effectiveness, testing could be limited to sequential testing using: Screen 1, employing commercial methods that
target specific mutations; [260,280–282] Screen 2, employing MLPA [274,275,283] (Fig. 10); (4) individuals with a
DLCNS ≥ 6 could be offered more comprehensive sequential testing: Screen 1, employing commercial methods that
target specific mutations; [250,270–272] Screen 2, employing MLPA; [274,275,283] Screen 3, employing sequential
EBESA of at least the LDLR gene (the recommended order
for EBESA is LDLR > ABOB > PCSK9) [3,66] (Fig. 10). All
results from commercial chip or kit technology that identify
a gene variant as being present should be confirmed using
the second validated testing method. This is a requirement of
good laboratory practice [264,266] and relates to the potential
analytical errors with the commercial methods.
It should be noted that the protocol in Fig. 10 refers to
diagnostic testing (mutation searching) for FH in a phenotypically defined ‘index case’. In a predictive testing setting
the laboratory will already know the mutation in the index
case and there is no need to screen for mutations other than
directly testing for the one identified in the family [8,66].
245
To increase acceptability, genetic testing for FH in children
should be available using DNA extracted from either blood
or buccal samples [263].
12.3. Assessing the significance of gene variants
detected in index cases
Various pieces of evidence are used to determine the significance of an identified gene variant [264] and this clearly
also applies to genetic testing for FH [66]. These include the
published literature (including database entries), epidemiologic studies, in vitro and (preferably) in vivo demonstration
that the variant causes a functional abnormality, and in silico
(molecular software) assessment [284,285]. For practical reasons, in silico assessment coupled with search of the literature
and established databases [267,286] are the usual primary
sources used for assessing the significance of a gene variant.
Unfortunately, published literature may in general contain
errors, so careful assessment is required. In silico data are
also variable in quality, particularly when it comes to assessing splicing mutations [287]. Patients and their managing
clinicians should also be aware that a laboratory interprets
each genetic test result in accordance with the best information available at the time of the test, and that it is possible
that new information may emerge which results in a change
in how a genetic test is interpreted.
Particular care needs to be taken when classifying a gene
variant as a pathogenic mutation, since it may be used in
predictive genetic testing of asymptomatic individuals. If a
variant of uncertain significance (either previously reported
or novel) is detected, it is not appropriate to regard it as
pathogenic. Clinical management of the individual and their
family should be based on the plasma lipid phenotype (and
other clinical criteria) and not on the genetic test result
[12,13,26,38].
Fig. 11 provides a general approach for assessing and
reporting the outcome of genetic testing for FH in a phenotypically defined index case. The significance of a gene
variant should be assessed as recommended above. The formal laboratory report of the FH variant/mutation should detail
the standardised methods used and the evidence supporting
the classification [264]. If a pathogenic variant (mutation) is
identified the report should also make the recommendation
that further relatives be genetically screened for FH [264].
Where no gene variant or pathogenic mutation is detected,
the report should include the caveat that such a result does
not definitively exclude the diagnosis of FH.
13. The web of care for FH: the optimal service
model
Fig. 12 encapsulates the multiple components that should
ideally comprise a comprehensive healthcare model for FH.
The recommendations and MoC described in this document
provide detailed pathways for the principal clinical compo-
246
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Dutch Lipid Clinic Score > 3 in Index Case
Prioritise >8 definite, 6 – 8 probable, 3 – 5 possible
Consents to Genetic
testing
Definite and
Probable FH
Possible FH
Screen 1
Commercial method
for detecting specific
pathogenic variants
Screen 1
Commercial method
for detecting specific
pathogenic variants
Variant absent
Variant absent
Screen 2
MLPA ‡
Variant present
Confirm by
sequencing and / or
appropriate
alternative method if
available
Variant absent
Issue report
Screen 3
Comprehensive
exon by exon
sequencing
Variant present
Assess significance
of gene variant (See
Figure 11)
Screen 2
MLPA ‡
Variant absent
Issue report
Include caveat that
FH due to rare gene
variants/mutations
cannot be excluded
Fig. 10. A laboratory protocol for genetic testing an ‘Index Case’ considered to have phenotypic FH. ‡ MLPA – Multiplex Ligation Probe Amplification.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
247
‘Index case’ consents to genetic test
Genetic testing
as per Figure 10
Variant detected
No variant detected
Variant of uncertain
significance
Issue Report
with caveat that FH
cannot be excluded
Assess significance
of variant *
Benign variant
Pathogenic variant
(mutation)
Issue Report
With appropriate recommendation for
testing relatives (See Figure 8 & 9)
Fig. 11. Assessment and reporting of the outcome of genetic testing in an ‘Index Case’. *See text for further details.
nents shown in Fig. 12. This MoC represents an overarching
system that significantly extends and updates other published
service models [12,13,25–28].
health service provision for FH should be to identify most
people with the condition in the community, aiming to
achieve realistic targets in given time-frame, e.g. 30% detection rate after 3 years of service implementation.
13.1. Focus, aims and objectives
13.2. Co-ordination and integration of care
Health service provision should focus on three main areas:
patient care services, laboratory services, and research and
clinical audit. The aims, objectives, expectations, outcomes
and key performance indicators for each of these should
be clearly identified and documented. Research and audit
agendas for FH have been published elsewhere [12,37,288].
Care pathways for the seamless flow of patients between
relevant specialties and other health providers, including primary care, should be specified and developed. The general
aim of clinical care should be to identify, assess, manage and treat children and adults with FH to the highest
level of clinical excellence. The primary therapeutic objective is not only to reduce cardiovascular risk related to
elevation in plasma LDL-cholesterol [1,2,12,13], but also
to treat other cardiovascular risk factors including obesity, hypertension, type 2 diabetes, metabolic syndrome
and smoking [17a,32,33,43,55–57,74,125,127]; psychological and psychosocial factors must also be addressed
[8,83–86,124,136,289]. A major long-term objective of
To achieve the clinical aims and objectives of the healthcare model for FH requires a dedicated multidisciplinary
service [110] that is best co-ordinated by a lipid disorders
clinic [26,71]. This service should be managed by suitably
credentialled personnel operating out of departments of internal medicine, endocrinology or cardiology. Physicians who
regularly manage patients with FH should have specialist
training in clinical lipidology and competencies in the prevention of CVD [26,72], and be credentialled to supervise the
training of junior physicians. Uncomplicated patients could
be referred back to their GPs for long-term care but should be
reviewed annually in a lipid disorders clinic. Existing clinics
may need to up-skill their approaches to cascade screening
for FH [9,290]. GPs should actively seek index cases in their
practice by recognizing the importance of the family history
of CVD and marked hypercholesterolaemia [68]. However,
the optimal method for systematic identification of FH in
primary care needs to be defined and a specific MoC devised
248
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Influencers & Stakeholders
Department of Health
Policy Makers
Health Networks
Atherosclerosis Association
Family Support Group
National Heart Foundation
Audit & Research
Program
Registry, Clinical &
basic Science, Clinical
trials, Epidemiology &
Health Economics
Specialist & Primary Care
Physicians, Physiciansin-training.
Training,
Credentialing,
Professional development
Structured Clinical
Management Program
Shared between specialist
clinics and primary care
Structured Education
Program
For community and health
providers, multidisciplinary
case conferences, journal
clubs, accreditation
Specialised AdultPaediatric Service
Family Clinics
Patient & Family
Support Groups
Consultation,
Education,
Advocacy
Administrative, Secretarial &
Information Technology
Services
Support for Clinics, Outreach
services and FH Registry
Cardiac & Imaging
Facilities
Stress testing
Ultrasonography
Echocardiography
Cardiac CT Scanning
Clinical Liaison
Cardiology
Cardiac Rehabilitation
Cardiothoracic Surgery
Stroke unit
Vascular Surgery
Specialist Nurses & Allied
Health Support
Pharmacy / Medication support
Nutrition
Psychology
Exercise
Lay counselling
Training & Professional
Development
Medical Laboratory
Services
Routine and
Specialising in lipids and
lipoproteins
Specialised
Laboratory for
Genetic Testing
Service and
research
Clinical Genetics,
Family & Genetic
Counselling
Defined clinical
pathways
Fig. 12. The web of care for FH: the optimal service model.
and tested. Decentralization of the management of FH should
be a major objective of future developments. As FH services
develop further in the community, the specific roles of general practice and other members of the primary care team
will become clearer and inform future MoCs. Outreach lipid
clinics in the community would be a useful first step, but clinical protocols need developing and testing. Telehealth may
have a role in the management of FH in Australia, [291]
but its cost-effectiveness remains to be evaluated. Patients
aged <16 years should be seen in a paediatric clinic run
by a specialist in metabolic medicine; dietetic counselling
is essential in this age group. An integrated adult–paediatric
clinic may be useful for certain families. Nurses should be
trained in managing patients and families with FH, and preferably have some experience in the prevention of CVD and
competency in genetic counselling [72,110,121]. Cascade
screening, including risk notification, should be co-ordinated
by a health practitioner or specialised nurse working out
of a central FH clinic or a clinical genetics service, and
should include (where required) provision of an outreach
service. With appropriate specialist training, nurses could
effectively administer genetic counselling [121]. Beyond cascade screening, FH nurses may be involved in several other
areas. These include the clinical care of patients, medication
support, education and training, audit and research, enhancing multidisciplinary links and working with a patient support
group; nurse co-ordinated clinics and multidisciplinary case
discussions should be integral to the service. By example,
the effectiveness of nurse-led interventions has been demonstrated for management of non-cholesterol CVD risk factors,
such as hypertension and diabetes [292,293]. The role of
the dietician is essential, for dietary management and weight
regulation is a cornerstone of treatment [14–17a,32,33,133].
Additional input from health and adolescent psychologists
and exercise physiologists [136,209] may be required in special circumstances. Health literacy needs to be appropriately
addressed [172,173]. Suitably trained nurses may have the
best overall skills for co-ordinating the workflow necessary
for the optimal care of patients with FH. Clinical pharmacists can have a special role in managing non-adherence to
medication and in liaising with GPs [166–168]. The use of
lay counselling [252], particularly in patients who lack understanding of their condition and are non-adherent to treatment,
is another promising resource that requires evaluating in the
context of FH.
Inter-specialty links
Beyond the lipid disorders clinic strong links are essential
with departments of clinical genetics in respect of the need for
family and genetic counselling [66,110,115,121,237], particularly during the cascade screening process. A genetics
centred service may be appropriate for cascade screening,
but it may be more effective if genetic counsellors are
based in and work out of the lipid disorders clinic. Not all
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
families or patients with FH require genetic counselling,
but some exposure and basic training in the principles of
genetic counselling is important for both physicians and
nurses who manage FH [72,121]. The general skills of a
genetic counsellor may, on the other hand, be also more
broadly utilised in an FH service when dealing with certain clinical issues [121], such as adherence with medication
[170,208].
Many patients with FH may be diagnosed amongst those
attending departments of cardiology, cardiothoracic surgery,
stroke medicine and vascular surgery [22,38,60], and close
liaison with all these is essential. At least one in twenty people
with premature CVD cared for by these specialties may have
FH [22,38]. A structured system for the routine screening and
accurate detection of FH in these clinical settings is strongly
recommended.
An FH service should also have close links with laboratory medicine and access to routine and specialised laboratory
and clinical tests [71]. These include routine lipids and
lipoproteins, apoB, Lp(a), homocysteine and high sensitivity
C-reactive protein (CRP). Genetic testing should be carried
out in a NATA/RCPA accredited laboratory [266] that can
screen for all the major genes that cause FH. These laboratories must comply with the standards set by the NPAAC [264].
The clinical assessment of patients with FH also involves
access to cardiac and imaging facilities, including treadmill
testing, myocardial perfusion scanning, CUS and echocardiography [13,76–78]. Close links with the department of
cardiology are essential for the referral of symptomatic, or
high risk cases, that will require coronary angiography and
invasive interventions.
Links with a transfusion medicine unit is important for
administering and monitoring LDL-apheresis [215–217].
LDL-apheresis should be offered to all patients with homozygous (or compound heterozygous) FH and to heterozygotes
who are not at target LDL-cholesterol and have progressive
CHD or are intolerant of cholesterol lowering medications
[215–217].
13.3. Administrative and information technology
support
The clinical service should be underpinned by appropriate
administrative and secretarial support [25,26]. A specialised
database for storing clinical and family data and information technology support systems are essential for effective
provision of services [25]. Computerised programs should
have several key capabilities for dealing with pedigree drawing, work flow management, production of template letters,
archiving data, clinical audits and research. Other important requirements include a high level of data confidentiality
and security, and compatibility with other healthcare software systems. Favourable experience with one such software
program [243] has been reported [294], but requires further evaluation in full clinical service mode. A secure, high
quality software system is required to establish and develop
249
a national registry of FH patients that links family members across states. The International Cholesterol Foundation
(InterChol) is an integrator and facilitator of communications between clinical services worldwide; its webpage
(www.interchol.org/links) provides links to several international bodies that deal with FH. Details of websites that could
be useful for clinical services caring for FH are given in
Appendix 8.
13.4. Clinical governance: audit, education, training
Efficient clinical governance is fundamental to all aspects
of an FH service [37,295]. Clinical governance should
entail holding regular multidisciplinary conferences, focusing on the quality and efficiency of all aspects of the
service. Retrospective and ongoing clinical audits are essential for monitoring the efficiency of service delivery. A
well-designed and comprehensive clinical registry can provide invaluable information for improving the quality of
care for FH [296], consistent with requirements for all
inherited cardiovascular conditions [110,297]. Continuing
education and training of all healthcare providers is essential for professional development and service improvement.
Regular journal clubs can inform on recent advances in
the diagnosis and care of patients with FH. Essential for
clinical governance is a clearly identifiable management
structure headed by a medical director and a clinic or nurse
manager [71]. Standard operating procedures, staff responsibilities and credentialling, documentation and essential
resources must all be clearly defined. Marketing the clinic,
increasing the number of referrals, devising an effective business plan and generating income for the service, as well
as promoting collaborative research, are all highly desirable requirements for developing an effective clinic service
for FH.
13.5. Patient and family support groups
Every effort should be made to initiate and sustain an
active association for patients and families affected by FH
via a support group [250,251]. This forum provides a network for individuals and families to form an advocacy group
to develop and promote services within the community, as
well as a nucleus for learning and sharing information that
is critical for the detection, management and reduction of
risk in families with FH. The perceptions and views of
patients and their families are clearly important in developing and improving health services for FH [110]. Patient
support for families and the raising of community and government awareness about FH is an important function of the
support groups. A national patient support organisation could
afford the best means of establishing a national registry of
both patients and services, including clinics undertaking cascade screening. Such an FH registry could be incorporated
into the Australian National Genetic Heart Disease Registry
[297].
250
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
13.6. Into the future: chronic care model,
commissioning, evaluation
Future design and development of health provision for
FH needs to take place within the framework of the
Chronic Care Model [298], and hence in a positive policy environment [299]. This will require interacting and
establishing partnerships with a wide spectrum of stakeholders, including patient support groups, heart foundations
and related non-government organizations, health networks,
health economists, policy makers and health ministers. In
time, the care of FH could be integrated into national and state
services for inherited cardiovascular conditions [110]. There
is a major need to increase public and health provider awareness of FH through continuing education programs [288].
Academic health service systems may be ideally suited for
co-ordinating and integrating the health care of FH and other
inherited cardiovascular disorders, but these need to be developed in Australia [300]. Publishing guidance on the care of
FH is one thing, but effectively implementing recommendations is another, a gap well emphasised by a recent report
from the UK [301]. Addressing the national and international
gaps in the management of hypercholesterolaemia at a community level will benefit the detection and care of patients
with FH [174–178]. Programs that are successful in delivering high quality services for FH also have an obligation in
advising on and supporting the development of such services
at a national and international level. We emphasize that an
optimal FH service provides a classical paradigm and standard for the detection and treatment of other disorders that
cause premature atherosclerosis that need attention in their
own right. Finally, we strongly recommend that our MoC for
FH be commissioned and incorporated into healthcare delivery and prevention strategies in our community with a very
high level of priority. It should also be subjected to regular
auditing and health economic evaluation, thereby allowing
the MoC to grow into a standard of excellence for the care of
all patients with FH.
Appendix 1. FH Australasia Network Consensus
Group and Process
Chair
Winthrop Prof. Gerald F. Watts (Lipid Disorders Clinic,
Metabolic Research Centre and Department of Internal
Medicine, Royal Perth Hospital, University of Western
Australia, Perth, Western Australia).
Writing committee
Winthrop Prof. Gerald F. Watts, Associate Prof. David
R Sullivan (Department of Biochemistry and Lipid Clinic,
Royal Prince Alfred Hospital, University of Sydney), Dr
Nicola Poplawski (SA Clinical Genetics Service, Women’s
and Children’s Hospital, North Adelaide, and Genetics and
Molecular Pathology Directorate, SA Pathology (WCH site),
North Adelaide), Prof. Frank van Bockxmeer (Cardiovascular Genetics Laboratory, Royal Perth Hospital, University of
Western Australia).
Steering committee
Winthrop Prof. Gerald F. Watts, Associate Prof. David
Sullivan, Prof. Frank M. van Bockxmeer, Prof. Ian HamiltonCraig (Preventive Cardiology and Lipid Clinic, Gold Coast
Hospital, Griffith University, Queensland), Prof. Peter M.
Clifton (Baker IDI Heart and Diabetes Institute, South
Australia), Associate Prof. Richard O’Brien (University of
Melbourne, Victoria), Dr Warrick Bishop (Department of
Cardiology, Calvary Cardiac Centre, Calvary Health Care,
Tasmania), Prof. Peter George (Biochemistry and Pathology,
Canterbury Health Laboratories, Lipid Clinic, Christchurch
Hospital, Christchurch, New Zealand).
Contributors
Prof Phillip J. Barter (Director, Heart Research Institute,
Sydney), Dr Timothy Bates (Lipid Disorders Clinic, Internal Medicine, Royal Perth Hospital), Clinical Prof. John
R. Burnett (Core Clinical Pathology & Biochemistry, PathWest Laboratory Medicine WA, Lipid Disorders Clinic,
Royal Perth Hospital, University of Western Australia), Dr
John Coakley (Paediatrics and Biochemistry, The Children’s
Hospital Westmead, Sydney), Prof. Patricia Davidson (Cardiovascular and Chronic Care, Curtin University, and Nursing
Research, St Vincent’s Hospital, Sydney), Winthrop Prof.
Jon Emery (School of Primary, Aboriginal and Rural Health
Care, University of Western Australia), Dr Andrew Martin (Department of Paediatric and Adolescent Medicine,
Princess Margaret Hospital, Perth), Dr Waleed Farid (FH
Patient Support Group of Western Australia), Ms Lucinda
Freeman (Department of Molecular and Clinical Genetics, Royal Prince Alfred Hospital, Sydney), Prof. Elizabeth
Geelhoed (Health Economics and Policy, School of Population Health, University of Western Australia), Ms Amanda
Juniper (Office of Population Health Genomics, Department of Health, Government of Western Australia), Dr
Alexa Kidd (Clinical Genetics, Canterbury Health Laboratories, Christchurch Hospital, New Zealand), Associate Prof.
Karam Kostner (Cardiac Imaging Group, Department of Cardiology, Mater Hospital, University of Queensland), Prof.
Ines Krass (Pharmacy Practice, Faculty of Pharmacy, University of Sydney), Mr Michael Livingston (International
Cholesterol Foundation, Sutton-Courtney, Oxfordshire, UK),
Ms Suzy Maxwell (Office of Population Health Genomics,
Department of Health, Government of Western Australia),
Prof. Peter O’Leary (Office of Population Health Genomics,
Department of Health, Government of Western Australia),
Ms Amal Owaimrin (Department of Dietetics, Familial
Hypercholesterolaemia Clinical Support Service, Auburn
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
251
Hospital, Sydney), Emeritus Prof. Trevor Redgrave (Lipid
Disorders Clinic, Royal Perth Hospital, Department of Physiology, School of Medicine and Pharmacology, University
of Western Australia), Ms Nicola Reid (Cardiovascular Prevention and Lipid Disorders Clinic, Christchurch Hospital,
New Zealand), Ms Lynda Southwell (FH Western Australia,
Royal Perth Hospital, University of Western Australia), Dr
Graeme Suthers (SA Clinical Genetics Service, Genetics
& Molecular Pathology Directorate, Women’s & Children’s
Hospital, Adelaide), Prof. Andrew Tonkin (Cardiovascular
Research Unit, Monash University, Melbourne, Victoria),
Prof. Simon Towler (Chief Medical Officer, Health Networks,
Department of Health, Government of Western Australia),
Prof. Ronald Trent (Department of Molecular & Clinical Genetics, Royal Prince Alfred Hospital, University of
Sydney).
PD, PJB), Abbott (GFW, DS, AT, TB, IHC, PJB, ROB),
Boehringer-Ingelheim (GFW, AT, TB, WB, ROB), SanofiAventis (GFW, DS, TB, WB, IHC), Novartis (GFW, AT,
IHC, PJB, ROB), GlaxoWellcome (GFW, IHC, ROB), Bristol Myers Squibb (AT, ROB), Servier (TB, WB, IHC, PMC,
ROB), Roche (GFW, PJB, DS).
Consensus process
Dutch Lipid Clinic Network Criteria for making a diagnosis of FH in adults [34].
The Steering Committee met three times in Adelaide,
Sydney and Brisbane, organised and chaired by GFW. A variable number of other contributors attended these meetings or
were invited to comment on evolving drafts of the paper via
email or telephone. The first meeting discussed a preliminary model of care for FH from Western Australia funded by
the Australia Better Health Initiative and based on material
initially developed by FH Australasia (GFW, DS). The second meeting reviewed the core evidence on FH (published
in the English language on PubMed and webaddresses) and
discussed and critically commented on a first draft of the
consensus paper written by GFW. GFW, DS, NP and FvB
then re-drafted sections of the paper, after reviewing further
comments by all members of the group. At the third meeting,
the Steering Committee re-examined a pre-final draft of the
paper and reached consensus on the principal recommendations of the model of care. GFW then prepared a final draft of
the paper. All members approved the final document before
submission.
Funding
The meetings of the FH Australasia Network Consensus Group were supported by educational grants to the
Australian Atherosclerosis Society from Pfizer, Abbott,
Astra-Zeneca and MSD. These companies were not present
at the Consensus Panel meetings and made no contributions to the design, content or final approval of the
document.
Disclosures
Some members of the Consensus Group have received
lecture honoraria, consultancy fees, and/or research funding from: Pfizer (GFW, DS, AT, TB, WB, IHC, PD, PMC,
PJB, ROB), Astra Zeneca (GFW, DS, AT, TB, WB, IHC,
NR, PMC, ROB), MSD (GFW, DS, AT, TB, WB, IHC, NR,
Acknowledgements
We thank Jennifer Seabrook from Meetings First and
the Australian Atherosclerosis Society for co-ordinating the
meetings of the FH Australasia Network Consensus Group.
Appendix 2. Dutch Lipid Clinic Network Criteria for
FH
Criteria
Family history
First degree relative with known premature coronary
and/or vascular disease (Men <55 years, Females <60
years), OR
First degree relative with known LDL-cholesterol
>95th percentile for age and sex
First degree relative with tendon xanthomata and/or
arcus cornealis, OR
Children aged <18 years with LDL-cholesterol
>95th percentile for age and sex
Clinical history
Patient with premature coronary artery disease (age
as above)
Patient with premature cerebral or peripheral vascular
disease (age as above)
Physical examination
Tendon xanthomata
Arcus cornealis at age <45 years
LDL-cholesterol (mmol/L)
LDL-C ≥8.5
LDL-C 6.5–8.4
LDL-C 5.0–6.4
LDL-C 4.0–4.9
DNA analysis—functional mutation in the LDLR,
APOB or PCSK9 gene
Score
1
2
2
1
6
4
8
5
3
1
8
Stratification
Total score
Definite FH
Probable FH
Possible FH
Unlikely FH
>8
6–8
3–5
<3
252
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Appendix 3. Simon Broome Criteria for FH
Simon Broome Criteria for the diagnosis of FH [12,41].
Definite FH
Raised cholesterol:
In children (<16years): total cholesterol > 6.7 mmol/L OR LDL-C > 4.0 mmol/L
In adults (>16 years): Total cholesterol > 7.5 mmol/L OR LDL-C > 4.9 mmol/L
AND
Tendon xanthomata in the patient or in a first or second degree relative
OR
DNA based evidence of a LDL-receptor, familial defective apo B-100 or PCSK9 mutation
Possible FH
Raised cholesterol:
In children (<16 years): total cholesterol > 6.7 mmol/L OR LDL-C > 4.0 mmol/L
In adults (> 16 years): total cholesterol > 7.5 mmol/L OR LDL-C > 4.9 mmol/L
AND one of the following:
Family history of premature myocardial infarction
MI at <50 years in second degree,
MI at <60 years in first degree relatives.
OR
Family history of raised cholesterol
In adult (>16 years), first or second degree relatives: total cholesterol >7.5 mmol/L
In child (<16 years), first degree relatives: total cholesterol >6.7 mmol/L
Appendix 4. MEDPED Criteria for FH
MEDPED Criteria for the diagnosis of FH [42].
Age (years) Total and LDL-cholesterol (mmol/L) criteria for Diagnosing Probable Heterozygous Familial Hypercholesterolaemia (FH)
<20
20–29
30–39
≥40
1st degree relative with FH
TC (LDL-C)
2nd degree relative with FH
TC (LDL-C)
3rd degree relative with FH
TC (LDL-C)
General population
TC (LDL-C)
5.7 (4.0)
6.2 (4.4)
7.0 (4.9)
7.5 (5.3)
5.9 (4.3)
6.5 (4.6)
7.2 (5.2)
7.8 (5.6)
6.2 (4.4)
6.7 (4.8)
7.5 (5.4)
8.0 (5.8)
7.0 (5.2)
7.5 (5.7)
8.8 (6.2)
9.3 (6.7)
TC = total cholesterol.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Appendix 5. Typical examination features of FH
Illustrations showing typical examination features of FH
253
254
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Appendix 6. Hypothetical Pedigree
I
Index case
II
III
Male
Female
FH phenotype expressed; note vertical
transmission of phenotype over each
succeeding generation
Hypothetical pedigree tree depicting dominantly inherited phenotype in FH.
Appendix 7. ‘Real case’ Pedigree
I
South African
Died age 50
II
High cholesterol & Angina
High cholesterol & Angina
Fatal MI age 60
Fatal MI age 50
Fatal MI age 50
Index case
III
IV
TC 9.2
Age 38
TC 9.8
Age 24
High cholesterol
3 x CABG
Age 46
High cholesterol
Cholesterol
unknown
Cholesterol
unknown
1 x CABG
Fatal MI age 50
4 x CABG
By age 40
High cholesterol
Age 26
2 x CABG
Fatal MI age 40
TC 10.2
Age 28
Cholesterol
unknown
Cholesterol
unknown
High cholesterol
2 x CABG, 2 x MI
Age 38
Cholesterol
unknown
Carriers of LDL-R gene mutation
exhibiting the FH clinical phenotype
Male
Female
Pedigree of actual family with a mutation in the LDL-receptor gene. This pedigree illustrates the dominant inheritance of
hypercholesterolaemia and premature CHD in a family with FH (LDL-R gene = c.681C > G (Asp227glu) in exon 4). TC = total
plasma cholesterol (mmol/L), MI = Myocardial Infarction, CABG = Coronary Artery Bypass Grafting.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
Appendix 8. Selected websites for clinical services
Selected websites that may be useful for clinical services
caring for patients with FH.
• International Cholesterol Foundation
www.interchol.org
New foundation, formed from the merger of
MEDPED-International and HEART-EU, that has a strong focus on
FH and provides useful links to international websites of interest to
patients, researchers and health professionals.
• HEART UK
www.heartuk.org.uk
Leading UK cholesterol charity that provides extensive resources
for health professionals, patients and families on all aspects of the
detection and management of FH.
• Public Health Genomics Foundation, UK
www.phgfoundation.org
International foundation that publishes authoritative reports on the
role of advances in genomics in health care, and has a particularly
excellent document on services for inherited cardiovascular
conditions.
• National Heart Foundation, Australia
www.heartfoundation.org.au
Leading Australian charity that provides a wealth of resources for
health professionals and the community on all aspects of primary
and secondary prevention of cardiovascular disease.
• British Heart Foundation
www.bhf.org.uk
Leading British foundation provides excellent resources for health
professionals and patients, including informative videos on a wide
spectrum of conditions and risk factors.
• Centre for Genetics Education, New South Wales Health
www.genetics.com.au
Educational arm of NSW Genetic Service that provides genetic
information of individuals and families affected by genetic
conditions and health professionals who work with them. Activities
include workshops and training programs.
• Human Genetics Society of Australasia
www.hgsa.org.au
Premier Australasian society that provides educational materials,
training, polices, guidelines and position statements on all aspects
of human genetics.
• Lipids Online, Baylor College of Medicine
www.lipidsonline.org
Established on-line facility, coordinated by Baylor College of
Medicine (Houston, Texas, USA), providing resources (slides,
visual meetings, commentaries), for clinicians, researches and
educators on several aspects of dyslipidaemia, atherosclerosis and
cardiovascular disease.
• National Lipids Association (NLA), USA
www.lipid.org
US based multidisciplinary specialty society providing education,
training, guidelines and position statements on all aspects of the
detection and management of dyslipidaemia and related disorders.
• Learn Your Lipids, NLA
www.learnyourlipids.com
Information for patients with dyslipidaemia, including FH, as
provided by the foundation of the National lipid Association in the
US.
• New Zealand Guidelines Group
www.nzgg.org.nz
New Zealand group of experts that specialises in developing and
implementing guidelines for best clinical practice; excellent
resources on the assessment and management of all cardiovascular
risk factors.
255
• Rational Assessment of Drugs and Research (RADAR),
National Prescribing Service (NPS)
www.nps.org.au/health professionals/publications/nps radar
Evidence based assessment of all new drugs, PBS listings and latest
research for health professionals provided by the NPS, an
independent organisation funded by the Australian Government
Department of Health and Ageing.
• Make Early Diagnosis Prevent Early Death (MEDPED) FH
www.medped.org
US based website of the original MEDPED Project coordinated by
the University of Utah School of Medicine (Salt Lake City, UT,
USA) focusing on all aspects of the management of FH, including
education of patients and families and the first attempt at
establishing a US registry.
• Office of Population Genomics, FH Pilot Cascade Screening
Program, Western Australia
www.genomics.health.wa.gov.au/fh
State funded office that aims to translate genomic knowledge into
health benefits for WA health; resources provided relevant to the
detection and management of FH
• Wales FH Testing Service, Cardiff University
www.fhwales.co.uk
Leading FH service in the UK that provides useful information and
resources for clinical practice, including activities of FH Family
Forum.
• FH Support Group of Western Australia
www.fhfamilysupportgroup.websyte.com.au
Website of the first support group in Australia for families with FH;
provides relevant information, communication and support services.
• FH Guideline Implementation Team Toolkit
www.heartuk.org.uk/FHToolkit
Invaluable resource for implementing the seminal NICE guideline
71 on identification and management of FH.
• FH Australasia Network, Australian Atherosclerosis Society
www.athero.org.au/FH
Website of the FH Australasian Network, updated in 2011
References
[1] Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic
causes of monogenic heterozygous familial hypercholesterolaemia: a HuGE prevalence review. Am J Epidemiol 2004;160:
407–20.
[2] Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia:
new insights in pathogenesis and treatment. J Clin Invest
2003;111:1795–803.
[3] Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes
of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med
2007;4:214–25.
[4] Marks D, Thorogood M, Neil HAW, Wonderling D, Humphries SE.
Comparing costs and benefits over a 10 year period for strategies
for familial hypercholesterolaemia screening. J Public Health Med
2003;25:47–52.
[5] World Health Organization. Familial hypercholesterolaemia: report
of a WHO consultation. Paris: World Health Organisation;
1997.
[6] Marks D, Thorogood M, Humphries S, Neil HAW. Cascade screening
for familial hypercholesterolaemia: implications of a pilot study for
national screening programmes. J Med Screen 2006;13: 156–9.
[7] Leren TP. Cascade genetic screening for familial hypercholesterolemia. Clin Genet 2004;66:483–7.
[8] Hadfield SG, Humphries SE. Implementation of cascade testing for
the detection of familial hypercholesterolaemia. Curr Opin Lipidol
2005;16:428–33.
256
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[9] Hadfield SG, Horara S, Starr BJ, et al. Family tracing to identify
patients with familial hypercholesterolaemia: the second audit of the
Department of Health familial hypercholesterolaemia cascade testing
project. Ann Clin Biochem 2009;46:24–32.
[10] Australian and New Zealand Horizon Scanning Unit Australian
Government. Genetic screening for familial hypercholesterolaemia.
Canberra: Department of Health and Ageing; 2007.
[11] Umans-Eckenhausen MAW, Defesche JC, Sijbrands EJG, Scheerder
RLJM, Kastelein JJP. Review of first 5 years of screening for
familial hypercholesterolaemia in the Netherlands. Lancet 2001;357:
165–8.
[12] National Institute for Health and Clinical Excellence, The
National Collaborating Centre for Primary Care. NICE clinical
guideline 71: identification and management of familial hypercholesterolaemia; 2008. Available from: www.nice.org.uk/nicemedia/
pdf/CG071NICEGuideline.pdf [2011].
[13] Civeira F. Guidelines for the diagnosis and management of
heterozygous familial hypercholesterolaemia. Atherosclerosis
2004;173:55–68.
[14] National Cholesterol Education Program (NCEP). Third report of
the National Cholesterol Education Program (NCEP) Expert Panel
on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation
2002;106:3143–421.
[15] Tonkin A, Barter P, Best J, et al. National Heart Foundation of
Australia and the Cardiac Society of Australia and New Zealand:
position statement on lipid management—2005. Heart Lung Circ
2005;14:275–91.
[16] Graham I, Atar D, Borch-Johnsen K, et al. European guidelines on
cardiovascular disease prevention in clinical practice: executive summary. Eur Heart J 2007;28:2375–414.
[17] (a) Brunzell JD, Davidson M, Furberg CD, et al. Lipoprotein
management in patients with cardiometabolic risk. Diabetes Care
2008;31:811–22;
(b) Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS
Guidelines for the management of dyslipidaemias. Eur Heart J
2011;32:1769–818.
[18] Neil HAW, Hammond T, Huxley R, Matthews DR, Humphries
SE. Extent of underdiagnosis of familial hypercholesterolaemia in
routine practice: prospective registry study. Br Med J 2000;321:
148.
[19] Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol
lowering treatment of patients with familial hypercholesterolemia:
a large cross-sectional study in the Netherlands. Atherosclerosis
2010;209:189–94.
[20] Huijgen R, Kindt I, Verhoeven S, et al. Two years after molecular
diagnosis of familial hypercholesterolemia: majority on cholesterollowering treatment but a minority reaches treatment goal. PLoS ONE
2010;5:e9220.
[21] Hamilton-Craig I. Case-finding for familial hypercholesterolemia in
the Asia-Pacific region. Semin Vasc Med 2004;4:87–92.
[22] Bates TR, Burnett JR, van Bockxmeer FM, Hamilton S, Arnolda
L, Watts GF. Detection of familial hypercholesterolaemia: a major
treatment gap in preventative cardiology. Heart Lung Circ 2008;17:
411–3.
[23] Muir LA, George PM, Laurie AD, Reid N, Whitehead L. Preventing
cardiovascular disease: a review of the effectiveness of identifying
the people with familial hypercholesterolaemia in New Zealand. N Z
Med J 2010;123:97–102.
[24] Davidson PM, Halcomb EJ, Hickman LD, Phillips JL, Graham B.
Beyond the rhetoric: what do we mean by a ‘model of care’? Aust J
Adv Nurs 2006;23:47–55.
[25] HEART UK. FH guideline implementation team toolkit; 2010. Available from: www.heartuk.org.uk/FHToolkit.
[26] Datta BN, McDowell IF, Rees A. Integrating provision of specialist
lipid services with cascade testing for familial hypercholesterolaemia.
Curr Opin Lipidol 2010;21:366–71.
[27] Stephenson SH, Larrinaga-Shum S, Hopkins PN. Benefits of the
MEDPED treatment support program for patients with familial hypercholesterolemia. J Clin Lipidol 2009;3:94–100.
[28] Hayden MR, Josephson R. Development of a program for identification of patients with familial hypercholesterolaemia in British
Columbia: a model for prevention of coronary disease. Am J Cardiol
1993;72:25D–9D.
[29] Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and
adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol
2011;5:133–40.
[30] Wierzbicki AS, Humphries SE, Minhas R, on behalf of the Guideline
Development Group. Familial hypercholesterolaemia: summary of
NICE guidance. Br Med J 2008;337:a1095.
[31] Minhas R, Humphries SE, Qureshi N, Neil HAW, on behalf of
the NGDG. Controversies in familial hypercholesterolaemia: recommendations of the NICE guideline development group for the
identification and management of familial hypercholesterolaemia.
Heart 2009;95:584–7.
[32] National Heart Foundation of Australia, Cardiac Society of Australia
and New Zealand. Reducing risk in heart disease 2007: guidelines
for preventing cardiovascular events in people with coronary heart
disease; 2008. Available from: www.heartfoundation.org.au/Professional Information/Clinical Practice/Prevention/Pages/default.aspx.
[33] New Zealand Guidelines Group. The assessment and management of
cardiovascular risk. Wellington: NZGG; 2003.
[34] World Health Organization. Familial hypercholesterolaemia. Report
of a second WHO consultation. Geneva: World Health Organization;
1999.
[35] Burnett JR, Ravine D, van Bockxmeer FM, Watts GF. Familial
hypercholesterolaemia: a look back, a look ahead. Med J Aust
2005;182:552–3.
[36] Sullivan D, Members of the CSANZ Cardiovascular Genetics Working Group. Guidelines for the diagnosis and management of familial
hypercholesterolaemia. Heart Lung Circ 2007;16:25–7.
[37] Familial Hypercholesterolaemia Western Australia Program Committee. Model of care familial hypercholesterolaemia; 2008. Available
from: www.genomics.health.wa.gov.au/fh/index.cfm [cited 28 March
2011].
[38] Watts GF, van Bockxmeer FM, Bates T, Burnett JR, Juniper A,
O’Leary P. A new model of care for familial hypercholesterolaemia
from Western Australia: closing a major gap in preventive cardiology.
Heart Lung Circ 2010;19:419–22.
[39] National Health and Medical Research Council (NHMRC). NHMRC
additional levels of evidence and grades for recommendations for
developers of guidelines. Pilot program 2005–2007; 2005.
[40] Watts GF, Lewis B, Sullivan DR. Familial hypercholesterolaemia: a
missed opportunity in preventive medicine. Nat Clin Pract Cardiovasc
Med 2007;4:404–5.
[41] Scientific Steering Committee, Simon Broome Register Group. Risk
of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register
group. Br Med J 1991;303:893–6.
[42] Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria
validated by molecular genetics. Am J Cardiol 1993;72:171–6.
[43] Marks D, Thorogood M, Neil HAW, Humphries SE. A review on the
diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168:1–14.
[44] Damgaard D, Larsen ML, Nissen PH, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in
a Danish population. Atherosclerosis 2005;180:155–60.
[45] Hopkins PN, Toth PP, Ballantyne CM, Rader DJ. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening
recommendations from the National Lipid Association Expert Panel
on Familial Hypercholesterolemia. J Clin Lipidol 2011;5:S9–17.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[46] Starr B, Hadfield G, Hutton BA, et al. Development of sensitive and specific age-and gender-specific low-density lipoprotein
cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin Chem Lab Med
2008;46:791–803.
[47] Tiyyagura SR, Smith DA. Standard lipid profile. Clin Lab Med
2006;26:707–32.
[48] Daniels SR, Greer FR, and the Committee on Nutrition. Lipid
screening and cardiovascular health in childhood. Pediatrics
2008;122:198–208.
[49] Freedman DS, Wang YC, Dietz WH, Xu J-H, Srinivasan SR, Berenson
GS. Changes and variability in high levels of low-density lipoprotein
cholesterol among children. Pediatrics 2010;126:266–73.
[50] Chow CK, Islam S, Bautista L, et al. Parental history and myocardial
infarction risk across the world: the interheart study. J Am Coll Cardiol
2011;57:619–27.
[51] Emery JD, Walter FM, Ravine D. Family history: the neglected risk
factor in disease prevention. Med J Aust 2010;192:677–8.
[52] Walter FM, Emery J. ‘Coming down the line’—patients’ understanding of their family history of common chronic disease. Arch Fam Med
2005;3:405–14.
[53] O’Neill SM, Rubinstein WS, Wang C, et al. Familial risk for common
diseases in primary care: the Family HealthwareTM Impact Trial. Am
J Prev Med 2009;36:506–14.
[54] Bates TR, Poulter EB, Van Bockxmeer FM, Watts GF. Family history: the neglected risk factor in disease prevention. Med J Aust
2010;193:429–30 [Letter].
[55] Hopkins PN, Stephenson S, Wu LL, Riley WA, Xin Y, Hunt SC. Evaluation of coronary risk factors in patients with heterozygous familial
hypercholesterolemia. Am J Cardiol 2001;87:547–53.
[56] Jansen ACM, van Aalst-Cohen ES, Tanck MW, et al. The contribution of classical risk factors to cardiovascular disease in
familial hypercholesterolaemia: data in 2400 patients. J Intern Med
2004;256:482–90.
[57] De Sauvage Nolting PRW, Defesche JC, Buirma RJA, Hutten BA,
Lansberg PJ, Kastelein JJP. Prevalence and significance of cardiovascular risk factors in a large cohort of patients with familial
hypercholesterolaemia. J Intern Med 2003;253:161–8.
[58] Sniderman A, Couture P, de Graaf J. Diagnosis and treatment
of apolipoprotein B dyslipoproteinemias. Nat Rev Endocrinol
2010;6:335–46.
[59] Genest J, McPherson R, Frohlich J, et al. 2009 Canadian Cardiovascular Society/Canadian guidelines for the diagnosis and treatment
of dyslipidemia and prevention of cardiovascular disease in the
adult—2009 recommendations. Can J Cardiol 2009;25:567–79.
[60] Dorsch MF, Lawrance RA, Durham NP, Hall AS. Familial
hypercholesterolaemia is underdiagnosed after AMI. Br Med J
2001;322:111.
[61] McKenney JM, Ganz P, Wiggins BS, Saseen JS. Statins. In:
Ballantyne CM, editor. Clinical lipidology: a companion to Braunwald’s heart disease. Philadelphia: Saunders, Elsevier; 2009. p. 253–
280.
[62] Ritchie SK, Murphy EC-S, Ice C, et al. Universal versus targeted blood
cholesterol screening among youth: the cardiac project. Pediatrics
2010;126:260–5.
[63] Wald DS, Bestwick JP, Wald NJ. Child-parent screening for familial
hypercholesterolaemia: screening strategy based on a meta-analysis.
Br Med J 2007;335:599.
[64] (a) Daniels SR, Gidding SS, de Ferranti SD. Pediatric aspects of familial hypercholesterolemias: recommendations from the National Lipid
Association Expert Panel on Familial Hypercholesterolemia. J Clin
Lipidol 2011;5:S30–7;
(b) Descamps OS, Tenoutasse S, Stephenne X, et al. Management
of familial hypercholesterolemia in children and young adults: Consensus paper developed by a panel of lipidologists, cardiologists,
paediatricians, nutritionists, gastroenterologists, general practitioners
and a patent organization. Atherosclerosis 2011 (in press).
257
[65] Marks D, Wonderling D, Thorogood M, Lambert H, Humphries
SE, Neil HAW. Cost effectiveness analysis of different approaches
of screening for familial hypercholesterolaemia. Br Med J
2002;324:1303–9.
[66] Humphries SE, Norbury G, Leigh S, Hadfield SG, Nair D. What is
the clinical utility of DNA testing in patients with familial hypercholesterolaemia? Curr Opin Lipidol 2008;19:362–8.
[67] Gray J, Jaiyeola A, Whiting M, Modell M, Wierzbicki AS. Identifying patients with familial hypercholesterolaemia in primary care:
an informatics-based approach in one primary care centre. Heart
2008;94:754–8.
[68] Qureshi N, Humphries SE, Seed M, Rowlands P, Minhas R, NICE
Guideline Development Group. Identification and management of
familial hypercholesterolaemia: What does it mean to primary care?
Br J Gen Pract 2009;59:773–8.
[69] Australian Government Department of Health and Ageing.
A27 health assessment provided for people aged 45–49 years
who are at risk of developing chronic disease; 2011. Available
from:
www9.health.gov.au/mbs/fullDisplay.cfm?type=note&qt=
NoteID&q=A27 [9th March 2011].
[70] Peterson G, Fitzmaurice K, Kruup H, Jackson S, Rasiah R. Cardiovascular risk screening program in Australian community pharmacies.
Pharm World Sci 2010;32:373–80.
[71] Mason CM. Development and management of a lipid clinic. In:
Davidson MH, Toth PP, Maki KC, editors. Contemporary cardiology: therapeutic lipidology. Totowa, NJ: Human Press Inc; 2007. p.
441–80.
[72] Bairey Merz CN, Alberts MJ, Balady GJ, et al. ACCF/AHA/ACP
2009 competence and training statement: a curriculum on prevention
of cardiovascular disease. J Am Coll Cardiol 2009;54:1336–63.
[73] Austin MA, Hutter CM, Zimmern RL, Humphries SE. Familial hypercholesterolemia and coronary heart disease: a HuGE association
review. Am J Epidemiol 2004;160:421–9.
[74] Oosterveer DM, Versmissen J, Schinkel AF, Langendonk JG, Mulder
M, Sijbrands EJ. Clinical and genetic factors influencing cardiovascular risk in patients with familial hypercholesterolemia. Clin Lipidol
2010;5:189–97.
[75] Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J 2010;31:2844–53.
[76] Taylor AJ, Merz CNB, Udelson JE. Executive summary—can
atherosclerosis imaging techniques improve the detection of patients
at risk for ischemic heart disease? J Am Coll Cardiol 2003;41:
1860–2.
[77] Naghavi M, Falk E, Hecht HS, et al. From vulnerable plague to vulnerable patient—part III: executive summary of the Screening for Heart
Attack Prevention and Education Task Force Report. Am J Cardiol
2007;99:1481–2.
[78] Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a
report of the American College of Cardiology Foundation/American
Heart Association Task Force on Practice Guidelines. Circulation
2010;122:2748–64.
[79] Michos ED, Nasir K, Rumberger JA, et al. Relation of family history
of premature coronary heart disease and metabolic risk factors to risk
of coronary arterial calcium in asymptomatic subjects. Am J Cardiol
2005;95:655–7.
[80] Hutter CM, Austin MA, Humphries SE. Familial hypercholesterolemia, peripheral arterial disease and stroke: a HuGE minireview.
Am J Epidemiol 2004;160:430–5.
[81] Wiegman A, de Groot E, Hutten BA, et al. Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia.
Lancet 2004;363:369–70.
[82] Dogan S, Duivenvoorden R, Grobbee DE, et al. Ultrasound protocols to measure carotid intima-media thickness in trials; comparison
of reproducibility, rate of progression, and effect of intervention in
subjects with familial hypercholesterolemia and subjects with mixed
dyslipidemia. Ann Med 2010;42:447–64.
258
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[83] Andersen LK, Jensen HK, Juul S, Faergeman O. Patients’ attitudes
toward detection of heterozygous familial hypercholesterolemia.
Arch Intern Med 1997;157:553–60.
[84] Agard A, Bolmsjo I, Hermeren G, Wahlstom J. Familial hypercholesterolemia: ethical, practical and psychological problems from
the perspectives of patients. Patient Educ Couns 2005;57:162–7.
[85] Claassen L, Henneman L, Kindt I, Marteau TM, Timmermans DRM.
Perceived risk and representations of cardiovascular disease and
preventive behaviour in people diagnosed with familial hypercholesterolemia. J Health Psychol 2010;15:33–43.
[86] Marteau T, Senior V, Humphries SE, et al. Psychological impact of
genetic testing for familial hypercholesterolemia within a previously
aware population: a randomized controlled trial. Am J Med Genet
2004;128A:285–93.
[87] Australian Bureau of Statistics. 4233.0—health literacy,
Australia; 2006. Available from: www.abs.gov.au/AUSSTATS/
abs@.nsf/DetailsPage/4233.02006.
[88] von Wagner C, Steptoe A, Wolf MS, Wardle J. Health literacy and
health actions: a review and a framework from health psychology.
Health Educ Behav 2009;36:860–77.
[89] Frich JC, Ose L, Malterud K, Fugelli P. Perceived vulnerability to heart
disease in patients with familial hypercholesterolemia: a qualitative
interview study. Arch Fam Med 2006;4:198–204.
[90] Walter FM, Emery J, Braithwaite D, Marteau TM. Lay understanding
of familial risk of common chronic diseases: a systematic review and
synthesis of qualitative research. Arch Fam Med 2004;2:583–94.
[91] Mark
DB,
Berman
DS,
Budoff
MJ,
et
al.
ACCF/ACR/AHA/NASCI/SAIP/SCAI/SCCT 2010 expert consensus
document on coronary computed tomographic angiography: a report
of the American College of Cardiology Foundation Task Force on
Expert Consensus Documents. J Am Coll Cardiol 2010;55:2663–99.
[92] Lloyd-Jones DM. Cardiovascular risk prediction: basic concepts, current status, and future directions. Circulation 2010;121:1768–77.
[93] Dunstan D, Zimmet P, Welborn T, et al. Diabesity & associated disorders in Australia—2000: the accelerating epidemic. Melbourne: The
Australian Diabetes, Obesity and Lifestyle Study; 2001.
[94] Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb
Vasc Biol 2008;28:629–36.
[95] Thompson GR, HEART-UK LDL Apheresis Working Group. Recommendations for the use of LDL apheresis. Atherosclerosis
2008;198:247–55.
[96] Rodenburg J, Vissers MN, Wiegman A, Trip MD, Bakker HD,
Kastelein JJP. Familial hypercholesterolaemia in children. Curr Opin
Lipidol 2004;15:405–11.
[97] Kavey R-EW, Allada V, Daniels SR, et al. Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the
American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the
Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research. Cardiovascular
Nursing, and the Kidney in Heart disease; and the interdisciplinary
working group on quality of care and outcomes research. Circulation
2006;114:2710–38.
[98] McCrindle BW, Urbina EM, Dennison BA, et al. Drug therapy of
high risk lipid abnormalities in children and adolescents. Circulation
2007;115:1–20.
[99] van der Graaf A, Kastelein J, Wiegman A. Heterozygous familial
hypercholesterolaemia in childhood: cardiovascular risk prevention.
J Inherit Metab Dis 2009;32:699–705.
[100] O’Gorman C, Higgins M, O’Neill M. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in
children: evaluation of cholesterol changes and side effects. Pediatr
Cardiol 2009;30:482–9.
[101] de Jongh S, Lilien MR, Bakker HD, Hutten BA, Kastelein JJP,
Stroes ESG. Family history of cardiovascular events and endothelial dysfunction in children with familial hypercholesterolemia.
Atherosclerosis 2002;163:193–7.
[102] Steinberg D, Glass CK, Witztum JL. Evidence mandating earlier
and more aggressive treatment of hypercholesterolemia. Circulation
2008;118:672–7.
[103] Obarzanek E, Kimm SY, Barton BA, et al. Long term safety
and efficacy of a cholesterol lowering diet in children with elevated low density lipoprotein cholesterol: seven year results of the
dietary intervention study in children (DISC). Paediatrics 2001;107:
256–64.
[104] de Jongh S, Lilien MR, op’t Roodt J, Stroes ESG, Bakker HD,
Kastelein JJP. Early statin therapy restores endothelial function in
children with familial hypercholesterolemia. J Am Coll Cardiol
2002;40:2117–21.
[105] Avis HJ, Vissers MN, Stein EA, et al. A systematic review and
meta-analysis of statin therapy in children with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2007;27:1803–10.
[106] Arambepola C, Farmer AJ, Perera R, Neil HAW. Statin treatment
for children and adolescents with heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. Atherosclerosis
2007;195:339–47.
[107] Rodenburg J, Vissers MN, Wiegman A, et al. Statin treatment in
children with familial hypercholesterolemia: the younger, the better.
Circulation 2007;116:664–8.
[108] Vuorio A, Kuoppala J, Kovanen PT, et al. Statins for children with
familial hypercholesterolemia. Cochrane Database Syst Rev 2010;(7).
Art. No.: CD006401.
[109] van Maarle MC, Stouthard MEA, Bonsel GJ. Quality of life in a family
based genetic cascade screening programme for familial hypercholesterolaemia: a longitudinal study among participants. J Med Genet
2003;40, e3–e.
[110] Burton H, Alberg C, Stewart A. Heart to heart: inherited cardiovascular conditions services. A needs assessment
and service review. PHG Foundation; 2009. Available from:
www.phgfoundation.org/reports/4986/.
[111] Humphries SE, Galton D, Nicholls P. Genetic testing for familial hypercholesterolaemia: practical and ethical issues. Q J Med
1997;90:169–81.
[112] Humphries SE, Whittall RA, Hubbart CS, et al. Genetic causes of
familial hypercholesterolaemia in patients in the UK: relation to
plasma lipid levels and coronary heart disease risk. J Med Genet
2006;43:943–9.
[113] World Health Organization. Review of ethical issues in medical genetics. Geneva: World Health Organisation; 2003.
[114] European Society of Human Genetics. Genetic testing in asymptomatic minors: recommendations of the European Society of Human
Genetics. Eur J Hum Genet 2009;17:720–1.
[115] Human Genetics Society of Australasia. Position statement: presymptomatic and predictive testing in children and young people;
2008. Available from: www.hgsa.org.au/2009/12/pre-symptomaticand-predictive-testing-in-children-and-young-people/ [25 January
2011].
[116] Australian Government Department of Health and Ageing.
PBS-eligibility criteria for lipid lowering drugs fact sheet; 2006. Canberra. Available from: www.health.gov.au/internet/main/publishing.
nsf/content/lipid eligibilitycriteria.htm [March 2010].
[117] Benlian P, Turquet A, Carrat F, et al. Diagnosis scoring for clinical
identification of children with heterozygous familial hypercholesterolemia. J Pediatr Gastroenterol Nutr 2009;48:456–63.
[118] van der Graaf A, Avis HJ, Kusters DM, et al. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort
of hypercholesterolemic children. Circulation 2011;123:1167–73.
[119] Newson AJ, Humphries SE. Cascade testing in familial hypercholesterolaemia: How should family members be contacted? Eur J Hum
Genet 2005;13:401–8.
[120] Suthers GK, Armstrong J, McCormack J, Trott D. Letting the
family know: balancing ethics and effectiveness when notifying relatives about genetic testing for a familial disorder. J Med Genet
2006;43:665–70.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[121] Human Genetics Society of Australasia. Process of genetic counselling; 2008. Available from: www.hgsa.org.au/2009/12/process-ofgenetic-counselling/ [25 January 2011].
[122] Urbina EM, Williams RV, Alpert BS, et al. Noninvasive assessment of subclinical atherosclerosis in children and adolescents:
recommendations for standard assessment for clinical research: a scientific statement from the American Heart Association. Hypertension
2009;54:919–50.
[123] Ose L. Diagnostic, clinical, and therapeutic aspects of familial hypercholesterolemia in children. Semin Vasc Med 2004;4:51–7.
[124] Tonstad S. Stratification of risk in children with familial hypercholesterolemia with focus on psychosocial issues. Nutr Metab Cardiovasc
Dis 2001;11(Suppl 5):64–7.
[125] American Diabetes Association. Standards of medical care in
diabetes—2010. Diabetes Care 2010;33:S11–61.
[126] Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the
joint national committee on prevention, detection, evaluation, and
treatment of high blood pressure. Hypertension 2003;42:1206–52.
[127] National Heart Foundation of Australia (National Blood Pressure
and Vascular Disease Advisory Committee). Guide to management
of hypertension 2008; 2010. Updated December. Available from:
www.heartfoundation.org.au/SiteCollectionDocuments/A Hypert
Guidelines2008 2010Update FINAL.pdf.
[128] Robinson JG, Goldberg AC. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from
the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011;5:S18–29.
[129] Ito MK, McGowan MP, Moriarty PM. Management of familial
hypercholesterolemias in adult patients: recommendations from the
National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011;5:S38–45.
[130] Cooper A, O’Flynn N, on behalf of the Guideline Development Group.
Risk assessment and lipid modification for primary and secondary
prevention of cardiovascular disease: summary of NICE guidance. Br
Med J 2008;336:1246–8.
[131] Connor WE, Connor SL. Importance of diet in the treatment of familial hypercholesterolemia. Am J Cardiol 1993;72:D42–53.
[132] Lichtenstein AH, Appel LJ, Brands M, et al. Diet and lifestyle recommendations revision 2006: a scientific statement from the American
Heart Association Nutrition Committee. Circulation 2006;114:82–96.
[133] Gidding SS, Lichtenstein AH, Faith MS, et al. Implementing American Heart Association pediatric and adult nutrition guidelines: a
scientific statement from the American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity and
Metabolism, Council on Cardiovascular Disease in the Young, Council on Arteriosclerosis, Thrombosis and Vascular Biology, Council
on Cardiovascular Nursing, Council on Epidemiology and Prevention, and Council for High Blood Pressure Research. Circulation
2009;119:1161–75.
[134] Moruisi KG, Oosthuizen W, Opperman AM. Phytosterols/stanols
lower cholesterol concentrations in familial hypercholesterolemic
subjects: a systematic review with meta-analysis. J Am Coll Nutr
2006;25:41–8.
[135] Artinian NT, Fletcher GF, Mozaffarian D, et al. Interventions to promote physical activity and dietary lifestyle changes for cardiovascular
risk factor reduction in adults: a scientific statement from the American Heart Association. Circulation 2010;122:406–41.
[136] Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors
on the pathogenesis of cardiovascular disease and implications for
therapy. Circulation 1999;99:2192–217.
[137] Pharmaceutical Management Agency (PHARMAC). Pharmaceutical schedule. 2011. Available from: www.pharmac.govt.
nz/2011/03/01/Schedule.pdf.
[138] Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of
cholesterol-lowering treatment: prospective meta-analysis of data
from 90,056 participants in 14 randomised trials of statins. Lancet
2005;366:1267–78.
259
[139] Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people
without established cardiovascular disease but with cardiovascular
risk factors: meta-analysis of randomised controlled trials. Br Med J
2009;338:b2376.
[140] Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and
safety of more intensive lowering of LDL cholesterol: a meta-analysis
of data from 170 000 participants in 26 randomised trials. Lancet
2010;376:1670–81.
[141] Ward S, Lloyd-Jones M, Pandor A, et al. A systematic review and
economic evaluation of statins for the prevention of coronary events.
Health Technol Assess 2007;11:1–160.
[142] Scientific Steering Committee, Simon Broome Register Group.
Mortality in treated heterozygous familial hypercholesterolaemia:
implications for clinical management. Atherosclerosis 1999;142:
105–12.
[143] Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of
statins in familial hypercholesterolaemia: a long term cohort study.
Br Med J 2008;337:a2423.
[144] Neil A, Cooper J, Betteridge J, et al. Reductions in all-cause, cancer,
and coronary mortality in statin-treated patients with heterozygous
familial hypercholesterolaemia: a prospective registry study. Eur
Heart J 2008;29:2625–33.
[145] Harada-Shiba M, Sugisawa T, Makino H, et al. Impact of statin
treatment on the clinical fate of heterozygous familial hypercholesterolemia. J Atheroscler Thromb 2010;17:667–74.
[146] Smilde TJ, van Wissen S, Wollersheim H, Trip MD, Kastelein
JJ, Stalenhoef AF. Effect of aggressive versus conventional lipid
lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet
2001;357:577–81.
[147] Alonso R, Fernandez de Bobadilla J, Mendez I, Lazaro P, Mata
N. Cost-effectiveness of managing familial hypercholesterolemia
using atorvastatin-based preventive therapy. Rev Esp Cardiol
2008;61:382–93.
[148] Nherera L, Calvert NW, DeMott K, et al. Cost-effectiveness analysis
of the use of a high-intensity statin compared to a low-intensity statin
in the management of patients with familial hypercholesterolaemia.
Curr Med Res Opin 2010;26:529–36.
[149] National Institute for Health and Clinical Excellence. Ezetimibe for the treatment of primary (heterozygous-familial and
non-familial) hypercholesterolaemia; 2010. Available from: guidance.nice.org.uk/nicemedia/live/11886/38213/38213.doc.
[150] Hamilton-Craig I, Kostner K, Colquhoun D, Woodhouse S. Combination therapy of statin and ezetimibe for the treatment of
familial hypercholesterolaemia. Vasc Health Risk Manag 2010;6:
1023–37.
[151] Toth PP. Drug treatment of hyperlipidaemia: a guide to the rational
use of lipid-lowering drugs. Drugs 2010;70:1363–79.
[152] Chapman MJ, Redfern JS, McGovern ME, Giral P. Niacin and fibrates
in atherogenic dyslipidemia: pharmacotherapy to reduce cardiovascular risk. Pharmacol Ther 2010;126:314–45.
[153] Guyton JR, Goldgerg AC. Bile acid sequestrants. In: Ballantyne CM,
editor. Clinical lipidology: a companion to Braunwald’s heart disease.
Philadelphia: Saunders, Elsevier; 2009. p. 281–314.
[154] Huijgen R, Abbink EJ, Bruckert E, et al. Colesevelam added to combination therapy with a statin and ezetimibe in patients with familial
hypercholesterolemia: a 12-week, multicenter, randomized, doubleblind, controlled trial. Clin Ther 2010;32:615–25.
[155] Kane JP, Malloy MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ.
Regression of coronary atherosclerosis during treatment of familial
hypercholesterolemia with combined drug regimens. J Am Med Assoc
1990;264:3007–12.
[156] Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol 2007;99:S3–18.
[157] Watts GF, Karpe F. Triglycerides and atherogenic dyslipidaemia:
extending treatment beyond statins in the high-risk cardiovascular
patient. Heart 2011;97:350–6.
260
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[158] Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart
Association. Circulation 2011;123(May):2292–333.
[159] Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-rich
lipoproteins and high-density lipoprotein cholesterol in patients at
high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J 2011.
[160] Saravanan P, Davidson NC, Schmidt EB, Calder PC. Cardiovascular
effects of marine omega-3 fatty acids. Lancet 2010;376:540–50.
[161] Bays H. Rationale for prescription omega-3-acid ethyl ester therapy
for hypertriglyceridemia: a primer for clinicians. Drugs Today (Barc)
2008;44:205–46.
[162] Yamashita S, Matsuzawa Y. Where are we with probucol: a new life
for an old drug? Atherosclerosis 2009;207:16–23.
[163] Bottorff MB. Statin safety and drug interactions: clinical implications.
Am J Cardiol 2006;97:S27–31.
[164] McKenney JM, Davidson MH, Jacobson TA, Guyton JR. Final conclusions and recommendations of the National Lipid Association
Statin Safety Assessment Task Force. Am J Cardiol 2006;97:S89–94.
[165] Guyton JR, Bays HE. Safety considerations with niacin therapy. Am
J Cardiol 2007;99:S22–31.
[166] Lee JK, Grace KA, Taylor AJ. Effect of a pharmacy care program
on medication adherence and persistence, blood pressure, and lowdensity lipoprotein cholesterol. J Am Med Assoc 2006;296:2563–71.
[167] Elliott R, Barber N, Clifford S, Horne R, Hartley E. The cost
effectiveness of a telephone-based pharmacy advisory service to
improve adherence to newly prescribed medicines. Pharm World Sci
2008;30:17–23.
[168] Eussen SR, van der Elst ME, Klungel OH, et al. A pharmaceutical
care program to improve adherence to statin therapy: a randomized
controlled trial. Ann Pharmacother 2010;44:1905–13.
[169] Choudhry NK, Fischer MA, Avorn J, et al. The implications of therapeutic complexity on adherence to cardiovascular medications. Arch
Intern Med 2011:E1–9.
[170] Bates TR, Connaughton VM, Watts GF. Non-adherence to statin
therapy: a major challenge for preventive cardiology. Expert Opin
Pharmacother 2009;10:2973–85.
[171] Senior V, Marteau T, Weinman J. Self-reported adherence to
cholesterol-lowering medication in patients with familial hypercholesterolaemia: the role of illness perceptions. Cardiovasc Drugs
Ther 2004;18:475–81.
[172] Scott TL, Gazmararian JA, Williams MV, Baker DW. Health literacy and preventive health care use among Medicare enrollees in a
managed care organization. Med Care 2002;40:395–404.
[173] Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patients
with inadequate health literacy. Fam Med 2004;36:588–94.
[174] Roth GA, Fihn SD, Mokdad AH, Aekplakorn W, Hasegawa T, Lim
SS. High total serum cholesterol, medication coverage and therapeutic
control: an analysis of national health examination survey data from
eight countries. Bull World Health Organ 2011;89:92–101.
[175] Simons LA, Oritz M, Calcino G. Long term persistence with statin
therapy: experience in Australia 2006–2010. Aust Fam Physician
2011;40:319–22.
[176] Carrington MJ, Retegan C, Johnston CI, Jennings GL, Stewart S.
Cholesterol complacency in Australia: time to revisit the basics of
cardiovascular disease prevention. J Clin Nurs 2009;18:678–86.
[177] Carrington MJ, Stewart S. Australia’s cholesterol crossroads: an
analysis of 199,331 GP patient records. Melbourne, Australia:
Baker IDI Heart and Diabetes Institute; 2011. Available from:
www.bakeridi.edu.au/Assets/Files/Australia’s%20Cholesterol%20
Crossroads%20Report FINAL.pdf.
[178] Kotseva K, Wood D, De Backer G, et al. Euroaspire III. Management
of cardiovascular risk factors in asymptomatic high-risk patients in
general practice: cross-sectional survey in 12 European countries. Eur
J Cardiovasc Prev Rehabil 2010;17:530–40.
[179] Vale M, Jelinek MV, Best JD, on behalf of the COACH study group.
How many patients with coronary heart disease are not achieving
[180]
[181]
[182]
[183]
[184]
[185]
[186]
[187]
[188]
[189]
[190]
[191]
[192]
[193]
[194]
[195]
[196]
their risk-factor targets? Experience in Victoria 1996–1998 versus
1999–2000. Med J Aust 2002;176:211–5.
Kotseva K, Wood D, De Backer G, De Bacquer D, Pyörälä K, Keil
U. Euroaspire III. A survey on the lifestyle, risk factors and use of
cardioprotective drug therapies in coronary patients from 22 European
countries. Eur J Cardiovasc Prev Rehabil 2009;16:121–37.
Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to
moderate muscular symptoms with high-dosage statin therapy in
hyperlipidemic patients the PRIMO Study. Cardiovasc Drugs Ther
2005;19:403–14.
Bays H, Dujovne C. Colesevelam HCl: a non-systemic lipid altering
drug. Expert Opin Pharmacother 2003;4:779–90.
Spence JD, Hackam DG. Treating arteries instead of risk factors: a paradigm change in management of atherosclerosis. Stroke
2010;41:1193–9.
Rothwell PM. Endarterectomy for symptomatic and asymptomatic
carotid stenosis. Neurol Clin 2008;26:1079–97.
Brott TG, Halperin JL, Abbara S, Bacharach JM, Barr JD, Bush
RL, et al. 2011 ASA/ACCF/AHA/AANN/AANS/ACR/ASNR/CNS/
SAIP/SCAI/SIR/SNIS/SVM/SVS guideline on the management of
patients with extracranial carotid and vertebral artery disease: a report
of the American College of Cardiology Foundation/American Heart
Association Task Force on Practice Guidelines, and the American
Stroke Association, American Association of Neuroscience Nurses,
American Association of Neurological Surgeons, American College
of Radiology, American Society of Neuroradiology, Congress of
Neurological Surgeons, Society of Atherosclerosis Imaging and Prevention, Society for Cardiovascular Angiography and Interventions,
Society of Interventional Radiology, Society of NeuroInterventional
Surgery, Society for Vascular Medicine, and Society for Vascular
Surgery. Circulation 2011;123:1–77.
Thorogood M, Seed M, De Mott K, Guideline Development Group.
Management of fertility in women with familial hypercholesterolaemia: summary of NICE guidance. Br J Obstet Gynaecol
2009;116:478–9.
van der Graaf A, Hutten BA, Kastelein JJ, Vissers MN. Premature cardiovascular disease in young women with heterozygous
familial hypercholesterolemia. Expert Rev Cardiovasc Ther 2006;4:
345–51.
Nenseter MS, Lindvig HW, Ueland T, et al. Lipoprotein(a) levels in
coronary heart disease-susceptible and -resistant patients with familial
hypercholesterolemia. Atherosclerosis 2011. March 2 [Epub ahead of
print].
Avis HJ, Hutten BA, Twickler MT, et al. Pregnancy in women suffering from familial hypercholesterolemia: a harmful period for both
mother and newborn? Curr Opin Lipidol 2009;20:484–90.
Kusters DM, Homsma SJM, Hutten BA, et al. Dilemmas in treatment
of women with familial hypercholesterolaemia during pregnancy.
Neth J Med 2010;68:299–303.
van der Graaf A, Vissers MN, Gaudet D, et al. Dyslipidemia of mothers with familial hypercholesterolemia deteriorates lipids in adult
offspring. Arterioscler Thromb Vasc Biol 2010;30:2673–7.
Geraedts JPM, De Wert GMWR. Preimplantation genetic diagnosis.
Clin Genet 2009;76:315–25.
Barlow-Stewart K, Saleh M, Parasivam G. FS17(c): prenatal
testing—CVS & amniocentesis. Sydney: Centre for Genetics Education; 2007 [18 January 2011] www.genetics.com.au/pdf/factsheets/
fs17c.pdf.
Amundsen AL, Ose L, Nenseter MS, Ntanios FY. Plant sterol esterenriched spread lowers plasma total and LDL cholesterol in children
with familial hypercholesterolemia. Am J Clin Nutr 2002;76:338–44.
Amundsen AL, Ntanios F, van der Put N, Ose L. Long-term compliance and changes in plasma lipids, plant sterols and carotenoids in
children and parents with FH consuming plant sterol ester-enriched
spread. Eur J Clin Nutr 2004;58:1612–20.
de Jongh S, Kerckhoffs MC, Grootenhuis MA, Bakker HD, Heymans
HSA, Last BF. Quality of life, anxiety and concerns among statin-
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[197]
[198]
[199]
[200]
[201]
[202]
[203]
[204]
[205]
[206]
[207]
[208]
[209]
[210]
[211]
[212]
[213]
[214]
[215]
[216]
treated children with familial hypercholesterolaemia and their parents.
Acta Paediatr 2003;92:1096–101.
van der Graaf A, Cuffie-Jackson C, Vissers MN, et al. Efficacy and
safety of coadministration of ezetimibe and simvastatin in adolescents
with heterozygous familial hypercholesterolemia. J Am Coll Cardiol
2008;52:1421–9.
Clauss S, Wai K-M, Kavey R-EW, Kuehl K. Ezetimibe treatment of pediatric patients with hypercholesterolemia. J Pediatr
2009;154:869–72.
Stein EA, Marais AD, Szamosi T, et al. Colesevelam hydrochloride:
efficacy and safety in pediatric subjects with heterozygous familial
hypercholesterolemia. J Pediatr 2010;156, 231-6.e3.
Monthly Index of Medical Specialities Australia. MIMS
online 2010. (data version: March 2010), New South Wales,
Australia; 2010. Available from: www-mimsonline-comau.rplibresources.health.wa.gov.au.
Wiegman A, Hutten BA, de Groot E, et al. Efficacy and safety of statin
therapy in children with familial hypercholesterolemia: a randomized
controlled trial. J Am Med Assoc 2004;292:331–7.
Avis HJ, Hutten BA, Gagne C, et al. Efficacy and safety of rosuvastatin
therapy for children with familial hypercholesterolemia. J Am Coll
Cardiol 2010;55:1121–6.
McCrindle BW, Ose L, Marais AD. Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or
severe hyperlipidemia: a multicenter, randomized, placebo-controlled
trial. J Pediatr 2003;143:74–80.
Engler M, Engler M, Malloy M, et al. Docosahexaenoic acid restores
endothelial function in children with hyperlipidemia: results from the
early study. Int J Clin Pharmacol Ther 2004;42:672–9.
Wierzbicki AS, Viljoen A. Hyperlipidaemia in paediatric patients:
the role of lipid-lowering therapy in clinical practice. Drug Saf
2010;33:115–25.
Juonala M, Magnussen CG, Venn A, et al. Influence of age on
associations between childhood risk factors and carotid intimamedia thickness in adulthood: the Cardiovascular Risk in Young
Finns Study, the Childhood Determinants of Adult Health Study, the
Bogalusa Heart Study, and the Muscatine Study for the International
Childhood Cardiovascular Cohort (i3C) Consortium. Circulation
2010;122:2514–20.
Pletcher MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipids
commonly present in young adults and coronary calcium later in life:
the CARDIA (Coronary Artery Risk Development in Young Adults)
Study. Ann Intern Med 2010;153:137–46.
Dawood OT, Izham M, Ibrahim M, Palaian S. Medication compliance
among children. World J Pediatr 2010;6:200–2.
Hill M, Pawsey M, Cutler A, Holt JL, Goldfield SR. Consensus standards for the care of children and adolescents in Australian health
services. Med J Aust 2011;194:78–82.
Hudgins LC, Kleinman B, Scheuer A, White S, Gordon BR. Longterm safety and efficacy of low-density lipoprotein apheresis in
childhood for homozygous familial hypercholesterolemia. Am J Cardiol 2008;102:1199–204.
Viner R. Barriers and good practice in transition from paediatric to
adult care. J R Soc Med 2001;94:2–4.
Thompson GR. LDL apheresis. Atherosclerosis 2003;167:1–13.
Thompsen J, Thompson PD. A systematic review of LDL apheresis in the treatment of cardiovascular disease. Atherosclerosis
2006;189:31–8.
Hudgins LC, Gordon BR, Parker TS, Saal SD, Levine DM,
Rubin AL. LDL apheresis: an effective and safe treatment for
refractory hypercholesterolemia. Cardiovasc Drug Rev 2002;20:
271–80.
Thompson GR, Barbir M, Davies D, et al. Efficacy criteria and cholesterol targets for LDL apheresis. Atherosclerosis 2010;208:317–21.
Thompson GR, Catapano A, Saheb S, et al. Severe hypercholesterolaemia: therapeutic goals and eligibility criteria for LDL apheresis in
Europe. Curr Opin Lipidol 2010;21:492–8.
261
[217] Szczepiorkowski Z, Bandarenko N, Kim H, et al. Guidelines on
the use of therapeutic apheresis in clinical practice—evidence-based
approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apheresis 2007;22:106–75.
[218] Mabuchi H, Koizumi J, Shimizu M, et al. Long-term efficacy of lowdensity lipoprotein apheresis on coronary heart disease in familial
hypercholesterolemia. Am J Cardiol 1998;82:1489–95.
[219] Aengevaeren WRM, Kroon AA, Stalenhoef AFH, Uijen GJH, Van
Der Werf T. Low density lipoprotein apheresis improves regional
myocardial perfusion in patients with hypercholesterolemia and
extensive coronary artery disease: the LDL-Apheresis Atherosclerosis
Regression Study (LAARS). J Am Coll Cardiol 1996;28:1696–704.
[220] Marais AD, Firth JC, Blom DJ. Homozygous familial hypercholesterolemia and its management. Semin Vasc Med 2004;4(43):50.
[221] Cashin-Hemphill L, Noone M, Abbott JF, Waksmonski CA, Lees RS.
Low-density lipoprotein apheresis therapy during pregnancy. Am J
Cardiol 2000;86:1160.
[222] Anedda S, Mura S, Marcello C, Pintus P. HELP LDL-apheresis in
two cases of familial hypercholesterolemic pregnant women. Transfus
Apher Sci 2011;44:21–4.
[223] HEART UK. Lipid Specialist Sub-Committee. LDL apheresis; 2009.
Available from: www.heartuk.org.uk/ldl/.
[224] Nemati MH, Astaneh B. Optimal management of familial hypercholesterolemia: treatment and management strategies. Vasc Health
Risk Manag 2010;6:1079–88.
[225] Kakaei F, Nikeghbalian S, Kazemi K, et al. Liver transplantation for
homozygous familial hypercholesterolemia: two case reports. Transplant Proc 2009;41:2939–41.
[226] Moini M, Mistry P, Schilsky ML. Liver transplantation for inherited metabolic disorders of the liver. Curr Opin Organ Tran
2010;15:269–76.
[227] Goldberg AC. Novel therapies and new targets of treatment for familial hypercholesterolemia. J Clin Lipidol 2010;4:350–6.
[228] Visser ME, Kastelein JJ, Stroes ES. Apolipoprotein B synthesis inhibition: results from clinical trials. Curr Opin Lipidol 2010;21:319–23.
[229] Duff CJ, Hooper NM. PCSK9: an emerging target for treatment of
hypercholesterolemia. Expert Opin Ther Targets 2011;15:157–68.
[230] Moghadasian M, Frohlich J, Saleem M, Hong J, Qayumi K, Scudamore C. Surgical management of dyslipidemia: clinical and
experimental evidence. J Invest Surg 2001;14:71–8.
[231] Buchwald H, Rudser KD, Williams SE, Michalek VN, Vagasky J,
Connett JE. Overall mortality, incremental life expectancy, and cause
of death at 25 years in the program on the surgical control of the
hyperlipidemias. Ann Surg 2010;251:1034–40.
[232] Al-Allaf F, Coutelle C, Waddington S, David A, Harbottle R, Themis
M. LDLR-gene therapy for familial hypercholesterolaemia: problems, progress, and perspectives. Int Arch Med 2010;3:36.
[233] Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE,
Neil HAW. Screening for hypercholesterolaemia versus case finding
for familial hypercholesterolaemia: a systematic review and costeffectiveness analysis. Health Technol Assess 2000;4:1–123.
[234] Wonderling D, Umans-Eckenhausen MAW, Marks D, Defesche JC,
Kastelein JJP, Thorogood M. Cost-effectiveness analysis of the
genetic screening program for familial hypercholesterolaemia in the
Netherlands. Semin Vasc Med 2004;4:97–104.
[235] Oliva J, López-Bastida J, Moreno SG, Mata P, Alonso R. Costeffectiveness analysis of a genetic screening program in the close
relatives of Spanish patients with familial hypercholesterolemia. Rev
Esp Cardiol 2009;62:57–65.
[236] Marang-van de Mheen PJ, ten Asbroek AHA, Bonneux L, Bonsel GJ,
Klazinga NS. Cost-effectiveness of a family and DNA based screening
programme on familial hypercholesterolaemia in the Netherlands. Eur
Heart J 2002;23:1922–30.
[237] Human Genetics Society of Australasia. Presymptomatic and
predictive testing for genetic disorders; 2005. Available from:
www.hgsa.org.au/2009/12/presymptomatic-and-predictive-testingfor-genetic-disorders/ [25 January 2011].
262
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[238] Forrest LE, Delatycki MB, Skene L, Aitken M. Communicating
genetic information in families—a review of guidelines and position
papers. Eur J Hum Genet 2007;15:612–8.
[239] National Health and Medical Research Council (NHMRC),
Office of the Privacy Commissioner. Use and disclosure of
genetic information to a patient’s genetic relatives under section 95AA of the Privacy Act 1988 (cth): guidelines for health
practitioners in the private sector. NHMRC; 2009. Available
from: www.nhmrc.gov.au/ files nhmrc/file/publications/synopses/
e96.pdf.
[240] Royal College of Physicians, Royal College of Pathologists, British
Society for Human Genetics. Consent and confidentiality in genetic
practice: guidance on genetic testing and sharing genetic information.
In: Report of the Joint Committee on Medical Genetics. London:
RCP/RCPath/BSHG; 2005.
[241] Saleh M, Barlow-Stewart K, Meiser B, Muchamore I. Challenges
faced by genetics service providers’ practicing in a culturally and
linguistically diverse population: an Australian experience. J Genet
Couns 2009;18:436–46.
[242] Progeny. Progeny Clinical 8. 2011. Available from:
www.progenygenetics.com/clinical.
[243] PASS Clinical. PASS Clinical vascular. Rijswijk, Netherlands; 2011.
Available from: www.pass-software.com.
[244] Maxwell SJ, Molster CM, Poke SJ, O’leary P. Communicating familial hypercholesterolemia genetic information within families. Gen
Test Mol Bioma 2009;13:301–6.
[245] Clarke A, Martin R, Kerzin-Storrar L, et al. Genetic professionals’
reports of nondisclosure of genetic risk information within families.
Eur J Hum Genet 2005;13:556–62.
[246] Suthers GK, McCusker EA, Wake SA. Alerting genetic relatives to
a risk of serious inherited disease without a patient’s consent. Med J
Aust 2011;194:385–6.
[247] Greco KE, Salveson C. Identifying genetics and genomics nursing
competencies common among published recommendations. J Nurs
Educ 2009;48:557–65.
[248] Leren TP, Finborud TH, Manshaus TE, Ose L, Berge KE. Diagnosis of
familial hypercholesterolemia in general practice using clinical diagnostic criteria or genetic testing as part of cascade genetic screening.
Community Genet 2008;11:26–35.
[249] Burke W, Emery J. Genetics education for primary-care providers.
Nat Rev Genet 2002;3:561–6.
[250] Cardiff University. Wales familial hypercholesterolaemia testing service: FH family forum; 2010. Available from: www.fhwales.co.uk [17
February 2010].
[251] FH Family Support Group. Available from: www.fhfamilysupportgroup.websyte.com.au [31 March 2011].
[252] Fleury J, Keller C, Perez A, Lee S. The role of lay health advisors
in cardiovascular risk reduction: a review. Am J Community Psychol
2009;44:28–42.
[253] Australian Government. Privacy Legislation Amendment Act 2006;
2006. Available from: www.comlaw.gov.au/Details/C2006A00099 [6
April 2011].
[254] Marang-van de Mheen PJ, van Maarle MC, Stouthard MEA. Getting
insurance after genetic screening on familial hypercholesterolaemia;
the need to educate both insurers and the public to increase adherence
to national guidelines in the Netherlands. J Epidemiol Community
Health 2002;56:145–7.
[255] Otlowski MFA, Stranger MJA, Barlow-Stewart K, Taylor S, Treloar
S. Investigating genetic discrimination in the Australian life insurance
sector: the use of genetic test results in underwriting, 1999–2003. J
Law Med 2007;14:367–96.
[256] Human Genetics Society of Australasia. Position statement:
genetic testing and life insurance in Australia; 2008. Available
from: www.hgsa.org.au/2009/12/genetic-testing-and-life-insurancein-australia/ [25 January 2011].
[257] Neil HA, Mant D. Cholesterol screening and life assurance. Br Med
J 1991;302:891–3.
[258] Neil HAW, Hammond T, Mant D, Humphries SE. Effect of statin
treatment for familial hypercholesterolaemia on life assurance: results
of consecutive surveys in 1990 and 2002. Br Med J 2004;328:
500–1.
[259] Financial Services Council. FSC standard no. 11. Genetic testing
policy; 2005 , www.ifsa.com.au/downloads/file/FSCStandards/11S
GeneticTestingPolicy.pdf.
[260] Alonso R, Defesche JC, Tejedor D, et al. Genetic diagnosis of familial hypercholesterolemia using a DNA-array based platform. Clin
Biochem 2009;42:899–903.
[261] Taylor A, Wang D, Patel K, et al. Mutation detection rate and spectrum
in familial hypercholesterolaemia patients in the UK pilot cascade
project. Clin Genet 2010;77:572–80.
[262] Lombardi MP, Redeker EJW, van Gent DHM, Smeele KL, Weederstein R, Mannens MM. Molecular genetic testing for familial
hypercholesterolaemia in the Netherlands: a stepwise screening strategy enhances the mutation detection rate. Genet Test 2006;10:77–84.
[263] DNA Genotek. Oragene DNA. Ontario, Canada: Kanata; 2011 ,
www.dnagenotek.com [15 March 2011].
[264] National Pathology Accreditation Advisory Council (NPAAC). Draft
requirements for medical testing of human nucleic acids-public consultation. 1st ed. 20xx Canberra: Australian Government, Department
of Health and Ageing; 2010.
[265] Brice P. PGD for familial hypercholesterolaemia approved. London:
PHG Foundation; 2007 , www.phgfoundation.org/news/3947/ [25
January 2011].
[266] National Association of Testing Authorities Australia (NATA).
Medical testing accreditation 2009; 2009. Available from:
www.nata.asn.au/types-of-accreditation [25 January 2011].
[267] Leigh SEA, Foster AH, Whittall RA, Hubbart CS, Humphries SE.
Update and analysis of the University College London Low Density
Lipoprotein Receptor Familial Hypercholesterolemia Database. Ann
Hum Genet 2008;72:485–98.
[268] Dedoussis GVZ, Schmidt H, Genschel J. LDL-receptor mutations in
Europe. Hum Mutat 2004;24:443–59.
[269] Marduel M, Carrié A, Sassolas A, et al. Molecular spectrum of
autosomal dominant hypercholesterolemia in France. Hum Mutat
2010;31:E1811–24.
[270] Laurie AD, Scott RS, George PM. Genetic screening of patients with
familial hypercholesterolaemia (FH): a New Zealand perspective.
Atherosclerosis Suppl 2004;5:13–5.
[271] Civeira F, Jarauta E, Cenarro A, et al. Frequency of low-density
lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J Am
Coll Cardiol 2008;52:1546–53.
[272] Kwiterovich Jr PO. Clinical implications of the molecular basis of
familial hypercholesterolemia and other inherited dyslipidemias. Circulation 2011;123:1153–5.
[273] Defesche JC, Lansberg PJ, Umans-Eckenhausen MA, Kastelein JJ.
Advanced method for the identification of patients with inherited
hypercholesterolemia. Semin Vasc Med 2004;4:59–65.
[274] Taylor A, Martin B, Wang D, Patel K, Humphries SE, Norbury G.
Multiplex ligation-dependent probe amplification analysis to screen
for deletions and duplications of the LDLR gene in patients with
familial hypercholesterolaemia. Clin Genet 2009;76:69–75.
[275] Holla ØL, Teie C, Berge KE, Leren TP. Identification of deletions and
duplications in the low density lipoprotein receptor gene by MLPA.
Clin Chim Acta 2005;356:164–71.
[276] Chiou K-R, Charng M-J, Chang H-M. Array-based resequencing
for mutations causing familial hypercholesterolemia. Atherosclerosis
2011. March 2 [Epub ahead of print].
[277] Dusková L, Kopecková L, Jansová E, et al. An apex-based genotyping
microarray for the screening of 168 mutations associated with familial
hypercholesterolemia. Atherosclerosis 2011;216:139–45.
[278] Mamotte C, van Bockxmeer F. A robust strategy for screening and
confirmation of familial defective apolipoprotein B-100. Clin Chem
1993;39:118–21.
G.F. Watts et al. / Atherosclerosis Supplements 12 (2011) 221–263
[279] Newton CR, Graham A, Heptinstall LE, et al. Analysis of any point
mutation in DNA. The amplification refractory mutation system
(ARMS). Nucleic Acids Res 1989;17:2503–16.
[280] Taylor A, Tabrah S, Wang D, et al. analysis to detect 13 common
mutations in familial hypercholesterolaemia. Clin Genet 2007;71:
561–8.
[281] Progenika. LIPOchip. Vizcaya, Spain; 2011. Available from:
www.progenika.com/eu/index.php?option=com content&task=
view&id=138&Itemid=18.
[282] Gen-Probe. Elucigene FH20. Abingdon, Oxfordshire, United Kingdom; 2011. Available from: www.gen-probe.com/global/productsservices/cardiovascular.aspx.
[283] MRC-Holland. SALSA MLPA kit P062. Amsterdam, Netherlands;
2011. Available from: www.mlpa.com.
[284] CLC-Bio. CLC Main Workbench. Aarhus, Denmark; 2011. Available
from: www.clcbio.com/index.php?id=92.
[285] Interactive Biosoftware. Alamut. Rouen, France; 2011. Available
from: www.interactive-biosoftware.com/alamut.html.
[286] University College London. Low density lipoprotein receptor
familial hypercholesterolemia database. London, United Kingdom;
2011. Available from: www.ucl.ac.uk/ldlr/Current/index.php?select
db=LDLR.
[287] Bourbon M, Duarte MA, Alves AC, Medeiros AM, Marques L, Soutar
AK. Genetic diagnosis of familial hypercholesterolaemia: the importance of functional analysis of potential splice-site mutations. J Med
Genet 2009;46:352–7.
[288] Goldberg AC, Robinson JG, Cromwell WC, Ross JL, Ziajka PE.
Future issues, public policy, and public awareness of familial
hypercholesterolemias: recommendations from the National Lipid
Association Expert Panel on Familial Hypercholesterolemia. J Clin
Lipidol 2011;5:S46–51.
[289] Aatre RD, Day SM. Psychological issues in genetic testing for inherited cardiovascular diseases. Circ Cardiovasc Genet 2011;4:81–90.
[290] Hadfield SG, Horara S, Starr BJ, et al. Are patients with familial hypercholesterolaemia well managed in lipid clinics? An audit of eleven
clinics from the Department of Health familial hypercholesterolaemia
cascade testing project. Ann Clin Biochem 2008;45:199–205.
263
[291] Neubeck L, Redfern J, Fernandez R, Briffa T, Bauman A, Freedman
SB. Telehealth interventions for the secondary prevention of coronary
heart disease: a systematic review. Eur J Cardiovasc Prev Rehabil
2009;16:281–9.
[292] Clark CE, Smith LFP, Taylor RS, Campbell JL. Nurse led interventions to improve control of blood pressure in people with
hypertension: systematic review and meta-analysis. Br Med J
2010;341:c3995.
[293] Loveman E, Frampton GK, Clegg AJ. The clinical effectiveness of
diabetes education models for type 2 diabetes: a systematic review.
Health Technol Assess 2008;12:1–116.
[294] Haralambos K. “PASS Clinical” software for familial hypercholesterolaemia (FH): evaluation report including 3 month clinical pilot;
2009. Available from: www.heartuk.org.uk/FHToolkit/.
[295] Braithwaite J, Travaglia JF. An overview of clinical governance
policies, practices and initiatives. Aust Health Rev 2008;32:
10–22.
[296] Evans SM, Bohensky M, Cameron PA, McNeil J. A survey of Australian clinical registries: can quality of care be measured? Intern Med
J 2011;41:42–8.
[297] Ingles J, McGaughran J, Vohra J, et al. Establishment of an Australian
national genetic heart disease registry. Heart Lung Circ 2008;17:
463–7.
[298] World Health Organization. Building blocks for action innovative care
for chronic conditions: global report; 2002.
[299] Coleman K, Austin BT, Brach C, Wagner EH. Evidence on the
chronic care model in the new millennium. Health Aff (Millwood)
2009;28:75–85.
[300] Fisk NM, Wesselingh SL, Beilby JJ, et al. Academic health
science centres in Australia: let’s get competitive. Med J Aust
2011;194:59–60.
[301] Pedersen KMV, Humphries SE, Roughton M, Besford JS. The
national audit of the management of familial hypercholesterolaemia 2010: full report. Clinical Standards Department, Royal
College of Physicians; 2010. Available from: www.rcplondon.ac.uk/
node/2584.