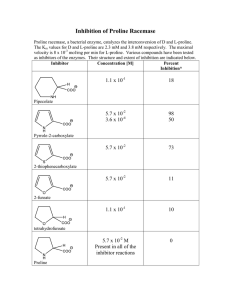
Conformational Studies on Peptides with Proline in the Right-Handed a-Helical Region
advertisement

Conformational Studies on Peptides with Proline
in the Right-Handed a-Helical Region
R. SANKARARAMAKRISHNAN and SARASWATHI VISHVESHWARA"
Molcwilar Bio-Physics Unit, Indian Institute of Science, Bangalore 560 01 2, India
SYNOPSIS
The proline residues in proteins are known to play an important structural role. Recently,
the role of a proline residue in the middle of right-handed a-helical segments of peptides
has been the focus of attention. This role seems to be particularly important in the case
of membrane proteins and in the tight packing of globular proteins. In the present study
the right-handed a-helical region of the Ala-Pro dipeptide and of polypeptides containing
this group have been investigated. Crystal structures of proline-containing a-helices from
some proteins have been analyzed and energy minimization studies on some model fragments
containing Ala-Pro in the right-handed a-helical conformation have been carried out using
flexible geometry. The present calculations indicate that the right-handed a-helical region
of conformational space is an energetically favored region that can also accommodate AlaPro in longer segments of right-handed a-helix. This is achieved due to minor variations
in some of the internal parameters. Deviations in the backbone parameters of proline in
the right-handed a-helix lead to a kink of about 23" in the helix axis. These deviations
have been characterized and a set of standard values has been suggested for producing such
a kink. These values can be used for model building and as starting points for further
minimization studies. Previous energy minimization studies have been done using rigid
geometry. This may explain why the minimum for Ala-Pro in the right-handed a-helical
region has not been recognized thus far.
INTRODUCTION
T h e special role of proline in the secondary structure
of proteins has long been recognized. It is known t o
occur at the bends, a t t h e beginning of t h e helix,
a n d is also known t o break helical structure^.'-^
These properties of proline have been attributed5 t o
t h e rigidity of t h e pyrrolidine ring a n d t o t h e lack
of a hydrogen o n t h e amide nitrogen, which otherwise would have participated in hydrogen-bond formation. T h e right-handed a-helical conformation
for residues ( other t h a n glycine) preceding proline
in t h e polypeptide chain has been characterized a s
energetically
It has, however been
observed that about 8% of proline occur in t h e middle of helices.' Recent crystal structure analysisg of
C 1990 ,John Wiles & Sons, Inc.
CCC 000fi-3:,~.5/~0/~-40287
12
$04.00
Biopolymers, \'()I 30, 287-298 (1990)
* T o whoin correspondence should be addressed.
a number of globular proteins has shown t h a t t h e
proline residue is indeed present in right-handed N helical segments where all t h e residues approximately adopt t h e parameters of a right-handed ahelix. I n such situations t h e proline residue introduces a bend of N 23" in t h e a-helix. T h e bending
of helices on t h e surface of globular proteins may
help in tight packing of t h e molecule. Also, a number
of membrane proteins have right-handed a-helices
containing proline residues,, which span t h e membrane." T h e detailed structure of such helices is not
known a n d computer modeling of membrane proteins such as bacteriorhodopsin has been limited
due t o t h e lack of information on t h e helical structure of peptides containing proline. Hence in t h e
present study, a detailed structural analysis of t h e
right-handed a-helical region of peptides containing
proline has been carried out. Flexible geometry
minimization has been carried out o n di- and polypeptides containing proline. Interesting information
regarding the structure and geometries of these units
''
287
288
SANKARARAMAKRISHNAN AND VISHVESHWARA
Table I Analysis of the Geometrical Parameters in Crystal Structures Containing Proline
in the Right-Handed a-Helical Segment
Protein"
Codeb
Myoglobin 82-96 (88)
3MBN
Citrate synthase (5-27)
(15)
Citrate synthase
(166-195) (183)
Citrate synthase
(166-195) (183)
Alcohol dehydrogenase
(324-336) (329)
Average values
Range of parameters
4p-4
4 - 3
4p-2
4p-1
4P
+p-4
*P-3
+P-2
+p-
+P
up-4
up-3
up-2
up- 1
-71.8
-59.7
-38.9
-32.6
154.4
-170.2
lMBO
-55.2
-69.1
-41.3
-37.3
-174.6
-169.4
2MBN
-81.6
-86.9
-25.8
-29.2
173.1
179.8
lMBD
-63.6
-63.9
-42.4
-42.6
178.6
-171.6
1MB5
-35.4
-76.8
-57.2
-35.4
-175.3
-176.9
-64.7
4CTS
-81.1
-18.5
-40.1
163.3
-169.8
lCTS
-58.2
-84.2
-31.7
-42.9
-177.7
-175.5
2CTS
-62.3
-85.3
-25.1
-38.9
-175.9
-178.9
-76.8
3CTS
-56.5
-32.1
-48.4
174.2
- 173.5
-62.3
4CTS
-65.4
-25.6
-54.1
171.9
-159.9
-64.6
lCTS
-86.8
-19.3
-22.5
167.3
-175.6
-62.4
-63.9
2CTS
-37.2
-43.1
172.2
-177.0
-62.0
3CTS
-60.3
-40.1
-40.1
173.5
-171.5
4ADH
-79.3
-69.2
-24.9
-51.3
170.0
-166.5
-65.2
5ADH
-67.1
-46.9
-14.9
-177.9
-176.3
-62.3
-73.8
-38.2
-33.8
174.5
-172.8
(-60, -85)
(-55, -75)
(-30, -45)
(-25, -45)
(-175, 165) (-180, -165)
-87.7
-39.4
-179.2
-80.8
-34.4
-176.9
-77.6
-40.9
-177.6
-76.7
-28.5
-170.8
-81.9
-24.8
179.7
-74.0
-51.2
-176.5
-65.4
-32.2
-174.0
-65.7
-55.8
-175.7
-65.2
-43.1
-177.0
-92.9
-29.3
173.1
-93.9
-20.2
177.7
-71.2
-41.5
-169.2
-83.9
-29.2
-178.4
-70.7
-55.7
-171.0
-120.2
-42.9
179.7
-80.5
-37.9
-177.1
(-65, -90)
(-25, -45)
(-180, -170)
1
-38.8
-67.8
176.9
-53.9
-46.9
-179.7
-45.5
-59.7
179.6
-64.2
-38.1
174.1
-67.0
-47.5
179.5
-50.0
-54.9
169.8
-53.4
-56.1
-179.8
-43.5
-48.6
-176.6
-52.7
-67.9
177.2
-31.3
-49.7
-170.6
-47.9
-55.7
173.1
-38.9
-56.1
177.6
-39.9
-59.8
178.1
-40.1
-49.2
176.3
-38.3
-46.1
-175.4
-47.0
-53.6
178.7
(-35, -55)
(-45, -65)
(170, -175)
UP
-51.6
-44.0
178.4
-67.7
-38.8
-176.6
-56.2
-41.8
178.7
-70.4
-35.0
-177.2
-65.9
-33.7
179.1
-49.4
-55.1
-169.4
-60.1
-27.8
-176.5
-67.2
-33.9
-178.9
-63.2
-35.4
179.7
-80.2
-20.7
- 176.8
-48.2
-47.8
179.8
-64.0
-18.4
171.8
-57.0
-25.3
177.9
-58.8
-35.2
179.1
-65.1
-47.3
179.8
-61.7
-36.0
-179.4
(-50, -70)
(-30, -45)
(175, -175)
p
Kink
Angle
72.2
26.2
70.6
29.3
67.3
23.3
72.4
29.6
66.2
41.0
60.5
26.2
76.1
25.5
67.8
25.7
69.6
26.2
85.2
25.1
79.5
25.9
94.6
24.2
84.0
25.4
69.0
22.4
74.1
11.8
The a-helical region and the position of proline residue is given for each protein.
The coordinates of the proteins in this table are taken from Brookhaven Protein Data Bank and the codes corresponding to these
proteins are given here.
a
PROLINE IN T H E RIGHT-HANDED a-HELICAL REGION
289
Table IIa Minimized Energies and Geometries for Ace-Ala-Pro-NHMe
Dipeptide
Proline
Puckering
Structure I (right-handed
a-helical region)
Structure I1 (left-handed
a-helical region)
Structure I11 (extended region)
Down
UP
Down
UP
Down
UP
Planar
Ala-Ala (right-handed
a-helical region)
h - 1
-38.9
-44.9
44.8
29.6
-64.6
-96.5
-111.3
-50.8
-53.9
-41.8
56.5
67.8
123.7
114.2
119.1
-34.8
@P
$P
-70.0
-56.9
-67.5
-52.8
-70.8
-51.9
-60.7
-59.3
-12.2
-28.2
-39.1
-44.8
-37.2
-43.6
-46.2
-28.2
27.6
-25.5
26.5
-28.5
27.4
-28.3
0.9
-
x2
O1
O2
Energy
(kcal/
mole)
-36.5
36.1
-33.7
36.5
-33.5
35.8
0.1
-
115.2
115.0
113.1
114.7
109.4
108.9
108.9
112.9
121.0
120.9
118.9
119.2
119.7
119.9
119.7
117.2
-6.52
-7.54
-3.18
-2.56
-5.01
-4.12
-1.24
-23.54
Table IIb Minimized Energies and Geometries for Ace-Ala-Pro-NHMe with Crystal Structures
as Starting Points
Protein"
Myoglobin (82-96) (88)
Citrate synthase (5-27) (15)
Citrate synthase (166-195) (183)
Alcohol dehydrogenase (324-336)
(329)
a
Codeb
BMBN
lMBO
BMBN
lMBD
1MB5
4CTS
lCTS
2CTS
3CTS
4CTS
lCTS
2CTS
3CTS
4ADH
5ADH
#p-l
-44.5
-44.8
-46.1
-46.3
-40.5
-45.6
-45.6
-46.2
-46.2
-46.0
-45.5
-45.8
-45.5
-45.8
-44.4
-43.3
-42.1
-41.2
-41.5
-53.1
-41.8
-41.8
-42.0
-41.5
-42.0
-42.7
-41.5
-41.8
-41.9
-43.1
4~
-57.3
-57.5
-55.3
-56.4
-71.4
-55.4
-57.2
-57.6
-56.3
-56.1
-56.6
-56.2
-57.1
-56.3
-56.8
*P
-26.3
-25.1
-27.8
-27.4
-10.8
-28.9
-24.6
-26.8
-26.9
-26.3
-27.7
-26.8
-25.9
-27.4
-28.1
-25.8
-26.2
-27.1
-26.4
28.5
-27.0
-27.1
-25.1
-26.5
-26.8
-26.2
-26.7
-25.7
-26.4
-26.1
xz
H1
Hz
Energy
(kcal/
mole)
36.5
37.0
36.9
36.9
-36.5
36.9
37.9
35.6
36.6
36.8
36.5
37.0
36.3
36.6
36.7
115.3
115.0
115.0
115.0
115.2
114.9
114.9
115.2
115.0
115.0
114.8
115.0
115.0
115.1
115.1
121.0
120.9
120.9
120.9
121.1
121.0
120.8
121.0
120.9
120.9
120.9
120.9
120.9
120.9
120.9
-7.51
-7.54
-7.60
-7.59
-6.54
-7.59
-7.55
-7.52
-7.60
-7.59
-7.51
-7.59
-7.57
-7.58
-7.54
See footnote a of Table I.
See footnote b of Table I.
has emerged. This was hitherto unrecognized because previous con formational studies have used
rigid geometrical parameters and varied only the dihedral angles. In the present paper an attempt has
also been made to provide standard parameters for
right-handed a-helices with proline in the middle,
based on crystal structure analysis and the flexible
geometry minimization studies. These parameters
can be used for model building studies, especially of
membrane protein segments containing proline and
can be used as starting geometry for further energy
minimization studies.
METHODS
In order to characterize the kink produced by proline
in the right-handed a-helix, two types of studies were
Table I11 Minimized Energies for Various
Conformational States of Ace-(Ala)4-Pro-NHMe
Conformational
State'
AAAAA
AAADA~
ACGDA
Proline
Puckering
Energy
(kcal/mole)
Down
UP
Down
UP
Down
UP
-42.12
-41.78
-40.13
-39.32
-35.66
-35.31
a The notation refers to the conformational states of five residues of peptide. A, G = right-handed a-helical regions; C, D
= extended regions. For further details see Ref. 7.
The lowest energy conformation as found in Ref. 8 and is
about 16 kcal/mole lower than the all right-handed a-helical conformer.
290
SANKARARAMAKRISHNAN AND VISHVESHWARA
Table IV Minimized Energies and Geometrical Parameters in the Hexapeptide Ace-(Ala)4-Pro-AlaNHMe with Crystal Structures as Starting Points
Protein"
Myoglobin (88)
@p-4
4p-3
4p-2
4p-1
4P
+p-4
1cP-3
1cp-2
1cP-1
1cP
Codeb
wp-4
up-3
up-2
wp-1
UP
3MBN
-47.5
-48.2
178.7
-52.4
-40.2
176.9
-48.6
-43.1
178.4
-49.8
-41.4
177.8
-50.8
-35.7
177.7
-54.4
-33.5
174.9
-49.1
-43.3
177.7
-53.5
-35.0
175.8
-57.0
-29.7
172.4
-47.8
-43.1
178.6
-49.9
-44.9
177.4
-48.8
-43.9
178.3
-47.8
-45.7
178.3
-47.7
-43.4
178.0
-48.9
-37.2
178.2
-50.2
-40.6
177.3
(-45, -55)
(-35, -45)
(175, 178)
-65.5
-35.1
-176.4
-66.5
-35.0
-172.8
-68.8
-36.1
-174.2
-67.5
-36.8
-173.7
-72.7
-35.9
-173.0
-74.7
-42.8
-172.3
-65.7
-38.8
-174.5
-71.5
-36.9
-172.6
-75.1
-56.8
-172.3
-61.6
-38.8
-174.7
-63.2
-34.1
-177.8
-62.1
-38.3
-176.7
-62.2
-36.9
-176.7
-70.9
-39.5
-170.6
-64.2
-38.4
-171.8
-67.5
-38.7
-174.0
(-63, -75)
(-35, -43)
(-172, -177)
-74.0
-45.9
-175.1
-81.2
-53.6
-167.8
-77.4
-48.9
-175.7
-78.9
-50.8
-175.2
-83.1
-52.3
-177.3
-70.6
-51.7
-175.2
-74.6
-51.2
-174.7
-78.5
-56.7
-174.3
-58.0
-50.2
-174.7
-81.9
-50.2
-172.7
-78.6
-39.1
-178.8
-78.4
-41.3
-176.2
-77.3
-44.7
--175.7
-77.8
-51.9
-175.6
-93.2
-49.7
-173.5
-77.6
-49.2
-174.8
(-70, -85)
(-40, -55)
(-173, -177)
lMBO
2MBN
lMBD
lMB5
Citrate synthase (15)
4CTS
lCTS
2CTS
3CTS
Citrate synthase
(183)
4CTS
lCTS
2CTS
Citrate synthase
(183)
3CTS
Alcohol
dehydrogenase
(329)
4ADH
5ADH
Average values
Range of values
* The position of the proline residue in the a-helical region is given for each protein
See footnote b of Table I.
-35.9
-59.5
-179.2
-46.6
-49.8
177.8
-38.6
-57.3
-179.4
-35.8
-60.1
-176.4
-37.1
-58.9
-177.3
-46.5
-57.6
179.9
-34.4
-62.2
-175.7
-39.5
-58.9
-177.4
-50.8
-62.2
178.5
-33.4
-61.3
-174.5
-33.6
-57.1
179.1
-33.9
-61.2
-178.0
-31.9
-60.8
-176.8
-40.9
-56.2
179.9
-38.0
-54.8
-178.1
-38.5
-58.4
-178.5
(-35, -45)
(-55, -65)
(-177, 179)
-65.8
-30.5
177.9
-53.4
-34.7
179.7
-65.7
-31.3
177.0
-63.5
-22.3
174.9
-60.7
-26.3
176.2
-65.3
-36.5
178.8
-61.7
-21.7
175.6
-63.3
-21.7
175.4
-66.4
-36.4
179.8
-63.9
-17.5
173.5
-59.7
-34.1
174.8
-63.2
-25.6
173.9
-59.8
-24.2
175.3
-63.5
-31.9
176.4
-65.3
-29.6
175.1
-62.8
-28.3
176.3
(-60, -65)
(-20, -35)
(175, 180)
Energy
(kcal/mole)
-55.0
-53.6
-54.1
-53.8
-53.4
-54.4
-54.1
-52.9
-54.5
-53.8
-55.1
-54.5
-54.8
-54.2
-53.5
PROLINE IN THE RIGHT-HANDED a-HELICAL REGION
carried out: ( a ) crystal structure analysis and ( b )
energy minimization.
Crystal Structure Analysis
Right-handed a-helical segments containing proline
are found in myoglobin, 12-15 citrate synthase,16,17and
alcohol d e h y d r o g e n a ~ e ' ~(Table
' ~ ~ I ) . The coordinates of these segments of proteins were taken from
Brookhaven Protein Data Bank.20 T h e backbone
conformational parameters of the four residues preceding proline were analyzed. The virtual dihedral
angles made by consecutive C a atoms were also
evaluated. T h e bend due to the proline residue was
characterized as the angle between the two helical
axes: one from the beginning of the helix to proline
and the other from the proline t o the end of the
helix. The algorithm suggested by Chou et a1.21was
used to find the helical axis and the program was
written for and run on an IBM compatible PC/AT.
The kink angle in the segments given in Table I and
in other proposed average structures described below
was evaluated using this program.
Energy Minimization Studies
Energy minimization was carried out using the AMBER (assisted model building with energy refinement)
adapted to run on a DEC-1090
computer. The partial atomic charges are those suggested by Singh and Kollman 24 and the various constants to evaluate the energy were from Weiner et
a1.2s All atoms including the hydrogens were considered in the calculations. A distance-dependent
dielectric constant t = Ril was employed in the evaluation of electrostatic interaction energies. All nonbonded interactions were calculated. The structure
was refined until the rms gradient of energy was less
than 0.1 kcal/ mole.
R
H
Dipeptides and polypeptides of varying lengths
containing Ala-Pro fragments were studied by this
energy minimization technique allowing all the parameters t o vary. For minimization of the peptides
Ace-Ala-Pro-NHMe, Ace- ( Ala)4-Pro-NHMe, and
Ace- ( Ala),-Pro-Ala-NHMe, a steepest descent
method was used for the first 500 cycles and then a
conjugate gradient method was used until convergence. For the polypeptide Ace- ( Ala)ll-Pro-( Ace)11NHMe, the steepest descent method was used until
convergence.
Minimization was carried out on Ace-Ala-ProNHMe using the set of (6,I
)), corresponding to the
ideal right-handed a-helical region, extended conformation, and left-handed a-helical region in the
conformational space as starting points. Standard
geometries for the bond lengths and bond angles
were chosen.26All of the three favored proline conformations (up, down, and planar) 27 were investigated. The results of these studies are given in Table
IIa. T h e geometries of the dipeptide X-Pro from the
crystal structures given in Table I were minimized,
replacing the residue "X" by "Ala," the results corresponding to these studies are presented in T a ble IIb.
The pentapeptide Ace- ( Ala)4-Pro-NHMe has
previously been studied' using constrained bond
lengths and bond angles. Some of the minimized
geometries from this study were considered as starting points (Table 111) and have been reexamined
allowing all the parameters t o vary during minimization.
For the energy minimization studies of the hexapeptide Ace- ( Ala),-Pro-Ala-NHMe, crystal structures mentioned in Table I were taken as starting
points. Four residues before proline and one residue
after proline were replaced by "Ala." The results
corresponding to these studies are presented in Table IV.
H
Figure 1. The conformational parameters of poly ( L-Ala) containing a proline residue
in the middle. " R ' represents the alanine residue. The virtual dihedral angle is represented
by broken lines. The symbol p in Table I represents the virtual dihedral angle
c;-3-c;-*-c;-,-c;.
291
,
,oo
90
90
h;l (GGLOBIN
{Residues 82-96)
i
1
2
3
5
4
6
.
8
9
10
I1
CITRATE SYNTHASE
(Residues 5 - 2 7 )
90
1
0
4CTS
t
l i T 5
3
XTS
C
3CTS
Figure 2. Plots of virtual dihedral angles corresponding to the right-handed a-helical
segments of some proteins [ ( a ) myoglobin, ( b and c ) citrate synthase, ( d ) alcohol dehydrogenase] containing proline in the middle. The numbers in the x axis represent the
virtual dihedral angles sequentially and the corresponding values of these virtual dihedral
angles are plotted in the y axis. In the x axis the symbol “1” represents the first virtual
of the helical segment, where C: is the first C“ carbon
dihedral angle ( Cp-Cp+1-Cp+2-Cp+3)
atom of the helical segment. The virtual dihedral angle ( Cg-3-CE-2-Cg-l-Cg) (denoted as
p in Table I ) corresponds to the marking “4” in ( a ) , “8” in ( b ) , “15” in ( c ) , and “3” in ( d ).
PROLINE IN T H E RIGHT-HANDED a-HELICAL REGION
CI T R AT E SY Id THAS E
(Residues 166-195)
100
90
80
70
60
50
3
-1 0
3 $3
24 1
ALCOHOL DEHY D ROGENASE
(Residues 324-336)
100
90
80
70
60
50
40
30
20
1
2
3
5
4
0
Figure 2.
4ADH
7
6
+
8
5ADH
(Continued from the previous page. )
9
10
293
294
SANKARARAMAKRISHNAN AND VISHVESHWARA
Finally, the polypeptide Ace- ( Ala ,-Pro- ( Ala)l,NHMe was considered for geometry minimization.
Four starting conformations were chosen. In one
case the backbone internal parameters of residues
172-193 reported by S. Remington et al.I7 in the
crystal structure of citrate synthase (code: 2CTS ) '"
were used. In the second case, the structure of residues 7-20 in the polypeptide was taken from the
backbone internal parameters of alcohol dehydrogenase (code: 4ADH) 2o reported by Eklund et al., l8
and the remaining residues were considered to have
values corresponding to an idealized a-helix ( 9
= -57.5", $ = -47.5' ) ." In the third and the fourth
cases the parameters for three residues before proline and up to proline were taken as the average
value of the crystal structures given in Table I, and
the idealized a-helical values were taken for other
parameters. The proline residue was considered in
the up conformation in the third and down conformation in the fourth case.
RESULTS AND DISCUSSION
Crystal Structure Analysis
Previous a n a l y ~ e s ' , of
~ ,crystal
~~
structures were unable to locate directly the parameters responsible
for the production of the kink in right-handed a helical polypeptides containing proline. Further
analysis of proteins from Protein Data Bank files2"
was therefore taken up. T h e parameters analyzed
are shown in Figure 1 and the results are given in
Table I. The data shows that the bond angles
( Np-l-CF-l-Cb-l) and ( C;-l-Cb-l-Np) are generally
widened. The dihedral angles (9, $ ) from dP-:3to $p
(Table I ) show considerable deviation from the
standard values [ idealized theoretical values of
(-57.5", -47.5") 2R and average values obtained from
protein crystal structures (-63.8', -41.0")29]. The
range and average values listed for @p-3 and @ p - 2 are
generally more negative than the standard values,
whereas #p-l is less negative. Also, the values of $p-3,
$p'p-2, and $p are generally less negative than the
standard value, and $p-l is more negative. The tabulated o values show that the peptide groups have
deviated t o some extent from planarity. The
and
are generally around -175". Although
and wp values deviate from planarity by a small
amount, no definite pattern can be observed.
T h e virtual bond lengths (C: -C:+l ) , bond angles
(Cp-Cp+,-Cp+2), and the dihedral angles ((2;C p+I-Cz2-Cp+3) in the crystal structures as indicated
in Figure 1 were analyzed. When compared with a
standard right-handed a-helix, there was no significant change in any of the parameters except the
virtual dihedral angle ( C;-:3-C&-CF-l-C;), denoted
as p (Table I ) . The average value of the virtual dihedral angle"' ( C:-CF+l-C:+2-C:+3) in a standard
right-handed a-helix is about 50". The p values that
are given in Table I range from 65" to go', indicating
the distortion introduced by proline. Plots of virtual
dihedral angles of the helical segments containing
proline in various proteins are shown in Figure 2ad. T h e virtual dihedral angle p corresponds to the
markings 4, 3, 8, and 15 on the x axis in Figure 2a,
b, c, and d, respectively. From these plots, it is clear
that the value of p in all cases is higher than the
other virtual dihedral angles. T h e distortion introduced by proline in the right-handed a-helix can
also be characterized by the kink angle. The kink
angles that are computed as given in methods section
are also presented in Table I. With a few exceptions
this value is between 20' and 30".
Thus, by a detailed analysis of the crystal structures, it has been possible to identify the backbone
parameters and the virtual dihedral angle that are
responsible for the kink produced by proline in the
right-handed a-helical region. A significant change
in any of the parameters listed in Table I leads to a
different value for the kink angle.
Energy Minimization Studies
Ace-Ah-Pro-NHMe. Energy minimization was car-
ried out on Ace-Ala-Pro-NHMe as described in the
methods section. The dipeptide and the relevant
conformational parameters are represented in Figure
3. The results presented in Table IIa show that along
with the extended and the left-handed a-helical regions the right-handed a-helical region for Ala-Pro
is also energetically favorable. This is contrary to
the previous studies, which considered the righthanded a-helical region to be energetically unfavorable due to steric contacts that fall outside the
Ramachandran limit.26 These earlier studies used
rigid geometrical parameters. The present study reveals that small adjustments in the bond lengths
and angles can greatly offset the energy penalty,
which is ascribed to a proline residue in the righthanded a-helical region. The results given in Table
IIa also show that the puckered ( u p or down) conformations of proline are preferred and the planar
conformation may be stable ( a local minimum) only
when Ala-Pro takes up a conformation in the extended region.
The geometrical parameters that give rise to the
minimum energy conformations are also given in
'z7
PROLINE IN T H E RIGHT-HANDED 0-HELICAL REGION
295
Figure 3. The conformational parameters of Ace-Ala-Pro-NHMe. "R" represents the
alanine residue.
Table IIa. A comparison of structure I of Ace-AlaPro-NHMe, with the minimized structure of AceAla-Ala-NHMe in the right-handed a-helical conformat ion, indicates a few differences. T h e values
of g p . and 1c,. are significantly different in structure I , the & p - , value being less negative and lc,-l
being more negative than the corresponding values
in the Ala-Ala dipeptide. T h e bond angles N-C"-C'
( 0 , ) and C"-C'-N (0,) are wider in Ace-Ala-ProNHMe. These trends are consistent with the analysis of crystal structures (Table I ) . These differences
relieve the short contact between the -CH3 side
group of alanine and the -C*HH,
group attached to
the irriido nitrogen t h a t was responsible for the high
energy of this conformation in the earlier rigid geomet ry minimization studiess T h e x values, X I and
x2, represent the orientation of the pyrrolidine ring
( u p and down). In structure I the dipeptide with
the proline ring in the up conformation is more stable t)v about 1 kcal/mole whereas in structures I1
and 111. the down conformation is more favored.
T h e results presented in Table IIb show that
when diEerent crystal structure parameters for the
Ala-Pro dipeptide are taken as the starting points,
all minimize to the same point. Since different.
starting geometries minimize to the same structure,
the parameters obtained for structure I (Table IIa
,
and b ) can be considered as standard ones for the
dipeptide in the right-handed a-helical conformation.
Ace- (Ah),-Pro-NHMe. Energy
minimization in
which bond lengths and bond angles were allowed
to vary was carried out on Ace- ( Ala),-Pro-NHMe
using the minimized structures reported by Piela et
al.' a s starting points. T h e different conformations
and energies are given in Table 111. In the lowest
energy conformation (-42.1 kcal/mole 1 , Ala-Pro
takes right-handed a-helical conformation. This
conformation for Ala-Pro was 16 kcal/mole above
the minimum in the minimization studies performed
using rigid geometry, indicating the importance of
flexible geometry minimization in these systems.
The geometrical features of the minimized structure
are consistent with that of the Ala-Pro dipeptide in
structure I (Table IIa) and with the crystal structure
analysis (Table I ) . The q5p-l has become less negative (-40.4' for down and -44.2' for u p ) and the
angles N-C"-C' and C"-C'-N have widened in comparison to that in Ala-Ala. T h e up-* value, in the
rigid geometry minimization, was distorted to wP-,
= - 1:3 1' for pro= - 138" for proline down and
line up. In the present case, the peptide unit is almost
planar ( wPpI = -178' ) . T h e values of x I and X L rep-
Table V Minimized Energies of Ace-(Ala), I-Pro-(Ala),I-NHMe in the Right-Handed
a-Helical Conformation
Polypeptide
Ace- (Ala),,-Pro-( A h ) -NHMe
Ace-(Ala),,,-NHMe
Starting Conformation
Citrate synthase
Alcohol dehydrogenase
Average values of @p-3 to up taken from Tables I1
and Iii for proline-up
Average values of @ p p - 3 to u ptaken from Tables I1
and I11 for proline-down
Standard right-handed a-helix
Energy
(in kcal/mole)
-3 12.03
- 3 1:3.28
-313.72
-313.71
- 34 2.25
296
SANKARARAMAKRISHNAN AND VISHVESHWARA
resenting the proline up and down conformations
are also similar to those given for structure I in Table
IIa. Thus the right-handed a-helical region for AlaPro appears to be the preferred conformation, even
when it is part of a polypeptide. To achieve this,
minor variations of a definite nature takes place in
the geometrical parameters of proline and the residues close to the proline.
Minimization of Ace- (Ala)4-Pro-Ala-NHMe. The
crystal structure analysis has shown that the backbone parameters in the right-handed a-helix containing proline deviated from the standard value by
a small but significant amount and that the observed
deviations extend over a pentapeptide unit that
contains proline and four other preceding residues
(Table I ) . This stretch of helix is represented by
the dihedral angles beginning from 4pp-4
and ending
with +bP as shown in Figure 1. Therefore the hexapeptide Ace- ( Ala )4-Pro-Ala-NHMewith proline as
the fifth residue was considered for energy minimization studies. The backbone and the proline parameters observed in the crystal structures were
taken as the staking points. The minimized energies
of the hexapeptide Ace- ( Ala)4-Pro-Ala-NHMe
range from -53 to -55 kcal/mole (see Table IV).
The range and the average minimized parameters
are also given in Table IV. The results generally
Table VIa Standard"Bond Angles for the
Proline Region in the Right-Handed a-Helical
Segment Containing a Proline Residue
Bond Angles
Values
(in degrees)
111.5
117.2
124.5
112.3
120.5
121.7
111.7
118.2
122.1
127.9
106.0
103.6
106.5
104.4
111.9
a Derived by averaging the values obtained on systems mentioned in Table IV and V.
Table VIb Standard"Bond Angles and Dihedral
Angles for the Proline Region (for both ProlineDown and -Up Puckerings) in the Right-Handed
a-Helical Segment
Values (in degrees)
Angles
a
Proline-Down
Proline-Up
116.9
123.9
114.8
119.0
122.1
112.7
-62.5
-42.8
-172.9
-70.3
-48.6
-171.1
-43.5
-55.6
-178.4
-64.2
-35.2
177.3
23.9
-31.3
-68.9
-37.9
-172.4
-75.9
-42.5
-174.3
-55.3
-50.6
171.7
-57.2
-43.9
179.9
-17.1
27.7
See footnote a of Table VIa.
agree with the crystal structure analysis with 4pp-3,
4p-2being more negative and dpPlless negative than
the standard values. The q P - 3 , # p - p , #p are less negative, and #p-l more negative, than the standard
values. The values of 4pp-4
and # p - 4 , however, do not
follow the crystal structure pattern. This is because
the conformation of the regular structure of the helix
is truncated in the hexapeptide. The u p - 3 and wp-2
values follow the same trend as the crystal structure
values, deviating from planarity by about -5" to
-10". The
and wp are nonplanar to the extent
of about -5" and + 5 O , respectively, a trend that was
not clear from the crystal structure analysis. The
range of the minimized values is less in comparison
with the crystal structure range. The average values
for 4pp-3
to # p , with the exception of #p-2 and $ p - l ,
are similar in the crystal structures (Table I ) and
in the minimized structures (Table IV) .
Studies on Large Fragments of Right-Handed a-Helix with Proline The above studies clearly show that
a right-handed a-helical conformation can be taken
up by X-Pro ( X = nonglycine residue) in a polymer.
However, small distortions from the ideal values
take place in the bond angles in the region of proline
PROLINE IN T H E RIGHT-HANDED a-HELICAL REGION
and in the backbone dihedral angles in the regions
preceding proline. T o arrive a t a set of geometrical
parameters for a polymer containing proline, Ace( Ala)ll-Pro-( Ala)ll-NHMe in the right-handed ahelical conformation was examined. Four starting
conformations, the details of which are given in the
methods section, were considered for minimization.
These parameters along with the energies are given
in Table V. T h e minimized energies are in the range
of 312.5 k 1kcal/mole. T h e geometrical parameters
obtained are also very close to each other and the
average values of some of the parameters are given
in Table VI. T h e overall helical structure with a
kink angle of about 23" as shown in Figure 4 is retained. When compared with a standard righthanded a-helix two hydrogen bonds are lost in these
structures. Due t o the absence of the amide proton
in proline the hydrogen bond between the atoms Np
and Q p - , (Figure 4) is lost, and the hydrogen bond
between atoms N,,, and Op-3is lost due to the kink.
Our studies on the hexapeptide fragment have shown
that some of the backbone parameters depend on
whether the proline is in the up or down conformation. T h e last two starting points in Table V correspond to the average structures given in Table I
and IY for the (Ala)3-Pro-Ala region, the proline
taking a n up conformation in one case and the down
conformation in the other. For the parameters # p - 2
and
the crystal structure average differs considerably from the hexapeptide-minimized values
(Table I and IV) . Both cases were considered for
minimization.
Based on the above studies, a set of average parameters for a right-handed a-helix with proline has
been obtained and are presented in Table VIa and
b. Notable features are the widening of the backbone
angles N,. ,-C;.-,-Cb-l and C;-l-Cb-l-Np by about
4
' and so,respectively, and the shortening of the
bond angle involving the proline ring carbon atoms.
Three backbone angles depend on the conformation
of the proline ring ( u p or down). The dihedral angles
X 1 and X 2 represent the orientation of the proline,
and these values are given in Table VIb. Since these
values represent the parameters of proline in the
right-handed a-helical region, they differ slightly
from those previously suggested l5 based on the average of all types of proline structures. The deviation
in the backbone dihedral angles 4p-3to wp show that,
although the trends as observed in the crystal structures are maintained, a different set of values are
obtained for the up and down conformations of proline. Particularly noticeable is the
parameter,
which deviates from planarity by +1.6" in the down
conformation and by -8" in the up conformation.
297
I
I
Figure 4. The minimized structure of Ace- ( Ala),,-Pro( Ala),,-NHMe with its kink. The intrahelical hydrogen
bonds are represented by broken lines. The hydrogen
bonds NP+l * * Op-3 and NP * * * Op-4 are abs'ent. The
details are given in the text.
T h e set of parameters given in Table VI gives rise
to a kink value of 22.1" and 20.5", respectively, for
the up and down conformations, and the p values
are 67" and 72", respectively. A variation in these
parameters o f f lo or 2" will not significantly change
the kink angle.
SUMMARY
By flexible geometry minimization studies, the righthanded a-helical conformation for X-Pro ( X = any
298
SANKARARAMAKRISHNAN AND VISHVESHWARA
nonglycyl residue) has been shown to be a n energyminimum conformation. Previous studies could not
identify this minimum since rigid geometry was used
in the minimization studies. In the right-handed a helix some of the bond angles deviate slightly from
the standard values in order to relieve steric contacts
between the side chain of the preceding residue of
proline and the methylene group attached to the
imido nitrogen of proline. Small variations with a
definite trend also occur in the backbone parameters.
The right-handed &-helical conformation is also
possible when the X-Pro unit is in the middle of a
right-handed a-helical segment. The present energy
studies support the crystal structure data analysis
and two-dimensional 'H-nmr ~ t u d i e s . ~The
' insertion of proline in the right-handed a-helical segment
causes the helix axis to bend by about 20"-30". Also,
the virtual dihedral angle involving four C" atoms( C;-3-C&-C;-l-C;)
is in the range of 65"-85" (average value of a virtual dihedral angle in a n a-helix
is = 50" ) . Such a bend distorts the backbone parameters from the ideal helical values. These deviations extend t o 3-4 residues before proline, the parameters of the preceding residue being affected the
most. The nature of deviations are characterized by
the crystal structure data analysis and by energy
minimization studies.
The proline up and down conformations give rise
to energetically equivalent a-helical structures involving slightly different sets of geometrical parameters. A set of standard parameters has been arrived
a t for the bend region in a right-handed a-helix containing proline. These parameters can be used in
model building and further energy minimization
studies.
We wish to thank Professor P. A. Kollman and his group
for providing us the AMBER program, and Dr. N. Sreerama for helping us in adapting the AMBER onto the DEC1090 computer of our institute.
REFERENCES
1. Anfinsen, C. B. & Scheraga, H. A. (1975) Adu. Protein
Chem. 29, 205-300.
2. Robson, B. & Suzuki, E. (1976) J. Mol. Biol. 1 0 7 ,
327-356.
3. Chou, P. Y. & Fasman, G. D. (1974) Biochemistry 13,
211-222.
4. Richardson, J. S. & Richardson, D. C. (1988) Science
240, 1648-1652.
5. Schimmel, P. R. & Flory, P. J. (1968) J. Mol. Biol.
34, 105-120.
6. Pincus, M. R., Klausner, R. D. & Scheraga, H. A.
(1982) Proc. Natl. Acad. Sci. U S A 79,5107-5110.
7. Zimmerman, S. S. & Scheraga, H. A. (1977) Biopolymers 16, 811-843.
8. Piela, L., Nemethy, G. & Scheraga, H. A. (1987) Biopolymers 26, 1587-1600.
9. Barlow, D. J. & Thornton, J. M. (1988) J. Mol. Biol.
201,601-619.
10. Brandl, C. J. & Deber, C. M. ( 1986) Proc. Natl. Acad.
Sci. U S A 83,917-921.
11. Sankararamakrishnan, R. & Vishveshwara, S. ( 1989)
J . Biomol. Struc. Dynam. 7, 187-205.
12. Takano, T. (1977) J. Mol. Biol. 110, 569-584.
13. Phillips, S. E. V. (1980) J . Mol. Biol. 142, 531-554.
14. Phillips, S. E. V. & Schoenborn, B. P. (1981) Nature
292,81-82.
15. Hanson, J. C. & Schoenborn, B. P. ( 1981 ) J . Mol.
Biol. 153, 117-146.
16. Wiegand, G., Remington, S., Diesenhofer, J. & Huber,
R. (1984) J . Mol. Biol. 174, 205-219.
17. Remington, S., Wiegand, G. & Huber, R. (1982) J.
Mol. Biol. 158, 111-152.
18. Eklund, H., Nordstrom, B., Zeppezauer, E., Soderlund,
G., Ohlsson, I., Boiwe, T., Soderberg, O., Tapia, O.,
Branden, C.-I. & Akeson, A. (1976) J . Mol. Biol. 102,
27-59.
19. Plapp, B. V., Eklund, H., Jones, T. A. & Branden,
C.-I. (1983) J . Biol. Chem. 258,5537-5547.
20. Bernstein, F. C., Koetzle, T. F., Williams, G. J. B.,
Meyer, E. F., Jr., Brice, M. D., Rodgers, J. R., Kennard,
O., Shimanouchi, T. & Tausmi, M. (1977) J. Mol.
Biol. 112, 535-542.
21. Chou, K. C., Nemethy, G. & Scheraga, H. A. (1984)
J . Am. Chem. SOC.106, 3161-3170.
22. Weiner, P. & Kollman, P. A. (1981) J. Comp. Chem.
2, 287-303.
23. Weiner, P. K., Singh, U. C., Kollman, P. A., Caldwell,
J. & Case, D. A. ( 1984) A Molecular Mechanics and
Dynamics Program-AMBER, University of California, San Francisco.
24. Singh, U. C. & Kollman, P. A. ( 1984) J . Comp. Chem.
5, 129-145.
25. Weiner, S . J., Kollman, P. A., Nguyen, D. T. & Case,
D. A. (1986) J. Comp. Chem. 7 , 230-252.
26. Ramachandran, G. N. & Sasisekharan, V. (1968) Adv.
Protein Chem. 23, 283-438.
27. Momany, F. A., McGuire, R. F., Burgess, A. W. &
Scheraga, H. A. (1975) J. Phys. Chem. 79,2361-2381.
28. Arnot, S. & Wonacott, A. J. (1966) J . Mol. Biol. 21,
371-383.
29. Presta, L. G. & Rose, G. D. (1988) Science 240,16321641.
30. Ramakrishnan, C. & Soman, K. V. (1982) Znt. J. Peptide Protein Res. 20, 218-237.
31. Arseniev, A. S., Maslennikov, I. V., Bystrov, V. F.,
Kozhich, A. T., Ivanov, V. T. & Ovchinnikov, Yu. A.
(1988) F E B S Lett. 231, 81-88.
Received August 10, I989
Accepted January 18, 1990