Document 13313560
advertisement

Leucine Rich Repeat Kinase 2 in the pathogenesis of Parkinson’s Disease by Tatiana D. Papkovskaia A thesis submitted for the degree of Doctor of Philosophy Department of Clinical Neuroscience, Institute of Neurology University College London 2013 1 Declaration I, Tatiana D. Papkovskaia confirm that the work presented in this thesis is my own. The help and contribution of others to this thesis is specified in the acknowledgements section. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 To my parents 3 Abstract The Leucine Rich Repeat kinase 2 (LRRK2) G2019S mutation is the most common genetic cause of Parkinons's disease (PD) which is clinically and pathologically indistinguishable from idiopathic PD. The effects of the G2019S mutation were explored in primary fibroblasts and SHSY5Y cells expressing wild type or G2019S LRRK2. LRRK2 was predominantly in the cytosol and small vesicular fraction of lymphoblasts and SHSY5Y cells with some localized to the mitochondria in overexpressing cells. While we could detect LRRK2 in various mouse and marmoset brain regions LRRK2 protein levels were higher in fibroblast and lymphoblast cultures. LRRK2 cellular distribution, mRNA and protein expression were not affected by the mutation. Mitochondrial abnormalities are a common feature in PD. To determine whether mitochondrial function is compromised in mutant cells, a detailed bioenergetic assessment was carried out on both G2019S cell models. An increase in basal and oligomycin inhibited respiration rates, reduced mitochondrial membrane potential and cellular ATP levels was observed for G2019S fibroblasts with similar changes observed in the neuroblastoma G2019S model. Respiratory rates and membrane potential were restored with LRRK2 kinase inhibition. Our data is consistent with reversable uncoupling of oxidative phosphorylation. Investigating transcriptional levels of mitochondrial uncoupling proteins (UCP) identified a G2019S dependent increase in UCP2 and 4 in fibroblasts and SHSY5Y cells. Upstream of this transcriptional event, an interaction between LRRK2 and the negative regulator of PGC1α expression, HDAC5 was investigated for both the wild type and G2019S protein. We have identified a role for endogenous LRRK2 in regulating mitochondrial bioenergetics with a kinase dependent gain of function for the G2019S mutation 4 consistent with partial uncoupling of oxidative phosphorylation. LRRK2 G2019S enhanced the HDAC5 association which may be responsible for increased PGC1α expression and the downstream UCP transcription linked with the observed mitochondrial phenotype. 5 Acknowledgements First, I would like to thank my supervisor Prof. Tony Schapira for giving me the opportunity to work at the Clinical Neuroscience Department on a stimulating project and in such a pleasant environment. Thank you for your guidance, support and the freedom to express my thoughts and ideas. I would also like to express my gratitude to Dr. Mark Cooper for his dedication to my work, for the time, effort, ideas and concerns throughout the last three years. Thank you for teaching me how to think, to write and to love what I do. I would also like to thank my third supervisor Prof. John Hardy for integrating me into his department at the Institute of Neurology and teaching me to appreciate genetics. Thank you to all my colleagues at the Royal Free Hospital, in particular Lydia Alvarez and Matthew Gegg who have shared this experience with me, for their continuous support and ideas which helped move the project forward. Thank you to Michael Cleeter and David Chau for their management of the department, the technical support and rapid response to all demands and queries, I don’t know how you do it. Thank you to the more recent lab members; Anna Migdalska, Joana Magalhaes and Catharina Joana Casper as well as visiting staff; Dominic Piston, Davor Ivankovic, Tanja Hering and Marine Bourlin who integrated and expanded our wonderful science family. Thank you for every pub and staff night out, they were all amazing! A special thank you to the Parkinson’s disease patients and their relatives as well as Prof. Dan Healy, Dr. Jan Willem Taanman and Dr. Susannah Horan involved in the generation of the fibroblast and lymphoblast cultures. Thank you to Dr. Mark Cooper and Dr. David Chau for generating the LRRK2 stable SHSY5Y cells, to Dr. Zhi Yao and Dr. Gyorgy Szabadkai for the DJ1 knockdown SHSY5Y cells. Thank you to Prof. Dario Alessi and Dr. Franscisco Inesta Vaquera for the LRRK2 antibodies, LRRK2 6 inhibitors, their help with the recombinant UCP2 production, the LRRK2 kinase assays and my wonderful experience in Dundee. Thank you to Dr. Rina Bandopadhyay for the help with the immunohistochemistry and the time spent at the Queen Square Brain Bank. Thank you to Prof. Michael Duchen for his help with the confocal analysis. Thank you to Prof. Dmitri Papkovsky (my dad) and his wonderful lab in Ireland for their collaboration on the phosphorescent analysis, I really enjoyed being home and working with you. Thank you to Janet North for her help with the FACS and data analysis. A special thank you to Eisai Limited who funded the work. In particular, thank you to Dr. James Staddon and Dr. Malcolm Roberts for their continuous support and intellectual input into the project. Thank you also for the LRRK2 antibodies, the Cellomics equipment and the wonderful time spent at Eisai in UCL. I would also like to thank those involved in the UKDPC Consortium for giving me the opportunity to present my work, share ideas and experience Parkinson’s research from a different perspective. Thank you to my parents, Dmitri and Natalia Papkovsky who have supported and encouraged me at every stage of this experience. A special thank you to my sister Katia Papkovskaia who has been a wonderful friend and housemate throughout my PhD. Thank you to all my friends, the ones from my past and also the many new ones I have made over the last three years. You have all contributed to my wonderful memories. 7 Table of contents List of tables……………………………………………………………………………………………………….. 17 List of figures………………………………………………………………………………………………………..18 List of Abbreviations……………………………………………………………………………………………..23 Chapter 1: Introduction 1.1.1 Basal ganglia and Parkinson’s disease……………………………………………………… 29 1.1.2 Pathology in PD……………………………………………………………………………………………30 1.2 Causative factors in PD…………………………………………………………………………………..32 1.2.1.1 Autosomal dominant PD genes: α-synuclein………………………………………….. 32 1.2.1.2 Autosomal dominant PD genes: Leucine Rich Repeat Kinase 2……………… 33 1.2.1.3 Autosomal recessive PD genes: Parkin……………………………………………………. 35 1.2.1.4 Autosomal recessive PD genes: PINK1…………………………………………………….. 35 1.2.1.5 Autosomal recessive PD genes: DJ1………………………………………………………… 36 1.2.1.6 Autosomal recessive PD genes: Glucocerebrosidase……………………………… 37 1.2.2 PD genetic models……………………………………………………………………………………… 37 1.2.3 Environmental factors in PD………………………………………………………………………..39 1.2.4 Mitochondria……………………………………………………………………………………………….39 1.2.4.1 Mechanism of action of mitochondrial toxins………………… ……………............ 42 1.2.4.2 Electron transport chain and PD……………………………………………………………… 42 1.2.4.3 Mitochondrial turnover and dynamics…………………………… ………………………. 44 1.2.4.4 Mitochondrial transcriptional regulation…………………………………................ 45 1.2.4.5 Mitochondrial and oxidative stress ………………………………………………………….46 1.2.4.6 Oxidative stress and mitochondrial uncoupling proteins………………………… 47 1.2.5 Lysosomal function and α-synuclein………………………………………………………….. 50 1.3 LRRK2……………………………………………………………………………………………………………..51 1.3.1 LRRK2 protein structure……………………………………………………………………………… 51 8 1.3.2 LRRK2 kinase ……………………………………………………………………………………………… 53 1.3.3 LRRK2 PD mutations ………………………………………………………………………………….. 54 1.3.4 LRRK2 PD mutations and neurotoxicity……………………………………………………… 55 1.3.5 LRRK2 models and PD………………………………………………………………………………… 55 1.3.6 LRRK2 detection antibodies……………………………………………………………………….. 57 1.3.7 LRRK2 mRNA and protein distribution………………………………………………………. 58 1.3.8 LRRK2 cellular distribution…………………………………………………………………………. 58 1.3.9 LRRK2 function…………………………………………………………………………………………… 59 1.3.9.1 LRRK2 and vesicles………………………………………………………………………………….. 59 1.3.9.2 LRRK2 and microtubules…………………………………………………………………………..60 1.3.9.3 LRRK2 transcription and translation ………………………………………………………. 60 1.3.9.4 LRRK2 and mitochondria ………………………………………………………………………… 62 1.4 Hypothesis …………………………………………………………………………………………………….64 1.5 Aims……………………………………………………………………………………………………………… 64 Chapter 2: Materials and Methods 2.1 Plasmid constructs………………………………………………………………………………………… 66 2.2 Brain samples……………………………………………………………………………………………….. 66 2.3 Cell lines…………………………………………………………………………………………………………67 2.4 Cell culture……………………………………………………………………………………………………. 69 2.5 Transformation of DH5a cells and DNA extraction……………………………………… 70 2.6 Concentration and purity of nucleic acids…………………………………………………….. 72 2.7 Restriction enzyme digest of pET-DEST51……………………………………………………. 72 2.8 Agarose gel electrophoresis………………………………………………………………………….. 73 2.9 Transfection of mammalian cells…………………………………………………………………. 73 2.10 Reverse transcriptase……………………………………………………………………… …………..75 2.11 Real Time PCR……………………………………………………………………………………………… 76 9 2.12 LRRK2 mutation screen in fibroblasts and SHSY5Y cells……………………………. 78 2.13 Whole cell and tissue extractions……………………………………………………………….. 78 2.14 Immunoprecipitation………………………………………………………………………………….. 79 2.15 Protein quantification…………………………………………………………………………………. 80 2.16 SDS PAGE……………………………………………………………………………………………………..80 2.17 Western blot analysis………………………………………………………………………………….. 81 2.18 Native gel electrophoresis…………………………………………………………………………… 81 2.19 Immunofluorescence of cultured cells……………………………………………………….. 82 2.20 Immunohistochemistry on human and mouse brains………………………………… 83 2.21 LRRK2 kinase activity assays……………………………………………………………………….. 84 2.22 Isolation of PMBCs and cell sorting……………………………………………………………... 85 2.23 Cellular fractionation…………………………………………………………………………………… 87 2.24 Affinity purification of mitochondria………………………………………………………….. 87 2.25 Mitochondrial sub-fractionation…………………………………………………………………. 88 2.26 Proteinase K digestion of mitochondria……………………………………………………… 89 2.27 Single cell analysis by confocal microscopy……………………………………………….. 89 2.27.1.1 Mitochondrial membrane potential: TMRM…………………………………………. 89 2.27.1.2 Method validation for TMRM……………………………………………………………….. 90 2.27.2 Mitochondrial content……………………………………………………………………………… 92 2.27.3 GFP…………………………………………………………………………………………………………… 93 2.28.1.1 Cellular respiration: oxygen electrode…………………………………………………… 94 2.28.1.2 Method validation for the oxygen electrode………………………………………… 94 2.28.2.1 Cellular respiration: phosphorescent analysis…………………………… …………..96 2.28.2.2 Method validation of phosphorescent analysis…………………………………….. 97 2.29.1 Extracellular acidification………………………………………………………………………… 99 2.29.2 Method validation for extracellular acidification measurements… …………..100 10 2.30 Cellular ATP content……………………………………………………………………………………. 103 2.31 Cellular ROS production: Array Scanner……………………………………………………… 103 Chapter 3: Results Characterization of LRRK2 in cell models and brain tissue 3.1. Western blot analysis of LRRK2 protein expression in cell models and brain tissue………………………………………………………………………………………………………………….. 105 3.2 Characterization of LRRK2-V5 over-expressing SHSY5Y cells……………………… 108 3.2.1 Western blot analysis of LRRK2 protein in SHSY5Y cells………………………….. 108 3.2.2 LRRK2 mutation screen in SHSY5Y cells………………………………………… …………..109 3.2.3 Western blot analysis of LRRK2 sub-cellular distribution in SHSY5Y cells…………………………………………………………………………………………………………………….. 111 3.2.4 Immunocytochemistry of LRRK2 sub-cellular distribution in SHSY5Y cells…………………………………………………………………………………………………………………….. 113 3.3 Characterization of endogenous LRRK2 in lymphoblasts……………………………... 115 3.3.1 Western blot analysis of LRRK2 protein in lymphoblasts……………… …………..115 3.3.2 Western blot analysis of LRRK2 sub-cellular distribution in lymphoblasts..…………………………………………………………………………………………………….. 118 3.4 LRRK2 immunoprecipitation…………………………………………………………………………..120 3.4.1 Characterization of LRRK2 immunoprecipitation for quantitative comparison…………………………………………………………………………………………………………..120 3.4.2 LRRK2 kinase activity in lymphoblasts and SHSY5Y cells…………………………… 123 3.5 Characterization of fibroblast cell lines …………………………………………… …………..126 3.5.1 Immunoprecipitation of LRRK2 protein from fibroblasts…………………………… 126 3.5.2 LRRK2 mutation screen in control and G2019S fibroblasts……………………….. 127 3.5.3 LRRK2 mRNA expression in control and G2019S fibroblasts………… …………..127 3.6 Analysis of LRRK2 knockdown in SHSY5Y cells……………………………………………… 129 11 3.7 Immunoprecipitation of endogenous LRRK2 protein from brain………………….. 132 3.8 Comparison of LRRK2 expression in cell models and brain tissue…… …………..136 3.9 Analysis of LRRK2 protein expression by blue native PAGE electrophoresis…………………………………………………………………………………………………….137 3.10 Immunohistochemical analysis of LRRK2 protein in brain…………………………. 141 Chapter 4: Results LRRK2 and mitochondrial function 4.1 Mitochondrial function in fibroblast cell lines………………………………………………. 145 4.1.1 The analysis of mitochondrial respiration in control and G2019S fibroblasts…………………………………………………………………………………………………………… 145 4.1.2 The analysis of mitochondrial content in control and G2019S LRRK2 fibroblasts…………………………………………………………………………………………………………… 147 4.1.3 The analysis of mitochondrial membrane potential in control and G2019S LRRK2 fibroblasts………………………………………………………………………………………………… 149 4.1.4 The analysis of cellular ATP content in control and G2019S LRRK2 fibroblasts…………………………………………………………………………………………………………… 150 4.1.5 The analysis of extracellular acidification rates in control and G2019S LRRK2 fibroblasts…………………………………………………………………………………………………………… 151 4.1.6 The analysis of cellular ROS in control and G2019S LRRK2 fibroblasts…….. 152 4.1.7 The analysis of mitochondrial proton leak in control and G2019S LRRK2 fibroblasts…………………………………………………………………………………………………………… 153 4.1.8 The analysis of mitochondrial morphology in control and G2019S LRRK2 fibroblasts…………………………………………………………………………………………………………… 154 4.2 Mitochondrial dysfunction models………………………………………………………………. 156 4.2.1 The use of control fibroblasts treated with rotenone or antimycin A as a model of respiratory chain dysfunction…………………………………………………………………………. 156 12 4.2.2 The use of control fibroblasts treated with rotenone and oligomycin to evaluate the influence of ETC inhibition on the mitochondrial ATPase……………… 157 4.2.3 The use of control fibroblasts cultured in galactose rich conditions as a model of limited cellular substrate supply………………………………………………………… …………. 159 4.3 The analysis of respiratory chain function in the SHSY5Y LRRK2-V5 overexpressing model………………………………………………………………………………………………… 161 4.4 LRRK2 kinase activity…………………………………………………………………………………….. 163 4.4.1 The analysis of LRRK2 kinase inhibition in the SHSY5Y LRRK2-V5 overexpressing model ………………………………………………………………………………………………..163 4.4.1.1 LRRK2-IN1……………………………………………………………………………………………….. 164 4.4.1.2 CZC25146………………………………………………………………………………………………... 164 4.4.1.3 TAE-648…………………………………………………………………………………………………… 165 4.4.2 The use of LRRK2 kinase inhibitors to evaluate effects of LRRK2 kinase inhibition on mitochondrial function in the SHSY5Y LRRK2-V5 over-expressing model…………………………………………………………………………………………………………………. 167 4.4.2.1 Evaluation of the effects of LRRK2 kinase inhibition on mitochondrial respiration………………………………………………………………………………………………………….. 167 4.4.2.2 Evaluation of the effects of LRRK2 kinase inhibition on the mitochondrial membrane potential…………………………………………………………………………………………… 167 4.5 LRRK2 mitochondrial localization……………………………………………………… …………. 169 4.5.1 Mitochondrial affinity purification……………………………………………………………… 169 4.5.1.1 Mitochondrial affinity purification versus differential centrifugation……. 171 4.5.1.2 Mitochondrial affinity purification from G2019S LRRK2-V5 over-expressing SHSY5Y cells………………………………………………………………………………………………………… 171 4.5.1.3 Mitochondrial affinity purification, LRRK2 kinase inhibition………………….. 172 4.5.1.4 Mitochondrial affinity purification from lymphoblasts…………………………… 172 13 4.5.2.1 Sub-fractionation of mitochondrial enriched fractions from SHSY5Y LRRK2-V5 over-expressing SHSY5Y cells………………………………………………………………………………. 175 4.5.2.2 Sub-fractionation of affinity purified mitochondria from SHSY5Y LRRK2-V5 over-expressing SHSY5Y cells………………………………………………………………………………. 176 4.5.3.1 Sub-fractionation of mitochondria to determine LRRK2 membrane association………………………………………………………………………………………………………….. 178 Chapter 5: Results LRRK2 regulation of mitochondrial uncoupling: mechanisms 5.1.1 The analysis of SHSY5Y cells over-expressing murine GFP-UCP2…………….. 182 5.1.2.1 The analysis of relative UCP mRNA expression in fibroblasts and SHSY5Y cells.. …………………………………………………………………………………………………………………..184 5.1.2.2 The analysis of relative UCP mRNA expression in G2019S fibroblasts and SHSY5Y cells………………………………………………………………………………………………………… 185 5.1.3 Western blot analysis of UCP2 protein in fibroblasts and SHSY5Y cells……. 186 5.1.4.1 Evaluation of the influence of genipin on UCP induced mitochondrial depolarization in SHSY5Y cells…………………………………………………………………………….. 188 5.1.4.2 Evaluation of the influence of genipin on UCP induced mitochondrial depolarization in fibroblasts……………………………………………………………………………….. 190 5.1.5 LRRK2 kinase activity assays with recombinant UCP2 protein……………………..192 5.1.6 The impact of UCP4 knockdown upon UCP4 mRNA levels and mitochondrial membrane potential in SHSY5Y cells……………………………………………………… …………..194 5.1.7 The influence of LRRK2 kinase inhibition upon UCP mRNA expression in SHSY5Y cells…………………………………………………………………………………………………………………….. 197 5.1.8 The impact of LRRK2 knockdown upon UCP4 mRNA levels and mitochondrial membrane potential in SHSY5Y cells………………………………………………………………….. 199 5.2.1 Characterization of DJ1 knockdown SHSY5Y cells……………………………………… 200 14 5.2.2 The analysis of UCP mRNA expression in DJ1 knockdown SHSY5Y cells…… 201 5.3 The influence of LRRK2 expression upon PGC1α, SOD2 and catalase mRNA in SHSY5Y cells ……………………………………………………………………………………………………….. 203 5.4.1 The analysis of HDAC5 cellular distribution by immunohistochemistry in SHSY5Y cells………………………………………………………………………………………………………… 204 5.4.2 The analysis of HDAC5 by Western blot in SHSY5Y cells ……………… …………. 206 5.4.3 The analysis of HDAC5 and LRRK2 interaction………………………………………….. 207 5.5.1 The analysis of DLP1 protein in SHSY5Y cells………………………………… …………..211 5.5.2 The analysis of LRRK2 and DLP1 interaction in SHSY5Y cells…………… …………..213 Chapter 6: Discussion 6.1 LRRK2 tissue distribution………………………………………………………………………………. 216 6.2 LRRK2 detection……………………………………………………………………………………………. 218 6.2.1 LRRK2 detection by Immunohistochemistry………………………………………………. 218 6.2.2 LRRK2 detection by Western blotting………………………………………………………… 219 6.2.3 LRRK2 detection by BN PAGE…………………………………………………………………….. 221 6.2.4 LRRK2 detection by immunoprecipitation and Western blotting…… …………. 223 6.3 Evaluation of LRRK2 protein expression in cell models and brain tissue……… 225 6.4 Evaluation of LRRK2 G2019S protein and mRNA expression……………………….. 228 6.5 LRRK2 cellular localization………………………………………………………………… …………..230 6.5.1 Wild type and G2019S LRRK2 cellular distribution……………………………………. 230 6.5.2 Wild type and G2019S LRRK2 mitochondrial localization………………………….. 232 6.5.3 LRRK2 localization in the mitochondria……………………………………………………… 233 6.6 Wild type and G2019S LRRK2 kinase activity……………………………………………….. 234 6.7 LRRK2 and mitochondrial function………………………………………………………………… 236 6.8 LRRK2 and mitochondrial permeability…………………………………………………………. 239 6.9 LRRK2 regulation of mitochondrial uncoupling proteins………………………………. 241 15 6.9.1.1 LRRK2 regulation of UCP mRNA expression………………………………… …………. 241 6.9.1.2 LRRK2 regulation of UCP protein…………………………………………………………….. 242 6.9.1.3 LRRK2 regulation of PGC1α expression ……………………………………… …………..244 6.9.1.4 LRRK2 regulation of HDAC5 ……………………………………………………………………. 246 6.9.1.5 LRRK2 GTPase mutations and mitochondrial function………………… …………..250 6.9.2 LRRK2 regulation of DJ1 …………………………………………………………………………….. 252 6.9.3 LRRK2 regulation of NFκβ …………………………………………………………………………. 254 6.9.4 UCP4 knockdown and LRRK2 regulation of Argonaute 2………………………….. 254 6.9.5 LRRK2 regulation of UCP phosphorylation………………………………………………….. 256 6.10 LRRK2 regulation of mitochondrial morphology…………………………………………. 256 6.11 LRRK2, mitochondrial uncoupling and cellular disturbances implicated in PD………………………………………………………………………………………………………………………..260 6.11.1 LRRK2 regulation of ATP, mitochondrial biogenesis and calcium…………… 260 6.11.2 LRRK2 regulation of autophagy and mitophagy………………………………………. 261 6.11.3 LRRK2 regulation of synaptic function and tubulin dynamics………………….. 263 6.12 Mitochondrial uncoupling proteins and disease………………………………………….. 264 6.13 Endogenous LRRK2 regulation of UCPs………………………..……………………………… 266 List of references references ……………………..………………………………………………………. 268 Appendix ……………………..……………………………………………………..………………………………301 16 List of Tables Table 2.1 Brain sample information describing the available tissue regions, source and post mortem (PM) delay of samples. Table 2.2 Information relating to the sources of punch skin biopsies and peripheral blood mononuclear cells from which the fibroblast and lymphoblast cell lines were derived. Table A1 List of siRNA and shRNA sequences used in siRNA knockdown experiments. Table A2 List of primers used for PCR analyses. Table A3 List of antibodies 17 List of Figures Figure 1.1 Schematic summary of the basal ganglia model. Figure 1.2 Schematic summary of the Braak model of pathological spread in PD. Figure 1.3 Schematic representation of the mitochondrial respiratory chain. Figure 1.4 Schematic diagram of the LRRK2 structure. Figure 2.1 Optimization of siRNA delivery into SHSY5Y cells by the method of transient transfection. Figure 2.2 Analysis of gene expression by quantitative real time PCR. Figure 2.3 Analysis of the mitochondrial membrane potential by TMRM staining. Figure 2.4 Analysis of mitochondrial content by TMRM staining. Figure 2.5 Analysis of cellular respiration by the Clark type oxygen electrode. Figure 2.6 Analysis of cellular respiration by phosphorescent oxygen probes. Figure 2.7 Analysis of cellular acidification by phosphorescent pH sensitive probes. Figure 2.8 The analysis of ROS by measuring the rate of dihydroethidium oxidation. Figure 3.1 LRRK2 antibody screen. Figure 3.2 Characterization of SHSY5Y cells over-expressing LRRK2. Figure 3.3 LRRK2 sub-cellular distribution analysed by differential centrifugation and Western blot analysis. Figure 3.4 LRRK2 sub-cellular distribution by immunocytochemistry in SHSY5Y cells. Figure 3.5 Expression of endogenous LRRK2 protein in EBV transformed lymphoblasts and lymphocytes. Figure 3.6 LRRK2 sub-cellular distribution in control lymphoblasts. Figure 3.7 Validation of LRRK2 immunoprecipitation (IP) for use in quantitative analysis of protein levels. Figure 3.8 Analysis of LRRK2 kinase activity in EBV transformed lymphoblasts and SHSY5Y cells. 18 Figure 3.9 Characterization of control and G2019S LRRK2 fibroblasts. Figure 3.10 Analysis of LRRK2 knockdown in SHSY5Y cells. Figure 3.11 Analysis of LRRK2 expression in the human brain by immunoprecipitation. Figure 3.12 Analysis of LRRK2 expression in rodent and non human primate brains by immunoprecipitation. Figure 3.13 Comparison of LRRK2 expression in cell models and brain tissue. Figure 3.14 Analysis of LRRK2 protein expression by blue native PAGE electrophoresis. Figure 3.15 Analysis of LRRK2 protein expression in human brain by blue native page electrophoresis. Figure 3.16 Immunohistochemical analysis of LRRK2 protein in mouse brain. Figure 3.17 Immunohistochemical analysis of LRRK2 protein in human brain. Figure 4.1 The analysis of mitochondrial respiration in control and G2019S LRRK2 fibroblasts. Figure 4.2 The analysis of mitochondrial content in control and G2019S LRRK2 fibroblasts. Figure 4.3 Use of TMRM to measure the mitochondrial membrane potential in control and G2019S LRRK2 fibroblasts. Figure 4.4 The analysis of cellular ATP content in control and G2019S LRRK2 fibroblasts. Figure 4.5 The analysis of extracellular acidification rates in control and G2019S LRRK2 fibroblasts. Figure 4.6 The analysis of cellular ROS in control and G2019S LRRK2 fibroblasts. Figure 4.7 The use of oligomycin to measure the mitochondrial proton leak in control and G2019S LRRK2 fibroblasts. 19 Figure 4.8 The use of TMRM to analyse mitochondrial morphology in control and G2019S LRRK2 fibroblasts. Figure 4.9 The use of control fibroblasts treated with rotenone or antimycin A as a model of respiratory chain dysfunction. Figure 4.10 The use of control fibroblasts treated with rotenone and oligomycin to evaluate the influence of ETC inhibition on the mitochondrial ATPase. Figure 4.11 The use of control fibroblasts cultured in galactose rich conditions as a model of limited cellular substrate supply. Figure 4.12 The analysis of respiratory chain function in the SHSY5Y LRRK2-V5 overexpressing model. Figure 4.13 The analysis of LRRK2 kinase inhibition in the SHSY5Y LRRK2-V5 overexpressing model. Figure 4.14 The use of LRRK2 kinase inhibitors to evaluate effects of LRRK2 kinase inhibition on mitochondrial function in the SHSY5Y LRRK2-V5 over-expressing model. Figure 4.15 Affinity purification of mitochondria from LRRK2-V5 over-expressing SHSY5Y cells and lymphoblasts. Figure 4.16 Mitochondrial sub-fractionation to identify LRRK2 mitochondrial localization. Figure 5.1 SHSY5Y cells over-expressing murine GFP-UCP2. Figure 5.2 Analysis of relative UCP mRNA expression fibroblasts and SHSY5Y cells. Figure 5.3 Expression of UCP mRNA in G2019S fibroblasts and SHSY5Y cells. Figure 5.4 Western blot analysis of UCP2 protein expression in fibroblasts and SHSY5Y cells. Figure 5.5 Evaluation of the influence of genipin on UCP induced mitochondrial depolarization in SHSY5Y cells. 20 Figure 5.6 Evaluation of the influence of genipin on UCP induced mitochondrial depolarization in fibroblasts. Figure 5.7 The analysis of UCP2 as a LRRK2 kinase target. Figure 5.8 The analysis of UCP4 knockdown in SHSY5Y cells using UCP4 siRNA. Figure 5.9 Influence of UCP4 levels on the mitochondrial membrane potential in SHSY5Y cells. Figure 5.10 The influence of LRRK2 kinase inhibition upon UCP mRNA expression in SHSY5Y cells. Figure 5.11 The impact of LRRK2 knockdown upon UCP4 mRNA levels and mitochondrial membrane potential in SHSY5Y cells. Figure 5.12 Characterization of DJ1 knockdown SHSY5Y cells. Figure 5.13 The analysis of UCP mRNA expression in DJ1 knockdown SHSY5Y cells. Figure 5.14 The influence of LRRK2 expression upon PGC1α, SOD2 and catalase expression in SHSY5Y cells. Figure 5.15 The analysis of HDAC5 cellular distribution by immunohistochemistry in SHSY5Y cells. Figure 5.16 The analysis of HDAC5 cellular levels by Western blot in SHSY5Y cells. Figure 5.17 The analysis of HDAC5 and LRRK2 interaction. Figure 5.18 The analysis of DLP1 protein in SHSY5Y cells. Figure 5.19 The analysis of LRRK2 and DLP1 interaction in SHSY5Y cells. Figure A1 Chemical structures of LRRK2 kinase inhibitors Figure A2 Agarose gel electrophoresis of RT PCR products. Figure A3 Analysis of LRRK2 expression in fibroblasts by immunoprecipitation. Figure A4 Agarose gel electrophoresis visualizing PCR products of the amplified LRRK2 exon 41 gene. 21 22 List of Abbreviations 4E-BP - eIF4E-binding protein Ψm – mitochondrial membrane potential AB – anode buffer AD – Alzheimer’s disease Ago2 – Argonate 2 AmpR – ampicillin resistance ANT - adenine nucleotide translocator ARE - antioxidant responsive element ATP – adenosine triphosphate ATP13A2 - ATPase type 13A2 AV – autophagic vacuole BCA - b BN PAGE – blue native polyacrylamide gel electrophoresis BSA – bovine serum albumin CAMKK2 – calmodulin like kinase kinase 2 CB – cathode buffer cDNA – coding DNA CHEK2 - checkpoint kinase 2 CMA – chaperone mediated autophagy CMV - cytomegalovirus COR - C-terminal of ROC CT – cycle threshhold DA - dopamine DAB - 3,3' -diaminobenzidine DAPI - 4',6-diamidino-2-phenylindole 23 DAT – dopamine transporter DDM - n-dodecyl-β-D-maltopyranoside DHE - dihidroethidium DMSO - dimethylsulfoxide DMEM - Dulbecco's Modified Eagle Medium DLB – dementia with Lewy bodies dLRRK2 – Drosophila melanogaster homologue of LRRK2 DLP1 - dynamin like protein 1 dNTP - deoxyribonucleotide triphosphate DTT - dithiothreitol EBV - Epstein-Barr Virus EDTA - ethylenediaminetetraacetic acid EF-1α – elongation factor 1 alpha EF2α – elongation factor 2 alpha ER – endoplasmic reticulum FADD - Fas-Associated protein with Death Domain FADH2 - flavin adenine dinucleotide hydroquinone FCCP - trifluorocarbonylcyanide Phenylhydrazone Fis1 - mitochondrial fission 1 protein FSD – full scale deflection GAK - Cyclin G-associated kinase GCase - glucocerebrosidase GSH - glutathione GSK3β - Glycogen synthase kinase 3 beta GST - LRRK2- 1326 to 2527 residues of the LRRK2 sequence conjugated to GST GTP - guanosine-5'-triphosphate 24 [H+] - proton H2O2 - hydrogen peroxide HDAC5 – histone deacetylase 5 IFNγ – interferon gamma IP – immunoprecipitation IPD – idiopathic Parkinson’s disease Keap1 - kelch-like ECH-associated protein 1 KLH sequence - keyhole limpet hemocyanin sequence KmR – kanamycin resistance LPS - lipopolysaccharide Lrk-1 – Carnobaelis elegans homologue of LRRK2 LRRK1 – Leucine Rich Repeat Kinase 1 LRRK2 - Leucine Rich Repeat Kinase 2 MAOB – monoamine oxidase B MAPK7 – mitogen activated kinase 7 MAPKKK – mitogen activated kinase kinase kinase MBP – myosin basic protein MEF – mouse embryonic fibroblasts MEF2 - myocyte enhancer factor-2 MeOH - methanol Mfn - mitofusin MVB – multivesicular bodies MYLK - myosin light chain kinase NAD(P)H - nicotinamide adenine dinucleotide (phosphate) NFAT - nuclear factor of activated T cells NFκB - nuclear factor kappa B 25 NGS - normal goat serum NMDA - N-methyl-D-aspartic acid NO – nitric oxide NP-40 - nonidet P40 Nrf2 - nuclear erythroid related factor-2 NRON - repressor of the nuclear factor of activated T cells O2.- - superoxide anion OCT – optimal cutting temperature OH.- - hydroxyl radical OONO- - peroxynitrate Opa1 - optic atrophy type 1 PBS – phosphate buffer saline PBS-T – phosphate buffer saline supplemented with Tween PCR – polymerase chain reaction PD – Parkinson’s disease PFA – paraformaldehyde PGC1α - peroxisome proliferator-activated receptor-γ coactivator 1-α PINK1 - PTEN-induced putative kinase 1 PKC – protein kinase C PKA – protein kinase A PMA - phorbol 12-myristate 13-acetate PMBC – peripheral blood mononuclear cells PMSF - phenylmethanesulfonylfluoride PNS – post nuclear supernatant PLK4 - polo-like kinase 4 26 PPAR - peroxisome proliferator-activated receptor PSF - polypyrimidine tract-binding protein associated splicing factor PTEN - phosphatase and tensin homolog RFU – relative fluorescent units RISC - RNA-induced silencing complex ROC - Ras of complex proteins ROS – reactive oxygen species RPU – relative phosphorscent units RPMI - Roswell park memorial institute medium RT PCR – real time PCR RXR - retinoid X receptor SA - specific activity SDHA - succinate dehydrogenase complex, subunit A SDS - sodium dodecyl sulphate SDS PAGE - sodium dodecyl sulfate polyacrylamide gel electrophoresis SNARE - soluble N-ethylmaleimide sensitive factor attachment protein receptor SNpc – substantia nigra pars compacta SOD2 – superoxide dismutase 2 TFAM – mitochondrial transcription factor TMRM - tetramethyl rhodamine methyl ester TR-F – time resolved fluorescence 27 UCP – uncoupling protein VDAC - voltage-dependent anion channels 28 Chapter 1: Introduction 1.1.1 Basal Ganglia and Parkinson’s Disease Parkinson’s disease (PD) is the second most common neurological disorder affecting approximately 2% of the population over the age of 60 and 4% over 80 years [1]. Clinical motor PD abnormalities are thought to result predominantly from the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Dopamine (DA) is a catecholamine, and the primary function of this modulatory neurotransmitter is to serve as the reinforcement signal for the learning and maintenance of adaptive behaviours. Introducing a stimulus to the striatum, nigral dopamine regulates striatal output to the thalamus through the intricate circuitry of the basal ganglia [2] (Fig 1.1, adapted from [3]).The pathophysiology and resulting clinical manifestation in PD is thought to arise as a result of striatal dopamine depletion, increased neuronal activity in the output nuclei of the basal ganglia and inhibition of thalamocortical responses. Suppression of thalamic neurons is required to maintain control of unwanted physical movements. Typical symptoms of hypokinesia, akinesia and bradykinesia develop due to this shift in basal ganglia function towards inhibitory cortically aided movements. The resulting loss of regulatory control leads to deterioration of simultaneous and repetitive movements with a decrease in amplitude of repetitions [4]. Tremor and rigidity seem to result from altered oscillatory activity in the basal ganglia circuitry, due to desynchronization of positive and negative regulatory pathways [5]. At the time of clinical manifestation it has been estimated that about 50-80 % of nigral dopaminergic neurons have been lost potentially accounting for the motor deficit. As well as regulation of motor function, the basal ganglia have been proposed to have a role in cognitive control [6], potentially explaining the cognitive malfunction in later stages of PD, although this could also be due to spread of PD 29 pathology to cortical areas (section 1.1.2). Cognitive symptoms are linked to the deterioration of cortical neurons in progressive disease stages. As well as motor and cognitive defects, affected subjects often develop autonomic disturbances. Gastrointestinal, cardiovascular and urogenital systems, under the control of cholinergic, noradrenergic and serotonergic nuclei can suffer, emphasising the contribution of other cell populations to the disease [5]. 1.1.2 Pathology in PD The pathological hallmarks of Parkinson’s disease are Lewy bodies and dystrophic neurites. Lewy bodies are proteinaceous, cytoplasmic inclusion composed primarily of aggregated α-synuclein protein and positively immunoreactive for ubiquitin [7] while dystrophic neurites appear as damaged projections of affected cells. According to the Braak hypothesis of PD transmission, the progressive pathology in PD is thought to occur as a result of pathological spread 30 from the brain stem to cortical regions potentially accounting for the motor deficit and delayed cognitive decline. This transmission has been subdivided into six stages, characterized by the appearance and spread of the neuropathological hallmarks (Fig 1.2, adapted from [8]). Pathology is initially confined to the olfactory bulb and lower brainstem regions, spreading up to the locus coeruleus in stage II of PD. Clinical symptoms are thought to occur in stage III where Lewy neurites, α-synuclein aggregates and Lewy bodies can be detected in SNpc. Stage IV comes with nigral cell loss and spread to the hippocampal and basal forebrain areas. Neocortical Lewy body pathology occurs in stage V, confined primarily to the temporal regions and continues to spread in stage VI of the disease [5]. Pathological diagnosis in PD requires depigmentation of the substantia nigra as a result of neuronal cell loss and presence of Lewy bodies in post mortem brain tissue to accompany the clinical motor abnormalities [9]. However, ‘parkinsonian’ phenotypes are not solely restricted to PD with features of idiopathic parkinsonism (IPD) found in a wider spectrum of movement disorders [10]. Furthermore, a clinical diagnosis of PD does not always correlate with the pathological requirements. In particular, juvenile onset recessive forms of the disease (section 1.2) present with more atypical clinical features that closely resemble dystonia [11]. In these cases, disease progression is slow and pathology variable. Autosomal dominant PD (section 1.2.) can differ in pathology even between family members carrying the same genetic mutation with Lewy body positive cases reported as well as those with tau, β-amyloid deposits (hallmark of Alzheimer’s disease (AD)) and ubiquitin positive inclusions [12]. Variable neuropathology in PD indicates the potential involvement of multiple cellular pathways in the disease pathogenesis which may or may not converge to produce a common clinical phenotype. 31 1.2 Causative factors in PD The etiology of the disease is unknown. Idiopathic forms of PD are thought to result from variable contributions of genetic and environmental origin, with rare cases caused solely by one or the other factors. Gene linkage analyses and positional cloning have identified specific mutations which confer a familial link in approximately 10 % of all PD cases [13]. The genes identified can be subdivided into two groups based on their inheritance pattern; autosomal dominant and autosomal recessive. 1.2.1.1 Autosomal dominant PD genes: α-synuclein The genetic link between α-synuclein and Parkinson’s disease was first identified in 1996 for a large Greek/Italian kindred with a mean age of onset of 55 years [14]. A year later, the G209C mutation (A53T) was described in the α-synuclein 32 gene located on chromosome 4 for this cohort [15]. Additional familial linkages in the α-synuclein gene were found as point mutations at positions 88 (G to C substitution, A30P mutation) in a German kindred and position 188 in a Spanish, Basque family (G to A substitution, E46K mutation) [16]. In addition, families carrying duplications (French cohort) [17] and triplications (Iowa cohort) [18] of the αsynuclein gene have been reported suggesting that cellular levels of the protein correlate with PD onset, progression and severity. Furthermore, genome-wide association studies generated from large cohorts of idiopathic PD patients suggest that α-synuclein sequence polymorphisms are a risk factor for the disease, in support of the gene linkage data [19-21]. Inherited α-synuclein mutations result in similar clinical phenotypes of Ldopa responsive parkinsonism but appear more aggressive when compared to idiopathic cases. Clinical features and pathology of point mutation and duplication α-synuclein cases fall into the spectrum of PD variations, clinically diagnosed as dementia with Lewy bodies (DLB) due to their progressive cognitive deficits [16, 22]. Lewy body pathology was identified for each familial α-synuclein case. α-synuclein is an intrinsically disordered 14 kDa protein highly abundant in the brain [23, 24]. Structurally, α-synuclein is capable of conformational changes which govern its membrane binding properties and aggregation propensity [25, 26]. 1.2.1.2 Autosomal dominant PD genes: Leucine Rich Repeat Kinase 2 Sequence analysis of large Japanese families identified a heritable component in the PARK8 locus on chromosome 12 (12p11.2-q13.1) [27]. Subsequently, Leucine Rich Repeat Kinase 2 (LRRK2) pathogenic mutations within linked families were mapped for North American (R1441C) and European (Y1699C) kindreds as well as a small German family (I2020T) [28]. In parallel, families and sporadic PD cases from the Basque country in Spain were identified to harbour 33 LRRK2 R1441G mutations while British families were linked to Y1699C substitutions by the same group [29]. Subsequent identification of the G2019S mutation spurred interest in the PD research field due to its high frequency in specific populations and association with the ‘classical’, late onset sporadic PD. First reports of G2019S came from two unrelated Caucasian families of North American-British and Ashkenazi Jewish ancestry [30]. Reports of 13 individuals from a variety of ethnic backgrounds (North America, Norway, Ireland and Poland) sharing a common disease associated phenotype, confirmed G2019S occurence in sporadic PD [31]. Population studies determined greatest G2019S prevalence in the Ashkenazi Jew and North African Arabs, identified in 27.9 %, and 30 % of all PD cases from these regions respectively [32, 33] with lower incidence in North American (7 % [34]) and European (6.6 % [35]) cohorts. In control groups, the highest frequency of G2019S was reported in Israel (2.4 %) and Arab Berber Tunisians (2 %) [32, 36]. Haplotype analysis of demographically distinct populations identified a common ancestrial founder [37]. More recently LRRK2 has been implicated in the disease through genome-wide association studies of idiopathic PD patients in the UK and Japanese cohorts [19, 20]. In humans and rodents a paralogous gene to LRRK2 has been identified sharing 26 % sequence identity termed Leucine Rich Repeat Kinase 1 [38]. However, PD associated mutations in the LRRK1 gene have not been described. The number of reported autopsies for all the mutant LRRK2 PD cases totals 38 [12]. Lewy bodies and dopaminergic cell loss have been identified in association with all established pathogenic mutations. Lewy bodies are generally described in the brain stem, cortex and the limbic system. A number of studies reported pure nigral degeneration free from Lewy bodies associated with LRRK2 [39] however, these cases are rare. Additional features include neurofibrillary tangles with the 34 degree of plaques and tangles in G2019S cases ranging from levels compatible with normal ageing to densities observed in demented Alzheimer’s disease patients [40]. Clearcut tauopathies have been noted for some R1441C and G2019S cases [12]. Although heterogeneity can be observed in mutant LRRK2 PD, in general, pathological hallmarks are comparable to those found in idiopathic disease [41]. 1.2.1.3 Autosomal recessive PD genes: Parkin Japanese families found to contain deletions in the PARK2 gene on chromosome 6, led to the discovery of parkin as a causative risk gene for autosomal recessive juvenile PD [42]. Loss of parkin protein function due to autosomal recessive familial gene deletions have been found in European families however, missense mutations, resulting in partial or complete loss of function, are more common among the Caucasian haplotypes [43]. Single allele mutations, inherited in an autosomal dominant manner have also been identified and seem to infer a risk factor for the disease [44]. Unlike sporadic PD, PD cases associated with loss of parkin function have an early onset and slow disease progression with good responses to dopamine replacement therapy [42-44]. Furthermore autopsies of parkin PD patients rarely show evidence of Lewy body pathology suggesting a distinctive cause for this subtype of PD [45]. The 50 kDa protein encoded by the parkin gene is a ubiquitin protein ligase with a RING finger domain [42]. 1.2.1.4 Autosomal recessive PD genes: PINK1 First identified in three consanguineous Sicilian families, loss of function mutations in the Phosphatase and Tensin (PTEN) induced kinase (PINK1) gene located on chromosome 1 were shown to cause autosomal recessively inherited PD [46]. Subsequently, the PINK1 homozygous L347P and a compound heterozygous 35 G240L/L489P were identified in Filipino families [47]. Mutations in PINK1 are infrequent when compared to familial parkin cases. Similar to parkin, heterozygous PINK1 mutations have been identified as a risk factor for PD [48]. In addition, PINK1 mutations have been found in sporadic PD subjects [49]. Clinical features of PINK1 PD present with early onset parkinsonism with slow disease progression, good levodopa response and bradykinetic symptoms mimicking that of parkin PD. Although only a few cases have been presented at autopsy, PD patients with PINK1 mutations have been described with Lewy body deposits [50]. Intracellular proteolytic processing of full length 63 kDa PINK1 generates two additional forms of the protein; 54 kDa and 10 kDa respectively [51]. The enzymatic domain in PINK1 has putative kinase characteristics although physiological phosphorylation substrates have not yet been identified. 1.2.1.5 Autosomal recessive PD genes: DJ1 PD associated mutations in DJ1 were first identified in the PARK7 gene, located on chromosome 1 in two consanguineous families from Italy and the Netherlands. Patients from the Dutch family carried a gene deletion while Italian subjects encoded a single base substitution (L166P) resulting in a loss of function protein [52]. DJ1 mutations are rare in PD, however, the clinical phenotype of DJ1 patients resemble those found in other early onset, recessively linked PD cases [45]. No neuropathology in PARK7 cases has been reported to date. The 20 kDa DJ1 protein exists as a dimer formed by a disulphide bond between two monomeric molecules. The potential for free radical oxidation of DJ1 cysteine residues labelled the protein as a redox sensing molecule [53]. 36 1.2.1.6 Autosomal recessive PD genes: Glucocerebrosidase The glucocerebrosidase (GCase) gene located on 1q21-22, spanning 11 exons, has been reported to have >300 mutations associated with Type 1-3 Gaucher’s disease, a lysosomal storage disorder. In addition, heterozygous mutations within the GCase are the most common risk factor for sporadic Parkinson’s disease [54]. Mutation penetrance can vary between ethnic groups observed at high frequency for Ashkenazi Jews [55] and low frequency in European cohorts [56]. Initially regarded as a risk factor rather than a mendelian form, identification of familial links has provided the criteria to label GCase as an autosomal recessive PD gene [57, 58]. Clinically, parkinsonian symptoms in GCase PD cases occur with early onset relative to idiopathic disease and typical Lewy body pathology has been described for the limited number of cases inspected at autopsy [59]. The GCase protein, synthesised in endoplasmic reticulum (ER) polyribosomes is targeted to the lysosome following peptide cleavage and N linked glycosylation. Polyubiquitination of GCase facilitates ER exit. In the lysosomal compartment, GCase functions to break down the lipid glucoceramide into glucose and ceramide [60]. Some of the most common GCase mutations are reported to trap GCase in the ER depleting the lysosomal GCase pool [61]. 1.2.2 PD genetic models The discovery of familial PD genes led to the development of genetic PD animal models in an attempt to recapitulate the clinical features and pathology of PD and dissect the mechanism of disease pathogenesis. Although α-synuclein transgenic mouse models have frequently been shown to develop motor abnormalities correlating with a reduction in dopamine transmission and αsynuclein positive inclusions, these protein aggregates were reported to be distinct 37 from Lewy bodies described in PD patients [62-65]. In addition, dopaminergic neuronal loss was not observed in the animals. Defects in mitochondrial morphology and mitochondrial dysfunction have been described in two studies [66, 67] suggesting a potential role of mitochondria in the α-synuclein pathology and motor disturbances associated with α-synuclein over-expression in mice. Although C.elegans and D.melanogaster lack α-synuclein orthologues, over-expression of the human wild type and mutant α-synuclein protein in flies has shown to result in age dependent motor abnormalities, dopamine depletion and the formation of αsynuclein positive aggregates [68, 69]. Slowness of movement has been described in C.elegans over-expressing wild type and mutant α-synuclein consistent with a perturbation in dopaminergic function [70, 71] and correlating with the loss of dopaminergic neurons [71] however, α-synuclein aggregates were not reported. Recessive PD has been modelled in animals through deletions of DJ1, PINK1 and parkin genes. Although PINK1, parkin and DJ1 knockout mice have signs of abnormal dopamine metabolism, nigral dopaminergic cell death is not a feature and Lewy bodies have not been observed in these animals [72-74]. Drosophila melanogaster models of loss of PINK1 and parkin function have revealed motor defects and dramatic defects in mitochondrial morphology in energy intensive tissues [75] as well as degeneration of dopaminergic neurons [76]. Although DJ1 knockout flies also show mitochondrial structural alterations, these appear to be distinct from those identified in PINK1 and parkin knockouts [77]. Mild mitochondrial abnormalities have been observed in PINK1 and parkin knockout mice however these features are less pronounced than the corresponding fly models [78]. Although none of the animal models recapitulate the full pathological spectrum of PD, some overlapping features indicate the involvement of distinctive cellular pathways in the disease pathogenesis. In particular, the pathways involved 38 in α-synuclein synthesis and degradation mediating steady state levels of the protein, synaptic function regulating dopamine metabolism and mitochondrial function contributing to cellular energy homeostasis. 1.2.3 Environmental factors in PD Familial mutations account for about 10 % of all PD cases suggesting additional factors may be involved in the disease pathogenesis. Population based studies have shown that head trauma may accelerate PD onset [79] while meta analysis [80] of studies in humans and rodents [81] have demonstrated a protective role for cigarette smoking and neuroprotection from nicotine. Studies looking at physical activity, caffeine use and non steroidal anti inflammatory drugs have all shown negative correlations with respect to the disease [82-84]. In addition, exposure to environmental toxins and pesticides has been shown to correlate with sporadic forms of PD [85, 86]. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was identified to induce Parkinsonian like symptoms and pathology in humans and non-human primates [85, 87]. Chronic, systemic administration of the pesticide rotenone in rodents led to the development of PD like symptoms and α-synuclein aggregates [88]. Paraquat, a quaternary ammonium herbicide, structurally resembles MPTP and resulted in selective loss of dopaminergic neurons, dopamine depletion and α-synuclein aggregation in rodents [89, 90]. The subsequent characterization of the molecular mechanisms associated with these compounds further emphasised the role of mitochondrial function in PD pathogenesis. The link between mitochondrial dysfunction and α-synuclein aggregation in toxic animal and human PD models was also noted. 1.2.4 Mitochondria Mitochondria are double membrane bound organelles consisting of a relatively permeable, cholesterol rich outer membrane and an impermeable, protein 39 rich inner membrane. The outer membrane houses different families of proteins facilitating mitochondrial import, organelle dynamics and apoptotic cascades. The cristae folds of the inner membrane allow for high concentrations of proteins involved in processes such as oxidative phosphorylation to meet the metabolic demands of the cell. The mitochondrial matrix incorporates mitochondrial transcriptional machinery working in concert with nuclear genes to regulate mitochondrial function. In addition, the mitochondrial matrix facilitates the tricarboxylic acid cycle (TCA) supplying reducing equivalents to the electron transport chain (ETC). In terms of mitochondrial involvement in PD pathogenesis, a considerable amount of attention has been focused on the mitochondrial respiratory chain. Structurally, the mitochondrial respiratory chain consists of 5 multi subunit complexes where Complexes I, III, IV and V span the inner mitochondrial membrane while Complex II contacts the membrane at the matrix side (Fig 1.3, adapted from [91]). Reducing equivalents (NAD(P)H and FADH2) from the TCA and β-oxidation function as electron donors feeding into Complexes I and II. Redox cofactors bound to their respective enzymes transfer electrons to the mobile electron carrier coenzyme Q 10 which in turn is oxidised by Complex III to allow for electron transfer to cytochrome C. Shuttling of reduced cytochrome C to Complex IV facilitates the reduction of oxygen by this enzyme. Electron transfer by Complexes I, III and IV results in the movement of protons [H+] across the inner membrane to the intermembrane space. The generated proton gradient is referred to as the mitochondrial membrane potential. The thermodynamic tendency of [H+] to flow back into the matrix generates energy, the driving force for Complex V (ATP synthase complex). Respiratory chain defects often lead to reduced electron flow, mirrored by a drop in 40 the mitochondrial membrane potential and a build-up of reducing equivalents, a source of potentially damaging reactive oxygen species (ROS). The reduction of oxygen by one electron at a time produces a range of relatively stable intermediates. One electron reduction generates the superoxide anion (O2.-) and spontaneous or enzyme induced dismutation of O2.- produces hydrogen peroxide (H2O2) which in turn may be partially reduced to hydroxyl radicals (OH.-). OH.- can be re-reduced by O2.- propagating the process [92]. Hydroxyl radicals can directly lead to lipid peroxidation and react with nucleic acids of mitochondrial DNA to cause DNA damage. The superoxide anion, although relatively non-toxic can react with nitric oxide (NO.-) to generate the highly reactive peroxynitrate (OONO-), damaging to lipids and DNA. 41 1.2.4.1 Mechanism of action of mitochondrial toxins MPTP selectivity for dopaminergic neurons is conferred by its affinity for the dopamine transporter (DAT) [93]. Lipophilic properties target it to the mitochondrial matrix where MPTP is metabolized by the enzyme monoamine oxidase B (MAO-B) to MPP+. MPP+ blocks electron transfer through Complex I of the ETC by adhering in close proximity to the coenzyme Q 10 binding site [94]. Additional effects on TCA cycle enzymes results in an energy crisis believed to lead to acute dopaminergic cell death. Rotenone is a well characterized inhibitor of mitochondrial Complex I. Similar to MPTP, its lipophilic nature allows it to cross the blood brain barrier, cellular membranes and accumulate in the mitochondria. Unlike MPTP, rotenone uptake is not restricted to dopaminergic neurons however, the enhanced vulnerability of these neurons to rotenone toxicity links mitochondrial function to PD pathology. Rotenone induced Complex I inhibition has been reported to enhance ROS production implicating ROS in the disease mechanism. Paraquat uptake is not mediated by DAT hence the molecular selectivity for dopaminergic neurons is different to that of MPTP [95]. Paraquat is believed to kill cells through failure of antioxidant defences and oxidative damage to cytosolic proteins rather than the primary defect in Complex I function as is the case for rotenone and MPP+ [96], further emphasising the role of ROS in disease pathogenesis. 1.2.4.2 Electron transport chain and PD The link between mitochondrial toxins and PD pathology led to the identification of mitochondrial respiratory defects in idiopathic PD patients. Complex I functional abnormalities were identified in the substantia nigra of IPD subjects and confirmed by multiple groups [97-100]. Altered Complex I enzyme assembly has 42 been reported in IPD patient brains [101]. Subsequently, Complex II defects were reported as well as Complex II in combination with Complex III or Complex IV abnormalities of mitochondria extracted from skeletal muscle, lymphocytes and patelets of PD subjects [102-106]. Although substantial variability was reported between groups, the association between respiratory chain defects and disease were consistent. Oxidative stress was another prominent feature identified in PD patient brain tissue similar to the effects observed in rotenone and paraquat models. Increased levels of lipid oxidation [107], oxidative protein damage [108, 109] and oxidative DNA damage [108, 110] have been reported in the SNpc of idiopathic PD patients. Oxidative stress has also been noted in peripheral tissues [110-114] suggesting that increased ROS generation is not restricted to the affected brain areas. Respiratory chain defects have also been noted in genetic models of PD. Primary cells from recessive PD patients as well as DJ1, PINK1 and parkin knockout models show evidence of respiratory chain abnormalities, particularly in Complex I activity [115-122] and increased levels of oxidative stress [118, 120, 121]. αsynuclein association with the inner mitochondrial membrane, specifically Complex I of the ETC is believed to modulate the bioenergetics changes associated with reduced activity of Complex I and increased levels of oxidative stress in α-synuclein over-expressing primary neurons [123]. Furthermore, the mitochondrial association and Complex I inhibition is thought to be exacerbated by the toxic oligomeric and aggregated α-synuclein [124], further emphasising the link between α-synuclein aggregation and PD pathogenesis. 43 1.2.4.3 Mitochondrial turnover and dynamics Discovery of the functional roles of the recessively linked PD proteins, PINK1 and parkin, has led to the mechanistic insight into a novel pathway linking the fundamental processes involved in mitochondrial homeostasis- mitochondrial dynamics and turnover. Mitochondria are now recognised as dynamic structures continuously undergoing fission and fusion events. The structural re-organisation of the mitochondrial network involves both the inner and outer mitochondrial membranes. Mitochondrial fusion events can dilute out mitochondrial DNA mutations and metabolic insufficiencies [125]. Outer membrane fusion events require the action of mitofusin 1 and 2 (Mfn) proteins while the inner membrane is fused through the action of Optic Atrophy 1 (Opa1) protein. Fission of both membranes is facilitated by mitochondrial recruitment of the cytosolic proteins dynamin like protein 1 (DLP1) and mitochondrial fission protein (Fis1). Oligomerization of DLP1 forms membrane boundaries and allows for mitochondrial budding and fragmentation [126]. Mitophagy is the process by which the mitochondrial network gets degraded. Parkin has been shown to be involved in the removal of damaged mitochondrial structures [127]. Depolarization of the mitochondrial membrane in response to chemical uncoupling or electron transport chain inhibitors results in parkin targeting from its native cytosolic location to the damaged regions of the mitochondrial outer membrane [128]. At the outer membrane, parkin promotes ubiquitin mediated degradation of the mitofusins and is thought to induce mitophagy by localized prevention of mitochondrial fusion events [129]. Isolated mitochondrial fragments targeted for mitophagic removal are subsequently incorporated into double membrane vesicles and shuttled to the lysosome for proteolytic processing [126]. PD linked parkin mutations interfere with various 44 aspects of this mechanism, affecting parkin translocation, ubiquitin ligase activity and mitophagic membrane formation [130]. Functionally PINK1 has been shown to act upstream of parkin in the pathway of mitochondrial turnover. PINK1 contains a mitochondrial localization motif importing the protein in a membrane potential dependent mechanism where it is rapidly cleaved by mitochondrial proteases generating the truncated PINK1 forms [131]. Mitochondrial depolarization leads to the accumulation of full length PINK1 at the outer surface, believed to act as the signal for parkin recruitment [132]. Mutations in PINK1 tend to either destabilize the protein structure or occur in the enzymatic domain, leading to a loss of function phenotype [46, 47]. The ETC and mitochondrial morphology defects described for PD linked PINK1 and parkin mutations are thought to result from insufficient removal of damaged organelles [129, 133]. In addition, parkin is believed to mediate GCase polyubiquitination in the ER and facilitate GCase exit [134]. Excessive parkin occupancy of trapped mutant GCase is believed to deplete the pool required for mitophagy resulting in the build up of damaged organelles [134]. Altered mitochondrial membrane binding properties of α-synuclein mutants are thought to account for the mitochondrial fragmentation in cells [135, 136]. α-synuclein membrane binding could thus influence mitochondrial fusion by interfering with the mitochondrial fission fusion machinery. Hence dysregulation of mitophagy has been linked to most of the genetic PD models and appears to contribute to PD pathogenesis. 1.2.4.4 Mitochondrial transcriptional regulation In addition to homeostatic maintenance of healthy mitochondria through turnover, fission and fusion events, recessive PD proteins have been implicated in an upstream pathway of mitochondrial control through transcriptional regulation. 45 Mitochondrial biogenesis is the process by which the mitochondrial pool is replenished. The main signalling pathway associated with its induction is regulated by the nuclear factor (erythroid-derived 2), kelch-like ECH-associated protein 1 (Nrf2/Keap1) complex stimulating a battery of approximately 100 genes under the inducible control of antioxidant response element (ARE) and myocyte enhancer element-2 (MEF2) [137]. In addition to the induction of expression of respiratory chain components, transport molecules, proteosomal proteins and chaperones, global regulatory factors; peroxisome proliferator-activated receptor gamma (PPARγ), retinoid X receptor-alpha (RXRα) and PPARγ co-activator 1 alpha (PGC1α) are induced to regulate the expression of mitochondrial lipid pathways integral to bioenergetic metabolism [138]. Nuclear transcription is coupled to mitochondrial RNA synthesis through the expression of a mitochondrial transcription factor (TFAM) [139]. The overall outcome is increased mitochondrial content, cellular respiratory capacity and improved maintenance of existing organelles. Transcription and replication of mitochondrial DNA was found to be enhanced by parkin over-expression [140], while down regulation of PINK1 correlated with reduced mitochondrial content [122], potentially contributing to the bioenergetic defect. A functional association between parkin and TFAM has been described in neuroblastoma cells, linking the catabolic and anabolic pathways of mitochondrial homeostasis [140] further implicating this pathway in the pathogenesis of PD. 1.2.4.5 Mitochondria and oxidative stress Mitochondria respond to oxidative stress by promoting synthesis of antioxidant enzymes. The antioxidant response can be stimulated as part of the Nrf2 signalling pathway, generally mediated by PGC1α regulated expression [141, 142] of antioxidant enzymes such as superoxide dismutase 2 (SOD2), glutathione (GSH) and 46 catalase. The Nrf2/Keap1 complex has been shown to be sensitive to cellular redox status governing PGC1α transcription [143]. As well as induction of expression by Keap1/Nrf2, PGC1α transcription is negatively regulated by nuclear levels of histone deacetylase 5 (HDAC5), a transcriptional regulator shuttling between the nucleus and cytosol [144]. The involvement of this branch of the Nrf2/Keap1 pathway in PD pathogenesis is supported by functional association of the downstream PGC1α with the recessively linked DJ1 protein [145]. In the nucleus, DJ1 inhibits sumolation of pyrimidine tract binding protein-associated splicing factor (PSF), which binds PGC1α and mediates its transcriptional activity [145]. DJ1 has been shown to have an altered redox state in response to oxidative stress and acidic conditions which may in turn influence the protein’s cellular localization with its nuclear translocation likely to govern the redox transcriptional response [146]. PD linked DJ1 mutations can affect protein cellular distribution and disrupt the PSF modulation resulting in reduced transcription of the antioxidant enzymes [147]. In addition, PGC1α levels respond to parkin silencing and over-expression potentially as a result of the oxidative stress associated with the loss of parkin phenotype [148, 149]. Furthermore, increased activity of PGC1α co-regulator PPARγ has been reported to be protective against dopaminergic cell loss in PD models [150, 151]. Although PGC1α is not regarded as a familial PD protein, polymorphisms in the gene sequence link it to sporadic disease [152], further emphasising the role of oxidative stress and mitochondria in PD pathogenesis. 1.2.4.6 Oxidative stress and mitochondrial uncoupling proteins In addition to redox maintenance by antioxidant enzymes, ROS levels can be regulated through the action of mitochondrial uncoupling proteins (UCPs). This family is comprised of five isoforms with structural homology between family 47 members. While UCP2 is expressed ubiquitously, UCP1 is predominantly expressed in brown fat, mediating thermogenesis and UCP4 and 5 are the neuronal isoforms with poorly defined functional roles. UCP3 is expressed in some but not all tissues and the specificity for its distribution has not yet been determined [153]. UCPs exist as dimers on the inner mitochondrial membrane forming a pore which allows protons to flow into the mitochondrial matrix. Proton flow through the UCP channel provides an alternative route from the mitochondrial ATPase and partially dissipates the mitochondrial membrane potential in the form of heat release [154, 155]. Mild uncoupling of oxidative phosphorylation upregulates ETC function and limits the amount of reducing equivalents available for ROS production. Regulated transcriptionally [156, 157], translationally [158] and by post translational modification [159], UCPs provide a rapid response to the demands of the electron transport chain. The UCPs have a relatively short half life of 40 minutes with the levels maintained through basal and inducible transcriptional regulation [160]. In particular, transcriptional activation occurs by PGC1α mediated activation or as part of the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway [156, 157, 161, 162]. Hence UCP transcription is potentially linked to the PD associated parkin and DJ1 proteins. Mitochondrial uncoupling proteins have been shown to play a protective role in PD models. Neuroprotective properties of UCPs have been demonstrated in rodents treated with MPTP where UCP2 over-expression reduced dopaminergic cell loss [163]. In addition, chronic MPTP treatment resulted in a time and dose dependent induction of UCP2, 4 and 5 in neuronal cells [164]. Mechanistically, superoxide generated during respiration can result in lipid peroxidation which in turn activates UCP expression to reduce the superoxide production, effects presumably exacerbated in MPTP models [165]. Although both neuronal and non 48 neuronal UCPs possess properties that can potentially protect cells against various stresses, toxic effects linking UCP over-expression and cell death have been reported [166, 167]. In addition to the protective role in PD models, a genetic link has been reported for the neuronal UCP isoforms and the PD associated DJ1 protein. As well as a role in induction of antioxidant enzymes DJ1 has recently been linked to a specific function regulating the autonomous pacemaker activity of dopaminergic neurons in the SNpc [168]. Pacemaking is unique to SNpc DA neurons and facilitates sustained release of dopamine necessary for the proper function of target structures in the striatum [169]. Mitochondrial regulation of this function can potentially explain the vulnerability of this cell population in PD. Pacemaker activity is regulated by long lasting (L) type calcium channels where cytosolic calcium oscillations come at a metabolic cost contributed to by oxidative phosphorylation as channel opening requires hyperpolarized membrane potentials [170]. Mitochondrial calcium uptake is coupled to rapid depolarization and repolarization of mitochondrial membranes assumed to be facilitated through the action of UCPs modulating the proton gradient [168]. DJ1 knockout animals showed a reduced amplitude and frequency of polarization events correlating with reduced expression of UCP mRNA. Furthermore superoxides can affect mitochondrial free calcium by regulating the expression of UCPs suggesting a potential regulatory loop involving the redox sensing DJ1 protein. Of note, mitochondrial calcium dysregulation has been reported in PINK1 knockout models suggesting a potential overlap between the recessive PD genes [172]. In support of the role of calcium in PD, MPTP and rotenone are associated with diminished mitochondrial calcium uptake and increased cytosolic free calcium in cultured cells [173-175]. Considering the genetic association between DJ1 and PD, 49 SNpc dopaminergic cell loss and the regulatory control of mitochondrial function, it is possible to postulate the involvement of the UCPs in PD pathogenesis. 1.2.5 Lysosomal function and α-synuclein Familial α-synuclein gene duplications and triplications as well as Lewy body deposits in PD brains suggest increased levels of α-synuclein protein are a key pathological factor in PD. Evidence of the role for mitochondrial dysfunction in αsynuclein aggregation came from studies identifying α-synuclein aggregates in MPTP and rotenone mouse models of PD [176-179]. Steady state α-synuclein levels are determined by the balance between protein synthesis and degradation pathways. Increased α-synuclein mRNA expression has been shown in SNpc of IPD patients [180] suggesting increased αsynuclein expression can potentially increase cellular concentrations of the protein. In addition, lysosomal function was determined as the rate limiting step in the clearance of protein aggregates [181] suggesting this process may contribute to increased α-synuclein levels in cells. α-synuclein degradation involves chaperone mediated autophagy (CMA) which requires binding of chaperones to an intrinsic pentapeptide motif in α-synuclein and the subsequent receptor mediated internalization of the protein into the lysosomal lumen where it is degraded by lysosomal proteases [182]. α-synuclein point mutations inhibit CMA [182] and may account for the build up in protein levels. Inhibition of CMA can stimulate another branch of the lysosomal degradation pathway, autophagy, which involves the formation of a double membrane vesicle around the target cargo and subsequent fusion with the lysosomal membrane [183]. Enhanced autophagy can partially compensate for the lack of CMA mediated degradation and has been shown to reduce protein aggregation in a number of cellular models of neurodegenerative diseases [184- 50 187]. However enhanced autophagy can also induce cell death under stress conditions [188]. As well as mutant α-synuclein inhibition of chaperone mediated autophagy, lysosomal dysfunction has been linked to wild type α-synuclein accumulation. Reduced levels of a lysosomal protease, cathepsin D, correlated with increased αsynuclein accumulation [189]. Clinical features of PD have been noted in patients with lysosomal storage disorders accompanied by α-synuclein accumulation in neurons and glia [190-192]. In addition, genome-wide association studies linked two lysosomal enzymes to an increased risk for developing PD, GCase [54, 193] and a lysosomal membrane protein probable cation-transporting ATPase 13A2 (ATP13A2). In support of the genetic studies mutant GCase has consistently correlated with αsynuclein accumulation in cell culture models potentially providing a link to the associated Lewy body pathology observed in GCase mutant PD cases [194, 195]. ATP13A2 deficiency is associated with impaired lysosomal degradation capacity, αsynuclein accumulation and toxicity [196, 197]. Analyses of brain tissue from subjects with lysosomal storage disorders have also identified morphologically and biochemically deficient mitochondria [198, 199] suggesting mitochondrial function and lysosomal regulation are intrinsically linked. As lysosomes are also the final destination for mitophagic cargo, lysosomal abnormalitites influence mitochondrial turnover providing a potential link to PINK1 and parkin mediated mitochondrial regulation. 1.3 LRRK2 1.3.1 LRRK2 protein structure High incidence of LRRK2 mutations, the pathological and clinical similarities with idiopathic disease suggest that LRRK2 plays an important mechanistic role in PD pathogenesis. 51 The 1.4 Mb LRRK2 gene consists of 51 exons encoding a protein of 2527 amino acids. Structurally the 286 kDa LRRK2 harbours multiple functional domains; ankyrin repeats (ANK), leucine-rich repeat domain (LRR), Ras in complex proteins domain (ROC), C-terminal of ROC region (COR), mitogen activated protein kinase kinase kinase (MAPKKK) domain and a WD40 domain (Fig 1.4, adapted from [200]). Based on sequence predictions the kinase domain consists of 2 structurally homologous lobes connected through an active hinge region. The ROC domain contains a guanosine triphosphate (GTP) binding site while the LRRK2 COR segment is thought to be a regulator of the ROC GTPase activity [201]. In vitro kinase and GTPase activity measurements confirmed LRRK2 as a catalytically active molecule [202, 203]. There is evidence to suggest the two enzymatic domains are intrinsically linked as GTP binding to the ROC domain is important for LRRK2 kinase activity [203]. However, GTP activity of kinase deficient LRRK2 are comparable to those of the wild type protein and GTP concentrations do not seem to affect kinase activity [203-206] suggesting the kinase is likely the modulator rather than output of enzymatic activity. The flanking ankyrin repeats, LRR and WD40 are domains regulating protein-protein interactions. WD40 contribution to the regulation of LRRK2 enzymatic activity is supported by the lack of detectable kinase activity in C terminally truncated LRRK2 mutants [202, 208]. The quaternary structure of LRRK2 is believed to be that of a kinase inactive oligomer. GTP binding leads to the dissociation of the complex and heterodimer formation with a concominant activation of the kinase. The ROC-COR region of LRRK2 has been mapped to the dimer interface and GTPase activity reported to mediate steady state dimer levels [209, 210]. 52 1.3.2 LRRK2 kinase As well as its inherent autophosphorylation capacity [203, 204, 211], LRRK2 can in vitro phosphorylate myelin basic protein (MBP) [203, 204] and an artificial peptide LRRKtide, based on predicted phosphorylation sites of the LRRK2 substrates ezrin, radixin and moesin [202]. Recently, a number of ATP competitive LRRK2 kinase inhibitors have been developed, varying in their structure (Appendix Fig A1), selectivity properties and potencies both in in vitro and ex vivo studies. LRRK2 IN1 [212] is a LRRK2 kinase inhibitor with IC50=13 and 6 nM for recombinant human wild type and G2019S LRRK2 respectively. Kinase selectivity profiling identified 12 additional targets for LRRK2 IN1 including IC50=160 nM for mitogen activated protein kinase 7 (MAPK7 or ERK5) and IC50>1 µM for CHK2 checkpoint homolog (CHEK2), myosin light chain kinase (MYLK) and calmodulin binding kinase kinase 2 (CAMKK2). CZC25146 [213] has an IC50=1-5 nM and 2-7 nM for the recombinant human wild type and G2019S LRRK2 enzymes respectively. Kinase selectivity profiling identified 5 additional target kinases with an IC50=10 nM-2 µM which include Polo-like kinase 4 (PLK4) and cyclin G 53 associated kinase (GAK) with no common LRRK2 IN1 targets identified in this IC50 range. TAE-684 [214] has a biochemical potency of IC50=7.8 nM and 6.1 nM for recombinant human wild type and G2019S LRRK2. TAE-684 has been described as a potent binder of many kinases with an IC50<100 nM for CHK2 and CAMKK2 and considerably less selective than LRRK2 IN1 and CZC25146. Identification of 14-3-3 as a LRRK2 interacting substrate and characterization of the phospho serine residues required for 14-3-3 binding has revealed an inherent relationship between kinase inhibition, 14-3-3 association and serine phosphorylation [215, 216]. These characteristics have been used to determine the effective inhibitor doses for LRRK2 in cells and animal models [213, 214, 216]. 1.3.3 LRRK2 PD mutations PD associated mutations are located in the enzymatic domains of LRRK2. The glycine residue at position 2019 forms part of a kinase activation motif conserved among LRRK2 kinase homologues [217]. Substitution of glycine with a bulkier alanine residue is thought to perturb the activation loop and produce an overactive kinase with greater affinity for ADP. In support of the structural predictions in vitro studies of LRRK2 enzymatic activities have consistently reported an increase in kinase activity of the protein associated with the G2019S mutation [201-203, 218]. Overall GTPase mutations seem to modestly raise the kinase activity of LRRK2, however, variable effects on LRRK2 kinase function have been reported [201-203, 218]. Mutations within the ROC-COR domain seem to induce both structural and activity associated changes. R1441G/C and Y1699C GTPase mutations have consistently been shown to lower the GTPase activity of the molecule [219, 220]. In addition R1441G/C but not Y1699C GTPase mutations perturb the LRRK2 dimer structural integrity [209, 210]. Kinase domain mutants appear not to impact on the 54 structure of the dimeric species [208, 221] but G2019S LRRK2 increases GTP hydrolysis [222]. 1.3.4 LRRK2 PD mutations and neurotoxicity In vitro studies implicate LRRK2 enzymatic changes in nigral dopaminergic cell death of LRRK2 PD patients. Neurotoxic effects of LRRK2 have been modelled in cell culture systems. Neuroblastoma cells, primary rat and mouse cortical cultures expressing mutant LRRK2 often present with nuclear shrinkage, reduced viability and enhanced sensitivity to toxic insult [203, 223, 224]. Mechanistically, over-expressed LRRK2 has been shown to interact with the apoptotic machinery. LRRK2 association with Apaf1, the main component of the apoptosome - a reported initiator of the cell death cascade [225] positions the protein downstream of a cytochrome c mediated death pathway. LRRK2 association with the death adaptor Fas associated protein (FADD) further emphasises the role of caspases in the toxic events associated with the mutant protein. An additional link to PD pathology in cell culture systems is shown by the ability of mutant LRRK2 to aggregate in HEK293 and SHSY5Y cells over-expressing the protein [215, 223, 224]. LRRK2 aggregates are positive for ubiquitin and could potentially represent a form of toxic species [226]. Some but not all the pathogenic mutants are reported to enhance aggregate formation [215, 223, 224]. However, LRRK2 aggregates are not always observed in over-expressing SHSY5Y and HEK293 cell systems [227, 228] and identification of equivalent species in animal models and human G2019S subjects warrants further investigation. 1.3.5 LRRK2 models and PD Some of the pathogenic hallmarks observed with mutant LRRK2 in cell culture systems have been extended to animal models of LRRK2 toxicity. Drosophila melanogaster and Caenorhabditis elegans contain a single homologue of the LRRK2 55 gene (dLRRK and lrk-1), with approximately 20% sequence identity to the human LRRK2 protein. The relevant LRRK2 homologues contain a conserved leucine rich core kinase (LRCK) sequence and the predicted conservation of LRRK2 tertiary structure bodes in favour of conserved cellular roles. Rodent LRRK2 homologues share 82 % sequence identity with the human LRRK2. Over-expression of the G2019S C.elegans lrk-1 homologue results in early larval arrest suggesting mutant LRRK2 is potentially toxic [229]. Although overexpression of human LRRK2 is less aggressive, the mutant protein increases the sensitivity of C. elegans dopaminergic neurons to mitochondrial toxins, suggesting the DA neurons are less viable [230]. Motor abnormalities, alterations in brain dopamine levels and dopaminergic cell defects are pronounced in Drosophila LRRK2 PD models [231, 232]. Defective dopamine transmission is consistently observed in mutant human LRRK2 transgenic (G2019S and R1441G/C) rodents correlating with an age dependent decrease in locomotor activity, responsive to L-Dopa treatment [233-235]. Mitochondrial abnormalities were noted in LRRK2 transgenic mouse models, associated with the over-expression of wild type and G2019S protein [233, 236]. Degeneration of axonal projections in rodent SNpc dopaminergic neurons is similar to the axonal demise observed in cell culture and C.elegans models. These axonal defects are thought to account for the striatal dopamine depletion and potentially reflect the human pathological scenario [233]. The severity of the mutant phenotype is dose dependent as mice expressing higher levels of the mutant LRRK2 protein present with more pronounced PD pathology [233]. Dopaminergic cell loss is rarely observed and α-synuclein deposits have never been reported in any LRRK2 models. Locomotor dysfunction, dopamine deficiency, cell toxicity and mitochondrial abnormalities observed in LRRK2 transgenic animal models are comparable to those 56 observed for other genetic PD models, suggesting overlapping pathways in PD pathogenesis. 1.3.6 LRRK2 detection antibodies Although low levels of ectopic wild type LRRK2 expression do not seem to affect cell viability, high expression has been linked to toxicity in cell culture systems, albeit milder relative to G2019S LRRK2 [227]. When this project was initiated, LRRK2 protein characterization in the brain, peripheral tissues and cell culture models relied on a library of in house and commercial antibodies raised against the LRRK2 sequence. Both polyclonal and monoclonal antibodies have been generated, targeting C and N terminal regions as well as the core domains of the protein. Early reports utilized over-expressing systems for antibody characterization struggling to detect low levels of endogenous LRRK2 [237]. Subsequent validation of commercial antibodies with LRRK2 knockout tissue identified non specific species of comparable molecular mass to full length LRRK2 protein [237]. Additional lower molecular weight immunoreactivity is common in LRRK2 detection by SDS PAGE, however, a limited number of these immunoreactive bands have been characterized [238, 239]. To address the problem of endogenous LRRK2 detection, enzyme purification for LRRK2 activity measurements and facilitate studies of LRRK2 function, Alessi et al have reported an immunoprecipitation protocol using a combination of two commercial LRRK2 antibodies to detect endogenous LRRK2 [202, 215, 216]. This method has not been applied to human brain samples or for quantitative comparison of LRRK2 protein expression. 57 1.3.7 LRRK2 mRNA and protein distribution In terms of mRNA expression, LRRK2 has consistently been shown to occur at high levels in kidneys, lungs and lymph nodes [240-243] with lower mRNA abundance in the brain [244]. In rodent and primate regional brain distribution studies, the striatum expressed significant levels of LRRK2 mRNA while nigral LRRK2 mRNA appeared to be of considerably lower abundance [245-248]. In an attempt to correlate LRRK2 protein distribution to early reports of LRRK2 mRNA, relative LRRK2 expression has been described for rodent and primate tissues [244, 248, 249]. LRRK2 antibodies have been utilized for immunological detection. In general immunohistochemical data on LRRK2 protein distribution appears to correlate with the mRNA literature with high LRRK2 abundance in striatum, cortex and cerebellum [244, 248, 249]. More recent reports, utilizing validated antibodies of better quality have confirmed high striatal LRRK2 expression providing confidence for localization and functional data published in earlier studies [250]. In general, LRRK2 brain distribution involves regions of the dopaminergic pathway. Regional and cell specific expression may have functional implications for the LRRK2 protein in neuronal subpopulations. 1.3.8 LRRK2 cellular distribution In addition to tissue distribution studies LRRK2 antibodies have been utilized to determine LRRK2 cellular localization. Cellular fractionation analyses have identified LRRK2 in mitochondrial, lysosomal, ER and vesicular compartments further supported by immunohistochemical and EM studies [201, 218, 251]. LRRK2 dimerization has been confirmed for protein immunoprecipitated from cells with membrane associated dimers described to exhibit enhanced kinase activity, adding to the initial in vitro studies [218, 239, 252]. Assuming accuracy in quantitative immunological assessment there is no evidence to suggest PD associated mutations 58 altered LRRK2 protein levels or cellular distribution in any of the cell models analyzed [218, 239, 252]. 1.3.9 LRRK2 function LRRK2 detection and characterization of its intracellular distribution in combination with the phenotypic changes associated with wild type, mutant and LRRK2 knockdown models have contributed to the functional studies and investigation of intracellular pathways potentially involving the LRRK2 protein. 1.3.9.1 LRRK2 and vesicles LRRK2 abundance in synaptosomes and vesicle enriched fractions combined with changes in dopamine transmission reported for mutant and LRRK2 deficient mouse models suggested a role for LRRK2 in synaptic function. Knockdown of LRRK2 correlated with morphological abnormalities at the pre synapse [253] and altered vesicle recycling dynamics [254]. The G2019S hyperactive kinase LRRK2 impaired synaptic vesicle endocytosis potentially through its association with the vesicular machinery [255]. The sub-cellular distribution of α-synuclein closely mimics that of LRRK2 [24]. In addition, α-synuclein has been shown to interact with the soluble Nethylmaleimide sensitive factor (NSF) attachment protein receptor (SNARE) complex, mediating neurotransmitter uptake and release [256] potentially suggesting a functional link between α-synuclein and LRRK2. Another subset of vesicles thought to functionally require LRRK2 are involved in endocytic-lysosomal trafficking. A membrane association with multivesicular bodies (MVBs) and autophagic vacuoles (AVs), prime vesicle components of this pathway, has been demonstrated by electron microscopy [257]. Knockdown of LRRK2 increased flux through the pathway while the R1441C mutant LRRK2 was described to cause an autophagic imbalance resulting in accumulation of 59 AVs and MVBs. Autophagic clearance of mutant and excess α-synuclein may therefore involve LRRK2. 1.3.9.2 LRRK2 and microtubules Axonal degeneration, neurite defects and tau pathology commonly associated with mutant LRRK2 models and PD patients implicated microtubule dynamics in PD pathogenesis [258-260]. The subsequent identification of the interaction between LRRK2 and β-tubulin, the main component of microtubules, and kinase dependent regulation of tubulin polymerization events [261, 262] supported a direct mechanistic role for LRRK2 in the observed phenotype. More recently, another microtubule related protein Tau, the main component of neurofibrillary tangles in PD and AD, was reported as an integral part of the LRRK2 β-tubulin complex [263] potentially providing an additional link between LRRK2 mutations and Tau pathology. Microtubule regulation has also linked LRRK2 to the canonical Wnt signalling pathway where a genetic interaction with α-synuclein is thought to take place. Tau phosphorylation is believed to be enhanced by the interaction of LRRK2 with the dishevelled protein (Dvl) [264], a downstream target of GSK3β which also induces αsynuclein dependent tau phosphorylation [265]. 1.3.9.3 LRRK2 transcription and translation Multiple protein interaction domains present in the LRRK2 sequence as well as its cytosolic abundance implicate the protein’s involvement in signalling cascades transcriptional and translational regulation. Microarray analyses of wild type, mutant LRRK2 over-expression and knockdown LRRK2 models implicated transcriptional contribution to the regulation of pathways involved in axonal guidance, actin cytoskeleton dynamics, calcium signalling, cell cycle and differentiation as LRRK2 mediated events [266]. Furthermore, mRNA expression of 60 genes involved in neuronal maintenance appeared to be mediated by LRRK2 levels in mouse embryonic stem cells [267]. The link between dysregulation of gene expression and PD pathology is demonstrated by increased α-synuclein mRNA levels in PD patient SNpc [268]. A number of reports suggest LRRK2 regulates α-synuclein steady state mRNA levels, potentially altering intracellular protein concentrations which may lead to aggregation [269, 270]. In support of the microarray data, mechanistic analyses implicated LRRK2 in transcriptional activation, albeit in peripheral tissues whereby LRRK2 was found to mediate the nuclear factor of activated T-cells (NFAT) cytosolic to nuclear translocation and gene regulation in response to inflammation [271]. LRRK2 interaction with the dishevelled protein (Dvl) is associated with β-catenin mediated transcriptional regulation in the canonical Wnt signalling pathway [272]. Translational repression and or degradation of mRNA inhibiting, miRNA particles through LRRK2 interaction with human Argonaute 2 protein (Ago2) and the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP) was shown to impact on PD associated genes in a LRRK2 kinase dependent mechanism [273]. In addition aberrant phosphorylation of 4E-BP by mutant LRRK2 correlated with dopaminergic cell death and morphological defects in the post synapse [231]. LRRK2 was also shown to mediate steady state 4E-BP protein levels suggesting a possible negative feedback loop [274]. A genetic interaction has been described between LRRK2, PINK1, parkin and 4E-BP. Knockdown of PINK1 or parkin affected cell viability of dopaminergic neurons in Drosophila, a phenotype exacerbated by the loss of the 4E-BP protein [275]. Knockdown of LRRK2 rescued this dopaminergic phenotype suggesting a potential interplay between PD assicated proteins and the 4E-BP pathway. 61 In addition, LRRK2 has been shown to mediate cellular stress responses through participation in canonical NFκB and MAPK cascades [203, 276, 277], pathways previously implicated in PD pathogenesis [278, 279], further emphasising altered transcriptional response in LRRK2 linked PD. 1.3.9.4 LRRK2 and mitochondria Mitochondrial involvement in LRRK2 pathogenesis is supported by a number of pathological, morphological and functional observations. In addition, cellular localization studies have confirmed a mitochondrial interaction [251, 280, 281] estimating approximately 10 % of the LRRK2 cellular pool at the outer mitochondrial membrane. LRRK2 appears to regulate survival of dopaminergic neurons by conferring resistance to oxidative stress [282] while LRRK2 toxicity induced by protein over-expression is exacerbated in rotenone and paraquat treated cells [283] potentially due to increased sensitivity to Complex I inhibition [284]. Mitochondrial respiratory chain defects have been described in mutant LRRK2 expressing cells correlating with structural alterations to the mitochondrial network [280, 281, 285]. Dopaminergic cell death has been linked to LRRK2 interaction with the mitochondrial apoptotic machinery [227, 286]. Although mechanistic links have been proposed for some of the LRRK2 associated cellular pathways with respect to other PD related genes, the strongest genetic association has involved the mitochondria. Defects in mitochondrial cristae observed in PINK1 deficient C.elegans were rescued by dLRRK2 knockdown suggesting an antagonistic mechanism of gene regulation [229]. In addition, rotenone sensitivity of LRRK2 mutant flies is suppressed by parkin, PINK1 or DJ1 coexpression [284, 287]. Furthermore, excessive mitochondrial fragmentation, reported in LRRK2, parkin, PINK1 or DJ1 deficient cells can be rescued by the artificial introduction of mitochondrial fusion machinery Mfn2 or a dominant 62 negative form of DLP1 which promotes fusion [280, 281, 284, 287], potentially linking the converging mechanisms. 63 1.4 Hypothesis We hypothesised a role for LRRK2 in the regulation of mitochondrial function, proposing the G2019S mutation would impact on the electron transport chain. We predicted, if confirmed, a detailed evaluation of upstream regulatory pathways could reveal overlapping and novel signalling events potentially reflecting previously unexplored mechanisms in idiopathic PD. 1.5 Aims 1. To characterize primary and transformed cell culture models expressing endogenous and ectopic wild type or G2019S LRRK2 protein. Characterization will require screening of commercially available antibodies for their ability to detect native, denatured and immunoprecipitated LRRK2 by Western blot and immunohistochemistry. 2. To determine whether the previously validated immunoprecipitation protocol can be used for quantitative evaluation of LRRK2 protein levels in order to assess relative expression levels in the cell models and brain tissue. 3. To use the primary and transformed cells as a LRRK2 model system to look at the impact of the G2019S mutation on mitochondrial function. A LRRK2 siRNA knockdown model will be used to address whether the ETC is regulated by endogenous wild type LRRK2. 4. To determine whether the functional state of the mitochondria in control, wild type and G2019S LRRK2 over-expressing cells or LRRK2 siRNA knockdown system correlates with morphological changes in the mitochondrial network. 5. To characterize the effects of LRRK2 kinase inhibitors in the over-expressing neuroblastoma model and determine the optimal parameters for subsequent evaluation of LRRK2 kinase contribution to mitochondrial function. 64 6. To explore the functional role of LRRK2 in upstream mechanisms regulating mitochondrial function. 65 Chapter 2: Materials and Methods All chemicals were obtained from Sigma or Merck & Co. unless otherwise stated. 2.1 Plasmid constructs pET-DEST51 plasmids (Invitrogen) containing the cDNA encoding full length human wild type or G2019S LRRK2 with a C terminal V5 tag, AmpR gene (ampicillin resistance for bacterial selection) and bsr gene (blasticidin resistance for mammalian selection) were kindly provided by Mark Cookson (Bethesda, USA), [221]. Mammalian gene expression in pET-DEST51 was driven by the elongation factor 1 (EF-1α) promoter. pEGFPN1 plasmid (Clontech) with an N terminally tagged GFP UCP2 insert, KmR gene (kanamycin resistance for bacterial selection) was provided by Koji Nishio (Nagoya, Japan) [288]. Mammalian gene expression in pEGFPN1 was driven by the cytomegalovirus (CMV) promoter. pGIPZ (Thermo Scientific) vector (scrambled shRNA: ATCTCGCTTGGGCGAGAGTAAG) or pGIPZ containing shRNA targeting the 3‘ UTR of the DJ1 gene (CCTACAAATTGTGTCTATA), sequence encoding GFP, AmpR and Pac (puromycin resistance for mammalian selection) genes were obtained from the University College London (UCL) shRNA library (Open Biosystems) and provided by Gyorgy Szabadkai (UCL). Mammalian gene expression in pGIPZ was driven by the CMV promoter. 2.2 Brain samples Human brain samples from two control subjects of 24 hour post mortem delay were obtained from the Queen Square Brain Bank. Cases were collected using a protocol approved by the London Multicentre Research Ethics Committee and stored under a license issued by the Human Tissue Authority. Human brain samples of 5 individuals (3 control, 1 IPD case, 1 Alzheimer’s disease case) of short post mortem delay times were obtained from Jose Obeso (Pamplona, Spain) and studied 66 with the approval of the Ethics Committee. Details of brain samples are described in (Table 2.1). Brain stem, olfactory lobe, mid brain, cerebellum, striatum and cortical regions were dissected from fresh brains of control male 6 week old mice (C57BL/6 strain). Equivalent tissue regions of one marmoset brain were obtained from Peter Jenner (King’s College, London). The marmoset brain was from a 2 year old female animal that received daily subcutaneous MPTP (2.0 mg/kg) injections for 5 consecutive days. LRRK2 knockout mouse brains obtained from C57BL/6 strain animals with a partial deletion of exon 39 and complete deletion of exon 40 of the LRRK2 gene [289] were kindly provided by D. Alessi (Dundee). 2.3 Cell lines Punch skin biopsies from PD patients carrying G2019S LRRK2 mutations and age matched controls (Table 2.2) were collected by Dr. Daniel G Healy (Royal Free Hospital, UCL). Human skin fibroblasts were established by Dr. Susannah Horan and Dr. Jan Willem Taanman (Clinical Neuroscience, UCL). 67 Venous blood (8 mls) was collected from the same subjects with a hypermic needle as part of a vacutainer in ethylenediaminetetraacetic acid (EDTA) vacuum tubes (Beckton Dickinson and Co). Peripheral blood mononuclear cells were isolated and commercially transformed into lymphoblastoid cell lines by Epstein-BarrVirus (EBV) (ECACC, Salisbury, UK). SHSY5Y cell lines stably expressing pGIPZ vectors containing shRNA targeting the 3’UTR of the DJ1 gene or scrambled shRNA were generated and kindly provided by Dr. Zhi Yao (Physiology Department, UCL). 68 2.4 Cell culture All cell culture medium was supplemented with 10 % (v/v) foetal calf serum (FCS, BioSera), penicillin (100 U/mL), streptomycin (100 mg/mL) and sodium pyruvate (100 mg/mL) unless stated otherwise. Adherent cells were grown on 10 cm plates while suspension cultures were maintained in surface-ventilated 75 cm flasks. Cells were cultured in a temperature-controlled, humidified incubator with 5 % CO2 at 37 oC (Hera Cell 240, Thermo Scientific, Essex UK). Adherent cell lines were passaged or harvested by trypsinization in versene (Invitrogen) containing 0.1 % trypsin enzyme. Briefly, culture medium was aspirated, cells washed once in phosphate buffer saline (PBS) and 1 ml of trypsin solution applied to each 10 cm plate with cells monitored until all of the culture detached from the surface in the trypsin suspension. Trypsin was neutralised with 10 ml culture medium and cells re-plated at 1:4 dilutions. Unless otherwise stated cell lines were cultured up to passage 25. Suspension cells or trypsinized adherent lines were harvested by centrifugation (1000 g, 10 min, RT). Cell counts were performed using a phase contrast Olympus CK2 Microscope (London, UK) on Trypan blue stained cells mounted on Fastread slides (Immune Systems Ltd., Devon, UK). Cell stocks were frozen slowly to -80 oC in culture medium containing 10 % dimethyl sulfoxide (DMSO) and stored in liquid nitrogen. Fibroblasts were cultivated under standard conditions in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) [290]. Skin biopsies were stored in sterile flasks containing fibroblast culture medium and processed on the same day. Following removal of subcutaneous fat tissue, skin sections were dissected into ~1mm2 explants and placed skin side up on 5 cm petri dishes containing 2 ml pre warmed growth medium. Surface tension of the medium maintained biopsies 69 adhered to the plate. Explants were cultured under standard growth conditions with the addition of 0.5 ml growth medium every 2 days. Upon appearance of epithelial monolayers and migrating fibroblasts the medium was replaced by 3 mls of fresh solution. Once confluent, fibroblasts were separated from the epithelial cells by trypsinization, re-plated in 10 cm dishes and expanded for stocks. Experiments were restricted to passages 4-12 to avoid cell senescence. In studies performed under substrate limiting conditions glucose free DMEM (Invitrogen) was supplemented with 4.5 g/L galactose and cells equilibrated in galactose medium for 24 hours prior to analysis. Lymphoblast cells were commercially provided as frozen stocks of 1 million cells per vial. Cells were defrosted at 37 oC, washed once (1000 g, 10 min, RT) in 10 ml of Roswell Park Memorial Institute (RPMI) 1640 culture medium (Invitrogen) containing 20 mM HEPES buffer (Invitrogen) and pelleted cells resuspended in 10 mls of fresh growth medium before transferring cell suspensions to 75 cm flasks [291]. SHSY5Y neuroblastoma cells were cultivated in 50/50 DMEM/F12 medium (Invitrogen) containing 0.1 % uridine and 1 % MEM non essential amino acids (Invitrogen) [292]. 2.5 Transformation of DH5α cells and DNA extraction pET-DEST51 and pGIPZ plasmid DNA (approximately 2 µg) was supplied spotted on Whatman filter paper. Marked regions containing the dried plasmid were excised under sterile conditions, placed in a sterile eppendorf tube and plasmids recovered by reconstitution in 50 µl TE buffer (10 mM Tris base, 1 mM EDTA, pH 8.0). Reconstituted plasmids (100 ng) were transformed into competent DH5α Escherichia coli cells (50 µL, Invitrogen) by incubation on ice (30 min) and heat shock (2 min, 42 oC) to close the cellular pores. Transformed cells were equilibrated in 1 ml 70 of Lysogeny broth (Sigma) (1 % tryptone, 0.5 % yeast extract, 1 % NaCl, 0.1 % glucose) on a temperature controlled shaker (37 o C, 2 hrs, 180 rpm). Isolated colonies were expanded overnight (37 o C) by plating on 10 cm agar (Sigma) plates containing appropriate antibiotic (ampicillin or kanamycin at 150 mg/ml where stated). Mini plasmid preparations were carried out for DNA sequencing. Single colonies were inoculated in 5 ml of Lysogeny broth containing antibiotic and expanded overnight in aerated flasks (37 o C, 180 rpm). Glycerol stocks (15 % glycerol, 85 % culture) were prepared for each colony, immediately flash frozen in liquid nitrogen and stored at -80 o C for subsequent culture expansions. Plasmids were isolated from the remaining 4 ml of pelleted culture (4 o C, 10 min, 1000g) with the Plasmid Miniprep kit (Quiagen, Chatsworth, CA, USA) according to manufacturer’s instructions and sequenced to confirm the integrated plasmid product. Large scale plasmid production (Maxi prep) was required to generate concentrated plasmid stocks for mammalian cell transfections. Pre cultures in the exponential growth phase (10 hrs, 37 o C, 180 rpm) were transferred to aerated conical flasks containing 50 ml broth with appropriate antibiotic and expanded for a further 8-10 hours. Plasmids were isolated from pelleted cultures (4 o C, 10 min, 1000g) with the Wizard miniprep kit (Promega, Hampshire, UK) or Maxiprep kit (Quiagen, Chatsworth, CA, USA) according to manufacturer’s instructions. The Wizard miniprep kit was specifically used for purification of LRRK2 containing pETDEST51 plasmids as it generated the best yield for this plasmid (1 mg of plasmid per 10 ml of culture). Quiagen maxiprep kit was used to purify the pEGFPN1 plasmid containing the UCP-GFP construct. 71 2.6 Concentration and purity of nucleic acids Concentration and purity of DNA and RNA samples was determined using a NanoDrop 1000 spectrophotometer (Thermo Scientific). Briefly, 1.5 µL of the sample was spotted onto the pedestal of the machine with sample absorbance determined at 260 nm (nucleic acid) and 280 nm (protein). The concentration of nucleic acids was calculated electronically by the NanoDrop 1000 software based on the equation A=εcl; where A is the absorbance of the sample, ε is the extinction coefficient, c is the concentration and l is the path length of sample. The 260/280 absorbance ratio was subsequently used to determine the purity of the DNA/RNA sample whereby values >1.8 for DNA and >2.0 for RNA were acceptable. Lower ratios suggested the presence of protein, phenol and other contaminants. 2.7 Restriction enzyme digest of pET-DEST51 To increase the probability of integration of the full length LRRK2 gene in mammalian cells LRRK2 containing plasmids were enzymatically digested targeting a region outside the LRRK2 coding sequence. The Fsp1 restriction enzyme (restriction site 5’tgc^gca3’, 3’acg^cgt5’, 5000 U/ml, New England Biolabs, Hitchin, UK) was chosen targeting position 6886 of the 7464 base pair pET-DEST51 plasmid. pETDEST51 plasmids (25 µg) purified as described in section 2.5 were digested (37 oC, 2 hours) with Fsp1 in a final volume of 200 µl containing 20µl NEB buffer 2 (New England Biolabs). Digestion was terminated by enzymatic inactivation (65 oC, 20 min). Digested constructs were recovered by ethanol precipitation (10 % NaAc, 250 % v/v ethanol, -20 oC, o/n) and pelleted by centrifugation (4 oC, 30 min, 17,000 g). Pellets were washed in 80 % ethanol, air dried and resuspended in 20 µl deionised, sterile TE buffer. 72 2.8 Agarose gel electrophoresis Electrophoresis of PCR products, intact and digested plasmids was carried out on agarose gels. Briefly, 1 µg of sample was combined with 6X loading buffer (Fermentas) and electrophoresed (60 V) on 0.8 – 2 % Agarose gels prepared in Trisacetate-EDTA buffer ((TAE), 40 mM Tris, 20 mM acetic acid, 1 mM EDTA, pH 8) containing 0.005 % SYBR safe DNA gel stain (Life technologies) for visualization of separated band products. Bands were visualised by ultraviolet illumination with relative sizes estimated based on migration of adjacent reference markers (BioLabs). UV illuminated bands were captured on an Olympus digital camera. 2.9 Transfection of mammalian cells SHSY5Y cells stably expressing wild type and G2019S LRRK2-V5 containing pET-DEST51 plasmids were generated and kindly donated by Dr. JM Cooper and Dr. K Chau (Clinical Neurosciences, UCL). Transfections of SHSY5Y cells with the linearized pEF-DEST51 plasmids containing the LRRK2 gene were carried out as previously described [293] using Superfect reagents (Quiagen, Hilden, Germany) according to manufacturer’s instructions. Plasmids (25 µg) were complexed with transfection reagent by incubation (15 min, RT) in serum free cell culture medium. Serum was omitted to prevent potential interference with complex formation. SHSY5Y Cells grown at 40 % confluency on 10 cm plates were treated with the mixture for 4 hours allowing for endocytosis of the plasmid, cultured overnight under standard conditions and subjected to blasticisdin (400 µg/ml) or puromycin selection (400 µg/ml) over a 6 week period. Culture medium was replaced every 2 days to remove dead cells with the formation of antibiotic resistant colonies monitored over this time course. Isolated colonies grown to a radius of 0.5 mm were separated by polystyrene microchambers (Sigma), trypsinized and transferred to 2 cm plates. Clones were grown to confluency and expanded for frozen stocks. 73 For transient protein expression purified plasmids were introduced into cells by electroporation. Cells (2.5 x 105) were harvested, washed once in PBS and resuspended in a mixture of 9 µl of reagent R and 1 µl of plasmid DNA (1 µg/ µl). The mixture was electroporated (Neon, Invitrogen, 1000 V, 30 seconds, 2 pulses), transferred to 1 ml growth medium without antibiotic and plated (1:5) on 6 well plates in a final volume of 2 ml. Cells cultured for a further two days under standard growth conditions. Gene silencing in SHSY5Y cells was achieved by introduction of small interfering RNA (siRNA) by the method of transient transfection. Details of siRNA sequences are described in (Appendix Table A1). Optimal siRNA delivery conditions for SHSY5Y cells were determined using fluorescently labelled siRNA constructs (Ambion). Transfection reagents; HiPerfect (Qiagen, Hilden, Germany), TransIT-TKO (Mirus ML015), TransIT-siQUEST (Mirus ML033), TransIT-2020 (Mirus MIR5400) were tested to determine which reagent achieved the most efficient siRNA delivery in the SHSY5Y cell line. Two volumes of transfection reagent (1 and 4 µl) were tested to determine whether the concentration would affect the efficiency of siRNA delivery. Transfection reagent was incubated with 10 nM fluorescently labelled siRNA in a final volume of 100 µl serum free culture medium (15 min, RT). SHSY5Y cells were plated at a density of 9 x 10-4 per well (24 well plate) in 500 µL of complete culture medium. Transfection mix was immediately added to the plated cells which were subsequently cultured overnight under standard culturing conditions. Transfected cells were identified by immunofluorescence 24 hours post transfection by red fluorescent punctate staining indicating the uptake of siRNA (Fig 2.1). Red punctuate staining was observed for SHSY5Y cells transfected with 4 µL of HiPerfect, ML015 and MIR5400 transfection reagents (Fig 2.1). 4 µL of HiPerfect reagent were chosen as the optimal siRNA delivery conditions as all the transfected 74 SHSY5Y cells contained red punctuate structures, unlike ML015 and MIR5400 where 30 % and 60 % of the cells were positive for red fluorescence. 2.10 Reverse transcriptase Total RNA was extracted from fibroblasts or SHSY5Y cells (1-5x104) with the RNeasy Mini kit (Qiagen) according to manufacturer’s instructions and quantified spectraphotometrically as described in Section 2.6. Purified RNA template (0.5 µg) was denatured and annealed (65 oC, 5 min) to random nanoamine primers (1 µL) in final volume of 10 µl (Primer design). Samples were cooled on ice (5 min) prior to addition of deoxyribonuclease 1 (1 µL), deoxynucleaotide triphosphate mix (dNTPs, 0.5 mM), reducing agent dithiothreitol (DTT, 10 mM) (Primer Design) and sterile water to a final volume of 20 µl. The reverse transcriptase extension step (25 oC, 5 min, 55 oC, 20 min), initiated at the primer binding sites, reverse transcribed the RNA template to produce complementary DNA (cDNA). Following reverse transcription, the enzyme was heat inactivated (75 oC , 15 min). 75 2.11 Real Time PCR For quantification of gene expression, 1 µL of the first strand cDNA sample was used as template in quantitative real-time PCR amplification with the TaqMan system or SybrGreen assay (Applied Biosystems, Paisley, UK). In TaqMan reactions an initial polymerase activation step (95 oC, 10 min) was followed by 40 cycles of PCR as described in the supplier’s protocol (15 sec at 95 oC, 1 min at 60 oC). Annealing temperatures for SybrGreen primers are described in (Appendix, Table A2), determined by subtracting 5 oC from the melting temperature (Tm) of the primer strand as calculated by the manufacturer (Eurogenetech), based on the length and nucleotide composition of the primer. Standard denaturation (95 °C, 15 sec) and extention (72 o C, 30sec) conditions were maintained across all SybrGreen experiments. SybrGreen primers were validated by cDNA template titration with a valid primer concentration range for PCR requiring one amplification cycle increase with doubling of template concentration (Fig 2.2A). RT PCR product melting curves were generated in a temperature range of 60-90 oC at 0.3 oC increments for each SybrGreen reaction to ensure specificity of amplifications for the gene target. Melting points of amplicons were considered appropriate in the range from 80 oC to 85 oC with additional peaks in the lower temperature range (60-80 oC) assigned to primer dimers (Fig 2.2B, C). Furthermore, TaqMan and SybrGreen PCR product sizes were checked by agarose gel electrophoresis with relative sizes estimated based on migration of adjacent reference markers (BioLabs) (Appendix, FigA2). The comparative cycle threshold (CT) method was used to quantify mRNA expression, correcting gene amplification to GAPDH CTs determined by the equation: Relative mRNA expression = (2-[∆∆Ct], ∆∆Ct = Ctgene-CtGAPDH) where the Ct value is the cycle number at which the fluorescence signal crosses the threshold 76 (0.01), (Fig 2.2D). Threshold values were chosen based on the first detectable increase in fluorescence from RT-PCR products and maintained across all experiments. 77 2.12 LRRK2 mutation screen in fibroblasts and SHSY5Y cells cDNA from control or G2019S fibroblasts, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells was screened for the presence of the G2019S mutation at position 6055 of the LRRK2 gene. Primers were designed targetting the LRRK2 sequence flanking a 700 bp fragment on exon 41 of the LRRK2 gene containing the 6055 nucleotide as part of the codon encoding the G2019A/S residue (Table A2). Primers (0.5 µM) were combined with 2 µL of total cDNA, and Phusion Flash High Fidelity PCR mastermix (Thermo Fisher Scientific, Loughborough, UK) in a final volume of 50 µl. The targeted region of LRRK2 was amplified by 30 cycles of a three step PCR amplification protocol as suggested by the manufacturer (denaturation 98 o C 10 min, annealing 20 sec, elongation 72 oC 30 sec). Annealing temperatures were determined by subtracting 5 oC from the melting temperature (Tm) of the primer strand as calculated by the manufacturer (Eurogenetech), based on the length and nucleotide composition of the primer. A final 10 minute extention step (72 oC) completed the PCR reaction. The approximate size of PCR products was determined by agarose gel electrophoresis (Section 2.8). Products were sequenced at the Scientific Support Unit (Wolfson Institute for Biomedical Research, University College London). Sequence comparison was ncarried out by BLAST alignment (PubMed) on the human LRRK2 fasta entry registered in the Uniprot Database (NP_940980) with sequence profiles generated by Sequencher 5.0. 2.13 Whole cell and tissue extractions SHSY5Y, fibroblast, lymphoblast cell pellets (1x106) or brain tissue (1 mg dry weight) were prepared for Western blot analysis by solubilization in 50 µl 0.1 % sodium dodecyl sulphate (SDS), 10 mM Tris HCL containing a cocktail of protease inhibitors (Thermo Scientific, targeting serine, cysteine, aspartic acid-proteases and amino peptidases). Brain tissue was homogenized in buffer with a Potter type 78 homogeniser, cleared by centrifugation (4oC, 17,000 g, 30 min) and sonicated (2 x 15 s pulses on ice (Decon Fs 100b)). Chromosomal DNA in whole cell extracts was digested by treating samples with DNAse1 (Promega, 1/10th the total reaction volume) in enzyme buffer (1/10th the total reaction volume, 45 min, 37 oC). Following DNAse treatment cell extracts were centrifuged (4oC, 17,000 g, 10 min) to remove insoluble and aggregated components. 2.14 Immunoprecipitation Immunoprecipitation procedures (IP) essentially followed those published by [215]. Cell pellets (6x106 cells per 500 µl buffer) were extracted (10 min, on ice) in lysis buffer (LB: 50 mM Tris HCl, 0.27 M sucrose, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 50 mM NaF supplemented with 1 % Triton X100 (IP) or 1 % nonidet P-40 (NP-40) (for co-IP), 1 mM benzamidine, 0.1 mM phenylmethanesulfonylfluoride (PMSF) and protease inhibitor cocktail), retaining detergent soluble fractions following centrifugation (4 o C, 17,000g, 10 min). For brain IP, samples were processed as described in the Abcam IP datasheet in LB buffer (2.5 mg frozen tissue per 1 ml buffer) by 30 strokes with a Potter type homogenizer, pulsed twice by sonication and cleared by centrifugation (4oC, 17000 g, 30 min). In co-IP experiments sonication steps were substituted by 10 passages through a 27 gauge needle (Terumo) to maintain integrity of protein complexes. Immunoprecipatation required 1 mg of protein, incubated with 20 µL of Protein G agarose beads (Invitrogen) pre coupled to antibody (4 °C, 30 min). 1 mg of lysate with 20 µ agarose beads without the addition of antibody were included in parallel control experiments to discount potential binding of immunoprecipitated proteins to beads. Samples were incubated with rotation (4 °C, 2 hours), beads 79 collected by centrifugation (17,000 g, 1 min) and non specific interactions removed by washes in salt (500 mM NaCl for IP, 150 mM for co-IP) containing LB followed by two washes in LB buffer (17,000 g, 1 min). Pulled down proteins were eluted from the beads by solubilisation in 50 µl 2 X LDS sample buffer (Invitrogen) supplemented with 2X reducing agent (Invitrogen) with 25 µl of the eluted sample used for SDS PAGE. An un-cropped Western blot of LRRK2 immunoprecipitates from control fibroblasts is shown in (Appendix Fig A3) where a single 280 kDa 3514-1 immunoreactive species was observed. 2.15 Protein quantification Protein quantification was carried out using the bicinchoninic acid (BCA) protocol (Pierce, Cramlington, UK) [294]. 2 and 4 µl of SDS (Section 2.13) or Triton X100 (Section 2.14) cell and tissue extracts were mixed with PBS to a final volume of 25 µl in a 96 well plate (Starsted). Blank samples were those containing sample extraction buffer and PBS while bovine serum albumin (BSA, 0-2000 µg/ml) was used as protein standard. Kit components A and B were mixed in a 1:50 ratio and 200 µL of the mixture added to each well. Samples were incubated at 37 oC for 30 minutes and assessed spectraphotometrically by quantifying the light absorbance at 562 nm (Synergy HT BioTEK spectrophotometer). Protein concentrations were determined from the linear absorbance plot generated by the known concentrations of bovine serum albumin (BSA) protein standards for each measurement. 2.16 SDS PAGE Western blot analysis of SDS soluble lysates (Section 2.13) or Triton X100 extracts (Section 2.14) required 25 µg of protein denatured in NuPAGE LDS sample buffer (Invitrogen) supplemented with NuPAGE sample reducing agent (Invitrogen). Proteins were heated (65 °C, 5 min) and electrophoresed on precast NuPAGE Novex 80 12 % or 4-12 % polyacrylamide Bis-Tris gels (Invitrogen) in the presence of MES (50 mM MES, 50 mM Tris Base, 0.1% SDS, 1 mM EDTA, pH 7.3 for higher molecular weight proteins, >150 kDa) or MOPS (50 mM MOPS, 50 mM Tris Base, 0.1% SDS, 1 mM EDTA, pH 7.3, for lower molecular weight species, <150 kDa) SDS running buffer (Invitrogen) (RT, 45 min, 200 V) using the BioRad system (Hertfordshire, UK). For proteins of molecular weight >150 kDa, electrophoresis was extended to 60 minutes to improve the separation of higher molecular weight species [244]. 2.17 Western blot analysis SDS PAGE separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes (GE Healthcare) (RT, 90 min, 30 V, BioRad system). Transfers were extended to 120 minutes for higher molecular weight proteins (>150 kDa). Membranes were subsequently blocked (RT, 1 hr) with 5 % w/v semi skimmed milk containing PBS with tween (PBS-T 0.3 % v/v) and incubated with primary antibodies under specified conditions (Appendix Table A3). Following washes in PBS-T (3 X 10 min), horseradish peroxidase labelled secondary antibodies (Dako) were applied at 1:3000 dilution in 5 % milk (RT, 1 hr). Membranes were washed again with PBS-T and protein bands visualized by chemiluminescence (Thermo Pierce) on X-ray film (GE Healthcare) estimating band molecular weights relative to standard protein markers of 10-250 kDa range (Precision Plus Protein Standards, BioRad). Protein bands were quantified by densitometry using Image J software. 2.18 Native gel electrophoresis Blue native PAGE (BN PAGE) electrophoresis was carried out to investigate the native structure of LRRK2 complexes, according to manufacturer’s instructions based on the protocol developed by [295]. To recover soluble proteins, flash frozen brain tissue (1 mg) or freshly prepared cell pellets (8x106 cells) were homogenised (Potter homogeniser) in 50 µl volume of 4X Native PAGE sample buffer (50 mM 81 BisTris, 6N HCl, 50 mM NaCl, 10 % w/v glycerol, 0.001 % Ponceau S, pH 7.2, Invitrogen) with the addition of a cocktail of protease inhibitors (Thermo scientific). Samples were further processed with a 27 gauge needle (20 strokes) and cleared by centrifugation (4oC, 30 min, 20,000 g). Protein samples (20 µg) were loaded on to 412 % Native-PAGE Novex Bis-Tris gels (Invitrogen) in the presence of 0.25 % v/v coomassie blue loading additive (Invitrogen). Electrophoresis was carried out in anode buffer (AB, outer tank chamber: 50 mM BisTris, 50 mM Tricine pH 6.8) and cathode buffer (inner tank chamber, AB containing 0.02 % v/v coomassie brilliant blue) at 150 V. Once approximately 1/3 of the coomassie blue border migrated down the gel, the cathode buffer was replaced by AB containing 0.002 % v/v coomassie buffer and electrophoresed for a further 1 hour (150 V). Separated proteins were transfer to PVDF membranes (30 V, 120 min). Membranes were equilibrated in absolute methanol to remove excess coomassie dye. NativeMark Unstained Protein Standards (20-1236 kDa, Invitrogen) were visualized by incubating in Ponceau S staining solution as reference markers to approximate the molecular weight of resolved protein complexes. 2.19 Immunofluorescence of cultured cells Cells plated up to 40 % confluency on 22 mm coverslips were fixed in 4 % paraformaldehyde (PFA, room temperature, 15 min) and permeabilized in absolute methanol (MeOH, -20 oC, 20 min) [296]. For visualization of nuclear proteins, paraformaldehyde fixation was followed by permeabilization with 0.1 % Triton (room temperature, 30 min) [297]. Nuclear staining required detergent concentrations to be maintained throughout the immunostaining procedure. Immunocytochemistry for LRRK2 labelling omitted PFA and required MeOH (-20 oC, 20 min), acetone (-20 oC, 5 min) permeabilization/fixation conditions [298]. During protocol optimization to determine optimal conditions for LRRK2 staining; PFA 82 fixation followed by sodium citrate (95 oC, 20 min), 1 % Triton X 100 (RT, 30 min) or MeOH permeabilization (as above) were also tested on control versus wild type LRRK2-V5 over-expressing SHSY5Y cells. Permibilized cells were subsequently processed in parallel using the same conditions. PBS washes (3X times) were preformed after each stage of the staining procedure. Following permeabilization, non specific binding was blocked by 10 % normal goat serum (NGS, Life Technologies) (RT, 30 min) and primary antibodies applied at appropriate dilutions (Appendix, Table A3) in 10 % NGS (RT, 2 hrs), followed by incubation with fluorescently labelled secondary antibodies (1:200, Molecular Probes) in 2 % NGS. Coverslips were mounted on glass slides with Citifluor containing Hoechst trihydrochloride trihydrate (1 mg/ml, Invitrogen) for nuclear counterstain. Fluorescent images were acquired using the same exposure settings with an Axiophot fluorescent microscope, KS400 software (Zeiss, Welwyn Garden City, UK) equipped with 40X and 100X PlanNeoFluar lenses and the appropriate fluorescent filters. 2.20 Immunohistochemistry on human and mouse brains Flash frozen mouse brain hemispheres embedded in optimum cutting temperature medium (OCT, Sakura) were cut into 12 µm sagittal slices by cryostat (OTF5000, Bright, Huntingdon, UK) sectioning and mounted on glass slides. Sections were dried (37 oC, 15 min) and frozen (-80 oC) until further processing. On the day of the staining, sections were thawed (RT, 15 min) and fixed in 4 % PFA (RT, 15 min) [299]. PBS washes (3X, 5min) were preformed after each stage of the procedure. Endogenous peroxidases were blocked (MeOH, 1 % H2O2, 10 min), followed by blocking of non specific binding (10 % goat serum, 30 min) and incubation with 83 primary antibodies (Table A3, 4 oC, o/n). The following day, sections were incubated with biotin labelled anti-rabbit secondary antibodies (Gibco, 1:200, RT, 30 min) and positive immunoreactivity visualized by 3’,3’diaminobenzidine (DAB) oxidation induced by hydrogen peroxide (1% in PBS solution). DAB brown colour development was monitored at 1 minute intervals to obtain the optimal signal to noise staining for each set of experiments. DAB oxidation was terminated by washing slides in PBS solution. Nuclei were counterstained with haematoxyline (5 min) and differentiated in acid alcohol (0.3 %) to remove background colouration. Differentiation was arrested by rinsing slides in warm tap water where the alkaline environment converted the histone bound haematin to a blue colour. Sections were dehydrated in ethanol baths of increasing ethanol concentration (70 – 100 %) followed by xyline to prepare slides for mounting with organic based neutral mounting medium (DPX, Leica). Images of relevant brain regions were captured on the Leica opitical imaging system (DM4000 B, Wetzlar, Germany) at 20X, 100X and 200X magnifications. 2.21 LRRK2 kinase activity assays In vitro LRRK2 kinase activity assays were performed on LRRK2 immunoprecipitated from 1 mg of cell lysate or 0.5 µg of recombinant GST-LRRK2 (with the help of Francisco Inesta-Vaquera (Dundee)). Recombinant GST-LRRK2, GSTMoesin and GST-UCP2 proteins were prepared and purified by Francisco InestaVaquera (Dundee) as previously described [202]. Briefly, 5 µg of pEBG-2T encoding residue 1326-end of LRRK2, full length Moesin or full length UCP2 was transiently transfected into HEK293 cells with 60 µl polyethylenimine in 3 mls of media, as suggested by the manufacturer (Sigma). Cells were cultured for 36 hours following transfection and extracted in LB (50 mM Tris HCl, 0.27 M sucrose, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 84 50 mM NaF) supplemented with 0.5 % NP40 and 150 mM NaCl. GST tagged proteins were purified from cell lysates by chromatography on glutathione-sepharose and bound proteins eluted in buffer containing 20 mM glutathione and 0.27 M sucrose. Kinase assays [215] were carried out in a volume of 50 µL containing kinase reaction buffer (RB, 50 mM Tris, 0.1 mM EGTA, pH 7.5), 10mM MgCl2, 0.1 M DTT, 20 µM of LRRKtide substrate (RLGRDKYKTLRQIRQ) and 0.1 mM [32P] adenosine triphosphate (ATP) (300 cpm/pmol). LRRK2 bound to IP beads was in a volume of 15 µL with the final reaction volume obtained by the addition of deionised H2O. Reactions were initiated with the addition of the LRRK2 kinase and incubated for 20 minutes at 30 oC. Reactions were terminated by addition of 12.5 µL EDTA (50 mM), centrifuged (17,000 g, 1 min) and 40 µL of the supernatant spotted on phosphocellulose p81 paper (Whatman). Samples were air dried and washed sequentially in 50 mM ortho-phosphoric acid (20 min, 3 times). Following acid treatment, samples were rinsed with 100 % acetone, dried and the amount of incorporated radioactivity assessed by Cerenkov counting in the presence of scintillation fluid. The LRRK2 kinase specific activity was calculated as: SA= cpm from reaction/[(SA of [32P] ATP in cpm/pmol)*(reaction time (min))*(enzyme amount in µg)]*[(reaction volume)*(spot volume)]. LRRK2 bound to beads in the pelleted fraction was eluted by addition of 4X sample buffer (Invitrogen) and analysed by Western blotting to correct for LRRK2 input. 2.22 Isolation of PMBCs and cell sorting Peripheral blood mononuclear cells (PMBCs) were isolated from 50 ml of whole blood by density gradient centrifugation (Axis Shield, Dundee, UK), as described by the manufacturer. Briefly, blood (diluted 1:1 in isolation buffer (IB: PBS, 85 0.5 % bovine serum albumin (BSA), 2 mM EDTA, pH7.2), layered onto a buffy coat gradient was fractionated by centrifugation (8,000 g, RT, 20 min). The white PMBC enriched layer located below the serum fraction was extracted with a 3 ml pasteur pipette into a 50 ml falcon tube. PMBCs were subsequently washed in cold IB (4 oC, 800 g, 5 min), counted as described in section 2.4 and incubated with magnetically labelled anti CD4, CD8 or CD19 antibodies (10 µl/105 cells) (Miltenyi, Surrey, UK, 4 o C, 1hr). Labelled cells were purified by positive magnetic selection (MiniMACS separator, Miltenyi) on LS columns (Miltenyi). Purified cells were re-suspended at a concentration of 1x106cells/ml in HBSS (156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2, 10 mM glucose, 10 mM HEPES, pH 7.35) and 100 µl of the cell suspension added to 4ml polystyrene round bottomed tubes (Sigma). Mouse anti human conjugated monoclonal antibodies used to analyse purified cells were supplied by Becton Dickinson Biosciences (Oxford, UK) and used at a volume of 10 µl/105 cells. 10 µl of CD4 (FITC), CD8 (FITC), CD19 (PC) and CD21 (CytC) were added to the tubes, samples vortexed and incubated at room temperature in the dark for 15 minutes. Cells were then washed free of excess antibody by adding 2mls of HBSS and centrifuged (200 g, 5 min). Supernatants were decanted and cells re-suspended in 500 µl of HBSS prior to flow cytometry acquisition. FACS analysis was carried out by Janet North (Royal Free Hospital, UCL). All readings were acquired on a BD FACSAria II analyser as list mode data files using FACSDiva software. A forward scatter (FSC) versus side scatter (SSC) dot plot was created to gate on the live lymphocyte like region of cells (P1) and to exclude cell debris. A second dot plot was created to show expression of cells from P1 region for CD19 against FSC. Region P2 was drawn to include all live cells from P1. A third dot plot was generated to show CD19 versus CD21 which included cells from P1 and P2 86 regions. A fourth dot plot was created to look at the expression of CD4 and CD8 on P1 and P2 regions to identify any contaminating T cells. Purity of CD8 and CD4 T cells was analysed in a similar manner. Cell populations of >90 % purity were used for Western blot analysis. 2.23 Cellular fractionation Lymphoblasts or SHSY5Y cells were harvested as described in Section 2.4. Cell pellets (20x106) were resuspended in homogenization buffer (2 ml, HB: 320 mM sucrose, 10 mM Tris HCl, 1 mM EDTA pH 7) containing protease inhibitors (Thermo Scientific). Disruption of plasma membranes was achieved by nitrogen cavitation (Parr Instrument Company, Moline Illinois) at 1200 psi (4 oC, 20 min). Sub-cellular fractions were obtained by sequential centrifugation at; 1000 g (P1); 12,000 g (P2); 27,000 g (P3) (4 oC, 10 min, Sorvall Super T21, DuPont); 47,000 g (P4) (4 oC, 15 min, Kontron, P4, T124, Centrikon, Zurich, Switzerland) and 200,000 g (P5) (4 oC, 90 min, P5, Beckman Coulter, High Wycombe, UK) respectively [300]. Pellets were washed once in HB buffer while cytosolic fractions were concentrated in 30 kDa cut off concentrator columns (Millipore) to a final volume of 50 µL. Fractions were solubilised in 50 µl of SDS sample buffer and 10 % of each sample analysed by Western blotting. 2.24 Affinity purification of mitochondria Lymphoblasts or SHSY5Y cells were harvested as described in Section 2.4. Cells (4x106) were lysed by repeated passage through a 27 gauge needle. Optimal homogenization conditions were determined by visual inspection of intact cell densities in trypan blue stained cell sample following increasing strokes with a 27 gauge needle in 1 ml buffer supplied with the mitochondrial isolation kit (Miltenyi, Surrey, UK). Conditions (25 strokes for lymphoblasts, 30 strokes for SHSY5Y cells) achieving >90 % disruption of cells were chosen for subsequent experiments. 87 Extracted cell suspensions were centrifuged to remove unbroken cells and nuclei (10 min, 4 oC, 1000 g). 10 % of the post nuclear supernatant (PNS) was retained to evaluate mitochondrial recovery. Magnetically tagged Tom22 antibodies labelled mitochondria in cell extracts (4 oC, 1 hr with rotation) and immunoreactive products were isolated by positive magnetic selection (MiniMACS separator, Miltenyi) on isolation columns (LS columns with a 30 µM cut-off, Miltenyi) [301]. Magnetically bound products were washed three times in buffer supplied with the kit and eluted in a final volume of 1 ml by removing samples from the magnet and applying the plunger supplied with the column. Affinity purified mitochondria were pelleted by centrifugation (12,000 g, 2 min). The PNS sample and affinity purified mitochondria were solubilised in equivalent volumes of SDS sample buffer and analysed by Western blotting as 1/10 PNS and 9/10 mitochondria. 2.25 Mitochondrial sub-fractionation Mitochondrial enriched fractions (MEF) isolated by differential centrifugation (12,000 g, as described in section 2.23) or mitochondria isolated by affinity purification from (4x106 for each extraction condition) SHSY5Y cells were incubated (30 min, 4 oC) in HB buffer, HB supplemented with increasing concentration of digitonin (0.5 – 3 mg/ml), hypotonic buffer (10 mM Tris HCl, 100 mM NaCl, pH 7.6) or 100 mM Na2CO3 (pH 11.5 followed by 5 min sonication) [302]. Samples were separated into soluble and insoluble components by centrifugation (4 °C, 20,000 g, 30 min) and pellets washed in appropriate buffers. Final pellets and soluble fractions were solubilised in 50 µl of SDS sample buffer and 10 % of each sample analysed by Western blotting. 88 2.26 Proteinase K digestion of mitochondria Affinity purified mitochondria (12x106 SHSY5Y cells) resuspended in proteinase K buffer (0.6 M sorbitol, 20 mM HEPES K-OH, pH 7.4) were digested with 0.5 or 1 µg/ml freshly prepared proteinase K enzyme (stock 50 µg/ml in buffer PB) for increasing incubation periods (0-30 min) [302]. Digestion was terminated by inhibiting proteinase K with phenyl methyl sulphonyl fluoride (PMSF, 1 µM, 4 °C, 10 min). Samples were subsequently separated into soluble and insoluble components by centrifugation (4 °C, 17,000 g, 10min), solubilised in 50 µl of SDS sample buffer and 10 % of each sample analysed by Western blotting. 2.27 Single cell analysis by confocal microscopy 2.27.1.1 Mitochondrial membrane potential: TMRM Tetramethylrhodamine Methyl Ester (TMRM) is a cationic lipophilic fluorescent dye incorporated into the mitochondrial matrix specifically by the electrochemical gradient generated by the mitochondrial membrane potential (Ψm) and can be used to quantify the Ψm in live cells. Fibroblasts or SHSY5Y cells were seeded on 20 mm coverslips at 30 % confluency and cultured over night under standard culturing conditions. The following day, mitochondrial membrane potential was assessed by staining cells with 25 nM TMRM (Molecular probes, 45 min, RT) in respiration buffer (HBSS: 156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2, 10 mM glucose, 10 mM HEPES, pH 7.35) [303]. TMRM intensity was measured in the presence of oligomycin (2 mg/ml), trifluorocarbonyl cyanide phenylhydrazone (FCCP, 0.5 M), LRRK2 IN-1 inhibitor (1 µM 90 min treatment prior to rate measurements) and genipin (1.375-10 nM) where stated. Fluorescent images were acquired on an inverted laser scanning confocal microscope (LSM510 Zeiss, Oberkochen, Germany) by exciting samples at 543 nm with maximum laser power directing speciments with the HFT 485/540 short pass 89 filter. Emitted red fluorescence was captured by 40x magnification through an oil objective with numerical aperture of 1.2 where a 560 nm long pass filter transmitted all light above 560 nm. Pinhole diameter was set at 244 to eliminate out of focus fluorescence reaching the detector. The detector gain was adjusted to 464 allowing for amplification of the fluorescent image and the amplifier offset set to 0.035 as the cut off thresh hold below which pixels were displayed as black. 12 Bit images were collected as the average of four scans (scan speed 7); 255 x 255 pixels for normal resolution increasing the pixel depth to 1024 x 1024 for high resolution images. In time course measurements images were acquired by a single image scan as 8 bit; 255 x 255 pixels at 5 or 8 minute intervals to reduce laser exposure to cells. Images were projected through the 1.52 µm Z stacks for a maximum intensity two dimentional TMRM image. TMRM fluorescence was quantified using Metamorph software calculating the average signal intensity per cell with a set threshold value to eliminate background noise determined independently for the daily set of TMRM measurements. Approximately 30 individual SHSY5Y cells or 15 fibroblasts were analysed for each imaged field, four fields per coverslip in two (SHSY5Y) or four (fibroblasts) independent experiments (n=200 cells per cell line). Data was expressed as the mean ± SEM, relative TMRM fluorescence (RFU) per cell. 2.27.1.2 Method validation for TMRM TMRM staining of control fibroblasts resulted in accumulation of the red dye within the mitochondrial compartment ((↓)Fig 2.3A). The TMRM fluorescence was found to be stable for at least one hour after the initial incubation period and all subsequent measurements were confined to this time frame (Fig 2.3A). Trifluorocarbonyl cyanide phenylhydrazone (FCCP) is a chemical uncoupler of oxidative phosphorylation known to dissipate the mitochondrial membrane potential. FCCP addition (1 µM) resulted in a 2 fold reduction of the TMRM signal 90 intensity accompanied by redistribution of the dye to the cytosolic compartment (*↓) consistent with dissipation of the proton gradient (Fig 2.3A, B). TMRM response to FCCP treatment validated the specificity of the dye for mitochondrial structures in pre treated cells. Oligomycin inhibits the mitochondrial ATPase, complex V of the mitochondrial respiratory chain resulting in the accumulation of protons in the mitochondrial matrix and increased mitochondrial membrane potential. Monitoring TMRM intensity over time in the presence of oligomycin (4 mg/ml) detected a gradual 75 % increase in TMRM fluorescence when compared to the stable signal of untreated cells (Fig 2.3C, D). Increased TMRM intensity reflected changes to the mitochondrial proton gradient consistent with the accumulation of protons in the mitochondrial matrix due to inhibited ATPase activity. TMRM response to FCCP and oligomycin suggested the methodology could be manipulated in a predictable way. 91 2.27.2 Mitochondrial content Mitochondrial content was assessed by TMRM staining of fibroblasts seeded at 40 % confluency on 20 mm cover slips. Live cell TMRM images were collected before (↓) and after (*↓) addition of 1 µM FCCP releasing the TMRM dye into the cytosol to provide an image of the cell contour (Fig 2.4). Maximal intensity Z projections of the TMRM signal were binarized in Image J and the proportion of the binarized signal quantified relative to the cell contour (**↓). Quantification of the binarized TMRM signal in Image J generated four values; minimum = 0, black image, 92 maximum = 255 white image, cell area and the mean TMRM intensity representing the proportion of the highlighted area as white. The % TMRM density per area of cell was calculated as [(mean TMRM intensity/255)*100]. 2.27.3 GFP SHSY5Y cells transiently transfected with GFP-UCP2 constructs (1 µg) were seeded on 20 mm coverslips at 40 % confluency and assessed by confocal microscopy 48 hours post transfection for the presence of green fluorescence. Fluorescent images were acquired on an inverted laser scanning confocal microscope (LSM510 Zeiss, Oberkochen, Germany) by exciting samples at 488 nm with 10 % laser power directing speciments with the HFT 405/488/543 short pass filter. Emitted red fluorescence was captured by 40x magnification through an oil objective with numerical aperture of 1.2, a 515 nm long pass filter transmitted all light above 515 nm. Pinhole diameter was set at 244 eliminating out of focus fluorescence reaching the detector. The detector gain was adjusted to 688 allowing for amplification of the fluorescent image and the amplifier offset set to 0.015 as the 93 cut off threshold below which pixels were displayed as black. Image resolution parameters; pixel depth, scan speed and time course acquisition specifications are described in Section 2.27.1.1. 2.28.1.1 Cellular respiration: oxygen electrode Cellular oxygen utilization rates were analyzed polarographically using a micro-Clark-type oxygen electrode (YSI, Hampshire, UK) [304]. The electrode was calibrated with air saturated respiration buffer (HBSS) thermostatically maintained at 37 °C. Whole fibroblasts or SHSY5Y cells (4x106) were resuspended in 250 µL of respiration medium (HBSS) and extracellular oxygen levels monitored in a sealed chamber over a period of 10 min. Uncoupled rates and mitochondrial proton leak were measured in the presence of FCCP (0.5-2 µM) or oligomycin (1-4 mg/ml) respectively. In experiments involving LRRK2 IN1, cells were treated with the inhibitor under standard culturing conditions (1 µM, 90 min) with LRRK2 IN1 maintained in solution throughout the harvesting steps and respiration measurements. Rates were calculated as the % decrease in the full scale deflection (FSD) per minute with 0 % FSD determined by addition of 1 μM cyanide. The rates of change of FSD were converted to changes in oxygen concentration assuming 406 nmol O per ml at the given temperature and corrected for protein content. 2.28.1.2 Method validation for the oxygen electrode Extracellular oxygen concentrations measured by the oxygen electrode for control fibroblasts showed a linear decrease in the signal over the course of the 10 minute measurement consistent with oxygen depletion as a result of cellular respiration (Fig 2.5A). Oxygen levels were stable in the presence of the respiratory chain complex IV inhibitor cyanide, reflecting inhibition of cellular oxygen consumption and validating the specificity of our linear rate signals. 94 FCCP was used to investigate maximal respiratory capacity by the Clark type oxygen electrode. Polarographic measurements of oxygen consumption in control fibroblasts showed that the same concentration of FCCP (1 µM) used to dissipate the mitochondrial membrane potential (Fig 2.3B) resulted in a two fold increase in the rate of cellular oxygen consumption consistent with an uncoupling effect on the ETC (Fig 2.5B). Titrating in more FCCP to 2 µM resulted in a reduced respiratory rate indicative of mitochondrial toxicity as has previously been described for this compound [305]. Oligomycin was used to investigate mitochondrial proton leak by the Clark type oxygen electrode. Polarographic measurements of oxygen consumption in control fibroblasts showed that addition of oligomycin at 4 mg/ml stabilized extracellular oxygen levels (Fig 2.5C) consistent with inhibition of cellular respiration. Lower oligomycin concentrations (1-3 mg/ml) showed a partial (70 %) decrease in the linear rate of oxygen depletion rate signal potentially due to incomplete inhibition of the mitochondrial ATPase. 4 mg/ml oligomycin were chosen for subsequent experiments as the concentration required to inhibit the mitochondrial ATPase in fibroblasts. Inhibition of mitochondrial respiration by cyanide and oligomycin as well as mitochondrial uncoupling by FCCP suggests the methodology could be manipulated in a predictable way. 95 2.28.2.1 Cellular respiration: phosphorescent analysis Phosphorescent analysis of cellular oxygen utilization is an assay based on the ability of oxygen to quench the excited state (lifetime) of the porphyrin based extracellular MitoXpress probe. As cellular respiration occurs oxygen is depleted in the surrounding environment correlating with an increase in the probe phosphorescent signal. Phosphorescent analysis of oxygen utilization required 50,000 fibroblasts seeded per well on standard 96-well plates (Sarstedt) pre-coated with 0.007 % collagen IV (Sigma) [306]. Measurements were conducted in 100 µl of air- 96 equilibrated DMEM supplemented with 1 mM pyruvate, 10 mM glucose (or 10 mM galactose where stated), 20 mM HEPES (pH 7.4) and 100 nM of phosphorescent MitoXpress-Xtra probe (Luxel Biosciences). Compounds used in the manipulation of respiratory chain function (antimycin A (1 µM), rotenone (1 µM), FCCP and oligomycin) were added at this point. Medium was overlaid with 50 µl of highly viscous oil to prevent oxygen diffusion. The plate was monitored at 37 °C on a Victor 2 time resolved fluorescent (TR-F) reader (PerkinElmer, Cambridge, UK) with a Samarium filter set (340 nm excitation, 642 nm emission). Duplicate TR-F intensity (F1, F2) readings were recorded in a gated (100 µs) measurement window at two delay (t1=30, t2 =70 µs) intervals. Measured TR-F intensity signals (F1, F2) for each well were converted into phosphorescence life-time values as follows: τ = (t1–t2)/ln(F1/F2). To determine the rate of change of probe phosphorescence in the presence of respiring cells lifetime values were determined at 2 min intervals over 90 min. An initial temperature equilibration phase of the probe lasting approximately 15 minutes was excluded from the rate measurement. Oxygen consumption rates were calculated as the rate of phosphorescent life intensity change in relative phosphorescent units (RPU) by taking the slope of the background subtracted linear signal. Rates were corrected for protein content. 2.28.2.2 Method validation of phosphorescent analysis Phosphorescent analysis of control fibroblasts determined a linear increase in the probe phosphorescent lifetime consistent with oxygen depletion in the extracellular medium (Fig 2.6A). The rate of change of phosphorescent lifetime was reduced to 30 % of the basal rate in untreated fibroblasts with the addition of rotenone (1 µM) consistent with inhibition of mitochondrial respiration (Fig 2.6B). 97 Control fibroblasts were treated with increasing concentrations (0-5 µM) of the chemical uncoupler. FCCP at 1 µM, a concentration shown to dissipate the mitochondrial membrane potential resulted in a 50 % increase in phosphorescent lifetime rate values with respiration rates reaching a maximum of 170 % at 1.5 µM relative to untreated cells (Fig 2.6C). Rates were comparable to basal values at higher FCCP concentrations. FCCP induced reduction in oxygen consumption at higher concentrations are indicative of mitochondrial toxicity [305]. The phosphorescent response to rotenone and FCCP suggested the methodology could be manipulated in a predictable way. 98 2.29.1 Extracellular acidification The pH-Xtra probe monitors cellular acid extrusion. The pH-Xtra phosphorescence signal is modulated by pH changes such that increased acidification causes increased phosphorescence. Extracellular acidification is an indirect measure of the conversion of pyruvate to lactic acid in cells and provides information on rates of glycolytic activity. 99 Rates of extracellular acidification were monitored as previously described [305]. Fibroblasts (50, 000 cells per well) were seeded on standard 96- well plates (Starstedt) pre-coated with 0.007 % collagen IV. Cells were maintained in buffer free DMEM supplemented with 1 mM pyruvate, 10 mM glucose (or 10 mM galactose where stated), 20 mM HEPES (pH 7.4) in a CO2 free incubator at 37 °C for 3 hours prior to analysis. Medium was replaced with 100 µL of fresh solution containing 1 µM of pH-Xtra probe. Compounds used in the manipulation of respiratory chain function (antimycin A (1 µM) and FCCP) were added at this point. Time resolved fluorescence (TR-F) was recorded on a Victor 2 plate reader thermostatically maintained at 37 °C utilizing a europium filter set (340 ± 35 nm excitation, 615 ± 8.5 nm emission). Duplicate TR-F intensity (F1, F2) readings were recorded in a gated (30 µs) measurement window at two delay (t 1=100, t2 =300 µs) intervals allowing for calculations of phosphorescence lifetime values τ = (t1– t2)/ln(F1/F2). For calculations of extracellular acidification τ was converted to pH = (1893.4 - τ )/227.54 and [H+] = (ln(- pH)) (Fig 2.7A, B, C). To determine the rate of change of probe phosphorescence, pH and [H+] lifetime values were acquired at 2 min intervals over 90 min. An initial temperature equilibration phase of the probe lasting approximately 15 minutes was excluded from the rate measurement. Extracellular acidification rates were calculated as the rate of change in [H+] per minute by taking the slope of the background subtracted linear signal and corrected for protein content. 2.29.2 Method validation for extracellular acidification measurements Fluorescent analysis of the pH sensitive probe in the presence of control fibroblasts determined a linear increase in the lifetime of probe phosphorescence consistent acidification of the extracellular medium (Fig 2.7A). 100 FCCP effects on extracellular acidification rates showed a dose dependent increase in the phosphorescent signal (Fig 2.7D) suggesting faster extracellular acidification as a result of increased rates of anaerobic glycolysis. The titration reached maximum rates, 50 % above basal acidification at 1 µM, comparable to rates observed at higher FCCP concentrations consistent with glycolysis functioning at maximum capacity despite mitochondrial toxicity. Inhibition of the mitochondrial respiratory chain by antimycin A increased the basal rate of extracellular acidification by 50 % (Fig 2.7E), mimicking the response to FCCP treatment and consistent with upregulated glycolytic flux in response to complex III inhibition. The probe phosphorescent lifetime response to FCCP and antimycin A suggested the methodology could be manipulated in a predictable way. 101 102 2.30 Cellular ATP content Fibroblasts were seeded at a density of 10, 000 cells per well on opaquewalled 96 well plates (Nunc) and total cellular ATP was quantified in luminescence units using CellTiter-Glo assay (Promega) according to manufacturer’s instructions. Briefly, following removal of culture medium and a PBS wash, 100 µL of CellTiter-Glo reagent containing renilla luciferase was added to each well. Cells were incubated with the reagent (10 min, RT) allowing for cell lysis to occur. Plates were incubated on a built in orbital shaker (Victor 2 plate reader) for 2 minutes following which end point luminescence readings were acquired (excitation 375 nm, emission 495 nm). Luminescence values were corrected for protein content. 2.31 Cellular ROS production: Array Scanner Oxidised forms of dihydroethidium dye (DHE) have high affinity for DNA. Upon oxidation, the fluorescent characteristics of reduced DHE (blue) change and the red signal accumulates in the nucleus. Fluorescent properties of DHE can be exploited to measure cellular ROS production. Fibroblasts seeded at a density of 5000 cells/well on 96 well plates (Nunc) were stained with 4’,6-diamidino-2-phenylindole (DAPI) in culture medium (1 µM, 37 °C, 20 min). Medium was replaced with HBSS containing 100 nM dihydroethidium (Molecular probes). Generation of reactive oxygen species was monitored by exciting samples with LED light source (543 nm (DHEox) and 361 nm (DAPI)) at 37 °C, 5% CO2 at 2 minute intervals for 1 hr (Array Scan VTI HCS Reader, Thermo Scientific, Pittsburg, USA) [307]. Emitted Red fluorescence was captured within the Dapi stained nuclear contour by a 20 X LD Plan-Neofluar objective through an automated filter wheel monitoring the increase in red fluorescence over the course of the measurement (Fig 2.8). Images were analyzed with the Cellomics Target Activation 103 Bio Application to assess the average DHE intensity per cell (n=100 per cell line) for each time point allowing for rates of DHE oxidation to be quantified in real time. 104 Chapter 3: Results Characterization of LRRK2 in cell models and brain tissue Western blot analysis of LRRK2 expression has previously been used to detect over-expressed LRRK2 in cultured cells as well as endogenous LRRK2 in cells of the immune system and tissues like kidneys, lungs and lymph nodes. However, previous reports using in house and commercial antibodies have struggled to detect low levels of endogenous LRRK2, suggesting better antibodies or more sensitive detection methods are required. Immunohistochemistry has been used to detect LRRK2 in mouse and human brain tissue while immunoprecipitation has been reported to detect endogenous brain LRRK2 protein and is often utilized for LRRK2 isolation from cells for use in enzymatic activity assays, however its use for quantitative comparison of LRRK2 expression has not been fully assessed. This chapter explored Western blotting, BN PAGE, immunohistochemistry and a combined immunoprecipitation and Western blot approach as methods to characterize LRRK2 protein expression in SHSY5Y cells over-expressing wild type and G2019S LRRK2-V5, control and G2019S PD patient fibroblasts, lymphoblasts and brain tissue, addressing the problem of endogenous LRRK2 detection. Furthermore, the protein detection methods were extended to characterization of LRRK2 cellular distribution and enzymatic activity measurements to determine the properties of the wild type LRRK2 protein as well as effects of the G2019S mutation on LRRK2 expression, localization and enzymatic activity. 3.1. Western Blot analysis of LRRK2 protein expression in cell models and brain tissue To determine the sensitivity of LRRK2 detection by Western blot in our endogenous and over-expressing LRRK2 cell model systems, four commercially available antibodies (NT2, 100-500, C terminal, 3154-1) raised to different regions of 105 the LRRK2 protein (Fig 3.1A) were screened for their ability to detect LRRK2. The polyclonal NT2 antibody was raised against a synthetic peptide encoding amino acids 100-200 of the human LRRK2 sequence. The 100-500 monoclonal antibody was raised against a synthetic peptide encoding amino acids 100-500 of the human LRRK2 sequence. The C terminal monoclonal antibody was raised against a synthetic peptide encoding amino acids 2079-2100 of human LRRK2 conjugated to a keyhole limpet hemocyanin (KLH) sequence. The 3514-1 monoclonal antibody was raised against a synthetic peptide encoding amino acids 970-2527 of the human LRRK2 sequence. All antibodies were raised in rabbits with target LRRK2 sequences 100 % conserved in mouse and rat LRRK2 homologues. Antibodies were evaluated for their ability to detect endogenous and over-expressed LRRK2 in human whole cell lysates of untransfected SHSY5Y cells, fibroblasts, lymphoblasts, human brain cortex and wild type LRRK2-V5 over-expressing SHSY5Y cells. Human cell lines c1 (fibroblasts) and c2 (lymphoblasts) described in Table 2.2 were used for the analysis. All four antibodies tested were able to detect a band at 280 kDa corresponding to the full length protein in the LRRK2-V5 over-expressing SHSY5Y cells as shown by the immunoreactive band in the (WT) lanes with the corresponding signal absent in the untransfected cell line (Fig 3.1B). NT2 and 3514-1 antibodies produced an immunoreactive band of appropriate molecular weight for full length LRRK2 in the lymphoblast cell line. None of the antibodies tested were able to clearly detect 280 kDa immunoreactivity in fibroblast lysates (Fig 3.1B). Weak 280 kDa immunoreactivity was also observed in control SHSY5Y cells (Fig 3.1B) and brain tissue for the 3514-1 but not 100-500 antibody (Fig 3.1C). Additional immunoreactive species of lower molecular weight were detected by NT2, 100-500 and 3514-1 in the over-expressing cell line (Fig 3.1B). Some bands, 20-75 kDa, (i) were of equal intensity to those in untransfected control 106 cells while others, 100-150 kDa (ii) were present in the over-expressing cells only suggesting species (ii) corresponded to the over-expressed LRRK2-V5 protein. Lower molecular weight bands (30-100 kDa) were also detected in lymphoblast, fibroblast, control SHSY5Y cells and human brain lysates (NT2, 100-500, 3514-1 (iii)) comigrating with those in the LRRK2-V5 over-expressing SHSY5Y cells potentially representing endogenous forms of LRRK2. Lower molecular weight bands (30-150 kDa) absent in SHSY5Y cells were also detected in lymphoblast, fibroblast and human brain lysates by the NT2 and 100-500 antibodies (iv), consistent with cell and tissue specific antibody immunoreactivity (Fig 3.1B,C). As we did not have the appropriate LRRK2 knockdown or knockout controls for fibroblast and lymphoblast cell lines we were unable to rule out non-specific binding of antibodies in these cell lines. The lower molecular weight band patterns were unique to each antibody potentially representing the position of antibody epitopes and the corresponding LRRK2 fragment. In addition, variability was observed between blots as shown by differences between the band pattern for wild type LRRK2 over-expressing SHSY5Y cells, comparing 100-500 and 3514-1 immunoreactivities in (Fig. 3B and C), complicating the identification of the true LRRK2 species. Our data suggests Western blot detection of the LRRK2 protein can identify over-expressed full length LRRK2, however, most antibodies identify additional immunoreactive bands of lower molecular weight questioning their specificity. Lower molecular weight immunoreactive species were also observed in cells and tissues expressing endogenous LRRK2 while immunoreactivity for the full length endogenous protein was weak. 107 3.2 Characterization of LRRK2-V5 over-expressing SHSY5Y cells 3.2.1 Western blot analysis of LRRK2 protein in SHSY5Y cells SHSY5Y cells stably expressing LRRK2-V5 were generated by J.M Cooper and D. Chau, and positive clones expressing the wild type or G2019S LRRK2-V5 were 108 obtained. Western blot analysis of untransfected, wild type or G2019S LRRK2-V5 over-expressing SHSY5Y whole cell lysates revealed a single 280 kDa band immunoreactive for V5 in SHSY5Y cells transfected with wild type and G2019S LRRK2-V5 absent in control SHSY5Y cell extracts (Fig 3.2A). The band intensity in wild type extracts was 2 fold higher relative to the G2019S clone when corrected for protein loading by GAPDH immunoreactivity. To ensure consistent expression of the LRRK2 constructs, protein levels were assessed with increasing passage number. Positive V5 immunoreactivity was detected by Western blot for both wild type (Fig 3.2B) and G2019S (Fig 3.2C) clones for passages 7-22. LRRK2 expression was reduced by 40 % in G2019S LRRK2-V5 over-expressing cells at passages 21 and 22 when corrected for protein loading by GAPDH immunoreactivity suggesting the stability of over-expressed G2019S LRRK2 protein was affected at these passages. All functional analyses were carried out for passages 8-20 to compare the effects of overexpressed wild type and G2019S LRRK2. 3.2.2 LRRK2 mutation screen in SHSY5Y cells To determine whether the LRRK2 pathogenic G2019S mutation was expressed in wild type and G2019S LRRK2-V5 over-expressing cell lines, mRNA was extracted from each clone, reverse transcribed and the corresponding coding DNA analysed for the presence of the G2019S mutation in exon 41 of the LRRK2 gene. Successful amplification of the 700 base pair fragment from exon 41 of LRRK2 cDNA in SHSY5Y cells was confirmed by the presence of a single DNA band co-migrating with a fragment amplified from the LRRK2 plasmid absent in the water blank reaction (Appendix Fig A4). DNA sequencing of wild type PCR products confirmed 100 % base pair complementarity to the human LRRK2 gene registered on the UniProt database (Fig 3.2D). cDNA from SHSY5Y cells stably over-expressing mutant LRRK2 protein was identified to have the adenine nucleotide at base pair position 109 6055 with endogenous wild type LRRK2 (guanine at position 6055) not resolved by sequencing potentially due to the differential level of expression (Fig 3.2E). The guanine to adenine substitution results in a codon encoding the serine (S) amino acid, confirming expression mutant LRRK2 protein in SHSY5Y cells. 110 3.2.3 Western blot analysis of LRRK2 sub-cellular distribution in SHSY5Y cells Having demonstrated wild type and G2019S LRRK2-V5 over-expression in SHSY5Y cells, LRRK2 cellular distribution was evaluated in these cells lines. SHSY5Y cells over-expressing LRRK2 were ruptured by nitrogen cavitation and fractionated by differential centrifugation. Western blot analysis of wild type LRRK2-V5 expressing cell fractions identified V5 immunoreactivity in all the isolated SHSY5Y compartments with highest abundance (47 %) in the 250,000 g and cytosolic fractions (Fig 3.3A). Quantification of LRRK2 immunoreactive bands by densitometry approximated 5 % (n=1) of cellular LRRK2 in each of the 10-47,000 g fractions. Compartmental characterization identified lysosomal abundance in the 47,000 g fraction as determined by the double immunoreactive GCase band of approximately 60 kDa representing the glycosylated and non glycosylated form of the lysosomal enzyme. Mitochondrial enrichment was found in the 47,000 g (60 %), 27,000 g (30 %) and 1,000 g (10 %) fractions, as determined by immunoreactivity to the Core Complex III mitochondrial marker. Immunoreactivity for the endoplasmic reticulum marker, calreticulin, was detected in 27,000 g (55 %), 12,000 g (40 %) and 47,000 g (5 %) fractions. Synaptophysin immunoreactivity, representing synaptic vesicle abundance were identified in 250,000 g (35 %) and cytosolic (55 %) compartments with the remaining 10 % synaptophysin immunoreactivity distributed equally between fractions 1-45,000 g. These data shows that wild type, over-expressed LRRK2-V5 can be found abundant in the cytosolic and vesicle enriched cellular compartments, with lesser abundance in fractions containing ER, lysosomal and mitochondrial markers. To determine whether G2019S expression is associated with altered LRRK2 cellular distribution in G2019S LRRK2-V5 over-expressing SHSY5Y cells, differential centrifugation and Western blot analysis was performed on the mutant cell line. 111 G2019S LRRK2 compartmental distribution in SHSY5Y cells reflected that of the wild type protein with highest LRRK2 abundance identified in 250,000 g (60 %) and cytosolic (30 %) fractions with 6 % (n=1) of the total V5 immunoreactivity distributed equally between the fractions 10-47,000 g (Fig 3.3B). Unlike wild type overexpressed LRRK2, 3 % of G2019S LRRK2 was identified in the 1,000 g fraction, corresponding to unbroken cells and nuclear components, potentially representing incomplete cellular disruption. Compartmental characterization of G2019S LRRK2 expressing fractions identified lysosomal abundance (95 %) in the 12,000 g fraction as determined by the double immunoreactive GCase band of approximately 60 kDa. Mitochondrial enrichment was found in the 47,000 g (30 %), 27,000 g (60 %) and 1,000 g (10 %) fractions, as determined by immunoreactivity to the Core Complex III mitochondrial marker. Calreticulin immunoreactivity, representing the endoplasmic reticulum was abundant in 27,000 g (55 %) and 12,000 g (35 %) fractions, while synaptophysin immunoreactivity was identified in 250,000 g (60 %) and cytosolic (35 %) fractions as well as 5 % of the total synaptophysin immunoreactivity distributed equally between fractions 1-47,000 g. Lysosomal and mitochondrial markers in fractionated G2019S LRRK2-V5 over-expressing SHSY5Y cells were detected in fractions isolated at lower centrifugation speeds (12,000 g) relative to the wild type LRRK2-V5 over-expressing SHSY5Y cells (47,000 g) potentially reflecting the smaller size of mitochondrial and lysosomal organelles. ER and synaptic markers were identified in fractions isolated by comparable centrifugation speeds (12,000 and 27,000 g) and (250,000 g respectively) for both wild type and G2019S LRRK2-V5 over-expressing cells. Our data shows that over-expressed LRRK2 V5 can be found abundant in the cytosolic and vesicle enriched cellular compartments, with smaller amounts present in fractions containing ER, lysosomal and mitochondrial markers. In addition G2019S 112 LRRK2-V5 cellular localization mirrors the cellular distribution of the wild type LRRK2-V5 protein in SHSY5Y cells. However, the difference in centrifugation speeds required to isolate the majority of the mitochondrial and lysosomal pools may reflect a difference in organelle boyant density between the wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells. 3.2.4 Immunocytochemistry of LRRK2 sub-cellular distribution in SHSY5Y cells In addition to Western blot analysis, wild type LRRK2-V5 over-expression and cellular distribution in SHSY5Y cells was characterized by immunocytochemistry. In an attempt to visualise LRRK2 over-expression by imunocytochemistry four permeabilization and fixation conditions were tested; methanol and acetone, paraformaldehyde and Triton X100, paraformaldehyde and methanol, paraformaldehyde and sodium citrate. Immunocytochemistry utilizing methanol and 113 acetone conditions confirmed LRRK2-V5 over-expression in wild type expressing SHSY5Y cells as determined by V5 immunoreactivity absent in untransfected cells (Fig 3.4A). Immunostaining conditions utilizing paraformaldehyde in combination with Triton X100 and methanol permeabilization or boiling in sodium citrate produced V5 immunoreactive signals in control SHSY5Y cells of comparable intensities to that observed in wild type LRRK2 over-expressing SHSY5Y cells. V5 positive immunoreactivity in paraformaldehyde fixed control SHSY5Y cells was likely due to non specific binding of the antibody under these conditions. Methanol acetone processing of SHSY5Y cells identified V5 positive immunoreactivity in wild type LRRK2 over-expressing SHSY5Y cells as spread diffusely throughout the cell body with some immunoreactive punctate structures detected (Fig 3.4B). Co-localization with compartmental markers was not assessed due to this diffuse nature of the LRRK2 signal. G2019S LRRK2-V5 over-expression in SHSY5Y cells could not reliably be identified by immunocytochemistry this may reflect the 2 fold lower expression of the G2019S LRRK2-V5 protein relative to wild type LRRK2-V5 over-expressing cells, questioning the sensitivity of this detection method. 114 3.3 Characterization of endogenous LRRK2 in lymphoblasts 3.3.1 Western blot analysis of LRRK2 protein in lymphoblasts Western blot analysis of LRRK2 protein detected endogenous expression in lymphoblasts with the NT2 antibody (Fig 3.1B) consistent with high LRRK2 abundance as previously reported for this cell type [244]. Lymphoblast cells are 115 derived from peripheral blood mononuclear cells (PMBCs) isolated from whole blood. To determine whether LRRK2 expression in control lymphoblasts reflects LRRK2 protein abundance in PMBCs, LRRK2 expression was compared by Western blot analysis. It was not possible to obtain PMBCs from any of the lymphoblast donors and hence PMBCs from 5 healthy control subjects of different ages (23-62 years) were screened for LRRK2 expression. A lymphoblast cell line (control subject, 56 years) was used for comparison. Positive NT2 immunoreactivity in the 280 kDa region confirmed LRRK2 expression in PMBCs with LRRK2 levels comparable between transformed and primary cells relative to GAPDH immunoreactivity (Fig 3.5A). LRRK2 PMBC expression was similar between the five control donors suggesting LRRK2 lymphoblast expression reflects levels in untransformed cells. PMBCs purified by density gradient isolation consist of a combination of T and B lymphocytes of differential abundance [308] while lymphoblasts are derived primarily from Epstein-Barr virus transformed B cells. To determine which lymphocyte subpopulations express LRRK2 protein PMBC cells (control subject, 49 years) were used for affinity purification and characterization of T (CD4, CD8) and B (CD19) lymphocyte cell subtypes. PMBCs were fractionated by positive selection using magnetically tagged antibodies specific for CD4, CD8 and CD19 lymphocytes and characterised for LRRK2 protein expression by Western blotting. Western Blot analysis of purified T and B cell extracts produced a strong NT2 immunoreactive band in the CD19 cells, 10 fold lower intensity immunoreactive signal in CD8 relative to GAPDH when corrected for protein loading but no clear LRRK2 immunoreactivity in CD4 cells (Fig 3.5B) consistent with LRRK2 protein abundance in B cells and substantially lower expression in T cells. Having shown that LRRK2 protein expression in transformed lymphoblasts reflects the protein levels of CD19 positive B cells from which the lymphoblasts were 116 derived, we set out to determine whether LRRK2 protein abundance is altered with G2019S expression. LRRK2 levels were compared between control and G2019S expressing lymphoblasts. Western blot analysis of control and G2019S lymphoblast cell lines was carried out by Susannah Horan (Clinical Neurosciences, UCL). As the NT2 antibody was not available at the time, the previously validated rabbit polyclonal NB300-268 was used (refer to diagram in 3.1A for epitope details). Western blot analysis of NB300-268 immunoreactivity indicated variability in LRRK2 levels between the cell lines within the control and G2019S lymphoblast groups (Fig 3.5C). When immunoreactivities were quantified, variability of 10 fold intensity difference was noted between highest and lowest LRRK2 expressing lymphoblast lines. The variability of LRRK2 expression in lymphoblasts complicated the assessment of G2019S expression effects on LRRK2 abundance. 117 3.3.2 Western blot analysis of LRRK2 sub-cellular distribution in lymphoblasts Western blot analysis of LRRK2 protein expression with the NT2 antibody determined that LRRK2 levels were approximately 3 fold lower in a high LRRK2 expressing lymphoblast cell line (c2) relative to wild type LRRK2-V5 over-expression 118 in SHSY5Y cells (Fig 3.1B). To compare the cellular distribution of endogenous LRRK2 in lymphoblasts to the data obtained for wild type and G2019S LRRK2-V5 SHSY5Y cells, differential centrifugation and Western blot analysis were carried out on the c2 lymphoblast cell line (Fig 3.6). 280 kDa NT2 immunoreactivity was abundant in the cytosolic (60 %) and 250,000 g (35 %) compartments with 5% of the total NT2 immunoreactivity distributed equally between 1-47,000 g compartments (Fig 3.6), comparable to the V5 immunoreactive pattern observed for wild type and G2019S LRRK2-V5 expressing SHSY5Y cells (Fig 3.3A, B). Higher molecular weight NT2 immunoreactive species (approximately 290 and 300 kDa) were detected in 12,000 g and 250,000 g fractions, absent in the 1,000, 27,000, 47,000 g and cytosolic fractions. Characterization of cellular compartmental distribution identified lysosomal abundance in the 12,000 g (60 %), 27,000 g (30 %) and 1,000 g (10 %) fractions as determined by GCase immunoreactivity. A single immunoreactive band was detected by the GCase antibody in these fractions suggesting lymphoblast derived GCase exists in a single 60 kDa form, potentially corresponding to the unglycosylated enzyme. Mitochondrial abundance was identified in 1,000 g (45 %) and 12,000 g (50 %) fractions with additional bands in 27,000 g and 47,000 g (5 %) compartments, as determined by Core CIII immunoreactivity. Calreticulin (ER) markers were present in the 47,000 g (40 %) fractions where a double band was detected potentially representing a post translationally modified form of calreticulin [309]. A single band (*) immunoreactive for the calreticulin antibody was identified in the 27,000 g fraction accounting for 60 % of the total calreticulin immunoreactivity, potentially representing the unglycosylated form of the protein. Similar to the results obtained in the LRRK2-V5 over-expressing SHSY5Y system, endogenous LRRK2 in lymphoblasts can be found abundant in the cytosolic 119 compartment, as well as minor levels in fractions containing ER, lysosomal and mitochondrial markers. These results suggest that LRRK2 cellular distribution was not affected by LRRK2 over-expression. 3.4 LRRK2 immunoprecipitation 3.4.1 Characterization of LRRK2 immunoprecipitation for quantitative comparison We addressed the problem of endogenous LRRK2 detection in fibroblasts, SHSY5Y cells and brain tissue. The immunoprecipitation protocol, based on methodology described by Alessi et al, was extended to the detection of endogenous LRRK2 protein [215]. Additional characterization of methodology was carried out to validate the use of this method for quantitative comparison. LRRK2 immunoprecipitated from control and LRRK2 knockout mouse brain cortex or SHSY5Y cells over expressing wild type LRRK2-V5 was assessed by Western blot analysis. Successful immunoprecipitation of full length LRRK2 from mouse 120 cortex was confirmed by the presence of a 280 kDa band undetected in the corresponding LRRK2 knockout brain immunoprecipitates and the no antibody control lane as determined by Western blot analysis with 3514-1 antibody (Fig 3.7A). LRRK2 immunoreactive bands co-migrated with full length wild type LRRK2-V5 in whole cell extracts and immunoprecipitates from SHSY5Y over-expressing cells. A 250 kDa species (*) was also present in immunoprecipitates of wild type LRRK2-V5 over-expressing SHSY5Y cells but absent in mouse brain and no antibody control immunoprecipitates suggesting the presence of an additional LRRK2 immunoreactive species in this cell type (Fig 3.7A). The solubility of LRRK2 in buffer used for immunoprecipitation was assessed in SHSY5Y cells over-expressing LRRK2 by Western blot analysis of detergent soluble and insoluble fractions. Non ionic detergent (1 % Triton X100 and NP-40) containing buffers retained little of the cellular LRRK2 in the insoluble pellet with >95 % of LRRK2 found in the soluble fraction as determined by V5 immunoreactivity (Fig 3.7B). Substitution of non ionic with ionic (2 % CHAPS, 1 % Cholate) or zwitterionic (1 % DDM) detergents recovered comparable levels (>95 %) of LRRK2 in the soluble faction. An additional band immunoreactive for the V5 antibody was detected in all soluble fractions (*). The relative size of the band (250 kDa) was comparable to the species identified in the LRRK2-V5 over-expressing SHSY5Y cells by immunoprecipitation (Fig 3.7A). Our data suggests >95 % of the cellular LRRK2 pool can be recovered in IP buffer supplemented with ionic, non ionic and zwitter ionic detergents in LRRK2 over expressing SHSY5Y cells. The linearity of the IP signal was assessed by immunoprecipitation of endogenous LRRK2 from increasing concentrations (100-600 µg) of fibroblast lysate extracted in 1 % Triton X100 containing IP buffer. 3514-1 immunoreactivity was detected in all fibroblast immunoprecipitates (Fig 3.7C) consistent with the presence 121 of LRRK2 protein in fibroblasts. The lower molecular weight species detected by IP and Western blot in wild type LRRK2-V5 over-expressing SHSY5Y cells (Fig 3.7A) was absent in the fibroblast IP. The 3514-1 band intensity was determined to be in linear proportion to the concentration of fibroblast input lysate as shown by plotting the band intensity relative to µg of lysate (Fig 3.7D). Our data demonstrated the specificity of the LRRK2 immunoprecipitation procedure as determined by analysis of wild type and LRRK2 knockout brain tissue, efficient LRRK2 solubilization (>95 %) using the IP buffer conditions in LRRK2 over expressing SHSY5Y cells and signal linearity based on the IP LRRK2 recovery from fibroblast input lysates confirming the suitability of IP for quantitative comparison of LRRK2 protein expression. 122 3.4.2 LRRK2 kinase activity in lymphoblasts and SHSY5Y cells Immunoprecipitation of LRRK2 from lymphoblasts and LRRK2 overexpressing HEK293 cells has previously been shown to produce a LRRK2 protein with sufficient purity for enzymatic activity measurements in vitro [202, 215]. LRRK2 was immunoprecipitated from 1 mg of 1 % Triton X100 lysates of control lymphoblasts, 123 wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells and assessed for kinase activity. Six lines of control lymphoblasts were used to determine the potential correlation between LRRK2 expression and kinase activity. Successful immunoprecipitation of LRRK2 was determined by the single 280 kDa 3514-1 immunoreactive band identified for all samples. The intensity of 3514-1 immunoreactivity was found to be variable for the lymphoblastoid cells (Fig 3.8A), consistent with the variable LRRK2 protein expression identified by Western blot analysis of whole cell lysates (Fig 3.5C). The 3514-1 band intensity was stronger for immunoprecipitates of wild type and G2019S LRRK2-V5 SHSY5Y cells consistent with greater LRRK2 protein expression in this cell type. Following immunoprecipitation, LRRK2 isolated from control lymphoblasts and over-expressing SHSY5Y cells was assessed for kinase activity by rate measuments of radiolabelled 32P incorporation into the LRRKtide substrate. Kinase activity measurements were carried out in Dundee with the help of Fransisco InestaVaquera. GST-LRRK2 isolated from HEK293 cells was used as a positive control for the reaction [202]. LRRKtide phosphorylation by the purified GST-LRRK2 was identified by detection of incorporated radioactivity as counts per minute abolished in the presence of a LRRK2 kinase inhibitor LRRK2-IN1, suggesting GST-LRRK2 was active and confirming LRRKtide as a GST-LRRK2 substrate (Fig 3.8B). Calculating specific activities from cpm values of the LRRK2 enzyme immunoprecipitated from lymphoblasts determined substantial variability between the control lymphoblast cell lines, with values ranging from 0.6-1 µmol/min/mg protein (Fig 3.8C). Due to variability in LRRK2 specific activity for control lymphoblasts, LRRK2 kinase activity was not assessed in G2019S expressing lymphoblast cells. 124 The specific activity of wild type and G2019S LRRK2-V5 immunoprecipitated from SHSY5Y over-expressing cells was 4-8 fold lower relative to the lymphoblast cells (Fig 3.8C) suggesting the over-expressed LRRK2 kinase is potentially less active in the SHSY5Y cells. The specific activity of immunoprecipitated G2019S LRRK2-V5 was approximately 2 fold greater relative to the wild type LRRK2 consistent with higher kinase activity associated with G2019S LRRK2-V5 (Fig 3.8C). Our data show that the active LRRK2 enzyme can be effectively immunoprecipitated from LRRK2-V5 over-expressing SHSY5Y cells and lymphoblasts. In addition, we have demonstrated that the kinase activity of LRRK2 is greater for LRRK2 immunoprecipitated from lymphoblasts relative to LRRK2-V5 over-expressed in SHSY5Y cells while the G2019S LRRK2 mutation correlates with the characteristic increase in the kinase activity of the mutant protein in our SHSY5Y LRRK2-V5 overexpressing system. 125 3.5 Characterization of fibroblast cell lines 3.5.1 Immunoprecipitation of LRRK2 protein from fibroblasts Six control and eight G2019S patient fibroblast cell lines were generated with details about the age of donor and age at collection described in Table 2.2. Having demonstrated endogenous LRRK2 protein expression in fibroblasts (Fig 3.7C) and evaluated the IP procedure for use in quantitative comparison of LRRK2 protein expression, we set out to determine whether the G2019S mutation correlated with 126 altered LRRK2 protein abundance in patient derived fibroblasts. Endogenous LRRK2 was immunoprecipitated from equivalent amounts of input Triton X100 lysates (1 mg) of 3 control and 3 G2019S patient fibroblast cell lines. The relative band intensity of the LRRK2 immunoreactive signal was comparable for wild type and G2019S fibroblast cell lines when corrected for IP input by quantifying the GAPDH immunoreactivity (Fig 3.9A). Unlike control and G2019S lymphoblasts (Fig 3.5C), LRRK2 protein expression in control and G2019S patient fibroblasts was less variable, despite differences in the age at sample collection of the donors. 3.5.2 LRRK2 mutation screen in control and G2019S fibroblasts To confirm expression of the pathogenic G2019S mutation, mRNA was extracted from each fibroblast cell line (6 control, 8 G2019S), reverse transcribed and the corresponding coding DNA analysed for the presence of the G2019S mutation on exon 41 of the LRRK2 gene. Successful amplification of the 700 base pair fragment from exon 41 of LRRK2 cDNA in fibroblasts confirmed by the presence of a single DNA band co-migrating with a fragment amplified from the LRRK2 plasmid absent in the water blank reaction (Appendix Fig A3). DNA sequencing of wild type PCR products confirmed 100 % base pair complementarity to the human LRRK2 gene registered on the UniProt database for all control cell lines (Fig 3.9B). All clinically diagnosed PD G2019S cases were found to contain the heterozygous mutant allele as determined by 50 % adenine abundance at base pair position 6055 of the LRRK2 gene (Fig 3.9C). 3.5.3 LRRK2 mRNA expression in control and G2019S fibroblasts To determine whether LRRK2 expression was altered in cells expressing the G2019S mutation, quantitative real time PCR analysis was carried out on 6 control and 8 G2019S fibroblast cell lines. Amplification of LRRK2 cDNA produced a PCR product in cycles 22-24 (equivalent GAPDH in cycles 14-16) of the RT PCR reaction 127 indicative of low levels of endogenous LRRK2 expression (Fig 3.9D). Quantification of LRRK2 cycle threshold relative to the cDNA loading control marker GAPDH for control versus G2019S fibroblast lines revealed a similar level of LRRK2 mRNA abundance (Fig 3.9E), consistent with equivalent LRRK2 mRNA expression in wild type and G2019S LRRK2 fibroblasts. Our data is consistent with comparable LRRK2 mRNA and protein abundance in control and G2019S patient fibroblasts. 128 3.6 Analysis of LRRK2 knockdown in SHSY5Y cells In addition to the quantitative comparison of LRRK2 protein expression between control and G2019S patient fibroblasts, LRRK2 protein detection by the combined immunoprecipitation and Western blot approach was extended to the 129 characterization of the LRRK2 siRNA knockdown model in SHSY5Y cells. LRRK2 siRNA was introduced into control SHSY5Y cells by the method of transient transfection. For prolonged LRRK2 gene silencing, cells were re-transfected with siRNA at day 3. SHSY5Y cells were analysed after 3 and 6 days of silencing for LRRK2 mRNA expression by quantitative real time PCR and LRRK2 protein abundance by immunoprecipitation and Western blot analysis. Following siRNA treatment, mRNA was extracted from SHSY5Y cells, reverse transcribed to cDNA and LRRK2 expression quantified by real time PCR. Data was expressed as % LRRK2 expression relative to untreated cells. Quantification of LRRK2 amplification relative to GAPDH CT values identified a 70 % reduction in LRRK2 mRNA expression in SHSY5Y cells treated with siRNA for 3 and 6 days when compared to untreated cells (Fig 3.10A) consistent with a reduction in LRRK2 gene expression. LRRK2 was immunoprecipitated from 1mg of 1 % Triton X100 lysates of control SHSY5Y cells and cells treated with LRRK2 siRNA. Western blot analysis of immunoprecipitates revealed a 65 % reduction in 3514-1 280 kDa immunoreactivity 3 days post transfection relative to untreated cells when corrected for GAPDH immunoreactivity representing IP input (Fig 3.10B, C). The observed 65 % reduction in 3514-1 band intensity was consistent with reduced LRRK2 protein expression in LRRK2 siRNA treated SHSY5Y cells. Extending the LRRK2 siRNA treatment to 6 days reduced the 3514-1 immunoreactive LRRK2 band to 85 % of the untreated SHSY5Y cells suggesting SHSY5Y LRRK2 protein levels were less abundant following prolonged incubations with LRRK2 siRNA. An additional 3514-1 immunoreactive band at 250 kDa (*) was identified for endogenous LRRK2 immunoprecipitated from SHSY5Y cells with the approximate molecular weight comparable to the species observed in the IP of the wild type LRRK2-V5 over-expressing SHSY5Y cells (Fig 3.7A). 130 The intensity of this band was reduced following LRRK2 siRNA treatment suggesting the band represents LRRK2 protein. Analysis of the LRRK2 siRNA SHSY5Y knockdown model not only revealed an efficient reduction of endogenous LRRK2 protein level but also further validated the specificity of the IP procedure as the intensity of the 280 kDa LRRK2 immunoreactive bands was sensitive to LRRK2 siRNA. 131 3.7 Immunoprecipitation of endogenous LRRK2 protein from brain Using the combined immunoprecipitation and Western blot protocol we analysed LRRK2 expression in two control human brains of 24 hour post mortem delay. LRRK2 was immunoprecipitated from equivalent amounts of Triton X100 lysates (1 mg) of human cerebellum, caudate nucleus, medial putamen and A10 region of the cortex. Western blot analysis revealed a weak 3514-1 280 kDa immunoreactive band in cerebellum 2 immunoprecipitate (←), co-migrating with the 280 kDa immunoreactive species detected in the fibroblast IP. This band was only seen following prolonged blot exposure, potentially representing low abundant full length LRRK2 protein. LRRK2 immunoreactivity was not readily detected in human brain caudate nucleus, medial putamen or cortical immunoprecipitates using this approach (Fig 3.11A). LRRK2 was immunoprecipitated from equivalent amounts of Triton X100 lysates (1 mg) of control human cortex and amygdala brain regions with post mortem delay times of 3h30-8h30 as well as an idiopathic PD brain of 1h30 and Alzheimer’s disease brain of 1h45 post mortem delay times respectively. We were unable to obtain control brains of 1h30 and 1h45 post mortem delays. Western blot analysis of LRRK2 immunoprecipitates identified weak 3514-1 280 kDa immunoreactivity for the 1h30 cortical extract of an idiopathic PD case (←). No clear bands were detected in the other cortical samples or amygdala immunoprecipitates suggesting low LRRK2 human brain abundance or a problem with the immunoprecipitation procedure (Fig 3.11B). 132 As LRRK2 could be immunoprecipitated from fresh mouse brain cortical extracts (Fig 3.7A), we assessed LRRK2 brain distribution in different regions of the mouse brain. Immunoprecipitation and Western blot analysis of LRRK2 from brain stem, olfactory lobe, mid brain, striatum, cortex and cerebellum regions of the mouse brains revealed a 3514-1 immunoreactive 280 kDa band in all brain regions analysed (Fig. 3.12A). This 280 kDa band co-migrated with the 3514-1 immunoreactivity of LRRK2 fibroblast immunoprecipitates and was absent in the IP where the LRRK2 antibody was omitted. LRRK2 immunoprecipitation from mouse brain produced strongest immunoreactivity in the striatum, two fold greater than that of the cerebellum and cortex, 5 fold higher when compared to the band intensity in the brain stem and 10 fold greater than that of the 280 kDa band in the midbrain and olfactory lobe (Fig 3.12A). Comparable 3514-1immunoreactive patterns were detected in the equivalent regions of the rat brain (Fig 3.12B) with 133 striatal band intensity 10 fold greater when compared to that detected in the midbrain. 3514-1 immunoreactivity was 2 fold greater in the cortex when compared to the striatum with a higher molecular weight band (←) consistently detected in immunoprecipitates from this region but absent in the equivalent mouse brain areas. 280 kDa immunoreactivities could not be detected in the rat olfactory lobe or brain stem at these exposure times potentially due to lower LRRK2 protein abundance. Extending these analyses to non human primates, LRRK2 was immunoprecipitated from equivalent regions of MPTP treated marmoset brains. We were unable to obtain brains from untreated control animals. Western blot analysis of LRRK2 immunoprecipitates produced a single 280 kDa 3514-1 immunoreactive band in the striatum and a band of 3 fold greater intensity in the cortex (Fig 3.12C). Weaker (10 fold) 3514-1 immunoreactivity was detected in the marmoset brain stem, olfactory lobe and midbrain, reflecting the LRRK2 protein distribution pattern identified in the equivalent brain regions of the rodent brains. Although the pattern of LRRK2 protein distribution is comparable between the mouse, rat and marmoset brains, we cannot rule out the potential effects of MPTP on LRRK2 expression. Our data shows that endogenous LRRK2 can be readily immunoprecipitated from rodent and non human primate brain tissue in contrast to the human brain tissue raising the question whether the contents of the human brain extracts could potentially be interfering with the IP procedure. To test this fibroblast IP lysates were spiked with equal concentrations of mouse or human brain cortex IP lysates and subject to LRRK2 immunoprecipitation. LRRK2 readily immunoprecipitated from fibroblast and mouse cortical extracts as determined by 3514-1 280 kDa immunoreactivity (Fig 3.12D). The relative band intensity in fibroblast immunoprecipitates spiked with a mouse brain extract was equivalent to the sum of the band intensities detected in the individual fibroblast and mouse brain samples. 134 No clear immunoreactivity was detected for the LRRK2 immunoprecipitates from a human cortical extract while a 3 fold reduction in signal intensity was observed for the fibroblast immunoprecipitate spiked with the human lysate relative to the band intensity detected in the pure fibroblast immunoprecipitate (Fig 3.12D) suggesting the composition of the human brain lysate may be affecting immunoprecipitation. 135 3.8 Comparison of LRRK2 expression in cell models and brain tissue Due to potential interference in the human LRRK2 IP, mouse brains were used for comparative analysis of LRRK2 protein expression with fibroblasts, lymphoblasts, control and LRRK2-V5 over-expressing SHSY5Y cells. LRRK2 immunoprecipitated from equal concentrations (1 mg) of input lysates confirmed highest endogenous LRRK2 abundance in lymphoblasts. Levels were 10 fold lower in fibroblasts and 50 fold lower in control SHSY5Y cells (Fig 3.13) based on quantification of 3514-1 280 kDa immunoreactivity. In mouse brains, 3514-1 immunoreactivity was greatest in the striatum and cortex, consistent with previous findings (Fig 3.12A) comparable to the relative band intensities detected in SHSY5Y cells and 7 fold lower than those in fibroblast immunoprecipitates. Wild type LRRK2V5 over-expression in SHSY5Y cells was quantified as 3.6 fold higher than in control lymphoblast and approximately 25 fold greater than in fibroblast immunoprecipitates as determined by 3514-1 280 kDa immunoreactivity (Fig 3.13). Additional lower molecular bands of 250 kDa were identified in over-expressing cells and lymphoblasts (←) while longer exposures revealed equivalent immunoreactive bands in fibroblast IPs (←) resembling the bands previously identified by Western blotting in LRRK2-V5 over-expressing SHSY5Y cells (Fig 3.7B). These species were also identified in control SHSY5Y cells and reduced upon LRRK2 siRNA treatment (Fig 3.10B, C) suggesting the 250 kDa species is found in both the LRRK2 over-expressing cell lines and systems expressing endogenous LRRK2. Our results show that control SHSY5Y cells and fibroblasts express LRRK2 protein at levels similar to those detected in the brain while the LRRK2 overexpressing SHSY5Y model has comparable LRRK2 expression relative to endogenous LRRK2 abundance in lymphoblasts. 136 3.9 Analysis of LRRK2 protein expression by blue native PAGE electrophoresis As an alternative analysis of LRRK2 in the human brain samples blue native PAGE combined with Western blotting was carried out with the 3514-1 LRRK2 antibody. To determine whether preserving the native structure of LRRK2 would improve the 3514-1 antibody specificity, blue native PAGE electrophoresis was carried out on soluble cell extracts of SHSY5Y cells over-expressing wild type LRRK2V5. In parallel, blots were probed with the V5 antibody to compare the specificities of the two antibodies. Cells were solubilised in extraction buffer in the absence and presence of detergent (1 % DDM or Triton X100) to determine whether inclusion of detergent would improve LRRK2 recovery. A single V5 280 kDa immunoreactive band (i) was detected by Western blot in soluble extracts of LRRK2-V5 overexpressing SHSY5Y cells. A 3514-1 280 kDa immunoreactive band of comparable size and signal intensity was also identified (Fig 3.14A) suggesting the two antibodies recognised the same species. Longer exposures revealed 900 kDa immunoreactive 137 species for both the V5 and 3514-1 antibodies (ii), potentially corresponding to a less abundant higher molecular weight wild type LRRK2-V5. Solubilisation of wild type LRRK2-V5 over-expressing SHSY5Y cells in buffer containing 1 % DDM, revealed a poorly resolved V5 immunoreactive smear by Western blot with 280 kDa (i) and 900 kDa (ii) bands visible at short exposures. Solubilisation of wild type LRRK2-V5 over-expressing SHSY5Y cells in buffer containing 1 % Triton X100 also produced some V5 immunoreactive smearing and a 280 kDa band at low exposure (i) with a weak 900 kDa band resolving at longer exposures (ii). Inclusion of DDM and Triton detergent appeared to promote recovery of monomeric (280 kDa) LRRK2 as the relative band intensity of the 280 kDa species was greater for both Triton and DDM containing samples relative to detergent free extracts. In addition, DDM produced a V5 immunoreactive 900 kDa species at short blot exposures (bands absent in detergent free extracts at these exposure times) with the detergent possibly promoting the recovery of the higher molecular weight LRRK2 species. However due to the sample smearing encountered at longer blot exposures we concluded that the conditions obtaining optimal signal to noise ratios in LRRK2-V5 over-expressing SHSY5Y cells were those which omitted the use of detergent. BN PAGE and Western blot analysis of detergent free, soluble extracts of human and wild type but not LRRK2 knockout mouse cerebellum revealed a weak 280 kDa 3514-1 immunoreactive band (←) co-migrating with wild type LRRK2-V5 in SHSY5Y over-expressing cells (Fig 3.14B). Higher molecular weight (900 kDa) 3514-1 immunoreactive species were not detected on this blot, suggesting variability related to sample processing or the Western blot procedure, as has previously been described for the 3514-1 antibody (Fig 3.1B,C). 138 BN PAGE electrophoresis of cerebellum, cortex, caudate nucleus and medial putamen regions of the human brain revealed a 3514-1 immunoreactive band of approximately 280 kDa (i) in all regions analysed, co-migrating with the 280 kDa band in wild type mouse cerebellum extracts (Fig 3.15, long exposure blots). At long 139 exposures, a 280 kDa 3514-1 immunoreactive band was also detected in the LRRK2 knockout mouse cerebellum, albeit at lower intensity relative to the wild type mouse cerebellum extract suggesting the antibody detected a non specific species at the molecular weight of monomeric LRRK2 under the conditions tested. Lower molecular weight (55-250 kDa) 3514-1 immunoreactive bands of weaker intensities (ii) were observed in all human brain regions as well as the wild type and LRRK2 knockout mouse cerebellum. Higher molecular weight, 900 kDa 3514-1 immunoreactive bands were also identified in all human and mouse brain samples including the mouse LRRK2 knockout cerebellum (iii), potentially corresponding to non-specific binding of the antibody. Multiple poorly resolved bands in both the mouse and human brain extracts prevented quantitative comparison of endogenous LRRK2 expression (Fig 3.15). In addition, the specificity of antibodies made it difficult to identify the true native LRRK2 species in the samples analysed. 140 3.10 Immunohistochemical analysis of LRRK2 protein in brain Endogenous LRRK2 protein has also previously been detected by immunohistochemical analysis of wild type mouse and control human brains. To determine whether the 3514-1 antibody can detect LRRK2 in the human brain by immunohistochemistry the antibody was initially tested on wild type and LRRK2 knockout mouse cerebellum brain sections. We focused on the cerebellum region of the mouse brain as it was easy to identify and based on our previous finding of high cerebellar LRRK2 protein abundance determined by the combined IP and Western 141 blot analysis (Fig 3.12A). Immunohistochemistry was subsequently used to address the problem of LRRK2 detection in the human brain rather than to study LRRK2 brain distribution hence other brain regions were not evaluated. The mouse cerebellum showed the characteristic pattern of neuronal haematoxyline staining of intricate patterns of folds and fissures. Paraformaldehyde fixation and subsequent 3514-1 antibody staining of flash frozen wild type mouse brain tissue revealed positive DAB immunoreactivity in the Purkinje neurons of the cerebellum (Fig 3.16A, B). Equivalent regions in the LRRK2 knockout mouse brain did not show DAB positivity when processed in parallel to wild type mouse brain sections suggesting DAB staining represented LRRK2. Diluting the concentration of the 3514-1 antibody (1:50-500) revealed the most intense DAB immunoreactivity for the 1:50 antibody dilution with lower intensity DAB staining a lower antibody concentrations (Fig 3.16 C). The pattern of 3514-1 DAB staining in the cerebellar sections, with purkinje neuron positivity was maintained across all antibody concentrations (1:50-500). 142 3514-1 staining (1:100 dilution) of flash frozen human cerebellar sections produced DAB positive staining in the purkinje neurons, comparable to the staining observed for wild type mouse cerebellum suggesting that LRRK2 protein is present in this cell population (Fig 3.17). Our results confirm that LRRK2 protein is expressed in 143 the human cerebellum at least, despite our inability to detect LRRK2 by Western blot or by the immunoprecipitation and Western blot approach. 144 Chapter 4: Results LRRK2 and mitochondrial function Mitochondrial abnormalitites that have previously been linked to PD in genetic and environmental PD models include reduced respiratory chain enzyme activity [97-100], limited substrate supply to oxidative phosphorylation due to defects in the glucose transport system [172] and ATPase reversal in the presence of gross mitochondrial respiratory chain defects [172]. These reported defects have correlated with reduced activity of the mitochondrial respiratory chain and elevated ROS generation [107, 172]. Furthermore, mitochondrial localization of PD associated proteins; PINK1, parkin, DJ1 and α-synuclein have been linked to mitochondrial functional changes associated with their respective mutant proteins. Having characterised LRRK2 expression in fibroblast, lymphoblast and SHSY5Y cell models in Chapter 3, this chapter evaluated mitochondrial function in wild type and G2019S LRRK2 expressing fibroblasts and SHSY5Y cells by measuring cellular oxygen consumption, mitochondrial membrane potential, glycolytic metabolism, basal ATP content and ROS generation. In addition, the underlying mechanisms of compromised mitochondrial energetics were explored; the relationship between mitochondrial changes, LRRK2 mitochondrial localization and kinase activity was evaluated in wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells in the presence and absence of LRRK2 kinase inhibition. The potential correlation between LRRK2 levels and mitochondrial function were assessed in LRRK2 over-expressing cells and the LRRK2 siRNA knockdown SHSY5Y model. 4.1 Mitochondrial function in fibroblast cell lines 4.1.1 The analysis of mitochondrial respiration in control and G2019S fibroblasts Cellular oxygen consumption was evaluated in intact control and G2019S fibroblasts by measuring extracellular oxygen concentration using two methods; 145 phosphorescent oxygen probes and polarographically with the Clark type oxygen electrode. Phosphorescent measurements were carried out on fibroblasts cultured under standard conditions (37 ˚C, 5 % CO2) in glucose rich medium with respiration rates measured over the period of one hour. Phosphorescent analysis of oxygen consumption in control fibroblasts revealed comparable respiratory rates (0.16-0.18 RFU/min/µg protein) for the six cell lines (Fig 4.1A) while substantial variability in cellular respiration rates was observed in the 8 G2019S LRRK2 fibroblast lines (0.180.6 RFU/min/µg protein). Comparing the fibroblast cell lines as control versus G2019S groups revealed a 128 % (p<0.0005) increase in cellular respiration rates for the G2019S cohort (Fig 4.1B). Polarographic measurements of oxygen consumption in the presence of glucose identified a 76 % (p<0.05) increase in respiration for the G2019S fibroblast group (n=8) relative to control (n=6) cell lines (Fig 4.1C). The increase in respiration identified in G2019S LRRK2 fibroblasts by the phosphorescent analysis and polarographic measurements is consistent with an up-regulation of mitochondrial function in these cell lines relative to control cells. Polarographic methods were also used to assess maximal respiratory rates in control and G2019S fibroblasts in response to addition of the chemical uncoupler, FCCP. FCCP induced a two fold increase in the basal oxygen consumption for control cells (Fig 4.1C). In G2019S cell lines FCCP addition increased the rate of oxygen consumption to that seen in FCCP treated control fibroblasts indicating no change in the maximal mitochondrial capacity. However, ratios of maximal to basal respiration were reduced compared to control cells suggesting mitochondria may be more uncoupled in LRRK2 G2019S cells. 146 4.1.2 The analysis of mitochondrial content in control and G2019S LRRK2 fibroblasts Cellular mitochondrial content can impact on cellular respiratory capacity therefore interpretation of oxygen consumption measurements required assessment of mitochondrial abundance. Mitochondrial content was quantified by Western blot analysis of various mitochondrial proteins in whole cell lysates of control and G2019S LRRK2 fibroblasts and the proportion of cells stained with TMRM. Relative to control fibroblasts the levels of inner mitochondrial membrane 147 localized Complex III (Core CIII) and II (SDHA) subunits as well as the mitochondrial transcription factor (TFAM) were similar in G2019S cells as determined by Core Complex III, SDHA and TFAM immunoreactivities (Fig 4.2A). Actin immunoreactivity demonstrated equivalent protein loading. These results are consistent with comparable electron transport chain subunit densities and abundance of mitochondrial DNA transcriptional regulators. Confocal analysis and quantification of the cellular proportion of control and G2019S LRRK2 fibroblasts stained with TMRM revealed comparable TMRM volumes per cell (20%) consistent with similar levels of mitochondria in control and G2019S cell lines (Fig 4.2B). Both analyses suggest LRRK2 G2019S expression in fibroblasts did not affect mitochondrial levels in fibroblasts. 148 4.1.3 The analysis of mitochondrial membrane potential in control and G2019S LRRK2 fibroblasts Mitochondrial membrane potential was assessed in 6 control and 8 G2019S fibroblasts cultured in glucose rich respiration buffer by TMRM staining and confocal microscopy. The intensity of TMRM staining appeared reduced in G2019S fibroblasts relative to control cells (Fig 4.3A). Quantification of TMRM intensity revealed comparable average values (1150-1200 RFU) for all the cell lines in the control group (Fig 4.3B). TMRM fluorescence was decreased for all G2019S fibroblast lines by 20 % (p<0.0001) relative to control cells (Fig 4.3C), consistent with a reduction in the mitochondrial membrane potential. 149 4.1.4 The analysis of cellular ATP content in control and G2019S LRRK2 fibroblasts Basal ATP content was assessed in 6 control and 4 G2019S cell lines by measuring luciferase luminescence in cellular extracts. Considerable variability was observed in luciferase luminescence for control (3.7x106 -4.8x106 RFU/µg protein) 150 and G2019S mutant fibroblasts (3.6 x106 – 4.4 x106 RFU/µg protein) (Fig 4.4A). Statistical comparison of control and G2019S fibroblasts identified a 10 % (p<0.05) decrease in average luminescence for the G2019S cells (Fig 4.4B), consistent with a reduced basal ATP content. The reduction in ATP content for the four G2019S cell lines did not correlate with changes in oxygen consumption rates (Fig 4.1A). 4.1.5 The analysis of extracellular acidification rates in control and G2019S LRRK2 fibroblasts Extracellular acidification rates were assessed in 6 control and 8 G2019S LRRK2 fibroblasts. Mesurements were carried out on whole cells in glucose rich, buffer free culturing conditions (37 ˚C) using pH sensitive phosphorescent probes over the period of one hour. Analysis of pH probe fluorescence revealed considerable variation in phosphorescent lifetime rate measurements for the control (1.2 x10-10-2.4 x10-10 [H+]/min/µg protein) and G2019S (0.3 x10-10 -4 x10-10 151 [H+]/min/µg protein) fibroblast groups (Fig 4.5A) suggesting variable rates of anaerobic glycolysis. Statistical comparison of control versus G2019S cell groups showed comparable extracellular acidification rates despite the variability observed among the individual cell lines (Fig 4.5B). Data shown indicates there was no obvious perturbation in glycolytic rates or correlation with changes in oxygen consumption in the G2019S fibroblasts. 4.1.6 The analysis of cellular ROS in control and G2019S LRRK2 fibroblasts To determine whether ROS levels were altered in G2019S expressing fibroblasts relative to control cells, ROS generation was evaluated by measuring dihydroethidium oxidation. Dihydroethidium oxidation was monitored in 3 control and 3 G2019S fibroblast cell lines as the rate of increase in red fluorescence. The average rate of nuclear accumulation of red DHE fluorescence was 36 % (p<0.005) 152 slower in G2019S cell lines relative to control fibroblasts (Fig 4.6) consistent with reduced rates of reactive oxygen species generation in the LRRK2 mutant cell lines. 4.1.7 The analysis of mitochondrial proton leak in control and G2019S LRRK2 fibroblasts To further investigate the possibility of mitochondrial uncoupling, mitochondrial proton leak was evaluated by measuring oxygen consumption rates polarographically in the presence of oligomycin for control and G2019S fibroblasts. Polarographic measurements of oxygen consumption rates in 4 control fibroblasts in the presence of glucose detected a respiratory rate comparable to the previous measurement (Fig 4.1A). The addition of oligomycin resulted in ablation of mitochondrial respiration in control cells (Fig 4.7), an effect that has previously been associated with complex V inhibition. In contrast, respiration rates detected in oligomycin treated G2019S fibroblasts were 14 % of untreated rates (p<0.05) consistent with a mitochondrial proton leak. 153 4.1.8 The analysis of mitochondrial morphology in control and G2019S LRRK2 fibroblasts Mitochondrial morphology has previously been reported to correlate with functional changes of the mitochondrial respiratory chain, [119, 121]. Mitochondrial morphology was assessed in 6 control and 8 G2019S LRRK2 cells by confocal microscopy focusing on the pattern of TMRM staining. Fibroblasts were imaged in respiration buffer following incubation with TMRM. In control fibroblast cells TMRM stained mitochondrial structures consisted of a web of interconnected mitochondrial processes, dense around the nuclear contour and stretching throughout the cell body (Fig 4.8). Both long (0.5 µm, ↓) and short (0.025 µm, ↓*) mitochondrial fragments could be identified representing elongated and fragmented regions of the mitochondrial network. 154 The G2019S fibroblast cell lines presented with a TMRM staining pattern resembling that of control cells consistent with a similar architecture of mitochondrial networks (Fig 4.8). The pattern of TMRM staining was similar for the individual cells in each control and G2019S fibroblast cell line, consistent with a uniform mitochondrial distribution and structure. 155 4.2 Mitochondrial dysfunction models To exclude other potential causes that could account for the observed G2019S changes, control fibroblast cell lines were used to model; respiratory chain enzymatic activity defects, limited substrate supply and mitochondrial ATPase reversal to determine how the bioenergetic changes associated with these models compare to changes identified in G2019S expressing fibroblasts. 4.2.1 The use of control fibroblasts treated with rotenone or antimycin A as a model of respiratory chain dysfunction Respiratory chain enzymatic activity defects, such as those reported in idiopathic PD patients, α-synuclein over-expressing SHSY5Y cells and PINK1 mutant fibroblasts from PD patients were modelled by treating control fibroblasts with inhibitors of Complex I (rotenone 1 µM) and complex III (antimycin A, 1 µM). Oxygen consumption rates of control fibroblasts, treated or untreated with ETC inhibitors were assessed polarographically in the presence of glucose. A 70 % reduction in phosphorescent lifetime rate values was detected for control fibroblasts treated with 1 µM rotenone relative to untreated cells, reflecting reduced respiratory chain activity (Fig 4.9). Oxygen consumption was not completely inhibited, as shown by the 30 % basal respiration still taking place in fibroblasts treated with rotenone, potentially reflecting compensatory effects of increased substrate supply through complex II of the respiratory chain. The rate of change of phosphorescent lifetime was reduced to 30 % of the basal rate for untreated fibroblasts with the addition of antimycin A, comparable to the response of fibroblasts to rotenone (Fig 4.9). 156 4.2.2 The use of control fibroblasts treated with rotenone and oligomycin to evaluate the influence of ETC inhibition on the mitochondrial ATPase Mitochondrial ATPase reversal has previously been associated with PINK1 and parkin linked PD as well as electron transport chain response to enzymatic inhibition. When complex I is inhibited the mitochondrial membrane potential is maintained by the reversal of the ATPase [310]. Control fibroblasts were treated with rotenone (1 µM) and the mitochondrial proton gradient monitored in respiration buffer by confocal microscopy of TMRM stained cells. Quantification of TMRM intensity of rotenone treated control fibroblast revealed fluorescent intensity values comparable to those of untreated cells 3 minutes after administration of the drug (Fig 4.10A). Addition of oligomycin (4 mg/ml) resulted in a time dependent increase in TMRM fluorescence in untreated control fibroblasts consistent with an increase in the mitochondrial membrane potential as a result of ATPase inhibition and accumulation of protons in the mitochondrial matrix. In contrast, oligomycin 157 addition to rotenone treated fibroblasts resulted in an immediate 30 % decrease in TMRM fluorescent intensity consistent with the dissipation of the mitochondrial proton gradient. TMRM fluorescent intensity in G2019S fibroblast cell lines was stable for 6 minutes following addition of oligomycin (4 mg/ml), comparable to oligomycin effects on control cells. Hence there is no evidence to suggest the mitochondrial membrane potential is being maintained in these cells by reversal of the ATPase (Fig 4.10B). 158 4.2.3 The use of control fibroblasts cultured in galactose rich conditions as a model of limited cellular substrate supply Oxygen consumption rates were assessed in control fibroblasts by phosphorescent analysis under glucose or galactose culture conditions (37 ˚C, 5 % CO2) over the course of one hour. As the rate limiting enzyme in galactose metabolism has slow first order rate kinetics, glycolytic flux is restricted. Phosphorescent probe lifetime rate measurements of control fibroblasts cultured in 159 galactose medium were 1.5 fold (p<0.0005) greater relative to rates recorded for control fibroblasts cultured in glucose consistent with an increase in cellular oxygen consumption when galactose was supplied as a substrate source (Fig 4.11A). Extracellular acidification rates were assessed for control fibroblasts in glucose or galactose rich, buffer free culture conditions (37 ˚C, 5 % CO2) using pH sensitive phosphorescent probes over the period of one hour. Control fibroblasts cultured in glucose showed a linear rate of increase in extracellular proton concentrations, consistent with a steady rate of anaerobic glycolysis and lactate generation (Fig 4.11B). Monitoring extracellular acidification rates in control fibroblasts cultured in galactose, revealed 10 fold slower (p <0.0005) extracellular acidification rates relative to fibroblasts cultured on glucose consistent with a decreased rate of glucose metabolism. The mitochondrial membrane potential was assessed in control fibroblasts cultured in glucose or galactose rich respiration buffer by confocal microscopy of TMRM stained cells. Quantification of TMRM fluorescent intensity identified a 10 % (p<0.0001) increase in TMRM fluorescence of fibroblasts cultured in galactose medium when compared to cells cultured under glucose conditions (Fig 4.11C) consistent with an increase in the mitochondrial membrane potential. Basal ATP levels were evaluated in control fibroblasts cultured in glucose or galactose rich conditions by quantification of luciferase luminescence in lysed cells. Luminescence was comparable in cells cultured on glucose or galactose (Fig 4.11D), suggesting basal ATP levels were unaffected by restriction in glycolytic substrate supply. 160 4.3 The analysis of respiratory chain function in the SHSY5Y LRRK2-V5 overexpressing model To confirm the defect in fibroblasts was replicated in SHSY5Y cells overexpressing G2019S LRRK2 mitochondrial function was evaluated in the SHSY5Y cell model. Control, wild type and G2019S LRRK2-V5 over expressing SHSY5Y cells were assessed for oxygen consumption rates by polarographic measurements, mitochondrial membrane potential by TMRM staining and confocal microscopy and 161 reactive oxygen species by spectrophotometric quantification of cellular aconitase activity. Polarographic measurements of oxygen consumption by the Clark type oxygen electrode revealed basal respiration in control SHSY5Y cells (4 nmol O/min/µg protein) comparable to rates determined for wild type LRRK2-V5 overexpressing SHSY5Y cells (Fig 4.12A). In contrast SHSY5Y cells over-expressing G2019S LRRK2-V5 had an 80 % (p<0.05) increase in the rate of oxygen consumption relative to control and wild type LRRK2-V5 over-expressing SHSY5Y cells (Fig 4.12A). Quantification of TMRM intensity determined comparable intensity measurements for control and wild-type LRRK2-V5 over-expressing SHSY5Y cells while a 20 % (p<0.0001) reduction in TMRM intensity was identified for G2019S LRRK2-V5 overexpressing SHSY5Y cells (Fig 4.12B), consistent with a decrease in the mitochondrial membrane potential. Comparable aconitase activities were determined for control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cell lines consistent with the absence of oxidative stress (Fig 4.12C). 162 4.4 LRRK2 kinase activity 4.4.1 The analysis of LRRK2 kinase inhibition in the SHSY5Y LRRK2-V5 overexpressing model LRRK2 kinase activity was previously shown to be greater for G2019S LRRK2V5 immunoprecipitated from SHSY5Y over-expressing SHSY5Y cells relative to the wild type protein (Fig 3.8C). To determine the influence of LRRK2 kinase inhibition on Serine 935 phosphorylation and mitochondrial function in SHSY5Y cells, LRRK2 163 over-expressing SHSY5Y cells were subject to treatment with LRRK2 specific kinase inhibitors. LRRK2 was immunoprecipitated from the cells and assessed by Western blot analysis using phospho Serine 935 specific LRRK2 antibodies. Mitochondrial function was assessed polarographically and by TMRM staining following LRRK2 kinase inhibition. 4.4.1.1 LRRK2-IN1 LRRK2 was immunoprecipitated from wild type LRRK2-V5 over-expressing SHSY5Y cells treated with increasing concentrations of LRRK2 IN1 (0-2 µM). Western blot analysis of immunoprecipitates revealed a dose dependent reduction of phospho Serine 935 immunoreactivity relative to the 3514-1 immunoreactive band corresponding to the full length immunoprecipitated LRRK2-V5 (Fig 4.13A). Quantification of the phospho Serine 935 band intensity relative to 3514-1 determined a 90 % reduction in the signal intensity for wild-type LRRK2-V5 SHSY5Y cells treated with 1µM LRRK2-IN1 when compared to untreated cells, consistent with a marked inhibition of LRRK2 kinase activity [215]. The effective LRRK2-IN1 concentration (1 µM) was within the range used to dephosphorylate Serine 935 in HEK293 and lymphoblast cell culture models [215]. Time course analysis (0-90 min) of wild type LRRK2-V5 over-expressing SHSY5Y cells with 1 µM LRRK2-IN1 treatment revealed a time dependent reduction in phospho Serine 935 immunoreactivity with a 90 % decrease in phospho signal relative to 3514-1 band intensity detected following 90 minutes of treatment when compared to untreated cells (Fig 4.13B), in line with previous reports. 4.4.1.2 CZC25146 LRRK2 was immunoprecipitated from wild type LRRK2-V5 over-expressing SHSY5Y cells treated with increasing concentrations of CZC25146 (0-1 µM). Western blot analysis of immunoprecipitates revealed a dose dependent reduction of 164 phospho Serine 935 immunoreactivity relative to the 3514-1 immunoreactive band corresponding to the full length immunoprecipitated LRRK2-V5 (Fig 4.13C). Quantification of the phospho Serine 935 band intensity relative to 3514-1 determined a 90 % reduction in the signal intensity for wild-type LRRK2-V5 SHSY5Y cells treated with 0.3 µM CZC25146 when compared to untreated cells, consistent with a marked inhibition of LRRK2 kinase activity. The effective CZC25146 concentration (0.3 µM) was within the range used to dephosphorylate Serine 935 in HEK293 and lymphoblast cell culture models [216]. In addition, the 0.3 µM CZC25146 dose resulted in comparable levels of phospho Serine 935 dephosphorylation as the 1 µM dose of LRRK2-IN1 suggesting CZC25146 was more potent than LRRK2-IN1 in our SHSY5Y cell culture model. 4.4.1.3 TAE-648 LRRK2 was immunoprecipitated from wild type LRRK2-V5 over-expressing SHSY5Y cells treated with increasing concentrations of TAE-684 (0-1 µM). Western blot analysis of immunoprecipitated LRRK2 revealed a dose dependent reduction of phospho Serine 935 immunoreactivity relative to the 3514-1 immunoreactive band corresponding to the full length immunoprecipitated LRRK2-V5 (Fig 4.13D). Quantification of the phospho Serine 935 band intensity relative to 3514-1 determined a 90 % reduction in the signal intensity for wild-type LRRK2-V5 SHSY5Y cells treated with 0.3 µM TAE-684 when compared to untreated cells, consistent with a marked inhibition of LRRK2 kinase activity. The effective TAE-684 concentration (0.3 µM) was within the range used to dephosphorylate Serine 935 in HEK293 and lymphoblast cell culture models [214]. In addition, the 0.3 µM TAE-684 dose resulted in comparable levels of phospho Serine 935 dephosphorylation, relative to total LRRK2 as the 1 µM dose of LRRK2-IN1 suggesting TAE-684 was a more potent LRRK2 kinase inhibitor in our cell culture model. 165 166 4.4.2 The use of LRRK2 kinase inhibitors to evaluate effects of LRRK2 kinase inhibition on mitochondrial function in the SHSY5Y LRRK2-V5 over-expressing model 4.4.2.1 Evaluation of the effects of LRRK2 kinase inhibition on mitochondrial respiration Polarographic measurements of cellular oxygen consumption in the presence of glucose carried out on control, wild type and G2019S LRRK2-V5 cells or cells treated with LRRK2-IN1 (1 µM, 90 min) revealed comparable respiration rates for control and wild type LRRK2-V5 over-expressing SHSY5Y cells in the presence and absence of LRRK2-IN1 (Fig 4.14A) suggesting LRRK2 kinase inhibition did not influence mitochondrial respiration. In contrast, a 25 % (p<0.05) reduction in oxygen consumption rate was reported for G2019 LRRK2-V5 over-expressing SHSY5Y cells following LRRK2 kinase inhibition with LRRK2-IN1. The inhibited rates were comparable to untreated control and wild type LRRK2-V5 over-expressing SHSY5Y cells (Fig 4.14A). LRRK2 Serine 935 phosphorylation status was evaluated following the respiratory measurements. Phospho Serine 935 immunoreactivity relative to 3514-1 band intensity, corresponding to total LRRK2-V5 levels, was reduced by >80 % for LRRK2-IN1 treated wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells relative to untreated cells (Fig 4.14B), consistent with a marked inhibition of LRRK2 kinase activity. 4.4.2.2 Evaluation of the effects of LRRK2 kinase inhibition on the mitochondrial membrane potential Control, wild type and G2019S LRRK2-V5 cells were treated with LRRK2-IN1 (1 µM, 90 min), CZC25146 (0.3 µM) or TAE-684 (0.3 µM) following which the mitochondrial membrane potential was assessed in untreated or inhibitor treated cells by confocal microscopy of TMRM stained cells in glucose rich respiration buffer. 167 LRRK2-IN1 treatment of wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells resulted in 10 % and 40 % increases (p<0.0001) in TMRM fluorescence relative to untreated cells (Fig 4.14C) suggesting LRRK2 kinase inhibition increased the mitochondrial membrane potential. CZC25146 (0.3 µM) treatment of control and wild type LRRK2-V5 overexpressing SHSY5Y cells resulted in a 10 % (p<0.0001) decrease in the TMRM fluorescence relative to untreated cells consistent with a reduction in the mitochondrial membrane potential. CZC25146 effects on the TMRM intensity for wild type LRRK2-V5 over-expressing SHSY5Y cells contrasted the 10 % TMRM intensity increase observed with LRRK2-IN1 treatment (Fig 4.14D), potentially due to the different off target effects described for the two compounds. In contrast, CZC25146 treatment of G2019S LRRK2-V5 over-expressing cells resulted in a 10 % (p<0.0001) increase in the intensity of TMRM staining consistent with an increase in the mitochondrial membrane potential. Although opposite effects were observed for control and wild type LRRK2-V5 over-expressing SHSY5Y cells in response to LRRK2-IN1 and CZC25146 treatment, the increase in TMRM intensity for the G2019S LRRK2-V5 expressing cells following kinase inhibition suggest LRRK2 kinase inhibition potentially restores the mitochondrial abnormality associated with G2019S LRRK2V5 protein. TAE-684 (0.3 µM) treatment of control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells resulted in a 60 % (p<0.0001) decrease in TMRM fluorescence relative to untreated cells suggesting TAE-684 could potentially inhibit mitochondrial function, possibly due to the broader selectivity profile of the drug (Fig 4.14D). 168 4.5 LRRK2 mitochondrial localization 4.5.1 Mitochondrial affinity purification Cellular fractionation experiments carried out in chapter 1 identified 10 % of the total LRRK2 cellular pool in the mitochondrial enriched fractions (MEF) for wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells as well as endogenous LRRK2 in lymphoblasts. 169 To determine whether LRRK2 localizes to the mitochondria and quantify the mitochondrial LRRK2 pool, mitochondrial affinity purification was carried out on wild type, G2019S LRRK2-V5 over-expressing cells and lymphoblasts. Affinity purified mitochondria were isolated from the post nuclear supernatant (PNS) of cells and assessed by Western blot analysis. A fraction (1/10th) of the PNS sample was retained for analysis of LRRK2 and mitochondrial input with all Western blot quantifications corrected for sample loading: PNS (1/10) and purified mitochondria (9/10) unless stated otherwise. Affinity purified mitochondria were identified by Western blot analysis of the 39 kDa immunoreactive band for Core Complex III marker of the inner mitochondrial matrix, quantified as 11 fold greater signal intensity relative to the Core Complex III in the PNS input fraction (Fig 4.15A). Correcting the Core Complex III immunoreactive signal for sample loading calculated >90 % recovery of mitochondria from the PNS input. Western blot immunoreactivity for cytochrome C (12 kDa), a marker of the inter membrane space, was 2 fold lower relative to the signal detected in the PNS input suggesting a portion of cytochrome C was lost during the affinity purification procedure. Western blot analysis detected calreticulin and GCase immunoreactivities in the PNS fraction of SHSY5Y cells but were absent in extracts of affinity purified mitochondria suggesting they were generally free from lysosomal and ER contaminants. V5 immunoreactivity was detected in the PNS and mitochondrial affinity purified fractions. Quantification of the mitochondrial V5 band intensity relative to PNS and corrected for sample loading determined 1-2 % (n=3) of the post-nuclear V5 immunoreactivity in the affinity purified fraction consistent with a mitochondrial localization of LRRK2-V5. 170 4.5.1.1 Mitochondrial affinity purification versus differential centrifugation Direct comparison of LRRK2 recovery from mitochondria isolated by differential centrifugation or affinity purification identified a 39 kDa immunoreactive band of 10 fold greater signal intensity relative to the PNS input lysates of wild type LRRK2-V5 over-expressing SHSY5Y cells, suggesting comparable mitochondrial recoveries by the two purification methods (Fig 4.15B). While mitochondrial enriched fractions isolated by differential centrifugation were immunoreactive for GCase, affinity purified preparations did not reveal clear GCase immunoreactivity in the same exposure time consistent with low lysosomal abundance. V5 immunoreactivity in mitochondria isolated by differential centrifugation was of 2 fold lower intensity relative to V5 in PNS input consistent with 7 % (n=1) of the PNS LRRK2 pool present in this mitochondrial isolation when corrected for sample loading (1/15th of the PNS was retained for Western blot analysis in this experiment). A V5 immunoreactive signal of 6 fold lower intensity was detected in affinity purified preparations consistent with 1% of the PNS LRRK2 pool in this mitochondrial fraction, consistent with previous observations (Fig 4.15A). Our data suggests the LRRK2 signal in the mitochondrial enriched fraction of crude preparations is potentially overestimated. 4.5.1.2 Mitochondrial affinity purification from G2019S LRRK2-V5 over-expressing SHSY5Y cells The 280 kDa immunoreactive V5 band was detected in mitochondrial affinity purifications from wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells. Quantification of wild type or G2019S V5 immunoreactivity in the affinity purified mitochondrial fraction relative to PNS and corrected for sample loading determined 1.5 and 1.6 % (n=2) of the V5 signal in the purified mitochondrial compartment consistent with comparable levels of LRRK2 mitochondria isolated from wild type 171 and G2019S LRRK2-V5 over-expressing SHSY5Y cells (Fig 4.15C). Mitochondrial affinity preparations from SHSY5Y cells over-expressing wild type or G2019S LRRK2V5 were free of lysosomal contaminants as determined by the absence of a clear GCase immunoreactive signal as detected in the PNS input. 4.5.1.3 Mitochondrial affinity purification, LRRK2 kinase inhibition To determine whether LRRK2 phosphorylation status affects its mitochondrial localization, SHSY5Y wild type over-expressing LRRK2-V5 cells were treated with LRRK2 IN1 (1 µM, 90 min) and affinity purified mitochondrial fractions assessed for relative LRRK2-V5 abundance by Western blot. Inhibition of LRRK2 kinase activity and Serine 935 de-phosphorylation did not affect the efficiency of mitochondrial recovery as determined by comparable Core Complex III immunoreactivities for LRRK2-IN1 treated and untreated wild type LRRK2-V5 overexpressing SHSY5Y cells relative to the PNS when corrected for sample loading (Fig 4.15D). The 280 kDa immunoreactive V5 band was detected in mitochondrial affinity purifications from LRRK2-IN1 treated and untreated wild type LRRK2-V5 cells. Quantification of the V5 band intensity relative to PNS input and sample loading determined 1.6 and 1.7 % (n=2) of V5 immunoreactivity in untreated and LRRK2-IN1 treated SHSY5Y cells consistent with comparable LRRK2 mitochondrial abundance in the presence and absence of LRRK2 kinase inhibition and Serine 935 dephosphorylation. 4.5.1.4 Mitochondrial affinity purification from lymphoblasts Affinity purified mitochondria were isolated from lymphoblasts to determine the proportion of endogenous protein in the mitochondrial compartment of this cell type. Successful purification of mitochondria was confirmed by the presence of a 39 kDa immunoreactive Core Complex III band in the affinity purified mitochondrial preparation (Fig 4.15E). Correcting the Core Complex III 172 immunoreactivity for sample loading relative to Core Complex III band intensity in the PNS input determined >90 % mitochondrial recovery by this purification method, comparable to purification recovery obtained with SHSY5Y cells (Fig 4.15A, C). Weak 3514-1 280 kDa immunoreactivity was detected by Western blot analysis of lymphoblast PNS input, however, 3514-1 immunoreactivity could not be clearly identified in affinity purified mitochondrial preparations (Fig 4.15E). In an attempt to increase the sensitivity of endogenous LRRK2 detection, LRRK2 was immunoprecipitated from the PNS input and affinity purified mitochondria. A clear 280 kDa 3514-1 immunoreactive band was identified in the lymphoblast PNS input (Fig 4.15E). LRRK2 immunoprecipitation from mitochondrial isolates containing 10 times the volume of the PNS input detected a faint 3514-1 immunoreactive band comigrating with the band identified in the PNS input consistent with low levels mitochondrial LRRK2 in lymphoblasts. The background noise did not allow for quantification of the relative LRRK2 mitochondrial abundance in lymphoblasts. 173 174 4.5.2.1 Sub-fractionation of mitochondrial enriched fractions from SHSY5Y LRRK2V5 over-expressing SHSY5Y cells To determine the intra mitochondrial distribution of LRRK2, mitochondrial fractionation was carried out in wild type LRRK2-V5 over-expressing SHSY5Y cells. Mitochondrial sub-fractionation was initially carried out on mitochondria isolated by differential centrifugation before extending the studies to affinity purified mitochondria. The outer mitochondrial membrane of mitochondria was selectively removed by digitonin treatment. Following treatment, mitochondrial enriched fractions were separated into soluble and pellet components by centrifugation and the corresponding extracts analysed by Western blotting. Western blot analysis of untreated MEFs identified porin (outer mitochondrial membrane marker) and Core Complex III (inner mitochondrial membrane marker) immunoreactivities in the pelleted mitochondrial compartment absent in the corresponding soluble fraction (Fig 4.16A). V5 immunoreactivity was also detected in the untreated pellet of MEFs in line with our previous findings (Fig 4.15B). MEFs isolated from wild type LRRK2-V5 over-expressing SHSY5Y cells treated with 2 mg/ml digitonin revealed 20 % of the total porin immunoreactivity in the soluble fraction with 80 % retained in the mitochondrial pellet. At 3 mg/ml porin immunoreactivity in the soluble fraction was comparable to the band intensity detected in the mitochondrial pellet consistent with the removal of 50 % of the outer mitochondrial membrane. Core Complex III immunoreactivity was absent in the soluble fraction following 2 mg/ml digitonin treatment but 50 % of the total Core Comlex III immunoreactivity was found in the soluble compartment at 3 mg/ml digitonin, consistent with the increased solubilisation of the inner mitochondrial membrane at this digitonin concentration. At 3 mg/ml, when the inner mitochondrial membrane of MEF preparations was detected in the soluble fraction 175 60 % of total porin immunoreactivity was retained in the MEF pellet suggesting a portion of the outer membrane was still associated with the inner membrane even though the integrity of the inner membrane was compromised. Western blot analysis of untreated MEFs showed that V5 immunoreactivity was absent in the soluble fraction while MEFs treated with 1 mg/ml digitonin had 50 % of the total V5 immunoreactivity in the soluble fraction consistent with solubilisation of half of the mitochondrial LRRK2 pool. Mitochondria treated with 2 mg/ml digitonin revealed 60 % and 40 % V5 immunoreactivities in the soluble and mitochondrial pellet fractions while at 3 mg/ml digitonin the V5 immunoreactive signal was equally distributed between the two fractions. Although we have previously demonstrated 90 % of the LRRK2 immunoreactivity in mitochondria isolated by differential centrifugation potentially represents signal contamination from other cellular fractions, data shown suggests LRRK2 response to digitonin mimics that of porin consistent with LRRK2 localization to the outer mitochondrial membrane of MEFs. 4.5.2.2 Sub-fractionation of affinity purified mitochondria from SHSY5Y LRRK2-V5 over-expressing SHSY5Y cells To determine LRRK2 localization in affinity purified mitochondria from wild type LRRK2-V5 over-expressing SHSY5Y cells, affinity purified mitochondrial preparations were subject to digitonin treatment. Western blot analysis of untreated affinity purified mitochondria identified 60 kDa porin and 39 kDa Core Complex III and V5 immunoreactivities in the mitochondrial pellet, absent in the soluble fraction, consistent with intact mitochondrial inner and outer membranes (Fig 4.16B). Digitonin at 1 mg/ml resulted in 10 % of total porin immunoreactivity in the soluble fraction while 2 mg/ml digitonin the soluble pool was estimated to contain approximately 20 % porin immunoreactivity. Higher concentrations, 3 and 4 176 mg/ml produced 40 % porin immunoreactivity in the soluble fraction with 60 % immunoreactivity retained in the mitochondrial pellet consistent with a dose dependent solubilisation of the outer mitochondrial membrane. Western blot analysis of affinity purified mitochondria treated with digitonin at 1 mg/ml detected 5 % of total Core Complex III immunoreactivity in the soluble fraction (Fig 4.16B). Digitonin at 2 mg/ml resulted in 20 % of the Core Complex III signal intensity present in the soluble fraction, while 3 and 4 mg/ml increased the soluble Core Complex III signal intensity to 50 % consistent with a dose dependent solubilization of the inner mitochondrial membrane. Similar to the data obtained for sub-cellular fractionation of mitochondria isolated by differential centrifugation, the digitonin titration did not appear to selectively target the outer mitochondrial membrane in highly purified mitochondrial preparations as the inner membrane was solubilised while a large portion of the outer membrane appeared intact. Despite the unpredictable inner and outer mitochondrial membrane marker behaviour, LRRK2 V5 immunoreactivity signal reflected the pattern of porin immunoreactive distribution with 10 %, 10 % and 20 % of the total V5 signal intensity identified in the soluble fraction for 1, 2 and 3 mg/ml digitonin, consistent with an outer mitochondrial membrane localisation in affinity purified mitochondrial preparations from wild type LRRK2-V5 over-expressing SHSY5Y cells. Attempts to sub-fractionate mitochondrial preparations with digitonin did not give the predicted data even after numerous attempts hence an alternative approach was tested. Affinity purified mitochondria were digested with proteinase k (5 and 10 µg/ml) for increasing incubation periods (0-20 min) following which mitochondria were pelleted by centrifugation and assessed by Western blot analysis. At 5 µg/ml proteinase, 39 kDa immunoreactivity for Core Complex III was 2 fold lower relative to untreated mitochondria for all the digestion times, consistent with 177 a partially digested inner mitochondrial membrane (Fig 4.16C). Increasing proteinase K to 10 µg/ml resulted in a 4 fold reduction of Core Complex III immunoreactivity for all time points analysed consistent with a correlation between enzyme concentration and the rate of mitochondrial inner membrane digestion by proteinase K. Porin immunoreactivity was present in untreated mitochondria but was absent in the presence of proteinase K for all concentrations and time points tested with a faint immunoreactive band detected following 20 minute treatment with 5 µg/ml proteinase K. The absence of a porin immunoreactive signal suggested the outer mitochondrial membrane was completely digested even after 5 minutes of proteinase K incubation at 5 µg/ml. An additional 15 kDa immunoreactive porin species was identified in affinity purified mitochondria treated with 5 and 10 µg/ml proteinase K for all time points (0-20 min) analysed. The 15 kDa porin immunoreactivity in the presence of the proteinase K was 3 fold greater relative to the band intensity in untreated mitochondrial preparations, suggesting the band could represent a porin degradation product. 4.5.3.1 Sub-fractionation of mitochondria to determine LRRK2 membrane association Existing data suggests LRRK2 is more abundant in a membrane associated form, preferentially binding to cholesterol rich membranes such as those present in lipid rafts. To determine whether mitochondrial LRRK2 in wild type LRRK2 overexpressing SHSY5Y cells is associated with mitochondrial membranes mitochondria were fractionated in hypotonic and sodium carbonate buffers. Initially, mitochondrial sub-fractionation was carried out on MEFs isolated by differential centrifugation. Following treatment, MEFs were separated into soluble and pellet components by centrifugation and the corresponding extracts analysed by Western blotting. Western blot analysis of mitochondria isolated by differential 178 centrifugation identified an immunoreactive signal for the inner membrane marker, SDHA, in the pelleted MEF while cytochtome C immunoreactivity, representing a soluble protein in the intermembrane space, was detected in both soluble fraction (20 %) and pellet (80 %) MEFs (Fig 4.16D). The presence of cytochrome C in the soluble compartment prior to fractionation suggested the integrity of mitochondria isolated by differential centrifugation may be partially compromised. LRRK2 was detected in the mitochondrial pellet as determined by 280 kDa V5 immunoreactivity. MEFs subject to treatment in hypotonic buffer resulted in a 3 fold increase in cytochrome C immunoreactivity in the soluble fraction relative to untreated MEFs consistent with cytochrome C release from the mitochondrial inter membrane space (Fig 4.16D). The distribution and relative intensity of the SDHA immunoreactivity in MEFs after the hypotonic treatment resembled that of untreated MEFs suggesting SDHA remained associated with the mitochondria and the inner mitochondrial membrane retained intact. V5 immunoreactivity following hypotonic treatment of MEFs was comparable to that of untreated cells and SHDA suggesting LRRK2 was not readily released from the mitochondria in hypotonic conditions. MEFs subject to sodium carbonate treatment resulted in a 4 fold decrease in the pellet SDHA immunoreactivity with the appearance of an immunoreactive band in the soluble fraction consistent with the release of the integral mitochondrial membrane protein in alkaline conditions. LRRK2 immunoreactive distribution in the presence of sodium carbonate resembled that of SDHA with approximately 30 % of the V5 signal identified in the soluble fraction characteristic of a membrane associated protein. Mitochondrial membrane sub-fractionation of affinity purified mitochondria was also investigated. V5, SDHA and cytochrome C immunoreactive bands were identified in the pellet of the untreated affinity purified mitochondria, absent in the 179 soluble fraction consistent with a better preserved mitochondrial integrity relative to the MEF isolated by differential centrifugation (Fig 4.16E). Cytochrome C was readily released from the mitochondria under hypotonic conditions as determined by the appearance of a cytochrome C immunoreactive signal in the soluble fraction with a corresponding 60 % decrease in the cytochrome C band intensity of the pellet relative to untreated mitochondria. Mitochondrial SDHA and V5 immunoreactivities resembled those of untreated mitochondria in hypotonic conditions as shown by the presence of immunoreactive bands in the pellet extracts only, suggesting both proteins were still associated with the mitochondria. A weak SDHA and V5 immunoreactive band was detected following sodium carbonate treatment of mitochondria in the soluble extracts suggesting SDHA and LRRK2 were partially solubilised from the affinity purified mitochondria in alkaline conditions consistent with the characteristics of membrane associated proteins. A large portion of the SDHA and V5 immunoreactive signals were retained in the pellet fraction. In addition cytochrome C was not fully solubilized with hypotonic treatment for both affinity purified mitochondria and MEF preparations (Fig 4.16D, E) as determined by the immunoreactive band in the pelleted fraction suggesting some mitochondria were still intact. The incomplete solubilisation of membrane proteins under alkaline conditions and intact mitochondrial membranes made it difficult to definitively map LRRK2 to mitochondrial membranes. 180 181 Chapter 5: Results LRRK2 regulation of mitochondrial uncoupling: mechanisms Mitochondrial permeability to protons can be influenced by the opening of the mitochondrial transition pore coupled with the release of free radicals [311] or through reduced integrity of the mitochondrial membrane [312]. Free radical involvement, mitochondrial morphology and LRRK2 mitochondrial association in G2019S cells were assessed in Chapter 4. Mitochondrial uncoupling can also occur through the action of mitochondrial uncoupling proteins [313] which can be regulated transcriptionally and in the mitochondria by post translational modification. The PD associated DJ1 protein has been implicated in UCP transcriptional regulation [168], a pathway which could potentially involve LRRK2. In addition, mitochondrial bioenergetics have previously been shown to be influenced by the DLP1 protein, a putative LRRK2 interacting partner [280], suggesting LRRK2 mitochondrial DLP1 association could contribute to the regulation of mitochondrial uncoupling reported in fibroblasts and SHSY5Y cells. This chapter explores the potential involvement of UCPs, DJ1 and DLP1 in the regulation of mitochondrial bioenergetics by LRRK2 in fibroblasts and SHSY5Y cells. 5.1.1 The analysis of SHSY5Y cells over-expressing murine GFP-UCP2 To determine how increased UCP2 expression influences mitochondrial bioenergetics in SHSY5Y cells, a GFP tagged murine UCP2 construct was transiently transfected into the cells. Confocal microscopy detected GFP fluorescence in transfected cells (Fig 5.1A) consistent with GFP-UCP2 protein expression. Co-staining the cells with the mitochondrial dye, TMRM, confirmed GFP expression in all cells and the superimposed GFP/TMRM fluorescence showed complete co-localization of the two fluorophores consistent with mitochondrial localization of the GFP tagged UCP2 molecule. The pattern of TMRM fluorescence resembled that of untransfected 182 SHSY5Y cells (Fig 5.1B) with no obvious change in mitochondrial morphology associated with GFP-UCP2 over-expression. Quantification of the TMRM intensity relative to control cells identified a 20 % (p<0.0001) reduction in GFP-UCP2 expressing cells (Fig 5.1C). 183 5.1.2.1 The analysis of relative UCP mRNA expression in fibroblasts and SHSY5Y cells Having identified changes in the mitochondrial membrane potential in response to UCP2 over-expression, the G2019S expressing fibroblast and SHSY5Y cell models were screened to quantify relative levels of UCP mRNA and protein. mRNA was extracted from control SHSY5Y cells and fibroblasts cultured under normal growth conditions, reverse transcribed to cDNA and UCP expression quantified by real time PCR analysis as the cycle threshold (CT) value relative to GAPDH CT. Sizes of all UCP PCR products were confirmed by agarose gel electrophoresis (Appendix Fig A2). In control fibroblasts UCP1 mRNA was not readily detected while UCP2 and 3 mRNA expression was approximately 5 and 10 fold lower relative to UCP4 and UCP5 mRNA levels (Fig 5.2A) suggesting UCP5 is the abundant mRNA isoform. In contrast, UCP2 was the most abundant mRNA isoform in control SHSY5Y cells (Fig 5.2B). UCP3 levels were approximately 6 fold lower when compared to UCP2 expression (Fig 5.2B). UCP4 mRNA abundance was 10 and 3 fold lower relative to UCP2 and 3. Expression of UCP5 mRNA was 5 and 1.5 fold lower relative to UCP2 and 3 and 2 fold higher relative to UCP4. Our data confirmed UCP2, 3, 4 and 5 expression in both fibroblasts and SHSY5Y cells and showed a cell specific pattern of UCP mRNA isoform distribution. 184 5.1.2.2 The analysis of relative UCP mRNA expression in G2019S fibroblasts and SHSY5Y cells To determine whether UCP2, 3, 4, 5 mRNA expression was altered in cells expressing G2019S LRRK2, mRNA levels were quantified and compared between control and G2019S fibroblasts and control, wild type and G2019S LRRK2-V5 expressing SHSY5Y cells. Data was expressed as percentage CT value relative to control cells. UCP mRNA abundance was higher in the G2019S fibroblasts relative to control cells (Fig 5.3A) with 300 %, 200 %, 20 % and 15 % increases in the levels of isoforms 2, 3, 4 and 5 respectively. However, only the UCP2 mRNA increase was statistically significant. SHSY5Y cells over-expressing wild type LRRK2-V5 had comparable levels of UCP mRNA to control SHSY5Y cells for all UCP isoforms analysed (Fig 5.3B), suggesting wild type LRRK2-V5 over-expression did not influence UCP mRNA levels. However, G2019S LRRK2-V5 over-expressing SHSY5Y cells showed a 300 % increase in the mRNA levels of the UCP4 isoform with transcript levels of UCPs 2, 3, 5 expressed at similar abundances relative to control and wild type LRRK2 over-expressing cells (Fig 5.3B) consistent with increased UCP4 mRNA expression. 185 5.1.3 Western blot analysis of UCP2 protein in fibroblasts and SHSY5Y cells To determine whether increased UCP mRNA expression correlated with UCP protein, Western blot analysis was carried out on whole cell lysates of fibroblasts and SHSY5Y cells. UCP2 antibody validation was initially preformed on GFP-UCP2 over-expressing SHSY5Y cells. A UCP2 immunoreactive band of approximately 120 kDa (i) was identified in the UCP2 GFP transfected cells absent in untransfected control SHSY5Y cells (Fig 5.4A) potentially corresponding to a GFP tagged UCP2 dimer (predicted UCP monomer 33 kDa, GFP 30 kDa). In addition, a 60 kDa (ii) immunoreactive band of four fold weaker intensity was detected in both the GFPUCP2 transfected and control SHSY5Y cells equivalent to the molecular weight of an untagged UCP2 dimer (Fig 5.4A, B). Additional higher and lower molecular bands at 20 kDa, 55 kDa, 80 kDa and 150 kDa were also present. These bands could not definitively be assigned to a particular UCP2 species and may represent truncated 186 endogenous protein, post-translationally modified UCP2, oligomeric forms or other UCP isoforms as has previously been reported for this antibody [158]. The 66 kDa (ii) immunoreactive species previously assigned to the endogenous untagged UCP2 dimer was detected in GFP UCP2, wild type and G2019S LRRK2-V5 SHSY5Y cell extracts in similar abundance with the equivalent band 3 fold lower in control SHSY5Y cells (Fig 5.4B). The level of the 66 kDa immunoreactive species was three fold lower for both control and G2019S LRRK2 patient fibroblasts relative to control SHSY5Y cells consistent with lower UCP2 mRNA expression in these cell lines. An additional band, common to both cell types, was detected at approximately 60 kDa (iv) with 2-4 fold higher immunoreactivity in fibroblasts relative to SHSY5Y cells. A UCP2 immunoreactive band at 125 kDa (iii) was identified in all fibroblast extracts potentially represented the endogenous UCP2 tetramer [314]. In addition, fibroblast extracts were immunoreactive for 75 kDa and 90 kDa UCP2 species, while SHSY5Y cells produced additional 70 kDa and 73 kDa UCP2 immunoreactive bands. As we were unable to definitively characterise the UCP2 species in UCP2-GFP over-expressing SHSY5Y cells it was not possible to quantify UCP2 levels in fibroblasts and SHSY5Y cells. 187 5.1.4.1 Evaluation of the influence of genipin on UCP induced mitochondrial depolarization in SHSY5Y cells To further investigate the potential role of UCP protein acting downstream of LRRK2 G2019S, the effects on mitochondrial bioenergetics by the previously characterized UCP inhibitor, genipin were tested [315]. The optimal concentration for this compound in SHSY5Y cells was determined by titrating increasing amounts of genipin (1.375-5.5 nM) into control and GFP-UCP2 over-expressing cells, monitoring the change in TMRM intensity over the course of 30 minutes (Fig 5.5A). Untransfected control SHSY5Y cells treated with genipin (1.375 nM) showed a mild, 5 % increase in TMRM intensity potentially due to the influence on endogenous UCPs. Genipin (1.375 nM) increased the TMRM fluorescent intensity in GFP-UCP2 cells by 20 % after 5 minutes of drug addition to levels comparable to those of untransfected control SHSY5Y cells consistent with a rescue of the reduced 188 mitochondrial membrane potential in GFP-UCP2 cells. The increase in the TMRM intensity was maintained for the remainder of the 30 minute time course. There was no obvious change in the TMRM or GFP distribution with cell morphology unperturbed at the lower dose of the drug (Fig 5.5B). GFP-UCP2 over-expressing SHSY5Y cells treated with 2.75 nM genipin showed a linear increase in TMRM intensity over the 30 minutes of the measurement with a 45 % increase in TMRM fluorescence recorded at the last time point (Fig 5.5A). Higher genipin concentrations (5.5 nM) caused a more rapid increase in TMRM intensity but made it difficult to image the cells as the genipin treatment resulted in cell mobility and nuclear shrinkage indicative of toxic side effects (Fig 5.5C, cells marked □). Control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells were treated with 1.375 nM genipin, the concentration of the drug which restored the mitochondrial membrane potential in UCP2-GFP expressing cells, and the mitochondrial membrane potential monitored by TMRM staining over 20 minutes. Consistent with previous measurements, TMRM intensity in control cells increased by approximately 5 % (Fig 5.5D) 5 minutes following genipin (1.375 nM) treatment with a stable TMRM signal intensity maintained for the duration of the time course. The TMRM intensity of wild type LRRK2-V5 over-expressing cells was not affected by genipin treatment while the TMRM intensity of G2019S LRRK2-V5 over-expressing SHSY5Y cells gradually increased in response to genipin, with a 5-10 % increase in TMRM fluorescence after 5 minutes and a 20 % increase after 10 minutes following treatment relative to untreated cells consistent with a restoration of the mitochondrial membrane potential to values resembling those observed for control and wild type LRRK2-V5 over-expressing SHSY5Y cells (Fig 5.5D). 189 5.1.4.2 Evaluation of the influence of genipin on UCP induced mitochondrial depolarization in fibroblasts Two cell lines of control (c1, c7) and three G2019S (p1, p2, p7) patient fibroblast lines were treated with genipin (1.375 nM) and the mitochondrial 190 membrane potential monitored by the intensity of TMRM staining over the course of 30 minutes. In control cells an increase in TMRM fluorescence was recorded over the course of the measurement with the TMRM signal measured to 110 % at 30 minutes relative to that of untreated fibroblasts (Fig 5.6A). However, 30 minutes after genipin addition, a 20 % drop in the TMRM intensity was observed for both cell lines accompanied by nuclear shrinkage and excessive mobility (cells marked □ in Fig 5.6B) in some of the treated control fibroblasts. Three lines of G2019S LRRK2 fibroblasts showed a more pronounced increase in TMRM intensity with a linear increase in TMRM fluorescence from the basal 80 % to 130 % for two of the G2019S lines and to 100 % (Fig 5.6A) for the third G2019S line when compared to untreated control fibroblasts. In contrast to the control fibroblasts, there was no detectable TMRM intensity drop observed for the G2019S cells over the course of the measurement and the cellular morphology remained unchanged following genipin treatment (Fig 5.6C). 191 5.1.5 LRRK2 kinase activity assays with recombinant UCP2 protein UCP activity has previously been shown to be regulated by phosphorylation [159] and the mitochondrial fractionation experiments carried out in Chapter 4 identified LRRK2 associated with mitochondrial membranes. To determine whether 192 LRRK2 could mediate UCP activity directly, in vitro LRRK2 kinase activity assays were carried out on wild type GST-LRRK2 immunoprecipitated from HEK293 cells and recombinant GST-UCP2 protein. Recombinant proteins were generated and LRRK2 kinase assays carried out by Francisco Inesta-Vaquera (Dundee). SDS PAGE and coomassie stain of the components of the kinase activity reaction identified wild type GST-LRRK2 (residues 1326-2527) as the 164 kDa coomassie stained band, absent in control reactions where LRRK2 was omitted (Fig 5.7). GST-moesin, a previously characterised substrate for the LRRK2 kinase was detected as the 110 kDa coomassie stained species absent in GST-UCP2 containing reactions. LRRK2 kinase activity reactions contained increasing GST-UCP2 (2-10 µM) substrate concentrations, as shown by the increase in 54 kDa coomassie band intensity corresponding to GST tagged UCP2 monomer. Additional higher and lower molecular weight species were identified in GST-UCP2 containing reactions, absent in the GST-Moesin lanes (*), however the identity of these species was not investigated. Autoradiography analysis of LRRK2 kinase activity assays containing wild type GST-LRRK2, GST-Moesin or GST-UCP2 detected a 164 kDa band corresponding to the truncated LRRK2 protein consistent with LRRK2 autophosphorylation. In addition, in LRRK2 kinase activity assays containing GST-Moesin, autoradiography detected a 110 kDa band corresponding to radiolabelled GST-Moesin consistent with phosphorylation by GST-LRRK2. In contrast, no clear bands were detected in LRRK2 kinase activity reactions containing GST-UCP2 protein by autoradiography, suggesting LRRK2 was not phosphorylating UCP2 in vitro under the conditions tested. 193 5.1.6 The impact of UCP4 knockdown upon UCP4 mRNA levels and mitochondrial membrane potential in SHSY5Y cells To examine the role of UCP4 up-regulation in G2019S LRRK2-V5 overexpressing SHSY5Y cells we investigated the influence of UCP4 knockdown. Control SHSY5Y cells were treated with UCP4 siRNA or scrambled siRNA for 3 days following which mRNA was extracted, reverse transcribed to cDNA and evaluated by real time PCR analysis. Control SHSY5Y cells treated with 10 nM and 5 nM UCP4 siRNA had 42 % and 46 % UCP4 mRNA levels relative to untreated cells when corrected for GAPDH 194 expression consistent with a marked reduction in UCP4 mRNA levels. Control SHSY5Y cells treated with scrambled siRNA had 21 % higher UCP4 mRNA transcript levels relative to untreated cells (Fig 5.8C). UCP4 mRNA levels in wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells treated with UCP4 siRNA (5 nM) were 53 % and 75 % relative to the corresponding untreated clones consistent with a lower efficiency of UCP4 mRNA silencing relative to that achieved in control SHSY5Y cells (Fig 5.9A). As commercial UCP4 antibodies were not available at the time of the study we were unable to quantify levels of UCP4 protein in UCP4 siRNA treated SHSY5Y cells. However, UCPs have previously been reported to have relatively short half lives [160] hence we investigated whether the reduction in UCP4 mRNA expression was sufficient to alter the mitochondrial membrane potential in these cells. Mitochondrial membrane potential was assessed in untreated and UCP4 siRNA (5 nM) treated control, wild type and G2019S LRRK2 V5 over-expressing SHSY5Y cells in glucose rich respiration buffer by TMRM staining and confocal microscopy. TMRM 195 intensities of control SHSY5Y cells treated with UCP4 siRNA were comparable to those of untreated SHSY5Y cells (Fig 5.9B). UCP4 siRNA treated wild type LRRK2-V5 SHSY5Y cells had 10 % (p<0.0001) lower TMRM fluorescence relative to untreated cells, indicative of a reduced mitochondrial membrane potential. The reason for this decrease is not known. G2019S LRRK2-V5 over-expressing SHSY5Y cells treated with UCP4 siRNA had 6 % (p<0.0001) higher TMRM intensities when compared to untreated cells, consistent with a higher mitochondrial membrane potential (Fig 5.9B) potentially due to lower UCP4 mRNA expression. However due to the low UCP4 siRNA knockdown efficiency in wild type and G2019S expressing cells and the effect of UCP4 siRNA treatment on the mitochondrial membrane potential in wild type LRRK2 over-expressing SHSY5Y cells, we could not fully evaluate the effect of UCP4 knockdown in the SHSY5Y system. 196 5.1.7 The influence of LRRK2 kinase inhibition upon UCP mRNA expression in SHSY5Y cells Bioenergetic assessment of wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells in Chapter 4 indicated the mitochondrial membrane potential and oxygen consumption abnormalities associated with G2019S expression could be partially reversed upon LRRK2 kinase inhibition. To determine whether these changes correlated with UCP mRNA transcript levels mRNA was extracted from 197 control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells following treatment with LRRK2 IN1 (1µM, 90 min), reverse transcribed to cDNA and UCP4 mRNA expression quantified by real time PCR. Data was expressed as the percentage change in UCP4 CT values relative to untreated control SHSY5Y cells. UCPs 2, 4 and 5 were reduced by 15-30 % in control SHSY5Y cells treated with LRRK2 IN1 (Fig 5.10). Wild type LRRK2-V5 over-expressing SHSY5Y cells treated with LRRK2IN1 showed a 10 % decrease in UCP2 mRNA levels but expression of UCP mRNA for isoforms 3-5 showed a considerable amount of variability between experiments making it difficult to evaluate the effect of LRRK2-IN1 treatment on the levels of these UCP isoforms (Fig 5.10). G2019S LRRK2-V5 expressing SHSY5Y cells treated with LRRK2-IN1 had similar UCP abundance for isoforms 2, 3 and 5 relative to untreated control cells. Basal UCP4 mRNA levels were 700 % higher in G2019S LRRK2-V5 over-expressing SHSY5Y cells when compared to control cells but were reduced to 150 % in cells treated with LRRK2 IN1 consistent with lower UCP4 mRNA abundance following LRRK2 kinase inhibition. The reduction in UCP4 mRNA expression in G2019S expressing SHSY5Y cells upon LRRK2 kinase inhibition correlated with a reduced rate of mitochondrial respiration and increased mitochondrial membrane potential (Fig 4.14 A,C). 198 5.1.8 The impact of LRRK2 knockdown upon UCP4 mRNA levels and mitochondrial membrane potential in SHSY5Y cells To investigate the influence of endogenous LRRK2 on the regulation of UCP expression, UCP4 mRNA levels were evaluated in the SHSY5Y LRRK2 siRNA knockdown model. mRNA was extracted from SHSY5Y cells treated with LRRK2 siRNA for 6 days, reverse transcribed to cDNA and assessed for UCP4 mRNA levels by quantitative real time PCR. SHSY5Y cells treated with LRRK2 siRNA showed a 53 % decrease in UCP4 transcript levels relative to untreated cells (Fig 5.11A) supporting a role for endogenous LRRK2 in influencing UCP4 mRNA abundance. Mitochondrial membrane potential was assessed in control and LRRK2 siRNA treated SHSY5Y cells in glucose rich respiration buffer by TMRM staining and confocal microscopy. The 53 % reduction in UCP4 mRNA expression in siRNA treated SHSY5Y cells correlated with a 10 % increase in TMRM intensity consistent a higher mitochondrial membrane potential relative to untreated cells (Fig 5.11B). 199 5.2.1 Characterization of DJ1 knockdown SHSY5Y cells Having shown UCP mRNA transcript levels are affected by changes in LRRK2 kinase activity as well as levels of endogenous protein, we set out to investigate potential upstream events which may contribute to the UCP mRNA expressional changes. DJ1 has previously been implicated in the UCP mRNA regulatory pathway in dopaminergic neurons [168]. To determine whether DJ1 plays a role in regulating UCP mRNA expression in SHSY5Y cells, 2 clones of SHSY5Y cells stably expressing DJ1 shRNA and 1 clone expressing scrambled shRNA were generated by Zhi Yao (UCL). mRNA was extracted from control, DJ1 shRNA and scrambled shRNA expressing SHSY5Y cells, reverse transcribed to cDNA and evaluated for DJ1 mRNA expression by quantitative real time PCR analysis. DJ1 mRNA levels were similar between control and scrambled shRNA treated SHSY5Y cells while DJ1 shRNA expression correlated with 85 % (clone x) and 95 % (clone y) lower DJ1 mRNA transcript levels relative to control SHSY5Y cells (Fig 5.12A). Western blot analysis of whole cell 200 lysates of control, DJ1 shRNA and scrambled shRNA expressing SHSY5Y cells showed comparable 30 kDa DJ1 immunoreactivities in control and scrambled shRNA treated cells (Fig 5.12 B, C) while an 80 % (clone x) and 90 % (clone y) decrease in the 30 kDa immunoreactivity was determined for DJ1 shRNA expressing cells relative to control cells consistent with reduced levels of DJ1 protein (Fig 5.12B, C). 5.2.2 The analysis of UCP mRNA expression in DJ1 knockdown SHSY5Y cells To determine if DJ1 could play a role in the G2019S LRRK2 up regulation of UCP4 in SHSY5Y cells, DJ1 knockdown cells were screened for UCP2-5 mRNA expression. mRNA was extracted from SHSY5Y cells, reverse transcribed to cDNA and UCP expression evaluated by quantitative real time PCR analysis. UCP2 mRNA levels were comparable between control and DJ1 shRNA expressing SHSY5Y cells and reduced by 40 % in the scrambled shRNA expressing cells relative to control cells, 201 although this value was not statistically significant (Fig 5.13A). UCP3 mRNA expression was significantly lower in DJ1 shRNA expressing SHSY5Y cells (40-50 %, p<0.05) relative to untreated cells suggesting the loss of DJ1 is affecting the expression of the UCP3 isoform (Fig 5.13B). UCP4 mRNA expression was comparable between control cells and DJ1 shRNA clone x while a 56 % increase in UCP4 mRNA expression was identified for DJ1 shRNA clone y (Fig 5.13C). UCP4 mRNA levels were 147 % more abundant in scrambled shRNA cells relative to control SHSY5Y cells, although this value was not statistically significant. UCP5 mRNA transcript levels were comparable between control, scrambled shRNA and DJ1 shRNA clone x cells with a 20 % reduction in UCP5 mRNA levels determined for DJ1 shRNA expressing clone y (Fig 5.13D). Due to the lack of a clear role for DJ1 in the regulation of UCP4 mRNA levels we did not pursue with knocking DJ1 down in G2019S LRRK2-V5 expressing SHSY5Y cells. 202 5.3 The influence of LRRK2 expression upon PGC1α, SOD2 and catalase mRNA in SHSY5Y cells UCP mRNA expression has previously been shown to be regulated by PGC1α/PPARγ mediated transcriptional activation by increased PGC1α levels [316]. PGC1α mRNA expression was quantified by real time PCR in control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells. PGC1α mRNA levels were comparable between control and wild type LRRK2-V5 over-expressing SHSY5Y cells while a 70 % increase in PGC1α mRNA was identified for the G2019S LRRK2-V5 cells consistent with increased PGC1α expression (Fig 5.14A). As PGC1α levels have also been shown to regulate expression of antioxidant defence genes including SOD2 and 203 catalase, we evaluated the mRNA levels of SOD2 and catalase in these cells. SOD2 (Fig 5.14C) and catalase (Fig 5.14B) mRNA levels were comparable between the control, wild type and G2019S LRRK2-V5 over-expressing cells. 5.4.1 The analysis of HDAC5 cellular distribution by immunohistochemistry in SHSY5Y cells HDAC5 involvement in PGC1α regulation was investigated by measuring nuclear and cytosolic HDAC5 protein by immunocytochemistry. Control SHSY5Y cells were fixed in paraformaldehyde and permeabilised in 0.1 % Triton X100. Titrating HDAC5 antibody concentrations (50-200 nM) revealed a dose dependent increase in the green fluorescence in SHSY5Y cells (Fig 5.15A). This increase in fluorescence was observed for large nuclear punctate structures (nuclear HDAC5 pool), diffuse cytosolic staining (cytoplasmic HDAC5 pool) and punctate cytosolic structures potentially representing HDAC5 polymers (complexes). Concentrations (200 µM) 204 where the nuclear HDAC5 punctate structures were best resolved were chosen for subsequent experiments. Phorbol myristate acetate (PMA) has previously been shown to induce HDAC nuclear export [317] and was used to test the HDAC5 antibody specificity in SHSY5Y cells monitoring the nuclear to cytosolic HDAC5 redistribution. The PMA dose (100 nM) was chosen based on concentrations previously shown to induce HDAC5 nuclear export in SHSY5Y cells [317]. Control SHSY5Y cells treated with PMA (100 nM, 90 min) resulted in a decrease in HDAC5 immunofluorescent intensity of nuclear immunoreactive punctae with a weak increase in cytosolic HDAC5 immunoreactivity consistent with a partial redistribution of the HDAC5 molecule from the nucleus into the cytosolic compartment. Cells subject to prolonged PMA treatment (3 hours) showed a further reduction in the intensity of the nuclear HDAC5 signal with the nuclear punctate structures appearing less well defined to the ones observed in the untreated SHSY5Y cells (Fig 5.15B). The intensity and frequency of the cytosolic punctate structures were comparable between untreated and PMA treated cells. As we did not observe gross changes in cellular HDAC5 distribution in response to PMA we did not extend the immunohistochemical analysis to wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells. 205 5.4.2 The analysis of HDAC5 by Western blot in SHSY5Y cells HDAC5 protein levels were evaluated in SHSY5Y cells by Western blot analysis of whole cell lysates. The HDAC5 antibody showed strong immunoreactivity for the 100 kDa species on Western blot of control SHSY5Y cells corresponding to 206 the predicted full length HDAC5 protein (Fig 5.16). Comparable 100 kDa HDAC5 immunoreactivities were identified following PMA treatment (100 nM, 90 min) suggesting PMA does not influence cellular HDAC5 levels (Fig 5.16). Comparison of HDAC5 immunoreactivity between control, wild type and G2019S LRRK2-V5 overexpressing SHSY5Y cells revealed similar HDAC5 band intensities relative to GAPDH immunoreactivity correcting for equivalent protein loading, in the absence and presence of PMA consistent with equivalent levels of cellular HDAC5 protein. 5.4.3 The analysis of HDAC5 and LRRK2 interaction HDAC5 immunoprecipitations were carried out on 1 % Triton X100 extracts of LRRK2 over-expressing SHSY5Y cells, control and LRRK2 knockout mouse brain cortex to investigate whether LRRK2 interacts with HDAC5. In addition, HDAC5 immunoprecipitations were carried out on LRRK2 over-expressing SHSY5Y cells treated with PMA in an attempt to enhance the cytosolic HDAC5 levels. Western blot analysis of HDAC5 immunoprecipitates from wild type and LRRK2 knockout mouse brains detected a HDAC5 immunoreactive species of approximately 100 kDa in all brain IPs, absent in IPs where HDAC5 antibodies were omitted (Fig 5.17A). The 207 100 kDa band co-migrated with the HDAC5 immunoreactivity detected in brain IP input lysate consistent with a successful pull-down of a full length HDAC5. Western blot analysis of HDAC immunoprecipitates revealed 3514-1 weak 280 kDa immunoreactivity detected in the wild type brains absent in LRRK2 knockout brain samples. 3514-1 280 kDa immunoreactivity could not be detected in the brain IP input lysate due to low LRRK2 levels, as has previously been shown in Chapter 3. An additional band of 170 kDa (*) was detected by the 3514-1 antibody in HDAC5 IP from mouse brains as well as a weaker intensity signal in the input fraction. As the signal was present in both control and LRRK2 knockout brains it was possible to assume non specific binding of the antibody. The co-IP of LRRK2 with HDAC5 suggested LRRK2 interacted with full length HDAC5 in the mouse brain. To determine whether the LRRK2 HDAC5 association occurs in SHSY5Y cells, HDAC5 was immunoprecipitated from 1 % Triton X100 extracts of control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells. Positive immunoreactivity for the HDAC5 antibody was detected in the IP input sample of SHSY5Y cells, consistent with full length HDAC5 protein (Fig 5.17B). Equivalent HDAC5 100 kDa immunoreactive bands were detected in immunoprecipitates from control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells consistent with comparable efficiencies in the HDAC5 IP (Fig 5.17B). An additional lower molecular weight species immunoreactive for HDAC5 was detected at approximately 90-95 kDa in the IP samples, absent in the IP input lysate (Fig 5.17B), control and LRRK2 knockout mouse brains (Fig 5.17A). The nature of this species was not evaluated. Probing the IP Western blot for V5 immunoreactivity detected a weak immunoreactive band in the wild type LRRK2-V5 over-expressing samples and a 280 kDa species of approximately two fold greater intensity in the G2019S LRRK2-V5 co-IP. No 208 equivalent bands were detected in untransfected control SHSY5Y cells or PMA treated samples suggesting PMA influenced the LRRK2 HDAC5 association. The G2019S LRRK2-V5 protein appeared to be more associated with HDAC5 when compared to wild type LRRK2 as shown by the increase in the V5 band intensity of HDAC5 co-IPs (Fig 5.17B), suggesting LRRK2 kinase activity may play a role in HDAC5 association. To investigate the effects of LRRK2 kinase inhibition on the LRRK2 HDAC5 co-IP, immunoprecipitation was carried out following treatment of wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells with LRRK2 kinase inhibitors (LRRK2-IN1 (1 µM, 90 min) and CZC25146 (0.3 µM, 60 min)). LRRK2 kinase inhibition by LRRK2-IN1 and CZC25146 and potentially PMA was evaluated by immunoprecipitating LRRK2 from 1 % Triton X100 lysates and probing for phosphorylation at Serine 935 by Western blot. LRRK2 Serine 935 phosphorylation was reduced by 80-90 % in both wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells following LRRK2-IN1 and CZC25146 treatment as determined by the band intensity of phospho Serine 935 relative to total LRRK2 (3514-1 antibody) consistent with the inhibition of the LRRK2 kinase activity (Fig 5.17C). In contrast, the phospho Serine 935 immunoreactive LRRK2 band from PMA treated wild type and G2019S LRRK2-V5 SHSY5Y cells was of comparable intensity relative to untreated cells suggesting PMA did not influence the LRRK2 Serine 935 phosphorylation status (Fig 5.17C). HDAC5 was immunoprecipitated from 1 % Triton X100 lysates of untreated, LRRK2 IN1 or CZC25146 treated wild type or G2019S LRRK2-V5 over-expressing SHSY5Y cells. Western blot analysis of HDAC5 immunoprecipitates revealed comparable immunoreactivities in the presence and absence of LRRK2 kinase inhibition consistent with equivalent HDAC5 IP efficiencies (Fig 5.17D). Probing HDAC5 IP Western blots for V5 immunoreactivity revealed an immunoreactive band 209 of approximately 2 fold greater intensity for the untreated G2019S LRRK2-V5 cell line relative to untreated wild type LRRK2-V5 SHSY5Y cells, in line with the previous experiment (Fig 5.17B). The 280 kDa V5 immunoreactive signal was of comparable intensity for wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells treated with both LRRK2-IN1 and CZC25146 LRRK2 kinase inhibitors relative to untreated cells suggesting LRRK2 kinase inhibition does not influence the LRRK2 HDAC5 interaction (Fig 5.17D) as detected by co-IP. An additional 300 kDa band immunoreactive for the 3514-1 antibody (*) was detected in the IP input lysates of both wild type and G2019S LRRK2 over-expressing SHSY5Y cells, the identity of this band was not investigated. 210 5.5.1 The analysis of DLP1 protein in SHSY5Y cells A LRRK2 DLP1 interaction has recently been described [280, 281] correlating with changes in mitochondrial function described for wild type and G2019S expressing SHSY5Y cells. To evaluate the potential role of DLP1 in the regulation of 211 mitochondrial uncoupling in G2019S expressing fibroblasts and SHSY5Y cells, DLP1 levels were assessed in whole cell lysates of control and G2019S LRRK2 fibroblasts by Western blot analysis. 90 kDa DLP1 immunoreactivity was detected in fibroblast extracts corresponding to the predicted molecular weight of the monomeric full length DLP1 (Fig 5.18A). Quantification of the DLP1 immunoreactivity relative to GAPDH confirmed equivalent levels of DLP1 abundance in control and G2019S fibroblasts (Fig 5.18B). Furthermore, DLP1 protein levels were also comparable between control, wild type and G2019S LRRK2-V5 over-expressing SHSY5Y cells as determined by DLP1 90 kDa immunoreactivity relative to GAPDH in whole cell extracts (Fig 5.18C). Mitochondrial levels of DLP1 were evaluated by Western blot analysis of affinity purified mitochondria isolated from wild type and G2019S LRRK2-V5 overexpressing SHSY5Y cells. The band intensity of Core Complex III markers was comparable for wild type and G2019S LRRK2-V5 mitochondrial isolations suggesting similar mitochondrial loading (Fig 5.18D). DLP1 90 kDa immunoreactive bands in extracts of purified mitochondria were of similar intensities for wild type and G2019S LRRK2-V5 expressing cells with comparable DLP1/Core Complex III ratios suggesting similar mitochondrial DLP1 levels. Furthermore, affinity purified mitochondria from wild type and G2019S LRRK2-V5 over-expressing cells treated with LRRK2-IN1 (1 µM, 90 min) revealed comparable DLP1 Core Complex III immunoreactivities. Although mitochondrial DLP1 quantifications were only carried out once (n=1) our data suggested LRRK2 G2019S and LRRK2 kinase inhibition did not impact on steady state mitochondrial DLP1 abundance (Fig 5.18E). 212 5.5.2 The analysis of LRRK2 and DLP1 interaction in SHSY5Y cells To determine whether LRRK2 interacts with DLP1 in SHSY5Y cells LRRK2 was immunoprecipitated from 1 % Triton X100 extracts of wild type and G2019S LRRK2V5 over-expressing SHSY5Y cells. Western blot analysis of immunoprecipitates revealed a 3514-1 immunoreactive 280 kDa band corresponding to the approximate molecular weight of the full length LRRK2 protein, co-migrating with the 280 kDa 213 immunoreactive species in the input fraction and absent in immunoprecipitates of samples where the primary antibody was omitted (Fig 5.19). Western blot analysis of LRRK2 immunoprecipitates detected a 90 kDa DLP1 immunoreactive band corresponding to the full length protein in the input sample, representing approximately 5 % of the total IP input. DLP1 immunoreactivity was not clearly detected in immunoprecipitates where the LRRK2 antibody was omitted, control, wild type or G2019S LRRK2-V5 IP samples at the equivalent exposure times. However, longer exposures detected a faint 90 kDa immunoreactive band corresponding to the molecular weight of the full length DLP1 protein in immunoprecipitates of G2019S LRRK2-V5 expressing SHSY5Y cells absent in control, wild type LRRK2-V5 immunoprecipitates and IPs where DLP1 antibodies were omitted consistent with a small portion of the LRRK2 G2019S mutant pool interacting with the DLP1 molecule. 214 215 Chapter 6: Discussion 6.1 LRRK2 tissue distribution Idiopathic PD is characterized by the loss of dopaminergic neurons in the SNpc while the Braak hypothesis of the pathological spread in PD suggests progressive disease transmission from the lower brain stem regions, midbrain and to the neocortex. The motor dysfunction and cognitive decline associated with the deterioration of these tissues suggests the function of neurons in these brain regions may play an important role in the disease pathogenesis. As LRRK2 mutations are clinically and pathologically indistinguishable from idiopathic PD, LRRK2 tissue distribution may provide an insight into the function of the protein in the context of PD pathology. Previous studies have demonstrated ubiquitous LRRK2 expression, with low levels of LRRK2 mRNA and protein detected in the rodent brain relative to peripheral tissues such as the kidneys lungs and lymph nodes [245]. High LRRK2 expression in peripheral tissues has been linked to cell specific processes such as microbial elimination in bone derived macrophages and RAW264.7 cell lines [241, 243], antibody secretion in B lymphocytes [242] and autophagic control of lung and kidney homeostasis [238, 240]. Although similar processes may be relevant in the brain potentially playing a role in PD pathogenesis, lower levels of LRRK2 mRNA imply neurons are less dependent on these LRRK2 regulated mechanisms, supported by a lack of a neuronal phenotype in LRRK2 knockout animals where kidney atrophy and poor immune responses have been noted [238, 240, 271]. Furthermore, the G2019S mutation does not appear to have an aberrant effect on the LRRK2 functions described in peripheral tissues. The immune response in T cells [271] and microbial elimination by macrophages [241] were unaffected by G2019S transgene expression. Mice expressing bacterial artificial chromosome (BAC) derived wild type and G2019S 216 LRRK2 constructs had normal kidney and lung function [238] suggesting the mutation does not functionally impair these cell types. Proposed brain specific functions for LRRK2 include maintenance of dopamine homeostasis which relies on the function of dopaminergic cells while the dysregulation of this pathway is linked to PD pathogenesis [234]. Striatal dopamine release in mice was enhanced by wild type LRRK2 over-expression and reduced in G2019S transgenic animals, however, dopaminergic cell loss was not observed questioning the suitability of the LRRK2 mouse model in the study of PD [234]. Although LRRK2 expression has been noted in dopaminergic brain areas, with immunohistochemical analysis detecting LRRK2 in the striatum, hippocampus and cortex of rodent and non human primates, substantially lower LRRK2 protein levels were detected in the substantia nigra [244, 249, 250, 318]. The G2019S mutation appears to be linked to the death of dopaminergic neurons in substantia nigra of PD patients which seems to correlate with low SNpc LRRK2 expression suggesting a specific functional role for LRRK2 in this cell type or the preferential vulnerability of these cells to a G2019S LRRK2 associated insult. Our analysis of LRRK2 mouse, rat and marmoset brain distribution by the combined immunoprecipitation and Western blot approach supported previous reports describing the LRRK2 brain expression pattern, detecting greatest LRRK2 levels in the striatum, cortex and cerebellum with lower LRRK2 abundance in the midbrain, olfactory lobe and brain stem. Comparing LRRK2 striatal and cortical expression to that of lymphoblasts detected 50 fold higher LRRK2 levels in the immune cells in support of the expressional differences between peripheral cells and brain tissue. 217 6.2 LRRK2 detection 6.2.1 LRRK2 detection by Immunohistochemistry Methods for LRRK2 protein detection in rodent and primate brains have previously included Western blotting, BN PAGE and immunohistochemistry using in house and commercially available polyclonal LRRK2 antibodies. Although generally a common distribution pattern has been noted (described in section 6.1), discrepancies between groups exist, most pronounced in immunohistochemical studies where LRRK2 expression in neuronal sub populations and glial cells has been debated. In particular immunohistochemical analysis of LRRK2 expression in Purkinje cerebellar granular cells have yielded mixed reports in both human and rodent tissues. Three studies reported positive LRRK2 immunoreactivity [206, 319, 320] while two others failed to detect LRRK2 in this cell subtype [246, 247]. Our characterization of the 3514-1 antibody in wild type mouse cerebellar sections revealed positive staining in this cell subtype absent in the equivalent LRRK2 knockout brain regions. Utilizing the same protocol we were able to detect LRRK2 immunoreactivity in human purkinje neurons of the cerebellum suggesting LRRK2 is indeed present in this cell type in the human brain. LRRK2 expression in brain microglia has also been debated with some reports suggesting glial cells do not express LRRK2 [249] while other studies using the 3514-1 and NB300-268 antibodies detected strong immunoreactivity in activated mouse microglia, human microglia and astrocytes [320, 321]. LRRK2 glial staining was not observed in control mouse or human cerebellar sections described here potentially due to the young age of the animals and absence of immunoinfiltration in the human cerebellum, a point that was not addressed. Considering the emerging literature on the role of LRRK2 in activated macrophages [241] and adhesive monocytes [322], it is possible that 218 LRRK2 does indeed have a role in brain immunoprotection which may be relevant to PD. Although immunohistochemical assessment can distinguish between cell populations in different brain regions, this technique is semi quantitative and cannot be used to compare LRRK2 brain expression between cell types, brain regions and species. In addition, although the specificity of the antibody can be validated by LRRK2 knockout tissue, the size of the immunoreactive LRRK2 species detected cannot be defined. Western blotting on the other hand is quantitative and provides molecular weight estimations for the immunoreactive species. 6.2.2 LRRK2 detection by Western blotting Western blot analysis for LRRK2 detection has been limited by the availability of antibodies that can detect low levels of endogenous LRRK2 protein and poor characterization of the <280 kDa LRRK2 immunoreactive species detected by these antibodies. Biskup et al, [237] [251] screened 8 in house and commercially available LRRK2 antibodies in SHSY5Y cells, M17, PC12, HEK293 cells and brain tissue. All of the antibodies detected multiple bands in both wild type and LRRK2 knockout mouse brains including a 280 kDa band corresponding to the full length LRRK2. The lack of specificity questioned the use of these antibodies [237, 251]. 280 kDa LRRK2 immunoreactivity in HEK293 cells was observed for two of the 8 antibodies and although the data was not shown, the authors stated that they had difficulty detecting endogenous LRRK2 in SHSY5Y, M17 and PC12 cells. The antibody screen described in this report detected 280 kDa LRRK2 immunoreactive species by Western blot in SHSY5Y cells over-expressing LRRK2 absent in control cells corresponding to the predicted molecular weight of the full length protein for all four antibodies analyzed. However, the four antibodies could not definitively identify endogenous 280 kDa LRRK2 in SHSY5Y cells, fibroblasts or brain extracts. In 219 addition to the 280 kDa LRRK2 immunoreactive species, lower molecular weight immunoreactive bands were also detected in all cells and tissues analysed in line with previous observations [237]. Similar to the <280 kDa LRRK2 immunoreactive species in the Biskup et al report, these bands could represent non specific binding of the antibodies however truncated versions of the protein and splice variants of endogenous LRRK2 cannot be ruled out. A number of studies validated in house antibodies for detection of endogenous full length LRRK2 species by siRNA knockdown of the protein in cell culture systems [224, 239, 243]. siRNA knockdown of LRRK2 confirmed endogenous LRRK2 detection in Hela cells [239] and Raw264.7 macrophages [243] potentially reflecting higher LRRK2 abundance in these cell types or improved antibody specificity for conditions used in the study involving HEK293 cells [224]. In all cases, the intensity of the 280 kDa band was reduced in siRNA treated cells consistent with reduced LRRK2 abundance. However, the effect of siRNA on the band intensity of <280 kDa immunoreactive LRRK2 species was not provided with the published data making it difficult to address the nature of these species. Analysis of the LRRK2 coding sequence derived from human SNpc predicted the 280 kDa size of the multidomain protein. When cloned in carcinoma cells, the LRRK2 immunoreactive band resolved at 280 kDa on SDS PAGE consistent with the expression of full length monomeric LRRK2 [203]. As cDNA was cloned into the HEK293 expression system, potential LRRK2 splice isoforms were not investigated. The possibility of tissue specific regulation required to initiate transcription or generate spliced and truncated forms of LRRK2 cannot be ruled out. In particular, BAC LRRK2 derived transcripts (containing the complete LRRK2 gene) appear to undergo post transcriptional or post-translational processing in mouse kidneys to generate a lower molecular weight protein observed by SDS PAGE absent in 220 equivalent extracts of brain or lung tissues and LRRK2 knockout controls [238]. Characterization by mass spectrometry to identify the exact nature of the lower molecular weight species confirmed the 160 kDa band as N terminally truncated LRRK2 [238]. Western blot analysis of LRRK2 protein expression described in this thesis revealed 160 kDa immunoreactivity for the 100-500 and 3514-1 antibodies in lymphoblasts and human brain tissue. As the 100-500 antibody was raised against the LRRK2 N terminus, the 160 kDa N terminally truncated LRRK2 would not bind this antibody, hence the 100-500 160 kDa immunoreactive bands are likely to be non specific. However, the 3514-1 antibody epitope spans the core domain of LRRK2 suggesting the 160 kDa LRRK2 and 3514-1 160 kDa immunoreactivity in lymphoblasts and human brain tissue could represent the LRRK2 N terminally truncated species. In addition, 160 kDa immunoreactivity was identified by 100-500 and 3514-1 antibodies in SHSY5Y cells over-expressing LRRK2, absent in control cells. Alternative LRRK2 splicing was ruled out in this cDNA expressing system, however post translational cleavage of full length LRRK2 may account for these immunoreactive species. LRRK2 Immunoreactive bands >280 kDa have been assigned to SDS stable dimers [244]. These > 280 kDa species were not detected in our analyses and were absent in a range of other antibody screens [237, 249] under a variety of solubilisation conditions tested questioning the reproducibility of these findings. Although phosphorylation of LRRK2 has been confirmed by mass spectrometry the species to size correlation has not yet been fully clarified [202, 211]. 6.2.3 LRRK2 detection by BN PAGE LRRK2 detection by BN PAGE has previously been used to look at the native structure of the protein. In lymphoblasts and HEK293 cells over-expressing LRRK2, LRRK2 was shown to exist predominantly as a dimer and in higher molecular weight 221 oligomeric forms while monomeric LRRK2 could not be detected in these systems under native conditions [228, 323]. The lack of native monomeric LRRK2 was challenged by Berger et al, [252] who demonstrated the 280 kDa as the predominant native species in mouse brain, lymphoblasts and HEK293 over-expressing cells. We investigated the native state of LRRK2 and the specificity of the 3514-1 LRRK2 antibody in over-expressing SHSY5Y cells and endogenous brain tissue using this methodology. LRRK2 V5 immunoreactivity confirmed the equivalent species detected by the 3514-1 antibody represented over-expressed LRRK2 in SHSY5Y cells. A 280 kDa species was the dominant form of LRRK2 detected in over-expressing SHSY5Y cells, human and rodent brains by BN PAGE electrophoresis in line with reports by Berger et al. This may in part be due to the preferential extraction of the soluble pool of LRRK2 using the extraction conditions for BN PAGE, as LRRK2 dimers have previously been shown to be enriched in the membrane fraction [252]. In support of this, inclusion of DDM and Triton X100 detergents during solubilisation of SHSY5Y cells over-expressing LRRK2 increased the recovery of the higher molecular weight LRRK2 species. Although detergents appeared to improve recovery of LRRK2, the resolution and LRRK2 migration on BN PAGE resulted in the formation of immunoreactive smears, previously assigned to higher molecular weight LRRK2 species [228, 323]. Both the V5 and 3514-1 antibodies showed comparable specificities for the monomeric and oligomeric forms of LRRK2 in contrast to SDS PAGE and Western blot analysis where multiple lower molecular weight species were detected by 35141, potentially due to improved specificity of the 3514-1 antibody for LRRK2 under native conditions. Assessment of human and rodent samples by BN PAGE produced a range of LRRK2 immunoreactive species not resolved in the over-expressing SHSY5Y cells 222 potentially due to low levels of tissue soluble LRRK2, supported by the higher ratio of membrane versus soluble LRRK2 in rodent tissue relative to lymphoblasts and LRRK2 over-expressing HEK293 cells [252]. In addition, a 280 kDa immunoreactive band was detected in LRRK2 knockout brain extracts suggesting non specific binding of the antibody may occur under these solubilisation conditions. As mouse LRRK2 knockout tissue has not previously been evaluated by BN PAGE, the true nature of the bands observed in lymphoblasts and brain tissue warrants further analysis. The poor recovery and resolution on Western blot of the LRRK2 immunoreactive species in mouse and human brain extracts did not allow for quantitative comparison of LRRK2 protein levels between different regions of human and rodent brain samples. 6.2.4 LRRK2 detection by immunoprecipitation and Western blotting The immunoprecipitation protocol utilized throughout this study demonstrated specificity and improved sensitivity of antibody detection when compared to Western blot analysis and BN PAGE. The combination of two LRRK2 antibodies consistently produced a 280 kDa species in all cells, rodent and non human primate brain tissue. The LRRK2 immunoreactive band could not be detected in knockout mouse brain tissue and the signal intensity was markedly reduced in SHSY5Y cells upon siRNA treatment. Solubilization conditions producing efficient >90 % LRRK2 recovery and the linear correlation for the LRRK2 signal intensity change relative to protein input made it comparable to the quantitative assessment of whole cell lysates by SDS PAGE [244]. IP assessment suggested the 280 kDa species was the dominant isoform in all models analyzed and that the truncated LRRK2 protein previously validated by mass spectrometry [238] was not detected by this methodology. As the 100-500 antibody was raised against the N terminus of LRRK2, N terminally truncated 160 kDa LRRK2 would not bind the 100-500 antibody. The absence of additional bands implied enhanced specificity for the 250-300 kDa 223 protein or reduced sensitivity for the lower molecular weight species suggesting the combination of IP and Western blot preferentially detects the full length LRRK2. In contrast to the immunohistochemical analyses where LRRK2 was readily detected in both human and mouse cerebellum [248, 251, 319, 320], immunoreactivity was either absent or extremely weak in Western blot analysis of immunoprecipitates from human brain lysates of the frontal cortex, amygdala, caudate nucleus and cerebellum. Even when analysing samples with post mortem delay times as short as 1h30. Due to the lack of a suitable control for the human tissue, the weak intensity identified as LRRK2 immunoreactivity in immunoprecipitates from one human cerebellum extract cannot be definitively confirmed as a true LRRK2 species, an issue which has been encountered in other studies [318]. Analysis of LRRK2 recovery from fibroblast lysates in the presence of human brain samples revealed a reduction in the signal intensity suggesting the human brain sample was interfering with the IP procedure. This phenomenon was not observed in the presence of mouse brain extracts suggesting quantifications of LRRK2 distribution in the rodent brain are not underestimated and the problem with LRRK2 detection in immunoprecipitates from human brain extracts is likely to be influenced by the contents of the human brain lysate. Based on our evaluation of immunohistochemistry, Western blotting, BN PAGE and immunoprecipitation in combination with Western blot analysis as methods for LRRK2 detection and quantification of protein expression, the combined immunoprecipitation and Western blot approach was chosen for quantitative comparison of the relative LRRK2 expression in fibroblast, lymphoblast, SHSY5Y cell lines and dopaminergic mouse brain areas in an attempt to evaluate the suitability of our cell systems as models to study LRRK2 function and the effects of the G2019S mutation in relation to PD pathogenesis. 224 6.3 Evaluation of LRRK2 protein expression in cell models and brain tissue LRRK2 protein was detected in fibroblasts, lymphoblasts, SHSY5Y cells and mouse brain tissue with the greatest expression reported in SHSY5Y LRRK2 overexpressing cells. Although fibroblasts have previously been shown to express LRRK2 mRNA [324], LRRK2 protein expression in this cell type was not demonstrated. Using the combined immunoprecipitation and Western blot approach, LRRK2 protein was detected in control human fibroblasts at 10 and 20 fold lower abundances relative to lymphoblasts and over-expressing SHSY5Y cells respectively. LRRK2 expression in fibroblasts was quantified as 10 fold higher relative to the striatal and cortical mouse brain regions as well as endogenous protein in SHSY5Y cells. Considering fibroblasts express low levels of endogenous LRRK2 and the availability of 8 cell lines from PD patients harbouring the G2019S mutation this cell model was determined as an attractive system to study the generic cellular consequences of G2019S LRRK2 in relation to PD pathogenesis. Lymphoblasts and B lymphocytes, cells from which the lymphoblasts were derived have previously been shown to express high levels of endogenous LRRK2 protein [242, 244] detectable by Western blot analysis of whole cell lysates. However, LRRK2 expression was reported as variable when 5 lymphoblast cell lines were evaluated [228]. We detected endogenous LRRK2 in lymphoblasts by Western blot (NT2, NB300-268 antibodies) and confirmed LRRK2 expression by the combined immunoprecipitation and Western blot approach. Western blot analysis detected variability in LRRK2 expression in 6 lines from control and 8 lines from G2019S LRRK2 PD patients, in line with previous observations [228]. Endogenous expression of LRRK2 protein in the highest expressing lymphoblast cell line was comparable to LRRK2 levels detected in primary PMBCs suggesting LRRK2 expression in lymphoblasts was physiologically relevant. PMBC fractionation revealed high LRRK2 225 expression in B cells and lower abundance in the T cell sub-types consistent with previous observations [242, 325]. Although LRRK2 protein levels were comparable between lymphoblasts and primary PMBCs, the variability in LRRK2 levels between cell lines would complicate the interpretation of the effect of LRRK2 function in relation to PD. However, the ability to detect endogenous LRRK2 by Western blot in lymphoblasts prompted us to study endogenous LRRK2 sub-cellular distribution in this cell type. SHSY5Y cells were chosen as a neuronal cell model to investigate LRRK2 function. Although endogenous LRRK2 expression in SHSY5Y cells was similar to protein levels detected in mouse striatum and cortex, we were limited to endogenous wild type LRRK2 expression in this cell type and were unable to investigate the effects of endogenous G2019S LRRK2. Hence SHSY5Y cells were utilized as a system to look at LRRK2 knockdown. LRRK2 has previously been shown to have a long half life 24-36 hours [201] in HEK293 cells and 3 day LRRK2 siRNA silencing of endogenous protein in RAW264.7 cells reduced the levels of the 280 kDa species by >90 % [241]. SHSY5Y cells described here treated with LRRK2 siRNA for a period of 3 days were shown to have 60 % less endogenous LRRK2 protein as determined by the combined immunoprecipitation and Western blot approach. Extending the LRRK2 siRNA treatment to 6 days achieved an 85 % reduction in endogenous LRRK2 protein in SHSY5Y cells. Although previous reports [257, 326, 327] have used 3 day silencing protocols, the greater knockdown efficiency following 6 days of LRRK2 silencing convinced us to utilize the 6 day silencing protocol to address the functional relevance of the loss of LRRK2 protein in the context of molecular pathways implicated in Parkinson’s disease. The LRRK2 over-expressing SHSY5Y model was generated to address the effects of the G2019S mutation on LRRK2 function in a neuronal system. The relative 226 level of LRRK2 expression in the over-expressing SHSY5Y cells was 2-3 fold greater than levels detected in the highest expressing lymphoblast lines. Considering LRRK2 protein levels in RAW624.7 cells and B lymphocytes have been shown to respond to IFNγ and LPS rising to 10 fold levels of magnitude relative to basal LRRK2 expression [241, 243], the level of LRRK2 over-expression in SHSY5Y cells can be considered high but within the physiological range. Using the combined immunoprecipitation and Western blot approach quantification of endogenous LRRK2 expression in control SHSY5Y cells and rodent striatum determined wild type LRRK2 over-expression in SHSY5Y cells to be 100 fold greater relative to endogenous levels of LRRK2 in the striatum and control SHSY5Y cells. Wild type and mutant LRRK2 over-expression has previously been associated with induction of apoptosis in HEK293 and HeLa cells [254]. Furthermore, overexpression of both the wild type and mutant LRRK2 protein has previously been shown to compromise cell viability in SHSY5Y cells [227] suggesting increased levels of LRRK2 are potentially toxic and questioning the suitability of a LRRK2 overexpressing cell model system. Although dopaminergic cell death is observed in SNpc of PD patients there is no evidence to suggest LRRK2 levels are altered in the PD brain [328]. Furthermore there was no evidence of cell death in brains or peripheral tissues of wild type and mutant LRRK2 transgenic mice suggesting LRRK2 overexpression in animal tissues does not result in apoptosis [240, 329, 330]. In addition, induction of LRRK2 protein expression in peripheral tissues leading to increased LRRK2 protein levels have been linked to functional roles of microbial elimination [243], T cell activation and cytokine secretion [271] with no correlation to apoptosis. Cell viability was not directly assessed in the SHSY5Y over-expressing cells however there was no obvious evidence of cell death or differences in growth rates between control, wild type and G2019S LRRK2 over-expressing cells. The 227 discrepancy between the SHSY5Y system described here and by Iaccarino et al may be due to the differences in the level of over-expression. LRRK2 over-expression in SHSY5Y cells described by Iaccarino et al was under the control of a CMV promoter potentially driving higher levels of over-expression. As the SHSY5Y cells over- expressing LRRK2 presented here have comparable LRRK2 protein levels to those detected in lymphoblasts, greater LRRK2 expression may be required to induce toxicity. A number of studies have reported cellular phenotypes associated with induced or over-expressed wild type LRRK2 protein suggesting functions governed by increased LRRK2 levels which may be independent of G2019S associated processes and relevant to the SHSY5Y LRRK2 over-expressing model. Microbial elimination by bone marrow derived macrophages involved the up-regulation of endogenous wild type LRRK2 protein [241]. Synaptic vesicle endocytosis was impaired by wild type LRRK2 over-expression and LRRK2 siRNA knockdown in primary neuronal cultures [255]. LRRK2 over-expression appeared to deplete the soluble β-tubulin pool in wild type and G2019S transgenic mouse brains consistent with enhanced tubulin polymerization [262]. To help identify artefacts associated with LRRK2 over-expression, the SHSY5Y over-expressing model and fibroblasts expressing endogenous wild type and G2019S LRRK2 were assessed in parallel. 6.4 Evaluation of LRRK2 G2019S protein and mRNA expression As previously described (section 6.3) LRRK2 function can be regulated by induction of expression by LPS, IFNγ and microbial infiltration with subsequent changes in steady state LRRK2 mRNA and protein levels. We set out to determine whether the G2019S mutation correlated with altered LRRK2 expression. LRRK2 mRNA levels have previously been assessed as part of a microarray analysis carried out on the same 6 control and 8 G2019S fibroblast cell lines used in this study, 228 finding comparable LRRK2 mRNA levels in control and G2019S cells [324]. Quantitative real time PCR analysis described here confirmed comparable levels of endogenous LRRK2 mRNA for wild type and mutant fibroblast cell lines. In addition, similar levels of LRRK2 mRNA have been described for control and G2019S LRRK2 PD brains as noted in the amygdala, putamen, cerebellum and frontal cortex regions further suggesting the pathogenic mutation does not correlate with differential LRRK2 mRNA expression [331]. LRRK2 polymorphisms have previously been shown to affect the stability of the LRRK2 protein. The T2397M polymorphism prevalent in patients suffering from inflammatory bowel disease has been demonstrated to result in lower levels of LRRK2 protein in B lymphocytes without affecting LRRK2 mRNA expression [271] suggesting single base amino acid substitutions can affect the half life of the molecule. In addition, engineered kinase dead forms of the LRRK2 protein (D1994S mutant) have consistently been shown to be present at lower levels despite comparable mRNA expression [238], suggesting the protein is less stable. LRRK2 G2019S has been demonstrated to have a half life comparable to the wild type molecule in HEK293 LRRK2 over-expressing cells [201] suggesting the G2019S mutation does not impact on LRRK2 stability. Furthermore, there is no evidence of altered LRRK2 protein levels in G2019S LRRK2 PD brain tissue, although this evaluation has been limited to semi quantitative immunohistochemical analysis of amygdala, putamen, cerebellum and frontal cortex regions [331]. In support of these immunohistochemical findings, the combined immunoprecipitation and Western blot approach determined similar LRRK2 protein levels in control and G2019S fibroblast lines. Although the level of LRRK2 expression was found to be two fold lower in the G2019S LRRK2 over-expressing SHSY5Y cells relative to wild type LRRK2 229 levels, this difference is most likely due to clone to clone variation rather than the processing of the mutant protein. 6.5 LRRK2 cellular localization 6.5.1 Wild type and G2019S LRRK2 cellular distribution As well as having a broad tissue distribution, LRRK2 appears to have a broad sub-cellular localization. Sub-cellular fractionation studies have detected LRRK2 in mitochondria, lysosomal, ER and Golgi enriched compartments with greatest proportion of cellular LRRK2 in the cytosolic and vesicular fraction as described for ectopic LRRK2 in SHSY5Y cells [239], HEK293 cells and endogenous protein in rat whole brain tissue homogenates [201, 251]. The proposed LRRK2 cellular distribution is in line with the data obtained here where LRRK2 was detected in all sub-cellular fractions with the greatest levels identified in the cytosolic and vesicle enriched compartments of SHSY5Y over-expressing cells. Endogenous LRRK2 protein in lymphoblasts had a comparable distribution pattern suggesting LRRK2 overexpression does not lead to gross changes in the localization of the protein. LRRK2 has been identified in synaptosomal isolations from mouse brain tissue with a proposed role in vesicle handling [239, 254]. Identification of LRRK2 in the vesicle enriched fraction in SHSY5Y cells provides a potential link between LRRK2 localization in the brain and synaptic function. Analysis by immunogold labelling and electron microscopy of endogenous LRRK2 in rat basal ganglia identified LRRK2 immunoreactivity in mitochondria, autophagosomes, endosomal and Golgi transport vesicles [251] supported by immunocytochemical analysis of over-expressed LRRK2 co-localization with ER, Golgi, lysosomal and mitochondrial markers in HEK293 and SHSY5Y cells [218, 227, 239, 251]. However, the immunogold analysis was performed without the corresponding LRRK2 knockout controls hence the specificity of the antibody labelling is questionable. Immunohistochemical evaluation of wild 230 type LRRK2-V5 over-expression in SHSY5Y cells described in this report identified a comparable pattern of diffuse antibody staining. Co-localization studies were not carried out, however lysosomal, mitochondrial and ER localization was supported by differential centrifugation analysis identifying 5 % of LRRK2 in cellular compartments containing these organelle markers. In line with these observations, the direct interaction of LRRK2 with multi vesicular bodies and mitochondria has been linked to autophagic regulation of the endocytic lysosomal pathway in BAC-LRRK2 transfected HEK293 cells, as well as mediating mitochondrial function and morphology in SHSY5Y cells and primary neurons [280, 332]. Pathological forms of LRRK2, including kinase domain (G2019S, I2020T) and GTPase (R1441G, R1441C) mutants have previously been linked to the formation of LRRK2 aggregates in HEK293 [215, 228, 298, 333] and SHSY5Y cells [224] overexpressing the protein. Aggregates in wild type LRRK2 over-expressing HEK293 and SHSY5Y cells have also been noted albeit at lower levels. LRRK2 aggregation has been linked to reduced cell viability [333] suggesting aberrant LRRK2 cellular localization may be a feature of G2019S LRRK2 PD. Cellular fractionation experiments identified similar LRRK2 localization for wild type and G2019S LRRK2 in HEK293 and SHSY5Y cells [201, 239]) comparable to the data obtained in our study comparing cellular distribution of wild type and G2019S LRRK2 by differential centrifugation. In addition, although limited to analysis of the wild type protein, our immunocytochemical evaluation of LRRK2 cellular localization did not reveal any LRRK2 positive aggregates in SHSY5Y over-expressing cells. Aggregated LRRK2 structures have not yet been described in PD brains or cell models expressing physiologically relevant levels of LRRK2 suggesting the observed aggregates could occur as a result of LRRK2 over-expression above a certain threshold. The level of 231 LRRK2 over-expression relative to endogenous LRRK2 was not evaluated in any of the studies reporting LRRK2 aggregates [215, 224, 228, 298, 333]. 6.5.2 Wild type and G2019S LRRK2 mitochondrial localization Previously, it has been estimated that 10 % of endogenous LRRK2 can be found in the mitochondrial enriched fraction of rat brain mitochondria isolated by glycerol density gradient [251]. LRRK2 mitochondrial localization has also been evaluated in affinity purified mitochondrial preparations [280, 281] however estimations of LRRK2 mitochondrial abundance were not carried out in these studies. In addition, an accurate comparison of wild type and G2019S LRRK2 mitochondrial levels has not yet been described. 10 % of total cellular LRRK2 was identified in the mitochondrial enriched compartment in the sub-cellular fractionation experiments. Although affinity purification and removal of lysosomal and ER contaminants confirmed that LRRK2 was present in highly purified mitochondria, the level of mitochondrial LRRK2 may well be very low. As a large portion of cytochrome C was lost during the mitochondrial affinity isolation, it is possible that the integrity of affinity purified mitochondria was compromised and a fraction of the outer membrane bound LRRK2 [251] was lost. Nonetheless comparing levels of wild type and G2019S LRRK2 in affinity purified preparations identified similar portions (1-2 %) of the total LRRK2 pool in the mitochondrial fraction further in support of comparable LRRK2 intracellular distribution for the wild type and G2019S protein. ER and lysosomal contamination was not investigated in mitochondria isolated from rat brain and may therefore contribute to the estimated 10 % of the LRRK2 mitochondrial pool [251]. However, mitochondrial LRRK2 levels in the rat brain may also be different to the level of mitochondrial LRRK2 in the over-expressing SHSY5Y cells. Cell specific LRRK2 mitochondrial localization is supported by the barely detectable levels of endogenous LRRK2 in the mitochondrial enriched fractions 232 isolated from lymphoblasts. The presence of LRRK2 in highly purified mitochondria from over-expressing SHSY5Y cells could relate to high LRRK2 levels, however these were only 2-3 fold higher than endogenous expression in lymphoblasts, comparable to the difference in LRRK2 cellular levels between wild type and G2019S overexpressing cells where similar levels of mitochondrial LRRK2 were identified. The differences in mitochondrial LRRK2 levels between lymphoblasts and LRRK2 overexpressing SHSY5Y cells are probably not due to antibody sensitivity problems as the combined immunoprecipitation and Western blot approach could detect LRRK2 immunoreactivity in diluted (1:10) fibroblast lysates containing 100 fold lower levels of LRRK2 protein relative to the equivalent amount of undiluted lymphoblast input. Rather the differences in mitochondrial LRRK2 abundance between the cell types could indicate a cell specific functional regulation. LRRK2 lacks an obvious mitochondrial localization sequence as has been described for the PINK1 protein [46]. The mechanism of LRRK2 mitochondrial targeting has not been investigated. Interactions with 14-3-3 proteins are recognized mechanisms regulating the mitochondrial localization of certain proteins including Bax [334]. LRRK2 is known to interact with 14-3-3 which is disrupted when LRRK2 kinase is inhibited [216]. However, inhibition of LRRK2 kinase activity disrupts 14-3-3 binding but did not influence mitochondrial LRRK2 levels suggesting 14-3-3 binding was not regulating its mitochondrial localization. 6.5.3 LRRK2 localization in the mitochondria 25-50 % of the total LRRK2 pool is believed to be membrane associated suggesting membrane association could play a role in the LRRK2 mitochondrial interaction [252]. LRRK2 association with the outer mitochondrial membrane in isolated rat brain mitochondria has previously been described [251]. In support of this LRRK2 was shown to behave like a mitochondrial membrane bound protein as 233 determined by the partial release of LRRK2 from the mitochondria under alkaline conditions. Hypotonic conditions did not appear to affect mitochondrial LRRK2 abundance further suggesting that mitochondrial LRRK2 was membrane associated as the membrane unbound protein cytochrome C was released under these conditions. Sterically, an interaction between LRRK2 and the outer mitochondrial membrane is most plausible. In addition, the full length 280 kDa molecule is too large for import through transmembrane channels [335]. Our limited data supported the LRRK2 association with the outer mitochondrial membrane however attempts to accurately map the LRRK2 mitochondrial localization were hampered by a number of technical issues. The shift of the inner membrane markers to the soluble fraction while the outer membrane markers were present in the pellet suggested the outer membrane was still intact when the inner membrane was already compromised, observed for both crude and pure preparation. We also experienced problems with the proteinase K digestion and the preferential removal of the outer membrane. Loss of mitochondrial integrity, as determined by cytochrome C release during mitochondrial affinity purifications and MEF isolations may have prematurely exposed the inner membrane to digitonin and proteinase K. As both methods have previously been successfully carried out on SHSY5Y cells [336, 337], additional optimization of the technique may be required focusing on preserving the integrity of mitochondrial isolations. 6.6 Wild type and G2019S LRRK2 kinase activity LRRK2 has previously been demonstrated as an active kinase in in vitro assays utilizing full length immunoprecipitated LRRK2 protein from HEK293 cells and lymphoblasts as well as a truncated version of LRRK2 lacking the N terminus purified from HEK293 cells (GST-LRRK2 1362-2527) [202], monitoring the incorporation of 32P 234 ATP into artificial substrates (LRRKtide) and LRRK2 interacting partners MBP [202]. In the same assays the pathological G2019S LRRK2 mutant has consistently been shown to have higher kinase activity when compared to the wild type protein [338] while inhibition of LRRK2 kinase activity by LRRK2-IN1, CZC25146 and TAE-684 has been shown to correlate with dephosphorylation of LRRK2 at Serine 910 and 935 [214, 215, 339]. LRRK2tide phosphorylation was detected in in vitro kinase assays of LRRK2 immunoprecipitated from lymphoblasts and over-expressing SHSY5Y cells, confirming these cell models expressed an active LRRK2 molecule. LRRK2 specific activity was 3-5 times lower for LRRK2 immunoprecipitated from over-expressing SHSY5Y cells relative to lymphoblasts suggesting LRRK2 is less active in this cell type. Although the discrepancy may be due to cell specific differences, the C terminal V5 tag may have impacted on the kinase activity of the protein. In relation to this, truncations of 7 C terminal residues in the LRRK2 WD40 domain have previously been shown to ablate the kinase activity of LRRK2 suggesting the C terminus is potentially involved in enzymatic regulation of the protein [202]. However, Serine 935 phosphorylation in wild type and G2019S LRRK2 over-expressing SHSY5Y cells was sensitive to LRRK2 kinase inhibition as determined by Western blot analysis of the immunoprecipitated protein further suggesting the tagged enzyme was active in this cell type. In addition, the inhibitor doses that were required to reduce LRRK2 Serine phosphorylation by >90 % were comparable to those shown for enzymatically active, N-terminally tagged LRRK2 immunoprecipitated from HEK293 cells [215] suggesting LRRK2 kinase activities were similar in the two systems. The kinase activity of G2019S LRRK2 immunoprecipitated from SHSY5Y overexpressing cells was 2 fold greater relative to the wild type protein in line with previous observations [338]. The potency of LRRK2 IN1 was greater for G2019S relative to wild type LRRK2 immunoprecipitated from SHSY5Y cells, implying G2019S 235 LRRK2 has greater affinity for ATP as LRRK2 IN1 is an ATP competitive inhibitor, consistent with enzymatic hyperactivation associated with the pathogenic mutant. The kinase activity of LRRK2 expressed in control and G2019S patient fibroblasts was not evaluated due to the low level of LRRK2 expression in this cell type. LRRK2 kinase activity in fibroblasts has not been reported in the literature, however the effects of the G2019S mutation on LRRK2 kinase activity have been described for kidney and immune cells lines [215] for endogenous and over-expressed LRRK2 suggesting the increased kinase activity associated with the mutant protein is not dependent on the cell type and G2019S fibroblasts are likely to have increased LRRK2 kinase activity relative to control cells. 6.7 LRRK2 and mitochondrial function Mitochondrial respiratory chain abnormalities are a common feature of idiopathic, genetic PD and experimental models of the disease; reduced Complex I activity has been described for idiopathic PD substantia nigra, platelets and skeletal muscle [99, 340] suggesting respiratory function was compromised. The parkinsonian phenotype induced by systemic MPTP injections in mice correlated with a reduction in mitochondrial respiration [341, 342]. Various genetic causes of PD have compromised mitochondrial function; Complex I defects have been noted in fibroblasts harbouring PD associated PINK1 mutations [343] while reduced mitochondrial respiration and mitochondrial membrane potential correlated with the loss of PINK1 in mouse embryonic fibroblasts [344]. Parkin deficiency in Drosophila melanogaster and PD patient derived parkin mutant fibroblasts were associated with reduced oxygen consumption rates [345, 346]. α-synuclein overexpression in primary cells was shown to inhibit Complex I activity [123]. Mitochondrial membrane potential and ATP levels were reduced in SHSY5Y cells, primary cortical cultures [280, 281] and fibroblasts [285] expressing G2019S LRRK2 236 consistent with abnormal respiratory chain function, suggesting respiratory chain dysfunction may be a common feature in different forms of PD and potentially involved in the disease pathogenesis. Although variability was detected in ATP measurements for control and G2019S fibroblasts described in this report, as a group G2019S cells had reduced ATP levels, consistent with the previous observations [280, 281, 285]. In addition, G2019S LRRK2 expression correlated with a 20 % reduction in mitochondrial membrane potential in SHSY5Y and fibroblast cell models. The reduction in mitochondrial membrane potential in SHSY5Y cells was comparable to what has previously been described [280, 281], while fibroblast showed a more modest defect in comparison to the 60 % decrease determined in the previous report [285]. Of note, Mortiboys et al, determined an 85 % reduction in the mitochondrial membrane potential in control fibroblasts treated with parkin siRNA [347], a decrease much greater than those generally reported for viable cells. Reductions of 15-20 % in the mitochondrial membrane potential have previously been observed for fibroblasts derived from patients with PINK1 mutations and PINK1 deficient mouse embryonic fibroblasts [172, 348] consistent with the magnitude of changes reported for our G2019S expressing fibroblasts. In addition, fibroblasts have previously been shown to adjust to gross mitochondrial defect such as complex I enzymatic deficiency by reversing the mitochondrial ATPase [310]. We have modelled this in control fibroblasts exposed to rotenone and demonstrated ATPase reversal by the response of the rotenone treated cells to oligomycin which led to the dissipation of the mitochondrial membrane potential. Hence a 60 and 85 % decrease in mitochondrial membrane potential is physiologically unlikely as the fibroblasts would most likely attempt to adjust to such a massive perturbation in the mitochondrial proton gradient. The discrepancy in the mitochondrial membrane 237 potential measurements is therefore likely due to an over-estimation of the magnitude of the defect. Nonetheless a common trend was observed among all the LRRK2 studies detecting a reduction in the mitochondrial membrane potential and ATP levels associated with G2019S LRRK2. Oxygen consumption measurements detected a 1.5-2 fold increase in cellular respiration rates in G2019S expressing fibroblasts and SHSY5Y cells. The increase in cellular respiration in G2019S expressing cells opposed the respiratory deficiencies reported for PINK1 deficient mouse embryonic fibroblasts [348], MPTP and rotenone treated mice [342] and parkin deficient Drosophila melanogaster [345]. The reduction in basal ATP levels and mitochondrial membrane potential have previously been assigned to a mitochondrial defect associated with G2019S LRRK2 expression consistent with the respiratory chain abnormalities linked with the genetic and experimental PD models [280, 281, 285]. Although the decrease in ATP levels and mitochondrial membrane potential detected in G2019S expressing fibroblasts and SHSY5Y cells mirrored the characteristics of bioenergetically compromised cells, increased oxygen utilization argued against a respiratory chain defect. Measurements of chemically uncoupled respiration revealed comparable cellular respiratory capacities for control and G2019S fibroblasts, further arguing against a mitochondrial dysfunction. Mitochondrial respiration of fibroblasts cultured under substrate limiting conditions was comparable to those observed in G2019S expressing cells suggesting mutant LRRK2 expressing cells could suffer from deficient glycolytic input, as has previously been shown for PINK1 deficient neuroblastoma cells and mouse primary dopaminergic neurons [172]. However, mitochondrial membrane potential was increased in fibroblasts cultured on galactose consistent with an increased flux 238 though the mitochondrial respiratory chain, different to the effect on the mitochondrial membrane potential observed in G2019S expressing fibroblasts and SHSY5Y cells. In addition, the respiratory chain defect described in PINK1 and parkin deficient cells has been associated with mitochondrial ATPase reversal in primary cortical neurons and SHSY5Y cells [172] as determined by the dissipation of the mitochondrial membrane potential in the presence of oligomycin. Oligomycin treatment produced comparable effects on the mitochondrial membrane potential in control and G2019S fibroblasts consistent with a normal functioning ATPase. As there was no evidence for limitation in glycolytic substrate supply or mitochondrial ATPase reversal in G2019S LRRK2 expressing fibroblasts and SHSY5Y cells these systems are likely to have a different underlying cause to the mitochondrial bioenergetic changes associated with mutant LRRK2. In fact, the combination of reduced mitochondrial membrane potential and enhanced respiration supported the presence of a mitochondrial proton leak [349]. Bioenergetically, a mitochondrial proton leak has previously been characterized by oxygen utilization measurements in the presence of oligomycin; a respiration rate was detected in oligomycin treated thymocytes over-expressing UCP2 [350] absent in control cells. Oligomycin inhibited respiration rates have also been shown to contribute to skeletal contractions in mice [351]. In support of a mitochondrial proton leak, oxygen utilization was taking place in G2019S expressing fibroblasts in the presence of oligomycin, absent in control cells. 6.8 LRRK2 and mitochondrial permeability The decrease in mitochondrial membrane potential combined with increased oxygen utilization rates associated with G2019S expression implied an increased permeability of the mitochondrial inner membrane to protons. This may involve opening of the mitochondrial permeability pore [352], the presence of 239 proteins on mitochondrial forming a pore [353] or the up-regulation and activation of uncoupling proteins [354]. Opening of the mitochondrial permeability pore is associated with increased reactive oxygen species (ROS), increased mitochondrial calcium levels and apoptosis [352]. Respiratory chain Complex I defects, a prominent feature in PD patient brains and skeletal muscle [99] as well as PINK1 deficient fibroblasts [343], have correlated with increased oxidative stress, apoptosis and mitochondrial permeability pore opening [348]. Wild type and mutant LRRK2 over-expression in SHSY5Y cells has previously been shown to promote PTP opening through the interaction between adenine nucleotide translocator (ANT) and porin [286]. In addition, SHSY5Y cells over-expressing the G2019S LRRK2 protein showed increased cytochrome C release, the activation of the caspase cascade and increased cell death [227]. However, there was no evidence of cell death in our G2019S expressing fibroblasts or SHSY5Y cells. In agreement with this thesis, oxidative stress levels were comparable between wild type and G2019S over-expressing SHSY5Y cells in one report [280] but in another study greater for G2019S expressing SHSY5Y cells [281]. Oxidative stress was also reported in G2019S expressing fibroblasts relative to control cells [285] however the rate of ROS generation in G2019S fibroblasts described in this report were lower relative to control cells. Hence the effects of G2019S expression on ROS generation are not consistent and may vary depending on cell type and assay conditions. The absence of cell death and increased ROS levels suggested the mitochondrial permeability transition pore was unlikely to cause the mitochondrial uncoupling in G2019S expressing fibroblasts and SHSY5Y cells. Regarding any direct interaction of LRRK2 with mitochondria leading to increased proton permeability of the inner membrane, we confirmed that a small proportion of LRRK2 was present in highly purified mitochondria. Further 240 mitochondrial sub-fractionation suggested LRRK2 associated with mitochondrial membranes, however, we were not able to restrict this to either the outer or inner membranes. The mitochondrial uncoupling in G2019S SHSY5Y cells was not likely to be an artefact of over-expressed LRRK2 localizing to the mitochondria as uncoupling was also observed in fibroblasts expressing endogenous G2019S LRRK2 and absent in SHSY5Y cells over-expressing the wild type protein. The mitochondrial uncoupling associated with G2019S mutant fibroblasts and SHSY5Y cells closely resemble what has previously been described for cells and tissues with increased uncoupling protein expression. Increased oxygen consumption rates, reduced mitochondrial membrane potential and ROS production have been reported for mitochondrial isolations from mouse brains, livers and kidneys of UCP2 transgenic mice [355] as well as CHO cells over expressing UCP1 [356]. Reduced membrane potential has been demonstrated for mitochondria in UCP4 over-expressing cells [357] and UCP4/5 reconstituted in liposomes [358]. In line with these observations, we observed a reduced mitochondrial membrane potential in SHSY5Y cells over-expressing the UCP2-GFP protein. 6.9 LRRK2 regulation of mitochondrial uncoupling proteins 6.9.1.1 LRRK2 regulation of UCP mRNA expression Mitochondrial uncoupling has been demonstrated to be regulated by specific UCP isoforms in different cell types; UCP1 is the dominant isoform in brown fat reported to regulate thermogenesis [359], while the UCP2 isoform is found in pancreatic β-cells and mediates insulin secretion through ATP mediated opening of potassium channels [360]. Expression of four UCP isoforms (UCP2-5) was detected in control fibroblasts and SHSY5Y cells, suggesting these proteins may have a functional role in these cell lines. UCP2, 4 and 5 mRNA expression has been reported in fibroblasts, although UCP3 mRNA expression was not investigated [361]. UCP2 has 241 previously been identified as the abundant UCP isoform in the rodent brain, expression of UCP4 and 5 was also described consistent with the pattern of UCP expression detected in SHSY5Y cells [362]. The LRRK2 G2019S mutation correlated with an increase in UCP2-5 mRNA in fibroblasts and increased UCP4 expression in SHSY5Y cells. The UCP mRNA changes observed in G2019S expressing fibroblasts and SHSY5Y cells suggested the LRRK2 associated mitochondrial uncoupling was regulated at the transcriptional level. Increased UCP4 mRNA expression has previously been demonstrated in SHSY5Y cells [363] and brain in experimental stroke models [364] while UCP2 expressional changes in response to PGC1α over-expression have been described in myotubes, confirming that these isoforms can be transcriptionally regulated. Of note, increased UCP4 levels in G2019S LRRK2 SHSY5Y cells suggested the artificial UCP2 overexpressing system used in this study may not be an accurate model of uncoupling for this cell type. The fold changes in UCP4 mRNA expression were comparable to 5 fold changes reported for UCP1 expression in brown fat comparing tissue isolated from warm acclimated mice to those exposed to cold temperatures [365]. A similar 2-3 fold induction in UCP3 mRNA has been described in adipocytes and skeletal muscle associated with reduced body weight in mice suggesting small changes in UCP mRNA levels are sufficient to induce phenotypic changes [366]. 6.9.1.2 LRRK2 regulation of UCP protein Changes in UCP2 and 3 mRNA have previously been shown to affect UCP protein levels in pancreatic β cells [367]. While ectopic UCP2 expression in the UCP2GFP SHSY5Y model was confirmed by confocal microscopy and Western blot, Western blot analysis detected additional higher and lower molecular weight immunoreactive species making it difficult to identify and quantitively evaluate 242 endogenous UCP2 protein in wild type and G2019S fibroblasts and SHSY5Y cells. Cross reactive bands of 28 and 50 kDa have previously been described for UCP2 over-expressing insulinoma cells [368] and in a variety of mouse and rodent tissues [368] suggesting antibody specificity may not be reliable enough to distinguish between true endogenous species. To determine the potential contribution of UCP protein to the mitochondrial uncoupling observed in G2019S fibroblasts and SHSY5Y cells, UCP activity was inhibited by genipin. Although genipin is often described as a general UCP inhibitor, the characterization of the drug has been limited to in vitro studies involving UCP2 protein [315] and its selectivity profile for the other UCP family members has not been fully elucidated. Studies of pancreatic islet cells have previously shown genipin to increase the mitochondrial membrane potential, ATP levels and promote insulin secretion, a function governed by UCP3 [315] while inhibition of mitochondrial proton leak in leukemic MX2 cells correlated with lower ROS levels and reduced drug resistance of the cancer cells [369] linking UCP2 protein activity and inhibition to respiratory chain function. Consistent with UCP mRNA expression in control SHSY5Y cells and fibroblasts, a mild 10 % increase in mitochondrial membrane potential was observed upon genipin treatment suggesting a role for UCP proteins in the regulation of respiratory chain function. In addition, restoration of the mitochondrial membrane potential in UCP2-GFP cells with genipin suggested the drug could counteract the phenotype of UCP2 protein over-expression. The specificity of genipin for the UCP2 isoform was questioned by the effects of the compound on the G2019S LRRK2 overexpressing SHSY5Y cells, where the UCP4 mRNA expression was preferentially induced. As UCP3 and 5 were found to be expressed in SHSY5Y cells and UCP2 mRNA was expressed at greater levels relative to UCP4 mRNA, the observed effect could be 243 due to genipin inhibition of all the UCPs in SHSY5Y cells. UCP2 mRNA levels were comparable for control, wild type and G2019S over-expressing clones however, we cannot rule out the possibility of increased UCP2 protein in SHSY5Y cells expressing G2019S LRRK2. Although it was not possible to specifically inhibit UCP2 and UCP4 in fibroblasts and SHSY5Y cells respectively, restoration of the mitochondrial membrane potential in G2019S expressing cells upon genipin treatment supported the role of UCP protein in the mitochondrial uncoupling associated with mutant LRRK2. Of note, effective genipin concentrations reported here were considerably lower with respect to ranges described in the literature. Genipin at 5 µM was required to restore the UCP dependent contractile response in cardiac myocytes [370]. Genipin at 10 µM was determined as the effective dose for toxicity in leukemic cells [369] while 200 µM genipin induced toxicity in hepatoma cells [371]. Although toxicity was not apparent in our short term genipin incubations for SHSY5Y cells, the mitochondrial membrane potential in control fibroblasts was reduced by 20 % in prolonged treatments (30 min) suggesting this cell type is more sensitive to the drug. The cell toxicity associated with genipin treatment is likely due to the off target effects associated with the drug. In support of this, there is no evidence to suggest siRNA knockdown of UCPs 2 and 4 lead to reduced cell viability [363, 372]. Effective genipin dose requirements for UCP inhibition in different cell types could potentially correlate with the levels of expression of the different UCP isoforms. 6.9.1.3 LRRK2 regulation of PGC1α expression The difference in the relative mRNA abundance between the UCP isoforms in SHSY5Y cells and fibroblasts as well as induction of specific UCPs (2 and 4) in these cells may reflect cell specific regulatory roles such as those described for adipose tissue and the regulation of thermogenesis by UCP1 and pancreatic β cells in the 244 modulation of insulin secretion by UCP2 [359, 360]. Tissue specific UCP4 and 5 regulation has also been described for dopaminergic neurons in the SNpc, required for the regulation of pacemaking activity [168]. Therefore LRRK2 regulation of different UCP isoforms in fibroblasts and SHSY5Y cells may be required for cell specific mitochondrial uncoupling mediated functions. Cell specific UCP mRNA expression has previously been linked to PGC1α mediated transcriptional regulation. Stimulation of PGC1α expression was described for brown adipocytes undergoing differentiation and developing muscle, processes requiring induction of UCP1 and UCP2 transcription [373, 374]. In these studies UCP1-3 expression was detected in both cell types however only UCP1 or 2 mRNA was preferentially stimulated in the two studies [373, 374]. Increased UCP4 mRNA levels correlated with a two fold increase in PGC1α expression in G2019S LRRK2 over-expressing cells suggesting PGC1α may play a role in the specific regulation of UCP4 expression. PGC1α mediated UCP2 regulation has previously correlated with an increase in catalase and SOD2 mRNA in endothelial cells undergoing oxidative stress [141]. In addition, increased mitochondrial densities in white adipose tissue [375] and muscle cells [376, 377] correlated with PGC1α mediated UCP1, 2, TFAM and Nrf1, 2 expression in line with stimulation of mitochondrial biogenesis suggesting the increase in UCP2 and UCP4 expression could be a part of a biosynthetic or antioxidant gene response. The relative expression of antioxidant enzymes SOD2 and catalase were comparable between control, wild type and G2019S expressing cells suggesting the increased UCP4 expression associated with the G2019S mutation was not a part of a global antioxidant response. Furthermore comparable abundance of mitochondrial proteins, TFAM and mitochondrial structures in control and mutant fibroblasts and SHSY5Y cells implied rates of mitochondrial biogenesis 245 were not perturbed. The absence of an oxidative stress response and changes to mitochondrial abundance implies the induction of UCP expression in the presence of G2019S is unlikely a stress response and selectively targets the UCP gene. The selectivity for UCP4 induction could potentially be accounted for by PGC1α interacting molecules in the relevant cell types. PGC1α mediated UCP response in skeletal and cardiac muscle as well as brown adipose tissue has previously been shown to be dependent on the isoform of its interacting gene regulator PPAR. UCP3 expression in skeletal muscle is more responsive to the PPARδ than PPARα isoform [378, 379] and UCP3 mRNA induction in cardiac muscle requires PPARα [380]. While induction of UCP2 expression in skeletal muscle and heart is responsive to PPAR related stimuli in a similar manner to UCP3, additional regulatory mechanisms exist [381]. PGC1α in muscle cells requires co-activators MyoD and p300 for UCP2 gene stimulation [382]. In this study, the direct regulation of the UCP promoter by PGC1α was not investigated and warrants further analysis to determine whether a specific PPAR isoform and additional regulators are involved. The selective changes in UCP4 mRNA further emphasise the importance of UCP regulated mitochondrial uncoupling in G2019S LRRK2 expressing fibroblasts and SHSY5Y cells. 6.9.1.4 LRRK2 regulation of HDAC5 Based on the sub-cellular localization data for both endogenous and overexpressed LRRK2, a large portion of the full length protein pool appeared to exist in the cytosolic compartment. The multiple structural domains of LRRK2 implicate a scaffold function in signalling cascades and the reported association with 14-3-3 [215, 216, 383] suggested LRRK2 may potentially be involved in compartmental targeting of 14-3-3 interacting molecules while 14-3-3 could in turn regulate LRRK2 function. LRRK2 mediated regulation of nuclear to cytosolic shuttling has been 246 described for the nuclear factor of activated T cells (NFAT) in T cells where LRRK2 was identified as a crucial component of the NRON complex maintaining NFAT in its inactive state in the cytosol [271]. Loss of LRRK2 was linked to the translocation of NFAT to the nucleus and hyperactivation of transcription. HDAC5 mediated PGC1α transcription has previously been shown to also involve nuclear translocation in response to protein kinase A (PKA) and protein kinase C (PKC) [317]. Hypothesising a mechanistic conservation, LRRK2 association with HDAC5 was investigated to determine what effect LRRK2 kinase activity has on the cellular localization and integrity of this complex and how the potential LRRK2 HDAC5 interaction could be involved in PGC1α transcription and UCP expression. In an attempt to determine what effect LRRK2 G2019S has on HDAC5 nuclear translocation, HDAC5 subcellular localization was investigated. Under basal conditions HDAC5 has been shown to localize to the nucleus and cytoplasm with the relative nuclear to cytosolic ratios depending on the cell system in question [317, 384]. HEK293 and HeLa cell lines have nuclear HDAC5 abundance while the localization of HDAC5 in H9C2 rat heart cell line was shown to vary depending on the differentiation status [385]. In addition, PMA treatment of cells in these studies has been shown to mobilize the HDAC5 pool and result in the complete redistribution of HDAC5 to the cytoplasm. HDAC5 nuclear export in response to PMA has been demonstrated in Cos7 cells [317, 384]. The antibodies used for immunocytochemistry have been limited to HDAC5 over-expressing systems with no reports of endogenous HDAC5 localization in SHSY5Y cells yet described [317, 384]. We detected endogenous HDAC5 in the nucleus and cytosol of SHSY5Y cells by immunocytochemistry, however PMA treatment of wild type cells did not grossly affect the nuclear HDAC5 pool suggesting antibody specificity may be an issue as the same PMA conditions have consistently been shown to lead to HDAC5 nuclear 247 export in SHSY5Y cells [317]. As we did not have a suitable HDAC5 knockout system to validate the HDAC5 antibody for immunohistochemistry, analysis of HDAC5 cellular distribution in wild type and G2019S LRRK2 over-expressing SHSY5Y cells was not carried out. Immunoprecipitation of the HDAC5 protein has previously been carried out on Flag tagged, over-expressed species in COS7 cells [386] and GFP tagged HDAC5 in SHSY5Y cells [387], detecting 120 kDa immunoreactive bands corresponding to the full length tagged molecule. Although the relative molecular mass of the HDAC5 immunoreactive band detected in HDAC5 immunoprecipatates matched the molecular weight of the predicted and previously described full length HDAC5, an additional lower molecular weight species was also detected which has not previously been reported for IPs of the HDAC5 protein. To determine whether the 80 kDa species detected here is an artefact associated with the HDAC5 antibody, additional characterization of the antibody should be carried out on HDAC5 knockout tissue. Nevertheless LRRK2 was detected in HDAC5 immunoprecipitated from wild type and G2019S LRRK2 over-expressing SHSY5Y cells and wild type but not LRRK2 knockout mouse brain cortex. Immunoprecipitation of LRRK2 has previously identified NFAT in T cells [271], Ago2 in the aged fly brain [273], 14-3-3 in HEK293 cells and lymphoblasts [216], DLP1 in SHSY5Y cells and primary cortical neurons [280, 281] as potential LRRK2 interacting molecules. LRRK2 interaction with DLP1 was enhanced for the G2019S LRRK2 mutant in SHSY5Y cells [280], the human Ago2 protein pulled down more of the G2019S LRRK2 than the wild type [273], LRRK2 kinase inhibition has previously been shown to correlate with the amount of bound 14-3-3 [215, 216] suggesting LRRK2 kinase activity may play a role in complex formation. However, although 14-3-3 binding to LRRK2 was affected by LRRK2 kinase inhibition, the 248 G2019S mutation did not impact on the amount of 14-3-3 associated with LRRK2 [215] suggesting additional regulatory factors may be involved in this interaction and the regulation of downstream signalling cascades. Association of NFAT with the NRON complex required the LRRK2 protein but the affinity of the interaction did not seem to be affected by the G2019S mutation [271] suggesting the integrity of complexes involving LRRK2 may not always depend on LRRK2 kinase status. The intensity of the LRRK2 immunoreactive species detected in the HDAC5 pulldowns was consistently stronger in G2019S LRRK2 over-expressing SHSY5Y lysates relative to wild type LRRK2. The efficiency of HDAC5 pulldown was similar between control, wild type and G2019S LRRK2 over-expressing suggesting that the G2019S LRRK2 protein was more associated with HDAC5, similar to the effects described for G2019S LRRK2 association with Ago2 and DLP1 proteins. Although not investigated in the studies involving Ago2 and DLP1, LRRK2 kinase inhibition did not seem to affect the integrity of the HDAC5 interaction, in contrast to what has previously been shown for 14-3-3 suggesting Serine 910 and 935 phosphorylation is not required for HDAC5 binding to LRRK2. We reasoned that any potential interaction with LRRK2 could be enhanced by enriching the cytosolic pool of HDAC5. PMA treatment of wild type and G2019S LRRK2 over-expressing SHSY5Y cells failed to detectibly enhance cytosolic HDAC5 as determined by Western blot analysis of IP input lysates, further questioning the antibody specificity. PMA treatment of wild type and G2019S LRRK2 over-expressing cells resulted in the loss of LRRK2 immunoreactivity in the HDAC IPs suggesting a dissociation of the HDAC5 LRRK2 complex. LRRK2 interaction with 14-3-3 has previously been shown to be disrupted upon de-phosphorylation of Serine residues [215, 216] in a PKA dependent mechanism [383]. As PMA activates a number of intracellular kinases and phosphatases including PKA [388], it is possible that the 249 intrinsic phosphorylation status of LRKR2 could be affected in the presence of the drug. Although LRRK2 phosphorylation at Serine 935 was not affected by PMA treatment, dephosphorylation of other serine residues [202, 383] cannot be ruled out. Identification of LRRK2 specific phosphatases and determination of their mechanism of action may provide a better understanding of LRRK2 interacting partners and the associated complex formation. LRRK2 association with HDAC5 suggested a potential role for LRRK2 in HDAC5 mediated transcriptional regulation of PGC1α and UCP4 in SHSY5Y cells. Increased association between G2019S LRRK2 and HDAC5 could potentially account for increased PGC1α and UCP4 mRNA in SHSY5Y cells. Although LRRK2 kinase inhibition did not influence the HDAC5 association, UCP4 expression was reduced in G2019S expressing SHSY5Y cells suggesting UCP4 transcription was altered while the LRRK2 HDAC5 association was still intact. An additional level of HDAC5 regulation could involve post translational modification [384] as has been previously described for NFAT and DLP1 proteins [389, 390]. Although DLP1 phosphorylation was comparable for HeLa cells transfected with wild type and G2019S LRRK2, specific phospho residues were not examined and site specific modification by G2019S LRRK2 cannot be ruled out. Furthermore, the contribution of LRRK2 GTPase activity could also contribute to the regulation of the HDAC5 interaction. 6.9.1.5 LRRK2 GTPase mutations and mitochondrial function LRRK2 GTPase mutations have been described in North American, Basque, English and Italian cohorts with the clinical features and pathology resembling that of idiopathic and G2019S LRRK2 linked PD [28, 29, 391]. Similar to the LRRK2 G2019S mutation, transgenic mice expressing R1441C do not have a neurodegenerative or kidney phenotype [233, 240]. Studies investigating the interplay between the two enzymatic domains are inconsistent with some groups describing GTPase LRRK2 250 mutations to decrease LRRK2 kinase activity in HEK293 and neuro2A cells [203, 204] while studies carried out in HEK293 cells in two other studies suggest GTPase mutations have no effect on the kinase activity of the protein [201, 202]. GTPase and kinase mutations have been shown to increase LRRK2 affinity for GTP in HEK293 cells [203], however kinase deficient LRRK2 has normal GTPase activity [204]. The effect of LRRK2 kinase inhibition on GTPase function has not besn studied. In addition the Serine 935 phosphorylation status of LRRK2 is reduced in GTPase but not LRRK2 G2019S mutants [215]. The enzymatic crosstalk between the kinase and GTPase domains as well as the pathological and clinical similarities associated with GTPase and kinse LRRK2 mutant proteins suggest the existence of converging pathways in the disease pathogenesis. Although over-expression of mutant kinase and GTPase LRRK2 proteins in SHSY5Y, HEK293 cells and rodent primary neurons is associated with increased toxicity [222] [203, 227, 260], functional studies have often struggled to identify a common converging phenotype; reduced complexity of dopaminergic midbrain neurons was described for G2019S but not R1441C transgenic mice [233], GTPase mutations enhanced filament formation and promoted association with microtubules in HEK293 cells, an effect not observed for LRRK2 G2019S [333], kinase but not GTPase mutations increased Tau phosphorylation [263]. These studies implicated the existence of pathways which are kinase independent but require LRRK2 GTPase activity and visa versa, questioning their contribution to the common PD pathology observed for the kinase and GTPase mutations. Mitochondrial fragmentation and the reduced mitochondrial membrane potential described [280, 281] was extended to the R1441C GTPase mutation suggesting mitochondrial function is affected by kinase and GTPase mutant LRRK2 proteins. In addition, GTPase LRRK2 mutants showed increased affinity for the DLP1 251 protein at the mitochondrial membrane of SHSY5Y cells [280] suggesting the GTPase mutation can affect the affinity of complex formation in a similar manner to G2019S LRRK2. Mitochondrial uncoupling associated with G2019S LRRK2 and the increase in UCP4 mRNA expression was described as a LRRK2 kinase dependent mechanism based on in vitro kinase activity assays characterizing G2019S LRRK2 as a hyperactive kinase and the effects of LRRK2 kinase inhibitors on UCP4 levels in SHSY5Y cells. Whether LRRK2 GTPase activity contributes to the regulation of UCP expression was not investigated. However, the proposed mechanism of HDAC5 contribution to the regulation of PGC1α and UCP4 mRNA levels by LRRK2 demonstrated increased affinity of G2019S LRRK2 for HDAC5 suggesting a structural modulation of the complex, comparable to the effects of G2019S and R1441C on the DLP1 interaction. LRRK2 kinase inhibition did not seem to disrupt the LRRK2 HDAC5 association. As LRRK2 kinase deficient mutants retain GTPase activity [204] GTPase function may be sufficient to maintain the interaction. As the PD associated GTPase R1441C mutation has been shown to contribute to mitochondrial abnormalities in cell models of PD, the effects of GTPase mutants on the regulation of the UCP4 transcription, PGC1α expression and HDAC5 association warrants further investigation. 6.9.2 LRRK2 regulation of DJ1 Mitochondrial uncoupling is believed to mediate the pacemaker activity of SNpc dopaminergic neurons while the loss of the PD associated DJ1 protein results in the dysregulation of pacemaking in mouse SNpc [168]. The proposed mechanism of regulation of the pacemaker activity was shown to involve UCP4 and 5. The level of these isoforms correlated with oscillations in the mitochondrial membrane potential. UCP4 and 5 levels were reduced in DJ1 knockout mice while the oscillatory activity of pacemaking was inhibited in these animals. As LRRK2 was shown to increase UCP2 and 4 in fibroblasts and SHSY5Y cells, it was possible to speculate that 252 LRRK2 regulated UCP mRNA through a genetic interaction with DJ1 potentially contributing to the regulation of pacemaking in SNpc. In addition to UCP mRNA regulation, DJ1 has also been shown to be involved in PGC1α mediated transcription by binding to upstream regulatory elements of PGC1α target genes [145] in line with PGC1α involvement in UCP4 regulation in G2019S LRRK2 SHSY5Y cells. Similar to the UCP transcriptional changes in G2019S expressing SHSY5Y cells, UCP gene regulation by DJ1 appeared specific as SOD2 and catalase expression was unperturbed in the SNpc of DJ1 knockout mice, [168], further implicating LRRK2, DJ1 and PGC1α in a common regulatory pathway involving UCP transcription. To investigate whether the regulation of UCP4 expression in SHSY5Y cells was mediated by DJ1, DJ1 knockdown cell lines were studied. Genetic knockdown of DJ1 in SHSY5Y cells did not affect UCP4 or 5 expression in contrast to what has previously been described for SNpc of DJ1 knockout animals [168] suggesting LRRK2 regulation of UCP4 in SHSY5Y cells is unlikely to be DJ1 dependent. As the functional role of UCP4 and 5 was assigned to the regulation of the pacemaker activity in SNpc neurons, the absence of pacemaking in SHSY5Y cells could eliminate the need for this regulatory process. In support of this UCP4 and 5 expression in the neighbouring ventral tegmental neurons lacking intrinsic pacemaker function was not affected by the loss of DJ1 in mice [168]. UCP3 levels were reduced by 50 % in both lines of DJ1 knockdown SHSY5Y cells suggesting a potential regulatory role of DJ1 for this UCP isoform in the neuroblastoma cells. UCP3 specific regulation implies a specific functional response as has previously been described for UCP3 expression in cardiac muscle cells mediating contractile responses and adipose tissue regulating body mass [359, 392]. As the G2019S mutation was associated with changes in UCP4 expression, we did not pursue with UCP transcriptional rescue experiments involving DJ1 and LRRK2 in 253 the SHSY5Y cells. However, a converging pathway of UCP mRNA regulation by LRRK2 and DJ1 in SNpc neurons should not be ruled out and warrant further investigation. 6.9.3 LRRK2 regulation of NFκB As well as transcriptional regulation of UCP mRNA through the PGC1α pathway, UCP mRNA expression has been demonstrated to be regulated through activation of the NFκB pathway. NFκB mediated UCP4 regulation has been shown in SHSY5Y cells in response to oxidative stress [357] suggesting UCP levels can be mediated by NFκB. In support of this an NFκB response element has been described on the UCP gene [357]. NFκB activation of UCP4 transcription in SHSY5Y cells occurs in parallel with SOD2 [393, 394] as a mechanism to protect against increased levels of cellular ROS [357, 393, 394]. LRRK2 has been demonstrated to regulate NFκB mediated transcription; over-expression of LRRK2 in HEK293 cells increased NFκB dependent transcription [243], shRNA knockdown of LRRK2 reduced NFκB mediated transcription [395] while wild type, and G2019S LRRK2 equally enhanced NFκB mediated transcription [395]. Although NFκB regulation of UCP4 was not investigated in SHSY5Y cells, SOD2 levels between control, wild type and G2019S LRRK2 over-expressing cells were similar suggesting NFκB mediated regulation was not involved. However UCP4 gene regulation by NFκB independent of SOD2 cannot be ruled out. 6.9.4 UCP4 knockdown and LRRK2 regulation of Argonate 2 In an attempt to reduce UCP4 levels in SHSY5Y, UCP4 siRNA knockdown was carried out. UCP4 knockdown has previously been linked to perturbations in calcium homeostasis in SHSY5Y cells [363] suggesting reduced UCP4 levels can affect the coupling state of mitochondria, in line with what was reported in SHSY5Y cells treated with LRRK2 siRNA. A similar efficiency of UCP4 siRNA knockdown (60%) in SHSY5Y cells was described here correlating with an increase in TMRM intensity 254 suggesting a potential link between UCP4 mRNA expression and the mitochondrial membrane potential. UCP4 protein levels were not evaluated in either study due to the lack of effective antibodies. Although the stability of the UCP4 protein has not been investigated, the UCP2 and 3 homologues have been reported to have a short half life of 1-4 hours [354, 396, 397] suggesting short term silencing protocols reduce UCP protein and affect cellular functions mediated by UCP levels. In support of this, a 55 % reduction in UCP2 mRNA in islet cells correlated with a 25 % decrease in mitochondrial membrane potential, 30 % reduction in respiration rates and a 20 % increase in ATP levels [372] suggesting small changes in UCP mRNA expression can have pronounced effects on respiratory chain function. The G2019S mutation has previously been shown to impact on LRRK2 regulation of the Ago2 containing RNA-induced silencing complex (RISC) mediating microRNA regulation of mRNA translation [273]. As this complex is also required for the siRNA interaction with its target genes it is possible to speculate that the efficiency of knockdown in the LRRK2 over-expressing cells is affected by LRRK2 involvement in the transcriptional regulation pathway. Similar LRRK2 siRNA conditions resulted in a knockdown of lower efficiency in wild type (40 %) and G2019S (25 %) LRRK2 over-expressing SHSY5Y cells. siRNA knockdown efficiencies in G2019S LRRK2 cell culture models have not previously been described and warrant further investigation. LRRK2 association with the Ago2/RISC complex and the proposed effects of G2019S on microRNA regulation could also potentially be involved in microRNA mediated regulation of UCP mRNA levels. In support of this, microRNA regulation of UCP2 mRNA expression has been described in cardiac and skeletal muscle required for muscle development [398]. However, the influence of this pathway on UCP expression in fibroblasts and SHSY5Y cells was not investigated. 255 6.9.5 LRRK2 regulation of UCP phosphorylation As well as regulation through transcription, UCP activity can be regulated at the protein level; increased mitochondrial uncoupling has correlated with increased phosphorylation of UCP1 in brown adipose tissues of cold acclimatised animals [159] while glutathionylation of cysteine residues in UCP2 and 3 has been shown to regulate mitochondrial respiration in thymocytes [399]. Although phosphorylation mediated regulation has been described for the UCP1 isoform, the serine residue is conserved among the family members [159] suggesting other UCP isoforms could potentially be regulated in a similar fashion. The kinase associated with UCP phosphorylation has not yet been described. LRRK2 has previously been shown to phosphorylate tau and 4E-BP functionally linked to mediating tubulin polymerization and induction of autophagy [231, 261]. Identification of LRRK2 in the mitochondrial enriched fraction of SHSY5Y cells suggested the protein could potentially mediate UCP activity by post translational regulation. To directly test LRRK2 phosphorylation of UCP2 in vitro kinase assays were carried out using recombinant proteins but failed to produce any detectable UCP2 phosphorylation. However, as UCP functional activity in vitro is enhanced by protein incorporation into liposomes [358, 400], it is also possible that the crude assay conditions presented in this study require modification. 6.10 LRRK2 regulation of mitochondrial morphology Changes in mitochondrial bioenergetics associated with LRRK2 G2019S expression in fibroblasts and SHSY5Y cells have previously been shown to correlate with mitochondrial morphological changes; mitochondria in LRRK2 G2019S patient derived fibroblasts were described as elongated although no quantitative assessment was carried out in the study [285]. In contrast mitochondrial fragmentation was reported for primary neurons and SHSY5Y cells over-expressing 256 wild type and more so mutant LRRK2 [280, 281] where quantifications were limited to neuronal processes with no calibration for the methodology. Visually, mitochondria in cell bodies appeared un-fragmented when compared to stress induced fragmentation in a number of other studies [135, 401]. Mitochondrial fragmentation in the neuronal processes reported for LRRK2 mutants could potentially relate to the mutant effects on neurite length and branching [259] more so than the bioenergetic changes, an issue not addressed by either of the studies. Although bioenergetic alterations were reported for both G2019S mutant fibroblasts and SHSY5Y cells, mitochondrial uncoupling did not appear to perturb the mitochondrial morphology. Intact mitochondrial networks described in patient fibroblasts reported here and by Mortiboys et al suggest LRRK2 associated mitochondrial fragmentation could result from LRRK2 over-expression or be limited to neuronal models as fragmented structures were reported for both wild type and mutant LRRK2 over-expressing SHSY5Y cells and primary cultures [280, 281]. Although difficult to assess without a direct comparison, the level of ectopic expression of LRRK2 in the SHSY5Y model presented here may not be sufficient to fragment the network. In terms of a cell specific phenotype, comparable discrepancies have been reported for studies involving PINK1. Mitochondrial fragmentation was reported in fibroblasts derived from patients with familial PINK1 mutations, suggesting primary skin cells are capable of this phenomenon [119]. In contrast genetic ablation of PINK1 in Cos7 cells increased mitochondrial size through tubulation [402] while HeLa and SHSY5Y PINK1 knockdown models presented with fragmented mitochondria [119, 121]. Swollen mitochondria were described in human dopaminergic neurons and primary mouse neuronal cultures from PINK1 knockout animals [403]. Electron microscopy studies of recessive PD animal models revealed additional 257 inconsistencies where flies lacking PINK1 were reported to have swollen mitochondria with disrupted cristae [404, 405] while genetic ablation in mice produced only a mild mitochondrial phenotype with no obvious morphological defects. Condensed and aggregated mitochondrial structures were also observed in G2019S LRRK2 transgenic mice [233]. While Lin et al, reported mitochondria with denser mitochondrial cristae in their G2019S LRRK2 transgenic mouse [235]. Animal studies investigating mitochondrial morphology in PINK1 and LRRK2 PD associated mutant did not reflect fragmented structures observed in the SHSY5Y cell models [280, 281]. Although chemical uncouplers can induce mitochondrial fragmentation [406], there is no evidence to suggest mild mitochondrial uncoupling correlates with mitochondrial fragmentation. Uncoupled HtrA2 deficient mouse embryonic fibroblasts and HeLa cells treated with HtrA2 siRNA showed elongated mitochondria suggesting mitochondrial uncoupling in these cells correlated with mitochondrial fusion [407]. UCP2-GFP over-expression in SHSY5Y cells did not appear to impact on the mitochondrial morphology while mitochondrial uncoupling in G2019S expressing fibroblasts and SHSY5Y cells did not correlate with mitochondrial fragmentation. In addition to chemical uncouplers mitochondrial fragmentation can be induced by a variety of cellular stressors such as nitric oxide (NO), FeSo4, N-methy-D-aspartic acid (NMDA) agonists, extracellular β-amyloid aggregates [408] suggesting this process may be a general feature of stressed cells. In support of this a genetic screen in C.elegans showed that individual gene ablation of 80 % of all genes leads to mitochondrial fragmentation [409]. Hence mitochondrial fragmentation is unlikely to correlate with specific respiratory chain alterations or contribute to mitochondrial uncoupling observed in G2019S expressing fibroblasts and SHSY5Y cells. 258 The bioenergetic changes and mitochondrial fragmentation associated with mutant LRRK2 over-expression have been linked to increased cellular and mitochondrial DLP1 levels [280, 281]. DLP1 mediated fragmentation has also been described in the PINK1 and parkin PD models as well as for fragmented neuronal cells following treatment with rotenone [410] suggesting defects in mitophagy and Complex I inhibition can influence the fission/fusion machinery. The trigger for mitochondrial DLP1 recruitment in mitochondrial toxin studies has been proposed to be the defective electron transport chain. However, whether the mechanism is membrane potential dependent or requires an enzyme stimulus has not been addressed. A direct interaction between DLP1 and LRRK2 has been described [280, 281]. This association was enhanced for the G2019S mutant LRRK2 suggesting mitochondrial LRRK2 could be the trigger for the DLP1 recruitment. A weak association was detected for G2019S LRRK2 immunoprecipitated from SHSY5Y cells absent for the wild type protein, potentially demonstrating enhanced mitochondrial DLP1 recruitment by the mitochondrial G2019S LRRK2 pool. However, mitochondrial LRRK2 and DLP1 abundance was comparable between control and G2019S expressing SHSY5Y cells, in contrast to previous observations, suggesting increased affinity for the G2019S LRRK2 DLP1 interaction which could take place in the cytosol, a point not addressed in any of the studies. Kinase inhibition did not appear to affect mitochondrial DLP1 recruitment further suggesting mitochondrial LRRK2 levels or kinase activity did not influence DLP1 translocation in SHSY5Y cells. Our data suggests that mitochondrial fragmentation and DLP1 levels are unlikely to contribute to the mitochondrial uncoupling described in G2019S expressing fibroblasts and SHSY5Y cells. 259 6.11 LRRK2, mitochondrial uncoupling and cellular disturbances implicated in PD 6.11.1 ATP, mitochondrial biogenesis and calcium ATP depletion and bioenergetic insufficiency have been proposed to contribute to cellular dysfunction and the death of dopaminergic neurons in PD. Reduced ATP levels have been reported for PINK1 deficient mouse embryonic fibroblasts [411], fibroblasts from patients with parkin mutations [346] and αsynuclein over-expression in HEK293 cells [412], correlating with respiratory chain defects. Reduced ATP levels were also described in the HtrA2 deficient mouse embryonic fibroblasts where mitochondrial uncoupling was described [413]. Reduced ATP levels have been described for G2019S expressing SHSY5Y cells [280] and G2019S LRRK2 mutant fibroblasts [285]. Consistent with these observations, a 15 % reduction in basal ATP levels was reported for G2019S patient fibroblasts relative to control cells described in this study suggesting reduced ATP levels are a common feature of uncoupled cells and cells with respiratory chain defects, potentially explaining some of the over-lapping pathology and clinical features observed in genetically linked PD cases. UCP levels have been shown to correlate with changes in cellular ATP and mitochondrial biogenesis, a pathway associated with the recessively linked PD genes suggesting dysregulated mitochondrial biogenesis may play a role in PD. Downregulation of parkin correlated with reduced mitochondrial content [122] while parkin over-expression was linked to enhanced mitochondrial biogenesis [140]. Increased cellular ATP correlated with UCP1 expression in brown adipocytes [414] correlating with increased mitochondrial biogenesis [415] while reduced mitochondrial DNA content was linked to lower levels of UCP1 expression [416]. Increased UCP2 expression in muscle cells also enhanced mitochondrial biogenesis and increased the cellular ATP content [373]. However, other studies have identified 260 mitochondrial uncoupling independent of mitochondrial biogenesis in UCP overexpressing systems; UCP2 over-expression resulted in mitochondrial uncoupling and reduced basal ATP levels in pancreatic β-cells [417]. ATP levels were reduced in skeletal muscle expressing high levels of UCP1 protein, relative to mice with lower UCP1 expression [418]. As mitochondrial mass, respiratory chain subunit and TFAM levels were comparable between control and G2019S LRRK2 expressing fibroblasts we ruled out the potential contribution of mitochondrial biogenesis to mitochondrial uncoupling in these cells. As well as UCP regulation of the mitochondrial membrane potential and ATP levels described in G2019S expressing fibroblasts and SHSY5Y cells, UCPs have a role in the regulation of cellular calcium homeostasis. Calcium dysregulation is a noted feature in PD; the loss of PINK1 protein has previously been shown to affect mitochondrial calcium handling resulting in a decreased threshold for calcium dependent cell death in primary neurons and neuroblastoma cells [172]. The influence of UCPs on the mitochondrial membrane potential has previously been related to their action as calcium uniporters, in line with the mechanism proposed to regulate SNpc pacemaking [168]. In support of this, UCP2 and UCP3 over-expression in human endothelial cells was associated with increased concentrations of free mitochondrial calcium [419]. LRRK2 influence on UCP mRNA expression in fibroblasts and SHSY5Y cells suggests that it is likely to play a role in the regulation of mitochondrial calcium homeostasis which in turn may contribute to PD pathogenesis. 6.11.2 LRRK2 regulation of autophagy and mitophagy LRRK2 regulation of autophagy has been linked to the regulation of steady state levels of misfolded proteins such as α-synuclein with the deregulation of this pathway in G2019S expressing induced pluripotent dopaminergic stem cells 261 resulting in increased α-synuclein levels [420], potentially providing the link to the pathological Lewy bodies observed in PD. However the mechanism of LRRK2 autophagic regulation is still under debate. Induced pluripotent stem cells generated from PD patients with LRRK2 G2019S mutations were compromised at the level of autophagic clearance [421] while autophagic flux was enhanced in G2019S patient fibroblasts [422]. An increase in autophagic vacuoles was observed in differentiated G2019S SHSY5Y cells [259] but whether these changes occurred as a result of increased autophagic activation or reduced autophagic clearance was not investigated. Tong et al, [240] reported a bi-phasic effect on autophagy in kidneys of knockout animals where autophagic flux was enhanced at 7 months and perturbed at 20 months suggesting age may contribute to the LRRK2 regulation of the autophagy pathway and account for some of the discrepancies observed in the experimental cell models. A number of studies emphasise unperturbed autophagic flux associated with G2019S [271] and LRRK2 siRNA knockdown in T lymphocytes and bone marrow derived macrophages [241]. The discrepancy may occur at the tissue or cell source as differentiated neuronal cells, proliferating fibroblasts and phagocytic immune cells have variable autophagic demands correlating with their respective functional roles [423, 424]. A recent study has demonstrated that UCP2 over-expression in pancreatic adrenocarcinoma cells correlated with reduced autophagy suggesting a potential link between mitochondrial uncoupling and the autophagy cascade [425]. Whether these findings can be replicated in the SHSY5Y system and extend to the UCP4 isoform warrants further investigation. Although LRRK2 regulation of autophagy was not evaluated in G2019S fibroblasts and SHSY5Y cells, mitochondrial removal by mitophagy was explored in the context of LRRK2. Mitochondrial depolarization is thought to act as the trigger for PINK1 mediated Parkin recruitment and stimulate mitophagy [426]. As the 262 mitochondrial membrane potential was reduced in G2019S expressing fibroblasts and SHSY5Y cells, it was possible to speculate that excessive mitophagy may occur in these cells. Although the process of mitochondrial turnover was not directly evaluated, mitochondrial content and levels of respiratory chain subunits were comparable between control and G2019S LRRK2 fibroblasts suggesting mitochondria were not depleted. The levels of mitochondrial transcription factor, SOD2 and catalase, genes that are part of the mitochondrial biosynthetic pathway were comparable between control, wild type and G2019S expressing SHSY5Y cells suggesting mitochondrial biosynthesis was not taking place. As mitochondrial synthesis and number were comparable between control and G2019S cells, excessive mitochondrial removal is unlikely to contribute to the mitochondrial uncoupling observed in mutant fibroblasts and SHSY5Y cells. 6.11.3 LRRK2 regulation of synaptic function and tubulin dynamics As well as mitochondrial morphology and autophagy, LRRK2 has been implicated in the regulation of synaptic function [254, 255] and tubulin dynamics [262, 427]. Mitochondria play a central role in meeting the demands of synapses for ATP and regulating calcium homeostasis which in turn controls synaptic vesicle endocytosis. UCP4 has previously been demonstrated to regulate calcium homeostasis at synapses [428]. siRNA knockdown of LRRK2 in SHSY5Y correlated with a 50 % reduction of UCP4 levels while LRRK2 silencing in presynaptic primary cortical mouse neurons reduced synaptic transmission [254] and enhanced synaptic vesicle recycling [255], both calcium dependent mechanisms [429, 430]. Overexpression of wild type LRRK2 impaired synaptic vesicle recycling in primary rat hippocampal neurons, while the effect was more pronounced for the G2019S mutant protein. The 5 fold increase in UCP4 mRNA in G2019S expressing SHSY5Y 263 cells is likely to result in reduced cytosolic calcium concentrations [419], which could potentially account for the impaired vesicle recycling at the synapse [430]. Mitochondrial trafficking to and from the synapse requires the interaction between glycogen synthase kinase 3β (GSK3β) and the Tau protein, forming microtubules [431]. The speed of this process is dependent on GSK3β levels and the subsequent phosphorylation of its Tau substrate [431]. Microtubule destabilization correlates with reduced mitochondrial membrane potential and visa versa [432]. Hence UCP4 over-expression and the reduced mitochondrial membrane potential in G2019S expressing SHSY5Y cells could impact on the mitochondrial association with tubulin. In addition, the cellular and mitochondrial G2019S LRRK2 pools could directly affect the phosphorylation status of tubulin which would in turn result in reduced affinity of tubulin for mitochondria. In support of this LRRK2 has been implicated in the regulation of microtubule assembly in mouse brain [262] with interactions between LRRK2 and tubulin reported in mouse brain and HEK293 cells [262]. Microtubule destabilization results in reduced mitochondrial trafficking to synaptic terminals [433] reflecting how functional changes in the mitochondria could affect mitochondrial delivery to the synapse and alter synaptic function in G2019S expressing neuronal systems. In support of this, reduced mitochondrial numbers have been noted in neuronal processes of G2019S expressing primary cortical cultures and SHSY5Y cells [280, 281]. 6.12 Mitochondrial uncoupling proteins and disease Mitochondrial uncoupling has been associated with the loss of function of the PD linked HtrA2 protein. Mouse embryonic fibroblasts generated from HtrA2 knockout animals had greater basal and oligomycin inhibited respiration rates relative to their respective wild type counterparts [413] consistent with the observations in G2019S LRRK2 expressing fibroblasts. The study reporting 264 mitochondrial uncoupling in HtrA2 knockout animals investigated the involvement of UCP2 protein. Although the data was not shown, the authors claimed no changes in UCP2 levels. The involvement of other UCP isoforms was not described hence the involvement of these UCP family members in the regulation of HtrA2 knockout mitochondrial phenotype cannot be ruled out. Alternative mechanisms for HtrA2 mediated mitochondrial uncoupling were not investigated. Although mitochondrial uncoupling in disease is not common, increased respiration has also been described for Luft’s disease. Although the etiology of the disorder is unknown the muscle atrophy associated with the disease has been linked to hypermetabolism and mitochondrial uncoupling in skeletal muscle [434, 435], potentially involving the UCPs. HtrA2 linked PD and Luft’s disease emphasise the correlation between mitochondrial uncoupling, neurodegeneration and skeletal pathology. In addition, the genetic link between DJ1, recessively inherited PD and the UCP mediated regulation of pacemaking activity specifically in the SNpc where cell death is observed in PD patients further implicated the potential role of mitochondrial uncoupling and UCPs in PD pathogenesis [168]. In support of this, pacemaker activity involves the action of voltage dependent L type calcium channel with inhibitors targeting this channel demonstrated to have a protective effect against PD in a Danish cohort [436]. Mechanistically, mitochondrial uncoupling proteins have previously been implicated in Parkinson’s disease as a protective mechanism against ROS [437-439]. Their protective effects have also been demonstrated on a background of respiratory chain dysfunction models but the effects of chronic UCP upregulation have not been investigated. In addition to protection from disease associated ROS, UCP have been proposed to play a general role in ageing; over-expression of UCP5 in Drosophila [440] correlated with increased lifespan, while UCP2 levels regulate 265 lifespan in mice [355]. However, the protective role of chronic UCP expression and activation correlating with increased lifespan has been challenged. UCP activation by fatty acids in fibroblasts was associated with premature senescence [441], as assessed by β-galactosidase staining. Over-expression of UCP2 in COS7 cells had no effect on lifespan but seemed to correlate with induction of apoptosis [167]. In addition, although transient transfections of UCPs are well tolerated, prolonged over-expression has been shown to result in death of HeLa, endothelial and fibroblast cell lines [166]. The potential link of mitochondrial uncoupling and cell death in PD has also been demonstrated by chronic exposure to low concentrations of FCCP correlating with reduced lifespan in cell culture models as a result of premature senescence [441]. Hepatic cells exposed to another uncoupler, dinitrophenol retarded proliferation [442]. Although LRRK2 G2019S expression has previously correlated with apoptotic induction [227, 286] senescence and cell death were not observed in G2019S fibroblasts and SHSY5Y cells described here, however compromised cell viability cannot be ruled out. 6.13 Endogenous LRRK2 regulation of UCPs The G2019S mutation can either lead to a novel function or hyperactivate a pathway regulated by wild type LRRK2. Two studies support the latter hypothesis; siRNA knockdown of endogenous LRRK2 increased autophagic activity in HEK293 cells while expression of a BAC G2019S construct impaired autophagy [257] suggesting this process is mediated by endogenous LRRK2 and dysregulated in the presence of the mutant protein. The effects of wild type BAC LRRK2 expression were not addressed. Ago2 mediated regulation of microRNA in Drosophila melanogaster appeared to involve endogenous LRRK2 as siRNA knockdown of LRRK2 correlated with reduced levels of dominant-negative dsRNA dependent protein kinase (DP1) and eukaryotic translation initiation factor 2 alpha (EIF2α) proteins, the steady state 266 levels of which are regulated in a microRNA mediated fashion [273]. While wild type LRRK2 over-expression in Drosophila melanogaster did not appear to grossly affect DP1 or EIF2α levels, G2019S LRRK2 expression increased the levels of both of these microRNA regulated proteins. In support of these findings G2019S over-expression in SHSY5Y cells led to mitochondrial uncoupling correlating with increased UCP4 levels. siRNA knockdown of LRRK2 was associated with increased mitochondrial membrane potential and reduced UCP4 expression suggesting endogenous LRRK2 in SHSY5Y cells may be required for UCP4 transcriptional regulation. In line with the report by Gehrke et al, wild type LRRK2 over-expression did not appear to impact on respiratory chain function or UCP levels. If both mechanisms are dependent on the levels of the associated LRRK2 substrate, increased levels of wild type protein would not affect downstream signalling, reduced LRRK2 levels would reduce the LRRK2 substrate interaction and downstream signalling while G2019S would compete with endogenous LRRK2 for the substrate and the effects of increased LRRK2 kinase activity could be implemented. This model is supported by LRRK2 kinase inhibitor effects in control SHSY5Y cells where a mild effect on UCP2, 4 and 5 mRNA expression was observed. UCP levels were unperturbed in wild type over-expressing cells (kinase inhibition was not 100 % effective suggesting some active LRRK2 remained in the cells) while UCP4 levels in G2019S expressing cells were partially restored. Therefore the G2019S mutation is likely to perturb LRRK2 regulation of UCPs and impact on different aspects of cellular function, the cumulative effect of these disturbances could contribute to PD pathogenesis. 267 References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. Gasser T: Genetics of Parkinson's disease. Curr Opin Neurol 2005, 18(4):363-369. Gibb WR: Functional neuropathology in Parkinson's disease. Eur Neurol 1997, 38 Suppl 2:21-25. Lanciego JL: Basal Ganglia Circuits: What's Now and Next? Front Neuroanat 2012, 6:4. Berardelli A, Rothwell JC, Thompson PD, Hallett M: Pathophysiology of bradykinesia in Parkinson's disease. Brain 2001, 124(Pt 11):2131-2146. Fahn S, Lang AE, Schapira AHV: Movement disorders 4. Philadelphia, PA: Saunders/Elsevier; 2010. Alexander GE, DeLong MR, Strick PL: Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986, 9:357-381. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M: Alpha-synuclein in Lewy bodies. Nature 1997, 388(6645):839-840. Blumberg MS, Freeman JH, Robinson SR: Oxford handbook of developmental behavioral neuroscience. New York: Oxford University Press; 2010. Forno LS: Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 1996, 55(3):259-272. Kahle PJ: alpha-Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol 2008, 115(1):87-95. Elia AE, Lalli S, Albanese A: Differential diagnosis of dystonia. Eur J Neurol 2010, 17 Suppl 1:1-8. Wider C, Dickson DW, Wszolek ZK: Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis 2010, 7(1-3):175-179. Tan LC, Venketasubramanian N, Jamora RD, Heng D: Incidence of Parkinson's disease in Singapore. Parkinsonism Relat Disord 2007, 13(1):40-43. Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT, Schaffer AA et al: Mapping of a gene for Parkinson's disease to chromosome 4q21q23. Science 1996, 274(5290):1197-1199. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R et al: Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997, 276(5321):2045-2047. Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B et al: The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004, 55(2):164-173. 268 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M et al: Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004, 364(9440):1167-1169. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R et al: alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003, 302(5646):841. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A et al: Genomewide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 2009, 41(12):1303-1307. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG et al: Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009, 41(12):1308-1312. Chiba-Falek O, Nussbaum RL: Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet 2001, 10(26):3101-3109. Shin CW, Kim HJ, Park SS, Kim SY, Kim JY, Jeon BS: Two Parkinson's disease patients with alpha-synuclein gene duplication and rapid cognitive decline. Mov Disord 2010, 25(7):957-959. Uversky VN: A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn 2003, 21(2):211234. Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T: The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14(2):467-475. Kim HY, Heise H, Fernandez CO, Baldus M, Zweckstetter M: Correlation of amyloid fibril beta-structure with the unfolded state of alpha-synuclein. Chembiochem 2007, 8(14):1671-1674. Davidson WS, Jonas A, Clayton DF, George JM: Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998, 273(16):9443-9449. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F: A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 2002, 51(3):296-301. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB et al: Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44(4):601-607. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N et al: Cloning of the gene containing mutations that cause PARK8linked Parkinson's disease. Neuron 2004, 44(4):595-600. 269 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, Dickson D, Wavrant Devrieze F, Hardy J, Singleton A: The dardarin G 2019 S mutation is a common cause of Parkinson's disease but not other neurodegenerative diseases. Neurosci Lett 2005, 389(3):137-139. Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J et al: Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 2005, 76(4):672-680. Orr-Urtreger A, Shifrin C, Rozovski U, Rosner S, Bercovich D, Gurevich T, Yagev-More H, Bar-Shira A, Giladi N: The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: is there a gender effect? Neurology 2007, 69(16):1595-1602. Change N, Mercier G, Lucotte G: Genetic screening of the G2019S mutation of the LRRK2 gene in Southwest European, North African, and Sephardic Jewish subjects. Genet Test 2008, 12(3):333-339. Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP et al: A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet 2005, 365(9457):415-416. Di Fonzo A, Rohe CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N et al: A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet 2005, 365(9457):412-415. Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, Prinjha RK, Upmanyu R, Kefi M, Zouari M et al: LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol 2008, 7(7):591-594. Zabetian CP, Morino H, Ujike H, Yamamoto M, Oda M, Maruyama H, Izumi Y, Kaji R, Griffith A, Leis BC et al: Identification and haplotype analysis of LRRK2 G2019S in Japanese patients with Parkinson disease. Neurology 2006, 67(4):697-699. Greggio E, Lewis PA, van der Brug MP, Ahmad R, Kaganovich A, Ding J, Beilina A, Baker AK, Cookson MR: Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem 2007, 102(1):93-102. Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E: G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies. J Neurol Neurosurg Psychiatry 2007, 78(6):626-628. Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ et al: Lrrk2 and Lewy body disease. Ann Neurol 2006, 59(2):388-393. Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M et al: Mutations in the gene LRRK2 encoding dardarin (PARK8) cause 270 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 2005, 128(Pt 12):27862796. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392(6676):605-608. Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW et al: Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000, 342(21):1560-1567. Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Adel S, Gonzales-McNeal M, Hilker R et al: Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 2005, 58(3):411-422. Houlden H, Singleton AB: The genetics and neuropathology of Parkinson's disease. Acta Neuropathol 2012, 124(3):325-338. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al: Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004, 304(5674):1158-1160. Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R et al: Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol 2004, 61(12):1898-1904. Valente EM, Brancati F, Caputo V, Graham EA, Davis MB, Ferraris A, Breteler MM, Gasser T, Bonifati V, Bentivoglio AR et al: PARK6 is a common cause of familial parkinsonism. Neurol Sci 2002, 23 Suppl 2:S117-118. Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR: PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 2004, 56(3):336-341. Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J et al: PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 2010, 133(Pt 4):1128-1142. Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA: Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012, 13(4):378-385. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M et al: Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299(5604):256-259. Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR: The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A 2004, 101(24):9103-9108. 271 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A et al: Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009, 361(17):1651-1661. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R: Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004, 351(19):1972-1977. Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E: Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 2004, 81(1):70-73. Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, Rudolph A, Wojcieszek J, Pfeiffer RF, Foroud T: Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009, 72(4):310-316. Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, Fukuda Y, Date H, Iwata A, Yamamoto M et al: Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009, 66(5):571-576. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, Li A, Holton J, Guerreiro R, Paudel R et al: Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009, 132(Pt 7):1783-1794. Chen M, Wang J: Gaucher disease: review of the literature. Arch Pathol Lab Med 2008, 132(5):851-853. Sawkar AR, Adamski-Werner SL, Cheng WC, Wong CH, Beutler E, Zimmer KP, Kelly JW: Gaucher disease-associated glucocerebrosidases show mutation-dependent chemical chaperoning profiles. Chem Biol 2005, 12(11):1235-1244. Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L: Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 2000, 287(5456):1265-1269. Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Muller V, Odoy S, Fujiwara H, Hasegawa M et al: Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest 2002, 110(10):1429-1439. Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM: Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34(4):521-533. Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ: Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol 2002, 175(1):35-48. Nuber S, Petrasch-Parwez E, Winner B, Winkler J, von Horsten S, Schmidt T, Boy J, Kuhn M, Nguyen HP, Teismann P et al: Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J Neurosci 2008, 28(10):2471-2484. 272 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL: Human alpha-synucleinharboring familial Parkinson's disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A 2002, 99(13):8968-8973. Feany MB, Bender WW: A Drosophila model of Parkinson's disease. Nature 2000, 404(6776):394-398. Takahashi M, Kanuka H, Fujiwara H, Koyama A, Hasegawa M, Miura M, Iwatsubo T: Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alphasynuclein transgenic Drosophila. Neurosci Lett 2003, 336(3):155158. Kuwahara T, Koyama A, Gengyo-Ando K, Masuda M, Kowa H, Tsunoda M, Mitani S, Iwatsubo T: Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem 2006, 281(1):334-340. Lakso M, Vartiainen S, Moilanen AM, Sirvio J, Thomas JH, Nass R, Blakely RD, Wong G: Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alphasynuclein. J Neurochem 2003, 86(1):165-172. Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ et al: Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 2003, 278(44):43628-43635. Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L et al: Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 2003, 12(18):2277-2291. Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM: Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A 2004, 101(29):10744-10749. Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ: Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 2003, 100(7):4078-4083. Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ: Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A 2005, 102(22):8024-8029. Hao LY, Giasson BI, Bonini NM: DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A 2010, 107(21):9747-9752. Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Muller WE, Kudin AP et al: Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One 2009, 4(6):e5777. 273 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. Rugbjerg K, Ritz B, Korbo L, Martinussen N, Olsen JH: Risk of Parkinson's disease after hospital contact for head injury: population based case-control study. BMJ 2008, 337:a2494. Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ: A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol 2002, 52(3):276-284. Quik M, O'Neill M, Perez XA: Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci 2007, 28(5):229-235. Frigerio R, Elbaz A, Sanft KR, Peterson BJ, Bower JH, Ahlskog JE, Grossardt BR, de Andrade M, Maraganore DM, Rocca WA: Education and occupations preceding Parkinson disease: a population-based case-control study. Neurology 2005, 65(10):15751583. Saaksjarvi K, Knekt P, Rissanen H, Laaksonen MA, Reunanen A, Mannisto S: Prospective study of coffee consumption and risk of Parkinson's disease. Eur J Clin Nutr 2008, 62(7):908-915. McGeer PL, Itagaki S, Boyes BE, McGeer EG: Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1988, 38(8):1285-1291. Langston JW, Ballard P, Tetrud JW, Irwin I: Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219(4587):979-980. Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR et al: Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect 2011, 119(6):866-872. Porras G, Li Q, Bezard E: Modeling Parkinson's Disease in Primates: The MPTP Model. Cold Spring Harb Perspect Med 2012, 2(3):a009308. Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT: Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 2000, 3(12):1301-1306. Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ: Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res 1999, 823(1-2):1-10. McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA: Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis 2002, 10(2):119-127. Cuperus R, Leen R, Tytgat GA, Caron HN, van Kuilenburg AB: Fenretinide induces mitochondrial ROS and inhibits the mitochondrial respiratory chain in neuroblastoma. Cell Mol Life Sci 2010, 67(5):807-816. Turrens JF: Mitochondrial formation of reactive oxygen species. J Physiol 2003, 552(Pt 2):335-344. 274 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. 106. Chiba K, Trevor AJ, Castagnoli N, Jr.: Active uptake of MPP+, a metabolite of MPTP, by brain synaptosomes. Biochem Biophys Res Commun 1985, 128(3):1228-1232. Nicklas WJ, Vyas I, Heikkila RE: Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6tetrahydropyridine. Life Sci 1985, 36(26):2503-2508. Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW: Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci 2005, 88(1):193-201. Bonneh-Barkay D, Langston WJ, Di Monte DA: Toxicity of redox cycling pesticides in primary mesencephalic cultures. Antioxid Redox Signal 2005, 7(5-6):649-653. Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD: Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain 1992, 115 ( Pt 2):333-342. Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD: Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989, 1(8649):1269. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD: Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 1990, 54(3):823-827. Janetzky B, Hauck S, Youdim MB, Riederer P, Jellinger K, Pantucek F, Zochling R, Boissl KW, Reichmann H: Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson's disease. Neurosci Lett 1994, 169(1-2):126-128. Keeney PM, Xie J, Capaldi RA, Bennett JP, Jr.: Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 2006, 26(19):5256-5264. Bindoff LA, Birch-Machin MA, Cartlidge NE, Parker WD, Jr., Turnbull DM: Respiratory chain abnormalities in skeletal muscle from patients with Parkinson's disease. J Neurol Sci 1991, 104(2):203-208. Shinde S, Pasupathy K: Respiratory-chain enzyme activities in isolated mitochondria of lymphocytes from patients with Parkinson's disease: preliminary study. Neurol India 2006, 54(4):390-393. Yoshino H, Nakagawa-Hattori Y, Kondo T, Mizuno Y: Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson's disease. J Neural Transm Park Dis Dement Sect 1992, 4(1):27-34. Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, Shults CW: Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol 1995, 37(6):714-722. Benecke R, Strumper P, Weiss H: Electron transfer complexes I and IV of platelets are abnormal in Parkinson's disease but 275 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. normal in Parkinson-plus syndromes. Brain 1993, 116 ( Pt 6):14511463. Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD: Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem 1989, 52(2):381-389. Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B: A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem 1997, 69(3):1326-1329. Floor E, Wetzel MG: Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem 1998, 70(1):268-275. Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K: Oxidative damage in nucleic acids and Parkinson's disease. J Neurosci Res 2007, 85(5):919-934. Selley ML: (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson's disease. Free Radic Biol Med 1998, 25(2):169-174. Kikuchi A, Takeda A, Onodera H, Kimpara T, Hisanaga K, Sato N, Nunomura A, Castellani RJ, Perry G, Smith MA et al: Systemic increase of oxidative nucleic acid damage in Parkinson's disease and multiple system atrophy. Neurobiol Dis 2002, 9(2):244-248. Abe T, Isobe C, Murata T, Sato C, Tohgi H: Alteration of 8hydroxyguanosine concentrations in the cerebrospinal fluid and serum from patients with Parkinson's disease. Neurosci Lett 2003, 336(2):105-108. Buhmann C, Arlt S, Kontush A, Moller-Bertram T, Sperber S, Oechsner M, Stuerenburg HJ, Beisiegel U: Plasma and CSF markers of oxidative stress are increased in Parkinson's disease and influenced by antiparkinsonian medication. Neurobiol Dis 2004, 15(1):160-170. Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, Wolburg H, Gizatullina Z, Gellerich FN, Woitalla D et al: Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One 2010, 5(2):e9367. Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR: DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 2011, 20(1):40-50. Heo JY, Park JH, Kim SJ, Seo KS, Han JS, Lee SH, Kim JM, Park JI, Park SK, Lim K et al: DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: involvement of mitochondrial complex I assembly. PLoS One 2012, 7(3):e32629. 276 118. 119. 120. 121. 122. 123. 124. 125. 126. 127. 128. 129. 130. 131. Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J: Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 2004, 279(18):18614-18622. Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T et al: Lossof-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 2007, 27(45):1241312418. Hoepken HH, Gispert S, Morales B, Wingerter O, Del Turco D, Mulsch A, Nussbaum RL, Muller K, Drose S, Brandt U et al: Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis 2007, 25(2):401411. Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT: Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009, 284(20):13843-13855. Gegg ME, Cooper JM, Schapira AH, Taanman JW: Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One 2009, 4(3):e4756. Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK: Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 2008, 283(14):9089-9100. Chinta SJ, Mallajosyula JK, Rane A, Andersen JK: Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett 2010, 486(3):235-239. Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ: Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A 2010, 107(26):11835-11840. Youle RJ, van der Bliek AM: Mitochondrial fission, fusion, and stress. Science 2012, 337(6098):1062-1065. Baker MJ, Tatsuta T, Langer T: Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol 2011, 3(7). Narendra D, Tanaka A, Suen DF, Youle RJ: Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008, 183(5):795-803. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW: Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010, 19(24):4861-4870. Narendra D, Tanaka A, Suen DF, Youle RJ: Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 2009, 5(5):706-708. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ: Mitochondrial membrane potential regulates PINK1 import and 277 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. proteolytic destabilization by PARL. J Cell Biol 2010, 191(5):933942. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ: PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8(1):e1000298. Liu W, Acin-Perez R, Geghman KD, Manfredi G, Lu B, Li C: Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A 2011, 108(31):12920-12924. Ron I, Rapaport D, Horowitz M: Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet 2010, 19(19):3771-3781. Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K et al: Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 2010, 29(20):3571-3589. Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J et al: Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem 2011, 286(23):20710-20726. Kobayashi M, Yamamoto M: Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal 2005, 7(3-4):385-394. Sykiotis GP, Bohmann D: Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell 2008, 14(1):76-85. Piantadosi CA, Suliman HB: Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem 2006, 281(1):324-333. Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T: Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 2006, 15(6):883-895. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M: PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res 2005, 66(3):562-573. Liang H, Ward WF: PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ 2006, 30(4):145-151. Li W, Kong AN: Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog 2009, 48(2):91-104. Czubryt MP, McAnally J, Fishman GI, Olson EN: Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha ) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A 2003, 100(4):1711-1716. Zhong N, Xu J: Synergistic activation of the human MnSOD promoter by DJ-1 and PGC-1alpha: regulation by SUMOylation and oxidation. Hum Mol Genet 2008, 17(21):3357-3367. 278 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM: Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J Neurosci Res 2009, 87(1):123-129. Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, Pifl C, Gygi SP, Geula C, Yankner BA: The Parkinson's diseaseassociated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum Mol Genet 2005, 14(9):1231-1241. Castillo-Quan JI: Parkin' control: regulation of PGC-1alpha through PARIS in Parkinson's disease. Dis Model Mech 2011, 4(4):427-429. Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM: PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 2011, 144(5):689-702. Sadeghian M, Marinova-Mutafchieva L, Broom L, Davis JB, Virley D, Medhurst AD, Dexter DT: Full and partial peroxisome proliferation-activated receptor-gamma agonists, but not delta agonist, rescue of dopaminergic neurons in the 6-OHDA parkinsonian model is associated with inhibition of microglial activation and MMP expression. J Neuroimmunol 2012, 246(12):69-77. Friling S, Bergsland M, Kjellander S: Activation of Retinoid X Receptor increases dopamine cell survival in models for Parkinson's disease. BMC Neurosci 2009, 10:146. Clark J, Reddy S, Zheng K, Betensky RA, Simon DK: Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson's disease. BMC Med Genet 2011, 12:69. Ledesma A, de Lacoba MG, Rial E: The mitochondrial uncoupling proteins. Genome Biol 2002, 3(12):REVIEWS3015. Ricquier D, Bouillaud F: The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J 2000, 345 Pt 2:161-179. Porter RK: Mitochondrial proton leak: a role for uncoupling proteins 2 and 3? Biochim Biophys Acta 2001, 1504(1):120-127. Medvedev AV, Snedden SK, Raimbault S, Ricquier D, Collins S: Transcriptional regulation of the mouse uncoupling protein-2 gene. Double E-box motif is required for peroxisome proliferatoractivated receptor-gamma-dependent activation. J Biol Chem 2001, 276(14):10817-10823. Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, Stepkowski SM, Davies PJ, Taegtmeyer H: Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J 2001, 15(3):833-845. Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B: Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 2001, 276(12):8705-8712. 279 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. 170. 172. 173. Carroll AM, Porter RK, Morrice NA: Identification of serine phosphorylation in mitochondrial uncoupling protein 1. Biochim Biophys Acta 2008, 1777(7-8):1060-1065. Azzu V, Affourtit C, Breen EP, Parker N, Brand MD: Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim Biophys Acta 2008, 1777(10):1378-1383. Derdak Z, Fulop P, Sabo E, Tavares R, Berthiaume EP, Resnick MB, Paragh G, Wands JR, Baffy G: Enhanced colon tumor induction in uncoupling protein-2 deficient mice is associated with NF-kappaB activation and oxidative stress. Carcinogenesis 2006, 27(5):956-961. Shilo S, Aharoni-Simon M, Tirosh O: Selenium attenuates expression of MnSOD and uncoupling protein 2 in J774.2 macrophages: molecular mechanism for its cell-death and antiinflammatory activity. Antioxid Redox Signal 2005, 7(1-2):276286. Jaburek M, Miyamoto S, Di Mascio P, Garlid KD, Jezek P: Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. J Biol Chem 2004, 279(51):53097-53102. Ho PW, Chan DY, Kwok KH, Chu AC, Ho JW, Kung MH, Ramsden DB, Ho SL: Methyl-4-phenylpyridinium ion modulates expression of mitochondrial uncoupling proteins 2, 4, and 5 in catecholaminergic (SK-N-SH) cells. J Neurosci Res 2005, 81(2):261-268. Echtay KS, Pakay JL, Esteves TC, Brand MD: Hydroxynonenal and uncoupling proteins: a model for protection against oxidative damage. Biofactors 2005, 24(1-4):119-130. Dikov D, Aulbach A, Muster B, Drose S, Jendrach M, BereiterHahn J: Do UCP2 and mild uncoupling improve longevity? Exp Gerontol 2010, 45(7-8):586-595. McDonald RB, Walker KM, Warman DB, Griffey SM, Warden CH, Ramsey JJ, Horwitz BA: Characterization of survival and phenotype throughout the life span in UCP2/UCP3 genetically altered mice. Exp Gerontol 2008, 43(12):1061-1068. Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ: Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468(7324):696-700. Albin RL, Young AB, Penney JB: The functional anatomy of basal ganglia disorders. Trends Neurosci 1989, 12(10):366-375. Lipscombe D: L-type calcium channels: highs and new lows. Circ Res 2002, 90(9):933-935. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW et al: PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 2009, 33(5):627-638. Frei B, Richter C: N-methyl-4-phenylpyridine (MMP+) together with 6-hydroxydopamine or dopamine stimulates Ca2+ release from mitochondria. FEBS Lett 1986, 198(1):99-102. 280 174. 175. 176. 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. Sousa SC, Maciel EN, Vercesi AE, Castilho RF: Ca2+-induced oxidative stress in brain mitochondria treated with the respiratory chain inhibitor rotenone. FEBS Lett 2003, 543(1-3):179-183. Wang XJ, Xu JX: Possible involvement of Ca2+ signaling in rotenone-induced apoptosis in human neuroblastoma SH-SY5Y cells. Neurosci Lett 2005, 376(2):127-132. Vila M, Vukosavic S, Jackson-Lewis V, Neystat M, Jakowec M, Przedborski S: Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J Neurochem 2000, 74(2):721-729. Meredith GE, Totterdell S, Petroske E, Santa Cruz K, Callison RC, Jr., Lau YS: Lysosomal malfunction accompanies alphasynuclein aggregation in a progressive mouse model of Parkinson's disease. Brain Res 2002, 956(1):156-165. Yu WH, Matsuoka Y, Sziraki I, Hashim A, Lafrancois J, Sershen H, Duff KE: Increased dopaminergic neuron sensitivity to 1methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in transgenic mice expressing mutant A53T alpha-synuclein. Neurochem Res 2008, 33(5):902-911. Nieto M, Gil-Bea FJ, Dalfo E, Cuadrado M, Cabodevilla F, Sanchez B, Catena S, Sesma T, Ribe E, Ferrer I et al: Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol Aging 2006, 27(6):848-856. McLean JR, Hallett PJ, Cooper O, Stanley M, Isacson O: Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson's disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol Cell Neurosci 2012, 49(2):230-239. St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ: Dopaminergic neuron loss and upregulation of chaperone protein mRNA induced by targeted overexpression of alpha-synuclein in mouse substantia nigra. J Neurochem 2007, 100(6):1449-1457. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D: Impaired degradation of mutant alpha-synuclein by chaperonemediated autophagy. Science 2004, 305(5688):1292-1295. Sridhar S, Botbol Y, Macian F, Cuervo AM: Autophagy and disease: always two sides to a problem. J Pathol 2012, 226(2):255273. Yamamoto A, Cremona ML, Rothman JE: Autophagy-mediated clearance of huntingtin aggregates triggered by the insulinsignaling pathway. J Cell Biol 2006, 172(5):719-731. Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ et al: Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 2006, 15(3):433-442. Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC: Lithium induces autophagy by 281 187. 188. 189. 190. 191. 192. 193. 194. 195. 196. 197. inhibiting inositol monophosphatase. J Cell Biol 2005, 170(7):11011111. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ et al: Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004, 36(6):585-595. Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L: Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 2009, 4(5):e5515. Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, Xie ZL, Speake LD, Parks R, Crabtree D et al: Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008, 1:17. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM et al: Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 2004, 82(3):192-207. Saito Y, Ruberu NN, Sawabe M, Arai T, Kazama H, Hosoi T, Yamanouchi H, Murayama S: Lewy body-related alphasynucleinopathy in aging. J Neuropathol Exp Neurol 2004, 63(7):742-749. Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, Boston H, Saftig P, Woulfe J, Feany MB et al: Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009, 2:5. Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VM, Trojanowski JQ, Sidransky E: Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006, 67(5):908-910. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D: Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146(1):37-52. Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL et al: CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A 2011, 108(29):1210112106. Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E: Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 2012, 109(24):9611-9616. Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D: Deficiency of ATP13A2 leads to lysosomal dysfunction, alphasynuclein accumulation, and neurotoxicity. J Neurosci 2012, 32(12):4240-4246. 282 198. 199. 200. 201. 202. 203. 204. 205. 206. 208. 209. 210. 211. Jolly RD, Brown S, Das AM, Walkley SU: Mitochondrial dysfunction in the neuronal ceroid-lipofuscinoses (Batten disease). Neurochem Int 2002, 40(6):565-571. Luiro K, Kopra O, Blom T, Gentile M, Mitchison HM, Hovatta I, Tornquist K, Jalanko A: Batten disease (JNCL) is linked to disturbances in mitochondrial, cytoskeletal, and synaptic compartments. J Neurosci Res 2006, 84(5):1124-1138. Lee BD, Dawson VL, Dawson TM: Leucine-rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson's disease. Trends Pharmacol Sci 2012, 33(7):365-373. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM: Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 2005, 102(46):16842-16847. Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR: LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J 2007, 405(2):307-317. West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL et al: Parkinson's disease-associated mutations in LRRK2 link enhanced GTPbinding and kinase activities to neuronal toxicity. Hum Mol Genet 2007, 16(2):223-232. Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T: GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry 2007, 46(5):1380-1388. Liu M, Dobson B, Glicksman MA, Yue Z, Stein RL: Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry 2010, 49(9):2008-2017. Taymans JM, Vancraenenbroeck R, Ollikainen P, Beilina A, Lobbestael E, De Maeyer M, Baekelandt V, Cookson MR: LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS One 2011, 6(8):e23207. Jorgensen ND, Peng Y, Ho CC, Rideout HJ, Petrey D, Liu P, Dauer WT: The WD40 domain is required for LRRK2 neurotoxicity. PLoS One 2009, 4(12):e8463. Deng J, Lewis PA, Greggio E, Sluch E, Beilina A, Cookson MR: Structure of the ROC domain from the Parkinson's diseaseassociated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci U S A 2008, 105(5):1499-1504. Klein CL, Rovelli G, Springer W, Schall C, Gasser T, Kahle PJ: Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J Neurochem 2009, 111(3):703-715. Greggio E, Taymans JM, Zhen EY, Ryder J, Vancraenenbroeck R, Beilina A, Sun P, Deng J, Jaffe H, Baekelandt V et al: The Parkinson's disease kinase LRRK2 autophosphorylates its GTPase 283 212. 213. 214. 215. 216. 217. 218. 219. 220. 221. 222. 223. domain at multiple sites. Biochem Biophys Res Commun 2009, 389(3):449-454. Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR et al: Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 2011, 7(4):203-205. Ramsden N, Perrin J, Ren Z, Lee BD, Zinn N, Dawson VL, Tam D, Bova M, Lang M, Drewes G et al: Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson's disease-related toxicity in human neurons. ACS Chem Biol 2011, 6(10):1021-1028. Zhang J, Deng X, Choi HG, Alessi DR, Gray NS: Characterization of TAE684 as a potent LRRK2 kinase inhibitor. Bioorg Med Chem Lett 2012, 22(5):1864-1869. Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR et al: 143-3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem J 2010, 430(3):393-404. Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ: Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J 2010, 430(3):405-413. Di Fonzo A, Tassorelli C, De Mari M, Chien HF, Ferreira J, Rohe CF, Riboldazzi G, Antonini A, Albani G, Mauro A et al: Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease. Eur J Hum Genet 2006, 14(3):322-331. Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M: The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 2006, 15(2):223-232. Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR: The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun 2007, 357(3):668-671. Daniels V, Vancraenenbroeck R, Law BM, Greggio E, Lobbestael E, Gao F, De Maeyer M, Cookson MR, Harvey K, Baekelandt V et al: Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J Neurochem 2011, 116(2):304-315. Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, Lewis P, Jain S, Ding J, Syed A et al: The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem 2008, 283(24):16906-16914. Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA: Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 2006, 9(10):1231-1233. Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA: Leucine-rich repeat kinase 2 (LRRK2) 284 224. 225. 226. 227. 228. 229. 230. 231. 232. 233. 234. 235. 236. interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A 2005, 102(51):18676-18681. Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ et al: Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 2006, 23(2):329-341. Xiong H, Qiu H, Zhuang L, Jiang R, Chen Y: Effects of 5-AzaCdR on the proliferation of human breast cancer cell line MCF-7 and on the expression of Apaf-1 gene. J Huazhong Univ Sci Technolog Med Sci 2009, 29(4):498-502. Lichtenberg M, Mansilla A, Zecchini VR, Fleming A, Rubinsztein DC: The Parkinson's disease protein LRRK2 impairs proteasome substrate clearance without affecting proteasome catalytic activity. Cell Death Dis 2011, 2:e196. Iaccarino C, Crosio C, Vitale C, Sanna G, Carri MT, Barone P: Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet 2007, 16(11):1319-1326. Sen S, Webber PJ, West AB: Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem 2009, 284(52):36346-36356. Samann J, Hegermann J, von Gromoff E, Eimer S, Baumeister R, Schmidt E: Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J Biol Chem 2009, 284(24):16482-16491. Wolozin B, Saha S, Guillily M, Ferree A, Riley M: Investigating convergent actions of genes linked to familial Parkinson's disease. Neurodegener Dis 2008, 5(3-4):182-185. Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B: Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 2008, 27(18):2432-2443. Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T et al: A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A 2008, 105(7):2693-2698. Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L et al: Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 2011, 6(4):e18568. Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z: Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci 2010, 30(5):1788-1797. Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J et al: Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-diseaserelated mutant alpha-synuclein. Neuron 2009, 64(6):807-827. Daher JP, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL et al: 285 237. 238. 239. 240. 241. 242. 243. 244. 245. 246. 247. 248. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet 2012, 21(11):2420-2431. Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB: Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci 2007, 8:102. Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S et al: LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet 2011, 20(21):4209-4223. Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N: Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet 2007, 16(6):678-690. Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, Cai H, Bonventre JV, Shen J: Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener 2012, 7:2. Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR et al: Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm 2011, 118(5):795-808. Kubo M, Kamiya Y, Nagashima R, Maekawa T, Eshima K, Azuma S, Ohta E, Obata F: LRRK2 is expressed in B-2 but not in B-1 B cells, and downregulated by cellular activation. J Neuroimmunol 2010, 229(1-2):123-128. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ et al: LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol 2010, 185(9):5577-5585. Melrose HL, Kent CB, Taylor JP, Dachsel JC, Hinkle KM, Lincoln SJ, Mok SS, Culvenor JG, Masters CL, Tyndall GM et al: A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience 2007, 147(4):1047-1058. Melrose H, Lincoln S, Tyndall G, Dickson D, Farrer M: Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience 2006, 139(3):791-794. Simon-Sanchez J, Herranz-Perez V, Olucha-Bordonau F, PerezTur J: LRRK2 is expressed in areas affected by Parkinson's disease in the adult mouse brain. Eur J Neurosci 2006, 23(3):659666. Higashi S, Biskup S, West AB, Trinkaus D, Dawson VL, Faull RL, Waldvogel HJ, Arai H, Dawson TM, Moore DJ et al: Localization of Parkinson's disease-associated LRRK2 in normal and pathological human brain. Brain Res 2007, 1155:208-219. Higashi S, Moore DJ, Colebrooke RE, Biskup S, Dawson VL, Arai H, Dawson TM, Emson PC: Expression and localization of 286 249. 250. 251. 252. 253. 254. 255. 256. 257. 258. 259. 260. 261. Parkinson's disease-associated leucine-rich repeat kinase 2 in the mouse brain. J Neurochem 2007, 100(2):368-381. Lee H, Melrose HL, Yue M, Pare JF, Farrer MJ, Smith Y: Lrrk2 localization in the primate basal ganglia and thalamus: a light and electron microscopic analysis in monkeys. Exp Neurol 2010, 224(2):438-447. Mandemakers W, Snellinx A, O'Neill MJ, de Strooper B: LRRK2 expression is enriched in the striosomal compartment of mouse striatum. Neurobiol Dis 2012, 48(3):582-593. Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ et al: Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol 2006, 60(5):557-569. Berger Z, Smith KA, Lavoie MJ: Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 2010, 49(26):5511-5523. Lee S, Liu HP, Lin WY, Guo H, Lu B: LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci 2010, 30(50):16959-16969. Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt-Weisenhorn DM, Wurst W et al: LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci 2011, 31(6):2225-2237. Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J et al: LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res 2008, 314(10):2055-2065. Lundblad M, Decressac M, Mattsson B, Bjorklund A: Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A 2012, 109(9):3213-3219. Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R: LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet 2009, 18(21):4022-4034. Heo HY, Kim KS, Seol W: Coordinate Regulation of Neurite Outgrowth by LRRK2 and Its Interactor, Rab5. Exp Neurobiol 2010, 19(2):97-105. Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT: Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 2008, 105(3):1048-1056. MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A: The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52(4):587-593. Gillardon F: Interaction of elongation factor 1-alpha with leucinerich repeat kinase 2 impairs kinase activity and microtubule bundling in vitro. Neuroscience 2009, 163(2):533-539. 287 262. 263. 264. 265. 266. 267. 268. 269. 270. 271. 272. 273. 274. 275. Gillardon F: Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability--a point of convergence in parkinsonian neurodegeneration? J Neurochem 2009, 110(5):1514-1522. Kawakami F, Yabata T, Ohta E, Maekawa T, Shimada N, Suzuki M, Maruyama H, Ichikawa T, Obata F: LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS One 2012, 7(1):e30834. Sancho RM, Law BM, Harvey K: Mutations in the LRRK2 RocCOR tandem domain link Parkinson's disease to Wnt signalling pathways. Hum Mol Genet 2009, 18(20):3955-3968. Duka T, Duka V, Joyce JN, Sidhu A: Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson's disease models. FASEB J 2009, 23(9):2820-2830. Habig K, Walter M, Poths S, Riess O, Bonin M: RNA interference of LRRK2-microarray expression analysis of a Parkinson's disease key player. Neurogenetics 2008, 9(2):83-94. Schulz C, Paus M, Frey K, Schmid R, Kohl Z, Mennerich D, Winkler J, Gillardon F: Leucine-rich repeat kinase 2 modulates retinoic acid-induced neuronal differentiation of murine embryonic stem cells. PLoS One 2011, 6(6):e20820. Chiba-Falek O, Lopez GJ, Nussbaum RL: Levels of alphasynuclein mRNA in sporadic Parkinson disease patients. Mov Disord 2006, 21(10):1703-1708. Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W et al: Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491(7425):603-607. Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ: Leucinerich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal 2010, 22(5):821-827. Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ: The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol 2011, 12(11):1063-1070. Berwick DC, Harvey K: LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane-localized LRP6. Hum Mol Genet 2012, 21(22):4966-4979. Gehrke S, Imai Y, Sokol N, Lu B: Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466(7306):637-641. Pons B, Armengol G, Livingstone M, Lopez L, Coch L, Sonenberg N, Ramon y Cajal S: Association between LRRK2 and 4E-BP1 protein levels in normal and malignant cells. Oncol Rep 2012, 27(1):225-231. Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ: Rapamycin activation of 4E-BP prevents 288 276. 277. 278. 279. 280. 281. 282. 283. 284. 285. 286. 287. 288. parkinsonian dopaminergic neuron loss. Nat Neurosci 2009, 12(9):1129-1135. Chen CY, Weng YH, Chien KY, Lin KJ, Yeh TH, Cheng YP, Lu CS, Wang HL: (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ 2012, 19(10):1623-1633. White LR, Toft M, Kvam SN, Farrer MJ, Aasly JO: MAPKpathway activity, Lrrk2 G2019S, and Parkinson's disease. J Neurosci Res 2007, 85(6):1288-1294. Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC: Nuclear translocation of NFkappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci U S A 1997, 94(14):75317536. Iwata A, Maruyama M, Kanazawa I, Nukina N: alpha-Synuclein affects the MAPK pathway and accelerates cell death. J Biol Chem 2001, 276(48):45320-45329. Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X: LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 2012, 21(9):1931-1944. Niu J, Yu M, Wang C, Xu Z: Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J Neurochem 2012, 122(3):650-658. Lee SB, Kim W, Lee S, Chung J: Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun 2007, 358(2):534-539. Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K et al: LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci 2009, 29(29):9210-9218. Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL: Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci 2009, 29(36):11257-11262. Mortiboys H, Johansen KK, Aasly JO, Bandmann O: Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75(22):20172020. Cui J, Yu M, Niu J, Yue Z, Xu Z: Expression of Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibits the Processing of uMtCK to Induce Cell Death in cell culture model system. Biosci Rep 2011. Venderova K, Kabbach G, Abdel-Messih E, Zhang Y, Parks RJ, Imai Y, Gehrke S, Ngsee J, Lavoie MJ, Slack RS et al: LeucineRich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson's disease. Hum Mol Genet 2009, 18(22):4390-4404. Nishio K, Qiao S, Yamashita H: Characterization of the differential expression of uncoupling protein 2 and ROS 289 289. 290. 291. 292. 293. 294. 295. 296. 297. 298. 299. 300. 301. 302. production in differentiated mouse macrophage-cells (Mm1) and the progenitor cells (M1). J Mol Histol 2005, 36(1-2):35-44. Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L et al: Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4phenyl-1,2,3,6-tetrahydropyridine). J Neurosci 2009, 29(50):1584615850. Bradley JL, Homayoun S, Hart PE, Schapira AH, Cooper JM: Role of oxidative damage in Friedreich's ataxia. Neurochem Res 2004, 29(3):561-567. Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH: Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann Neurol 1999, 45(1):25-32. Chau KY, Cooper JM, Schapira AH: Rasagiline protects against alpha-synuclein induced sensitivity to oxidative stress in dopaminergic cells. Neurochem Int 2010, 57(5):525-529. Chau KY, Korlipara LV, Cooper JM, Schapira AH: Protection against paraquat and A53T alpha-synuclein toxicity by cabergoline is partially mediated by dopamine receptors. J Neurol Sci 2009, 278(1-2):44-53. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC: Measurement of protein using bicinchoninic acid. Anal Biochem 1985, 150(1):76-85. Schagger H: Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol 2001, 65:231-244. Rahman S, Lake BD, Taanman JW, Hanna MG, Cooper JM, Schapira AH, Leonard JV: Cytochrome oxidase immunohistochemistry: clues for genetic mechanisms. Brain 2000, 123 Pt 3:591-600. Waltregny D, North B, Van Mellaert F, de Leval J, Verdin E, Castronovo V: Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. Eur J Histochem 2004, 48(3):273-290. Waxman EA, Covy JP, Bukh I, Li X, Dawson TM, Giasson BI: Leucine-rich repeat kinase 2 expression leads to aggresome formation that is not associated with alpha-synuclein inclusions. J Neuropathol Exp Neurol 2009, 68(7):785-796. Lillie RD: Histopathologic technic and practical histochemistry, 3d edn. New York: McGraw-Hill; 1965. Graham JM: Isolation of mitochondria from tissues and cells by differential centrifugation. Curr Protoc Cell Biol 2001, Chapter 3:Unit 3 3. Hornig-Do HT, Gunther G, Bust M, Lehnartz P, Bosio A, Wiesner RJ: Isolation of functional pure mitochondria by superparamagnetic microbeads. Anal Biochem 2009, 389(1):1-5. Pon LA, Schon EA, American Society for Cell Biology.: Mitochondria, 2nd edn. San Diego, Calif.: Academic Press; 2007. 290 303. 304. 305. 306. 307. 308. 309. 310. 311. 312. 313. 314. 315. 316. Kouri K, Duchen MR, Lemmens-Gruber R: Effects of beauvericin on the metabolic state and ionic homeostasis of ventricular myocytes of the guinea pig. Chem Res Toxicol 2005, 18(11):16611668. Wilson-Fritch L, Burkart A, Bell G, Mendelson K, Leszyk J, Nicoloro S, Czech M, Corvera S: Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol Cell Biol 2003, 23(3):1085-1094. Hynes J, Natoli E, Jr., Will Y: Fluorescent pH and oxygen probes of the assessment of mitochondrial toxicity in isolated mitochondria and whole cells. Curr Protoc Toxicol 2009, Chapter 2:Unit 2 16. Zhdanov AV, Favre C, O'Flaherty L, Adam J, O'Connor R, Pollard PJ, Papkovsky DB: Comparative bioenergetic assessment of transformed cells using a cell energy budget platform. Integr Biol (Camb) 2011, 3(11):1135-1142. Gullbo J, Fryknas M, Rickardson L, Darcy P, Hagg M, Wickstrom M, Hassan S, Westman G, Brnjic S, Nygren P et al: Phenotypebased drug screening in primary ovarian carcinoma cultures identifies intracellular iron depletion as a promising strategy for cancer treatment. Biochem Pharmacol 2011, 82(2):139-147. Delves PJ: Roitt's essential immunology, 11th edn. Malden, Mass. ; Oxford: Blackwell; 2006. Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M: Calreticulin: one protein, one gene, many functions. Biochem J 1999, 344 Pt 2:281-292. Chinopoulos C, Gerencser AA, Mandi M, Mathe K, Torocsik B, Doczi J, Turiak L, Kiss G, Konrad C, Vajda S et al: Forward operation of adenine nucleotide translocase during F0F1-ATPase reversal: critical role of matrix substrate-level phosphorylation. FASEB J 2010, 24(7):2405-2416. Vercesi AE, Kowaltowski AJ, Grijalba MT, Meinicke AR, Castilho RF: The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep 1997, 17(1):43-52. Rasmussen HN, Andersen AJ, Rasmussen UF: Optimization of preparation of mitochondria from 25-100 mg skeletal muscle. Anal Biochem 1997, 252(1):153-159. Krauss S, Zhang CY, Lowell BB: The mitochondrial uncouplingprotein homologues. Nat Rev Mol Cell Biol 2005, 6(3):248-261. Kajimoto K, Yamazaki N, Kataoka M, Terada H, Shinohara Y: Identification of possible protein machinery involved in the thermogenic function of brown adipose tissue. J Med Invest 2004, 51(1-2):20-28. Zhang CY, Parton LE, Ye CP, Krauss S, Shen R, Lin CT, Porco JA, Jr., Lowell BB: Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab 2006, 3(6):417427. Puigserver P, Spiegelman BM: Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): 291 317. 318. 319. 320. 321. 322. 323. 324. 325. 326. 327. 328. 329. transcriptional coactivator and metabolic regulator. Endocr Rev 2003, 24(1):78-90. Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA: Protein kinases C and D mediate agonistdependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 2004, 24(19):8374-8385. Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L: LRRK2 expression linked to dopamine-innervated areas. Ann Neurol 2006, 59(4):714-719. Taymans JM, Van den Haute C, Baekelandt V: Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem 2006, 98(3):951-961. Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM: Biochemical and pathological characterization of Lrrk2. Ann Neurol 2006, 59(2):315-322. Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB: LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 2012, 32(5):16021611. Thevenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ: Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One 2011, 6(6):e21519. Dusonchet J, Kochubey O, Stafa K, Young SM, Jr., Zufferey R, Moore DJ, Schneider BL, Aebischer P: A rat model of progressive nigral neurodegeneration induced by the Parkinson's diseaseassociated G2019S mutation in LRRK2. J Neurosci 2011, 31(3):907-912. Devine MJ, Kaganovich A, Ryten M, Mamais A, Trabzuni D, Manzoni C, McGoldrick P, Chan D, Dillman A, Zerle J et al: Pathogenic LRRK2 mutations do not alter gene expression in cell model systems or human brain tissue. PLoS One 2011, 6(7):e22489. Maekawa T, Kubo M, Yokoyama I, Ohta E, Obata F: Agedependent and cell-population-restricted LRRK2 expression in normal mouse spleen. Biochem Biophys Res Commun 2010, 392(3):431-435. Milosevic J, Schwarz SC, Ogunlade V, Meyer AK, Storch A, Schwarz J: Emerging role of LRRK2 in human neural progenitor cell cycle progression, survival and differentiation. Mol Neurodegener 2009, 4:25. Chan D, Citro A, Cordy JM, Shen GC, Wolozin B: Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2). J Biol Chem 2011, 286(18):16140-16149. Cho HJ, Liu G, Jin SM, Parisiadou L, Xie C, Yu J, Sun L, Ma B, Ding J, Vancraenenbroeck R et al: MicroRNA-205 regulates the expression of Parkinson's disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet 2012. Melrose HL, Dachsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L et al: Impaired dopaminergic neurotransmission and microtubule-associated 292 330. 331. 332. 333. 334. 335. 336. 337. 338. 339. 340. 341. 342. 343. protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis 2010, 40(3):503-517. Maekawa T, Mori S, Sasaki Y, Miyajima T, Azuma S, Ohta E, Obata F: The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol Neurodegener 2012, 7:15. Sharma S, Bandopadhyay R, Lashley T, Renton AE, Kingsbury AE, Kumaran R, Kallis C, Vilarino-Guell C, O'Sullivan SS, Lees AJ et al: LRRK2 expression in idiopathic and G2019S positive Parkinson's disease subjects: a morphological and quantitative study. Neuropathol Appl Neurobiol 2011, 37(7):777-790. Alegre-Abarrategui J, Wade-Martins R: Parkinson disease, LRRK2 and the endocytic-autophagic pathway. Autophagy 2009, 5(8):1208-1210. Kett LR, Boassa D, Ho CC, Rideout HJ, Hu J, Terada M, Ellisman M, Dauer WT: LRRK2 Parkinson disease mutations enhance its microtubule association. Hum Mol Genet 2012, 21(4):890-899. Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y: JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J 2004, 23(8):1889-1899. Schwartz MP, Matouschek A: The dimensions of the protein import channels in the outer and inner mitochondrial membranes. Proc Natl Acad Sci U S A 1999, 96(23):13086-13090. Lin W, Kang UJ: Structural determinants of PINK1 topology and dual subcellular distribution. BMC Cell Biol 2010, 11:90. Badugu R, Garcia M, Bondada V, Joshi A, Geddes JW: N terminus of calpain 1 is a mitochondrial targeting sequence. J Biol Chem 2008, 283(6):3409-3417. Melrose H: Update on the functional biology of Lrrk2. Future Neurol 2008, 3(6):669-681. Hermanson SB, Carlson CB, Riddle SM, Zhao J, Vogel KW, Nichols RJ, Bi K: Screening for novel LRRK2 inhibitors using a high-throughput TR-FRET cellular assay for LRRK2 Ser935 phosphorylation. PLoS One 2012, 7(8):e43580. Parker WD, Jr., Boyson SJ, Parks JK: Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 1989, 26(6):719-723. Tieu K, Ischiropoulos H, Przedborski S: Nitric oxide and reactive oxygen species in Parkinson's disease. IUBMB Life 2003, 55(6):329-335. Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R et al: D-betahydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 2003, 112(6):892-901. Grunewald A, Gegg ME, Taanman JW, King RH, Kock N, Klein C, Schapira AH: Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp Neurol 2009, 219(1):266-273. 293 344. 345. 346. 347. 348. 349. 350. 351. 352. 353. 354. 355. 356. 357. Gautier CA, Kitada T, Shen J: Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 2008, 105(32):1136411369. Vincent A, Briggs L, Chatwin GF, Emery E, Tomlins R, Oswald M, Middleton CA, Evans GJ, Sweeney ST, Elliott CJ: parkininduced defects in neurophysiology and locomotion are generated by metabolic dysfunction and not oxidative stress. Hum Mol Genet 2012, 21(8):1760-1769. Grunewald A, Voges L, Rakovic A, Kasten M, Vandebona H, Hemmelmann C, Lohmann K, Orolicki S, Ramirez A, Schapira AH et al: Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS One 2010, 5(9):e12962. Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR et al: Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol 2008, 64(5):555-565. Gautier CA, Giaime E, Caballero E, Nunez L, Song Z, Chan D, Villalobos C, Shen J: Regulation of mitochondrial permeability transition pore by PINK1. Mol Neurodegener 2012, 7:22. Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD: Mitochondrial proton and electron leaks. Essays Biochem 2010, 47:53-67. Krauss S, Zhang CY, Lowell BB: A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci U S A 2002, 99(1):118-122. Rolfe DF, Brand MD: Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am J Physiol 1996, 271(4 Pt 1):C1380-1389. Rasola A, Bernardi P: The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12(5):815-833. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM: p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149(7):1536-1548. Rousset S, Alves-Guerra MC, Mozo J, Miroux B, CassardDoulcier AM, Bouillaud F, Ricquier D: The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53 Suppl 1:S130-135. Andrews ZB, Horvath TL: Uncoupling protein-2 regulates lifespan in mice. Am J Physiol Endocrinol Metab 2009, 296(4):E621-627. Casteilla L, Blondel O, Klaus S, Raimbault S, Diolez P, Moreau F, Bouillaud F, Ricquier D: Stable expression of functional mitochondrial uncoupling protein in Chinese hamster ovary cells. Proc Natl Acad Sci U S A 1990, 87(13):5124-5128. Ho PW, Ho JW, Tse HM, So DH, Yiu DC, Liu HF, Chan KH, Kung MH, Ramsden DB, Ho SL: Uncoupling protein-4 (UCP4) increases ATP supply by interacting with mitochondrial Complex II in neuroblastoma cells. PLoS One 2012, 7(2):e32810. 294 358. 359. 360. 361. 362. 363. 364. 365. 366. 367. 368. 369. 370. Hoang T, Smith MD, Jelokhani-Niaraki M: Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4, and UCP5. Biochemistry 2012, 51(19):4004-4014. Cannon B, Nedergaard J: Brown adipose tissue: function and physiological significance. Physiol Rev 2004, 84(1):277-359. Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, Tsushima RG, Pennefather PS, Salapatek AM, Wheeler MB: Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes 2001, 50(6):1302-1310. Mori S, Yoshizuka N, Takizawa M, Takema Y, Murase T, Tokimitsu I, Saito M: Expression of uncoupling proteins in human skin and skin-derived cells. J Invest Dermatol 2008, 128(8):18941900. Lengacher S, Magistretti PJ, Pellerin L: Quantitative rt-PCR analysis of uncoupling protein isoforms in mouse brain cortex: methodological optimization and comparison of expression with brown adipose tissue and skeletal muscle. J Cereb Blood Flow Metab 2004, 24(7):780-788. Wu Z, Zhang J, Zhao B: Superoxide anion regulates the mitochondrial free Ca2+ through uncoupling proteins. Antioxid Redox Signal 2009, 11(8):1805-1818. Liu Y, Jiang H, Xing FQ, Huang WJ, Mao LH, He LY: Uncoupling protein 2 expression affects androgen synthesis in polycystic ovary syndrome. Endocrine 2012. Madsen L, Pedersen LM, Lillefosse HH, Fjaere E, Bronstad I, Hao Q, Petersen RK, Hallenborg P, Ma T, De Matteis R et al: UCP1 induction during recruitment of brown adipocytes in white adipose tissue is dependent on cyclooxygenase activity. PLoS One 2010, 5(6):e11391. Emilsson V, O'Dowd J, Wang S, Liu YL, Sennitt M, Heyman R, Cawthorne MA: The effects of rexinoids and rosiglitazone on body weight and uncoupling protein isoform expression in the Zucker fa/fa rat. Metabolism 2000, 49(12):1610-1615. Li Y, Maedler K, Shu L, Haataja L: UCP-2 and UCP-3 proteins are differentially regulated in pancreatic beta-cells. PLoS One 2008, 3(1):e1397. Rupprecht A, Brauer AU, Smorodchenko A, Goyn J, Hilse KE, Shabalina IG, Infante-Duarte C, Pohl EE: Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during T-cell proliferation. PLoS One 2012, 7(8):e41406. Mailloux RJ, Adjeitey CN, Harper ME: Genipin-induced inhibition of uncoupling protein-2 sensitizes drug-resistant cancer cells to cytotoxic agents. PLoS One 2010, 5(10):e13289. Huntgeburth M, Tiemann K, Shahverdyan R, Schluter KD, Schreckenberg R, Gross ML, Modersheim S, Caglayan E, MullerEhmsen J, Ghanem A et al: Transforming growth factor beta(1) oppositely regulates the hypertrophic and contractile response to 295 371. 372. 373. 374. 375. 376. 377. 378. 379. 380. 381. beta-adrenergic stimulation in the heart. PLoS One 2011, 6(11):e26628. Kim BC, Kim HG, Lee SA, Lim S, Park EH, Kim SJ, Lim CJ: Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of mitochondrial pathway. Biochem Pharmacol 2005, 70(9):1398-1407. Diao J, Allister EM, Koshkin V, Lee SC, Bhattacharjee A, Tang C, Giacca A, Chan CB, Wheeler MB: UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc Natl Acad Sci U S A 2008, 105(33):12057-12062. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC et al: Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98(1):115-124. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP: Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 2000, 106(7):847-856. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM: A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92(6):829-839. Evans MJ, Scarpulla RC: NRF-1: a trans-activator of nuclearencoded respiratory genes in animal cells. Genes Dev 1990, 4(6):1023-1034. Virbasius CA, Virbasius JV, Scarpulla RC: NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev 1993, 7(12A):2431-2445. Nagase I, Yoshida S, Canas X, Irie Y, Kimura K, Yoshida T, Saito M: Up-regulation of uncoupling protein 3 by thyroid hormone, peroxisome proliferator-activated receptor ligands and 9-cis retinoic acid in L6 myotubes. FEBS Lett 1999, 461(3):319-322. Son C, Hosoda K, Matsuda J, Fujikura J, Yonemitsu S, Iwakura H, Masuzaki H, Ogawa Y, Hayashi T, Itoh H et al: Up-regulation of uncoupling protein 3 gene expression by fatty acids and agonists for PPARs in L6 myotubes. Endocrinology 2001, 142(10):41894194. Brun S, Carmona MC, Mampel T, Vinas O, Giralt M, Iglesias R, Villarroya F: Activators of peroxisome proliferator-activated receptor-alpha induce the expression of the uncoupling protein-3 gene in skeletal muscle: a potential mechanism for the lipid intakedependent activation of uncoupling protein-3 gene expression at birth. Diabetes 1999, 48(6):1217-1222. Chevillotte E, Rieusset J, Roques M, Desage M, Vidal H: The regulation of uncoupling protein-2 gene expression by omega-6 polyunsaturated fatty acids in human skeletal muscle cells involves multiple pathways, including the nuclear receptor peroxisome proliferator-activated receptor beta. J Biol Chem 2001, 276(14):10853-10860. 296 382. 383. 384. 385. 386. 387. 388. 389. 390. 391. 392. 393. Solanes G, Pedraza N, Iglesias R, Giralt M, Villarroya F: Functional relationship between MyoD and peroxisome proliferator-activated receptor-dependent regulatory pathways in the control of the human uncoupling protein-3 gene transcription. Mol Endocrinol 2003, 17(10):1944-1958. Li X, Wang QJ, Pan N, Lee S, Zhao Y, Chait BT, Yue Z: Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson's disease. PLoS One 2011, 6(3):e17153. Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG: PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 2010, 107(35):15467-15472. Sucharov CC, Langer S, Bristow M, Leinwand L: Shuttling of HDAC5 in H9C2 cells regulates YY1 function through CaMKIV/PKD and PP2A. Am J Physiol Cell Physiol 2006, 291(5):C1029-1037. McKinsey TA, Zhang CL, Olson EN: Identification of a signalresponsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 2001, 21(18):6312-6321. Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS: Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res 2004, 64(3):1079-1086. Baillie G, MacKenzie SJ, Houslay MD: Phorbol 12-myristate 13acetate triggers the protein kinase A-mediated phosphorylation and activation of the PDE4D5 cAMP phosphodiesterase in human aortic smooth muscle cells through a route involving extracellular signal regulated kinase (ERK). Mol Pharmacol 2001, 60(5):11001111. Yang TT, Xiong Q, Enslen H, Davis RJ, Chow CW: Phosphorylation of NFATc4 by p38 mitogen-activated protein kinases. Mol Cell Biol 2002, 22(11):3892-3904. Chang CR, Blackstone C: Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci 2010, 1201:34-39. Criscuolo C, De Rosa A, Guacci A, Simons EJ, Breedveld GJ, Peluso S, Volpe G, Filla A, Oostra BA, Bonifati V et al: The LRRK2 R1441C mutation is more frequent than G2019S in Parkinson's disease patients from southern Italy. Mov Disord 2011, 26(9):1733-1736. Nabben M, Hoeks J: Mitochondrial uncoupling protein 3 and its role in cardiac- and skeletal muscle metabolism. Physiol Behav 2008, 94(2):259-269. Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K: Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res 1997, 49(6):681-697. 297 394. 395. 396. 397. 398. 399. 400. 401. 402. 403. 404. 405. 406. 407. Lee FY, Li Y, Zhu H, Yang S, Lin HZ, Trush M, Diehl AM: Tumor necrosis factor increases mitochondrial oxidant production and induces expression of uncoupling protein-2 in the regenerating mice [correction of rat] liver. Hepatology 1999, 29(3):677-687. Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe EH: Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One 2012, 7(4):e34693. Azzu V, Mookerjee SA, Brand MD: Rapid turnover of mitochondrial uncoupling protein 3. Biochem J 2010, 426(1):13-17. Azzu V, Jastroch M, Divakaruni AS, Brand MD: The regulation and turnover of mitochondrial uncoupling proteins. Biochim Biophys Acta 2010, 1797(6-7):785-791. Chen X, Wang K, Chen J, Guo J, Yin Y, Cai X, Guo X, Wang G, Yang R, Zhu L et al: In vitro evidence suggests that miR-133amediated regulation of uncoupling protein 2 (UCP2) is an indispensable step in myogenic differentiation. J Biol Chem 2009, 284(8):5362-5369. Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME: Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem 2011, 286(24):21865-21875. Breen EP, Gouin SG, Murphy AF, Haines LR, Jackson AM, Pearson TW, Murphy PV, Porter RK: On the mechanism of mitochondrial uncoupling protein 1 function. J Biol Chem 2006, 281(4):2114-2119. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G et al: Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008, 27(2):433-446. Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B: Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A 2008, 105(19):7070-7075. Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AY, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E et al: PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS One 2008, 3(6):e2455. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M: Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441(7097):1162-1166. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM et al: Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441(7097):11571161. Lyamzaev KG, Izyumov DS, Avetisyan AV, Yang F, Pletjushkina OY, Chernyak BV: Inhibition of mitochondrial bioenergetics: the effects on structure of mitochondria in the cell and on apoptosis. Acta Biochim Pol 2004, 51(2):553-562. Kieper N, Holmstrom KM, Ciceri D, Fiesel FC, Wolburg H, Ziviani E, Whitworth AJ, Martins LM, Kahle PJ, Kruger R: 298 408. 409. 410. 411. 412. 413. 414. 415. 416. 417. 418. Modulation of mitochondrial function and morphology by interaction of Omi/HtrA2 with the mitochondrial fusion factor OPA1. Exp Cell Res 2010, 316(7):1213-1224. Nakamura T, Lipton SA: Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer's and Parkinson's diseases. Apoptosis 2010, 15(11):1354-1363. Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T: An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem 2008, 143(4):449-454. Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J et al: Nitric oxide-induced mitochondrial fission is regulated by dynaminrelated GTPases in neurons. EMBO J 2006, 25(16):3900-3911. Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V, Debyser Z, Callewaert G, Koopman WJ, Willems PH et al: Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci 2011, 124(Pt 7):1115-1125. Shavali S, Brown-Borg HM, Ebadi M, Porter J: Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett 2008, 439(2):125-128. Plun-Favreau H, Burchell VS, Holmstrom KM, Yao Z, Deas E, Cain K, Fedele V, Moisoi N, Campanella M, Miguel Martins L et al: HtrA2 deficiency causes mitochondrial uncoupling through the F(1)F(0)-ATP synthase and consequent ATP depletion. Cell Death Dis 2012, 3:e335. Srivastava RA, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS: AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases: Thematic Review Series: New Lipid and Lipoprotein Targets for the Treatment of Cardiometabolic Diseases. J Lipid Res 2012, 53(12):2490-2514. Lee JY, Takahashi N, Yasubuchi M, Kim YI, Hashizaki H, Kim MJ, Sakamoto T, Goto T, Kawada T: Triiodothyronine induces UCP-1 expression and mitochondrial biogenesis in human adipocytes. Am J Physiol Cell Physiol 2012, 302(2):C463-472. Rodriguez de la Concepcion ML, Yubero P, Domingo JC, Iglesias R, Domingo P, Villarroya F, Giralt M: Reverse transcriptase inhibitors alter uncoupling protein-1 and mitochondrial biogenesis in brown adipocytes. Antivir Ther 2005, 10(4):515-526. Patane G, Anello M, Piro S, Vigneri R, Purrello F, Rabuazzo AM: Role of ATP production and uncoupling protein-2 in the insulin secretory defect induced by chronic exposure to high glucose or free fatty acids and effects of peroxisome proliferator-activated receptor-gamma inhibition. Diabetes 2002, 51(9):2749-2756. Han DH, Nolte LA, Ju JS, Coleman T, Holloszy JO, Semenkovich CF: UCP-mediated energy depletion in skeletal muscle increases glucose transport despite lipid accumulation and mitochondrial 299 419. 420. 421. 422. 423. 424. 425. 426. 427. 428. 429. 430. 431. dysfunction. Am J Physiol Endocrinol Metab 2004, 286(3):E347353. Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF: Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 2007, 9(4):445-452. Yagi T, Kosakai A, Ito D, Okada Y, Akamatsu W, Nihei Y, Nabetani A, Ishikawa F, Arai Y, Hirose N et al: Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS One 2012, 7(7):e41572. Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, JimenezDelgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A et al: Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol Med 2012, 4(5):380-395. Gomez-Suaga P, Luzon-Toro B, Churamani D, Zhang L, BloorYoung D, Patel S, Woodman PG, Churchill GC, Hilfiker S: Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 2012, 21(3):511-525. Kuballa P, Nolte WM, Castoreno AB, Xavier RJ: Autophagy and the immune system. Annu Rev Immunol 2012, 30:611-646. Lee JA: Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival? Exp Neurobiol 2012, 21(1):1-8. Dando I, Fiorini C, Pozza ED, Padroni C, Costanzo C, Palmieri M, Donadelli M: UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim Biophys Acta 2012. Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M et al: PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 2012, 3:1016. Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL: The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res 2008, 86(8):17111720. Mattson MP, Liu D: Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem Biophys Res Commun 2003, 304(3):539-549. Cousin MA, Robinson PJ: Ca(2+) influx inhibits dynamin and arrests synaptic vesicle endocytosis at the active zone. J Neurosci 2000, 20(3):949-957. Marks B, McMahon HT: Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr Biol 1998, 8(13):740-749. Llorens-Martin M, Lopez-Domenech G, Soriano E, Avila J: GSK3beta is involved in the relief of mitochondria pausing in a Tau-dependent manner. PLoS One 2011, 6(11):e27686. 300 432. 433. 434. 435. 436. 437. 438. 439. 440. 441. 442. Maldonado EN, Lemasters JJ: Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J Pharmacol Exp Ther 2012, 342(3):637-641. Sheng ZH, Cai Q: Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 2012, 13(2):77-93. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B: A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest 1962, 41:1776-1804. Sjostrand FS: Molecular pathology of Luft disease and structure and function of mitochondria. J Submicrosc Cytol Pathol 1999, 31(1):41-50. Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S: L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 2010, 67(5):600-606. Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL: Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci 2005, 25(1):184-191. Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC, Paneda C et al: Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J Neurochem 2005, 93(2):493-501. Islam R, Yang L, Sah M, Kannan K, Anamani D, Vijayan C, Kwok J, Cantino ME, Beal MF, Fridell YW: A neuroprotective role of the human uncoupling protein 2 (hUCP2) in a Drosophila Parkinson's disease model. Neurobiol Dis 2012, 46(1):137-146. Sanchez-Blanco A, Fridell YW, Helfand SL: Involvement of Drosophila uncoupling protein 5 in metabolism and aging. Genetics 2006, 172(3):1699-1710. Stockl P, Zankl C, Hutter E, Unterluggauer H, Laun P, Heeren G, Bogengruber E, Herndler-Brandstetter D, Breitenbach M, JansenDurr P: Partial uncoupling of oxidative phosphorylation induces premature senescence in human fibroblasts and yeast mother cells. Free Radic Biol Med 2007, 43(6):947-958. Desquiret V, Loiseau D, Jacques C, Douay O, Malthiery Y, Ritz P, Roussel D: Dinitrophenol-induced mitochondrial uncoupling in vivo triggers respiratory adaptation in HepG2 cells. Biochim Biophys Acta 2006, 1757(1):21-30. 301 Appendix 302 303 304 305


![Anti-LRRK2 (phospho T1491) antibody [MJFR5-88-3] ab140106](http://s2.studylib.net/store/data/012652944_1-a796b37db6668ca261b6522aff828f23-300x300.png)
![Anti-LRRK2 (phospho T1503) antibody [MJF-R6 (227-1a)]](http://s2.studylib.net/store/data/012652945_1-9387a32ca76e64f94e7ec341ad96103b-300x300.png)
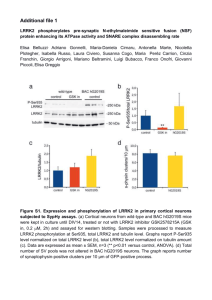

![Anti-LRRK2 (phospho T2483) antibody [MJF-R8 (21-2e)]](http://s2.studylib.net/store/data/012652946_1-7877eadb0a19f3ab2942203a17eadb7f-300x300.png)
