22.00J/1.021J/2.030J/3.021J/10.333J/18.361J/HST558J Introduction to Modeling and Simulation (IM/S) (Spring 2005)
advertisement
 (Spring 2005)")
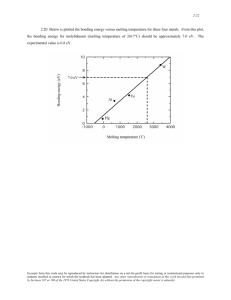
22.00J/1.021J/2.030J/3.021J/10.333J/18.361J/HST558J Introduction to Modeling and Simulation (IM/S) (Spring 2005) __________________________________________________________________ Lecture MD3 (4/15/05) Direct Simulation of Melting This lecture discusses an application of molecular dynamics simulation to the study of melting of a single crystal. We introduce the concept of phase transition as viewed from two different points of view, thermodynamics and kinetics. When applied to melting, the former is concerned with the coexistence of the crystal and liquid states of matter in terms of their free energies; this defines the melting temperature, a fundamental property associated with every known solid. The latter deals with how the process of melting actually occurs - what is the mechanism by which the crystal lattice loses structural integrity; in this context one can ask about the importance of a surface as a nucleation site for the structural transformation. We discuss a case study of melting in Silicon. We show that he thermodynamic and kinetic pictures, made possible by atomistic simulations, are in fact quite compatible, with each offering particular insights that the other does not. Our conclusion is that melting exists in two forms, the everyday phenomenon that is governed by the consideration of free energy (we call this thermodynamic melting), and a less well-known process which occurs in the absences of surfaces or other crystal defects (we call this mechanical melting). The Mystery of Melting It is said there are three equilibrium states (phases) of matter, solid, liquid, and vapor. Sometimes the plasma, a system of charged particles (ions and electrons), is called the fourth state. But this is not universal. Another form of matter, the glassy state, has been given special status in some scientific circles. This too is not as universally recognized as the crystal, liquid, and vapor phases. The conditions which determine which phase that a particular substance should have are typically the temperature, density, and pressure. These are called state variables. For compound substances, the composition is another relevant state variable. It is known from thermodynamics that for a simple (one-component) substance there are only two independent state variables. Thus, if the pressure and temperature are specified for the material, the density is known from a relation called the equation of state. A useful way of displaying the three phases of a substance is the phase diagram, an example of which is shown in Fig. 1. What is shown is the projection of the phase regions in three variables, pressure, temperature and density, onto a plane of two of the variables, in this case pressure and temperature. Another projection is shown in Fig. 2 for the noble-gas element argon. The field of study of equilibrium phases of matter is called phase equilibria. What determines the equilibrium phase of a substance is the free energy, a fundamental property which one would like to be able to calculate. This is a highly nontrivial problem except in very special cases, like an ideal gas or a system of coupled harmonic oscillators. Practical calculations often involve introducing simplifying 1 assumptions. The most powerful method to calculate free energies is by molecular dynamics or Monte Carlo simulation. Fig.1. Phase diagram of a simple substance projected on the pressure-temperature plane. Point A denotes the triple point, where the three phases come together, and point C is the critical point beyond which there is no distinction between liquid and vapor (fluid phase). Crossing the phase boundary AB means the substance is melting or freezing, while crossing AC it is boiling or condensing. The dashed curve indicates a phase transition in the solid state, such as going from one crystal structure to another. Fig.2. Phase diagram of argon in the temperature-density plane determined by MD simulation. Tt denotes the triple point temperature (87oK). The points indicate experimental data. Suppose a substance is in equilibrium at some condition and one of the state variables, say temperature, is suddenly increased so that the system has to evolve to another phase in order to be in equilibrium with the new condition. The study of how the transformation from one phase to another takes place is another field called phase transition. In this lecture we are concerned with a special case, the melting of a perfect crystal, or the crystal-to-liquid transition. Since this is just a common process, one that must be studied exhaustively since the earliest days of scientific inquiry of natural phenomena, we may ask what, if anything, is still not known about this phenomenon. While it is true that much is known about melting, the student may be surprised to learn that melting remains a challenge to our fundamental understanding in the sense that we still do not have a first-principles theory of this phase transition. What this means is that for a given substance we cannot predict all the melting characteristics of this material without resorting to experimental measurements. Another open question is the effect of 2 surfaces and other defects in the solid on the melting process. Still another mystery is the consistency between different models of melting that have been proposed in the literature. As we will see, part of the difficulties of studying a macroscopic phenomenon such as melting is that we do not have detailed information of the process at the molecular level. Once such details become available, the prospects of clarification improve considerably. If the results we will discuss here are any indications, molecular dynamics simulations can help to bring about a better understanding of this basic physical phenomenon. Thermodynamics vs. Kinetics In thermodynamics melting is the coexistence of two equilibrium phases, the crystal and the liquid. For a substance in any phase, there are different energies one can associate with the system, the internal energy E, which is the sum of kinetic and potential energies, and two free energies, the Helmholtz and Gibbs free energies, F and G, respectively, F = E − TS (1) G = E − TS + PV (2) where T is the system temperature, S the entropy, P the pressure, and V the volume. Among these quantities the most difficult to evaluate is entropy. The calculation of entropy for a system in solid or liquid state is a highly nontrivial problem in thermodynamics and statistical mechanics. Fortunately our discussion does not require us to delve into the calculation of entropy. Since the equilibrium state is the state of minimum Gibbs free energy, the mathematical expression of coexistence of crystal and liquid at a certain temperature is the condition, GC (Tm ) = GL (Tm ) (3) As illustrated in Fig. 3, for the crystal and liquid one can calculate the variation of the Gibbs free energy with temperature. When plotted together, it is no surprise that GC(T) lies below GL(T) when T is below the melting point Tm, since below Tm the equilibrium phase is the crystal. For T greater then Tm, GL(T) is now greater. Thus, the two phases have the same value of the Gibbs free energy at Tm; in other words, (3) is the definition of the melting point of the substance. (It is understood that both phases at at the same pressure so melting occurs as an isobaric process.) 3 Fig. 3. The Gibbs free energy of crystal and liquid Si calculated using the StillingerWeber three-body interatomic potential by means of MD simulation [2]. Crossing of the two free energies gives the melting point Tm at 1691 K, while the experimental value is 1683 K. What we have just said is that thermodynamics tells us how to determine the melting temperature Tm, which is another way of saying that thermodynamics specifies when the system should melt. But it says nothing about how the melting process should occur. For example, it says nothing about whether a free surface is required for the melting process to occur at Tm. How a process takes place in the sense of a mechanism by which the phenomenon occurs is the topic of kinetics. The best way to obtain such information is to directly observe the melting process on the relevant length and time scale. What if we want to know how melting begins on the molecular level. In the absence of direct experimental measurements, we will resort to molecular dynamics simulation. Melting in Silicon: A Case Study We continue our discussion of melting in the case of silicon for which the consistency between thermodynamics and kinetics was investigated [1]. In 1985 an empirical three-body interatomic potential was constructed for this important material and used shortly thereafter to calculate the Gibbs free energies for crystal and liquid Si [2]. The results of this calculation, shown in Fig. 3, predicted a melting point of 1691 ± 30 K, which turns out to be surprisingly close to the experimental value of 1683 K. We can say more about this remarkable agreement, but that is not the point of this discussion. What we want to emphasize is that in the case of Si we have a potential model for which the thermodynamic definition of melting, (3), has been applied and a melting point Tm predicted. The expectation therefore is that if the same potential were used in a molecular dynamics simulation of melting, one should see melting occurring at 1691. This was not what happened at first in the direct simulation. What we want to describe now is the subsequent attempt to resolve this apparent inconsistency, and the lessons that one learned from this process. 4 To observe melting we take a simulation cell in the form of a crystal lattice of Si which has the diamond cubic structure and apply periodic boundary conditions in all three dimensions, thus simulating a perfect crystal with no surfaces. We allow the atoms to interact through the same potential function that give the free energy results shown in Fig. 2, and gradually raised the temperature of the crystal. When the temperature reached 1691K nothing happened, so the simulation was continued to higher and higher temperature. Even at 2200K the crystal seemed to be stable. Finally at T ~ 2500K the ordered lattice started to collapse, the onset of disorder seemed to occur uniformly throughout the system. How can we tell when an atom or a small group of atoms becomes disordered? One way is to look at the coordination number, defined to be the number of nearest neighbors (atoms within a certain interatomic distance to the atom in question). The coordination number is 4 for atoms situated on the Si lattice, whereas it would be 6 on the average for atoms in liquid Si. By visualizing the atoms during simulation through a color-coded display, one can readily determine that the system is changing from a ordered local environment (crystal) to a disordered environment (liquid). The observation of collapse of the crystal structure at 2500K in the MD simulation using the same potential that produced a melting point of 1691K seemed to be a contradiction with our understanding of melting. Two questions may be asked. Why didn't the crystal melt at 1691K as predicted by thermodynamics? What kind of transition took place at 2500K? After some consideration of what could be wrong, it was decided to perform another simulation, this time with a free surface. In this second series of simulations, the periodic boundary condition was turned off in one direction, so in that direction one has two free surfaces, as shown in Fig. 4. It was then observed that so long as the Fig. 4. Schematic of the simulation cell with periodic boundary conditions in all three directions (left). Simulation using this cell shows collapse of the crystal structure at 2500 K. The periodic condition has been removed in the cell on the right so that the top and bottom are now free surfaces. Several simulations in the temperature range 1800 - 2200 K show a liquid layer first formed on the free surfaces and subsequently moved toward the bulk at a speed of v(T). 5 temperature was above 1700K, the free surface invariably became disordered first during the simulation (we will call the disorded part the melt), and the extent of the disordering then propagated into the bulk crystal as time evolved. We were able to track the front of the disordering, the melt-crystal interface, by calculating an order parameter for each layer of the interface, S (k ) = 1 N N ∑e 2 i k ⋅r j (4) j=1 where k is a vector determined by a particular set of crystalline planes in the diamond cubic lattice, and r j is the position of particle j. S is called the static structure factor. It is normalized such that it has a value close to unity when the particles are ordered and k is properly chosen, and a value close to zero when the particles are disordered. By limiting the summation to only atoms in a particular slice of the crystal, one can monitor the structural order in the system layer by layer. When this is applied to the onset of surface one can follow the moving interface as the surface disordering grows into the bulk, and in this way extract a speed for the moving front v(T), with T being the temperature at which the simulation is being conducted. The fact that the free surface disordering or melting always preceded melting into the bulk was considered to be a significant indication of the mechanism of melting. But how can one make contact with the thermodynamic prediction of 1691K? The answer to this came in the next step of our analysis. When we plotted the propagation speed v(T) as a function of T, as shown in Fig. 5, we noticed that if we extrapolated the curve to zero speed, that would give a temperature, 1710K, which is close to the predicted value of Tm. It dawned on us that this is just the right thing to do, since extrapolation to zero speed is a way of determining the temperature of coexistence, when the crystal-melt interface would not move in either direction. Another lesson here is that one does not have to be sitting right on the melting point, which can be quite difficult to manage. Instead one can approach it (from either side) by extrapolation, which can be made more and more precise if one desires. Fig. 5. Schematic of variation of the speed of the moving melt-crystal interface with temperature as determined by MD simulation and analysis. Data are shown as closed circles, the scale being the highest point is at 2200K and 100 m/s and the lowest point is at about 1800K and 20 m/s. Linear extrapolation gives an estimated melting temperature of Tm ~ 1710K. What have we learned? 6 We have found an answer to the first question of why didn't the crystal melt at the predicted temperature of 1691K. The answer is that although our model of Si crystal, based on the same potential that gave the prediction, knows the melting point is at 1691K, it cannot nucleated the melting at that temperature because it has no free surfaces or any other defect that could provide the necessary nucleation sites. We have demonstrated that this is the case by using two other defects, a grain boundary and a 13vacancy void [1]. In each case, melting nucleated at the defect sites and extended into the bulk, and extrapolating of the propagation speed gave essentially the same result as the free surface simulations. Put in a different way, we have shown that melting at Tm = 1691K, which we will call thermodynamic melting, is a heterogeneous process, involving the nucleation and growth of a liquid layer in a crystalline environment of neighboring atoms. We give another demonstration of our understanding of the melting process we normally observe either in the natural environment at large or in a laboratory measurement, namely what is conventionally regarded as melting is what we now call thermodynamic melting. Fig. 6 shows a comparison of the predicted melting curve - a line of melting points in the pressure-temperature plane of the phase diagram - with experimental data reported in the literature [5]. In this case, the substance is the noblegas element argon. Fig. 6. Comparison of predicated melting curve for argon (curve) with experimental data (points). Upper inset show the intersection of the Gibbs free energy curves for the solid and liquid, thus defining the melting point at a particular pressure (50 MPa). Lower inset shows the corresponding differences in entropy S and enthalpy H (H=E+PV) between the liquid and the solid. This gives the further insight that melting is driven by the more rapid increase in entropy in the liquid, a result that is intuitively quite reasonable but difficult to demonstrate without the quantitative details that the free-energy calculation made possible by atomistic simulation [5]. As to the second question of what is the process that occurred at 2500K, we have found that the behavior of uniform structural disordering is triggered by the loss of mechanical stability. One can in fact predict when this would occur by deriving elastic stability criteria [4]. Our simulations show that when there are no free surfaces or any other defects in the system, thermodynamic melting would be suppressed beyond Tm. Then one can heat the crystal (superheat) up to limit of its mechanical stability, the point at which the system collapses in a homogeneous manner. This is what happened at 2500K. We call this process mechanical melting. Normally this is not observed since thermodynamic melting always sets in at a lower temperature. 7 In conclusion, we have demonstrated that there exist two kinds of melting, the one that occurs first is thermodynamic melting which is heterogeneous and requires the presence of nucleation sites. The second kind is mechanical melting which is instability of the entire lattice, and this occurs uniformly. Additional Comments: An analogous problem to what we have discussed here is to ask how do crystals break under stress. This problem also can be studied by atomistic simulation [5]. There the distinction between homogeneous and heterogeneous processes is again relevant. We defer further discussions until a future opportunity. In the meantime, any student interested in pursuing this kind of investigation may consider doing a term project on the topic. MD Overview The literature on MD simulation studies is very large. For an entry one might start with a brief commentary that I wrote about a year ago [6], a pdf file of which is posted on the MIT server following this Lecture Notes. Another local source of information is a set of notes I wrote for a short course. At the end of the present lecture we will show a few short videos to illustrate a number of Current applications. At MIT I have been involved with a group of students and collaborators over some 20 years using atomistic simulation techniques to understand fundamental properties and behavior of condensed matter. You are welcome to browse our website: mmm.mit.edu. References cited [1] S. R. Phillpot, S. Yip, D. Wolf, "How Do Crystals Melt?", Computers In Physics, Nov/Dec, p. 20 (1989). [2] J. Q, Broughton and X. P. Li, "Phase Diagram of Silicon by Molecular Dynamics", Physical Review B35, 9120 (1987). [3] M. de Koning, A. Antonelli, S. Yip, "Single-simulation Determination of Phase Boundaries: A Dynamic Clausisus-Clapeyron Method", Journal of Chemical Physics 115, 11025 (2001). [4] J. Wang, J. Li, S. Yip, D. Wolf, S. phillpot, "Unifying Two Criteria of Born: Elastic Instability and Melting of Homogeneous Crystals", Physica A240, 396 (1997). [5] J. Li and S. Yip, "Atomistic Measures of Materials Strength", Computer Modelling in Engineering and Science 3, 219 (2002). [6] S. Yip, "Synergistic Science", Nature Materials 2, 3 (2003). 8