Molecular Biology Structure of human β2-adrenergic receptor highlighting the cholesterolbinding site in the middle of the receptor. Work done in the laboratory of Raymond Stevens, Ph.D., professor. Pick-Wei Lau, Graduate Student, and Ian MacRae, Ph.D., Assistant Professor MOLECULAR BIOLOGY DEPAR TMENT OF MOLECULAR BIOLOGY S TA F F Peter E. Wright, Ph.D.* Professor and Chairman Cecil H. and Ida M. Green Investigator in Medical Research Ruben Abagyan, Ph.D. Professor Carlos F. Barbas III, Ph.D.** Professor Janet and W. Keith Kellogg II Chair, Molecular Biology Rajesh Belani, Ph.D. Adjunct Assistant Professor aTyr Pharma La Jolla, California Michael N. Boddy, Ph.D. Assistant Professor Charles L. Brooks III, Ph.D.*** University of Michigan Ann Arbor, Michigan David A. Case, Ph.D.*** Rutgers University Piscataway, New Jersey Geoffrey Chang, Ph.D.* Associate Professor Eli Chapman, Ph.D. Assistant Professor of Molecular Biology Jerold Chun, M.D., Ph.D. Professor Lluis Ribas De Pouplana, Ph.D. Adjunct Assistant Professor Omnia Molecular Barcelona, Spain Ashok Deniz, Ph.D. Associate Professor H. Jane Dyson, Ph.D. Professor James Arthur Fee, Ph.D. Professor of Research 2008 Elizabeth D. Getzoff, Ph.D.**** Professor THE SCRIPPS RESEARCH INSTITUTE 203 Ian MacRae, Ph.D. Assistant Professor Robyn L. Stanfield, Ph.D. Assistant Professor Clare McGowan, Ph.D.***** Associate Professor Raymond C. Stevens, Ph.D.† Professor David S. Goodsell, Jr., Ph.D. Associate Professor of Molecular Biology Duncan E. McRee, Ph.D. Adjunct Associate Professor Sorrento Technology San Diego, California Charles D. Stout, Ph.D. Associate Professor Joel M. Gottesfeld, Ph.D. Professor David P. Millar, Ph.D. Professor Jennifer Harris, Ph.D. Assistant Professor of Biochemistry Louis Noodleman, Ph.D. Associate Professor David B. Goodin, Ph.D. Associate Professor Christian A. Hassig, Ph.D. Adjunct Assistant Professor Kalypsis, Inc. San Diego, California Peter B. Hedlund, M.D., Ph.D. Assistant Professor of Molecular Biology John E. Johnson, Ph.D. Professor Gerald F. Joyce, M.D., Ph.D.** Professor Dean, Faculty Ehud Keinan, Ph.D. Adjunct Professor Technion-Israel Institute of Technology Haifa, Israel Richard A. Lerner, M.D., Ph.D.** President, The Scripps Research Institute Lita Annenberg Hazen Professor of Immunochemistry Cecil H. and Ida M. Green Chair in Chemistry Scott Lesley, Ph.D. Assistant Professor of Biochemistry Tianwei Lin, Ph.D.*** Assistant Professor University of Hong Kong Hong Kong, China Arthur J. Olson, Ph.D. Professor Takanori Otomo, Ph.D. Assistant Professor Vijay Reddy, Ph.D. Associate Professor Steven I. Reed, Ph.D.***** Professor Paul Russell, Ph.D.***** Professor Michel Sanner, Ph.D. Associate Professor Harold Scheraga, Ph.D. Adjunct Professor Cornell University Ithaca, New York Paul R. Schimmel, Ph.D.** Ernest and Jean Hahn Professor of Molecular Biology and Chemistry Anette Schneemann, Ph.D. Associate Professor Subhash C. Sinha, Ph.D. Associate Professor of Medicinal Chemistry Gary Siuzdak, Ph.D. Adjunct Professor Director, Center for Mass Spectrometry Vaughn V. Smider, Ph.D. Assistant Professor of Molecular Biology Peiqing Sun, Ph.D. Associate Professor J. Gregor Sutcliffe, Ph.D. Professor John A. Tainer, Ph.D.* Professor Fujie Tanaka, Ph.D. Associate Professor Elizabeth Anne Thomas, Ph.D. Assistant Professor James R. Williamson, Ph.D.** Professor Dean, Kellogg School of Science and Technology Ian A. Wilson, D.Phil.* Professor Curt Wittenberg, Ph.D.***** Professor Kurt Wüthrich, Ph.D.** Cecil H. and Ida M. Green Professor of Structural Biology Xiang-Lei Yang, Ph.D.* Associate Professor of Molecular Biology Todd O. Yeates, Ph.D. Adjunct Professor University of California Los Angeles, California Qinghai Zhang, Ph.D. Assistant Professor SERVICE FACILITIES Gerard Kroon Manager, Nuclear Magnetic Resonance Facilities 204 MOLECULAR BIOLOGY Michael E. Pique Director, Computer Graphics Development 2008 THE SCRIPPS RESEARCH INSTITUTE Kelly Lee, Ph.D. Michael Baksh, Ph.D. Gerald Edward Dodson, Ph.D. Brian Paegel, Ph.D. Manidipa Banerjee, Ph.D. Claire Louise Dovey, Ph.D. Jefferson Perry, Ph.D. Christine Beuck, Ph.D. Richard R. Rivera, Ph.D. David Boehr, Ph.D. Jerome Dupuy, Ph.D.*** Université Mediterranée Marseille, France David S. Shin, Ph.D. Yannick Bomble, Ph.D.*** National Renewable Energy Laboratory Golden, Colorado Michelle Duquette-Huber, Ph.D. Giovanni Bottegoni, Ph.D.*** Instituto Italiano de Tecnologia Genova, Italy Susanna V. Ekholm-Reed, Ph.D. S E N I O R S TA F F SCIENTISTS Marc Elsliger, Ph.D. Quansheng Zhou, Ph.D. S TA F F S C I E N T I S T S Philip Arno Venter, Ph.D. Xiaoqin Ye, M.D., Ph.D.*** University of Georgia Athens, Georgia Svitlana Berezhna, Ph.D. Stephen Edgcomb, Ph.D. Venkata Reddy Erigala, Ph.D. Vadim Cherezov, Ph.D. R E S E A R C H A S S O C I AT E S Reto Horst, Ph.D. Tommy Bui, Ph.D. Maria Martinez-Yamout, Ph.D. Melanie Ann Adams, Ph.D.*** University of Toronto Toronto, Ontario, Canada Rosa Maria Cardoso, Ph.D.†† Allan Chris Merrera Ferreon, Ph.D. Garrett M. Morris, Ph.D. Fabio Agnelli, Ph.D. Andrew Barry Carmel, Ph.D. Josephine Chu Ferreon, Ph.D. Chiaki Nishimura, Ph.D. Hanna-Stina Martinsson Ahlzén, Ph.D. Juan Jovel Castillo, Ph.D. Stefano Forli, Ph.D. Amarnath Chatterjee, Ph.D. Chi-Yu Fu, Ph.D. Susana Chaves, Ph.D. †† Zara Fulton, Ph.D. Yen-Ju Chen, Ph.D.*** National Synchrotron Radiation Research Center Hsinchu, Taiwan Yann Gambin, Ph.D. Zhiyong Chen, Ph.D.*** Trius Therapeutics San Diego, California Joshua Gill, Ph.D. Srinivas Chittaboina, Ph.D.†† Rajib Kumar Goswami, Ph.D. Ji Woong Choi, Ph.D. Arsen Grigoryan, Ph.D. Chung Jen Chou, Ph.D. Bettina Groschel, Ph.D.*** Ardea Biosciences, Inc. San Diego, California Daniel Felitsky, Ph.D. Jeffrey Speir, Ph.D. Reetesh Raj Akhouri, Ph.D. Mutsuo Yamaguchi, Ph.D. Xueyong Zhu, Ph.D. SENIOR RESEARCH Alexander Ivanov Alexandrov, Ph.D.*** Takeda San Diego, California A S S O C I AT E S Stephen G. Aller, Ph.D. Ryan Burnett, Ph.D.*** Tocagen, Inc. San Diego, California Beatriz Gonzalez Alonso, Ph.D. Adrienne Elizabeth Dubin, Ph.D. Li Fan, Ph.D. Maria Alejandra GamezAbascal, Ph.D.*** Universidad Autonoma de Madrid Madrid, Spain Juliano Alves, Ph.D.*** Genomics Institute of the Novartis Research Foundation San Diego, California William Anderson, Ph.D. Andrew James Annalora, Ph.D. Munehito Arai, Ph.D. Li-Chiou Chuang, Ph.D. †† Linda Maria Columbus, Ph.D.*** University of Virginia Charlottesville, Virginia Julia Gavrilyuk, Ph.D. Charles Gersbach, Ph.D. Edith Caroline Glazer, Ph.D. Hai-Ming Guo, Ph.D.*** Hanan Normal University Hanan, China Min Guo, Ph.D. Elsa Garcin, Ph.D.*** University of Maryland Baltimore, Maryland Guillermo Asmar-Rovira, Ph.D. Julio Kovacs, Ph.D.*** SeaSpace Corporation Poway, California Wojciech Augustyniak, Ph.D.*** Max-Planck Institute Leipzig, Germany Sandro Cosconati, Ph.D.*** University of Naples Naples, Italy Sung-Hun Bae, Ph.D. Robert De Bruin, Ph.D. Byung Woo Han, Ph.D. Krishna Mohan Bajjuri, Ph.D. Pritilekha Deka, Ph.D. Rodney Harris, Ph.D. Brent Krueger, Ph.D.*** Hope College Holland, Michigan Stephen Connelly, Ph.D. Adam Corper, Ph.D. Rey-Ting Guo, Ph.D. Mahender Gurram, Ph.D.*** AMRI Albany, New York Peter Haberz, Ph.D. MOLECULAR BIOLOGY 2008 David M. Herman, Ph.D. Edward Lemke, Ph.D. Deron Herr, Ph.D. Vasco Liberal, Ph.D.*** Alert Life Sciences Computing Villa Novade Gaia, Portugal Kenichi Hitomi, Ph.D. Minsun Hong, Ph.D. Wen-Xu Hong, Ph.D. Fang Hu, Ph.D. William M. Lindstrom, Ph.D.*** Acelot, Inc. Santa Barbara, California Veli-Pekka Jaakola, Ph.D. Bin Liu, Ph.D. Thamara Janaratne, Ph.D. Hsiao-Wei Liu, Ph.D. Christine Jespersen, Ph.D. Wei Liu, Ph.D. Margaret Alice Johnson, Ph.D. Kunheng Luo, Ph.D. Steven Johnson, Ph.D. Susanna Juraja, Ph.D. Sachin Kale, Ph.D. Tse Siang Kang, Ph.D. Darly Joseph Manayani, Ph.D.*** Pax Vax San Diego, California Mili Kapoor, Ph.D. Santiago Cavero Martinez, Ph.D. †† Ananta Karmakar, Ph.D. Tsutomu Matsui, Ph.D. Andrey Aleksandrovich Karyakin, Ph.D. Derrick Meinhold, Ph.D. THE SCRIPPS RESEARCH INSTITUTE Wataru Nomura, Ph.D.*** Tokyo Medical and Dental University Tokyo, Japan Amy Odegard, Ph.D. Akira Onoda, Ph.D.*** Osaka University Osaka, Japan Bill Francesco Pedrini, Ph.D.*** ETH Zurich Zürich, Switzerland Jennifer S. Scorah, Ph.D.*** CovX La Jolla, California Alexander Perryman, Ph.D. Pedro Serrano-Navarro, Ph.D. Suzanne Peterson, Ph.D. Zahra Shajani, Ph.D. David A. Shore, Ph.D. Andrew Mercer, Ph.D. Eda Koculi, Ph.D. †† Jonathan Mikolosko, Ph.D. Ron Piran, Ph.D. Bethany Koehntop, Ph.D. Mauro Mileni, Ph.D. Irina Kufareva, Ph.D. Sharon Kwan, Ph.D. Maki Minakawa, Ph.D.*** Riken, Wako Institute Saitama, Japan William Placzek, Ph.D.*** Burnham Institute for Medical Research La Jolla, California Bianca Lam, Ph.D. Satoshi Mizuta, Ph.D. Goran Pljevaljc̆ić, Ph.D. Stephanie Pond, Ph.D.*** Prognosys Biosciences, Inc. La Jolla, California John Prudden, Ph.D. Joachim Latzer, Ph.D. Tetsuji Mutoh, Ph.D. Chang-Wook Lee, Ph.D. Mir Hussain Nawaz, Ph.D. Chul Won Lee, Ph.D. Tuan Nguyen, Ph.D. Young-Tae Lee, Ph.D. George Nicola, Ph.D. Sophie Lefebvre, Ph.D. †† Kyoko Noguchi, M.D., Ph.D. Andre Schiefner, Ph.D. Vladimir Pelmenschikov, Ph.D.*** Vrije Universiteit Amsterdam, the Netherlands Dae Hee Kim, Ph.D. Samrat Mukhopadhyay, Ph.D. Gor Sarkisyan, Ph.D. Lauren J. Schwimmer, Ph.D. †† Elena Menichelli, Ph.D. Jason Lanman, Ph.D. Mariana Santa-Marta, Ph.D. Robert Pejchal, Ph.D. Reza Khayat, Ph.D. Davide Moiani, Ph.D. Manuel Rueda, Ph.D. Riturparna Sinha Roy, Ph.D.*** Brigham and Women’s Hospital Boston, Massachusetts Jessica Petrillo, Ph.D.*** Office of Chemical and Biological Weapons, Threat Reduction Washington, D.C. Emma Langley, Ph.D. †† Christopher Roth, Ph.D. Sung-Jean Park, Ph.D. Donald Keidel, Ph.D. Biswaranjan Mohanty, Ph.D. Rae Robertson, Ph.D. Alim Seit-Nebi, Ph.D. †† Eva Mejia Ramirez De Arellano, Ph.D. Petra Langerak, Ph.D. Gabriela Ring, Ph.D. So-Jung Park, Ph.D. Stephanie Pebernard, Ph.D.†† 205 Kimberly A. Reynolds, Ph.D.*** University of Texas Dallas, Texas Jin-Kyu Rhee, Ph.D.*** Department of Chemistry, Scripps Research Errin Rider, Ph.D. Daniela Andrea Slavin, Ph.D.*** Ligand Pharmaceuticals San Diego, California Elisabetta Soragni, Ph.D. Surya Venkata Sripada, Ph.D.*** Syngene International, Ltd. Bangalore, India Pawel Stanczak, Ph.D. Thomas Steinbrecher, Ph.D. Pal Stenmark, Ph.D.*** Karolinska Institutet Stockholm, Sweden Shih-Che Su, Ph.D. Sebastian Sudek, Ph.D. Salahuddin Syed, Ph.D. Michael T. Sykes, Ph.D. Blair R. Szymczyna, Ph.D. 206 MOLECULAR BIOLOGY 2008 Bin Tang, Ph.D. Sung-Il Yoon, Ph.D. Rebecca E. Taurog, Ph.D. Naoto Yoshizuka, M.D., Ph.D. Ewan Richardson Taylor, Ph.D. Haile Zhang, Ph.D. Leonardo Teixeira, Ph.D. Mauricio Carrillo Tripp, Ph.D. Oleg Trott, Ph.D Ulrich Tschulena, Ph.D.*** German Cancer Research Center Heidelberg, Germany Julie L. Tubbs, Ph.D. Qing Zhang, Ph.D.*** Glaxo Smith Kline Shanghai, China Wei Zhang, Ph.D.*** aTyr Pharma La Jolla, California Qiang Zhao, Ph.D. Yong Zhao, Ph.D.*** Cambridge Soft Cambridge, Massachusetts THE SCRIPPS RESEARCH INSTITUTE Tammy Dwyer, Ph.D. San Diego State University San Diego, California Wayne A. Fenton, Ph.D. Yale University New Haven, Connecticut Astrid Graslund, Ph.D. Stockholm University Stockholm, Sweden Arne Holmgren, M.D., Ph.D. Karolinska Institutet Stockholm, Sweden Barry Honig, Ph.D. Columbia University New York, New York Hisatoshi Uehara, Ph.D. Naoto Utsumi, Ph.D.*** Otsuka Pharmaceutical Co., Ltd. Tokushima, Japan S C I E N T I F I C A S S O C I AT E S Ajay Vashisht, Ph.D.*** University of California Los Angeles, California Dennis Carlton, B.S. Sangita Venkataraman, Ph.D. Xiaoping Dai, Ph.D. Petra Verdino, Ph.D. Marc Deller, D.Phil. My Vo, Ph.D. Gye Won Han, Ph.D. Jun Wang, Ph.D. Wenge Han, Ph.D. Jessica Williams, Ph.D. Michael Allen Hanson, Ph.D. Robert Scott Williams, Ph.D. Vance Wong, Ph.D. Hope Johnson, Ph.D.*** California State University Fullerton, California Ulrich Wuellner, Ph.D. Teresa Jones, Ph.D. Wei Xie, Ph.D.*** Salk Institute for Biological Studies La Jolla, California Lin Wang, Ph.D.*** MedImmune, L.L.C. Gaithersburg, Maryland Chunping Xu, Ph.D. V I S I T I N G I N V E S T I G AT O R S Rui Xu, Ph.D. Yoshiki Yamada, Ph.D. †† Tohru Yamagaki, Ph.D.*** Suntory Institute for Bioorganic Research Osaka, Japan Kye Sook Yi, Ph.D. Enrique Abola, Ph.D. Andrew S. Arvai, M.S. Ellen Yu-Lin Tsai Chien, Ph.D. Stephen J. Benkovic, Ph.D. Pennsylvania State University University Park, Pennsylvania Tobias Cramer, Ph.D. CINECA Supercomputer Center HPC Europe Bologna, Italy Arthur Horwich, M.D. Yale University New Haven, Connecticut Tai-Huang Huang, Ph.D. Academica Sinica Taipei, Taiwan Michael Johnson, Ph.D. University of Chicago Chicago, Illinois Joseph David Ng, Ph.D. University of Alabama Huntsville, Alabama Mary Jo Ondrechen, Ph.D. Northeastern University Boston, Massachusetts Victoria A. Roberts, Ph.D. University of California San Diego, California Robert D. Rosenstein, Ph.D. Lawrence Berkeley National Laboratory Berkeley, California * Joint appointment in The Skaggs Institute for Chemical Biology ** Joint appointments in the Department of Chemistry and The Skaggs Institute for Chemical Biology *** Appointment completed; new location shown **** Joint appointments in the Department of Immunology and The Skaggs Institute for Chemical Biology ***** Joint appointment in the Department of Cell Biology † †† Joint appointment in the Department of Chemistry Appointment completed MOLECULAR BIOLOGY 2008 Chairman’s Overview olecular biology forms the cornerstone of biological and biomedical research. Research in the Department of Molecular Biology encompasses a broad range of disciplines, extending from structural and computational biology at one extreme to molecular genetics at the other. Our scientists continue to make rapid progress toward a deeper understanding of the fundamental molecular events that underlie the processes of life. Major Peter E. Wright, Ph.D. advances have been made in elucidating the structural biology of signal transduction, receptor recognition, and viral assembly; understanding mechanisms of viral infectivity; determining the structures of membrane proteins and multidrug transporters; understanding the molecular basis of nucleic acid recognition and DNA repair; and determining the mechanisms of protein folding and ribosome assembly. Progress has been made in elucidating the molecular events involved in regulation of the cell cycle, tumor development, induction of sleep, the molecular origins of neuronal development and of CNS disorders, the regulation of transcription, and decoding of genetic information in translation. Finally, new advances have been made in the design of novel low molecular weight compounds that can specifically regulate genes and in biomolecular engineering, building novel functions into viruses, antibodies, zinc finger proteins, RNA, and DNA. Progress in these and other areas is described in detail on the following pages, and only a few highlights are mentioned here. In a landmark achievement, ranked as 1 of the top 10 breakthroughs of 2007 by Science magazine, Raymond Stevens and his collaborators determined the 3- dimensional structure of the β2-adrenergic receptor. This structure, which represents the culmination of 15 years of research in the Stevens laboratory, promises to revolutionize research on G protein–coupled receptors (GPCRs) and have a major impact on drug discovery. More than 800 GPCRs have been identified, making them the largest M THE SCRIPPS RESEARCH INSTITUTE 207 family of membrane protein receptors, yet previously, the structure of only 1 GPCR was available, for the light receptor rhodopsin. The structure of the β 2 -adrenergic receptor provides the first insights into the large class of GPCRs that regulate signal transduction processes by binding diffusible ligands. The GPCRs control a broad spectrum of physiologic responses, activating intracellular signaling pathways in response to stimuli from outside the cell. GPCRs regulate cell growth and differentiation; control cardiovascular function, metabolism, and the immune response; and play a key role in neurotransmission. Approximately half of currently used drugs function by binding to and regulating receptors from the GPCR family. The structure of the β2-adrenergic receptor provides unprecedented insights into the mechanism by which GPCRs interact with their natural ligands and with drugs and paves the way to design of new and more effective pharmaceutical agents with fewer side effects. Research in the laboratory of John Tainer has led to a detailed understanding of the mechanism by which mutations in a critical enzyme involved in DNA repair lead to 3 distinct disease phenotypes. Dr. Tainer and coworkers determined the structure of the XPD helicase, which is absolutely required for nucleotide excision repair of damaged DNA, and measured the effects of diseasecausing mutations on the enzymatic activity of the helicase. Mutations associated with xeroderma pigmentosum impair the DNA helicase activity of XPD and greatly predispose patients to skin cancer. Mutations associated with Cockayne syndrome also reduce helicase activity and, in addition, cause the XPD enzyme to become stuck on the DNA that is undergoing repair. Finally, trichothiodystrophy mutants cause framework defects that disrupt the integrity of the repair machinery. Related structural work in the Tainer laboratory provided important insights into the mechanism of base excision repair of damaged DNA. A molecular level knowledge of mechanisms of DNA repair is of central importance to understanding cancer, developmental diseases, and aging, and pathogen-specific DNA repair enzymes are potential targets for novel antibacterial and antifungal agents. A collaboration between Curt Wittenberg and Paul Russell of our department and John Yates, Department of Cell Biology, has led to novel insights into the molecular mechanism by which cells respond to errors in DNA replication. During normal cell division, a protein named Nrm1 binds to DNA and represses the expression of key genes during the G1 phase of the cell cycle. Under conditions of stress, replication stalls, and repression of 208 MOLECULAR BIOLOGY 2008 these G 1-phase genes by Nrm1 is blocked, resulting in expression of proteins needed to correct the problem that caused the stall. Understanding the molecular mechanisms responsible for checkpoint control during the cell cycle is critical for understanding oncogenesis and may eventually facilitate development of novel cancer therapeutics that target the replication checkpoints. Recent work by Paul Schimmel and members of his group has revealed the mechanism by which alanyl-tRNA synthetase edits mischarged tRNAAla to correct errors of protein synthesis. Mistranslation, which occurs when an incorrect amino acid binds to tRNA and becomes incorporated into a protein, leads to synthesis of proteins containing errors. Such errors of protein synthesis are associated with numerous diseases. The research by Dr. Schimmel and coworkers provides novel insights into the checkpoints that guard against misincorporation of amino acids and greatly extends our understanding of how cells avoid errors during protein synthesis. Research by Nick Boddy, Clare McGowan, John Tainer, and their coworkers has led to the identification of a previously unknown and completely unexpected family of ubiquitin ligases that mediate cross talk between the sumoylation and ubiquitination pathways. These SUMOtargeted ubiquitin ligases specifically target SUMOmodified proteins for ubiquitination and subsequent proteasomal degradation. The ligases bind specifically to sumoylated proteins and catalyze ubiquitination of the proteins, thereby playing a central role in regulation of sumoylation pathways and in the homeostasis of SUMOmodified proteins. SUMO-targeted ubiquitin ligases are involved in the regulation of genome stability, and evidence exists that they play a central role in controlling cancer metastasis, making them potential targets for novel therapeutics designed to inhibit cancer growth. Molecular biology remains a field of enormous opportunity and excitement. The scientists in this department are taking full advantage of powerful new technologies to advance our understanding of fundamental biological processes at the molecular level. Their discoveries will ultimately be translated into new advances in biotechnology and medicine. THE SCRIPPS RESEARCH INSTITUTE MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 209 Investigators’ Reports Structural Biology of Viral Proteins, Molecular Assemblies, and the Immune System I.A. Wilson, R.L. Stanfield, J. Stevens, X. Zhu, M.A. Adams, C.H. Bell, R.M.F. Cardoso, J. Carlson, P.J. Carney, S. Connelly, A.L. Corper, T. Cross, X. Dai, E.W. Debler, W.L. Densley, B.J. Droese, D.C. Ekiert, M.-A. Elsliger, S. Ferguson, Z. Fulton, B.W. Han, G.W. Han, M. Hong, M.J. Jimenez-Dalmaroni, R.N. Kirchdoerfer, J.R. Mikolosko, R. Pejchal, G.P. Porter, A. Schiefner, D.A. Shore, R.S. Stefanko, J.A. Vanhnasy, P. Verdino, R. Xu, X. Xu, S.I. Yoon o understand the immune response to invading pathogens, such as bacteria and viruses, we focus on the structure-function relationships of immune molecules and their microbial targets. These structural results are especially useful in the design of drugs and vaccines that target the pathogens and protect the host. T T H E I N N AT E I M M U N E S Y S T E M To enhance our understanding of the molecular biology of innate immune receptors, we are investigating the activation requirements of γδ T cells. In collaboration with W. Havran, Department of Immunology and Microbial Science, we determined the structure of junctional adhesion molecule-like (JAML), the γδ T cell–specific costimulatory molecule, in complex with coxsackievirusadenovirus receptor, its endogenous ligand (Fig. 1). The structure revealed an unusually hydrophilic complex interface that suggests potential mechanisms for receptor triggering. Upon JAML engagement, different kinases are then recruited at the JAML intracellular domain to activate kinase signaling cascades, production of cytokines and chemokines, and, ultimately, proliferation of γδ T cells. Toll-like receptors are cell-surface receptors that detect invading microbes by recognizing a variety of pathogen-associated molecular patterns, including bacterial cell walls and viral nucleic acids. To reveal structural mechanisms involved in activation and regulation of these receptors, we have expressed the extracellular domain of human Toll-like receptor 4 with myeloid differentiation protein-2 for structural studies. This immune complex binds bacterial lipopolysaccharides, ultimately leading to sepsis. F i g . 1 . Crystal structure of JAML in complex with coxsackievirus- adenovirus receptor (CAR). JAML and the receptor interact with their membrane-distal D1 immunoglobulin domains in a face-to-face β-sheet interaction. Among the pattern-recognition molecules, the intracellular Nod-like receptors also act as key mediators of innate immunity and of the inflammatory response to microbial infections. Mutations in the genes for these receptors are associated with chronic inflammatory barrier diseases, such as Crohn’s disease and bronchial asthma. We have expressed and purified Nod1 and Nod2 proteins for crystallization. The studies on the Toll- and Nod-like receptors are collaborations with R. Ulevitch, Department of Immunology and Microbial Science, and B. Beutler, Department of Genetics. N O N M A M M A L I A N I N N AT E A N D A D A P T I V E I M M U N I T Y Variable lymphocyte receptors (VLRs) play a key role in recognition of antigens in the adaptive immune response of jawless vertebrates. In collaboration with M.D. Cooper, Emory University School of Medicine, Atlanta, Georgia, we determined the crystal structure of the lamprey 210 MOLECULAR BIOLOGY 2008 VLR2913 ectodomain to 2.1-Å resolution. The VLR folds into a horseshoe-shaped, solenoidal assembly of 5 leucine-rich repeats. Although the antigen for VLR2913 is unknown, the highly similar VLR4 interacts with the anthrax antigen, bacillus collagen-like protein of anthracis (BclA). We have modeled VLR4 with Modeller, on the basis of the VLR2913 structure, and then used Autodock 4.0 software to dock the VLR4 model to BclA, in collaboration with A.J. Olson and G.M. Morris, Department of Molecular Biology (Fig. 2). The docking results suggest that the concave surface of VLR4 is the recognition site for BclA. THE SCRIPPS RESEARCH INSTITUTE ion hole at P9 that is present only in the diabetes-associated MHC I-Ag7. We isolated a TCR hybridoma (21.30) F i g . 3 . Crystals of the MHC class I–peptide–CD8αβ single- chain complex. F i g . 2 . Model of the complex formed by lamprey VLR4 and BclA. The VLR4 was modeled by using the computer program Modeller, with the lamprey VLR2913 crystal structure as a template. The VLR4 model was then tested for the lowest energy docking orientation with BclA by using Autodock software. CLASSICAL AND NONCLASSICAL MHC AND T-CELL RECEPTOR SIGNALING T-cell receptors (TCRs) recognize peptide antigen displayed on the surface of antigen-presenting cells by MHC molecules. Coreceptor molecules, such as CD8αβ and CD4, provide costimulatory signals that are required for full T-cell activation. To ascertain the structural basis for the coreceptor function of CD8αβ and understand the mechanisms that underlie T-cell activation, we used a single-chain MHC-CD8αβ construct to determine the structure of the CD8αβ-MHC-peptide complex (Fig. 3). To explore potential mechanisms of diabetogenesis, we are investigating whether TCRs recognize the oxyan- that is sensitive to the presence or absence of a negatively charged P9 peptide residue and determined the crystal structure to 3.5-Å resolution of TCR 21.30– I-Ag7–HEL9-27, where P9 is a glycine. Surprisingly, the structure revealed that TCR 21.30 does not directly contact the I-Ag7 P9 pocket. Experiments are under way to ascertain the TCR sensitivity to this residue position. MHC molecules play a critical role in initiating cellmediated immunity by presenting both foreign antigens and self-antigens. Efficient loading of peptide antigens on MHC class I molecules requires proteins in the endoplasmic reticulum collectively known as the peptideloading complex. The complex consists of the transporter associated with antigen processing, tapasin, calreticulin, calnexin, ERp57, and an MHC class I molecule. Currently, we are focusing on the structural and biochemical characterization of the transporter, tapasin, and the peptide-free form of MHC class I molecules. Recombinant expression of tapasin from several species, as well as recombinant expression of the transporter, have provided valuable tools for analyzing the structure, function, and assembly of the peptide-loading complex. Biochemical and biophysical techniques are being used to provide structural models for MHC class Ia folding and antigen presentation. The MHC and TCR studies are a collaboration with L. Teyton, Department of Immunology and Microbial Science. CD1 molecules are MHC class I antigen-presenting molecules that present lipids, glycolipids, and lipopeptides to effector T cells. CD1 molecules are involved in host defense and also have immunoregulatory functions. Glycolipids presented by CD1d stimulate natural killer MOLECULAR BIOLOGY 2008 T cells, which are of clinical interest because they rapidly secrete cytokines that either promote or suppress different immune responses. On the basis of our structural studies, C.-H. Wong and his group, Department of Chemistry, synthesized a series of glycolipids that are more potent than other glycolipids tested previously and have increased efficacy in T-cell assays. Structures for 2 of the most stimulating glycolipids in complex with CD1d have revealed that loading and anchoring of the lipids are the key determinants for effective lipid presentation and subsequent T-cell stimulation. INFLUENZA VIRUS To aid in design of vaccines and drugs to prevent future influenza pandemics, we are studying proteins from different influenza strains as part of a consortium funded by the National Institute of Allergy and Infectious Diseases. The 1918 flu pandemic was the most devastating epidemic in recorded world history, and efforts are ongoing to target the neuraminidase of the 1918 influenza virus in structure-based drug design. We have determined crystal structures for the 1918 N1 neuraminidase from crystals with an unusual defect called a “lattice translocation.” Although the successful use of twinned data for structure determination has become relatively routine in recent years, structure determination with lattice-translocation defects has only been previously reported for 5 structures. In addition, structures of the 1918 neuraminidase in complex with zanamivir (Relenza) and oseltamivir (Tamiflu) have revealed new cavities for drug binding (Fig. 4) and how the presence F i g . 4 . Molecular surface of the 1918 influenza virus N1 neuraminidase active site. Zanamivir (ball-and-stick model) has been docked into the unliganded neuraminidase structure on the basis of the drug’s position in the zanamivir-neuraminidase complex. This superposition reveals a large, unoccupied cavity close to the zanamivir binding site that may be an excellent target for the design of inhibitors. THE SCRIPPS RESEARCH INSTITUTE 211 or absence of different ions can affect the overall assembly of the neuraminidase tetramer. The H5N1 avian influenza viruses currently circulating in Asia, Europe, and Africa are extremely virulent in humans, causing severe disease and often death. H5N1 viruses are not readily transmitted among humans, possibly because of differences in the receptor specificity of the hemagglutinin viral fusion protein. To investigate structural changes critical for receptor switching from avian to human specificity, we have developed a baculovirus display platform that will enable us to test large libraries of hemagglutinin mutants for binding to immobilized glycans or human bronchial cell monolayers. The receptor specificity of the selected variants will be determined by using glycan arrays, and structural changes associated with receptor switching will be characterized by using x-ray crystallography. In collaboration with G.J. Tobin, Biological Mimetics, Inc., Frederick, Maryland, the full-length hemagglutinin from an outbreak of influenza in Wyoming and HA2 hemagglutinin fragments from outbreaks in South Carolina in 1918 (H1H1) and Vietnam in 2003 (H5N1) are being expressed for structural studies of conformations before and after fusion. In addition, many collaborations are ongoing to investigate the neutralization of H1N1 and H5N1 viruses by monoclonal antibodies. H I V T Y P E 1 VA C C I N E The need for an effective HIV vaccine is greater than ever as the virus continues to devastate areas of the world such as sub-Saharan Africa. As a part of our vaccine development efforts, we are studying the viral envelope “spikes” composed of heterotrimeric complexes of gp120 and gp41. Upon binding receptors CD4 and CXCR4/CCR5, the trimer undergoes as yet uncharacterized conformational changes that lead to fusion of the viral membrane with the target cell, initiating infection. Crystallization of the trimer in the prefusion state will enable a detailed understanding of its antibody epitope landscape and reveal how neutralizing antibodies can recognize this evolutionarily moving target. We recently determined the crystal structure of a human monoclonal antibody, F425-B4e8 (B4e8), that cross-reacts with the gp120 V3 region of primary viral isolates from subtypes B, C, and D. The B4e8 Fab in complex with the 24mer V3 peptide RP142 showed that the antibody recognizes a novel V3 loop conformation with a 5-residue α-turn around the conserved GPGRA apex of the β-hairpin loop (Fig. 5). The Fab interacts primarily through main-chain interactions with major 212 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE found that the prolonged luminescence is due to a charge-transfer excited complex of an anionic stilbene and a cationic, parallel π-stacked tryptophan. Upon charge recombination, this complex generates exceptionally bright blue light. Formation of the complex is supported by a deep ligand-binding pocket, which in turn is due to a noncanonical interface between the 2 variable antibody subunits. These studies are collaborations with R.A. Lerner, K.D. Janda, and P.G. Schultz, Department of Chemistry; D.P. Millar, Department of Molecular Biology; and H.B. Gray, California Institute of Technology, Pasadena, California. HISTONE DEACETYLASES F i g . 5 . Structure of F425-B4e8 Fab in complex with a peptide (red) representing the V3 region of HIV type 1 gp120. B4e8 is unusually broad in its neutralization of different HIV type 1 viral isolates, and the peptide conformation recognized has an unusual α-turn around the tip of the loop. contacts to only 2 V3 peptide side chains, explaining how B4e8 can accommodate sequence variation within V3 and hence can neutralize different isolates of HIV type 1. Our research on HIV is done in collaboration with D.R. Burton, Department of Immunology and Microbial Science; P.E. Dawson, Department of Cell Biology; C.-H. Wong, Department of Chemistry; J.K. Scott, Simon Fraser University, Burnaby, British Columbia; J. Moore, Weill Medical College of Cornell University, New York, New York; H. Katinger, R. Kunert, and G. Stiegler, University für Bodenkultur, Vienna, Austria; R. Wyatt and P. Kwong, Vaccine Research Center, National Institutes of Health, Bethesda, Maryland; W. Olson, and K. Kang, Progenics Pharmaceuticals, Inc., Tarrytown, New York; the National Institutes of Health, Bethesda, Maryland; and the Neutralizing Antibody Consortium of the International AIDS Vaccine Initiative, New York, New York. BLUE FLUORESCENT ANTIBODIES EP2-19G2, an antibody to a trans-stilbene, has bright blue luminescence and has been used as a biosensor in various applications. By extensive biophysical characterization of the stilbene-antibody complex, we Histone deacetylases catalyze removal of the acetyl group from amino-terminal lysine residues in histones, resulting in chromatin condensation and transcriptional repression. Inhibitors of histone deacetylases are a widely used treatment for many types of cancer. In recent years, these compounds have been emerging as a potential therapy for neurodegenerative disorders, such as Friedreich ataxia, an inherited disease that affects the nervous system and results in muscle weakness, heart disease, and speech difficulties. In collaboration with J.M. Gottesfeld, Department of Molecular Biology, and with support from the Friedreich’s Ataxia Research Alliance, Springfield, Virginia, we are expressing several histone deacetylases for determinations of crystal structures of the enzymes in complex with inhibitors. JOINT CENTER FOR STRUCTURAL GENOMICS The Joint Center for Structural Genomics is a large consortium of scientists from Scripps Research; the Stanford Synchrotron Radiation Laboratory; the University of California, San Diego; the Burnham Institute for Medical Research; and the Genomics Institute of the Novartis Research Foundation. The center is funded by the Protein Structure Initiative of the National Institute of General Medical Sciences. Its purpose is highthroughput structure determination of large families of proteins with no or limited structural representatives, biologically important targets that are conserved as the central machinery of life, the complete proteome from Thermotoga maritima, metagenomic and human microbiome targets, and other targets suggested by the community. To date, the members of the consortium have pioneered many novel high-throughput methods and technologies applicable to structural biology and have determined more than 665 unique structures, including more than 200 novel structures in the past year. MOLECULAR BIOLOGY 2008 PUBLICATIONS Astronomo, R.D., Lee, H.K., Scanlan, C.N., Pantophlet, R., Huang, C.Y., Wilson, I.A., Blixt, O., Dwek, R.A., Wong, C.-H., Burton, D.R. A glycoconjugate antigen based on the recognition motif of a broadly neutralizing human immunodeficiency virus antibody, 2G12, is immunogenic but elicits antibodies unable to bind to the self glycans of gp120. J. Virol. 82:6359, 2008. Bell, C.H., Pantophlet, R., Schiefner, A., Cavacini, L.A., Stanfield, R.L., Burton, D.R., Wilson, I.A. Structure of antibody F425-B4e8 in complex with a V3 peptide reveals a new binding mode for HIV-1 neutralization. J. Mol. Biol. 375:969, 2008. Burley, S.K., Joachimiak, A., Montelione, G.T., Wilson, I.A. Contributions to the NIH-NIGMS Protein Structure Initiative from the PSI Production Centers. Structure 16:5, 2008. Burton, D.R., Wilson, I.A. Immunology: square-dancing antibodies. Science 317:1507, 2007. Debler, E.W., Müller, R., Hilvert, D., Wilson, I.A. Conformational isomerism can limit antibody catalysis. J. Biol. Chem. 283:16554, 2008. Debler, E.W., Kaufmann, G.F., Meijler, M.M., Heine, A., Mee, J.M., Pljevaljcic, G., Di Bilio, A.J., Schultz, P.G., Millar, D.P., Janda, K.D., Wilson, I.A., Gray, H.B., Lerner, R.A. Deeply inverted electron-hole recombination in a luminescent antibody-stilbene complex. Science 319:1232, 2008. Huang, C.C., Lam, S.N., Acharya, P., Tang, M., Xiang, S.H., Hussan, S.S., Stanfield, R.L., Robinson, J., Sodroski, J., Wilson, I.A., Wyatt, R., Bewley, C.A., Kwong, P.D. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317:1930, 2007. Huang, S., Romanchuk, G., Pattridge, K., Lesley, S.A., Wilson, I.A., Matthews, R.G., Ludwig, M. Reactivation of methionine synthase from Thermotoga maritima (TM0268) requires the downstream gene product TM0269. Protein Sci. 16:1588, 2007. Johnson, S.M., Connelly, S., Wilson, I.A., Kelly, J.W. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J. Med. Chem. 51:260, 2008. Kozbial, P., Xu, Q., Chiu, H.J., et al. Crystal structures of MW1337R and lin2004: representatives of a novel protein family that adopt a four-helical bundle fold. Proteins 71:1589, 2008. Krishna, S.S., Tautz, L., Xu, Q., et al. Crystal structure of NMA1982 from Neisseria meningitidis at 1.5 Å resolution provides a structural scaffold for nonclassical, eukaryotic-like phosphatases. Proteins 69:415, 2007. Mathews, I.I., McMullan, D., Miller, M.D., et al. Crystal structure of 2-keto-3deoxygluconate kinase (TM0067) from Thermotoga maritima at 2.05 Å resolution. Proteins 70:603, 2008. Menendez, A., Calarese, D.A., Stanfield, R.L., Chow, K.C., Scanlan, C.N., Kunert, R., Katinger, H., Burton, D.R., Wilson, I.A., Scott, J.K. A peptide inhibitor of HIV-1 neutralizing antibody 2G12 is not a structural mimic of the natural carbohydrate epitope on gp120. FASEB J. 22:1380, 2008. Premkumar, L., Rife, C.L., Sri Krishna, S., et al. Crystal structure of TM1030 from Thermotoga maritima at 2.3 Å resolution reveals molecular details of its transcription repressor function. Proteins 68:418, 2007. Sanguineti, S., Centeno Crowley, J.M., Lodeiro Merlo, M.F., Cerutti, M.L., Wilson, I.A., Goldbaum, F.A., Stanfield, R.L., de Prat-Gay, G. Specific recognition of a DNA immunogen by its elicited antibody. J. Mol. Biol. 370:183, 2007. Saphire, E.O., Montero, M., Menendez, A., van Houten, N.E., Irving, M.B., Pantophlet, R., Zwick, M.B., Parren, P.W., Burton, D.R., Scott, J.K., Wilson, I.A. Structure of a high-affinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J. Mol. Biol. 369:696, 2007. Slabinski, L., Jaroszewski, L., Rodrigues, A.P., Rychlewski, L., Wilson, I.A., Lesley, S.A., Godzik, A. The challenge of protein structure determination—lessons from structural genomics. Protein Sci. 16:2472, 2007. Slabinski, L., Jaroszewski, L., Rychlewski, L., Wilson, I.A., Lesley, S.A., Godzik, A. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics 23:3403, 2007. THE SCRIPPS RESEARCH INSTITUTE 213 Stoll, R., Lee, B.M., Debler, E.W., Laity, J.H., Wilson, I.A., Dyson, H.J., Wright, P.E. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol. 372:1227, 2007. Structural Genomics Consortium; China Structural Genomics Consortium; Northeast Structural Genomics Consortium; Gräslund, S., Nordlund, P., Weigelt, J., et al. Protein production and purification. Nat. Methods 5:135, 2008. Wei, C.J., Xu, L., Kong, W.P., Shi, W., Canis, K., Stevens, J., Yang, Z.Y., Dell, A., Haslam, S.M., Wilson, I.A., Nabel, G.J. Comparative efficacy of neutralizing antibodies elicited by recombinant hemagglutinin proteins from avian H5N1 influenza virus. J. Virol. 82:6200, 2008. Xu, Q., Kozbial, P., McMullan, D., et al. Crystal structure of an ADP-ribosylated protein with a cytidine deaminase-like fold, but unknown function (TM1506), from Thermotoga maritima at 2.70 Å resolution. Proteins 71:1546, 2008. Xu, Q., Saikatendu, K.S., Krishna, S.S., et al. Crystal structure of MtnX phosphatase from Bacillus subtilis at 2.0 Å resolution provides a structural basis for bipartite phosphomonoester hydrolysis of 2-hydroxy-3-keto-5-methylthiopentenyl-1-phosphate. Proteins 69:433, 2007. Zajonc, D.M., Savage, P.B., Bendelac, A., Wilson, I.A., Teyton, L. Crystal structures of mouse CD1d-iGb3 complex and its cognate Vα14 T cell receptor suggest a model for dual recognition of foreign and self glycolipids. J. Mol. Biol. 377:1104, 2008. Zajonc, D.M., Wilson, I.A. Architecture of CD1 proteins. Curr. Top. Microbiol. Immunol. 314:27, 2007. Zubieta, C., Joseph, R., Krishna, S.S., et al. Identification and structural characterization of heme binding in a novel dye-decolorizing peroxidase, TyrA. Proteins 69:234, 2007. Zubieta, C., Krishna, S.S., Kapoor, M., et al. Crystal structures of two novel dyedecolorizing peroxidases reveal a β-barrel fold with a conserved heme-binding motif. Proteins 69:223, 2007. Zubieta, C., Krishna, S.S., McMullan, D., et al. Crystal structure of homoserine Osuccinyltransferase from Bacillus cereus at 2.4 Å resolution. Proteins 68:999, 2007. Protein Structures, Activities, and Regulation: The Functioning of Molecular Machines E.D. Getzoff, A.S. Arvai, E.D. Garcin, C. Hitomi, K. Hitomi, M.D. Kroeger, M.E. Pique, A.J. Pratt, D.S. Shin o understand how proteins function as molecular machines, we use structural molecular biology to characterize and control proteins. We apply the tools of structural, molecular, and computational biology to analyze proteins of biological and biomedical interest, especially proteins that work synergistically with coupled chromophores, metal ions, or other cofactors. T NITRIC OXIDE SYNTHASE AND A NEW APPROACH TO DESIGN OF SELECTIVE INHIBITORS Nitric oxide synthase (NOS) enzymes synthesize nitric oxide, a signal for vasodilation and neurotransmission at low levels and a defensive cytotoxin at higher levels. Synthesis of nitric oxide by NOS requires calmodulin-coordinated interactions between the catalytic, oxy- 214 MOLECULAR BIOLOGY 2008 genase, and electron-supplying reductase modules of the enzyme. NOS uses heme, zinc, tetrahydrobiopterin, calcium, NADPH, FMN, and FAD cofactors. The goals of our structure-based drug design projects are to selectively inhibit inducible NOS, to prevent inflammatory disorders, or neuronal NOS, to prevent migraines, while maintaining blood pressure regulation by endothelial NOS. Our x-ray crystallographic structures of wild-type and mutant NOS oxygenase dimers with substrate, intermediate, inhibitors, cofactors, and cofactor analogs, determined in collaboration with J.A. Tainer, Department of Molecular Biology, and D. Stuehr, the Cleveland Clinic, Cleveland, Ohio, provide insights into both catalytic mechanism and inhibitor selectivity. The nearly complete sequence and structural conservation in the active sites of the 3 NOS isozymes is a significant challenge in the design of isozyme-specific inhibitors. Nevertheless, our latest results indicate that plasticity of distant isozyme-specific residues modulates conformational changes of invariant residues in the substrate-binding site. We applied these results to develop the anchored plasticity approach for the structure-based design of selective inhibitors (Fig. 1). In this approach, conserved binding sites are used to anchor the core of an inhibitor, while distant sequence differences are exploited to provide selectivity. Our structure of the neuronal NOS reductase reveals new insights into the complex regulatory mechanisms of F i g . 1 . Diagram illustrating the anchored-plasticity approach to selective drug design. THE SCRIPPS RESEARCH INSTITUTE this enzyme family. We integrated biochemical data with our structures of NOS oxygenase, NOS reductase, and calmodulin in complex with NOS peptides to propose assembly and mechanistic hypotheses for the holoenzyme. We obtained promising results in support of these hypotheses by using solution small-angle x-ray scattering, which can provide molecular envelopes for macromolecules and macromolecular complexes in solution. We have proposed a moving-domain mechanism for controlling the rate-limiting flow of electrons from the flavin cofactors of NOS reductase to the catalytic NOS oxygenase heme. Our assembly and mechanistic hypotheses also explain the kinetics of regulatory sitespecific phosphorylation and dephosphorylation events that both activate and inactivate synthesis of nitric oxide in vivo. In ongoing research, we are using complementary biochemical, biophysical, and computational methods to define, describe, and understand how NOS chemistry, structure, assembly, dynamics, and protein-protein interactions regulate production of nitric oxide in vivo. PHOTOACTIVE PROTEINS AND CIRCADIAN CLOCKS To understand in atomic detail how chromophorebound proteins translate sunlight into defined conformational changes for biological functions, we are exploring the reaction mechanisms of the blue-light receptors photoactive yellow protein (PYP), photolyase, and cryptochrome. PYP is the prototype for the Per-Arnt-Sim domain proteins of circadian clocks, whereas FAD-containing proteins of the photolyase/cryptochrome family catalyze DNA repair or act in circadian clocks. To understand the photocycle of PYP and to propose a common mechanism for signaling by Per-Arnt-Sim domains, we combined ultra-high-resolution and time-resolved crystallographic structures of the PYP dark state and 2 photocycle intermediates with site-directed mutagenesis; spectroscopy; deuterium-hydrogen exchange mass spectrometry, in collaboration with V. Woods, University of California, San Diego; and quantum mechanical and electrostatic computational methods, in combination with L. Noodleman, Department of Molecular Biology. Cryptochrome flavoproteins function as blue-light receptors in plants and as components of circadian clocks in animals. We determined the first crystallographic structure of a cryptochrome; the structure revealed commonalities with the homologous photolyases in DNA binding and redox-dependent function but showed differences in active-site and interactionsurface features. We found that this cryptochrome binds MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 215 the same antenna cofactor found in a photolyase homolog but uses different amino acid residues to form the cofactor-binding site. Our new structures and spectroscopy, obtained in collaboration with S. Weber, Universität Freiburg in Germany, of cryptochromes and of photolyases from 2 other branches of the photolyase/ cryptochrome family that repair cyclobutane pyrimidine dimers and (6-4) photoproducts help us decipher the cryptic structure, function, and evolutionary relationships of these fascinating redox-active proteins. Furthermore, the (6-4) photolyase enzyme provides an excellent model for human cryptochrome. A simple, but functional, circadian clock can be reconstituted in vitro from the 3 cyanobacterial proteins KaiA, KaiB, and KaiC alone. Yet, the structure and dynamics of the functional assembly are not understood. Our crystallographic, dynamical light scattering and smallangle x-ray scattering studies revealed that KaiB selfassembles into a tetramer. We also study clock proteins with PYP-like Per-Arnt-Sim domains that bind to mammalian cryptochromes. Our goal is to determine the detailed chemistry and atomic structure of these proteins, define their mechanisms of action and interaction, and use our results to understand and regulate biological function. Roberts, B.R., Tainer, J.A., Getzoff, E.D., Malencik, D.A., Anderson, S.R., Bomben, V.C., Meyers, K.R., Karplus, P.A., Beckman, J.S. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol. 373:877, 2007. S U P E R O X I D E D I S M U TA S E S M A L L - A N G L E X - R AY S C AT T E R I N G I N S O L U T I O N The superoxide radical is a central player in the biology of reactive oxygen and nitrogen intermediates, which mediate signaling and oxidative damage, a key factor in aging and cancer, in vivo. Mutations in human copper zinc superoxide dismutase (SOD), the enzyme that converts superoxide to molecular oxygen and hydrogen peroxide, cause the fatal neurodegenerative disease familial amyotrophic lateral sclerosis, or Lou Gehrig disease. We are analyzing the structural chemistry of SOD to help bridge the gap from protein structures to enzyme stability and activities in vivo. We designed a zinc-free variant of human SOD to help test the role of zinc binding and loss in disease. Our results, obtained in collaboration with J.A. Tainer, support the importance of the stable SOD core structure in preventing amyloid formation and toxic effects. For comparison, we determined the structure and stability of the SOD from the most extreme eukaryotic thermophile known: the deep-sea hydrothermal vent worm Alvinella pompejana. PUBLICATIONS Garcin, E.D., Arvai, A.S., Rosenfeld, R.J., Kroeger, M.D., Crane, B.R., Andersson, G., Andrews, G., Hamley, P.J., Mallinder, P.R., Nicholls, D.J., St-Gallay, S.A., Tinker, A.C., Gensmantel, N.P., Mete, A., Cheshire, D.R., Connolly, S., Stuehr, D.J., Åberg, A., Wallace, A.V., Tainer, J.A., Getzoff, E.D. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol., in press. Yamamoto, J., Tanaka, Y., Hitomi, K., Getzoff, E.D., Iwai, S. Spectroscopic studies on a novel intramolecular hydrogen bond within the (6-4) photoproduct. Nucleic Acids Symp. Ser. (Oxf). Issue 51:79, 2007. Structural Biology of Molecular Interactions and Design J.A. Tainer, A.S. Arvai, B.R. Chapados, L. Fan, E. Garcin, G. Guenther, C. Hitomi, K. Hitomi, M.D. Kroeger, J.J. Perry, M.E. Pique, D.S. Shin, J.L. Tubbs, R.S. Williams e are developing new technologies and systems to close the gaps from proteins to pathways and from interaction networks to biological outcomes in cells. We focus on molecular mechanisms and relationships for proteins that control DNA damage responses, reactive oxygen species, protein modifications, and pathogenesis. Our results have relevance for improved understanding and therapeutic approaches for cancer, aging, and degenerative diseases and for bacterial pathogens. W C O M B I N E D W I T H C R Y S TA L L O G R A P H Y A N D C O M P U TAT I O N Aided by our synchrotron facility SIBLYS, we are developing and applying technologies to develop accurate structures of protein conformation, assembly, and interactions in solution by combining x-ray scattering with x-ray crystallography and computation. We recently developed methods for high-throughput analyses via small-angle x-ray scattering. Small-angle x-ray scattering, crystallography, and computation together allow multiscale modeling and fundamental insights to allosteric mechanisms, self-assemblies, and dynamic molecular machines acting as master keys to cell biology. P R O T E I N M O D I F I C AT I O N S A N D F U N C T I O N The ubiquitin-like protein family of posttranslational modifications consists primarily of ubiquitin and the small ubiquitin modifier SUMO. In collaboration with M.N. Boddy, Department of Molecular Biology, we have helped discover an intriguing family of proteins, SUMOtargeted ubiquitin ligases (STUbLs), that directly connect the ubiquitination and sumoylation pathways. Uniquely, STUbLs use SUMO interaction motifs to recognize their sumoylated targets. STUbLs act as global regulators of protein sumoylation levels, and cells lacking STUbLs 216 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE have associated genomic instability and hypersensitivity to genotoxic stress; the human STUbL RNF4 is implicated in cancer. REACTIVE OXYGEN CONTROL ENZYMES Superoxide dismutases (SODs) and nitric oxide synthases are master regulators for reactive oxygen species involved in injury, pathogenesis, aging, and degenerative diseases. We tested if Alvinella pompejana, a deepsea hydrothermal vent worm, could be a eukaryotic source for thermostable and humanlike proteins and whether information on SOD mechanism and stability could be obtained by examining A pompejana SOD. We discovered that the worm SOD has a remarkably high sequence identity with other mammalian SOD enzymes but is substantially more stable than human SOD. Moreover, crystals from initial conditions diffracted just beyond 1-Å resolution. These results extend knowledge of SOD stability and catalysis and also suggest that A pompejana may be a unique resource of macromolecules of enhanced stability for science and technology. For human copper, zinc SOD, we are examining single-site mutations that cause the neurodegeneration in Lou Gehrig disease or familial amyotrophic lateral sclerosis. Our structures show a key role for the zinc ion in the defects associated with the disease. For the nitric oxide synthases, our combined solution scattering and crystallographic methods are revealing regulatory mechanisms responsible for controlling nitric oxide levels, which act as an important signal and as a cytotoxin, with implications for inflammatory and neurodegenerative diseases. Our structures of the synthases, determined in collaboration with E.D. Getzoff, Department of Molecular Biology, are enabling us to design new inhibitors to directly control nitric oxide levels for the treatment of human diseases. DNA REPAIR AND GENETIC EVOLUTION Structural knowledge allows possible selective inhibition of certain DNA repair pathways for new cancer therapies. Endonuclease IV is an archetype for an endonuclease superfamily critical for DNA-base excision repair. Our structures of endonuclease IV revealed a mechanism for binding to and incising areas of DNA damage that involves 3 metal ions and explained how the chemistry avoids the release of toxic and mutagenic repair intermediates (Fig. 1). Mutations in XPD helicase cause 3 distinct phenotypes: cancer-prone xeroderma pigmentosum and the aging disorders Cockayne syndrome and trichothiodystrophy. To clarify molecular differences that underlie F i g . 1 . Endonuclease IV E261Q DNA substrate–bound and DNA- free x-ray structures. A, Apurinic-apyrimidinic (AP)-DNA complex stereo shows the 3-metal-ion active site (green spheres); residues R37, Y72, and Q261 (pink); and bound DNA substrate with both the AP site sugar and phosphate moieties and the cognate nucleotide (orange) flipped out from normal duplex DNA. The 2Fo-Fc electron density map is contoured at 1 σ (blue mesh). B, DNA-free structure with active-site phosphate and 3 zinc ions coordination. Omit map is contoured at 2 (light blue) and 4 (dark blue) σ for the bound phosphate group. C, DNA substrate complex binding to active-site metal ions. High-quality omit map (contoured at 2 σ, pink mesh) shows the intact phosphodiester bond (black arrow) that constrains the Zn3 to Cyt6 O3′ distance to 2.7 Å. Based on Garcin, E.D., Hosfield, D.J., Desai, S.A., et al., DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat. Struct. Mol. Biol. 15:515, 2008. these diseases, we determined crystal structures of the XPD helicase catalytic core from Sulfolobus acidocaldarius and measured mutant enzyme activities. Mutations associated with xeroderma pigmentosum map along the ATP-binding edge and DNA-binding channel and impair helicase activity essential for nucleotide excision repair. Mutations associated with xeroderma pigmentosum and Cockayne syndrome both impair helicase activity and likely affect functional movement. Mutations association with trichothiodystrophy lose or retain helicase activity but map to sites in all 4 domains expected to MOLECULAR BIOLOGY 2008 cause framework defects affecting the integrity of transcription factor IIH. These new results broaden our understanding of how structural changes in the XPD helicase might affect cancer risks or result in developmental or aging phenotypes (Fig. 2). In general, the structural biology of pro- THE SCRIPPS RESEARCH INSTITUTE 217 sion to host cells, and natural transformation. Because they are prominently exposed on bacterial surfaces, pili are attractive targets for the host immune response and for vaccines and therapeutic reagents. Our studies are providing an integrated understanding of the assembly and disassembly of type IV pili. This understanding suggests new approaches to drug and vaccine design for bacterial pathogens, including Francisella tularensis, a highly virulent microorganism that causes tularemia. Because of its high infectivity and potential airborne transmission, F tularensis is designated a category A bioterrorism agent. PUBLICATIONS Chrencik, J.E., Brooun, A., Zhang, H., Mathews, I.I., Hura, G.L., Foster, S., Perry, J.J.P., Streiff, M., Ramage, P., Widmer, H., Bokoch, G.M., Tainer, J.A., Weckbecker, G., Kuhn, P. Structural basis of guanine nucleotide exchange mediated by the T-cell essential Vav1. J. Mol. Biol. 380:828, 2008. Fan, L., Fuss, J.O., Cheng, Q.J., Arvai, A.S., Hammel M., Roberts, V.A., Cooper, P.K., Tainer, J.A. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133:789, 2008. Garcin, E.D., Hosfield, D.J., Desai, S.A., Haas, B.J., Björas, M., Cunningham, R.P., Tainer, J.A. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat. Struct. Mol. Biol. 15:515, 2008. Perry, J.J., Tainer, J.A., Boddy, M.N. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem. Sci. 33:201, 2008. Perry, J.J.P., Tainer, J.A. Structural biology of Cockayne syndrome proteins, their interactions and insights into DNA repair mechanisms. In: Molecular Mechanisms of Cockayne Syndrome. Ahmad, S.I. (Ed.). Landes Bioscences, Austin, TX, in press. F i g . 2 . Structural placement of disease-causing mutations in XPD helicase. Mapping the 3 classes of mutations onto the SaXPD structure reveals patterns associated with each disease defect. A, Stereo pair mapping the distribution of disease-causing mutations on a XPD C α trace. Disease-causing mutation sites (Cα colored sphere): red (XP), greenish yellow (XP/CS), and purple (TTD). Residue F136 is also shown (cyan). B, XPDcc fold and domain architecture (ribbons) with labeled disease-causing mutation sites as spheres colored as in A. C, XP mutations affect DNA- and ATP-binding regions. D, XP/CS mutations affect HD1-HD2 conformational changes. E, TTD mutations affect the overall framework stability. Reprinted from Fan, L., Fuss, J.O., Cheng, Q.J., Arvai, A.S., Hammel M., Roberts, V.A., Cooper, P.K., Tainer, J.A. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133:789, 2008. Copyright 2008, with permission from Elsevier. teins such as XPD, which control reactive oxygen species and DNA repair, may provide master keys to brain abnormalities, cancer, and aging. Putnam, C.D., Hammel, M., Hura, G.L., Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40:191, 2007. Prudden, J., Pebernard, S., Raffa, G., Slavin, D.A., Perry, J.J., Tainer, J.A., McGowan, C.H., Boddy, M.N. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 26:4089, 2007. Roberts, B.R., Tainer, J.A., Getzoff, E.D., Malencik, D.A., Anderson, S.R., Bomben, V.C., Meyers, K.R., Karplus, P.A., Beckman, J.S. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol. 373:877, 2007 Tubbs, J.L., Pegg, A.E., Tainer, J.A. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair (Amst.) 6:1100, 2007. Structural Biology of Integral Membrane Proteins G. Chang, S. Aller, X. He, A. Karyakin, S. Lieu, P. Szewczk, BACTERIAL PILI AND INFECTIOUS DISEASES Type IV pili are essential virulence factors for many bacterial pathogens and therefore act in many important infectious diseases. Functions of type IV pili include motility, formation of microcolonies and biofilms, adhe- T. Tuan, A. Ward, J. Yu S tudy of the structure of membrane proteins is important for understanding their function. We are interested in 4 areas: (1) the molecular struc- 218 MOLECULAR BIOLOGY 2008 tural basis for the transport of lipids and drugs across the cell membrane by multidrug resistance (MDR) transporters, (2) the crystallography of mammalian MDR transporters and the structural basis of their inhibition, (3) the discovery and rational design of potent MDR reversal agents, and (4) the development and application of a cell-free system capable of producing large quantities of integral membrane proteins. We use several experimental methods, and we collaborate with scientists in other laboratories to achieve our goals. MDR in the treatment of cancer and infectious disease is often caused by an upregulation of drug efflux pumps imbedded in the cell membrane. The molecular basis of multispecificity and drug efflux by these transporters is not well understood. Through our structural studies, we are elucidating the mechanisms for the transport of amphipathic substrate across the cell membrane in several families of transporters: ATP-binding cassette, small multidrug resistance, major facilitator superfamily, and multiple antimicrobial extrusion. In collaboration with M.G. Finn, Department of Chemistry, and Q. Zhang, Department of Molecular Biology, we are discovering and designing potent inhibitors to be used synergistically with established antibiotics and cancer chemotherapeutics. In collaboration with R.A. Milligan, Department of Cell Biology, we are using electron cryomicroscopy to visualize transporter structures. We have determined 4 x-ray structures of the ATPbinding cassette transporter MsbA trapped in different conformations: 2 with nucleotide bound and 2 with no nucleotide. Comparisons of the nucleotide-free conformations of MsbA revealed a flexible hinge formed by extracellular loops 2 and 3. The hinge allows the nucleotide-binding domains to disassociate while the ATPbinding half sites remain facing each other. The binding of nucleotide causes a packing rearrangement of the transmembrane helices and changes the accessibility of the transporter from cytoplasmic (inward) facing to extracellular (outward) facing. The inward and outward openings are mediated by 2 different sets of transmembrane helix interactions. Altogether, the conformational changes between these structures suggest that large ranges of motion may be required for substrate transport. EmrE, an MDR transporter from the small multidrug resistance family, functions as a homodimer of a small 4-transmembrane protein. The membrane insertion topology of the 2 monomers is controversial. EmrE was reported to have a unique orientation in the membrane. Models based on electron microscopy and on several THE SCRIPPS RESEARCH INSTITUTE biochemical studies posit an antiparallel dimer. The structures of EmrE in complex with a transport substrate are highly similar to the electron microscopy structure. The first 3 transmembrane helices from each monomer surround the substrate-binding chamber, whereas the fourth helix participates only in dimer formation. Selenomethionine markers clearly indicate an antiparallel orientation for the monomers, supporting a “dual topology” model. EmrD, an MDR transporter from the major facilitator superfamily, expels hydrophobic compounds across the inner membrane. The x-ray structure reveals an interior composed of hydrophobic residues, a finding consistent with the role of EmrD in transporting amphipathic molecules that uncouple the proton gradient across the cell membrane. Two long loops extend into the inner leaflet side of the cell membrane and may recognize and bind substrate directly from the lipid bilayer. On the basis of the structure, we propose that multisubstrate specificity, binding, and transport are facilitated by these loop regions and the internal cavity. Structure and Function of Membrane-Bound Enzymes C.D. Stout, M. Yamaguchi, R. Akhouri, V.M.M. Luna, A. Annalora e focus on the structure and function of membrane-bound enzymes and the development of methods for crystallizing membrane proteins. We study the mechanism of transhydrogenase, a mitochondrial respiratory enzyme complex that couples proton translocation with hydride transfer. This enzyme is essential for maintaining NADPH levels in mitochondria, and it plays a critical role in insulin secretion in beta cells of the pancreas. We use x-ray crystallography, biochemical and spectroscopic methods, electron microscopy in collaboration with M. Yeager, Department of Cell Biology, and nuclear magnetic resonance in collaboration with H.J. Dyson, Department of Molecular Biology. Currently, further progress in understanding the structure and function of transhydrogenase is stymied by lack of a 3-dimensional structure; therefore, our primary effort now is to obtain diffraction-quality crystals of the enzyme in its membrane-bound configuration. Crystallization experiments with transhydrogenase, cytochrome ba 3 oxidase, and cytochrome P450s are W MOLECULAR BIOLOGY 2008 further enabled by use of novel detergents synthesized by Q. Zhang, Department of Molecular Biology. In collaboration with J.A. Fee, Department of Molecular Biology, we are studying the mechanism of cytochrome ba3 oxidase, a homolog of the terminal enzyme of respiration in mitochondria. We are using crystal structure analysis, mutagenesis, and spectroscopy to visualize intermediates in the reduction of oxygen to water, to define the pathways for protons and oxygen into the active site, and to understand the coupling of reduction potential to proton translocation across the membrane. We have also used protein engineering to improve the crystallization behavior of this membrane protein. In collaboration with E.F. Johnson, Department of Molecular Biology; J.R. Halpert, University of California, San Diego; and I. Pikuleva, University of Texas Medical Branch, Galveston, Texas, we are characterizing the structure and function of mammalian microsomal cytochrome P450s. These membrane-associated enzymes specifically metabolize a wide diversity of exogenous compounds and drugs. High-resolution structures have been determined for the principal drug-metabolizing microsomal P450s in the liver and lung in humans: 1A2, 2A6, 2A13, 2C5, 2C8, 2C9, 2C19, 3A4, and 2B4. For 2B4, 5 structures of the enzyme in markedly different conformations provide insight to substrate binding and membrane insertion. A structure of the brain-specific, cholesterol-metabolizing P450 CYP46 has been determined, and recently the first structure of the mitochondrial P450 CYP24A1 has been determined at 2.0-Å resolution (Fig. 1). This class of P450s is involved in the metabolism of lipophilic hormones. In collaboration with P.E. Dawson, Department of Cell Biology, we are developing the assembly of synthetic peptides and phospholipids into discs that contain lipid bilayers. Nanodiscs composed of human apolipoprotein A-I and phospholipids self-assemble into discrete, watersoluble, bilayer-containing particles. Integral membrane proteins incorporated into these particles retain their enzymatic activity, are amenable to biochemical assays, and may have superior properties for crystallization in the absence of detergents. A major effort to determine the basis of HIV resistance to antiviral drugs is ongoing in collaboration with A.J. Olson and J.E. Elder, Department of Molecular Biology; B.E. Torbett, Department of Molecular and Experimental Medicine; M.G. Finn, Department of Chemistry; and D.E. McRee, ActiveSight, San Diego, California. One aspect of this project entails determining the crystal THE SCRIPPS RESEARCH INSTITUTE 219 F i g . 1 . Electron density for mitochondrial cytochrome P450 CYP24A1, the enzyme responsible for stereospecific, 6-step oxidation of vitamin D3. The active form of vitamin D3 regulates calcium and phosphate homeostasis and stimulates cellular differentiation but inhibits proliferation, making CYP24A1 an attractive target for development of cancer therapeutics. structure of mutant proteases from drug-resistant HIV in complex with broad-spectrum inhibitors. Currently, 220 MOLECULAR BIOLOGY 2008 we are using fragment-based screening to discover new classes of inhibitors as lead compounds for drug discovery. In these experiments, we use high-throughput structural genomics technologies to acquire hundreds of high-resolution data sets to discover specific binding of druglike small molecules. PUBLICATIONS Liu, B., Luna, V.M., Chen, Y., Stout, C.D., Fee, J.A. An unexpected outcome of surface engineering an integral membrane protein: improved crystallization of cytochrome ba3 from Thermus thermophilus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63:1029, 2007. Luna, V.M., Chen, Y., Fee, J.A., Stout, C.D. Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry 47:4657, 2008. Mast, N., White, M.A., Bjorkhem, I., Johnson, E.F., Stout, C.D., Pikuleva, I.A. Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U. S. A. 105:9546, 2008. THE SCRIPPS RESEARCH INSTITUTE pathways whereby oxygen arrives at the heme a 3–CuB center, which is deeply buried within the transmembrane domain of these enzymes. Using xenon- and krypton-pressurized crystals of cytochrome ba 3 , we identified the hydrophobic cavities in the oxidase. The location and characteristics of this channel led us to believe that it is a ligand-transport channel. To gain further insight into transport channels in cytochrome c oxidases, we have also analyzed hydrophobic channels in cytochrome c oxidases of known structure. The locations of the xenon-binding sites indicate a continuous, bifurcated channel that opens from 2 points on the protein exterior and leads directly to the dinuclear center (Fig. 1A). Opening into the bilayer (Fig. 1B) Sansen, S., Hsu, M.-H., Stout, C.D., Johnson, E.F. Structural insight into the altered substrate specificity of human cytochrome P450 2A6 mutants. Arch. Biochem. Biophys. 464:197, 2007. Schoch, G.A., Yano, J.K., Sansen, S., Dansette, P.M., Stout, C.D., Johnson, E.F. Determinants of cytochrome P450 2C8 substrate binding: structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J. Biol. Chem. 283:17227, 2008. White, M.A., Mast, N., Bjorkhem, I., Johnson, E.F., Stout, C.D., Pikuleva, I.A. Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr. D Biol. Crystallogr. 64:487, 2008. Zhao, Y., Sun, L., Muralidhara, B.K., Kumar, S., White, M.A., Stout, C.D., Halpert, J.R. Structural and thermodynamic consequences of 1-(4-chlorophenyl)imidazole binding to cytochrome P450 2B4. Biochemistry 46:11559, 2007. Structural Analysis of Oxygen Transport Channels in Thermus thermophilus Cytochrome ba3 Oxidase J.A. Fee, V.M. Luna, B. Liu, Y. Chen, C.D. Stout ytochrome c oxidase is the principal terminal oxidase in the aerobic metabolism of all animals, plants, and yeasts and in some bacteria. In the thermophillic bacterium Thermus thermophilus, cytochrome ba3 oxidase is the preferred respiratory enzyme that catalyzes the flow of electrons from reduced cytochrome c to dioxygen, forming water concomitant with the formation of a proton gradient under low oxygen tensions. Dioxygen reduction occurs at the high-spin, heme a 3–CuB dinuclear center. Although enzymatic turnover has been studied extensively, little is known about the C F i g . 1 . Location and characteristics of the oxygen channel in cyto- chrome ba 3. For both panels, subunit I is pale green, subunit II is magenta, and subunit IIa is turquoise. A, Top view of the oxidase shows the computationally derived shape of the oxygen channel (yellow surface). Xenon and krypton molecules completely line the hydrophobic channel from the dinuclear center to the boundary of the protein. The CuA domain of subunit II was removed to illustrate the unique Y shape of the channel. B, Transmembrane view of the oxidase shows the oxygen channel opening into the lipid bilayer. MOLECULAR BIOLOGY 2008 allows the channel to use the higher oxygen concentration in lipid bilayers. Compared with the other cytochrome c oxidases of known structure, the structure of cytochrome ba 3 with its Y-shaped channel is unique. The channel has 2 openings and allows unimpeded access to the enzymatic center of the protein. On the basis of its location and relative occupancy (highest), the xenon 1 site (Fig. 2) serves as the portal THE SCRIPPS RESEARCH INSTITUTE 221 by a hydrophilic path. We hypothesize that the newly formed water molecules are repelled by the hydrophobic surface of the oxygen channel but attracted to the hydrophilic area around the heme a3 propionates, where they exit the dinuclear center in a hydrophilic “vent” (Fig. 2). This spatially separated vectorial transfer from substrate (oxygen) to product (water) cavities during turnover would increase the overall rate of the reaction. PUBLICATIONS Liu, B., Luna, V.M., Chen, Y., Stout, C.D., Fee, J.A. An unexpected outcome of surface engineering an integral membrane protein: improved crystallization of cytochrome ba3 from Thermus thermophilus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63:1029, 2007. Luna, V.M., Chen, Y., Fee, J.A., Stout, C.D. Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry 47:4657, 2008. Chemistry and Membrane Protein Structural Biology Q. Zhang, M.M. Baksh, W.-X. Hong, Y. Weng he structural characterization of integral membrane proteins has been an important challenge for decades, and the structures of only a tiny fraction of these proteins have been solved to date. An integral membrane protein with built-in detergents can be regarded as the equivalent of a soluble protein, and the many challenges in the study of membrane proteins are associated with the use of the spherical micelleforming detergents. Currently, a pressing need exists for new types of cell-membrane mimicking reagents to advance the field of membrane protein structural biology. We designed a new class of steroid-based molecules with facial amphiphilicity instead of the end polarity of the conventional head-to-tail detergents. The facial amphiphiles should provide a better membrane-mimicking environment than the spherical micelle-forming detergents do and thus shield the transmembrane hydrophobic surface of integral membrane proteins. The results of using these amphiphiles to stabilize several integral membrane proteins in functional states and in the protein structural characterization as well have been promising. In collaborative studies with C.D. Stout and D.B. Goodin, Department of Molecular Biology, use of our facial amphiphiles led to the successful crystallization of the mitochondrial cytochrome P450 CYP24A1 at 2.0-Å resolution. In collaboration with Dr. Stout, T F i g . 2 . Environment around the dinuclear center of cytochrome ba 3. Surface renditions of the pocket encompassing the dinuclear center are primarily hydrophobic (salmon). No direct access exists from the xenon 1 (Xe1) site (slate; top) to the CuB (blue; bottom) of the dinuclear center (sky-blue arrow). We hypothesize that after the conversion of oxygen to water, the water exits the dinuclear center through a hydrophilic area (surface representation in pale yellow) surrounding the propionates of heme a3 (marine-blue arrow). Although not present in our structures, ordered waters found at the heme a 3 propionates in a higher resolution structure of cytochrome ba 3 are shown as orange spheres. into the dinuclear center for oxygen. Oxygen initially binds to CuB before being transferred to heme a 3, but there is no straight-line access from the xenon 1 site to CuB. Although oxygen favors the hydrophobic environment surrounding the dinuclear center, during enzymatic turnover, the water produced does not and must exit 222 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE R.C. Stevens, and G. Chang, Department of Molecular Biology, and M. Yeager, Department of Cell Biology, we are attempting to crystallize a variety of other targets. Meanwhile, we are combining different strategies to overcome the many challenges in membrane protein structural biology. Systematic screening of a large number of detergents is desirable in many circumstances, because selection of detergents is still empirical. In collaboration with K. Wüthrich, Department of Molecular Biology, we have developed a strategy for efficient screening of a large detergent library for the reconstitution of integral membrane proteins. Using this approach, we identified several new detergents that yielded high-quality nuclear magnetic resonance spectra of the membrane proteins OmpX and OmpW. PUBLICATIONS Zhang, Q., Horst, R., Geralt, M., Ma, X., Hong, W.-X., Finn, M.G., Stevens, R.C., Wüthrich, K. Microscale NMR screening of new detergents for membrane protein structural biology. J. Am. Chem. Soc. 130:7357, 2008. Zhang, Q., Ma, X., Ward, A., Hong, W.-X., Jaakola, V.-P., Stevens, R.C., Finn, M.G., Chang, G. Designing facial amphiphiles for the stabilization of integral membrane proteins. Angew. Chem. Int. Ed. 46:7023, 2007. G Protein–Coupled Receptors R.C. Stevens, E.E. Abola, A.I. Alexandrov, L.K. Allin, G.A. Asmar-Rovira, K.A. Baker, R.R. Benoit, M.H. Bracey, Q. Chai, V.G. Cherezov, E.Y.T. Chien, E. Chun, S. Daudenarde, J. Dupuy, A. Gámez, J. Gatchalian, M.T. Griffith, M.A. Hanson, V.-P. Jaakola, J.S. Joseph, T.S. Kang, J. Liu, K. Masuda, M. Michino, M. Mileni, C.B. Roth, K.S. Saikatendu, I. Slaymaker, P. Stenmark, T. Trinh, J. Velasquez, L. Wang, B. Wu, Q. Zhao protein–coupled receptors (GPCRs) are the largest family of proteins in the human genome, with more 1000 receptors. These receptors are a key signaling component throughout the human body and the target for more than 50% of all drugs. In 2007, we reported for the first time the 3-dimensional structure of a human GPCR, the β2-adrenergic receptor (Fig. 1). Since then, we have made rapid progress on additional cocrystal structures with different drug molecules bound and variants to probe receptor function. In parallel with the structural studies, we are also conducting biochemical and biological experiments to further understand how the receptors transmit signals across the cell membrane and influence so many different events within the cell (Fig. 2). G F i g . 1 . Structure of human β2-adrenergic receptor highlighting the cholesterol-binding site in the middle of the receptor. Recently, we found that cholesterol, a molecule typically associated with membrane fluidity and curvature, is a key stabilizing molecule for GPCRs that bind in the “cholesterol consensus motif.” The discovery of this unique binding site provides a potential new avenue for modulating receptor function and for showing, for the first time, the direct binding interaction between cholesterol and a membrane protein. Almost certainly, this discovery is a more general event in biology. To further compliment our studies, in collaboration with K. Wüthrich, Department of Molecular Biology, we are using nuclear magnetic resonance to understand the dynamics of the receptor and the influence of ligands. Last, we are working to decipher the protein-protein interactions in GPCR systems between the receptors and G proteins, arrestin, and G protein receptor kinases. STRUCTURAL PROTEOMICS OF GPCRS Perhaps as exciting as the first human GPCR structure is our development of technologies and a pipeline that enable us to solve multiple GPCR structures more rapidly and to tackle representative members of the entire GPCR family (Fig. 3). Our development of a MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 223 F i g . 3 . Family tree of class A human GPCRs. To date, only rho- dopsin and one member of the amine family have been structurally characterized. F i g . 2 . Analysis of the increase in helical packing and thermal stability of the β 2 -adrenergic receptor due to cholesterol binding. A, Receptor is colored by normalized occluded surface area. Red thick lines indicate the compact areas of the receptor; blue thin lines are the least compact. Helix IV has the lowest packing of the 7 helices in the tertiary structure, particularly on the cytoplasmic end. Cholesterol binding stabilizes the receptor by increasing packing constraints, especially in the vicinity of the cytoplasmic end of helix IV. From 10% to 70% of the total available surface area is involved in packing interactions. B, Differences in the normalized occluded surface area of the receptor due to cholesterol binding. The increase in packing of available surface area due to cholesterol binding is 0% to 15%; the most significant increases are for residues on helices II and IV. C, Molecular surface representation of the receptor and cholesterol. Green corresponds to atoms on both cholesterol and the receptor that are within 4 Å of each other. Blue corresponds to atoms on the receptor that are 4 and 5 Å from the cholesterol molecules. On the right, the cholesterol molecules have been lifted out of the binding groove to better show the interactions and the groove. D, Isothermal denaturation curves with 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin dye were used to determine the half-life of the β2-adrenergic recepter in the presence of 1 M guanidine hydrochloride with and without both chloesterol hemisuccinate (CHS) and timolol (Tim). The thickness of the line represents the 95% confidence interval over 3 replicates; the fitted half-lives are indicated next to the respective curves. Both timolol and cholesterol cause an approximate 5-fold increase in half-life under these conditions. In combination, the effect is almost 16-fold relative to apo. Reprinted from Hanson, M.A., Cherezov, V., Griffith, M.T., et al. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16:897, 2008, with permission from Elsevier. structural proteomics approach to understanding entire protein families came out of frustration with the pace at which information on structural biology became available. Although we are now focusing on the biological aspects of GPCRs, during the past 10 years, we have developed new tools to change the field of structural biology by accelerating the rate of determination of protein structures, an endeavor that includes pioneering microliter expression/purification for structural studies, nanovolume crystallization, and automated image collection. These technologies were initially tested at the Joint Center for Structural Genomics (http://www.jcsg.org), in collaboration with I.A. Wilson, Department of Molecular Biology, where the power of the new tools was demonstrated. Although the Joint Center for Structural Genomics 2 has continued as a successful production for the second phase of the Protein Structure Initiative of the National Institute of General Medical Sciences, in collaboration with P. Kuhn, Department of Cell Biology, we have created 2 new centers funded by the National Institutes of Health that focus on technologic innovations in structural biology. The first center is the Joint Center for Membrane Protein Technologies (http://jcimpt.scripps.edu). Here, in collaboration with K. Wüthrich, Q. Zhang, and G. Chang, Department of Molecular Biology; M.G. Finn, Department of Chemistry; and P. Kuhn and M. Yeager, Department of Cell Biology, we do research exclusively on membrane proteins, including GPCRs. The second center is the Accelerated Technologies Center for Gene to 3D Structure (http://www.atcg3d.org). Here we are collaborating with Dr. Kuhn and with researchers from deCODE biostructures, Bainbridge Island, Washington; 224 MOLECULAR BIOLOGY 2008 Lyncean Technologies, Palo Alto, California; and the University of Chicago, Chicago, Illinois, on novel crystallization and x-ray methods. THE SCRIPPS RESEARCH INSTITUTE Serrano, P., Johnson, M.A., Almeida, M.S., Horst, R., Hermann, T., Joseph, J.S., Neuman, B.W., Subramanian, V., Saikatendu, K.S., Buchmeier, M.J., Stevens, R.C., Kuhn, P., Wüthrich, K. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J. Virol. 81:12049, 2007. STRUCTURE-BASED DRUG DISCOVERY Over the years, we have been involved in the basic science and development of several therapeutic agents to treat neurologically related disorders (e.g., phenylketonuria, pain, dystonias). With the structure determination of a GPCR now in hand and the basic mechanistic research in this area progressing, we will turn our attention to developing improved drugs to treat human disease. Slaymaker, I.M., Bracey, M., Mileni, M., Garfunkle, J., Cravatt, B.F., Boger, D.L., Stevens, R.C. Correlation of inhibitor effects on enzyme activity and thermal stability for the integral membrane protein fatty acid amide hydrolase. Bioorg. Med. Chem. Lett., in press. Stevens, R.C. Generation of protein structures for the 21st century. Structure 15:1517, 2007. Structural Genomics Consortium; China Structural Genomics Consortium; Northeast Structural Genomics Consortium, Gräslund, S., Nordlund, P., Weigelt, J., et al. Protein production and purification. Nat. Methods 5:135, 2008. PUBLICATIONS Alexandrov, A.I., Mileni, M., Chien, E.Y.T., Hanson, M.A., Stevens, R.C. Microscale fluorescent thermal stability assay for membrane proteins. Structure 16:351, 2008. Wang, L., Gámez, A., Archer, H., Abola, E.E., Sarkissian, C.N., Fitzpartick, P., Wendt, D., Zhang, Y., Vellard, M., Bliesath, J., Bell, S.M., Lemontt, J.F., Scriver, C.R., Stevens, R.C. Structural and biochemical characterization of the therapeutic Anabaena variabilis phenylalanine ammonia lyase. J. Mol. Biol. 380:623, 2008. Asmar-Rovira, G.A., Asseo-García, A.M., Quesada, O., Hanson, M.A., Nogueras, C., Lasalde-Dominicci, J.A., Stevens, R.C. Biophysical and ion channel functional characterization of the Torpedo californica nicotinic acetylcholine receptor in varying detergent-lipid environments. J. Membr. Biol. 223:13, 2008. Zhang, Q., Horst, R., Geralt, M., Ma, X., Hong, W.X., Finn, M.G., Stevens, R.C., Wüthrich, K. Microscale NMR screening of new detergents for membrane protein structural biology. J. Am. Chem. Soc. 130:7357, 2008. Cherezov, V., Rosenbaum, D.M., Hanson, M.A., Rasmussen, S.G.F., Thian, F.S., Kobilka, T.S., Choi, H.-J., Kuhn, P., Weis, W.I., Kobilka, B.K., Stevens, R.C. High-resolution crystal structure of an engineered human β2-adrenergic G proteincoupled receptor. Science 318:1258, 2007. Gámez, A., Wang, L., Sarkissian, C.N., Wendt, D., Fitzpatrick, P., Lemontt, J.F., Scriver, C.R., Stevens, R.C. Structure-based epitope and PEGylation sites mapping of phenylalanine ammonia-lyase for enzyme substitution treatment of phenylketonuria. Mol. Genet. Metab. 91:325, 2007. Zhang, Q., Ma, X., Ward, A., Hong, W.X., Jaakola, V.P., Stevens, R.C., Finn, M.G., Chang, G. Designing facial amphiphiles for the stabilization of integral membrane proteins. Angew. Chem. Int. Ed. 46:7023, 2007. Zurflüh, M.R., Zschocke, J., Linder, M., Feillet, F., Chery, C., Burlina, A., Stevens, R.C., Thony, B., Blau, N. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum. Mutat. 29:1079, 2008. Hanson, M.A., Brooun, A., Baker, K.A., Jaakola, V.P., Roth, C., Chien, E., Alexandrov, A., Velasquez, J., Davis, L., Griffith, M., Moy, K., Ganser-Pornillos, B., Kuhn, P., Ellis, S., Yeager, M., Stevens, R.C. Profiling of membrane protein variants in a baculovirus system by coupling cell-surface detection with small-scale parallel expression. Protein Expr. Purif. 56:85, 2007. High-Throughput Approaches to Protein Structure and Function Hanson, M.A., Cherezov, V., Griffith, M.T., Roth, C.B., Jaakola, V.-P., Chien, E.Y.T., Velasquez, J., Kuhn, P., Stevens, R.C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16:897, 2008. S.A. Lesley, H. Johnson, S. Sudek, T. Janaratne, S. Kale, M. Deller, D. Carlton, T. Clayton, A. Grzechnik, C. Farr Jauch, R., Ng, C.K., Saikatendu, K.S., Stevens, R.C., Kolatkar, P.R. Crystal structure and DNA binding of the homeodomain of the stem cell transcription factor Nanog. J. Mol. Biol. 376:758, 2008. Mathews, I.I., McMullan, D., Miller, M.D., et al. Crystal structure of 2-keto-3deoxygluconate kinase (TM0067) from Thermatoga maritima at 2.05 Å resolution. Proteins 70:603, 2008. Neuman, B.W., Joseph, J.S., Saikatendu, K.S., Serrano, P., Chatterjee, A., Johnson, M.A., Liao, L., Klaus, J.P., Yates, J.R. III, Wüthrich, K., Stevens, R.C., Buchmeier, M.J., Kuhn, P. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 82:5279, 2008. Ng, J.D., Clark, P.J., Stevens, R.C., Kuhn, P. In situ x-ray analysis of protein crystals in low-birefringent and x-ray transmissive plastic microchannels. Acta Crystallogr. D Biol. Crystallogr. 64:189, 2008. Ng, J.D., Stevens, R.C., Kuhn, P. Protein crystallization in restricted geometry: advancing old ideas for modern times in structural proteomics. Methods Mol. Biol. 426:363, 2008. Rosenbaum, D.M., Cherezov, V., Hanson, M.A., Rasmussen, S.G.F., Thian, F.S., Kobilka, T.S., Choi, H.-J., Yao, X.-J., Weis, W.I., Stevens, R.C., Kobilka, B.K. GPCR engineering provides high-resolution structural insights into β2-adrenergic receptor function. Science 318:1266, 2007. Roth, C.B., Hanson, M.A., Stevens, R.C. Stabilization of the human β2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu-1223.41, a critical residue in GPCR structure. J. Mol. Biol. 376:1305, 2008. valuating protein structure and function is of primary importance for understanding the basic biology of the cell and is a challenge because of the constantly expanding wealth of genomic information. To address this challenge, we have established highthroughput approaches for evaluating structural and functional diversity of proteins as part of a structural genomics effort with the Joint Center for Structural Genomics. We use these same tools to characterize the molecular basis of the specificity of enzyme substrates and to probe the roles the enzymes play in cellular and metabolic pathways. The goal of the Joint Center for Structural Genomics is to develop a high-throughput and cost-effective structure pipeline and to use the pipeline to determine novel protein folds and explore protein structure-function relationships. We have used this approach in an extensive study of the thermophilic bacterium Thermotoga maritima and for targets from mouse and more E MOLECULAR BIOLOGY 2008 than 100 other bacterial genomes. Our technologies have enabled us to perform comprehensive structural studies of these proteomes. To date, these efforts have resulted in more than 700 novel protein structures from the center. We continue to develop technology in membrane protein crystallization and protein complexes. We are also using this approach to study commensal bacteria in human health and disease. Awareness is increasing that both normal and disease states are influenced by the composition and byproducts of the microbial communities in our bodies. We are using highthroughput discovery methods to screen for new roles and influences of these microbes in disease and metabolism. Combining metagenomic studies with protein characterization and cell-based assays, we are screening large panels of secreted proteins from the human gut in a number of basic pathways involved in inflamation, cell death, and drug metabolism. Understanding how genome sequences are related to the biology of an organism requires correct annotation of gene function. Many computational approaches are available for assigning a putative gene function on the basis of similarity to known activities. However, as the evolutionary distances extend and the number of putative homologs increases, the reliability of such unvalidated predictions becomes suspect. We use direct experimentation to test putative predictions and focused ligand screens to identify new activities. Structural Biology and Structural Genomics With Nuclear Magnetic Resonance Spectroscopy K. Wüthrich, W. Augustyniak, A. Chatterjee, M. Geralt, R. Horst, M. Johnson, B. Pedrini, W.J. Placzek, J.K. Rhee, P. Serrano, P. Stanczak e are developing methods to improve the efficiency and reliability of nuclear magnetic resonance (NMR) structure determination of proteins in solution and are applying the methods to target proteins selected within the framework of the Joint Center for Structural Genomics. In addition, as part of the research of the Center for Functional and Structural Proteomics of SARS-CoV (FSPS; http://visp.scripps.edu/ W THE SCRIPPS RESEARCH INSTITUTE 225 SARS/default.aspx), we are characterizing the proteome of the coronavirus (SARS-CoV) that causes severe acute respiratory syndrome (SARS). SARS-COV STRUCTURAL GENOMICS Although a 2003 SARS pandemic was contained by public health measures, no vaccine or effective treatment for SARS is available, and the basic mechanisms of coronavirus infections are not yet understood. SARSCoV contains a 29-kb positive-stranded RNA genome. About two-thirds of the genome is devoted to encoding a replicase polyprotein, which is cleaved by 2 viral proteases to release the mature nonstructural proteins. These proteins are responsible for the enzymatic functions that allow the virus to replicate in infected cells and therefore are potential targets for drug development. The FSPS project was started with the expectation that structure-based functional studies will reveal new functional features that are not detectable when only the amino acid sequence is known. S T R U C T U R E D E T E R M I N AT I O N O F T H E NONSTRUCTURAL PROTEIN 3c The region of the SARS-CoV nonstructural protein 3 (nsp3) that spans residues 366–722 is a functional domain termed the SARS-unique domain (SUD) because it is not present in other known coronaviruses. Expression in Escherichia coli indicated that SUD does not form a single globular structure, and NMR studies showed that it consists of at least 3 distinct structural domains, which may provide for multiple functions (Fig. 1). A central globular domain, SUD-M (M stands for middle), spans residues 527–651. The NMR structure of this protein shows a macrodomain fold with similarity to that of proteins that bind the important regulatory molecule ADP-ribose in eukaryotic cells (Fig. 2). Structure-based attempts to determine the function of SUD-M started with a search of the Protein Data Bank for structural homologs of SUD-M. The closest 3-dimensional structural homolog was the protein nsp3b, which is located immediately N-terminal to the SUD region in the SARS-CoV proteome and functions as an ADP-ribose1′′-phosphatase. This finding was a surprise, because the sequence identity between the 2 domains is only 6%. Tests for binding of a variety of different ligands, based on NMR chemical-shift perturbation measurements, revealed that SUD-M recognizes single-stranded polyadenosine RNA. A possible function suggested by this observation is in viral genome replication or transcription, which may involve the recognition of polyadenylated tails of viral RNA by one or more viral proteins. SUD-M might thus be a 226 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE F i g . 1 . Structural coverage of nsp3. The horizontal black line represents the polypeptide segment 1–1318; the initially annotated functional domains are indicated above the line. Globular domain structures determined so far are shown in ribbon representation, along with color-coded information on the structure determination method used and the new, structure-based functional annotation. In between the globular domains, blue lines represent flexibly disordered segments as determined by NMR spectroscopy, and black lines indicate unstructured segments implicated by the absence of x-ray diffraction in protein crystals. Regions of the protein with unknown structures are colored green; these regions extend to the C terminus of nsp3 at residue 1922. F i g . 2 . A, Ensemble of 20 conformers representing the solution structure of the protein domain SUD-M (see also Fig. 1). The conformers were superimposed for minimal root-mean-square deviation of the backbone N, C α , and C′ atoms of the residues 527–651. Selected sequence positions relative to the intact nsp3 (Fig. 1) are indicated by numbers. Helical secondary structures are red, β-strands are green, and segments with no regular secondary structure are gray. B, Surface view of SUD-M in the same orientation as in A, with the regions affected by the binding of single-stranded polyadenosine RNA in magenta to highlight the probable RNA-binding site. potential target for the development of antiviral drugs that disrupt viral replication. On the basis of phylogenetic and bioinformatics analyses, nsp3 was initially predicted to consist of 7 functional domains: nsp3a–nsp3g. The NMR structure determination of SUD-M was just one of many steps toward elucidating the structure of the much larger nsp3 polypeptide. The overall structural characterization of the region nsp3a–nsp3e now provides a detailed picture of the domain organization (Fig. 1), and new functions have already been identified. As illustrated in Figure 1, using NMR spectroscopy and x-ray crystallography with polypeptide constructs of variable lengths makes it possible not only to determine the structures of the individual segmentally arranged globular domains but also to characterize the intervening linker regions. With this strategy, which was adapted from target selection in structural genomics projects, we can obtain a comprehensive view of the multidomain protein, which shows that the protein has overall a predominantly extended shape. The structural information thus obtained enabled us to immediately predict different functions of nsp3. We are following up our structural findings with biomedical and physiologic studies. PUBLICATIONS Almeida, M.S., Johnson, M.A., Herrmann, T., Geralt, M., Wüthrich, K. Novel β-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 81:3151, 2007. Chatterjee, A., Johnson, M.A., Serrano, P., Pedrini, B., Wüthrich, K. NMR assignment of the domain 513-651 from the SARS-CoV nonstructural protein nsp3. Biomol. NMR Assign. 1:191, 2007. Horst, R., Fenton, W.A., Englander, W.S., Wüthrich, K., Horwich A.L. Folding trajectories of human dihydrofolate reductase inside the GroEL GroES chaperonin cavity and free in solution. Proc. Natl. Acad. Sci. U. S. A. 104:20788, 2007. Johnson, M.A., Southworth, M.W., Herrmann, T., Brace, L., Perler, F.B., Wüthrich, K. NMR structure of a KlbA intein precursor from Methanococcus jannaschii. Protein Sci. 16:1316, 2007. Johnson, M.A., Southworth, M.W., Perler, F.B., Wüthrich K. NMR assignment of a KlbA intein precursor from Methanococcus jannaschii. Biomol. NMR Assign. 1:19, 2007. MOLECULAR BIOLOGY 2008 Pedrini, B., Placzek, W.J., Koculi, E., Alimenti, C., LaTerza, A., Luporini, P., Wüthrich, K. Cold-adaptation in sea-water-borne signal proteins: sequence and NMR structure of the pheromone En-6 from the Antarctic ciliate Euplotes nobilii. J. Mol. Biol. 372:277, 2007. Placzek, W.J., Almeida, M.S., Wüthrich, K. NMR structure and functional characterization of a human cancer-related nucleoside triphosphatase. J. Mol. Biol. 367:788, 2007. Placzek, W.J., Etezady-Esfarjani, T., Herrmann, T., Pedrini, B., Peti, W., Alimenti, C., Luporini, P., Wüthrich, K. Cold-adapted signal proteins: NMR structures of pheromones from the Antarctic ciliate Euplotes nobilii. IUBMB Life 59:578, 2007. Serrano, P., Johnson, M.A., Almeida, M.S., Horst, R., Herrmann, T., Joseph, J.S., Neuman, B.W., Subramanian, V., Saikatendu, K.S., Buchmeier, M.J., Stevens, R.C., Kuhn, P., Wüthrich, K. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J. Virol. 81:12049, 2007. Nuclear Magnetic Resonance of 3-Dimensional Structure and Dynamics of Proteins in Solution P.E. Wright, H.J. Dyson, M. Arai, R. Burge, P. Deka, J. Ferreon, T.-H. Huang, B.B. Koehntop, M. Kostic, B. Lee, C.W. Lee, M. Landes, M. Martinez-Yamout, T. Nishikawa, K. Sugase, J. Wojciak, M. Zeeb, E. Manlapaz, L.L. Tennant, D.A. Case, J. Gottesfeld e use multidimensional nuclear magnetic resonance (NMR) spectroscopy to investigate the structures, dynamics, and interactions of proteins in solution. Such studies are essential for understanding the mechanisms of action of these proteins and for elucidating structure-function relationships. The focus of our current research is protein-protein and protein–nucleic acid interactions involved in the regulation of gene expression. W TRANSCRIPTION FACTOR–NUCLEIC ACID COMPLEXES NMR methods are being used to determine the 3-dimensional structures and intramolecular dynamics of zinc finger motifs from several eukaryotic transcriptional regulatory proteins, both free and complexed with target nucleic acid. Zinc fingers are among the most abundant domains in eukaryotic genomes. They play a central role in the regulation of gene expression at both the transcriptional and the posttranscriptional level, mediated through their interactions with DNA, RNA, or protein components of the transcriptional machinery. The C2H 2 zinc finger, first identified in transcription factor IIIA (TFIIIA), is used by numerous transcription factors to achieve sequence-specific recognition of DNA. THE SCRIPPS RESEARCH INSTITUTE 227 Growing evidence, however, indicates that some C 2H 2 zinc finger proteins control gene expression both through their interactions with DNA regulatory elements and, at the posttranscriptional level, through binding to RNA. The best-characterized example of a C2H2 zinc finger protein that binds specifically to both DNA and RNA is TFIIIA, which contains 9 zinc fingers. We showed previously that different subsets of zinc fingers are responsible for high-affinity binding of TFIIIA to DNA (fingers 1–3) and to 5S RNA (fingers 4–6). To obtain insights into the mechanism by which the TFIIIA zinc fingers recognize both DNA and RNA, we have used NMR methods to determine the structures of the complex formed by zf1-3 (a protein consisting of fingers 1–3) with DNA and by zf4-6 (a protein consisting of fingers 4–6) with a fragment of 5S RNA. Three-dimensional structures were determined previously for the complex of zf1-3 with the cognate 15-bp oligonucleotide duplex. The structures contain several novel features and reveal that prevailing models of DNA recognition, which assume that zinc fingers are independent modules that contact bases through a limited set of amino acids, are outmoded. In addition to its role in binding to and regulating the 5S RNA gene, TFIIIA also forms a complex with the 5S RNA transcript. NMR structures of the complex formed by zinc fingers 4–6 with a truncated form of 5S RNA have been completed and give important insights into the structural basis for 5S RNA recognition. Finger 4 of the protein recognizes both the structure of the RNA backbone and the specific bases in the loop E motif of the RNA, in a classic lock-and-key interaction. Fingers 5 and 6, with a single residue between them, undergo mutual induced-fit folding with the loop A region of the RNA, which is highly flexible in the absence of the protein. NMR studies of 2 alternate splice variants of the Wilms tumor zinc finger protein (WT1) are in progress. These proteins differ only through insertion of 3 additional amino acids (the tripeptide lysine-threonine-serine) in the linker between fingers 3 and 4, yet have marked differences in their DNA-binding properties and subcellular localization. 15N relaxation measurements indicate that the insertion increases the flexibility of the linker between fingers 3 and 4 and abrogates binding of the fourth zinc finger to its cognate site in the DNA major groove, thereby modulating DNA-binding activity. X-ray and NMR structures of the complexes of the WT1 zinc fingers with 14- and 17-bp DNA oligonucleo- 228 MOLECULAR BIOLOGY 2008 tides have been determined. Zinc fingers 2–4 are inserted deeply into the DNA major groove, making sequence-specific contacts with bases. The structure provides insights into the mechanism by which diseasecausing mutations in the zinc finger domain interfere with DNA binding. In contrast to fingers 2–4, zinc finger 1 has mostly nonspecific interactions with the DNA. High-affinity DNA binding is mediated by fingers 2–4; incorporation of additional amino acids in the linker by alternate splicing disrupts the finger 4 interactions and abrogates DNA binding. NMR structural studies of a complex of the 4 WT1 zinc fingers with an RNA aptamer are nearing completion. In contrast to DNA binding, the RNA interaction is dominated by zinc fingers 1–3, which bind in the widened major groove formed in the vicinity of a bulged base. The interactions of zinc finger 4 with the RNA loop make only a secondary contribution to binding affinity. We have also determined the structure of a novel double-stranded RNA-binding zinc finger protein and have commenced experiments to define the mechanism of binding to adenovirus VA1 RNA. We recently determined the structure of a novel zinc finger protein named Churchill that is involved in regulation of neural induction during embryogenesis. At the time of its discovery, it was suggested that the protein contained 2 zinc fingers of the C4 type and functioned as a DNA-binding transcription factor. Our NMR structure shows that far from containing canonical C4 zinc fingers, Churchill contains 3 bound zinc ions in novel coordination sites, including an unusual binuclear zinc cluster, which jointly stabilize a single-layer β-sheet (Fig. 1). We showed further that Churchill does not bind DNA and suggest that it may function in embryogenesis by mediating protein-protein interactions. PROTEIN-PROTEIN INTERACTIONS IN T R A N S C R I P T I O N A L R E G U L AT I O N Transcriptional regulation in eukaryotes relies on protein-protein interactions between DNA-bound factors and coactivators that, in turn, interact with the basal transcription machinery. The transcriptional coactivator CREB-binding protein (CBP) and its homolog p300 play an essential role in cell growth, differentiation, and development. Understanding the molecular mechanisms by which CBP and p300 recognize their various target proteins is of fundamental biomedical importance. CBP and p300 have been implicated in diseases such as leukemia, cancer, and mental retardation and are novel targets for therapeutic intervention. THE SCRIPPS RESEARCH INSTITUTE F i g . 1 . Structure of Churchill. We previously determined the structure of the kinaseinducible activation domain of the transcription factor CREB bound to its target domain (the KIX domain) in CBP. Ongoing work is directed toward mapping the interactions between KIX and the transcriptional activation domains of the proto-oncogene c-Myb and of the mixed-lineage leukemia protein. The solution structure of the ternary complex between KIX, c-Myb, and the mixed-lineage leukemia protein has been completed and provides insights into the structural basis for the ability of the KIX domain to interact simultaneously and allosterically with 2 different effectors. Our work has also provided new understanding of the thermodynamics of the coupled folding and binding processes involved in interaction of KIX with transcriptional activation domains. We used R 2 relaxation dispersion experiments to elucidate the mechanism by which folding of the kinaseinducible activation domain of CREB is coupled to binding to its KIX target domain. These experiments revealed formation of an ensemble of transient and largely unfolded encounter complexes at multiple sites on the surface of KIX. The encounter complexes are stabilized primarily by nonspecific hydrophobic contacts and evolve via an intermediate to the fully bound state without dissociation from KIX. The C-terminal helix of the kinase-inducible domain is only partially folded in the intermediate and becomes stabilized by intermolecular interactions formed in the final bound state. Future applications of our method will provide new understanding of the molecular mechanism by which intrinsically disordered proteins perform their diverse biological functions. MOLECULAR BIOLOGY 2008 Recently, we determined the structure of the complex between the hypoxia-inducible factor Hif-1α and the TAZ1 domain of CBP. The interaction between Hif-1α and CBP/p300 is of major therapeutic interest because of the central role Hif-1α plays in tumor progression and metastasis; disruption of this interaction leads to attenuation of tumor growth. A protein named CITED2 functions as a negative feedback regulator of the hypoxic response by competing with Hif-1α for binding to the TAZ1 domain of CBP. By determining the structure of the complex, we showed that the intrinsically unstructured Hif-1α and CITED2 domains use partly overlapping surfaces of the TAZ1 motif to achieve high-affinity binding and compete effectively with each other for CBP/p300. To further elucidate the molecular and structural basis for CBP-dependent coordinated gene expression, we have determined the solution structures of the complexes formed by the transactivation domains of the transcription factors STAT2 and STAT1 with CBP TAZ1 and TAZ2 domains, respectively. Despite the overall topological similarity of the CBP TAZ domains, the structures reveal 2 very different modes of complex formation. Our findings suggest that TAZ1 may bind activation domains capable of contacting multiple surface grooves simultaneously in preference to smaller activation motifs that are restricted to a single, contiguous binding surface. The latter mode of binding is sufficient for stable complex formation with TAZ2. Binding of both STAT activation domains involves coupled folding and binding processes. We are continuing to map the multiplicity of interactions between CBP/p300 domains and their numerous biological targets. Our goal is to understand the complex interplay of interactions that mediate key biological processes in health and disease. PUBLICATIONS Ebert, M.-O., Bae, S.-H., Dyson, H.J., Wright, P.E. NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry 47:1299, 2008. Lee, B.M., Buck-Koehntop, B.A., Martinez-Yamout, M.A. Dyson, H.J., Wright, P.E. Embryonic neural inducing factor Churchill is not a DNA-binding zinc finger protein: solution structure reveals a solvent-exposed β-sheet and zinc binuclear cluster J. Mol. Biol. 371:1274, 2007. Stoll, R., Lee, B.M., Debler, E.W., Laity, J.H., Wilson, I.A., Dyson, H.J., Wright, P.E. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol. 372:1227, 2007. Sugase, K., Landes, M.A., Wright, P.E., Martinez-Yamout, M.A. Overexpression of post-translationally modified peptides in Escherichia coli by co-expression with modifying enzymes. Protein Expr. Purif. 57:108, 2008. Sugase, K., Lansing, J.C., Dyson, H.J., Wright, P.E. Tailoring relaxation dispersion experiments for fast-associating protein complexes J. Am. Chem. Soc. 129:13406, 2007. THE SCRIPPS RESEARCH INSTITUTE 229 Folding of Proteins and Protein Fragments P.E. Wright, H.J. Dyson, D. Meinhold, C. Nishimura, D. Felitsky, M. Kostic, S.J. Park, J. Chung, L.L. Tennant, V. Bychkova,* T. Yamagaki * Institute of Protein Research, Puschino, Russia he molecular mechanism by which proteins fold into their 3-dimensional structures remains one of the most important unsolved problems in structural biology. Nuclear magnetic resonance (NMR) spectroscopy is uniquely suited to provide information on the structure of transient intermediates formed during protein folding. Previously, we used NMR methods to show that many peptide fragments of proteins have a tendency to adopt folded conformations in water solution. The presence of transiently populated folded structures, including reverse turns, helices, nascent helices, and hydrophobic clusters, in water solutions of short peptides has important implications for initiation of protein folding. Formation of elements of secondary structure probably plays an important role in the initiation of protein folding by reducing the number of conformations that must be explored by the polypeptide chain and by directing subsequent folding pathways. T A P O M Y O G L O B I N F O L D I N G PAT H WAY A major program in our laboratory is directed toward a structural and mechanistic description of the apomyoglobin folding pathway. Previously, we used quenched-flow pulse-labeling methods in conjunction with 2-dimensional NMR spectroscopy to map the kinetic folding pathway of the wild-type protein. With these methods, we showed that an intermediate in which the A, G, and H helices and part of the B helix adopt hydrogen-bonded secondary structure is formed within 6 milliseconds of the initiation of refolding. Folding then proceeds by stabilization of additional structure in the B helix and in the C and E helices. We are using carefully selected myoglobin mutants and both optical stopped-flow spectroscopy and NMR methods to further probe the kinetic folding pathway. For some of the mutants studied, the changes in amino acid sequence resulted in changes in the folding pathway of the protein. These experiments are providing novel insights into both the local and long-range interactions that stabilize the kinetic folding intermediate. Of particular importance, long-range interactions have been observed that indi- 230 MOLECULAR BIOLOGY 2008 cate nativelike packing of some of the helices in the kinetic molten globule intermediate. However, folding is impeded by local nonnative helix packing; the H helix is translocated relative to the G helix by a single helical turn, and folding cannot proceed until this defect is repaired. Apomyoglobin provides a unique opportunity for detailed characterization of the structure and dynamics of a protein-folding intermediate. Conditions were previously identified under which the apomyoglobin molten globule intermediate is sufficiently stable for acquisition of multidimensional heteronuclear NMR spectra. Analysis of 13C and other chemical shifts and measurements of polypeptide dynamics provided unprecedented insights into the structure of this state. The A, G, and H helices and part of the B helix are folded and form the core of the molten globule. This core is stabilized by relatively nonspecific hydrophobic interactions that restrict the motions of the polypeptide chain. Fluctuating helical structure is formed in regions outside the core, although the amount of helix is low and the chain retains considerable flexibility. The F helix acts as a gate for heme binding and only adopts stable structure in the fully folded holoprotein. The acid-denatured (unfolded) state of apomyoglobin is an excellent model for the fluctuating local interactions that lead to the transient formation of unstable elements of secondary structure and local hydrophobic clusters during the earliest stages of folding. NMR data indicated substantial formation of helical secondary structure in the acid-denatured state in regions that form the A and H helices in the folded protein and also revealed nonnative structure in the D and E helix regions. Because the A and H regions adopt stabilized helical structure in the earliest detectable folding intermediate, these results lend strong support to folding models in which spontaneous formation of local elements of secondary structure plays a role in initiating formation of the A-[B]-G-H molten globule folding intermediate. In addition to formation of transient helical structure, formation of local hydrophobic clusters has been detected by using 15N relaxation measurements. Significantly, these clusters are formed in regions where the average surface area buried upon folding is large. In contrast to acid-denatured unfolded apomyoglobin, the urea-denatured state is largely devoid of structure, although residual hydrophobic interactions have been detected by using relaxation measurements. We have measured residual dipolar couplings for unfolded states of apomyoglobin by using partial align- THE SCRIPPS RESEARCH INSTITUTE ment in strained polyacrylamide gels. These data provide novel insights into the structure and dynamics of the unfolded polypeptide chain. We have shown that the residual dipolar couplings arise from the well-known statistical properties of flexible polypeptide chains. Residual dipolar couplings provide valuable insights into the dynamic and conformational propensities of unfolded and partly folded states of proteins and hold great promise for charting the upper reaches of protein-folding landscapes. To probe long-range interactions in unfolded and partially folded states of apomyoglobin, we introduced spin-label probes at several sites throughout the polypeptide chain. These experiments led to the surprising discovery that transient structures with nativelike longrange contacts between hydrophobic clusters exist within the ensemble of conformations formed by the aciddenatured state of apomyoglobin. They also indicated that the packing of helices in the molten globule state is similar to that in the native folded protein. The relative amounts of the transiently collapsed states formed in the apomyoglobin polypeptide chain are determined by the entropic cost of loop closure. The specificity of the long-range contacts in the most structured of these states suggests that the contacts play a key role in directing chain collapse and initiating folding. The view of protein folding that results from our work on apomyoglobin is one in which collapse of the polypeptide chain to form increasingly compact states leads to progressive accumulation of secondary structure and increasing restriction of fluctuations in the polypeptide backbone. Chain flexibility is greatest at the earliest stages of folding, when transient elements of secondary structure and local hydrophobic clusters are formed. As the folding protein becomes increasingly compact, backbone motions become more restricted, the hydrophobic core is formed and extended, and nascent elements of secondary structure are progressively stabilized. The ordered tertiary structure characteristic of the native protein, with well-packed side chains and relatively low-amplitude local dynamics, appears to form rather late in folding. We recently introduced a variation on the classic quench-flow technique, which makes use of the capabilities of modern NMR spectrometers and heteronuclear NMR experiments, to study the proteins labeled along the folding pathway in an unfolded state in an aprotic organic solvent. This method allows detection of many more amide proton probes than in the classic MOLECULAR BIOLOGY 2008 method, which requires formation of the fully folded protein and the measurement of the protein’s NMR spectrum in water solution. This method is particularly useful in documenting changes in the folding pathway that result in the destabilization of parts of the protein in the molten globule intermediate. We recently showed that self-compensating mutations designed to change the amino acid sequence such that the average area buried upon folding is significantly changed while the 3-dimensional structure of the final folded state remains the same. These studies showed that the average area buried upon folding is an accurate predictor of those parts of the apomyoglobin molecule that will fold first and participate in the molten globule intermediate. Quench-flow hydrogen exchange experiments performed on a series of hydrophobic core mutants indicated that the overall helix-packing topology of the kinetic folding intermediate is like that of the native protein, despite local nonnative interactions in packing of the G and H helices (Fig. 1). Finally, using a rapid mixing device, THE SCRIPPS RESEARCH INSTITUTE 231 mechanism known as quorum sensing; bacteria release extracellular signal molecules, called autoinducers, for cell-cell communication within and between bacterial species. A number of bacteria appear to use quorum sensing for regulation of gene expression in response to fluctuations in cell population density. Processes regulated in this way include symbiosis, virulence, competence, conjugation, production of antibiotics, motility, sporulation, and formation of biofilms. We determined the 3-dimensional solution structure of a complex composed of the N-terminal 171 residues of the quorum-sensing protein SdiA of Escherichia coli and an autoinducer molecule, N-octanoyl-L-homoserine lactone (HSL). The SdiA-HSL system shows the “folding switch” behavior associated with quorum-sensing factors produced by other bacterial species. In the presence of HSL, SdiA is stable and folded and can be produced in good yields from an E coli expression system. In the absence of the autoinducer, the SdiA is expressed into inclusion bodies. Samples of the SdiAHSL complex can be denatured but cannot be refolded in aqueous buffers. The solution structure of the complex provides a likely explanation for this behavior. The autoinducer molecule is tightly bound in a deep pocket in the hydrophobic core and is bounded by specific hydrogen bonds to the side chains of conserved residues. The autoinducer thus forms an integral part of the hydrophobic core of the folded SdiA. CHAPERONE–COCHAPERONE–CLIENT PROTEIN INTERACTIONS F i g . 1 . Schematic representation of the amide proton occupancies in the kinetic intermediate state formed in the burst phase of apomyoglobin folding (solid ribbons), mapped onto the structure of fully folded myoglobin. Areas of the protein that are not folded until later stages are shown as dotted lines. we have reduced the dead time of the kinetic refolding experiments and have shown that a compact helical intermediate is formed within 400 microseconds after initiation of apomyoglobin refolding. The new measurements reveal that folding occurs by a hierarchical process: the A, G, and H helices fold rapidly to form a compact core, and the other helices fold more slowly by docking onto the preformed core. FOLDING-UNFOLDING TRANSITIONS IN CELLULAR M E TA B O L I S M Many species of bacteria sense and respond to their own population density by an intricate autoregulatory Understanding the role of unfolded states in cellular processes will require an understanding of the structural basis of their interactions, but unfolded proteins are impossible to characterize structurally by x-ray crystallography, and spectroscopic methods of all kinds are limited. Unfolded proteins must be explored under conditions that approximate the proteins’ physiologic milieu: in solution, at physiologic pHs and salt concentrations, and in the presence of specific cofactors. Structural insights will be obtained not only from the delineation of 3-dimensional structures but also from the description of conformational ensembles and of the motions of polypeptide chains under various conditions. To gain new insights into the structural basis for the ability of unfolded and partly folded proteins to function in living systems, we study the interactions of “client” proteins and cochaperones with a well-known eukaryotic chaperone, Hsp90. Some of the protein components are much larger than have traditionally been 232 MOLECULAR BIOLOGY 2008 studied by using solution NMR. However, we have designed a set of experiments that will allow us to draw valid conclusions about the extent and role of disorder in Hsp90 interactions. In particular, we will apply techniques recently developed in our laboratory for analyzing hydrogen-deuterium exchange from unstable partially folded proteins by trapping the 2H-labeled species in the aprotic solvent dimethyl sulfoxide. This powerful new technique will be used to probe the structure, stability, and interactions of client proteins and cochaperones with Hsp90. PUBLICATIONS Felitsky, D.J., Lietzow, M.A., Dyson, H.J., Wright, P.E. Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc. Natl. Acad. Sci. U. S. A. 105:6278, 2008. Nishimura, C., Dyson, H.J., Wright, P.E. The kinetic and equilibrium molten globule intermediates of apoleghemoglobin differ in structure. J. Mol. Biol. 378:715, 2008. Schwarzinger, S., Mohana-Borges, R., Kroon, G.J.A., Dyson, H.J., Wright, P.E. Structural characterization of partially folded intermediates of apomyoglobin H64F. Protein Sci. 17:313, 2008. Nuclear Magnetic Resonance Studies of the Structure and Dynamics of Enzymes H.J. Dyson, P.E. Wright, S.H. Bae, G. Bhabha, D. Boehr, C. Cervantes,* G. Kroon, M. Martinez-Yamout, S.C. Sue, L.M. Tuttle, L.L. Tennant, C.L. Brooks, S.J. Benkovic,** A. Holmgren,*** E.A. Komives* * University of California, San Diego, California ** Pennsylvania State University, University Park, Pennsylvania *** Karolinska Institutet, Stockholm, Sweden e use site-specific information on structure and dynamics, obtained from nuclear magnetic resonance (NMR), to further the understanding of protein function. We focus on the mechanism of enzymes and the relationship between dynamics and function in a number of medically important systems. W DYNAMICS IN ENZYME ACTION Dynamic processes are implicit in the catalytic function of all enzymes. We use state-of-the-art NMR methods to elucidate the dynamic properties of several enzymes. New methods have been developed for analysis of NMR relaxation data for proteins that tumble anisotropically and for analysis of slow timescale motions. Dihydrofolate reductase (DHFR) plays a central role in folate metabolism and is the target enzyme for a number of antibacterial and anticancer agents. 15N THE SCRIPPS RESEARCH INSTITUTE relaxation experiments on DHFR from Escherichia coli have revealed a rich diversity of backbone dynamical features for a broad range of timescales (picoseconds to milliseconds). A major focus is the characterization of all intermediates in the DHFR reaction cycle. We have identified functionally important motions in loops that control access to the active site of the reductase. These motions differ in amplitude and timescale depending on the presence of substrate and/or cofactor in the active site. In addition, measurements of the population distribution of aliphatic side-chain rotamers provide evidence for coupled motion of active-site side chains that could enhance the catalytic process. Most recently, we used relaxation dispersion measurements to obtain direct information on microsecond to millisecond timescale motions in DHFR, allowing us to characterize the structures of excited states involved in some of these catalysis-relevant processes. Each intermediate in the catalytic cycle samples low-lying excited states whose conformations resemble the ground-state structures of the preceding and following intermediates. Fluctuations between these states occur on a timescale that is directly relevant to the structural transitions involved in progression through the catalytic cycle. Substrate and cofactor exchange occurs through these excited substates. The maximum hydride transfer and steady-state turnover rates are governed by the dynamics of transitions between the ground and excited states of the intermediates. The modulation of the energy landscape by the bound ligands funnels the enzyme through its reaction cycle along a preferred kinetic path. DHFR is also the test system for a series of experiments to address the question, If all of the chemistry goes on at the active site, what is the purpose of the rest of the enzyme? We are using chimeric mutants, synthesized by our collaborator S.J. Benkovic, Pennsylvania State University, by using a library approach. The purpose of these experiments is to test the hypothesis that local variations in amino acid sequence, 3-dimensional structure, and polypeptide chain dynamics strongly influence the local interactions that mediate enzyme catalysis and may constitute the essential circumstance that allows enzymes to achieve high turnover rates as well as exquisite specificity in their reactions. A combination of NMR structure and dynamics measurements, single-molecule fluorescence measurements, and analysis of the catalytic steps in these mutant proteins provides new insights into the role of the protein in enzyme catalysis. MOLECULAR BIOLOGY 2008 S T R U C T U R E A N D D Y N A M I C S O F P R I O N VA R I A N T S Onset of prion diseases is caused by conversion of the cellular prion protein PrPC into an abnormally folded isoform, PrPSc, that has the same primary structure as PrPC but a totally different 3-dimensional conformation. The abnormally folded (“scrapie”) form of the protein is associated with several diseases, including scrapie in sheep, bovine spongiform encephalopathy (mad cow disease), and human Creutzfelt-Jakob disease and other inherited prion diseases. We are gathering information on the mechanism of PrPSc formation that can be obtained from structural and dynamic studies of mutant prion proteins corresponding to inherited prion diseases. Individuals carrying familial mutations such as P102L (P101L in our study) are more susceptible to prion disease. On the other hand, sheep or humans carrying Q167R and/or Q218K mutations are resistant to scrapie and Creutzfelt-Jakob disease, respectively. We are using the protease-resistant cores of wild-type and mutant mouse prion proteins to study the structural and dynamic basis of PrPC-to-PrPSc conversion in inherited prion diseases. The core is sufficient to transmit infectivity. DYNAMICS AND THE FUNCTION OF IκBα It is becoming increasingly clear that the function of many systems in living cells depends not only on the structures of the components but also on the structures’ flexibility. Numerous examples exist in which components of an important biological interaction are unstructured or partly structured. In addition, even those interacting molecules that can be classified as “folded” have areas of mobility. Often, these areas are located precisely in the active site of an enzyme or in the binding site of an interacting molecule. A central molecular interaction in cellular control is the interaction between the nuclear transcription factor NF-κB and its inhibitor IκBα. IκBα consists of a series of ankyrin repeats, which appear to have differential mobility. Using hydrogen-deuterium exchange and mass spectrometry, our collaborator, E.A. Komives, University of California, San Diego, found that the second, third, and fourth ankyrin repeats of IκBα are well folded, whereas the fifth and sixth repeats, apparently with exactly the same structure, are highly dynamic. These observations prompt a number of questions: Are the motions inferred from the hydrogen-deuterium mass spectrometry experiments also reflected in the backbone and side-chain dynamics of the protein, as measured by NMR relaxation? Are the motions still present in the IκBα–NF-κB complex? Are they necessary for complex formation, so that if they are damped out, for THE SCRIPPS RESEARCH INSTITUTE 233 example by site-directed mutagenesis at appropriate positions, is the formation of the complex disfavored? To answer these questions, we are doing a series of NMR experiments on IκBα and its complexes with NF-κB. PUBLICATIONS Boehr, D.D., Dyson, H.J., Wright, P.E. Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry 47:9227, 2008. Sue, S.C., Cervantes, C., Komives, E.A., Dyson, H.J. Transfer of flexibility between ankyrin repeats in IκBα upon formation of the NF-κB complex. J. Mol. Biol. 380:917, 2008. Chaperonin-Mediated Protein Folding A.L. Horwich, E. Chapman, S.M. Johnson, E. Koculi uring the past year, we have continued to investigate the mechanism of action of the large molecular machines known as chaperonins that assist in protein folding in the cell. These megadalton-sized double-ring assemblies are found in the cytosol of all organisms, where they assist in the folding of many newly translated proteins, effectively carrying out the final step of information transfer. Chaperonins are also present in the matrix of mitochondria and the stroma of chloroplasts, where they assist in the folding of proteins imported from the cytosol. In all contexts, chaperonins assist folding by 2 sequential actions involving the central cavity of their rings: (1) binding nonnative proteins in the cavity of an open ring, forestalling aggregation that could occur if the proteins were free in solution, and (2) folding, triggered by association of ATP with the substrate-bound ring, producing release of the substrate protein into the now-encapsulated cavity, where the substrate folds in isolation. D F O L D I N G T R A J E C T O R Y O F A P R O T E I N S U B S T R AT E I N S I D E T H E E N C A P S U L AT E D C AV I T Y O F B A C T E R I A L GroEL-GroES VS FOLDING FREE IN SOLUTION In collaboration with K. Wüthrich, Department of Molecular Biology, we used hydrogen-deuterium exchange and nuclear magnetic resonance (NMR) analysis to compare the folding trajectory of the substrate protein human dihydrofolate reductase (DHFR) inside the GroES-encapsulated chaperonin cavity of a single-ring version of GroEL vs folding free in solution (Fig. 1). For both situations, folding was started in aqueous buffer, and at various times an excess of deuterium oxide was added and the folding reaction was allowed to continue to completion (the half-time of the reaction is about 2 min- 234 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE cavity it was 80%–90%. When DHFR was free in solution, more than half of the protein was lost to aggregation. Thus, folding to the native state in the chaperonin cavity occurs by the same route as in solution, but multimolecular aggregation is forestalled in the case of the chaperonin reaction by the “solitary confinement” of the folding substrate protein. The results of additional studies with larger substrate proteins under conditions in which the substrates can reach native form either free in solution or inside the chaperonin cavity (so-called permissive conditions) supported our earlier findings; that is, aggregation occurred in solution but not in the chaperonin reaction. Thus, the major action of the encapsulated chaperonin cavity appears to be provision of a site for confining the folding protein in a hydrophilic chamber with polar “nonstick” walls, where the protein can fold in isolation, without the possibility of aggregation. S T R U C T U R A L S TAT E S O F A G R O U P 2 C H A P E R O N I N F i g . 1 . Experimental protocol for using amide proton (H)-deuterium (D) exchange monitored by NMR to analyze the refolding trajectory of human DHFR while the reductase is inside the chaperonin cavity (left) or free in solution (right). T is the refolding time in water before addition of deuterium oxide (DO2). Methotrexate (Mtx) was added to both reactions to stabilize the native state of DHFR. Reprinted from Horst et al. Proc. Natl. Acad. Sci. U. S. A. 104:20788, 2007. Copyright 2007 National Academy of Sciences U.S.A. utes). The refolded native DHFR was then recovered and analyzed by using NMR. A total of 51 amide proton positions in native DHFR are well protected and served as “probes.” Any amide proton involved in a hydrogen bond at the time deuterium oxide is added would be expected to be relatively protected from hydrogen-deuterium exchange and should be visible in the native protein on NMR analysis, whereas amide protons in regions lacking structure at the time the deuterium oxide was added could be exchanged to deuterons and would be invisible. We found that the acquisition of protection at the probe positions followed single exponential kinetics, and the patterns of protection and rates of acquisition were virtually identical for DHFR folding in the cavity or free in solution. That is, the folding trajectories were identical. The extent of recovery of native DHFR free in solution, however, was about 40%, whereas in the Rather than use a detachable cochaperonin (e.g., bacterial GroES), the chaperonins in the cytosol of eukaryotes and in archaea have a built-in “lid” structure composed of α-helical apical domain protrusions that come together to enclose the cavity. The nature of subunit movements during the ATP-driven reaction cycle of these machines is not well understood. In collaboration with B. Carragher and C. Potter, Department of Cell Biology, we used a homo-oligomeric archaeal chaperonin from Methanococcus maripaludis, overproduced and purified from Escherichia coli, to address the nature of such movements. The apo form of the chaperonin had poorly resolving wide-open apical domains, essentially pointing straight upward from the stable equatorial “base” into the bulk solution, without side-by-side contacts. The different attitudes of these domains suggested flexible positioning in this apo state. We think that this state is the substrate-binding proficient state of the machine. The complex composed of the chaperonin, ADP, and aluminum fluoride populated 3 states: an open state resembling the apo form, a state with one ring partially closed and the other wide open, and a fully closed state like that of a crystal structure of the archaeal thermosome (Fig. 2). The apo form now had regular apical density with the helical protrusions pointing straight upward. The partially closed state presented clockwise-rotated apical domains on 1 ring that could partially enclose a substrate of considerable size, up to about 80 kD. This state may comprise a folding-active state for larger proteins. MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 235 cules that can be programmed by chemical synthesis to recognize specific DNA sequences, and (2) histone deacetylase (HDAC) inhibitors, compounds that alter the postsynthetic modification states of chromosomal proteins and thereby modulate gene expression. Our goals are to develop polyamides as therapeutics for human cancer and HDAC inhibitors as therapeutics for neurodegenerative diseases and cystic fibrosis. B L O C K I N G C A N C E R C E L L P R O L I F E R AT I O N W I T H A P O LYA M I D E - C H L O R A M B U C I L C O N J U G AT E F i g . 2 . Three-dimensional image reconstructions of the 3 ADP·AIFx states of the M maripaludis chaperonin. Left panels, Open state; center panels, partially closed state (top ring; bottom ring is open); right panels, closed state, with fitted domains from the homologous type 2 thermosome chaperonin. Top panels, End views, viewing down the 8-fold symmetry axis; bottom panels, side views. The fully closed state included only approximately half as much volume, produced by inward rotation of subunits. Whether the fully closed state remains an obligate step in the reaction cycle remains to be addressed. It may be a folding chamber dedicated solely to smaller protein substrates. PUBLICATIONS Clare, D.K., Stagg, S., Quispe, J., Farr, G.W., Horwich, A.L., Saibil, H.R. Multiple states of a nucleotide-bound group 2 chaperonin. Structure 16:528, 2008. Horst, R., Fenton, W.A., Englander, S.W., Wüthrich, K., Horwich, A.L. Folding trajectories of human dihydrofolate reductase inside the GroEL-GroES chaperonin cavity and free in solution. Proc. Natl. Acad. Sci. U. S. A. 104:20788, 2007. Chemical Regulation of Gene Expression J.M. Gottesfeld, R. Burnett, J. Chou, D. Herman, S. Ku, C. Xu, E. Soragni, E.A. Thomas, D. Hutt,* W.E. Balch,* S. Tsai,** M. Farkas,** P.B. Dervan,** S.L. Perlman,*** G. Coppola,*** D. Geschwind,*** M. Rai,**** M. Pandolfo,**** T. O’Hare,***** B. Druker***** * Department of Cell Biology, Scripps Research ** California Institute of Technology, Pasadena, California *** University of California, Los Angeles, California **** Universite Libre de Bruxelles-Hospital Erasme, Brussels, Belgium ***** Oregon Health and Sciences University, Portland, Oregon W e focus on 2 classes of small molecules that can alter gene expression in human cells: (1) pyrrole-imidazole polyamides, a class of mole- DNA alkylators, such as the nitrogen mustard chlorambucil, are among the most common agents used to treat cancer in humans and act by damaging DNA. Because chlorambucil alkylates DNA at all available guanine residues in cellular DNA, coupling chlorambucil to a sequence-specific polyamide decreases the number of sites in the genome that are damaged and reduces unwanted side effects while retaining the ability of the compound to kill cancer cells. We recently found that a polyamide-chlorambucil conjugate called 1R-Chl blocks proliferation of multiple cancer cell lines in culture by causing the cells to arrest at the G2/M stage of the cell cycle. The compound blocks tumor growth in immunocompromised mice, including cells derived from colon, prostate, chronic myelogenous leukemia, and lung cancers, and no apparent toxic effects occur at doses required for a therapeutic effect. Using microarray analysis, we discovered that the gene target of 1R-Chl is the gene for histone H4c, a member of the gene family that encodes a critical component of cellular chromatin and a gene that is highly expressed in a wide range of cancer cells. Reduction in histone H4 protein by polyamide treatment was confirmed in cells treated with 1R-Chl, which caused chromatin decondensation. Small interfering RNAs to H4c mRNA also had this effect, providing target validation for the effects of 1R-Chl. Recent studies indicated that 1R-Chl causes growth arrest of chronic myelogenous leukemia cells harboring both wild-type BCR-ABL, the gene translocation that causes the leukemia, and 3 tyrosine kinase inhibitor–resistant strains, suggesting that 1R-Chl could be used in combination therapy for chronic myelogenous leukemia or in cases of relapsed disease. We are using additional animal and cellular models for human cancer and proliferative diseases to determine the therapeutic potential of 1R-Chl. G E N E R E G U L AT I O N I N N E U R O L O G I C D I S E A S E S The neurodegenerative disease Friedreich ataxia is caused by gene silencing through expansion of GAA•TTC 236 MOLECULAR BIOLOGY 2008 triplet repeats in the first intron of a nuclear gene that encodes the essential mitochondrial protein frataxin. We used antibodies to the various modification states of the core histones and chromatin immunoprecipitation methods to examine the chromatin structure of the gene frataxin in normal cells and in cells derived from patients with Friedreich ataxia. We found that gene silencing at expanded frataxin alleles is accompanied by hypoacetylation of histones H3 and H4 and methylation of histone H3, consistent with a heterochromatin-mediated repression mechanism. These findings suggest that HDAC inhibitors, compounds that reverse heterochromatin, might activate frataxin. On the basis of the structure of a commercial HDAC inhibitor that partially reverses frataxin silencing, we synthesized and assayed a series of derivatives of the inhibitor and identified novel pimelic diphenylamide HDAC inhibitors that reverse frataxin silencing in primary lymphocytes from patients with Friedreich ataxia. One molecule, 4b, acts directly on the histones associated with frataxin, increasing acetylation at particular lysine residues on histones H3 and H4. A chemical proteomics approach revealed that HDAC3 is the likely cellular target of our inhibitors. Of note, the HDAC inhibitors cross the blood-brain barrier and increase levels of frataxin mRNA in the brain and other organs in a mouse model of the human disease. Genome-wide microarray studies indicated that our compounds partially correct the transcription pattern of genes affected by Friedreich ataxia in the brains of these mice and in lymphocytes from patients with Friedreich ataxia to the transcription pattern of healthy animals or individuals. The pharmacokinetic and toxic properties of the compounds are under investigation. HUNTINGTON’S DISEASE Transcriptional dysregulation is a core pathologic feature of Huntington’s disease, one of several CAG triplet repeat disorders characterized by movement deficits and cognitive dysfunction. Recent studies have shown therapeutic effects of commercially available HDAC inhibitors in several models of Huntington’s disease; however, the therapeutic value of the compounds is limited by their toxic effects. Chronic oral administration of the HDAC inhibitor 4b, beginning after the onset of motor deficits in R6/2 300Q transgenic mice, which have about 300 CAG repeats, significantly improved motor performance, overall appearance, and body weight. These effects were associated with significant attenuation of gross decline in brain size and striatal atrophy. THE SCRIPPS RESEARCH INSTITUTE Microarray studies revealed that 4b treatment ameliorated, in part, deficits in gene expression caused by the presence of the mutant huntingtin protein in the striatum, cortex, and cerebellum of the transgenic mice. For selected genes, we found that 4b treatment reversed histone H3 hypoacetylation that occurs in the presence of mutant huntingtin, in association with correction of the expression levels of various mRNAs. These findings suggest that 4b, and possibly related HDAC inhibitors, may be clinically beneficial for patients with Huntington’s disease. CYSTIC FIBROSIS Cystic fibrosis, a disease that affects the lungs and organs of the digestive tract, is caused by the loss of the chloride channel, cystic fibrosis transmembrane conductance regulator (CFTR), on the cell surface. The resulting loss of ionic homeostasis leads to a thickening of the airway surface liquid, which clogs the lungs and the pancreatic duct. Although numerous mutations have been identified in CFTR, the gene for CFTR, most patients express a form of the protein that is misfolded and is therefore retained in the endoplasmic reticulum. We found that inhibition of HDAC7, either with HDAC inhibitors or short interfering RNA, resulted in rescue of this trafficking mutant and restoration of its surface chloride channel activity. These data suggest that HDAC7 is an attractive target for the development of small-molecule correctors of CFTR-trafficking defects. The clinical use of general HDAC inhibitors as antitumor agents suggests that HDAC7-specific inhibitors would have beneficial effects greater than any adverse side effects and could be useful in the treatment of cystic fibrosis. PUBLICATIONS Chou, C.J., Farkas, M.E., Tsai, S.M., Alvarez, D., Dervan, P.B., Gottesfeld, J.M. Small molecules targeting histone H4 as potential therapeutics for chronic myelogenous leukemia. Mol. Cancer Ther. 7:769, 2008. Gottesfeld J.M. Small molecules affecting transcription in Friedreich ataxia. Pharmacol. Ther. 116:236, 2007. Rai, M., Soragni, E., Jenssen, K., Burnett, R., Herman, D., Coppola, G., Geschwind D.H., Gottesfeld, J.M., Pandolfo, M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE 3: e1958, 2008. Nucleic Acid Dynamics D.P. Millar, J. Gill, G. Pljevaljcić, R. Robertson, J. Wang, E.J.C. Van der Schans T he focus of our research is the assembly and conformational dynamics of nucleic acid–based macromolecular machines and assemblies. We MOLECULAR BIOLOGY 2008 use single-molecule fluorescence methods to investigate a range of systems, including ribonucleoprotein complexes and DNA polymerases. Our studies reveal the dynamic structural rearrangements that occur during the assembly and function of these complex macromolecular assemblies. R I B O N U C L E O P R O T E I N A S S E M B LY The Rev protein from HIV type 1 is a key regulatory protein that controls the transition from early to late patterns of viral gene expression. Rev binds to a highly structured region within the viral mRNA, known as the Rev response element (RRE), where it forms an oligomeric ribonucleoprotein complex. The formation of this complex inhibits splicing and facilitates export of the viral RNA from the nucleus to the cytoplasm. Because of its critical role in the viral life cycle, the Rev-RRE complex provides a novel target for development of therapeutic drugs. To dissect the mechanism of assembly of ribonucleoproteins, we use single-molecule fluorescence imaging methods to monitor the formation of oligomeric complexes of Rev on individual RRE molecules immobilized on a solid surface. We found that a single Rev monomer binds initially to a high-affinity site in stem loop IIB of the RRE and that assembly subsequently proceeds by the stepwise addition of additional Rev monomers. The elementary rate constants for each step of assembly were also obtained from the single-molecule data. We also use single-pair Förster or fluorescence resonance energy transfer (FRET) to probe changes in the conformation of the RRE during the assembly process. We are using the results of these mechanistic studies to develop novel fluorescence-based methods for high-throughput screening of libraries of chemical compounds. The new screening tools are being used to identify small molecules that block initial binding of Rev to the RRE or prevent the subsequent Rev-Rev oligomerization. Another example of ribonucleoprotein assembly under study is the signal recognition particle. This particle is a fascinating molecular machine responsible for the cotranslational targeting of secretory or membrane proteins to the endoplasmic reticulum. This large complex, composed of a 300-nucleotide RNA and 6 proteins, interacts with both the ribosome, during translational arrest, and a membrane-bound receptor. We are developing novel spectroscopic techniques (based on multicolor FRET) to dissect the assembly pathway of the particle, focusing on the temporal order of protein-binding events and the associated RNA-folding transitions. THE SCRIPPS RESEARCH INSTITUTE 237 In parallel, we are developing methods to label large RNA molecules with donor and acceptor probes for FRET measurements. These reagents and methods are being used to monitor assembly reactions of signal recognition particle proteins on individual immobilized RNA molecules by means of single-molecule FRET microscopy. The interplay between protein-binding and RNA-folding events revealed in our studies is providing general insights into the mechanism of assembly of ribonucleoproteins. D N A P O LY M E R A S E S DNA polymerases are remarkable for their ability to synthesize DNA at rates of several hundred base pairs per second while maintaining an extremely low frequency of errors. To elucidate the origin of polymerase fidelity, we are using single-molecule fluorescence methods to examine the dynamic interactions that occur between a DNA polymerase and its DNA and nucleotide substrates. The FRET method is being used to observe conformational transitions of the enzyme-DNA complex that occur during selection and incorporation of an incoming nucleotide substrate. Similar methods are being used to monitor the proofreading step after synthesis. Our results reveal that binding of a correct nucleotide substrate induces a slow conformational change within the polymerase, causing the “fingers” domain to close over the DNA primer terminus and incoming nucleotide. Our studies are also providing new insights into the mechanisms used to transfer a DNA substrate from the synthesis site to the exonuclease active site during proofreading. The advantage of single-molecule observations is that they eliminate the need to synchronize a population of molecules, allowing these dynamic processes to be observed directly. PUBLICATIONS Bailey, M.F., Van der Schans, E.J.C., Millar, D.P. Dimerization of the Klenow fragment of Escherichia coli DNA polymerase I is linked to its mode of DNA binding. Biochemistry 46:8085, 2007. Debler, E.W., Kaufmann, G.F., Meijler, M.M., Heine, A., Mee, J.M., Pljevaljcić, G., Di Bilio, A.J., Schultz, P.G., Millar, D.P., Janda, K.D., Wilson, I.A., Gray, H.B., Lerner, R.A. Deeply inverted electron-hole recombination in an antibody-stilbene complex. Science 319:1232, 2008. Pljevaljcić, G., Millar, D. Single-molecule fluorescence methods for the analysis of RNA folding and ribonucleoprotein assembly. Methods Enzymol., in press. Stengel, G., Gill, J.P., Sandin, P., Wilhelmsson, M., Albinsson, B., Nordén, B., Millar, D. Conformational dynamics of DNA polymerase probed with a novel fluorescent DNA base analogue. Biochemistry 46:12289, 2007. 238 MOLECULAR BIOLOGY 2008 Single-Molecule Biophysics: Folding, Assembly, and Function A.A. Deniz, S.Y. Berezhna, A.C.M. Ferreon, Y. Gambin, E. Lemke, H.-W. Liu, C. Moran, S. Mukhopadhyay e develop and use state-of-the-art single-molecule fluorescence methods and high-sensitivity ensemble methods to address key biological questions about the structure, dynamics, and function of biomolecules. These methods offer key advantages over traditional measurements, allowing us to directly observe the behavior of individual subpopulations in mixtures of molecules and to measure the kinetics of structural transitions during stochastic processes under equilibrium conditions. A major goal is to apply single-molecule methods to studies of protein folding and aggregation. For example, partially folded or misfolded protein structures are thought to play important cellular roles, and these states can be studied by using single-molecule methods. In one instance, we are studying the structural properties and aggregation of α-synuclein, a protein implicated in the pathogenesis of Parkinson’s disease and other neurodegenerative diseases. We used single-molecule fluorescence to study the folding induced in this protein by binding of lipid and membrane mimics. Single-molecule fluorescence resonance energy transfer (FRET) experiments with sodium dodecyl sulfate, which is used to denature proteins, revealed an extraordinarily complex binding-induced folding landscape composed of several folded states and transitions. We found that not only membranes but also small molecules can trigger these transitions, significant for the biological function and disease relevance of this protein. In collaboration with S.L. Lindquist, Whitehead Institute, Cambridge, Massachusetts, we are examining the dynamic interplay between folding and aggregation of the prion domain (NM) of Sup35, a yeast translational termination factor whose activity is modulated by conversion to a prion form. Using a combination of single-molecule methods, we previously showed that monomeric native NM populates a collapsed and rapidly fluctuating ensemble of disordered structures. Recently, using a combination of fluorescence intensity and polarization measurements during NM aggregation, we observed rapid formation of oligomeric species followed by a reaction phase in which conformation and size change in concert; both occur before the fibril growth phase detected in W THE SCRIPPS RESEARCH INSTITUTE standard aggregation experiments. Our observations shed new light on how amyloid conformation is sequestered in the context of oligomers rather than monomers, behavior that may be key in aggregation and biological function of Sup35 and other amyloidogenic proteins. The detailed structural and dynamical mapping of protein binding-folding and aggregation landscapes provided by single-molecule measurements will be applicable to other natively unfolded and amyloid-forming proteins. Thus, in collaboration with J.W. Kelly, Department of Chemistry, we are exploring the conformational and aggregation properties of the amyloid β-peptide involved in Alzheimer’s disease to better understand the role of the peptide in biology and disease. Important events on the folding landscapes of proteins and other biomolecules occur on millisecond or faster timescales. To facilitate understanding of such fast processes, in collaboration with A. Groisman, University of California, San Diego, we are developing rapid microfluidic mixing methods. Experiments with singlemolecule resolution are providing us with new information about how different α-synuclein states are connected on the energy landscape. Additionally, we are continuing to develop and use multicolor single-molecule FRET methods to study assembly mechanisms of larger multicomponent molecular machines. For example, 3-color FRET experiments done in collaboration with J.R. Williamson, Department of Molecular Biology, are providing novel insights into the coupling between folding of 2 RNA junctions in the 30S ribosomal subunit. Finally, we are using multicolor fluorescence imaging to study the cellular pathways of RNA interference. In collaboration with P.G. Schultz, Department of Chemistry, we probed the time-dependent cellular locations of short interfering RNA and a key RNA interference protein, Ago2, and found correlations with sites of viral replication. We have started ensemble and single-molecule FRET measurements to better understand the formation of RNA-induced silencing complexes in vitro and in living cells. PUBLICATIONS Deniz, A.A., Mukhopadhyay, S., Lemke, E.L. Single-molecule biophysics: at the interface of biology, physics and chemistry. J. R. Soc. Interface 5:15, 2008. Mukhopadhyay, S., Deniz, A.A. Fluorescence from diffusing single molecules illuminates biomolecular structure and dynamics. J. Fluoresc. 17:775, 2007. Wang, H., Duennwald, M.L., Roberts, B.E., Rozeboom, L.M., Zhang, Y.L., Steele, A.D., Krishnan, R., Su, L.J., Griffin, D., Mukhopadhyay, S., Hennessy, E.J., Weigele, P., Blanchard, B.J., King, J., Deniz, A.A., Buchwald, S.L., Ingram, V.M., Lindquist, S., Shorter, J. Direct and selective elimination of specific prions and amyloids by 4,5dianilinophthalimide and analogs. Proc. Natl. Acad. Sci. U. S. A. 105:7159, 2008. MOLECULAR BIOLOGY Computer Modeling of Proteins and Nucleic Acids D.A. Case, Y. Bomble, J Lätzer, V. Pelmenschikov, T. Steinbrecher, S. Tang, V. Wong omputer simulations offer an exciting approach to the study of many aspects of biochemical interactions. We focus primarily on molecular dynamics simulations (in which Newton’s equations of motions are solved numerically) to model the solution behavior of biomacromolecules. Recent applications include detailed analyses of electrostatic interactions in short peptides (folded and unfolded), proteins, and oligonucleotides in solution. In addition, molecular dynamics methods are useful in refining solution structures of proteins by using constraints derived from nuclear magnetic resonance (NMR) spectroscopy, and we are continuing to explore new methods in this area. Our developments are incorporated into the Amber molecular modeling package, designed for large-scale biomolecular simulations, and into other software, including Nucleic Acid Builder, for developing 3-dimensional models of unusual nucleic acid structures; SHIFTS, for analyzing chemical shifts in proteins and nucleic acids; RNAmotif, for finding structural motifs in genomic sequence databases; and DOCK, for placing inhibitors into enzyme active sites. C NMR AND THE STRUCTURE AND DYNAMICS OF PROTEINS AND NUCLEIC ACIDS Our overall goal is to extract the maximum amount of information about biomolecular structure and dynamics from NMR experiments. To this end, we are studying the use of direct refinement methods for determining biomolecular structures in solution on the basis of NMR data, going beyond distance constraints to generate closer connections between calculated and observed spectra. We are also using quantum chemistry to study chemical shifts and spin-spin coupling constants. Other types of data, such as chemical shift anisotropies, direct dipolar couplings in partially oriented samples, and analysis of cross-correlated relaxation, are also being used to guide structure refinement. In recent structural studies, we focused on the binding of small ligands to DNA and on models for chemically damaged DNA. In studies on dynamics, we have concentrated on the effects of overall rotational diffusion on NMR relaxation. We have looked at the extent to which molecular dynamics simulations show the 2008 THE SCRIPPS RESEARCH INSTITUTE 239 expected rotational diffusion behavior, and we have developed novel models to determine relaxation parameters in the presence of time-varying rotational diffusion coefficients. NUCLEIC ACID MODELING Another project centers on the development of novel computer methods to construct models of “unusual” nucleic acids that go beyond traditional helical motifs. We are using these methods to study circular DNA, small RNA fragments, and nucleosome core particles. We continue to develop efficient computer implementations of continuum solvent methods to allow simplified simulations that do not require a detailed description of the solvent (water) molecules; this approach also provides a useful way to study salt effects. Recent efforts have made second derivatives of these energies available, so that normal mode analyses of nucleic acids with dozens to hundreds of nucleotides can be analyzed and the predictions compared with those of simpler, elastic continuum models. These efforts provide a new avenue for developing and testing lowresolution models that can be used for large molecular assemblies. Currently, we are applying these methods to nucleosome core particles and to bending and twisting flexibility of linear and circular duplex DNA. D Y N A M I C S A N D E N E R G E T I C S O F N AT I V E A N D N O N N AT I V E S TAT E S O F P R O T E I N S Analysis methods similar to those described for nucleic acids are also being used to estimate electrostatic and thermodynamic properties of proteins. A key feature is the development of computational methods that can be used to model pH and salt dependence of complex conformational transitions such as unfolding events. A second aspect of this research is a detailed interpretation of NMR results for proteins through molecular dynamics simulations and the construction of models for molecular motion and disorder. All of these modeling activities are based on molecular mechanics force fields, which provide estimates of energies as a function of conformation. We continue to work on improvements in force fields; recently, we focused on adding aspects of electronic polarizability, going beyond the usual fixed-charge models, and on methods for handling arbitrary organic molecules that might be considered potential inhibitors in drug discovery efforts. Overall, the new models should provide a better picture of the noncovalent interactions between peptide groups and the groups’ surroundings, leading ultimately to more faithful simulations. 240 MOLECULAR BIOLOGY 2008 PUBLICATIONS Bomble, Y., Case, D.A. Multiscale modeling of nucleic acids: insights into DNA flexibility. Biopolymers 89:722, 2008. THE SCRIPPS RESEARCH INSTITUTE Chen, J., Dupradeau, F.-Y., Case, D.A., Turner, C.J., Stubbe, J. DNA oligonucleotides with A, T, G or C opposite an abasic site: structure and dynamics. Nucleic Acid Res. 36:253, 2008. Quantum Chemical Analysis for Redox-Active Metalloenzymes and for Photochemistry Dupradeau, F.Y., Pissard, S., Coulhon, M.P., Cadet, E., Foulon, K., Fourcade, C., Goossens, M., Case, D.A., Rochette, J. An unusual case of hemochromatosis due to a new compound heterozygosity in HFE (p.[Gly43Asp;His63Asp]+[Cys282Tyr]): structural implications with respect to binding with transferrin receptor 1. Hum. Mutat. 29:206, 2008. L. Noodleman, D.A. Case, W.-G. Han, V. Pelmenschikov, J.A. Fee, L. Hunsicker-Wang,* T. Liu,** M. Ullmann,*** D. Bashford**** * Trinity University, San Antonio, Texas Graves, A.P., Shivakumar, D.M., Boyce, S.E., Jacobson, M.P., Case, D.A., Shoichet, B. Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J. Mol. Biol. 377:914, 2008. Guo, Y., Wang, H., Xiao, Y., Vogt, S., Thauer, R.K., Shima, S., Volkers, P.I., Rauchfuss, T.B., Pelmenschikov, V., Case, D.A., Alp, E.E., Sturhahn, W., Yoda, Y., Cramer, S.P. Characterization of the Fe site in iron-sulfur cluster-free hydrogenase (Hmd) and of a model compound via nuclear resonance vibrational spectroscopy (NRVS). Inorg. Chem. 47:3969, 2008. Hou, T., Zhang, W., Case, D.A., Wang, W. Characterization of domain-peptide interaction interface: a case study on the amphiphysin-1 SH3 domain. J. Mol. Biol. 376:1201, 2008. Lukoyanov, D., Pelmenschikov, V., Maeser, N., Laryukhin, M., Yang, T.C., Noodleman, L., Dean, D.R., Case, D.A., Seefeldt, L.C., Hoffman, B.M. Testing if the interstitial atom, X, of the nitrogenase molybdenum-iron cofactor is N or C: ENDOR, ESEEM, and DFT studies of the S = 3/2 resting state in multiple environments. Inorg. Chem. 46:11437, 2007. Payne, R.J., Ficht, S., Tang, S., Brik, A., Yang, Y.-Y., Case. D.A., Wong, C.H. Extended sugar-assisted glycopeptide ligations: development, scope, and application. J. Am. Chem. Soc. 129:13527, 2007. Pelmenschikov, V., Case, D.A., Noodleman, L. Ligand-bound S = 1/2 FeMo-cofactor of nitrogenase: hyperfine interaction analysis and implication for the central ligand X identity. Inorg. Chem. 47:6162, 2008. Seabra, G.M., Walker, R.C., Elstner, M., Case, D.A., Roitberg, A.E. Implementation of the SCC-DFTB method for hybrid QM/MM simulations within the Amber molecular dynamics package. J. Phys. Chem. A 111:5655, 2007. Steinbrecher, T., Mobley, D.L., Case, D.A. Nonlinear scaling schemes for LennardJones interactions in free energy calculation. J. Chem. Phys. 127:214108, 2007. Tang, S., Case, D.A. Vibrational averaging of chemical shifts anisotropies in model peptides. J. Biomol. NMR 38:255, 2007. Thielges, M.C., Case, D.A., Romesberg, F.E. Carbon-deuterium bonds as probes of dihydrofolate reductase. J. Am. Chem. Soc. 130:6597, 2008. Walker, R.C., Crowley, M.F., Case, D.A. The implementation of a fast and accurate QM/MM potential method in Amber. J. Comput. Chem. 28:1019, 2008. Wong, V., Case, D.A. Comparing MD simulations and NMR relaxation parameters. Annu. Rep. Comput. Chem., in press. Wong, V., Case, D.A. Evaluating rotational diffusion from protein MD simulations. J. Phys. Chem. B 112:6013, 2008. ** University of Maryland, College Park, Maryland *** University of Bayreuth, Bayreuth, Germany **** St. Jude Children’s Research Hospital, Memphis, Tennessee e are using a combination of modern quantum chemistry (density functional theory, DFT) and classical electrostatics to describe the energetics, reaction pathways, and spectroscopic properties of metalloenzymes. In addition, we are analyzing other systems that have novel catalytic, photochemical, or photophysical properties. Critical biosynthetic and regulatory processes may involve catalytic transformations of fairly small molecules or groups by transition-metal centers. The ironmolybdenum cofactor center of nitrogenase catalyzes the multielectron reduction of molecular nitrogen to 2 molecules of ammonia plus molecular hydrogen. We are continuing our research on the catalytic cycle of this enzyme, following up on our earlier work on the structure and oxidation state of the cofactor complex in the “resting enzyme” before multielectron reduction and nitrogen binding. On the basis of DFT calculated vs experimental physical properties, including redox potentials, cluster geometries, and Mössbauer isomer shifts, the core cluster has a (MoFe7S9X ) prismane active site, where the central X likely is nitride and the “resting cluster oxidation state” is Mo(IV)Fe(II)4Fe(III)3. A central carbide is also a possibility, because no clear central ligand hyperfine signal has been detected for the paramagnetic resting state. We have now examined the 2-electron reduced iron-molybdenum cofactor cluster with the bound alternative product complex (allylic alcohol generated from propargyl alcohol plus 2 electrons plus 2 protons). Three different ligand-binding structures are feasible (Fig. 1), and all are expected to yield a substantial central ligand hyperfine signal for either 14,15 N or 13 C. This finding would be the first confirmed use of nitride or carbide in any enzyme. Our current structural and redox evidence strongly argues against the presence of a central oxide. W MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 241 F i g . 2 . Proposed active-site structure of methane monooxyge- nase peroxo intermediate P. F i g . 1 . DFT-optimized model structures of allyl alcohol bound at Fe6 of the iron-molybdenum cofactor center of nitrogenase. The calculated relative energies and hyperfine couplings for central nitrogen atom are indicated. Methane monooxygenase catalyzes the oxidation of methane to methanol. This reaction is extremely important in the biosphere because methane is a greenhouse gas. Further, the monooxygenase can catalyze reactions with alternative substrates, a characteristic that has implications for detoxification of organic pollutants. We have been examining various steps in the catalytic cycle, comparing predictions based on DFT with those of Mössbauer spectroscopy. We have focused particularly on the critical intermediate Q, which performs the oxygen insertion reaction. Recently, we evaluated the reaction pathway from the earlier peroxo intermediate P (Fig. 2) to the dioxo intermediate Q. The iron complex at the active site of methane monooxygenase resembles the complex in ribonucleotide reductases (RNRs; see following); the resemblance is particularly close for RNRs in some pathogenic bacteria, including Chlamydia, in which the tyrosine near the ironoxo dimer complex (commonly found in Escherichia coli and mammalian RNRs) is replaced by phenylalanine. Evidence now indicates that in RNRs from Chlamydia the di-iron center is replaced by a manganese-iron center. The altered RNR mechanism in these pathogens is of great interest for exploring feasible drug treatments. Class I RNRs are aerobic enzymes that catalyze the reduction of ribonucleotides to deoxyribonucleotides, providing the required building blocks for DNA replication and repair. These enzymes are targets for anticancer, antiviral, and antibacterial drugs. These ribonucleotideto-deoxyribonucleotide reactions occur via a long-range radical (or proton-coupled electron transfer) propagation mechanism initiated by a fairly stable tyrosine radical, “the pilot light.” When this pilot light goes out, the tyrosine radical is regenerated by a high-oxidation-state Fe(III)-Fe(IV)-oxo enzyme intermediate called X. We are using DFT and electrostatics calculations in combination with analysis of Mössbauer, electron nuclear double resonance, and magnetic circular dichroism spectroscopies to search for a proper structural and electronic model for intermediate X. In collaboration with K. Hahn, University of North Carolina at Chapel Hill, we have examined the physical and spectroscopic properties of novel solvent-sensitive fluorescent dyes of interest for imaging live cells. These dyes can act as biosensors for changes in conformational state in cell-signaling proteins, with potential uses in high-throughput drug screening. With J.A. Fee, Department of Molecular Biology, we are exploring the mechanism of 4-electron reduction of molecular oxygen to 2 molecules of water by cytochrome oxidase (complex IV of mitochondria and the related cytochrome ba3 from Thermus thermophilus). The cop- 242 MOLECULAR BIOLOGY 2008 per-iron-heme complex links molecular oxygen reduction to proton pumping across the mitochondrial membrane. PUBLICATIONS Fee, J.A., Case, D.A., Noodleman, L. Toward a chemical mechanism of proton pumping by the B-type cytochrome c oxidases: application of density functional theory to cytochrome ba3 of Thermus thermophilus. J. Am. Chem. Soc. 130:15002, 2008. Han, W.-G., Noodleman, L. Structural model studies for the high-valent intermediate Q of methane monooxygenase from broken-symmetry density functional calculations. Inorganica Chim. Acta 361:973, 2008. Han, W.-G., Noodleman, L. Structural model studies for the peroxo intermediate P and the reaction pathway from P → Q of methane monooxygenase using brokensymmetry density functional calculations. Inorg. Chem. 47:2975, 2008. Lukoyanov, D., Pelmenschikov, V., Maeser, N., Laryukhin, M., Yang, T.C., Noodleman, L., Dean, D.R., Case, D.A., Seefeldt, L.C., Hoffman, B.M. Testing if the interstitial atom, X, of the nitrogenase molybdenum-iron cofactor is N or C: ENDOR, ESEEM, and DFT studies of the S = 3/2 resting state in multiple environments. Inorg. Chem. 46:11437, 2007. Noodleman, L., Case, D.A. Broken symmetry states of iron sulfur clusters. In: Computational Inorganic and Bioinorganic Chemistry. Solomon, E.I., King, R.B., Scott, R.A. (Eds.) Wiley & Sons, New York, in press. Noodleman, L., Pique, M.E., Roberts, V.A. Properties and functions of iron-sulfur clusters. In: Wiley Encyclopedia of Chemical Biology. Begley, T.P. (Ed.). Wiley Interscience, New York, in press. Pelmenschikov, V., Case, D.A., Noodleman, L. Ligand-bound S = 1/2 FeMo-cofactor of nitrogenase: hyperfine interaction analysis and implication for the central ligand X identity. Inorg. Chem. 47:6162, 2008. Toutchkine, A., Han, W.-G., Ullmann, M., Liu, T., Bashford, D., Noodleman, L., Hahn, K.M. Experimental and DFT studies: novel structural modifications greatly enhance the solvent sensitivity of live cell imaging dyes. J. Phys. Chem. A 111:10849, 2007. Computation and Visualization in Structural Biology A.J. Olson, D.S. Goodsell, M.F. Sanner, M. Chang, S. Cosconati,* S. Dallakyan, S. Forli, A. Gillet, R. Harris, R. Huey, J. Huntoon, G. Johnson, D. Keidel, W. Lindstrom,** G.M. Morris, A. Omelchenko, A. Perryman, M. Pique, R. Rosenstein, M. Utsintong,*** G. Vareille, Q. Zhang, Y. Zhao**** THE SCRIPPS RESEARCH INSTITUTE and research settings, methods for predicting biomolecular interactions and using these interactions in structurebased inhibitor design, analyzing biomolecular structure and function, and presenting the biomolecular world in education and outreach. C O M P O N E N T - B A S E D V I S U A L I Z AT I O N E N V I R O N M E N T S To facilitate the integration and interoperation of computational models and techniques from a wide variety of scientific disciplines, we continue to expand our component-based software environment. The environment is centered on Python, a high-level, object-oriented, interpretive programming language. This approach allows the compartmentalization and reuse of software components. Python provides a powerful “glue” for assembling computational components and, at the same time, a flexible language for the interactive scripting of new applications. In 2007, we released 4 versions of our software tools: 1.4.4, 1.4.5, 1.4.6, and 1.5.0. We are able to release new versions so quickly because of the stateof-the-art overnight software testing environment we have developed. Starting with version 1.4.6, we modified our installation mechanism to allow the simultaneous installation of multiple versions of the software. All software tools have been markedly improved. For example, we enhanced ADT in supporting the new AutoDock4 (described in more detail later); and in the Python Molecular Viewer, ADT modules no longer require a graphical user interface, rather they can now also function as stand-alone scripts or as nodes in Vision, our graphical network editor. Users can now load their own code as nodes in Vision. We have improved APBSCommands.py to use local installations of APBS (a software package for the numerical solution of the Poisson-Boltzmann equation) or remote APBS Web services through the Opal toolkit. We have added support for ambient occlusion for polygonal meshes in the visualization component, making it easier to visualize cavities on molecular surfaces (Fig. 1). * University of Naples “Federico II,” Naples, Italy ** Acelot, Inc., Santa Barbara, California *** Mahidol University, Bangkok, Thailand **** CambridgeSoft, Cambridge, Massachusetts n the Molecular Graphics Laboratory, we develop novel computational methods to analyze, understand, and communicate the structure and interactions of complex biomolecular systems. Within our componentbased simulation and visualization environment, we continue to develop 3-dimensional molecular models as a tangible human-computer interface in educational I F i g . 1 . A molecular surface for HIV protease, with traditional shading on the left and ambient occlusion on the right. Ambient occlusion simulates the reduced lighting in sheltered areas and gives useful cues to the shape of complex molecular surfaces. Image generated with the Python Molecular Viewer. MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 243 This software has been widely distributed. We had more than 132,000 downloads during the past 12 months, and 1761 unique users voluntarily registered in our user database (a sustained average of 5 users per day). Most recently, we created an articulated model of MsbA, a bacterial ABC transporter that undergoes a large and complex conformational change in the course of its transport cycle. TA N G I B L E I N T E R FA C E S I N S T R U C T U R A L B I O L O G Y AND INTEGRASE We have continued to develop autofabricated physical models (“solid printing”) of biological molecules in the context of an augmented reality environment, with the goal of using the models in both research and education. In collaboration with E. Keinan, Department of Molecular Biology, we used tangible models to design self-assembling chemical structures that mimic the selfassembly of viral capsids. Our hypothesis was first developed and tested by using tangible models and then further characterized by molecular dynamics simulation. The models allowed direct physical testing of different possibilities for chemical complementarity of subunits, for instance, identifying 2 alternative binding modes for the self-assembled corannulene currently under development (Fig. 2). We have also begun work on larger models that capture the dynamic characteristics of biomolecules. We are using several approaches in our continuing work on the development of inhibitors of HIV protease. After analyzing the evolution of drug resistance against previously approved inhibitors, we developed a method to predict mutations associated with drug resistance that might be induced by new HIV protease inhibitors. With this technique, we were able to detect more than half of the major mutations in drug-resistant HIV proteases. Site-directed mutagenesis validated resistance of 3 predicted mutations (I47V, F53L, and I84V) for AB2, a novel inhibitor of HIV protease, and the clinically approved drug amprenavir. In collaboration with ActiveSite, San Diego, California, and C.D. Stout, Department of Molecular Biology, we are developing methods to interpret electron density maps from crystallographic fragment screens. In our method, a combination of peak characterization and computational docking is used to deconvolute peaks in the crystallographic electron density maps that correspond to fragment binding in crystals soaked with a mixture of fragments. The method is very rapid, because docking is only performed in the immediate vicinity of the identified peaks, and so can be easily incorporated into the high-throughput work flow in use at ActiveSite. To improve the efficiency of finding promising chemical lead compounds, the so-called hit rate in drug discovery, we developed a method that combines highthroughput screening and virtual screening. We performed a virtual screen of 1476 compounds obtained from a cell-based anti-HIV inhibitor screen. We used a novel selection procedure to choose compounds that bound preferentially either to the active site or to a putative allosteric exosite, but not to both. A total of 5 compounds showed micromolar binding constants; 3 were predicted to bind in the active site and 2 in the exosite. Kinetic analysis showed noncompetitive binding for the predicted exosite inhibitors, which are now being further characterized. We are also collaborating with researchers at Pfizer, Inc., Sandwich, England, and with J.A. McCammon and his group at the University of California, San Diego, to explore the structure and function of HIV integrase (Fig. 3). We have modeled a form of integrase with a F i g . 2 . Physical models were used to study self-assembly of viruses and a designed corannulene complex. Left, Three frames from a movie showing self-assembly of a virus model. The subunits were fabricated on the basis of the molecular structure, and magnets were placed at the interfaces. When the models are shaken in a small bottle, the intact virus self-assembles. Right, Using these same structural model abstractions, a corannulene complex was designed. Two possible modes of assembly were discovered by model building and subsequent computational simulation. DEVELOPMENT OF INHIBITORS FOR HIV PROTEASE 244 MOLECULAR BIOLOGY 2008 F i g . 3 . Computational modeling and molecular dynamics were used to generate models of HIV integrase with a closed active-site loop and 2 hydrated magnesium ions (green). Two conformations of the enzyme are shown with ribbons, and water molecules and catalytic residues that coordinate the magnesium ions are shown with bonds. Image generated with the Python Molecular Viewer. closed loop and 2 magnesium ions that is expected to be relevant to structure-based drug design. We are extending this research to model several drug-resistant mutants and explore how these mutations modify the dynamics of the enzyme. When combined with quantum mechanical calculations, these models will be useful for studying the mechanism of catalysis. Long-term goals are to characterize the interaction of the catalytic domain with DNA and ultimately use this information to characterize binding of DNA to the intact enzyme. PROTEIN-LIGAND DOCKING WITH AUTODOCK According to recent reports, the computer program AutoDock is the most widely cited docking method, and we have continued to make it a central tool for predicting biomolecular interactions. AutoDock4 was released in 2007 and is available through a convenient opensource license. AutoDock was the first docking code to be used in a public, Internet-based distributed computing project, and we have continued to use this massive computing resource to perform experiments that are beyond the reach of traditional computing. We are currently using the World Community Grid, a large Internet distributed computing project sponsored by IBM, to support FightAIDS@Home on more than 800,000 client computers. Personal computers are used by the program when the computers are not in use by their owners, providing an enormous, and largely untapped, computational resource. THE SCRIPPS RESEARCH INSTITUTE To support our growing user community, we have hosted tutorials, workshops, and lectures presenting basic methods in AutoDock and specific application to virtual screening. We have also continued to expand the capabilities of AutoDock and the graphical user interface AutoDockTools, for instance, exploring the use of gradient information in the search, predicting covalent complexes between ligands and proteins, and streamlining all aspects of virtual screening. Most recently, we tested a method for using results from reiterated docking experiments to evaluate an empirical vibrational entropy of binding in ligand-protein complexes in collaboration with R.K. Bele, University of California, San Diego, and K.S. Carroll, University of Michigan, Ann Arbor. In addition, we have added new support for the relaxed complex method, in which snapshots are taken from a molecular dynamics simulation and used to sample the range of conformations available to a protein target. We have continued development and application of AutoLigand, a program used to identify and quantify optimal binding sites in proteins. We showed that the method is effective for identifying binding sites in proteins and for optimizing the binding of drugs to protein targets. We have used the method to design exosite inhibitors of HIV protease; inhibitors for histidine deacetylase, in collaboration with J.M. Gottesfeld, Department of Molecular Biology; and exosite inhibitors for p38 MAP kinase, in collaboration with J.A. Tainer, Department of Molecular Biology. PROTEIN FLEXIBILITY AND PROTEIN-PROTEIN DOCKING In collaboration with C. Bajaj, University of Texas, Austin, we have continued the development of a new protein-protein docking technique that is now implemented in a software program called F2Dock (which stands for Fast Fourier-transform–based docking). This program is being tested on a set of protein-protein complexes to evaluate its performance and identify its shortcomings. We have continued the development and testing of a new automated docking program called FLIPDock that uses a hierarchical and multiresolution representation of the flexibility of biological macromolecules to model protein motion and induced fit. We have further developed this software tool and applied it to a variety of docking problems in which receptor flexibility is known to cause the failure of rigid receptor-based docking simulations. We have optimized FLIPDock and fixed several problems in the software; we released version 0.1 beta to a selected set of users. MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 245 To test the hypothesis that low-resolution surfaces can be used to improve the success of protein-protein docking, we did a systematic study of the effects of multiresolution blurred surfaces on shape complementarity in a set of 66 protein-protein complexes for which complexed and unbound structures are available. We found that medium-resolution smoothing can reproduce about 88% of the shape complementarity of atomic resolution surfaces, and complexes formed from the free component structures show many overlaps and gaps with atomic resolution surfaces, which are improved by smoothing the surfaces to low resolution. C O M P U TAT I O N A L M O D E L I N G O F E X T R A C E L L U L A R INTERACTIONS OF TISSUE FACTOR Signaling through protease-activated receptor 2 (PAR-2) by the complex composed of tissue factor and coagulation factor VIIa regulates gene transcription and protein translation, cell proliferation and survival, and cell motility and integrin activation. This signaling involves cleavage of the PAR-2 extracellular N-terminal tail between arginine at position 36 and serine at position 37 by the protease domain of factor VIIa. However, it is unclear how the protease domain of factor VIIa recognizes and binds the PAR-2 tail to facilitate the cleavage. We used molecular modeling and molecular dynamics simulations to derive the interactions between PAR-2 and factor VIIa. We found 3 types of key interactions at noncatalytic sites of the factor VIIa protease domain. Four of the key factor VIIa residues were then experimentally shown to be involved in PAR-2 activation. V I S U A L M E T H O D S F R O M AT O M S T O C E L L S Understanding structural molecular biology is essential to foster progress and critical decision making among students, policy makers, and the general public. In the past year, we continued our long-standing commitment to science education and outreach with a combination of presentations, popular and professional illustrations and animations, 3-dimensional tangible models, and a presence on the World Wide Web. In these projects, we use the diverse visualization tools developed in the Molecular Graphics Laboratory to disseminate results that range from atomic structure to cellular function. In the past year, the “Molecule of the Month” at the Protein Data Bank entered its ninth year of providing an accessible introduction to the central database of biomolecular structure (Fig. 4). In collaboration with T. Herman, Milwaukee School of Engineering in Wisconsin, we continued work on “Protein Active Learning Modules” that provide educational materials for the F i g . 4 . The adrenergic receptor was presented as the 100th installment of the “Molecule of the Month” at the Protein Data Bank (http://www.pdb.org). Each month, a new molecule is presented with a description of its structure, function, and relevance to health and welfare. Visitors are then given suggestions on to how to begin their own exploration of the structures in the Protein Data Bank. high school and undergraduate level. We are also extending our modeling efforts from the realm of molecules and complexes into the realm of whole cells. Building on previous illustrative work, we are developing methods to model significant parts of living cells (Fig. 5). Currently, we are building these tools in the context of high-end computer animation software, to allow easy creation of educational materials. F i g . 5 . A simulated 3-dimensional model of part of a cell, with a cell membrane at the top and generic cytoplasm at the bottom. 246 MOLECULAR BIOLOGY 2008 PUBLICATIONS Amaro, R., Minh, D.L., Cheng, L., Lindstrom, W.M., Jr., Olson, A.J., Lin, J.-H., Li, W.W., McCammon, J.A. Remarkable loop flexibility in avian influenza N1 and its implications for antiviral drug design. J. Am. Chem. Soc. 129:7764, 2007. Beuscher, A.E., Olson, A.J. Iterative docking strategies for virtual ligand screening. In: Computational and Structural Approaches to Drug Discovery: Ligand-Protein Interactions. Stroud, R.M., Finer-Moore, J. (Eds.). RSC Publishing, Cambridge, England, 2007, p. 242. A volume in the series RSC Biomolecular Sciences. THE SCRIPPS RESEARCH INSTITUTE T a b l e 1 . New drug leads and inhibitors. Lead or inhibitor Condition affected Androgen receptor antagonist (Fig. 1) Cancer Chang, M.W., Lindstrom, W., Olson, A.J., Belew, R.K. Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries. J. Chem. Info. Model. 47:1258, 2007. Evans, M.J., Morris, G.M., Wu, J., Olson, A.J., Sorensen, E.J., Cravatt, B.F. Mechanistic and structural requirements for active site labeling of phosphoglycerate mutase by spiroepoxides. Mol. Biosyst. 3:495, 2007. J. Dalton, Ohio State University, Columbus, Ohio α1-Antitrypsin polymerization blocker α1-Antitrypsin deficiency, emphysema D.A. Lomas, University of Cambridge, Cambridge, England Serotonin N-acetyltransferase inhibitor Sleep and mood disorders P.A. Cole, Johns Hopkins School of Medicine, Baltimore, Maryland Enoyl reductase inhibitor Malaria D.A. Fidock, Albert Einstein College of Medicine, Bronx, New York Goodsell, D.S. Making the step from chemistry to biology and back. Nat. Chem. Biol. 3:681, 2007. J.C. Sacchettini, Texas A&M University, College Station, Texas Goodsell, D.S., Johnson, G.T. Filling in the gaps: artistic license in education and outreach. PloS Biol. 5:e308, 2007. Harris, R., Olson, A.J., Goodsell, D.S. Automated prediction of ligand-binding sites in proteins. Proteins 70:1506, 2008. Illingworth, C.J.R., Morris, G.M., Parkes, K.E.B., Snell, C.R., Reynolds, C.A. Assessing the role of polarization in docking. J. Phys. Chem. A, in press. Morris, G.M., Huey, R., Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinformatics, in press. Telomeric G-quadruplex Cancer K.Y. Wong, Hong Kong Polytechnic University, Hong Kong, China Melaninconcentrating hormone receptor 1 Obesity F. Monsma, Schering Plough Research Institute, Kenilworth, New Jersey A. Orry, C. Cavasotto, MolSoft L.L.C., La Jolla, California Olson, A.J., Hu, Y.H.U., Keinan, E. Chemical mimicry of viral capsid self-assembly. Proc. Natl. Acad. Sci. U. S. A. 104: 20731, 2007. Zhao, Y., Sanner, M.F. FLIPDock: docking flexible ligands into flexible receptors. Proteins 68:726, 2007. Zhao, Y., Sanner, M.F. Protein-ligand docking with multiple flexible side chains. J. Comput. Aided Mol. Des., in press. Computational Structural Proteomics for Drug Discovery X.K. Zhang, Burnham Institute, La Jolla, California P. Sexton, A. Christopoulos, Monash University, Victoria, Australia Bongini, L., Fanelli, D., Piazza, F., De Los Rios, P., Sanner, M., Skoglund, U. A dynamical study of antibody-antigen encounter reactions. Phys. Biol. 4:172, 2007. Chang, M.W., Belew, R.K., Carroll, K.S., Olson, A.J., Goodsell, D.S. Empirical entropic contributions in computational docking: evaluation in APS reductase complexes. J. Comput. Chem. 29:1753, 2008. Collaborators Ubiquitin-like poxvirus proteinase I7L Smallpox V. Katritch, D. Hruby, SIGA Technologies, Inc., Corvallis, Oregon tural models of inhibition of sirtuin 2. This foundation may be helpful in the further development of sirtuin 2 inhibitors for the treatment of Parkinson’s disease. R. Abagyan, G. Bottegoni, A. Grigoryan, J. Kovacs, I. Kufareva, G. Nicola, S.-J. Park, K. Reynolds, M. Rueda e focus on developing and implementing new mathematical and computational methods to improve structure prediction and docking methods for structure-based drug design. W DE NOVO DISCOVERY OF DRUG LEADS Our improved structure-based methods led to the discovery of new drug leads and inhibitors in collaborative studies with many scientists (Table 1). A D VA N C E D A P P L I C AT I O N S O F M O L E C U L A R MODELING AND DOCKING In collaborations with A.G. Kazantsev, Harvard Medical School, Charlestown, Massachusetts, we built struc- F i g . 1 . A novel androgen receptor antagonist repurposed from an antipsychotic drug. MOLECULAR BIOLOGY 2008 We used the multiple receptor conformation approach to elucidate the structural mechanism of inhibition of the 3-phosphoinositide-dependent protein kinase-1 by 3-hydroxyanthranilic acid, a tryptophan metabolite. We found that 3-hydroxyanthranilic acid inhibited activation of the transcription factor NF-κB upon engagement of T-cell receptors by specifically targeting the kinase. Our docking models helped rationalize this interaction, which appears to play a key role in natural regulation of NF-κB activity. Collaboration with M. Yeager, Department of Cell Biology, led to the development of techniques and models for complex multisubunit transmembrane proteins. The detailed atomic models of the gap junction channel and several other membrane proteins have been assigned to low-resolution electron density and refined by using the internal coordinate mechanics protocols. We also used the internal coordinate mechanics protocols in collaboration with J. Gertsch, ETH Zurich, Zürich, Switzerland, to develop models of supramolecular aggregates of cannabinomimetics. J. Fernandez-Recio and colleagues, University of Zaragoza, Zaragoza, Spain, collaborated with us to improve the prediction for transient proteinprotein complexes. Finally, in collaboration with L.J. Miller, Mayo Clinic Scottsdale, Scottsdale, Arizona; P.C. Lam, MolSoft; and P.M. Sexton and A. Christopoulos, University of Monash; we designed a series of protocols designed for predicting realistic atomic models of a ternary complex of the secretin peptide and 2 domains of its receptor, a family B G protein–coupled receptor. CHALLENGES IN STRUCTURE-BASED DOCKING AND SCREENING Receptor flexibility is a critical issue in structurebased virtual screening methods. Most of the docking protocols rely on a fixed conformation of the receptor or on prior knowledge of multiple conformations representing the variation of the pocket. We have developed an induced-fit docking protocol called SCARE (SCan Alanines and Refines) that requires only a single initial pocket conformation for ligand docking and no prior knowledge about the location of the binding site. To overcome the error of binding pose and binding score of ligands with single fixed receptors, we have introduced multiple receptor conformation, which will improve the docking calculation overall and account for receptor flexibility. We used this approach to screen large ligand databases against p38α kinase and the nuclear receptor peroxisome proliferator–activated receptor γ and found dramatic improvement in database enrichment factors against both receptors. THE SCRIPPS RESEARCH INSTITUTE 247 We also devised a method for evaluating the druggability (i.e., the potential to be targeted by a small-molecule drug) of protein targets at the level of structural proteomes. The method includes evaluating the structural feasibility of targeting the given protein with a small molecule (the presence of a sufficiently large and sufficiently buried binding pocket), predicting the target genomic instability and escape mutations (evolutionary conservation of the binding site across the genome of interest and related genomes), and predicting possible drug off-target activities and toxicities (the lack of homology with human proteins). This method has been applied to studies of the malarial proteome. PUBLICATIONS Bisson, W.H., Cheltsov, A.V., Bruey-Sedano, N., Lin, B., Chen, J., Goldberger, N., May, L.T., Christopoulos, A., Dalton, J.T., Sexton, P.M., Zhang, X.K., Abagyan, R. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc. Natl. Acad. Sci. U. S. A. 104:11927, 2007. Bottegoni, G., Kufareva, I., Totrov, M., Abagyan, R. A new method for ligand docking to flexible receptors by dual alanine scanning and refinement (SCARE). J. Comput. Aided Mol. Des. 22:311, 2008. Cavasotto, C.N., Orry, A.J., Murgolo, N.J., Czarniecki, M.F., Kocsi, S.A., Hawes, B.E., O’Neill, K.A., Hine, H., Burton, M.S., Voigt, J.H., Abagyan, R.A., Bayne, M.L., Monsma, F.J., Jr. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 51:581, 2008. Chrencik, J.E., Brooun, A., Recht, M.I., Nicola, G., Davis, L.K., Abagyan, R., Widmer, H., Pasquale, E.B., Kuhn, P. Three-dimensional structure of the EphB2 receptor in complex with an antagonistic peptide reveals a novel mode of inhibition. J. Biol. Chem. 282:36505, 2007. Dong, M., Lam, P.C., Gao, F., Hosohata, K., Pinon, D.I., Sexton, P.M., Abagyan, R., Miller, L.J. Molecular approximations between residues 21 and 23 of secretin and its receptor: development of a model for peptide docking with the amino terminus of the secretin receptor. Mol. Pharmacol. 72:280, 2007. Harikumar, K.G., Lam, P.C., Dong, M., Sexton, P.M., Abagyan, R.,Miller, L.J. Fluorescence resonance energy transfer analysis of secretin docking to its receptor: mapping distances between residues distributed throughout the ligand pharmacophore and distinct receptor residues. J. Biol. Chem. 282:32834, 2007. Hayashi, T., Mo, J.H., Gong, X., Rossetto, C., Jang, A., Beck, L., Elliott, G.I., Kufareva, I., Abagyan, R., Broide, D.H., Lee, J., Raz, E. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc. Natl. Acad. Sci. U. S. A. 104:18619, 2007. Katritch, V., Byrd, C.M., Tseitin, V., Dai, D., Raush, E., Totrov, M., Abagyan, R., Jordan, R., Hruby, D.E. Discovery of small molecule inhibitors of ubiquitin-like poxvirus proteinase I7L using homology modeling and covalent docking approaches. J. Comput. Aided Mol. Des. 21:549, 2007. Kovacs, J.A., Baker, K.A., Altenberg, G.A., Abagyan, R., Yeager, M. Molecular modeling and mutagenesis of gap junction channels. Prog. Biophys. Mol. Biol. 94:15, 2007. Kovacs, J.A., Yeager, M., Abagyan, R. Computational prediction of atomic structures of helical membrane proteins aided by EM maps. Biophys. J. 93:1950, 2007. Lee, H.S., Choi, J., Kufareva, I., Abagyan, R., Filikov, A., Yang, Y., Yoon, S. Optimization of high throughput virtual screening by combining shape-matching and docking methods. J. Chem. Inf. Model. 48:489, 2008. Ma, D.L., Lai, T.S., Chan, F.Y., Chung, W.H., Abagyan, R., Leung, Y.C., Wong, K.Y. Discovery of a drug-like G-quadruplex binding ligand by high-throughput docking. ChemMedChem 3:881, 2008. 248 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE Mallya, M., Phillips, R.L., Saldanha, S.A., Gooptu, B., Brown, S.C., Termine, D.J., Shirvani, A.M., Wu, Y., Sifers, R.N., Abagyan, R., Lomas, D.A. Small molecules block the polymerization of Z α1-antitrypsin and increase the clearance of intracellular aggregates. J. Med. Chem. 50:5357, 2007. Medina, M., Abagyan, R., Gómez-Moreno, C., Fernandez-Recio, J. Docking analysis of transient complexes: interaction of ferredoxin-NADP+ reductase with ferredoxin and flavodoxin. Proteins 72:848, 2008. Nicola, G., Smith, C.A., Abagyan, R. New method for the assessment of all druglike pockets across a structural genome. J. Comput. Biol. 15:231, 2008. Nicola, G., Smith, C.A., Lucumi, E., Kuo, M.R., Karagyozov, L., Fidock, D.A., Sacchettini, J.C., Abagyan, R. Discovery of novel inhibitors targeting enoyl-acyl carrier protein reductase in Plasmodium falciparum by structure-based virtual screening. Biochem. Biophys. Res. Commun. 358:686, 2007. Outeiro, T.F., Kontopoulos, E., Altmann, S.M., Kufareva, I., Strathearn, K.E., Amore, A.M., Volk, C.B., Maxwell, M.M., Rochet, J.C., McLean, P.J., Young, A.B., Abagyan, R., Feany, M.B., Hyman, B.T., Kazantsev, A.G. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317:516, 2007. Raduner, S., Bisson, W., Abagyan, R., Altmann, K.H., Gertsch, J. Self-assembling cannabinomimetics: supramolecular structures of N-alkyl amides. J. Nat. Prod. 70:1010, 2007. F i g . 1 . A novel nonlinear approach that incorporates both ana- lytical and bioinformatics technologies for quantitative comparisons of metabolites. data analysis approaches to identify and structurally characterize metabolites of physiologic importance. N A N O S T R U C T U R E - I N I T I AT O R M A S S S P E C T R O M E T R Y A N A LY S I S , A C T I V I T Y, A N D I M A G I N G To develop ultra-high-sensitivity approaches in mass spectrometry (Fig. 2), we are using nanostruc- Szewczuk, L.M., Saldanha, S.A., Ganguly, S., Bowers, E.M., Javoroncov, M., Karanam, B., Culhane, J.C., Holbert, M.A., Klein, D.C., Abagyan, R., Cole, P.A. De novo discovery of serotonin N-acetyltransferase inhibitors. J. Med. Chem. 50:5330, 2007. Totrov, M., Abagyan, R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr. Opin. Struct. Biol. 18:178, 2008. Mass Spectrometry: Metabolomics Profiling, Activity, and Imaging G. Siuzdak, J. Apon, H.P. Benton, B.O. Crews, K. Harris, L. Hoang, E. Kalisiak, A. Nordström, T. Northen, M. Sonderegger, S. Trauger, W. Uritboonthai, W. Webb, W. Wikoff, D. Wong, H.K. Woo, O. Yanes M E TA B O L O M I C S ndogenous metabolites, ubiquitous in biofluids, tissues, and organisms of every kind, are crucial elements in understanding fundamental biochemistry, disease diagnosis, and drug toxicity. The inherent advantage of monitoring small molecules rather than proteins is the relative ease of quantitative analysis with mass spectrometry. We are implementing novel mass spectrometry and bioinformatics techniques (Fig. 1) to investigate the profile of small-molecule metabolites. The purpose of this effort is to develop mass-based metabolomics technology for a better understanding of fundamental biochemistry, such as in stem cell differentiation, as well as a diagnostic research tool. Our ultimate goal is to create analytical and chemical technologies and E F i g . 2 . Illustration of the Nimzyme assay: immobilization of metab- olites in the fluorous ‘‘clathrate’’ phase of the nanostructure-initiator mass spectrometry surface (A), incubation of the surface with the sample to screen for enzymatic activity (B), and laser irradiation, resulting in vaporization of the fluorous phase, efficiently transferring the immobilized substrate and products into the gas phase. Based in part on Northen, T.R., Lee, J.-C., Hoang, L., Raymond, J., Hwang, D.-R, Yannone, S.M., Wong, C.-H, Siuzdak, G. A nanostructure-initiator mass spectrometry-based enzyme activity assay. Proc. Natl. Acad. Sci. U. S. A. 105:3678, 2008. Copyright 2008 National Academy of Sciences U.S.A. tured clatherates to facilitate vaporization and ionization of biomolecules. Using this technology, termed nanostructure-initiator mass spectrometry, we can analyze a wide range of molecules with unprecedented sensitivity, in the yoctomole (10-24 moles) range. The method is also being developed as a platform for biomolecular tissue imaging. PUBLICATIONS Benton, H.P., Wong, D.M., Trauger, S.A., Siuzdak, G. XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal. Chem., in press. Nordström, A., Want, E., Northen, T., Lehtiö, J., Siuzdak, G. Multiple ionization mass spectrometry strategy used to reveal the complexity of metabolomics. Anal. Chem. 80:421, 2008. Northen, T.R., Lee, J.-C., Hoang, L., Raymond, J., Hwang, D.-R., Yannone, S.M., Wong, C.-H, Siuzdak, G. A nanostructure-initiator mass spectrometry-based enzyme activity assay. Proc. Natl. Acad. Sci. U. S. A. 105:3678, 2008. MOLECULAR BIOLOGY 2008 Northen, T.R., Woo, H.K., Northen, M.T., Nordström, A., Uritboonthai, W., Turner, K., Siuzdak, G. High surface area of porous silicon drives desorption of intact molecules. J. Am. Soc. Mass Spectrom. 18:1945, 2007. THE SCRIPPS RESEARCH INSTITUTE 249 at an RNA structure called the Rev responsive element (RRE; Fig. 1). Binding of Rev to the RRE directs mRNAs Northen, T.R., Yanes, O., Northen, M.T., Marrinucci, D., Uritboonthai, W., Apon, J., Golledge, S.L., Nordström, A., Siuzdak, G. Clathrate nanostructures for mass spectrometry [letter]. Nature 449:1033, 2007. Trauger, S.A., Kalisiak, E., Kalisiak, J., Morita, H., Weinberg, M.V., Menon, A.L., Poole, F.L. II, Adams, M.W.W., Siuzdak, G. Correlating the transcriptome, proteome, and metabolome in the environmental adaptation of a hyperthermophile. J. Proteome Res. 7:1027, 2008. Vallon, V., Eraly, S.A., Wikoff, W.R., Rieg, T., Kaler, G., Truong, D.M., Ahn, S.Y., Mahapatra, N.R., Mahata, S.K., Gangoiti, J.A., Wu, W., Barshop, B.A., Siuzdak, G., Nigam, S.K. Organic anion transporter 3 contributes to the regulation of blood pressure. J. Am. Soc. Nephrol., in press. Wikoff, W.R., Gangoitti, J.A., Barshop, B.A., Siuzdak, G. Metabolomics identifies novel perturbations in human disorders of propionate metabolism. Clin. Chem. 53:2169, 2007. Wikoff, W.R., Pendyala, G., Siuzdak, G., Fox H.S. Metabolomic analysis of the cerebrospinal fluid reveals changes in phospholipase expression in the CNS of SIVinfected macaques. J. Clin. Invest. 118:2661, 2008. Woo, H.-K., Northen, T.R., Yanes, O., Siuzdak, G. Nanostructure-initiator mass spectrometry (NIMS): a protocol for preparing and applying NIMS surfaces for high sensitivity mass analysis. Nat. Protoc., in press. RNA-Protein Complexes Mediating Nuclear Transport of HIV mRNAs J.R. Williamson, F. Agnelli, W. Anderson, A. Beck, C. Beuck, F i g . 1 . Nuclear transport of HIV Rev. The Rev protein shuttles in and out of the nucleus by sequentially interacting with a series of human factors in 3 key complexes. First, an import complex is formed by association of Rev (yellow) with the nuclear transport factor importin-β (blue). Once in the nucleus, Rev forms an oligomeric RNA complex by binding to the RRE RNA. Multiple copies of Rev bind to the RRE to promote efficient export, but the details of the oligomeric structure are not known. Rev recruits the nuclear transport factor CRM-1 (orange), which facilitates transport of the RevRRE complex back to the cytoplasm. Other factors associate with this complex, including the helicases DDX1 and DDX3, to form the export complex. DDX1 (red) interacts directly with Rev, whereas A. Bunner, A. Carmel, S. Chen, S., Edgcomb, D. Kerkow, DDX3 (blue) interacts with Rev by indirect binding to CRM-1. The S. Kwan, E. Menichelli, W. Ridgeway, G. Ring, H. Schultheisz, Z. Shajani, E. Sperling, M.T. Sykes, B. Szymczyna, J. Wu helicases may dissociate proteins from the viral RNA after export to the cytoplasm (lower right) to facilitate translation of the viral mRNA or packaging of the viral genome. Each of these complexes may be a new target for intervention against HIV. evelopment of novel therapeutic strategies against HIV infection is a pressing need and requires elucidation of the fundamental mechanisms of HIV replication. Currently prescribed anti-HIV drugs inhibit the viral protease, viral reverse transcriptase, or viral integrase, but these enzymes are only a small fraction of the viral proteins responsible for some key steps in viral replication. To develop new therapeutic strategies, we need to understand additional steps in viral replication and to identify new potential targets. Rev is an essential HIV protein that is required to mediate transport of HIV viral mRNAs from the nucleus, where they are transcribed, into the cytoplasm, where they are either translated or packaged into new virions. Early in infection, viral mRNAs are fully processed in the nucleus and exported to the cytoplasm, where several small regulatory proteins, including Rev, are synthesized. The Rev protein itself is imported back into the nucleus, where it interacts with newly transcribed viral mRNAs D from the nucleus to the cytoplasm before full RNA processing takes place. These longer mRNAs code for the structural proteins necessary to assemble a new virus. Thus, binding of Rev to the RRE changes the pattern of gene expression from production of the early regulatory genes to production of the late structural genes, and this binding is therefore a potential point of therapeutic intervention. Several human cellular proteins act in concert with Rev to mediate the nuclear transport of viral mRNAs. In collaboration with L.R. Gerace and J.R. Yates, Department of Cell Biology, using a proteomic method to investigate Rev-RRE–associated factors, we identified dozens of such potential cofactor proteins. In particular, we discovered several so-called DEAD-box helicases, named after the conserved sequence signature, that associate with Rev during viral mRNA transport. The helicase DDX1 250 MOLECULAR BIOLOGY 2008 is thought to associate with Rev while Rev is bound to the RRE RNA in the nucleus; the helicase DDX3 interacts indirectly with Rev through the transport protein CRM-1. The presence of helicases in the ribonucleoprotein complex for viral mRNA export raises some interesting questions about how Rev functions. Helicases are ATPdependent motors that can unwind duplex RNAs or displace proteins from RNA-protein complexes. Possibly these helicases play critical roles in either assembling the proper RNA complex for nuclear transport or in disassembling the complex in the cytoplasm to allow translation or packaging of the virus. We are using biochemical and structural biology approaches to investigate the interactions of Rev with the helicases DDX1 and DDX3. The normal human substrates for these helicases are not known, and we must develop binding and functional assays based on Rev and Rev-RRE complexes. The protein-protein interactions are monitored by using fluorescence assays or isothermal titration calorimetry; formation of RNA-protein complexes is monitored by using polyacrylamide electrophoretic mobility shift assays. In addition, we are developing helicase ATPase assays with a variety of substrates to determine how the helicase activity is modulated in the presence of Rev and RRE. Finally, we are working toward structure determination of protein-protein and protein-RNA complexes to understand the molecular basis for this interaction. Our results will provide the basis for understanding potentially new targets for antiviral therapy. In addition, although helicases are widespread in human cells, the authentic substrates for these enzymes are known in only a few instances. Studying of Rev as a cargo for nuclear transport and as an authentic substrate for helicases will provide important insights into helicase function. The biochemical and structural work may lead to assays for the discovery of inhibitors of Rev function by a novel mechanism with therapeutic potential. PUBLICATIONS Edgcomb, S.P., Aschrafi, A., Kompfner, E., Williamson, J.R., Gerace, L., Hennig, M. Protein structure and oligomerization are important for the formation of exportcompetent HIV-1 Rev-RRE complexes. Protein Sci. 17:420, 2008. Hennig, M., Scott, L.G., Sperling, E., Bermel, W., Williamson, J.R. Synthesis of 5-fluoropyrimidine nucleotides as sensitive NMR probes of RNA structure. J. Am. Chem. Soc. 129:14911, 2007. Naidoo, N., Harrop, S.J, Sobti, M., Haynes, P.A., Szymczyna, B.R., Williamson, J.R., Curmi, P.M., Mabbutt, B.C. Crystal structure of Lsm3 octamer from Saccharomyces cerevisae: implications for Lsm ring organisation and recruitment. J. Mol. Biol. 377:1357, 2008. Sperling, E., Bunner, A.E., Sykes, M.T., Williamson, J.R. Quantitative analysis of isotope distributions in proteomic mass spectrometry using least-squares Fourier transform convolution. Anal. Chem. 80:4906, 2008. THE SCRIPPS RESEARCH INSTITUTE Vallurupalli, P., Scott, L., Williamson, J.R., Kay, L.E. Strong coupling effects during X-pulse CPMG experiments recorded on heteronuclear ABX spin systems: artifacts and a simple solution. J. Biomol. NMR 38:41, 2007. Structure and Biology of RNA Interference Machinery I.J. MacRae, P.-W. Lau, A.J. Pratt NA interference is a widespread eukaryotic mechanism of gene silencing that plays a fundamental role in many aspects of animal biology, including developmental timing, stem cell division, memory, and learning. On the cellular level, RNA interference is mediated by a family of ribonucleoprotein complexes called RNA-induced silencing complexes (RISCs), which silence genes by mediating translational repression and degradation of targeted mRNAs. The versatility and power of RNA interference arises from the fact that RISCs can be programmed to target any nucleic acid sequence for silencing. RISC programming is therefore a critical cellular function, requiring the action of a specialized macromolecular assembly called the RISCloading complex (RLC). We are studying the structure and catalytic mechanism of the human RLC. We hope that insight into the function of this complex will facilitate the development of therapeutic agents based on RNA interference. On the molecular level, RLCs program RISCs with target sequence information by mediating the noncovalent binding, or “loading,” of an RNA of approximately 22 nucleotides into the protein Argonaute. Argonaute forms the core subunit of RISCs, in which the small RNA functions as a guide for gene silencing through base-pairing recognition of target mRNAs. One of our major long-term goals is to visualize RISC programming by obtaining high-resolution crystal structures of the RLC at multiple steps in loading. Thus, we have established an efficient method for reconstituting human RLCs from individually purified recombinant preparations of the 3 RLC subunits: Dicer, Argonaute 2, and the TARRNA binding protein. The recombinant nature of the reconstitution will also allow us to produce mutant RLCs that stall at key steps in loading. We will then use the mutants to probe the mechanism of RISC loading. We anticipate that crystallization of the RLC will be technically challenging and may take years to accomplish. To obtain structural information in the earlier stages of this study, we have established a collabora- R MOLECULAR BIOLOGY 2008 tion with B. Carragher and C. Potter at Scripps Research in the National Resource for Automated Molecular Microscopy. Our goal is to generate structures of the RLC by using electron cryomicroscopy and single-particle analysis. These structures will reveal the overall architecture of the RLC and enable us to visualize largescale conformational changes associated with different steps of RISC loading. Modeling the available crystal structures of Giardia Dicer, which represents the conserved catalytic core structure of human Dicer, and Pyrococcus Argonaute, an archeal ortholog of human Argonaute 2, into electron microscopy maps will answer some of the basic questions that shape our thinking about the RLC mechanism. Ideas and hypotheses derived from structural studies will then be tested biochemically by using reconstituted mutant and chemically modified RLCs. The combined structural and functional studies will provide new insights into RNA interference in humans and may suggest new approaches for using RNA interference for the treatment of human disease. Development of the Genetic Code and Its Connection to Human Disease P. Schimmel, X.-L. Yang, R. Belani, E. Chong, M. Guo, R.T. Guo, M. Hanan, W.-W. He, I.L. Jung, M. Kapoor, J. Liu, E. Merriman, M.H. Nawaz, R. Shapiro, M. Vo, W. Zhang, Q. Zhou e focus on aminoacyl tRNA synthetases and how their role as catalysts of aminoacylation is connected to disease and to broader biological systems through expanded functions that were developed over the long evolutionary history of the enzymes. The enzymes are ancient and are thought to have appeared in the transition from the putative RNA world to the theater of proteins. By linking amino acids to tRNAs that bear anticodon triplets encoding the attached amino acids, the synthetases establish the algorithm of the genetic code. Thus, alanyl-tRNA synthetases catalyze formation of Ala-tRNAAla, glycyl-tRNA synthetase catalyzes production of Gly-tRNAGly, and isoleucyl-tRNA synthetase produces Ile-tRNAIle. Altogether there are 20 tRNA synthetases, 1 for each amino acid, and the tRNA to which the amino acid is attached bears the triplet of the code for that amino acid. It is, therefore, this reaction that establishes the code. W THE SCRIPPS RESEARCH INSTITUTE 251 Recent research established that heritable mutations in these 20 enzymes are causally linked to specific human diseases. This linkage occurs in one of two ways. First is the connection to mistranslation (Fig. 1). F i g . 1 . Effects of mistranslation. This connection occurs because many of the enzymes have an editing activity (encoded by a distinct active site) that clears amino acids when the amino acids are attached to the wrong tRNA. For example, alanyltRNA synthetase occasionally confuses glycine or serine for alanine, so Gly-tRNAAla or Ser-tRNAAla is produced. However, the occasional mischarged tRNA is cleared by a specific editing activity that can distinguish serine and glycine from alanine in the context of tRNAAla. Were these mischarged amino acids not cleared away, mistranslation would occur. Recently, we found that disruption of the editing activity not only is toxic to bacteria but also leads to serious pathologic changes in mammalian cells, in a trans-dominant way. Even a mild mutation, which produces only a 2-fold decrease in the activity for editing, leads to a heritable condition in mice, characterized by ataxia and neurodegeneration. We also discovered that mistranslation is mutagenic in aging bacteria. We are now exploring the possibility that mistranslation is mutagenic in mammalian cells in a way that could lead to oncogenic transformation. How does alanyl-tRNA synthetase distinguish mischarged Ser-tRNA Ala from the correctly charged SertRNASer; that is, how does it use the context of the tRNA to strip serine from tRNAAla but not from tRNASer? The enzyme has distinct domains for aminoacylation (formation of charged tRNAAla) and editing (clearing of 252 MOLECULAR BIOLOGY 2008 mischarged tRNAAla). These domains are encoded by polypeptides arranged in tandem along the sequence of alanyl-tRNA synthetase. For aminoacylation, tRNAAla synthetases throughout evolution are marked for aminoacylation with alanine by a single guanine-uracil base pair (G3:U70; located in the acceptor stem; Fig. 2). F i g . 2 . tRNA Ala acceptor stems. Transfer of this base pair into non-tRNAAla synthetases converts the enzymes into alanine-accepting tRNAs. The recognition of a single G3:U70 base pair is through determinants in the N-terminal aminoacylation domain of the protein. Remarkably, and to our surprise, this same base pair is used by the editing domain to pick out mischarged tRNAAla. Thus, the same base pair is recognized by distinct domains within the same protein. Last, in the human and mouse genomes, in addition to being encoded as part of alanyl-tRNA synthetase, the editing domain is separately encoded as a standalone fragment known as AlaXp. This fragment can also clear mischarged tRNAAla (Fig. 3). Thus, mammalian F i g . 3 . Alanyl-tRNA synthetase (AlaRS) and artificial and natural editing-proficient fragments. cells have developed 3 ways to prevent confusion of serine and glycine for alanine: (1) reasonably accurate (about 99%) selection of alanine in the aminoacylation step, (2) clearance of occasional errors, of confusing serine or glycine for alanine, by the editing domain of THE SCRIPPS RESEARCH INSTITUTE the enzyme, and (3) clearance of any residual mischarged tRNAAla by the free-standing AlaXp. In summary, we started with the observation of how a single mutation in the site for editing led to disease in mice. From there, we uncovered the first example of 2 distinct domains in the same protein being able to recognize the same base pair. The second connection of tRNA synthetases to heritable human diseases is through the expanded functions of the enzymes. Many human tRNA synthetases have functions beyond aminoacylation. For example, when activated, tyrosyl-tRNA synthetase and tryptophanyl-tRNA synthetase have opposing activities in angiogenesis. Lysyl-tRNA synthetase is incorporated into HIV virions for bringing in tRNALys, which is used as a primer for reverse transcription of the viral RNA genome during infection of a host cell. These extratranslation activities link aminoacyl-tRNA synthetases with boarder biological systems. Two specific tRNA synthetases, tyrosyl- and glycyltRNA synthetase, are linked to Charcot-Marie-Tooth (CMT) disease, the most common heritable peripheral neuropathy; 1 of 2000 persons in the United States and Europe has the disease. Eleven different mutant alleles of GARS, the gene that encodes glycyl-tRNA synthetase, are reported to cause an axonal form of CMT in a dominant way. About half of the 11 CMTcausing mutants of glycyl-tRNA synthetase are unaffected in their aminoacylation activities. Similarly, dominant mutations in YARS, the gene that encodes tyrosyl-tRNA synthetase, have also been detected as the genetic cause of the disease in patients with CMT. Again, the mutations are not correlated with an aminoacylation defect. The lack of correlation of CMT mutations with aminoacylation activity suggests additional, expanded functions for glycyl- and tyrosyl-tRNA synthetases in neuronal cells. The 11 CMT-linked mutations are spread out on the primary sequence of glycyl-tRNA synthetase. However, as revealed in our crystal structure of the synthetase, all of the CMT-associated residues are located on or near the dimerization interface of the α2 homodimer (Fig. 4). Strikingly, 2 CMT-causing mutations, each from a different subunit, are associated with residues in the wildtype enzyme that make contact with each other across the dimer interface. This finding suggests that the dimer interface is critical for the potential expanded function of glycyl-tRNA synthetase. Because both monomer and dimer forms of the synthetase coexist in equilibrium, we hypothesized that the monomer form, which is inactive MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 253 Kapoor, M., Zhou, Q., Otero, F., Myers, C.A., Bates, A., Belani, R., Liu, J., Luo, J.-K., Tzima, E., Zhang, D.-E., Yang, X.-L., Schimmel, P. Evidence for annexin IIS100A10 complex and plasmin in mobilization of cytokine activity of human TrpRS. J. Biol. Chem. 283:2070, 2008. Waas, W.F., Druzina, Z., Hanan, M., Schimmel, P. Role of a tRNA base modification and its precursors in frameshifting in eukaryotes. J. Biol. Chem. 282:26026, 2007. Waas, W.F., Schimmel, P. Evidence that tRNA synthetase-directed proton transfer stops mistranslation. Biochemistry 46:12062, 2007. Yang, X.-L., Kapoor, M., Otero, F.J., Slike, B.M., Tsuruta, H., Frausto, R., Bates, A., Ewalt, K.L., Cheresh, D.A., Schimmel, P. Gain-of-function mutational activation of a human tRNA synthetase procytokine. Chem. Biol. 14:1323, 2007. Zhou, Q., Kiosses, W.B., Liu, J., Schimmel, P. Tumor endothelial cell tube formation model for determining anti-angiogenic activity of a tRNA synthetase cytokine. Methods 44:190, 2008. Directed Evolution of Nucleic Acid Enzymes F i g . 4 . Crystal structure of human glycyl-tRNA synthetase homodimer. for aminoacylation, has a distinct biological role specific for neuronal cells. Disruption of this expanded role links glycyl-tRNA synthetase to the etiology of CMT. Interestingly, when transfected into a mouse neuroblastoma N2a cell line, genes encoding each of the CMT-causing glycyl-tRNA synthetase mutants were defective in distribution into neurite terminals. This distribution defect is reminiscent of the muscle weakness around the terminal nerves in patients with CMT. We have speculated that the CMT-associated mutations of glycyl-tRNA synthetase affect transportation of the synthetase into the neurites via a mechanism linked to the dimerization interface. Our awareness of the connections of tRNA synthetases, which are intimately associated with the development of the genetic code, to diseases is only beginning. We anticipate that many more connections will be found. PUBLICATIONS Bacher, J., Waas, W.F., Metzgar, D., de Crécy-Lagard, V., Schimmel, P. Genetic code ambiguity confers a selective advantage on Acinetobacter baylyi. J. Bacteriol. 189:6494, 2007. Beebe, K., Mock, M., Merriman, E., Schimmel, P. Distinct domains of tRNA synthetase recognize the same base pair. Nature 451:90, 2008. Beebe, K., Waas, W., Druzina, Z., Guo, M., Schimmel, P. A universal plate format for increased throughput of assays that monitor multiple aminoacyl transfer RNA synthetase activities. Anal. Biochem. 368:111, 2007. Greenberg, Y., King, M., Kiosses, W.B., Ewalt, K., Yang, X.-L., Schimmel, P., Reader, J.S., Tzima, E. The novel fragment of tyrosyl-tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J. 22:1597, 2008. Guo, M., Ignatov, M., Musier-Forsyth, K., Schimmel, P., Yang, X.-L. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. U. S. A. 105:2331, 2008. G.F. Joyce, S.E. Hamilton, D.P. Horning, T.A. Lincoln, B.J. Lam, B.M. Paegel, K.L. Petrie, S.B. Voytek he scientific community will soon celebrate the 200th anniversary of the birth of Charles Darwin and the 150th anniversary of the publication of his seminal work On the Origin of Species by Means of Natural Selection. The principles of darwinian evolution are fundamental to understanding biological organization at the level of populations of organisms and for explaining the development of biological genomes and macromolecular function. In our laboratory, darwinian evolution has become a chemical tool for discovering and optimizing functional macromolecules in the test tube. We have developed powerful methods for the in vitro evolution of nucleic acids and are applying those methods to the discovery of molecules of biochemical and biomedical importance. In addition, we are studying the processes of darwinian evolution itself, carried out at the level of molecules rather than at the level of cells or organisms. T CONTINUOUS IN VITRO EVOLUTION We have devised a system for the continuous in vitro evolution of RNA enzymes that have RNA-joining activity. The system operates at a constant temperature within a common reaction vessel. RNA enzymes in a population of trillions are challenged to attach themselves to an RNA substrate, and as a consequence, the reacted enzymes become amplified by polymerase proteins (also present in the reaction mixture) to generate progeny. The progeny enzymes in turn have the opportunity to perform the reaction, causing the population to expand exponentially. Whenever the supply of sub- 254 MOLECULAR BIOLOGY 2008 strates becomes exhausted, a fresh supply of reactants can be provided, allowing exponential amplification to continue indefinitely. Until recently, all continuous in vitro evolution experiments were done with a single species of RNA enzyme derived from the CL1 ligase. We recently established a second continuously evolving enzyme based on descendants of the DSL ligase. This new enzyme was propagated for hundreds of successive generations to optimize its catalytic activity. Recently, we challenged the 2 distinct species of continuously evolving enzymes to operate within the same environment (Fig. 1). Initially, variants of the CL1 ligase dominated the mixture, prompting us to add an F i g . 1 . Continuous coevolution of 2 distinct species of RNA enzymes with RNA-joining activity. Zigzag lines indicate the concentration of the CL1 (blue) and DSL (red) enzymes before and after each cycle of exponential growth and dilution (based on the concentration of the corresponding cDNA). inhibitory molecule to modulate their growth. Subsequently, variants of the DSL ligase became dominant, and these too were kept in check by adding a speciesspecific inhibitor. Eventually, we succeeded in maintaining the 2 species without the use of inhibitors by supplying 5 different RNA substrates. Each of the 2 enzymes evolved to use different substrates as its preferred resource, exploiting distinct niches within the common environment. This situation is a demonstration of niche formation at the molecular level, analogous to processes of biological evolution essential for maintaining species diversity within natural ecosystems. S E L F - S U S TA I N E D R E P L I C AT I O N O F R N A The continuous in vitro evolution system depends on 2 protein enzymes, a retroviral reverse transcriptase and a bacteriophage RNA polymerase, to bring about the amplification of reacted RNA enzymes. Recently, we developed a system in which the RNA enzymes THE SCRIPPS RESEARCH INSTITUTE catalyze their own replication. We began with the R3C ligase developed previously in our laboratory. This molecule has a simple architecture amenable to various rearrangements. Previously, the R3C ligase was configured so that it would join 2 pieces of RNA to produce additional copies of itself, thus achieving RNA-catalyzed self-replication. Next, the enzyme was converted to a cross-catalytic format whereby 2 RNA enzymes brought about each other’s synthesis from a total of 4 component RNA substrates. During the past year, we used in vitro evolution to enhance the activity of the cross-replicating RNA enzymes, improving their catalytic rate by more than 20-fold and increasing their extent of reaction from 15% to 90%. The resulting enzymes are able to undergo self-sustained exponential amplification at a constant temperature, achieving nearly billion-fold amplification in 30 hours. The cross-replicating RNA enzymes can amplify themselves indefinitely in the absence of proteins or any other biological materials. As in the continuous evolution system, we allow the molecules to expand exponentially until the supply of substrates is exhausted and then provide fresh reactants to allow exponential amplification to continue indefinitely. We have prepared several versions of the cross-replicating enzymes that differ with respect to their genotype and corresponding phenotype. The genotype specifies the identity of the cross-replicating partners, and the phenotype is reflected in the catalytic properties of the molecules. In this way, we are striving to construct an artificial genetic system that can undergo self-sustained darwinian evolution. Thus far, we have shown selection among various crossreplicators that undergo exponential amplification within a common reaction mixture. Occasionally, a novel recombinant arises that amplifies more efficiently than either of its parents. With this system, it may be possible to explore alternative solutions to different environmental constraints, as occur in the natural evolution of biological organisms. PUBLICATIONS Joyce, G.F. Forty years of in vitro evolution. Angew. Chem. Int. Ed. 46:6420, 2007. Paegel, B.M., Joyce, G.F. Darwinian evolution on a chip. PLoS Biol. 6:e85, 2008. Voytek, S.B., Joyce, G.F. Emergence of a fast-reacting ribozyme that is capable of undergoing continuous evolution. Proc. Natl. Acad. Sci. U. S. A. 104:15288 2007. MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 255 Studies at the Interface of Molecular Biology, Chemistry, and Medicine C.F. Barbas III, K. Albertshofer, T. Bui, R.P. Fuller, C. Gersbach, B. Gonzalez, R. Gordley, J. Guo, D.H. Kim, R.A. Lerner, W. Nomura, A. Onoda, S.S.V. Ramasastry, M. Santa Marta, L.J. Schwimmer, D. Shabat,* F. Tanaka, U. Tschulena, N. Utsumi, K.S. Yi, H. Zhang * Tel Aviv University, Tel Aviv, Israel e are concerned with problems in molecular biology, chemistry, and medicine. Many of our studies involve learning or improving Nature’s strategies to prepare novel molecules that perform specific functional tasks, such as regulating a gene, destroying cancer, or catalyzing a reaction with enzymelike efficiency. We hope to apply these novel insights, technologies, methods, and their products to provide solutions to human diseases, including cancer, HIV disease, and genetic diseases. W D I R E C T I N G T H E E V O L U T I O N O F C ATA LY T I C F U N C T I O N Using reactive immunization, we have developed antibodies that catalyze aldol as well as retro-aldol reactions of a wide variety of molecules. The catalytic proficiency of the best of these antibodies is almost 1014, a value 1000 times that of the best catalytic antibodies reported to date and overall the best of any synthetic protein catalyst. We have shown the efficient asymmetric synthesis and resolution of a variety of molecules, including tertiary and fluorinated aldols, and have used these chiral synthons to synthesize natural products (Fig. 1). These results highlight the potential synthetic usefulness of catalytic antibodies as artificial enzymes in addressing problems in organic chemistry that are not solved by using natural enzymes or more traditional synthetic methods. Other advances in this area include the development of the first peptide aldolase enzymes. By using both design and selection, we have created small peptide catalysts that recapitulate many of the kinetic features of large enzyme catalysts. These smaller enzymes allow us to address the relationship between the size of natural proteins and the proteins’ catalytic efficiency. O R G A N O C ATA LY S I S : A B I O O R G A N I C A P P R O A C H T O C ATA LY T I C A S Y M M E T R I C S Y N T H E S I S To further explore the principles of catalysis, we are studying amine catalysis as a function of catalytic F i g . 1 . A variety of compounds synthesized with the world’s first commercially available catalytic antibody, 38C2, produced at Scripps Research. scaffold. Using insights garnered from our studies of aldolase antibodies, we determined the efficacy of simple chiral amines and amino acids for catalysis of aldol and related imine and enamine chemistries such as Michael, Mannich, Knoevenagel, and Diels-Alder reactions. Although aldolase antibodies are superior catalysts in terms of the kinetic parameters, these more simple catalysts are enabling us to quantify the significance of pocket sequestration in catalysis. Furthermore, many of these catalysts are cheap, environmentally friendly, and practical for large-scale synthesis. With this approach, we showed the scope and usefulness of the first efficient amine catalysts of direct asymmetric aldol, Mannich, Diels-Alder, and Michael reactions. The organocatalyst approach is a direct outcome of our studies of catalytic antibodies and provides an effective alternative to organometallic reactions that use severe reaction conditions and oftentoxic catalysts. In extensions of these concepts, we designed novel amino acid derivatives that direct the stereochemical outcome of reactions in ways not possible with proline. In other studies, we created the first asymmetric smallmolecule aldol catalysts that are highly effective with water and seawater as solvent. We think that our results are also relevant to the prebiotic synthesis of the molecules of life. For example, we have shown that our amino acid strategy can be used to synthesize carbohydrates directly, thereby providing a provocative prebiotic route to the sugars essential for life. 256 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE I S P R I M O R D I A L A S Y M M E T R I C O R G A N O C ATA LY S I S A N E X TA N T B I O S Y N T H E T I C M E C H A N I S M ? We think that in time, research will show that organocatalysts or “aminozymes” (chiral amines or amino acids with biosynthetic roles) constitute components of an unseen biosynthetic apparatus at work in cells today. As we begin to appreciate the fascinating chemical transformations that are now possible through organocatalysis, and amino acid catalysis in particular, we need to look at cellular metabolism and biosynthesis in a new light. Classically, we are trained to search for a “protein” enzyme for each and every step in the synthesis of a natural product in vivo. We suggest that many of the more elusive metabolic enzymes are likely to be organocatalysts and, in many instances, simple amino acids. Because intracellular concentrations of amino acids can exceed 1 M, many wonderful and diverse exotic natural products may actually be synthesized in vivo with the aid of aminozymes and other forms of organocatalysts more complicated than amino acids (Fig. 2). ANTIBODY ENGINEERING: THERAPEUTIC ANTIBODIES, IN AND OUT OF CELLS We developed the first human antibody phage display libraries and the first synthetic antibodies and methods for the in vitro evolution of antibody affinity. The ability to manipulate large libraries of human antibodies and to evolve such antibodies in the laboratory provides tremendous opportunities to develop new medicines. Laboratories and pharmaceutical companies around the world now apply the phage display technology that we developed for antibody Fab fragments. In our laboratory, we are targeting cancer and HIV disease. One of our antibodies, IgG1-b12, protects animals against primary challenge with HIV type 1 (HIV-1) and has been further studied by many researchers. We improved this antibody by developing in vitro evolution strategies that enhanced its neutralization activity. By coupling laboratory-evolved antibodies with potent toxins, we showed that immunotoxins can effectively kill infected cells. We are also developing genetic methods to halt HIV by gene therapy. We created unique human antibodies that can be expressed inside cells to make the cells resistant to HIV infection. In the future, these antibodies might be delivered to the stem cells of patients infected with HIV-1, allowing the development a disease-free immune system that would obviate the intense regimen of antiviral drugs now required to treat HIV disease. Using our increased understanding of antibody-antigen interactions, we extended our efforts in cancer F i g . 2 . Potential roles of aminozymes in the biosynthesis of the Daphniphyllum alkaloids (A) and the potential anticancer compound FR182877 (B). therapy and developed rapid methods for creating human antibodies from antibodies derived from other species. We produced human antibodies that should enable us to selectively starve a variety of cancers by inhibiting angiogenesis and antibodies that will be used to deliver radioisotopes to colon cancers to destroy the tumors. We hope that these antibodies will be used in clinical trials done by our collaborators at the SloanKettering Cancer Center, New York, New York. On the basis of our studies on HIV-1, we used intracellular expression of antibodies directed against angiogenic receptors to create a new gene-based approach to cancer. Our results indicate that this type of gene therapy can be successfully applied to the treatment of cancer. T H E R A P E U T I C A P P L I C AT I O N S O F C ATA LY T I C ANTIBODIES The development of highly efficient catalytic antibodies opens the door to many practical applications. One of the most fascinating is the use of such antibodies in human therapy. We think that use of this strategy can improve chemotherapeutic approaches to diseases such as cancer and AIDS. Chemotherapeutic regimens are typically limited by nonspecific toxic effects. To address this problem, we developed a novel and broadly applicable drug-masking chemistry that operates in conjunction with our unique broad-scope catalytic antibodies. This masking chemistry is applicable to a wide range of drugs because it is compatible with virtually any heteroatom. We showed that generic drug-masking groups can be selectively removed by sequential retro-aldol-retro-Michael reactions catalyzed by antibody 38C2 (Fig. 3). This reaction cascade is not catalyzed by any known natural enzyme. Application of this masking chemistry to the anticancer drugs doxorubicin, camptothecin, and etoposide produced prodrugs with substantially reduced toxicity. MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 257 F i g . 3 . Targeting cancer and HIV with prodrugs activated by catalytic antibodies. A bifunctional antibody is shown targeting a cancer cell for destruction. A nontoxic analog of doxorubicin, prodoxorubicin, is being activated by an aldolase antibody to the toxic form of the drug. These prodrugs are selectively unmasked by the catalytic antibody when the antibody is applied at therapeutically relevant concentrations. The efficacy of this approach has been shown in in vivo models of cancer. Currently, we are developing more potent drugs and novel antibodies that will allow us to target breast, colon, and prostate cancers as well as cells infected with HIV-1. On the basis of our preliminary findings, we think that our approach can become a key tool in selective chemotherapeutic strategies. To see a movie illustrating this approach, visit http://www.scripps.edu/mb/barbas/antibody/antibody.mov. C H E M I C A L LY P R O G R A M M E D A N T I B O D I E S : T H E ADVENT OF CHEMOBODIES We think that combining the chemical diversity of small synthetic molecules with the immunologic characteristics of antibody molecules will lead to therapeutic agents with superior properties. Therefore, we developed a conceptually new device that equips small synthetic molecules with both the immunological effector functions and the long serum half-life of a generic antibody molecule. For a prototype, we developed a targeting device based on the formation of a covalent bond of defined stoichiometry between (1) a 1,3-diketone derivative of an arginine–glycine–aspartic acid peptidomimetic that targets the integrins αvβ3 and αvβ5 and (2) the reactive lysine of aldolase antibody 38C2 (Fig. 4). The resulting complex spontaneously assembled in vitro and in vivo, selectively retargeted antibody 38C2 to the surface of cells expressing the integrins αvβ3 and αvβ5, dramatically increased the circulatory half-life of the peptidomimetic, and effectively reduced tumor growth in animal models of human Kaposi sarcoma, colon cancer, and melanoma. Three novel chemically programmed antibodies, an entirely new class of drugs, are now in phase 1 clinical studies. F i g . 4 . Designed small-molecule targeting agents (SCS-873 is shown) program the specificity of the antibody 38C2 (A). The resulting chemobodies (cp38C2, B) have characteristics that are often superior to those of either the small molecule or the antibody alone. ZINC FINGER GENE SWITCHES AND ENZYMES The solutions to many diseases might be simply turning genes on or off in a selective way or adding or deleting genes. To accomplish all of these aims, we are studying molecular recognition of DNA by zinc finger proteins and methods of creating novel zinc finger DNAbinding proteins. We showed that proteins that contain zinc fingers can be selected or designed to recognize novel DNA sequences. These studies are aiding the elucidation of rules for sequence-specific recognition within this family of proteins. We selected and designed specific zinc finger domains that will constitute an alphabet of 64 domains that will allow any DNA sequence to be bound selectively. The prospects for this “second genetic code” are fascinating and may have a major impact on basic and applied biology. We showed the potential of this approach in multiple mammalian and plant cell lines and in whole organisms. With the use of characterized modular zinc finger domains, polydactyl proteins capable of recognizing an 18-nucleotide site can be rapidly constructed (see www.zincfingertools.org). Our results suggest that zinc finger proteins might be useful as genetic regulators for a variety of human ailments and provide the basis for a new strategy of gene therapy. Our goal is to develop this class of therapeutic proteins to inhibit or enhance the synthesis of proteins, providing a direct strategy for fighting diseases of either somatic or viral origin. We are also developing proteins that will inhibit the growth of tumors and others that will inhibit the expres- 258 MOLECULAR BIOLOGY 2008 sion of a protein known as CCR5, which is a key to infection of human cells by HIV-1. We developed an HIV-1–targeting transcription factor that strongly suppresses HIV-1 replication and another transcription factor that upregulates fetal hemoglobin as a treatment for sickle cell anemia. More recently, we have focused on evolving zinc finger enzymes that modify the genome. These studies have led to the development of programmable zinc finger recombinases (Fig. 5) that promise to THE SCRIPPS RESEARCH INSTITUTE Zhang, H., Ramasastry, S.S.V., Tanaka, F., Barbas, C.F. III. Organocatalytic antiMannich reactions with dihydroxyacetone and acyclic dihydroxyacetone derivatives: a facile route to amino sugars. Adv. Synth. Catal. 350:791, 2008. Catalytic Antibodies, Synthetic Enzymes, Biomolecular Computing, and Synthetic Capsids E. Keinan, O. Reany, N. Metanis, E. Kossoy, M. Soreni, R. Piran, M. Sinha, I. Ben-Shir, T. Shekhter, T. Ratner, T. Mejuch, E. Solel, S. Shoshani, R. Gershoni, A. Karmakar, D. Pappo, G. Parvari C ATA LY T I C A N T I B O D I E S F i g . 5 . Model of a zinc finger recombinase with programmable specificity created by using rational design and directed molecular evolution. reshape the way scientists manipulate the genome for study and therapy of disease. PUBLICATIONS Barbas, C.F. III. Organocatalysis lost: modern chemistry, ancient chemistry, and an unseen biosynthetic apparatus. Angew. Chem. Int. Ed. 47:42, 2008. Blancafort, P., Tschan, M.P., Bergquist, S., Guthy, D., Brachat, A., Sheeter, D.A., Torbett, B.E., Erdmann, D., Barbas, C.F. III. Modulation of drug resistance by artificial transcription factors. Mol. Cancer Ther. 7:688, 2008. Jiang, L., Althoff, E.A., Clemente, F.R, Doyle, L., Röthlisberger, D., Zanghellini, A., Gallaher, J.L., Betker, J.L., Tanaka, F., Barbas, C.F. III, Hilvert, D., Houk, K.N., Stoddard, B.L., Baker D. De novo computational design of retro-aldol enzymes. Science 319:1387, 2008. Magnenat, L., Schwimmer, L.J., Barbas, C.F. III. Drug-inducible and simultaneous regulation of endogenous genes by single-chain nuclear receptor-based zinc-finger transcription factor gene switches. Gene Ther. 15:1223, 2008. Nomura, W., Barbas, C.F. III. In vivo site-specific DNA methylation with a designed sequence-enabled DNA methylase. J. Am. Chem. Soc. 129:8676, 2007. Ramasastry, S.S.V., Albertshofer, K., Utsumi, N., Barbas, C.F. III. Water-compatible organocatalysts for direct asymmetric syn-aldol reactions of dihydroxyacetone and aldehydes. Org. Lett.10:1621, 2008. Ramasastry, S.S.V., Albertshofer, K., Utsumi, N., Tanaka, F., Barbas, C.F. III. Mimicking fructose and rhamnulose aldolases: organocatalytic syn-aldol reactions with unprotected dihydroxyacetone. Angew. Chem. Int. Ed. 46:5572, 2007. Tanaka, F., Fuller, R.P., Asawapornmongkol, L., Warsinke, A., Gobuty, S., Barbas, C.F. III. Development of a small peptide tag for covalent labeling of proteins. Bioconjug. Chem. 18:1318, 2007. relatively unexplored opportunity in the science of catalytic antibodies is modifying the phenotype of an organism by incorporating the gene for a catalytic antibody into the genome of that organism. An attractive application of this concept would be the expression of such a catalyst in transgenic plants to provide a beneficial trait. For example, introduction of a herbicide-resistance trait in commercial plants is highly desirable because plants with the trait could be grown in the presence of a nonselective herbicide that affects only weeds and other undesired plant species. We have shown that herbicide-resistant plants can be engineered by designing both a herbicide and a catalytic antibody that destroys the herbicide within the plants. Such a transgenic plant was achieved via a 3-step maneuver: (1) development of a new carbamate herbicide, one that can be catalytically destroyed by the aldolase antibody 38C2; (2) separate expression of the light chain and half of the heavy chain (Fab) of the catalytic antibody in the endoplasmic reticulum of 2 plant lines of Arabidopsis thaliana; and (3) crosspollination of these 2 transgenic plants to produce a herbicide-resistant F1 hybrid (Fig. 1). In vivo expression of catalytic antibodies could become a useful, general strategy to achieve desired phenotype modifications not only in plants but also in other organisms. A SYNTHETIC ENZYMES Utsumi, N., Imai, M., Tanaka, F., Ramasastry, S.S.V., Barbas, C.F. III. Mimicking aldolases through organocatalysis: syn-selective aldol reactions with protected dihydroxyacetone. Org. Lett. 9:3445, 2007. Zhang, H., Mitsumori, S., Utsumi, N., Imai, M., Garcia-Delgado, M., Mifsud, M., Albertshofer, K., Tanaka, F., Barbas, C.F. III. Catalysis of 3-pyrrolidinecarboxylic acid and related pyrrolidine derivatives in enantioselective anti-Mannich-type reactions: importance of the 3-acid group on pyrrolidine for stereocontrol. J. Am. Chem. Soc. 130:875, 2008. Selenoenzymes have a central role in maintaining cellular redox potential. These enzymes have selenylsulfide bonds in their active sites that catalyze the reduction of peroxides, sulfoxides, and disulfides. The selenol-disulfide exchange reaction is common to all of the enzymes, and the active-site redox potential reflects MOLECULAR BIOLOGY 2008 F i g . 1 . Influence of herbicide (4) on the rooting and development of seedlings of F 1 hybrids and control A thaliana plants. The control plants are shown in A and C; the hybrid plant lines (F1) expressing both light and heavy chains of catalytic antibody 38C2, in B and D. Plantlets grown on medium without the herbicide are shown in A and B; those grown with the herbicide are shown in C and D. the ratio between the forward and reverse rates of this reaction. The preparation of enzymes containing selenocysteine is experimentally challenging. As a result, little is known about the kinetic role of selenols in enzyme active sites, and the redox potential of a selenylsulfide or diselenide bond in a protein has not been experimentally determined. To fully evaluate the effects of selenocysteine on oxidoreductase redox potential and kinetics, we chemically synthesized glutaredoxin 3 (Grx3) and all 3 selenocysteine variants of its conserved 11CXX14C active site and determined their redox potentials. The position of redox equilibrium between Grx3(C11U-C14U) (–308 mV) and thioredoxin (–270 mV) suggests a possible role for diselenide bonds in biological systems. Kinetic analysis showed that the lower redox potentials of the selenocysteine variants are due primarily to the greater nucleophilicity of the active-site selenium. The 102- to 104-fold increase in the rate of thioredoxin reduction by the selenoGrx3 analogs indicates that oxidoreductases containing either selenylsulfide or diselenide bonds can have physiologically compatible redox potentials and enhanced reduction kinetics in comparison with their sulfide counterparts. This research was done in collaboration with P.E. Dawson, Department of Cell Biology. BIOMOLECULAR COMPUTING DEVICES In fully autonomous molecular computing devices, all components, including input, output, software, and THE SCRIPPS RESEARCH INSTITUTE 259 hardware, are specific molecules that interact with each other through a cascade of programmable chemical events, progressing from the input molecule to the molecular output signal. DNA molecules and DNA enzymes have been used as convenient, readily available components of such computing devices because the DNA materials have highly predictable recognition patterns, reactivity, and information-encoding features. Furthermore, DNA-based computers can become part of a biological system, generating outputs in the form of biomolecular structures and functions. Our previously reported 2-symbol–2-state finite automata computed autonomously, and all of their components were soluble biomolecules mixed in solution. The hardware consisted of 2 enzymes, an endonuclease and a ligase, and the software and the input were double-stranded DNA oligomers. More recently, we designed and created 3-symbol–3-state automata that can carry out more complex computations. In addition, we found that immobilization of the input molecules on chips allowed parallel computation, a system that can be used encrypt information. The main advantage of autonomous biomolecular computing devices compared with electronic computers is the ability of the devices to interact directly with biological systems. No interface is required because all components of molecular computers, including hardware, software, input, and output, are molecules that interact in solution along a cascade of programmable chemical events. We showed for the first time that the output of a molecular finite automaton can be a visible bacterial phenotype. Our 2-symbol–2-state finite automaton uses linear double-stranded DNA inputs prepared by inserting a string of 6-bp symbols into the lacZ gene on plasmid pUC18. The computation resulted in a circular plasmid that differed from the original pUC18 by either a 9-bp (accepting state) or an 11-bp (unaccepting state) insert within the lacZ gene. Upon transformation and expression of the resultant plasmids in Escherichia coli, either blue colonies or white colonies, respectively, were formed (Fig. 2). SYNTHETIC CAPSIDS Stable structures of icosahedral symmetry can have numerous functional roles, including chemical microencapsulation and delivery of drugs and biomolecules, a way to observe encapsulated reactive intermediates, presentation of epitopes for efficient immunization, synthesis of nanoparticles of uniform size, and formation of structural elements for molecular supramolecular con- 260 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE Macromolecular Interactions: Evolution, Engineering, and Detection V.V. Smider, C.C. Liu, J. Mills, B. Hutchins, B. Leonard e are involved in several projects related to molecular recognition. These include understanding the evolution and biochemical characteristics of the human germ-line antibody repertoire, chemically enhancing antibody properties, analyzing RNA hairpin interactions that regulate DNA replication, and developing new chemical technologies to detect interactions between macromolecules. W ENHANCED AND NOVEL INTERACTIONS: ENGINEERED ANTIBODIES F i g . 2 . Computation with aaba input in the presence (A) and absence (B) of transition molecules results in white bacteria when the transition molecules are present. Computation with abba input in the presence (C) and absence (D) of transition molecules results in blue bacteria when the transition molecules are present. structs and molecular computing. By examining physical models of spherical virus assembly, we developed a general synthetic strategy for producing chemical capsids at size scales between fullerenes and spherical viruses. Such capsids can be formed by self-assembly from a class of novel symmetric molecules developed from a pentagonal core. By designing chemical complementarity into the 5 interface edges of the molecule, we can produce self-assembling stable structures of icosahedral symmetry. We considered 3 different binding mechanisms: hydrogen bonding, metal binding, and formation of disulfide bonds. These structures can be designed to assemble and disassemble under controlled environmental conditions. We have conducted molecular dynamics simulation on a class of corannulene-based molecules to demonstrate the characteristics of selfassembly and to aid in the design of the molecular subunits. This research was done in collaboration with A.J. Olson, Department of Molecular Biology. Antibodies bind their antigens noncovalently. However, for certain diagnostic or therapeutic applications, antibodies that irreversibly bind or modify their antigens would be useful. We recently succeeded in engineering a metal-dependent antibody that irreversibly binds its antigen, TNF-α, perhaps through an exchange inert cobalt complex (Fig. 1). In collaboration with P.G. Schultz, Department of Chemistry, we are engineering other metalloantibodies and are incorporating amino acids with unique side chains (unnatural amino acids) into antibodies. Similarly, we are creating unique multivalent protein constructs. This technology should allow both genetic and chemical expansion of the antibody repertoire for therapeutic and diagnostic applications. R N A H A I R P I N S R E G U L AT I N G D N A R E P L I C AT I O N RNA molecules are now recognized as important regulators of many cellular properties via RNA-RNA interactions. One of the simplest and evolutionarily ancient mechanisms of RNA-mediated regulation occurs at plasmid origins of replication. We are applying molecular evolution techniques to these regions and using simple copy number and compatibility assays to ascertain the regulatory and recognition features of these hairpins in Escherichia coli. B I O M O L E C U L A R D E T E C T I O N : O X A L AT E E S T E R CHEMIFLUORESCENCE PUBLICATIONS Kossoy, E., Lavid, N., Soreni-Harari, M., Shoham, Y., Keinan, E. A programmable biomolecular computing machine with bacterial phenotype output. Chembiochem 8:1255, 2007. Olson, A.J., Hu, Y.H.E., Keinan, E. Chemical mimicry of viral capsid self-assembly. Proc. Natl. Acad. Sci. U. S. A. 104:20731, 2007. Sensitive methods such as fluorescence can be used to detect multiple macromolecular interactions simultaneously (multiplex detection). An alternative approach is detection via chemifluorescence, in which a fluorescent dye is activated through chemical transfer of energy. We recently synthesized aqueous oxalate MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 261 F i g . 2 . A water-soluble oxalate ester can activate fluorescently labeled protein. Top, The postulated reaction between an oxalate ester and hydrogen peroxide to produce a dioxetane intermediate, which can transfer energy to a fluorescent dye to produce carbon dioxide and light emission at the dye’s characteristic wavelength. Bottom, Samples of fluorescently labeled bovine serum albumin (BSA-ROX, BSA-JOE, BSA-Cy3, and BSA-Cy5) adsorbed to a membrane and exposed to activated water-soluble oxalate ester emit light of characteristic wavelengths. F i g . 1 . An antibody scFv that binds its antigen irreversibly. Top, Western blot shows a complex between an engineered scFv 21H9 and its TNF antigen at 44 kD (lanes 6 and 9) that only forms in the presence of cobalt. Both scFv and TNF are in the complex, as indicated by staining with antibody to TNF (lane 9a) and antibody to scFv (lane 9b). The parental scFv RA72, which binds TNF nonco- valently, does not form the 44-kD complex (lanes 1–4). Bottom, Close-up model of the 21H9 scFv (green) with its metal binding site (red) in proximity to mapped linkage regions (pink) of TNF. The TNF trimer is shown as an outset with linkage residues in cyan and pink. ester compounds capable of exciting fluorescent dyes conjugated to biomolecules (Fig. 2). Unlike fluorescence, chemifluorescence allows integration of the emission signal over time, a situation that may result in highly sensitive detection of molecular interactions. Applications include microarrays, blots, and solid-phase immunoassays in research and diagnostic settings. PUBLICATIONS Grünewald, J., Tsao, M.L., Perera, R., Dong, L., Niessen, F., Wen, B.G., Kubitz, D.M., Smider, V.V., Ruf, W., Nasoff, M., Lerner, R.A., Schultz, P.G. Immunochemical termination of self-tolerance. Proc. Natl. Acad. Sci. U. S. A. 105:11276, 2008. Trisler, K., Looger, L.L., Sharma, V., Baker, M., Benson, D.E., Trauger, S., Schultz, P.G., Smider, V.V. A metalloantibody that irreversibly binds a protein antigen. J. Biol. Chem. 282:26344, 2007. Functional Characterization of Proteases via Combinatorial Libraries J.L. Harris, J. Alves he human genome encodes more than 500 proteases. These enzymes play important roles in all aspects of biology, from the beginning of life (fertilization, development, differentiation) to the end of life (cell death, apoptosis, degradation) and most biological processes in between. Proteases are also implicated in many pathologic states, such as cancer, inflammation, infectious disease, and neurodegeneration. Proteases exert their activity by cleaving peptide bonds in proteins. These cleavage events can be exquisitely selective, depending on the substrate specificity of the protease. Identifying the substrate specificity of a protease aids in understanding the role of the protease in a specific biological process. T 262 MOLECULAR BIOLOGY 2008 To define the substrate specificity of proteases, we have developed peptide nucleic acid (PNA)–encoded substrate and inhibitor libraries. Encoding substrate and inhibitor libraries with PNA tags allows not only for capture of the synthetic history of the library in the resulting molecule but also for spatial deconvolution of the molecules on DNA microarrays. The PNA tag is covalently cosynthesized with the small-molecule compound that can interact with enzymes in biological samples. With this approach, we can efficiently synthesize thousands of molecules in the same time it would take to synthesize a single individual molecule. Although the resulting library of thousands of compounds is screened as a mixture, the specific molecules within the library can be easily identified on a DNA microarray by using the hybridization properties of the encoded PNA tag. Another technique for defining the substrate specificity of proteases includes the use of positional scanning combinatorial libraries with fluorogenic latent molecules based on the coumarin or rhodamine scaffold. Using this technique, we have developed substrates that can be used to monitor not only the substrate specificity of proteases in enzymatic assays but also proteolytic activity in live cells for the development of diagnostic agents or inhibitors. For example, we developed a series of rhodamine-based substrates to monitor the specific action of cathepsin C, a protease implicated in immunity and immunologic diseases, in live cells by using flow cytometry. Another example of functional characterization of protease activity is the profiling of the substrate specificity of kallikreins. In collaboration with E.P. Diamandis, University of Toronto, Toronto, Ontario, we used a substrate library of approximately 160,000 fluorogenic substrates to characterize the structure-activity relationship of kallikreins, a protease class that has been strongly linked to cancer progression. PUBLICATIONS Borgoño, C.A., Gavigan, J.A., Alves, J., Bowles, B., Harris, J.L., Sotiropoulou, G., Diamandis, E.P. Defining the extended substrate specificity of kallikrein 1-related peptidases. Biol. Chem. 388:1215, 2007. Denault, J.B., Drag, M., Salvesen, G.S., Alves, J., Heidt, A.B., Deveraux, Q., Harris, J.L. Small molecules not direct activators of caspases [letter]. Nat. Chem. Biol. 3:519, 2007. Hampton, E.N., Knuth, M.W., Li, J., Harris, J.L., Lesley, S.A., Spraggon, G. The self-inhibited structure of full-length PCSK9 at 1.9 Å reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 104:14604, 2007. Li, J., Petrassi, H.M., Tumanut, C., Masick, B.T., Trussell, C., Harris, J.L. Substrate optimization for monitoring cathepsin C activity in live cells. Bioorg. Med. Chem., in press. THE SCRIPPS RESEARCH INSTITUTE Li, J., Tumanut, C., Gavigan, J.A., Huang, W.J., Hampton, E.N., Tumanut, R., Suen, K.F., Trauger, J.W., Spraggon, G., Lesley, S.A., Liau, G., Yowe, D., Harris, J.L. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem. J. 406:203, 2007. Anticancer Agents: Synthesis and Selective Delivery S.C. Sinha, R.A. Lerner, R. Goswami, V. Erigala, D. Xue, K.M. Bajjuri, Z. Chen, Z.-Z. Huang ur main research interests include antibody catalysis, synthesis of natural and unnatural molecules, drug development, and selective drug delivery. This year, we mainly focused on the construction and evaluation of novel antibody conjugates for the development of prodrug therapy and chemically programmed antibody approaches to cancer therapy. In addition, we synthesized the naturally occurring anticancer adjacent bis-tetrahydrofuran acetogenins and their analogs. O S Y N T H E S I S O F C E L L - TA R G E T I N G A N T I B O D I E S For the development of the prodrug therapy approach, we used the catalytic aldolase antibody 38C2 and a small-molecule inhibitor of integrin α v β 3 to prepare 2 cell-targeting catalytic antibodies (Fig. 1A). In these conjugates, the small molecules combined with 38C2 through the surface lysine residues or the sulfide groups obtained by reduction of the disulfide bridge in the antibody hinge region. Mass spectrometry indicated that each conjugate had approximately 2 molecules of the small molecule. In flow cytometry assay, both conjugates bound cells expressing αvβ3 with high affinity. Because the small molecule used for the conjugates also had weak affinity for the integrin α v β 5 , the resulting conjugates also bound to cells expressing that integrin. We found that the conjugates efficiently catalyzed the activation of several doxorubicin prodrugs, and the released drug was cytotoxic for MDA-MB-231 breast cancer cells in vitro. We have also designed a series of doxorubicin prodrugs (Fig. 1C) and evaluated them in vitro. Now we are using the prodrugs and antibody conjugates to evaluate the antibody-prodrug therapy approach in vivo. Using αvβ3 antagonists, we have also prepared and evaluated noncatalytic 38C2 conjugates or the chemically programmed 38C2 (cp38C2) in which the compound reacted to the antibody-binding sites through a diketone or the vinylketone linker (Fig. 1A). We are now MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 263 the most cytotoxic compounds that target the mitochondrial enzyme complex I. We have prepared 10 stereoisomeric asimicin analogs and a new naturally occurring acetogenin, 27-hydroxybullatacin (Fig. 1D), and its 15-epimer. To explore the binding pocket of these acetogenins in complex I, we also prepared a fluorescent analog of 15-epi-rolliniastatin. In preliminary studies, these compounds appeared to bind in the complex I binding pocket, additional studies are required to confirm the results. The in vitro evaluations and complex I binding studies are carried out in collaboration with F. Valeriote, Henry Ford Health System, Detroit, Michigan, and M. Verkhovskaya, University of Helsinki, Finland. B I S - T E T R A H Y D R O F U R A N D E R I VAT I V E S The demands for novel chemical entities with unknown biological functions are ever growing. Compounds with tetrahydrofuran fragments in the molecules have important biological properties, including antitumor and antibacterial activities. However, a library of these compounds had never been attempted. In collaboration with P. Wipf, University of Pittsburgh, Pennsylvania, we are developing multicomponent reaction products based on mono- and bis-tetrahydrofurans. Using the tetrahydrofurans and their multicomponent reaction products, we are also synthesizing a library of the azidealkyne “click” products. These compounds will undergo systematic biological evaluations. F i g . 1 . Structures of compounds that target integrins αvβ3/αvβ5 (A) and αvβ3/αvβ6 (B), doxorubicin prodrugs (C), and the adjacent bis-tetrahydrofuran 27-hydroxybullatacin (D). focusing on compounds and antibody conjugates that would target other integrins, including αvβ6, α5β1, and αIIbβ3, which may be useful in the treatment and diagnosis of various human diseases. For example, we prepared noncatalytic 38C2 conjugates by using the diketone and vinylketone derivatives of an analog of peptoids (Fig. 1B) that are known to bind integrins αvβ3 and αvβ6. We are evaluating these conjugates in vitro and in vivo. These studies are carried out in collaboration with C.F. Barbas, Department of Molecular Biology, B. Felding-Habermann, Department of Molecular and Experimental Medicine, and C. Liu, Department of Immunology and Microbial Science. SYNTHESIS OF ANNONACEOUS ACETOGENIN ANALOGS We are developing a library of annonaceous acetogenin analogs, chemical entities based on the novel mono- and bis-tetrahydrofurans, and of protease inhibitors. Asimicin-type annonaceous acetogenins are among PROTEASE INHIBITORS Numerous proteases are overexpressed in several cancer cell lines and are highly implicated in tumor growth and metastases. Inhibition of such proteases has potential application in cancer therapy. In collaboration with Dr. Liu, we are developing inhibitors of legumain protease, which is highly expressed in several cancer cell lines, including prostate and breast cancer cells. Although our main goal is to discover a novel protease with previously unknown structure, we are also determining the structure-activity relationship of the already known nanomolar inhibitors of the legumain protease. PUBLICATIONS Chen, Z., Sinha, S.C. Total synthesis of 27-hydroxy-bullatacin and its C-15 epimer, and studies on their inhibitory effect on bovine heart mitochondrial complex I functions. Tetrahedron 64:1603, 2008. 264 MOLECULAR BIOLOGY 2008 Chemical Transformations of Small Molecules and Proteins and Reactions Between Them F. Tanaka, K. Albertshofer, C.F. Barbas III, R.P. Fuller, H.-M. Guo, M. Minakawa, S.S.V. Ramasastry, N. Utsumi, H. Zhang e create and develop molecules and methods that contribute to basic biomedical research and to the development of drug candidates and therapeutic strategies. One aspect of our research is the development of new strategies and methods for labeling proteins with small molecules at certain positions. Covalent labeling reactions are required for preparing protein–small molecule conjugates that are useful in research and medicine. For example, protein-drug conjugates are often safer and more effective therapeutics than is drug or protein alone. Small molecules conjugated to proteins have been used to direct the localization of conjugates in living cells according to the binding specificity of the small molecules. We are developing synthetic labeling molecules that selectively and covalently react with tyrosine. Labeling reactions specific for tyrosine can be used to label a tyrosine naturally present in a protein and/or a tyrosine within a tag fused to a protein. Tyrosine residues occur often less on the surface of folded proteins than do residues containing lysine or carboxylic acid. Thus, accessible tyrosine is an attractive covalent conjugation site. For proteins that do not have accessible tyrosine on the surface, the phenol group is orthogonal; these proteins should not be affected by reagents or compounds that react with phenols or tyrosine. When a tag that contains accessible tyrosine is fused to a protein, the tyrosine within the tag can be used as a selective labeling site. Whereas incorporation of unusual amino acids into proteins often requires special conditions and reagents, preparation of protein fusions with tyrosine-containing tags does not. Therefore, reactions at tyrosines will be a convenient way to specifically label proteins. We have developed cyclic imine derivatives that react with phenols, including tyrosine-containing peptides, in water over a wide pH range without the need for additional catalysts at room temperature to 37°C. The reaction is the formation of a carbon-carbon bond W THE SCRIPPS RESEARCH INSTITUTE at the carbon of the phenol benzene ring; bonds formed via this reaction were stable. The imine derivatives can be conjugated with fluorescent molecules and drug molecules by using chemical synthesis. Therefore, through reactions at tyrosines with these molecules, a variety of molecules can be covalently and selectively attached to a protein of interest. When a labeling molecule contains a moiety that selectively and covalently reacts with a specific amino acid residue and a moiety that noncovalently interacts with a target protein, the labeling molecule can be used to tag naturally occurring proteins of interest in natural environments, including cells. When a labeling molecule contains a moiety that binds to the surface of a protein, the protein can be labeled without affecting the protein’s function. On the other hand, when the specificity of a labeling molecule is imparted through a ligand that binds to the active site of an enzyme, the resulting molecule can be an efficient, selective, irreversible inhibitor of the enzyme. Accordingly, we are developing imine-derived molecules for these types of uses. We are also developing protein and peptide tags that selectively form covalent bonds with designed synthetic molecules; these tags are generated by using reaction-based selections from combinatorial libraries of proteins and peptides. When the tag is fused to a protein of interest, the fusion protein can be modified at the tag sequence through the reaction with the designed synthetic compound. We are also developing synthetic methods for concise access to functionalized molecules in regioselective, diastereoselective, and enantioselective fashions. These methods are useful for synthesizing bioactive molecules, ligands of proteins, and candidates of these molecules. Overall, our research provides insight into the construction of functional molecules and into biological functions of naturally occurring molecules such as proteins and enzymes. PUBLICATIONS Guo, H.-M., Minakawa, M., Tanaka, F. Fluorogenic imines for fluorescent detection of Mannich-type reactions of phenols in water. J. Org. Chem. 73:3964, 2008. Hayashi, I., Mizuno, H., Tong, K.I., Furuta, T., Tanaka, F., Yoshimura, M., Miyawaki, A., Ikura, M. Crystallographic evidence for water-assisted photo-induced peptide cleavage in the stony coral fluorescent protein Kaede. J. Mol. Biol. 372:918, 2007. Jiang, L., Althoff, E.A., Clemente, F.R., Doyle, L., Röthlisberger, D., Zanghellini, A., Gallaher, J.L., Betker, J.L., Tanaka, F., Barbas, C.F. III, Hilvert, D., Houk, K.N., Stoddard, B., Baker, D. De novo computational design of retro-aldol enzymes. Science 319:1387, 2008. Ramasastry, S.S.V., Albertshofer, K., Utsumi, N., Tanaka, F., Barbas, C.F. III. Mimicking fructose and rhamnulose aldolases: organocatalytic syn-aldol reactions with unprotected dihydroxyacetone. Angew. Chem. Int. Ed. 46:5572, 2007. MOLECULAR BIOLOGY 2008 Tanaka, F., Hu, Y., Sutton, J., Asawapornmongkol, L., Fuller, R., Olson, A.J., Barbas, C.F. III, Lerner, R.A. Selection of phage-displayed peptides that bind to a particular ligand-bound antibody. Bioorg. Med. Chem. 16:5926, 2008. Utsumi, N., Imai, M., Tanaka, F., Ramasastry, S.S.V., Barbas, C.F. III. Mimicking aldolases through organocatalysis: syn-selective aldol reactions with protected dihydroxyacetone. Org. Lett. 9:3445, 2007. Zhang, H., Mitsumori, S., Utsumi, N., Imai, M., Garcia-Delgado, N., Mifsud, M., Albertshofer, K., Cheong, P.H.-Y., Houk, K.N., Tanaka, F., Barbas, C.F. III. Catalysis of 3-pyrrolidinecarboxylic acid and related pyrrolidine derivatives in enantioselective anti-Mannich-type reactions: importance of the 3-acid group on pyrrolidine for stereocontrol. J. Am. Chem. Soc. 130:875, 2008. THE SCRIPPS RESEARCH INSTITUTE 265 linker length, and chemical composition. The structures reveal a significant structural plasticity of the distal substrate-binding channel of the enzyme that arises from combinations of several modes of movements in the F, G, and B′ helices. Changes in the hydrogen-bonding interactions between wire and protein affected ligand orientation more than did binding affinity (Fig. 1). The linker length Zhang, H., Ramasastry, S.S.V., Tanaka, F., Barbas, C.F. III. Organocatalytic antiMannich reactions with dihydroxyacetone and acyclic dihydroxyacetone derivatives: a facile route to amino sugars. Adv. Synth. Catal. 350:791, 2008. Metalloenzyme Engineering D.B. Goodin, C.D. Stout, H.B. Gray,* E.C. Glazer, R.F. Wilson, A. Annalora, S. Vetter * California Institute of Technology, Pasadena, California he primary purposes of our research are to understand the diversity of metalloenzyme catalysts and to develop methods for directed evolution of enzymes with novel function. Our goal is to gain sufficient control over substrate-enzyme interactions and the oxidative chemistry catalyzed at the active site to allow directed evolution of catalysts capable of regiospecific and stereospecific oxidation of a given target substrate. We use a number of techniques in structural biology and spectroscopy and strategies of rational protein redesign and molecular evolution. In the past year, we focused on developing synthetic molecular wires to probe the active site and function of the heme enzymes P450cam and nitric oxide synthase (NOS). These wires, which consist of substrate analogs tethered to a reporter or sensitizer, are designed to bind specifically to the active-site channel of a given enzyme and are effective tools for inhibitor discovery, phototriggered enzyme turnover, and molecular evolution strategies. We have made significant progress in obtaining a detailed structural characterization of a library of tethered substrate wires that bind the substrate access channel of P450 cam . In the past, we showed that alkyl adamantane wires bind to the active site of P450cam and induce a large conformational change at the enzyme’s active site. On the basis of these findings, we have designed a new generation of P450 wires, and we recently solved the crystal structures of P450cam in complex with more than 12 variants that differ from each other in the position of hydrogen-bonding groups, T F i g . 1 . Crystal structures of 3 tethered substrate wires bound to the active site of P450cam. The linkers differ in the location of an amide that provides a critical hydrogen-bonding interaction with the substrate. and hydrophobicity have a significant effect on the conformation disorder in both the wire and the protein. We have also made significant progress in characterizing photoactive probes specific for the pterin site of murine inducible NOS. Addressing the pterin site through a molecular wire will help define the role of the cofactor in catalysis by allowing photochemical triggering of enzyme turnover. We have synthesized a series of ruthenium(II)-pterin wires and have characterized their interactions with murine inducible NOS. Binding was confirmed by heme-induced quenching of either pterin or ruthenium(II) fluorescence upon interaction with the heme domain of inducible NOS, and this quenching was reversed by binding of the natural pterin cofactor. Time-resolved emission experiments showed bound and free forms of the wire and allowed estimation of the affinity and the distance between the ruthenium(II) and heme. Our findings are consistent with the modeled geometry of the wire within the pterin-binding site. We recently observed photoinduced reduction of ferric NOS after 450-nm illumination of the ruthenium(II) center in the presence of reductive quenchers. These wires are being examined for their potential to generate unstable intermediates in the NOS reaction cycle. To explore the potential of generating novel P450like catalysts, we have introduced thiolate coordination into the heme of the lipocalin nitrophorin 1. UV and visible light, resonance Raman spectroscopy, and magnetic 266 MOLECULAR BIOLOGY 2008 circular dichroism spectra suggest weak thiolate coordination only in the ferric state of the H60C mutant of nitrophorin 1. Two crystal structures of the mutant in complex with imidazole and histamine were solved to 1.7- and 1.96-Å resolution, respectively. Both structures show that the H60C mutation is well tolerated by the protein scaffold and suggest that heme-thiolate coordination in the mutant nitrophorin 1 requires some movement of the heme within its binding cavity. PUBLICATIONS Contakes, S.M., Nguyen, Y.H.L., Gray, H.B., Glazer, E.C., Hays, A.-M., Goodin, D.B. Conjugates of heme-thiolate enzymes with photoactive metal-diimine wires. Struct. Bond. 123:177, 2007. Glazer, E.C, Nguyen, Y.H., Gray, H.B., Goodin, D.B. Probing inducible nitric oxide synthase with a pterin-ruthenium(II) sensitizer wire. Angew. Chem. Int. Ed. 47:898, 2008. Structure, Function, and Applications of Virus Particles J.E. Johnson, M. Banerjee, C.-Y. Fu, I. Gertsman, R. Huang, R. Khayat, G. Lander, J. Lanman, K.K. Lee, T. Matsui, A. Odegard, J. Speir, R. Taurog e investigate model virus systems that provide insights for understanding viral assembly, maturation, entry, localization, and replication. We have also developed viruses as reagents for applications in nanomedicine, nanochemistry, and nanobiology. We investigate viruses that infect bacteria, insects, plants, and the extreme thermophile Sulfolobus. These viruses have genomes of single-stranded RNA and double-stranded DNA. We use a variety of physical methods to investigate structure-function relationships, including single-crystal x-ray diffraction, static and time-resolved solution x-ray diffraction, electron cryomicroscopy and image reconstruction, mass spectrometry, structure-based computational analyses, and methods associated with thermodynamic characterization of virus particles and the particles’ transitions. Biological methods we use include the genetic engineering of viral genes and their expression in Escherichia coli, mammalian cells, insect cells, and yeast and the characterization of these gene products by physical methods. For cytologic studies of viral entry and infection, we use fluorescence and electron microscopy and particles assembled in heterologous expression systems. Our studies depend on extensive consultations and collaborations with others at Scripps W THE SCRIPPS RESEARCH INSTITUTE Research, including the groups led by B. Carragher, M.G. Finn, M. Manchester, R.A. Milligan, C. Potter, V. Reddy, A. Schneemann, G. Siuzdak, and J.R. Williamson, and a variety of groups outside of Scripps. DOUBLE-STRANDED DNA VIRUSES HK97 is a double-stranded DNA virus similar to bacteriophage λ. It undergoes a remarkable morphogenesis in its assembly and maturation, and this process can be recapitulated in vitro. We determined the atomic resolution structure of the 650-Å mature head II particle and discovered the mechanism used to concatenate the subunits of the particle into a chain-mail structure similar to that seen in armor of medieval knights. In the past year, we focused on the structures of prohead I and prohead II, the first and second intermediates in the assembly pathway. The prohead II structure is at 3.7-Å resolution, and the subunit fold and the location of many side chains have been determined. The tertiary structure of the subunit in prohead II differs from that of the subunit in head II, an unexpected result. At lower resolution, the transition from prohead II to head II appeared to be rigid body motions. It is now clear that contacts near the 3-fold particle axes are fixed and that a dramatic change occurs in the subunit structure, with a twist about 3 β-strands and the bending of a long helix, although the domains remain largely rigid. The change in tertiary structure may be the energy storage mechanism that propels the maturation of the particle. We used electron cryomicroscopy to study bacteriophage λ in the prohead and head states. The crystal structure of the HK97 bacteriophage capsid fits most of the T = 7 λ particle density with only minor adjustment. A prominent surface feature at the 3-fold axes corresponds to the cementing protein glycoprotein D, necessary for stabilization of the capsid shell. The position of the glycoprotein coincides with the location of the covalent cross-link formed in the docked HK97 crystal structure, suggesting an evolutionary replacement of this gene product in bacteriophage λ by autocatalytic chemistry in HK97. SINGLE-STRANDED RNA VIRUSES Flock House virus is a T = 3, single-stranded RNA virus that infects Drosophila. Infectivity of Flock House virus requires the autocatalytic cleavage of the capsid protein at residue 363, liberating the C-terminal 44-residue γ peptides that remain associated with the particle. In vitro studies indicated that the amphipathic-helical part (residues 364–385) is membrane active, suggesting MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE 267 its role in RNA membrane translocation during infection. We have now shown that a maturation-defective mutant of Flock House virus can be rescued by viruslike particles that lack the genome but undergo maturation cleavage in a baculovirus expression system. We propose that colocalization of the 2 defective particle types in an entry compartment allows the rescue by γ peptides. We used time-resolved electron cryomicroscopy for structural studies of the T = 4 tetravirus Nudaurelia capensis ω virus. We found that a large-scale structural change induced by lowering the pH from 7 to 5 occurs in less than 100 milliseconds, but the annealing of the polypeptide chains to form active autocatalytic sites varies dramatically with the position of the subunit in the surface lattice. Subunits adjacent to 5- and 3-fold axes form active sites in less than 3 minutes, whereas the other 2 quasi-equivalent subunits are much slower. One of the latter subunits forms active sites in 30 minutes; the other requires more than 2 hours. These data explain well the unusual kinetics of the cleavage reaction. Speir, J.A., Johnson, J.E. Tetravirus structure. In: Encyclopedia of Virology, 3rd ed. Mahy, B.W.J., van Reganmortel, M.H.V. (Eds.). Academic Press/Elsevier, New York, 2008, Vol. 5, p. 27. PUBLICATIONS Banerjee, M., Johnson, J.E. Activation, exposure and penetration of virally encoded, membrane-active, polypeptides during non-enveloped virus entry. Curr. Protein Pept. Sci. 9:16, 2008. e are interested in identifying and understanding the structural underpinnings and requirements for the self-assembly, stability, and targeting specificities of viral capsids. We use this information to design novel protein shells that polyvalently display multiple copies of peptides or proteins of interest. We use structural, computational, bioinformatics, and genetic methods. Viruses are highly evolved macromolecular assemblages that perform a variety of functions during their life cycle, including self-assembly to form uniform capsids, selective packaging of the genome, binding to susceptible host cells, and delivering the genetic material to the targeted cells Simple viruses, such as nonenveloped viruses, form capsids with homogeneous composition and quaternary architecture. Hence, these viruses are useful for structural and functional analyses. We recently determined the structure of Seneca Valley virus (Fig. 1), which belongs to a new genus (Senecavirus) of the Picornaviridae family. Seneca Valley virus is the first known naturally occurring nonpathogenic picornavirus shown be selectively cytotoxic to tumor cells with features of neuroendocrine cancer. This research was done in collaboration with scientists at Neotropix, Inc., Malvern, Pennsylvania. In collaboration with G.R. Nemerow, Department of Immunology, we have made progress in determining the structure of the entire human adenovirus at 5.5-Å Gan, L., Johnson, J.E. An optimal exposure strategy for cryoprotected virus crystals with lattice constants greater than 1000 Å. J. Synchrotron Radiat. 15:223, 2008. Johnson, J.E. Multi-disciplinary studies of viruses: the role of structure in shaping the questions and answers. J. Struct. Biol., in press. Johnson, J.E., Chiu, W. DNA packaging and delivery machines in tailed bacteriophages. Curr. Opin. Struct. Biol. 17:237, 2007. Johnson, J.E., Speir, J.A. Principles of virus structure. In: Encyclopedia of Virology, 3rd ed. Mahy, B.W.J., van Reganmortel, M.H.V. (Eds.). Academic Press/Elsevier, New York, 2008, Vol. 5, p. 393. Kang, S., Lander, G., Johnson, J.E., Prevelige, P.E. Development of bacteriophage P22 as a platform for molecular display: genetic and chemical modifications of the procapsid exterior surface. Chembiochem 9:514, 2008. Lanman, J., Crum, J., Deerinck, T.J., Gaietta, J.M., Schneemann, A., Sosinsky, G., Ellisman, M.H., Johnson, J.E. Visualizing Flock House virus infection in Drosophila cells with correlated fluorescence and electron microscopy. J. Struct. Biol. 161:439, 2008. Lee, J., Doerschuk, P.C., Johnson, J.E. Exact reduced-complexity maximum likelihood reconstruction of multiple 3-D objects from unlabeled unoriented 2-D projections and electron microscopy of viruses. IEEE Trans. Image Process. 16:2865, 2007. Lee, K.K., Gan, L., Tsuruta, H.C.M., Conway, J.F., Duda, R.L., Hendrix, R.W., Steven, A.C., Johnson, J.E. Virus capsid expansion driven by the capture of mobile surface loops. Structure, in press. Sosinsky, G.E., Crum, J., Jones, Y.Z., Lanman, J., Smarr, B., Terada, M., Martone, M.E., Deerinck, T.J., Johnson, J.E., Ellisman, M.H. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. J. Struct. Biol. 161:359, 2008. Speir, J.A., Johnson, J.E. Nonenveloped virus structure. In: Encyclopedia of Virology, 3rd ed. Mahy, B.W.J., van Reganmortel, M.H.V. (Eds.). Academic Press/Elsevier, New York, 2008, Vol. 5, p. 380. Steinmetz, N.F., Lin, T., Lomonossoff, G. P., Johnson, J.E. Structure-based engineering of an icosahedral virus for nanomedicine and nanotechnology. In: Viruses as Nanomaterials for Biomedicine and Bioengineering. Manchester, M., Steinmetz, N.F. (Eds.). Springer, New York, in press. Szymczyna, B.R., Gan, L., Johnson, J.E., Williamson, J.R. Solution NMR studies of the maturation intermediates of a 13 MDa viral capsid. J. Am. Chem. Soc. 129:7867, 2007. Walukiewicz, H.E., Banerjee, M., Schneemann, A., Johnson, J.E. Rescue of maturation-defective Flock House virus infectivity with noninfectious, mature, viruslike particles. J. Virol. 82:2025, 2008. Wickner, R.B., Tang, J., Gardner, N., Johnson, J.E. The yeast double-stranded RNA virus L-A resembles mammalian dsRNA virus cores. In: Segmented DoubleStranded RNA Viruses: Structure and Molecular Biology. Patton, J.T. (Ed.). Caister Academic Press, Portland, OR, 2007, p. 105. Structure, Informatics, and Design in Virology V.S. Reddy, M. Tripp, S. Venkataraman W 268 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE enables the attachment of peptides or proteins of interest to the C terminus in a linear fashion and their display on the capsid surface. Such reagents act as multivalent decoys of the pathogenic molecules and so can be used as potential vaccines. PUBLICATIONS Carrillo-Tripp, M., Brooks, C.L. III, Reddy, V.S. A novel method to map and compare protein-protein interactions in spherical viral capsids. Proteins, in press. F i g . 1 . Protomer structure (left) and capsid surface (right) of Seneca Valley virus, an oncolytic picornavirus representative of a new genus. resolution by x-ray crystallography. Adenoviruses are common human pathogens and major causative agents of acute respiratory and ocular infections. Currently, adenoviruses are being used as vectors in gene transfer studies. The structure of an adenovirus at high resolution will provide insight into reengineering of advenovirus vectors to improve vaccine or gene delivery to specific host cell types. Data acquisition to 4-Å resolution is under way. We have also determined the x-ray structures of 2 fiber knobs of adenovirus serotypes 35 and 16 and have identified structural requirements for binding of the knobs to the cellular receptor CD46. We continue to maintain and expand the virus structure database, VIPERdb (http://viperdb.scripps.edu), a Web portal for structures and associated structural properties of viral capsids. The capsid structures in the database were analyzed in terms of protein-protein interactions, contacting residue pairs, association energies, individual residue contributions, and surface characteristics by using computational methods. We recently developed a novel method to map and compare protein-protein interactions in viral capsids irrespective of the size and the architecture of the capsids. The resultant maps can be used as road maps to visualize the extent and distribution of interactions required for the formation of viral capsids. The results of the analysis are stored in the database. VIPERdb is being developed and maintained as part of the National Institutes of Health research resource Multiscale Modeling Tools for Structural Biology, headed by C.L. Brooks, University of Michigan, Ann Abor. We are also actively involved in generating novel vaccines against cytotoxins such as ricin and against pathogens by expressing antigenic regions of pathogenic molecules on the surfaces of viral capsids. Tomato bushy stunt virus–like capsids are our display platform of choice. A unique subunit fold of the viral subunit Lad, S.P., Yang, G., Scott, D.A., Wang, G., Nair, P., Mathison, J., Reddy, V.S., Li, E. Chlamydial CT441 Is a PDZ domain-containing tail-specific protease that interferes with the NF-κB pathway of immune response. J. Bacteriol. 189:6619, 2007. Manayani, D.., Thomas, D., Dryden, K.A., Reddy, V., Siladi, M.E., Marlett, J.M., Rainey, G.J., Pique, M.E., Scobie, H.M., Yeager, M., Young, J.A., Manchester, M., Schneemann, A. A viral nanoparticle with dual function as an anthrax antitoxin and vaccine. PLoS Pathog. 3:1422, 2007. Pache, L., Venkataraman, S., Nemerow, G.R., Reddy, V.S. Conservation of fiber structure and CD46 usage by subgroup B2 adenoviruses. Virology 375:573, 2008. Venkataraman, S., Reddy, S.P., Loo, J., Idamakanti, N., Hallenbeck, P.L., Reddy, V.S. Crystallization and preliminary x-ray diffraction studies of Seneca Valley virus001, a new member of the Picornaviridae family. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64:293, 2008. Biology and Applications of Icosahedral Viral Capsids A. Schneemann, N. Brunn, J. Jovel, J.E. Petrillo, P.A. Venter oat proteins of nonenveloped, icosahedral viruses perform multiple functions during viral infection, including capsid assembly, specific encapsidation of the viral genome, binding to a cellular receptor, and uncoating. In some viruses, a single type of protein is sufficient to carry out these functions; we are interested in the determinants that endow a polypeptide chain with such versatility. We seek to harness this versatility for novel applications of viruses in biotechnology and nanotechnology. We focus on a structurally and genetically well-characterized virus family, the T = 3 nodaviruses. Nodaviruses are composed of 180 copies of a single coat protein and 2 strands of positive-sense RNA. Currently, we are elucidating the mechanism by which the 2 genomic RNAs are packaged into a single virion. Our data indicate that the 2 viral RNAs are recognized separately, but it is not yet known whether packaging occurs sequentially and whether one or more coat protein subunits are involved in this process. Interestingly, we found that RNA genome packaging is coupled to genome replication and that specific packaging of the viral genome also requires coat protein translated from newly synthesized viral RNA. The C MOLECULAR BIOLOGY 2008 coupling of these processes may be a safety mechanism for the virus to ensure efficient and accurate formation of progeny virions in infected cells. We are also investigating the mechanism by which nodaviral protein B2 suppresses RNA silencing. We are identifying the double-stranded RNAs that serve as substrates for B2 binding during nodavirus infection, and we are correlating these results with data from confocal microscopy and immunoprecipitation studies. We have identified amino acid residues in B2 that are critical for the protein’s function as an RNA-binding protein; preliminary data suggest that B2 may have a second function during the viral replication cycle that is unrelated to its function as a suppressor of RNA silencing. We are also using nodavirus particles as a novel platform to develop improved vaccines. In collaboration with researchers at Scripps Research, the Salk Institute, La Jolla, California, and Harvard University, Boston, we displayed the VWA domain of capillary morphogenesis protein 2, the cellular receptor for anthrax toxin, in a multivalent fashion on the surface of the virion. Two insertion sites yielding different patterns of 180 copies of the VWA domain were selected on the basis of computational modeling of the high-resolution crystal structure of Flock House virus, an insect nodavirus. The resulting chimeric viruslike particles functioned as a potent anthrax antitoxin in cell culture and protected rats from challenge with lethal toxin. This research is important because it shows that protein domains containing more than 150 amino acids can be displayed on Flock House virus in a biologically functional form, suggesting numerous additional applications. Moreover, chimeric particles decorated with anthrax protective antigen elicited a potent neutralizing antibody response against the antigen that protected rats from challenge with lethal toxin 4 weeks after a single immunization without adjuvants. This chimeric particle platform is a dually acting reagent for the treatment of and protection against anthrax. PUBLICATIONS Lanman, J., Crum, J., Deerinck, T.J., Gaietta, G.M., Schneemann, A., Sosinsky, G.E., Ellisman, M.H., Johnson, J.E. Visualizing Flock House virus infection in Drosophila cells with correlated fluorescence and electron microscopy. J. Struct. Biol. 161:439, 2008. Manayani, D.J., Thomas, D., Dryden, K.A., Reddy, V., Siladi, M.E., Marlett, J.M., Rainey, G.J.A., Pique, M.E., Scobie, H.M., Yeager, M., Young, J.A.T., Manchester, M., Schneemann, A. A viral nanoparticle with dual function as an anthrax antitoxin and vaccine. PLoS Pathog. 3:1422, 2007. Walukiewicz, H.E., Banerjee, M., Schneemann, A., Johnson, J.E. Rescue of maturation-defective Flock House virus infectivity with noninfectious, mature, viruslike particles. J. Virol. 82:2025, 2008. THE SCRIPPS RESEARCH INSTITUTE 269 Molecular Mechanism of Autophagosome Formation T. Otomo, C. Otomo utophagy is a conserved degradative mechanism triggered in response to various stresses in eukaryotic cells. Cells use autophagy to break down cytoplasmic fractions in bulk and large cytotoxic components such as protein aggregates, organelles, and intracellular pathogens. The hallmark of autophagy is de novo formation of a large double-membrane–enclosed transport vesicle called an autophagosome, which sequesters a fraction of cytoplasm and delivers the fraction to a lysosome for degradation. Formation of autophagosomes is thought to involve new molecular mechanisms that differ from those in classical vesicular transport systems. We are interested in the molecular basis of the membrane dynamics and the cargo recognition rules in autophagy. Our goals are to explain the mechanisms by revealing atomic structures of relevant protein complexes and establishing in vitro biochemical reconstitutions and to predict potential cargos on the basis of structural data. Targeting autophagy in therapies and drug development for various diseases such as cancer, neurodegeneration, and infectious disease, has been discussed. Our studies on the mechanistic basis would facilitate development of such strategies. Among many autophagy-related proteins, we are currently focusing on 2 ubiquitin-like protein modifiers, Atg12 and Atg8, which are autophagy specific. These molecules are important because they play key roles in membrane dynamics. Atg12 is conjugated to another autophagy protein, Atg5, and Atg8 is conjugated to phosphatidylethanolamine through ubiquitin-like enzymatic reactions. The Atg12-Atg5 conjugate has 3 functions. First, it acts as an E3 ligaselike enzyme in the conjugation of Atg8 to phosphatidylethanolamine. Second, the Atg12-Atg5 conjugate forms large oligomers when bound to a coiled coil protein, Atg16. The resulting Atg12-Atg5-Atg16 complex may be the membrane scaffold for autophagosomes. Third, Atg12-Atg5 interacts with a bacterial protein, VirG, from Shigella flexneri, which is the key for autophagosomes to recognize this shigella species. We have succeeded in generating the Saccharomyces cerevisiae and human Atg12-Atg5 conjugates by developing bacterial coexpression systems, A 270 MOLECULAR BIOLOGY 2008 which will allow us to investigate structural mechanisms of the functions of the conjugates. The Atg8-phosphatidylethanolamine conjugate functions in cargo recognition and membrane tethering. Membrane tethering is intriguing because it may explain how autophagosomal membranes elongate into doublemembrane vesicles. Biophysical and structural characterization of the conjugate would open the door to understanding this mysterious membrane dynamics. Currently, we are producing proteins critical for these studies. Overall, the combination of structural study and biochemical reconstitution would answer the mechanistic questions in autophagosome formation. Control of Cell Division THE SCRIPPS RESEARCH INSTITUTE late the proteasome. Proteasomes are complex proteases that target ubiquitylated proteins, including important cell-cycle regulatory proteins. Surprisingly, we found that Cks1/Cdk1 regulates a nonproteolytic function of proteasomes, the transcriptional activation of the gene CDC20. Specifically, Cks1 and Cdk1 are required to recruit proteasomes to CDC20 for efficient transcriptional elongation. Our investigations of CDC20 have led to the conclusion that Cks1 and Cdk1 are required for recruitment of proteasomes to and transcriptional elongation of many other genes as well. Currently, we are elucidating the mechanism whereby the Cks1/Cdk1proteasome complex facilitates transcriptional elongation. Our most recent results suggest that Cks1 and proteasomes in conjunction with Cdk1 mediate remodeling of chromatin by removing nucleosomes. CONTROL IN MAMMALIAN CELLS S.I. Reed, L.-C. Chuang, M. Henze, V. Liberal, K. Luo, S. Ekholm-Reed, L. Teixeira, H.-S. Martinsson-Ahlzén iological processes of great complexity can be approached by beginning with a systematic genetic analysis in which the relevant components are first identified and then the consequences of selectively eliminating the components via mutation are investigated. We have used yeast, which is uniquely tractable to this type of analysis, to investigate control of cell division. In recent years, it has become apparent that the most central cellular processes throughout the eukaryotic phylogeny are highly conserved in terms of both the regulatory mechanisms used and the proteins involved. Thus, it has been possible in many instances to generalize from yeast cells to human cells. B CONTROL IN YEAST We have focused on the role and regulation of the Cdc28 protein kinase (Cdk1). Initially identified by means of a mutational analysis of the yeast cell cycle, this protein kinase and its analogs are ubiquitous in eukaryotic cells and central to a number of aspects of control of cell-cycle progression. One current area of interest is regulation of cellular morphogenesis by Cdk1. The activity of Cdk1 driven by mitotic cyclins modulates polarized growth in yeast cells. Specifically, these activities depolarize growth by altering the actin cytoskeleton. We found that several proteins that modulate actin structure are targeted by Cdk1, and we are investigating how these phosphorylation events control actin depolarization and cell shape. While investigating mitosis in yeast, we found that Cks1, a small Cdk1-associated protein, appears to regu- We showed previously that the human homologs of the Cdc28 protein kinase are so highly conserved, structurally and functionally, relative to the yeast protein kinase, that they can function and be regulated properly in a yeast cell. Analyzing control of the cell cycle in mammalian cells, we produced evidence for the existence of both positive and negative regulatory schemes, similar to those elucidated in yeast. A principal research focus is the positive regulator of Cdk2, cyclin E. Cyclin E is often found overexpressed and/or deregulated in human cancers. Using a tissue culture model, we showed that deregulation of cyclin E confers genomic instability, probably explaining the link to carcinogenesis. The observation that deregulation of cyclin E confers genomic instability has led us to hypothesize a mechanism of cyclin E–mediated carcinogenesis based on accelerated loss of heterozygosity at tumor suppressor loci. We are testing this hypothesis in transgenic mouse models. We showed that a cyclin E transgene expressed in mammary epithelium significantly increases loss of heterozygosity at the p53 locus, leading to enhanced mammary carcinogenesis. We are extending these investigations by using mouse prostate, testis, and skin models. In an attempt to understand cyclin E–mediated genomic instability, we are investigating how deregulation of cyclin E affects both S phase and mitosis. Recent data suggest that deregulation of cyclin E impairs DNA replication by interfering with assembly of the prereplication complex. We found that cells deregulated for cyclin E expression progress through S phase abnormally slowly and tend to enter mitosis prematurely. Cyclin E deregula- MOLECULAR BIOLOGY 2008 tion also impairs the metaphase-anaphase transition by promoting the accumulation of inhibitors of anaphase. Our interest in cyclin E deregulation in cancer led us to investigate the pathway for turnover of cyclin. We showed that phosphorylation-dependent proteolysis of cyclin E depends on a protein-ubiquitin ligase known as SCFhCdc4. The F-box protein hCdc4 is the specificity factor that targets phosphorylated cyclin E. We are investigating how ubiquitylation of cyclin E is coordinated with other processes required for its degradation, including prolyl isomerization. We are also investigating SCFhCdc4 ubiquitylation of other important cellular proteins. Recently, we began determining the role of SCFhCdc4 in neurodegenerative disease. We found that parkin, a protein often mutated in hereditary Parkinson’s disease, regulates the stability of hCdc4, possibly leading to neuropathologic changes. We discovered an SCFhCdc4 substrate, peroxisome proliferator–activated receptor γ coactivator 1, that may be the effector of hCdc4 deregulation in Parkinson’s disease. In addition, we showed that SCF hCdc4 regulates the turnover of presenilins in the brain, proteins strongly implicated in Alzheimer’s disease. Another area of interest is the role of Cks proteins in mammals, complementing our research in yeast. Mammals express 2 paralogs of yeast Cks1, known as Cks1 and Cks2. Experiments in mice lacking the genes for Cks1 and Cks2 revealed that each paralog has a specialized function. Cks1 is required as a cofactor for Skp2-mediated ubiquitylation and turnover of inhibitors p21, p27, and p130. Cks2 is required for the transition from metaphase to anaphase in both male and female meiosis I. Nevertheless, mice nullizygous at the individual loci are viable. However, doubly nullizygous mice have not been observed because embryos die at the morula stage, a finding consistent with an essential redundant function. We found that this function is probably is involved in regulation of transcription and linked to chromatin remodeling, as in yeast. Furthermore, we found that deregulation of Cks1 and Cks2, which often occurs in cancer, results in override of the intra–S phase checkpoint, which protects cells from DNA damage while the cells are undergoing DNA replication. Failure of the checkpoint would most likely make cells cancer prone. PUBLICATIONS Akhoondi, S., Sun, D., von der Lehr, N., Apostolidou, S., Klotz, K., Maljukova, A., Cepeda, D., Fiegl, H., Dafou, D., Marth, C., Mueller-Holzner, E., Corcoran, M., Dagnell, M., Nejad, S.Z., Nayer, B.N., Zali, M.R., Hansson, J., Egyhazi, S., Petersson, F., Sangfelt, P., Nordgren, H., Grander, D., Reed, S.I., Widschwendter, M., Sangfelt, O., Spruck, C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 67:9006, 2007. THE SCRIPPS RESEARCH INSTITUTE 271 Baskerville, C., Segal, M., Reed, S.I. The protease activity of yeast separase (esp1) is required for anaphase spindle elongation independently of its role in cleavage of cohesin. Genetics 178:2361, 2008. Keck, J.M., Summers, M.K., Tedesco, D., Ekholm-Reed, S., Chuang, L.-C., Jackson, P.K., Reed, S.I. Cyclin E overexpression impairs progression through mitosis by inhibiting APCCdh1. J. Cell Biol. 178:371, 2007. Martinsson-Ahlzén, H.S., Liberal, V., Grünenfelder, B., Chaves, S.R., Spruck, C.H., Reed, S.I. Cyclin-dependent kinase-associated proteins Cks1 and Cks2 are essential during early embryogenesis and for cell cycle progression in somatic cells. Mol. Cell. Biol. 28:5698, 2008. Olson, B.L., Hock, M.B., Ekholm-Reed, S., Wohlschlegel, J.A., Dev, K.K., Kralli, A., Reed, S.I. SCFCdc4 acts antagonistically to the PGC-1α transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 22:252, 2008. Reed, S.I. Deathproof: new insights on the role of skp2 in tumorigenesis. Cancer Cell 13:88, 2008. Sangfelt, O., Cepeda, D., Maljukova, A., van Drogen, F., Reed, S.I. Both SCFCdc4α and SCFCdcrγ are required for cyclin E turnover in cell lines that do not overexpress cyclin E. Cell Cycle 7:1, 2008. Control of Gene Expression During the Cell Cycle and in Response to DNA Replication Stress C. Wittenberg, R.A.M. de Bruin, M. Guaderrama, T.I. Kalashnikova he ability of cells to proliferate and to respond to internal and environmental stimuli requires the capacity to rapidly change the abundance and activity of the proteins that mediate those processes. That regulation depends mainly on the ability to modulate gene expression. Recently, we have focused on the mechanisms by which cells exert control over gene expression to regulate cell proliferation and intracellular and environmental signals. In most cells, the initiation of a new round of cell division during the G1 phase of the cell cycle is accompanied by the activation of a large family of genes that encode activities involved in the duplication and segregation of cellular components. G1-specific (also known as G1/S) genes also encode regulatory factors that promote subsequent events in the cell cycle. In the budding yeast Saccharomyces cerevisiae, G1-specific genes are regulated by the transcription factors SBF and MBF. SBF acts as a transcriptional activator and promotes gene expression during the G1 interval. In contrast, MBF acts primarily as a transcriptional repressor and limits transcription of target genes to the G1 phase. Together T 272 MOLECULAR BIOLOGY 2008 SBF and MBF regulate the expression of nearly 200 genes that promote cell proliferation. Using mass spectrometry–based multidimensional protein identification technology, in collaboration with J.R. Yates, Department of Cell Biology, we have identified novel regulators of SBF and MBF. We established that promoter-bound SBF associates with the Whi5 repressor during early G1 phase and that Whi5 is inactivated via phosphorylation by a G1-specific cyclin-dependent protein kinase, thereby activating transcription. This regulation is analogous to the regulation of genes dependent on the transcription factor E2F by the retinoblastoma tumor suppressor in humans. In addition, we identified Nrm1, a novel MBF-associated corepressor. When expressed as an MBF target during late G1 phase, Nrm1 associates with MBF at target promoters and represses expression as cells enter S phase. In addition to the Whi5 and Nrm1 transcriptional coregulators, we identified Msa1, Msa2, and Stb1 as modulators of the transcriptional activity of SBF- and MBF-regulated genes. We have now extended our studies of cell-cycle regulated transcription to the distantly related fission yeast Schizosaccharomyces pombe, in collaboration with P. Russell, Department of Molecular Biology. G1/S transcription in fission yeast is mediated by a single transcription factor called MBF that, like MBF in budding yeast, acts primarily as a transcriptional repressor of G1/S genes. We are studying fission yeast homologs of budding yeast Nrm1 and Whi5. The Nrm1 homolog, SpNrm1, is a target of MBF and, like budding yeast Nrm1, acts as a corepressor with MBF as cells exit G1 phase. Inactivation of fission yeast Nrm1, unlike inactivation of its budding yeast homolog, has a dramatic effect on G1/S gene expression that results in significant cellular phenotypes. In addition to being regulated during the cell cycle, the G1-specific transcriptional machinery is regulated by checkpoints that monitor the integrity of cellular structures and processes. In fission yeast, when DNA replication forks are stalled during S phase, repression of MBF-regulated transcription is disrupted, and expression of MBF target genes is induced. We have shown that SpNrm1 is a direct target for phosphorylation by the checkpoint protein kinase Cds1, a functional homolog of human Chk1, and that its phosphorylation leads to dissociation from MBF and activation of G1/S gene expression (Fig. 1). That response requires the ATM/ATRlike checkpoint kinase Rad3 and other elements of DNA replication checkpoint signaling. We also found that THE SCRIPPS RESEARCH INSTITUTE F i g . 1 . Regulation of G 1-specific transcription by the DNA replication checkpoint in fission yeast, budding yeast, and humans. In budding and fission yeast, activation of the DNA replication checkpoint by stalling the progression of DNA replication prevents the repression of G 1 /S transcription as cells enter S phase. Similarly, in human cells, the Chk1 checkpoint kinase is required for maintenance of G1 /S transcription. This mechanism promotes the repair and reactivation of DNA replication forks and prevents the loss of genome integrity. derepression of MBF target genes occurs in budding yeast after activation of the DNA replication checkpoint. As in fission yeast, transcriptional activation occurs via inactivation of Nrm1 (Fig. 1). G1/S transcription in mammalian cells is regulated by the E2F family of transcription factors. Although the components of the G1-specific transcriptional regulatory system in mammalian cells are apparently unrelated to the yeast regulators, as indicated by amino acid sequence, the architecture of the mammalian and the yeast systems is strikingly similar. We hypothesized that a checkpoint regulatory cascade like that we discovered in the yeast systems controls G1-specific genes via E2F family members in human cells and that the cascade might therefore play a crucial role in the maintenance of genome integrity. We are testing that hypothesis in collaboration with C.H. McGowan, Department of Molecular Biology. We found that human G 1 -specific genes, like those in budding and fission yeasts, remain active during S phase when DNA replication forks stall (Fig. 1). That response, which promotes the expression of genes required for DNA replication and repair, is abrogated when the Cds1 homolog Chk1 is inactivated by chemical inhibitors or short interfering RNA. We are investigating the mechanism by which human G1specific genes are repressed upon entry into S phase cells and how the DNA replication checkpoint machinery interrupts that regulation. Together, our recent findings establish that the mechanisms that govern the expression of G1/S gene expression are conserved among eukaryotes. Furthermore, we have shown that DNA replication stress leads to the activation of those genes during S phase, rein- MOLECULAR BIOLOGY 2008 forcing the cells’ ability to cope with that stress and avoid genomic instability. We expect our continuing research on both yeasts and human cells to yield information critical for our understanding of the cellular response to replication stress, a process central to both the genesis and the treatment of human cancer. PUBLICATIONS Ashe, M., de Bruin, R.A.M., Kalashnikova, T.I., McDonald, W.H., Yates, J.R. III, Wittenberg, C. The SBF- and MBF-associated protein Msa1 is required for proper timing of G1-specific transcription in Saccharomyces cerevisiae. J. Biol. Chem. 283:6040, 2008. de Bruin, R.A.M., Kalashnikova, T.I., Aslanian, A., Wohlschlegel, J.A., Chahwan, C., Yates, J.R. III, Russell, P., Wittenberg, C. DNA replication checkpoint promotes G1/S transcription by inactivating the MBF repressor Nrm1. Proc. Natl. Acad. Sci. U. S. A. 105:11230, 2008. de Bruin, R.A.M., Kalashnikova, T.I., Wittenberg, C. Stb1 collaborates with other regulators to modulate the G1-specific transcriptional circuit. Mol. Cell. Biol., in press. Limbo, O., Chahwan, C., Yamada, Y., de Bruin, R.A.M., Wittenberg, C., Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 28:134 2007. Cell-Cycle Checkpoints, DNA Damage, and Cytotoxic Stress Responses P. Russell, S. Cavero, C. Dovey, G. Dodson, P. Kennedy, O. Limbo, E. Mejia, S. Rozenzhak, A. Vashisht, J. Williams, Y. Yamada NA damage and cytotoxic stress elicit cellular responses that are remarkably similar in yeast and humans. Consequently, studies of genetically tractable microorganisms such as the fission yeast Schizosaccharomyces pombe are highly relevant to understanding these cellular processes in more complex multicellular organisms. We study DNA damage and cytotoxic stress responses because defects in these mechanisms underlie a wide range of human diseases, including cancer. D THE SCRIPPS RESEARCH INSTITUTE 273 but also mental retardation, neurodegeneration, premature aging, immunologic defects, and infertility. The mechanism used to repair double-strand breaks depends on the circumstances in which the damage occurs. Breaks that arise in the postreplicative (G 2 ) phase of the cell cycle are most effectively repaired by homologous recombination, an error-free mechanism in which the undamaged sister chromatid is used as a template for repair of the broken chromosome. In the prereplicative G1 phase, the only option for repair of double-strand breaks is the error-prone mechanism of nonhomologous end joining. In contrast, homologous recombination is required for repair when double-strand breaks are formed by collapse of replication forks during S phase. We recently investigated how cells regulate repair of double-strand breaks during the cell cycle. Because S phase marks the point in the cell cycle at which homologous recombination can repair these breaks, we hypothesized that expression of a homologous recombination factor might coincide with the onset of S phase. Metaanalysis of genome-wide expression profiling studies indicated candidate genes. We found that one of these genes, which we named ctp1, is required for repair of double-strand breaks by homologous recombination. Genetic and biochemical studies revealed that ctp1 mutants cannot resect double-strand breaks, a critical first step of homologous recombination. Other studies indicated that the protein Ctp1 is a cofactor of the Mre11-Rad50-Nbs1 complex, a tumor suppressor in humans (Fig. 1). Ctp1 is so named because it is related to the human protein CtIP. Interestingly, CtIP can interact CHECKPOINTS AND DNA DAMAGE RESPONSES DNA double-strand breaks are among the most lethal and genome-destabilizing types of DNA damage. The breaks arise from exogenous sources such as ionizing radiation, from endogenous sources such as processing of DNA damaged by free radicals, and through errors involving DNA replication. Rapid and accurate repair of double-strand breaks is essential for preserving genome integrity. Indeed, defects in DNA repair are responsible for a multitude of human diseases, most notably cancer, F i g . 1 . Regulation of Ctp1 expression controls repair of double- strand breaks during the cell cycle. Ctp1 acts with the Mre11-Rad50Nbs1 (MRN) complex to promote the resection of double-strand breaks, which is required for repair by homologous recombination (HR). Expression of Ctp1 commences at the transition from G1 to S phase, thereby favoring homologous recombination as the preferred mode of repair during S and G2 phases. Ctp1 is not expressed in G1 phase, leaving nonhomologous end joining (NHEJ) as the only option for repair of double-strand breaks. 274 MOLECULAR BIOLOGY 2008 with BRCA1. The function of BRCA1 has remained an enigma since its discovery as a tumor suppressor that is mutated in a large fraction of hereditary breast cancers. Our findings suggest that interactions between CtIP and BRCA1 may preserve genome stability and prevent cancer by controlling repair of double-strand breaks by homologous recombination. In another study, we investigated the role of Mus81 in repair of single-end breaks. When we discovered Mus81 several years ago, we proposed that it is a specialized DNA-cutting enzyme known as a Holliday junction resolvase. Holliday junctions are the cruciformshaped DNA structures that form at sites of DNA crossovers. Holliday junctions are also proposed to form when broken replication forks are restored. Using a yeast strain in which replication forks break at a specific site on a chromosome, we collaborated with B. Arcangioli, Institut Pasteur, Paris, France, to show that Mus81 DNA cleavage activity is essential for the survival of a broken fork (Fig. 2). We also found that Mus81 specifically associates with the chromosomal region where the Holliday junctions form. These data strengthen the evidence that Mus81 is a Holliday junction resolvase. Interestingly, in human THE SCRIPPS RESEARCH INSTITUTE cells, Mus81 is specifically required for survival in the presence of cisplatin, a drug used to treat various types of cancers such as sarcomas (e.g., bone cancer), some carcinomas (e.g., ovarian cancer), and lymphomas. Drugs that target Mus81 may be useful when used in combination with cisplatin. CYTOTOXIC STRESS RESPONSE AND SYSTEMS BIOLOGY Production of reactive oxygen species (ROS) is a normal byproduct of aerobic metabolism. Intracellular ROS can also arise through exposure to environmental toxicants, such as heavy metals or metalloids (e.g., cadmium and arsenic) and some pesticides. Oxidative stress in the form of ROS can be highly toxic, causing damage to proteins, lipids, and nucleic acids. Indeed, the cumulative effects of exposure to ROS are thought to be a causative factor in many of the most widespread and debilitating human diseases, such as atherosclerosis, Alzheimer’s disease, Parkinson’s disease, and cancer, and in the aging process itself. In the past year, we used systems biology to define all the nonessential genes required for tolerance to cadmium and arsenic in fission yeast. Using the fission yeast haploid deletion library, we have defined approximately 250 genes involved in cadmium or arsenic tolerance. Prominent among these are the gene required for biosynthesis of coenzyme Q10. Coenzyme Q10 is an antioxidant commonly used as a dietary supplement for treatment of hypertension, Parkinson’s disease, and other maladies. Our studies are helping us understand the role of coenzyme Q10 in the physiologic response to environmental toxicants. PUBLICATIONS de Bruin, R.A.M., Kalashnikova, T.I., Aslanian, A., Wohlschlegel, J., Chahwan, C., Yates, J.R. III, Russell, P., Wittenberg, C. The DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc. Natl. Acad. Sci. U. S. A. 105:11230, 2008. Dovey, C.L., Russell, P. Mms22 preserves genomic integrity during DNA replication in Schizosaccharomyces pombe. Genetics 177:47, 2007. Kennedy, P.J., Vashisht, A.A., Hoe, K.-L., Kim, D.U., Park, H.O., Hayles, J., Russell, P. A genome-wide screen of genes involved in cadmium tolerance in Schizosaccharomyces pombe. Toxicol. Sci. 106:124, 2008. F i g . 2 . Mus81 is required for recovery from breakage of replication forks. A, Replication fork encounters a nick in the leading strand template, leading to breakage of the replication fork. B, Breakage of the replication fork creates a single-end break. C, The single-end break is resected, leaving a single-strand tail that invades the sister chromatid. Strand invasion creates a D-loop that in principle can be cleaved by the Mus81-Eme1 complex. D, Alternatively, strand invasion can lead to reformation of the replication fork before cleavage of the resulting Holliday junction by Mus81-Eme1. E, Either pathway can lead to restoration of the replication fork. Limbo, O., Chahwan, C., Yamada, Y., de Bruin, R.A.M., Wittenberg, C., Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 28:134, 2007. Martin, V., Du, L.-L., Rozenzhak, S., Russell, P. Protection of telomeres by a conserved Stn1-Ten1 complex. Proc. Natl. Acad. Sci. U. S. A. 104:14038, 2007. Noguchi, E., Ansbach, A.B., Noguchi, C., Russell, P. Assays used to study the DNA replication checkpoint in fission yeast. Methods Mol. Biol., in press. Rodríguez-Gabriel, M.A., Russell, P. Control of mRNA stability by SAPKs. Top. Curr. Genet. 20:159, 2008. MOLECULAR BIOLOGY 2008 Roseaulin, L., Yamada, Y., Tsutsui, Y., Russell, P., Iwasaki, H., Arcangioli, B. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 27:1378, 2008. Williams, R.S., Moncalian, G., Williams, J.S., Yamade, Y., Limbo, O., Shin, D.S., Groocock, L.M., Cahill, D., Hitomi, C., Guenther, G., Moiani, D., Carney, J.P., Russell, P., Tainer, J.A. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135:97, 2008. DNA Damage Responses in Human Cells C.H. McGowan, M. Duquette, E. Langley, J. Scorah, D. Slavin, E. Taylor n long-lived animals, including humans, the advantages of being able to replace damaged or aged cells are offset by the inherent susceptibility of dividing cells to acquire mutations and become cancerous. DNA is inherently vulnerable to many sorts of chemical and physical modifications; thus, as cells duplicate and divide, they can acquire mutations. Cells have evolved with a complex network of DNA repair processes and cell-cycle checkpoint responses that ensure that damaged DNA is repaired before it is replicated and becomes fixed in the genome. Our overall objective is to understand how mammalian cells protect themselves from DNA damage and thus from cancer. We are especially interested in understanding basic cellular responses to clinically relevant agents. We use a combination of molecular, cellular, and genetic techniques to determine how these DNA repair pathways operate in human cells. Checkpoints control the order and timing of events in the cell cycle; they ensure that independent processes are appropriately coupled. In addition, checkpoints promote the use of the most appropriate repair pathway. We used genetic models to identify 2 checkpoint kinases in humans that limit progression of the cell cycle when DNA is damaged. We are studying the function of both of these kinases and of a number of their substrates. One of the kinases, Chk2, is activated in response to DNA damage. Chk2 physically interacts with Mus81-Eme1, a conserved DNA repair protein that has endonuclease activity against structure-specific DNA substrates, including Holliday junctions. Enzymatic analysis, immunofluorescence studies, and the use of RNA interference have all contributed to the conclusion that Mus81-Eme1 is required for recombination repair in human cells. We are also using gene targeting to study the function of the Mus81-Eme1 endonu- I THE SCRIPPS RESEARCH INSTITUTE 275 clease in mice. Inactivation of Mus81 in mice increases genomic instability and sensitivity to DNA damage but does not promote tumorigenesis. In addition, we have shown that Mus81-Eme1 is specifically required for survival after exposure to cisplatin, mitomycin C, and other commonly used anticancer drugs. Using laseractivated psoralens to create DNA damage in specific subnuclear regions, we are defining the mechanism by which Mus81-Eme1 and other enzymes function in the repair of chemotherapeutic agents. Anticancer therapy is mainly based on the use of genotoxic agents that damage DNA and thus kill dividing cells. Coordination of cell-cycle checkpoints and DNA repair is especially important when unusually high amounts of DNA damage occur after radiation or genotoxic chemotherapy. Hence, a detailed understanding of cellular responses to DNA damage is essential in understanding both the development and the treatment of disease in humans. PUBLICATIONS Prudden, J., Pebernard, S., Raffa, G., Slavin, D.A., Perry, J.J., Tainer, J.A., McGowan, C.H., Boddy, M.N. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 26:4089, 2007. Scorah, J., Dong, M.Q., Yates, J.R. III, Scott, M., Gillespie, D., McGowan, C.H. A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J. Biol. Chem. 283:17250, 2008. Taylor, E.R., McGowan, C.H. Cleavage mechanism of human Mus81-Eme1 acting on Holliday-junction structures. Proc. Natl. Acad. Sci. U. S. A. 105:3757, 2008. DNA Repair and the Maintenance of Genomic Stability M.N. Boddy, J. Heideker, S. Pebernard, J. Prudden NA repair pathways have evolved to protect the genome from ever-present genotoxic agents. Highlighting the importance of the pathways, defects in DNA repair mechanisms strongly predispose the host to cancer and to neurologic and developmental disorders. The DNA repair systems we study in fission yeast are evolutionarily conserved, and therefore our studies provide a valuable framework for understanding genome maintenance in human cells. Although many DNA repair mechanisms have been described, information on how they are coordinated with necessary changes in chromatin structure is limited. D 276 MOLECULAR BIOLOGY 2008 THE SMC5-SMC6 COMPLEX In collaboration with J.R. Yates, Department of Cell Biology, we purified the structural maintenance of chromosomes (SMC) complex Smc5-Smc6 to identify its core components. The holocomplex consists of the Smc5Smc6 heterodimer and 6 additional non-SMC elements, Nse1–Nse6 (Fig. 1A). We showed that Smc5-Smc6 F i g . 1 . Smc5-Smc6 holocomplex and STUbLs. A, Nse1, Nse3, and Nse4 form a stable heterotrimer that associates with Smc5. Nse2 interacts directly with Smc5 in the absence of the other Nse proteins. Smc6 interacts directly with Smc5 but with none of the other components. Nse5 and Nse6 form a stable heterodimer that also binds directly to Smc5. Double-headed arrows indicate interactions between subcomplexes. Nse5 and Nse6 may recruit the holocomplex to stalled replication forks and certain DNA damage sites. B, A target protein is sumoylated, an event that leads to recruitment of STUbLs via interactions with SUMO and the substrate. For Rad60 or NIP45, STUbLs interact via the SUMO-like domains. STUbLs then ubiquitinate the substrate and target it for degradation. prevents the deleterious engagement of an ordinarily beneficial DNA repair pathway called homologous recombination. Smc5-Smc6 either prevents initiation of homologous recombination or separates physically linked chromosomes that arise late in the process. Spontaneous DNA damage in Smc5-Smc6 mutant cells is due to the attempted separation of chromosomes into daughter cells while the chromosomes are still physically linked. Such defective chromosome separation in humans could result in cancer and other diseases. A N U N P R E C E D E N T E D S U M O - TA R G E T E D U B I Q U I T I N LIGASE The covalent attachment of ubiquitin and the small ubiquitin-like protein SUMO to target proteins plays key roles in genome stability; each of the 2 moieties (i.e., ubiquitin and SUMO) has physiologically distinct effects on the function of target proteins. We have identified the SUMO-targeted ubiquitin ligase (STUbL) family, which provides a novel and unanticipated regulatory link between the ubiquitination and sumoylation pathways. Members of the STUbL family include Slx8-Rfp1 THE SCRIPPS RESEARCH INSTITUTE in fission yeast, RNF4 in humans, MIP1 in slime molds, and SLX5/8 in budding yeast. STUbLs are recruited to sumoylated proteins and proteins containing SUMOlike domains to mediate ubiquitination and degradation of these proteins (Fig. 1B). Cells with mutations in Slx8-Rfp1 accumulate sumoylated proteins, have genomic instability, and are hypersensitive to genotoxic stress. These Slx8-Rfp1 mutant phenotypes are suppressed by concomitant deletion of the major SUMO ligase Pli1, demonstrating the specificity of STUbLs as regulators of sumoylated proteins. Expression of human RNF4 restores homeostasis of the SUMO pathway in fission yeast that lack Slx8Rfp1, underscoring the evolutionary functional conservation of STUbLs. The DNA repair factor Rad60 (an accessory factor of the Smc5-Smc6 complex) and its human homolog NIP45, which contain SUMO-like domains, are candidate STUbL targets. Consistently, mutations in Rad60 and Slx8-Rfp1 cause similar DNA repair defects. PUBLICATIONS Pebernard, S., Perry, J.J., Tainer, J.A., Boddy, M.N. Nse1 RING-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol. Biol. Cell 19:4099, 2008. Perry, J.J., Tainer, J.A., Boddy, M.N. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem. Sci. 33:201, 2008. Prudden, J., Pebernard, S., Raffa, G., Slavin, D.A., Perry, J.J., Tainer, J.A., McGowan, C.H., Boddy, M.N. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 26:4089, 2007. Signal Transduction Pathways Mediating Cellular Responses to Oncogenic Mutations P. Sun, J. Kwong, R. Liao, A. Seit-Nebi, N. Yoshizuka evelopment of cancer is a result of multiple oncogenic genetic alterations, including activation of oncogenes and inactivation of tumor suppressors. Although these oncogenic mutations contribute to tumorigenic phenotypes, normal cells usually respond to oncogenic changes by initiating tumor-suppressing defense mechanisms such as apoptosis and premature senescence (a stable form of growth arrest). Consequently, tumor development requires additional mutations that compromise these antioncogenic responses. Our main interests are to delineate the signal transduction pathways that mediate these tumor-suppress- D MOLECULAR BIOLOGY 2008 ing responses and to determine how these responses are evaded during cancer development. Currently, we are focusing on 2 well-known oncogenes: ras and mdm2. The oncogene ras encodes a family of small GTPbinding proteins that are often activated in human tumors and contribute to tumor development. In normal cells, however, the initial response to ras activation is premature senescence. Recent studies have shown that like apoptosis, oncogene-induced senescence is a bona fide tumor-suppressing mechanism in vivo that must be compromised in order for cancer to develop. However, the signaling pathways responsible for this important antitumorigenic response are poorly understood. We have shown that ras induces senescence through sequential activation of 2 MAP kinase pathways. Initially, ras activates the MAP kinase kinase (MEK)–extracellular signal–regulated kinase (ERK) pathway. Sustained activation of MEK-ERK turns on the stress-induced p38 pathway, which subsequently causes senescence. These studies have revealed a novel, tumor-suppressing function of p38, in addition to its known roles in inflammation and stress responses. The functions of p38 are mediated by its downstream substrates, including a family of serine/threonine protein kinases. We found that one of these substrate kinases of p38, p38-regulated/activated protein kinase (PRAK), mediates senescence upon activation by p38 in response to oncogenic ras. In mice, PRAK deficiency enhances skin carcinogenesis induced by the environmental mutagen 7,12-dimethylbenz(a)anthracene, coinciding with compromised induction of senescence. In primary cells, inactivation of PRAK prevents senescence and promotes oncogenic transformation. Moreover, PRAK activates p53 by direct phosphorylation of the serine at position 37 in p53. We propose that phosphorylation of p53 by PRAK after activation of p38 MAP kinase by ras plays an important role in rasinduced senescence and tumor suppression. Experiments are under way to characterize additional downstream substrates and upstream regulators of PRAK that contribute to the induction of senescence and tumor suppression. In addition, we are attempting to systematically analyze the signaling components of the p38 pathway to determine their roles in different cellular functions involving p38, including inflammation and tumor suppression. Results from these studies will provide the basis for developing anti-inflammatory and anticancer drugs that target the p38 pathway. Another focus of our research is mdm2, an oncogene that can mediate transformation primarily through THE SCRIPPS RESEARCH INSTITUTE 277 inactivation of the tumor suppressor p53. Previously, we found that MDM2, the protein encoded by mdm2, confers resistance to cell-cycle arrest induced by transforming growth factor β (TGF-β), a growth-inhibitory cytokine. In studies on the molecular mechanism that underlies MDM2-mediated resistance to TGF-β, we found that MDM2 makes cells refractory to the cytokine by overcoming a TGF-β–induced arrest of the cell cycle in G1. Investigation of the structure-function relationship of MDM2 revealed 3 elements essential for MDM2 to confer resistance to TGF-β. One element was the C-terminal half of the p53-binding domain, which at least partially retained p53 binding and inhibitory activity. Second, the ability of MDM2 to mediate resistance to TGF-β was disrupted by mutation of the nuclear localization signal. Finally, mutations of the zinc coordination residues of the RING finger domain abrogated resistance to TGF-β but not the ability of MDM2 to inhibit p53 activity or to bind MDMX, another p53 regulator. These data suggest that RING finger–mediated p53 inhibition and MDMX interaction are not sufficient to cause TGF-β resistance and imply a crucial role of the E3 ubiquitin ligase activity of this domain in MDM2-mediated TGF-β resistance. PUBLICATIONS Han, J., Sun, P. The pathways to tumor suppression via route p38. Trends Biochem. Sci. 32:364, 2007. Liu, E., Lee, A.Y., Chiba, T., Olson, E., Sun, P., Wu, X. The ATR-mediated S phase checkpoint prevents DNA rereplication in mammalian cells when licensing control is disrupted. J. Cell Biol. 179:643, 2007. Wang, K., Cheng, C., Li, L., Liu, H., Huang, Q., Xia, C.H., Yao, K., Sun, P., Horwitz, J., Gong, X. γD-crystallin-associated protein aggregation and lens fiber cell denucleation. Invest. Ophthalmol. Vis. Sci. 48:3719, 2007. Genetic Modifiers of Behavioral Despair as Targets for New Antidepressants J.G. Sutcliffe, P.B. Hedlund, F.E. Bloom,* B.S. Hilbush* * ModGene, L.L.C., La Jolla, California he forced-swim test and the tail-suspension test are mouse models with high value for predicting the antidepressant activity of a drug. In both tests, a state of behavioral despair is created in which mice cease attempts to escape and become immobile because of the adverse situation. Known human antidepressants T 278 MOLECULAR BIOLOGY 2008 increase the length of time the mice attempt to escape, thus decreasing the immobility times. Significant behavioral differences exist among inbred mouse strains in these tests. For example, during a 4-minute forced-swim test, unmedicated, inbred C57BL/6J mice spent 240 seconds or 63% of the swim immobile. In contrast, inbred DBA/2J mice spent only 47 seconds or 20% of the time immobile. In collaboration with researchers at ModGene, L.L.C., we used these strain differences to identify genes whose activities contribute to relative basal despair status. We measured baseline immobility times of both males and females of 27 strains of recombinant inbred mice that were produced from C57 x DBA matings. Each strain had a characteristic immobility time, ranging from 2% (5 seconds) to 70% (168 seconds) of the swim, and differences between males and females were detected within strains. We correlated these data with the haplotype data from these mouse strains and detected despair modifier genes on chromosome 4 in all mice and additionally on chromosomes 11 and 13 in female mice and chromosome 18 in male mice, indicating some sexual dimorphism in determinants of this behavioral despair, as is known for depression in humans. In analogous studies with the tail-suspension test, we detected the same chromosome 4 genes in both males and females and the same chromosome 11 and 13 genes in females, suggesting that these 3 genes are directly related to the despair behavior rather than to the ability to perform in one of the behavioral tests. We then tested the hypothesis that at least some of the strain differences occur because of the amount of mRNA that accumulates from a single gene in each of the mapped chromosomal regions. We examined the concentrations of each of more than 30,000 mRNAs in several regions of the brains of the recombinant inbred mice. We identified a single gene within the chromosome 4 region for which the amount of mRNA was high in all strains that inherited the C57 genotype and low in all strains that inherited the DBA genotype. Similarly, we found a single chromosome 11 gene and a single chromosome 13 gene for which the mRNA output was inherited in the same mendelian fashion as the corresponding despair modifier gene. The identities of the genes responsible for modifying the despair phenotype offer a powerful point of departure, serving as targets for the development of new pharmaceutical agents to treat depression. The studies used to detect the genes provide evidence that THE SCRIPPS RESEARCH INSTITUTE differences in the activities of the protein products of the genes directly contribute to differences in phenotype. Thus, a drug that altered the activity of a protein encoded by a specific gene and did not have other deleterious side effects would be suitable candidate to test for antidepressant activity. The target gene on chromosome 4 encodes a previously known protein that has never been associated with brain disorders. The activity of the protein is regulated by phosphorylation by a specific protein kinase. The concentration of the mRNA of the target gene is higher in C57 mice (the mice that exhibit greater despair) than in DBA mice. Therefore, we sought a way to reduce the activity of the gene to make the behavior of the C57 mice more like that of the DBA mice. Compounds that inhibit the activity of the specific kinase in cultured tumor cells have recently been synthesized. These compounds also inhibit target phosphorylation in the brains of treated mice. When these compounds were administered to C57 or DBA mice, immobility times in the forced-swim test and the tailsuspension test were reduced in a dose-dependent fashion, indicative of an antidepressant-like effect. The compounds had no effects in anxiety or locomotor tests. These observations suggest that these compounds can be considered as leads for testing as antidepressants. PUBLICATIONS Desplats, P.A., Denny, C.A., Kass, K.E., Gilmartin, T., Head, S.R., Sutcliffe, J.G., Seyfried, T.N., Thomas, E.A. Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol. Dis. 27:265, 2007. Semenova, S., Geyer, M.A., Sutcliffe, J.G., Markou, A., Hedlund, P.B. Inactivation of the 5-HT7 receptor partially blocks phencyclidine-induced disruption of prepulse inhibition. Biol. Psychiatry 63:98, 2008. Molecular Neurobiology of CNS Disorders E.A. Thomas, B. Tang, J.G. Sutcliffe MOLECULAR BASIS FOR DISEASE PROGRESSION IN SCHIZOPHRENIA chizophrenia is a devastating mental illness that occurs in 1% of the general population. The molecular factors that influence the course of illness in schizophrenia and how treatment modifies these factors are areas of interest in our group. We have generated genome-wide RNA expression profiles from tissue samples obtained at autopsy from the prefrontal cortex of patients who had had schizophrenia for various S MOLECULAR BIOLOGY 2008 lengths of time, including a cohort of patients who died within 5 years after the initial diagnosis. We found that the early stages of disease are associated with the greatest derangement in gene expression and that these genes are associated with diverse systems and pathways. In particular, we are focusing on genes that encode proteins with transcriptional regulatory activity, because these proteins may drive further pathologic or compensatory changes detected later in illness. We are also interested in genes correlated with age in humans who have schizophrenia and in those regulated by antipsychotic drug treatment in mouse models. The identification of genes associated with the early vs later stages of schizophrenia will be important for understanding disease progression and might lead to the development of agents that modify the course of disease. H U N T I N G T O N ’ S D I S E A S E : T R E AT M E N T A P P R O A C H E S T H AT TA R G E T G E N E T R A N S C R I P T I O N Huntington’s disease is an autosomal-dominant neurologic disorder caused by a CAG repeat expansion within the coding region of the gene for the disease, resulting in a mutant protein with an expanded polyglutamine tract. Mutant huntingtin protein can disrupt transcription by diverse mechanisms, including loss of function of transcription factors and chromatin-mediated repression. We are exploring mechanisms for transcriptional dysregulation in Huntington’s disease and testing therapeutic strategies aimed at improving transcriptional output via modulation of chromatin structure. We have identified target genes for 2 transcriptional regulatory proteins, Bcl11b and Foxp1, which have highly enriched expression in the striatum, the brain region most affected in Huntington’s disease. We found that these transcription factors interact with huntingtin protein, suggesting a role in the dysregulation of striatal gene expression in patients with Huntington’s disease. In collaboration with J.M. Gottesfeld, Department of Molecular Biology, we are testing the therapeutic effects of novel histone deacetylase inhibitors in mouse models of Huntington’s disease. One of these inhibitors, HDACi 4b, prevents the disease phenotypes in R6/2 transgenic mice. Using microarray analysis, we found that treatment with HDACi 4b ameliorated abnormalities in gene expression in these mice and caused complete normalization of expression of subsets of genes, which may be considered biomarkers for treatment effectiveness. Our findings suggest that HDACi 4b treatment may be useful in slowing the progression of symptoms in patients with Huntington’s disease. THE SCRIPPS RESEARCH INSTITUTE 279 PUBLICATIONS Dean, B., Digney, A., Sundram, S., Thomas, E.A., Scarr E. Plasma apolipoprotein E is decreased in schizophrenia spectrum and bipolar disorder. Psychiatry Res. 158:75, 2008. Desplats, P.A., Denny, C.A., Kass, K.E., Gilmartin, T., Head, S.R., Sutcliffe, J.G., Seyfried, T.N., Thomas, E.A. Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol. Dis. 27:265, 2007. Desplats, P.A., Lambert, J.R., Thomas, E.A. Functional roles for the striatalenriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington’s disease. Neurobiol. Dis., in press. Narayan, S., Head, S.R., Gilmartin, T.J., Dean, B., Thomas, E.A. Evidence for disruption of sphingolipid metabolism in schizophrenia. J Neurosci Res., in press. Narayan, S., Tang, B., Head, S.R., Gilmartin, T.J., Sutcliffe, J.G., Dean, B., Thomas, E.A. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res., in press. Stuart Gibbons, A., Scarr, E., McOmish, C., Hannan, A.J., Thomas, E.A., Dean, B. Regulator of G-protein signalling 4 expression is not altered in the prefrontal cortex in schizophrenia. Aust. N. Z. J. Psychiatry 42:740, 2008. Thomas, E.A., Coppola, G., Desplats, P.A., Tang, B., Soragni, E., Burnett, R., Gao, F., Fitzgerald, K.M., Borok, J.F., Herman, D., Geschwind, D.H., Gottesfeld, J.M. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 105:15564, 2008. The 5-HT7 Receptor in Neuropsychiatric Disorders P.B. Hedlund, G. Sarkisyan, P.E. Danielson, J.G. Sutcliffe nterest in the serotonin 5-HT7 receptor as a putative target for the treatment of neuropsychiatric disorders has been growing continually. The interest was prompted by the finding that several classes of drugs used to treat disorders such as depression and schizophrenia have high affinity for this receptor. We have established evidence that supports a role for the 5-HT7 receptor in depression, obsessive-compulsive disorder, and schizophrenia. I DEPRESSION The forced swim test and the tail suspension test are animal models of behavioral despair that have high value for predicting the antidepressant efficacy of drugs. The tests can also be used to characterize animals in which genes have been inactivated. In both of these tests, we showed that mice lacking the 5-HT7 receptor have a behavioral profile similar to that of mice treated with antidepressants. We replicated these findings by using a compound that acts as a selective antagonist at the receptor. Thus, both blockade and inactivation of the 5-HT 7 receptor yield the same result. New evidence suggests that the effects of antidepressants that act as either serotonin reuptake inhibitors or norepi- 280 MOLECULAR BIOLOGY 2008 THE SCRIPPS RESEARCH INSTITUTE nephrine reuptake inhibitors are potentiated by selective antagonism of the 5-HT7 receptor. These synergistic interactions provide valuable new insights into the mechanism of action of antidepressants and open up new possibilities for the treatment of mood disorders. consequences of aneuploidy in the brain. While continuing to characterize the extent and regional variations of aneuploidy in the brains of healthy humans and normal mice, we are determining the link between neural aneuploidy and human brain disorders. OBSESSIVE-COMPULSIVE DISORDER LY S O P H O S P H O L I P I D S I G N A L I N G Obsessive-compulsive disorder is commonly treated with antidepressants and thus is related to depression. In an animal model of obsessive-compulsive disorder (marble burying), we showed that blockade or inactivation of the 5-HT7 receptor results in less stereotypic behavior. These findings further support the hypothesis that the 5-HT7 receptor is an important alternative or supplemental target for antidepressants. Lysophospholipids are classically known as metabolites in the biosynthesis of cell membranes; however, their newly discovered functions in a diverse array of biological processes have highlighted their importance in health and disease. We discovered the first lysophospholipid receptor, LPA1, and we now know of 10 receptors. We have continued to explore the cellular and physiologic functions of receptor-mediated lysophospholipid signaling, primarily by generating and examining mice that lack the receptors. In ongoing studies, we are deciphering the downstream signaling cascades that mediate the different cellular functions of lysophospholipid receptor activation, determining the contribution of receptor-mediated lysophospholipid signaling to mammalian brain development, analyzing the neuroanatomic and neurobehavioral phenotypes of mice that lack lysophospholipid receptors, and examining how lysophospholipids and their receptors are involved in myelination and myelination-related diseases, including multiple sclerosis. SCHIZOPHRENIA Prepulse inhibition (PPI) of the acoustic startle reflex is a well-characterized model of schizophrenia. The model is especially relevant because similar responses can be observed in patients with schizophrenia. We showed that PPI per se is not altered in mice lacking the 5-HT7 receptor but that when PPI is disrupted by phencyclidine, these mice are significantly less affected than are mice that have the receptor. Phencyclidineinduced disruption involves a glutamatergic component of PPI that is relevant for the action of atypical antipsychotic agents such as clozapine. Clozapine is a drug with relatively high affinity for the 5-HT7 receptor. NEURAL ANEUPLOIDY PUBLICATIONS Semenova, S., Geyer, M.A., Sutcliffe, J.G., Markou, A., Hedlund, P.B. Inactivation of the 5-HT7 receptor partially blocks phencyclidine-induced disruption of prepulse inhibition. Biol. Psychiatry 63:98, 2008. Lysophospholipid Signaling and Neural Aneuploidy J. Chun, S. Barral, J. Choi, A. Dubin, S. Gardell, D. Herr, G. Kennedy, M. Kingsbury, C.W. Lee, D. Letourneau, D. Lin, M. Lu, T. Mutoh, K. Noguchi, C. Paczkowski, S. Peterson, R. Rivera, S. Teo, S. Tunaru, W. Westra, X. Ye, Y. Yung, L. Zhu indings in the past year led to new directions in our long-term projects. In our studies on lysophospholipid signaling, we discovered roles for lysophosphatidic acid (LPA) receptors in lung and kidney fibrosis and continued our studies on the functions of the receptors for sphingosine 1-phosphate (S1P) in the immune, cardiovascular, and nervous systems. In our research on neural aneuploidy, we are moving toward a deeper understanding of the functional F Since our initial discovery that many cells in the brain are aneuploid, we have been developing new methods to investigate aneuploidy to address its anatomic and functional significance. During the past year, we extended analyses of the genomic complement of neural cells to the neural cells of teleost fishes, showing the evolutionary conservation of aneuploidy. We are continuing to investigate how neural aneuploidy affects brain function at both cellular and system-wide levels, including the possible contribution to dysfunction in neurologic disorders. PUBLICATIONS Chan, L.C., Peters, W., Xu, Y., Chun, J., Farese, R.V., Jr., Cases, S. LPA3 receptor mediates chemotaxis of immature murine dendritic cells to unsaturated lysophosphatidic acid (LPA). J. Leukoc. Biol. 82:1193, 2007. Chun, J. Extracellular lipid signals. In: Wiley Encyclopedia of Chemical Biology. Begley, T.P. Wiley-Interscience, New York, in press. Chun, J. Genomic disorder and gene expression in the developing CNS. In: The New Encyclopedia of Neuroscience. Squire, L.R. (Ed. in Chief). Elsevier, Philadelphia, in press. Chun, J. How the lysophospholipid got its receptor. Scientist 21:48, September 2007. Chun, J. The sources of a lipid conundrum. Science 316:208, 2007. MOLECULAR BIOLOGY 2008 Hama, K., Aoki, J., Inoue, A., Endo, T., Amano, T., Motoki, R., Kanai, M., Ye, X., Chun, J., Matsuki, N., Suzuki, H., Shibasaki, M., Arai, H. Embryo spacing and implantation timing are differentially regulated by LPA3-mediated lysophosphatidic acid signaling. Biol. Reprod. 77:954, 2007. Herr, D., Chun, J. Effects of LPA and S1P on the nervous system and implications for their involvement in disease. Curr. Drug Targets 8:155, 2007. Herr, D., Grillet, N., Schwander, M., Rivera, R., Müller, U., Chun, J. Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. J. Neurosci. 27:1474, 2007. Keller, C.D., Gil, P.R., Tölle, M., van der Giet, M., Chun, J., Radeke, H.H., Schäfer-Korting, M., Kleuser, B. Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. Am. J. Pathol. 170:281, 2007. Means, C.K., Xiao, C.Y., Li, Z., Zhang, T., Omens, J.H., Ishii, I., Chun, J., Brown, J.H. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 292:H2944, 2007. Peterson, S., Rehen, S., Westra, W., Yung, Y., Chun, J. Spectral karyotyping and fluorescent in situ hybridization. In: Human Stem Cell Manual: A Laboratory Guide. Loring, J.F., Schwartz, P.H., Wesselschmidt, R.l. (Eds.). Academic Press Elsevier, New York, 2007, p. 71. Pradère, J.-P., Klein, J., Grès, S., Guigné, C., Neau, E., Valet, P., Calise, D., Chun, J., Bascands, J.-L., Saulnier-Blache, J.-S., Schanstra, J.P. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 18:3110, 2007. Rajendran, R.S., Zupanc, M.M., Lösche, A., Westra, J., Chun, J., Zupanc, G.K.H. Numerical chromosome variation and mitotic segregation defects in the adult brain of teleost fish. Dev. Neurobiol. 67:1334, 2007. Rivera, R., Chun, J. Potential therapeutic roles of lysophospholipid signaling in autoimmune-related disease. Future Lipidol. 2:535, 2007. Serriere-Lanneau, V., Teixeira-Clerc, F., Li, L., Schippers, M., de Wries, W., Julien, B., Tran-Van-Nhieu, J., Manin, S., Poelstra, K., Chun, J., Carpentie, S., Levade, T., Mallat, A., Lotersztajn, S. The sphingosine 1-phosphate receptor S1P2 triggers hepatic wound healing. FASEB J. 21:2005, 2007. Walter, D.H., Rochwalsky, U., Reinhold, J., Seeger, F., Aicher, A., Urbich, C., Spyridopoulos, I., Chun, J., Brinkmann, V., Keul, P., Levkau, B., Zeiher, A.M., Dimmeler, S., Haendeler, J. Sphingosine-1-phosphate stimulates the functional capacity of progenitor cells by activation of the CXCR4-dependent signaling pathway via the S1P3 receptor. Arterioscler. Thromb. Vasc. Biol. 27:275, 2007. THE SCRIPPS RESEARCH INSTITUTE 281
 0
0
advertisement
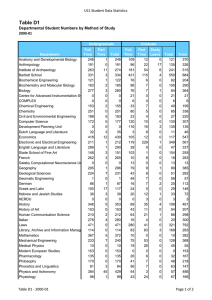
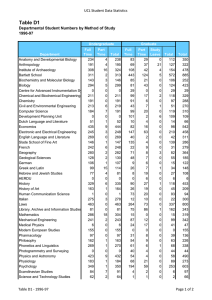
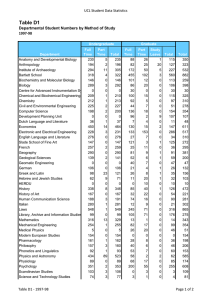

Download
advertisement
Add this document to collection(s)
You can add this document to your study collection(s)
Sign in Available only to authorized usersAdd this document to saved
You can add this document to your saved list
Sign in Available only to authorized users