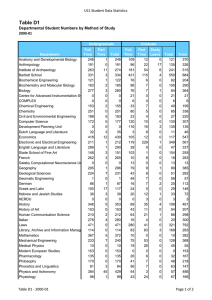
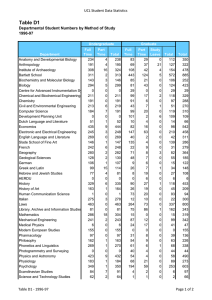
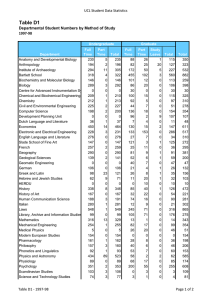
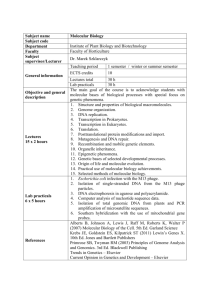
Molecular Biology
advertisement

Molecular Biology Conservation of Regulation of G1-to-S transcription in eukaryotes. Eukaryotes from yeast to humans coordinately express a family of genes during the G1 phase in preparation for traversing the cell cycle. Although these transcriptional regulators in yeast and humans have no primary sequence homology, their function is conserved. The transcriptional repressor Whi5 and the corepressor Nrm1 in yeast perform functions analogous to the mammalian pocket proteins Rb and p107, respectively. Nrm1, which represses transcription as cells enter S phase, is a target for the DNA replication checkpoint, and p107 may be similarly regulated to ensure genome integrity. Work done by Rob de Bruin, Ph.D., research associate, in the laboratory of Curt Wittenberg, Ph.D., professor. Paul R. Schimmel, Ph.D., Ernest and Jean Hahn Professor of Molecular Biology and Chemistry, and Wei Zhang, Ph.D., Research Asssociate MOLECULAR BIOLOGY DEPAR TMENT OF MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE Lluis Ribas De Pouplana, Ph.D. Adjunct Assistant Professor Barcelona Science Park Barcelona, Spain Ehud Keinan, Ph.D. Adjunct Professor Technion-Israel Institute of Technology Haifa, Israel Ashok Deniz, Ph.D. Associate Professor Richard A. Lerner, M.D., Ph.D.** President, Scripps Research Lita Annenberg Hazen Professor of Immunochemistry Cecil H. and Ida M. Green Chair in Chemistry S TA F F Peter E. Wright, Ph.D.* Professor and Chairman Cecil H. and Ida M. Green Investigator in Medical Research Ruben Abagyan, Ph.D. Professor Carlos F. Barbas III, Ph.D.** Professor Janet and W. Keith Kellogg II Chair, Molecular Biology Rajesh Belani, Ph.D. Adjunct Assistant Professor Scripps Mercy Hospital San Diego, California Ola Blixt, Ph.D. Assistant Professor of Molecular Biology Michael N. Boddy, Ph.D. Assistant Professor Charles L. Brooks III, Ph.D. Professor David A. Case, Ph.D. Professor Geoffrey Chang, Ph.D.* Associate Professor Eli Chapman, Ph.D. Assistant Professor of Molecular Biology Jerold Chun, M.D., Ph.D. Professor Aymeric Pierre De Parseval, Ph.D.*** Assistant Professor of Molecular Biology Public Health Research Institute Newark, New Jersey H. Jane Dyson, Ph.D. Professor John H. Elder, Ph.D.**** Professor Martha J. Fedor, Ph.D.* Associate Professor James A. Fee, Ph.D. Professor of Research Scott Lesley, Ph.D. Assistant Professor of Biochemistry Tianwei Lin, Ph.D. Assistant Professor Elizabeth D. Getzoff, Ph.D.***** Professor Ian MacRae, Ph.D. Assistant Professor David B. Goodin, Ph.D. Associate Professor Clare McGowan, Ph.D. † Associate Professor David S. Goodsell Jr., Ph.D. Associate Professor of Molecular Biology Duncan E. McRee, Ph.D. Adjunct Associate Professor ActiveSight San Diego, California Joel M. Gottesfeld, Ph.D. Professor Jennifer Harris, Ph.D. Assistant Professor of Biochemistry Christian A. Hassig, Ph.D. Adjunct Assistant Professor Kalypsis, Inc. San Diego, California Peter B. Hedlund, M.D., Ph.D. Assistant Professor of Molecular Biology John E. Johnson, Ph.D. Professor Gerald F. Joyce, M.D., Ph.D.** Professor Dean, Faculty David P. Millar, Ph.D. Associate Professor Louis Noodleman, Ph.D. Associate Professor 183 Harold Scheraga, Ph.D. Adjunct Professor George W. and Grace L. Todd Professor of Chemistry, Emeritus Cornell University Ithaca, New York Paul R. Schimmel, Ph.D.** Ernest and Jean Hahn Professor of Molecular Biology and Chemistry Anette Schneemann, Ph.D. Associate Professor Subhash C. Sinha, Ph.D.* Associate Professor of Medicinal Chemistry Gary Siuzdak, Ph.D. Adjunct Associate Professor Director, Center for Mass Spectrometry Vaughn V. Smider, Ph.D. Assistant Professor of Molecular Biology Robyn L. Stanfield, Ph.D. Assistant Professor James Stevens, Ph.D.*** Assistant Professor Centers for Disease Control and Prevention Atlanta, Georgia Arthur J. Olson, Ph.D. Professor Raymond C. Stevens, Ph.D. ††† Professor James C. Paulson, Ph.D. †† Professor Charles D. Stout, Ph.D. Associate Professor Vijay Reddy, Ph.D. Associate Professor Peiqing Sun, Ph.D. Associate Professor Steven I. Reed, Ph.D. † Professor J. Gregor Sutcliffe, Ph.D. Professor Paul Russell, Ph.D. † Professor John A. Tainer, Ph.D.* Professor Michel Sanner, Ph.D. Associate Professor Fujie Tanaka, Ph.D. Associate Professor 184 MOLECULAR BIOLOGY Elizabeth A. Thomas, Ph.D. Assistant Professor James R. Williamson, Ph.D.***** Professor Associate Dean, Kellogg School of Science and Technology 2007 Peter Sobieszcsuk, Ph.D.†††† Core Manager, Consortium for Functional Glycomics THE SCRIPPS RESEARCH INSTITUTE Kelly Lee, Ph.D. Joseph W. Arndt, Ph.D.*** Biogen Idec Cambridge, Massachusetts Richard R. Rivera, Ph.D. Karunesh Arora, Ph.D. Kenji Sugase, Ph.D.*** Suntory Institute for Bioorganic Research Osaka, Japan Mabelle Ashe, Ph.D. †††† Philip Arno Venter, Ph.D. Jamie Mitchell Bacher, Ph.D.*** Rincon Pharmaceuticals La Jolla, California Julio Kovacs, Ph.D. S E N I O R S TA F F SCIENTIST Quansheng Zhou, Ph.D. Ian A. Wilson, D.Phil.* Professor S TA F F S C I E N T I S T S Curt Wittenberg, Ph.D. † Professor Kurt Wüthrich, Ph.D. Cecil H. and Ida M. Green Professor of Structural Biology Svitlana Berezhna, Ph.D. Reto Horst, Ph.D. Sung-Hun Bae, Ph.D. Ying Chuan Lin, Ph.D. Maria Martinez-Yamout, Ph.D. Xiang-Lei Yang, Ph.D. Assistant Professor of Molecular Biology Xiaoqin Ye, M.D., Ph.D.*** University of Georgia Athens, Georgia Wojciech Augustyniak, Ph.D. Garrett M. Morris, Ph.D. Chiaki Nishimura, Ph.D. Todd O. Yeates, Ph.D. Adjunct Professor University of California Los Angeles, California Jeffrey Speir, Ph.D. Qinghai Zhang, Ph.D. Assistant Professor Xueyong Zhu, Ph.D. Mutsuo Yamaguchi, Ph.D. R E S E A R C H A S S O C I AT E S Melanie Ann Adams, Ph.D. Fabio Agnelli, Ph.D. Hanna-Stina Martinsson Ahlzén, Ph.D. Reetesh Raj Akhouri, Ph.D. Alexander Ivanov Alexandrov, Ph.D. Stephen G. Aller, Ph.D. SENIOR RESEARCH Manidipa Banerjee, Ph.D. Konstantinos Beis, Ph.D.*** Imperial College London London, England Christine Beuck, Ph.D. David Boehr, Ph.D. Yannick Bomble, Ph.D. David Bostick, Ph.D. Giovanni Bottegoni, Ph.D. SERVICE FACILITIES Beatriz Gonzalez Alonso, Ph.D. A S S O C I AT E S John Chung, Ph.D.*** Manager, Nuclear Magnetic Resonance Facilities Juniper Networks, Inc. Sunnyvale, California Gerard Kroon Manager, Nuclear Magnetic Resonance Facilities David Barondeau, Ph.D.*** Texas A&M University College Station, Texas Juliano Alves, Ph.D.*** Genomics Institute of the Novartis Research Foundation San Diego, California Tommy Bui, Ph.D. Yu An, Ph.D.** Millipore Temecula, California Justin E. Carlson, Ph.D. †††† Brigitte Anliker, Ph.D.*** University of Düsseldorf Düsseldorf, Germany Qing Chai, M.D., Ph.D.*** Applied Molecular Evolution San Diego, California Andrew James Annalora, Ph.D. Ramya Thirumalai Chakravarthy, Ph.D. Maria Alejandra GamezAbascal, Ph.D. Munehito Arai, Ph.D. Amarnath Chatterjee, Ph.D. Elsa Garcin, Ph.D. Roger Armen, Ph.D. Susana Chaves, Ph.D. Kirk Beebe, Ph.D.*** Metabolon, Inc. Durham, North Carolina Ryan Burnett, Ph.D. Bo Ma, Ph.D. Core Manager, Consortium for Functional Glycomics Michael E. Pique Director, Computer Graphics Development Nahid Razi, Ph.D. Assistant Core Manager, Consortium for Functional Glycomics Lintao Bu, Ph.D. Zhongguo Chen, Paul Card, Ph.D. †††† Rosa Maria Cardoso, Ph.D. Andrew Barry Carmel, Ph.D. Ph.D. †††† Adrienne Elizabeth Dubin, Ph.D. Li Fan, Ph.D. MOLECULAR BIOLOGY Jianhan Chen, Ph.D.*** Kansas State University Manhattan, Kansas Weihsu Claire Chen, Ph.D. Yen-Ju Chen, Ph.D. Ying Chen, M.D. Zhiyong Chen, Ph.D.*** Trius Therapeutics, Inc. San Deigo, California Srinivas Chittaboina, Ph.D. 2007 Claire Louise Dovey, Ph.D. Zhanna Druzina, Ph.D.*** SGX Pharmaceuticals, Inc. San Diego, California Li-Lin Du, Ph.D.*** National Institute of Biological Sciences Beijing, China Michelle Duquette-Huber, Ph.D. THE SCRIPPS RESEARCH INSTITUTE Shoufa Han, Ph.D.*** Xiamen University Xiamen, China Rodney Harris, Ph.D. Ilja V. Khavrutskii, Ph.D.*** University of California San Diego, California Joreg Hinnerwisch, Ph.D.*** ALTANA Chemie AG Wesel, Germany Reza Khayat, Ph.D. Dae Hee Kim, Ph.D. Stephen Edgcomb, Ph.D. Wen-Xu Hong, Ph.D. Chung Jen Chou, Ph.D. Susanna V. Ekholm-Reed, Ph.D. Li-Chiou Chuang, Ph.D. Daniel Felitsky, Ph.D. Yunfeng Hu, Ph.D.*** Tanabe Research Laboratories U.S.A., Inc. San Diego, California Linda Maria Columbus, Ph.D.*** University of Virginia Charlottesville, Virginia Allan Chris Merrera Ferreon, Ph.D. Gladys Completo, Ph.D. Yann Gambin, Ph.D. Stephen Connelly, Ph.D. Shannon E. Gardell, Ph.D. †††† Josephine Chu Ferreon, Ph.D. Adam Corper, Ph.D. Charles Gersbach, Ph.D. Carla P. Da Costa, Ph.D. †††† Surya Kanta De, Ph.D.*** Burnham Institute for Medical Research La Jolla, California Jennifer Knight, Ph.D. Minsun Hong, Ph.D. Eda Koculi, Ph.D. Zheng-Zheng Huang, Ph.D. Kwan Hoon Hyun, Ph.D.*** CrystalGenomics Seoul, Korea Masanori Imai, Ph.D.*** Hokkaido Pharmaceutical University Hokkaido, Japan Irina Kufareva, Ph.D. Shantanu Kumar, Ph.D.*** University of California San Diego, California Sharon Kwan, Ph.D. Bianca Lam, Ph.D. Emma Langley, Ph.D. Joshua Gill, Ph.D. Thamara Janaratne, Ph.D. Chang-Wook Lee, Ph.D. Edith Caroline Glazer, Ph.D. Kai Jenssen, Ph.D.*** Merz Pharma Frankfurt, Germany Chul Won Lee, Ph.D. Rajib Kumar Goswami, Ph.D. Bettina Groschel, Ph.D. Margaret Alice Johnson, Ph.D. Hai-Ming Guo, Ph.D. June Hyung Lee, Ph.D.*** New York University New York, New York Sophie Lefebvre, Ph.D. Steven Johnson, Ph.D. Edward Lemke, Ph.D. Susanna Juraja, Ph.D. Robert de Bruin, Ph.D. Mahender Gurram, Ph.D. Gerald Edward Dodson, Ph.D. Milka Kostic, Ph.D.*** Cell Press Cambridge, Massachussetts Jason Lanman, Ph.D. Min Guo, Ph.D. Paula Desplats, Ph.D.*** University of California San Diego, California Bethany Koehntop, Ph.D. Veli-Pekka Jaakola, Ph.D. Sandro Cosconati, Ph.D. Qizhi Cui, Ph.D. National Research Council Ottawa, Ontario, Canada Yang Khandogin, Ph.D.*** University of Oklahoma Norman, Oklahoma Deron Herr, Ph.D. Scott Eberhardy, Ph.D.*** Schering-Plough Corporation Union, New Jersey Ji Woong Choi, Ph.D. Andrey Aleksandrovich Karyakin, Ph.D. David M. Herman, Ph.D. Kenichi Hitomi, Ph.D. Jungwoo Choe, Ph.D.*** University of Seoul Seoul, Korea 185 Liao Liang, Ph.D. Bong Kwan Han, Ph.D.*** Cornell University Ithaca, New York Christian Kannemeier, Ph.D.*** PrimeCell Therapeutics L.L.C. Irvine, California Byung Woo Han, Ph.D. Mili Kapoor, Ph.D. Vasco Liberal, Ph.D. William M. Lindstrom, Ph.D. Bin Liu, Ph.D. 186 MOLECULAR BIOLOGY Kunheng Luo, Ph.D. Ann MacLaren, Ph.D.*** Biogen Idec San Diego, California 2007 THE SCRIPPS RESEARCH INSTITUTE Wataru Nomura, Ph.D.*** Tokyo Medical and Dental University Tokyo, Japan Sanjay Adrian Saldanha, Ph.D.*** Translational Research Institute, Scripps Florida Rebecca E. Taurog, Ph.D. Ewan Richardson Taylor, Ph.D. Amy Odegard, Ph.D. Mariana Santa-Marta, Ph.D. Leonardo Teixeira, Ph.D. Darly Joseph Manayani, Ph.D. Lisa Renee Olano, Ph.D. Andre Schiefner, Ph.D. Hua Tian, Ph.D. Jeff Mandell, Ph.D.*** Ionian Technologies, Inc. San Diego, California Brian L. Olson, Ph.D.*** St. Cloud University St. Cloud, Minnesota Lauren J. Schwimmer, Ph.D. Mauricio Carrillo Tripp, Ph.D. Jennifer S. Scorah, Ph.D. Oleg Trott, Ph.D Santiago Cavero Martinez, Ph.D. Akira Onoda, Ph.D. Alim Seit-Nebi, Ph.D. Mary O’Reilly, Ph.D. Pedro Serrano-Navarro, Ph.D. Ulrich Tschulena, Ph.D.*** German Cancer Research Center Heidelberg, Germany Brian Paegel, Ph.D. Craig McLean Shepherd, Ph.D.*** Burnham Institute for Medical Research La Jolla, California Tsutomu Matsui, Ph.D. Derrick Meinhold, Ph.D. Eva Mejia Ramirez De Arellano, Ph.D. Elena Menichelli, Ph.D. Jonathan Mikolosko, Ph.D. Mauro Mileni, Ph.D. Maki Minakawa, Ph.D. Marissa Mock, Ph.D.*** Biocatalytics Pasadena, California Biswaranjan Mohanty, Ph.D. Samrat Mukhopadhyay, Ph.D. Tetsuji Mutoh, Ph.D. Sujatha Narayan, Ph.D. †††† Mir Hussain Nawaz, Ph.D. Hung Nguyen, Ph.D. Tuan Nguyen, Ph.D. Sung-Jean Park, Ph.D. Sandeep Patel, Ph.D.*** University of Delaware Newark, Delaware Stephanie Pebernard, Ph.D. Bill Francesco Pedrini, Ph.D. Robert Pejchal, Ph.D. Vladimir Pelmenschikov, Ph.D. Jefferson Perry, Ph.D. Suzanne Peterson, Ph.D. Jessica Petrillo, Ph.D. David S. Shin, Ph.D. Develeena Shivakumar, Ph.D.*** University of Chicago Chicago, Illinois David A. Shore, Ph.D. Daniela Andrea Slavin, Ph.D. Elisabetta Soragni, Ph.D. Kyoko Noguchi, M.D., Ph.D. Sorin Tunaru, Ph.D. Naoto Utsumi, Ph.D. Frank van Drogen, Ph.D.*** Institute für Biochemie Zürich, Switzerland Ajay Vashisht, Ph.D. Sangita Venkataraman, Ph.D. Petra Verdino, Ph.D. My Vo, Ph.D. Pawel Stanczak, Ph.D. William Frederick Waas, Ph.D.*** Defined Health Florham Park, New Jersey Thomas Steinbrecher, Ph.D. Shun-ichi Wada, Ph.D. †††† Shih-Che Su, Ph.D. Jun Wang, Ph.D. Sebastian Sudek, Ph.D. Jessica Williams, Ph.D. Magnus Sundstrom, Ph.D. Robert Scott Williams, Ph.D. Michael T. Sykes, Ph.D. Eric L. Wise, Ph.D.*** Wayne State University Detroit, Michigan Surya Venkata Sripada, Ph.D. Goran Pljevaljc̆ić, Ph.D. Stephanie Pond, Ph.D. John Prudden, Ph.D. Christopher L. Reyes, Ph.D.*** Biogen Idec San Diego, California George Nicola, Ph.D. Tadateru Nishikawa, Ph.D.*** Bruker BioSpin Tsukuba, Japan Julie L. Tubbs, Ph.D. Kimberly A. Reynolds, Ph.D. Blair R. Szymczyna, Ph.D. Jin-Kyu Rhee, Ph.D. Hiroaki Tateno, Ph.D.*** National Institute of Advanced Industrial Sceince and Technology Tsukuba, Japan Gabriela Ring, Ph.D. Riturparna Sinha Roy, Ph.D. Jonathan Wojciak, Ph.D.*** Lpath, Inc. San Diego, California Vance Wong, Ph.D. MOLECULAR BIOLOGY Timothy I. Wood, Ph.D.*** Walter Reed Army Medical Center Washington, D.C. 2007 Ognian V. Bohorov, Ph.D. Dennis Carlton, B.S. Vadim Cherezov, Ph.D. Wei Xie, Ph.D. Ellen Yu-Lin Tsai Chien, Ph.D. Lan Xu, Ph.D.*** BioVerdant San Diego, California Xiaoping Dai, Ph.D. Marc Deller, D.Phil Yoshiki Yamada, Ph.D. THE SCRIPPS RESEARCH INSTITUTE Joseph David Ng, Ph.D. University of Alabama Huntsville, Alabama Victoria A. Roberts, Ph.D. University of California San Diego, California Robert D. Rosenstein, Ph.D. Lawrence Berkeley National Laboratory Berkeley, California Gye Won Han, Ph.D. Tohru Yamagaki, Ph.D. Wenge Han, Ph.D. Atsushi Yamagata, Ph.D.*** Tokyo University Tokyo, Japan Michael Allen Hanson, Ph.D. Debbie Tahmassebi, Ph.D. University of San Diego San Diego, California Hope Johnson, Ph.D. Yongjun Ye, Ph.D.*** VIRxSYS Corporation Gaithersburg, Maryland Marcy A. Kingsbury, Ph.D.*** Indiana University Bloomington, Indiana Kye Sook Yi, Ph.D. Padmaja Natarajan, Ph.D.†††† Yong Yin, Ph.D. †††† Lin Wang, Ph.D. Sung-Il Yoon, Ph.D. Kenji Yoshimoto, Ph.D. Naoto Yoshizuka, M.D., Ph.D. Yuan Yuan, Ph.D.*** Millipore Temecula, California Markus Zeeb, Ph.D.*** Boehringer Ingelheim GmbH Biberach an der Riss, Germany VISITING I N V E S T I G AT O R S Stephen J. Benkovic, Ph.D. Pennsylvania State University University Park, Pennsylvania Wayne A. Fenton, Ph.D. Yale University New Haven, Connecticut Astrid Graslund, Ph.D. Stockholm University Stockholm, Sweden Arne Holmgren, M.D., Ph.D. Karolinska Institutet Stockholm, Sweden Qing Zhang, Ph.D. Wei Zhang, Ph.D. Barry Honig, Ph.D. Columbia University New York, New York Yong Zhao, Ph.D. S C I E N T I F I C A S S O C I AT E S Enrique Abola, Ph.D. Andrew S. Arvai, M.S. ** Joint appointments in the Department of Chemistry and The Skaggs Institute for Chemical Biology *** Appointment completed; new location shown **** Joint appointment in the Department of Molecular and Integrative Neurosciences * **** Joint appointments in the Department of Immunology and The Skaggs Institute for Chemical Biology † †† Ying Zeng, Ph.D. Haile Zhang, Ph.D. * Joint appointment in The Skaggs Institute for Chemical Biology Arthur Horwich, M.D. Yale University New Haven, Connecticut Tai-Huang Huang, Ph.D. Academica Sinica Taipei, Taiwan Joint appointment in the Department of Cell Biology Joint appointment in the Department of Molecular and Experimental Medicine ††† Joint appointment in the Department of Chemistry †††† Appointment completed 187 188 MOLECULAR BIOLOGY 2007 Peter E. Wright, Ph.D. Chairman’s Overview esearch in the Department of Molecular Biology encompasses a broad range of disciplines, extending from structural and computational biology at one extreme to molecular genetics at the other. During the past year, our scientists have continued to make rapid progress toward understanding the fundamental molecular events that underlie the processes of life. Major advances have been made in elucidating the structural biology of signal transduction, receptor recognition, and viral assembly; understanding mechanisms of viral infectivity; determining the structures of membrane proteins and multidrug transporters; understanding the molecular basis of nucleic acid recognition and DNA repair; and determining the mechanisms of protein folding and ribosome assembly. Progress has been made in elucidating the molecular events involved in regulation of the cell cycle, tumor development, induction of sleep, the molecular origins of neuronal development and CNS disorders, the regulation of transcription, and decoding of genetic information in translation. Finally, new advances have been made in the design of novel low molecular weight compounds that can specifically regulate genes and in biomolecular engineering, building novel functions into viruses, antibodies, zinc finger proteins, RNA, and DNA. Progress in these and other areas is described in detail on the fol- R THE SCRIPPS RESEARCH INSTITUTE lowing pages, and only a few highlights are mentioned here. The Department of Molecular Biology is also home to 2 major National Institutes of Health initiatives: the Joint Center for Structural Genomics and the Consortium for Functional Glycomics. Research in the laboratory of Raymond Stevens has revealed the structural basis for the high-affinity interaction of botulinum toxin with nerve cell receptors. Botulinum toxin is a potent bacterial toxin that causes paralysis by binding at neuromuscular junctions at femtomolar concentrations to block neurotransmitter release. Dr. Stevens and coworkers determined the x-ray structure of type B botulinum toxin bound to the recognition motif of synaptotagmin II, an integral membrane protein found in synaptic vesicles. In addition to binding synaptotagmin, the botulinum toxin binds simultaneously to ganglioside molecules in the presynaptic membrane to form a very high-affinity multivalent complex in which the toxin is oriented optimally to facilitate the formation of pores. The molecular insights gained from this structural analysis may form the basis for designing new small-molecule therapeutic agents to treat botulism or for generating novel cross-reactive antibodies that can bind with high affinity to several botulinum toxin subtypes. Dr. Stevens and his colleagues have generated such cross-reactive antibodies and have determined structures of the antibodies complexed with toxin to develop new strategies for evolving antibodies with broadened specificity. New advances were made in understanding the behavior of natively disordered proteins. It is now recognized that many eukaryotic proteins contain long unstructured regions or are entirely unfolded in their native state. Ashok Deniz and coworkers applied single-molecule fluorescence and fluorescence correlation methods to characterize the prion protein Sup35, which functions as a translation termination factor in yeast. Sup35 can switch to a self-replicating amyloid state under physiologic conditions, a process analogous to formation of amyloid fibrils by misfolded human proteins in many neurodegenerative diseases. Dr. Deniz and coworkers showed that the monomeric collapsed state of Sup35 consists of an ensemble of rapidly fluctuating conformations. Assembly of the monomers into oligomeric intermediates is an essential first step for nucleating the formation of amyloid fibers. The novel fluorescence techniques developed by scientists in the Deniz laboratory hold exceptional promise for characterizing the structure and dynamics of the toxic intermediates formed MOLECULAR BIOLOGY 2007 during the aggregation processes that lead to Alzheimer’s and other neurodegenerative diseases. In my laboratory, Jane Dyson and I provided the first insights into the mechanism by which a natively disordered transcription activation domain recognizes its target and folds into a compact structure upon binding. Recent work by Arthur Horwich and colleagues has provided important insights into the topology of substrate proteins folding within the cavity of the chaperonin GroEL-GroES. Recent research by Peiqing Sun and coworkers has provided novel insights into a signaling pathway that mediates cellular senescence and, when activated, inhibits tumor development. These studies have indicated that the p38-regulated/activated protein kinase (PRAK) is an essential component of the senescence pathway. Inactivation of PRAK in normal human cells prevents senescence and makes them more susceptible to oncogenic transformation. Mice lacking the PRAK gene have increased susceptibility to skin cancer and have accelerated development of lymphoma. Mechanistically, PRAK appears to function by phosphorylating and activating the tumor suppressor p53. These studies suggest a potential new approach to cancer therapy: using molecules that can activate the PRAK pathway. Carlos Barbas, Subhash Sinha, Richard Lerner, and colleagues have reported a major advance in the synthesis of therapeutic antibodies. These scientists have developed catalytic antibodies that effectively target integrin αvβ3 expressed by human breast cancer cells and catalyze the activation of doxorubicin prodrugs. These studies lay the basis for development of a new class of therapeutic antibodies with both targeting and drug-activating functions. Despite the advent of high-throughput screening and library technologies, the success rate for generation of novel drugs from new chemical entities is depressingly low. Ruben Abagyan and coworkers have developed a novel computational method for “drug repurposing.” With this method, they optimize leads to side-effect targets of marketed drugs while eliminating activity at the original target and maintaining acceptable bioavailability and toxicity profiles. Because these repurposed compounds are derived from safe and established drugs, they are likely to enter clinical trials at lower cost than would lead compounds based entirely on new chemical entities. Molecular biology remains a field of enormous opportunity and excitement. The scientists in this department are taking full advantage of powerful new technologies THE SCRIPPS RESEARCH INSTITUTE 189 to advance our understanding of fundamental biological processes at the molecular level. Their discoveries will ultimately be translated into new advances in biotechnology and in medicine. 190 MOLECULAR BIOLOGY 2007 INVESTIGATORS’ R EPORTS Structural Biology of Viral Proteins, Molecular Assemblies, and the Immune System THE SCRIPPS RESEARCH INSTITUTE Z13 bind to conserved and overlapping epitopes on the membrane-proximal region on the viral envelope protein gp41. We have determined multiple 4E10 structures in complex with peptide epitopes and have tested the peptides for immunogenicity. In addition, we have grafted this epitope into the gp120 of HIV-1 (Fig. 1) to investigate whether these engineered antigens can elicit HIV-neutralizing antibodies. I.A. Wilson, R.L. Stanfield, J. Stevens, X. Zhu, M.A. Adams, Y. An, K. Beis, C.H. Bell, D.A. Calarese, R.M.F. Cardoso, J. Carlson, P.J. Carney, J.-W. Choe, S. Connelly, A.L. Corper, T.A. Cross, X. Dai, E.W. Debler, W.L. Densley, D.C. Ekiert, M.-A. Elsliger, S. Ferguson, B.W. Han, G.W. Han, M. Hong, M.J. Jimenez-Dalmaroni, J.R. Mikolosko, R. Pejchal, A. Schiefner, D.A. Shore, R.S. Stefanko, J.A. Vanhnasy, P. Verdino, L. Xu, X. Xu, S.I. Yoon, D.M. Zajonc e use x-ray crystallography to investigate the structure and function of many different receptors in the innate and adaptive immune systems. We are also studying antigens from influenza virus and HIV type 1 (HIV-1) to determine how these viruses are neutralized by the immune system. W INFLUENZA VIRUS F i g . 1 . HIV-1 vaccine design by insertion of the HIV-1 gp41 The 1918 influenza pandemic remains the most devastating single infectious outbreak on record; at least 40 million people died of the disease. Influenza has 2 surface glycoproteins: hemagglutinin and neuraminidase. Hemagglutinin plays a key role in viral infection by binding to sialylated receptors on target cells, resulting in viral entry and membrane fusion. Neuraminidase facilitates release of newly formed viral particles from the host cell, enabling spread of the virus to neighboring cells. As part of a consortium funded by the National Institute of Allergy and Infectious Diseases to understand the pathogenicity of the 1918 virus and the high mortality rate, we have determined crystal structures of the H1 hemagglutinin from 1918 as well as the more recent H5N1 hemagglutinin from 2004 and the 1918 N1 neuraminidase to aid in design of vaccines to prevent future influenza pandemics. membrane-proximal region into gp120. The red helix represents the 4E10 epitope that has been inserted into the V1/V2 region of NEUTRALIZING ANTIBODIES TO HIV-1 Successful eradication of HIV-1 depends on an effective vaccine. Unfortunately, this virus excels in evading the immune system, thus hindering vaccine development. By studying a handful of broadly neutralizing antibodies to HIV-1 in complex with their viral antigens, we hope to elucidate vulnerable sites for antibody neutralization. The antibodies 4E10 and gp120 for testing as a potential immunogen for an HIV-1 vaccine. The gp41 and gp120 viral envelope proteins form trimeric spikes on the intact virus. After binding human CD4 and chemokine receptors, the trimer undergoes conformational changes that lead to fusion of the viral and target cell membranes, initiating infection. We are characterizing the structure of the trimer to understand membrane fusion and why antibodies can only neutralize intact envelope trimers. Our research on HIV is done in collaboration with D. Burton, Department of Immunology; P. Dawson, Department of Cell Biology; C.-H. Wong, Department of Chemistry; J.K. Scott, Simon Fraser University, Burnaby, British Columbia; J. Moore, Weill Medical College of Cornell University, New York, New York; H. Katinger, R. Kunert, and G. Stiegler, University für Bodenkultur, Vienna, Austria; R. Wyatt and P. Kwong, Vaccine Research Center, National Institutes of Health, Bethesda, Maryland; W. Olson, and K. Kang, Progenics Pharmaceuticals, Inc., Tarrytown, New York; the National Institutes of Health, Bethesda, Maryland; and the Neutralizing Antibody Consortium of the International AIDS Vaccine Initiative, New York, New York. MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 191 G PROTEIN–COUPLED RECEPTORS G protein–coupled receptors (GPCRs) are a large family of integral membrane proteins central to numerous biological processes. Because of their involvement in many diseases, GPCRs collectively account for more than 50% of current chemotherapeutic targets. We are developing recombinant expression systems for numerous chemokine receptor GPCRs. Although yield and protein stability continue to challenge structural investigation of these proteins, early results in protein production appear promising. CLASSICAL AND NONCLASSICAL MHC AND T-CELL RECEPTOR SIGNALING MHC class I molecules enable the immune system to monitor cells for infection and cancer via presentation of antigenic peptides on the cell surface. Efficient presentation of a diverse repertoire of peptides by MHC class I molecules depends on the peptide loading complex, of which tapasin is an essential component. We have expressed soluble tapasin in Escherichia coli for structural studies to further elucidate the mechanism of MHC class I peptide loading. We have a long-term interest in the structural aspects of MHCs and T-cell receptor (TCR) recognition. Recent results include the crystal structure of rat MHC class Ib BM1 in complex with peptide Qdm at 2.15 Å. This structure reveals how BM1 differs from other MHC homologs and hence defines the structural determinants of natural killer cell regulation in innate immunity. We are also studying the TCR KRN that recognizes the MHC class II allele I-Ag7 in complex with a self-peptide derived from glucose-6-phosphate isomerase that initiates rheumatoid arthritis in KRN transgenic mice. Crystals of the complex composed of KRN, I-Ag7, and the glucose-6-phosphate isomerase peptide have been obtained, and structure determination is ongoing. To provide insights into potential mechanisms of diabetogenesis, we are also investigating whether TCRs recognize an oxyanion hole in I-Ag7. To this end, a crystal structure for the TCR HEL, isolated from nonobese diabetic mice immunized against the peptide HEL9-27, in complex with I-Ag7–HEL9-27 has been determined (Fig. 2). This work is a collaboration with L. Teyton, Department of Immunology. The T-cell response also depends on the CD3 complex, which consists of 4 subunits (ε, δ, γ, and ζ) that form pairs of dimers that assemble on the TCR to facilitate transmembrane signal transduction. Although structures of CD3εδ and CD3εγ are now available, the F i g . 2 . Crystal structure of the HEL TCR–I-A g7–HEL 9-27 com- plex. The TCR is shown in pink, the MHC in olive green, and the peptide in yellow. mechanism coupling TCR recognition of antigen to T-cell activation has yet to be determined. Thus, we have developed mutant TCRs with high affinity for CD3 subunits for structural analysis of the intact TCRCD3 complex. We are also studying the TCR coreceptor CD8, an essential element in the cytotoxic T-cell response to peptide antigen, to determine the molecular basis of the function of the CD8 coreceptor. Mycobacterial phosphatidylinositol tetramannosides (PIMs) are a major component of the mycobacterial outer membrane leaflet and when presented by CD1d stimulate invariant human Vα24 and mouse Vα14 natural killer T cells. In collaboration with W. Severn and G. Painter, Industrial Research Ltd., Upper Hut, New Zealand, we determined the crystal structure of mouse CD1d in complex with synthetic PIM2, which 192 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE represents the most complex headgroup of all CD1d ligand structures to date (Fig. 3). Compared with other CD1d ligands, PIM2 has an increased number of polar interactions between its headgroup and CD1, but reduced specificity for the diacylglycerol backbone. F i g . 4 . Crystal structure of JAML in complex with the mitogenic antibody 4E10. JAML has 2 immunoglobulin domains (D1 and D2) arranged in a unique orientation for tandem immunoglobulin domains. Fab complementarity-determining region loops (L1, L2, L3, H1, H2, H3) contact primarily the D2 domain. F i g . 3 . Comparison of the different ligands bound to mouse CD1d. A, PIM2. B, Sulfatide. C, The highly potent α-galactosylceramide. T H E I N N AT E I M M U N E S Y S T E M The enigmatic γδ T cells link innate and adaptive immunity. These cells carry out functions such as tumor cell recognition, maintenance of tissue homeostasis, and tissue repair and act as the first line of defense against bacterial and viral infections. In collaboration with W. Havran, Department of Immunology, we determined the crystal structures of JAML, a costimulatory molecule specific for γδ T cells, alone and in complex with a mitogenic antibody (Fig. 4). The structures revealed a unique assembly of tandem immunoglobulin domains. Conformational changes in the ectodomain of JAML on activation may be the trigger for initiating kinase signaling cascades, cytokine and chemokine production, and, ultimately, cell proliferation. Toll-like receptors (TLRs) are cell-surface receptors in humans that detect microbes invading through the skin or intestinal mucosa. TLRs play a key role in initiating immune responses by recognizing a variety of pathogen-associated molecular patterns, including components of bacterial cell walls and viral nucleic acids. Since determining the structure of TLR3 , we are working on other human TLRs in complex with their ligands. The nucleotide-binding oligomerization domains (NODs) 1 and 2 are intracellular receptors that recognize bacterial peptidoglycans. NOD2 mutations have been associated with inflammatory Crohn’s disease. We aim to crystallize the putative binding leucine-rich repeat domain of NOD2 and also the intracellular caspase-recruitment domain involved in signal transduction. The TLR and NOD studies are collaborations with B. Beutler and R. Ulevitch, Department of Immunology. N O N M A M M A L I A N I N N AT E A N D A D A P T I V E I M M U N I T Y Sharks and other cartilaginous fish diverged from mammals over 5 million years ago and are the most ancient vertebrates with an adaptive immune system that involves antibodies, TCRs, MHC molecules, and recombination-activating genes. In collaboration with M. Flajnik and H. Dooley, University of Maryland, Baltimore, we have determined structures for germ-line and somatically mutated variable domains from a MOLECULAR BIOLOGY 2007 nurse shark “new antigen receptor ” antibody in unliganded and antigen-complexed forms. These primitive antibody binding domains share many features with mammalian antibodies, such as flexibility of the H3 complementarity-determining region, and correlation between affinity, increased contacts in the binding site, and somatic mutations. Furthermore, sequence comparisons indicate that shark proteins Saac-UAA*01 and Sqac-UAA*NC1 are members of the classical and nonclassical class I MHCs, respectively. Currently, in collaboration with C. Dascher, Mount Sinai School of Medicine, New York, New York, we are optimizing expression of these proteins in baculovirus and Drosophila systems for crystallization trials to explore the proteins’ structure and function. Jawless fish are thought to have an adaptive immune system based not on antibodies, but on variable lymphocyte receptors (VLRs). VLRs are composed of variable numbers of leucine-rich repeats that can be rearranged in a combinatorial fashion to recognize a number of diverse antigens. In collaboration with M.D. Cooper, University of Alabama, Birmingham, we have produced and are crystallizing several different constructs of VLR4, VLR5, and VLR2913. C ATA LY T I C A N T I B O D I E S Antibodies can catalyze a myriad of interesting, often difficult, enzymatic reactions. Currently, we are working with D. Hilvert, ETH, Zurich, Switzerland, to determine structures of the catalytic antibodies 34E4 and 13G5 that carry out proton transfer, one of the most fundamental chemical reactions in chemistry and biology (Fig. 5). With K.D. Janda and P. Wentworth, Department of Chemistry, we are studying antibodies 38H10 and 29G12, which catalyze the 1,3-dipolar cycloaddition reaction that is extremely useful for synthesis of diverse chiral heterocyclic compounds and is not efficiently catalyzed by any natural enzyme. F i g . 5 . Crystal structures of antibody 13G5 show how bifunctional catalysis of the proton-transfer reaction is realized by AspH35 as the general base and a water molecule as the acid catalyst. THE SCRIPPS RESEARCH INSTITUTE 193 GREEN FLUORESCENT ANTIBODIES Scientists in the laboratory of P.G. Schultz, Department of Chemistry, have generated antibodies to the donor-acceptor substituted stilbene trans-4-N,N-dimethylamino-4′-cyanostilbene (DCS) that yield bright blue to green fluorescence upon illumination. The relatively long excitation and emission wavelengths of the antibody-stilbene complexes may make them useful for in vitro and in vivo applications as fluorescent biosensors. To investigate the photophysics of DCS in atomic detail, we have determined the crystal structure of Fab 11G10 in complex with hapten at 2.75 Å. The charge distribution and aromatic ring systems in the combining site are well suited to stabilize charge separation in the excited state of DCS and thus provide a structural basis for emission at longer wavelengths than those associated with other antibodies to stilbene. THERAPEUTIC ANTIBODIES Gram-negative bacteria can use N-acyl homoserine lactones (AHLs) as signaling molecules in quorum sensing, a population density–dependent mechanism to coordinate gene expression. K.D. Janda, Department of Chemistry, has used a lactam mimetic of AHL as an immunogen to generate antibody RS2-1G9, which also recognizes the naturally occurring AHL with high affinity. Because of this cross-reactivity, RS2-1G9 shows remarkable inhibition of quorum-sensing signaling in Pseudomonas aeruginosa, a common opportunistic pathogen in humans. The crystal structure of Fab RS21G9 in complex with a lactam analog (Fig. 6) revealed complete encapsulation of the polar lactam and can now aid in further development of an antibody-based therapy against bacterial pathogens that interferes with quorum sensing. F i g . 6 . The crystal structure of the quorum-quenching RS2-1G9 Fab in complex with a lactam analog. The polar lactam moiety is totally encapsulated in the antibody-combining site. 194 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE N-formyl peptides, such as formyl-methionyl-leucylphenylalanine (fMLF), are potent chemotactic factors for leukocytes. Unwanted inflammation should be suppressed by removing the N-formyl peptides through binding to antibodies. F. Tanaka, Department of Chemistry, has developed antibody 6G5 that selectively binds fMLF. Crystal structures of the Fab′ and its complex with fMLF at 2.7 and 1.95 Å, respectively, revealed the mechanism of the discrimination between N-formyl and nonformylated peptides and now provide information for further generation of humanized antibodies as antiinflammatory drugs. Didonato, M., Krishna, S.S., Schwarzenbacher, R., et al. Crystal structure of 2-phosphosulfolactate phosphatase (ComB) from Clostridium acetobutylicum at 2.6 Å resolution reveals a new fold with a novel active site. Proteins 65:771, 2006. JOINT CENTER FOR STRUCTURAL GENOMICS Kosloff, M., Han, G.W., Krishna, S.S., et al. Comparative structural analysis of a novel glutathione S-transferase (ATU5508) from Agrobacterium tumefaciens at 2.0 Å resolution. Proteins 65:527, 2006. The Joint Center for Structural Genomics is a large consortium of scientists from Scripps Research; the Stanford Synchrotron Radiation Laboratory; the University of California, San Diego; the Burnham Institute for Medical Research; and the Genomics Institute of the Novartis Research Foundation. The center is funded by the Protein Structure Initiative of the National Institute of General Medical Sciences. Its purpose is high-throughput structure determination of large families of protein sequences with no structural representatives, biologically important targets that are conserved as the central machinery of life; the complete proteome from Thermotoga maritima; metagenomic and human gut microbiome targets; and other targets suggested by the community. To date, the members of the consortium have pioneered many novel high-throughput methods and technologies applicable to structural biology and have determined more than 400 novel structures. PUBLICATIONS Binley, J.M., Ngo-Abdalla, S., Moore, P., Bobardt, M., Chatterji, U., Gallay, P., Burton, D.R., Wilson, I.A., Elder, J.H., de Parseval, A. Inhibition of HIV Env binding to cellular receptors by monoclonal antibody 2G12 as probed by Fc-tagged gp120 [published correction appears in Retrovirology 4:23, 2007]. Retrovirology 3:39, 2006. Cardoso, R.M., Brunel, F.M., Ferguson, S., Zwick, M., Burton, D.R., Dawson, P.E., Wilson, I.A. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J. Mol. Biol. 365:1533, 2007. Cheng, T.Y., Relloso, M., Van Rhijn, I., Young, D.C., Besra, G.S., Briken, V., Zajonc, D.M., Wilson, I.A., Porcelli, S., Moody, D.B. Role of lipid trimming and CD1 groove size in cellular antigen presentation. EMBO J. 25:2989, 2006. Cooper, Z.D., Narasimhan, D., Sunahara, R.K., Mierzejewski, P., Jutkiewicz, E.M., Larsen, N.A., Wilson, I.A., Landry, D.W., Woods, J.H. Rapid and robust protection against cocaine-induced lethality in rats by the bacterial cocaine esterase. Mol. Pharmacol. 70:1885, 2006. Han, G.W., Sri Krishna, S., Schwarzenbacher, R., et al. Crystal structure of the ApbE protein (TM1553) from Thermotoga maritima at 1.58 Å resolution. Proteins 64:1083, 2006. Jiang, Z., Georgel, P., Li, C., Choe, J., Crozat, K., Rutschmann, S., Du, X., Bigby, T., Mudd, S., Sovath, S., Wilson, I.A., Olson, A., Beutler, B. Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 103:10961, 2006. Kinjo, Y., Tupin, E., Wu, D., Fujio, M., Garcia-Navarro, R., Benhnia, M.R., Zajonc, D.M., Ben-Menachem, G., Ainge, G.D., Painter, G.F., Khurana, A., Hoebe, K., Behar, S.M., Beutler, B., Wilson, I.A., Tsuji, M., Sellati, T.J., Wong, C.-H., Kronenberg, M. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat. Immunol. 7:978, 2006. Law, M., Cardoso, R.M., Wilson, I.A., Burton, D.R. Antigenic and immunogenic study of membrane-proximal external region-grafted gp120 antigens by a DNA prime-protein boost immunization strategy. J. Virol. 81:4272, 2007. Lazoura, E., Lodding, J., Farrugia, W., Ramsland, P.A., Stevens, J., Wilson, I.A., Pietersz, G.A., Apostolopoulos, V. Enhanced major histocompatibility complex class I binding and immune responses through anchor modification of the noncanonical tumour-associated mucin 1-8 peptide. Immunology 119:306, 2006. Mathews, I.I., Krishna, S.S., Schwarzenbacher, R., et al. Crystal structure of phosphoribosylformyl-glycinamidine synthase II, PurS subunit (TM1244) from Thermotoga maritima at 1.90 Å resolution. Proteins 65:249, 2006. Muller, R., Debler, E.W., Steinmann, M., Seebeck, F.P., Wilson, I.A., Hilvert, D. Bifunctional catalysis of proton transfer at an antibody active site. J. Am. Chem. Soc. 129:460, 2007. Nelson, J.D., Brunel, F.M., Jensen, R., Crooks, E.T., Cardoso, R.M., Wang, M., Hessell, A., Wilson, I.A., Binley, J.M., Dawson, P.E., Burton, D.R., Zwick, M.B. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol. 81:4033, 2007. Rudolph, M.G., Stanfield, R.L., Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24:419, 2006. Schwarzenbacher, R., McMullan, D., Krishna, S.S., et al. Crystal structure of a glycerate kinase (TM1585) from Thermotoga maritima at 2.70 Å resolution reveals a new fold. Proteins 65:243, 2006. Stanfield, R.L., Dooley, H., Verdino, P., Flajnik, M.F., Wilson, I.A. Maturation of shark single-domain (IgNAR) antibodies: evidence for induced-fit binding. J. Mol. Biol. 367:358, 2007. Stevens, J., Blixt, O., Paulson, J.C., Wilson, I.A. Glycan microarray technologies: tools to survey host specificity of influenza viruses. Nat. Rev. Microbiol. 4:857, 2006. Tian, F., Debler, E.W., Millar, D.P., Deniz, A.A., Wilson, I.A., Schultz, P.G. The effects of antibodies on stilbene excited-state energetics. Angew. Chem. Int. Ed. 45:7763, 2006. Weekes, D., Miller, M.D., Krishna, S.S., et al. Crystal structure of a transcription regulator (TM1602) from Thermotoga maritima at 2.3 Å resolution. Proteins 67:247, 2007. Debler, E.W., Kaufmann, G.F., Kirchdoerfer, R.N., Mee, J.M., Janda, K.D., Wilson, I.A. Crystal structures of a quorum-quenching antibody. J. Mol. Biol. 368:1392, 2007. Xu, L., Chong, Y., Hwang, I., D’Onofrio, A., Amore, K., Beardsley, G.P., Li, C., Olson, A.J., Boger, D.L., Wilson, I.A. Structure-based design, synthesis, evaluation and crystal structures of transition state analogue inhibitors of inosine monophosphate cyclohydrolase. J. Biol. Chem. 282:13033, 2007. DeMartino, J.K., Hwang, I., Xu, L., Wilson, I.A., Boger, D.L. Discovery of a potent, nonpolyglutamatable inhibitor of glycinamide ribonucleotide transformylase. J. Med. Chem. 49:2998, 2006. Xu, Q., Krishna, S.S., McMullan, D., et al. Crystal structure of an ORFan protein (TM1622) from Thermotoga maritima at 1.75 Å resolution reveals a fold similar to the Ran-binding protein Mog1p. Proteins 65:777, 2006. MOLECULAR BIOLOGY 2007 Xu, Q., Schwarzenbacher, R., Krishna, S.S., et al. Crystal structure of acireductone dioxygenase (ARD) from Mus musculus at 2.06 Å resolution. Proteins 64:808, 2006. Zajonc, D.M., Ainge, G.D., Painter, G.F., Severn, W.B., Wilson, I.A. Structural characterization of mycobacterial phosphatidylinositol mannoside binding to mouse CD1d. J. Immunol. 177:4577, 2006. Structure and Function of Proteins as Molecular Machines E.D. Getzoff, P. Aoto, A.S. Arvai, D.P. Barondeau, R.M. Brudler, T. Cross, E.D. Garcin-Hosfield, C. Hitomi, K. Hitomi, M.E. Pique, J.L. Tubbs, T.I. Wood ur goal is to understand how proteins function as molecular machines. We apply the tools of structural, molecular, and computational biology to proteins of biological and biomedical interest, especially proteins that work synergistically with coupled chromophores, metal ions, or other cofactors. O NITRIC OXIDE SYNTHASES Nitric oxide synthase (NOS) enzymes synthesize nitric oxide, a signal for vasodilation and neurotransmission at low levels and a defensive cytotoxin at higher levels. Synthesis of nitric oxide by NOS requires calmodulin-orchestrated interactions between the catalytic, heme-containing oxygenase module and the electronsupplying reductase module of the enzyme. Our x-ray crystallographic structures of wild-type and mutant NOS oxygenase dimers with substrate, intermediate, inhibitors, cofactors, and cofactor analogs, determined in collaboration with J. Tainer, Department of Molecular Biology, and D. Stuehr, the Cleveland Clinic, Cleveland, Ohio, provide insights into the catalytic mechanism and dimer stability. The goals of our structure-based drug design projects are to selectively inhibit inducible NOS, to prevent inflammatory disorders, or neuronal NOS, to prevent migraines, while maintaining blood pressure regulation by endothelial NOS. The nearly complete sequence and structural conservation in the active sites of the 3 NOS isozymes is a significant challenge in the design of isozyme-specific inhibitors. Nevertheless, our latest results indicate that plasticity of distant isozyme-specific residues modulates conformational changes of invariant residues in the substrate-binding site. These differences in residue plasticity can be exploited to create inhibitors that are 3000 times more selective for one isozyme than for another despite binding-site conservation. THE SCRIPPS RESEARCH INSTITUTE 195 Our structure of the neuronal NOS reductase reveals new insights into the complex regulatory mechanisms of this enzyme family. We integrated biochemical data with our structures of dimeric NOS oxygenase, dimeric NOS reductase, and calmodulin in complex with peptides derived from NOS to propose a model for the assembled holoenzyme. We have obtained promising results in support of this assembly model by using solution small-angle x-ray scattering, which can provide molecular envelopes for macromolecules and macromolecular complexes in solution. On the basis of our NOS research, we have proposed a moving-domain mechanism for controlling the rate-limiting flow of electrons from the flavin cofactors of NOS reductase to the catalytic NOS oxygenase heme. Our assembly and mechanistic hypotheses also explain the kinetics of regulatory site-specific phosphorylation and dephosphorylation events, as defined by our collaborators G. Rameau, Johns Hopkins School of Medicine, Baltimore, Maryland, and E. Ziff , New York University, New York City, that both activate and inactivate nitric oxide synthesis in vivo (Fig. 1). PHOTOACTIVE PROTEINS AND CIRCADIAN CLOCKS To understand in atomic detail how proteins translate sunlight into defined conformational changes for biological functions, we are exploring the reaction mechanisms of the blue-light receptors photoactive yellow protein (PYP), photolyase, and cryptochrome. PYP is the prototype for the Per-Arnt-Sim domain proteins of circadian clocks, whereas proteins of the photolyase/ cryptochrome family catalyze DNA repair or act in circadian clocks. To understand the protein photocycle of PYP and propose a common mechanism for signaling by Per-Arnt-Sim domains, we combined ultra-high-resolution and time-resolved crystallographic structures of the PYP dark state and 2 photocycle intermediates with site-directed mutagenesis; ultraviolet-visible spectroscopy; time-resolved Fourier transform infrared spectroscopy; deuterium-hydrogen exchange mass spectrometry, in collaboration with V. Woods, University of California, San Diego; and quantum mechanical and electrostatic computational methods, in collaboration with L. Noodleman, Department of Molecular Biology. Cryptochrome flavoproteins are homologs of lightdependent DNA repair photolyases that function as blue-light receptors in plants and as components of circadian clocks in animals. We determined the first crystallographic structure of a cryptochrome; the structure revealed commonalities with photolyases in DNA 196 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE F i g . 1 . Structural mechanism for the temporal sequence of neuronal NOS (nNOS) phosphorylations. binding and redox-dependent function but showed differences in active-site and interaction-surface features. Recently, we showed that this cryptochrome binds the same antenna cofactor found in a photolyase homolog but uses different amino acid residues to form the cofactor-binding site. Our new structures and spectroscopy, in collaboration with S. Weber, Freie Universität Berlin in Germany, of cryptochromes and of photolyases from 2 other branches of the photolyase/cryptochrome family that repair cyclobutane pyrimidine dimers and (6-4) photoproducts help us decipher the cryptic structure, function, and evolutionary relationships of these fascinating redox-active proteins. Furthermore, the (6-4) photolyase enzyme provides an excellent model for evaluating the functions of human cryptochrome (Fig. 2). A simple, but functional, circadian clock can be reconstituted in vitro from the 3 cyanobacterial proteins KaiA, KaiB, and KaiC alone. Yet, the structure and dynamics of the functional assembly are not understood. Our crystallographic, dynamical light scattering, and small-angle x-ray scattering studies revealed that KaiB self-assembles into a tetramer. We also study clock proteins with PYP-like Per-Arnt-Sim domains that bind to mammalian cryptochromes. Our goal is to determine the detailed chemistry and atomic structure of these proteins, define their mechanisms of action and F i g . 2 . The FAD binding sites in the crystallographic structure of cryptochrome (left) and a homology model for (6-4) photolyase (right) were used to design single-site mutants of the 2 histidine residues evolved to function catalytically in DNA repair. We differentiated distinct functions for these catalytic histidines (His354 and His358) by examining pulsed electron nuclear double resonance spectra of the wild-type and mutant proteins under different pH conditions. interaction, and use our results to understand and regulate their biological function. P O S T T R A N S L AT I O N A L M O D I F I C AT I O N C H E M I S T R Y Green fluorescent protein (GFP) and the homologous red fluorescent protein (RFP) spontaneously self-modify their polypeptide chains to form their characteristic green and red fluorophores. We discovered that the architecture of GFP and RFP promotes a remarkable MOLECULAR BIOLOGY 2007 range of other posttranslational modification chemistry. Our high-resolution crystallographic studies of GFP and RFP intermediates in chromophore cyclization and oxidation provide a novel mechanism for the spontaneous synthesis of this tripeptide fluorophore within the protein scaffold. Remarkably, the same protein architectural features that favor peptide cyclization can drive peptide hydrolysis at 4 consecutive positions along the polypeptide backbone and red-shift the spectral properties of the chromophore. Decarboxylation and cleavage of carbon-carbon bonds in designed GFP variants further support a role for the GFP protein environment in facilitating formation of radicals and 1-electron chemistry. Together, our results elucidate the natural mechanism of fluorophore formation and provide the groundwork for the design of proteins with novel catalytic or reporter properties. THE SCRIPPS RESEARCH INSTITUTE 197 S M A L L - A N G L E X - R AY S C AT T E R I N G I N S O L U T I O N C O M B I N E D W I T H C R Y S TA L L O G R A P H Y A N D C O M P U TAT I O N Aided by our advanced synchrotron facility SIBLYS, we are developing tools and technologies to combine x-ray scattering in solution with x-ray crystallography and computation. These paired x-ray techniques can create the complete and accurate images of macromolecules in solution often required to address critical structural questions in biology. Small-angle x-ray scattering, crystallography, and computation together allow multiscale modeling of and fundamental insights to allosteric mechanisms, self-assemblies, and dynamic molecular machines acting in diverse processes ranging from eukaryotic DNA replication, recombination, and repair to microbial membrane secretion and assembly systems (Fig. 1). PUBLICATIONS Barondeau, D.P., Kassmann, C.J., Tainer, J.A., Getzoff, E.D. The case of the missing ring: radical cleavage of a carbon-carbon bond and implications for GFP chromophore biosynthesis. J. Am. Chem. Soc. 129:3118, 2007. Hill, N.J., Stotland, A., Solomon, M., Secrest, P., Getzoff, E., Sarvetnick, N. Resistance of the target islet tissue to autoimmune destruction contributes to genetic susceptibility in type 1 diabetes. Biol. Direct 2:5, 2007. Panda, K., Haque, M.M., Garcin-Hosfield, E.D., Durra, D., Getzoff, E.D., Stuehr, D.J. Surface charge interactions of the FMN module govern catalysis by nitric-oxide synthase. J. Biol. Chem. 281:36819, 2006. Rameau, G.A., Tukey, D.S., Garcin-Hosfield, E.D., Titcombe, R.F., Misra, C., Khatri, L., Getzoff, E.D., Ziff, E.B. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 27:3445, 2007. Schleicher, E., Hitomi, K., Kay, C.W., Getzoff, E.D., Todo, T., Weber, S. Electron nuclear double resonance differentiates complementary roles for active site histidines in (6-4) photolyase. J. Biol. Chem. 282:4738, 2007. Structural Biology of Molecular Interactions and Design J.A. Tainer, A.S. Arvai, D.P. Barondeau, B. R. Chapados, T.H. Cross, L. Fan, C. Hitomi, K. Hitomi, J.J. Perry, M.E. Pique, D.S. Shin, R.S. Williams, A. Yamagata e focus on molecular mechanisms and relationships of protein regulators and effectors of DNA damage responses, reactive oxygen species, and pathogenesis. To help understand multidomain macromolecules with conformational changes and functionally important flexibility in these processes, we are combining solution methods with high-resolution structures and advanced computation. W F i g . 1 . Structures of the Gsp ATPase superfamily for microbial secretion and assembly based on the results of a combination of small-angle x-ray scattering and crystallography. A, Side view of the 2 alternating configurations of the ATPase GspE monomer bound to the nonhydrolyzable ATP analog adenylylimidodiphosphate (AMPPNP) in the hexameric structure determined by crystallography. B, Top views of the crystal structure and 2 models proposed on the basis of modifying subunits to adopt either all-open (brown) or all-closed (green) conformations. C, In solution, the ATPase in excess AMPPNP adopts a conformation most similar to the all-closed model, whereas a solution with excess ADP is best described as a mixture of all the models. Adapted from Yamagata, A., Tainer, J.A. Hexameric structures of the archaeal secretion ATPase GspE and implications for a universal secretion mechanism. EMBO J. 26:878, 2007. 198 MOLECULAR BIOLOGY 2007 P R O T E I N M O D I F I C AT I O N S A N D F U N C T I O N The number of protein-coding genes in the human genome is much smaller than the number of proteins found in human cells. This difference is due to the increase in protein diversity caused by alternative splicing and posttranslational modification of proteins. We are collaborating with E. Getzoff, Department of Molecular Biology, to understand the spontaneous cyclization of the peptide backbone and the oxidation chemistry that convert 3 amino acids into a fluorophore for the family of green fluorescent proteins. We recently discovered how the structural chemistry of green fluorescent protein can accomplish many different types of posttranslational modification, including the surprising radical cleavage of a carbon-carbon bond. E N Z Y M E S T H AT C O N T R O L R E A C T I V E O X Y G E N SPECIES Superoxide dismutases and nitric oxide synthases are master regulators for reactive oxygen species involved in injury, pathogenesis, aging, and degenerative diseases. For human copper, zinc superoxide dismutase, we are examining single-site mutations that cause the neurodegeneration in Lou Gehrig disease or familial amyotrophic lateral sclerosis. We recently solved high-resolution structures that reveal a key role for the zinc ion in the structural defects associated with the disease. For nitric oxide synthases, we are using our combined solution x-ray scattering and crystallographic methods to examine electron transfer and regulatory mechanisms that control levels of nitric oxide, which acts as an important signal and cytotoxin with implications for inflammatory and neurodegenerative diseases. DNA REPAIR AND GENETIC EVOLUTION The genetic information for heredity is encoded in DNA molecules that are constantly under attack from sunlight, ionizing radiation, and other environmental carcinogens. Surprisingly, however, most DNA damage is due to chemical reactions and free radicals that arise from normal cellular metabolism and oxidative responses to infectious disease. In fact, life is impossible without strong DNA repair responses. As a result, mutations that cause defects in DNA repair systems may cause cancer and degenerative diseases associated with aging. However, selective inhibition of certain DNA repair pathways may offer new methods for cancer therapy. We are solving structures of the O 6 -alkylguanineDNA alkyltransferase to aid in the design of inhibitors for cancer chemotherapy. Our structures of several DNAbase repair enzymes are revealing the detailed structural THE SCRIPPS RESEARCH INSTITUTE chemistry of base-excision repair machinery. Our structures of the enzyme that cuts the DNA backbone for the second step of base repair suggest how the enzyme holds the DNA product and hands the product off to the polymerase to replace the removed nucleotide. We are also examining the interface exchange that allows dynamic complexes to form on proliferating cell nuclear antigen when it is loaded onto double-stranded DNA. We found that it binds the DNA minor groove and can cooperate with replication and repair enzymes in their interactions with the adjacent DNA double helix to process and rejoin DNA ends. To understand the initial response to DNA double-strand breaks, we are solving new structures of the Rad50 ABC ATPase and the partner Mre11 nuclease with bound DNA that reveal how this complex holds and processes DNA ends to repair DNA double-strand breaks (Fig. 2). In general, F i g 2 . Models for assembly of the Mre11-Rad50-Nbs1 complex (MRN). Intracomplex zinc hook minimizes unproductive, intercomplex interactions in the absence of DNA. DNA binding (1) straightens the Rad50 coiled coils, a condition that favors intercomplex tethering via Rad50 zinc hooks with extended and parallel coils (2). K indicates kink regions that intersperse regions with strong potential for forming coiled coils (cc). we think that the structural biology of the proteins that control reactive oxygen species and DNA repair may provide master keys to understanding of and therapeutic interventions for brain abnormalities, cancer, and aging. BACTERIAL PILI AND INFECTIOUS DISEASES Type IV pili are essential virulence factors for many bacterial pathogens. Pili play key roles in surface motility, adhesion, formation of microcolonies and biofilms, natural transformation, and signaling. We are characterizing the system machinery of type IV pili, including structures of type IV pilin subunits, assembly of the subunits into pilus fibers, the pilus membrane protein partners, and the assembly ATPases (Fig. 1). We are MOLECULAR BIOLOGY 2007 developing an integrated understanding of assembly and disassembly of type IV pili by a pistonlike pushpull mechanism revealed by our structures of the assembly machinery and assembled type IV pilus fibers. This understanding suggests new approaches to drug and vaccine design for bacterial pathogens. PUBLICATIONS Barondeau, D.P., Kassmann, C.J., Tainer, J.A., Getzoff E.D. The case of the missing ring: radical cleavage of a carbon-carbon bond and implications for GFP chromophore biosynthesis. J. Am. Chem. Soc. 129:3118, 2007. Blaine, R., Roberts, B.R., Tainer, J.A., Getzoff, E.D., Malencik, D.A., Anderson, S.R., Bomben, V.C., Meyers, K.R., Karplus, A., Beckman, J.S. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol., in press. Hitomi, K., Iwaia, S., Tainer, J.A. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst.) 6:410, 2007. Ivanov, I., Chapados, B.R., McCammon, J.A., Tainer, J.A. Proliferating cell nuclear antigen loaded onto double-stranded DNA: dynamics, minor groove interactions and functional implications. Nucleic Acids Res. 34:6023, 2006. Ivanov I., Tainer, J.A., McCammon, J.A. Unraveling the three-metal-ion catalytic mechanism of the DNA repair enzyme endonuclease IV. Proc. Natl. Acad. Sci. U. S. A. 104:1465, 2007. Perry, J.J., Fan, L., Tainer, J.A. Developing master keys to brain pathology, cancer and aging from the structural biology of proteins controlling reactive oxygen species and DNA repair. Neuroscience 145:1280, 2007. Putnam, C.D., Hammel, M., Hura, G.L., Tainer, J.A. Solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys., in press. Tsutakawa, S.E., Hura, G.L., Frankel, K.A., Cooper, P.K., Tainer, J.A. Structural analysis of flexible proteins in solution by small angle x-ray scattering combined with crystallography. J. Struct. Biol. 158:214, 2007. Tubbs, J.L., Pegg, A.E., Tainer,J.A. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair (Amst.), in press. Vijayakumar, S., Chapados, B.R., Schmidt, K.H., Kolodner, R.D., Tainer, J.A., Tomkinson, A.E. The C-terminal domain of yeast PCNA is required for physical and functional interactions with Cdc9 DNA ligase. Nucleic Acids Res. 35:1624, 2007. Williams, R., Sengerova, B., Osborne, S., Syson, K., Ault, S., Kilgour, A., Chapados, B.R., Tainer, J.A., Sayers, J.R., Grasby, J.A. Comparison of the catalytic parameters and reaction specificities of a phage and an archaeal flap endonuclease. J. Mol. Biol. 371:34, 2007. Williams, R.S., Tainer, J.A. Learning our ABCs: Rad50 directs MRN repair functions via adenylate kinase activity from the conserved ATP binding cassette. Mol Cell 25:789, 2007. Williams, R.S., Williams, J.S., Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 85:509, 2007. Yamagata, A., Tainer, J.A. Hexameric structures of the archaeal secretion ATPase GspE and implications for a universal secretion mechanism. EMBO J. 26:878, 2007. THE SCRIPPS RESEARCH INSTITUTE 199 Structural Biology of Integral Membrane Proteins G. Chang, S. Aller, Y. Chen, X. He, A. Karyakin, S. Lieu, C.L. Reyes, P. Szewczyk, T. Tuan, A. Ward, J. Yu ur studies of membrane proteins encompass 5 areas: (1) the molecular structural basis for lipid and drug transport across the cell membrane by multidrug resistance (MDR) transporters, (2) the crystallography of mammalian MDR transporters, (3) signal transduction by receptors, (4) the discovery and design of potent MDR reversal agents, and (5) the development of a cell-free system capable of producing large quantities of integral membrane proteins. We use several experimental methods, including detergent/lipid protein biochemistry, 3-dimensional crystallization of integral membrane proteins, fluorescence anisotropy, calorimetry, protein x-ray crystallography, and functional analysis of transporters. We are addressing the molecular basis of MDR in the treatment of infectious disease and cancer. MDR can be caused by drug efflux pumps imbedded in the cell membrane. Through our structural studies on MDR transporters, we delineated the molecular mechanics for the transport of amphipathic substrate across the cell membrane. We hope that these structures can be used to design more potent inhibitors to be used synergistically with established chemotherapeutic agents. We are combining chemistry and biology with structures in collaboration with M.G. Finn, Department of Chemistry, and Q. Zhang, Department of Molecular Biology. In collaboration with R.A. Milligan, Department of Cell Biology, we are using electron cryomicroscopy to visualize the structures of our transporters. We recently reanalyzed our x-ray diffraction data for the lipid ATP-binding cassette transporter MsbA and determined 4 structures trapped in different conformations: 2 with nucleotide bound and 2 with no nucleotide. Comparisons of the 2 types of conformations revealed that MsbA has a flexible hinge formed by extracellular loops 2 and 3. The hinge allows the nucleotide-binding domains to disassociate while the ATP-binding half sites remain facing each other. The binding of nucleotide causes a packing rearrangement of the transmembrane helices and changes the accessibility of the transporter from cytoplasmic (inward) facing to extracellular (outward) facing. Interestingly, O 200 MOLECULAR BIOLOGY 2007 the inward and outward openings are mediated by 2 different sets of transmembrane helix interactions. We have also reanalyzed our x-ray data on the MDR transporter EmrE. EmrE functions as a homodimer of a small 4-transmembrane protein. The membrane insertion topology of the 2 monomers is controversial. EmrE was reported to have a unique orientation in the membrane. Models based on electron microscopy and on recent biochemical studies posit an antiparallel dimer. The corrected structures in complex with a transport substrate are similar to the electron microscopy structure and indicate an antiparallel orientation for the monomers, supporting a “dual topology” model. Previously, we determined the x-ray structure of EmrD, an MDR transporter from the major facilitator superfamily. EmrD expels amphipathic compounds across the inner membrane of Escherichia coli. The structure reveals an interior that is composed of hydrophobic residues, a finding consistent with the role of EmrD in transporting amphipathic molecules. Two long loops extend into the inner leaflet side of the cell membrane and may recognize and bind substrate directly from the lipid bilayer. We propose that multisubstrate specificity, binding, and transport are facilitated by these loop regions and the internal cavity. Structure and Function of Membrane-Bound Enzymes C.D. Stout, M. Yamaguchi, R. Akhouri, V.M.M. Luna, A. Annalora, H.A. Heaslet, J. Chartron e focus on the structure and function of membrane-bound enzymes and the development of methods for crystallizing membrane proteins. We study the mechanism of transhydrogenase, a mitochondrial respiratory enzyme complex that couples proton translocation with hydride transfer. This enzyme is essential for maintaining NADPH levels in mitochondria, and it plays a critical role in insulin secretion in beta cells of the pancreas. We use x-ray crystallography, biochemical and spectroscopic methods, electron microscopy studies in collaboration with M. Yeager, Department of Cell Biology, and nuclear magnetic resonance in collaboration with J. Dyson, Department of Molecular Biology. Currently, further progress in understanding the structure and function of transhydrogenase is stymied by the lack of a 3-dimensional structure; W THE SCRIPPS RESEARCH INSTITUTE therefore, our primary effort now is to obtain diffraction-quality crystals of the enzyme in its membranebound configuration. In collaboration with J.A. Fee, Department of Molecular Biology, we are studying the mechanism of cytochrome ba3 oxidase, a homolog of the terminal enzyme of respiration in mitochondria. We are using high-resolution crystal structures, mutagenesis, and spectroscopy to visualize intermediates in the reduction of oxygen to water, to define the pathways for protons and oxygen into the active site of the oxidase (Fig. 1), and to understand the coupling of reduction potential to proton translocation across the membrane. F i g . 1 . The pathway for oxygen molecules from the membrane into the active site of cytochrome ba 3 oxidase, as identified by pressurization of crystals with xenon gas. The enzyme contains a forked, hydrophobic channel (green mesh) leading from the lipid bilayer into the active site where oxygen binds between iron and copper atoms and is reduced to water. In collaboration with P. Dawson, Department of Cell Biology, we are developing the assembly of synthetic peptides and phospholipids into discs that contain lipid bilayers. Nanodiscs composed of human apolipoprotein A-I and phospholipids self-assemble into discrete, water-soluble, bilayer-containing particles. Integral membrane proteins incorporated into these particles retain their enzymatic activity, are amenable to biochemical assays, and may have superior properties for crystallization in the absence of detergents. Both transhydrogenase and cytochrome ba 3 oxidase have been incorporated into nanodiscs. In collaboration with E.F. Johnson, Department of Molecular Biology; J.R. Halpert and I. Pikuleva, University of Texas Medical Branch, Galveston, Texas; and MOLECULAR BIOLOGY 2007 others, we are characterizing the structure and function of mammalian cytochrome P450s. These membraneassociated enzymes are involved in the biosynthesis of lipophilic hormones and specifically metabolize a wide variety of exogenous compounds and drugs. High-resolution structures have been determined for the principal drug-metabolizing microsomal P450s in liver and lung in humans: 1A2, 2A6, 2A13, 2C8, 2C9, 2C19, 3A4, and 2B4. For 2B4, 4 structures of the enzyme in markedly different conformations provide insight to substrate binding and membrane insertion. A structure of the brain-specific, cholesterol-metabolizing P450 CYP46 has been determined, and the first mitochondrial P450, CYP24A1, has been crystallized (Fig. 2). THE SCRIPPS RESEARCH INSTITUTE 201 PUBLICATIONS Chartron, J., Shiau, C., Stout, C.D., Carroll, K.S. 3′-Phosphoadenosine-5′-phosphosulfate reductase in complex with thioredoxin: a structural snapshot in the catalytic cycle. Biochemistry 46:3942, 2007. Heaslet, H., Lin, Y.-C., Tam, K., Torbett, B.E., Elder, J.E., Stout, C.D. Crystal structure of an FIV/HIV chimeric protease complexed with the broad-based inhibitor, TL-3. Retrovirology 4:1, 2007. Heaslet, H., Rosenfeld, R., Giffin, M., Lin, Y.-C., Tam, K., Torbett, B.E., Elder, J.H., McRee, D.E., Stout, C.D. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr. D Biol. Crystallogr. 63(Pt. 8):866, 2007. Sansen, S., Hsu, M.-H., Stout, C.D., Johnson, E.F. Structural insight into the altered substrate specificity of human cytochrome P450 2A6 mutants. Arch. Biochem. Biophys., in press. Sansen, S., Yano, J.K., Reynald, R.L., Schoch, G.A., Griffin, K.J., Stout, C.D., Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 282:14348, 2007. Smith, B.D., Sanders, J.L., Porubsky, P.R., Lushington, G.H., Stout, C.D., Scott, E.E. Structure of the human lung cytochrome P450 2A13. J. Biol. Chem. 282:17306, 2007. F i g . 2 . Crystals of mitochondrial cytochrome P450 CYP24A1, the enzyme responsible for stereospecific 6-step oxidation of 1,25-dihydroxy vitamin D3. The active form of vitamin D3 regulates calcium and phosphorus homeostasis and also stimulates cellular differentiation while inhibiting proliferation. Thus, CYP24A1 is an attractive target for the development of cancer therapeutics. A major effort to determine the basis of HIV resistance to antiviral drugs is ongoing in collaboration with A. Olson and J.E. Elder, Department of Molecular Biology; B.E. Torbett, Department of Molecular and Experimental Medicine; M.G. Finn, Department of Chemistry; and D.E. McRee, ActiveSight, San Diego, California. One aspect of this project entails determining the crystal structures of mutant proteases from drug-resistant HIV in complex with broad-spectrum inhibitors; another is the use of fragment-based screening to discover new classes of inhibitors as lead compounds for drug discovery. Additional research projects involve crystal structures and mechanistic studies of an iron-sulfur enzyme, adenosine-5′-phosphosulfate reductase, and its homolog, 3′-phosphoadenosine-5′-phosphosulfate reductase, in collaboration with K.S. Carroll, University of Michigan, Ann Arbor, Michigan. Effect of Surface Engineering on the Crystallization Properties of the Integral Membrane Protein Cytochrome ba3 From Thermus thermophilus J.A. Fee, B. Liu, V.M. Luna, C.D. Stout, Y. Chen e are using the cytochrome ba3 from Thermus thermophilus to increase our understanding of how cytochrome c oxidases function. Cytochrome c oxidases catalyze the following deceptively simple reaction: W 4 cytochrome c2+ + O2 + 8 H+in g 4 cytochrome c3+ + 2 H2O + 4 H+out where the subscripts in and out refer, respectively, to the cytoplasm and the periplasmic space of prokaryotic cells. The free energy of dioxygen reduction is thus captured as a proton gradient; the out side is positive and the in side is negative-proton pumping. Recently, we altered the amino acid sequence of cytochrome ba 3 from T thermophilus and made the unexpected finding that the modified protein readily and rapidly crystallizes, with x-ray diffraction to approximately 2.6–2.5 Å. Our long-range goals are to obtain structures of intermediates that occur during dioxygen 202 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE reduction and to relate the structures to the important, unsolved problem of proton pumping. For these experiments, we will trap intermediates in crystals of cytochrome ba 3 at subzero temperatures. Central to achieving these goals is our homologous expression system for cytochrome ba3 that permits easy purification of the enzyme in amounts needed for physicochemical experimentation. The recombinant enzyme crystallizes in the P43212 space group with considerable difficulty, and the crystals are not suitable for the kinds of studies we envision. Native protein molecules disposed in the unit cell are shown in Figure 1A. Careful F i g . 2 . Lattice contacts of the structure of wild-type (A) and double- mutant (B, I-Lys258Arg and II-Glu4Gln) cytochrome ba3 oxidase. In the wild-type enzyme, 2 salt bridges occur between the symmetry mates I-Lys258–I-Glu510 and I-Glu510–I-Lys258; the hydrogen bonds involving II-Glu4 and I-Lys258 are also evident. In the mutant, 2 possible new hydrogen bonds are found between II-Gln4 and symmetrical II-Glu96 and between I-R258 and symmetrical II-Asn93. Subunit I and subunit II in the original molecule are pink and yellow, respectively. For the symmetrical mate, they are green and blue, respectively. F i g . 1 . Packing contacts of the wild-type (A) and double-mutant (B, I-Lys258Arg and II-Glu4Gln) cytochrome ba3 oxidase. Native protein molecules can be viewed as “linear dimers” in the unit cell (see text), whereas doubly mutated protein molecules are seen as “angled dimers” forming contacts with one another in a springlike structure. Yellow indicates subunit I; magenta, subunit II; and pink, subunit IIa. The transmembrane parts are defined by the barrels of transmembrane helices. The paucity of hydrophilic contacts is evident. examination of the protein-protein contacts revealed a possibility for a unique symmetry-related protein-protein ineraction within the dimeric pair of molecules related by a 2-fold rotation (left-to-right at top and bottom in Fig. 1A). As shown in Figure 2A, the positively charged residue I-Lys258 and the negatively charged residue I-Glu510 make a symmetrical salt bridge across the dimer interface. However, the negatively charged residue II-Glu4 is also in close contact with I-Lys258, adding an unbalanced negative charge that would be expected to weaken the primary interaction between the symmetry mates. We hypothesized that mutagenic conversion of I-Lys258 to I-Arg258 and of II-Glu4 to II-Gln4 may result in 2 strong, intermolecular salt bridges between I-Arg258 and I-Glu510 and eliminate the unbalancing charge of II-Glu4. The double-mutant protein (containing I-Arg258 and II-Gln4), obtained by now-standard techniques of molecular biology, was fully active as a cytochrome c oxidase and crystallized in 2–3 days compared with approximately 1 month for recombinant cytochrome ba3. X-ray crystallographic studies yielded a new struc- MOLECULAR BIOLOGY 2007 ture of the enzyme. Remarkably, crystallization of the double-mutant occurred in a different space group, P41212, with distinctively different protein-protein contacts (Figs. 1B and 2B). Thus, I-Arg258 no longer interacts with I-Glu510; instead it forms a hydrogen bond with the carbonyl atom of II-Asn93, and II-Gln4 may form a hydrogen bond to II-Glu96. Although none of our predictions were fulfilled, the double-mutant protein provides an easy source of crystals for x-ray diffraction, thereby opening many possibilities for novel mechanistic studies. Developing Reagents for Studies of Membrane Proteins Q. Zhang, M.G. Finn,* W.-X. Hong, R.S. Roy * Department of Chemistry, Scripps Research ntegral membrane proteins tend to lose stability and activity outside the membrane bilayer, a situation that makes their biophysical and biochemical characterization difficult. We have developed new membrane-mimicking systems to stabilize integral membrane proteins for structural and functional studies. We designed and synthesized a type of facial amphiphile derived from cholic acid that features increased facial amphiphilicity and a hydrophobic skeleton tailored to mimic the properties of cholesterol components of real lipids (Fig. 1). Our structurally unique facial amphi- I THE SCRIPPS RESEARCH INSTITUTE 203 Crystallization of a variety of targets is being attempted, in collaboration with Drs. Chang and Stevens and M. Yeager, Department of Cell Biology. Last, in collaborative studies with K. Wüthrich and colleagues, Department of Molecular Biology, we have characterized new reagents useful for the solution nuclear magnetic resonance examination of integral membrane proteins. A library of newly synthesized detergents has been evaluated for support on refolding of the β-barrel membrane protein OmpX. One class of lipidlike zwitterionic detergents appears to be superior, producing structure-quality nuclear magnetic resonance spectra. In each of these efforts, different examples of new amphiphiles appear to be useful, emphasizing the need for more diverse reagents to be developed for this important branch of structural biology. PUBLICATIONS Bieschke, J., Zhang, Q., Bosco, D.A., Lerner, R.A., Powers, E.T., Wentworth, P., Jr., Kelly, J.W. Small molecule oxidation products trigger disease-associated protein misfolding. Acc. Chem. Res. 39:611, 2006. Stewart, C.R., Wilson, L.M., Zhang, Q., Pham, C.L.L., Waddington, L., Staples, M.K., Stapleton, D., Kelly, J.W., Howlett, G.J. Oxidized cholesterol metabolites found in human atherosclerotic lesions promote apolipoprotein C-II amyloid fibril formation. Biochemistry 46:5552, 2007. Zhang, Q., Ma, X., Ward, A., Hong, W.-X., Jaakola, V.-P., Stevens, R.C., Finn, MG., Chang, G. Designing facial amphiphiles for the stabilization of integral membrane proteins. Angew. Chem. Int. Ed., in press. Structural Neurobiology and Development of Protein Therapeutic Agents R.C. Stevens, E.E. Abola, A.I. Alexandrov, H.M. Archer, J.W. Arndt, G.A. Asmar-Rovira, R.R. Benoit, M.H. Bracey, A. Brooun, Q. Chai, V.G. Cherezov, E. Chien, S. Daudenarde, J. Dupuy, A. Gámez, M.T. Griffith, M.A. Hanson, V.-P. Jaakola, F i g . 1 . New design of facial amphiphiles derived from cholic J.S. Joseph, K. Masuda, M. Mileni, K. Moy, J. Ng, C. Roth, K.S. Saikatendu, V. Subramanian, J. Velasquez, L. Wang, acid and their proposed binding mode by integral membrane proteins. The membranous sector in deep blue is shielded by the flat hydrophobic surface of facial amphiphiles. B. Wu, Q. Zhao philes have binding properties distinct from those of the classical head-to-tail detergents, as evidenced by the formation of smaller and stronger membrane protein complexes by the synthetic amphiphiles. In collaboration with G. Chang and R.C. Stevens, Department of Molecular Biology, we have shown that the designed facial amphiphiles substantially stabilize several well-known integral membrane protein systems. n the past, out of frustration with the rate at which information on structural biology was emerging, we focused on developing new tools to change the field by accelerating the rate of determination of protein structures. This endeavor included pioneering microliter expression/purification for structural studies, nanovolume crystallization, automated image collection, and robotics for collecting synchrotron beamline data. These tech- HIGH-THROUGHPUT STRUCTURAL BIOLOGY I 204 MOLECULAR BIOLOGY 2007 nologies were initially tested by the team at the Joint Center for Structural Genomics (http://www.jcsg.org) in collaboration with I. Wilson, Department of Molecular Biology, where the power of the new tools was demonstrated. To advance the technologies toward more challenging protein complexes and membrane proteins, in collaboration with P. Kuhn, Department of Cell Biology, we have created 2 new technology-focused centers funded by the National Institutes of Health. The first center is the Joint Center for Innovative Membrane Protein Technologies (http://jcimpt.scripps.edu). Here, in collaboration with K. Wüthrich, Q. Zhang, and G. Chang, Department of Molecular Biology; M.G. Finn, Department of Chemistry; and P. Kuhn and M. Yeager, Department of Cell Biology, we do research exclusively on membrane proteins, including G protein–coupled receptors. The second center is the Accelerated Technologies Center for Gene to 3D Structure (http://www .atcg3d.org). Here we are doing collaborative work with Dr. Kuhn and with researchers from deCODE biostructures, Bainbridge Island, Washington; Lyncean Technologies, Palo Alto, California; and the University of Chicago, Chicago, Illinois. In 2005, we showed that high-resolution electron density maps and refined models can be obtained from in situ diffraction of crystals grown in microcapillaries. In 2008, the first laboratory-sized synchrotron will be installed at Scripps Research. The synchrotron has performance characteristics comparable to those of a synchrotron beam in terms of intensity and tunability and will enable us to use direct diffraction analysis of ongoing in situ crystallization experiments to accelerate determination of macromolecular structures. STRUCTURAL NEUROBIOLOGY Although we have developed high-throughput methods to accelerate the determination of protein structures, our primary interest is using these tools to study the chemistry and biology of neurotransmission and of diseases that affect neurons, particularly childhood neurologic disorders. Our goals are to understand how neuronal cells function on a molecular level and, on the basis of that understanding, create new molecules and materials that mimic neuronal signal transduction and recognition. BIOSYNTHESIS OF NEUROTRANSMITTERS For neuronal signal transduction, the presynaptic cell synthesizes neurotransmitters that then traverse the synaptic cleft. We are using the high-throughput methods to determine the inclusive structures of complete biochemical pathways. Specifically, we are inter- THE SCRIPPS RESEARCH INSTITUTE ested in determining the structures of all the enzymes in the biosynthesis pathways of neurotransmitters in order to understand the mechanistic details of each individual enzymatic reaction at the atomic level. This approach also allows us to determine the best path of drug discovery for the biosynthesis of neurotransmitters. T H E R A P E U T I C A G E N T S F O R T R E AT M E N T O F CHILDHOOD PHENYLKETONURIA In addition to the basic questions under investigation about neurotransmitter biosynthesis, recent clinical studies suggest that some patients with the metabolic disorder phenylketonuria are responsive to (6R)- L erythro-5,6,7,8-tetrahydrobiopterin, the natural cofactor of phenylalanine hydroxylase. We are collaborating with scientists at BioMarin Pharmaceuticals Inc., Novato, California, to correlate how structure can be used to predict which patients with phenylketonuria most likely will respond to treatment with this cofactor. Phase 3 clinical trials for the treatment of phenylketonuria with the cofactor, called Kuvan, have been completed. For classical phenylketonuria, we are developing an enzyme replacement therapeutic agent that is currently in preclinical development. The therapy is based on administration of a modified form of phenylalanine ammonia lyase discovered in our structural studies (Fig. 1). Last, we are determining the structural basis of diseases caused by several other enzymes involved in the biosynthesis of neurotransmitters. Many of these disorders are rare or occur during childhood. BOTULINUM NEUROTOXINS Clostridial neurotoxins are responsible for disrupting neurotransmission. They include tetanus toxin and the 7 serotypes of botulinum toxin. We are determining the molecular events involved in the binding, pore formation, translocation, and catalysis of botulinum neurotoxin. Although botulinum toxin is most known for its deadly effects, it is now being used therapeutically to treat involuntary muscle disorders such as cerebral palsy and neuromuscular dystonias. Previously, we determined the structures of the 150-kD holotoxin form, the holotoxin bound to antibodies, and the catalytic domains of several serotypes (A, B, D, F, G). Recently, we determined the structure of the cell-surface receptor toxin complex (Fig. 2). These structures are being used to understand and redesign the toxin’s mechanism of action and to determine additional therapeutic applications of the toxin. CANNABINOID SIGNALING In collaboration with B.F. Cravatt, Department of Cell Biology, we solved the structure of fatty acid amide MOLECULAR BIOLOGY 2007 F i g . 1 . Crystal structures of phenylalanine ammonia lyase (PAL) tetramers. The structure of wild-type Rhodosporidium toruloides PAL (RtPAL) and the Anabaena variabilis PAL (AvPAL) C503S/C563S double mutant were determined at 1.6- and 2.2-Å resolution, respectively. A, Tetramer structures of RtPAL and AvPAL with each monomer in a different color. B, The bottom view (90° rotation of the view in A) of the same structures with the prosthetic group 4-methylideneimidazole-5-one in the 5 active sites as red sticks. These proteins were engineered and chemically modified as potential once-a-week injectable therapeutic agents for treatment of phenylketonuria. hydrolase, a degradative integral membrane enzyme responsible for setting intracellular levels of endocannabinoids, to 2.8 Å. Fatty acid amide hydrolase is intimately associated with CNS signaling processes such as retrograde synaptic transmission, a process that is also modulated by the illicit substance 9∆-tetrahydrocannabinol. With our knowledge of the 3-dimensional structure, we are trying to understand how the enzyme works at a basic level and how it might be the basis for potential drug discovery. THE SCRIPPS RESEARCH INSTITUTE 205 F i g . 2 . A, Model of binding of type B botulinum neurotoxin via both Syt-II and ganglioside receptors at the presynaptic membrane. The cytoplasm domain of synaptotagmin is presented as 2 C2 domains (light pink) bound with calcium ions (yellow spheres), modeled by 1TJX and 1DQV. Reprinted from Chai, Q., Arndt, J.W., Dong, M., Tepp, W.H., Johnson, E.A., Chapman, E.R., Stevens, R.C. Structural basis of cell surface receptor recognition by botulinum neurotoxin B. Nature 444:1096, 2006. B, Overview of type A1 botulinum neurotoxin (yellow) in complex with the CR1 antibody (antibody heavy and light chains in magenta and green, respectively). Reprinted from Garcia-Rodriguez, C., Levy, R., Arndt, J.W., Forsyth, C.M., Razai, A., Lou, J., Geren, I., Stevens, R.C., Marks, J.D. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat. Biotechnol. 25:107, 2007. G PROTEIN–COUPLED RECEPTORS We are also trying to determine the 3-dimensional structure of G protein–coupled receptors. These receptors are the largest mammalian protein family known and are key signaling molecules for neuronal signal transmission, viral entry, vision, and smell. Using novel technologies that we have developed, we hope to produce a critical breakthrough in the coming year. Abola, E., Kuhn, P., Stevens, R.C. Miniaturization of the structural biology technologies: from expression to biophysical analyses. In: Structural Genomics on Membrane Proteins. Lundstrom, K.H. (Ed.). CRC Press, Boca Raton, FL, 2006, p. 261. PUBLICATIONS Abola, E., Carlton, D.D., Kuhn, P., Stevens, R.C. Five years of increasing structural biology throughput: a retrospective analysis. In: Structure-based Drug Discovery. Jhoti, H., Leach, A. (Eds.). Springer, New York, 2007, p. 1. Gámez, A., Wang, L., Sarkissian, C.N., Wendt, D., Fitzpatrick, P., Lemontt, J.F., Scriver, C.R., Stevens, R.C. Structure-based epitope and PEGylation sites mapping of phenylalanine ammonia-lyase for enzyme substitution treatment of phenylketonuria Mol. Genet. Metab., in press. Chai, Q., Arndt, J.W., Dong, M., Tepp, W.H., Johnson, E.A., Chapman, E.R., Stevens, R.C. Structural basis of cell surface receptor recognition by botulinum neurotoxin B. Nature 444:1096, 2006. DiDonato, M., Krishna, S.S., Schwarzenbacher, R., et al. Crystal structure of 2-phosphysulfolactate phosphatase (ComB) from Clostridium acetobutylicum at 2.6 Å resolution reveals a new fold with a novel active site. Proteins 65:771, 2006. 206 MOLECULAR BIOLOGY 2007 Garcia-Rodriguez, C., Levy, R., Arndt, J.W., Forsyth, C.M., Razai, A., Lou, J., Geren, I., Stevens, R.C., Marks, J.D. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat. Biotechnol. 25:107, 2007. Gerdts, C.J., Tereshko, V., Yadav, M.K., Dementieva, I., Collart, F., Joachimiak, A., Stevens, R.C., Kuhn, P., Kossiakoff, A., Ismagilov, R.F. Time-controlled microfluidic seeding in nL-volume droplets to separate nucleation and growth stages of protein crystallization. Angew. Chem. Int. Ed. 45:8156, 2006. Hanson, M.A., Brooun, A., Baker, K.A., Jaakola, V.-P., Roth, C., Chien, E., Alexandrov, A., Velasquez, J., Davis, L., Griffith, M., Moy, K., Ganser-Pornillos, B., Kuhn, P., Ellis, S., Yeager, M., Stevens, R.C. Profiling of membrane protein variants in a baculovirus system by coupling cell-surface detection with small-scale parallel expression. Prot. Expr. Purif., in press. Joseph, J.S., Saikatendu, K.S., Subramanian, V., Neuman, B.W., Buchmeier, M.J., Stevens, R.C., Kuhn, P. Crystal structure of a monomeric form of severe acute respiratory syndrome coronavirus endonuclease nsp15 suggests a role for hexamerization as an allosteric switch. J. Virol. 81:6700, 2007. Kosloff, M., Han, G.W., Krishna, S.S., et al. Comparative structural analysis of a novel glutathione S-transferase (ATU5508) from Agrobacterium tumefaciens at 2.0 Å resolution. Proteins 65:527, 2006. Saikatendu, K.S., Joseph, J.S., Subramanian, V., Neuman, B.W., Buchmeier, M.J., Stevens, R.C., Kuhn, P. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 8:3913, 2007. Stevens, R.C. SPINE Forward. Acta Crystallogr. D62:0-0, 2006. Swaminathan, S., Stevens, R.C. Three-dimensional protein structures of light chains of botulinum neurotoxin serotypes A, B, and E and tetanus neurotoxin. In: Treatments From Toxins: The Therapeutic Potential of Clostridial Neurotoxins. Foster, K.A., Hambleton, P., Shone, C.C. (Eds.). CRC Press, Boca Raton, FL, 2007, p. 19. Wang, L., Surendran, S., Michals-Matalon, K., Bhatia, G., Tanskley, S., Koch, R., Grady, J., Tyring, S.K., Stevens, R.C., Guttler, F., Matalon, R. Mutations in the regulatory domain of phenylalanine hydroxylase and response to tetrahydrobiopterin. Genet. Test. 11:174, 2007. Xu, Q., Krishna, S.S., McMullan, D., et al. Crystal structure of an ORFan protein (TM1622) from Thermotoga maritima at 1.75 Å resolution reveals a fold similar to the Ran-binding protein Mog1p. Proteins 65:777, 2006. High-Throughput Approaches to Protein Structure and Function S.A. Lesley, L. Columbus, H. Johnson, S. Sudek, T. Janaratne, M. Deller, D. Carlton, Y. Elias, T. Clayton, T. Trout, A. Grzechnik xamining protein structure and function is of primary importance for understanding the basic biology of the cell and is a challenge because of the constantly expanding wealth of genomic information. To address this challenge, we have established high-throughput approaches for evaluating structural and functional diversity of proteins as part of a structural genomics effort with the Joint Center for Structural Genomics. We use these same tools to characterize the molecular basis of the specificity of enzyme substrates. The goals of the Joint Center for Structural Genomics are to develop a high-throughput and cost-effective E THE SCRIPPS RESEARCH INSTITUTE structure pipeline and to use the pipeline to determine novel protein folds and explore protein structure-function relationships. We have used this approach in an extensive study of the thermophilic bacterium Thermotoga maritima and for targets from mouse and more than 100 other bacterial genomes. Our technologies have enabled us to perform comprehensive structural studies of these proteomes. To date, these efforts have resulted in more than 450 novel protein structures from the center. We have also used structural data and performed biochemical assays to validate predicted activities and determine function for numerous targets for which no function could be predicted. In order to understand how genome sequences are related to the biology of an organism, correct annotation of gene function is essential. Many computational approaches are available for assigning a putative gene function on the basis of similarity to known activities. However, as the evolutionary distances extend and the number of putative homologs increases, the reliability of such unvalidated predictions becomes suspect. Likewise, although enzyme classes and broad activities can be predicted with some certainty, much less can be inferred about substrate specificity. This specificity defines the role of an enzyme at a functional level, and understanding enzyme selectivity is essential to understanding the enzyme in the biology of an organism. In collaboration with A. Osterman, the Burnham Institute for Medical Research, La Jolla, California, and B. Geierstanger, Genomics Institute of the Novartis Research Foundation, San Diego, we are undertaking an approach to such characterization that can be applied generally and in a high-throughput fashion to proteins with unknown or putative functions. The approach consists of a bioinformatic platform to analyze gene relationships and propose putative functions for testing, use of an existing high-throughput platform to produce thousands of proteins in milligram quantities for analysis, ligand screening technologies combined with metabolite-focused compound libraries to define specificity profiles of enzymes, and focused experimental validation of proposed functions of key target genes and pathways. PUBLICATIONS Columbus, L., Lipfert, J., Klock, H., Millett, I., Doniach, S., Lesley, S.A. Expression, purification, and characterization of Thermotoga maritima membrane proteins for structure determination. Protein Sci. 15:961, 2006. Eshaghi, S., Niegowski, D., Kohl, A., Martinez Molina, D., Lesley, S.A., Nordlund, P. Crystal structure of a divalent metal ion transporter CorA at 2.9 angstrom resolution [published correction appears in Science 313:1389, 2006]. Science 313:354, 2006. MOLECULAR BIOLOGY 2007 Mason, A., Agrawal, N., Washington, M.T., Lesley, S.A., Kohen, A. A lag-phase in the reduction of flavin dependent thymidylate synthase (FDTS) revealed a mechanistic missing link. Chem. Commun. (Camb.) 1781, Issue 16, 2006. Nuclear Magnetic Resonance Spectroscopy, Chaperonins, and Structural Genomics K. Wüthrich, W. Augustyniak, A. Chatterjee, M. Geralt, R. Horst, M. Johnson, B. Pedrini, W.J. Placzek, J.K. Rhee, THE SCRIPPS RESEARCH INSTITUTE 207 on a fully deuterated background, this experiment resulted in backbone resonance assignments and the identification of conformational constraints for GroES in the 472-kD SR1-GroES complex. We currently are extending the research on the GroEsystem of E coli, which must be studied at temperatures near 25°C, to the GroEL system and its substrate proteins in the extreme thermophile Thermus thermophilus, which can be studied at 60°C or possibly even at higher temperatures. This change led to a remarkable improvement of the spectral resolution (Fig. 1), and we are now P. Serrano e focus on 2 areas of research: chaperonins and structural genomics. For both areas, we use and develop methods of nuclear magnetic resonance (NMR) spectroscopy. W CHAPERONINS In collaboration with A. Horwich, Yale University and Howard Hughes Medical Institute, New Haven, Connecticut, who is a visiting scientist at Scripps Research, we are investigating structural and mechanistic aspects of the function of GroE-type chaperonin systems in Escherichia coli. This research concerns protein folding in healthy and diseased organisms and thus is directly related to the currently extensively discussed protein misfolding diseases. We used transverse relaxation-optimized spectroscopy (TROSY) and cross-correlated relaxation-enhanced polarization transfer (CRINEPT) to study complexes composed of the stable isotope-labeled cochaperonin GroES (72 kD) with the unlabeled chaperonin GroEL (800 kD) or its single-ring variant SR1 (400 kD). We found that informative [15N,1H]-correlation spectra can be obtained for GroES in these complexes; the molecular weights were 872 kD for the GroES-GroEL complex and 472 kD for the GroES-SR1 complex. We then started work on sequence-specific resonance assignments for GroES in these complexes, which are mandatory for further, more detailed analysis. This research required the development of new NMR approaches because routine procedures are not sufficiently sensitive for these large structures. We found that 3-dimensional [1H,1H]nuclear Overhauser effect–[15N, 1H]–CRINEPT–heteronuclear multiple quantum correlation spectroscopy could be used to detect proton-proton nuclear Overhauser effect connectivities for the 2 H, 15 N-labeled GroES in 1:1 complexes with the chaperonins. In combination with uniform and residue-selective 15N labeling of GroES F i g . 1 . Two-dimensional [15N,1H]-TROSY correlation spectrum of T thermophilus GroEL recorded with a sample volume of 300 µL on a Bruker AVANCE 800 spectrometer at 60°C and a 5-mm roomtemperature probehead. The overall measuring time was 2 hours. working on the sequence-specific NMR assignment of the 800-kD T thermophilus GroEL. Our results suggests that previously inaccessible detailed information on chaperonin systems may result from studies of the T thermophilus proteins. STRUCTURAL GENOMICS In our studies in structural genomics, we participate in the Joint Center for Structural Genomics, the Joint Center for Innovative Membrane Protein Technologies, and the consortium for Functional and Structural Proteomics Analysis of SARS-CoV–Related Proteins. Our activities with the use of microcoil NMR equipment combined with microexpression of proteins for quality screening of recombinant proteins are illustrated here with studies of membrane proteins. We previously established a miniaturized pipeline in which 1-dimensional 1H NMR spectroscopy is used for automated screening of newly synthesized polypeptides for the presence of globular domains. Although this approach is now in routine use for soluble proteins with 208 MOLECULAR BIOLOGY molecular weights up to about 30 kD, initial experiments with detergent-solubilized membrane proteins showed that the use of this type of spectroscopy is limited by the background signals from the nondeuterated detergents, which partially overlap with the protein signals. Furthermore, it turned out that the high molecular weight of the mixed protein-detergent micelles requires the use of TROSY-based NMR experiments in combination with uniform 2H,15N-labeling of the membrane proteins. Using this approach and new detergents synthesized by Q. Zhang, Department of Molecular Biology, we solubilized the E coli outer-membrane protein X (OmpX). We then used the resulting samples to obtain [15N,1H]NMR correlation maps as diagnostic fingerprints of the conformational state of OmpX reconstituted in mixed micelles with these detergents. Figure 2 shows NMR 2007 THE SCRIPPS RESEARCH INSTITUTE aforementioned technology, we expect to contribute to a future widening of this bottleneck. PUBLICATIONS Almeida, M.S., Johnson, M.A., Wüthrich, K. NMR assignment of the SARS-CoV protein nsp1. J. Biomol. NMR 36(Suppl.1):46, 2006. Baker, K.A., Hilty, C., Peti, W., Prince, A., Pfaffinger, P.J., Wider, G., Wüthrich, K., Choe, S. NMR-derived dynamic aspects of N-type inactivation of a Kv channel suggest a transient interaction with the T1 domain. Biochemistry 45:1663, 2006. Etezady-Esfarjani, T., Herrmann, T., Horst, R., Wüthrich, K. Automated protein NMR structure determination in crude cell-extract. J. Biomol. NMR 34:3, 2006. Etezady-Esfarjani, T., Placzek, W.J., Herrmann, T., Wüthrich, K. Solution structures of the putative anti-σ-factor antagonist TM1442 from Thermotoga maritima in the free and phosphorylated states. Magn. Reson. Chem. 44(Spec. No.):S61, 2006. Horst, R., Wider, G., Fiaux, J., Bertelsen, E.B., Horwich, A.L., Wüthrich, K. Proton-proton Overhauser NMR spectroscopy with polypeptide chains in large structures. Proc. Natl. Acad. Sci. U. S. A. 103:15445, 2006. Johnson, M.A., Peti, W., Herrmann, T., Wilson, I.A., Wüthrich, K. Solution structure of As11650, an acyl carrier protein from Anabaena sp. PCC 7120 with a variant phosphopantetheinylation-site. Protein Sci. 15:1030, 2006. Placzek, W.J., Almeida, M.A., Wüthrich, K. NMR assignment of a human cancerrelated nucleoside triphosphatase. J. Biomol. NMR 36(Suppl. 1):59, 2006. Serrano, P., Almeida, M.S., Johnson, M.A., Wüthrich, K. NMR assignment of the protein nsp3a from SARS-CoV. J. Biomol. NMR 36(Suppl.1):45, 2006. F i g . 2 . Two-dimensional [ 15N, 1H]-TROSY correlation spectra of 2H, 15N-labeled OmpX reconstituted in different detergents: A, 138Fos. B, 179-Fos. C, 34-Fos. D, 185-Fos. The spectra were recorded with sample volumes of 7 µL on a Bruker DRX-700 spectrometer at 25°C with a 1-mm room-temperature microcoil probehead. The overall measurement time per experiment was 9 hours. spectra recorded with 7-µL samples of solutions containing OmpX in mixed micelles with the detergents 138-Fos and 179-Fos (panels A and B), where the protein is uniformly well folded, and with 34-Fos and 185-Fos (panels C and D), where the protein forms nonspecific aggregates. As a result of this study, we have identified 2 new detergents with promising properties for the preparation of membrane proteins for structural studies. The preparation of integral membrane proteins for structural biology and structural genomics is a bottleneck that has limited structure determinations by x-ray crystallography or NMR spectroscopy to only about 100 structures, as compared with over 30,000 structures of soluble proteins. By systematic screening of new membrane protein–detergent combinations with the Nuclear Magnetic Resonance of 3-Dimensional Structure and Dynamics of Proteins in Solution P.E. Wright, H.J. Dyson, M. Arai, R. Burge, J. Ferreon, T.-H. Huang, B.A. Buck-Koehntop, M. Kostic, B. Lee, C.W. Lee, M. Landes, M. Martinez-Yamout, T. Nishikawa, K. Sugase, J. Wojciak, M. Zeeb, E. Manlapaz, L.L. Tennant, J. Chung, D.A. Case, J. Gottesfeld, R. Evans,* M. Montminy* * Salk Institute for Biological Studies, La Jolla, California e use multidimensional nuclear magnetic resonance (NMR) spectroscopy to investigate the structures, dynamics, and interactions of proteins in solution. Such studies are essential for understanding the mechanisms of action of these proteins and for elucidating structure-function relationships. The focus of our current research is protein-protein and protein–nucleic acid interactions involved in the regulation of gene expression. W T R A N S C R I P T I O N FA C T O R – N U C L E I C A C I D C O M P L E X E S NMR methods are being used to determine the 3-dimensional structures and intramolecular dynamics of zinc finger motifs from several eukaryotic transcriptional regulatory proteins, both free and complexed with MOLECULAR BIOLOGY 2007 target nucleic acid. Zinc fingers are among the most abundant domains in eukaryotic genomes. They play a central role in the regulation of gene expression at both the transcriptional and the posttranscriptional level, mediated through their interactions with DNA, RNA, or protein components of the transcriptional machinery. The C2H 2 zinc finger, first identified in transcription factor IIIA (TFIIIA), is used by numerous transcription factors to achieve sequence-specific recognition of DNA. Growing evidence, however, indicates that some C 2H 2 zinc finger proteins control gene expression both through their interactions with DNA regulatory elements and, at the posttranscriptional level, through binding to RNA. The best-characterized example of a C2H2 zinc finger protein that binds specifically to both DNA and to RNA is TFIIIA, which contains 9 zinc fingers. We showed previously that different subsets of zinc fingers are responsible for high-affinity binding of TFIIIA to DNA (fingers 1–3) and to 5S RNA (fingers 4–6). To obtain insights into the mechanism by which the TFIIIA zinc fingers recognize both DNA and RNA, we have used NMR methods to determine the structures of the complex formed by zf1-3 (a protein containing fingers 1–3) with DNA and by zf4-6 (a protein consisting of fingers 4–6) with a fragment of 5S RNA. Three-dimensional structures were determined previously for the complex of zf1-3 with the cognate 15-bp oligonucleotide duplex. The structures contain several novel features and reveal that prevailing models of DNA recognition, which assume that zinc fingers are independent modules that contact bases through a limited set of amino acids, are outmoded. In addition to its role in binding to and regulating the 5S RNA gene, TFIIIA also forms a complex with the 5S RNA transcript. NMR structures of the complex formed by zinc fingers 4–6 with a truncated form of 5S RNA have been completed and give important insights into the structural basis for 5S RNA recognition. Finger 4 of the protein recognizes both the structure of the RNA backbone and the specific bases in the loop E motif of the RNA, in a classic lock-and-key interaction. Fingers 5 and 6, with a single residue between them, undergo mutual induced-fit folding with the loop A region of the RNA, which is highly flexible in the absence of the protein. NMR studies of 2 alternate splice variants of the Wilms tumor zinc finger protein (WT1) are in progress. These proteins differ only through insertion of 3 additional amino acids (the tripeptide lysine-threonine-serine) THE SCRIPPS RESEARCH INSTITUTE 209 in the linker between fingers 3 and 4, yet have marked differences in their DNA-binding properties and subcellular localization. 15N relaxation measurements indicate that the insertion increases the flexibility of the linker between fingers 3 and 4 and abrogates binding of the fourth zinc finger to its cognate site in the DNA major groove, thereby modulating DNA-binding activity. X-ray and NMR structures of the complexes of the WT1 zinc fingers with 14- and 17-bp DNA oligonucleotides have been determined. Zinc fingers 2–4 are inserted deeply into the DNA major groove, making sequence-specific contacts with bases. The structure provides insights into the mechanism by which disease-causing mutations in the zinc finger domain interfere with DNA binding. In contrast to fingers 2–4, zinc finger 1 has mostly nonspecific interactions with the DNA. High-affinity DNA binding is mediated by fingers 2–4; incorporation of additional amino acids in the linker by alternate splicing disrupts the finger 4 interactions and abrogates DNA binding (Fig. 1). F i g . 1 . Ribbon diagram of the composite NMR/x-ray structures of the complex of WT1 zinc fingers 1–4 and the 17-bp DNA duplex in solution. NMR structural studies of a complex of the 4 WT1 zinc fingers with an RNA aptamer are nearing completion. In contrast to DNA binding, the RNA interaction is dominated by zinc fingers 1–3, which bind in the widened major groove formed in the vicinity of a bulged 210 MOLECULAR BIOLOGY 2007 base. The interactions of zinc finger 4 with the RNA loop make only a secondary contribution to binding affinity. We have also determined the structure of a novel double-stranded RNA-binding zinc finger protein and have commenced experiments to define the mechanism of binding to adenovirus VA1 RNA. We recently determined the structure of a novel zinc finger protein termed Churchill that is involved in regulation of neural induction during embryogenesis. At the time of its discovery, it was suggested that the protein contained 2 zinc fingers of the C4 type and functioned as a DNA-binding transcription factor. Our NMR structure shows that far from containing canonical C 4 zinc fingers, Churchill contains 3 bound zinc ions in novel coordination sites, including an unusual binuclear zinc cluster, which jointly stabilize a single-layer β-sheet. We showed further that Churchill does not bind DNA and suggest that it may function in embryogenesis by mediating protein-protein interactions. THE SCRIPPS RESEARCH INSTITUTE experiments to elucidate the mechanism by which folding of the kinase-inducible activation domain of CREB is coupled to binding to its KIX target domain. These experiments revealed formation of an ensemble of transient and largely unfolded encounter complexes at multiple sites on the surface of KIX. The encounter complexes are stabilized primarily by nonspecific hydrophobic contacts and evolve via an intermediate to the fully bound state without dissociation from KIX. The C-terminal helix of pKID is only partially folded in the intermediate and becomes stabilized by intermolecular interactions formed in the final bound state. Future applications of our method will provide new understanding of the molecular mechanism by which intrinsically disordered proteins perform their diverse biological functions (Fig. 2). PROTEIN-PROTEIN INTERACTIONS IN T R A N S C R I P T I O N A L R E G U L AT I O N Transcriptional regulation in eukaryotes relies on protein-protein interactions between DNA-bound factors and coactivators that, in turn, interact with the basal transcription machinery. The transcriptional coactivator CREB-binding protein (CBP) and its homolog p300 play an essential role in cell growth, differentiation, and development. Understanding the molecular mechanisms by which CBP and p300 recognize their various target proteins is of fundamental biomedical importance. CBP and p300 have been implicated in diseases such as leukemia, cancer, and mental retardation and are novel targets for therapeutic intervention. We previously determined the structure of the phosphorylated kinase-inducible activation domain (pKID) of the transcription factor CREB bound to its target domain (the KIX domain) in CBP. Ongoing work is directed toward mapping the interactions between KIX and the transcriptional activation domains of the proto-oncogene c-Myb and of the mixed-lineage leukemia protein. The solution structure of the ternary complex between KIX, c-Myb, and the mixed-lineage leukemia protein has been completed and provides insights into the structural basis for the ability of the KIX domain to interact simultaneously and allosterically with 2 different effectors. Our work has also provided new understanding of the thermodynamics of the coupled folding and binding processes involved in interaction of KIX with transcriptional activation domains. We used R 2 relaxation dispersion F i g . 2 . 15N R 2 relaxation dispersion profile for Arg124 of pKID recorded at 800 MHz (filled circles) and 500 MHz (open circles). Dispersion curves for 1 mM [ 15N]-pKID in the presence of 0.95, 1.00, 1.05, and 1.10 mM KIX are shown. Recently, we determined the structure of the complex between the hypoxia-inducible factor Hif-1α and the TAZ1 domain of CBP. The interaction between Hif-1α and CBP/p300 is of major therapeutic interest because of the central role Hif-1α plays in tumor progression and metastasis; disruption of this interaction leads to attenuation of tumor growth. A protein named CITED2 functions as a negative feedback regulator of the hypoxic response by competing with Hif-1α for binding to the TAZ1 domain of CBP. By determining the structure of the complex, we showed that the intrinsically unstructured Hif-1α and CITED2 domains use partly overlapping surfaces of the TAZ1 motif to achieve highaffinity binding and compete effectively with each other for CBP/p300. To further elucidate the molecular and structural basis for CBP-dependent coordinated gene expression, we have determined the solution structures of the com- MOLECULAR BIOLOGY 2007 plexes formed by the transactivation domains of the transcription factors STAT2 and STAT1 with CBP TAZ1 and TAZ2 domains, respectively. Despite the overall topological similarity of the CBP TAZ domains, the structures reveal 2 very different modes of complex formation. Our findings suggest that TAZ1 may bind activation domains capable of contacting multiple surface grooves simultaneously in preference to smaller activation motifs that are restricted to a single, contiguous binding surface. The latter mode of binding is sufficient for stable complex formation with TAZ2. Binding of both STAT activation domains involves coupled folding and binding processes. We are continuing to map the multiplicity of interactions between CBP/p300 domains and their numerous biological targets. Our goal is to understand the complex interplay of interactions that mediate key biological processes in health and disease. PUBLICATIONS Lee, B.M., Buck-Koehntop, B.A., Martinez-Yamout, M.A., Dyson, H.J., Wright, P.E. Embryonic neural inducing factor Churchill is not a DNA-binding zinc finger protein: solution structure reveals a solvent-exposed β-sheet and zinc binuclear cluster. J. Mol. Biol., in press. Stoll, R., Lee, B.M., Debler, E.W., Laity, J.H., Wilson, I.A., Dyson, H.J., Wright, P.E. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol., in press. Sugase, K., Dyson, H.J., Wright, P.E. Mechanism of coupled folding and binding of an intrinsically unstructured protein. Nature 447:1021, 2007. Sun, P., Yoshizuka, N., New, L., Moser, B.A., Li, Y., Liao, R., Xie, C., Chen, J., Deng, Q., Yamout, M., Dong, M.Q., Frangou, C.G., Yates, J.R. III, Wright, P.E., Han. J. PRAK is essential for ras-induced senescence and tumor suppression. Cell 128:295, 2007. Folding of Proteins and Protein Fragments P.E. Wright, H.J. Dyson, D. Meinhold, C. Nishimura, D. Felitsky, M. Kostic, S.J. Park, J. Chung, L.L. Tennant, V. Bychkova,* T. Yamagaki * Institute of Protein Research, Puschino, Russia he molecular mechanism by which proteins fold into their 3-dimensional structures remains one of the most important unsolved problems in structural biology. Nuclear magnetic resonance (NMR) spectroscopy is uniquely suited to provide information on the structure of transient intermediates formed during protein folding. Previously, we used NMR methods to show that many peptide fragments of proteins tend to adopt folded conformations in water solution. The T THE SCRIPPS RESEARCH INSTITUTE 211 presence of transiently populated folded structures, including reverse turns, helices, nascent helices, and hydrophobic clusters, in water solutions of short peptides has important implications for initiation of protein folding. Formation of elements of secondary structure probably plays an important role in the initiation of protein folding by reducing the number of conformations that must be explored by the polypeptide chain and by directing subsequent folding pathways. A P O M Y O G L O B I N F O L D I N G PAT H WAY A major program in our laboratory is directed toward a structural and mechanistic description of the apomyoglobin folding pathway. Previously we used quenched-flow pulse-labeling methods in conjunction with 2-dimensional NMR spectroscopy to map the kinetic folding pathway of the wild-type protein. With these methods, we showed that an intermediate in which the A, G, and H helices and part of the B helix adopt hydrogen-bonded secondary structure is formed within 6 milliseconds of the initiation of refolding. Folding then proceeds by stabilization of additional structure in the B helix and in the C and E helices. We are using carefully selected myoglobin mutants and both optical stopped-flow spectroscopy and NMR methods to further probe the kinetic folding pathway. For some of the mutants studied, the changes in amino acid sequence resulted in changes in the folding pathway of the protein. These experiments are providing novel insights into both the local and long-range interactions that stabilize the kinetic folding intermediate. Of particular importance, long-range interactions have been observed that indicate nativelike packing of some of the helices in the kinetic molten globule intermediate. However, folding is impeded by local nonnative helix packing; the H helix is translocated relative to the G helix by a single helical turn, and folding cannot proceed until this defect is repaired. Apomyoglobin provides a unique opportunity for detailed characterization of the structure and dynamics of a protein-folding intermediate. Conditions were previously identified under which the apomyoglobin molten globule intermediate is sufficiently stable for acquisition of multidimensional heteronuclear NMR spectra. Analysis of 13C and other chemical shifts and measurements of polypeptide dynamics provided unprecedented insights into the structure of this state. The A, G, and H helices and part of the B helix are folded and form the core of the molten globule. This core is stabilized by relatively nonspecific hydrophobic 212 MOLECULAR BIOLOGY 2007 interactions that restrict the motions of the polypeptide chain. Fluctuating helical structure is formed in regions outside the core, although the amount of helix is low and the chain retains considerable flexibility. The F helix acts as a gate for heme binding and only adopts stable structure in the fully folded holoprotein. The acid-denatured (unfolded) state of apomyoglobin is an excellent model for the fluctuating local interactions that lead to the transient formation of unstable elements of secondary structure and local hydrophobic clusters during the earliest stages of folding. NMR data indicated substantial formation of helical secondary structure in the acid-denatured state in regions that form the A and H helices in the folded protein and also revealed nonnative structure in the D and E helix regions. Because the A and H regions adopt stabilized helical structure in the earliest detectable folding intermediate, these results lend strong support to folding models in which spontaneous formation of local elements of secondary structure plays a role in initiating formation of the A-[B]-G-H molten globule folding intermediate. In addition to formation of transient helical structure, formation of local hydrophobic clusters has been detected by using 15N relaxation measurements. Significantly, these clusters are formed in regions where the average surface area buried upon folding is large. In contrast to acid-denatured unfolded apomyoglobin, the urea-denatured state is largely devoid of structure, although residual hydrophobic interactions have been detected by using relaxation measurements. We measured residual dipolar couplings for unfolded states of apomyoglobin by using partial alignment in strained polyacrylamide gels. These data provide novel insights into the structure and dynamics of the unfolded polypeptide chain. We have shown that the residual dipolar couplings arise from the well-known statistical properties of flexible polypeptide chains. Residual dipolar couplings provide valuable insights into the dynamic and conformational propensities of unfolded and partly folded states of proteins and hold great promise for charting the upper reaches of protein folding landscapes. To probe long-range interactions in unfolded and partially folded states of apomyoglobin, we introduced spin-label probes at several sites throughout the polypeptide chain. These experiments led to the surprising discovery that transient structures with nativelike longrange contacts between hydrophobic clusters exist within the ensemble of conformations formed by the acid-denatured state of apomyoglobin. They also indi- THE SCRIPPS RESEARCH INSTITUTE cated that the packing of helices in the molten globule state is similar to that in the native folded protein. The view of protein folding that results from our work on apomyoglobin is one in which collapse of the polypeptide chain to form increasingly compact states leads to progressive accumulation of secondary structure and increasing restriction of fluctuations in the polypeptide backbone. Chain flexibility is greatest at the earliest stages of folding, when transient elements of secondary structure and local hydrophobic clusters are formed. As the folding protein becomes increasingly compact, backbone motions become more restricted, the hydrophobic core is formed and extended, and nascent elements of secondary structure are progressively stabilized. The ordered tertiary structure characteristic of the native protein, with well-packed side chains and relatively low-amplitude local dynamics, appears to form rather late in folding. We recently introduced a variation on the classic quench-flow technique, which makes use of the capabilities of modern NMR spectrometers and heteronuclear NMR experiments, to study the proteins labeled along the folding pathway in an unfolded state in an aprotic organic solvent. This method allows detection of many more amide proton probes than in the classic method, which requires formation of the fully folded protein and the measurement of the protein’s NMR spectrum in water solution. This method is particularly useful in documenting changes in the folding pathway that result in the destabilization of parts of the protein in the molten globule intermediate. We recently showed that self-compensating mutations designed to change the amino acid sequence such that the average area buried upon folding is significantly changed while the 3-dimensional structure of the final folded state remains the same. These studies showed that the average area buried upon folding is an accurate predictor of those parts of the apomyoglobin molecule that will fold first and participate in the molten globule intermediate. Quench-flow hydrogen exchange experiments performed on a series of hydrophobic core mutants indicated that the overall helix-packing topology of the kinetic folding intermediate is like that of the native protein, despite local nonnative interactions in packing of the G and H helices (Fig. 1). Finally, using a rapid mixing device, we have reduced the dead time of the kinetic refolding experiments and have shown that a compact helical intermediate is formed within 400 microseconds after initiation of apomyoglobin refolding. MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 213 CHAPERONE–COCHAPERONE–CLIENT PROTEIN INTERACTIONS F i g . 1 . Schematic representation of the amide proton occupancies in the kinetic intermediate state formed in the burst phase of apomyoglobin folding (solid ribbons), mapped onto the structure of fully folded myoglobin. Areas of the protein that are not folded until later stages are shown as dotted lines. FOLDING-UNFOLDING TRANSITIONS IN CELLULAR M E TA B O L I S M Many species of bacteria sense and respond to their own population density by an intricate autoregulatory mechanism known as quorum sensing; bacteria release extracellular signal molecules, called autoinducers, for cell-cell communication within and between bacterial species. A number of bacteria appear to use quorum sensing for regulation of gene expression in response to fluctuations in cell population density. Processes regulated in this way include symbiosis, virulence, competence, conjugation, production of antibiotics, motility, sporulation, and formation of biofilms. We determined the 3-dimensional solution structure of a complex composed of the N-terminal 171 residues of the quorum-sensing protein SdiA of Escherichia coli and an autoinducer molecule, N-octanoyl-1-homoserine lactone (HSL). The SdiA-HSL system shows the “folding switch” behavior associated with quorum-sensing factors produced by other bacterial species. In the presence of HSL, SdiA is stable and folded and can be produced in good yields from an E coli expression system. In the absence of the autoinducer, SdiA is expressed into inclusion bodies. Samples of the SdiA-HSL complex can be denatured but cannot be refolded in aqueous buffers. The solution structure of the complex provides a likely explanation for this behavior. The autoinducer molecule is tightly bound in a deep pocket in the hydrophobic core and is bounded by specific hydrogen bonds to the side chains of conserved residues. The autoinducer thus forms an integral part of the hydrophobic core of the folded SdiA. Understanding the role of unfolded states in cellular processes will require an understanding of the structural basis of their interactions, but unfolded proteins are impossible to characterize structurally by x-ray crystallography, and spectroscopic methods of all kinds are limited. Unfolded proteins must be explored under conditions that approximate their physiologic milieu: in solution, at physiologic pHs and salt concentrations, and in the presence of specific cofactors. Structural insights will be obtained not only from the delineation of 3-dimensional structures but also from the description of conformational ensembles and of the motions of polypeptide chains under various conditions. To gain new insights into the structural basis for the ability of unfolded and partly folded proteins to function in living systems, we study the interactions of “client” proteins and cochaperones with a well-known eukaryotic chaperone, Hsp90. Some of the protein components are much larger than have traditionally been studied by using solution NMR. However, we have designed a set of experiments that will allow us to draw valid conclusions about the extent and role of disorder in Hsp90 interactions. In particular, we will apply techniques recently developed in our laboratory for the analysis of hydrogen-deuterium exchange from unstable partially folded proteins by trapping the 2 H-labeled species in the aprotic solvent dimethyl sulfoxide. This powerful new technique will be used to probe the structure, stability, and interactions of client proteins and cochaperones with Hsp90. Nuclear Magnetic Resonance Studies of the Structure and Dynamics of Enzymes H.J. Dyson, P.E. Wright, S.H. Bae, G. Bhabha, D. Boehr, P. Card, C. Cervantes,* G. Kroon, M. Martinez-Yamout, S.C. Sue, L.M. Tuttle, L.L. Tennant, J. Chung, C.L. Brooks, S.J. Benkovic,** A. Holmgren,*** E.A. Komives* * University of California, San Diego, California ** Pennsylvania State University, University Park, Pennsylvania *** Karolinska Institutet, Stockholm, Sweden W e use site-specific information on structure and dynamics, obtained from nuclear magnetic resonance (NMR), to further the understand- 214 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE ing of protein function. We focus on the mechanism of enzymes and the relationship between dynamics and function in a number of medically important systems. DYNAMICS IN ENZYME ACTION Dynamic processes are implicit in the catalytic function of all enzymes. We use state-of-the-art NMR methods to elucidate the dynamic properties of several enzymes. New methods have been developed for analysis of NMR relaxation data for proteins that tumble anisotropically and for analysis of slow timescale motions. Dihydrofolate reductase plays a central role in folate metabolism and is the target enzyme for a number of antibacterial and anticancer agents. 15N relaxation experiments on dihydrofolate reductase from Escherichia coli revealed a rich diversity of backbone dynamical features for a broad range of timescales (picoseconds to milliseconds). A major focus is the characterization of all intermediates in the dihydrofolate reductase reaction cycle. We have identified functionally important motions in loops that control access to the active site of dihydrofolate reductase. These motions differ in amplitude and timescale depending on the presence of substrate and/or cofactor in the active site. In addition, measurements of the population distribution of aliphatic side-chain rotamers provide evidence for coupled motion of activesite side chains that could enhance the catalytic process. Most recently, we used relaxation dispersion measurements to obtain direct information on microsecond-millisecond timescale motions in dihydrofolate reductase, allowing us to characterize the structures of excited states involved in some of these catalysisrelevant processes. Each intermediate in the catalytic cycle samples low-lying excited states whose conformations resemble the ground-state structures of the preceding and following intermediates. Fluctuations between these states occur on a timescale that is directly relevant to the structural transitions involved in progression through the catalytic cycle (Fig. 1). Substrate and cofactor exchange occurs through these excited substates. The maximum hydride transfer and steady-state turnover rates are governed by the dynamics of transitions between the ground and excited states of the intermediates. The modulation of the energy landscape by the bound ligands funnels the enzyme through its reaction cycle along a preferred kinetic path (Fig. 1). Dihydrofolate reductase is also the test system for a series of experiments to address the question, If all F i g . 1 . Schematic diagram of the energy landscape of dihydrofo- late reductase catalysis. Ground-state (larger) and higher energy (smaller) structures of each intermediate in the cycle, modeled on published x-ray structures, are shown. For each intermediate in the catalytic cycle, the higher energy conformations detected in the relaxation dispersion experiments resemble the ground-state conformations of adjacent intermediates. Rate constants for the interconversion between the complexes, measured by pre–steady-state enzyme kinetics at 298 K, pH 6, are indicated with red arrows; the rates measured in relaxation dispersion experiments are indicated with black arrows. From Boehr et al., Science 313:1638, 2006. Reprinted with permission from AAAS. of the chemistry goes on at the active site, what is the purpose of the rest of the enzyme? We are using chimeric mutants, synthesized by our collaborator S.J. Benkovic, Pennsylvania State University, University Park, Pennsylvania, by using a library approach. The purpose of these experiments is to test the hypothesis that local variations in amino acid sequence, 3-dimensional structure, and polypeptide chain dynamics strongly influence the local interactions that mediate enzyme catalysis and may constitute the essential circumstance that allows enzymes to achieve high turnover rates as well as exquisite specificity in their reactions. A combination of NMR structure and dynamics measurements, single-molecule fluorescence measurements, and analysis of the catalytic steps in these mutant proteins provides new insights into the role of the protein in enzyme catalysis. S T R U C T U R E A N D D Y N A M I C S O F P R I O N VA R I A N T S Onset of prion diseases is caused by conversion of the cellular prion protein PrPC into an abnormally folded isoform, PrPSc, that has the same primary structure as PrPC but a totally different 3-dimensional conforma- MOLECULAR BIOLOGY 2007 tion. The abnormally folded (“scrapie”) form of the protein is associated with several diseases, including scrapie in sheep, bovine spongiform encephalopathy (mad cow disease), and human Creutzfelt-Jakob disease and other inherited prion diseases. We are gathering information on the mechanism of PrPSc formation that can be obtained from structural and dynamic studies of mutant prion proteins corresponding to inherited prion diseases. Individuals carrying familial mutations such as P102L (P101L in our study) are more susceptible to prion disease. On the other hand, sheep or humans carrying Q167R and/or Q218K mutations are resistant to scrapie and Creutzfelt-Jakob disease, respectively. We are using the protease-resistant cores of wild-type and mutant mouse prion proteins to study the structural and dynamic basis of PrPC-to-PrPSc conversion in inherited prion diseases. The core is sufficient to transmit infectivity. DYNAMICS AND THE FUNCTION OF IκBα It is becoming increasingly clear that the function of many systems in living cells depends not only on the structures of the components but also on the structures’ flexibility. Numerous examples exist in which components of an important biological interaction are unstructured or partly structured. In addition, even those interacting molecules that can be classified as “folded” have areas of mobility. Often, these areas are located precisely in the active site of an enzyme or in the binding site of an interacting molecule. A central molecular interaction in cellular control is the interaction between the nuclear transcription factor NF-κB and its inhibitor IκBα. IκBα consists of a series of ankyrin repeats, which appear to have differential mobility. Using hydrogen-deuterium exchange and mass spectrometry, our collaborator, E.A. Komives, University of California, San Diego, found that the second, third, and fourth ankyrin repeats of IκBα are well folded, whereas the fifth and sixth repeats, apparently with exactly the same structure, are highly dynamic. These observations prompt a number of questions: Are the motions inferred from the hydrogen-deuterium mass spectrometry experiments also reflected in the backbone and side-chain dynamics of the protein, as measured by NMR relaxation? Are the motions still present in the IκBα–NF-κB complex? Are they necessary for complex formation, so that if they are damped out, for example, by site-directed mutagenesis at appropriate positions, is the formation of the complex disfavored? THE SCRIPPS RESEARCH INSTITUTE 215 To answer these questions, we are doing a series of NMR experiments on IκBα and its complexes with NF-κB. Chaperonin-Mediated Protein Folding A.L. Horwich, E. Chapman, S.M. Johnson, E. Koculi uring the past year, we have continued to investigate the GroEL-GroES chaperonin system. This system assists the folding to native form of a large number of newly translated proteins. D GroEL ACTION IN VIVO In in vivo studies, we have isolated and characterized a new temperature-sensitive Escherichia coli GroEL mutant. To our knowledge, this mutant, E461K, is the most severe conditional mutant to date. The substitution at the ring-ring interface residue of GroEL abolishes the cooperative binding and hydrolysis of ATP within rings and anticooperativity between them and is strongly temperature sensitive for mediating folding in vitro. In culture, E coli containing the mutant protein grow normally at 23°C, but upon a shift to 37°C, their growth slows within 30 minutes and stops by 1–2 hours. During this time, translation continues, but misfolding and aggregation occur, for example, with the induced test protein ribulose-1,5-bisphosphate carboxylase/oxygenase. More generally, as revealed by pulse-chase studies, a broad collection of newly translated E coli proteins are misfolded and aggregate in the mutant cells. These abnormal proteins were identified by using proteolysis and mass spectrometry in collaboration with J.R. Yates, Department of Cell Biology, confirming the global effects of E461K on newly translated proteins. The abnormal proteins included many larger than 60 kD, too large to be encapsulated in the GroEL-GroES cavity. In vitro, 2 of these larger proteins, MetE (88 kD) and aconitase (92 kD), refolded to native form when GroEL alone was present, suggesting that rapid interaction with the open chaperonin ring can provide a form of folding assistance without encapsulation. Thus, these studies suggest a potentially broader role for GroEL in mediating folding, although we cannot entirely exclude a model in which the aggregation of stringent cis cavity-requiring substrate proteins leads to an avalanche of aggregation involving other newly translated proteins. Direct cross-linking studies between 216 MOLECULAR BIOLOGY 2007 GroEL and substrate proteins in E coli cells in culture are under way to resolve this question. T O P O L O G Y O F S U B S T R AT E P R O T E I N B O U N D T O A N OPEN GroEL RING In other studies, we have investigated the topology of substrate proteins bound to an open ring of GroEL. In collaboration with H. Saibil, Birkbeck College, London, England, we used electron cryomicroscopy image reconstructions of binary complexes of the substrate protein malate dehydrogenase (MDH) in complex with GroEL (Fig. 1). Additional studies of such binary com- THE SCRIPPS RESEARCH INSTITUTE ity (Fig. 1, left panels). Although parts of the substrate protein were observed here in physical association with the apical domains, other parts of the substrate not detected by electron microscopy could populate any location inside the central cavity, for example, down into the equatorial zone, as shown by disulfide crosslinking studies in which cysteine substitutions were “scanned” both inside the central cavity and across the outside surfaces of GroEL (the outer surfaces did not form cross-links with bound substrate). C O N F O R M AT I O N A L C H A N G E S O F S U B S T R AT E P R O T E I N D U R I N G F O L D I N G I N T H E C I S C AV I T Y F i g . 1 . Electron cryomicroscopy reconstructions of binary complexes of the substrate MDH (yellow, green, blue) with the chaperonin GroEL (gray). End views, top panels; tilted views, middle panels; cutaway side views, bottom panels. plexes, as well as type II chaperonins, are under way in collaboration with B. Carragher and C. Potter, Department of Cell Biology. In the studies of MDH-GroEL complexes, we found that the MDH subunit was bound to the apical domains of the GroEL ring in various topologies. In one topology, 3 consecutive apical domains were contacted by substrate localized at the rim of the central cavity (Fig. 1, right panels). In another topology, 3 consecutive domains were occupied by substrate localized across the middle part of the apical domains, where 2 α-helices (H and I) were present whose hydrophobic side chains have been implicated in binding by mutational studies (Fig. 1, center panels). In a third topology, the substrate was more deeply situated, interacting with consecutive apical domains via a surface that contains both helix I and an underlying hydrophobic extended segment, with the substrate pointing even more deeply into the central cav- In other studies in vitro, we have probed the conformations of substrate proteins both during binding by GroEL and during folding by GroEL-GroES. For example, we have used formation of disulfide bonds in the secretory protein trypsinogen, which behaves as a stringent (GroEL-GroES dependent) substrate during refolding in vitro after dilution from urea and reductant. In these studies, trypsinogen molecules were analyzed at various times by using proteolysis and mass spectrometry. We found that only native long-range disulfides are formed, pinning the 2 β-barrels together at the top and bottom of the active-site cleft, during folding of trypsinogen inside GroEL-GroES; that is, only a nativelike global topology is selected. However, shorter range nonnative bonds occur early in the reaction, formed within the 2 β-barrel domains, but these are “corrected” to native later during folding, suggesting a conformational annealing activity inside the cis cavity that occurs in the absence of any further ATP binding or hydrolysis. As another means of observing substrate proteins, in collaboration with K. Wüthrich, Department of Molecular Biology, we have been using nuclear magnetic resonance to study another substrate protein: rhodanese. We are examining behavior of this substrate bound to GroEL. We have also been examining nuclear magnetic resonance spectra of the GroEL chaperonin itself. PUBLICATIONS Chapman, E., Farr, G.W., Usaite, R., Furtak, K., Fenton, W.A., Chaudhuri, T.K., Hondorp, E.R., Matthews, R.G., Wolf, S.G., Yates, J.R., Pypaert, M., Horwich, A.L. Global aggregation of newly translated proteins in an Escherichia coli strain deficient of the chaperonin GroEL. Proc. Natl. Acad. Sci. U. S. A. 103:15800, 2006. Elad, N., Farr, G.W., Clare, D.K., Orlova, E.V., Horwich, A.L., Saibil, H.R. Topologies of a substrate protein bound to the chaperonin GroEL. Mol. Cell 26:415, 2007. Farr, G.W., Fenton, W.A., Horwich, A.L. Perturbed ATPase activity and not “close confinement” of substrate in the cis cavity affects rates of folding by tail-multiplied GroEL. Proc. Natl. Acad. Sci. U. S. A. 104:5342, 2007. Park, E.S., Fenton, W.A., Horwich, A.L. Disulfide formation as probe of topology during folding in GroEL-GroES reveals correct formation of long-range bonds and editing of incorrect short-ranges ones. Proc. Natl. Acad. Sci. U. S. A. 104:2145, 2007. MOLECULAR BIOLOGY 2007 Chemical Regulation of Gene Expression J.M. Gottesfeld, R. Burnett, C.J. Chou, D. Herman, S. Ku, S. Lefebvre, E. Soragni, S. Tsai,* M. Farkas,* P.B. Dervan,* S.L. Perlman,** G. Coppola,** D. Geschwind,** M. Rai,*** M. Pandolfo*** * California Institute of Technology, Pasadena, California ** University of California, Los Angeles, California *** Universite Libre de Bruxelles-Hospital Erasme, Brussels, Belgium he ability to control gene expression at will has been a longstanding goal in molecular biology and human medicine. We focus on 2 classes of small molecules that can alter gene expression in human cells: (1) pyrrole-imidazole polyamides, molecules that can be programmed by chemical synthesis to recognize a wide range of DNA sequences, and (2) histone deacetylase inhibitors, compounds that alter the postsynthetic modification states of major chromosomal proteins and thereby activate gene expression. Our recent efforts to develop polyamides as therapeutic agents for human cancer and novel histone deacetylase inhibitors that offer promise in the treatment of neurodegenerative diseases are summarized in the following sections. T B L O C K I N G C A N C E R C E L L P R O L I F E R AT I O N W I T H A P O LYA M I D E - C H L O R A M B U C I L C O N J U G AT E DNA alkylators, common agents used to treat cancer in humans, act by damaging DNA. A DNA alkylator used to treat a variety of lymphatic cancers is the nitrogen mustard chlorambucil. Because chlorambucil alkylates DNA at all available guanine residues in cellular DNA, coupling of chlorambucil to a more sequence-selective molecule, such as a polyamide, decreases the number of sites in the genome that are damaged and may decrease unwanted side effects while retaining the ability of the compound to kill cancer cells. We recently found that a specific polyamide-chlorambucil conjugate called 1R-Chl alters the morphology and growth characteristics of multiple cancer cell lines in culture and causes these cells to arrest in the G 2 /M stage of the cell cycle. The compound blocks proliferation of various cancer cell lines in immunocompromised mice, including cells derived from colon, prostate, and lung cancers and from chronic myelogenous leukemia, and no apparent toxic effects occur at doses required for a therapeutic effect. Using microarray analysis, we found that the gene target of 1R-Chl is histone H4c, a member of the gene THE SCRIPPS RESEARCH INSTITUTE 217 family that encodes a critical component of cellular chromatin and a gene that is highly expressed in a wide range of cancer cells. Reduction in histone H4 protein by polyamide treatment was confirmed in cells treated with 1R-Chl, which caused chromatin decondensation. Small interfering RNAs to H4c mRNA also have this effect, providing target validation for the effects of 1R-Chl. Another polyamide-chlorambucil conjugate that targets the H4c gene elicits the same cellular response; molecules of similar composition that do not target this gene are ineffective. Using gene chip expression analysis, we found that only 156 genes are affected by 1RChl in K562 chronic myelogenous leukemia cells and identified 2 genes of interest, which encode histones H4c and H4k/j in this cancer cell type. Pathway analysis suggests that DNA damage is the eventual outcome of 1R-Chl treatment, findings in agreement with a 2-hit model for the action of 1R-Chl. 1R-Chl–induced DNA damage is cell-type specific, indicating that downregulation of H4c and H4/j gene transcription is a prerequisite for eventual DNA damage and G 2 /M arrest of K562 and other cancer cell lines. These observations also explain why, unlike other conventional DNA alkylators, 1R-Chl has few or no toxic effects in cells in culture or in whole animals. The therapeutic potential of these molecules is being pursued through additional animal and cellular models for human cancer and proliferative diseases. P O LYA M I D E S A S A C T I VAT O R S O F G E N E E X P R E S S I O N The neurodegenerative disease Friedreich’s ataxia is caused by gene silencing through expansion of GAA•TTC triplet repeats in the first intron of a nuclear gene that encodes the essential mitochondrial protein frataxin. Normal frataxin alleles have 6–34 repeats, whereas alleles from patients with Friedreich’s ataxia have 66–1700 repeats. Longer repeats cause a more profound deficiency in the protein frataxin and are associated with earlier onset and increased severity of the disease. Two models have been proposed to account for gene silencing by expanded GAA•TTC repeats: unusual DNA structures and repressive heterochromatin. Molecules that reverse formation of unusual DNA structures and/or heterochromatin in the gene for frataxin most likely increase transcription through expanded GAA•TTC repeats, thereby relieving the deficiency in frataxin mRNA and protein in cells from patients with Friedreich’s ataxia. We found that polyamides targeting GAA•TTC repeats partially alleviated repression of 218 MOLECULAR BIOLOGY 2007 the gene frataxin in a cell line derived from white blood cells from a patient with Friedreich’s ataxia. These molecules also increased frataxin protein levels in these cells, and microarray studies showed that a limited number of genes in the human genome were affected by polyamides targeting GAA•TTC repeat DNA. We hypothesize that polyamides might act as a thermodynamic “sink” and lock GAA•TTC repeats into double-stranded B DNA. Such an event would disfavor duplex unpairing, which is necessary for formation of the unusual DNA structures associated with expanded triplet repeats. Alternatively, polyamides may relieve heterochromatin-mediated repression by opening the chromatin domain containing frataxin. To explore this latter hypothesis, we turned to another class of small molecules. H I S T O N E D E A C E T Y L A S E I N H I B I T O R S T H AT R E V E R S E F R ATA X I N S I L E N C I N G We used antibodies to the various modification states of the core histones and chromatin immunoprecipitation methods to examine the chromatin structure of the gene for frataxin in normal cells and in cell lines derived from patients with Friedreich’s ataxia. We found that gene silencing at expanded frataxin alleles was accompanied by hypoacetylation of histones H3 and H4 and methylation of histone H3 at lysine 9, consistent with a heterochromatin-mediated repression mechanism. These findings suggest that histone deacetylase inhibitors, compounds that reverse heterochromatin, might activate frataxin. We identified a commercial histone deacetylase inhibitor, BML-210, that partially reverses silencing in the Friedreich’s ataxia cell line. On the basis of the structure of this compound, we synthesized and assayed a series of derivatives of BML-210 and identified histone deacetylase inhibitors that reverse frataxin silencing in primary lymphocytes from patients with Friedreich’s ataxia. These molecules act directly on the histones associated with frataxin, increasing acetylation at particular lysine residues on histones H3 and H4. Of note, the histone deacetylase inhibitors cross the blood-brain barrier and increase levels of frataxin mRNA in the brain and other organs in a mouse model of the human disease. Genome-wide microarray studies indicated that our compounds partially correct the transcription pattern of genes affected by Friedreich’s ataxia in the brain of these mice and in lymphocytes from patients with the disease to the transcription pattern of healthy animals or individuals. We are investigating the pharmacokinetic and toxicity properties of the compounds. We are also exploring the usefulness THE SCRIPPS RESEARCH INSTITUTE of these compounds in related neurodegenerative and neuromuscular diseases, such as Huntington’s disease, spinal muscular atrophy, and myotonic dystrophy. PUBLICATIONS Tsai, S.M., Farkas, M.E., Chou, C.J., Gottesfeld, J.M., Dervan, P.B. Unanticipated differences between α- and γ-aminobutyric acid-linked hairpin polyamide-alkylator conjugates. Nucleic Acids Res. 35:307, 2007. Nucleic Acid Dynamics D.P. Millar, J. Gill, G. Pljevaljc̆ić, S. Pond, J. Wang, E.J.C. Van der Schans he focus of our research is the assembly and conformational dynamics of nucleic acid–based macromolecular machines and assemblies. We use single-molecule fluorescence methods to investigate a range of systems, including ribozymes, ribonucleoprotein complexes, and DNA polymerases. Our studies reveal the dynamic structural rearrangements that occur during the assembly and function of these complex macromolecular machines. T RIBOZYMES RNA conformation plays a central role in the mechanism of ribozyme catalysis. The hairpin ribozyme is a small nucleolytic ribozyme that serves as a model system for studies of RNA folding and catalysis. The hairpin ribozyme consists of 2 internal loops, 1 of which contains the scissile phosphodiester bond, displayed on 2 arms of a 4-way multihelix junction. To attain catalytic activity, the ribozyme must fold into a compact conformation in which the 2 loops become connected by a network of tertiary hydrogen bonds. We monitor the formation of this docked structure by using fluorescence resonance energy transfer (FRET) and ribozyme constructs labeled with donor and acceptor dyes within the loop-bearing arms. By measuring FRET at the level of single ribozyme molecules, we reveal subpopulations of compact and extended conformers that are not detected in ensemble experiments. Using this approach, we found that the ribozyme populates an intermediate state in which the 2 loops are in proximity but tertiary interactions have yet to form. This quasi-docked state forms rapidly (submillisecond timescale); however, the tertiary contacts between the 2 loops are established through a slow conformational search. The hairpin ribozyme is an ideal system for exploring this fundamental mechanism of the formation of RNA tertiary structure. MOLECULAR BIOLOGY 2007 R I B O N U C L E O P R O T E I N A S S E M B LY The Rev protein from HIV type 1 is a key regulatory protein that controls the transition from early to late patterns of viral gene expression. Rev binds to a highly structured region within the viral mRNA, known as the Rev response element (RRE), where it forms an oligomeric ribonucleoprotein complex. The formation of this complex inhibits splicing and facilitates export of the unspliced viral RNA from the nucleus to the cytoplasm. Because of its critical role in the viral life cycle, the Rev-RRE complex provides a novel target for the development of therapeutic drugs. To dissect the mechanism of assembly of ribonucleoprotein complexes, we use single-molecule fluorescence imaging methods to monitor the progressive formation of oligomeric complexes of Rev on individual RRE molecules immobilized on a solid surface. We also use single-pair FRET to probe changes in the conformation of the RRE during the assembly process. We are using the results of these mechanistic studies to develop novel fluorescence-based methods for high-throughput screening of libraries of chemical compounds. The new screening tools are being used to identify small molecules that block binding of Rev to the RRE or prevent the subsequent Rev-Rev oligomerization. D N A P O LY M E R A S E S DNA polymerases are remarkable for their ability to synthesize DNA at rates approaching several hundred base pairs per second while maintaining an extremely low frequency of errors. To elucidate the origin of polymerase fidelity, we are using single-molecule fluorescence methods to examine the dynamic interactions that occur between a DNA polymerase and its DNA and nucleotide substrates. The FRET method is being used to observe conformational transitions of the enzymeDNA complex that occur during selection and incorporation of an incoming nucleotide substrate. Our results reveal that binding of a correct nucleotide substrate induces a slow conformational change within the polymerase, causing the “fingers” subdomain to close over the DNA primer terminus and incoming nucleotide. Our studies are providing new insights into the dynamic structural changes responsible for nucleotide recognition and selection by DNA polymerases. The advantage of single-molecule observations is that they eliminate the need to synchronize a population of molecules, allowing these dynamic processes to be observed directly. THE SCRIPPS RESEARCH INSTITUTE 219 PUBLICATIONS Bailey, M.F., Van der Schans, E.J.C., Millar, D.P. Dimerization of the Klenow fragment of Escherichia coli DNA polymerase I is linked to its mode of DNA binding. Biochemistry, in press. Grover, R.K., Pond, S.J., Cui, Q., Subramanian, P., Case, D.A., Millar, D.P., Wentworth, P. O-Glycoside orientation is an essential aspect of base J recognition by the kinetoplastid DNA-binding protein JBP1. Angew. Chem. Int. Ed. 46:2839, 2007. Tian, F., Debler, E.W., Millar, D.P., Deniz, A.A., Wilson, I.A., Schultz, P.G. The effects of antibodies on stilbene excited-state energetics. Angew. Chem. Int. Ed. 45:7763, 2006. Single-Molecule Biophysics: Folding, Assembly, and Function A.A. Deniz, S.Y. Berezhna, A.C.M. Ferreon, Y. Gambin, E. Lemke, S. Mukhopadhyay e develop and use state-of-the-art single-molecule fluorescence methods and high-sensitivity ensemble methods to address key biological questions by probing multiple structures or reaction pathways during the folding, assembly, and function of biomolecules. These methods offer key advantages over traditional measurements, allowing us to directly observe the behavior of individual subpopulations in mixtures of molecules and to measure the kinetics of structural transitions of stochastic processes under equilibrium conditions. A major goal is to apply single-molecule methods to studies of protein folding and aggregation. For example, partially folded or misfolded protein structures are thought to play important cellular roles, and these states can be studied by using single-molecule methods. In this context, in collaboration with S.L. Lindquist, Whitehead Institute for Biomedical Research, Cambridge, Massachusetts, we are examining the interplay between folding and aggregation of Sup35, a yeast translational termination factor whose activity is modulated by conversion to a prion form. By using singlemolecule fluorescence resonance energy transfer (FRET) and subnanomolar concentrations of Sup35, we minimize potential artifacts due to protein aggregation. Using single-molecule dual-color fluorescence coincidence analysis, we have directly tested that the protein is monomeric at these concentrations. Our single-molecule FRET denaturation analysis of Sup35 indicated that this protein does not have significant stable structure under native conditions. Additionally, an analysis of signal fluctuations from a small number of Sup35 molecules (fluorescence correlation spectroscopy), W 220 MOLECULAR BIOLOGY 2007 revealed rapid structural fluctuations on the nanosecond timescale. Overall, the results show that native Sup35 is intrinsically disordered and populates a compact and rapidly fluctuating ensemble of structures, behavior that may be key in its aggregation and biological function. We are also studying the structural properties and aggregation of α-synuclein, a protein implicated in the pathogenesis of Parkinson’s disease and other neurodegenerative diseases. As a prelude to single-molecule studies, we have carried out a detailed thermodynamic study of the conformational properties of this protein in isolation, as well as in the presence of the detergent sodium dodecyl sulfate. Interestingly, sodium dodecyl sulfate, which is often used to denature proteins, can induce folding of α-synuclein. Our results indicate that depending on its environment, α-synuclein can occupy several different structural states. This structural plasticity could be important during the function of this protein in the cell. We are now using single-molecule methods to study in more detail both the monomeric structures and the early stages of aggregation in Sup35 and α-synuclein. Important events on the folding landscapes of proteins and other biomolecules occur on millisecond or faster timescales. To facilitate understanding of such fast folding processes, in collaboration with A. Groisman, University of California, San Diego, we are developing microfluidic mixing methods for both single-molecule and ensemble experiments. In particular, microfluidic mixing combined with single-molecule fluorescence detection will provide more detailed insights into folding landscapes of proteins and other biological molecules. To better study folding processes and the assembly and function of larger and multicomponent biological complexes, we are developing multicolor single-molecule FRET methods. We are using these novel methods to study the assembly mechanisms of fragments of the bacterial ribosome, in collaboration with J.R. Williamson, Department of Molecular Biology. Finally, we are also using multicolor fluorescence imaging to study the pathways of nuclear and cytoplasmic RNA interference. Recently, in collaboration with P.G. Schultz, Department of Chemistry, we probed the relative localizations of short interfering RNA; a key RNA interference protein, Ago2; and P-bodies, which are RNA-processing cytoplasmic bodies that may be involved in RNA interference. Our imaging and biochemical data indicate that P-bodies play only a minor role in RNA interference. We are beginning single-par- THE SCRIPPS RESEARCH INSTITUTE ticle tracking experiments to probe RNA interference mechanisms in greater detail. PUBLICATIONS Deniz, A.A., Mukhopadhyay, S., Lemke, E.L. Single-molecule biophysics: at the interface of biology, physics, and chemistry. J. R. Soc. Interface, in press. Ferreon, A.C.M., Deniz, A.A. α-Synuclein multistate folding thermodynamics: implications for protein misfolding and aggregation. Biochemistry 46:4499, 2007. Mukhopadhyay, S., Deniz, A.A. Fluorescence from diffusing single molecules illuminates biomolecular structure and dynamics. J. Fluoresc., in press. Mukhopadhyay, S., Krishnan, R., Lemke, E.A., Lindquist, S., Deniz, A.A. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. U. S. A. 104:2649, 2007. Tian, F., Debler, E.W., Millar, D.P., Deniz, A.A., Wilson, I.A., Schultz, P.G. The effects of antibodies on stilbene excited-state energetics. Angew. Chem. Int. Ed. 46:7763, 2006. Computer Modeling of Proteins and Nucleic Acids D.A. Case, Y. Bomble, M. Crowley, Q. Cui, F. Dupradeau,* S. Moon, V. Pelmenschikov, D. Shivakumar, T. Steinbrecher, R.C. Walker, V. Wong, W. Zhang * Université Jules Verne, Amiens, France omputer simulations offer an exciting approach to the study of many aspects of biochemical interactions. We focus primarily on molecular dynamics simulations (in which Newton’s equations of motions are solved numerically) to model the solution behavior of biomacromolecules. Recent applications include detailed analyses of electrostatic interactions in short peptides (folded and unfolded), proteins, and oligonucleotides in solution. In addition, molecular dynamics methods are useful in refining solution structures of proteins by using constraints derived from nuclear magnetic resonance (NMR) spectroscopy, and we continue to explore new methods in this area. Our developments are incorporated into the Amber molecular modeling package, designed for large-scale biomolecular simulations, and into other software, including Nucleic Acid Builder, for developing 3-dimensional models of unusual nucleic acid structures; SHIFTS, for analyzing chemical shifts in proteins and nucleic acids; RNAmotif, for finding structural motifs in genomic sequence databases; and DOCK, for placing inhibitors into enzyme active sites. C NMR AND THE STRUCTURE AND DYNAMICS OF PROTEINS AND NUCLEIC ACIDS Our overall goal is to extract the maximum amount of information about biomolecular structure and dynam- MOLECULAR BIOLOGY 2007 ics from NMR experiments. To this end, we are studying the use of direct refinement methods for determining biomolecular structures in solution, going beyond distance constraints to generate closer connections between calculated and observed spectra. We are also using quantum chemistry to study chemical shifts and spin-spin coupling constants. Other types of data, such as chemical shift anisotropies, direct dipolar couplings in partially oriented samples, and analysis of cross-correlated relaxation, are also being used to guide structure refinement. In recent structural studies, we focused on the binding of small ligands to DNA and on models for chemically damaged DNA. For example, Figure 1 shows an F i g . 1 . Structure of a part of DNA with a missing base. Left, Structure expected for an idealized B-form helix. Right, The actual structure, as determined by NMR spectroscopy. abasic site, a site in which the nucleic acid base has been removed. The left side shows what is expected if the regular, B-form helix is not modified. The right side shows the NMR structure, illustrating marked structural modifications that could be recognized by DNA repair enzymes. NUCLEIC ACID MODELING Another project centers on the development of novel computer methods to construct models of “unusual” nucleic acids that go beyond traditional helical motifs. We are using these methods to study circular DNA, small RNA fragments, and nucleosome core particles. We continue to develop efficient computer implementations of continuum solvent methods to allow simplified simulations that do not require a detailed description of the solvent (water) molecules; this approach also provides a useful way to study salt effects. Recent efforts have made second derivatives of these energies available, so that normal mode analyses of nucleic acids with dozens to hundreds of nucleotides can be analyzed and the predictions compared THE SCRIPPS RESEARCH INSTITUTE 221 with those of simpler, elastic continuum models. These efforts provide a new avenue for developing and testing low-resolution models that can be used for large molecular assemblies. A key current application is to nucleosome core particles. D Y N A M I C S A N D E N E R G E T I C S O F N AT I V E A N D N O N N AT I V E S TAT E S O F P R O T E I N S Analysis methods similar to those described for nucleic acids are also being used to estimate electrostatic and thermodynamic properties of proteins. A key feature is the development of computational methods that can be used to model pH and salt dependence of complex conformational transitions such as unfolding events. A second aspect of this work is a detailed interpretation of NMR results for proteins through molecular dynamics simulations and the construction of models for molecular motion and disorder. All of these modeling activities are based on molecular mechanics force fields, which provide estimates of energies as a function of conformation. We continue to work on improvements in force fields; recently, we focused on adding aspects of electronic polarizability, going beyond the usual fixed-charge models, and on methods for handling arbitrary organic molecules that might be considered potential inhibitors in drug discovery efforts. Overall, the new models should provide a better picture of the noncovalent interactions between peptide groups and the groups’ surroundings, leading ultimately to more faithful simulations. PUBLICATIONS Chen, J., Dupradeau, F.-Y., Case, D.A., Turner, C.J., Stubbe, J. NMR structural studies and molecular modeling of duplex DNA containing normal and 4′-oxidized abasic sites. Biochemistry 46:3096, 2007. Cui, Q., Tan, R.-K.Z., Harvey, S.C., Case, D.A. Low-resolution molecular dynamics simulations of the 30S ribosomal subunit. Multiscale Model. Simul. 5:1248, 2006. Grover, R.K., Pond, S.J., Cui, Q., Subramaniam, P., Case, D.A., Millar, D.P., Wentworth, P., Jr. O-Glycoside orientation is an essential aspect of base J recognition by the kinetoplastid DNA-binding protein JBP1. Angew. Chem. Int. Ed. 46:2839, 2007. Mongan, J., Simmerling, C., McCammon, J.A., Case, D.A., Onufriev, A. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 3:156, 2007. Moon, S., Case, D.A. A new model for chemical shifts of amide hydrogens in proteins. J. Biomol. NMR 38:139, 2007. Paesani, F., Zhang, W., Case, D.A., Cheatham, T.E. III, Voth, G.A. An accurate and simple quantum model for liquid water. J. Chem. Phys. 125:184507, 2006. Seabra, G.M., Walker, R.C., Elstner, M., Case, D.A., Roitberg, A.E. Implementation of the SCC-DFTB method for hybrid QM/MM simulations within the Amber molecular dynamics package. J. Phys. Chem. A, in press. Tang, S., Case, D.A. Vibrational averaging of chemical shifts anisotropies in model peptides. J. Biomol. NMR, in press. 222 MOLECULAR BIOLOGY 2007 Quantum Chemical Analysis for Redox-Active Metalloenzymes and for Photochemistry L. Noodleman, D.A. Case, W.-G. Han, V. Pelmenschikov, J.A. Fee, L. Hunsicker-Wang,* T. Lovell,** T. Liu,*** M. Ullmann,**** D. Bashford***** * Trinity University, San Antonio, Texas ** AstraZeneca R&D, Mölndal, Sweden *** University of Maryland, College Park, Maryland **** Universität Bayreuth, Bayreuth, Germany ***** St. Jude Children’s Research Hospital, Memphis, Tennessee e are using a combination of modern quantum chemistry (density functional theory, DFT) and classical electrostatics to describe the energetics, reaction pathways, and spectroscopic properties of metalloenzymes. In addition, we are analyzing other systems that have novel catalytic, photochemical, or photophysical properties. Critical biosynthetic and regulatory processes may involve catalytic transformations of fairly small molecules or groups by transition-metal centers. The ironmolybdenum cofactor center (Fig. 1) of nitrogenase catalyzes the multielectron reduction of molecular nitrogen to 2 molecules of ammonia plus molecular hydrogen. We are continuing our work on the catalytic cycle of this enzyme, following up on our earlier research on the structure and oxidation state of the cofactor complex in the “resting enzyme” before multielectron reduction and nitrogen binding. W THE SCRIPPS RESEARCH INSTITUTE On the basis of DFT calculated vs experimental physical properties, including redox potentials, cluster geometries, and Mössbauer isomer shifts, the core cluster has a (MoFe7S 9X) prismane active site, where the central X most likely is nitride and the “resting cluster oxidation state” is Mo(IV)Fe(II)4Fe(III)3. A central carbide is also a possibility, because no clear central ligand hyperfine signal has yet been detected for the paramagnetic resting state. Further exploration of different redox states, including bound alternative substrates and inhibitors studied by using electron nuclear double resonance and Mössbauer spectroscopies and compared with quantum chemical calculations, is under way to illuminate both the active-site structure and the reaction mechanism. Methane monooxygenase catalyzes the oxidation of methane to methanol. This reaction is extremely important in the biosphere because methane is a greenhouse gas. Further, the monooxygenase can catalyze reactions with alternative substrates, a characteristic that has implications for detoxification of organic pollutants. We have been examining various steps in the catalytic cycle, comparing predictions based on DFT findings with those of Mössbauer spectroscopy. We have focused particularly on the critical intermediate Q (Fig. 2), which F i g . 2 . The proposed active-site structure of methane monooxy- genase intermediate Q. F i g . 1 . The hyperfine signal from the interstitial X atom of the nitrogenase FeMoco cofactor is small (Aiso ~0.3 MHZ, X = 14N case studied) for the resting-state enzyme because of the symmetry conditions. On the basis of DFT calculations, we propose 2 principal modes of FeMoco symmetry perturbation via 2-electron–2-proton reduction at the metal-sulfide cluster and ligand binding: electronic (Aiso ~6.8 MHZ) and structural (Aiso ~22.9 MHZ). A bound allylic alcohol, derived from substrate propargyl alcohol plus 2 electrons plus 2 protons is shown. performs the oxygen insertion reaction. The iron complex at the active site of methane monooxygenase resembles the complex in ribonucleotide reductases (see following); the resemblance is particularly close for ribonucleotide reductases in some pathogenic bacteria, including Chlamydia, in which the tyrosine near the iron-oxo dimer complex is replaced by phenylala- MOLECULAR BIOLOGY 2007 nine. The altered mechanism of action of the ribonucleotide reductases in these pathogens is of great interest for exploring feasible drug treatments. Class I ribonucleotide reductases are aerobic enzymes that catalyze the reduction of ribonucleotides to deoxyribonucleotides, providing the required building blocks for DNA replication and repair. These enzymes are targets for anticancer, antiviral, and antibacterial drugs. These ribonucleotide-to-deoxyribonucleotide reactions occur via a long-range radical (or proton-coupled electron transfer) propagation mechanism initiated by a fairly stable tyrosine radical, “the pilot light.” When this pilot light goes out, the tyrosine radical is regenerated by a high-oxidation-state Fe(III)-Fe(IV)-oxo enzyme intermediate called X. Using DFT and electrostatics calculations in combination with analysis of Mössbauer, electron nuclear double resonance, and magnetic circular dichroism spectroscopies to search for a proper structural and electronic model for intermediate X, we have zeroed in a promising structure. Now the pathways to and from intermediate X can be properly examined. In collaboration with K. Hahn, University of North Carolina at Chapel Hill, we have examined the physical and spectroscopic properties of novel solvent-sensitive fluorescent dyes of interest for imaging live cells. These dyes can act as biosensors for changes in conformational state in cell signaling proteins, with potential uses in high-throughput drug screening. With J.A. Fee, Department of Molecular Biology, we are exploring the mechanism of 4-electron reduction of molecular oxygen to 2 molecules of water by cytochrome oxidase (complex IV of mitochondria and the related cytochrome oxidase ba 3 from Thermus thermophilus). The copper-iron-heme complex links molecular oxygen reduction to proton pumping across the mitochondrial membrane. PUBLICATIONS Han, W.-G., Noodleman, L. Structural model studies for the high-valent intermediate Q of methane monooxygenase from broken-symmetry density functional calculations. Inorg. Chim. Acta, in press. THE SCRIPPS RESEARCH INSTITUTE 223 Theoretical and Computational Molecular Biophysics C.L. Brooks III, R. Armen, I. Borelli, D. Bostick, D. Braun, L. Bu, J. Chen, M.F. Crowley, O. Guvench, R. Hills, W. Im,* J. Khandogin, I. Khavrutskii, J. Knight, J. Lee, R. Manige, M. Michino, A. Mitsutake,** H.D. Nguyen, S. Patel,*** V. Reddy, H.A. Scheraga,**** C. Shepard, I.F. Thorpe, M.C. Tripp, R, Wheeler,***** K. Yoshimoto * Kansas University, Lawrence, Kansas ** Kelo University, Tokyo, Japan *** University of Delaware, Newark, Delaware **** Cornell University, Ithaca, New York ***** University of Oklahoma, Norman, Oklahoma e wish to understand the forces that determine the structure of proteins, peptides, nucleic acids, and complexes containing these molecules and the processes by which these structures are adopted. To address these issues, we use statistical mechanics, molecular simulation, statistical modeling, and quantum chemistry. Creating atomic-level models to simulate biophysical processes (e.g., protein folding or binding of a ligand to a biological receptor) requires (1) development of new polarizable potential energy functions that accurately represent the atomic interactions and (2) use of quantum chemistry to aid in determining the parameters for the models. Characterization of thermodynamic and kinetic properties requires the development and implementation of new theoretical and computational approaches that connect averages over atomistic descriptions to experimentally measurable thermodynamic and kinetic properties. Interpreting experimental results at more microscopic levels is fueled by the development and investigation of theoretical models for the processes of interest. Massive computational resources are needed to realize these objectives, and this need motivates our efforts to achieve the efficient use of new computer architectures, including large supercomputers, Linux Beowulf clusters, computational grids, and Internet-based volunteer supercomputers. Each of the objectives and techniques mentioned represents an ongoing area of development in our research program in computational biophysics. The following are highlights of a few specific projects. W AMMONIUM TRANSPORT Uncovering the means by which cells transport ions and molecules across the lipid membrane via 224 MOLECULAR BIOLOGY 2007 channels and transporters will provide a deeper understanding of a range of diseases caused by the malfunction of these channels. The selective transport of ammonium (NH4+) across biological membranes is a homeostatic necessity in prokaryotic and eukaryotic cells. In humans and other animals, ammonium is transported by members of the Rhesus (Rh) family of proteins. The family consists of 2 types of protein: erythroid and nonerythroid. Erythroid Rh proteins are expressed on the surface of erythrocytes, where they perform immunogenic and structural roles. Nonerythroid Rh proteins are expressed in kidneys, liver, and testes, where they aid in disposal of ammonium and regulation of pH. Biophysical and structural characterization of a bacterial homolog of a human (kidney) ammonium channel, AmtB from Escherichia coli, has revealed that selective “sensing” of ammonium is achieved by forcing the deprotonation of ammonium and allowing only uncharged ammonia (NH3) to traverse the channel via a narrow hydrophobic lumen. The static x-ray structure alone, however, does not clearly indicate where and how along the pathway toward the cytoplasm ammonium becomes deprotonated on the periplasmic end of the channel and then reprotonated on the cytoplasmic end. Using detailed molecular dynamics sampling techniques, we investigated and clarified the protonation control mechanism of AmtB. Our calculations reveal that the “equivalence points” for deprotonation (and reprotonation) of ammonium occur at dehydrative phenylalanine landmarks along the transport pathway (Fig. 1). At these landmarks, ammonium is able to form only 3 (or fewer) hydrogen bonds with the protein or water molecules and has complete access to either periplasmic or cytoplasmic aqueous solution. According to our simulation studies, AmtB indirectly controls ammonium (de)protonation by directly controlling its hydration, effectively exploiting the propensity of ammonium to (de)protonate when only 3 hydrogen bonds are available. Ultimately, the only proton acceptor available at the (de)protonation regions (green bars in Fig. 1) is water. A S S E M B LY O F V I R U S PA R T I C L E S The controlled assembly of viruslike particles is a marked biomedical interest in the bionanotechnology of vaccine design, gene therapy, and medical imaging. For example, viruslike particles of human papillomavirus are used in the vaccine for cervical cancer. Applications of viruslike particles require an understanding of the principles that govern the spontaneous self-assembly of viral capsids. THE SCRIPPS RESEARCH INSTITUTE F i g . 1 . Transport of ammonium through the ammonium channel. In the AmtB ammonium channel monomer, ammonium (Am1 and Am5) and ammonia binding sites (Am2–Am4, the hydrophobic lumen) derived from diffraction (red spheres) and molecular dynamics umbrella sampling analysis (blue spheres) are shown (top, left). Ammonium (de)protonation regions (green horizontal bars) coincide with dehydrative phenylalanine residues. Ammonium equivalence points (top, right) along the transport pathway were determined by using free energy calculations for ammonium (dashed line) and ammonia (solid line) transport as a function of the respective positions of the 2 components along the transport axis. The equivalence points for deprotonation occur at the regions of intersection (green bars) of these free energy curves. Close-up (bottom, right) shows bound ammonia at each of the 3 binding positions within the hydrophobic lumen. Spherical (icosahedral) viral capsids typically are composed of multiple copies (e.g., 60, 180, 240) of a single protein capsid and have a geometric shape (Fig. 2). Using coarse-grained models for viral capsid proteins and specialized molecular dynamics techniques, we are exploring the nature of viral capsid assembly as a function of temperature and protein concentration. We found that the assembly of T = 1 icosahedral capsids occurs with high fidelity over only a small range of temperatures and protein concentrations (Fig. 2, top). Outside this range, particularly at low temperatures or high protein concentrations, large enclosed “monster particles” are produced. The dynamics of capsid assembly under optimal conditions is a nucleated process, with few assembly intermediates of any great size. The assembly of T = 3 capsids proceeds via a more complex mechanism. Three protein species, corresponding to the capsid protein in quasi-equivalent environments in the assembled protein, spontaneously assemble via a rich-phase diagram (Fig. 2, bottom). Under nearoptimal conditions for growth of T = 3 capsids, not only icosahedral capsids but also oblate, angular, twisted and tubular isometric forms occur. The presence and MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 225 Chen, J., Brooks, C.L. III. Critical importance of length-scale dependence in implicit modeling of hydrophobic interactions. J. Am. Chem. Soc. 129:2444, 2007. Hills, R.D., Jr., Brooks, C.L. III. Hydrophobic cooperativity as a mechanism for amyloid nucleation. J. Mol. Biol. 368:894, 2007. Khandogin, J., Brooks, C.L. III. Molecular simulations of pH-mediated biological processes. Annu. Rep. Comput. Chem., in press. Khandogin, J., Brooks, C.L. III. Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry 45:9363, 2006. Khandogin, J., Chen, J., Brooks, C.L. III. Exploring atomistic details of pH-dependent peptide folding. Proc. Natl. Acad. Sci. U. S. A. 103:18546, 2006. Khandogin, J., Raleigh, D.P., Brooks, C.L. III. Folding intermediate in the villin headpiece domain arises from disruption of a N-terminal hydrogen-bonded network. J. Am. Chem. Soc. 129:3056, 2007. Khavrutskii, I.V., Arora, K., Brooks, C.L. III. Harmonic Fourier beads method for studying rare events on rugged energy surfaces. J. Chem. Phys. 125:174108, 2006. Khavrutskii, I.V., Price, D.J., Lee, J., Brooks, C.L. III. Conformational change of the methionine 20 loop of Escherichia coli dihydrofolate reductase modulates pKa of the bound dihydrofolate. Protein Sci., in press. F i g . 2 . Viral capsid assembly. The assembled capsids of the sim- plest (T = 1) and next most complicated (T = 3) icosahedral viruses are highly organized spherical particles composed of geometrically regular copies of identical capsid proteins (top, left). For T = 1 particles, under optimal conditions for capsid growth, assembly occurs via a kinetic mechanism that involves the stepwise addition of small protein assemblies (i.e., monomers, dimers, and trimers), leading to complete capsids. Under conditions of lower temperature, assembly is nucleated too rapidly, and partial growth leads to the combining of many partial capsids to form monster particles. The assembly of T = 3 capsids under near-optimal conditions yields a range of closed capsid forms that are determined by the number of 5- to 6-fold symmetry dislocations that occur (top, right). Additionally, at lower temperatures, the assembly process yields quite open, spirallike monster particles that strongly resemble species seen in electron microscope imaging of viral assembly products for T = 3 and higher T-number capsids (e.g., human papillomavirus). abundance of these nonicosahedral closed structures are controlled by the proclivity for 5- to 6-fold symmetric dislocations. Thus, by controlling the conformational equilibrium of the capsid proteins, we can shift the nature and distribution of the assembled particles. Nguyen, H.D., Reddy, V.S., Brooks, C.L. III. Deciphering the kinetic mechanism of spontaneous self-assembly of icosahedral capsids. Nano Lett. 7:338, 2007. Shepherd, C.M., Borelli, I.A., Lander, G., Natarajan, P., Siddavanahalli, V., Bajaj, C., Johnson, J.E., Brooks, C.L. III, Reddy, V.S. VIPERdb: a relational database for structural virology. Nucleic Acids Res. 34(Database Issue):D386, 2006. Sherman, M.B., Guenther, R.H., Tama, F., Sit, T.L., Brooks, C.L. III, Mikhailov, A.M., Orlova, E.V., Baker, T.S., Lommel, S.A. Removal of divalent cations induces structural transitions in red clover necrotic mosaic virus, revealing a potential mechanism for RNA release. J. Virol. 80:10395, 2006. Tama, F., Ren, G., Brooks, C.L. III, Mitra, A.K. Model of the toxic complex of anthrax: responsive conformational changes in both the lethal factor and the protective antigen heptamer. Protein Sci. 15:2190, 2006. Thorpe, I.F., Brooks, C.L. III. Molecular evolution of affinity and flexibility in the immune system. Proc. Natl. Acad. Sci. U. S. A., in press. Zimmermann, J., Oakman, E.L., Thorpe, I.F., Shi, X., Abbyad, P., Brooks, C.L. III, Boxer, S.G., Romesberg, F.E. Antibody evolution constrains conformational heterogeneity by tailoring protein dynamics. Proc. Natl. Acad. Sci., U.S.A. 103:13722, 2006. Computation and Visualization in Structural Biology A.J. Olson, D.S. Goodsell, M.F. Sanner, S. Dallakyan, PUBLICATIONS Bostick, D.L., Brooks, C.L. III. Deprotonation by dehydration: the origin of ammonium sensing in the AmtB channel. PLoS Comput. Biol. 3:e22, 2007. Bostick, D.L., Brooks, C.L. III. On the equivalence point for ammonium (de)protonation during its transport through the AmtB channel. Biophys. J., in press. Bostick, D.L., Brooks, C.L. III. Selectivity in K+ channels is due to topological control of the permeant ion’s coordinated state. Proc. Natl. Acad. Sci. U. S. A., in press. Bu, L., Im, W., Brooks, C.L. III. Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys. J. 92:854, 2007. Chen, J., Brooks, C.L. III. Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins 67:922, 2007. A. Gillet, R. Harris, Y. Hu,* R. Huey, J. Huntoon, S. Karnati, W. Lindstrom, G.M. Morris, A. Omelchenko, M. Pique, B. Norledge, R. Rosenstein, M. Utsintong, G. Vareille, Q. Zhang, Y. Zhao * Tanabe Research Laboratories U.S.A., Inc., San Diego, California n the Molecular Graphics Laboratory, we develop novel computational methods to analyze, understand, and communicate the structure and interactions of complex biomolecular systems. In this past year, we developed a new method for docking mole- I 226 MOLECULAR BIOLOGY 2007 cules to flexible targets, showing its effectiveness with an important target for AIDS therapy. Using our distributed computing resource FightAIDS@Home, we also performed a massive virtual screen of potential drug compounds that might affect this target. Within our component-based visualization environment, we continue to develop 3-dimensional molecular models as a tangible human-computer interface in educational and research settings and methods for predicting biomolecular interactions, analyzing biomolecular structure and function, and presenting the biomolecular world in education and outreach. THE SCRIPPS RESEARCH INSTITUTE binding energies of a panel of 1800 ligands against 71 wild-type and mutant proteases (Fig. 1). This profile is PROTEIN FLEXIBILITY IN DOCKING Computational docking is an indispensable tool in the development of new drugs. However, protein motion and induced fit are major challenges in many medically relevant systems, such as the flexible HIV protease that is a major target for current AIDS therapy. To address this challenge, we have developed a hierarchical and multiresolution representation of the flexibility of biological macromolecules that can be used in computational simulations. This treelike structure enables the computationally tractable encoding of a small subset of a protein’s conformational subspace. We have developed a docking method, FLIPDock, in which Flexibility Tree is used with our AutoDock empirical free energy force field. With FLIPDock, we have reproduced a crossdocking experiment carried out earlier with AutoDock in which 20 inhibitors of HIV protease I were docked systematically into the 20 conformations of the receptor. We showed that by adding receptor flexibility, we could increase the rate of successful cross docking from 72% to 98%. I D E N T I F I C AT I O N O F S PA N N I N G M U TA N T S AutoDock was the first docking code to be used in a public, Internet-based, distributed computing project, and we have continued to use this massive computing resource to perform experiments that are beyond the reach of traditional computing. We are currently using the World Community Grid, a large Internet-distributed computing project sponsored by IBM, to support FightAIDS@Home on more than 500,000 clients. Personal computers are used by the program when the computers are not in use by their owners, providing an enormous, and largely untapped, computational resource. We recently completed an analysis of results from FightAIDS@Home; our goal was to identify a subset of mutant protease structures that span the ligand-binding diversity of the entire set. We analyzed the profile of F i g . 1 . Top, Profile of binding energies from a large virtual screen against a large collection of protease structures. Bottom, Representative structures highlighted in a Sammon mapping. like a fingerprint: proteases with similar characteristics have similar profiles, but differences in the profiles highlight significant structural differences between different proteases. For instance, some proteases may have strong binding to large compounds, and others may prefer smaller compounds. Principal component analysis was then used to simplify the data set and to identify “spanning” proteases that represent the unique features different subsets of proteases. We identified 1 wild-type protease as the structure that best characterizes the central tendency of the entire protease set. We also identified 7 additional structures, including 1 wild-type structure, 2 HIV type 2 protease structures, and 4 single and multiple mutants, as structures that represent distinct subclasses and thus would be our best choice for structures to use in future efforts to design anti-HIV drugs. TA N G I B L E I N T E R FA C E S I N S T R U C T U R A L B I O L O G Y We have continued to develop autofabricated physical models (“solid printing”) of biological molecules MOLECULAR BIOLOGY 2007 and their components and assemblies; our goal is to use them in both research and education. We integrated computer graphics and computation with these physical models by using augmented reality to create custom interfaces to facilitate exploration and computation of molecular interactions. We have begun to use a selfassisted protein-folding model to teach elements of protein structure and assembly to our graduate students. We are continuing to develop the software that will enable the control of interactive computations through manipulation of the tangible models. The responses to the models have been positive, and the research community is beginning to see how a solid 3-dimensional model can provide tangible, multimodal feedback that mouse, keyboard, and image behind a computer screen cannot. Building on the success of our flexible, articulated models of protein structure, we have begun work on larger models that illustrate the dynamic characteristics of biomolecules. We recently completed the design and fabrication of a model of clathrin (Fig. 2). The model THE SCRIPPS RESEARCH INSTITUTE 227 comprehensive thermodynamic model that allows incorporation of intramolecular energies into the predicted free energy of binding. It also incorporates a chargebased method for evaluating desolvation that is designed to use a typical set of atom types. The method has been calibrated by using a set of 188 diverse protein-ligand complexes of known structure and binding energy and has been tested by using a set of 100 complexes of ligands with retroviral proteases. AutoDock4 also allows the explicit modeling of flexibility in selected side chains, a characteristic that will greatly expand the systems that can be studied. AutoDockTools, the companion graphical user interface for AutoDock, has also been expanded and improved for use with AutoDock4. In particular, AutoDockTools now allows easy definition of flexible side chains in a macromolecule. We have also developed new tools for the analysis of docking results, including an advanced tool for visualizing protein-ligand interactions, and extensive tools for setting up and running large virtual screens. We are currently developing tools to analyze the results of virtual screens to create “probability maps” that indicate the binding characteristics of a large panel of small molecules. C O M P O N E N T - B A S E D V I S U A L I Z AT I O N E N V I R O N M E N T S F i g . 2 . A flexible physical model of clathrin. is composed of articulated triskelion models that can be assembled to form an entire clathrin coat. Magnets are used to model the major point of interaction during the assembly. A D VA N C E S I N C O M P U TAT I O N A L D O C K I N G According to recent reports, the computer program AutoDock is the world’s most widely cited docking method, and we have continued to make it a central tool for predicting biomolecular interaction. AutoDock4 was released in early 2007 and is available through a convenient open source license. AutoDock4 includes a new semiempirical free energy force field based on a To facilitate the integration and interoperation of computational models and techniques from a wide variety of scientific disciplines, we continue to expand our component-based software environment. The environment is centered on Python, a high-level, object-oriented, interpretive programming language. This approach allows the compartmentalization and reuse of software components. Python provides a powerful “glue” for assembling computational components and, at the same time, a flexible language for rapid prototyping and interactive scripting of new applications. We released version 1.4.4 of our software components in January 2007. This release contains substantial enhancements, including a completely rewritten interface to APBS, a software package for the numerical solution of the Poisson-Boltzmann equation, making it easy to produce high-quality pictures of electrostatic potentials on molecular surfaces. A new control panel provides a high-level interface for rapidly displaying molecular models in a variety of representations. This new release is also distributed with installer programs for computers running the Windows, Macintosh OS X, and Linux operating systems. This release of the MGLTools software suite also provides a new update mechanism, 228 MOLECULAR BIOLOGY 2007 allowing users to update their software interactively over the Internet and granting them access to “bug” fixes and enhancements on a nightly basis. This update mechanism is safe, because the user retains the ability to “roll back” and restore a previously installed version of the software. IDENTIFYING OPTIMAL BINDING SITES We have develop a method, termed AutoLigand, that can be used to identify and quantify optimal binding sites, yielding a chemically detailed prediction of the shape of a ligand within a predicted binding site. Using a grid-based description of the binding interaction, AutoLigand identifies a contiguous “envelope” of maximal affinity for the macromolecular structure (Fig. 3). F i g . 3 . Several low-energy AutoLigand envelopes are found with HIV protease, including the large envelope in the active site at the center and 2 exosites on either side. The envelopes are colored according to the optimal atom type at each point: gray for carbon, red for oxygen, and blue for nitrogen. The envelop is a contiguous region in space filled with points that represent potential atom centers for atoms in a ligand. In brief, with the AutoLigand method, an affinity-potential grid around the macromolecular structure is calculated and then a flood-fill and site optimization process is used to identify the contiguous envelope with the best summed interaction energy. We have shown that the method is effective for identifying binding sites in proteins and for optimizing the binding of drugs to protein targets. PROTEIN-PROTEIN DOCKING WITH SURFDOCK Protein-protein docking remains a major challenge in predicting the structure of protein complexes. In our protein-protein docking method, SurfDock, we recently developed a blurred surface and a scoring term that effectively conserve the shape complementarity of protein-protein complexes under small conformational changes. We are currently developing a novel electrostatic scoring term that uses the discrete charges on the blurred surfaces. A coarse function and discrete charges are generated to reproduce the electrostatic energies by solving the nonlinear Poisson-Boltzmann THE SCRIPPS RESEARCH INSTITUTE equation by using the APBS software program. The coarse function mimics the formula of either the Coulombic or the Debye-Hückel potential. We are testing different combinations of the coarse function and discrete charges to determine the optimal method for calculating electrostatic interaction energies for a set of known protein-protein complexes. C O M P U TAT I O N A L M O D E L I N G O F E X T R A C E L L U L A R INTERACTIONS OF TISSUE FACTOR In collaboration with W. Ruf, Department of Immunology, we have continued to study protein interactions in the blood coagulation pathway. Most recently, we focused on protease-activated receptors (PARs), G protein–coupled receptors that convert an extracellular proteolytic cleavage event into a transmembrane signal. We have built a homology structure of PAR-2 as the basis for studying PAR-2 activation mechanisms, and we have docked a series of peptides and chemical compounds to the model to derive its activation mechanisms. The docking shows that the 2 PAR-2 activating peptides have similar dominating binding modes and that both bind to the same side of the PAR-2 extracellular domain with a similar interaction between arginine and glutamic acid. The docking results agree with the experimental findings and have led to ideas for several PAR-2 mutations to test hypotheses about ligand binding. Dr. Ruf and his group have also shown that disulfide isomerization by protein disulfide isomerase (PDI) switches the complex composed of tissue factor and coagulation factor VIIa (TF-VIIa) from coagulation to cell signaling. To understand how the isomerase interacts with the complex, we built the homology model of human PDI and manually docked 3-dimensional physical models of PDI with the TF-VIIa complex. PDI is a large, flexible molecule, so computer docking is extremely difficult. However, 3-dimensional physical models allowed effective exploration of possible binding modes. The physical model of PDI has flexible joints between the N- and C-terminal tails and the central domains, and the model of the TF-VIIa complex has flexible joints between the EGF-1 and Gla domains of VIIa and the extracellular C-terminal strand of TF. With these highly articulated physical models, we found a reasonable binding mode that can use both catalytic sites of PDI and both forms of an important disulfide. On the basis of the binding mode, we suggested 6 mutations in TF that will affect PDI binding but not binding of coagulation factor X. MOLECULAR BIOLOGY V I S U A L M E T H O D S F R O M AT O M S T O C E L L S Understanding structural molecular biology is essential to foster progress and critical decision making among students, policy makers, and the general public. In the past year, we continued our long-standing commitment to science education and outreach with a combination of presentations, popular and professional illustrations and animations, 3-dimensional tangible models, and a presence on the World Wide Web. In these projects, we use the diverse visualization tools developed in the Molecular Graphics Laboratory to disseminate results that range from atomic structure to cellular function. We also continued several regular features that informally present molecular structure and function. The “Molecule of the Month” at the Protein Data Bank entered its eighth year of providing an accessible introduction to the central database of biomolecular structure. Each month, a new molecule is presented with a description of the molecule’s structure, function, and relevance to health and welfare (Fig. 4). Visitors are 2007 THE SCRIPPS RESEARCH INSTITUTE 229 of interest to clinical oncologists and provide a source of continuing education for physicians; “Recognition in Action,” a new series at the Journal of Molecular Recognition; and work with the Nanoscale Informal Science Network supported by the National Science Foundation to develop new materials for presenting the science of nanotechnology. PUBLICATIONS Chang, M., Lindstrom, W., Olson, A.J., Belew, R.K. Analysis of wild-type and mutant structures via in silico docking against diverse ligand libraries. J. Chem. Info. Model., in press. Eubanks, L.M., Rogers, C.J., Beuscher, A.E., Koob, G.F., Olson, A.J., Dickerson, T.J., Janda, K.D. A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol. Pharm. 3:773, 2006. Goodsell, D.S. The molecular perspective: alcohol. Oncologist 11:1045, 2006. Goodsell, D.S. The molecular perspective: estrogen sulfotransferase. Oncologist 11:418, 2006. Goodsell, D.S. The molecular perspective: tissue factor. Oncologist 11:849, 2006. Goodsell, D.S. Seeing the nanoscale. Nanotoday 1:44, August 2006. Goodsell, D.S. Toll-like receptors. J. Mol. Recognit. 19:387, 2006. Herman, T., Morris, J., Colton, S., Batiza, A., Patrick, M., Franzen, M., Goodsell, D.S. Tactile teaching: exploring protein structure/function using physical models. Biochem. Mol. Biol. Educ. 34:247, 2006. Huey, R., Morris, G.M., Olson, A.J., Goodsell, D.S. A semi-empirical free energy force field with charge-based desolvation. J. Comput. Chem. 28:1145, 2007. Jiang, Z., Georgel, P., Li, C., Choe, J., Crozat, K., Rutschmann, S., Du, X., Bigby, T., Mudd, S., Sovath, S., Wilson, I.A., Olson, A.J., Beutler, B. Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 103:10961, 2006. Morris, G.M., Huey, R., Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinformatics, in press. Rogers, J.P., Beuscher, A.E., Flajolet, M., McAvoy, T., Nairn, A.C., Olson, A.J., Greengard, P. Discovery of protein phosphatase 2C inhibitors by virtual screening. J. Med. Chem. 49:1658, 2006. Zhao, Y., Stoffler, D., Sanner, M. Hierarchical and multi-resolution representation of protein flexibility. Bioinformatics 22:2768, 2006. Structural Bioinformatics and Computer-Aided Drug Discovery R. Abagyan, G. Bottegoni, W. Bisson,* A. Cheltsov,* K. Hyun,** J. Kovacs, I. Kufareva, G. Nicola, A. Saldanha * Burnham Institute for Medical Research, La Jolla, California F i g . 4 . Luciferase, the protein that creates light in fireflies, was featured in the “Molecule of the Month.” then given suggestions about how to begin their own exploration of the structures in the data bank. Other projects include “The Molecular Perspective,” articles in the journal The Oncologist that present structures ** CrystalGenomics Inc., Seoul, Korea urrently, the Protein Data Bank contains more than 43,000 structures, providing a unique opportunity for computational studies and rational design of therapeutic agents. We continue to focus on developing and applying mathematical and computa- C 230 MOLECULAR BIOLOGY 2007 tional methods of molecular modeling and function prediction. In the past year, using virtual ligand docking and screening, we discovered novel inhibitors/antagonists of several therapeutically relevant protein targets. M AT H E M AT I C A L M E T H O D S Machine learning is widely used in protein annotation and cheminformatics. However, its performance is often hindered by computational bias caused by overrepresentation of particular subfamilies in the training sets. We developed novel measures of prediction quality based on inverse density weights assigned to data points. The advantage of the new approach was demonstrated by deriving a model in which a full set of known signal peptides was used to predict a single signal peptide. Two-dimensional image alignment is a key step in 3-dimensional single-particle reconstruction. Recently, we reported on “fast Bessel matching,” a real-space correlation-based approach for performing the alignment. The problem is reduced to the calculation of a single 3-dimensional fast Fourier transform. C O M P U TAT I O N A L M E T H O D S In our laboratory or in collaboration with researchers at Structural Bioinformatics Group, Madrid, Spain, we developed several automatic methods and tools for predicting protein flexibility, interactions, and large-complex geometries. A method called PIER (Protein IntErface Recognition; http://abagyan.scripps.edu/PIER/) is used to predict interfaces on an isolated protein structure and does not depend on evolutionary information. The accuracy and efficiency of the method were proved on a large and diverse benchmark. Our previous work on protein flexibility was complemented with a Web server, DFprot (http://sbg.cib.csic.es/ Software/DFprot). The server identifies the hinge regions of the uploaded structure by using normal-mode analysis. To fill the gap between atomic resolution models of protein domains and low-resolution density maps of larger molecular assemblies, efficient fitting tools are needed, ones that can handle partial and incomplete data. Our novel method, ADP_EM (http://sbg.cib.csic.es/ Software/ADP_EM), combines a fast rotational search based on spherical harmonics with a simple translational scanning. Use of the method produces accurate fits in times ranging from seconds to a few minutes. A P P L I C AT I O N S Methods we have developed were successfully applied to several proteins and complexes: G protein– coupled receptors, complement system proteins (CD59), gap junction channels, and a lipid-transfer protein. THE SCRIPPS RESEARCH INSTITUTE This work was done in collaboration with researchers from the Mayo Clinic, Scottsdale, Arizona; Medical University of South Carolina in Charleston; Memorial Sloan-Kettering Cancer Center, New York City; University of Minnesota in Austin; and Cornell University, Ithaca, New York. Activation of G protein–coupled receptors is thought to involve an agonist-induced conformational change. We used the software docking program Internal Coordinates Mechanics to build a 3-dimensional model of the agonist-bound cholecystokinin receptor. The model helped us rationalize the experimental data on the interaction between the conserved lysine at position 187 and aspartic acid at position 5; these residues had no influence on agonist binding but were crucial for agonist-stimulated signaling. Using peptide screens, functional assays, and computer modeling/docking studies, we identified a 6-residue sequence of human complement C9, which spans residues 365–371, as the primary recognition domain for the membrane glycoprotein CD59. Our docking experiments confirmed that the C9-binding site on CD59 is located at a hydrophobic pocket, as was putatively indicated by our previous studies. PIER prediction of the interaction propensity of mammalian glycolipid-transfer protein in apo- and ligand-bound forms was confirmed by the observed oligomerization states: the apo form is a monomer, whereas the sphingosine-bound complexes form crystallographic dimers. The increased propensity for protein-membrane interaction that occurs upon ligand binding might underlie a unique mechanism of regulation of lipid transfer between proteins and membranes. The internal coordinate sampling method was extended to incorporate low-resolution electron microscopy data, symmetry, and requirements for a membrane environment. The obtained protocol was used in collaboration with M. Yeager, Department of Cell Biology, to build a model of a large hexameric intermembrane gap junction channel. INHIBITOR DISCOVERY AND DRUG REPURPOSING This past year, we used virtual ligand docking and screening not only to discover inhibitors but also to study the side effects of marketed drugs and to find new indications for the drugs. Using a rational structurebased search, we found that members of the phenothiazine family of antipsychotic agents act as antagonists for the human androgen receptor. In collaboration with groups from the University of Melbourne and Monash MOLECULAR BIOLOGY 2007 University, Victoria, Australia; the Burnham Institute, La Jolla, California; and Ohio University, Athens, Ohio; we rationally “repurposed” one of the agents and arrived at a potent new androgen receptor antagonist without the original antipsychotic activity (Fig. 1). THE SCRIPPS RESEARCH INSTITUTE 231 introduction of an allosteric ligand. This work was done in collaboration with researchers at the University of California, San Diego, and the Howard Hughes Medical Institute, La Jolla, California. PUBLICATIONS Altmann, S.M., Muryshev, A., Fossale, E., Maxwell, M.M., Norflus, F.N., Fox, J., Hersch, S.M., Young, A.B., MacDonald, M.E., Abagyan, R., Kazantsev, A.G. Discovery of bioactive small-molecule inhibitor of poly ADP-ribose polymerase: implications for energy-deficient cells. Chem. Biol. 13:765, 2006. Bisson, W.H., Cheltsov, A.V., Bruey-Sedano, N., Lin, B., Chen, J., Goldberger, N., May, L.T., Christopoulos, A., Dalton, J.T., Sexton, P.M., Zhang, X.-K., Abagyan, R. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc. Natl. Acad. Sci. U. S. A., in press. Budagyan, L., Abagyan, R. Weighted quality estimates in machine learning. Bioinformatics 22:2597, 2006. Dong, M., Ding, X.Q., Thomas, S.E., Gao, F., Lam, P.C., Abagyan, R., Miller, L.J. Role of lysine 187 within the second extracellular loop of the type A cholecystokinin receptor in agonist-induced activation: use of complementary charge-reversal mutagenesis to define a functionally important interdomain interaction. Biochemistry 46:4522, 2007. Garzon, J.I., Kovacs, J., Abagyan, R., Chacon, P. ADP_EM: fast exhaustive multiresolution docking for high-throughput coverage. Bioinformatics 23:427, 2007. Garzon, J.I., Kovacs, J., Abagyan, R., Chacon, P. DFprot: a web tool for predicting local chain deformability. Bioinformatics 23:901, 2007. Huang, Y., Qiao, F., Abagyan, R., Hazard, S., Tomlinson, S. Defining the CD59-C9 binding interaction. J. Biol. Chem. 281:27398, 2006. Kovacs, J., Abagyan, R., Yeager, M. Fast Bessel matching. J. Comput. Theor. Nanosci. 4:84, 2007. Kovacs, J., Baker, K., Altenberg, G., Abagyan, R., Yeager, M. Molecular modeling and mutagenesis of gap junction channels. Prog. Biophys. Mol. Biol., in press. Kufareva I., Budagyan L., Raush E., Totrov M., Abagyan, R. PIER: protein interface recognition for structural proteomics. Proteins 67:400, 2007. F i g . 1 . Model of phenothiazine derivative, D4, docked into human androgen receptor. The native ligand, dihydrotestosterone, is shown in thin purple sticks for comparison. In collaboration with researchers from Harvard Medical School, Boston, we discovered a compound that inhibited the enzymatic activity of poly-(ADP-ribose) polymerase 1 in vitro and prevented ATP loss and cell death in a surrogate model of oxidative stress in vivo. The neuroprotective role of the polymerase inhibitors was confirmed in a model system of Huntington’s disease. In another project, we targeted the enoyl-acyl carrier protein reductase from a malaria parasite. Using structure-based virtual ligand screening, we identified 3 novel compounds with low micromolar activity in enzymatic and cell-based assays. The high affinity of protein-protein interactions makes them a difficult target for competitive small-molecule inhibitors. We devised an assay principle for detecting allosteric modulators of such interactions. In our assay based on this principle, a competitive fluorescent peptide probe is used to assess the integrity of the complex of type Iα cAMP-dependent protein kinase upon Malinina, L., Malakhova, M.L., Kanack, A.T., Lu, M., Abagyan, R., Brown, R.E., Patel, D.J. The liganding of glycolipid transfer protein is controlled by glycolipid acyl structure. PLoS Biol. 4:e362, 2006. Nicola, G., Smith, C.A., Lucumi, E., Kuo, M.R., Kavagyozov, L., Fidock, D.A., Sacchettini, J.C., Abagyan, R.A. Discovery of novel inhibitors targeting enoyl-acyl carrier protein reductase in Plasmodium falciparum by structure-based virtual screening. Biochem. Biophys. Res. Commun., in press. Saldanha, S.A., Kaler, G., Cottam, H.B., Abagyan, R., Taylor, S.S. Assay principle for modulators of protein-protein interactions and its application to non-ATP-competitive ligands targeting protein kinase A. Anal. Chem. 78:8265, 2006. Mass Spectrometry: Metabolomics and Imaging G. Siuzdak, J. Apon, H.P. Benton, E. Go, K. Harris, L. Hoang, E. Kalisiak, G. O’Maille, M. Sonderegger, S. Trauger, W. Uritboonthai, E. Want, W. Webb, W. Wikoff, H. Morita, D. Wong, A. Nordstrom, H.K. Woo, T. Northen, O. Yanes M E TA B O L O M I C S E ndogenous metabolites, ubiquitous in biofluids, tissues, and organisms of every kind, are crucial elements in understanding fundamental bio- 232 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE chemistry, disease diagnosis, and drug toxicity. The inherent advantage of monitoring small molecules rather than proteins is the relative ease of quantitative analysis of the molecules with mass spectrometry. We are implementing novel mass spectrometry and bioinformatics techniques (Fig. 1) to investigate the F i g . 1 . A novel nonlinear approach that incorporates both ana- lytical and bioinformatics technologies for quantitative comparisons of metabolites. profile of small-molecule metabolites. Our purposes are to correlate metabolite activity with protein regulation and to develop metabolite analysis as a diagnostic method. Our ultimate goal is to create analytical and chemical technologies and data analysis approaches to identify and structurally characterize metabolites of physiologic importance. N A N O S T R U C T U R E - I N I T I AT O R M A S S S P E C T R O M E T R Y A N A LY S I S A N D I M A G I N G We are also using nanostructured clatherates to facilitate vaporization and ionization of biomolecules to develop ultra-high-sensitivity approaches in mass spectrometry (Fig. 2). Using this technology, termed nanostructure-initiator mass spectrometry, we can analyze of a wide range of molecules with unprecedented sensitivity, in the yoctomole (10–24 moles) range. The method is also being developed as a platform for biomolecular tissue imaging. PUBLICATIONS Chace, D.H., Barr, J.R., Duncan, M.W., Matern, D., Morris, M.R., Palmer-Toy, D.E., Rockwood, A.L., Siuzdak, G., Urbani, A., Yergey, A.L., Chan, Y.M. Mass Spectrometry in the Clinical Laboratory: General Principles and Guidance. Clinical and Laboratory Standards Institute, Wayne, PA, 2007. CSLI Document C50-P, Vol. 27, No. 3. Cohen, L., Go, E.P., Siuzdak, G. Small-molecule desorption/ionization mass analysis. In: MALDI-MS: A Practical Guide to Instrumentation, Methods and Applications. Hillenkamp, F., Peter-Katalinic, J. (Eds.). Wiley-VCH, New York, 2007, p. 299. Go, E.P., Uritboonthai, W., Apon, J.A., Trauger, S.A., Nordstrom, A., O’Maille, G., Brittain, S., Peters, E.C., Siuzdak, G. Selective metabolite and peptide capture/mass detection using fluorous affinity tags. J. Proteome Res. 6:1492, 2007. Go, E.P., Wikoff, W.R., Shen, Z., O’Maille, G., Morita, H., Conrads, T.P., Nordstrom, A., Trauger, S.A., Uritboonthai, W., Lucas, D.A., Chan, K.C., Veenstra, T.D., Lewicki, H., Oldstone, M.B., Schneemann, A., Siuzdak, G. Mass spectrometry reveals specific and global molecular transformations during viral infection. J. Proteome Res. 5:2405, 2006. F i g . 2 . Nanostructure-initiator mass spectrometry is a new plat- form for mass spectrometry surface analysis and allows for sensitive biomolecule analysis from surfaces that also include array and tissue imaging. Mutch, D.M., O’Maille, G., Wikoff, W.R., Wiedmer, T., Sims, P.J., Siuzdak, G. Mobilization of pro-inflammatory lipids in obese Plscr3-deficient mice. Genome Biol. 8:R38, 2007. Nordstrom, A., He, L., Siuzdak, G. Desorption/ionization on silicon (DIOS). In: Ionization Methods. Gross, M.L., Caprioli, R.M. (Eds.). Elsevier, New York, 2007, p. 676. The Encyclopedia of Mass Spectrometry, Vol. 6. Gross, M.L., Caprioli, R.M. (Eds. in Chief). Pendyala, G., Want, E.J., Webb, W., Siuzdak, G., Fox, H.S. Biomarkers for neuroAIDS: the widening scope of metabolomics. J. Neuroimmune Pharmacol. 2:72, 2007. Shen, Z., Want, E.J., Chen, W., Keating, W., Nussbaumer, W., Moore, R., Gentle, T.M., Siuzdak, G. Sepsis plasma protein profiling with immunodepletion, threedimensional liquid chromatography tandem mass spectrometry, and spectrum counting. J. Proteome Res. 5:3154, 2006. Steenwyk, R.C., Hutzler, J.M., Sams, J., Shen, Z., Siuzdak, G. Atmospheric pressure desorption/ionization on silicon ion trap mass spectrometry applied to the quantitation of midazolam in rat plasma and determination of midazolam 1′-hydroxylation kinetics in human liver microsomes. Rapid Commun. Mass Spectrom. 20:3717, 2006. Want, E.J., Nordstrom, A., Morita, H., Siuzdak, G. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J. Proteome Res. 6:459, 2007. Want, E.J., Smith, C.A., Qin, C., Van Horne, K.C., Siuzdak, G. Phospholipid capture combined with non-linear chromatographic correction for improved metabolite profiling. Metabolomics 2:145, 2006. MOLECULAR BIOLOGY 2007 RNA-Protein Interactions in Germ-Line Development J.R. Williamson, F. Agnelli, A. Beck, C. Beuck, A. Bunner, A. Carmel, S. Edgcomb, E. Johnson, D. Kerkow, E. Kompfner, S. Kwan, E. Menichelli, W. Ridgeway, G. Ring, H. Schultheisz, E. Sperling, B. Szymczyna, J. Wu NA-protein interactions regulate gene expression at many different points, including transcription, mRNA processing, and translation. Translational regulation, whereby gene expression is modulated by controlling the synthesis of protein from the mRNA, is a complex processes involving many different protein factors. Often in eukaryotic mRNAs, translational regulation is mediated through RNA sequences that are immediately downstream of the actual coding region, but the molecular mechanism for this regulation is poorly understood. We are studying a set of RNA-protein complexes that mediate translational regulation of a particular mRNA from the nematode Caenorhabditis elegans. The tra-2 mRNA is a major control point for both somatic sex determination and germ-line development in this well-studied organism (Fig. 1A). Each adult nematode THE SCRIPPS RESEARCH INSTITUTE 233 The regulation of tra-2 expression is controlled by binding of proteins to the downstream noncoding region of the tra-2 mRNA. This region has 2 important protein binding sites: the tra-2 retention element (TRE) and the tra-2 gli element (TGE). Once tra-2 mRNA is transcribed in the nucleus, a regulatory complex is formed by binding of the nuclear retention factor NXF-2 to the TRE (Fig. 2A). This complex is retained in the R F i g . 1 . Germ-line development in the nemotode C elegans. A, Light micrograph of a nematode. Scale bar = 0.1 mm. B, Diagram of nematode anatomy, highlighting the gonads. C, Schematic of germ-line development in the late larval stage when sperm is produced. D, Schematic of germ-line development in the adult animal where eggs are produced. has 2 gonads that account for 75% of the mass of the animal (Fig. 1B). Caenorhabditis elegans is hermaphroditic and self-fertile because of a complex regulation of germ-line development. Late in the larval stage, the germ line produces sperm for a brief time that are stored for later use (Fig. 1C). The germ line switches to production of eggs in the adult nematode (Fig. 1D). A key gene in this developmental switch from sperm production to egg production is tra-2. F i g . 2 . Regulation of tra-2 translation by RNA-protein complexes. A, The tra-2 retention complex is formed in the nucleus by binding of the protein NXF-2 to the TRE, preventing normal mRNA export. B, Export of tra-2 mRNA from the nucleus to the cytoplasm is mediated by binding of protein TRA-1 to the TRE. C, In the cytoplasm, expression of the protein TRA-2 is repressed by binding of GLD-1 and FOG-2 to the TGE element. D, After dissociation of the GLD-1–FOG-2 complex, the tra-2 mRNA can be translated to produce the TRA-2 required for normal production of eggs in the adult nematode. nucleus, preventing translation of tra-2 mRNA. In order for the tra-2 mRNA to be exported, NXF-2 must be displaced by the protein TRA-1, which also binds to the TRE (Fig. 2B). This complex is competent to be exported into the cytoplasm, where the tra-2 mRNA is subjected to another level of translational regulation. In the cytoplasm, tra-2 mRNA can be bound by the proteins GLD-1 and FOG-2, which prevent translation (Fig. 2C) By an unknown mechanism, this repressive complex can be dissociated to release the tra-2 mRNA for translation of the TRA-2 protein at the appropriate time and place (Fig. 2D). We are trying to understand the RNA-protein interactions between NXF-2, TRA-1, GLD-1, FOG-2, and the tra-2 mRNA. We have expressed and purified these proteins and have developed biochemical assays for binding. Currently, we are determining the structures of these interesting and important RNA-protein complexes. All of these proteins have homologs in the human genome, although the details of the function of the homologs can be quite different than the function of the nemotode proteins. These studies are important because they will be models for interaction of these protein domains with 234 MOLECULAR BIOLOGY 2007 RNA that will be useful for many other systems. In addition, translational regulation by protein elements that bind downstream of the coding region in mRNAs is poorly understood. Our biochemical and structural approach will offer new insights into both RNA-protein recognition and regulation of translation. PUBLICATIONS Karnaukhov, A.V., Karnaukhova, E.V., Williamson, J.R. Numerical matrices method for nonlinear system identification and description of dynamics of biochemical reaction networks. Biophys. J. 92:3549, 2007. Leontis, N.B., Altman, R.B., Berman, H.M., Brenner, S.E., Brown, J.W., Engelke, D.R., Harvey, S.C., Holbrook, S.R., Jossinet, F., Lewis, S.E., Major, F., Mathews, D.H., Richardson, J.S., Williamson, J.R., Westhof, E. The RNA Ontology Consortium: an open invitation to the RNA community. RNA 12:533, 2006. Szymczyna, B.R., Gan, L., Johnson, J.E., Williamson, J.R. Solution NMR studies of the maturation intermediates of a 13 MDa viral capsid. J. Am. Chem. Soc. 129:7867, 2007. Vallurupalli, P., Scott, L., Hennig, M., Williamson, J.R., Kay, L.E. New RNA labeling methods offer dramatic sensitivity enhancements in 2H NMR relaxation spectra. J. Am. Chem. Soc. 128:9346, 2006. Development of the Genetic Code and Its Connection to Human Disease P. Schimmel, X.-L. Yang, J. Bacher, K. Beebe, R. Belani, E. Chong, Z. Druzina, P. Fanta, M. Guo, M. Hanan, M. Kapoor, E. Merriman, M. Mock, C. Motta, L. Nangle, F. Otero, R. Reddy, W. Waas, W. Xie, W. Zhang, Q. Zhou e focus on a group of enzymes known as aminoacyl tRNA synthetases. These enzymes arose early in evolution, as proteins emerged from a putative RNA world. The 20 synthetases (1 synthetase for each amino acid) make the connection between the nucleotide triplets of the code imbedded in tRNAs (anticodon triplets) and the cognate amino acids. This connection is made through the aminoacylation reactions, in which alanine is attached to yield tRNAAla, serine to yield tRNASer, valine to yield tRNAVal, and so on. Remarkably, this simple set of 20 enzymes is now understood to be connected to disease. This connection occurs in at least 2 ways. First, in all cells, from the simplest prokaryote to the highest eukaryote, one amino acid is often confused for another. Examples are the confusion of serine for alanine and threonine for valine. The result is the attachment of the wrong amino acid to a tRNA and then incorporation of the amino acid at the wrong codon W THE SCRIPPS RESEARCH INSTITUTE of an mRNA, such as the incorporation of serine at the codon for alanine. The resulting mistranslation can cause cell death. Even a small amount of mistranslation causes ataxia through the degeneration of Purkinje cells in the cerebellum. However, in the development of the code, these ancient proteins (tRNA synthetases) have acquired the capacity for editing, that is, an activity to prevent the incorporation of an amino acid at the wrong codon. This activity clears mischarged tRNAs, such as Ser-tRNAAla or Thr-tRNAVal, and thereby enforces the correct amino acid–nucleotide triplet relationships of the genetic code. At the same time, in mammals, even mild mutations in the editing domain of a tRNA synthetase can lead to disease. These mutations can be vertically transmitted to progeny, which are also put into a diseased state. (Stronger mutations are lethal and therefore are not transmitted.) Most recent research has shown that defects in editing are mutagenic in aging bacteria. These observations have raised the possibility of whether editing defects have a role in diseases of aging, including cancer, in humans. We showed the consequences of defects in editing in bacteria, mammalian cells, and mice. Our research has included the development of a special sensor that directly detects mistranslation in mammalian cells. With this sensor, created by using a special construction with green fluorescent protein, we can detect the confusion of threonine for valine. This confusion occasionally creates Thr-tRNA Val , so that threonine is inserted at the codons for valine. The fluorescent protein in the sensor is a mutated form that can generate a fluorescent signal only when threonine is mistakenly inserted at a specific codon for valine (Fig. 1). Using this sys- F i g . 1 . Top, Construction of a biosensor based on green fluores- cent protein that detects mistaken insertion of threonine at codons for valine. Bottom, Decrease in fluorescent signal when threonine is replaced by valine. MOLECULAR BIOLOGY 2007 tem, we were able to study how a defect in the editing activity of valyl-tRNA synthetase (which normally clears Thr-tRNAVal) caused mistranslation that had pathologic consequences. The second connection of aminoacyl tRNA synthetases to disease is through their expanded functions. During their long evolution, the enzymes have acquired other activities in cell signaling pathways. These additional functions connect the synthetases and translation to broad biological systems, such as the pathways for angiogenesis, inflammation, and neurogenesis. Our recent findings support the idea that these expanded functions were added in a stepwise way, as the tree of life grew from a common ancestor that split into the 3 great kingdoms: prokaryotes, archae, and eukaryotes. For eukaryotes, the building of biological systems for the vasculature and nervous system, for example, was paralleled by the increasing complexity of the synthetases. Understanding this process and how aminoacylation was adapted to the acquisition of new domains and motifs for cell signaling is one of our central goals. In addition, we are studying specific diseases that suggest alternative functions for tRNA synthetases (Table 1). T a b l e 1 . Examples of expanded functions of human aminoacyl tRNA synthetases Aminoacyl tRNA synthetase Expanded function TyrRS Inflammation and angiogenesis TrpRS Angiogenesis Glu-ProRS Inflammation GlnRS Antiapoptosis LysRS Regulation of transcription Charcot-Marie-Tooth disease (CMT) is an example of a tRNA-synthetase–related disease that suggests an alternative function for glycyl-tRNA synthetase (GlyRS). CMT is the most common heritable disease of the peripheral nervous system. Human GlyRS and one of its mutants, G526R, which is causally associated with CMT, have been crystallized and their structures determined at 2.95 and 2.85 Å, respectively (Fig. 2). Altogether, at least 10 disease-causing mutant alleles of GlyRS have been reported. These mutations are scattered broadly across the primary sequence and have no apparent unifying connection. Mapping mutations onto our structure of human GlyRS showed their proximity to the dimer interface. G526R has an overall struc- THE SCRIPPS RESEARCH INSTITUTE 235 F i g . 2 . Crystal structure of human GlyRS with location of the disease-causing mutation G526. ture similar to that of the wild-type enzyme but, through a long-range effect, has a greater dimer interface. Experimental analyses indicate that although the CMT phenotype did not correlate with aminoacylation activity, most mutations affect dimer formation, either enhancing (e.g., G526R) or weakening the dimer. Remarkably, all CMT mutant GlyRSs expressed in neuroblastoma cells were defective in their distribution into neurites. This defect may be connected in some way to a change in the dimer interface, which itself may interact with other specific partners in neurons. PUBLICATIONS Bacher, J, Schimmel, P. An editing-defective tRNA synthetase is mutagenic in aging bacteria via the SOS response. Proc. Natl. Acad. Sci. U. S. A. 104:1907, 2007. Bacher, J.M., Waas, W.F., Metzgar, D., de Crecy-Lagard, V., Schimmel, P. Genetic code ambiguity affords a selective advantage to Acinetobacter baylyi. J. Bacteriol., in press. Beebe, K., Waas, W., Druzina, Z., Guo, M., Schimmel, P. A universal plate format for high-throughput assays that monitor multiple aminoacyl tRNA synthetase activities. Anal. Biochem., in press. Nangle, L.A., Zhang, W., Xie, W., Yang, X.L., Schimmel, P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl. Acad. Sci. U. S. A. 104:1123, 2007. Schimmel, P., Yang, X.-L. Perfecting the genetic code with an RNP complex. Structure 14:1729, 2006. Xie, W., Nangle, L.A., Zhang, W., Schimmel, P., Yang, X.-L. Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 104:9976, 2007. Xie, W., Schimmel, P., Yang, X.-L. Crystallization and preliminary x-ray analysis of a native human tRNA synthetase whose allelic variants are associated with Charcot-Marie-Tooth disease. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 62(Pt. 12):1243, 2006. Yang, X.-L., Guo, M., Kapoor, M., Ewalt, K.L., Otero, F.J., Skene, R.J., McRee, D.E., Schimmel, P. Functional and crystal structure analysis of active site adaptations of a potent anti-angiogenic human tRNA synthetase. Structure 15:793, 2007. 236 MOLECULAR BIOLOGY 2007 Structure-Function Analysis of Expanded Activities of Human tRNA Synthetases X.-L. Yang, P. Schimmel, R. Belani, E. Chong, P. Fanta, M. Guo, M. Hanan, M. Kapoor, E. Merriman, M. Mock, C. Motta, L. Nangle, F. Otero, R. Reddy, W. Waas, W. Xie, W. Zhang, Q. Zhou e focus on a subgroup of the components of the translation apparatus that in some instances have functions beyond translation. This subgroup is known as the aminoacyl-tRNA synthetases, 20 enzymes (1 enzyme for each amino acid) that catalyze the first step of protein synthesis by aminoacylation of tRNAs. The alternative functions of these synthetases in humans suggest a broad connection of translation to signal transduction pathways in angiogenesis, inflammation, and neurogenesis. In mammalian cells, the synthetases are gathered together with accessory factors into a large complex known as the multisynthetase complex (MSC; Fig. 1). W THE SCRIPPS RESEARCH INSTITUTE facilitate protein synthesis and, more importantly, how it may allow the recruitment and the release of some or all components of the MSC in response to various cell signals. Mammalian aminoacyl-tRNA synthetases usually have acquired additional domains, which may provide a regulatory mechanism for noncanonical functions, as shown for human tyrosyl-tRNA synthetase (TyrRS) and tryptophanyl-tRNA synthetase (TrpRS). Human TyrRS has a C-terminal appended domain that is homologous to a known human cytokine, endothelial monocyte-activating polypeptide II. The C-terminal domain and the rest of the enzyme, mini-TyrRS, can be released by extracellular proteases such as plasmin. Interestingly, both mini-TyrRS and the C-terminal domain are active in cell signaling. A glutamic acid–leucine–arginine (ELR) tripeptide motif is essential for the proangiogenic activity of mini-TyrRS. The full-length TyrRS is inactive as a cytokine. We hypothesize that the steric block of the critical ELR with the C-terminal domain prevents elucidation of cytokine activity in native TyrRS. This idea is being tested by mutating a conserved tyrosine (Y341) that in our crystal structure of mini-TyrRS is tethered to the ELR tripeptide. This mutation could potentially open up the ELR motif in the full-length TyrRS structure and activate the cytokine function, consistent with the idea that the purpose of natural fragmentation is to relieve steric blocking of critical epitopes (Fig. 2). F i g . 2 . Potential mutational activation of the cytokine activity of human TyrRS. F i g . 1 . The multisynthetase complex formed by 9 human tRNA synthetases and 3 accessory factors. Other tRNA synthetases are thought to be loosely associated with the complex. A subset of 9 synthetases are more tightly associated in the complex than are others; 3 specific factors, p18, p38, and p43, serve as scaffolding. This organization into a complex may in some way facilitate protein synthesis. Additionally or alternatively, the complex itself may be a reservoir or depository of unactivated cytokines. We are working on understanding the structure and the organization of the MSC, in relation to how it may Human TrpRS, in contrast to TyrRS, has an N-terminal appended domain. Interestingly, it has an embedded antiangiogenic activity upon removal of the N-terminal domain. The activation occurs naturally both by alternative splicing and by proteolysis. We hypothesize that the activation is achieved by exposure of the active site of TrpRS, which allows the interaction with VE-cadherin as a receptor on endothelial cells. Removal of the N-terminal domain can expose the tryptophan binding site, which is closed in the full-length TrpRS as shown in our crystal structure. We are testing this hypothesis, which could link translation with angiogene- MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 237 sis via the amino acid binding pocket. Structural and functional analyses of human TrpRS have suggested a new way to form an active site, a way that appears to be an adaptation to accommodate the cytokine function of the enzyme. We think the adaptation creates selective pressure to retain the expanded function. PUBLICATIONS Nangle, L.A., Zhang, W., Xie, W., Yang, X.L., Schimmel, P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl. Acad. Sci. U. S. A. 104:11239, 2007. Schimmel, P., Yang, X.-L. Perfecting the genetic code with an RNP complex. Structure 14:1729, 2006. Xie, W., Nangle, L.A., Zhang, W., Schimmel, P., Yang, X.-L. Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 104:9976, 2007. Xie, W., Schimmel, P., Yang, X.-L. Crystallization and preliminary x-ray analysis of a native human tRNA synthetase whose allelic variants are associated with Charcot- Marie-Tooth disease. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 62(Pt. 12):1243, 2006. Yang, X.-L., Guo, M., Kapoor, M., Ewalt, K.L., Otero, F.J., Skene, R.J., McRee, D.E., Schimmel, P. Functional and crystal structure analysis of active site adaptations of a potent anti-angiogenic human tRNA synthetase. Structure 15:793, 2007. Yang, X.-L., Otero, F.J. , Ewalt, K.L., Liu, J., Swairjo, M.A., Kohrer, C., RajBhandary, U.L., Skene, R.J., McRee, D.E., Schimmel, P. Two conformations of a crystalline human tRNA synthetase-tRNA complex: implications for protein synthesis. EMBO J. 25:2919, 2006. Mechanisms of RNA Assembly and Catalysis M.J. Fedor, J.W. Cottrell, C.P. Da Costa, S. Daudenarde, E.M. Mahen, M. Roychowdhury-Saha ur goal is to generate fundamental insights into catalysis by RNA enzymes. Our results contribute to the basic knowledge of RNA structure and function in normal growth and development and provide a framework for developing technical and therapeutic applications involving RNAs as targets and reagents. The well-characterized structure of the hairpin ribozyme provides a valuable framework for investigating the contributions of individual active-site interactions to RNA structure and function. The hairpin ribozyme catalyzes a reversible self-cleavage reaction in which nucleophilic attack of a ribose 2′-hydroxyl on an adjacent phosphorus proceeds through a trigonal bipyramidal transition state that leads to the formation of 2′,3′cyclic phosphate and 5′-hydroxyl termini (Fig. 1). A network of stacking and hydrogen-bonding interactions align the reactive phosphate in the appropriate orientation for an SN2-type nucleophilic attack and orient O F i g . 1 . Chemical mechanism of RNA cleavage mediated by the family of small catalytic RNAs that includes the hairpin ribozyme. Cleavage of the phosphodiester bond occurs through an SN2-type mechanism that involves in-line attack of the 2′ oxygen nucleophile on the adjacent phosphorus to form a trigonal bipyramidal transition state. Breaking of the 5′ oxygen-phosphorus bond generates products with 5′-hydroxyl and 2′,3′-cyclic phosphate termini. nucleotide base functional groups near the reactive phosphate to facilitate catalytic chemistry (Fig. 2). G+1 is the first nucleotide on the 3′ side of the reactive phosphodiester, so interactions with G+1 stabilize the ribozyme-product complex. G+1 position has no direct contact with the reactive phosphodiester, but interactions between G+1 and nucleotides in loop B define the in-line trajectory of the reactive phosphodiester. Loss of G+1 virtually eliminates catalytic activity, highlighting the significant contribution of active-site architecture in lowering the activation barrier to catalysis. Remarkably, the structure of the ribozyme-product complex is virtually indistinguishable from the structure of the ribozyme complex with a transition-state mimic, making it difficult to distinguish structural contributions to ground- and transition-state stability. The purpose of our recent biochemical experiments was to learn if seemingly identical interactions make different contributions to the activation barrier to catalysis, the stability of ribozyme complexes in the ground state, and the internal equilibrium between cleavage and ligation. We found that all modifications of the G+1 binding pocket inhibited ligation more than they did cleavage. These results confirm our previous evidence that the stability of tertiary structure is the major determinant of the balance between cleavage and ligation. Substituting seemingly equivalent functional groups sometimes had quite different functional consequences. In one striking example, deletion of an active-site adenine, A38, increased the activation barrier to catalysis by more than +5 kcal/mol but reduced the groundstate stability of the ribozyme-product complex by just +1.6 kcal/mol. These quantitative functional studies provide one of the most detailed views yet of structurefunction relationships within a ribozyme active site. 238 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE Directed Evolution of Nucleic Acid Enzymes G.F. Joyce, S.E. Hamilton, D.P. Horning, T.A. Jackson, B.J. Lam, B.M. Paegel, K.L. Petrie, S.B. Voytek t has been 40 years since Spiegelman and colleagues first demonstrated how RNA molecules can be evolved in the test tube. Those early experiments involved the replication of certain viral RNAs by the corresponding viral replicase proteins. Since then, powerful methods have been developed for amplifying (and mutating) almost any RNA, including RNAs with catalytic function. We have been developing methods for the in vitro evolution of RNA and applying those methods to the discovery of novel RNAs of biochemical and biomedical significance. In addition, we are studying the processes of darwinian evolution itself, carried out at the level of molecules rather than in whole organisms. I A C O N T I N U O U S LY E V O LV I N G R N A E N Z Y M E F i g . 2 . Network of tertiary interactions formed with G+1 that create the hairpin ribozyme active site. A, Three-dimensional structure of the G+1 binding pocket in a ribozyme complex with a vanadate mimic of the transition state with the vanadate indicated in yellow. B, Three-dimensional structure of the G+1 binding pocket in a ribozyme complex with cleavage product RNA with the 2′,3′-cyclic phosphate terminus of the 5′ cleavage product instead of the vanadate transition-state mimic. In order to help distinguish loop A and loop B residues, G+1 and A–1 nucleotides in loop A are colored light blue, and carbon atoms of C25, G36, and A38 nucleotides in loop B are colored green. Tertiary interactions between the essential loop A and B domains of the hairpin ribozyme define the architecture of the hairpin ribozyme active site and fix the reacting groups in the in-line orientation appropriate for the SN2-type reaction mechanism. The interdomain interface is created by the extrusion of the G+1 nucleotide from loop A into a binding pocket in loop B. C, Diagram of interdomain hydrogen-bonding interactions formed with the G+1 nucleotide. Modifications of nucleotides that donate or accept interdomain hydrogen bonds were designed to probe the functional significance of the bonds. Reproduced with permission from Cottrell, J.W., Kuzmin, Y.I., Fedor M.J. Functional analysis of hairpin ribozyme active site architecture. J. Biol. Chem. 282:13498, 2007. Copyright © 2007 by the American Society for Biochemistry and Molecular Biology. PUBLICATIONS Da Costa, C.P., Fedor, M.J., Scott, L.G. 8-Azaguanine reporter of purine ionization states in structured RNAs. J. Am. Chem. Soc. 129:3426, 2007. Cottrell, J.W., Kuzmin, Y.I., Fedor M.J. Functional analysis of hairpin ribozyme active site architecture. J. Biol. Chem. 282:13498, 2007. Many RNA enzymes have been developed by using in vitro evolution, but special attention is directed to those that catalyze the RNA-templated joining of RNA. This reaction is chemically equivalent to the reaction carried out by an RNA polymerase protein. In some instances, RNA enzymes with simple RNA-joining activity have been further evolved to function as RNA-dependent RNA polymerases. Ultimately, such RNA enzymes might be evolved to catalyze the production of additional copies of the RNA enzyme itself. Then the in vitro evolution of RNA could be self-sustaining, without the need for any proteins. As both a step toward and a model for the selfsustained evolution of RNA, we have devised a system for the continuous in vitro evolution of RNA enzymes that have RNA-joining activity. Any RNA molecule in the population that performs the desired reaction becomes amplified to produce “progeny” molecules, which then have the opportunity to perform the reaction again. All of the events of continuous evolution take place within a common reaction mixture and occur repeatedly, so long as an adequate supply of reaction materials is maintained. When these materials are exhausted, a small aliquot of the mixture can be transferred to a new reaction vessel that contains a fresh supply of reagents. In this way, we have been able to propagate large populations of evolving RNA molecules for hundreds of successive “generations.” Until recently, all continuous in vitro evolution experiments were done with descendants of a particular RNA MOLECULAR BIOLOGY 2007 enzyme, the “class I” RNA ligase. Within the past year, we established a second continuously evolving RNA enzyme based on descendants of the “DSL” RNA ligase. Achieving this result required many rounds of stepwise in vitro evolution under stringent selection pressure. Using a quench-flow apparatus, we selected molecules that could perform the RNA-joining reaction in as little as 15 milliseconds. The resulting optimized variants were capable of initiating continuous evolution, and once continuous evolution began, it could be carried on indefinitely. The continuously evolving population has been carried through 80 successive transfers, maintained against an overall dilution of 10207-fold. The molecules can be maintained against such extraordinarily large dilutions because they are amplified exponentially, so long as they have the requisite catalytic properties. Because of the acquisition of numerous mutations, RNA enzymes isolated after the 80th transfer (Fig. 1) had F i g . 1 . Secondary structure of an RNA enzyme capable of under- going continuous in vitro evolution. The arrow indicates the site of the RNA-joining reaction between an RNA substrate and the RNA enzyme, which is the basis for selective amplification. A typical RNA enzyme that was obtained after 80 transfers of continuous evolution contains 24 mutations (highlighted in black) relative to the RNA that was present at the start of the evolution procedure. even better performance than did the enzymes used to initiate continuous evolution. Now that we have 2 distinct “species” of continuously evolving RNA enzymes, we can conduct in vitro evolution studies in which the 2 are made to operate within a common environment. These studies will allow us to explore the possibility of competition and cooperation among evolving molecular species. EVOLUTION ON A CHIP We recently developed a novel approach for the continuous evolution of RNA enzymes that uses microfluidic chip technology. With this approach, evolution is car- THE SCRIPPS RESEARCH INSTITUTE 239 ried out in an automated fashion under computer control, with continuous monitoring of the population size and precise manipulation of the reaction mixture. We have used the microfluidic device to conduct evolution experiments, beginning with a population of billions of RNA enzymes with RNA-joining activity and carrying out many successive rounds of RNA catalysis and selective amplification. The concentration of RNA is monitored by using the intercalating dye thiazole orange and a confocal laser fluorescence microscope. Whenever a predetermined threshold concentration is reached, the computer initiates a set of microvalve operations to isolate part of the reaction mixture and combine it with a fresh supply of reagents. In one microfluidic evolution experiment, we carried out 363 successive dilutions of 10-fold each, progressively reducing the concentration of substrate over time. As the continuously evolving RNA enzymes adapted to the reduced substrate concentration, they required less time to achieve 10-fold amplification, a situation that was reflected in a progressively reduced time between successive dilutions (Fig. 2). We are using this system F i g . 2 . Microfluidic-based evolution of an RNA enzyme with RNA- joining activity. The RNA, which had been adapted to a substrate concentration of 5 µM, was made to undergo 100 successive 10-fold dilutions in the presence of a substrate concentration of only 1 µM. Each dilution was triggered when the total concentration of RNA enzyme reached about 350 nM. for “evolution on a chip” to develop other evolved properties and to address fundamental questions about macromolecular evolution, such as the role of genetic diversity in escaping evolutionary bottlenecks. PUBLICATIONS Joyce, G.F. Forty years of in vitro evolution. Angew. Chem. Int. Ed., in press. Joyce, G.F. A glimpse of biology’s first enzyme. Science 315:1507, 2007. Paegel, B.M., Grover, W.H., Skelley, A.M., Mathies, R.A., Joyce, G.F. Microfluidic serial dilution circuit. Anal. Chem. 78:7522, 2006. 240 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE Studies at the Interface of Molecular Biology, Chemistry, and Medicine C.F. Barbas III, K. Albertshofer, T. Bui, S. Eberhardy, R.P. Fuller, C. Gersbach, B. Gonzalez, R. Gordley, J. Guo, D.H. Kim, R.L. Lerner, W. Nomura, A. Onoda, S.S.V. Ramasastry, M. Santa Marta, L.J. Schwimmer, D. Shabat,* F. Tanaka, U. Tschulena, N. Utsumi, Y. Ye, K.S. Yi, Y. Yuan, H. Zhang * Tel Aviv University, Tel Aviv, Israel e are concerned with problems in molecular biology, chemistry, and medicine. Many of our studies involve learning or improving Nature’s strategies to prepare novel molecules that perform specific functional tasks, such as regulating a gene, destroying cancer, or catalyzing a reaction with enzymelike efficiency. We hope to apply these novel insights, technologies, methods and their products to provide solutions to human diseases, including cancer, HIV disease, and genetic diseases. W D I R E C T I N G T H E E V O L U T I O N O F C ATA LY T I C F U N C T I O N Using reactive immunization, we have developed antibodies that catalyze aldol as well as retro-aldol reactions of a wide variety of molecules. The catalytic proficiency of the best of these antibodies is almost 10 14 , a value 1000 times that of the best catalytic antibodies reported to date and overall the best of any synthetic protein catalyst. We have shown the efficient asymmetric synthesis and resolution of a variety of molecules, including tertiary and fluorinated aldols, and have used these chiral synthons to synthesize natural products (Fig. 1). These results highlight the potential synthetic usefulness of catalytic antibodies as artificial enzymes in addressing problems in organic chemistry that are not solved by using natural enzymes or more traditional synthetic methods. Other advances in this area include the development of the first peptide aldolase enzymes. By using both design and selection, we have created small peptide catalysts that recapitulate many of the kinetic features of large enzyme catalysts. These smaller enzymes allow us to address the relationship between the size of natural proteins and the proteins’ catalytic efficiency. O R G A N O C ATA LY S I S : A B I O O R G A N I C A P P R O A C H T O C ATA LY T I C A S Y M M E T R I C S Y N T H E S I S To further explore the principles of catalysis, we are studying amine catalysis as a function of catalytic F i g . 1 . A variety of compounds synthesized with the world’s first commercially available catalytic antibody, 38C2, produced at Scripps Research. scaffold. Using insights garnered from our studies of aldolase antibodies, we determined the efficacy of simple chiral amines and amino acids for catalysis of aldol and related imine and enamine chemistries such as Michael, Mannich, Knoevenagel, and Diels-Alder reactions. Although aldolase antibodies are superior catalysts in terms of the kinetic parameters, these more simple catalysts are enabling us to quantify the significance of pocket sequestration in catalysis. Furthermore, many of these catalysts are cheap, environmentally friendly, and practical for large-scale synthesis. With this approach, we showed the scope and usefulness of the first efficient amine catalysts of direct asymmetric aldol, Mannich, Diels-Alder, and Michael reactions. The organocatalyst approach is a direct outcome of our studies of catalytic antibodies and provides an effective alternative to organometallic reactions that use severe reaction conditions and oftentoxic catalysts. We think that our discovery that simple naturally occurring amino acids such as L-proline and other amines can effectively catalyze a variety of enantioselective intermolecular reactions will change the way many reactions will be performed. As a testament to the mild nature of this approach, we developed the first catalytic asymmetric aldol, Mannich, Michael, and fluorination reactions involving aldehydes as nucleophiles. Previously, such reactions were considered out of the reach of traditional synthetic methods. In extensions of these concepts, we designed novel amino acid derivatives that direct the stereochemical MOLECULAR BIOLOGY 2007 outcome of reactions in ways not possible with proline (Fig. 2). In other studies, we created the first asymmet- F i g . 2 . A, Proline catalyst provides access to syn-Mannich and anti-aldol products. B, Design of (3R,5R)-5-methyl-3-pyrrolidinecarboxylic acid allows efficient access to anti-Mannich products. C, Design of a primary amine catalyst provides access to anti-Mannich and syn-aldol products involving hydroxyketones. ric small-molecule aldol catalysts that are highly effective with water and seawater as solvent. We think that our results are also relevant to the prebiotic synthesis of the molecules of life. For example, we have shown that our amino acid strategy can be used to synthesize carbohydrates directly, thereby providing a provocative prebiotic route to the sugars essential for life. We hypothesize that organocatalysis with amino acids plays an important role in the metabolism of living organisms today. ANTIBODY ENGINEERING: THERAPEUTIC ANTIBODIES, IN AND OUT OF CELLS We developed the first human antibody phage display libraries and the first synthetic antibodies and methods for the in vitro evolution of antibody affinity. The ability to manipulate large libraries of human antibodies and to evolve such antibodies in the laboratory provides tremendous opportunities to develop new medicines. Laboratories and pharmaceutical companies around the world now apply the phage display technology that we developed for antibody Fab fragments. In our laboratory, we are targeting cancer and HIV disease. One of our antibodies, IgG1-b12, protects animals against primary challenge with HIV type 1 (HIV-1) and has been further studied by many researchers. We improved this antibody by developing in vitro evolution THE SCRIPPS RESEARCH INSTITUTE 241 strategies that enhanced its neutralization activity. By coupling laboratory-evolved antibodies with potent toxins, we showed that immunotoxins can effectively kill infected cells. We are also developing genetic methods to halt HIV by gene therapy. We created unique human antibodies that can be expressed inside cells to make the cells resistant to HIV infection. In the future, these antibodies might be delivered to the stem cells of patients infected with HIV-1, allowing the development of a disease-free immune system that would obviate the intense regimen of antiviral drugs now required to treat HIV disease. Using our increased understanding of antibodyantigen interactions, we extended our efforts in cancer therapy and developed rapid methods for creating human antibodies from antibodies derived from other species. We produced human antibodies that should enable us to selectively starve a variety of cancers by inhibiting angiogenesis and antibodies that will be used to deliver radioisotopes to colon cancers to destroy the tumors. We hope that these antibodies will be used in clinical trials done by our collaborators at the SloanKettering Cancer Center in New York City. On the basis of our studies on HIV-1, we used intracellular expression of antibodies directed against angiogenic receptors to create a new gene-based approach to cancer. Our studies indicate that this type of gene therapy can be successfully applied to the treatment of cancer. T H E R A P E U T I C A P P L I C AT I O N S O F C ATA LY T I C ANTIBODIES The development of highly efficient catalytic antibodies opens the door to many practical applications. One of the most fascinating is the use of such antibodies in human therapy. We think that use of this strategy can improve chemotherapeutic approaches to diseases such as cancer and AIDS. Chemotherapeutic regimens are typically limited by nonspecific toxic effects. To address this problem, we developed a novel and broadly applicable drug-masking chemistry that operates in conjunction with our unique broad-scope catalytic antibodies. This masking chemistry is applicable to a wide range of drugs because it is compatible with virtually any heteroatom. We showed that generic drug-masking groups can be selectively removed by sequential retro-aldol–retro-Michael reactions catalyzed by antibody 38C2 (Fig. 3). This reaction cascade is not catalyzed by any known natural enzyme. 242 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE F i g . 3 . Targeting cancer and HIV with prodrugs activated by catalytic antibodies. A bifunctional antibody is shown targeting a cancer cell for destruction. A nontoxic analog of doxorubicin, prodoxorubicin, is being activated by an aldolase antibody to the toxic form of the drug. Application of this masking chemistry to the anticancer drugs doxorubicin, camptothecin, and etoposide produced prodrugs with substantially reduced toxicity. These prodrugs are selectively unmasked by the catalytic antibody when the antibody is applied at therapeutically relevant concentrations. The efficacy of this approach has been shown in in vivo models of cancer. Currently, we are developing more potent drugs and novel antibodies that will allow us to target breast, colon, and prostate cancers as well as cells infected with HIV-1. On the basis of our preliminary findings, we think that our approach can become a key tool in selective chemotherapeutic strategies. To see a movie illustrating this approach, visit http://www.scripps.edu/ mb/barbas/antibody/antibody.mov. C H E M I C A L LY P R O G R A M M E D A N T I B O D I E S : T H E ADVENT OF CHEMOBODIES We think that combining the chemical diversity of small synthetic molecules with the immunologic characteristics of antibody molecules will lead to therapeutic agents with superior properties. Therefore, we developed a conceptually new device that equips small synthetic molecules with both the immunologic effector functions and the long serum half-life of a generic antibody molecule. For a prototype, we developed a targeting device based on the formation of a covalent bond of defined stoichiometry between (1) a 1,3-diketone derivative of an arginine–glycine–aspartic acid peptidomimetic that targets the integrins αvβ3 and αvβ5 and (2) the reactive lysine of aldolase antibody 38C2 (Fig. 4). The resulting complex spontaneously assembled in vitro and in vivo, selectively retargeted antibody 38C2 to the surface of cells expressing the integrins αvβ3 and αvβ5, dramatically increased the circulatory half-life of the F i g . 4 . Designed small-molecule targeting agents (SCS-873 as shown) program the specificity of the antibody 38C2 (A). The resulting chemobodies (cp38C2, B) have characteristics that are often superior to those of either the small molecule or the antibody alone. peptidomimetic, and effectively reduced tumor growth in animal models of human Kaposi sarcoma, colon cancer, and melanoma. ZINC FINGER GENE SWITCHES AND ENZYMES The solutions to many diseases might be simply turning genes on or off in a selective way or adding or deleting genes. In order to accomplish all of these aims, we are studying molecular recognition of DNA by zinc finger proteins and methods of creating novel zinc finger DNA-binding proteins. We showed that proteins that contain zinc fingers can be selected or designed to recognize novel DNA sequences. These studies are aiding the elucidation of rules for sequence-specific recognition within this family of proteins. We selected and designed specific zinc finger domains that will constitute an alphabet of 64 domains that will allow any DNA sequence to be bound selectively. The prospects for this “second genetic code” are fascinating and may have a major impact on basic and applied biology. We showed the potential of this approach in multiple mammalian and plant cell lines and in whole organisms. With the use of characterized modular zinc finger domains, polydactyl proteins capable of recognizing an 18-nucleotide site can be rapidly constructed (see www.zincfingertools.org). Our results suggest that zinc finger proteins might be useful as genetic regulators for a variety of human aliments and provide the basis for a new strategy of gene therapy. Our goal is to develop this class of therapeutic proteins to inhibit or enhance MOLECULAR BIOLOGY 2007 the synthesis of proteins, providing a direct strategy for fighting diseases of either somatic or viral origin. We are also developing proteins that will inhibit the growth of tumors and others that will inhibit the expression of a protein known as CCR5, which is a key to infection of human cells by HIV-1. We developed an HIV-1–targeting transcription factor that strongly suppresses HIV-1 replication and another transcription factor that upregulates fetal hemoglobin as a treatment for sickle cell anemia. More recently, we have focused on evolving zinc finger enzymes that modify the genome. These studies have led to the development of programmable zinc finger recombinases (Fig. 5) that promise F i g . 5 . Model of a zinc finger recombinase with programmable specificity created by using rational design and directed molecular evolution. to reshape the way scientists manipulate the genome for study and therapy of disease. PUBLICATIONS Abraham, S., Guo, F., Li, L.-S., Rader, C., Liu, C., Barbas, C.F. III, Lerner, R.A., Sinha, S.C. Synthesis of the next-generation therapeutic antibodies that combine cell targeting and antibody-catalyzed prodrug activation. Proc. Natl. Acad. Sci. U. S. A. 104:5584, 2007. Albertshofer, K., Thayumanavan, R., Utsumi, N., Tanaka, F., Barbas, C.F. III. Amine-catalyzed Michael reactions of an aminoaldehyde derivative to nitroolefins. Tetrahedron Lett. 48:693, 2007. Chowdari, N.S., Ahmad, M., Albertshofer, K., Tanaka, F., Barbas, C.F. III. Expedient synthesis of chiral 1,2- and 1,4-diamines: protecting group dependent regioselectivity in direct organocatalytic asymmetric Mannich reactions. Org. Lett. 8:2839, 2006. Doppalapudi, V.R., Tryder, N., Li, L., Aja, T., Griffith, D., Liao, F.F., Roxas, G., Ramprasad, M.P., Bradshaw, C., Barbas, C.F. III. Chemically programmed antibodies: endothelin receptor targeting CovX-Bodies. Bioorg. Med. Chem. Lett. 17:501, 2007. Gordley, R.M., Smith, J.D., Graslund, T., Barbas, C.F. III. Evolution of programmable zinc finger-recombinases with activity in human cells. J. Mol. Biol. 367:802, 2007. Guo, F., Das, S., Mueller, B.M., Barbas, C.F. III, Lerner, R.A., Sinha, S.C. Breaking the one antibody-one target axiom. Proc. Natl. Acad. Sci. U. S. A. 103:11009, 2006. Mase, N., Watanabe, K., Yoda, H., Takabe, K., Tanaka, F., Barbas, C.F. III. Organocatalytic direct Michael reaction of ketones and aldehydes with β-nitrostyrene in brine. J. Am. Chem. Soc. 128:4966, 2006. Rader, C., Barbas, C.F. III. Synthetic antibodies prey on B cells. Blood 108:2889, 2006. Ramasastry, S.S.V., Zhang, H., Tanaka, F., Barbas, C.F. III. Direct catalytic asymmetric synthesis of anti-1,2-amino alcohols and syn-1,2-diols through organocatalytic anti-Mannich and syn-aldol reactions. J. Am. Chem. Soc. 129:288, 2007. THE SCRIPPS RESEARCH INSTITUTE 243 Segal, D.J., Crotty, J.W., Bhakta, M.S., Barbas, C.F. III, Horton, N.C. Structure of Aart, a designed six-finger zinc finger peptide, bound to DNA. J. Mol. Biol. 363:405, 2006. Shamis, M., Barbas, C.F. III, Shabat, D. A new visual screening assay for catalytic antibodies with retro-aldol retro-Michael activity. Bioorg. Med. Chem. Lett. 17:1172, 2006. Sinha, S.C., Das, S., Li, L.-S., Lerner, R.A., Barbas, C.F. III. Preparation of integrin αvβ3-targeting Ab 38C2 constructs. Nat. Protoc. 2:449, 2007. Tanaka, F., Barbas, C.F. III. Aldol and Mannich-type reactions. In: Enantioselective Organocatalysis: Reactions and Experimental Procedures. Dalko, P.I. (Ed.). WileyVCH, Weinheim, Germany, 2007, p. 19. Utsumi, N., Zhang, H., Tanaka, F., Barbas, C.F. III. A way to highly enantiomerically enriched aza-Morita-Baylis-Hillman-type products. Angew. Chem. Int. Ed. 46:1878, 2007. Zhang, H., Mifsud, M., Tanaka, F., Barbas, C.F. III. 3-Pyrrolidinecarboxylic acid for direct catalytic asymmetric anti-Mannich-type reactions of unmodified ketones. J. Am. Chem. Soc. 128:9630, 2006. Synthetic Enzymes, Catalytic Antibodies, Ozone Scavengers in Asthma, Molecular Motors, and Biomolecular Computing E. Keinan, C.H. Lo, N. Panda, O. Reany, C. Bauer, Y. Weiss, N. Metanis, E. Kossoy, M. Soreni, R. Piran, M. Sinha, I. Ben-Shir, T. Shekhter, T. Ratner, T. Mejuch, E. Solel, S. Shoshani, R. Gershoni e focus on synthetically modified enzymes, applications of antibody catalysis, anticancer and antiasthma agents, molecular rotary motors, and biomolecular computation, as illustrated in the following examples. W C ATA LY S I S W I T H S Y N T H E T I C A L LY M O D I F I E D ENZYMES Selenoenzymes have a central role in maintaining cellular redox potential. These enzymes have selenylsulfide bonds in their active sites that catalyze the reduction of peroxides, sulfoxides, and disulfides. The selenol-disulfide exchange reaction is common to all of these enzymes, and the active site of redox potential reflects the ratio between the forward and the reverse rates of this reaction. Preparation of enzymes containing selenocysteine is experimentally challenging. As a result, little is known about the kinetic role of selenols in enzyme active sites, and the redox potential of a selenylsulfide or diselenide bond in a protein has not been experimentally determined. To fully evaluate the effects of selenocysteine on oxidoreductase redox potential and kinetics, we chem- 244 MOLECULAR BIOLOGY ically synthesized glutaredoxin 3 and all 3 selenocysteine variants of its conserved 11CXX14C active site and determined their redox potentials. In particular, the position of redox equilibrium between glutaredoxin 3(C11U-C14U) (–308 mV) and thioredoxin (–270 mV) suggests a possible role for diselenide bonds in biological systems. Kinetic analysis indicated that the lower redox potentials of the selenocysteine variants are due primarily to the greater nucleophilicity of the active-site selenium. The 102- to 104-fold increase in the rate of thioredoxin reduction by the seleno-glutaredoxin 3 analogs indicates that oxidoreductases containing either selenylsulfide or diselenide bonds can have physiologically compatible redox potentials and enhanced reduction kinetics in comparison with their sulfide counterparts. This research on synthetic enzymes is being done in collaboration with P.E. Dawson, Department of Cell Biology. C ATA LY T I C A N T I B O D I E S Introduction of a herbicide-resistance trait in commercial plants is highly desirable because it allows novel herbicide management options, particularly those that allow the control of weed species closely related to the crop and of other undesired plant species. We recently showed that herbicide-resistant plants can be engineered by designing both a herbicide and a catalytic antibody that destroys the herbicide within the plants (Fig. 1). First, we developed a new carbamate F i g . 1 . Influence of herbicide (4) on the rooting and development of seedlings of F1 hybrids and control A thaliana plants. The control plants are shown in A and C; the hybrid plant lines (F1) expressing both light and heavy chains of catalytic antibody 38C2, in B and D. Plantlets grown on medium without the herbicide are shown in A and B; those grown with the herbicide are shown in C and D. 2007 THE SCRIPPS RESEARCH INSTITUTE herbicide that can be catalytically destroyed by the aldolase antibody 38C2. Then we separated expression of the light chain and half of the heavy chain (Fab) of the catalytic antibody in the endoplasmic reticulum of 2 plant lines of Arabidopsis thaliana. Finally, we used cross-pollination of these 2 transgenic plants to produce a herbicide-resistant F1 hybrid. O Z O N E S C AV E N G E R S W I T H A N T I A S T H M A A C T I V I T Y A new hypothesis we proposed for the mechanism of asthmatic inflammation has led to an ozone-scavenging compound that prevents bronchial obstruction in rats with asthma. Previously, scientists at Scripps Research discovered that ozone can be generated not only via the antibody-mediated water oxidation pathway but also by antibody-coated activated white blood cells during inflammatory processes. This finding led us to speculate that the pulmonary inflammation in asthma might be caused by ozone production by white blood cells in lungs and that inhalation of electronrich olefins, which are known ozone scavengers, might have antiasthmatic effects. In experiments in rats, inhalation of such a compound, limonene, caused a significant improvement in signs of asthma. These results could have consequences in the management of asthma. M O L E C U L A R R O TA R Y M O T O R S The hypothesis that molecular-scale rotary motors can be designed and constructed from synthetic components is based on the available structural information on the biological precedents, such as bacterial flagellar motors and ATP synthase, which interconvert chemical energy and coordinated mechanical motion. Synthetic motors could offer considerable advantages in the development of complex nanomachinery because the motors can tolerate a more diverse range of conditions than biological machines can. One of the most challenging design elements of molecular rotary motors is the need for high-speed rotation, which requires low “friction.” We suggest that in order to achieve minimal friction, the members of the rotor-stator couple should repel each other. We propose a new approach to the design of frictionless molecular rotary motors in which a rotaxanetype architecture in which a macrocyclic cucurbituril host serves as a stator and a rigid, polyyne guest serves as a rotor. The feasibility of the key design element, the repulsive interaction between these components, is supported by molecular mechanics calculations with model systems and has been experimentally confirmed MOLECULAR BIOLOGY 2007 by microcalorimetry and by x-ray crystallography with several synthetic host-guest complexes. The results all suggest that the diyne rod floats at the center of the macrocyclic host with no apparent van der Waals contacts between the rod and the host (Fig. 2). THE SCRIPPS RESEARCH INSTITUTE 245 design of a 3-state–3-symbol automaton, thus increasing the number of syntactically distinct programs from 765 to 1 billion. We have also amplified the applicability of this design by using surface-anchored input molecules and surface plasmon resonance technology to monitor the computation steps in real time. This technology allowed parallel computation and automatic, real-time detection with DNA chips that have multiple input molecules and can be used as pixel arrays for image encryption. PUBLICATIONS Kossoy, E., Lavid, N., Soreni-Harari, M., Shoham, Y., Keinan, E. A programmable bio-molecular computing machine with bacterial phenotype output. Chembiochem, in press. Lo, H.C., Han, H., D’Souza, L.J., Sinha, S.C., Keinan, E. Rhenium(VII) oxide catalyzed heteroacylative ring-opening dimerization of tetrahydrofuran. J. Am. Chem. Soc. 129:1246, 2007. Lo, H.C., Iron, M.A., Martin, J.M.L., Keinan, E. Proton walk in the aqueous platinum complex [TpPtMeCO] via a sticky σ-methane ligand. Chem. Eur. J. 13:2812, 2007. Metanis, N., Keinan, E., Dawson, P.E. Synthetic seleno-glutaredoxin 3 analogues are highly reducing oxidoreductases with enhanced catalytic efficiency. J. Am. Chem. Soc. 128:16684, 2006. Weiss, Y., Rubin, B., Shulman, A., Ben Shir, I., Keinan, E., Wolf, S. Determination of plant resistance to carbamate herbicidal compounds inhibiting cell division and early growth by seed and plantlets bioassays. Nat. Protoc. 1:2282, 2006. F i g . 2 . Solid-state structure of inclusion complexes of cucurbituril with 1 (A, side view; B, top view), 2 (C), and 3 (D). Red indicates oxygen; blue, nitrogen; gray, carbon; brown, sulfur; and green, chloride. Hydrogen atoms have been omitted for clarity. The com- plexes and part of their environment are shown. The structures were created by using the Oak Ridge Thermal Ellipsoid Plot computer program for illustrating crystal structures. Macromolecular Interactions: Evolution, Engineering, and Detection BIOMOLECULAR COMPUTING DEVICES Previously, we described the first nanoscale, programmable finite automaton with 2 symbols and 2 states that computed autonomously. All of the components of the device, including hardware, software, input, and output, were biomolecules mixed together in solution. The hardware consisted of a restriction nuclease and a ligase; the software (transition rules) and the input were double-stranded DNA oligomers. Computation was carried out by processing the input molecule via repetitive cycles of restriction, hybridization, and ligation reactions to produce a final-state output in the form of a double-stranded DNA molecule. Recently, we have taken the concept of molecular computing a step further by constructing computing devices in which the computation output is a specific biological function rather than a specific molecule. In addition, we markedly increased the levels of complexity and mathematical power of these automata by the V.V. Smider, C.C. Liu, J. Mills, B. Hutchins, B. Leonard e are involved in several projects related to molecular recognition. These include understanding the evolution and biochemical characteristics of the human germ-line antibody repertoire, chemically enhancing antibody properties, analyzing RNA hairpin interactions that regulate DNA replication, and developing new chemical technologies to detect interactions between macromolecules. W DEVELOPMENT AND BIOCHEMICAL PROPERTIES OF THE GERM-LINE IMMUNE REPERTOIRE Some germ-line antibodies recognize certain carbohydrate antigens with high avidity, suggesting that darwinian forces helped shape the antibody repertoire. We have developed techniques to rapidly combinatorially pair heavy and light chains. Using these techniques, we will be able to construct and analyze a large germline repertoire in a spatially addressed format. Libraries 246 MOLECULAR BIOLOGY 2007 will be produced and analyzed for binding to antigens of common human pathogens to provide a repertoire binding “snapshot.” Such data could lend insight into the evolutionary forces behind the development of certain V regions and VH-VL combinations. THE SCRIPPS RESEARCH INSTITUTE ENHANCED AND NOVEL INTERACTIONS: P.G. Schultz, Department of Chemistry, we are engineering other metalloantibodies and are incorporating amino acids with unique side chains (unnatural amino acids) into antibodies. This technology should allow both genetic and chemical expansion of the antibody repertoire for therapeutic and diagnostic applications. ENGINEERED ANTIBODIES R N A H A I R P I N S R E G U L AT I N G D N A R E P L I C AT I O N Antibodies bind their antigens noncovalently. However, for certain diagnostic or therapeutic applications, antibodies that irreversibly bind or modify their antigens would be useful. We recently succeeded in engineering a metal-dependent antibody that irreversibly binds its antigen, TNF-α, perhaps through an exchange inert cobalt complex (Fig. 1). In collaboration with RNA molecules are now recognized as important regulators of many cellular properties via RNA-RNA interactions. One of the simplest and evolutionarily ancient mechanisms of RNA-mediated regulation occurs at plasmid origins of replication. We are applying molecular evolution techniques to these regions and using simple copy number and compatibility assays to ascertain the regulatory and recognition features of these hairpins in Escherichia coli. B I O M O L E C U L A R D E T E C T I O N : O X A L AT E E S T E R CHEMIFLUORESCENCE Sensitive methods such as fluorescence can be used to detect multiple macromolecular interactions simultaneously (multiplex detection). An alternative approach is detection via chemifluorescence, in which a fluorescent dye is activated through chemical transfer of energy. We recently synthesized aqueous oxalate ester compounds capable of exciting fluorescent dyes conjugated to biomolecules (Fig. 2). Unlike fluorescence, chemi- F i g . 1 . An antibody scFv that binds its antigen irreversibly. Top, Western blot shows a complex between an engineered scFv 21H9 and its TNF antigen at 44 kD (lanes 6 and 9) that only forms in the presence of cobalt. Both scFv and TNF are in the complex, as indicated by staining with antibody to TNF (lane 9a) and antibody to scFv (lane 9b). The parental scFv RA72, which binds TNF noncovalently, does not form the 44-kD complex (lanes 1–4). Bottom, Close-up model of the 21H9 scFv (green) with its metal binding site (red) in proximity to mapped linkage regions (pink) of TNF. The TNF trimer is shown as an outset with linkage residues in cyan and pink. F i g . 2 . A water-soluble oxalate ester can activate fluorescently labeled protein. Top, The postulated reaction between an oxalate ester and hydrogen peroxide to produce a dioxetane intermediate, which can transfer energy to a fluorescent dye to produce carbon dioxide and light emission at the dye’s characteristic wavelength. Bottom, Samples of fluorescently labeled bovine serum albumin (BSA-ROX, BSA-JOE, BSA-Cy3, and BSA-Cy5) adsorbed to a membrane and exposed to activated water-soluble oxalate ester emit light of characteristic wavelengths. MOLECULAR BIOLOGY 2007 fluorescence allows integration of the emission signal over time, a situation that may result in highly sensitive detection of molecular interactions. Applications include microarrays, blots, and solid-phase immunoassays in research and diagnostic settings. PUBLICATIONS Leonard, B., Sharma, V., Smider, V. Co-expression of antibody Fab heavy and light chain genes from separate evolved compatible replicons in E. coli. J. Immunol. Methods. 317:56, 2006. Functional Characterization of Enzymes via Combinatorial Libraries THE SCRIPPS RESEARCH INSTITUTE 247 In addition to using the PNA-encoded libraries to discover new enzymes and enzyme functions, we have also used the libraries as an ultra-high-throughput screening platform for the identification of inhibitors and substrates for individual enzymes. A library of more than 500 inhibitors can be easily screened in just a few drops of solution. Another example of functional characterization of protein activity is the profiling of the substrate specificity of cysteine proteases, metacaspases, from Arabidopsis thaliana in collaboration with F. Van Breusegem, Ghent University, Ghent, Belgium. Using a substrate library of approximately 160,000 fluorogenic substrates, we characterized the structure-activity relationship of the metacaspases and facilitated the identification of the first reported endogenous plant protease-inhibitor interaction. J.L. Harris, J. Alves e are developing and using technologies based on small-molecule protein modifiers to profile the active state of enzymes in various biological environments. In collaboration with N. Winssinger, Université Louis Pasteur, Strasbourg, Germany, we have developed an encoding strategy that uses peptide nucleic acid (PNA) sequences. Encoding combinatorial libraries with PNA tags allows not only for capture of synthetic history of the library in the resulting molecule but also for spatial deconvolution of the molecules on DNA microarrays. The PNA tag is covalently cosynthesized with the smallmolecule compound that can interact with enzymes in biological samples. With this approach, we can efficiently synthesize thousands of molecules in the same time it would take to synthesize a single individual molecule. Although the resulting library of thousands of compounds is screened as a mixture, the specific molecules within the library can be easily identified on a DNA microarray by using the hybridization properties of the encoded PNA tag. We have developed PNA-encoded libraries to profile multiple enzyme classes, including cysteine proteases, serine proteases, and, most recently, kinases. The screening platform has also been adapted to profile samples in parallel by using readily available laboratory equipment. These libraries have been applied to various biological systems, including airborne allergens, malaria, and viral samples, and have resulted in the identification and characterization of enzymes within those systems that may result in potential new therapies to combat the associated diseases. W PUBLICATIONS Epple, R., Urbina, H.D., Russo, R., Liu, H., Mason, D., Bursulaya, B., Tumanut, C., Li, J., Harris, J.L. Bicyclic carbamates as inhibitors of papain-like cathepsin proteases. Bioorg. Med. Chem. Lett. 17:1254, 2007. Urbina, H.D., Debaene, F., Jost, B., Bole-Feysot, C., Mason, D.E., Kuzmic, P., Harris, J.L., Winssinger, N. Self-assembled small-molecule microarrays for protease screening and profiling. Chembiochem 7:1790, 2006. Vercammen, D., Belenghi, B., van de Cotte, B., Beunens, T., Gavigan, J.A., De Rycke, R., Brackenier, A., Inze, D., Harris, J.L., Van Breusegem, F. Serpin1 of Arabidopsis thaliana is a suicide inhibitor for metacaspase 9. J. Mol. Biol. 364:625, 2006. Prodrug and Targeting Therapies and Synthesis of Anticancer and Antibacterial Agents S.C. Sinha, R.A. Lerner, Z. Chen, S. De, Z.-Z. Huang ne of our main research interests is antibody catalysis, with emphasis on its applications in organic synthesis and selective drug delivery. In the past, using monoclonal aldolase antibodies 38C2 and 93F3, we synthesized numerous natural products, including highly potent cytotoxic agents such as epothilones and their analogs. Currently, we are developing these antibodies for use in selective cancer chemotherapy. We are using 2 related approaches. In the first, we are using a small-molecule antagonist of the integrin αvβ3 to redirect antibody 38C2 to cancer cells. In the second, we are using antibody 38C2 or 93F3 to catalyze prodrug activation in an “antibody prodrug therapy”; our aim with this method is to reduce indiscriminate toxic effects to normal cells. In addition, we O 248 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE are focusing on the synthesis of anticancer and antibacterial natural products and their analogs. S E L E C T I V E C H E M O T H E R A P Y W I T H C ATA LY T I C ALDOLASE ANTIBODIES Using integrin αvβ3–targeting small molecules and antibody 38C2, we have produced 2 types of 38C2 constructs: catalytic and noncatalytic (Fig. 1). The cata- F i g . 2 . Structure of integrin-targeting compounds for the 38C2- construct formation, doxorubicin prodrugs, the adjacent bis-tetrahydrofuran annonaceous acetogenins and their precursors, and sorangiolide and its macrocyclic lactone intermediates. ment of Molecular Biology, and B. Felding-Habermann, Department of Molecular and Experimental Medicine. F i g . 1 . Schematic drawing of the integrin αvβ3–targeting noncatalytic and catalytic antibody constructs. Abbreviations: Ab, antibody; TA, targeting agent. lytic construct is a classical conjugate of small molecules to 38C2 through the surface lysine residues or the sulfide groups obtained by reduction of the disulfide bridge in the antibody hinge region. In the noncatalytic construct, the compound conjugates in the 38C2 binding sites and redirects the antibody to cells. Using mass spectrometry, we found that both constructs have approximately 2 molecules of a small molecule. In flow cytometry assays, both constructs bound cells expressing integrin αvβ3 with high affinity. Because integrin αvβ3 is expressed on numerous primary and metastatic tumor cells as well as in the vasculature of the tumors, the resulting antibody constructs are expected to be highly useful in cancer therapy. Using MDA-MB-231 cells in a mouse model of breast cancer metastasis, we evaluated the efficacy of the noncatalytic constructs. The animals treated with these constructs had fewer metastases than those treated with the small molecule alone or the control group did. Similarly, the catalytic 38C2 construct (III in Fig. 1) activated doxorubicin prodrugs and caused cytotoxic effects in MDA-MB-231 breast cancer cells in vitro. For the studies with the catalytic construct, we also designed a series of doxorubicin prodrugs (Fig. 2) and evaluated them in vitro. Our selective chemotherapy studies are carried out in collaboration with C.F. Barbas, Depart- SYNTHESIS OF ANTICANCER AND ANTIBACTERIAL AGENTS During the past year, we mainly focused on the synthesis of the anticancer adjacent bis-tetrahydrofuran annonaceous acetogenins and total synthesis of the antibacterial agent sorangiolide (Fig. 2). Annonaceous acetogenins are highly cytotoxic compounds, and each acetogenin can have as many as 64 stereoisomers because of the bis-tetrahydrofuran fragment alone, which is flanked by a hydroxy group on each side. Understanding the structure-activity relationship of these compounds and carrying out comprehensive biological studies require total synthesis of all 64 stereoisomers of each acetogenin. These syntheses have been the focus of research for several years not only by us but also by many others. However, despite a continued effort, synthesis of all 64 stereoisomers of asimicin or bullatacin, which are among the most cytotoxic adjacent bis-tetrahydrofuran acetogenins, has not yet been achieved. We have now prepared 10 bis-tetrahydrofuran precursors that will yield all 64 asimicin stereoisomers. Sorangiolides A and B are 18-membered macrocyclic lactones isolated from Sorangium cellulosum strain So ce12. They have weak antibiotic activity against gram-positive bacteria (e.g., Staphylococcus aureus). Although the structure of these compounds was confirmed by an x-ray analysis of sorangiolide A, no synthetic studies have been reported. Moreover, the relative or absolute stereochemistry of the hydroxy group in MOLECULAR BIOLOGY sorangiolide B at C-6 is yet to be determined. Therefore, to synthesize the naturally occurring sorangiolides and their analogs for biological evaluations, we designed and produced advanced macrocyclic precursors (Fig. 2), which will be converted to the target compounds and their analogs. PUBLICATIONS Abraham, S., Guo, F., Li, L.-S., Rader, C., Liu, C., Barbas, C.F. III, Lerner, R.A. Sinha, S.C. Synthesis of the next-generation therapeutic antibodies that combine cell targeting and antibody-catalyzed prodrug activation. Proc. Natl. Acad. Sci. U. S. A. 104:5584, 2007. Das, S., Abraham, S., Sinha, S.C. Studies toward the total synthesis of sorangiolides and their analogs: a convergent stereoselective synthesis of the macrocyclic lactone precursors. Org. Lett. 9:2273, 2007. Lo, H.C., Han, H., D’Souza, L.J., Sinha, S.C., Keinan, E. Rhenium(VII) oxide-catalyzed heteroacylative ring-opening dimerization of tetrahydrofuran. J. Am. Chem. Soc. 129:1246, 2007. Sinha, S.C., Das, S., Li, L.-S., Lerner, R.A., Barbas, C.F. III. Preparation of integrin αvβ3-targeting Ab 38C2 constructs. Nat. Protoc. 2:449, 2007. Chemical Transformations of Small Molecules and Proteins and Reactions Between Them F. Tanaka, K. Albertshofer, L. Asawapornmongkol, C.F. Barbas III, R.P. Fuller, H.-M. Guo, M. Minakawa, S.S.V. Ramasastry, N. Utsumi, H. Zhang e create and develop molecules and methods that contribute to basic biomedical research and to the development of drug candidates and therapeutic strategies. One aspect of our research is the development of new strategies and methods for labeling of proteins with small molecules at certain positions. We are creating protein and peptide tags that selectively form covalent bonds with designed synthetic molecules; these tags are generated by using reaction-based selections from combinatorial libraries of proteins and peptides. When the tag is fused to a protein of interest, the fusion protein can be modified at the tag sequence through the reaction with the designed synthetic compound. Designed compounds can be conjugated with fluorescent molecules and drug molecules by chemical synthesis; therefore, through use of this labeling system, a variety of molecules can be covalently and selectively attached to a protein of interest. We are also developing synthetic labeling molecules that selectively and covalently react with target W 2007 THE SCRIPPS RESEARCH INSTITUTE 249 proteins without preengineering of proteins and without need for fusion to a tag sequence. In this strategy, labeling molecules contain a moiety that selectively and covalently reacts with a specific amino acid residue (e.g., tyrosine) and a moiety that noncovalently interacts with a target protein. Specificity for the target protein is imparted by noncovalent interaction. With this system, naturally occurring proteins of interest can be selectively tagged with synthetic molecules in natural environments, including cells and animals. When a labeling molecule contains a moiety that binds to the surface of a protein, the protein can be labeled without affecting the protein’s function. On the other hand, when the specificity of a labeling molecule is imparted through a ligand that binds to the active site of an enzyme, the resulting molecule can be an efficient, selective, irreversible inhibitor of the enzyme. For example, we are developing labeling molecules and irreversible inhibitors for carbonic anhydrases. These molecules will be useful for study of carbonic anhydrases and may also lead to the development of efficient inhibitors of these enzymes for treatment of diseases, including cancer. We are also developing synthetic methods for concise access to functionalized molecules in regioselective, diastereoselective, and enantioselective fashions. These methods are useful for syntheses of bioactive molecules, ligands of proteins, and candidates of these molecules. Overall, our research provides insight into the construction of functional molecules and into biological functions of naturally occurring molecules such as proteins and enzymes. PUBLICATIONS Albertshofer, K., Thayumanavan, R., Utsumi, N., Tanaka, F., Barbas, C.F. III. Amine-catalyzed Michael reactions of an aminoaldehyde derivative to nitroolefins. Tetrahedron Lett. 48:693, 2007. Chowdari, N.S., Ahmad, M., Albertshofer, K., Tanaka, F., Barbas, C.F. III. Expedient synthesis of chiral 1,2- and 1,4-diamines: protecting group dependent regioselectivity in direct organocatalytic asymmetric Mannich reactions. Org. Lett. 8:2839, 2006. Mase, N., Watanabe, K., Yoda, H., Takabe, K., Tanaka, F., Barbas, C.F. III. Organocatalytic direct Michael reaction of ketones and aldehydes with β-nitrostyrene in brine [published addition/correction appears in J. Am. Chem. Soc. 128:17153, 2006]. J. Am. Chem. Soc. 128:4966, 2006. Ramasastry, S.S.V., Zhang, H., Tanaka, F., Barbas, C.F. III. Direct catalytic asymmetric synthesis of anti-1,2-amino alcohols and syn-1,2-diols through organocatalytic anti-Mannich and syn-aldol reactions. J. Am. Chem. Soc. 129:288, 2007. Tanaka, F., Barbas, C.F. III. Aldol and Mannich-type reactions. In: Enantioselective Organocatalysis: Reactions and Experimental Procedures. Dalko, P.I. (Ed.). WileyVCH, Weinheim, Germany, 2007, p. 19. Tanaka, F., Fuller, R. Control of function of a small peptide by a protein. Bioorg. Med. Chem. Lett. 16:4059, 2006. 250 MOLECULAR BIOLOGY 2007 Tanaka, F., Fuller, R., Asawapornmongkol, L., Warsinke, A., Gobuty, S., Barbas, C.F. III. Development of a small peptide tag for covalent labeling of proteins. Bioconjug. Chem. 18:1318, 2007. Tanaka, F., Jones, T., Kubitz, D., Lerner, R.A. Anti-formyl peptide antibodies. Bioorg. Med. Chem. Lett. 17:1943, 2007. Utsumi, N., Zhang, H., Tanaka, F., Barbas, C.F. III. A way to highly enantiomerically enriched aza-Morita-Baylis-Hillman-type products. Angew. Chem. Int. Ed. 46:1878, 2007. Zhang, H., Mifsud, M., Tanaka, F., Barbas, C.F. III. 3-Pyrrolidinecarboxylic acid for direct catalytic asymmetric anti-Mannich-type reactions of unmodified ketones. J. Am. Chem. Soc. 128:9630, 2006. Structure, Function, and Applications of Virus Particles J.E. Johnson, M. Banerjee, Z. Chen, I. Gertsman, R. Huang, R. Khayat, G. Lander, J. Lanman, K.K. Lee, T. Matsui, P. Natarajan, A. Odegard, J. Speir, R. Taurog e investigate model virus systems that provide insights for understanding viral assembly, maturation, entry, localization, and replication. We have also developed viruses as reagents for applications in nanomedicine, chemistry, and biology. We investigate viruses that infect bacteria, insects, plants, and the extreme thermophile Sulfolobus. These viruses have genomes of single-stranded RNA, and double-stranded DNA. We use a variety of physical methods to investigate structure-function relationships, including singlecrystal x-ray diffraction, static and time-resolved solution x-ray diffraction, electron cryomicroscopy and image reconstruction, mass spectrometry, structure-based computational analyses, and methods associated with thermodynamic characterization of virus particles and their transitions. Biological methods we use include the genetic engineering of viral genes and their expression in Escherichia coli, mammalian cells, insect cells, and yeast and the characterization of these gene products by physical methods. For cytologic studies of viral entry and infection, we use fluorescence and electron microscopy and particles assembled in heterologous expression systems. Our studies depend on extensive consultations and collaborations with others at Scripps Research, including groups led by B. Carragher, M.G. Finn, M. Manchester, D.R. Millar, C. Potter, V. Reddy, A. Schneemann, G. Siuzdak, J.R. Williamson, and M.J. Yeager, and a variety of groups outside of Scripps. W DOUBLE-STRANDED DNA VIRUSES HK97 is a double-stranded DNA virus similar to bacteriophage λ. It undergoes a remarkable morpho- THE SCRIPPS RESEARCH INSTITUTE genesis in its assembly and maturation, and this process can be recapitulated in vitro. We determined the atomic resolution structure of the 650-Å mature, head II particle and discovered the mechanism used to concatenate the subunits of the particle into a chain-mail fabric similar to that seen in armor of medieval knights. In the past year, we focused on the dynamics of maturation. Prohead II is a 500-Å metastable intermediate at pH 7 that can be induced to begin maturation by lowering the pH to 4. Solution x-ray scattering and single-molecule fluorescence showed that the initial transition to a particle of about 560 Å occurs as a highly cooperative, stochastic event with no detectable intermediates that takes place in less than 1 second for an individual particle. A quorum of cross-links must form in this particle to generate the second expansion intermediate (about 650 Å), which also forms cooperatively with no detectable intermediates. At pH 4, formation of cross-links continues, with 360 formed per particle. The late stage of maturation is a classic Brownian ratchet in which pentameric subunits fluctuate like a piston through a radial trajectory of 15 Å and are trapped at the top of the trajectory by formation of the covalent cross-link. If the cross-link can not form, the maturation stops with the pentamers still sampling the trajectory. Bacteriophage P22 is the prototype of the Podoviridae that are characterized by a T = 7 capsid with a short tail structure incorporated into a unique 5-fold vertex. We determined an asymmetric reconstruction of this particle that revealed spooled DNA, the dodecameric portal, and the location of the 9 gene products known to be in the particle. Recently, structures of bacteriophage λ were determined at subnanometer resolution by electron cryomicroscopy. These structures showed that the fold of the capsid protein is the same as that of the HK97 subunit. Sulfolobus turreted icosahedral virus is an archaeal virus isolated from Sulfolobus, which grows in the acidic hot sulfur springs (pH 2–4, 72°C–92°C) in Yellowstone National Park. An electron cryomicroscopy reconstruction of the virus showed that the capsid has pseudo T = 31 quasi symmetry and is 1000 Å in diameter, including the pentons. The x-ray structure of the major capsid protein of the virus revealed a fold nearly identical to the folds of the major capsid proteins of the eukaryotic adenoviruses and PRD-1, a virus that infects bacteria. These findings indicate a viral phylogeny that spans the 3 domains of life. Difference electron density maps in which the x-ray model MOLECULAR BIOLOGY 2007 is subtracted from the electron cryomicroscopy density clearly show an internal membrane in which the capsid proteins are anchored. THE SCRIPPS RESEARCH INSTITUTE 251 Design and Informatics in Structural Virology SINGLE-STRANDED RNA VIRUSES Flock House virus is a T = 3, single-stranded RNA virus that infects Drosophila. We are studying viral entry and early expression and assembly of the capsid protein. Recently, studies on viral entry indicated the presence of an “eluted” particle early in infection that has initiated its disassembly program but is then eluted back into the medium. We did a phenotypic characterization of the particles, and we are using electron cryomicroscopy to study them. For studies on the expression and assembly of the capsid protein, we are using tetra-cysteine tags inserted genetically in the capsid protein that allow the freshly made proteins to be optically visualized with a fluorophore and in the electron microscope with photoconversion of the fluorophore. Recently, high-pressure freezing of infected cells revealed exceptionally detailed features of viral entry and regions of replication within the cell. Tomographs prepared with the micrographs show that translation of the RNA encoding the capsid protein and the assembly of virions takes place within chambers created by remodeled mitochondria. Refined atomic models of tetravirus structures and structure-based mutagenesis combined with highly sensitive assays for defining phenotypes have revealed the electrostatic principles of maturation for the T = 4 tetraviruses. PUBLICATIONS Cheung, C.L., Chung, S.W., Chatterji, A., Lin, T., Johnson, J.E., Hok, S., Perkins, J., De Yoreo, J.J. Physical controls on directed virus assembly at nanoscale chemical templates. J. Am. Chem. Soc. 128:10801, 2006. Gan, L., Speir, J.A., Conway, J.F., Lander, G., Cheng, N., Firek, B.A., Hendrix, R.W., Duda, R.L., Liljas, L., Johnson, J.E. Capsid conformational sampling in HK97 maturation visualized by x-ray crystallography and cryo-EM. Structure 14:1655, 2006. Johnson, J.E., Chiu, W. DNA packaging and delivery machines in tailed bacteriophages. Curr. Opin. Struct. Biol. 17:237, 2007. Maia, L.F., Soares, M.R., Valente, A.P., Almeida, F.C., Oliveira, A.C., Gomes, A.M., Freitas, M.S., Schneemann, A., Johnson, J.E., Silva, J.L. Structure of a membrane-binding domain from a non-enveloped animal virus: insights into the mechanism of membrane permeability and cellular entry. J. Biol. Chem. 281:29278, 2006. Martin, B.D., Soto, C.M., Blum, A.S., Sapsford, K.E., Whitley, J.L., Johnson, J.E., Chatterji, A., Ratna, B.R. An engineered virus as a bright fluorescent tag and scaffold for cargo proteins: capture and transport by gliding microtubules. J. Nanosci. Nanotechnol. 6:2451, 2006. Poliakov, A., van Duijn, E., Lander, G., Fu, C.Y., Johnson, J.E., Prevelige, P.E., Jr., Heck, A.J. Macromolecular mass spectrometry and electron microscopy as complementary tools for investigation of the heterogeneity of bacteriophage portal assemblies. J. Struct. Biol. 157:371, 2007. V.S. Reddy, G.V. Subbarao, S. Venkataraman, M. Tripp, P. Singh, R. Mannige, I. Borelli, J. Loo, S. Kumar e are interested in identifying and understanding the structural underpinnings and requirements for formation and function of viral capsids. We use this information to design novel protein shells that polyvalently display multiple copies of peptides or proteins of interest. We use structural, computational, bioinformatics, and genetic methods. Viruses are highly evolved macromolecular assemblages that perform a variety of functions during their life cycle, including self-assembly into uniform capsids, selective packaging of the genome, binding to host cells, and delivery of the genetic material to the targeted cells. Simple viruses, such as nonenveloped viruses, form capsids with homogeneous composition and quaternary architecture. Hence, these viruses are useful for structural and functional analyses. In collaboration with G.R. Nemerow, Department of Immunology, we are using x-ray crystallography to determine the structure of the entire human adenovirus particle, currently at 7.5-Å resolution. Acquisition of high-resolution data is under way. We recently determined the structure of Seneca Valley virus, which belongs to a new genus (Senecavirus) of the Picornaviridae family. Senecaviruses are of particular interest because they are selectively pathogenic to cancer cells. This research was done in collaboration with scientists at Neotropix, Inc., Malvern, Pennsylvania. We continue to maintain and expand the virus structural database, namely VIPERdb (http://viperdb .scripps.edu), a Web portal for structures and associated structural properties of viral capsids. The capsid structures in the database were analyzed in terms of protein-protein interactions, contacting residue pairs, association energies, individual residue contributions, and surface characteristics by using computational methods. The results of the analysis are stored in the database. VIPERdb is being developed and maintained as part of the Multiscale Modeling Tools for Structural Biology, a National Institutes of Health research resource headed by C.L. Brooks, Department of Molecular Biology. We are also actively involved in generating novel vaccines against cytotoxins such as ricin and against pathogens by expressing antigenic regions of patho- W 252 MOLECULAR BIOLOGY 2007 genic molecules on the surfaces of viral capsids. Currently, tomato bushy stunt virus–like capsids are our display platform of choice. A unique subunit fold of the viral subunit enables the attachment of peptides or proteins of interest to the C terminus in a linear fashion and their display on the capsid surface. Such reagents act as multivalent decoys of the pathogenic molecules and so can be used as potential vaccines. PUBLICATIONS Kumar, S., Ochoa, W., Kobayashi, S., Reddy, V.S. Presence of a surface-exposed loop facilitates trypsinization of particles of Sinsiro virus, a genogroup II.3 norovirus. J. Virol. 81:1119, 2007. Nguyen, H.D., Reddy, V.S., Brooks, C.L. III. Deciphering the kinetic mechanism of spontaneous self-assembly of icosahedral capsids. Nano Lett. 7:338, 2007. Biology and Applications of Icosahedral Viral Capsids A. Schneemann, B. Groschel, D.J. Manayani, D. Marshall, J.E. Petrillo, M.E. Siladi, P.A. Venter oat proteins of nonenveloped, icosahedral viruses perform multiple functions during the course of viral infection, including capsid assembly, specific encapsidation of the viral genome, binding to a cellular receptor, and uncoating. In some viruses, a single type of protein is sufficient to carry out these functions; we are interested in the determinants that endow a polypeptide chain with such versatility. We seek to harness this versatility for novel applications of viruses in biotechnology and nanotechnology. We focus on a structurally and genetically well-characterized virus family, the T = 3 nodaviruses. Nodaviruses are composed of 180 copies of a single coat protein and 2 strands of positive-sense RNA. Currently, we are elucidating the mechanism by which the 2 genomic RNAs are packaged into a single virion. Our long-term goal is to develop nodaviruses as RNA packaging and delivery vectors. Our data indicate that the 2 viral RNAs are recognized separately, but it is not yet known whether packaging occurs sequentially and whether one or more coat protein subunits are involved in this process. Interestingly, we found that RNA genome packaging is coupled to genome replication, suggesting potential approaches for packaging of foreign RNAs. Specific packaging of the viral genome also requires coat protein translated from newly synthesized viral RNA. Thus, genome replication, RNA packaging, and C THE SCRIPPS RESEARCH INSTITUTE viral assembly are tightly coupled processes. The coupling of these processes may be a safety mechanism for the virus to ensure efficient and accurate formation of progeny virions in infected cells. We are also investigating the mechanism by which nodaviral protein B2 suppresses RNA silencing in infected cells. We are identifying the double-stranded RNAs that serve as substrates for B2 binding during nodavirus infection, and we are correlating these results with data from confocal microscopy and immunoprecipitation studies. We have identified amino acid residues in B2 that are critical for the protein’s function as an RNA-binding protein; preliminary data suggest that B2 may have a second function during the viral replication cycle that is unrelated to its function as a suppressor of RNA silencing. We are also collaborating with several investigators at Scripps Research, the Salk Institute, La Jolla, California, and Harvard University, Boston, to develop nodaviruses as platforms for delivery of anthrax antitoxins. To this end, we are using particles to display the VWA domain of capillary morphogenesis protein 2, the cellular receptor for anthrax toxin, in a multivalent fashion on the surface of the virion. Two insertion sites yielding different patterns of 180 copies of the VWA domain were selected on the basis of computational modeling of the high-resolution crystal structure of Flock House virus, an insect nodavirus. The resulting chimeric viruslike particles functioned as a potent anthrax antitoxin in cell culture and protected rats from challenge with lethal toxin. This research is important because it shows that protein domains containing more than 150 amino acids can be displayed on Flock House virus in a biologically functional form, suggesting numerous additional applications. Moreover, chimeric particles decorated with anthrax protective antigen elicited a potent neutralizing antibody response against the antigen that protected rats from challenge with lethal toxin 4 weeks after a single immunization without adjuvants. This chimeric particle platform is a dually acting reagent for the treatment of and protection against anthrax. PUBLICATIONS Go, E.P., Wikoff, W.R., Shen, Z., O’Maille, G., Morita, H., Conrads, T.P., Nordstrom, A., Trauger, S.A., Uritboonthai, W., Lucas, D.A., Chan, K.C., Veenstra, T.D., Lewicki, H., Oldstone, M.B., Schneemann, A., Siuzdak, G. Mass spectrometry reveals specific and global molecular transformations during viral infection. J. Proteome Res. 5:2405, 2006. Maia, L.F., Soares, M.R., Valente, A.P., Almeida, F.C., Oliveira, A.C., Gomes, A.M., Freitas, M.., Schneemann, A., Johnson, J.E., Silva, J.L. Structure of a membranebinding domain from a non-enveloped animal virus: insights into the mechanism of membrane permeability and cellular entry. J. Biol. Chem. 281:29278, 2006. MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 253 ur research involves the molecular characterization of retroviruses and the development of ways to interfere with the retroviral life cycle. In particular, we use feline immunodeficiency virus (FIV) for the study of lentivirus infections. FIV causes an AIDSlike syndrome in domestic cats and has structural and functional similarities to HIV, the cause of AIDS in humans. Thus, developing ways to interfere with FIV infection may result in useful treatments for infections in both cats and humans. Our primary interests continue to be the molecular characterization of receptor interactions and the molecular basis for the development of drug resistance in the critical aspartic protease encoded as part of the enzyme cassette of all retroviruses. tionary conservation of the mechanism of infection by FIV and HIV, even though these 2 lentiviruses use different binding receptors. Results to date in both lentivirus systems support the notion that this mechanism protects certain epitopes on the surface glycoprotein from immune surveillance until the moment of virus binding and entry into the cell. We used a panel of neutralizing monoclonal antibodies to map the region of gp95 in which these CD134-dependent neutralizing epitopes reside. In more recent studies, we used synthetic peptides containing the antibody-reactive region to map the epitopes recognized by the neutralizing antibodies. A cluster of these epitopes resides in the variable loop 3 of FIV gp95, consistent with the notion that this region is central to CD134 receptor binding. We previously mapped regions of feline CD134 receptor involved in interaction with gp95 by using chimeric proteins consisting of feline and human CD134 (the human homolog does not bind FIV glycoprotein) and site-directed mutagenesis. During the past year, we did similar mapping studies on gp95 to further define regions critical to CD134 and CXCR4 binding. So far, the results are consistent with the idea that variable loop 3 is the contact region for binding to both receptors. Cocrystallization studies are in progress to determine the structure of the region surrounding the antibody-binding sites. We are also using these antibodies to develop specific agents that interfere with receptor binding and may be useful as therapeutic agents. RECEPTOR STUDIES P R O T E A S E D R U G R E S I S TA N C E Like acute strains of HIV, FIV uses the chemokine receptor CXCR4 to enter the target cell, the CD4 + T lymphocyte. However, both HIV and FIV have other primary binding receptors that bind the virus as a prelude to interaction with the entry receptor. We think that these other receptors increase the effective local concentration of the incoming virus and alter the conformation of the surface glycoprotein to increase the binding affinity of CXCR4. Whereas HIV uses the cellsurface protein CD4 as a primary binding receptor, FIV uses the activation antigen CD134. CD134 is expressed on activated CD4+ T cells, a finding that explains why FIV can infect and kill CD4+ T cells, even though the virus does not bind CD4. We have shown that interaction of the FIV surface glycoprotein gp95 with CD134 causes a conformational change in gp95. This conformational change increases the affinity of gp95 for CXCR4; similar changes occur when HIV gp120 binds CD4. Thus, there is an evolu- The aspartic protease of lentiviruses is responsible for processing the viral Gag and Pol polyproteins that must occur at the proper time and in the proper sequence in order to generate infectious virus. Drugs against the HIV protease are key components of highly active antiretroviral therapy, a treatment regimen used successfully to treat, but not cure, patients infected with HIV. Both FIV and HIV encode an aspartic protease and although the FIV and HIV proteases are structurally similar, the 2 enzymes have unique sequence-cleavage properties. We have used the parallels and differences between FIV and HIV proteases to better understand the molecular determinants that govern substrate/inhibitor selectivity. We hope that our results will define the limits of plasticity of the 2 enzymes and lead to insights into the development of drug resistance. As reported previously, we showed that the number of amino acid residues involved in the sensitivity of the proteases to drugs is limited, and we can markedly Thiéry, R., Cozien, J., Cabon, J., Lamour, F., Baud, M., Schneemann, A. Induction of a protective immune response against viral nervous necrosis in the European sea bass Dicentrarchus labrax by using betanodavirus virus-like particles. J. Virol. 80:10201, 2006. Venter, P.A., Schneemann, A. Assembly of two independent populations of Flock House virus particles with distinct RNA packaging characteristics in the same cell. J. Virol. 81:613, 2007. Venter, P.A., Schneemann, A. Nodaviridae. In: Encyclopedia of Virology, 3rd ed. Mahy, B.W.J., van Regenmortel, M. (Eds.). Academic Press, San Diego, in press. Molecular Biology of Retroviruses J.H. Elder, Y.C. Lin, M. Sundstrom, M. Giffin,* C.D. Stout, B.E. Torbett,* J. Gatchalian, I.A. Kim * Department of Molecular and Experimental Medicine, Scripps Research O 254 MOLECULAR BIOLOGY 2007 change the sensitivity of the FIV protease to be more like that of the HIV protease by changing as few as 4 amino acids around the active site. However, changing the substrate-cleavage specificity requires substantially more changes. These findings explain how the virus, when an infection is treated with a drug, can mutate to avoid the drug but retain sufficient substrate-cleavage specificity to allow proper Gag/Pol processing and generation of infectious virus. Critical to this process is maintaining the proper order of site cleavage in Gag/Pol, and changes in this order result in generation of noninfectious virus. We think that altering the order of cleavage, in addition to blocking protease activity, may be useful as a novel intervention strategy. PUBLICATIONS Heaslet, H., Lin, Y.C., Tam, K., Torbett, B.E., Elder, J.H., Stout, C.D. Crystal structure of an FIV/HIV chimeric protease complexed with the broad-based inhibitor, TL-3. Retrovirology 4:1, 2007. Lin, Y.C., Brik, A., de Parseval, A., Tam, K., Torbett, B.E., Wong, C.H., Elder, J.H. Altered Gag polyprotein cleavage specificity of feline immunodeficiency virus/human immunodeficiency virus mutant proteases as demonstrated in a cellbased expression system. J. Virol. 80:7832, 2006. Manuell, A.L., Beligni, M.V., Elder, J.H., Siefker, D.T., Tran, M., Weber, A., McDonald, T.L., Mayfield, S.P. Robust expression of a bioactive mammalian protein in Chlamydomonas chloroplast. Plant Biotechnol. J., 5:402, 2007. Whiting, M., Tripp, J.C., Lin, Y.C., Lindstrom, W., Olson, A.J., Elder, J.H., Sharpless, K.B., Fokin, V.V. Rapid discovery and structure-activity profiling of novel inhibitors of human immunodeficiency virus type 1 protease enabled by the copper(I)-catalyzed synthesis of 1,2,3-triazoles and their further functionalization. J. Med. Chem. 49:7697, 2006. THE SCRIPPS RESEARCH INSTITUTE tethered to a reporter or sensitizer, are designed to bind specifically to the active-site channel of a given enzyme. Wires specific for the substrate- or cofactorbinding sites of a given enzyme will be useful tools for inhibitor discovery, phototriggered enzyme turnover, and molecular evolution strategies. We are developing a series of tethered substrates with variable linkers and affinity tags as reporters of binding interactions within the substrate access channel of P450cam. In the past, we showed that these P450 wires induce a range of conformational changes in the helices adjacent to the enzyme’s active site. On the basis of these findings, we have designed a new generation of P450 wires, and we have recently solved the crystal structure of P450cam bound to a wire containing both substrate and a biotin affinity tag. In addition, we have successfully showed phage display of P450cam as a C-terminal fusion to the g3 phage protein. These results will enable an unprecedented structural view of phage display evolution experiments with P450. We have also made significant progress in designing photoactive probes specific for the pterin site of murine inducible NOS. Addressing the pterin site through a molecular wire may allow photochemical triggering of enzyme turnover and help define the role of the cofactor in catalysis. We have synthesized the series of Ru(II)-pterin wires (Fig. 1) and have shown that at least Metalloenzyme Engineering D.B. Goodin, C.D. Stout, E.C. Glazer, R.F. Wilson, A. Annalora, S. Vetter, A.-M. Hays he purposes of our research are to understand the diversity of metalloenzyme catalysts and to develop methods for directed evolution of enzymes with novel function. Our goal is to gain sufficient control over substrate-enzyme interactions and the subsequent oxidative chemistry catalyzed by hypervalent heme cofactors to allow directed evolution of catalysts capable of regiospecific and stereospecific oxidation of a given target substrate. We use a number of techniques in structural biology and spectroscopy and strategies of rational protein redesign and molecular evolution. In the past year, we focused our efforts on developing synthetic molecular wires to probe the active site and function of P450cam and nitric oxide synthase (NOS). These wires, which consist of substrate analogs T F i g . 1 . Pterin-Ru(II) wires synthesized as probes of the pterin site of NOS. one of the wires binds to murine inducible NOS. Binding was confirmed by heme-induced quenching of either pterin or Ru(II) fluorescence upon interaction with the heme domain of inducible NOS, and this quenching was reversed by binding of the natural pterin cofactor. Timeresolved emission experiments showed bound and free forms of the wire and allowed estimation of the affinity MOLECULAR BIOLOGY 2007 and the distance between the Ru(II) and heme. Our findings are consistent with the modeled geometry of the wire within the pterin-binding site (Fig. 2). Ongoing F i g . 2 . Model of a pterin-Ru(II) wire docked into the structure of inducible NOSheme. studies of the structure and phototriggered redox behavior of these wires bound to inducible NOS will provide a new approach to probing the mechanism of this important enzyme. PUBLICATIONS Contakes, S.M., Nguyen, Y.H.L., Gray, H.B., Glazer, E.C., Hays, A.-M.A., Goodin, D.B. Conjugates of heme-thiolate enzymes with photoactive metal-diimine wires. In: Structure and Bonding. Yam, V.W.W. (Ed.). Springer, New York, in press. Control of Cell Division THE SCRIPPS RESEARCH INSTITUTE 255 this protein kinase and its analogs are ubiquitous in eukaryotic cells and central to a number of aspects of control of cell-cycle progression. One area of interest is regulation of cellular morphogenesis by Cdk1. The activity of Cdk1 driven by mitotic cyclins modulates polarized growth in yeast cells. Specifically, these activities depolarize growth by altering the actin cytoskeleton. We found that several proteins that modulate actin structure are targeted by Cdk1, and we are investigating how these phosphorylation events control actin depolarization and cell shape. While investigating mitosis in yeast, we found that Cks1, a small Cdk1-associated protein, appears to regulate the proteasome. Proteasomes are complex proteases that target ubiquitylated proteins, including important cell-cycle regulatory proteins. Surprisingly, we found that Cks1 regulates a nonproteolytic function of proteasomes, the transcriptional activation of CDC20. Specifically, Cks1 is required to recruit proteasomes to the gene CDC20 for efficient transcriptional elongation. Our investigations of CDC20 have led to the conclusion that Cks1 is required for recruitment of proteasomes to and transcriptional elongation of many other genes as well. Currently, we are elucidating the mechanism whereby Cks1 recruits proteasomes and facilitates transcriptional elongation. Our most recent results suggest that Cks1 and proteasomes in conjunction with Cdk1 mediate remodeling of chromatin by removing nucleosomes. CONTROL IN MAMMALIAN CELLS S.I. Reed, C. Baskerville, L.-C. Chuang, M. Henze, J. Keck, V. Liberal, K. Luo, B. Olson, S. Ekholm-Reed, S. Rudyak, F. van Drogen, J. Wohlschlegel iological processes of great complexity can be approached by beginning with a systematic genetic analysis in which the relevant components are first identified and the consequences of selectively eliminating the components via mutation are investigated. We have used yeast, which is uniquely tractable to this type of analysis, to investigate control of cell division. It has been apparent for some time that the most central cellular processes throughout the eukaryotic phylogeny are highly conserved in terms of both the regulatory mechanisms used and the proteins involved. Thus, it has been possible in many instances to generalize from yeast cells to human cells. B CONTROL IN YEAST We have focused on the role and regulation of the Cdc28 protein kinase (Cdk1). Initially identified by means of a mutational analysis of the yeast cell cycle, We showed previously that the human homologs of the Cdc28 protein kinase are so highly conserved, structurally and functionally, relative to the yeast protein kinase, that they can function and be regulated properly in a yeast cell. Analyzing control of the cell cycle in mammalian cells, we produced evidence for the existence of regulatory schemes, similar to those elucidated in yeast, that use networks of both positive and negative regulators. A principal research focus is the positive regulator of Cdk2, cyclin E. Cyclin E is often overexpressed and/or deregulated in human cancers. Using a tissue culture model, we showed that deregulation of cyclin E confers genomic instability, probably explaining the link to carcinogenesis. The observation that deregulation of cyclin E confers genomic instability has led us to hypothesize a mechanism of cyclin E–mediated carcinogenesis based on accelerated loss of heterozygosity at tumor suppressor loci. We are testing this hypothesis in transgenic mouse models. We showed that a cyclin E trans- 256 MOLECULAR BIOLOGY 2007 gene expressed in mammary epithelium significantly increases loss of heterozygosity at the p53 locus, leading to enhanced mammary carcinogenesis. We are extending these investigations by using mouse prostate, testis, and skin models. In an attempt to understand cyclin E–mediated genomic instability, we are investigating how deregulation of cyclin E affects both S phase and mitosis. Recent data suggest that deregulation of cyclin E impairs DNA replication by interfering with assembly of the prereplication complex. Cyclin E deregulation also impairs the transition from metaphase to anaphase by promoting the accumulation of inhibitors of anaphase. Our interest in cyclin E deregulation in cancer led us to investigate the pathway for turnover of cyclin E. We showed that phosphorylation-dependent proteolysis of cyclin E depends on a protein-ubiquitin ligase known as SCFhCdc4. The F-box protein hCdc4 is the specificity factor that targets phosphorylated cyclin E. We are investigating how ubiquitylation of cyclin E is coordinated with other processes required for its degradation, including prolyl isomerization. We are also investigating SCFhCdc4 ubiquitylation of other important cellular proteins. Recently, we began determining the role of SCFhCdc4 in neurodegenerative disease. We found that parkin, a protein often mutated in hereditary Parkinson’s disease, regulates the stability of hCdc4, possibly leading to neuropathologic changes. We discovered an SCFhCdc4 substrate, peroxisome proliferator–activated receptor γ coactivator-1, that may be the effector of hCdc4 deregulation in Parkinson’s disease. In addition, we showed that SCFhCdc4 regulates the turnover of presenilins in the brain, proteins strongly implicated in Alzheimer’s disease. Another area of interest is the role of Cks proteins in mammals, complementing our research in yeast. Mammals express 2 paralogs of yeast Cks1, known as Cks1 and Cks2. Experiments in mice lacking the gene for Cks1 and Cks2 revealed that each paralog has a specialized function. Cks1 is required as a cofactor for Skp2-mediated ubiquitylation and turnover of inhibitors p21, p27, and p130. Cks2 is required for the transition from metaphase to anaphase in both male and female meiosis I. Nevertheless, mice nullizygous at the individual loci are viable. However, doubly nullizygous mice have not been observed because embryos die at the morula stage, a finding consistent with an essential redundant function. We found that this function probably is involved in regulation of transcription and linked to chromatin remodeling, as in yeast. THE SCRIPPS RESEARCH INSTITUTE PUBLICATIONS Keck, J.M., Summers, M.K., Tedesco, D., Ekholm-Reed, S., Chuang, L.-C., Jackson, P.K., Reed S.I. Cyclin E overexpression impairs progression through mitosis by inhibiting APCCdh1. J. Cell Biol., in press. Keller, U.B., Old, J.B., Dorsey, F.C., Nilsson, J.A., Nilsson, L., Maclean, K.H., Chung, L., Yang, C., Spruck, C., Boyd, K., Reed S.I., Cleveland, J.L. Myc targets Cks1 to provoke the suppression of p27Kip1, proliferation and lymphomagenesis. EMBO J. 26:2562, 2007. Smith, A.P.L., Henze, M., Lee, J.A., Osborn, K.G., Keck, J., Tedesco, D., Bortner, D.M., Rosenberg, M.P., Reed, S.I. Deregulated cyclin E promotes p53 loss of heterozygosity and tumorigenesis in the mouse mammary gland. Oncogene 25:7245, 2006. Control of Gene Expression During the Cell Cycle and in Response to Environmental Stimuli C. Wittenberg, R.A.M. de Bruin, M. Guaderrama, T.I. Kalashnikova he dynamism and plasticity of biological systems depend on the capacity to rapidly alter the abundance and activities of cellular constituents. That capacity depends, in large part, on the ability to rapidly modulate gene expression. Recently, we have focused on the mechanisms by which cells exert control over gene expression to regulate cell proliferation and to respond to changes in environmental conditions. T R E G U L AT I O N O F C E L L P R O L I F E R AT I O N In most cells, commitment to a new round of cell division during the G1 phase of the cell cycle is accompanied by the activation of a large family of genes that encode activities involved in the duplication and segregation of cellular components. G1-specific genes also encode regulatory factors that promote subsequent cell-cycle events. In the budding yeast Saccharomyces cerevisiae, G1-specific genes are regulated by either the SBF or the MBF transcription factor. In collaboration with J.R. Yates, Department of Cell Biology, we used mass spectrometry–based multidimensional protein identification technology to identify novel regulators of these transcription factors. SBF acts as a transcriptional activator and promotes expression of its targets specifically during the G1 interval. We established that promoter-bound SBF associates with the Whi5 repressor during early G 1 phase and that Whi5 is inactivated via phosphorylation by a G 1 -specific cyclin-dependent protein kinase, thereby activating transcription (Fig. 1). This regulation is analo- MOLECULAR BIOLOGY 2007 F i g . 1 . Model for G 1-specific transcriptional regulation in yeast and humans. The recent identification of novel regulators of G1-specific transcription in yeast confirms the striking functional homology between the apparently unrelated transcriptional regulatory networks in yeast and humans. In budding yeast, the SBF transcription factor is repressed during early G 1 phase by the transcriptional inhibitor Whi5. Phosphorylation of Whi5 by a cyclin-dependent kinase promotes its release from SBF, leading to activation of G1-specific transcription. In mammalian cells, the tumor suppressor Rb represses E2F-dependent transcription in early G1 phase. Rb, like Whi5, is inactivated by phosphorylation by a cyclin-dependent kinase, initiating the G1-to-S transition. A second class of G1-specific genes, regulated by MBF, is inactivated upon exit from the G1 phase. The corepressor Nrm1, which is conserved between the distantly related budding and fission yeasts, accumulates as cells progress into late G1 phase and binds to MBF at target promoters, thereby repressing transcription. Similarly, in mammalian cells, the expression of some G1-specific genes is inactivated by E2F4 in collaboration with p107, an E2F1 target. Phosphorylation of Nrm1 via the DNA replication checkpoint in response to replication stress results in dissociation of Nrm1 from MBF-regulated promoters and derepression of MBF targets. It remains to be established whether the DNA structure checkpoints regulate G1-specific transcription via corepressors of E2F in mammalian cells. gous to the regulation of E2F by the tumor suppressor Rb in humans. In contrast, MBF acts primarily as a transcriptional repressor and limits transcription of target genes to the G1 phase. We identified Nrm1, a novel MBF-associated corepressor. When expressed as an MBF target during late G1 phase, Nrm1 associates with MBF at target promoters and represses expression as cells enter S phase (Fig. 1). Similarly, the Nrm1 homolog, SpNrm1, in the fission yeast Schizosaccharomyces pombe regulates MBF, the sole G 1-specific transcription factor in this yeast. In addition to being regulated during the cell cycle, the G1-specific transcriptional machinery is regulated by checkpoints that monitor the integrity of cellular structures and processes. When replication forks are stalled during S phase in S pombe, repression of MBFregulated transcription is disrupted and expression of MBF target genes is induced. We have shown that THE SCRIPPS RESEARCH INSTITUTE 257 SpNrm1 is a direct target for phosphorylation by the checkpoint kinase Cds1, a homolog of human Chk2, and that its phosphorylation leads to dissociation from MBF and activation of gene expression (Fig. 1). That response requires the ATM/ATR-like checkpoint kinase Rad3. We recently found that derepression of MBF target genes in S cerevisiae also occurs via regulation of Nrm1 in response to activation of the DNA replication checkpoint. A similar regulatory cascade may control G1-specific genes via E2F family members in animal cells and thereby play a crucial role in the maintenance of genome integrity. A D A P TAT I O N T O E N V I R O N M E N TA L S T I M U L I Remodeling of the gene expression program also occurs in cells adapting to environmental changes. We have studied the regulation of the HXT genes, which encode hexose permeases. Those genes are induced by growth on glucose and repressed on most other carbon sources. Extracellular glucose interacts with cellsurface receptors that initiate a signaling cascade that culminates with the activation of HXT gene transcription. We have shown that signaling leads to the phosphorylation-dependent destruction of the transcriptional corepressor Mth1 by the E3 ubiquitin ligase SCF Grr1 . Destruction of Mth1 leads to phosphoryation of the transcriptional repressor Rgt1 and its dissociation from HXT gene promoters. Conversely, repression of HXT gene expression requires Mth1 and is associated with Rgt1 dephosphorylation. In collaboration with T. Hunter, Salk Institute for Biological Studies, La Jolla, California, we recently showed that a type 2A protein phosphatase complex, Pph3/Psy2, which associates with Mth1, promotes Rgt1 dephosphoryation. Consequently, Pph3/Psy2 is important for the reestablisment of repression of HXT genes when environmental glucose is limiting. Interestingly, SCFGrr1, the E3 ubiquitin ligase required for destruction of Mth1 during HXT gene induction, is also important for destruction of G1 cyclins, critical regulators of cell-cycle initiation. We are investigating the basis for discrimination between targets by SCFGrr1. We found that basic residues in the leucine-rich repeat and parts of the C terminus of the F-box protein Grr1 are important for recognition of phosphorylated substrates. Identification of additional Grr1 substrates, in collaboration with Dr. Yates, and additional characterization of Grr1 will facilitate those studies. PUBLICATIONS Limbo, O., Chahwan, C., Yamada, Y., de Bruin, R.A.M., Wittenberg, C., Russell, P. Ctp1 is a cell cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell, in press. 258 MOLECULAR BIOLOGY 2007 Cell-Cycle Checkpoints, DNA Damage, and Cytotoxic Stress Responses P. Russell, A. Amar, C. Dovey, L.-L. Du, P. Kennedy, O. Limbo, V. Martin, S. Rozenzhak, J. Williams, Y. Yamada NA damage and cytotoxic stress elicit cellular responses that are highly conserved throughout eukaryotic evolution. Consequently, studies of genetically tractable microorganisms such as the fission yeast Schizosaccharomyces pombe can provide a useful framework for the design and interpretation of experiments performed with more complex multicellular organisms. We use fission yeast to study cell-cycle checkpoints, DNA repair, and stress response mechanisms. Defects in these mechanisms underlie a number of human diseases, including cancer. D CHECKPOINTS AND DNA DAMAGE RESPONSES DNA double-strand breaks are among the most lethal and genome-destabilizing type of DNA damage. They can arise from exogenous sources, such as ionizing radiation, or through errors involving DNA replication. Rapid and accurate repair of double-strand breaks is essential for preserving genome integrity. The mechanism used to repair the breaks depends on the circumstances in which the DNA damage occurs. Double-strand breaks that arise in the postreplicative G2 phase of a haploid organism are most effectively repaired by homologous recombination, an error-free mechanism that uses the undamaged sister chromatid as a template for repair of the broken chromosome. In contrast, when double-strand breaks occur in the prereplicative G1 phase, the sister chromatid is unavailable as a template for DNA repair. In this circumstance, the only option for repair of the breaks in unique DNA sequences is nonhomologous end joining. This errorprone mechanism, which was originally discovered in mammalian cells, is broadly conserved among eukaryotes, including the budding yeast Saccharomyces cerevisiae and the fission yeast S pombe. Recently, we have been identifying novel DNA repair proteins in fission yeast. An example is the protein Xlf1, which we found through its sequence similarity to the human protein Cernunnos. Patients with defects in Cernunnos have a set of phenotypes, including microcephaly and immunodeficiency, that closely resemble the characteristics of patients with defects in DNA ligase THE SCRIPPS RESEARCH INSTITUTE IV, which is required for nonhomologous end joining. We found that Xlf1 is required for nonhomologous end joining in fission yeast. Notably, cells lacking Xlf1 cannot survive exposure to ionizing radiation during G1 phase. Xlf1 is 1 of 4 proteins required for nonhomologous end joining in fission yeast. Preservation of genome integrity in eukaryotic organisms also depends on telomeres, specialized chromatin structures that compose the ends of linear chromosomes. Telomeres cap and protect chromosome ends, distinguishing the ends from double-strand breaks elsewhere in the genome. Failure to properly protect chromosome ends leads to chromosome end-to-end fusions and other genome rearrangements, leading to forms of genomic instability typically associated with cancer. In the past year, we discovered 2 novel telomere proteins in fission yeast. Our interest in these proteins began when we discovered that they have predicted OB-fold domains, which are a compact structural motif found in a variety of proteins that interact with single-stranded DNA. We found that the proteins form a heterodimeric complex that binds to DNA ends. Bioinformatic studies revealed that the proteins are distant homologs of the proteins Stn1 and Ten1 that occur in budding yeast; Stn1 and Ten1 were heretofore thought to be restricted to closely related species of budding yeast. CYTOTOXIC STRESS RESPONSE Production of reactive oxygen species (ROS), such as hydroxyl radicals, superoxide anions, and hydrogen peroxide, is a normal byproduct of aerobic metabolism in all eukaryotic organisms. Elevation of intracellular ROS can also arise through exposure to environmental toxicants, such as heavy metals or metalloids (e.g., cadmium and arsenic) and some pesticides. Oxidative stress in the form of ROS can be highly toxic, causing damage to proteins, lipids, and nucleic acids. Indeed, the cumulative effects of exposure to ROS are thought to be a causative factor in many of the most widespread and debilitating human diseases, such as atherosclerosis, Alzheimer’s disease, Parkinson’s disease, and cancer, and in the aging process itself. Consequently, all eukaryotic organisms have multiple cellular mechanisms to prevent the excessive accumulation of ROS and protect against their harmful effects. Antioxidant defense mechanisms include use of both nonenzymatic molecules such as glutathione and several vitamins and ROS scavenger enzymes such as superoxide dismutase, catalase, and glutathione peroxidase. In the past year, we made the surprising discovery that the cellular response to oxidative stress substan- MOLECULAR BIOLOGY 2007 tially depends on Upf1, a component of the nonsensemediated messenger RNA decay system. Whole genome expression profiling studies showed that Upf1 controls the expression of more than 100 genes that are transcriptionally induced in response to oxidative stress; most of these are also controlled by the transcription factor Atf1. The unexpected connection between a factor in nonsense-mediated messenger RNA decay and the oxidative stress response in fission yeast may provide important new clues about the physiologic function of nonsense-mediated messenger RNA decay in other species. PUBLICATIONS Cavero, S., Chahwan, C., Russell, P. Xlf1 is required for DNA repair by nonhomologous end joining in Schizosaccharomyces pombe. Genetics 175:963, 2006. Martin, V., Du, L.-L., Rozenzhak, S., Russell, P. Protection of telomeres by a conserved Stn1-Ten1 complex. Proc. Natl. Acad. Sci. U. S. A., in press. DNA Damage Responses in Human Cells C.H. McGowan, M. Duquette, E. Langley, J. Scorah, D. Slavin, E. Taylor omplex multiceullar organisms, such as humans, contain large numbers of mitotically competent cells that are capable of renewal, repair, and, to some extent, regeneration. The advantages of being able to replace damaged or aged cells are offset by the inherent susceptibility of mitotic cells to mutate and become cancerous. DNA is inherently vulnerable to many sorts of chemical and physical modifications; thus, as cells duplicate and divide, they can acquire mutations. Both spontaneous and induced DNA damage must be repaired with minimal changes if growth, renewal, and repair are to be successful. Our overall objective is to understand how mammalian cells protect themselves from DNA damage and thus from cancer. We are especially interested in understanding basic cellular responses to clinically relevant agents. Eukaryotic cells have evolved with a complex network of DNA repair processes and cell-cycle checkpoint responses to ensure that damaged DNA is repaired before it is replicated and becomes fixed in the genome. These pathways are highly conserved throughout evolution, and much information about human responses to DNA damage has been gained from studies of simple, genetically tractable organisms such as yeast. We C THE SCRIPPS RESEARCH INSTITUTE 259 use a combination of molecular, cellular, and genetic techniques to determine how these pathways operate in human cells. Checkpoints control the order and timing of events in the cell cycle; they ensure that independent processes are appropriately coupled. In addition, checkpoints promote the use of the most appropriate repair pathway. We used genetic models to identify 2 checkpoint kinases in humans that limit progression of the cell cycle when DNA is damaged. We are studying the function of both of these kinases and of a number of their substrates. One of the kinases, Chk2, is activated in response to DNA damage. Chk2 physically interacts with Mus81-Eme1, a conserved DNA repair protein that has homology to the xeroderma pigmentosum F family of endonucleases. Xeroderma pigmentosum is a cancer-prone disorder that results from a failure to appropriately repair damaged DNA. Biochemical analysis indicates that Mus81-Eme1 has endonuclease activity against structure-specific DNA substrates, including Holliday junctions. Enzymatic analysis, immunofluorescence studies, and the use of RNA interference have all contributed to the conclusion that Mus81-Eme1 is required for recombination repair in human cells. We are also using gene targeting to study the function of the Mus81-Eme1 endonuclease in mice. Inactivation of Mus81 in mice increases genomic instability and sensitivity to DNA damage but does not promote tumorigenesis. In addition, we have shown that Mus81-Eme1 is specifically required for survival after exposure to cisplatin, mitomycin C, and other commonly used anticancer drugs. As a point of interaction between checkpoint control and DNA repair, the relationship between Mus8-Eme1 and Chk2 probably provides information critical to understanding the response to DNA damage as a whole. Anticancer therapy is largely based on the use of genotoxic agents that damage DNA and thus kill dividing cells. Coordination of cell-cycle checkpoints and DNA repair is especially important when unusually high amounts of DNA damage occur after radiation or genotoxic chemotherapy. Hence, a detailed understanding of cellular responses to DNA damage is essential in understanding both the development and the treatment of disease in humans. PUBLICATIONS Gueven, N., Becherel, O.J., Howe, O., Chen, P., Haince, J.F., Ouellet, M.E., Poirier, G.G., Waterhouse, N., Fusser, M., Epe, B., de Murcia, J.M., de Murcia, G., McGowan, C.H., Parton, R., Mothersill, C., Grattan-Smith, P., Lavin, M. A novel form of ataxia oculomotor apraxia characterized by oxidative stress and apoptosis resistance. Cell Death Differ. 14:1149, 2007. 260 MOLECULAR BIOLOGY 2007 DNA Repair and the Maintenance of Genomic Stability M.N. Boddy, S. Pebernard, J. Prudden NA repair pathways have evolved to protect the genome from ever-present genotoxic agents. Highlighting the importance of the pathways, defects in DNA repair mechanisms strongly predispose the host to cancer and to neurologic and developmental disorders. The DNA repair systems we study in fission yeast are evolutionarily conserved, and therefore our studies provide a valuable framework for understanding genome maintenance in human cells. Although many DNA repair mechanisms have been described, information on how they are coordinated with necessary changes in chromatin structure is limited. D THE SMC5-SMC6 COMPLEX In collaboration with J.R. Yates, Department of Cell Biology, we purified the structural maintenance of chromosomes (SMC) complex Smc5-Smc6 to identify its core components. The holocomplex consists of the Smc5-Smc6 heterodimer and 6 additional non-SMC elements, Nse1–Nse6 (Fig. 1A). We showed that Smc5- F i g . 1 . Smc5-Smc6 holocomplex and STUbLs. A, Nse1, Nse3, and Nse4 form a stable heterotrimer that associates with Smc5. Nse2 interacts directly with Smc5 in the absence of the other Nse proteins. Smc6 interacts directly with Smc5 but with none of the other components. Nse5 and Nse6 form a stable heterodimer that also binds directly to Smc5. Double-headed arrows indicate interactions between subcomplexes. Nse5 and Nse6 may recruit the holocomplex to stalled replication forks and certain DNA damage sites. B, A target protein is sumoylated, an event that leads to recruitment of STUbLs via interactions with SUMO and the substrate. For Rad60 or NIP45, the STUbLs interact via the SUMO-like domains. STUbLs then ubiquitinate the substrate, promoting regulation via altered localization, protein interaction, or degradation. THE SCRIPPS RESEARCH INSTITUTE Smc6 prevents the deleterious engagement of an ordinarily beneficial DNA repair pathway called homologous recombination. Smc5-Smc6 either prevents initiation of homologous recombination or separates physically linked chromosomes that arise late in this process. Spontaneous DNA damage in Smc5-Smc6 mutant cells is due to the attempted separation of chromosomes into daughter cells while the chromosomes are still physically linked. Such defective chromosome separation in humans could result in cancer and other diseases. A N U N P R E C E D E N T E D S U M O - TA R G E T E D U B I Q U I T I N LIGASE The covalent attachment of ubiquitin and the small ubiquitin-like protein SUMO to target proteins plays key roles in genome stability; each of the 2 moieties (i.e., ubiquitin and SUMO) has physiologically distinct effects on the function of target proteins. We have identified the SUMO-targeted ubiquitin ligase (STUbL) family, which provides a novel and unanticipated regulatory link between the ubiquitination and sumoylation pathways. Members of the STUbL family include Slx8-Rfp1 in fission yeast, RNF4 in humans, MIP1 in slime molds, and SLX5/8 in budding yeast. STUbLs are recruited to sumoylated proteins and proteins containing SUMOlike domains to mediate the ubiquitination and regulation of these proteins (Fig. 1B). Cells with mutations in Slx8-Rfp1 accumulate sumoylated proteins, display genomic instability, and are hypersensitive to genotoxic stress. These Slx8-Rfp1 mutant phenotypes are suppressed by concomitant deletion of the major SUMO ligase Pli1, demonstrating the specificity of STUbLs as regulators of sumoylated proteins. Expression of human RNF4 restores homeostasis of the SUMO pathway in fission yeast that lack Slx8-Rfp1, underscoring the evolutionary functional conservation of STUbLs. The DNA repair factor Rad60 (an accessory factor of the Smc5-Smc6 complex) and its human homolog NIP45, which contain SUMO-like domains, are candidate STUbL targets. Consistently, mutations in Rad60 and Slx8-Rfp1 cause similar DNA repair defects. MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 261 Signal Transduction Pathways Mediating Cellular Responses to Oncogenic Mutations P. Sun, C. Kannemeier, J. Kwong, R. Liao, A. Seit-Nebi, N. Yoshizuka evelopment of cancer is a result of multiple oncogenic genetic alterations, including activation of oncogenes and inactivation of tumor suppressors. Although these oncogenic mutations contribute to tumorigenic phenotypes, normal cells can respond to oncogenic changes by initiating tumor-suppressing defense mechanisms such as apoptosis and premature senescence (a stable form of growth arrest). As a result, tumor development requires additional mutations that compromise these antioncogenic responses. Our main interests are to delineate the signal transduction pathways that mediate these tumor-suppressing responses and to determine how these responses are evaded during cancer development. Currently, we are focusing on 2 well-known oncogenes: ras and mdm2. The oncogene ras encodes a family of small GTPbinding proteins that are often activated in human tumors and contribute to tumor development. In normal cells, however, the initial response to ras activation is premature senescence. Recent studies have shown that like apoptosis, oncogene-induced senescence is a bona fide tumor-suppressing mechanism in vivo that must be compromised in order for cancer to develop. However, the signaling pathways responsible for this important antitumorigenic response are poorly understood. We have shown that ras induces senescence through sequential activation of 2 MAP kinase pathways (Fig. 1). Initially, ras activates the MAP kinase kinase (MEK)–extracellular signal–regulated kinase (ERK) pathway. Sustained activation of MEK-ERK turns on the stress-induced p38 pathway, which subsequently causes senescence. These studies have revealed a novel, tumorsuppressing function of p38, in addition to its known roles in inflammation and stress responses. In other studies, we identified additional signaling components that mediate ras-induced senescence (Fig. 1), including p38-regulated/activated protein kinase (PRAK). PRAK is a p38 MAP kinase substrate whose physiologic functions are poorly understood. We found that PRAK mediates senescence upon activation by p38 in response to oncogenic ras. In mice, PRAK deficiency D F i g . 1 . Signal transduction pathways mediating oncogene-induced premature senescence. Oncogenic ras induces sequential activation of the tumorigenic Raf-MEK-ERK MAP kinase pathway and the stressinduced MKK3/6-p38-PRAK MAP kinase pathway. Activation of the p38 pathway may be mediated by increased intracellular levels of reactive oxygen species (ROS) induced by the ras-Raf-MEK-ERK signaling cascade. Activated components of the p38 pathway phosphorylate multiple residues on p53, including S33 and S46 (by p38), S37 (by PRAK), and others, leading to increased transcriptional activity of p53 and induction of p21WAF1, a transcriptional target of p53. Active p38 also induces the expression of p16INK4A and p14/p19ARF, which, together with the p53-p21 cascade, cause premature senescence that serves as a tumor-suppressing defense mechanism. All the oncogenic events are shown in red; the tumor-suppressing events are in blue. enhances skin carcinogenesis induced by the environmental mutagen 7,12-dimethylbenz(a)anthracene, coinciding with compromised induction of senescence. In primary cells, inactivation of PRAK prevents senescence and promotes oncogenic transformation. Moreover, PRAK activates p53 by direct phosphorylation at Ser37 of p53 (Fig. 1). We propose that phosphorylation of p53 by PRAK after activation of p38 MAP kinase by ras plays an important role in ras-induced senescence and tumor suppression. Experiments are under way to discover additional signaling components that regulate the induction of senescence and to determine whether the p38 pathway is disarmed during cancer development in humans. Another focus of our research is mdm2, an oncogene that can mediate transformation primarily through inactivation of the tumor suppressor protein p53. Previously, we found that MDM2, the protein encoded by mdm2, confers resistance to cell-cycle arrest induced by transforming growth factor β (TGF-β), a growthinhibitory cytokine. In studies on the molecular mechanism that underlies MDM2-mediated resistance to TGF-β, we found that MDM2 makes cells refractory to the cytokine by overcoming a TGF-β–induced arrest of 262 MOLECULAR BIOLOGY 2007 the cell cycle in phase G1. Because the TGF-β–resistant phenotype is reversible upon removal of MDM2, MDM2 probably confers resistance to TGF-β by directly targeting the cellular machinery involved in the growth inhibition mediated by the cytokine. In both mink lung epithelial cells and human mammary epithelial cells, 3 elements were required for MDM2-mediated resistance to TGF-β. One element was the C-terminal half of the p53 binding domain, which at least partially retained p53 binding and inhibitory activity. Second, the ability of MDM2 to mediate TGF-β resistance was disrupted by mutation of the nuclear localization signal but was restored by coexpression of MDMX, a relative of MDM2 and another p53 regulator. Finally, mutations of the zinc coordination residues of the RING finger domain abrogated TGF-β resistance but not the ability of MDM2 to inhibit p53 activity or to bind MDMX. These data suggest that RING finger–mediated p53 inhibition and MDMX interaction are not sufficient to cause TGF-β resistance and imply a crucial role for the E3 ubiquitin ligase activity of this domain in MDM2-mediated TGF-β resistance. PUBLICATIONS Kannemeier, C., Liao, R., Sun, P. The RING finger domain of MDM2 is essential for MDM2-mediated TGF-β resistance. Mol. Biol. Cell 18:2367, 2007. Sun, P., Yoshizuka, N., New, L., Moser, B.A., Li, Y., Liao, R., Xie, C., Chen, J., Deng, Q., Yamout, M., Dong, M.-Q., Frangou, C.G., Yates, J.R. III, Wright, P.E., Han, J. PRAK is essential for ras-induced senescence and tumor suppression. Cell 128:295, 2007. Genetic Modifiers of Behavioral Despair as Targets for New Antidepressants J.G. Sutcliffe, P.B. Hedlund, F.E. Bloom,* B.S. Hilbush* * ModGene, L.L.C., La Jolla, California ouse models have been developed that have high value for predicting the antidepressant activity of a drug. The 2 models used most often are the forced-swim test and the tail-suspension test. In both tests, a state of behavioral despair is created in which mice cease to struggle and become immobile when confronted with an adverse situation. Known human antidepressants increase the length of time the mice struggle, thus decreasing the immobility time. Significant behavioral differences exist among inbred mouse strains in these tests. For example, during the M THE SCRIPPS RESEARCH INSTITUTE last 4 minutes of a 6-minute forced swim test, unmedicated, inbred C57BL/6J mice spent 152 seconds of the possible 240 seconds (63%) immobile. In contrast, inbred DBA/2J mice spent only 47 seconds (20%) immobile. Thus, genetic differences between the 2 mouse strains influence behaviors in the test. In collaboration with researchers at ModGene, L.L.C., we used the differences in the responses of the 2 strains to identify genes whose activities contribute to relative basal despair status. We investigated the hypothesis that the differences between the 2 inbred mouse lines that result in different baseline immobility times in the forced-swim test are due to cumulative quantitative differences in the activities of several modifier genes, called quantitative trait loci (QTLs), that could be placed on the genetic map. We further investigated the hypothesis that at least some of the strain differences occur because of differences in the amount of mRNA that accumulates from a single gene in each of the mapped chromosomal regions. We measured baseline immobility times of C57BL/6J mice, DBA/2J mice, and 27 strains of recombinant inbred mice that were produced from C57 x DBA matings (10 mice of each strain for each sex). Each strain had a characteristic immobility time, ranging from 5 seconds to 165 seconds; this finding indicated that several genes must be contributing to the strain differences because the range was greater than the difference between the parental strains. We also found differences between males and females within strains. We correlated these data with the haplotype data from the mouse strains and detected QTL genes associated with despair on chromosome 4 in all mice and additionally on chromosomes 11 and 13 in female mice and chromosome 18 in male mice. These results indicated some sexual dimorphism in determinants of this behavioral despair, as is known for depression in humans. We then correlated the inheritance of each QTL with the concentration of each of more than 30,000 mRNAs in the brains of the recombinant inbred mice. We identified a 100% correlation between inheritance of each QTL and the heritability of the amount of mRNA that accumulated from a single chromosome 4 gene, a single chromosome 11 gene, and a single chromosome 13 gene. No chromosome 18 gene correlation was detected. In analogous studies with the tail-suspension test, we detected the same chromosome 4 genes in both males and females and the same chromosome 11 and 13 genes in females only, suggesting that these 3 genes are MOLECULAR BIOLOGY 2007 directly related to the despair behavior rather than to the ability to perform in one of the behavioral tests. The identities of the genes responsible for the quantitative traits provide a powerful point of departure for the development of new pharmaceutical agents to treat depression because the studies in which the genes were detected provide evidence that differences in the activities of the protein products of the genes directly contribute to differences in phenotype. Thus, a drug that altered the activity of a protein encoded by a specific gene in the beneficial direction (either inhibition or augmentation), and did not have other deleterious side effects, would be a suitable candidate to test for antidepressant activity. The chromosome 4 gene encodes a previously known protein (never associated with brain disorders) whose activity is regulated by phosphorylation by a specific protein kinase. Because the concentration of mRNA for the protein is higher in C57BL/6J mice (the mice that have the greater despair) than in DBA mice, we sought a way to reduce the activity of the protein and make the C57BL/6J mice behave more like the DBA/2J mice. A compound that inhibits the activity of the specific kinase in cultured tumor cells has recently been synthesized. When the compound was administered in a very low dose to C57BL/6J mice, their immobility was reduced to 78 seconds, indicative of an antidepressantlike effect. A 20-fold higher dose reduced immobility to 0 seconds. The compound also reduced immobility when administered to DBA mice and had analogous effects on tail-suspension immobility times. These data suggest that the compound can be considered a lead compound for testing as an antidepressant. PUBLICATIONS de Lecea, L., Sutcliffe, J.G. The Hypocretins (Orexins). In: Handbook of Biologically Active Peptides. Kastin, A. (Ed.). Academic Press, San Diego, 2006, p. 721. Desplats, P.A., Denny, C.A., Kass, K.E., Gilmartin, T., Head, S.R., Sutcliffe, J.G., Seyfried, T.N., Thomas, E.A. Glycolipid and ganglioside metabolism imbalances In Huntington’s disease. Neurobiol. Dis., in press. Hedlund, P.B., Sutcliffe, J.G. The 5-HT7 receptor influences stereotypic behavior in a model of obsessive-compulsive disorder. Neurosci. Lett. 414:247, 2007. Semenova, S., Geyer, M.A., Sutcliffe, J.G., Markou, A., Hedlund, P.B. Inactivation of the 5-HT7 receptor partially blocks phencyclidine-induced disruption of prepulse inhibition. Biol. Psychiatry, in press. Sutcliffe, J.G., de Lecea, L. The hypocretin/orexin system. In: Handbook of Contemporary Neuropharmacology. Sibley, D., et al. (Eds.). Wiley-Interscience, Hoboken, NJ, 2007, Vol. 3, p. 125. THE SCRIPPS RESEARCH INSTITUTE 263 Molecular Neurobiology of CNS Disorders E.A. Thomas, J.G. Sutcliffe, P.A. Desplats, E.L. Woodward DIVERSITY OF GENE EXPRESSION IN SCHIZOPHRENIA chizophrenia is a heterogeneous psychiatric disorder with variable signs and symptoms that change during the course of the illness. Additionally, the outcome after initial diagnosis varies widely. We have studied the molecular differences that occur during progression of schizophrenia and those that may be associated with its heterogeneity. We generated genome-wide RNA expression profiles from tissue samples obtained at autopsy from the prefrontal cortex of 32 patients who had schizophrenia for 2 to 53 years and from the prefrontal cortex of 32 age- and sex-matched controls (persons without schizophrenia). We found that patients with acute and chronic illness had major differences in gene expression profiles. Acute illness was associated with dramatic changes in gene expression, whereas chronic illness was associated with a stabilization of aberrant gene expression. Currently, we are focusing on genes correlated with disease progression and those regulated by antipsychotic drug treatment. Further, we have detected modules of coexpressed genes corresponding to subgroups of patients from all cohorts. We have delineated 4 biologically relevant subgroups of schizophrenia defined by different gene expression profiles; each subgroup is associated with distinct physiologic pathways. In particular, we have focused on alterations of genes associated with glycosphingolipid metabolism and myelination in the pathophysiologic changes associated with psychiatric disease. The distinct molecular portraits we identified might represent bona fide “subtypes” of schizophrenia. Each subtype might be associated with different signs and symptoms and have a different pattern of disease progression and outcome. S T R A N S C R I P T I O N A L D Y S R E G U L AT I O N I N H U N T I N G T O N ’ S D I S E A S E : S T R I ATA L S P E C I F I C I T Y Much evidence supports a role for transcriptional dysregulation in Huntington’s disease. Of particular interest is how these disturbances may be specifically manifested in the striatum, the area of the brain with the most striking neuropathologic deficits in this disease. Currently, we are investigating transcriptional regulatory proteins that have highly enriched expres- 264 MOLECULAR BIOLOGY 2007 sion in the striatum and the roles of the proteins in the pathologic changes associated with Huntington’s disease. We have focused on 2 particular transcription factors, Bcl11b and Foxp1, which we hypothesize play important roles in striatal gene expression. Both of these factors have decreased expression in mouse models of Huntington’s disease and in the caudate of patients with Huntington’s disease and can interact with the protein huntingtin. Sequestration of these factors into nuclear aggregates in Huntington’s disease resulting in loss of function may contribute to specific dysregulation of striatal gene expression. This mechanism may explain, in part, the specificity of the pathologic changes associated with Huntington’s disease. PUBLICATIONS Dean, B., Keriakous, B., Scarr, E., Thomas, E.A. Gene expression profiling in Brodmann’s area 46 from subjects with schizophrenia. Aust. N. Z. J. Psychiatry 41:308, 2007. Desplats, P.A., Denny, C.A., Kass, K.E., Gilmartin, T., Head, S.R., Sutcliffe, J.G., Seyfried, T.N., Thomas, E.A. Glycolipid and ganglioside metabolism imbalances In Huntington’s disease. Neurobiol. Dis., in press. Narayan, S., Kass, K.E., Thomas, E.A. Chronic haloperidol treatment results in a decrease in the expression of myelin/oligodendrocyte-related genes in the mouse brain. J. Neurosci. Res. 85:757, 2007. Sundram, S., Scarr, E., Digney, A., Thomas, E.A., Dean, B. Plasma apolipoprotein E is decreased in schizophrenia spectrum and bipolar disorder. Psychiatry Res., in press. Thomas, E.A. Molecular profiling of antipsychotic drug function: convergent mechanisms in the pathology and treatment of psychiatric disorders. Mol. Neurobiol. 34:109, 2006. Thomas, E.A., Yao, J.K. Clozapine specifically alters the arachidonic acid pathway in mice lacking apolipoprotein D. Schizophr. Res. 89:147, 2007. The 5-HT7 Receptor in Neuropsychiatric Disorders P.B. Hedlund, P.E. Danielson, S. Semenova, M.A. Geyer, A. Markou, J.G. Sutcliffe nterest in the serotonin 5-HT7 receptor as a putative target in neuropsychiatric disorders has been growing continually. The interest was prompted by the finding that several classes of drugs used to treat disorders such as depression and schizophrenia have high affinity for the 5-HT7 receptor. We have established evidence that supports a role for this receptor in depression, obsessive-compulsive disorder, and schizophrenia. I DEPRESSION The forced swim test and the tail suspension test are animal models of behavioral despair that have high value for predicting the antidepressant efficacy of drugs. THE SCRIPPS RESEARCH INSTITUTE The tests can also be used to characterize animals in which genes have been deleted. In both of these tests, we showed that mice lacking the 5-HT7 receptor have a behavioral profile similar to that of mice treated with antidepressants. We replicated these findings by using a compound that acts as a selective antagonist at the 5-HT7 receptor. Thus, both blockade and inactivation of the receptor yield the same result. New evidence suggests that the effects of antidepressants acting as selective serotonin reuptake inhibitors are potentiated by 5-HT 7 receptor antagonists. Taken together, our results suggest an important role for the 5-HT7 receptor in depression, and antagonists at this receptor should be evaluated as treatment for depression, either independently or in combination with existing antidepressants. OBSESSIVE-COMPULSIVE DISORDER Obsessive-compulsive disorder is commonly treated with antidepressants, and thus is related to depression. In an animal model of obsessive-compulsive disorder (marble burying), we showed that blockade or inactivation of the 5-HT7 receptor results in less compulsive behavior. These findings further support the hypothesis that the 5-HT7 receptor is an important alternative or supplemental target for antidepressants. SCHIZOPHRENIA Prepulse inhibition (PPI) of the acoustic startle reflex is a well-characterized model of schizophrenia. The model is especially relevant because similar responses can be observed in patients with schizophrenia. We showed that PPI per se is not altered in mice lacking the 5-HT7 receptor, but that when PPI is disrupted by phencyclidine, these mice are significantly less affected than are mice that have the receptor. Phencyclidineinduced disruption involves a glutamatergic component of PPI that is relevant for the action of atypical antipsychotic agents such as clozapine. Clozapine is a drug with relatively high affinity for the 5-HT7 receptor. PUBLICATIONS de Lecea, L., Sutcliffe, J.G. The hypocretins (orexins). In: Handbook of Biologically Active Peptides. Kastin, A. (Ed.). Elsevier, Philadelphia, 2006, p. 721. Hedlund, P.B., Sutcliffe, J.G. The 5-HT7 receptor influences stereotypic behavior in a model of obsessive-compulsive disorder. Neurosci. Lett. 414:247, 2007. Landry, E.S., Lapointe, N.P., Rouillard, C., Levesque, D., Hedlund, P.B., Guertin, P.A. Contribution of spinal 5-HT1A and 5-HT7 receptors to locomotor-like movement induced by 8-OH-DPAT in spinal cord-transected mice. Eur. J. Neurosci. 24:535, 2006. Semenova, S., Geyer, M.A., Sutcliffe, J.G., Markou, A., Hedlund, P.B. Inactivation of the 5-HT7 receptor partially blocks PCP-induced disruption of prepulse inhibition. Biol. Psychiatry, in press. Sutcliffe, J.G., de Lecea, L. The hypocretin/orexin system. In: Handbook of Contemporary Neuropharmacology. Sibley, D., et al. (Eds.). Wiley-Interscience, Hoboken, NJ, 2007, Vol. 3, p. 125. MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE 265 Lysophospholipid Signaling and Neural Aneuploidy J. Chun, B. Anliker, J. Choi, A. Dubin, S. Gardell, D. Herr, G. Kennedy, M. Kingsbury, C. Lee, D. Lin, M. Lu, T. Mutoh, K. Noguchi, C. Paczkowski, S. Peterson, R. Rivera, W. Westra, X. Ye, Y. Yung, L. Zhu n the past year, several discoveries have led to new directions in our long-term projects. In our studies on lysophospholipid signaling, we discovered a new biological role for sphingosine 1-phosphate (S1P) in hearing and vestibular function. We also identified and characterized a novel lysophosphatidic acid (LPA) receptor termed LPA5 and found new signaling properties of another LPA receptor, LPA4. We continue to identify signaling pathways and possible new lysophospholipid receptors; our aim is to understand key biological functions of lysophospholipid signaling. We made several technological advancements in our studies of neural aneuploidy, paving the way for a deeper understanding of the functional consequences of aneuploidy in the brain. While continuing to characterize the extent and regional variations of aneuploidy in the brains of healthy humans and normal mice, we are laying the groundwork to determine potential links between neural aneuploidy and human brain disorders. I LY S O P H O S P H O L I P I D S I G N A L I N G Lysophospholipids are classically known as metabolites in the biosynthesis of cell membranes; however, they also have extracellular effects. Researchers in our laboratory discovered the first lysophopholipid receptor, now known as LPA1, and showed that most of the extracellular effects could be mediated via cell-surface receptors (of the G protein–coupled receptor superfamily). We have continued to explore the cellular and physiologic functions of receptor-mediated lysophospholipid signaling, primarily by generating and examining mutant mice that lack lysophospholipid receptors. We currently have multiple lines of these mutant “knockout” mice that are missing one or more lysophospholipid receptors. Identifying degenerative hearing loss in S1P2/S1P3 double knockout mice (2 inoperable genes) adds another medically relevant indication to a growing list that reflects the potential medical relevance of lysophospholipid signaling. In mice lacking the S1P2 and S1P3 receptors, auditory and vestibular problems develop gradually, resulting in vestibular (related to balance) dysfunction and complete hearing loss by early adulthood (Fig. 1A). We found that these problems are due F i g . 1 . Auditory defects in S1P 2 /S1P 3 double knockout mice. A, The acoustic startle response, a measure of hearing function, is nearly absent in mice lacking both copies of S1P 2 , indicating that these mice are deaf. B, Histologic examination of cochlea from wild-type (left) and S1P2/S1P3 double knockout (right) adult mice reveals loss and disorganization of inner hair cells. to neurodegeneration of the hair cells that normally transduce auditory and vestibular signals to the brain (Fig. 1B). This result suggests that S1P receptors contribute to the survival of normal hair cells and may provide a new molecular target for preventing neurodegenerative hearing loss. NEURAL ANEUPLOIDY Cells in the brain can be genomically nonidentical because they have lost or gained chromosomes, a condition termed aneuploidy. Since discovering this new aspect of brain organization, we have continued to extend our knowledge. We are developing new methods to investigate the anatomic and functional importance of aneuploidy. In the past year, using optimized fluorescent in situ hybridization, we were able to analyze multiple chromosomes in individual postmitotic cells, providing a clearer look at the chromosomal complement present within single neurons (Fig. 2). In addition, we are devel- F i g . 2 . Multiple sequential hybridizations with fluorescently labeled DNA probes. In this image, a nucleus from a single postmitotic mouse neuron was probed for 10 different chromosome (Ch) pairs (mice have a total of 20 chromosome pairs). Three copies can be detected for chromosomes 8 and 14. 266 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE oping and optimizing techniques with increased throughput to determine the range of chromosomal aneuploidies in larger populations of cells. We continue to investigate how neural aneuploidy affects brain function at both cellular and system-wide levels, including the possible contribution of aneuploidy to neuropsychiatric disorders. Chemical Glycobiology PUBLICATIONS Birgbauer, E., Chun, J. New developments in the biological functions of lysophospholipids. Cell. Mol. Life Sci. 63:2695, 2006. P. Sobieszczuk, L. Stewart, H. Tian, Y. Zeng Chao, C., Herr, D., Chun, J., Xu, Y. Ser18 and Ser23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 25:2615, 2006. Chun, J., Rosen, H. Lysophospholipid receptors as potential drug targets in tissue transplantation and autoimmune diseases. Cur. Pharm. Des. 12:161, 2006. Fukushima, N., Shano, S., Moriyama, R., Chun, J. Lysophosphatidic acid stimulates neuronal differentiation of cortical neuroblasts through the LPA1-Gi/o pathway. Neurochem. Int. 50:302, 2007. Gardell, S.E., Dubin, A.E., Chun, J. Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 12:65, 2006. J.C. Paulson, O. Blixt, L.K. Allin, H. Andersson-Sand, O. Berger, O.V. Bohorov, J. Busch, R. Chakravarthy, W. Chen, G. Completo, H. Fang, D. Lebus, L. Liao, X. Liu, B. Ma, R. McBride, C. Nycholat, M. O’Reilly, N. Razi, e investigate the roles of glycan-binding proteins that mediate cellular processes central to immunoregulation and human disease. We work at the interface of biology and chemistry to understand how the interaction of glycan-binding proteins with their ligands mediates cell-cell interactions, endocytosis, and cell signaling. Our multidisciplinary approach is complemented by a diverse group of chemists, biochemists, cell biologists, and molecular biologists. W BIOLOGICAL ROLES OF SIGLECS Herr, D.R., Grillet, N., Schwander, M., Rivera, R., Muller, U., Chun, J. Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. J. Neurosci. 27:1474, 2007. Inoue, M., Yamaguchi, A., Kawakami, M., Chun, J., Ueda, H. Loss of spinal substance P pain transmission under the condition of LPA1 receptor-mediated neuropathic pain. Mol. Pain 2:25, 2006. Kingsbury, M.A., Yung, Y.C., Peterson, S.E., Westra, J.W., Chun, J. Aneuploidy in the normal and diseased brain. Cell. Mol. Life Sci. 63:2626, 2006. Lee, C.W., Rivera, R., Dubin, A.E., Chun, J. LPA4/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing Gs-, Gq/Gi-mediated calcium signaling and G12/13mediated Rho activation. J. Biol. Chem. 282:4130, 2007. Lee, C.-W., Rivera, R., Gardell, S., Dubin, A.E., Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 281:23589, 2006. Rehen, S.K., Kingsbury, M.A., Almeida, B.S.V., Herr, D., Peterson, S., Chun, J. A new method of embryonic culture for assessing global changes in brain organization. J. Neurosci. Methods 158:100, 2006. Rivera, R., Chun, J. Biological effects of lysophospholipids. Rev. Physiol. Biochem. Pharmacol. Published online August 10, 2006. doi:10.1007/112_0507. Theilmeier, G., Schmidt, C., Herrmann, J., Keul, P., Schäfers, M., Herrgott, I., Mersmann, J., Larmann, J., Hermann, S., Stypmann, J., Schober, O., Hildebrand, R., Schulz, R., Heusch, G., Haude, M., von Wnuck Lipinski, K., Herzog, C., Schmitz, M., Erbel, R., Chun, J., Levkau, B. High-density lipoproteins and their constituent sphingosine-1-phosphate directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114:1403, 2006. The siglecs are a family of 13 sialic acid–binding proteins that function as cell-signaling coreceptors. They are expressed on glial cells and on a variety of leukocytes that mediate acquired and innate immune functions, including B cells, eosinophils, macrophages, dendritic cells, and natural killer cells. Siglecs are a subfamily of the immunoglobulin superfamily that have a unique N-terminal Ig domain that confers the ability to bind sialic acid–containing carbohydrate groups (sialosides) of glycoproteins and glycolipids. The cytoplasmic domains of most siglecs contain tyrosine-based inhibitory motifs characteristic of accessory proteins that regulate transmembrane signaling and endocytosis of cell-surface receptor proteins. The diverse specificity for their sialoside ligands and their variable cytoplasmic regulatory elements provide siglecs with attributes for unique roles in the cell-surface biology of each cell that expresses them. The best understood siglec is CD22 (siglec-2), an accessory molecule of the B-cell receptor (BCR) complex that has both positive and negative effects on receptor signaling. The carbohydrate ligand recognized by CD22 is the sequence Siaα2-6Galβ1-4GlcNAc found on both neighboring glycoproteins of both B cells (cis ligands) and on cells that interact with B cells (e.g., T cells, trans ligands). Interactions of CD22 with cis or trans ligands regulate aspects of B-cell activation, proliferation, and development. We found that CD22 is predominately associated with clathrin-coated pits in resting B cells, whereas BCRs are minimally associated with clathrin domains. Mice MOLECULAR BIOLOGY 2007 deficient in the ligand for CD22 have greater colocalization of CD22 and the BCR in fused raft-clathrin domains than do mice that have the ligand, accounting for the immunosuppression in deficient mice. In wild-type mice, after antigen activation, the BCR is endocytosed via raftclathrin domains, a logical site for the dampening of B-cell signaling by CD22. In resting cells, CD22 undergoes constitutive endocytosis, which can result in internalization of high-affinity ligands of CD22 (Fig. 1). F i g . 1 . Relationship between microdomain localization of the BCR and CD22 (siglec-2), a regulator of BCR signaling that binds glycan ligands. A major barrier to studying the ligand-binding properties of siglecs and their role in siglec biology is the difficulty in creating synthetic probes that compete with endogenous (cis) ligands. Even highly multivalent polymers containing the natural glycan ligand will not bind to cell-surface siglecs unless cis ligands are first destroyed. However, we found that bifunctional molecules containing a high-affinity ligand of CD22 coupled to the antigen NP will dock an anti-NP IgM to CD22 on the surface of B cells. In effect, the IgM acts as a decavalent protein scaffold that promotes spontaneous assembly of an immune complex on the surface of B cells driven by the bifunctional ligand of CD22 (Fig. 2). Once THE SCRIPPS RESEARCH INSTITUTE 267 formed, IgM activates complement-mediated killing of the cell, suggesting a therapeutic approach for treatment of B-cell leukemia. Another siglec of current interest is myelin-associated glycoprotein (siglec-4). This siglec is expressed on glial cells and recognizes the sialoside Siaα-3Galβ1-3(Siaα26)GalNAc-R found on O-linked glycans of glycoproteins and glycolipids. Functionally, myelin-associated glycoprotein stabilizes interactions between glial cells and axons essential for normal organization of myelin and inhibits axonal regeneration, which is currently a target for pharmaceutical intervention to promote nerve regeneration. We have developed potent inhibitors of myelin-associated glycoprotein that reverse its ability to block growth of axons, and in collaborative studies with R. Schnaar, Johns Hopkins University, Baltimore, Maryland, we are investigating the potential of the inhibitors to promote nerve growth in vivo. With these successes, we have embarked on a major effort to identify high-affinity ligand analogs of each siglec to produce ligand-based tools to investigate the biological roles of the siglecs in innate and adaptive immunity. A major focus is investigation of multivalent protein scaffolds that provide favorable geometry for presentation of bifunctional ligands to cell-surface siglecs. In this regard, we are collaborating with M.G. Finn, Department of Chemistry, on the use of viral capsids that can be functionalized to carry variable numbers of synthetic ligands. S I A L O S I D E A N A L O G G LY C A N A R R AY S We have developed a robotically printed glycan array that displays sialoside analogs to assess the affinity of siglecs for unnatural substituents at the C-9 and C-5 positions of sialic acids. Even in the initial experiments with 65 acyl substituents at the C-9 position of sialic acid, the method was a powerful one for identifying substituents that increase the affinity of the natural ligand for siglecs by 100-fold or more (Fig. 3). In collaboration with K.B. Sharpless, Department of Chemistry, we have created another 80 analogs by using click chemistry to couple a library of alkynes to sialosides containing 9-azido-N-acetyl-neuraminic acid. Results from the array can be rapidly assimilated into the synthesis of high-affinity ligands and ligand-based probes of the corresponding siglec by using our flexible chemoenzymatic synthesis strategies. C O N S O R T I U M F O R F U N C T I O N A L G LY C O M I C S F i g . 2 . Bifunctional ligands of CD22 mediate binding of IgM to CD22 on the surface of B cells. Members of our laboratory also staff 2 scientific cores for the Consortium for Functional Glycomics, 268 MOLECULAR BIOLOGY 2007 THE SCRIPPS RESEARCH INSTITUTE influenza and related H1 avian influenza viruses and the more recent avian influenza virus (H5N1) to identify mutations required to switch specificity from avian receptors to human-type receptors. PUBLICATIONS Alvarez, R.A., Blixt, O. Identification of ligand specificities for glycan-binding proteins using glycan arrays. Methods Enzymol. 415:292, 2006. Blixt, O., Hoffmann, J., Svenson, S., Norberg, T. Pathogen-specific carbohydrate antigen microarrays: a chip for detection of Salmonella O-antigen-specific antibodies. Glycoconjugate, in press. Blixt, O. Razi, N. Chemoenzymatic synthesis of glycan libraries. Methods Enzymol. 415:137, 2006. Bohorov, O., Anderson-Sand, H., Hoffmann, J. Blixt, O. Arraying glycomics: a novel bi-functional spacer for one-step microscale derivatization of free reducing glycans. Glycobiology 16:21C, 2006. F i g . 3 . Sialoside analog glycan microarray reveals high-affinity ligands for CD22. A, Sialoside ligands of CD22 with amino-terminated linkers are printed on N-hydroxyl succinimide (NHS)–activated glass slides, resulting in a covalent amide bond. B, The natural ligand (3) with various substituents (1, 2, 4, 6) and a nonligand control (5) are printed in 10 replicates at 10 twofold diluted printing concentrations. Overlay with a fluorescence-labeled CD22-Ig chimera reveals the increased binding to various substituents compared with the natural ligand. organized to elucidate the mechanisms by which glycanbinding proteins mediate cell communication (http://www .functionalglycomics.org/). Scientists in the Mouse Transgenics Core, led by B. Ma, have created 8 novel mouse strains from C57Bl/6 embryonic stem cells that are deficient in genes for key glycan-binding proteins that affect immune function. Scientists in the Glycan Array Synthesis Core, led by O. Blixt, have produced a library of synthetic glycans by chemoenzymatic synthesis for use in numerous applications. In addition, scientists in the Scripps DNA Microarray Core, led by S. Head, designed and conducted investigator-initiated analysis with a custom-based microarray with genes of relevance for the consortium. A major achievement by staff in the Glycan Array Synthesis Core is the development of the world’s largest glycan microarray, which currently has more than 300 unique structures, mostly synthetic glycans produced by chemoenzymatic synthesis. Now produced in collaboration with the DNA Microarray Core, the microarray is widely used by investigators around the world to assess the specificity of glycan-binding proteins that mediate a broad scope of biological interactions. In an exemplary collaboration with I.A. Wilson and J. Stevens, Department of Molecular Biology, this array was used to investigate the specificity of the 1918 pandemic Crocker, P., Paulson, J.C., Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 4:255, 2007. Glaser, L., Conenello, G., Paulson, J.C., Palese, P. Effective replication of human influenza viruses in mice lacking a major α2-6 sialyltransferase. Virus Res. 126:9, 2007. Han, S., Collins, B.E., Paulson, J.C. Synthesis of 9-substituted sialic acids as probes for CD22-ligand Interactions on B cells. In: Frontiers in Modern Carbohydrate Chemistry. Demchenko, A.V. (Ed.). Oxford University Press, New York, 2007, p. 2. ACS Symposium Series 960. Taniguchi, N., Paulson, J.C. Frontiers in Glycomics: Bioinformatics and Biomarkers in Disease, September 11-13, 2006, Natcher Conference Center, NIH Campus, Bethesda, MD, USA. Proteomics 7:1360, 2007. Tateno, H., Li, H., Schur, M.J., Bovin, N., Crocker, P.R., Wakarchuk, W.E., Paulson, J.C. Distinct endocytic mechanisms of CD22 (siglec-2) and siglec-F reflect roles in cell signaling and innate immunity. Mol. Cell. Biol. 27:5699, 2007.