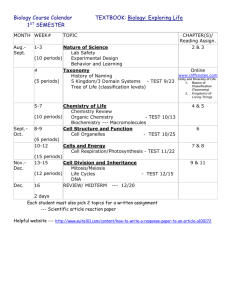
T S I C
advertisement

Z THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY S c i e n t i f i c R e p o r t 2 0 0 8 O n t h e c o v e r : A key structure of Ebola virus. Scientists at The Scripps Research Institute, led by Erica Ollmann Saphire, have determined the structure of a critical protein from Ebola virus. This image shows the virus spike protein (blue and white), which is necessary for entry of Ebola virus into human cells, bound to an immune system antibody (yellow) acting to neutralize the virus. The structure provides a major step forward in understanding how the deadly virus works and may be useful in developing vaccines against or treatments for Ebola virus infections. Image created by Christina Corbaci, administrative assistant, and Jeff Lee, Ph.D., senior research associate, in the laboratories of Erica Ollmann Saphire, Ph.D., associate professor, and Dennis Burton, Ph.D., professor. X-ray images courtesy of Dr. Lyle Conrad, Cynthia Goldsmith, and Dr. Fred Murphy, Centers For Disease Control and Prevention. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 TA B L E O F C O N T E N T S Engineering Eukaryotic Algal Chloroplasts for Production of Human Therapeutic Proteins and of Biofuels Skaggs Members 4 Skaggs Scholars 5 Board Members 6 President’s Introduction 7 Director’s Overview 9 Stephen P. Mayfield, Ph.D. Auditory Perception and Hearing Impairment: From Mouse Models to Human Genetic Disease Ulrich Müller, Ph.D. 37 Chemical Synthesis and Chemical Biology K.C. Nicolaou, Ph.D. I N V E S T I G AT O R S ’ R E P O R T S 36 38 Filling Space at the Molecular Level Julius Rebek, Jr., Ph.D. Catalysis, Cancer, and the Regulation of Genes: Inventing Molecules With Defined Functions Carlos F. Barbas III, Ph.D. 11 Role of Mistranslation in Disease Paul R. Schimmel, Ph.D. Cytokine Inflammatory Signaling in Obesity and Neurodegeneration Tamas Bartfai, Ph.D. 13 14 42 K. Barry Sharpless, Ph.D. 43 Molecular Biology of Olfaction 15 Structure and Biology of Multidrug Transporters Geoffrey Chang, Ph.D. Peter Schultz, Ph.D. Click Chemistry and Biological Activity Synthetic, Medicinal, and Bioorganic Chemistry Dale L. Boger, Ph.D. 41 New Amino Acid Building Blocks Training in Molecular and Experimental Medicine Ernest Beutler, M.D. 40 17 Lisa S. Stowers, Ph.D. 45 Macromolecular Master Keys for Genome integrity, Reactive Oxygen Control, and Pathogenesis John A. Tainer, Ph.D. 46 Chemical Physiology Benjamin F. Cravatt, Ph.D. 17 Chemical Approaches to Disease Paul Wentworth, Jr., Ph.D. 48 Fundamental Processes in Neural Development Gerald M. Edelman, M.D., Ph.D. 19 James R. Williamson, Ph.D. Chemical Etiology of Nucleic Acid Structure Albert Eschenmoser, Ph.D. 20 Intracellular RNA Assembly Martha J. Fedor, Ph.D. Pathway Engineering for Enzymatic Synthesis 50 X-ray Crystallographic Studies of Therapeutically Important Macromolecular Targets Ian A. Wilson, D.Phil. 51 21 Bioorganic and Synthetic Chemistry Click and Virus-Based Chemistry for Biological Discovery M.G. Finn, Ph.D. Chi-Huey Wong, Ph.D. 22 Insights Into Protein Chemistry and Biology From Protein Structure Elizabeth D. Getzoff, Ph.D. Structure-Based Design of Bioactive Agents M. Reza Ghadiri, Ph.D. 30 31 Understanding and Ameliorating Protein Misfolding Diseases Jeffery W. Kelly, Ph.D. Published by TSRI Press®. All rights reserved. © Copyright 2008, The Scripps Research Institute. 59 Staff Awards and Activities 61 Author Index 63 Subject Index 65 27 Synthetic Enzymes, Catalytic Antibodies, Biomolecular Computing, and Synthetic Capsids Ehud Keinan, Ph.D. Automation of Nuclear Magnetic Resonance Structure Determination of Proteins in Solution Kurt Wüthrich, Ph.D. Continuous In Vitro Evolution of RNA Enzymes Gerald F. Joyce, M.D., Ph.D. 56 26 Bacterial Quorum Sensing Kim D. Janda, Ph.D. Studies of Macromolecular Recognition by Multidimensional Nuclear Magnetic Resonance Peter E. Wright, Ph.D. 24 55 33 3 4 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY Elizabeth D. Getzoff, Ph.D.†† Professor MEMBERS M. Reza Ghadiri, Ph.D.* Professor Julius Rebek, Jr., Ph.D.* Professor and Director Carlos F. Barbas III, Ph.D.** Professor Janet and W. Keith Kellogg II Chair in Molecular Biology Tamas Bartfai, Ph.D.*** Professor Chairman, Molecular and Integrative Neurosciences Department, Scripps Research Director, Harold L. Dorris Neurological Research Institute Ernest Beutler, M.D. † Professor Chairman, Department of Molecular and Experimental Medicine, Scripps Research Dale L. Boger, Ph.D.* Richard and Alice Cramer Professor of Chemistry Geoffrey Chang, Ph.D.** Associate Professor Benjamin F. Cravatt, Ph.D.**** Professor Chairman, Department of Chemical Physiology Director, Helen L. Dorris Child and Adolescent Neuro-Psychiatric Disorder Institute Gerald M. Edelman, M.D., Ph.D.***** Professor Chairman, Department of Neurobiology, Scripps Research Albert Eschenmoser, Ph.D.* Professor Martha J. Fedor, Ph.D.** Associate Professor M.G. Finn, Ph.D.* Professor Kim D. Janda, Ph.D.* Professor Ely R. Callaway, Jr., Chair in Chemistry Director, Worm Institute for Research and Medicine Gerald F. Joyce, M.D., Ph.D. ††† Professor Dean, Faculty Ehud Keinan, Ph.D.** Adjunct Professor Jeffery W. Kelly, Ph.D.* Lita Annenberg Hazen Professor of Chemistry Chairman, Department of Molecular and Experimental Medicine, Scripps Research Richard A. Lerner, M.D. ††† President, Scripps Research Lita Annenberg Hazen Professor of Immunochemistry Cecil H. and Ida M. Green Chair in Chemistry Stephen P. Mayfield, Ph.D. †††† Professor Associate Dean, Kellogg School of Science and Technology Peter Schultz, Ph.D.* Professor Scripps Family Chair K. Barry Sharpless, Ph.D.* W.M. Keck Professor of Chemistry Lisa T. Stowers, Ph.D. †††† Assistant Professor John A. Tainer, Ph.D.** Professor Paul Wentworth, Jr., Ph.D.* Professor Henry Dube, Ph.D. Richard J. Hooley, Ph.D. ‡‡ University of California Riverside, California Jun-Li Hou, Ph.D. Seiji Kamioka, Ph.D. Lionel Moisan, Ph.D. ‡‡ CEA Gif-Sur-Yvette, France Severin Odermatt, Ph.D. ‡ Agustí Lledó Ponsati, Ph.D. James R. Williamson, Ph.D. ††† Professor Dean, Graduate and Post Graduate Studies Per Restorp, Ph.D. Ian A. Wilson, D.Phil.** Professor Siddhartha Shenoy, Ph.D. Chi-Huey Wong, Ph.D.* Ernest W. Hahn Professor and Chair in Chemistry Peter E. Wright, Ph.D.** Professor Cecil H. and Ida M. Green Investigator in Biomedical Research Chairman, Department of Molecular Biology, Scripps Research Ph.D. ††† Kurt Wüthrich, Cecil H. and Ida M. Green Professor of Structural Biology Michael Schramm, Ph.D. ‡‡ California State University Long Beach, California Craig Turner, Ph.D. Shengxiong Xiao, Ph.D. * Joint appointment in the Department of Chemistry ** Joint appointment in the Department of Molecular Biology *** Joint appointment in the Molecular and Integrative Neurosciences Department **** Joint appointment in the Departments of Chemical Physiology and Chemistry ***** Joint appointment in the Department of Neurobiology † †† Ulrich Müller †††† Professor R E B E K L A B O R A T O R Y ††††† ††† K.C. Nicolaou, Ph.D.* Aline W. and L.S. Skaggs Professor of Chemical Biology Darlene Shiley Chair in Chemistry Chairman, Department of Chemistry, Scripps Research Dariush Ajami, Ph.D. Assistant Professor of Molecular Assembly Paul R. Schimmel, Ph.D. †† Ernest and Jean Hahn Professor and Chair in Molecular Biology and Chemistry Fernando R. Pinacho Crisotomo, Ph.D. ‡‡ Burnham Institute for Medical Research La Jolla, California †††† ††††† R E S E A R C H A S S O C I AT E S Mark Ams, Ph.D. Elizabeth Barrett, Ph.D ‡ ‡ ‡‡ Deceased Joint appointments in the Departments of Molecular Biology and Immunology Joint appointments in the Departments of Chemistry and Molecular Biology Joint appointment in the Department of Cell Biology Rebek laboratory staff. Staff of the other members are listed in their respective departments Appointment completed Appointment completed; new location shown THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 THE SKAGGS SCHOLARS With a contribution from the Skaggs family, Scripps Research established the Skaggs Clinical Scholars Program in an effort to more closely integrate clinical and basic research within the Scripps organization. The program identifies research-oriented clinicians and funds meritorious collaborative research projects between the clinical scholar and appropriate scientists at Scripps Research. A broader goal is to expand the body of knowledge related to human disease and to develop effective therapeutic interventions. Faith Barnett, M.D., Ph.D. Division of Neurosurgery, Scripps Clinic Rajesh Belani, M.D. Division of Hematology and Oncology, Scripps Clinic Clifford W. Colwell, Jr., M.D. Shiley Center for Orthopaedic Research and Education at Scripps Clinic (SCORE) John W. Cronin, M.D. Division of Chest and Critical Care Medicine, Scripps Clinic George E. Dailey, M.D. Division of Diabetes and Endocrinology, Scripps Clinic Arthur Dawson, M.D. Division of Chest and Critical Care Medicine, Scripps Clinic J. Thomas Heywood, M.D. Heart Failure Clinic, Division of Cardiology, Scripps Clinic Ronald A. Simon, M.D. Division of Allergy, Asthma, and Immunology, Scripps Clinic Mary A. Kalafut, M.D. Division of Neurology, Scripps Clinic Richard A. Smith, M.D. Center for Neurologic Study, Scripps Memorial Hospital Andrew J. King, M.D. Division of Nephrology, Scripps Clinic Donald D. Stevenson, M.D. Division of Allergy, Asthma, and Immunology, Scripps Clinic Michael P. Kosty, M.D. Division of Hematology and Oncology, Scripps Clinic Daniel A. Nachtsheim, M.D. Division of Urology, Scripps Clinic Shirley M. Otis, M.D. Division of Neurology, Scripps Clinic Athena Philis-Tsimikas, M.D. Whittier Institute for Diabetes Paul S. Phillips, M.D. Interventional Cardiology, Scripps Mercy Hospital Paul J. Pockros, M.D. Division of Gastroenterology and Hepatology, Scripps Clinic Mathew J. Price, M.D. Division of Interventional Cardiology, Scripps Clinic Robert J. Russo, M.D., Ph.D. Division of Cardiovascular Diseases, Scripps Clinic Darlene J. Elias, M.D. Division of Chest and Critical Care Medicine, Scripps Clinic Farhad F. Shadan, M.D., Ph.D. Division of Chest and Critical Care Medicine, Scripps Clinic Sheila F. Friedlander, M.D. Pediatric and Adolescent Dermatology, Rady Children’s Hospital Alexander R. Shikhman, M.D., Ph.D. Division of Rheumatology, Scripps Clinic Williamson B. Strum, M.D. Division of Gastroenterology and Hepatology, Scripps Clinic Gary W. Williams, M.D., Ph.D. Department of Medicine, Scripps Clinic Katharine M. Woessner, M.D. Division of Allergy, Asthma, and Immunology, Scripps Clinic 5 6 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 THE SKAGGS INSTITUTE THE SCRIPPS FOR RESEARCH RESEARCH INSTITUTE BOARD OF TRUSTEES BOARD OF TRUSTEES Claudia S. Luttrell President Suzie Balukoff Thomas C. Beecher Douglas A. Bingham, Esq. The Most Reverend Robert H. Brom Ron L. Cutshall Kim D. Janda, Ph.D. Jeffery W. Kelly, Ph.D. Richard A. Lerner, M.D. George L. Moosman Don L. Skaggs Mark S. Skaggs Donna J. Weston John J. Moores Chair of the Board, Scripps Research Chairman, JMI and San Diego Padres Chair, Board of Trustees, the Carter Center Warren Beatty President, Mulholland Productions Incorporated A. Brent Eastman, M.D. Medical Director, Scripps Health Richard J. Elkus, Jr. Director, KLA-Tencor, Lam Research, Virage Logic Member, Board of Trustees, Palo Alto Medical Foundation Marjorie Fink Philanthropist Phillip Frost, M.D. Chairman and Chief Executive Officer, OPKO Health, Inc. President, The Frost Group Mrs. William McCormick Blair, Jr. Vice President, Albert and Mary Lasker Foundation Louis L. Gonda Chairman and Chief Executive Officer, Lexington Commercial Holdings Chairman, Lexington Ventures, L.L.C. Chairman, Lexington Realty, L.L.C. J. Gary Burkhead Retired, Vice-Chairman, Fidelity Investments Paul L. Herrling, Ph.D. Head, Corporate Research, Novartis International AG Gary N. Coburn Retired Senior Managing Director, Putnam Investments Lawrence C. Horowitz, M.D. President and Managing General Partner, Selby Lane Enterprises II, L.L.C. Vincent E. Benstead Former Partner, PricewaterhouseCoopers Gerald Cohn Retired Executive, Private Investor George H. Conrades Chairman and Chief Executive Officer, Akamai Technologies, Inc. J. Michael Cook Retired Chairman and Chief Executive Officer, Deloitte + Touche Rod Dammeyer President, CAC, L.L.C. John G. Davies, Esq. Of Counsel, Allen Matkins Judicial Appointments Advisor for Governor Arnold Schwarzenegger Thomas E. Dewey, Jr. Member, McFarland Dewey & Co., L.L.C. Alexander W. Dreyfoos Private Investor Chairman, Raymond F. Kravis Center for the Performing Arts Thomas H. Insley Vice President and Chief Financial Officer, SkinMedica, Inc. Amin Khoury Chairman and Chief Executive Officer, B/E Aerospace, Inc. Richard A. Lerner, M.D. President, The Scripps Research Institute Claudia S. Luttrell President, The Skaggs Institute for Research James R. Mellor Former Chairman and Chief Executive Officer, General Dynamics Corporation The Hon. Lynn Schenk Former Congresswoman, California Ralph J. Shapiro Chair, Avondale Investment Company Mark S. Skaggs Board Member, the ALSAM Foundation The Hon. Alice D. Sullivan (Ret.) California Superior Court Judge, Retired Andrew Viterbi, Ph.D. President, Viterbi Group, L.L.C. OFFICERS Richard A. Lerner, M.D. President and Chief Executive Officer Douglas A. Bingham, Esq. Executive Vice President and Chief Operating Officer; Secretary Donna J. Weston Senior Vice President and Chief Financial Officer; Treasurer Thomas E. Northrup, Esq., Ph.D. Chief Business Counsel; Assistant Secretary THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 President’s Introduction am proud to report on some of the many accomplishments at The Skaggs Institute for Chemical Biology at The Scripps Research Institute during the past year. I SCIENTIFIC BREAKTHROUGHS This past year’s scientific findings from scientists Richard at the Skaggs Institute are, as in earlier years, extraordinary. A. Lerner, M.D. • Professor Chi-Huey Wong and colleagues developed a new 2-punch strategy against HIV and successfully tested aspects of the strategy in the laboratory. The investigators created devices they call glycodendrons that are designed to do 2 things at once: (1) inhibit the transport of HIV from where it traditionally enters the body, preventing the virus from moving deeper inside where it can infect immune cells, and (2) set up an immune antibody response to a unique carbohydrate structure on the surface of the virus. • Professor Kim Janda, Associate Professor Eric Zorrilla (Scripps Research), and colleagues discovered a catalytic antibody that degrades a known appetite stimulant. The antibody works against the gastric hormone ghrelin, which has been linked to weight gain and fat storage. These findings may lead to a potentially novel treatment for obesity. • Using samples from survivors of the 2005–2006 “bird flu” outbreak in Turkey, an international team, including researchers at Sea Lane Biotechnologies, L.L.C., Atherton, California, and me, created the first comprehensive libraries of monoclonal antibodies against avian influenza virus (type H5N1). These antibody libraries may be useful in developing a therapy that could stop an influenza pandemic and provide treatment to the people infected and in pointing the way to the development of a universal flu vaccine. • Paul Schimmel, Ernest and Jean Hahn Professor and Chair in Molecular Biology and Chemistry, and colleagues uncovered 2 surprising new methods for correcting mistakes in protein production. This editing system is important because even small mistakes in protein production can have profound disease effects. • Jeffery Kelly, chair of the Department of Molecular and Experimental Medicine and Lita Annenberg Hazen Professor of Chemistry, and colleagues discovered that 2 widely available prescription drugs restore partial cellular folding, trafficking, and function to a variety of mutant enzymes responsible for 3 distinct lysosomal storage diseases, maladies involving failure of multiple organ systems. The team found that the calcium channel blockers diltiazem and verapamil, which are used to treat hypertension, increased the overall function of mutant lysosomal enzymes associated with Gaucher disease, α-mannosidosis, and type IIIA mucopolysaccharidosis in cell lines derived from tissues from patients with these diseases. • Professor John Tainer and colleagues revealed how tiny mutations in a single gene can produce 3 strikingly different childhood diseases. The scientists solved a crystal structure of the enzyme XPD helicase, which unwinds DNA to fix damage that regularly occurs. This research sheds light on 3 different inherited syndromes: xeroderma pigmentosum, which increases the risk for skin cancer by several thousandfold, and Cockayne syndrome and trichothiodystrophy, which are premature aging and developmental disorders. • Professor Elizabeth Getzoff and colleagues developed a new method for chemically targeting a single enzyme to block production of nitric oxide without limiting the beneficial production of this oxide by other closely related enzymes. The technique provides a general solution that should enable development of new drugs to treat medical problems linked to nitric oxide overproduction, such as arthritis, and may aid in the discovery of treatments for other conditions such as HIV disease and AIDS. • Professor Gerald Joyce, dean of the faculty, and colleagues demonstrated genetic adaptation to selective pressure in real time. Under the control of a computer, a population of billions of genes went through 500 cycles of forced adaptation to emerge as molecules that could grow faster and faster on a continually dwindling source of chemical fuel. • Professor Peter Schultz, who holds the Scripps Family Chair, and colleagues produced a powerful immune response in mice by incorporating an unnatural amino acid into a target protein. This novel 7 8 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 approach could be useful in developing new vaccines for cancer, infectious diseases, and other disorders. • Professor Benjamin Cravatt, chair of the Department of Chemical Physiology and director of the Helen L. Dorris Child and Adolescent Neuro-Psychiatric Disorder Institute, and colleagues did a protein survey that nearly tripled the number of proteins known to be involved in programmed cell death and refuted a long-held idea about the life cycle of proteins. The findings may open doors for the discovery of new drugs. PEOPLE NEWS It is with great sadness that I report the death, on October 5, 2008, of Professor Ernie Beutler, chair of the Department of Molecular and Experimental Medicine since 1978. His passing is a great loss to science, to the Skaggs Institute and Scripps Research, and to all who knew and worked with him over his long, brilliant career. It is difficult to adequately acknowledge his host of significant discoveries—among them X-inactivation and novel treatments for Gaucher disease and several forms of leukemia, including hairy cell leukemia—or to fully recognize his authorship of more than 1000 scientific articles in all the leading journals in his field, his numerous monographs and book chapters, and his editing of the widely used textbook Williams Hematology. He was an extraordinary man who led an exceptional life, and I am most thankful that he crossed our path and stayed with us for so long. Filling the position of chair of the Department of Molecular and Experimental Medicine is Jeffery Kelly, who also recently became chair of the Board of Trustees of the Skaggs Institute for Research. Assuming the deanship of the Kellogg School from Dr. Kelly is Professor Jamie Williamson. Dr. Williamson will build on his 7 years as associate dean to lead this top-ranked graduate program into the future. FA C U LT Y H O N O R S In 2008, members of the Skaggs Institute again received many honors and awards. • Professor Peter Wright, chair of the Department of Molecular Biology and Cecil H. and Ida M. Green Investigator in Biomedical Research, was acknowledged for his outstanding research achievements by election to the National Academy of Sciences. • Professor Albert Eschenmoser won the Benjamin Franklin Medal in Chemistry. Franklin Institute Awards are given for outstanding achievements that have enhanced the quality of human life and deepened our understanding of the universe. Dr. Eschenmoser was recognized for his research on the structure of nucleic acids, leading to the understanding of why RNA and DNA have the structures they do. • Professor Ian Wilson was showered with honors, including an honorary degree from the University of St. Andrews in Scotland in recognition of achievements “at the forefront of research to understand the immune system and influenza”; election as a Corresponding Fellow to the Royal Society of Edinburgh, Scotland’s National Academy of Science and Letters; and election to the Board of Directors of the Keystone Symposia. • Professor Carlos Barbas III received the 2009 Tetrahedron Young Investigator Award, Bioorganic and Medicinal Chemistry, an award for scientists less than 45 years old who have exhibited “exceptional creativity and dedication” in their fields. In addition, Dr. Barbas was chosen for the American Chemical Society Arthur C. Cope Scholar Award, which recognizes excellence in organic chemistry. • Jeffery Kelly won the American Peptide Society’s Vincent du Vigneaud Award, sponsored by Bachem, Inc., Torrance, California, for “fundamental, visionary research on folding and aggregation processes in peptides and proteins, and for courageous, insightful exploration of the biological and medical implications of his discoveries.” I am delighted to take this moment to appreciate the many, significant accomplishments that have brought us this far, through the extraordinary generosity and continuing support of the Skaggs family. Thank you also to the many members of the Scripps Research community, including donors, trustees, friends, faculty, staff, postdoctoral fellows, and students, for your dedication, hard work, and vision. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Director’s Overview he Skaggs Institute for Chemical Biology was established in 1996 by a spectacular gift from L.S. “Sam” Skaggs. During the past 12 years, more than 100 million dollars has been awarded in research support for members of the Skaggs Institute. Currently, the funding supports 31 prinJulius Rebek, Jr., Ph.D. cipal investigators, 99 postdoctoral fellows, and 61 graduate students. The mission of the institute is to conduct science that leads to new medicinal agents to relieve suffering. Here I describe some of the progress made by members toward these goals. More details can be found in the individual reports. Stephen Mayfield has used his genetically modified algae to produce carbon-neutral liquid biofuels, a splendid result at a time when fossil fuels reserves are dwindling. Algae can produce biomass at a rate higher than terrestrial plants do and can be used to synthesize therapeutic proteins. In short, algae are a versatile and renewable energy source. M. Reza Ghadiri has developed new cyclic peptide mimetics as scaffolds to present amino acid side chains involved in protein-protein interactions. Using the triazole as a peptide bond surrogate, he has developed useful bioactive probe molecules that imitate the 3-dimensional pharmacophore of naturally occurring tetrapeptides. M.G. Finn continues to modify the surfaces of intact viral capsids by using, among other methods, click chemistry. These modifications have been used to display carbohydrates on the exterior capsid surface as well as polycations that efficiently inhibit the action of heparin. Jeff Kelly, the new chairman of the Department of Molecular and Experimental Medicine, is studying the role of amyloidosis in diabetes. Deposits of amylin in the pancreas are related to the compromised function of these secretory cells that characterize the disease. Jamie Williamson has developed a powerful enzymatic synthesis of nucleotides such as adenosine triphosphate. The process involves 28 enzymes but can be carried out in 60% yield starting from glucose, carbon dioxide, ammonia, and serine. The synthesis is ideal for isotopically labeled products for use in nuclear T magnetic resonance analysis of the structure of proteins and nucleic acids. Ullrich Müller is studying the hair cells of the inner ear that are the principal mechanosensors for the detection of sound and head movement. He is unraveling the molecular composition of the mechanotransduction machinery in these cells by identifying the genes that control their functions. Ehud Keinan has proposed a general synthetic strategy of using a simple pentagonal core to produce chemical capsids that are approximately the size of spherical viruses. He has modeled the assembly and dissociation of these systems under controlled environmental conditions and has made progress in synthesizing the molecules that have the proper shapes and recognition surfaces. Dale Boger and his group work on inhibiting enzymes that control natural painkillers such as anandamide. They have developed synthetic molecules that are more efficient than ibuprofen and are similar to morphine in potency as analgesics in neuropathic pain. Carlos Barbas used a reductionist approach on catalytic antibodies to identify the key features of their catalytic abilities. He has shown that simple chiral amines can be nearly as effective in asymmetric catalysis for many reactions that make complex carbon-carbon bond arrays. Geoffrey Chang has developed x-ray crystallography to characterize molecules involved in multidrug resistance. These molecules transport small drug molecules from inside the cell to outside and are involved in the efflux of antibiotic compounds. The goal is to develop inhibitors of the process that can be used in the treatment of infections. Gerald Joyce, dean of the Scripps Research faculty, has developed “evolution on a chip.” This method combines a large population of RNA molecules and computer controlled microfluidic chips that allow adaptation to occur through hundreds of cycles in a few days. He has also developed small molecules that can trigger RNA enzymes to catalyze their own formation: molecular replication. Kim Janda is working to manipulate the chemical biology of cell-to-cell signaling known as quorum sensing. His findings have applications in controlling virulence and infectivity of bacterial and other microbial agents. Peter Schultz continues to add more amino acids to the repertoire of synthetic biology. Proteins made from amino acids with an expanded genetic code can confer an evolutionary advantage and improved pharmacologic properties. These proteins are directed to applications in biomedical technology. 9 10 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Ian Wilson continues to study those few potent but broadly neutralizing antibodies that recognize HIV type 1. The elusive goal is still to develop the structural information in these complexes for use in a vaccine. Lisa Stowers studies neural circuits that underlie innate behavior. She uses olfactory stimulus in rodents to identify the neurons involved. Her studies suggest that maternal-infant behavior in rodents is also triggered by olfactory mechanisms. In prebiotic chemistry, a debate continues on the relative importance of replication vs metabolism in the origins of life. Albert Eschenmoser is making progress on both of these fronts. He and his group make use of eversimplified backbones derived from glyceric acid for replication and explore the chemistry of glyoxylate for metabolism. Chi-Huey Wong has invented a new method for the ligation of peptides in which attached sugars are used as delivery vehicles. The intent is to optimize the methods to achieve the total synthesis of therapeutic glycoproteins as single isomers. In my own research group, we continue to explore the behavior of molecules in small spaces. These arrangements, known as encapsulation complexes, isolate molecules from the medium and expose unusual behaviors, shapes, and reaction intermediates that cannot be seen in solution. Among the honors bestowed on the Skaggs investigators, 2 were particularly noteworthy. Peter Wright, chairman of the Department of Molecular Biology, was elected to the National Academy of Science, and Tamas Bartfai, Chairman of the Molecular and Integrative Neurosciences Department, was elected to the Swedish Academy of Sciences. Members of the Skaggs Institute won numerous national and international prizes and earned many honorary degrees in the past year. My colleagues and I are grateful for the continued support of the Skaggs Institute for Research. They provide generous funding for basic science at the interface of chemistry and biology. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 INVESTIGATORS’ R EPORTS Catalysis, Cancer, and the Regulation of Genes: Inventing Molecules With Defined Functions C.F. Barbas III, K. Albertshofer, T. Bui, R.P. Fuller, T. Gaj, J. Gavrilyuk, C. Gersbach, B. Gonzalez, R.M. Gordley, J. Guo, S. Juraja, D.H. Kim, A. Mercer, S. Mizuta, A. Onoda, S. Salahuddin, M. Santa Marta, L.J. Schwimmer, F. Tanaka, H. Uehara, N. Utsumi, U. Wuellner, K.S. Yi, H. Zhang e are concerned with problems at the interface of molecular biology, chemistry, and medicine. Many of our studies involve learning or improving on Nature’s strategies to prepare novel molecules that perform specific functional tasks, Carlos F. Barbas III, Ph.D. Professor such as regulating a gene, Molecular Biology destroying cancer, or catalyzing a reaction with small molecules in an enzymelike manner. We hope to apply these novel insights, methods, and products to provide solutions to human diseases, including cancer, HIV disease, and genetic diseases. W C ATA LY T I C A N T I B O D I E S We are extending and refining approaches to catalytic antibodies by using novel recombinant strategies coupled with reactive immunization, chemical-event selections, and the design of unique multiturnover selection chemistries. We are developing in vitro selection and evolutionary strategies as routes for obtaining antibodies of defined biological and chemical activity. These strategies involve the directed evolution of human, rodent, and synthetic antibodies. Essentially, we are evolving proteins to function as efficient catalysts, a task that is naturally performed over eons, and one that we aim to complete in weeks. The approach is a blend of chemistry, enzymology, and molecular biology. A major focus of our research is the development of strategies to produce antibodies that efficiently form and break carbon-carbon bonds. In addition to fashioning new enzymatic function to study the chemistry of imines and enamines, we hope to apply these cata- 11 lysts in novel therapies against cancer and HIV type 1 infection that couple catalytic antibody activity with activation of designed prodrugs. O R G A N O C ATA LY S I S In studying how proteins catalyze reactions, we often examine how the constituent components react. These studies have led to a new green approach to catalytic asymmetric synthesis that can be applied to the synthesis of drugs and druglike molecules. Using insights garnered from our studies of aldolase antibodies, we prepared simple chiral amino acids and amines to catalyze aldol and related imine and enamine chemistries such as Michael and Mannich reactions. We also studied small amine-bearing peptides that are catalytic. Although aldolase antibodies are superior catalysts, simple chiral amino acids and amines are enabling us to determine the importance of pocket sequestration in catalysis. We showed that L-proline and other chiral amines can be efficient asymmetric catalysts of a variety of important imine- and enamine-based reactions. Studies from our laboratory and the contributions of others have produced advances toward one of the ultimate goals in organic chemistry: the catalytic asymmetric assembly of simple and readily available precursor molecules into stereochemically complex products under operationally simple and, in some instances, environmentally friendly experimental protocols. An important result of these studies is the development of catalysts that allow aldehydes, for the first time, to be used efficiently as nucleophiles in a wide variety of catalytic asymmetric reactions. Previously, only naturally occurring enzymes were thought capable of this chemical feat. With future efforts, small organic catalysts may match some of Nature’s other heretofore unmatched synthetic prowess. These catalysts might help explain the development of complex chemical systems in the prebiotic world and provide hints toward yet-to-be discovered mechanisms in extant biological systems. Using this method, we directly synthesized a wide variety of α and β amino acids, carbohydrates, and lactams. Stereochemically complex molecules can now be assembled by using small molecules in a manner analogous to that of natural enzymes. Novel catalyst designs have enabled us to synthesize particular diastereoisomers previously not accessible with proline, and we envision that this approach will largely replace the use of aldolase enzymes in synthesis (Fig. 1). New and exciting catalytic asymmetric reactions continue to emerge from these studies. 12 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 F i g . 1 . Organocatalysis with natural and designed amino acids leads to a variety of efficient asymmetric syntheses previously approachable only via enzyme catalysis. A–C, Design considerations for a family of organoaldolases that allow large families of carbohydrates to be readily synthesized. C H E M I C A L LY P R O G R A M M E D A N T I B O D I E S In targeting cancer, we take a multidisciplinary approach that involves gene regulation, catalytic antibodies, drug design, and combinatorial antibody libraries. Using a chemically programmed antibody strategy, we recently showed the power of combining small-molecule chemistry with immunochemistry. We designed smallmolecule integrin antagonists to self-assemble into a covalent complex with antibody 38C2 (Fig. 2). The resulting chemically programmed antibody had significant advantages compared with small molecules or antibody alone in studies of metastatic melanoma, colon cancer, and breast cancer. We recently developed a powerful new approach to a programmable vaccine strategy based on a universal vaccination that elicits programmable antibodies. DESIGNER TRANSCRIPTION FACTORS AND ENZYMES From the simplest to the most complex, proteins that bind nucleic acids are involved in orchestrating gene expression. DNA and RNA are the molecules that store the recipes of all life forms. The fertilized egg of a human contains the genetic recipe for the development and differentiation of a single cell into 2 cells, 4 cells, and so on, finally yielding a complete individual. The coordinated expression or reading of the recipes for life allows cells containing the same genetic information to perform different functions and to have distinctly different physical characteristics. Lack of coordination F i g . 2 . Combining the power of small-molecule chemistry with the power of protein chemistry and immunology, we have created a new and effective class of therapeutic molecules, chemically programmed antibodies. A covalently bound diketone is shown in the active site of an aldolase antibody. This covalent chemistry allows rapid antibody programming. due to genetic defects or to viral seizure of control of the cell results in disease. In one project, we are developing methods to produce proteins that bind to specific DNA sequences to control specified genes. As we showed earlier, these proteins can be used as specific genetic switches to turn on or turn off genes on demand, creating an operating system for genomes. To this end, we selected and designed specific zinc finger domains that will constitute an alphabet of 64 domains that will allow any DNA sequence to be bound selectively. The prospects for this “second genetic code” are fascinating and should have a major impact on basic and applied biology. Billions of transcription factors can now be prepared by using our approach. Our goal is to develop a new class of therapeutic proteins that inhibit or enhance the synthesis of proteins, providing a new strategy for fighting diseases of either somatic or viral origin. Using a novel library of transcription factors, we developed a strategy that effectively allows us to turn on and turn off every gene in the genome. We recently THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 extended this approach to enable us to endow a variety of enzymes with sequence specificity of our own design (Fig. 3). In the future, these new enzymes will enable us to insert, delete, or otherwise modify genes with surgical precision within any genome. 13 Zhang, H.L., Mitsumori, S., Utsumi, N., Imai, M., Garcia-Delgado, N., Mifsud, M., Albertshofer, K., Tanaka, F., Barbas, C.F. III. Catalysis of 3-pyrrolidinecarboxylic acid and related pyrrolidine derivatives in enantioselective anti-Mannichtype reactions: importance of the 3-acid group on pyrrolidine for stereocontrol. J. Am. Chem. Soc. 130:875, 2008. Cytokine Inflammatory Signaling in Obesity and Neurodegeneration T. Bartfai, B. Conti, I. Tabarean, M. Sanchez-Alavez, O. Osborn, I. Klein he “endogenous pyrogen,” the proinflammatory cytokine IL-1, which has been a major topic of our studies, is a key cytotoxic agent for the pancreatic beta cells that produce insulin. In obesity, increased insulin resistance necessitates the production Tamas Bartfai, Ph.D. and release of more and more Professor and Chairman Molecular and Integrative insulin to control blood glucose Neurosciences levels. We wish to know if blocking IL-1 signaling in obese individuals can protect the beta cells and thus postpone or block the development of type 2 diabetes and the need for insulin injection as the endogenous production of insulin decreases and finally stops. We plan to use AS-1 and EM163, blockers of signaling by Toll-like receptors that were developed in collaboration with J. Rebek, the Skaggs Institute. These low molecular weight blockers, which are mimetics of the adaptor protein myeloid differentiation factor 88, are ideal for oral administration and may be used for chronic treatment of obesity/type 2 diabetes. As proof of principle of the therapeutic value of blocking IL-1 signaling, we used high-affinity mouse monoclonal antibodies that neutralize IL-1. In obese animals with stressed, enlarged, and/or dying beta pancreatic cells, blockade of cytotoxic IL-1 signaling protected beta cells and prevented enlargement of the cells. These findings suggest that such blockade would likely postpone the development of type 2 diabetes, for which obesity, alongside age, is a major risk factor. This study opens the way for the use of molecules such AS-1 and EM 163 and their derivatives in treatment of obesity to postpone development of type 2 diabetes. IL-1 is also a major inflammatory and neurogenesispromoting signal in the developing brain. Blockade of T F i g . 3 . Through a combination of rational and evolutionary design, we created a variety of zinc finger enzymes that function in human cells. Novel enzymes such as recombinases, methylases, nucleases, and integrases are under development. A designed zinc finger recombinase enzyme is shown above the sequence on which it acts. PUBLICATIONS Alonso, D., Kitagaki, S., Utsumi, N., Barbas, C.F. III. Towards organocatalytic polyketide synthases with diverse electrophile scope: trifluoroethyl thioesters as nucleophiles in organocatalytic Michael reactions and beyond. Angew. Chem. Int. Ed. 47:4588, 2008. Blancafort, P., Tschan, M.P., Bergquist, S., Guthy, D., Brachat, A., Sheeter, D.A., Torbett, B.E., Edrmann, D., Barbas, C.F. III. Modulation of drug resistance by artificial transcription factors. Mol. Cancer Ther. 7:688, 2008. Gordley, R.M., Gersbach, C.A., Barbas, C.F. III. Synthesis of programmable integrases. Proc. Natl. Acad. Sci. U. S. A., in press. Jiang, L., Althoff, E.A., Clemente, F.R, Doyle, L., Röthlisberger, D., Zanghellini, A., Gallaher, J.L., Betker, J.L., Tanaka, F., Barbas, C.F. III, Hilvert, D., Houk, K.N., Stoddard, B.L., Baker D. De novo computational design of retro-aldol enzymes. Science 319:1387, 2008. Magnenat, L., Schwimmer, L.J., Barbas, C.F. III. Drug-inducible and simultaneous regulation of endogenous genes by single-chain nuclear receptor-based zinc-finger transcription factor gene switches [published correction appears in Gene Ther. 15:1246, 2008]. Gene Ther. 15:1223, 2008. Massa, A., Utsumi, U., Barbas, C.F. III. N-Tosylimidates in highly enantioselective organocatalytic Michael reactions. Tetrahedron Lett. 50:145, 2009. Ramasastry, S.S.V., Albertshofer, K., Utsumi, N., Barbas, C.F. III. Water-compatible organocatalysts for direct asymmetric syn-aldol reactions of dihydroxyacetone and aldehydes. Org. Lett. 10:1621, 2008. Tanaka, F., Hu, Y., Sutton, J., Asawapornmongkol, L., Fuller, R., Olson, A.J., Barbas, C.F. III, Lerner, R.A. Selection of phage-displayed peptides that bind to a particular ligand-bound antibody. Bioorg. Med. Chem. 16:5926, 2008. Utsumi, N., Kitagaki, S., Barbas, C.F. III. Organocatalytic Mannich-type reactions of trifluoroethyl thioesters. Org. Lett. 10:3405, 2008. Zhang, H., Ramasastry, S.S.V., Tanaka, F., Barbas, C.F. III. Organocatalytic antiMannich reactions with dihydroxyacetone and acyclic dihydroxyacetone derivatives: a facile route to amino sugars. Adv. Synth. Catal. 350:791, 2008. 14 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 IL-1 signaling during development blunts the division of nerve cells and thus produces cognitive deficits. This effect is an important caveat for using blockers of IL-1 signaling in nonadults. The protein transthyretin can produce plaques similar to the proteolytic degradation product of the amyloid precusor protein, which forms amyloid plaques, the hallmark of Alzheimer’s disease. It was assumed that transthyretin acts as “seed” in the formation of amyloid plaques and that it indirectly may influence the concentration and actions of the neurotoxic monomers amyloid β-peptide1-40/42. However, collaborative studies with J.N. Buxbaum, Scripps Research, in transgenic mice that overproduce the amyloid peptide indicated the opposite. Transthyretin can protect the brain from the neurotoxic effects of amyloid β-peptides, probably by promoting the clearance of the neurotoxic monomers, and thus may be part of endogenous neuroprotection mechanisms. These findings open a previously unknown therapeutic possibility for treatment of Alzheimer’s disease. PUBLICATIONS Buxbaum, J.N., Ye, Z., Reixach, N., Friske, L., Levy, C., Das, P., Golde, T., Masliah, E., Roberts, A.R., Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. U. S. A. 105:2681, 2008. Osborn, O., Brownell, S.E., Sanchez-Alavez, M., Salomon, D., Gram, H., Bartfai, T. Treatment with an interleukin 1 beta antibody improves glycemic control in dietinduced obesity. Cytokine 44:141, 2008. Spulber, S., Oprica, M., Bartfai, T., Winblad, B., Schultzberg, M. Blunted neurogenesis and gliosis due to transgenic overexpression of human soluble IL-1ra in the mouse. Eur. J. Neurosci. 27:549, 2008. Training in Molecular and Experimental Medicine E. Beutler strong relationship between the basic sciences of chemistry and biology and clinical medicine is essential for understanding the basic biology of disease and the directed development of therapeutic interventions. Ernest Beutler, M.D. Professor and Chairman This understanding requires Molecular and specific technical training that Experimental Medicine provides a perspective encompassing both sides. The Skaggs Institute for Chemical A Biology has attempted to provide such training by supporting young scientists in the Department of Molecular and Experimental Medicine. Christian Nievera, under the supervision of Xiaohua Wu, associate professor, is studying molecular mechanisms involved in the maintenance of genome stability and repair after DNA damage. Genome instability and aberrant DNA repair lead to gross chromosomal rearrangement, a major underlying cause for tumorigenesis. Dr. Nievera is determining the role of the Mre11/Rad50/ Nbs1 (MRN) complex and its interaction with replication protein A in modulating the S-phase checkpoint after DNA damage. He is examining the mechanism by which this interaction leads to the suppression of replication origin firing in response to DNA damage. Furthermore, he has found that MRN interacts directly with the breast cancer suppressor protein BRCA1. He is determining how BRCA1 works with MRN to repair DNA. Because much of natural resistance to malignant transformation due to mutational events appears to be related to the capacity of cells to preserve the integrity of DNA, an understanding of the mechanisms involved in these responses is critical. Jaroslav Truksa, a trainee in my laboratory, has been studying transcriptional regulation of hepcidin, a critical regulator of iron metabolism. Hepcidin appears to be particularly important in anemia of chronic inflammation and iron refractory anemia. Aberrant hepcidin expression is also associated with hemochromatosis. Dr. Truksa has used innovative methods—a luciferase reporter and in vivo bioluminescence—to study transcriptional regulation in intact animals. With this technology, he has defined an upstream region of the hepcidin promoter that is important in the response to ingested iron. Using tissue culture methods, he has defined a second, distinct region in the hepcidin promoter that responds to cytokine stimulation. In addition, he examined the role of Tmprss6, a novel protein associated with iron refractory anemia in humans, and found that it suppresses the total level of expression of hepcidin induced by inflammatory cytokines and bone morphogenic proteins. Dr. Truksa is also examining the repression of hepcidin by growth differentiation factor GDF15, which is highly expressed in thalassemia. He is investigating the intracellular mediators involved in signaling between the cell-surface modulators of iron (hemojuvelin, HFE, TfR2, and Tmprss6) and the hepcidin gene. Understanding the pathway of iron regulation by hepcidin will provide THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 insight into the future management both primary and secondary iron storage diseases. Each of the trainees has fulfilled the goals of the Skaggs program by applying basic scientific knowledge and techniques to disease-related biologic systems. They have each published several articles in outstanding journals and have made or are making contributions to the understanding of clinical disorders. 15 cycles) that are active in vivo in all rodent models of pain examined. The most extensively studied of the inhibitors to date (e.g., OL-135 at 20 mg/kg) are as efficacious as morphine (at 1–3 mg/kg), ibuprofen (at 100 mg/kg), and gabapentin (at 500 mg/kg) and, unlike ibuprofen and gabapentin, maintain activity across all rodent pain models (Fig. 1). This research constitutes Synthetic, Medicinal, and Bioorganic Chemistry D.L. Boger, E. Anderson, Y. Ando, K. Boyle, C. Burke, R. Clark, D. Colby, C. Crane, J. DeMartino, K. Duncan, C. Ezzili, J. Garfunkle, H. Ge, D. Hochstatter, I. Hwang, R. Jones, H. Kakei, D. Kato, F.S. Kimball, J. Lajiness, S. Lee, K. MacMillan, K. Otrubova, P. Patel, W. Robertson, R. Rodriguez, Y. Sasaki, M. Schnermann, S. Seto, H. Shimamura, C. Slown, S. Stamm, J. Stover, D. Swingle, S. Takizawa, A. Tam, P. Va, L. Whitby, A. Wolfe, J. Xie, A. Zuhl he research interests of our group include the total synthesis of biologically active natural products, the development of new synthetic methods, heterocyclic chemistry, bioorganic and medicinal chemistry, combinatorial Dale L. Boger, Ph.D. chemistry, the study of DNAProfessor agent interactions, and the Chemistry chemistry of antitumor antibiotics. We place a special emphasis on investigations to define the structure-function relationships of natural or designed agents in efforts to understand the origin of the biological properties. T FAT T Y A C I D A M I D E H Y D R O L A S E I N H I B I T O R S Inhibiting fatty acid amide hydrolase (FAAH) increases endogenous levels of anandamide, which in turn leads to analgesia in models of neuropathic and chronic pain. Extending studies conducted in collaboration with R.A. Lerner and B.F. Cravatt, the Skaggs Institute, that led to the identification, isolation, characterization, and delineation of the functional role of FAAH, we have developed a series of exceptionally potent and selective inhibitors of FAAH (α-ketohetero- F i g . 1 . Profile of a prototypical FAAH inhibitor. a well-defined translation of basic science (target discovery, target structure) into useful tools (inhibitors of enzyme for target validation) and their further development into potential therapeutic agents (optimization for in vivo efficacy). DNA-BINDING AGENTS Our continuing examination of naturally occurring antitumor agents that derive their biological properties through sequence-selective DNA binding resulted in a detailed study of yatakemycin. We defined the exceptional potency of the natural material; characterized its DNA alkylation properties, consisting of an adenine N3 alkylation central to a 5-bp adenine-thymine–rich site; and conducted first- and second-generation syntheses of the natural product in efforts that provided both the natural and the unnatural enantiomers. The unnatural enantiomer was just as effective and potent as the natural product itself. 16 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 These efforts not only provided a sufficient amount of the scarce natural product for detailed studies of its properties but also enabled the assignment of its unknown absolute configuration. Unexpectedly, these efforts also led to a reassignment of the structure of the natural product and revealed a new group (thiomethyl ester) in the molecule that contributes to the properties of yatakemycin. With the systematic preparation of more than 70 analogs, we have defined key structural features responsible for the biological properties of this antitumor agent. VA N C O M Y C I N A N D R E L AT E D G LY C O P E P T I D E ANTIBIOTICS Vancomycin and its family of related naturally occurring glycopeptides, which include teicoplanin and ristocetin, are potent antibiotics used to treat infections caused by microorganisms resistant to other antibiotics. Antibiotics in the vancomycin family inhibit the synthesis of bacterial cell walls by binding to the terminal D -Ala-D -Ala of the precursor cell wall peptidoglycan, thereby inhibiting the action of transpeptidases and transglycosylases required to complete the cell wall synthesis. As part of a program to define the structural features that contribute to the activity of members of the vancomycin family and to explore approaches to improve the biological properties of the drugs, we developed an efficient de novo synthesis of the aglycons of these antibiotics. The emergence of vancomycin resistance is associated with an alteration of the bacterial cell wall precursor to D -Ala-D -Lac, resulting in a 1000-fold loss in vancomycin binding affinity and antimicrobial activity. We showed experimentally that this loss in binding affinity is due primarily to destabilizing lone-pair interactions (100-fold) rather than to the simple loss of a single hydrogen bond (10-fold) in the bound complex (Fig. 2). This finding has consequences on the reengineering of vancomycin to bind D -Ala- D -Lac so that antimicrobial activity against vancomycin-resistant bacteria can be restored. We recently completed the total synthesis of the [ψ[CH 2NH]Tpg4]vancomycin aglycon in which the residue 4 carbonyl and its destabilizing lone pairs have been removed from the vancomycin structure. Examination of this aglycon revealed that such a reengineering of vancomycin provides antibiotics active against vancomycin-sensitive and vancomycin-resistant bacteria. Ramoplanin, a naturally occurring complex of 3 components, represents a new and potent antibiotic F i g . 2 . Origin of vancomycin-destabilized binding to D-Ala-D-Lac. effective against antibiotic-resistant bacteria, including vancomycin-resistant strains. As a complement to our studies on the reengineering of vancomycin, we are examining this new class of antibiotics. To date, this research has resulted in the total synthesis of the ramoplanin A2 aglycon (major component of the complex), the total synthesis and structural reassignment of the ramoplanin A1 and A3 aglycons (minor components), and the preparation of a series of key analogs. One analog, [Dap 2 ]ramoplanin A2 aglycon in which the labile depsipeptide ester is replaced by a stable amide, was slightly more potent and much more stable than the natural product and provided a stable template for detailed structure-function studies. PUBLICATIONS Eubanks, L.M., Hixon, M.S., Jin, W., Hong, S., Clancy, C.M., Tepp, W.H., Baldwin, M.R., Malizio, C.J., Goodnough, M.C., Barbieri, J.T., Johnson, E.A., Boger, D.L., Dickerson, T.J., Janda, K.D. An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists [published correction appears in Proc. Natl. Acad. Sci. U. S. A. 104:6490, 2008]. Proc. Natl. Acad. Sci. U. S. A. 104:2602, 2007. Hardouin, C., Kelso, M.J., Romero, F.A., Rayl, T.J., Leung, D., Hwang, I., Cravatt, B.F., Boger, D.L. Structure-activity relationships of the α-ketooxazole inhibitors of fatty acid amide hydrolase. J. Med. Chem. 50:3359, 2007. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Ishikawa, H., Boger, D.L. Total synthesis of (–)- and ent-(+)-4-desacetoxy-5desethylvindoline. Heterocycles 72:95, 2007. Jin, W., Trzupek, J.D., Rayl, T.J., Broward, M.A., Vielhauer, G.A., Weir, S.J., Hwang, I., Boger, D.L. A unique class of duocarmycin and CC-1065 analogues subject to reductive activation. J. Am. Chem. Soc. 129:15391, 2007. Lee, S.Y., Clark, R.C., Boger, D.L. Total synthesis, stereochemical reassignment, and absolute configuration of chlorofusin. J. Am. Chem. Soc. 129:9860, 2007. Nam, J., Shin, D., Rew, Y., Boger, D.L. Alanine scan of [L-Dap2]ramoplanin A2 aglycon: assessment of the importance of each residue. J. Am. Chem. Soc. 129:8747, 2007. Romero, F.A., Du, W., Hwang, I., Rayl, T.J., Kimball, F.S., Leung, D., Hoover, H.S., Apodaca, R.L., Breitenbucher, J.G., Cravatt, B.F., Boger, D.L. Potent and selective α-ketoheterocycle-based inhibitors of the anandamide and oleamide catabolizing enzyme, fatty acid amide hydrolase. J. Med. Chem. 50:1058, 2007. Tichenor, M.S., MacMillan, K.S., Stover, J.S., Wolkenberg, S.E., Pavani, M.G., Zanella, L., Zaid, A.N., Spalluto, G., Rayl, T.J., Hwang, I., Baraldi, P.G., Boger, D.L. Rational design, synthesis, and evaluation, of key analogues of CC-1065 and the duocarmycins. J. Am. Chem. Soc. 129:14092, 2007. Tichenor, M.S., MacMillan, K.S., Trzupek, J.D., Rayl, T.J., Hwang, I., Boger, D.L. Systematic exploration of the structural features of yatakemycin impacting DNA alkylation and biological activity. J. Am. Chem. Soc. 129:10858, 2007. Xu, L., Chong, Y., Hwang, I., D’Onofrio, A., Amore, K., Beardsley, G.P., Li, C., Olson, A.J., Boger, D.L., Wilson, I.A. Structure-based design, synthesis, evaluation, and crystal structures of transition state analogue inhibitors of inosine monophosphate cyclohydrolase. J. Biol. Chem. 282:13033, 2007. Structure and Biology of Multidrug Transporters 17 member of the ATP-binding cassette transporter family. We are also using electron cryomicroscopy, in collaboration with R. Milligan, Scripps Research, to study other conformations. In addition, we have determined the x-ray structures of proton-drug antiporters, including EmrE from the small multidrug resistance transporter family and EmrD from the major facilitator superfamily. We recently determined the x-ray structure of P-glycoprotein in complexes with drugs to understand the structural basis of polyspecificity. With support from the Skaggs Institute, we have focused on determining the structure of a bacterial MDR transporter from the multiple antimicrobial toxin extrusion family, which has important clinical relevance. This family is the last family of MDR transporters whose structure has not yet been characterized, and these transporters play an important role in the efflux of a variety of antibiotic compounds, including fluoroquinolones. MDR transporters from this family also efflux antimicrobial compounds in plants as part of the plants’ natural defense mechanism against microbes. A structure of the bacterial transporter would greatly aid the rational design of inhibitors to reverse MDR in the treatment of infection. Chemical Physiology G. Chang, S. Aller, Y. Chen, X. He, A. Karyakin, S. Lieu, T. Nguyen, M. Revin, P. Szewczyk, A. Ward, J. Yu ultidrug resistance (MDR) is a major clinical problem in the chemotherapy of infection and cancer. The structural characterization of MDR transporters is important for the development of future compounds to reverse drug resistance. We Geoffrey Chang, Ph.D. Associate Professor are interested in the molecular Molecular Biology basis of the transport of drugs and lipids across the cell membrane by bacterial and mammalian MDR transporters. We combine structure, function, and chemistry through collaborations with M.G. Finn, the Skaggs Institute, and Q. Zhang, Scripps Research. We use several techniques, including detergent/lipid protein chromatography, membrane protein crystallization, and x-ray crystallography. We have already solved the molecular structures of several conformations of the lipid flippase MsbA, a M B.F. Cravatt, D. Bachovchin, J. Blankman, M. Dix, S. Ji, F. Kopp, W. Li, J. Long, B. Martin, K. Masuda, M. McKinney, D. Nomura, G. Simon, J. Thomas, S. Tully, E. Weerapana e are interested in understanding complex physiology and behavior at the level of chemistry and molecules. At the center of cross talk between different physiologic processes are endogenous compounds that provide a molecular mode Benjamin F. Cravatt, Ph.D. Professor for intersystem communicaCell Biology tion. However, many of these molecular messages remain unknown, and even in the instances in which the participating molecules have been defined, the mechanisms by which these compounds function and their modes of regulation are for the most part still a mystery. One family of chemical messengers we study is the endogenous cannabinoids (“endocannabinoids”), a class W 18 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 of lipid signaling molecules that activate cannabinoid receptors in the nervous system and peripheral tissues. The levels and signaling function of endocannabinoids are tightly regulated by enzymes to maintain proper control over the influence of the endocannabinoids on brain and body physiology. One of our major goals is to identify endocannabinoid biosynthetic and degradative enzymes and develop selective genetic and pharmacologic tools to perturb the function of these enzymes in vivo. An example is fatty acid amide hydrolase (FAAH), which terminates the signaling function of the endocannabinoid anandamide, as well as several other amidated lipid transmitters. We are using transgenic and synthetic chemistry techniques to study the role of FAAH in regulating fatty acid amide levels in vivo. We found that transgenic mice that lack FAAH have highly elevated levels of fatty acid amide in the brain that correlate with reduced pain behavior, suggesting that FAAH may be a new therapeutic target for the treatment of pain and related neural disorders. In collaboration with R.C. Stevens, Scripps Research, we solved the first 3-dimensional structure of FAAH. We are using this information to design potent and selective inhibitors of the enzyme. In studies with D.L. Boger, the Skaggs Institute, we have identified potent FAAH inhibitors and using a functional proteomic screen developed by us, have shown that these inhibitors are highly selective for this enzyme. We are also interested in proteins responsible for the biosynthesis of endocannabinoids. A second major focus in the laboratory is the design and use of chemical probes for the global analysis of protein function. The evolving field of proteomics, defined as the simultaneous analysis of the complete protein content of a given cell or tissue, encompasses considerable conceptual and technical challenges. We hope to enhance the quality of information obtained from proteomics experiments by using chemical probes that indicate the collective catalytic activities of entire classes of enzymes. Using activity-based probes that target the serine and metallo hydrolases, we have identified several enzymes with altered activities in human cancers. Using a combination of pharmacologic and molecular biology approaches, we are now testing the role that these enzymes play in cancer pathogenesis. Finally, we are developing proteomic and metabolomic platforms to map endogenous substrates of enzymes in native biological systems. These large-scale technologies are intended to provide a global, unbiased portrait of the physiologic activities of enzymes, thereby aiding in the functional annotation and assessment of the enzymes as potential therapeutic targets for a range of human diseases. PUBLICATIONS Ahn, K., Johnson, D.S., Fitzgerald, L.R., Liimatta, M., Arendse, A., Stevenson, T., Lund, E.T., Nugent, R.A., Nomanbhoy, T.K., Alexander, J.P., Cravatt, B.F. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 46:13019, 2007. Ahn, K., McKinney, M.K., Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 108:1687, 2008. Blankman, J.L., Simon, G.M. Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 14:1347, 2007. Carlson, E.E., Cravatt, B.F. Enrichment tags for enhanced-resolution profiling of the polar metabolome. J. Am. Chem. Soc. 129:15780, 2007. Cravatt, B.F., Simon, G.M., Yates, J.R. III. The biological impact of mass-spectrometry-based proteomics. Nature 450:991, 2007. Cravatt, B.F., Wright, A.T., Kozarich, J.W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 77:383, 2008. Dix, M.M., Simon, G.M., Cravatt, B.F. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134:679, 2008. Salisbury, C.M., Cravatt, B.F. Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes. J. Am. Chem. Soc. 130:2184, 2008. Simon, G.M., Cravatt, B.F. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 283:9341, 2008. Weerapana, E., Simon, G.M., Cravatt, B.F. Disparate proteome reactivity profiles of carbon electrophiles. Nat. Chem. Biol. 4:405, 2008. Fundamental Processes in Neural Development G.M. Edelman, A. Atkins, D.C.Y. Koh, D. Matsuda, P. Panopoulos, J. Pilotte cientists in the Department of Neurobiology focus on the features of primary cellular processes that regulate the development of the vertebrate nervous system. In the past year, the emphasis has been on factors that control the translation of mRNA Gerald M. Edelman, M.D., Ph.D. into protein, including the speProfessor and Chairman cific regulation of local transNeurobiology lation at synapses. Equally important have been studies of RNA-binding proteins such as the cold-induced RNA-binding motif protein 3 S THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 (RBM3) and its influence on the differentiation and function of neurons at the level of both transcription and translation. Julie Pilotte has observed that RBM3 is highly expressed in proliferative zones and plastic regions of the brain in rats and is developmentally regulated. Within mature neurons, RBM3 is present in nuclei and in dendrites. Dr. Pilotte’s studies in neurons and other cells have shown that RBM3 localizes to leading edges of processes and to structures containing translation machinery that presage the formation of mature adhesion plaques. Functional studies indicate that RBM3 has a profound effect on cell motility and morphology, an effect that appears to extend to neurite outgrowth. These effects of RBM3 likely involve the potent regulatory influence of RBM3 on mRNA translation. Our previous work established that RBM3 greatly enhances mRNA translation rates, potentially by altering the production of microRNAs. Dr. Pilotte has conducted a comprehensive analysis of the relationship between RBM3 expression levels and the production of microRNAs. The data suggest several possible mechanisms underlying the effects of RBM3 on cell morphology and motility that involve microRNAs that target components of the translation machinery and cytoskeleton. These studies are ongoing. Overall, RBM3 appears to have a variety of functions critical for cell morphology, migration, and maturation that may be involved in the brain during neural development and in plasticity in adults. In the cytoplasm, RBM3 plays important roles in regulating mRNA transport and translation. In many cells, however, the protein is more strongly expressed in the nucleus, where its function is just beginning to be defined. Annette Atkins used immunoprecipitation of RBM3 from nuclear extracts and proteomic analyses in collaboration with L. Liao and J.R. Yates, Scripps Research, to show that the protein colocalizes with a subset of proteins from the splicing machinery. Her further studies have shown that the protein can influence the splicing of specific mRNAs. Moreover, she obtained convincing evidence that RBM3 regulates its own expression by splicing an exon with a premature termination codon from its message, preventing degradation of the mRNA via a process known as nonsense-mediated decay. These results add an important new dimension to the role of RBM3 and related RNA-binding proteins in regulating protein synthesis at multiple levels. Vincent Mauro and his colleagues have been studying basic mechanisms of translation initiation in eukaryotes. 19 Earlier studies by this group showed that a 9-nucleotide segment from the 5′ leader of the Gtx homeodomain mRNA facilitated translation initiation by base pairing to 18S rRNA, the RNA component of 40S ribosomal subunits. Although the Gtx element was tested in isolation in earlier studies, the results indicated that eukaryotic mRNAs could initiate translation much as the Shine-Dalgarno interaction does in bacteria. Studies by Panagiotis Panopoulos have now shown the physiologic relevance of this element in the context of 2 natural mRNAs that contain this sequence in their 5′ leaders: Gtx itself and fibroblast growth factor 2. For these studies, he used modified RNA oligonucleotides to block mRNA-rRNA base pairing by targeting complementary sequences in either the 18S rRNA or mRNAs and by mutating the Gtx element in the context of the natural mRNA sequences. Dora Koh has investigated the translation of the β-site amyloid precursor protein–cleaving enzyme 1 (BACE1) mRNA. The increased translation of this mRNA has been implicated in the etiology of Alzheimer’s disease. Her studies resolved an apparent discrepancy between various published studies by showing that the various results could be explained by the use of different expression systems and differences in interpretation. She showed that the translation of the BACE1 mRNA was affected by the expression system and that it occurred by a ribosomal shunting mechanism when the mRNA was expressed in the nucleus via RNA polymerase II. In other studies, she has probed the RNA conformation of 2 endogenous RNAs in living cells: RNase P, which was probed as a proof of concept, and BACE1. These studies revealed a strong correlation between nucleotide accessibility and the site of translation initiation in the BACE1 mRNA, supporting the tethering/clustering model of translation initiation that was previously postulated by Dr. Mauro’s group. Daiki Matsuda is using synthetic mRNAs with multiple potential initiation codons and various obstacles designed to block individual codons to further investigate the notion that the accessibility of the initiation codon is a key factor affecting its use. All of these studies are designed to help define the molecular and cellular events that regulate the development and function of the nervous system. Our focus on fundamental processes is based on the belief that understanding these processes can provide the necessary framework for defining the mechanisms underlying not just one but many diseases. Indeed, our studies 20 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 have already provided insights into aspects of a number of diseases, including Alzheimer’s disease and mental retardation syndromes. Chemical Etiology of Nucleic Acid Structure A. Eschenmoser, R. Krishnamurthy, G.K. Mittapalli, R.R. Kondreddi, Y. Osornio, V.S. Naidu not considered a potentially prebiotic system, in contrast to the oligomer system derived from glyceric acid and tagged via amide bonds with 5-aminopyrimidines. We have completed the synthesis of such a glyceric acid–derived oligomer containing six 5-aminouracil units (6-mer) and have studied its base-pairing properties with DNA, RNA, and α-L-threofuranosyl-(3′g2′) nucleic acid. Base pairing was strong between the 6mer and poly-d(A) (Fig. 2), was somewhat weaker with the corresponding poly-r(A), and even occurred with α-L-threofuranosyl-3′g2′) nucleic acid. n the general context of our project to map the landscape of potentially primordial informational oligomer systems, we have been working during the past year on the following topics. I OLIGOMERS BASED ON 5 - A M I N O P Y R I M I D I N E – TA G G E D 2′g3′-PHOSPHODIESTER– L I N K E D G LY C E R I C A C I D Albert Eschenmoser, Ph.D. Professor Chemistry BACKBONES We have undertaken the synthesis and study of the base-pairing properties of oligomers derived from a 2′g3′-phosphodiester–linked glyceric acid backbone that bears 2,4-disubsituted 5-aminopyrimidines, attached to the carboxyl group of glyceric acid via an amide bond with the 5-amino group, as recognition elements (Fig. 1). The structure of this oligomeric system is based on a structural simplification of the oligonucleotides containing lyxopyranosyl-(3′g4′)– and threofuranosyl-(2′g3′)–linked phosphodiester backbones, which we have studied previously. Among the oligomer systems depicted in Figure 1, the nucleic acid derived from the glycerol backbone is F i g . 1 . Structural simplification of α- L -threofuranosyl-(3′g2′) nucleic acid, which was inspired by studies on (3′g4′)-lyxopyranosyl nucleic acid, gives rise to acyclic informational oligomeric systems. Two examples are shown: glycerol nucleic acid and glyceric acid nucleic acid. F i g . 2 . UV (left) and circular dichroism (right) spectroscopic data for base pairing between 5-aminouracil–tagged 2-phosphoglycerate hexamer and DNA, poly(dA); c = 5+5 µM. Measurements were made in phosphate buffer. E X P L O R I N G T H E C H E M I S T R Y O F G LY O X Y L AT E A N D D I H Y D R O X Y F U M A R AT E A research project such as mapping the landscape of potentially primordial informational oligomer systems eventually demands the conception of, and the commitment to, a detailed chemical scenario for the type of organic chemistry that is supposed to have led to such oligomers under primordial conditions. Figure 3 depicts the chemical nature of the scenario we have decided to study experimentally. In the reaction cycle, glyoxylate would autocatalytically convert itself into its dimer dihydroxyfumarate. Dihydroxyfumarate is a known compound that we postulate can act as a common starting material for a large variety of biomolecules, such as sugars, α-amino acids, and pyrimidines, and for other organics of etiologic interest by reactions that are essentially unexplored thus far but are deemed compatible with the constraints of a primordial chemistry. We are conducting exploratory studies for assessing the chemistry of selected intermediates postulated to be formed from the chemistry of glyoxylate and dihydroxyfumarate. Some of the promising preliminary results include the formation of dihydroxyacetone from the reaction of dihydroxyfumarate with glyoxylate, conversion of 2,3-dioxobutanoic acid (one of the proposed products of the THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 F i g . 3 . Hypothetical autocatalytic cycle for the dimerization of glyoxylate to dihydroxyfumarate and the biomolecules to be derived from the constituents of that cycle. reaction between glyoxylate and dihydroxyfumarate) to alanine, and identification of hitherto undiscovered reaction pathways and intermediates in the reaction of dihydroxyfumarate with itself and with glyoxylate. PUBLICATIONS Eschenmoser, A. On a hypothetical generational relationship between HCN and constituents of the reductive citric acid cycle. Chem. Biodivers. 4:554, 2007. 21 tures to perform these functions, these nucleic acids tend to assemble into a mixture of properly folded and misfolded structures during assembly reactions carried out in vitro. We hope to learn how RNAs avoid misfolding and assemble into functional structures during biogenesis in vivo. We have developed a way to use self-cleaving ribozymes embedded in chimeric mRNAs and noncoding RNAs to probe RNA assembly in living cells. Ribozyme self-cleavage provides a sensitive, quantitative signal that functional ribozyme structures have formed. We began by engineering ribozyme variants with flanking inserts that have the potential to form complementary base pairs with parts of the ribozyme sequence. The inserts are located either upstream or downstream of the ribozyme sequence so they will be transcribed either before or after the ribozyme. This arrangement allows us to examine how the sequential nature of RNA synthesis affects folding outcomes (Fig. 1). Assembly of AltH1, the alternative helix, is incompatible with assembly of H1, the ribozyme helix, and prevents self-cleavage. If RNA helices assemble sequentially during transcription, an upstream insert would inhibit ribozyme assem- Eschenmoser, A. The search for the chemistry of life’s origin. Tetrahedron 63:12821, 2007. Koch, K., Schweizer, B., Eschenmoser, A. Reactions of the HCN-tetramer with aldehydes. Chem. Biodivers. 4:541, 2007. Intracellular RNA Assembly M.J. Fedor, J.W. Cottrell, L. Li, L. Liu, O. Tam, P. Watson, S. Zimmerman ur goal is to understand how RNAs fold into the correct 3-dimensional structures inside cells. The mRNAs have long been recognized as key intermediates in the transmission of information from the DNA sequence of genes to the amino acid sequence of proteins, and nonMartha J. Fedor, Ph.D. Associate Professor coding RNAs are now known Molecular Biology to perform several essential biological functions previously attributed to proteins. Although RNAs must adopt precise 3-dimensional struc- O F i g . 1 . Chimeric RNAs contain sequences with the potential to form self-cleaving ribozymes or nonfunctional structures, depending on the relative contributions of kinetics and thermodynamics to RNAfolding outcomes. The secondary structure of this self-cleaving ribozyme contains an essential base-paired helix, called H1, with 14 bp. A, A downstream insert (green) is complementary to the adjacent ribozyme sequence and has the potential to form an alternative helix with 12 bp, called 3′AltH1. During cotranscriptional RNA assembly in vitro and in vivo, the H1 structure that forms first resists competition from the downstream 3′AltH1 and allows assembly of a functional, self-cleaving ribozyme. B, In a second ribozyme variant, an upstream insert (red) is complementary to the ribozyme sequence and can form a helix with 12 bp, called 5′AltH1, which is incompatible with H1 assembly. Although the 5′AltH1 helix has lower thermodynamic stability than does H1, 5′AltH1 can form first during transcription to create a kinetic trap that blocks subsequent assembly of a functional ribozyme both in vitro and in vivo. 22 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 bly more than a downstream insert would. We also designed AltH1 and H1 with different numbers of stabilizing base pairs to learn how thermodynamic stability contributes to folding outcomes. In the first set of ribozyme variants we examined, upstream or downstream AltH1 sequences with 10 bp were able to compete with assembly of H1 sequences with 8 bp. This result suggested that thermodynamic stability, and not the sequential nature of RNA synthesis, was the major determinant of folding outcomes in vivo. In vitro, an H1 helix with 8 bp dissociates slowly, with a half-time on the order of days, so this evidence that a downstream AltH1 can block assembly of an upstream H1 implied that some feature of the intracellular environment accelerates exchange between upstream and downstream structures. In a second set of variants, H1 helices with 14 bp were combined with AltH1 helices with 12 bp. These changes slowed helix dissociation rates even further and reversed the relative thermodynamic stability of H1 and AltH1 so that H1 helices were now more stable. Strikingly, assembly of these variants did not always produce the most thermodynamically favored outcome. As expected, significant cleavage did occur in the presence of a downstream insert, evidence that stabilizing H1 by adding base pairs can prevent competition from a shorter downstream 3′AltH1 that has lower kinetic and thermodynamic stability. However, the upstream 5′AltH1 also inhibits self-cleavage, even though H1 is longer and more stable, in contrast to previous evidence that the most thermodynamically stable structure predominates in vivo. Thus, ribozymes with 8 bp in H1 and 10 bp in AltH1 seem to reach thermodynamic equilibrium during intracellular assembly, whereas ribozymes with 14 bp in H1 and 12 bp in 5′AltH1 seem to become trapped in an upstream 5′AltH1 with lower thermodynamic stability. Evidently, slow helix dissociation can trap RNAs in thermodynamically less favored structures in vivo, but kinetic trapping requires much longer helices with much slower dissociation kinetics than expected from the behavior of similar RNA structures in vitro. Current efforts focus on understanding the molecular basis of accelerated exchange between adjacent helices during RNA biogenesis in living cells. PUBLICATIONS Fedor, M.J. Comparative enzymology and structural biology of RNA self-cleavage. Annu. Rev. Biophys., in press. Click and Virus-Based Chemistry for Biological Discovery M.G. Finn, M. Baksh, D. Banerjee, J. Fiedler, V. Hong, A. Kislukhin, A. Udit irus capsids, the protein shells of virus particles that self-assemble in host cells, are a readily available biological material that we use for a variety of healthrelated applications. During the past several years, with M.G. Finn, Ph.D. crucial support from the Skaggs Professor Institute, we have developed Chemistry methods for the chemical derivatization of these structures, enabling us to bring the full power of both chemistry and molecular biology to bear in the creation of biologically active particles. The chemical methods are derived from click chemistry, the field of highly reliable synthetic methods developed and popularized by scientists in the laboratories of K.B. Sharpless, the Skaggs Institute, V.V. Fokin, Scripps Research, and in our laboratory. The following applications were pursued in the past year. V F U N D A M E N TA L C O N J U G AT I O N R E A C T I O N S O F PROTEINS AND VIRUS PAR TICLES We continue to develop new chemistry that allows the facile connections of biologically active molecules to capsid surfaces. Recently, we focused on connectors that become fluorescent when making bonds to the desired compounds and that can break those bonds at a predetermined rate (Fig. 1). This type of copper-free click chemistry is useful for the synthesis of protein conjugates in vivo and for the construction of delivery agents that release their cargoes at the desired site of action, such as a tumor. P O LY VA L E N T C A R B O H Y D R AT E C O N J U G AT E S We have extended our investigation of the immune response to carbohydrates displayed on the exterior surface of virus capsids. We found that the immunogenicity previously detected in chickens also occurs in mice and that both IgM and IgG antibodies are produced that are highly selective for the displayed glycans. This finding is important because many pathogens and disease states are marked by the display of unusual surface glycans. If immune responses can be raised against these glycans, the possibility exists to create novel vaccines. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 F i g . 1 . Top, Thiol-selective reagents that efficiently label proteins and peptides, providing a fluorescent signal upon bond formation. Bottom, Example of the generation of fluorescence upon the addition of a protein containing a free cysteine residue to a 100-µM solution of the thiol-selective reagent. C AT I O N I C PA R T I C L E S Polycations are of interest as cell-penetrating agents and as molecules that bind tightly to polyanions such as DNA and RNA. The polyanion heparin is used widely to inhibit blood clotting, but problems with heparin overdose are widespread. In the operating room, overdoses are corrected by administering cationic molecules that complex with heparin to reduce its effect, but the cationic molecules themselves are anticoagulants when used at too high a concentration. We created viruslike particles with enhanced levels of positive charge on their surfaces and found that these species efficiently inhibit the action of heparin (Fig. 2). Furthermore, the particles are not anticoagulants by themselves or in the presence of heparin and so appear to circumvent the major difficulty with current antiheparin agents in clinical use. Further testing of these particles in animal models is under way. PUBLICATIONS Bourne, C., Lee, S., Venkataiah, B., Lee, A., Korba, B., Finn, M.G., Zlotnick, A. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J. Virol. 82:10262, 2008. Hong, V., Udit, A.K., Evans, R.A., Finn, M.G. Electrochemically protected copper(I)-catalyzed azide-alkyne cycloaddition. ChemBioChem 9:1481, 2008. Kaltgrad, E., O’Reilly, M.K., Liao, L., Han, S., Paulson, J., Finn, M.G. On-virus construction of polyvalent glycan ligands for cell-surface receptor. J. Am. Chem. Soc. 130:4578, 2008. 23 F i g . 2 . Measurement of the time required for clotting of a standard sample of normal human plasma upon the administration of heparin and test antagonist. Control experiments indicated that clotting required approximately 55 seconds in the absence of heparin and 2 minutes in the presence of heparin. The active viruslike particles T18R and D14R and the wild-type particle to which 95 copies of cationic peptide 1 (WT-(1)95) were attached by click chemistry completely inhibited the anticoagulant activity of heparin without themselves inhibiting coagulation at higher concentrations. This finding contrasts sharply with the actions of peptide 1 and the clinical agent protamine. These molecules inhibit heparin at low concentrations but then give rise to strong anticoagulation when added at slightly higher concentrations, making them difficult to use in a clinical setting. The data points for protamine and peptide 1 at >400 seconds represent experiments in which clotting was not observed within that time. Miermont, A., Barnhill, H., Strable, E., Lu, X., Wall, K.A., Wang, Q., Finn, M.G., Huang, X. Cowpea mosaic virus capsid, a promising carrier for the development of carbohydrate based antitumor vaccines. Chem. Eur. J. 14:4939, 2008. Prasuhn, D.E., Jr., Singh, P., Strable, E., Brown, S., Manchester, M., Finn, M.G. Plasma clearance of bacteriophage Qβ particles as a function of surface charge. J. Am. Chem. Soc. 130:1328, 2008. Strable, E., Prasuhn, D.E., Jr., Udit, A.K., Brown, S., Link, A.J., Ngo, J.T., Lander, G., Quispe, J., Potter, C.S., Carragher, B., Tirrell, D.A., Finn, M.G. Unnatural amino acid incorporation into virus-like particles. Bioconjug. Chem. 19:866, 2008. Udit, A.K., Brown, S., Baksh, M.M., Finn, M.G. Immobilization of bacteriophage Qβ on metal-derivatized surfaces via polyvalent display of hexahistidine tags. J. Inorg. Biochem. 102:2142, 2008. Udit, A.K., Everett, C., Gale, A.J., Kyle, J.R., Ozkan, M., Finn, M.G. Heparin antagonism by polyvalent display of cationic motifs on virus-like particle. ChemBioChem, in press. Zhang, Q., Horst, R., Geralt, M., Ma, X., Hong, W.-X., Finn, M.G., Stevens, R.C., Wüthrich, K. Microscale NMR screening of new detergents for membrane protein structural biology. J. Am. Chem. Soc. 130:7357, 2008. 24 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Insights Into Protein Chemistry and Biology From Protein Structure E.D. Getzoff, A.S. Arvai, E.D. Garcin, C. Hitomi, K. Hitomi, M.D. Kroeger, M.E. Pique, D.S. Shin, J.L. Tubbs e investigate the chemistry and biology of proteins, starting from the determination and analysis of protein structures. In projects funded by the Skaggs Institute, we have focused on understanding catalysis and regulation of the redox-active superoxide dis- Elizabeth D. Getzoff, Ph.D. Professor mutase and nitric oxide synMolecular Biology thase (NOS) metalloenzymes that control reactive oxygen species and on proteincofactor interactions that regulate the response of photoactive proteins to light. We integrate high-resolution crystallographic results with those from spectroscopy, hydrogen-deuterium exchange mass spectrometry, and x-ray scattering to probe chemical, conformational, and dynamic changes in proteins and their cofactors. On the basis of our integrated results, we propose comprehensive mechanistic models that explain how proteins function as efficient catalysts and molecular machines. We test these hypotheses with biochemical and mutational analyses, to improve understanding of how proteins achieve and regulate their activities and to aid applications of this knowledge for the design of proteins and inhibitors. This year, we achieved major advances in isozymeselective inhibition of NOS, including the development of the anchored plasticity approach for the design of selective inhibitors. Our research on the light-activated DNA-repair of (6-4) photoproducts by the enzyme (6-4) photolyase also shed light on how human cryptochromes function in circadian clocks to control biological rhythms. W ISOZYME-SELECTIVE INHIBITION OF NITRIC OXIDE SYNTHASE The 3 human NOS isozymes offer key therapeutic targets for neurotransmission (neuronal NOS), regulation of blood pressure (endothelial NOS), and the immune response (inducible NOS). These highly similar, but differently regulated, isozymes all synthesize the diatomic molecule nitric oxide, which is both a molecular signal (at low concentrations) and a cytotoxin (at high concentrations). The aims of our ongoing cross-disciplinary mutational, biochemical, and structural investigations of NOS are to (1) determine the bases for functional domain interactions, cofactor recognition, and tuning for electron transfer and catalysis; (2) characterize the diverse regulatory mechanisms that differentially control the NOS isozymes; and (3) elucidate distinguishing features for isozyme-specific inhibitors. Isozyme-specific NOS inhibitors are sought for medicinal purposes and for advancing understanding of basic human physiology but present a huge challenge because of active-site conservation. Our comprehensive structural and mutagenesis analyses of NOS in complexes with isozyme-selective inhibitors revealed determinants for isozyme selectivity. In inducible but not endothelial NOS, bulky inhibitors promote a cascade of conformational changes up to 20 Å away from the substrate and inhibitor-binding site (Fig. 1). Correlated side-chain rotations accommo- F i g . 1 . Isozyme-selective inhibition of NOS. In both isozymes, the aminopyridine core of the inhibitor (orange) stacks above the heme and forms hydrogen bonds (black dots) mimicking those of the arginine substrate. In human endothelial NOS (left), the long tail of the inhibitor protrudes between the heme carboxylate groups (foreground). In inducible NOS (right), the inhibitor tail projects upward, inducing a cascade of side-chain conformational changes leading to the opening of a new selectivity pocket that enhances binding affinity. In endothelial NOS, conformational changes in the conserved glutamine (Q246) and arginine (R249) side chains are prevented by bulky isozymespecific amino acids distant from the substrate-binding pocket. date the rigid bulky tails of the selective inhibitors and expose a new specificity pocket for enhanced inhibitor binding in inducible NOS. Although first-shell (touching the inhibitor) and second-shell (touching the first shell) residues that begin this conformational cascade are invariant, in endothelial NOS their correlated rotations leading to the opening of the specificity pocket are precluded by bulky isozyme-specific residues at the far end. Thus, isozyme differences in the plasticity of THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 25 second- and third-shell residues modulate conformational changes of invariant first-shell residues to determine inhibitor selectivity. Our combined results allowed us to propose, and successfully test, the anchored plasticity approach for the design of selective inhibitors. With this approach, we exploit conserved binding sites coupled to distant isozyme-specific residues via cascades of conformational changes; the inhibitor core is designed to mimic the binding of a substrate or cofactor in a conserved binding site. This anchor is extended by rigid bulky substituents oriented along pathways leading to sequence or structural variations needed for selectivity. This anchored plasticity approach exemplifies general principles for the development of novel selective inhibitors that overcome active-site conservation. PROTEIN PAR TNERSHIP WITH FAD TO REPAIR DNA OR CONTROL BIOLOGICAL CLOCKS We are investigating how living things use cofactorprotein partnerships to transduce environmental changes into appropriate biological responses. Proteins of the cryptochrome/photolyase family share not only the same protein fold but also the redox-active FAD cofactor bound in an unusual U-shaped conformation beneath a positively charged groove designed for DNA binding. Through structural and functional studies of diverse members of the cryptochrome/photolyase families, we are deciphering how the similarities and differences in these molecules direct the same cofactor and protein fold to produce different biological responses to light: cryptochromes control biological rhythms, whereas photolyases repair DNA damage. To develop and test hypotheses for structure-function relationships in the cryptochrome/photolyase family, we determined the x-ray crystallographic structure of (6-4) photolyase, which confers UV protection to plants and has high sequence similarities (~50% identity) to human cryptochromes (Fig. 2). In humans and other vertebrates, cryptochromes are essential components of the circadian clock, which regulates sleep-wake cycles and other daily biological rhythms. The eukaryotic (6-4) photolyase structure revealed a substrate recognition site specific for the UV-induced DNA lesion, the (6-4) photoproduct, and cofactor binding sites different from those of the bacterial photolyase, consistent with distinct mechanisms for activities and regulation. The entrance to the activesite cavity above FAD is constricted by adjacent phosphate-binding and protrusion motifs that correlate with a phosphorylation site and nuclear localization sequence F i g . 2 . Structure of (6-4) photolyase. Ribbon diagram shows the N-terminal α/β domain (top), the helical domain with bound FAD cofactor (yellow), and the C-terminal extension (lower right) for a eukaryotic photolyase that also serves as an improved model for human cryptochromes. for cryptochrome. We coupled our structural studies with site-directed mutagenesis and functional assays. We tested (6-4) photolyase mutants for DNA-repair activity and tested mouse cryptochrome mutants for clock functions by transient transfection assays done in collaboration with L. DeHaro and S. Panda, the Salk Institute for Biological Studies. PUBLICATIONS Biskup, T., Schleicher, E., Okafuji, A., Link, G., Hitomi, K., Getzoff, E.D., Weber, S. Direct observation of a photoinduced radical pair in a cryptochrome blue-light photoreceptor. Angew. Chem. Int. Ed. 48:404, 2009. Garcin, E.D., Arvai, A.S., Rosenfeld, R.J., Kroeger, M.D., Crane, B.R., Andersson, G., Andrews, G., Hamley, P.J., Mallinder, P.R., Nicholls, D.J., St-Gallay, S.A., Tinker, A.C., Gensmantel, N.P., Mete, A., Cheshire, D.R., Connolly, S., Stuehr, D.J., Åberg, A., Wallace, A.V., Tainer, J.A., Getzoff, E.D. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol. 4:700, 2008. Shin, D.S., Didonato, M., Barondeau, D.P., Hura, G.L., Hitomi, C., Berglund, J.A., Getzoff, E.D., Cary, S.C., Tainer, J.A. Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. J. Mol. Biol., in press. 26 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Structure-Based Design of Bioactive Agents M.R. Ghadiri, J.M. Beierle, A. Chavochi, L. Leman, A. Montero, C.A. Olsen e are interested in advancing rational structure-based strategies for the design of bioactive agents. W G E N E R AT I O N O F T U R N MIMETICS Many protein-protein interactions are mediated through the recognition of β-turn sec- M. Reza Ghadiri, Ph.D. ondary structures. Consequently, Professor Chemistry small-molecule β-turn mimetics are valuable probes for assessing bioactive ligand conformations, establishing pharmacophoric requirements, and pursuing rational drug designs. Although effective drug scaffolds have been developed to precisely position up to 4 functional groups primarily in 2 dimensions, an analogous rigid scaffold capable of predictably juxtaposing 4 amino acid side chains in 3 dimensions has not been readily available. In order to meet this deficiency, diverse approaches have been taken to constrain peptides or peptidelike structures into turn conformations. One strategy for generating turn mimics is the use of cyclic tetrapeptides. Because of their appropriate size, shape, and useful synthetic modularity, cyclic tetrapeptides in principle offer an attractive platform to mimic β-turn regions. However, these tetrapeptides remain largely unexplored because of poor synthetic efficiency in constructing the strained 12-membered ring, an inability to control cis-trans backbone geometry, and the apparent requirement to sacrifice 1 of 4 amino acid residues to incorporate a proline or other turn-forming residue. To confront the limitations associated with cyclic tetrapeptides as β-turn mimics, we have designed and structurally analyzed 2 alternative classes of 13- or 14-membered ring pseudotetrapeptides containing either 1 or 2 triazole moieties, respectively (Fig. 1). Moreover, we have completed the design, syntheses, structural analyses, and determination of the somatostatin receptor binding activities of a library of all 16 possible strereoisomeric pseudotetramers incorporating F i g . 1 . Chemical and molecular structures of representative mem- bers of 2 classes of cyclic pseudotetrapeptide scaffolds. All 4 compounds had 1H NMR spectra consistent with a single conformational species in solution. Structures were determined by using multidimensional NMR (1–3) or x-ray crystallography (4). Dmb = dimethoxybenzyl. the somatostatin pharmacophore. In these studies, we exploited the 1,4-disubstituted 1,2,3-triazole as a trans peptide-bond surrogate. Structural analysis of the diastereomeric library with nuclear magnetic resonance (NMR) spectroscopy indicated that each peptide scaffold adopts a distinct, rigid, conformationally homogeneous turnlike structure in solution. The 3-dimensional pharmacophoric display of the pseudotetrapeptides is systematically altered by varying the stereochemistry around the otherwise constitutionally identical scaffolds, yielding both compounds with broad-spectrum activity against the 5 human somatostatin receptor subtypes and compounds with receptor selectivity. Our studies provide a basic set of scaffolds with subtle but predictable differences in the spatial display of amino acid side chains that are useful for rational, structure-based drug design. INHIBITORS OF HISTONE DEACETYLASES A fundamental strategy in rationally designing synthetic compounds to bind a protein of interest is to use a known ligand as a structural model to specify the precise conformational and pharmacophoric requirements for binding. Despite the remarkable success of this approach, a major difficulty is that compared to the receptor-bound structure, free ligands (in the absence of their cognate receptors) often adopt multiple conformations in solution or in the solid state. These occurrences can make design models based on the free ligand structure difficult to obtain or even misleading. Using the rigid scaffold strategy described earlier, we have gathered evidence that the more potent conformation of apicidin, an archetypal member of a fam- THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 ily of naturally occurring cyclic tetrapeptide inhibitors of histone deacetylases (HDACs), is not the previously believed all-trans (t-t-t-t) structure that predominates in solution, but rather a cis-trans-trans-trans (c-t-t-t) conformation (Fig. 2). Our approach relies on the design, synthesis, structural characterization, and functional analysis of a series of cyclic pseudotetrapeptides bearing 1,4- or 1,5-disubstituted 1,2,3-triazole amino acids that serve as trans- or cis-amide bond surro- 27 gates, respectively. We have shown that by replacing an amide bond with a triazole, we can fix the bond in question in either a trans- or a cis-like configuration, allowing us to individually probe the binding affinity of distinct peptide conformations. The heterocyclic compounds adopt conformations that overlay closely with the targeted conformations of apicidin and have potent HDAC inhibitory activities, in some instances equivalent to or better than those of the natural product. This study highlights the usefulness of triazole-modified cyclic peptides in constructing useful bioactive probe molecules, supports the c-t-t-t conformation as the bioactive conformation of the cyclic tetrapeptide HDAC inhibitors, and provides a useful 3-dimensional pharmacophoric model for use in advancing design principles for more selective HDAC inhibitors. Bacterial Quorum Sensing K.D. Janda, J. Ashley, K. Capková, S. De Lamo Marin, J. Denery, T.J. Dickerson, A. Di Mola, B. Ellis, L. Eubanks, K. Fukuchi, C. Hernandez, G. Kaufmann, C. Lowery, S. Mahajan, A. Mayorov, G. McElhaney, J. Mee, A. Moreno, Y. Nakai, A. Nguyen, A. Nunes, J. Park, A. Rohrbach, C. Saccavini, N. Salzameda, S. Steiniger, J.B. Treweek, A. Willis, Y. Xu, Y. Yoneda, B. Zhou, H. Zhou riginally described as a method of cell-to-cell signaling through which bacterial populations engaged in coordinated behavior, quorum sensing has since been shown to mediate several microbial processes, from formation Kim D. Janda, Ph.D. of biofilms and bioluminesProfessor cence to expression of viruChemistry lence factors, interspecies competition, and infectivity. Thus, the study of quorum sensing has both general relevance to the field of microbiology and medical implications in relation to combating bacterial infectivity. With respect to bacterial infectivity, interference of quorum sensing has been approached experimentally by designing antagonists against autoinducers, the signaling molecules secreted by bacteria, and by using immunopharmacotherapeutic agents to bind secreted quorum-sensing compounds, thereby preventing signal transmission between cells. O F i g . 2 . NMR structures for the triazole-modified apicidin analogs (side chains are omitted for clarity). A, Peptide 2 adopted a t-t-t-t conformation (top) that overlays well (bottom) on the lowest energy calculated conformation of apicidin (yellow; which reportedly closely matches the predominant conformation of apicidin in dimethylsulfoxide). B, Peptide 11 adopted a c-t-t-t conformation; 2 families of structures were observed that differ in the rotation of the Trp/Aoda amide relative to the backbone (top). The structures overlay well on the crystal structure of apicidin (bottom). C, Peptide 12 adopted a c-t-c-t conformation (top), which overlays well on the crystal structure of the natural product dihydrotentoxin (yellow; bottom). D, Overlay of Ca atoms for compounds 2 (magenta), 11 (2 structural families, green and cyan), and 12 (yellow). Because of the c-t-c-t conformation of 12, the Aoda, Trp, and Leu side chains of this peptide project in the same plane as the backbone ring rather than projecting upward and out of the plane as for the other peptides. E, Overlay of Ca and Cb atoms for compounds 2 (magenta) and 11 (2 structural families, green and cyan). The Ca atoms of the Ala and Ile/Leu residues are farther apart in 2, and the Ca-Cb vector of 2 directs the Ile side chain outward away from the ring rather than directly above the ring in 11. 28 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 M O D U L AT I O N O F Q U O R U M S E N S I N G T H R O U G H S Y N T H E T I C 4 , 5 - D I H Y D R O X Y - 2 , 3 - P E N TA N E D I O N E A ANALOGS The capacity for autoinducers to mediate a variety of interspecies and interkingdom interactions is achieved through the specificity of different bacterial species in secreting and recognizing quorum-sensing molecules within distinct structural classes. Traditionally, autoinducers have been classified within 2 major groups: acyl homoserine lactones (AHLs), used by gram-negative bacteria, and oligopeptides, used by gram-positive bacteria. It has since been realized that the signaling molecules used by bacteria span a much larger chemical space; more notable autoinducers include the Pseudomonas quinolone signal, bradyoxetin, and AI-2, a class of autoinducers derived from the precursor 4,5-dihydroxy2,3-pentanedione (DPD). The AI-2 quorum-sensing system is used by both gram-negative and gram-positive bacteria, and the DPD synthase is expressed by more than 70 bacterial species. The prevalence of AI-2 was suggestive of its role in interspecies signaling, and indeed the hypothesis that a common biosynthetic pathway exists among bacteria has been confirmed chemically. We chose Vibrio harveyi and Salmonella typhimurium as bacterial models for our investigations of AI-2–based quorum sensing because these are the only species in which the discrete structures of DPD-based autoinducers and their respective receptor proteins have been identified. We hypothesized that upon DPD secretion by one species, many DPD-based signaling molecules would be generated because of the reactivity and variable stereochemistry of the parent DPD compound. To probe the specificity of AI-2–based quorum sensing, we synthesized a panel of C1-substituted DPD analogs and evaluated them in 2 biological assays (Fig. 1). A goal of this experimental plan was to identify DPD-based agonists or antagonists that could be used in investigations of unknown AI-2 receptor proteins and the modulation of AI-2–based signaling. In the first assay, the effect of all DPD analogs on β-galactosidase production in S typhimurium was measured in the absence (agonist screen) and presence (antagonist screen) of DPD. Excitingly, all test compounds antagonized AI-2–based quorum sensing. In particular, the propyland butyl-substituted analogs caused potent inhibition of quorum sensing, with IC50 values 10-fold lower than the concentration of the natural DPD signal (Fig. 1B). B F i g . 1 . DPD analogs. A, Synthetic route for construction of DPD with alkyl groups introduced at the C-1 position. B, Effect of synthesized DPD-related compounds 5a–5g on quorum sensing as assessed in 2 biological assays: induction of β-galactosidase activity in S typhimurium and bioluminescence of V harveyi. Like S typhimurium, V harveyi is responsive to the AI-2 class of autoinducers. The second screen capitalized on the inability of V har veyi cells to luminesce through either the AHL pathway or the AI-2 pathway in the absence of exogenous DPD. Application of the DPD analogs to V harveyi cultures resulted in only mild agonist activity. However, addition of 1 µM DPD to these cultures had a dramatic synergistic effect: activation of bioluminescence with the DPD analogs was several-fold greater than that caused by 1 µM DPD alone (Fig. 1B). IMPEDING BACTERIAL INFECTION THROUGH ANTIBODIES AGAINST QUORUM-SENSING COMPOUNDS We have pioneered an antibody-based strategy to combat bacterial infectivity by disrupting transmission of quorum-sensing signals. Recently, we applied our antibody-based technology to disruption of the quorumsensing circuits of Pseudomonas aeruginosa. This gramnegative bacterium uses various quorum-sensing systems for more nefarious purposes with respect to interspecies communication. Namely, the 2-alkyl-4-quinolones and related AHLs (Fig. 2) secreted by P aeruginosa have antibacterial activity against gram-positive bacteria, allowing P aeruginosa to outcompete other bacterial species within a shared environment. The clinical relevance of quorum sensing by P aeruginosa includes the displacement of Staphylococcus aureus in the lungs of THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 29 Kaufmann, G.F., Park, J., Mee, J.M., Ulevitch, R.J., Janda, K.D. The quorum quenching antibody RS2-1G9 protects macrophages from the cytotoxic effects of the Pseudomonas aeruginosa quorum sensing signalling molecule N-3-oxo-dodecanoyl-homoserine lactone. Mol. Immunol. 45:2710, 2008. Kravchenko, V.V., Kaufmann, G.F., Mathison, J.C., Scott, D.A., Katz, A.Z., Grauer D.C., Lehmann, M., Meijler, M.M., Janda, K.D., Ulevitch, R.J. Modulation of gene expression via disruption of NF-κB signaling by a bacterial small molecule. Science 321:259, 2008. F i g . 2 . General chemical structure of ALHs (left), a major class of autoinducers, and a representative member of this class, 3-oxo-C12HSL (right). patients with cystic fibrosis and detrimental AHL-mediated immunomodulation in host cells, including an altered inflammatory response, a weakened host defense system, and induction of apoptosis. Therefore, removal of AHLs, which are thought to mediate the cytotoxicity in mammalian macrophages and neutrophils, may be advantageous to controlling this aspect of quorum sensing–related pathogenicity. To this end, we engineered monoclonal antibodies against AHL targets, and most notably, we showed that the monoclonal antibody RS2-1G9 had inhibitory activity in vitro against quorum sensing of P aeruginosa based on 3-oxo-C12-homoserine lactone (3-oxo-C12-HSL; Fig. 2). RS2-1G9 not only protected murine macrophages exposed to 3-oxo-C 12-HSL in a concentrationdependent manner but also prevented the downstream activation of cellular stress kinase pathways, indicating complete sequestration of 3-oxo-C 12-HSL. Thus, using this immunopharmacotherapy to quench expression of bacterial virulence factors and quorum sensing holds promise both in preventing infection and AHL-associated cytotoxicity and in developing therapies that will not promote the evolution of methicillin-resistant S aureus and future “superbugs.” PUBLICATIONS Brogan, A.P., Dickerson, T.J., Janda, K.D. Nornicotine-organocatalyzed aqueous reduction of α,β-unsaturated aldehydes. Chem. Commun. (Camb.) Issue 46:4952, 2007. Capková, K., Hixon, M.S., McAllister, L.A., Janda, K.D. Toward the discovery of potent inhibitors of botulinum neurotoxin A: development of a robust LC MS based assay operational from low to subnanomolar enzyme concentrations. Chem. Commun. (Camb.) Issue 30:3525, 2008. Capková, K., Yoneda, Y., Dickerson, T.J., Janda, K.D. Synthesis and structureactivity relationships of second-generation hydroxamate botulinum neurotoxin A protease inhibitors. Bioorg. Med. Chem. Lett. 17:6463, 2007. Debler, E.W., Kaufmann, G.F., Meijler, M.M., Heine, A., Mee, J.M., Pljevaljcic, G., Di Bilio, A.J., Schultz, P.G., Millar, D.P., Janda, K.D., Wilson, I.A., Gray, H.B., Lerner, R.A. Deeply inverted electron-hole recombination in a luminescent antibody-stilbene complex. Science 319:1232, 2008. Kaufmann, G.F., Park, J., Janda, K.D. Bacterial quorum sensing: a new target for anti-infective immunotherapy. Expert Opin. Biol. Ther. 8:719, 2008. Lowery, C.A., Dickerson, T.J., Janda, K.D. Interspecies and interkingdom communication mediated by bacterial quorum sensing. Chem. Soc. Rev. 37:1337, 2008. Lowery, C.A., Park, J., Kaufmann, G.F., Janda, K.D. An unexpected switch in the modulation of AI-2-based quorum sensing discovered through synthetic 4,5-dihydroxy-2,3-pentanedione analogues. J. Am. Chem. Soc. 130:9200, 2008. Park, J., Dickerson, T.J., Janda, K.D. Major sperm protein as a diagnostic antigen for onchocerciasis. Bioorg. Med. Chem. 16:7206, 2008. Park, J., Jagasia, R., Kaufmann, G.F., Mathison, J.C., Ruiz, D.I., Moss, J.A., Meijler, M.M., Ulevitch, R.J., Janda, K.D. Infection control by antibody disruption of bacterial quorum sensing signaling. Chem. Biol. 14:1119, 2007. Park, J., Kaufmann, G.F., Bowen, J.P., Arbiser, J.L., Janda K.D. Solenopsin A, a venom alkaloid from the fire ant Solenopsis invicta, inhibits quorum sensing signaling in Pseudomonas aeruginosa. J. Infect. Dis. 198:198, 2008. Richardson, H.N., Zhao, Y., Fekete, E.M., Funk, C.K., Wirsching, P., Janda, K.D., Zorrilla, E.P., Koob, G.F. MPZP: a novel small molecule corticotropin-releasing factor type 1 receptor (CRF1) antagonist. Pharmacol. Biochem. Behav. 88:497, 2008. Willis, B., Eubanks, L.M., Dickerson, T.J., Janda, K.D. The strange case of the botulinum neurotoxin: using chemistry and biology to modulate the most deadly poison. Angew. Chem. Int. Ed.47:8360, 2008. Willis, B., Eubanks, L.M., Wood, M.R., Janda, K.D., Dickerson, T.J., Lerner, R.A. Biologically templated organic polymers with nanoscale order. Proc. Natl. Acad. Sci. U. S. A. 105:1416, 2008. Xu, Y., Hixon, M.S., Dawson, P.E., Janda, K.D. Development of a FRET assay for monitoring of HIV gp41 core disruption. J. Org. Chem. 72:6700, 2007. Yoneda, Y., Steiniger, S.C., Capková, K., Mee, J.M., Liu, Y., Kaufmann, G.F., Janda, K.D. A cell-penetrating peptidic GRP78 ligand for tumor cell-specific prodrug therapy. Bioorg. Med. Chem. Lett. 18:1632, 2008. Zarebski, L.M., Vaughan, K., Sidney, J., Peters, B., Grey, H., Janda, K.D., Casadevall, A., Sette, A. Analysis of epitope information related to Bacillus anthracis and Clostridium botulinum. Expert Rev. Vaccines 7:55, 2008. Zhou, B., Carney, C., Janda, K.D. Selection and characterization of human antibodies neutralizing Bacillus anthracis toxin. Bioorg. Med. Chem. 16:1903, 2008. Zhou, B., Pellett, S., Tepp, W.H., Zhou, H., Johnson, E.A., Janda, K.D. Delineating the susceptibility of botulinum neurotoxins to denaturation through thermal effects. FEBS Lett. 582:1526, 2008. Zhou, H., Zhou, B., Ma, H., Carney, C., Janda, K.D. Selection and characterization of human monoclonal antibodies against Abrin by phage display. Bioorg. Med. Chem. Lett. 17:5690, 2007. 30 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Continuous In Vitro Evolution of RNA Enzymes G.F. Joyce, B.J. Lam, T.A. Lincoln, B.M. Paegel, K.L. Petrie e have devised methods for evolving nucleic acid enzymes in the test tube. These methods have enabled us to develop a variety of RNA and DNA enzymes, some of which have had applications in biomedicine. We continue both to Gerald F. Joyce, M.D., Ph.D. Professor advance the technology of Molecular Biology directed molecular evolution and to seek novel applications for our evolved enzymes. W A D VA N C E D D I R E C T E D E V O L U T I O N T E C H N O L O G Y Most in vitro evolution studies involve a powerful but laborious process in which a population of molecules is first challenged to perform a biochemical task, segregated on the basis of whether or not the molecules performed the task, and then amplified to produce “progeny” molecules that resemble but are not identical to their parents. This entire set of procedures is repeated until the population adapts to the task at hand, a process typically requiring weeks to months to complete. We have developed methods that allow us to carry out evolution in a continuous manner, within a single reaction mixture. The only required manipulation is to refresh periodically the supply of reagents, a task that is accomplished by either a serial transfer or serial dilution procedure. The continuous evolution method allows adaptation to occur within a period of only a few days. Recently, we developed a marked enhancement of continuous in vitro evolution in which the process is carried out within a computer-controlled microfluidic chip. The circuits on the chip contain a population of billions of RNA enzymes with RNA-joining activity, and these molecules can be challenged to adapt to various imposed selection constraints. The growth of the population is monitored continuously by using a laser confocal microscope. Whenever the population size reaches a predetermined threshold, chip-based operations are executed to isolate a fraction of the population and mix it with fresh reagents. In a recently published article, we described the first example of “evolution on a chip,” in which a population of RNA enzymes underwent 500 iterations of 10-fold exponential growth followed by 10-fold dilution, carried out during a period of 70 hours. During that time, the molecules evolved to use progressively lower concentrations of a required substrate; each step of that adaptation was observed in real time. We recently devised 2 further enhancements of the continuous evolution method. The first involves a controlled mutagenesis technique that can be applied throughout selective amplification. This technique allows us to maintain a diverse population of individuals, even in the face of stringent selection pressure, thus enabling a more comprehensive exploration of potentially advantageous variants. The second enhancement involves a method for isolating and then propagating individual RNA enzymes within water-in-oil compartments within a microfluidic chip. A novel multiport injector design allows us to produce millions of individual fluidic compartments of precisely controlled size (Fig. 1), ranging F i g . 1 . High-throughput production of water droplets in oil, carried out within a microfluidic chip. The liquid (containing a blue dye) enters the chip and fans out radially to meet the oil, where it forms microdroplets (here 100 µm in diameter), which are then collected. Each droplet contains, on average, 1 starting RNA enzyme and the materials necessary for that enzyme to undergo continuous evolution. from 20 to 100 µm in diameter (containing 4–500 pL). These microcompartments allow each enzyme to express a “cellular ” phenotype based on the enzyme’s catalytic function. L I G A N D - D E P E N D E N T E X P O N E N T I A L A M P L I F I C AT I O N OF RNA We previously developed an RNA enzyme that catalyzes its own replication by joining 2 RNA substrates THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 to form additional copies of itself. This enzyme was converted to a cross-catalytic format whereby 2 RNA enzymes catalyze each other’s synthesis from a total of 4 RNA substrates. We then used in vitro evolution to improve substantially the activity of the cross-replicating RNA enzymes. The enzymes now can undergo efficient exponential amplification, generating about a billion copies in 30 hours at a constant temperature of 42°C. Recently, we inserted a ligand-binding domain adjacent to the catalytic domain of the cross-replicating enzymes so that the enzymes undergo exponential amplification in the presence, but not the absence, of the corresponding ligand. The catalytic domain assumes its active conformation only when the ligand is present, resulting in a large signal that can be used to detect and quantify compounds of biomedical interest, such as proteins, drugs, and metabolites. For example, the cross-replicating enzymes were made dependent on theophylline, a drug commonly used to treat respiratory diseases, for which the dose must be carefully adjusted on the basis of its level in the serum. Strong exponential amplification occurred in the presence of theophylline, but amplification in the presence of caffeine was undetectable (Fig. 2), even though the 2 F i g . 2 . Exponential amplification of cross-replicating RNA enzymes dependent on the compound theophylline. Amplification occurs in the presence of 1 mM theophylline (filled circles) but not in the presence of 1 mM caffeine (open circles). compounds differ by only a single methyl group. Furthermore, the exponential growth rate of the enzymes depended on the concentration of theophylline, a characteristic that allowed us to construct standardized curves that could be used to determine the concentration of theophylline in an unknown sample. The method 31 is analogous to quantitative polymerase chain reaction for the detection of nucleic acids but can be generalized to a wide variety of targets relevant to medical diagnostics and environmental monitoring. PUBLICATIONS Paegel, B.M., Joyce, G.F. Darwinian evolution on a chip. PLoS Biol. 6:e85, 2008. Synthetic Enzymes, Catalytic Antibodies, Biomolecular Computing, and Synthetic Capsids E. Keinan, O. Reany, N. Metanis, E. Kossoy, M. Soreni, R. Piran, M. Sinha, I. Ben-Shir, T. Shekhter, T. Ratner, T. Mejuch, E. Solel, S. Shoshani, R. Gershoni, A. Karmakar, D. Pappo, G. Parvari SYNTHETIC ENZYMES fforts to generate new enzymatic activities from existing protein scaffolds may not only provide biotechnologically useful catalysts but also lead to a better understanding of the natural process of evolution. Enzymes are Ehud Keinan, Ph.D. usually characterized as catAdjunct Professor alyzing a specific reaction by a Molecular Biology unique chemical mechanism. However, small changes in the amino acid sequence of some enzymes can markedly alter the catalytic properties of the enzymes, affecting the substrate selectivity and subtle aspects of the catalytic mechanism. The catalytic promiscuity displayed in these enzymes may be an important factor in the natural evolution of new catalytic activities and in the development of new catalysts through protein engineering. We are particularly interested in selenoenzymes, which have a central role in maintaining cellular redox potential. These enzymes have selenylsulfide bonds in their active sites that catalyze the reduction of peroxides, sulfoxides, and disulfides. The selenol-disulfide exchange reaction is common to all of the enzymes, and the activesite redox potential reflects the ratio between the forward and reverse rates of this reaction. The preparation of enzymes containing selenocysteine is experimentally E 32 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 challenging. As a result, little is known about the kinetic role of selenols in enzyme active sites, and the redox potential of a selenylsulfide or diselenide bond in a protein has not been experimentally determined. To fully evaluate the effects of selenocysteine on oxidoreductase redox potential and kinetics, we chemically synthesized glutaredoxin 3 (Grx3) and all 3 selenocysteine variants of its conserved 11CXX14C active site and determined their redox potentials. In particular, the position of redox equilibrium between Grx3(C11UC14U) (–308 mV) and thioredoxin (–270 mV) suggests a possible role for diselenide bonds in biological systems. Kinetic analysis showed that the lower redox potentials of the selenocysteine variants are due primarily to the greater nucleophilicity of the active-site selenium. The 10 2 - to 10 4 -fold increase in the rate of thioredoxin reduction by the seleno-Grx3 analogs indicates that oxidoreductases containing either selenylsulfide or diselenide bonds can have physiologically compatible redox potentials and enhanced reduction kinetics in comparison with their sulfide counterparts. This research on synthetic enzymes is a collaboration with P.E. Dawson, Scripps Research. C ATA LY T I C A N T I B O D I E S A relatively unexplored opportunity in the science of catalytic antibodies is modifying the phenotype of an organism in vivo by incorporating the gene for a catalytic antibody into the genome of that organism. An attractive application of this concept would be the expression of such a catalyst in transgenic plants to provide a beneficial trait. For example, introduction of a herbicide-resistance trait in commercial plants is highly desirable because plants with the trait could be grown in the presence of a nonselective herbicide that affects only weeds and other undesired plant species. We have shown that herbicide-resistant plants can be engineered by designing both a herbicide and a catalytic antibody that destroys the herbicide within the plants. Such a transgenic plant was achieved via a 3-step maneuver: (1) development of a new carbamate herbicide, one that can be catalytically destroyed by the aldolase antibody 38C2; (2) separate expression of the light chain and half of the heavy chain (Fab) of the catalytic antibody in the endoplasmic reticulum of 2 plant lines of Arabidopsis thaliana; and (3) cross-pollination of these 2 transgenic plants to produce a herbicide-resistant F1 hybrid (Fig. 1). In vivo expression of catalytic antibodies could become a useful, general strategy to achieve desired phenotype modifications not only in plants but also in other organisms. F i g . 1 . Influence of a new herbicide (1) on the rooting and development of A thaliana plant lines. The control plants are shown in A, C, and E; the hybrid plants (F1) expressing both light and heavy chains, in B, D, and F. Plantlets grown on medium without the herbicide are shown in A and B; those grown with the herbicide, in C and F. BIOMOLECULAR COMPUTING DEVICES In fully autonomous molecular computing devices, all components, including input, output, software, and hardware, are specific molecules that interact with each other through a cascade of programmable chemical events, progressing from the input molecule to the molecular output signal. DNA molecules and DNA enzymes have been used as convenient, readily available components of such computing devices because the DNA materials have highly predictable recognition patterns, reactivity, and information-encoding features. Furthermore, DNA-based computers can become part of a biological system, generating outputs in the form of biomolecular structures and functions. Our previously reported 2-symbol–2-state finite automata computed autonomously, and all of their components were soluble biomolecules mixed in solution. The hardware consisted of 2 enzymes, an endonuclease and a ligase, and the software and the input were double-stranded DNA oligomers. More recently, we designed and created 3-symbol–3-state automata that can carry out more complex computations. In addition, we found that immobilization of the input molecules on chips allowed parallel computation, a system that can be to encrypt information. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 The main advantage of autonomous biomolecular computing devices compared with electronic computers is the ability of the devices to interact directly with biological systems. No interface is required because all components of molecular computers, including hardware, software, input, and output, are molecules that interact in solution along a cascade of programmable chemical events. We showed for the first time that the output of a molecular finite automaton can be a visible bacterial phenotype. Our 2-symbol–2-state finite automaton uses linear double-stranded DNA inputs prepared by inserting a string of 6-bp symbols into the lacZ gene on plasmid pUC18. The computation resulted in a circular plasmid that differed from the original pUC18 by either a 9-bp (accepting state) or an 11-bp (unaccepting state) insert within the lacZ gene. Upon transformation and expression of the resultant plasmids in Escherichia coli, either blue colonies or white colonies, respectively, were formed (Fig. 2). 33 constructs and molecular computing. By examining physical models of spherical virus assembly, we developed a general synthetic strategy for producing chemical capsids at sizes between fullerenes and spherical viruses. Such capsids can be formed by self-assembly from a class of novel symmetric molecules developed from a pentagonal core. By designing chemical complementarity into the 5 interface edges of the molecule, we can produce self-assembling stable structures of icosahedral symmetry. We considered 3 different binding mechanisms: hydrogen bonding, metal binding, and formation of disulfide bonds. These structures can be designed to assemble and disassemble under controlled environmental conditions. We have conducted molecular dynamics simulation on a class of corannulene-based molecules to demonstrate the characteristics of self-assembly and to aid in the design of the molecular subunits. This research was done in collaboration with A.J. Olson, Scripps Research. PUBLICATIONS Ben Shir, I., Sasmal, S., Mejuch, T., Sinha, M.K., Kapon, M., Keinan, E. Repulsive interaction can be a key design element of molecular rotary motors. J. Org. Chem. 73:8772, 2008. Pappo, D., Mejuch, T., Reany, O., Solel, E., Gurram, M., Keinan, E. Diverse functionalization of corannulene: easy access to pentagonal superstructures. Org. Lett., in press. Understanding and Ameliorating Protein-Misfolding Diseases J.W. Kelly, S. Choi, E. Culyba, M.T.A. Dendle, D. Du, C. Fearns, A.A. Fuller, T.-W. Mu, A. Murray, D. Ong, J. Paulsson, E.T. Powers, P. Rao, M. Saure, R. Simkovsky, S. Siegel, J. Solomon, K. Usui, Y. Wang, I. Yonemoto, Z. Yu F i g . 2 . Computation with aaba input in the presence (A) and absence (B) of transition molecules results in white bacteria when the transition molecules are present. Computation with abba input in the presence (C) and absence (D) of transition molecules results in blue bacteria when the transition molecules are present. SYNTHETIC CAPSIDS Stable structures of icosahedral symmetry can have numerous functional roles, including chemical microencapsulation and delivery of drugs and biomolecules, a way to observe encapsulated reactive intermediates, presentation of epitopes for efficient immunization, synthesis of nanoparticles of uniform size, and formation of structural elements for molecular supramolecular ur main goal is to gain insight into the mechanisms of proteome maintenance that can be used to develop new therapeutic strategies to ameliorate protein-misfolding diseases when deficiencies in protein maintenance occur. Maintenance of the proteome Jeffery W. Kelly, Ph.D. Professor, Chemistry (proteostasis) both inside and Chairman, Molecular and outside human cells is essential Experimental Medicine for development, reproduction, and successful aging. Deficiencies in proteostasis lead O 34 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 to many metabolic, oncologic, neurodegenerative, and cardiovascular disorders. We focus mostly on neurodegenerative diseases, and we benefit greatly from collaborations with J. Buxbaum, J.R. Yates, and W.E. Balch at Scripps Research and with A. Dillin at the Salk Institute for Biological Studies, La Jolla, California. A MODEL FOR PROTEIN EXPORT FROM THE ENDOPLASMIC RETICULUM About one-third of all eukaryotic proteins, including all membrane proteins and secreted proteins, are folded in and then exported from the endoplasmic reticulum. Proteins are initially unstructured and must fold into welldefined structures to become functional. Unfortunately, proteins can also misfold, leaving them trapped in nonfunctional, sometimes aggregated, structures. Because of this inherent inefficiency in protein folding, the endoplasmic reticulum has a set of pathways that regulate protein folding and export. These pathways include an export pathway, which recognizes and exports properly folded proteins; an endoplasmic reticulum–associated degradation pathway, which recognizes and degrades unfolded or misfolded proteins; and a chaperoning pathway, which recognizes and recovers misfolded proteins. The efficiency with which a given protein is exported, defined as the rate of synthesis divided by the rate of export, depends on the interplay between the activities of these 3 pathways and the thermodynamics and kinetics of the folding and misfolding processes of the protein. We recently described the FoldEx model of folding for export from the endoplasmic reticulum; this model was designed to semiquantitatively capture this interplay. In FoldEx, the activities of the export, degradation, and chaperone-mediated folding pathways are (most easily) controlled through the concentrations of the machineries that make up the pathways. The thermodynamics of folding are quantified by the equilibrium constant for folding, and the kinetics of folding or misfolding are quantified by the time required for folding or misfolding to reach its half-way point. The FoldEx model establishes that no single feature of protein folding energetics or endoplasmic reticulum biology dictates folding and transport efficiency. Instead, a network view provides insight into the basis for cellular diversity, disease origins, and protein homeostasis and predicts strategies for restoring protein homeostasis in protein-misfolding diseases. P R O T E O S TA S I S R E G U L AT O R S A N D P H A R M A C O L O G I C C H A P E R O N E S I N LY S O S O M A L S T O R A G E D I S E A S E S Lysosomal storage diseases are loss-of-function diseases often caused by a mutation in one of the lysosomal enzymes. The mutation results in excessive misfolding and degradation of the enzyme within the endoplasmic reticulum instead of proper folding and trafficking of the enzyme to the lysosome. The resulting deficiency in lysosomal enzyme activity leads to accumulation of the substrate of the mutant lysosomal enzyme. At least 40 distinct lysosomal storage diseases have been identified; the most prevalent is Gaucher disease, which is caused by a deficiency in the activity of lysosomal glucocerebrosidase. Previously, we showed that in fibroblasts derived from the tissue of a patient with Gaucher disease, novel pharmacologic chaperones enhanced glucocerebrosidase activity up to 7.2-fold by binding directly to the enzyme and thereby stabilizing it. More recently, we found that the innate proteostasis capacity of a cell can be enhanced with small molecules we call proteostasis regulators to fold mutated enzymes that would otherwise misfold and be degraded, resulting in increased trafficking of the mutated enzyme to the lysosome and increased function. We discovered that inhibiting L-type calcium channels with either diltiazem or verapamil partially restored enzyme homeostasis in 3 distinct lysosomal storage diseases: Gaucher disease, α-mannosidosis, and type IIIA mucopolysaccharidosis. The increased capacity of the endoplasmic reticulum to fold misfolding-prone proteins probably is due to a modest, calcium ion–mediated upregulation of a subset of molecular chaperones, including calnexin and calreticulin. C O R R E L AT I N G T H E F O L D I N G A N D A S S E M B LY ENERGETICS OF TRANSTHYRETIN WITH DISEASE PHENOTYPES Transthyretin, a tetrameric protein, is the primary transporter of retinol-binding protein and the secondary transporter of the thyroid hormone thyroxine. Transthyretin can dissociate, misfold, and aggregate, forming deposits that interfere with the normal functioning of several tissues or organs. Destabilized mutants of transthyretin are particularly prone to aggregation, but the precise energetic effects of the mutations are obscured by the linked folding and assembly equilibria of transthyretin. We used urea denaturation studies of transthyretin and several of its mutants to quantify the thermodynamically linked quaternary and tertiary structural stability to better understand the relationship between mutant folding energetics and amyloid disease phenotype. Using a method of analysis that simultaneously accounts for the 2-step denaturation (tetramer disso- THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 ciation followed by unfolding), we analyzed the stability of quaternary and tertiary structures of wild-type transthyretin and the V122I variant, which is linked to late-onset familial amyloid cardiomyopathy, the most common familial transthyretin amyloid disease. The results indicated that V122I transthyretin has a destabilized quaternary structure and a stable tertiary structure relative to wild-type transthyretin. We also examined 3 other variants of transthyretin: L55P, V30M, and A25T. We found that both the L55P mutant, associated with the most aggressive familial transthyretin amyloid disease, and the V30M mutant, the most common mutation associated with neuropathic forms of transthyretin amyloidosis, have complex denaturation pathways that cannot be fit to the 2-step denaturation model. Nevertheless, L55P transthyretin is clearly less stable than is wild-type transthyretin, primarily because the tertiary structure of L55P is unstable, although its quaternary structure is destabilized as well. Published data suggest that V30M transthyretin has stable quaternary structure but unstable tertiary structure. The A25T mutant, associated with CNS amyloidosis, is highly prone to aggregation and has drastically reduced quaternary and tertiary structural stability. The observed differences in stability among the disease-associated transthyretin variants highlight the complexity and the heterogeneity of transthyretin amyloid disease, an observation with important implications for the treatment of these diseases. A G G R E G AT I O N O F A M Y L I N A N D I T S P R O C E S S I N G 35 the amyloidogenicity of amylin and its processing intermediates is negatively correlated with net charge and charge at the C terminus. Although our conditions may not precisely reflect those of amyloidogenesis in vivo, the lower amyloidogenicity of the processing intermediates relative to amylin suggests that the presence of the intermediates in intracellular amyloid deposits in the increasingly stressed beta cells of patients with diabetes may be a consequence of general defects in protein homeostasis known to occur in diabetes rather than the result of the amylin processing intermediates acting as initiators of amyloid. PUBLICATIONS Balch, W.E., Morimoto, R.I., Dillin, A., Kelly, J.W. Adapting proteostasis for disease intervention. Science 319:916, 2008. Bieschke, J., Siegel, S.J., Fu, J., Kelly, J.W. Alzheimer’s Aβ peptides containing an isostructural backbone mutation afford distinct aggregate morphologies but analogous cytotoxicity: evidence for a common low-abundance toxic structure(s)? Biochemistry 47:50, 2008. Gao, J., Kelly, J.W. Toward quantification of protein backbone-backbone hydrogen bonding energies: an energetic analysis of an amide-to-ester mutation in an α-helix within a protein. Protein Sci. 17:1096, 2008. Hurshman Babbes, A.R., Powers, E.T., Kelly, J.W. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry 47:6969, 2008. Johnson, S.M., Connelly, S., Wilson, I.A., Kelly, J.W. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J. Med. Chem. 51:260, 2008. Liu, F., Du, D., Fuller, A.A., Davoren, J.E., Wipf, P., Kelly, J.W., Gruebele, M. An experimental survey of the transition between two-state and downhill protein folding scenarios. Proc. Natl. Acad. Sci. U. S. A. 105:2369, 2008. I N T E R M E D I AT E S Human amylin, or islet amyloid polypeptide, is a peptide cosecreted with insulin by the beta cells of the pancreatic islets of Langerhans. The 37-residue, C-terminally amidated human amylin peptide is derived from a proprotein that undergoes formation of disulfide bonds in the endoplasmic reticulum and then 4 enzymatic processing events in the immature secretory granule. Human amylin forms both intracellular and extracellular amyloid deposits in the pancreas of most patients with type 2 diabetes, likely reflecting compromised function of secretory cells. In addition, amylin-processing intermediates have been reported as components of intracellular amyloid in beta cells. We investigated the amyloidogenicity of amylin and its processing intermediates in vitro. Under conditions mimicking those in immature secretory granules (37°C, pH 6), amylin forms amyloid aggregates more rapidly than its processing intermediates and its reduced counterparts form aggregates. Our results indicate that Mu, T.-W., Fowler, D.M., Kelly, J.W. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PloS Biol. 6:e26, 2008. Mu, T.-W., Ong, D.S.T., Wang, Y.-J., Balch, W.E., Yates, J.R. III, Segatori, L., Kelly, J.W. Chemical and biological approaches synergize to ameliorate proteinfolding diseases. Cell 134:769, 2008. Reixach, N., Foss, T.R., Santelli, E., Pascual, J., Kelly, J.W. Human-murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic: a lesson in the generation of murine models of diseases involving oligomeric proteins. J. Biol. Chem. 283:2098, 2008. Wiseman, R.L., Powers, E.T., Buxbaum, J.N., Kelly, J.W., Balch, W.E. An adaptable standard for protein export from the endoplasmic reticulum. Cell 131:809, 2007. Yonemoto, I.T., Kroon, G.J.A., Dyson, H.J., Balch, W.E., Kelly, J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 47:9900, 2008. 36 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Engineering Eukaryotic Algal Chloroplasts for Production of Human Therapeutic Proteins and of Biofuels S.P. Mayfield, M. Tran, A. Manuell, J. Marin-Navarro, M. Muto, P. Pettersson, P. Lee, B. Rasala, M. Jager lgae offer a tremendous opportunity as a biotechnology platform both for the production of protein therapeutics and as a source of renewable energy. Because production of therapeutic proteins in algae can be achieved at a fraction of the cost of traStephen P. Mayfield, Ph.D. ditional mammalian cell culProfessor ture, algal production has the Cell Biology potential to dramatically reduce the cost of protein-based drugs. Algae can also produce biomass at 10 times the rate of terrestrial plants and can be grown in minimal media on land not suitable for food crop production, making algae a potential source of renewable biofuels. To realize the potential of algae for production of biofuels and therapeutic proteins, we must understand and control algal gene expression. In algae, both proteins and biofuel molecules are produced in chloroplasts, and understanding chloroplast gene expression will be essential to developing algae as a biotechnology platform. Using molecular, biochemical, genetic, and structural biology, we have identified key factors that control gene expression within the chloroplast and have used this knowledge to produce algae that have strong, regulated protein expression. With this basic understanding of the genetics and biology of algae, we can develop eukaryotic algae as a cost-effective system for biotechnology applications, including the production of human therapeutic proteins, industrial enzymes, and biofuels. A HUMAN THERAPEUTIC PROTEINS During the past several years, we have developed a system for the expression of recombinant proteins in the green alga Chlamydomonas reinhardtii. We now routinely obtain strong expression of complex mammalian proteins that are suitable as human therapeutic agents. We have expressed a number of proteins in algae, including monoclonal antibodies, growth factors, and a variety of other potential therapeutics. In addition to therapeutic proteins, we have also expressed eukaryotic protein toxins, an achievement that is possible because chloroplasts are naturally resistant to such toxins. We have now developed antibody-toxin fusion proteins, a class of recombinant protein molecules that can target and kill eukaryotic cells, including human cancer cells. The production of antibody-toxin fusion proteins is unique to our expression system; bacterial expression systems cannot efficiently produce these complex molecules, and mammalian cell cultures would be killed by the toxin during production. Thus, algal chloroplasts are the best system for the production of this type of superior cancer therapeutic agent. To examine the potential of algae to produce antibody-toxin fusion proteins as cancer therapeutics, we engineered an antibody to CD19 to fuse with an exotoxin-A protein domain to produce the antibody-toxin fusion protein anti-CD19–ETA, which targets human B-cell lymphomas. This recombinant fusion protein was expressed in algae, where it accumulated as a soluble protein. Using cell-based assays, we showed that isolated anti-CD19–ETA efficiently binds to CD19 + human B-cell lymphomas but does not bind to CD19– normal human cells. Once bound to the tumor cells, anti-CD19–ETA efficiently kills the cells. These cellbased assays are the first step in demonstrating the potential of these fusion proteins as human anticancer therapeutic agents. BIOFUELS With fossil fuel reserves dwindling, mandates requiring the reduction of carbon dioxide emissions, and a need for national energy independence, we face the formidable challenge of developing sustainable forms of carbon-neutral energy in an economically practical manner. Algae offer the potential to produce carbon-neutral liquid biofuels at a scale and cost that can be competitive with existing fossil fuel production. In addition, production of biofuels from algae will not compete with production of food crops for use of arable land. Economic production of biofuels from algae will require production of other molecules in addition to the biofuel molecules, because all of the biomass produced from algae will need to have a commercial value. The technology we developed for the production of therapeutic proteins in algae can be THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 used to produce algae with valuable protein coproducts as well as improved biofuel characteristics, and we are now developing algae as a renewable energy source. PUBLICATIONS Beligni, M.V., Mayfield, S.P. Arabidopsis thaliana mutants reveal a role for CSP41a and CSP41b, two ribosome-associated endonucleases, in chloroplast ribosomal RNA metabolism. Plant Mol. Biol. 67:389, 2008. Manuell, A.L., Beligni, M.V., Elder, J.H., Siefker, D.T., Tran, M., Weber, A., McDonald, T.L., Mayfield, S.P. Robust expression of a bioactive mammalian protein in Chlamydomonas chloroplast. Plant Biotechnol. J. 5:402, 2007. Auditory Perception and Hearing Impairment: From Mouse Models to Human Genetic Disease U. Müller, C. Barros, F. Conti, H. Elledge, S. Franco, N. Grillet, S. Harkins-Perry, P. Kazmierczak, I. Martinez-Garay, R. Radakovits, C. Ramos, A. Reynolds, M. Schwander, S. Webb, W. Xiong echanosensation, the transduction of mechanical force into an electrochemical signal, allows living organisms to detect touch, hear, register movement and gravity, and sense changes in cell volume and shape. The hair cells of the mammalian inner ear are the principal mechUllrich Müller, Ph.D. Professor anosensors for the detection Cell Biology of sound and head movement. We identify and study genes that control the development and function of hair cells. We are particularly interested in the molecules that form the mechanotransduction machinery of these cells. We also analyze the mechanisms that establish neuronal connections between the auditory sense organs and the cerebral cortex and the formation of cell layers and neuronal circuits within the cerebral cortex. M MOLECULAR COMPOSITION OF THE MECHANOTRANSDUCTION MACHINERY IN HAIR CELLS The mechanically sensitive organelle of a hair cell is the hair bundle, which consists of dozens of stereocilia that project from the apical cell surface. Mechanotransduction channels are localized close to the tips of 37 stereocilia. Tip-links, extracellular filaments that connect the tips of neighboring stereocilia and are visible by electron microscopy, are thought to transmit sound-induced tension force onto the transduction channels. The molecular identity of most components of the mechanotransduction complex is still largely unknown. To identify genes that control hair cell function, such as the transduction channel and the tip-links, we use genetic approaches. Approximately 1 child in 1000 children is born deaf, and a large part of the human population experiences age-related hearing loss. Many forms of hearing loss are of genetic origin, and mutations in more than 400 genes cause deafness. Recently, we discovered that some of the genes linked to Usher syndrome, the leading cause of deaf-blindness in humans, may encode components of the mechanotransduction machinery in hair cells. Using mouse model systems, we have shown that the Usher syndrome proteins cadherin 23 and protocadherin 15 interact to form tiplinks in hair cells. Intriguingly, some of the mutations in the genes for cadherin 23 and protocadherin 15 that cause deafness in humans disrupt interactions between the 2 proteins. Others leave interactions intact and probably change the mechanical properties of tip-links and affect the mechanotransduction process. Currently, we are defining the biophysical properties of cadherin 23 and protocadherin 15, investigating whether other genes linked to deafness encode components of the mechanotransduction machinery, and generating mouse models for diseases that affect hair cell function in mechanotransduction. MOUSE MODELS OF DEAFNESS Deafness has a profound effect on the quality of life of the affected individuals, yet few promising therapeutic approaches exist to help these individuals. To identify genes that control auditory perception and to provide animal models for the human disease, we carried out a genetic screen in mice. Using N-ethyl-N-nitrosourea, we introduced point mutations in the germ line of mice. Using phenotypic screens, we identified more than 20 mouse lines in which the mice inherit hearing defects as recessive traits. We have mapped many of the mutations to chromosomal intervals and have used DNA sequencing to identify mutations in single genes that cause some of the hearing defects. All of the genes that we have identified so far are expressed in hair cells. One of the genes encodes the unconventional motor protein myosin VIIa. Interestingly, mutations in myosin VIIa have been linked to 38 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Usher syndrome in humans. Our phenotypic analysis of the mice with the myosin VIIa mutation indicated that the mutation affects expression of the protein differentially in the ear and retina and provides a molecular explanation of why some mutations in the human gene for myosin VIIa affect the function of the protein in the ear and retina, whereas other mutations only affect the function of myosin VIIa in the ear. A mutation in a second mouse line maps to a gene that encodes an enzyme important for the control of neurotransmitter levels. We have identified a similar mutation in human families with deafness. Our studies show that genetic screens in mice are powerful tools for identifying mutations that control the function of hair cells and that mutations in mice can serve as models for deafness in humans. In future studies, we will define the structure and function of the normal and pathogenic variants of the involved proteins to gain insights into disease mechanisms at the cellular, molecular, and anatomic level. PUBLICATIONS Du, X., Schwander, M., Moresco, E.M., Viviani, P., Haller, C., Hildebrand, M.S., Pak, K., Tarantino, L., Roberts, A. Richardson, H., Koob, G., Najmabadi, H., Ryan, A.F., Smith, R.J., Müller, U., Beutler, B. A catechol-O-methyltransferase that is essential for auditory function in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 105:14609, 2008. Müller, U. Cadherins and mechanotransduction by hair cells. Curr. Opin. Cell Biol. 20:557, 2008. Müller, U., Gillespie, P. Silencing the cochlear amplifier by immobilizing prestin. Neuron 58:299, 2008. Chemical Synthesis and Chemical Biology K.C. Nicolaou, A. Agua, R. Aversa, W. Brenzovich, J. Chen, S. Dalby, D. Edmonds, S. Ellery, A. Estrada, B. Fraga, M. Frederick, M. Freestone, C. Gelin, J. Goodwin-Tindall, V. Gondi, M. Hesse, P. Huang, Z. Huang, V. Jeso, J. Jin, M. Kar, A. Krasovskiy, A. Lemire, R. Levin, A. Li, H. Li, Y. Lim, T. Lister, U. Majumder, C. Mathison, A. Morgan, A. Nold, A. Ortiz, N. Patil, B. Pratt, R. Reingruber, F. Rivas, A. Sanchez Ruiz, D. Sarlah, D. Shaw, A. Stepan, A. Talbot, Y. Tang, V. Trépanier, G. Tria, T. Umezawa, J. Wang, T. Wu, W. Zhan, H. Zhang uring the past year, we made considerable progress in a number of areas, including the total synthesis and biological investigation of neurotoxins, antitumor agents, and antibiotics (Fig. 1). In an effort to resolve a conK.C. Nicolaou, Ph.D. troversy over the structure of Professor and Chairman maitotoxin, the largest and most Chemistry potent marine neurotoxin, we synthesized the GHIJK and GHIJKMNO domains of the molecule and provided spectroscopic support for the originally assigned structure of this biomolecule. We also provided synthetic azaspiracids, another group of marine neurotoxins, to biologists who investigated the neurotoxic properties and mechanism of action of the molecules. During the same period, we completed the total synthesis of several members of the artochamin family of natural products, which have cytotoxic properties. We also developed an asymmetric synthesis of the uncialamycins and determined their DNA-cleaving properties and extremely high cytotoxic effects against tumor cells and drug-resistant bacteria. Our research in infectious diseases yielded a series of biyouyanagin analogs, some of which had anti-HIV/AIDS properties. In addition, we developed asymmetric total syntheses of platensimycin and platencin, 2 naturally occurring antibiotics that exert their activities against drug-resistant bacteria through a novel mechanism involving inhibition of fatty acid biosynthesis. Carbaplatensimycin, a carbon analog of platensimycin, was also synthesized and tested. In yet another area, we developed chemistry suitable for the eventual total synthesis of sporolide B, D THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 39 Nicolaou, K.C., Dethe, D.H., Leung, G.Y.C., Zou, B., Chen, D.Y.-K. Total synthesis of thiopeptide antibiotics GE2270A, GE2270T, and GE2270C. Chem. Asian J. 3:413, 2008. Nicolaou, K.C., Frederick, M.O., Aversa, R.J. The continuing saga of the marine polyether biotoxins. Angew. Chem. Int. Ed. 47:7182, 2008. Nicolaou, K.C., Frederick, M.O., Burtoloso, A.C.B., Denton, R.M., Rivas, F., Cole, K.P., Aversa, R.J., Gibe, R., Umezawa, T., Suzuki, T. Chemical synthesis of the GHIJKLMNO ring system of maitotoxin. J. Am. Chem. Soc. 130:7466, 2008. Nicolaou, K.C., Krasovskiy, A., Trépanier, V.É., Chen, D.Y.-K. An expedient strategy for the synthesis of tryptamines and other heterocycles. Angew. Chem. Int. Ed. 47:4217, 2008. Nicolaou, K.C., Leung, G.Y.C., Dethe, D.H., Guduru, R., Sun, Y.-P. Lim, C.S., Chen, D.Y.-K. Chemical synthesis and biological evaluation of palmerolide A analogues. J. Am. Chem. Soc. 130:10019, 2008. Nicolaou, K.C., Li, A. Total syntheses and structural revisions of α- and β-diversonolic esters and total syntheses of diversonol and blennolide C. Angew. Chem. Int. Ed. 47:6579, 2008. Nicolaou, K.C., Lister, T., Denton, R.M., Gelin, C.F. Total synthesis of artochamins F, H, I, and J through cascade reactions. Tetrahedron 64:4736, 2008. Nicolaou, K.C., Ortiz, A., Denton, R.M. Metathesis reactions in the synthesis of complex molecules. Chem. Today 25:70, 2007. Nicolaou, K.C., Pappo, D., Tsang, K.Y., Gibe, R., Chen, D.Y.-K. A chiral pool based synthesis of platensimycin. Angew. Chem. Int. Ed. 47:944, 2008. Nicolaou, K.C., Stepan, A.F., Lister, T., Montero, A., Tria, G.S., Turner, C.I., Tang, Y., Wang, J., Denton, R.M., Edmonds, D.J. Design, synthesis, and biological evaluation of platensimycin analogues with varying degrees of molecular complexity. J. Am. Chem. Soc. 130:13110, 2008. Nicolaou, K.C., Sun, Y.-P., Guduru, R., Banerji, B., Chen, D.Y.-K. Total synthesis of the originally proposed and revised structures of palmerolide A and isomers thereof. J. Am. Chem. Soc. 130:3633, 2008. F i g . 1 . Selected target molecules. a metabolite of a powerful enediyne antitumor antibiotic. Strides have also been made toward the total synthesis of nocathiacin III and lomaiviticin B, 2 potent antitumor antibiotics. Overall, our research programs continue to sharpen the tools of chemical synthesis and provide biologically active molecules, some natural and some designed, for chemical biology studies. By advancing the art of chemical synthesis, and through the preparation of biological tools and potential drug candidates, we strengthen the foundation of the drug discovery and development process. PUBLICATIONS Nicolaou, K.C., Chen, J.S., Edmonds, D.J., Estrada, A.A. Recent advances in the total synthesis and biology of naturally occurring antibiotics. Angew. Chem. Int. Ed., in press. Nicolaou, K.C., Chen, J.S., Zhang, H., Montero, A. Asymmetric synthesis and biological properties of uncialamycin and 26-epi-unicialamycin. Angew. Chem. Int. Ed. 47:185, 2008. Nicolaou, K.C., Cole, K.P., Frederick, M.O., Aversa, R.J., Denton, R.M. Chemical synthesis of the GHIJK ring system and further experimental support for the originally assigned structure of maitotoxin. Angew. Chem. Int. Ed. 46:8875, 2007. Nicolaou, K.C., Dethe, D.H., Chen, D.Y.-K. Total syntheses of amythiamicins A, B, and C. Chem. Commun. (Camb.) Issue 23:2632, 2008. Nicolaou, K.C., Sun, Y.-P., Peng, X.-S., Polet, D., Chen, D.Y.-K. Total synthesis of (+)-cortistatin. Angew. Chem. Int. Ed. 47:7310, 2008. Nicolaou, K.C., Toh, Q.-Y., Chen, D.Y.-K. An expedient asymmetric synthesis of platencin. J. Am. Chem. Soc. 130:11292, 2008. Nicolaou, K.C., Tria, G.S., Edmonds, D.J. Total synthesis of platencin. Angew. Chem. Int. Ed. 47:1780, 2008. Nicolaou, K.C., Wang, J., Tang, Y. Synthesis of the sporolide ring framework through a cascade sequence involving an intramolecular [4+2] cycloaddition reaction of an o-quinone. Angew. Chem. Int. Ed. 47:1432, 2008. Nicolaou, K.C., Wu, T.R., Sarlan, D., Shaw, D.M., Rowcliffe, E., Burton, D.R. Total synthesis, revised structure, and biological evaluation of biyouyanagin A and analogues thereof. J. Am. Chem. Soc. 130:11114, 2008. Vale, C., Gómez-Limia, B., Nicolaou, K.C., Frederick, M.O., Vieytes, M.R., Botana, L.M. The c-Jun-N-terminal kinase is involved in the neurotoxic effect of azaspiracid-1. Cell. Physiol. Biochem. 20:957, 2007. Vale, C., Wandscheer, C., Nicolaou, K.C., Frederick, M.O., Alfonso, C., Vieytes, M.R., Botana, L.M. Cytotoxic effect of azaspiracid-2 and azaspiracid-2-methyl ester in cultured neurons: involvement of the c-Jun N-terminal kinase. J. Neurosci. Res. 86:2952, 2008. Vilariño, N., Nicolaou, K.C., Frederick, M.O., Cagide, E., Alfonso, C., Alonso, E., Vieytes, M.R., Botana, L.M. Azaspiracid substituent at C1 is relevant to in vitro toxicity. Chem. Res. Toxicol. 21:1823, 2008. 40 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Filling Space at the Molecular Level J. Rebek, Jr., D. Ajami, M. Ams, E. Barrett, S. Beer, T.J. Dale, R.J. Hooley, J.-L. Hou, H. Van Anda, S. Xiao, spaces. They have revealed phenomena never before observed, such as coiled alkanes, stabilization of reactive intermediates, places where new forms of stereochemistry can emerge, and reaction chambers with welldefined shapes. We found that some capsules, such as shown in Figure 2, can incorporate spacer elements A. Lledo, H. Dube, P. Restorp, S. Kamioka MOLECULAR MIMICRY rotein-protein interactions are involved in many cell signaling events, and a large fraction of protein surfaces involve α-helices. This secondary structure presents side chains of the component amino acids along one face of the Julius Rebek, Jr., Ph.D. helix that are recognized by the Professor and Director The Skaggs Institute partner protein. To interfere for Chemical Biology with these protein-protein interactions, several research groups have made α-helix mimetics. Our efforts have gone into those that have amphiphilic behavior, that is, ones that are hydrophobic on one side and hydrophilic on the other. We have found rapid and efficient ways of assembling these mimics by using a pyridizine-based scaffold. The mimics have good solubility, and their synthesis can be easily scaled up. The pyridizines and the scheme for their assembly are shown in Figure 1. P F i g . 2 . Size and shape of the space inside a capsule show the tapered ends and the constricted center. Center, Encapsulated 1-hexadecene inside the assembly. Right, Encapsulated 1-hexadecyne. The thin alkene and alkyne groups penetrate deep into the bottom of the capsule. The central parts of the guests are in fully extended conformations, and compression of the alkane appears near the top. known as glycolurils in response to the presence of guests. The expanded capsules shown are present only when a suitable guest is able to fill the space inside. The shape of the space inside is shown in the figure, and narrow functional groups such as primary alkenes and acetylenes can fit in the tapered ends of the space. C O E N C A P S U L AT I O N With 2 different guests inside a capsule, the contact points between the guests can be mapped out by using nuclear magnetic resonance techniques. For example, with ethane and heptane coencapsulated, as shown in Figure 3, we found that the 2 ends of heptane are alternately in contact with the ethane. This contact is achieved by the flipping of the heptane F i g . 1 . A, Overlay of an idealized α-helix protein structure with a new scaffold. B, Synthetic scheme shows the component parts that present the amino acid side chains on the scaffold and the atoms that make up the hydrophilic surface. EXPANDED CAPSULES Reversible encapsulation complexes are synthetic receptors that more or less completely surround their target guests. They provide a window through which molecular behavior can be seen in extremely small F i g . 3 . Ethane (gray) and heptane (rainbow colored) are coen- capsulated. Tumbling motions of the heptane can be detected by nuclear magnetic resonance techniques that show contact between the 2 ends of the longer guest with ethane. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 inside the capsule, rather than by the exchange of places of the 2 guests inside. C AV I TA N D S W I T H I N T R O V E R T E D F U N C T I O N A L I T Y Cavitands are open-ended molecular vessels that allow relatively rapid motions of guests inside and out. Figure 4 shows a system held together by hydrogen 41 Role of Mistranslation in Disease P. Schimmel, X.-L. Yang, R. Belani, Y. Chong, Z. Druzina, J. Frater, M. Guo, R.-T. Guo, M. Hanan, W. He, I.L. Jung, M. Kapoor, S.H. Lee, J. Liu, E. Merriman, M.H. Nawaz, R. Shapiro, Y.Z. Song, M.-N. Vo, W. Zhang, Q. Zhou istranslation occurs when the wrong amino acid is inserted into a growing polypeptide chain during protein synthesis. Most usually, proteins are error-free; that is, each specific protein has its own, specific amino acid sequence that is defined by the gene that encodes the protein. Paul R. Schimmel, Ph.D. Professor (The process of protein synthesis Molecular Biology “translates” the sequence of a gene into the corresponding protein sequence.) When an error is made, so that the wrong amino acid occasionally appears at a specific location in the sequence of a protein, this aberration can lead to a protein with altered biological activity. Additionally or alternatively, the error-containing protein may misfold. Recently, we showed that mistranslation that causes altered protein structure and function is connected to disease. Normally, errors of translation are prevented by the editing activities of tRNA synthetases. These synthetases attach amino acids to tRNAs, matching each amino acid to its cognate tRNA partner. The attached amino acid is then carried to the ribosome, where it is inserted into a growing polypeptide chain at the position specified by the anticodon of the tRNA. If the wrong amino acid is accidentally attached to a particular tRNA and is not corrected by the editing activity of the appropriate tRNA synthetase, then that amino acid is carried in the same way to the ribosome and inserted into a growing polypeptide. This insertion is at the place normally occupied by the correct amino acid. Mutations in the editing centers of tRNA synthetases can thus lead to mistranslation. Recently, we showed that mistranslation arising from such mutations is deleterious to bacterial and mammalian cells. In addition, we found that a mild editing defect in a specific tRNA synthetase (alanyl-tRNA synthetase) led to neurodegeneration in mice. M F i g . 4 . A deep vaselike structure presents a cavity, and the carboxylic acid (red) is directed into the space. Isonitriles are captured in the cavity and react with the acid to give stabilized intermediates, such as A. bonding that features a seam of hydrogen bonds that maintain the vaselike shape. The cavitand is attached to an anthracene that delivers a carboxylic acid to the inside space. We have used this system to trap reactive intermediates such as those involved in reactions of isonitriles. The intermediates have only microsecond lifetimes in solution but are stabilized inside the cavitand for up to 15 minutes, long enough to characterize them by using nuclear magnetic resonance and infrared spectroscopic techniques. PUBLICATIONS Ajami, D., Rebek, J., Jr. Gas behavior in self-assembled capsules. Angew. Chem. Int. Ed. 47:6059, 2008. Ajami, D., Rebek, J., Jr. Longer guests drive the reversible assembly of hyperextended capsules. Angew. Chem. Int. Ed. 46:9283, 2007. Ajami, D., Rebek, J., Jr. Reversible encapsulation of terminal alkenes and alkynes. Heterocycles 76:169, 2008. Ajami, D., Schramm, M.P., Rebek, J., Jr. Translational motion inside self-assembled encapsulation complexes. Tetrahedron, in press. Mann, E., Moisan, L., Hou, J.-L., Rebek, J., Jr. Synthesis of pyridazines functionalized with amino acid side chains. Tetrahedron Lett. 49:903, 2008. Moisan, L., Odermatt, S., Gombosuren, N., Carella, A., Rebek, J., Jr. Synthesis of an oxazole-pyrrole-piperazine scaffold as an α-helix mimetic. Eur. J. Org. Chem. 10:1673, 2008. Restorp, P., Rebek, J., Jr. Reaction of isonitriles with carboxylic acids in a cavitand: observation of elusive isoimide intermediates. J. Am. Chem. Soc. 130:11850, 2008. Restorp, P., Rebek, J., Jr. Synthesis of α-helix mimetics with four side-chains. Bioorg. Med. Chem. Lett. 18:5909, 2008. 42 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Currently, we are focusing on the possibility that editing defects are causally connected to the etiology of some cancers. Because cancer is mostly a disease of aging, the random occurrence of mutations in the editing domains of tRNA synthetases in somatic cells can lead to mistranslation in those tissues. In other studies, we established that in aging bacteria, an editing-defective tRNA synthetase (the defect itself caused by a mutation in its editing domain) can lead to mutations in the error-prone DNA repair apparatus. When error-prone repair is perturbed, random errors spontaneously occur more frequently in the genome. We envision that a similar situation happens in mammalian cells; that is, an editing-defective tRNA synthetase can itself induce more mutational errors in the genome as the organism ages. Some of these mutations might occur in oncogenes that when activated lead to transformation to the oncogenic state. With this possibility in mind, we established a collaboration with P. Vogt, Scripps Research, to see whether, in a model system developed in Dr. Vogt’s laboratory, oncogenesis can be induced by an editing-defective tRNA synthetase. At the same time, we are investigating activities of enzymes associated with DNA repair to see if in mammalian cells carrying an editing defect, one or more of these enzymes is affected by mistranslation. A perturbation of one of the enzymes associated with DNA repair could lead to the fixing of random mutations into the genome. Some of these mutations might occur in oncogenes. PUBLICATIONS Beebe, K., Mock, M., Merriman, E., Schimmel, P. Distinct domains of tRNA synthetase recognize the same base pair. Nature 451:90, 2008. Cheng, G., Zhang, H., Yang, X.-L., Tzima, E., Ewalt, K.L., Schimmel, P., Faber, J.E. Effect of mini-tyrosyl-tRNA synthetase on ischemic angiogenesis, leukocyte recruitment, and vascular permeability. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295:R1138, 2008. Chong, Y.-E., Yang, X.-L., Schimmel, P. Natural homology of tRNA synthetase editing domain rescues conditional lethality caused by mistranslation. J. Biol. Chem. 283:30073, 2008. Greenberg, Y., King, M., Kiosses, W.B., Ewalt, K., Yang, X.-L., Schimmel, P., Reader, J. S., Tzima, E. The novel fragment of tyrosyl-tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J. 22:1597, 2008. Guo, M., Ignatov, M., Musier-Forsyth, K., Schimmel, P., Yang, X.-L. Crystal structure of novel tetrameric form of human lysyl-tRNA synthetase: implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. U. S. A. 105:2331, 2008. Kapoor, M., Zhou, Q., Otero, F., Myers, C.A., Bates, A., Belani, R., Liu, J., Luo, J.-K., Tzima, E., Zhang, D.-E., Yang, X.-L., Schimmel, P. Evidence for annexin IIS100A10 complex and plasmin in mobilization of cytokine activity of human TrpRS. J. Biol. Chem. 283:2070. 2008. Park, S.G., Schimmel, P., Kim, S. Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl. Acad. Sci. U. S. A. 105:11043. 2008. Schimmel, P. Development of tRNA synthetases and connection to genetic code and disease. Protein Sci. 17:1643, 2008. Schimmel, P. An editing activity that prevents mistranslation and connection to disease. J. Biol. Chem. 283:28777, 2008. Zhou, Q., Kiosses, W.B., Liu, J., Schimmel, P. Tumor endothelial cell tube formation model for determining anti-angiogenic activity of a tRNA synthetase cytokine. Methods 44:190, 2008. New Amino Acid Building Blocks P.G. Schultz, E.M. Brustad, J. Grünewald, J. Guo, H.S. Lee, C.C. Liu, F.B. Peters, T.S. Young lmost all processes of living cells, from gene regulation and information processing to photosynthesis, are carried out by proteins. These are large molecules synthesized from 20 amino acid building blocks. This set of 20 Peter Schultz, Ph.D. amino acids is the basis for the Professor Chemistry genetic code, the code that specifies the relationship between the nucleotide sequence of a gene and the amino acid sequence of a protein. This fact leads to rather interesting questions: Why does every form of life have the same set of building blocks? Why not 21 or more? Moreover, if we can add new amino acid building blocks to the genetic code, will we be able to create proteins or even cells with enhanced chemical, physical, or biological properties? We continue to address this issue by using chemical and molecular biological methods to add new components to the protein biosynthetic machinery of bacteria. Using this approach, we effectively expanded the genetic code of both prokaryotes and eukaryotes by genetically encoding new amino acids (including photoaffinity labels, chemically reactive amino acids, posttranslationally modified amino acids, and amino acids with altered electronic and steric properties) in response to unique 3and 4-base codons. Currently, we are exploring additional amino acids with novel biological and physicochemical properties, other organisms, and a number of biomedical applications of this technology. For example, in the past year, we showed that tolerance against the self-antigen TNF-α in mice can be broken by the selective introduction of p-nitrophenylalanine. This research promises to greatly A THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 facilitate the generation of cancer vaccines and vaccines against third-world pathogens; we are currently extending it to lung and breast cancer vaccines and vaccines against tuberculosis, malaria. and HIV disease. We also genetically encoded a boronate-containing amino acid for the scarless purification of proteins, selective protein modification, and the development of selective antibodies to both glycoproteins and serine proteases involved in viral infections and cancer. In addition, we genetically encoded a hydroxyquinoline metal ion–binding amino acid for heavy-atom phasing in protein structure determination and for the introduction of radioisotopes into antibodies for cancer therapy and imaging. In other studies, we used a phenylselenide-containing amino acid to generate proteins containing posttranslationally modified lysine residues to study the role of histone modification in the epigenetics of cancer and developmental biology. We also showed that phage display can be used to select anti-gp120 sulfotyrosinecontaining antibodies with enhanced affinities relative to the naturally sulfated anti-gp120 antibody. This result shows for the first time that an expanded genetic code can confer an evolutionary advantage and may lead to the generation of therapeutic proteins or peptides with improved biological or pharmacologic properties due to the presence of unnatural amino acids. We also genetically encoded the fluorophore prodan in mammalian cells to facilitate cellular studies of protein structure, function, and localization; genetically encoded an unnatural amino acid that can cleave the polypeptide backbone in a light-dependent fashion as a cell biological tool; and biosynthesized DNA-binding proteins that can oxidatively cleave the DNA backbone in a sequence-specific fashion as a probe of proteinDNA recognition. Furthermore, we showed that unnatural amino acids can be used to selectively introduce fluorescence resonance energy transfer pairs into proteins for single-molecule imaging studies, and we developed a high-yield Pichia expression system to produce therapeutic proteins containing unnatural amino acids. PUBLICATIONS Brustad, E., Bushey, M.L., Brock, A., Chittuluru, J., Schultz, P.G. A promiscuous aminoacyl-tRNA synthetase that incorporates cysteine, methionine, and alanine homologs into proteins. Bioorg. Med. Chem. Lett. 18:6004, 2008. Brustad, E., Bushey, M.L., Lee, J.W., Groff, D., Liu, W., Schultz, P.G. A genetically encoded boronate-containing amino acid. Angew. Chem. Int. Ed. 47:8220, 2008. Grünewald, J., Tsao, M.-L., Perera, R., Dong, L., Niessen, F., Wen, B.G., Kubitz, D.M., Smider, V.V., Ruf, W., Nasoff, M., Lerner, R.A., Schultz, P.G. Immunochemical termination of self-tolerance. Proc. Natl. Acad. Sci. U. S. A. 105:11276, 2008. 43 Guo, J., Wang, J., Anderson, J.C., Schultz, P.G. Addition of an α-hydroxy acid to the genetic code of bacteria. Angew. Chem. Int. Ed. 47:722, 2008. Guo, J., Wang, J., Lee, J.-S., Schultz, P.G. Site-specific incorporation of methyland acetyl-lysine analogues into recombinant proteins. Angew. Chem. Int. Ed. 47:6399, 2008. Lee, J., Hong, J., Nam, T.-G., Peters, E.C., Orth, A.P., Geierstanger, B.H., Goldfinger, L.E., Ginsberg, M.H., Cho, C.Y., Schultz, P.G. A small molecule inhibitor of α4 integrin-dependent cell migration. Bioorg. Med. Chem., in press. Lemke, E.A., Summerer, D., Geierstanger, B.H., Brittain, S.M., Schultz, P.G. Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat. Chem. Biol. 3:769, 2007. Tippmann, E.M., Liu, W., Summerer, D., Mack, A.V., Schultz, P.G. A genetically encoded diazirine photocrosslinker in Escherichia coli. ChemBioChem 8:2210, 2007. Click Chemistry and Biological Activity K.B. Sharpless, V.V. Fokin, J. Culhane, J. Fotsing, S. Grecian, N. Grimster, J. Hein, T. Horneff, J. Kalisiak, K. Korthals, S.-W. Kwok, S. Pitram, J. Raushel, B. Stump, J. Tripp, C. Valdez, T. Weide e aim to discover new chemical processes that allow rapid and efficient synthesis of molecules with a desired function from diverse building blocks. Since 1996, the support of the Skaggs Institute for Chemical Biology has been instrumental to the K. Barry Sharpless, Ph.D. Professor development of the concept Chemistry and the applications of click chemistry. Click chemistry embodies an attitude that function matters most and that tools that enable researchers to achieve function are to be prized. Thus, click chemistry relies on the use of a few near-perfect chemical reactions for the synthesis and assembly of specially designed building blocks. These building blocks have a high built-in energy content that drives a spontaneous, selective, and irreversible linking reaction with complementary sites in the reactive partners. The power of click chemistry lies in the ability to rapidly generate novel structures that may not necessarily resemble known biologically active compounds. For discovering compounds that may be useful as drugs, this strategy provides a means for the rapid exploration of the chemical space. For optimization of compounds, the method enables rapid structure-activity profiling W 44 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 through generation of analog libraries. Click chemistry does not replace existing methods for drug discovery; rather it complements and extends them. It works well in conjunction with structure-based design and combinatorial chemistry techniques. Click chemistry is both enabled and constrained by its reliance on a few nearly perfect reactions, and this characteristic raises concerns about limitations on the access of click chemistry to chemical diversity. However, the pool of druglike compounds may be as large as 1063. Currently, only a few million compounds that fulfill these criteria are known, implying that only an infinitesimal part of the potential medicinal chemistry universe has been explored so far. These facts have staggering implications for drug discovery. First and foremost, most molecules with useful properties remain to be discovered. Second, the majority of useful new compounds likely will be found in unconventional structure space. Thus, with click chemistry, we have the interesting proposition that greater diversity can be achieved with fewer reactions, because it is not the number of reactions that is important, but the reach of the reactions, which is determined by the tolerance to variations in the nature of their components. Click chemistry approaches have already proved themselves in biomedical research, ranging from synthetic chemistry to bioconjugation strategies, polymer chemistry, and materials science. R A P I D M O D I F I C AT I O N O F A N T I B I O T I C S T O O V E R C O M E R E S I S TA N C E Although macrolides, including erythromycin A have been widely prescribed for more than 50 years, the emergence of widespread bacterial resistance to these molecules is a serious and expanding problem. Thirdgeneration macrolides, such as telithromycin, have been developed in recent years as effective means to overcome resistant bacterial strains. However, despite these efforts, only a few macrolide candidates with activity against methicillin-resistant Staphylococcus aureus (MRSA) have been identified to date. Clearly, the medical need for new antibiotics to combat strains of MRSA is urgent. In our collaboration with scientists at the Kitasato Institute, Tokyo, Japan, we have reexamined the activity of various derivatives of erythromycin A against 12 types of gram-positive bacteria, including macrolide-resistant strains, and 1 gram-negative organism. We found that 11,12-di-O-iso-butyryl-8,9-anhydroerythromycin A 6,9hemiketal has moderate activity against 4 strains of MRSA and 2 strains of vancomycin-resistant entero- cocci (VRE). Further modification of an alkynylated derivative of this lead compound by using the copper-catalyzed azide-alkyne cycloaddition, the flagship click reaction, quickly led to identification of several triazole-containing erythromycin A analogs with improved activity against MRSA and VRE strains (Fig. 1). These promising antibacterials are currently undergoing further evaluation. F i g . 1 . Novel erythromycin A derivatives with activity against strains of MRSA and VRE. MIC = minimum inhibitory concentration. IN SITU CLICK CHEMISTRY Although click chemistry allows rapid assembly of diverse collections of molecules that may serve as lead structures, further evolution of the molecules is traditionally achieved by iterative cycles of screening for biological activity and synthetic modification. Can direct involvement of the target, usually a specific receptor or enzyme, in the selection and evolution of possible drug candidates accelerate this drug discovery cycle? The aim of using in situ click chemistry is to engage an enzyme in the selection and covalent assembly of its own “best-fitting” inhibitor. Although the concept has been previously tested by several researchers, the in situ click chemistry approach is unique because it relies on the completely bioorthogonal 1,3-dipolar cycloaddition of organic azides and alkynes. This highly exergonic reaction produces 5-membered nitrogen heterocycles, 1,2,3triazoles, which are exceedingly stable to acidic and basic hydrolysis and to severe reduction-oxidation conditions. At the same time, the triazoles produced can actively participate in hydrogen-bonding, dipole-dipole, and π-stacking interactions. The efficacy of in situ click chemistry has been demonstrated by the discovery of novel, highly potent inhibitors of acetylcholinesterase, carbonic anhydrase, and HIV protease. During the past year, we extended the approach to more challenging targets. Among these are the enzyme β-secretase, which is involved in the progression of Alzheimer’s disease; several members of the vast kinase family; metalloproteases; and nicotinic THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 acetylcholine receptors, the family of ligand-gated ion channels responsible for key neurotransmission events. PUBLICATIONS Finn, M.G., Kolb, H.C., Fokin, V.V., Sharpless, K.B. Concept and applications of click chemistry from the standpoint of advocates. Kagaku to Kogyo 60:976, 2007. Hawker, C.J., Fokin, V.V., Finn, M.G., Sharpless, K.B. Bringing efficiency to materials synthesis: the philosophy of click chemistry. Aust. J. Chem. 60:381, 2007. Kalisiak, J., Sharpless, K.B., Fokin, V.V. Efficient synthesis of 2-substituted-1,2,3triazoles. Org. Lett. 10:3171, 2008. Kwok, S.-W., Hein, J.E., Fokin, V.V., Sharpless, K.B. Regioselective synthesis of either 1H- or 2H-1,2,3-triazoles via Michael addition to α,β-unsaturated ketones. Heterocycles 76:1141, 2008. Liu, Y., Díaz, D.D., Accurso, A.A., Sharpless, K.B., Fokin, V.V., Finn, M.G. Click chemistry in materials synthesis, III: metal-adhesive polymers from Cu(I)-catalyzed azide-alkyne cycloaddition. J. Polym. Sci. A Polym. Chem. 45:5182, 2007. Radić, Z., Manetsch, R., Fournier, D., Sharpless, K.B., Taylor, P. Probing gorge dimensions of cholinesterases by freeze-frame click chemistry. Chem. Biol. Interact. 175:161, 2008. Sugawara, A., Sunazuka, T., Hirose, T., Nagai, K., Yamaguchi, Y., Hanaki, H., Sharpless, K.B., Omura, S. Design and synthesis via click chemistry of 8,9-anhydroerythromycin A 6,9-hemiketal analogues with anti-MRSA and -VRE activity. Bioorg. Med. Chem. Lett. 17:6340, 2007. Van der Eycken, E., Sharpless, K.B. Click chemistry. QSAR Comb. Sci. 26:1115, 2007. Vestberg, R., Malkoch, M., Kade, M., Wu, P., Fokin, V.V., Sharpless, K.B., Drockenmuller, E., Hawker, C.J. Role of architecture and molecular weight in the formation of tailor-made ultrathin multilayers using dendritic macromolecules and click chemistry. J. Polym. Sci. A Polym. Chem. 45:2835, 2007. Yoo, E.J., Ahlquist, M., Bae, I., Sharpless, K.B., Fokin, V.V., Chang, S. Mechanistic studies on the Cu-catalyzed three-component reactions of sulfonyl azides, 1alkynes and amines, alcohols, or water: dichotomy via a common pathway J. Org. Chem. 73:5520, 2008. Molecular Biology of Olfaction L. Stowers, P. Chamero, K. Flanagan, D. Logan, T. Martin, F. Papes, A. Kaur ppropriate behavior relative to the surroundings is necessary for survival; however, the neural mechanisms that detect important cues in the environment, process the meaning of the cues, and initiate the corresponding behavior are largely unknown. The neural Lisa S. Stowers, Ph.D. Assistant Professor code of behavior is difficult Cell Biology to study because most future behavior is determined by previous experience, which often differs subtly among individuals. These differences in experience lead to variable perceptions and unpre- A 45 dictable behavior outcomes across individuals placed in the same environment. The underlying neural circuits are therefore correspondingly variable and dynamic. To ensure experimental clarity and reproducibility, we study the neural circuits that underlie innate behavior. Olfactory stimuli are known to elicit innate behaviors in rodents. For example, when a male encounters another individual in his environment, he detects and processes the emitted chemical cues to determine the age and sex of the intruder. If the cues signal that the intruder is a juvenile, the male will respond appropriately and not alter his behavior. If the intruder is a female, he will court her, and if he detects a male, he will respond with aggression. Innate behaviors are strong and universal, suggesting they are driven by genetically programmed, invariant neural circuits. We are investigating innate mouse behaviors that are stereotyped and quantifiable. Just as a biochemical assay is used to map and elucidate a metabolic pathway, we use innate behavior as a functional assay to identify the corresponding ligand cues and mediating neurons. Although this approach requires an unconventional research plan, it can be used to sort out and identify the unique biological tools, ligands, and receptors essential for studies at the cellular and molecular levels, the neural circuit that detects the environment and generates appropriate behavior. We have been isolating the chemical ligands, pheromones, that specifically govern social behaviors such as aggression and mating. These ligands are detected in rodents by neurons and mechanisms that are active in all terrestrial vertebrates but are not functional in humans. To expand our investigation of innate neural circuits, we are also elucidating the ligands and responsive sensory mechanisms that promote maternal-infant behavior. This behavior is the defining behavior of mammals, including humans. We expect that identification of the neuronal code that generates this innate behavior in mice will enable us to directly investigate mechanisms of innate behavior in humans. We have isolated the source of pheromones that promote maternal-infant behavior, a step allows us now to specifically activate, and thereby identify, the population of sensory neurons dedicated to promoting this behavior. Our findings suggest that this fundamental social behavior is governed by a unique olfactory mechanism, unlike other pheromone-mediated behaviors, that may have orthologous counterparts that are present and functional in humans. To further investigate these mech- 46 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 anisms, we are manipulating activation of the sensory circuit by activating and inactivating ligand receptors, signal transduction elements, and ion channels to identify the neurons we find essential for behavior. We are also fractionating the natural source of pheromones to purify the ligands. Once purified, these molecules will allow us to investigate the kinetics of their response and manipulate their properties to validate their role in promoting behavior. We expect that our studies will provide the tools to expand our understanding of the logic of neuronal coding of innate behaviors in mice and, additionally, investigate the molecular mechanisms that underlie human social behavior. PUBLICATIONS Logan, D.W., Marton, T.F., Stowers, L. Species specificity in major urinary proteins by parallel evolution. PLoS ONE 3:e3280, 2008. Macromolecular Master Keys for Genome integrity, Reactive Oxygen Control, and Pathogenesis J.A. Tainer, A.S. Arvai, B.R. Chapados, L. Fan, C. Hitomi, K. Hitomi, J.J. Perry, M.E. Pique, D.S. Shin, J.L. Tubbs, R.S. Williams o date, funds from the Skaggs Institute for Chemical Biology has supported the training of 15 graduate students and 33 postdoctoral fellows and contributions to 176 publications. In particular, Skaggs funding is used for medically relevant structureJohn A. Tainer, Ph.D. function investigations of macro- Professor Molecular Biology molecular master keys for genome integrity, reactive oxygen control, and pathogenesis. Skaggs funding also supports our synchrotron beam line, at the Advanced Light Source, University of California, Berkeley/Lawrence Berkeley National Laboratory. The synchrotron is used to characterize macromolecular complexes, conformations, and interactions in solution via small-angle x-ray scattering (SAXS) and at high resolution via macromolecular x-ray crystallography. We are furthermore developing and using new data interpreta- T tion tools for the detailed visualizations of protein complexes and modified proteins that undergo functionally important changes in shape and assembly. Working with P. Kuhn, Scripps Research, for example, we used our SAXS technologies to characterize solution structures that explain guanine nucleotide exchange mediated by the T-cell essential Vav1. We are working to develop SAXS for drug discovery so that the technology can be used to identify small molecules that bind and inhibit enzymes. We are continuing research on pathogenic bacteria, including drug and vaccine design for the type IV pilin system. We have new structures for the fiberforming type IV pilin protein, the assembly ATPase, and associated machinery. Type IV pilin proteins are critical bacterial virulence factors for cholera, pneumonia, gonorrhea, meningitis, and severe diarrhea. Our combined SAXS and macromolecular x-ray crystallography methods are providing a mechanistic understanding relevant to controlling these virulence factors. In collaboration with M.N. Boddy, Scripps Research, we have identified the SUMO-targeted ubiquitin ligase family of proteins. These proteins provide communication between the sumoylation and ubiquitination pathways and act as master regulators of DNA damage responses. Thus, the proteins are of obvious value for the development of novel cancer drugs. We also defined new structure-function relationships for the multicomponent Smc5-Smc6 complex in genome stability. Our work on macromolecules with key roles in pathways controlling reactive oxygen species and DNA damage responses is important for preserving the nervous system and controlling cancer. In our structure-based design projects, we collaborate with E.D. Getzoff, Skaggs Institute, to characterize inhibitors of nitric oxide synthase. In this collaborative work, we have discovered a new general method for designing inhibitors to specific isozymes of nitric oxide synthase that promise to reduce unwanted side effects and allow targeted interventions for arthritis, stroke, and cancer. Defining master keys to regulate reactive oxygen species such as superoxide and nitric oxide help provide an informed basis to avoid the oxidative death of neurons in neurodegenerative diseases and to use reactive oxygen species to kill cells involved in cancer and pathogenesis. In other cancer related research, we characterized O6-alkylguanine-DNA alkyltransferase activity on x-linked DNA. This alkyltransferase is a target both for the prevention of cancer and for chemotherapy, because it THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 repairs mutagenic lesions in DNA and limits the effectiveness of alkylating chemotherapies. We also used high-resolution crystal structures and mutational analyses to explain how endonuclease IV uses 3 metal ions to remove a damaged DNA site and then hold the product to avoid the release of toxic repair intermediates. We similarly characterized the role of active-site metal ions for flap endonuclease-1, which removes DNA flaps generated during both replication and repair. In recent investigations on the nucleotide excision repair system, which repairs bulky lesions in DNA, we have focused on the helicase XPD. Our results define XPD helicase structures and activities to provide new insights into the cancer and aging phenotypes of XPD mutations (Fig. 1). Our findings also help explain the severe developmental problems associated with Cockayne syndrome, which includes defects in both DNA repair and transcription. Moreover, the effects of hijacking transcription factors and repair shielding associated with some XPD mutants may be the basis for the success of cisplatin as an anticancer agent. These results may therefore provide a mechanistic basis for the design if novel cancer agents that extend the therapeutic usefulness of cisplatin to other tumor types. To understand the Mre11-Rad50-Nbs1 (MRN) complex that initiates repair of DNA double-strand breaks and homologous recombination, we are collaborating with P. Russell, Scripps Research. Our results help show how MRN mutations cause the Nijmegen breakage syndrome and ataxia telangiectasia–like disorder, diseases in humans that predispose individuals to cancer. Our new Mre11-DNA structures and mutants reveal key Mre11 roles in DNA end synapsis and nuclease processing (Fig. 2). We also identified Rad50 mutants that form meiotic DNA double-strand breaks and revealed an essential structural role for Rad50 in axial element and synaptonemal complex formation. In related research on homologous recombination, we used our new SAXS methods to define the domain structure interactions for the homologous recombination protein BARD1. We are now investigating the structural basis for homologous recombination; our results may have implications for improving cancer interventions by reprogramming cells for death vs repair in response to double-strand breaks. PUBLICATIONS Acharya, S.N., Many, A.M., Schroeder, A.P., Kennedy, F.M., Savytskyy, O.P., Grubb, J.T., Vincent, J.A., Friedle, E.A., Celerin, M., Maillet, D.S., Palmerini, H.J., Greischar, M.A., Moncalian, G., Williams, R.S., Tainer, J.A., Zolan, M.E. Coprinus cinereus rad50 mutants reveal an essential structural role for Rad50 in axial element and synaptonemal complex formation, homolog pairing and meiotic recombination. Genetics 180:1889, 2008. 47 F i g . 1 . Structural placement of disease-causing mutations in XPD helicase. Mapping the 3 classes of mutations onto the SaXPD structure reveals patterns associated with each disease defect. A, Stereopair mapping the distribution of disease-causing mutations on a XPD Cα trace. Disease-causing mutation sites (Cα colored sphere): red (XP), greenish yellow (XP/CS), and purple (TTD). Residue F136 is also shown (cyan). B, XPDcc fold and domain architecture (ribbons) with labeled disease-causing mutation sites as spheres colored as in A. C, XP mutations affect DNA- and ATP-binding regions. D, XP/CS mutations affect HD1-HD2 conformational changes. E, TTD mutations affect the overall framework stability. Reprinted from Fan, L., Fuss, J.O., Cheng, Q.J., Arvai, A.S., Hammel, M., Roberts, V.A., Cooper, P.K., Tainer, J.A. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133:789, 2008, copyright 2008, with permission from Elsevier. Chrencik, J.E., Brooun, A., Zhang, H., Mathews, I.I., Hura, G.L., Foster, S., Perry, J.J., Streiff, M., Ramage, P., Widmer, H., Bokoch, G.M., Tainer, J.A., Weckbecker, G., Kuhn, P. Structural basis of guanine nucleotide exchange mediated by the T-cell essential Vav1. J. Mol. Biol. 380:828, 2008. Edwards, R.A., Lee, M.S., Tsutakawa, S.E., Williams, R.S., Tainer, JA., Glover, J.N.M. The BARD1 C-terminal domain structure and interactions with polyadenylation factor, CstF-50. Biochemistry 47:11446, 2008. Fan, L., Fuss, J.O., Cheng, Q.J., Arvai, A.S., Hammel, M., Roberts, V.A., Cooper, P.K., Tainer, J.A. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133:789, 2008. 48 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Chemical Approaches to Disease P. Wentworth, Jr., R.K. Grover, B.D. Song, M.M.R. Peram, J. Rogel, R.P. Troseth, D. Angrish, V. Dubrovskaya, A.D. Wentworth ur interdisciplinary research focus involves aspects of bioorganic, biophysical, physical organic, synthetic, and analytical chemistry coupled with biochemical techniques, cell-based assays, and animal models. Ongoing Paul Wentworth, Jr., Ph.D. projects include studies on Professor atherosclerosis, neurodegener- Chemistry ative diseases, ischemia-reperfusion injury, macular degeneration, cancer, inflammation, and infectious diseases. Currently, we are validating a new antileishmania drug target and increasing the scope of the role of inflammatory aldehydes in protein misfolding diseases. The genomic DNA of kinetoplastid parasites contains a unique modified base, 5-(β-D -glucopyranosyloxymethyl)-2′-deoxyuridine or base J. Recently, we reported the first in-depth analysis of the molecular recognition between the O-linked glycoside component of deoxyribose J (dJ) in telomeric dJ-containing double-stranded DNA and J-binding protein 1 (JBP1) of Crithidia fasciculata. Comparison between the molecular dynamics snapshots and the free energy of binding of JBP1 to a panel of duplex oligonucleotides containing telomeric modified dJ revealed that JBP1 binding to dJ-containing oligonucleotides occurs preferentially when the β-D-glucopyranosyl moiety adopts a conformation within the major groove wherein the C-2 and C-3 hydroxyl groups of the glucoside make hydrogen-bond contacts to the nonbridging pro-R phosphoryl oxygen of the J-1 nucleotide phosphate group. If this orientation is perturbed even slightly, JBP1 binding affinity drops to the level of replacement of dJ by deoxythymidine. In the past year, we have expanded this work and shown that this same edge-on conformation occurs in JBP1 in nonpathogenic Leishmania tarantolae. We also designed phosphorothioate probes to replace the J-1 residue in the duplex DNA. We hypothesized that replacement of the J-1 phosphoryl oxygen with sulfur O F i g . 2 . ATLD missense mutations W210C and N117S, which impair Nbs1 binding, cluster to a single Mre11 dimer surface opposite the DNA binding cleft. Fang, Q., Noronha, A.M., Murphy, S.P., Wilds, C.J., Tubbs, J.L., Tainer, J.A., Chowdhury, Q., Guengerich, F.P., Pegg, A.E. Repair of O6-G-alkyl-O6-G interstrand cross-links by human O6-alkylguanine-DNA alkyltransferase. Biochemistry 47:10892, 2008. Garcin, E.D., Arvai, A.S., Rosenfeld, R.J., Kroeger, M.D., Crane, B.R., Andersson, G., Andrews, G., Hamley, P.J., Malinder, P.R., Nicholis, D.J., St-Gallay, S.A., Tinker, A.C., Gensmantel, N.P., Mete, A., Cheshire, D.R., Connolly, S., Stuhr, D.J., Alberg, A., Wallace, A.V., Tainer, J.A., Getzoff, E.D. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol. 4:700, 2008. Garcin, E.D., Hosfield, D.J., Desai, S.A., Haas, B.J., Björas, M., Cunningham, R.P., Tainer, J.A. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat. Struct. Mol. Biol. 15:515, 2008. Pebernard, S., Perry, J.J., Tainer, J.A., Boddy, M.N. Nse1 RING-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol. Biol. Cell 19:4099, 2008. Perry, J.J., Tainer, J.A. Structural biology of Cockayne syndrome proteins, their interactions and insights into DNA repair mechanisms. In: Molecular Mechanisms of Cockayne Syndrome. Ahmad, S.I. (Ed.). Landes Biosciences, Austin, TX, in press. Perry, J.J.P., Tainer, J.A, Boddy, M.N. A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem. Sci. 33:201, 2008. Syson, K., Tomlinson, C., Chapados, B.R., Sayers, J.R., Tainer, J.A., Williams, N.H., Grasby, J.A. Three metal ions participate in the reaction catalyzed by T5 flap endonuclease. J. Biol. Chem. 283:28741, 2008. Williams, R.S., Moncalian, G., Williams, J.S., Yamada, Y., Limbo, O., Shin, D.S., Groocock, L.M., Cahill, D., Hitomi, C., Guenther, G., Moiani, D., Carney, J.P., Russell, P., Tainer, J.A. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135:97, 2008. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 would have a clear effect on the strength of the C-2 and C-3 hydrogen bonding. This hypothesis was proved by the finding that JBP1 does not bind to the phosphorothioate-containing duplex DNA as tightly as to normal duplex DNA. In conclusion, the use of new phosphorothioate J-1 DNA as a chemical tool has validated our proposed binding hypothesis for JBP1 to J-DNA. This study further emphasizes the importance of our earlier proposed hypothesis that glucose conformation in the major groove of DNA is due to hydrogen bonds between the quasiequatorial C-2 and C-3 hydroxyl groups of the sugar and the pro-R phosphoryl oxygen of the J-1 nucleotide. This understanding has clear ramifications for structure-based drug design of therapeutics against Leishmania parasites. Recently, we discovered a process that we are studying in the context of several disease-related sporadic amyloidoses. We have shown that in vitro, certain inflammatory-derived lipidic aldehydes, when adducted to proamyloidogenic proteins in the proteins’ native state, can induce misfolding and aggregation of the native protein sequences. This past year, we expanded our studies to aggregation of antibody light chains. In vivo, such aggregation leads to the systemic deposition of immunoglobulin light-chain domains in the form of either amyloid fibrils (AL-amyloidosis) or amorphous deposits (light-chain deposition disease), mainly in cardiac or renal tissue, and is a pathologic condition that is often fatal. Molecular factors that may contribute to the propensity of antibody light chains to aggregate in vivo, such as the protein primary structure or local environment, are intensive areas of study. We have now shown that aggregation of human antibody κ (κ-MJM) and λ (λ-L155) light chains can be accelerated in vitro when they are incubated under physiologically relevant conditions in the presence of a panel of biologically relevant lipid-derived aldehydes: 4-hydroxynonenal, malondialdehyde, glyoxal, atheronal-A, and atheronal-B. Thioflavin-T and Congo red binding assays coupled with turbidity studies revealed that this aldehyde-induced aggregation can be associated with alteration of protein secondary structure to an increased β-sheet conformation. We found that the nature of the conformational change depends primarily on the lipidic aldehyde, not the protein sequence. Thus, the cholesterol 5,6-seco-sterols, atheronal-A and atheronal-B, caused amorphous aggregations that did not bind thioflavin-T or Congo red for both light chains, whereas 49 4-hydroxynonenal, malondialdehyde, and glyoxal induced aggregates that bound both thioflavin-T and Congo red. Transmission electron microscopy revealed that amyloid fibrils were formed during the aggregation of κ-MJM and λ-L155 light chains mediated by 4-hydroxynonenal (Fig. 1), whereas aggregates induced by atheronal-B were amorphous. F i g . 1 . Transmission electron microscopy image of fibrillar aggre- gates of antibody light chains induced by 4-hydroxynonenal in vitro. Kinetic profiles of light-chain aggregation revealed clear differences between the aldehydes. Atheronal-A and atheronal-B caused a classic nucleated polymerization-type aggregation, with a lag phase (~150 hours) followed by a growth phase that plateaued, whereas 4-hydroxynonenal, malondialdehyde, and glyoxal triggered a seeded-type aggregation that has no lag phase. Studies of the accelerated aggregation of κ-MJM and λ-L155 induced by 4-hydroxynonenal revealed a clear dependence on the concentration of the aldehyde and a process that can be inhibited by the naturally occurring osmolyte trimethylamine N-oxide. On the basis of these data, we think that our recently discovered model of protein misfolding induced by inflammatory aldehydes may now extend to aggregation of antibody light chains. PUBLICATIONS Nieva, J., Shafton, A., Altobell, L.J. III, Tripurenani, S., Rogel, J.K., Wentworth, A.D., Lerner, R.A., Wentworth, P., Jr. Inflammatory aldehydes accelerate antibody light chain amyloid and amorphous aggregation. Biochemistry 47:7695, 2008. 50 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Scanlan, C.N., Ritchie, G.E., Baruah, K., Harvey, D.J., Crispin, M.D., Singer, B.B., Lucka, L., Wormald, M., Wentworth, P., Jr., Zitzmann, N., Rudd, P.M., Burton, D.R., Dwek, R.A. Inhibition of mammalian glycan biosynthesis produces non-self antigens for broadly neutralising, HIV-1 specific antibody. J. Mol. Biol. 372:16, 2007. Scheinost, J.C., Boldt, G.E., Wentworth, P., Jr. The chemical biology of protein misfolding. In: Encyclopedia of Chemical Biology. Wiley-VCH, New York, in press. Scheinost, J.C., Wang, H., Boldt, G.E., Offer, J., Wentworth, P., Jr. Cholesterol secosterol-induced aggregation of methylated amyloid β-peptides—insights into aldehydeinitiated fibrillization of amyloid-β. Angew. Chem. Int. Ed. 47:3919, 2008. Temperinini, C., Cecchi, A., Boyle, N.A., Scozzafava, A., Cabeza, J.E., Wentworth, P., Jr., Blackburn, G.M., Supuran, C.T. Carbonic anhydrase inhibitors. Interaction of 2-N,N-dimethylamino-1,3,4-thiadiazole-5-methanesulfonamide with 12 mammalian isoforms: kinetic and x-ray crystallographic studies. Bioorg. Med. Chem. Lett. 18:999, 2008. Wentworth, P., Jr., Witter, D. Antibody-catalyzed water-oxidation pathway. Pure Appl. Chem. 80:1849, 2008. Pathway Engineering for Enzymatic Synthesis J.R. Williamson, W. Anderson, F. Agnelli, A. Beck, C. Beuck, A. Bunner, A. Carmel, S. Chen, S. Edgcomb, D. Kerkow, S. Kwan, E. Menicelli, W. Ridgeway, G. Ring, H. Schultheisz, L.G. Scott, Z. Shajani, E. Sperling, M. Sykes, B. Szymczyna, J. Wu nzymes are the protein factors in cells that are responsible for effecting chemical transformations of metabolites and macromolecules. Enzymes are responsible for synthesizing the thousands of small molecules in a cell that are necessary for a functioning metabolism and for synthesizJames R. Williamson, Ph.D. ing diverse natural products Professor that can have important medi- Molecular Biology cinal properties. Enzymatic synthesis in the laboratory is a powerful alternative to organic chemical synthesis for certain types of molecules. Because enzymes have complex 3dimensional folds, they can bind specifically to substrates and catalyze complex chemical reactions that are sometimes difficult to achieve with organic synthesis. A series of enzymes can be used simultaneously to effect a series of chemical reactions without isolation of the intermediate products. Thus, enzymatic synthesis is a powerful tool that can be applied to certain synthetic problems. E We are interested in the structure of RNA molecules and RNA-protein complexes that are important for translation or regulation of protein expression. One of the structural biology tools we use is nuclear magnetic resonance (NMR) spectroscopy, which can be used to determine the structure of macromolecules in solution. Application of NMR requires the incorporation of the stable isotope labels 13C and 15N, which can be difficult with RNA molecules. One aspect of our research program is developing methods to incorporate these stable isotope labels into RNA molecules, to enable structural studies. We have developed a flexible and powerful enzymatic synthesis of the purine nucleotides ATP and GTP. The method is, to our knowledge, the most complex and intricately engineered enzymatic synthesis that has been carried out in a laboratory to date. The process requires 28 enzymes, each of which was overproduced in Escherichia coli and purified before synthesis. The longest linear series of reactions has 19 sequential steps, but the process can be carried out with a yield of about 60%. The scheme requires input of 3 types of reagents (Fig. 1). First, the starting material substrates are incorporated into the final nucleotide product. Second, trace amounts of catalytic cofactors, such as NAD, glutamine, aspartate, and tetrahydrofolate, are supplied. The cofactors are recycled by using metabolic enzymes. Third, excess amounts of fuel reagents such as creatine phosphate and α-ketoglutarate are added to drive the recycling reactions and to drive the overall synthesis from the starting materials to the products (ATP or GTP). Almost every one of the 19 key steps involves recycling of a cofactor and consumption of fuel molecules. The overall synthesis results in the efficient synthesis of ATP or GTP starting from glucose, carbon dioxide, ammonia, and serine. These precursors are available in isotopically labeled forms, and a variety of different labeling patterns can be constructed by using different combinations of the labeled precursors. Using this approach, we were able to synthesize isotopically labeled ATP and GTP with novel labeling patterns that have useful properties for NMR analysis of RNA structure. Although preparing all of the enzymes for this biosynthetic approach is time-consuming, the resulting labeled nucleotides cannot easily be synthesized in any other way. The enzymatic synthesis approach we have developed should have broad applicability in the synthesis of other high-value biochemicals that can be used for structural biology or metabolic profiling experiments. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 51 Szymczyna, B.R., Gan, L., Johnson, J.E., Williamson, J.R. Solution NMR studies of the maturation intermediates of a 13 MDa viral capsid. J. Am. Chem. Soc. 129:7867, 2007. Tahmassebi, D.C., Williamson, J.R. Balancing teaching and research in obtaining a faculty position at a predominantly undergraduate institution. ACS Chem. Biol. 2:521, 2007. Williamson, J.R. Biophysical studies of bacterial ribosome assembly. Curr. Opin. Struct. Biol. 18:299, 2008. Williamson, J.R. Cooperativity in macromolecular assembly. Nat. Chem. Biol. 4:458, 2008. X-ray Crystallographic Studies of Therapeutically Important Macromolecular Targets I.A. Wilson, M.A. Adams, C.H. Bell, R.M.F. Cardoso, S. Connelly, B.J. Droese, D.C. Ekiert, M.-A. Elsliger, Z. Fulton, B.W. Han, M. Hong, M.J. Jimenez-Dalmaroni, R.N. Kirchdoerfer, R. Pejchal, G.P. Porter, A. Schiefner, F i g . 1 . One-pot de novo enzymatic synthesis of purine nucleotides. Glucose, carbon dioxide (CO2), ammonia (NH3), and serine, the stoichiometrically consumed reagents incorporated into products, are highlighted in yellow. A, Enzymes from the glycolytic pathway and purine biosynthetic pathways convert glucose into ATP and GTP. Intermediates are shown in the vertical sequence, and the abbreviation for each enzyme is shown in italics. Cofactors (e.g., ATP, NAD, THF) are shown to the left or the right of the main reaction sequence. The circular arrow symbols indicate the enzymatic regeneration of the cofactors, which are color coded for the reactions shown in the cofactor regeneration schemes (B). Red = ATP recycling, green = NAD recycling, purple = glutamine recycling, orange = aspartate recycling, blue = folate recycling. PUBLICATIONS Edgcomb, S.P., Aschrafi, A., Kompfner, E., Williamson, J.R., Gerace, L., Hennig. M. Protein structure and oligomerization are important for the formation of exportcompetent HIV-1 Rev-RRE complexes. Protein Sci. 17:420, 2008. Hennig, M., Scott, L.G., Sperling, E., Bermel, W., Williamson, J.R. Synthesis of 5-fluoropyrimidine nucleotides as sensitive NMR probes of RNA structure. J. Am. Chem. Soc. 129:14911, 2007. Kiessling, L.L., Doudna, J.A., Johnsson, K., Mapp, A.K., Marletta, M.A., Seeberger, P.H., Williamson, J.R., Wedde, S.G. A higher degree of difficulty. ACS Chem. Biol. 2:197, 2007. Naidoo, N., Harrop, S.J., Sobti, M., Haynes, P.A., Szymczyna, B.R., Williamson, J.R., Curmi, P.M.G., Mabbut, B.C. Crystal structure of Lsm3 octamer from Saccharomyces cerevisae: implications for Lsm ring organisation and recruitment. J. Mol. Biol. 377:1357, 2008. Schultheisz, H.L., Szymczyna, B.R., Scott, L.G., Williamson, J.R. Pathway engineered de novo enzymatic purine nucleotide synthesis. ACS Chem. Biol. 3:499, 2008. Sperling, E., Bunner, A., Sykes, M.T., Williamson, J.R. Quantitative analysis of isotope distributions in proteomic mass spectrometry using least-squares Fourier transform convolution. Anal. Chem. 80:4906, 2008. R.L. Stanfield, R.S. Stefanko, J.A. Vanhnasy, R. Xu, X. Xu, S.I. Yoon, X. Zhu H I V T Y P E 1 VA C C I N E D E S I G N IV type 1 (HIV-1) continues to constitute a major worldwide health threat, with approximately 2.1 million HIV-related deaths and 2.5 million new HIV-1 infections in 2007. Currently, more than Ian A. Wilson, D.Phil. 20 anti-HIV drugs approved by Professor the Food and Drug Administration Molecular Biology are on the market. Although these drugs can be effective at lowering the levels of circulating virus, they cannot completely eliminate the virus, are expensive, and must be taken daily for life. Clearly, an effective vaccine against HIV-1 is needed to control the rampant spread of this devastating pandemic. An effective vaccine likely must elicit a vigorous antibody response to block or neutralize viral infection. However, in many studies of patients infected with HIV-1, only a handful of potent, broadly neutralizing antibodies have been discovered that recognize a large percentage of the circulating viral strains. Our goal has been to understand how these rare antibodies are able to combat the virus. We are elucidating the 3-dimensional structures of these rare antibodies in complexes with the antibodies’ H 52 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 viral epitopes from the envelope proteins gp120 and gp41. The antibodies under study include b12, which recognizes a highly conserved, but deeply recessed pocket on gp120 that is the receptor-binding site for CD4; 2G12, which binds to a mannose-rich carbohydrate cluster on the normally immunologically silent face of gp120; several antibodies that recognize the hypervariable V3 region of gp120; and 4E10 and Z13e1, which interact with overlapping epitopes on gp41, just proximal to its membrane-spanning domain. Our original structural studies of antibody 2G12 in complex with mannose sugars have led to design of many nonnatural carbohydrates, peptides, and smallmolecule mimics for testing as potential immunogens. Recently, in collaboration with B. Davis, University of Oxford, Oxford, England, we determined a 2.8-Å crystal structure for 2G12 in complex with a novel, nonself mimic of the D1 arm of Man4/Man9GlcNac2, the type of carbohydrate commonly found on the gp120 silent face. This nonnatural mannose variant contains a C-6 methyl substitution of the mannose at the terminus of the D1 arm and inhibits binding of 2G12 to gp120 better than does the D1 arm itself. This compound is the first nonself D1 arm derivative to demonstrate inhibition of 2G12/gp120 binding better than that of the natural D1 arm. Preliminary diffraction data have also been collected from crystals of 2G12 in complex with a C-6 methyl monosaccharide compound. Further optimization of crystallization conditions for both complexes to obtain higher resolution diffraction data are under way. We are currently refining the crystal structure for Fab Z13e1, a neutralizing antibody that recognizes an epitope in the gp41 membrane proximal external region that overlaps the Fab 4E10 epitope (Fig. 1). Z13e1 has been evolved to have higher affinity than the parent antibody Z13; the higher affinity is due to 5 mutations in complementarity-determining region L3 (residues L90–L97). Although Z13e1 and 4E10 recognize similar epitopes, the antibody-bound conformations for the epitopes differ markedly, perhaps giving some insight to conformational changes that may occur during viral entry into cells. The differences in the key contact residues also correlate with the breadth and neutralization potency of 4E10 vs Z13e1. Our studies on HIV are done in collaboration with D.R. Burton, M. Zwick, R. Pantophlet, P.E. Dawson, and C.-H. Wong, Scripps Research; B. Davis, University of Oxford; L. Cavacini and J.K. Scott, Simon Fraser Univer- F i g . 1 . Structure of Fab Z13e1 in complex with its epitope peptide on the HIV-1 gp41 membrane proximal external region. The Fab is shown in a solid surface representation, with the light and heavy chains in cyan and blue, respectively. The peptide, shown in a balland-stick representation, binds primarily to the heavy chain of the antibody and adopts a different conformation than do the corresponding residues bound to broadly neutralizing antibody 4E10. This epitope occurs in gp41 just before the protein enters the membrane, so Z13e1 may have to contact the membrane to bind its epitope. sity, Burnaby, British Columbia; J. Moore, Weill Medical College of Cornell University, New York, New York; H. Katinger, R. Kunert, and G. Stiegler, University für Bodenkultur, Vienna, Austria; R. Wyatt and P. Kwong, Vaccine Research Center, National Institutes of Health, Bethesda, Maryland; W. Olson and K. Kang, Progenics Pharmaceuticals, Inc., Tarrytown, New York; the National Institutes of Health; and the Neutralizing Antibody Consortium of the International AIDS Vaccine Initiative. I N F L U E N Z A V I R U S G LY C O P R O T E I N S The 1918 influenza pandemic, which was responsible for more than 20 million deaths worldwide, and the more recent “bird flu,” with its even higher mortality rate (about 60% of patients in whom it is diagnosed), are constant reminders of the potential devastation that could ensue if a new influenza pandemic were to occur. To aid in the design of a vaccine to protect against such highly virulent strains of influenza, we are carrying out structural and functional studies of envelope proteins of influenza virus in complex with neutralizing antibodies to the virus. All known antibodies that neutralize influenza virus recognize the hemagglutinin viral envelope protein. In general, antibodies to hemagglutinin generally recognize highly variable epitopes at the membrane distal end of the hemagglutinin trimer. However, a small proportion of the host repertoire of antibodies is directed against other sites on hemagglutinin, including several antibodies that bind on the side of the hemagglutinin THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 trimer and recognize highly invariant epitopes. Several of these unusual antibodies neutralize the hemagglutinin of different strains and subtypes of influenza virus, both in vivo and in vitro. To gain insight into the mechanism of virus neutralization and the nature of the epitopes recognized, we are investigating several of these broadly neutralizing antibodies in complex with hemagglutinins that represent different pandemic strains and subtypes (H1, H2, H3) of human influenza virus as well as avian H5N1 influenza viruses. Understanding how these more broadly neutralizing antibodies interfere with viral entry and subsequent infection, as well as the nature of the highly conserved epitopes, will provide insights into the functional and antigenic constraints on the hemagglutinin of influenza virus. Currently, we are working with 2 Fabs, H5M11 and H5M9, that neutralize the H5N1 avian influenza virus A/Goose/Guangdong/1/96 and more recent avian strains that have infected humans and with antibodies to the influenza virus that were isolated from survivors of the 1918 pandemic by J. Crowe, Vanderbilt University, Nashville, Tennessee. The structure of Fab H5M11 has been determined, and we are working toward crystallization of other Fabs in complex with hemagglutinins from the H5N1 and 1918 H1N1 viruses. Hemagglutinin facilitates cell fusion through interactions with host membranes. Although crystal structures of hemagglutinin ectodomains have been extensively studied, little is known about the conformation or function of the membrane-interacting regions. We are working toward the determination of crystal structures of the full-length hemagglutinin in its states before and after fusion. In collaborative research with G. Tobin, Biological Mimetics, Inc., Frederick, Maryland, crystallization of the full-length hemagglutinin from A/Wyoming/3/03 (H3 subtype) and bacterial expression of the postfusion form of the protein are under way. We also propose to isolate and structurally analyze the hemagglutinin from the pandemic A/Japan/305/57 (H2 subtype) virus. These studies will advance our understanding of the mechanism of hemagglutinin-induced fusion and provide novel targets for design of fusion inhibitors. The crystal structure of the neuraminidase of the 1918 H1N1 virus has been determined to 1.45 Å. A large cavity in the active site in the neuraminidase offers new opportunities for structure-based drug design. Crystal structures of the 1918 neuraminidase in complex with the antiviral drugs oseltamivir (Tamiflu) and zanamivir (Relenza) show that the loop bordering the cavity is 53 extremely flexible in binding substrates, a characteristic that may indicate that the 1918 neuraminidase can bind more chemically diverse ligands than can neuraminidases from some other subtypes of the virus. This high-resolution structural information is being used for rational design of inhibitors against influenza virus. Additional collaborators in the influenza research include our colleagues in the flu consortium funded by the National Institute of Allergy and Infectious Diseases; scientists at Crucell, Leiden, the Netherlands; J. Crowe, Vanderbilt University; A. Lanzavecchia, Institute for Research in Biomedicine, Bellinzona, Switzerland; and X. Che, Southern Medical University China, Guangzhou, China. T H E I N N AT E I M M U N E R E S P O N S E A G A I N S T M I C R O B I A L PAT H O G E N S Toll-like receptors (TLRs) are glycoproteins that are essential for innate immune recognition of microbial pathogens. The TLR extracellular domains are horseshoe-shaped molecules consisting of leucine-rich repeat domains that begin and end with an N- and a C-terminal cap domain. Recently, we have been studying the TLR4, which plays an essential role in recognition and signaling of bacterial lipopolysaccharide. Among the TLR family members, TLR4 is unique in requiring another molecule, myeloid differentiation protein-2 (MD-2), for its function. MD-2 directly binds lipopolysaccharide and induces dimerization and activation of TLR4 for signaling. To provide insights into the structural mechanism used by lipopolysaccharide to activate TLR4, we have expressed the extracellular domain of TLR4 (sTLR4) in complex with MD-2 in a baculovirus expression system. Biophysical studies suggest that purified 1:1 complexes of sTLR4–MD-2 homodimerize to form 2:2 complexes in the presence of lipopolysaccharide. X-ray crystallographic studies are being carried out to reveal the structural architecture of the sTLR4–MD-2 assembly induced by lipopolysaccharide. Jawless fish, such as the lamprey, do not have immune receptors, such as antibodies, T-cell receptors, or MHC molecules, yet the fish still have an adaptive immune response to antigen. Recently, it was shown that cell-surface molecules, termed variable lymphocyte receptors (VLRs) are responsible for the adaptive immune response in jawless fish. These receptors resemble the mammalian innate system TLRs, with an overall horseshoe shape made up of a variable number of different leucine-rich repeat domains. In collaboration with M. Cooper, Emory University, Atlanta, Geor- 54 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 gia, we recently determined the first crystal structure of a VLR-antigen complex, RBC36, in complex with the H trisaccharide derived from the H antigen of human type O erythrocytes (Fig. 2). This structure reveals for the first time the location and nature of the VLR antigen-binding site. F i g . 3 . Crystal structure of CD1d with designed agonists. The ligand-presenting α1α2 platform of mouse CD1d is shown in gray, overlaid by the transparent molecular surface, with the bound superimposed ligands C6Ph (red), C8Ph (yellow), C8PhF (green), and the short α-galactosylceramide agonist PBS-25 (blue). The phenyl substitutions protrude deeply into the A′ pocket (see inset on right for view of ligands only). The 2 main A′ and F′ pockets in CD1d for ligand binding are indicated. A spacer lipid (likely palmitic acid) also partially fills the A′ pocket when the glycolipid ligand itself does not fully occupy the pocket. The Cα positions of the ligands show an overall root mean square displacement of 0.42 Å. F i g . 2 . Structure of the lamprey VLR red blood cell in complex with its H trisaccharide antigen. Left, The VLR is shown as a solid surface, with the antigen depicted in a ball-and-stick representation. Right, The same view of the structure but highlighting the secondary structural elements of the VLR. The antigen binds to the concave side of the horseshoe-shaped VLR and is cradled on one side by a large fingerlike insert in the C-terminal leucine-rich repeat. The CD1 family of innate receptors consists of MHC class I–like, antigen-presenting molecules that present lipids, glycolipids, and lipopeptides to effector T cells. The receptors are expressed on antigen-presenting cells and are involved in host defense and in immunoregulatory functions. Glycolipids presented by CD1d are capable of stimulating natural killer T cells. Natural killer T cells are of clinical interest because when stimulated by CD1, they rapidly secrete a number of cytokines that either promote or suppress different immune responses. One of the most potent agonists for natural killer T cells is α-galactosylceramide. On the basis of our structural studies during the past 5 years, a series of glycolipids have been synthesized by scientists in the laboratory of C.-H. Wong, Skaggs Institute. These new ligands, which have phenyl ring substitutions in the fatty acid part of α-galactosylceramide, are more potent than the native ceramide and have an altered efficacy in T-cell assays. We have now determined the structures for 3 of the most stimulating glycolipids in complex with CD1d. Our analysis revealed that only minor structural changes occur in the A′ pocket (Fig. 3). The phenyl-ring derivatives have better packing than the native α-galactosyl- ceramide and thereby increase the overall stability of the CD1d-ligand complexes. PUBLICATIONS Astronomo, R.D., Lee, H.K., Scanlan, C.N., Pantophlet, R., Huang, C.Y., Wilson, I.A., Blixt, O., Dwek, R.A., Wong, C.H., Burton, D.R. A glycoconjugate antigen based on the recognition motif of a broadly neutralizing human immunodeficiency virus antibody, 2G12, is immunogenic but elicits antibodies unable to bind to the self glycans of gp120. J. Virol. 82:6359, 2008. Bell, C.H., Pantophlet, R., Schiefner, A., Cavacini, L.A., Stanfield, R.L., Burton, D.R., Wilson, I.A. Structure of antibody F425-B4e8 in complex with a V3 peptide reveals a new binding mode for HIV-1 neutralization. J. Mol. Biol. 375:969, 2008. Burton, D.R., Wilson, I.A. Immunology: square-dancing antibodies. Science 317:1507, 2007. Debler, E.W., Kaufmann, G.F., Meijler, M.M., Heine, A., Mee, J.M., Pljevaljcic, G., Di Bilio, A.J., Schultz, P.G., Millar, D.P., Janda, K.D., Wilson, I.A., Gray, H.B., Lerner, R.A. Deeply inverted electron-hole recombination in a luminescent antibody-stilbene complex. Science 319:1232, 2008. Debler, E.W., Müller, R., Hilvert, D., Wilson, I.A. Conformational isomerism can limit antibody catalysis. J. Biol. Chem. 283:16554, 2008. Demartino, J.K., Hwang, I., Connelly, S., Wilson, I.A., Boger, D.L. Asymmetric synthesis of inhibitors of glycinamide ribonucleotide transformylase. J. Med. Chem. 51:5441, 2008. Dhillon, A.K., Stanfield, R.L., Gorny, M.K., Williams, C., Zolla-Pazner, S., Wilson, I.A. Structure determination of an anti-HIV-1 Fab 447-52D-peptide complex from an epitaxially twinned data set. Acta Crystallogr. D Biol. Crystallogr. 64:792, 2008. Huang, C.C., Lam, S.N., Acharya, P., Tang, M., Xiang, S.H., Hussan, S.S., Stanfield, R.L., Robinson, J., Sodroski, J., Wilson, I.A., Wyatt, R., Bewley, C.A., Kwong, P.D. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317:1930, 2007. Johnson, S.M., Connelly, S., Wilson, I.A., Kelly, J.W. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J. Med. Chem. 51:260, 2008. Menendez, A., Calarese, D.A., Stanfield, R.L., Chow, K.C., Scanlan, C.N., Kunert, R., Katinger, H., Burton, D.R., Wilson, I.A., Scott, J.K. A peptide inhibitor of HIV1 neutralizing antibody 2G12 is not a structural mimic of the natural carbohydrate epitope on gp120. FASEB J. 22:1380, 2008. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Relloso, M., Cheng, T.Y., Im, J.S., Parisini, E., Roura-Mir, C., DeBono, C., Zajonc, D.M., Murga, L.F., Ondrechen, M.J., Wilson, I.A., Porcelli, S.A., Moody, D.B. pHdependent interdomain tethers of CD1b regulate its antigen capture. Immunity 28:774, 2008. Stevens, J., Blixt, O., Chen, L.M., Donis, R.O., Paulson, J.C., Wilson, I.A. Recent avian H5N1 viruses exhibit increased propensity for acquiring human receptor specificity. J. Mol. Biol. 381:1382, 2008. Verdino, P., Aldag, C., Hilvert, D., Wilson, I.A. Closely related antibody receptors exploit fundamentally different strategies for steroid recognition. Proc. Natl. Acad. Sci. U. S. A. 105:11725, 2008. Wei, C.J., Xu, L., Kong, W.P., Shi, W., Canis, K., Stevens, J., Yang, Z.Y., Dell, A., Haslam, S.M., Wilson, I.A., Nabel, G.J. Comparative efficacy of neutralizing antibodies elicited by recombinant hemagglutinin proteins from avian H5N1 influenza virus. J. Virol. 82:6200, 2008. Zajonc, D.M., Savage, P.B., Bendelac, A., Wilson, I.A., Teyton, L. Crystal structures of mouse CD1d-iGb3 complex and its cognate Vα14 T cell receptor suggest a model for dual recognition of foreign and self glycolipids. J. Mol. Biol. 377:1104, 2008. Zajonc, D.M., Wilson, I.A. Architecture of CD1 proteins. Curr. Top. Microbiol. Immunol. 314:27, 2007. Zhu, X., Xu, X., Wilson, I.A. Structure determination of the 1918 H1N1 neuraminidase from a crystal with lattice-translocation defects. Acta Crystallogr. D Biol. Crystallogr. 64:843, 2008. Bioorganic and Synthetic Chemistry C.-H. Wong, C. Bennett, S. Dean, S. Ficht, Y. Fu, W. Greenberg, R. Guy, S. Hanson, Z. Hong, D.-R. Hwang, J.-C. Lee, P.-H. Liang, R. Payne, M. Schelwies, S.-K. Wang e develop new chemical and enzymatic strategies for synthesis of bioactive small molecules and biomolecules. We use the methods to probe carbohydrate-mediated recognition events important in cancer, bacterial infections, and viral infections, including HIV disease and influenza. W Chi-Huey Wong, Ph.D. Professor Chemistry SYNTHETIC METHODS We have developed new methods for sugar-assisted ligation of glycopeptides for synthesis of homogenous glycoproteins. We have used the methods in conjunction with enzymatic glycosylation techniques to assemble complex glycopeptides by chemical synthesis, and we are optimizing the techniques to achieve the total synthesis of therapeutic glycoproteins. Glycoproteins are expressed in vivo as complex mixtures of glycoforms, a situation that hinders efforts to study the role of glyco- 55 sylation in protein folding, stability, and function. By synthesizing pure glycoforms, we can characterize in molecular detail the effects of glycans on protein function. Using chemical techniques such as programmable 1-pot oligosaccharide synthesis, as well as enzymatic synthesis, we create glycoarrays on glass slides for high-throughput quantitative analysis of protein-carbohydrate interactions. These arrays are being used to study the binding specificity of carbohydrate-binding receptors and antibodies. We have applied aldolases, glycosyltransferases, glycosidases, and other enzymes to develop practical new methods of synthesizing molecules such as iminocyclitols, which are inhibitors of glycosidases and other enzymes; glycopeptides; and other glycoconjugates. Using directed evolution, we are evolving these enzymes to catalyze new reactions and synthesize new molecules of pharmaceutical relevance. C A R B O H Y D R AT E - M E D I AT E D R E C O G N I T I O N I N D I S E A S E We are using our synthetic methods to discover inhibitors and therapeutic agents in several diseases related to carbohydrates. Current targets include bacterial transglycosidase, sulfatases, and glycoprocessing enzymes involved in the biosynthesis of carbohydrates that mediate cancer metastasis, inflammation, and viral infection. Enzymatically synthesized iminocyclitols are being investigated as treatments for osteoarthritis and Gaucher disease. Inspired by the broadly neutralizing anti-HIV antibody 2G12, which recognizes a dense array of oligomannose displayed on HIV gp120, we are designing dendrimeric oligomannose structures for development of an HIV vaccine. In collaboration with D.R. Burton, Scripps Research, we are testing the immunogenicity of these constructs. We have designed glycolipid ligands for CD1 that activate natural killer T cells and are a promising new immunotherapeutic approach for treatment of bacterial and viral infections and cancer. The ligands may also be useful as adjuvants in vaccine development. G LY C O P R O T E O M I C S A N D M O L E C U L A R G LY C O B I O L O G Y Using metabolic oligosaccharide engineering, we have developed methods for incorporating tagged sugars into glycans expressed on mammalian cells. The engineered glycans can be labeled with a variety of molecules by using click chemistry. One application is glycan-specific fluorescent labeling, which is used for fluorescent imaging to compare glycosylation patterns of different cells, such as normal vs cancer cells or cancer cells vs cancer stem cells. We found that protein fucosylation and sialylation are both elevated in cancer cell lines. 56 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 A second application of this chemistry is GIDmap, a new method for glycoproteomic analysis (Fig. 1). Hanson, S.R., Greenberg, W.A., Wong C.-H. Probing glycans with the copper(I)catalyzed [3+2] azide-alkyne cycloaddition. QSAR Comb. Sci. 26:1243, 2007. Kinjo, Y., Pei, B., Bufali, S., Raju, R., Richardson, S.K., Imamura, M., Fujio, M., Wu, D., Khurana, A., Kawahara, K., Wong, C.-H., Howell, A.R., Seeberger, P.H., Kronenberg, M. Natural Sphingomonas glycolipids vary greatly in their ability to activate natural killer T cells. Chem. Biol. 15:654, 2008. Liang, P.-H., Imamura, M., Li, X., Wu, D., Fujio, M., Guy, R.T., Wu, B.-C., Tsuji, M., Wong, C.-H. Quantitative microarray analysis of intact glycolipid-CD1d interaction and correlation with cell-based cytokine production. J. Am. Chem. Soc. 130:12348, 2008. Liang, P.-H., Wu, C.-Y., Greenberg, W.A., Wong C.-H. Glycan arrays: biological and medical applications. Curr. Opin. Chem. Biol. 12:86, 2008. Northen, T.R., Lee, J.-C., Hoang, L., Raymond, J., Hwang, D.-R., Yannone, S.M., Wong, C.-H., Siuzdak, G. A nanostructure-initiator mass spectrometry-based enzyme activity assay. Proc. Natl. Acad. Sci. U. S. A. 105:3678, 2008. Payne, R.J., Ficht, S., Greenberg, W.A., Wong, C.-H. Cysteine-free peptide and glycopeptide ligation by direct aminolysis. Angew. Chem. Int. Ed. 47:4411, 2008. Wang, S.-K., Liang, P.-H., Astronomo, R.D., Hsu, T.-L., Hsieh, S.-L., Burton, D.R., Wong, C.-H. Targeting the carbohydrates on HIV-1: interaction of oligomannose dendrons with human monoclonal antibody 2G12 and DC-SIGN. Proc. Natl. Acad. Sci. U. S. A. 105:3690, 2008. Whalen, L.J., Greenberg, W.A., Mitchell, M.L., Wong, C.-H. Iminosugar-based glycosyltransferase inhibitors. In: Iminosugars: From Synthesis to Therapeutic Applications. Compain, P., Martin, O.R. (Eds.). Wiley-VCH, Hoboken, NJ, 2007, p. 153. Wu, D., Fujio, M., Wong, C.-H. Glycolipids as immunostimulating agents. Bioorg. Med. Chem. 16:1073, 2008. F i g . 1 . GIDmap glycoproteomic analysis via metabolic oligosaccharide engineering. Whole cells are fed with tagged sugars, and after biochemical incorporation into cellular glycoproteins, click chemistry is used to attach a handle for purification of tagged proteins. Mass spectrometric proteomic methods are then used to identify proteins that are differentially glycosylated. We are using GIDmap to identify proteins that are aberrantly glycosylated in different stages of cancer. These cancer-associated glycoproteins may be useful as biomarkers for diagnostics or as targets for therapeutic intervention. PUBLICATIONS Astronomo, R.D., Lee, H.K., Scanlan, C.N., Pantophlet, R., Huang, C.Y., Wilson, I.A., Blixt, O., Dwek, R.A., Wong C.-H., Burton D.R. A glycoconjugate antigen based on the recognition motif of a broadly neutralizing human immunodeficiency virus antibody, 2G12, is immunogenic but elicits antibodies unable to bind to the self glycans of gp120. J. Virology 82:6359, 2008. Bennett, C.S., Dean, S.M., Payne, R.J., Ficht, S., Brik, A., Wong, C.-H. Sugarassisted glycopeptide ligation with complex oligosaccharides: scope and limitations. J. Am. Chem. Soc. 130:11945, 2008. Ficht, S., Payne, R.J., Guy, R.T., Wong, C.-H. Solid-phase synthesis of peptide and glycopeptide thioesters through side-chain-anchoring strategies. Chem. Eur. J. 14:3620, 2008. Giffin, M.J., Heaslet, H., Brik, A., Lin, Y.-C., Cauvi, G., Wong, C.-H., McRee, D.E., Elder, J.H., Stout, C.D., Torbett, B.E. A copper(I)-catalyzed 1,2,3-triazole azide-alkyne click compound is a potent inhibitor of a multidrug-resistant HIV-1 protease variant. J. Med. Chem. 51:6263, 2008. Studies of Macromolecular Recognition by Multidimensional Nuclear Magnetic Resonance P.E. Wright, H.J. Dyson, M. Martinez-Yamout, M. Arai, S.-H. Bae, D. Boehr, B. Buck-Koehntop, P. Deka, D. Felitsky, J. Ferreon, P. Haberz, C.W. Lee, D. Meinhold, S.-J. Park, M. Landes, E. Manlapaz pecific interactions between molecules are of fundamental importance in all biological processes. An understanding of how biological macromolecules such as proteins and nucleic acids recognize each other is essential for understanding the fundamental molecular events of life. Knowledge Peter E. Wright, Ph.D. of the 3-dimensional structures Professor and Chairman of biological macromolecules Molecular Biology is key to understanding their interactions and functions and also forms the basis for rational design of new drugs. A particularly powerful S THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 method for mapping the 3-dimensional structures and interactions of biological macromolecules in solution is multidimensional nuclear magnetic resonance (NMR) spectroscopy. We are using this method to study a number of protein-protein and protein–nucleic acid interactions of fundamental importance in health and disease. Transcriptional regulation in eukaryotes relies on protein-protein interactions between DNA-bound factors and coactivators that, in turn, interact with the basal transcription machinery. A major effort in our laboratory is focused on elucidating the structural principles that determine specificity of key protein-protein interactions involved in regulation of gene expression. The transcriptional coactivator CREB-binding protein (CBP) and its ortholog p300 play a central role in cell growth, differentiation, and development in higher eukaryotes. CBP and p300 mediate interactions between a number of gene regulatory proteins and viral proteins, including proteins from several tumor viruses and hepatitis B virus. Understanding the molecular mechanisms by which CBP recognizes its various target proteins is of fundamental biomedical importance. CBP has been implicated in diverse human diseases such as leukemia, cancer, and mental retardation and is a novel target for therapeutic intervention. We have initiated a major program to determine the structure of CBP and p300 and map their functional interactions with other components of the transcriptional machinery. Our research reveals that many regions of these coactivators are intrinsically disordered, as are many of the transcriptional regulatory proteins with which they interact. Indeed, our results have indicated that coupled folding and binding processes play a major role in transcriptional regulation. We have performed NMR relaxation experiments to elucidate the mechanism of coupled folding and binding processes and to identify “hot spots” in protein-protein interfaces that could potentially be targeted by smallmolecule inhibitors. We initially used these methods to investigate the interactions involved in the regulation of hypoxia, namely binding of the α-subunit of the hypoxiainducible transcription factor (HIF-1α) to the TAZ1 zinc finger motif of CBP/p300. We have now extended these relaxation measurements to the complex formed between the activation domain of the p160 nuclear receptor coactivator ACTR and the nuclear coactivator binding domain of CBP. Both proteins are intrinsically disordered and fold synergistically upon binding. Although the free proteins are highly flexible, the complex has the motional characteristics of a globular protein domain, 57 with no significant residual flexibility that might compensate for the loss of entropy incurred upon formation of a complex. Some years ago, we determined the 3-dimensional structure of the phosphorylated kinase inducible activation domain (pKID) of the transcription factor CREB bound to its target domain (the KIX domain) in CBP. The structure provides a starting point for design of small molecules that can inhibit the CREB-KIX interactions, an important goal in development of novel therapeutics for treatment of diabetes. We have developed a new method, using R2 relaxation dispersion experiments and NMR titrations, to investigate the pathway by which intrinsically disordered proteins fold into ordered structures upon binding to their biological targets. We have used this method to study the mechanism of pKID binding to KIX. The pKID first forms an ensemble of transient encounter complexes at multiple sites on the surface of KIX and then folds via a pathway involving a partially structured intermediate. Folding of the pKID helices occurs on the surface of KIX; the mechanism of recognition involves an induced protein folding event, rather than selection of a small population of prefolded helical structures from the solution conformational ensemble. We have also used the method to study mechanisms of binding of the hydroxylated HIF-1α transactivation domain to the TAZ1 domain of CBP and have commenced studies of the binding of the proto-oncogene cMyb to the KIX domain of CBP. This research is leading to a new understanding of the molecular mechanisms by which intrinsically disordered proteins perform their diverse biological functions. In the course of these studies, we have developed novel methods for measuring the affinities with which intrinsically disordered proteins bind to their targets (Fig. 1). F i g . 1 . Global fit of chemical-shift titration data to obtain accu- rate dissociation constants. 58 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 CBP and p300 contain several zinc-binding domains (ZZ domain, PHD motif, TAZ1 and TAZ2 domains) that mediate critical interactions with numerous transcriptional regulators. We have determined the structures of each of these domains during recent years. Our current efforts are focused on structural analysis of the complexes formed between the TAZ1 and TAZ2 domains and the activation domains of the numerous transcription factors with which the TAZ1 and TAZ2 domains interact. We have determined the structures of the complexes formed between the TAZ domains and the activation domains of the signal transducer and activator of transcription (STAT) family of transcriptional regulators. These interactions play a key role in cytokinedependent signal transduction. Structures have been determined for the complex of TAZ1 with the STAT2 activation domain and for TAZ2 bound to STAT1 (Fig. 2). phorylation of the p53 activation domain inhibits binding of HDM2 and enhances binding to CBP/p300, thereby stabilizing p53 and activating transcription of p53-regulated genes. Our findings provide novel insights into the mechanism of p53 regulation in response to DNA damage and genotoxic stress. In addition, we have determined the structures of the complexes formed between the KIX domain of CBP and the p53 activation domain and between the TAZ2 domain of CBP and the adenoviral oncoprotein E1A. Finally, we have made major advances in understanding the mechanism by which the zinc finger protein muscleblind recognizes both pathogenic doublestranded repeat RNA sequences and single-stranded regulatory RNA elements. Sequestration of muscleblind by CUG- and CCUG-repeat RNA disrupts alternate RNA splicing and is the underlying molecular cause of myotonic dystrophy, the most common form of adult-onset muscular dystrophy. We have determined the structure of the first 2 zinc fingers of muscleblind, which fold into a unique globular structure (Fig. 3), and we have mapped their interactions with single-stranded RNA. We have identified the specific RNA sequence required for high-affinity binding and are currently working on the structure of the RNA complex. F i g . 2 . Structures of the TAZ1-STAT2 complex (A) and the TAZ2- STAT1 complex (B). The protein backbones of the STAT activation domains are shown as pink ribbons; the backbones of the TAZ1 and TAZ2 domains, as blue and green ribbons, respectively. The STAT1 and STAT2 activation domains are intrinsically disordered and fold upon binding to the TAZ motifs, burying a large surface area and forming a hydrophobic intermolecular core. The different structural features of the TAZ1 and TAZ2 scaffolds dictate the conformation and sites of binding of the STAT2 and STAT1 motifs. CBP and p300 play a critical role in the regulation of the tumor suppressor p53. They interact directly with p53 and are required for p53-mediated transcriptional activation. They also function to regulate p53 stability. We have used NMR spectroscopy and isothermal titration calorimetry to investigate the binding interactions between the transcriptional activation domain of p53 and its target domains in CBP/p300. We found that the p53 activation domain can bind simultaneously to CBP/p300 and the ubiquitin ligase HDM2, which regulates p53 stability, to form a ternary complex. Phos- F i g . 3 . Ribbon representation of the structure of muscleblind zinc fingers. PUBLICATIONS Boehr, D.D., Dyson, H.J., Wright, P.E. Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry 47:9227, 2008. Boehr, D.D., Wright, P.E. How do proteins interact? Science 320:1429, 2008. Ebert, M.-O., Bae, S.-H. Dyson, H.J., Wright, P.E. NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry 47:1299, 2008. Felitsky, D.J., Lietzow, M.A., Dyson, H.J., Wright, P.E. Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc. Natl Acad. Sci. U. S. A. 105:6278, 2008. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Sugase, K., Landes, M.A., Wright, P.E., Martinez-Yamout, M.A. Overexpression of post-translationally modified peptides in Escherichia coli by co-expression with modifying enzymes. Protein Expr. Purif. 57:108, 2008. Sugase, K., Lansing, J.C., Dyson, H.J., Wright, P.E. Tailoring relaxation dispersion experiments for fast-associating protein complexes. J. Am. Chem. Soc. 129:13406, 2007. Automation of Nuclear Magnetic Resonance Structure Determination of Proteins in Solution 59 and further add to the reliability of the results obtained. To increase automation of NMR structure determination, a research team directed by me at the ETH Zürich, Zürich, Switzerland, developed new software and new NMR experiments. In the context of our work in structural genomics as part of the Joint Center for Structural Genomics (www.jcsg.org), we have now assembled these software modules into a new protocol for structure determination that includes extensive automation. In Figure 1 showing the newly introduced automated NMR protocol for structure determination, 2 key features of the procedure are in red. First, a novel approach K. Wüthrich, B. Pedrini, P. Serrano, B. Mohanty, R. Horst e use nuclear magnetic resonance (NMR) spectroscopy in solution for studies in structural biology and structural genomics. The following are 2 illustrations of current applications: structural characterization of the proteome of the coronavirus that causes severe Kurt Wüthrich, Ph.D. acute respiratory syndrome, Professor which is pursued under the Molecular Biology auspices of the Center for Functional and Structural Proteomics of the SARS Coronavirus (FSPS; http://visp.scripps.edu/SARS/default.aspx), and studies of chaperone-mediated protein folding, which is a collaboration with A. Horwich, a guest scientist at Scripps Research from Yale University, New Haven, Connecticut. In an effort to continually enhance the significance of the NMR observations and the efficiency with which NMR structures can be solved, developing methods is an important part of our activities. During the past year, the team members supported in part or entirely by funds from the Skaggs Institute have made important contributions to new and improved NMR approaches. Because of the important role of NMR in drug discovery and drug design, these developments bear directly on many aspects of biomedical research. Currently, NMR determinations of protein structure in solution are typically performed by experienced spectroscopists who use interactive informatics tools. Increased use of fully automated steps in structure determination promises to increase the efficiency of the procedure W F i g . 1 . Protocol for automated NMR structure determination. to quality assessment of the protein solutions intended for NMR structure determination is introduced in the form of the “NMR profile.” The NMR profile enables a quantitative assessment of the suitability of the sample for the use of different NMR techniques for assignments of the polypeptide backbone. Second, the protocol includes fully automated structure determination that leads reliably to an accurate determination of the polypeptide backbone fold. Two additional important aspects of the new protocol are as follows: Once the polypeptide backbone assignment has been obtained, the additional information needed for assigning the amino acid side chains and the structure calculation are obtained from the same heteronuclear-resolved [ 1H,1H]-nuclear Overhauser enhancement spectroscopy (NOESY) data sets, a step that ensures high internal consistency of the entire procedure. The protocol contains 2 important interactive steps to ensure (1) completeness of the polypeptide backbone assignments and (2) refinement and validation of the automatically solved structure. We have so far applied the protocol to a number of different target proteins. As an illustration, Figure 2 shows 60 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 the NMR structure of the hypothetical protein TM0212 from Thermotoga maritima. For this protein and several other proteins, the automated part of the structure determination, leading to the accurate description of the F i g . 2 . Stereoview of the protein TM0212 from T maritima determined by using the protocol of Figure 1. Color scheme: polypeptide backbone, gray; well-structured amino acid side chains, yellow; other amino acid side chains, blue. polypeptide backbone fold (see Fig. 1), was achieved within 1 week. PUBLICATIONS Johnson, M.A., Southworth, M.W., Herrmann, T., Brace, L., Perler, F.B., Wüthrich, K. NMR structure of a Klba intein precursor from Methanococcus jannaschii. Protein Sci. 16:1316, 2007. Pedrini, B., Placzek, W.J., Koculi, E., Alimenti, C., LaTerza, A., Luporini, P., Wüthrich, K. Cold-adaptation in sea-water-borne signal proteins: sequence and NMR structure of the pheromone En-6 from the antarctic ciliate Euplotes nobilii. J. Mol. Biol. 372:277, 2007. Placzek, W.J., Almeida, M.S., Wüthrich, K. NMR structure and functional characterization of a human cancer-related nucleoside triphosphatase. J. Mol. Biol. 367:788, 2007. Placzek, W.J., Etezady-Esfarjani, T., Herrmann, T., Pedrini, B., Peti, W., Alimenti, C., Luporini, P., Wüthrich, K. Cold-adapted signal proteins: NMR structures of pheromones from the antarctic ciliate Euplotes nobilii. IUBMB Life 59:578, 2007. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Staff Awards and Activities 61 Kelly, J.W.— Vincent du Vigneaud Award, American Pep- tide Society. Barbas, C.F. III— Arthur C. Cope Scholar Award, American Lerner, R.A.— Jesse W. Beams Memorial Lecture in Bio- Chemical Society; Tetrahedron Young Investigator Award, Bioorganic and Medicinal Chemistry; Fellow, American Association for the Advancement of Science; In-Cites Highly Cited Researcher, Thomson Scientific, Philadelphia, Pennsylvania; Member, Faculty in Chemical Biology, Faculty of 1000, Biology Reports, Ltd.; Editorial Boards, Bioorganic and Medicinal Chemistry Letters, Bioorganic and Medicinal Chemistry. physics, University of Virginia, Charlottesville, Virginia; K.T. Wang Bioorganic Chemistry Lectureship, Taipei, Taiwan; Editorial Boards, Bioorganic and Medicinal Chemistry, Bioorganic and Medicinal Chemistry Letters, Catalysis Technology, Drug Targeting and Delivery, Journal of Virology, Molecular Biology and Medicine, Molecular Medicine, Vaccine, Angewandte Chemie. Bartfai, T.—Member, Royal Swedish Academy of Sciences, Lifetime Achievement in Hematology, American Society of Hematology; AABB Karl Landsteiner Award and Lectureship; Donald I. Feinstein Distinguished Lecturer, University of Southern California, Los Angeles, California; Member, National Academy of Sciences, Institute of Medicine of the National Academies, American Academy of Arts and Sciences; Chairman, Scientific Advisory Board, Burnham Institute for Medical Research; Member, Scientific Advisory Boards, Edwards Lifesciences, iMetrikus, Optimer Pharmaceuticals; Contributing Editor, Blood Cells, Molecules and Diseases; Associate Editor, Acta Haematologica. Meeting, American Chemical Society; August-Wilhelmvon-Hofmann-Denkmünze Award; Marvel Lecturer, University of Illinois at Urbana, Champaign, Illinois; Creigee Lecturer, University of Karlsruhe, Germany; Behringer Simon Lecturer, ETH, Zürich, Switzerland; Co-Editor-in-Chief, Chemistr y and Biology; Editorial Boards, Tetrahedron Publications, Synthesis, Carbohydrate Letters, Chemistry—A European Journal, Perspectives in Drug Discovery and Design, Indian Journal of Chemistry, Section B, Combinatorial Chemistry HighThroughput Screening, Current Opinion in Bioorganic Chemistry, Current Organic Chemistry, Organic Letters, ChemBioChem, Chemistry and Biodiversity, Bulletin for the Chemical Society of Japan, Chemistry—An Asian Journal, International Journal of Oncology. Boger, D.L.—Nieuwland Lecturer, Notre Dame Univer- Rebek, J., Jr.—Ta-shue Chou Award, Academia Sinica, sity, Norte Dame, Indiana; Wyeth Lecturer, University of Strathclyde, Glasgow, Scotland; Editor-in-Chief, Bioorganic and Medicinal Chemistry Letters; Editorial Boards, Tetrahedron Publications, Organic Reactions, Current Opinion in Drug Discovery and Development, Current Drugs. Taiwan; Joullie Lecturer, University of Pennsylvania, Philadelphia, Pennsylvania; Marker Lecture, University of Maryland, College Park, Maryland; Member, Wittgenstein Prize Committee; Editorial Boards, Chemistry and Biology, Current Opinion in Chemistry and Biology, Journal of Supramolecular Chemistry. Chemistry Section. Beutler, E.— First Annual Wallace H. Coulter Award for Fedor, M.J.— Chair, Macromolecular Structure and Func- tion Study Section E, National Institutes of Health; Associate Editor, Journal of Biological Chemistry; Editorial Board, RNA. Janda, K.D.— Section Head, Faculty in Chemical Biol- ogy, Faculty of 1000, Biology Reports, Ltd.; American Regional Editor, Bioorganic and Medicinal Chemistry; Editorial Boards, Chemical Reviews, Journal of Medicinal Chemistry, Combinatorial Chemistry Research and Applications, Bioorganic and Medicinal Chemistry Letters, Combinatorial Chemistry High-Throughput Screening. Joyce, G.F.— Member, National Academy of Sciences; Member, Committee on International Security and Arms Control, National Academy of Sciences; Member, External Advisory Board, Beckman Institute, California Institute of Technology, Pasadena, California; Head, Faculty in Chemical Biology, Faculty of 1000, Biology Reports, Ltd.; Associate Editor, Evolutionary Computation, Origins of Life and Evolution of the Biosphere. Nicolaou, K.C.— Award of Excellence, Western Regional Sharpless, K.B.— Honorary Doctorate, Hong Kong Uni- versity of Science and Technology, Hong Kong, China; Nobel Laureate Lectures, Griffith University, Brisbane, Australia; Editorial Boards, Advanced Synthesis and Catalysis, Arkivoc, Beilstein Journal of Organic Chemistry, Bulletin of the Chemical Society of Japan, Chemistry—An Asian Journal, Chirality, Current Opinion in Drug Discovery and Development, Current Drug Discovery Technologies, Enantiomer, Organic Letters, Synlett, Topics in Stereochemistry. Williamson, J.R.—Associate Editor, Annual Reviews of Biophysics and Biomolecular Structure; Editorial Boards, ACS Chemical Biology, RNA, Molecular Cell, Chemistry and Biology, Structure. Wilson, I.A.— Honorary Doctorate of Science, University of St. Andrews, Scotland; Fellow, Royal Society of London; Corresponding Fellow, Royal Society of Edinburgh; Member, American Academy of Arts and Sciences; Member, Board of Directors, Keystone Symposia; Associate 62 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Editor, Journal of Molecular Biology, Immunity; Editorial Boards, Science, Journal of Experimental Medicine. Wong, C.-H.— F.A. Cotton Medal for Excellence in Chem- ical Research, American Chemical Society; Scientific Advisor, Max-Planck-Institute, Dortmund, Germany; Editor-in-Chief, Bioorganic and Medicinal Chemistry; Chair, Executive Board of Editors, Tetrahedron Publications; Editorial Boards, Current Opinion in Chemical Biology, Biocatalysis, Advanced Synthesis and Catalysis. Wright, P.E.— Leach Medal, Lorne Conference on Pro- tein Structure and Function, Lorne, Australia; Fellow, International Society of Magnetic Resonance; Member, National Academy of Sciences; Editor-in-Chief, Journal of Molecular Biology; Editorial Boards, Biochemistry, Current Opinion in Structural Biology, Journal of Biomolecular NMR. Wüthrich, K.— Doctor of Medicine honoris causa, Univer- sity of Pécs, Hungary; Doctor of Chemistry honoris causa, Universidad del Norte, Asunción, Paraguay; Doctor honoris causa, Lomonosov Moscow State University, Moscow, Russia; Laurea Specialistica in Biotechnology honoris causa, University of Verona, Italy; Docteur honoris causa, Université René Descartes, Paris, France; Irving L. Schwartz Lecture, Mount Sinai School of Medicine, New York, New York; Aline U. and James M. Orten Memorial Lecture, Wayne State University, Detroit, Michigan; Philip Handler Lecture, Duke University Medical Center, Durham, North Carolina; Editor-in-Chief, Journal of Biomolecular NMR; Associate Editor, Advanced Science Letters; Editorial Boards, Biochimie, Biomolecular NMR Assignments, Biopolymers, ChemBioChem, Chemical Physics Letters, Current Opinion in Structural Biology, IUBMB Life, Journal of Magnetic Resonance, Journal of Membrane Biology, Journal of Structural and Functional Genomics, Proteins, Structure. THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 AUTHOR INDEX Boldface entries indicate principal investigators who are members of the Skaggs Institute. The other authors are colleagues and support staff who work closely with the members. Adams, M.A. 51 Agnelli, F. 50 Agua, A. 38 Ajami, D. 40 Albertshofer, K. 11 Aller, S. 17 Ams, M. 40 Anderson, E. 15 Anderson, W. 50 Ando, Y. 15 Angrish, D. 48 Arai, M. 56 Arvai, A.S. 24, 46 Ashley, J. 27 Atkins, A. 18 Aversa, R. 38 Bachovchin, D. 17 Bae, S.-H. 56 Baksh, M. 22 Banerjee, D. 22 Barbas, C.F. III 11 Barrett, E. 40 Barros, C. 37 Bartfai, T. 13 Beck, A. 50 Beer, S. 40 Beierle, J.M. 26 Belani, R. 41 Bell, C.H. 51 Bennett, C. 55 Ben-Shir, I. 31 Beuck, C. 50 Beutler, E. 14 Blankman, J. 17 Boehr, D. 56 Boger, D.L. 15 Boyle, K. 15 Brenzovich, W. 38 Brustad, E.M. 42 Buck-Koehntop, B. 56 Bui, T. 11 Bunner, A. 50 Burke, C. 15 Capková, K. 27 Cardoso, R.M.F. 51 Carmel, A. 50 Chamero, P. 45 Chang, G. 17 Chapados, B.R. 46 Chavochi, A. 26 Chen, J. 38 Chen, S. 50 Chen, Y. 17 Choi, S. 33 Chong, Y. 41 Clark, R. 15 Colby, D. 15 Connelly, S. 51 Conti, B. 13 Conti, F. 37 Cottrell, J.W. 21 Crane, C. 15 Cravatt, B.F. 17 Culhane, J. 43 Culyba, E. 33 Dalby, S. 38 Dale, T.J. 40 De Lamo Marin, S. 27 Dean, S. 55 Deka, P. 56 DeMartino, J. 15 Dendle, M.T.A. 33 Denery, J. 27 Di Mola, A. 27 Dickerson, T.J. 27 Dix, M. 17 Droese, B.J. 51 Druzina, Z. 41 Du, D. 33 Dube, H. 40 Dubrovskaya, V. 48 Duncan, K. 15 Dyson, H.J. 56 Edelman, G.M. 18 Edgcomb, S. 50 Edmonds, D. 38 Ekiert, D.C. 51 Elledge, H. 37 Ellery, S. 38 Ellis, B. 27 Elsliger, M.-A. 51 Eschenmoser, A. 20 Estrada, A. 38 Eubanks, L. 27 Ezzili, C. 15 Fan, L. 46 Fearns, C. 33 Fedor, M.J. 21 Felitsky, D. 56 Ferreon, J. 56 Ficht, S. 55 Fiedler, J. 22 Finn, M.G. 22 Flanagan, K. 45 Fokin, V.V. 43 Fotsing, J. 43 Fraga, B. 38 Franco, S. 37 Frater, J. 41 Frederick, M. 38 Freestone, M. 38 Fu, Y. 55 Fukuchi, K. 27 Fuller, A.A. 33 Fuller, R.P. 11 Fulton, Z. 51 Gaj, T. 11 Garcin, E.D. 24 Garfunkle, J. 15 Gavrilyuk, J. 11 Ge, H. 15 Gelin, C. 38 Gersbach, C. 11 Gershoni, R. 31 Getzoff, E.D. 24 Ghadiri, M.R. 26 Gondi, V. 38 Gonzalez, B. 11 Goodwin-Tindall, J. 38 Gordley, R.M. 11 Grecian, S. 43 Greenberg, W. 55 Grillet, N. 37 Grimster, N. 43 Grover, R.K. 48 Grünewald, J. 42 Guo, J. 11 Guo, J. 42 Guo, M. 41 Guo, R.-T. 41 Guy, R. 55 Haberz, P. 56 Han, B.W. 51 Hanan, M. 41 Hanson, S. 55 Harkins-Perry, S. 37 He, W. 41 He, X. 17 Hein, J. 43 Hernandez, C. 27 Hesse, M. 38 Hitomi, C. 24, 46 Hitomi, K. 24, 46 Hochstatter, D. 15 Hong, M. 51 Hong, V. 22 Hong, Z. 55 Hooley, R.J. 40 Horneff, T. 43 Horst, R. 59 Hou, J.-L. 40 63 Huang, P. 38 Huang, Z. 38 Hwang, D.-R. 55 Hwang, I. 15 Jager, M. 36 Janda, K.D. 27 Jeso, V. 38 Ji, S. 17 Jimenez-Dalmaroni, M.J. 51 Jin, J. 38 Jones, R. 15 Joyce, G.F. 30 Jung, I.L. 41 Juraja, S. 11 Kakei, H. 15 Kalisiak, J. 43 Kamioka, S. 40 Kapoor, M. 41 Kar, M. 38 Karmakar, A. 31 Karyakin, A. 17 Kato, D. 15 Kaufmann, G. 27 Kaur, A. 45 Kazmierczak, P. 37 Keinan, E. 31 Kelly, J.W. 33 Kerkow, D. 50 Kim, D.H. 11 Kimball, F.S. 15 Kirchdoerfer, R.N. 51 Kislukhin, A. 22 Klein, I. 13 Koh, D.C.Y. 18 Kondreddi, R.R. 20 Kopp, F. 17 Korthals, K. 43 Kossoy, E. 31 Krasovskiy, A. 38 Krishnamurthy, R. 20 Kroeger, M.D. 24 Kwan, S. 50 Kwok, S.-W. 43 Lajiness, J. 15 Lam, B.J. 30 Landes, M. 56 Lee, C.W. 56 Lee, H.S. 42 Lee, J.-C. 55 Lee, P. 36 Lee, S. 15 Lee, S.H. 41 Leman, L. 26 Lemire, A. 38 Levin, R. 38 Li, A. 38 Li, H. 38 64 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 Li, L. 21 Li, W. 17 Liang, P.-H. 55 Lieu, S. 17 Lim, Y. 38 Lincoln, T.A. 30 Lister, T. 38 Liu, C.C. 42 Liu, J. 41 Liu, L. 21 Lledo, A. 40 Logan, D. 45 Long, J. 17 Lowery, C. 27 MacMillan, K. 15 Mahajan, S. 27 Majumder, U. 38 Manlapaz, E. 56 Manuell, A. 36 Marin-Navarro, J. 36 Martin, B. 17 Martin, T. 45 Martinez-Garay, I. 37 Martinez-Yamout, M. 56 Masuda, K. 17 Mathison, C. 38 Matsuda, D. 18 Mayfield, S.P. 36 Mayorov, A. 27 McElhaney, G. 27 McKinney, M. 17 Mee, J. 27 Meinhold, D. 56 Mejuch, T. 31 Menicelli, E. 50 Mercer, A. 11 Merriman, E. 41 Metanis, N. 31 Mittapalli, G.K. 20 Mizuta, S. 11 Mohanty, B. 59 Montero, A. 26 Moreno, A. 27 Morgan, A. 38 Mu, T.-W. 33 Müller, U. 37 Murray, A. 33 Muto, M. 36 Naidu, V.S. 20 Nakai, Y. 27 Nawaz, M.H. 41 Nguyen, A. 27 Nguyen, T. 17 Nicolaou, K.C. 38 Nold, A. 38 Nomura, D. 17 Nunes, A. 27 Olsen, C.A. 26 Ong, D. 33 Onoda, A. 11 Ortiz, A. 38 Osborn, O. 13 Osornio, Y. 20 Otrubova, K. 15 Paegel, B.M. 30 Panopoulos, P. 18 Papes, F. 45 Pappo, D. 31 Park, J. 27 Park, S.-J. 56 Parvari, G. 31 Patel, P. 15 Patil, N. 38 Paulsson, J. 33 Payne, R. 55 Pedrini, B. 59 Pejchal, R. 51 Peram, M.M.R. 48 Perry, J.J. 46 Peters, F.B. 42 Petrie, K.L. 30 Pettersson, P. 36 Pilotte, J. 18 Pique, M.E. 24, 46 Piran, R. 31 Pitram, S. 43 Porter, G.P. 51 Powers, E.T. 33 Pratt, B. 38 Radakovits, R. 37 Ramos, C. 37 Rao, P. 33 Rasala, B. 36 Ratner, T. 31 Raushel, J. 43 Reany, O. 31 Rebek, J., Jr. 40 Reingruber, R. 38 Restorp, P. 40 Revin, M. 17 Reynolds, A. 37 Ridgeway, W. 50 Ring, G. 50 Rivas, F. 38 Robertson, W. 15 Rodriguez, R. 15 Rogel, J. 48 Rohrbach, A. 27 Saccavini, C. 27 Salahuddin, S. 11 Salzameda, N. 27 Sanchez Ruiz, A. 38 Sanchez-Alavez, M. 13 Santa Marta, M. 11 Sarlah, D. 38 Sasaki, Y. 15 Saure, M. 33 Schelwies, M. 55 Schiefner, A. 51 Schimmel, P. 41 Schnermann, M. 15 Schultheisz, H. 50 Schultz, P.G. 42 Schwander, M. 37 Schwimmer, L.J. 11 Scott, L.G. 50 Serrano, P. 59 Seto, S. 15 Shajani, Z. 50 Shapiro, R. 41 Sharpless, K.B. 43 Shaw, D. 38 Shekhter, T. 31 Shimamura, H. 15 Shin, D.S. 24, 46 Shoshani, S. 31 Siegel, S. 33 Simkovsky, R. 33 Simon, G. 17 Sinha, M. 31 Slown, C. 15 Solel, E. 31 Solomon, J. 33 Song, B.D. 48 Song, Y.Z. 41 Soreni, M. 31 Sperling, E. 50 Stamm, S. 15 Stanfield, R.L. 51 Stefanko, R.S. 51 Steiniger, S. 27 Stepan, A. 38 Stover, J. 15 Stowers, L. 45 Stump, B. 43 Swingle, D. 15 Sykes, M. 50 Szewczyk, P. 17 Szymczyna, B. 50 Tabarean, I. 13 Tainer, J.A. 46 Takizawa, S. 15 Talbot, A. 38 Tam, A. 15 Tam, O. 21 Tanaka, F. 11 Tang, Y. 38 Thomas, J. 17 Tran, M. 36 Trépanier, V. 38 Treweek, J.B. 27 Tria, G. 38 Tripp, J. 43 Troseth, R.P. 48 Tubbs, J.L. 24, 46 Tully, S. 17 Udit, A. 22 Uehara, H. 11 Umezawa, T. 38 Usui, K. 33 Utsumi, N. 11 Va, P. 15 Valdez, C. 43 Van Anda, H. 40 Vanhnasy, J.A. 51 Vo, M.-N. 41 Wang, J. 38 Wang, S.-K. 55 Wang, Y. 33 Ward, A. 17 Watson, P. 21 Webb, S. 37 Weerapana, E. 17 Weide, T. 43 Wentworth, A.D. 48 Wentworth, P., Jr. 48 Whitby, L. 15 Williams, R.S. 46 Williamson, J.R. 50 Willis, A. 27 Wilson, I.A. 51 Wolfe, A. 15 Wong, C.-H. 55 Wright, P.E. 56 Wu, J. 50 Wu, T. 38 Wuellner, U. 11 Wüthrich, K. 59 Xiao, S. 40 Xie, J. 15 Xiong, W. 37 Xu, R. 51 Xu, X. 51 Xu, Y. 27 Yang, X.-L. 41 Yi, K.S. 11 Yoneda, Y. 27 Yonemoto, I. 33 Yoon, S.I. 51 Young, T.S. 42 Yu, J. 17 Yu, Z. 33 Zhan, W. 38 Zhang, H. 11 Zhang, H. 38 Zhang, W. 41 Zhou, B. 27 Zhou, H. 27 Zhou, Q. 41 Zhu, X. 51 Zimmerman, S. 21 Zuhl, A. 15 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 SUBJECT INDEX Aging 42 Algae 36 Amylin 35 Amyloid β-peptides 14 Amyloidogenesis 35 Amyloidoses 49 Anchored plasticity 25 Anemia 14 Antibiotics 16, 38, 44 Antibodies chemically programmed 12 Anticoagulants 23 Antitumor agents 38 Ataxia telangiectasia–like disorder 47 Autoinducers 28 Base J 48 Biofuels 36 Biomolecular computing 32 Cancer 12 Capsids 33 Catalytic antibodies 11 in transgenic plants 32 Cavitands 41 Chloroplasts 36 Click chemistry 22, 43 Cockayne syndrome 47 Cryptochromes 25 Cytokines 13 Deafness 37 Diabetes 13, 35 Directed evolution 30 DNA damage 14 DNA repair 46 DNA replication 14 DNA-binding agents 15 DNA-binding proteins 57 Drug design 51 Drug discovery 43 Drug resistance 44 Encapsulation 40 Endocannabinoids 17 Enzymatic synthesis 50 Enzymes synthetic 31 Fatty acid amide hydrolase 18 inhibitors of 15 Gaucher disease 34 Gene regulation 12 Genetic code 42 Glycobiology 55 Glycoproteins 55 Glycoproteomics 55 Hair cells 37 Hepcidin 14 Histone-deacetylase inhibitors 26 HIV vaccine 51 Hypoxia 57 Inflammation 13 Influenza virus 52 RNA structure 50 RNA-binding proteins 19 Self-assembly 40 Sensory neurons 45 Signaling 27 Innate behavior 45 Innate immunity 53 Iron metabolism 14 Leishmaniasis 48 Light-chain aggregation 49 Lysosomal storage diseases 34 Mechanotransduction 37 Metabolic engineering 50 Microfluidics 30 Mistranslation 41 Molecular mimicry 40 Mouse models of deafness 37 Multidrug resistance 16, 17 Multidrug transporters 17 Natural products 38 Neural development 18 Neurodegeneration 13 Neurotoxins 38 Neutralizing antibodies for HIV 51 for influenza virus 52 Nijmegen breakage syndrome 47 Nitric oxide synthases 24, 46 Nuclear magnetic resonance 56 Nucleic acids chemical etiology of 20 structure of 20 Obesity 13 Olfaction 45 Oncogenesis 42 Organocatalysis 11 Pain 8, 15 Pheromones 45 Photolyases 25 Protein structure 24, 46, 56 automated NMR determination of 59 Protein-misfolding diseases 33 Proteomics 18 Proteostasis 34 Quorum sensing 27 Ramoplanin 16 Receptors CD1 54 Toll-like 53 variable lymphocyte 53 Ribosomes 19 Ribozymes 21 RNA 18 RNA enzymes 30 RNA folding 21 Structure-based design 26 Sugar-assisted ligation 55 Therapeutic proteins 36 Transcription 57 Transcription factors 12 Translation 19 Transthyretin 14, 34 tRNA synthetases 41 Turn mimetics 26 Type IV pilin 46 Unnatural amino acids 42 Usher syndrome 37 Vancomycin 16 Virus particles 22 X-ray crystallography 51 Yatakemycin 16 Zinc fingers 12, 58 65 66 THE SKAGGS INSTITUTE FOR CHEMICAL BIOLOGY 2008 ACKNOWLEDGMENTS The scientists who have contributed sections to this report wish to acknowledge the dedication and hard work of the laboratory technicians who helped bring the research to fruition, the administrative assistants who made it presentable for publication, and the support personnel who provided critical specialized services and equipment. EDITOR Barbara L. Halliburton, Ph.D. PROJECT MANAGER Jann Coury Office of Communications P R I N T I N G A N D D U P L I C AT I O N Precision Litho Maryland Composition PHOTOGRAPHY Michael Balderas Biomedical Graphics Department, Scripps Research Mark Dastrup D E PA R T M E N TA L C O O R D I N AT I O N Janette Lundgren Beas The Skaggs Institute for Chemical Biology Ruby Santos Department of Molecular Biology Marcia McRae Molecular and Integrative Sciences Department Cheryl Negus Department of Cell Biology Department of Chemical Physiology Vicky Nielsen Department of Chemistry Lynn Oleski Department of Molecular and Experimental Medicine Michelle Platero Department of Neurobiology The Skaggs Institute for Chemical Biology scientific report is published annually by The Scripps Research Institute and is available on request from Office of Communications TPC-30 The Scripps Research Institute 10550 North Torrey Pines Road La Jolla, CA 92037 (858) 784-2171 e-mail: kevin@scripps.edu