Formal [2+2+2] Cycloaddition Strategy Based on an Intramolecular Propargylic Ene Reaction/Diels-Alder
advertisement

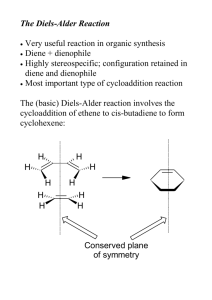
Formal [2+2+2] Cycloaddition Strategy Based on an Intramolecular Propargylic Ene Reaction/Diels-Alder Cycloaddition Cascade The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Robinson, Julia M. et al. “Formal [2 + 2 + 2] Cycloaddition Strategy Based on an Intramolecular Propargylic Ene Reaction/DielsAlder Cycloaddition Cascade.” Journal of the American Chemical Society 132 (2010): 11039-11041. Web. 16 Dec. 2011. © 2011 American Chemical Society As Published http://dx.doi.org/10.1021/ja1053829 Publisher American Chemical Society Version Author's final manuscript Accessed Wed May 25 21:49:09 EDT 2016 Citable Link http://hdl.handle.net/1721.1/67701 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Detailed Terms Formal [2 + 2 + 2] Cycloaddition Strategy Based on an Intramolecular Propargylic Ene Reaction/Diels-Alder Cycloaddition Cascade Julia M. Robinson, Takeo Sakai, Katsuhiko Okano, Takafumi Kitawaki, and Rick L. Danheiser* Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 RECEIVED DATE (automatically inserted by publisher); E-mail: danheisr@mit.edu [2 + 2 + 2] Cycloadditions provide rapid access to polysubstituted six-membered rings. The most prominent variant of this powerful strategy is the transition-metalcatalyzed process in which three alkynes combine to form benzenoid aromatic products. 1 Although the fully intramolecular metal-catalyzed cotrimerization is generally an efficient process, the bimolecular variant involving the reaction of an α,ω-diyne with a third acetylene suffers from several limitations. Often a large excess of the third alkyne must be employed due to its competitive trimerization, and reactions in which the third alkyne is unsymmetrical frequently proceed with poor regioselectivity.2 Herein we describe a formal, metal-free, bimolecular [2 + 2 + 2] cycloaddition strategy based on a cascade of two pericyclic processes (Scheme 1). The first step involves an intramolecular propargylic ene-type reaction of a 1,6-diyne to generate a vinylallene of type 2. The vinylallene is necessarily in an s-cis conformation, rendering it highly reactive in the second step, an inter- or intramolecular Diels-Alder reaction with an alkenyl or alkynyl dienophile.3 The efficacy of the propargylic-type ene reaction in this sequence is noteworthy. Although the intramolecular ene reaction is a well established process, 4 very few examples have been reported involving propargylic rather than allylic hydrogen atoms. 5,6 Overall, this two-stage pericyclic cascade sequence provides rapid access to highly functionalized alkylidenecyclohexane derivatives. When alkynes are employed as dienophiles, the initial products are isotoluene derivatives7 that are easily isomerized to aromatic products upon exposure to base. Scheme 1. Propargylic Ene Reaction/Diels-Alder Cycloaddition Cascade R1 H Z R1 Propargylic ene reaction dienophile and illustrating the effect of variations in the 1,6diyne tether on the facility of the propargylic ene reaction. 8 Good yields are obtained using only one equivalent of dienophile, in contrast to most cases of the metal-catalyzed bimolecular [2 + 2 + 2] process. Products are obtained as predominantly Z isomers, as expected for cycloadditions in which bond formation to the central carbon of the allene moiety occurs preferentially trans to the R1 substituent on the terminal carbon of the allene (Scheme 1).9 In the case of these ene reactions involving unactivated alkynes as enophiles, efficient reaction generally requires temperatures in the range 150-160 °C.10 Thermolysis in the absence of dienophiles leads to the formation of vinylallene dimers which are obtained as complex mixtures of regio- and stereoisomers. 11 Scheme 2. Formal [2 + 2 + 2] Cycloadditions with N-Methylmaleimide as Dienophile Z + H 1 2 G R2 [4 + 2] cycloaddition Z R3 3 G Scheme 2 presents representative examples of the formal [2 + 2 + 2] cycloaddition involving N-methylmaleimide as O N Me H O H O Pr H O H N Me O H O N Me TsN O H O 4 N Me H O 5 O 6 160 °C, 21 h 160 °C, 21 h 160 °C, 11 h 94% (Z:E = 91:9) 52% 75% (Z:E = 74:26) Pr Pr O H O N Me N Me PhO2S H O 7 R1 Z Pr PhO2S R3 G N Me H + H toluene (0.1M) O (1 equiv) R2 Z R O CH2R H O O 8 150 °C, 8 h 160 °C, 21 h 73% (Z:E = 69:31) 74% (Z:E = 81:19) Table 1. Formal [2 + 2 + 2] Cycloadditions with Alkenyl Dienophiles Bu toluene (0.1M) 110 or 160 °C R,H + O EWG G entry G (1 equiv) dienophile yield (%)a major product Pr 1 H 9 160 °C, 21 h Me O 72b,c Me O 10 O Pr O 2 CO2Me 11 H N Me 160 °C, 1 h O Table 2. Formal [2 + 2 + 2] Cycloadditions with Alkynyl Dienophiles O N Me O MeO2C H amounts of BHT as a radical inhibitor. No competition from [4 + 2] arenyne 12 or “ynone” 13 type cycloadditions were observed in cases with G = Ph or CO2Me (Table 1, entries 24). Reactions involving unsymmetrical alkenyl and alkynyl dienophiles proceed smoothly with good to excellent regioselectivity in the Diels-Alder step as observed previously for cycloadditions of vinylallenes.3 Diastereoselectivity in the Diels-Alder step is also high, and endo cycloadducts are observed as the exclusive products of the reaction (Table 1, entries 2-6). 14 Finally, in the case of alkynyl dienophiles (Table 2), [4 + 2] cycloaddition initially generates an isotoluene-type intermediate that isomerizes to the isolated aromatic product upon exposure to a catalytic amount of DBU at room temperature. 93d,e 12 O + O G Pr O 3 Ph 13 H N Me O O Ph H 65f entry G Me 11 reflux, 19 h 1 O O H 9 57f Me MeO2C reflux, 21 h Me O CO2Me 75 O CO2Me CO2Me 19 O H 9 160 °C, 21 h O Me O Me 20 40b O 68g Me O Bu Bu 2 C!CSi(i-Pr)3 16 yield (%)a major product 15 O Pr 5 then add 10 mol% DBU 25 °C, 3-17 h CO2Me 160 °C, 21 h CO2Me (1 equiv) dienophile 14 O Pr 4 EWG O N Me 160 °C, 12 h toluene 110 or 160 °C R,H Bu Bu CO2Me 17 3 C!CSi(i-Pr)3 16 reflux, 21 h Si(i-Pr)3 CO2Me 81 O CO2Me CO2Me 21 Pr Si(i-Pr)3 6 C!CSi(i-Pr)3 16 reflux, 21 h 63h O CO2Me CO2Me a Isolated yield of products purified by chromatography. regioisomeric Diels-Alder product was isolated in 6% yield. b The 18 Si(i-Pr)3 a Isolated yield of products purified by chromatography. b Mixture of Z and E isomers (86:14). c The regioisomeric Diels-Alder product was isolated in 11% yield. d Reaction at reflux for 21 h gave 12 in 68% yield. e Mixture of Z and E isomers (92:8). f Mixture of Z and E isomers (93:7). g Mixture of Z and E isomers (90:10). h Mixture of Z and E isomers (81:19). Further examples demonstrating the scope of the bimolecular version of this formal [2 + 2 + 2] cycloaddition strategy are shown in Tables 1 and 2. As expected, propargylic ene reactions involving diynes bearing electronwithdrawing groups proceed under milder conditions and in these cases the tandem ene reaction/Diels-Alder cascade can be carried out in refluxing toluene (Table 1, entries 2 and 4-6, Table 2, entry 3). In reactions involving MVK, methyl acrylate, and butynone, yields are slightly improved by carrying out the tandem process in the presence of small Fully intramolecular “cyclotrimerizations” of triynes constitute one of the most useful variants of the transitionmetal-catalyzed process, providing efficient access to angularly-fused polycyclic systems. Several examples of purely thermal [2 + 2 + 2] cycloadditions of triynes have been reported prior to our work. Notably, in 1999 Kociolek and Johnson reported that the gas phase thermolysis of 1,6,11dodecatriyne at 500-600 °C and 0.01 torr affords a hexahydroindacene [2 + 2 + 2] cycloadduct together with derived dehydrogenation products in 35% combined yield.15 Johnson proposed a stepwise mechanism to account for this process in which bond formation initially occurs between two alkynes to generate a 1,4-diradical which is then trapped by the third alkyne to directly produce an aromatic ring. An analogous diradical mechanism was proposed by Parsons et al. to account for an interesting cyclization of enediynes discovered in their laboratory. 16 More recently, Ley and coworkers have reported the cyclotrimerization of triyne 22 carried out in DMF either by heating at 200 °C or by using focused microwave heating.17 Ley et al. reported the isolation of an intermediate that they identified as 27, leading to the proposal of the mechanism outlined in Scheme 3 involving several unusual strained and reactive intermediates. We have repeated the thermolysis of 22 and obtained unequivocal evidence that the intermediate formed in this reaction is not 27, but rather the isomeric triene 28, 18 the product expected if the reaction proceeds via a sequential propargylic ene/[4 + 2] cycloaddition cascade mechanism rather than the pathway proposed by Ley and coworkers as outlined in Scheme 3. In addition, we have also found that cyclotrimerization of the related substrate 30 leads to the formation of diene 31 (the product predicted by our pericyclic cascade mechanism), rather than the isomeric diene 32 reported by Ley et al. as the product of this reaction.17 Scheme 3. Mechanism Proposed (Ref. 14) for Thermal Cyclotrimerization of 22 O or O O 23 22 O 24 O O 27 25 26 Scheme 4. Cyclotrimerization of Triyne 22 a O b O 22 O 28 29 a Conditions: (a) 160 °C, 14 h, toluene. (b) Add 0.1 equiv TsOH-H 2O, 60 °C, 3 h; 56% total overall yield after reduction of indene byproduct (H2, Pd/C, MeOH, rt). Scheme 5. Cyclotrimerization of 30 toluene 200 °C 1h O 90% 30 O O 31 32 In summary, this study establishes the utility of a bimolecular propargylic ene reaction/Diels-Alder cascade as a method for achieving formal, thermal [2 + 2 + 2] cycloadditions leading to functionalized polycyclic compounds. The mechanism of several earlier fully intramolecular related transformations appear to involve an analogous pericyclic cascade process rather than the diradicalmediated pathways proposed previously. Further studies are underway in our laboratory aimed at developing additional synthetically useful variants of this [2 + 2 + 2] strategy. Acknowledgment. We thank the National Institutes of Health (GM 28273), Merck Research Laboratories, Daiichi Sankyo Co., and Boehringer Ingelheim Pharmaceuticals for generous financial support. J. R. was supported in part by National Science Foundation and AstraZeneca Graduate Fellowships. T. S. was supported in part by a Research Fellowship of the Japan Society for the Promotion of Science (JSPS) for Young Scientists. Supporting Information Available: Experimental procedures, characterization data, and 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org. References 1 For recent reviews, see (a) Agenet, N.; Buisine, O.; Slowinski, F.; Gandon, V.; Aubert, C.; Malacria, M. Org. React. 2007, 68, 1. (b) Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741. (c) Chopade, P. R.; Louie, J. Adv. Synth. Catal. 2006, 348, 2307. (d) Galan, B. R.; Rovis, T. Angew. Chem. Int. Ed. 2009, 48, 2830. (e) Tanaka, K. Synlett 2007, 1977. 2 A few exceptions have appeared in the context of natural product syntheses. For example, see Nicolaou, K. C.; Tang, Y.; Wang, J. Angew. Chem. Int. Ed. 2009, 48, 3449. 3 For a review of Diels-Alder reactions of vinylallenes, see Murakami, M.; Matsuda, T. Cycloadditions of Allenes. In Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, 2004; Vol. 2, pp 727-815. 4 Reviews: (a) Oppolzer, W.; Snieckus, V. Angew. Chem. Int. Ed. 1978, 17, 476. (b) Snider, B. B. Ene Reactions with Alkenes as Enophiles. In Comprehensive Organic Synthesis; Paquette, L. A., Ed.; Pergamon Press: Oxford, 1991; Vol. 5, pp 1-28. 5 (a) Oppolzer, W.; Pfenninger, E.; Keller, K. Helv. Chim. Acta 1973, 56, 1807. (b) Shea, K. J.; Burke, L. D.; England, W. P. Tetrahedron Lett. 1988, 29, 407. (c) Jayanth, T. T.; Jeganmohan, M.; Cheng, M.-J.; Chu, S.-Y.; Cheng, C.-H. J. Am. Chem. Soc. 2006, 128, 2232. (d) Altable, M.; Filippone, S.; MartínDomenech, A.; Güell, M.; Solà, M.; Martín, N. Org. Lett. 2006, 8, 5959. (e) For a report of the isolation of dimers believed to be derived from the product of a propargylic ene reaction, see Peña, D.; Pérez, D.; Guitián, E.; Castedo, L. Eur. J. Org. Chem. 2003, 1238. 6 As our study neared completion, Roglans et al. reported three related examples of a fully intramolecular propargylic ene/intramolecular Diels-Alder process involving 15-membered macrocyclic triaza triynes and enediynes: González, I.; Pla-Quintana, A.; Roglans, A.; Dachs, A.; Solà, M.; Parella, T.; Farjas, J.; Roura, P.; Lloveras, V; Vidal-Gancedo, J. Chem. Commun. 2010, 46, 2944. 7 For the synthesis and thermal behavior of p-isotoluene, see Gajewski, J. J.; Gortva, A. M. J. Am. Chem. Soc. 1982, 104, 334 and references cited therein. 8 For full details on the synthesis of the 1,6-diyne cycloaddition substrates, see the Supporting Information. 9 E/Z stereochemistry was assigned by differential nOe experiments. See Supporting Information for details. 10 Attempts to extend the intramolecular propargylic ene reaction to 1,7-diynes were not successful. Unreacted starting material was recovered after prolonged heating at 200 °C (see Supporting Information for further details). Note that in contrast to intramolecular ene reactions of 1,6-dienes, similar reactions involving 1,7-dienes require higher temperatures and proceed in low yield.4 11 For the isolation and characterization of products generated by dimerization of the vinylallene produced by thermolysis of 16, see the Supporting Information. 12 Reviewed in Wessig, P.; Müller, G. Chem. Rev. 2008, 108, 2051. 13 Wills, M. S. B.; Danheiser, R. L. J. Am. Chem. Soc. 1998, 120, 9378. 14 Stereochemical assignments are based on nOe experiments and analysis of 1 H NMR coupling constants. For details, see Supporting Information. 15 Kociolek, M. G.; Johnson, R. P. Tetrahedron Lett. 1999, 40, 4141. 16 Parsons, P. J.; Waters, A. J.; Walter, D. S.; Board, J. J. Org. Chem. 2007, 72, 1395. 17 Saaby, S.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3365. 18 The structure of 28 was assigned based on NMR studies. For details, see Supporting Information. R1 R1 R1 R2 Z H Z H G propargylic ene reaction + R2 Z R3 R3 G [4 + 2] cycloaddition G A formal, metal-free, [2 + 2 + 2] cycloaddition strategy is described based on a cascade of two pericyclic processes. The first step involves an intramolecular propargylic ene reaction of a 1,6-diyne to generate a vinylallene, which then reacts in an inter- or intramolecular Diels-Alder reaction with an alkenyl or alkynyl dienophile. Reactions involving unsymmetrical alkenyl and alkynyl dienophiles proceed with good to excellent regioselectivity, and the diastereoselectivity in the DielsAlder step is also high, with endo cycloadducts produced as the exclusive products of the reaction. In the case of alkynyl dienophiles, [4 + 2] cycloaddition initially generates an isotoluene-type intermediate that isomerizes to the isolated aromatic product upon exposure to a catalytic amount of DBU at room temperature. The mechanism of several earlier fully intramolecular related transformations have been shown to involve an analogous process rather than the diradical-mediated pathways proposed previously.