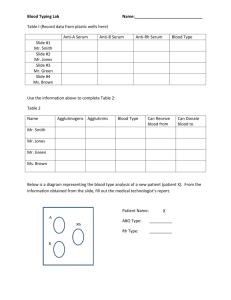
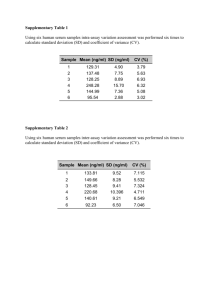
for the degree of Doctor of Philosophy i n Deryk Thomas Loo
advertisement

AN ABSTRACT OF THE THESIS OF Deryk Thomas Loo Biochemistry and Biophysics for the degree of presented on Doctor of Philosophy i n July 19. 1991 Title: Nonsenescent Serum-Free Mouse Proastroblast Cells: Extended Culture. Growth Responses In Vitro. and Application to the Culture of Human Embryonic Astrocytes. Abstract approved:4Redacted for Privacy David W. Barnes Mouse embryo cells cultured in vitro in serum-supplemented media undergo growth crisis, resulting in the loss of genomically normal cells prior to the appearance of established, aneuploid cell lines. I used the technique of serum-free cell culture to develop a serum-free mouse embryo (SFME) cell line in which serum was replaced by a set of defined supplements. SFME cells, cultured in a nutrient medium supplemented with insulin, transferrin, epidermal growth factor (EGF), high-density lipoprotein (HDL), and fibronectin, have maintained a diploid karyotype with no detectable chromosomal abnormalities for more than 200 generations. The cells did not undergo growth crisis and remain in culture today. SFME cells were dependent on EGF for survival and were reversibly growth inhibited by serum or platelet-free plasma. Treatment of SFME cells with serum or transforming growth factor beta led to the appearance of glial fibrillary acid protein (GFAP), a specific marker for astrocytes, identifying SFME cells as proastroblasts. Following the derivation of SFME cells my research focussed on (1) defining more precisely the growth response of SFME cells to medium supplements, (2) investigating the relationship between the nonsenescent nature of SFME cells and their responses to serum and EG1., and (3) applying the serum-free cell culture methods to the multipassage culture of human embryonic astrocytes. SFME cells in serum-containing medium arrested in the G1 phase of the cell cycle with greatly reduced DNA replication activity. A portion of the inhibitory activity of serum was extracted by charcoal, a procedure that removed steroid and thyroid hormones. However, the effect of serum on untransformed SFME cells could not be prevented by addition of antiglucocorticoid, and ras-transformed clones of SFME cells, which are not growth inhibited by serum, retained inhibitory responses to glucocorticoid and thyroid hormone T3. These results suggest that glucocorticoid or thyroid hormones may contribute to the inhibitory activity of serum on SFME cells, but additional factors are involved. SFME cell death resulting from EGF deprivation exhibited characteristics associated with apoptosis or programmed cell death. Ultrastructural analysis showed cells became small and vacuolated, with pyknotic nuclei. The cultures contained almost exclusively G1- phase cells. Chromatin exhibited a pattern of degradation into oligonucleosome-length fragments generating a regularly spaced ladder. I applied the serum-free approach used to derive SFME cells to the multipassage culture of human embryonic astrocytes. Cells were cultured in nutrient medium supplemented with insulin, transferrin, EGF, HDL, fibronectin, basic fibroblast growth factor and heparin. Cultures were maintained for a maximum of 70 population doublings before proliferation ceased. The cells synthesized GFAP. Nonsenescent Serum-Free Mouse Proastroblasts: Extended Culture, Growth Responses In Vitro, and Application to the Culture of Human Embryonic Astrocytes by Deryk Thomas Loo A THESIS Submitted to Oregon State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy Completed July 19, 1991 Commencement June 1992 APPROVED: Redacted for Privacy Professor of Biochemistry and Biophysics in charge of major Redacted for Privacy Head of Department of Biochemistry and Biophysics Redacted for Privacy Dean of GraduKte School Date thesis presented Typed by July. 19, 1991 Deryk Thomas Loo DEDICATION AND ACKNOWLEDGMENTS The path I followed to the degree of doctor of philosophy has been an enormous learning experience, one I will never forget. I am thankful for all the lessons I have been taught, both inside and outside of the laboratory. Without the love and support of my parents, Eddie and Diana, and my wife, Jenefer, this thesis would not have been possible. I dedicate my thesis to my parents and my wife. Thank you. I would also like to thank my sisters, Megan and Kelly, for tolerating their older brother, and my Godmother, Elaine Currie, for her support throughout my life. The research presented in my thesis resulted from a collaboration among the many members of the laboratory. I would like to thank everyone in the lab, especially Cathy Rawson, for their help and friendship. Finally, I would like to thank David Barnes for his financial support and for his unique six year course in molecular and cellular biology. TABLE OF CONTENTS CHAPTER 1 1 Introduction and Review of Literature INTRODUCTION 1 REVIEW OF LITERATURE 3 Animal cell culture 3 Brain cell diversity and mammalian cell culture 10 Cell death 13 Orientation to format 17 CHAPTER 2 18 Extended Culture of Mouse Embryo Cells Without Senescence: Inhibition by Serum SUMMARY 19 INTRODUCTION 20 METHODS AND RESULTS 21 DISCUSSION 25 ACKNOWLEDGMENTS 26 CHAPTER 3 35 Serum-Free Mouse Embryo Cells: Growth Responses In Vitro SUMMARY 36 INTRODUCTION 37 MATERIALS AND METHODS 38 Materials 38 Serum-free cell culture procedures 38 Serum-free culture medium 39 Experimental procedures 41 RESULTS 42 Effects of insulin and transferrin on SFME cells 42 Effects of EGF and FGFIHBGF on SFME cells 42 Effects of attachment proteins and HDL on SFME cells 44 DISCUSSION 46 ACKNOWLEDGMENTS 50 CHAPTER 4 60 Glucocorticoid and Thyroid Hormones Inhibit Proliferation of Serum-Free Mouse Embryo (SFME) Cells SUMMARY 61 INTRODUCTION 62 MATERIALS AND METHODS 64 Materials 64 SFME cell culture 64 Assay of cell growth 65 Seru-n treatments 66 RESULTS 67 Inhibition of SFME cells by glucocorticoid and thyroid hormone 67 Contribution of glucocorticoid and thyroid hormone to the 68 inhibitory activity of serum on SFME cells DISCUSSION 70 ACKNOWLEDGMENTS 73 CHAPTER 5 84 Serial Passage of Embryonic Human Astrocytes In Serum-Free, Hormone-Supplemented Medium SUMMARY 85 INTRODUCTION 86 MATERIALS AND METHODS 88 Cell culture 88 Western blots 89 GFAP culture immunoassay 90 RESULTS 92 Serial passage culture of embryonic human astrocytes 92 Growth characteristics of embryonic human astrocytes in vitro 93 Expression of GFAP by embryonic human astrocytes in vitro 94 DISCUSSION 95 ACKNOWLEDGMENTS 99 CHAPTER 6 107 Conclusion and Future Prospectives CONCLUSION 107 FUTURE PERSPECTIVES 112 BIBLIOGRAPHY 115 APPENDIX 1 129 Serum Inhibition of Proliferation of Serum-Free Mouse Embryo Cells SUMMARY 130 INTRODUCTION 131 MATERIALS AND METHODS 133 Cell culture 133 Flow cytometry 133 Assays 134 Karyotyping 134 RESULTS 135 Effects of serum on SFME cells 135 Variant SFME cells capable of growing in serum-containing medium 136 DISCUSSION 138 ACKNOWLEDGMENTS 140 APPENDIX 2 151 Death of Serum-Free Mouse Embryo Cells Caused by Epidermal Growth Factor Deprivation SUMMARY 152 INTRODUCTION 153 MATERIALS AND METHODS 155 Cell culture 155 Electron microscopy 155 DNA fragmentation analysis 156 Enzyme assay 157 Flow cytometry 157 Polysome profiles 158 Northern blot hybridization analysis 159 RESULTS 161 Ultrastructural changes associated with EGF deprivation 161 Biochemical and physiological changes associated with EGF deprivation 163 DISCUSSION 165 ACKNOWLEDGMENTS 168 LIST OF FIGURES Eau Figure 2.1 Growth of BALB/c mouse embryo cells upon successive 27 transfer in serum-containing or serum-free media 2.2 Karyotype of mouse embryo cell cultures 29 2.3 Effect of medium supplements on growth of mouse embryo 31 cells that were derived in serum-free medium 2.4 Growth inhibition of serum-free-derived mouse embryo 33 cells by plasma or serum 3.1 Growth response of SFME cells to insulin and IGF I 52 3.2 Growth response of SFME cells to transferrin and lactoferrin 53 3.3 Growth response of SFME cells to EGF and TGF alpha 54 3.4 Photomicrographs of SFME cells 55 3.5 Recovery of SFME cells from plating in growth-factor-deficient 56 medium 3.6 Growth response of SFME cells to acidic and basic FGF 57 in the absence of EGF 3.7 Growth response of SFME cells to FGF in the presence 58 and absence of EGF and heparin 3.8 Growth response of SFME cells to HDL 59 4.1 Partial removal of inhibitory activity of calf serum for 76 SFME cells by charcoal treatment 4.2 Steroid inhibition of SFME cell growth 77 4.3 Concentration dependence of hydrocortisone inhibition 78 of SFME cell growth 4.4 Concentration dependence of triiodothyronine inhibition 79 of SFME cell growth 4.5 Specificity of thyroid hormone inhibition of SFME cells 80 4.6 Concentration dependence of charcoal-treated calf serum for 81 glucocorticoid and T3 inhibition of SFME cell growth 4.7 Time course and reversibility of triiodothyronine and hydrocortisone 82 inhibition of SFME cell growth 4.8 Partial removal of inhibitory activity of calf serum by 83 anion exchange resin 5.1 Long-term culture of embryonic human astrocytes in serum-free 100 medium 5.2 Photomicrograph of embryonic human astrocytes in serum-free 101 medium. 5.3 Long-term culture of embryonic human astrocytes in medium 102 supplemented with low concentrations of calf serum 5.4 Growth of embryonic human astrocytes in serum-free and 103 serum-supplemented media 5.5 Response of embryonic human astrocytes to peptide growth factors 104 in serum-free medium 5.6 Expression of GFAP in embryonic human astrocytes 105 5.7 Western immunoblot of GFAP in extracts of embryonic human 106 astrocytes in serum-free medium LIST OF TABLES Table ragg 3.1 Effect of attachment factors on SFME cell growth 51 4.1 Calf serum inhibition of SFME cells in the presence of 74 antiglucocorticoid RU38486 4.2 Glucocorticoid and triiodothyronine inhibition of rastransformed SFME cells 75 LIST OF APPENDIX FIGURES Figure Page A1.1 Culture of SFME cells in serum-containing mediu 143 A1.2 Recovery of SFME cells from plating in serum-containing medium 144 A1.3 Flow cytometric analysis of SFME cells in serum-containing medium A1.4 Selection of variant SFME cells capable of growing in serum- 145 146 containing medium A1.5 Karyotype of variant SFME cells selected by culture in serum- 147 containing medium A1.6 Karyotype of variant SFME clones capable of growing in serum- 148 containing medium A1.7 Growth of variant SFME cells in serum-containing and serum-free 149 medium A1.8 Reduced inhibition of variant SFME cells by thyroid hormine, 150 glucocorticoid, and gamma-interferon A2.1 SFME cells cultured with EGF or without EGF for 4 hours 169 A2.2 Cell death and degeneration in SFME cells cultured for 8 hours 172 in the absence of EGF A2.3 SFME cell death and severe degeneration after 24 hours of EGF 175 deprivation. A2.4 DNA fragmentation in EGF-deprived SFME cells 177 A2.5 178 Release of cytoplasmic enzyme in culture medium by SFME cells in the absence of EGF A2.6 Flow cytometric size analysis of SFME cells 179 A2.7 Flow cytometric DNA analysis of SFME cells 180 A2.8 Polysome profiles of SFME cells 181 A2.9 Northern blot hybridization analysis of actin mRNA from SFME cells 182 LIST OF APPENDIX TABLES Ent Table A1.1 Synthetic and enzymatic activities of SFME cells in serum-free 141 serum-containing media A1.2 Karyotypic abnormalities of variant SFME clones capable of growing in serum-containing media 142 Nonsenescent Serum-Free Mouse Proastroblasts: Extended Culture, Growth Responses In Vitro, and Application to the Culture of Human Embryonic Astrocytes CHAPTER 1 Introduction and Review of Literature INTRODUCTION One of the fundamental problems pursued by cell and molecular biologists is the mechanism underlying the developmental regulation of cells in an organism. This is a great undertaking. The sheer number and diversity of cells, their complex organization and interactions, and the difficulty of controlling experimental variables in intact organisms and interpreting observations from these experiments increase the complexity of the whole animal system. One technique scientists have taken in order to study questions directed at complex systems is to break the system down into smaller, more definable pieces. The elucidation of small pieces of the large system may then be taken together to aid in describing the system as a whole. Biologists have used this approach in order to study the cellular and molecular mecnanisms involved in growth and development. This approach necessarily adopts the "cell theory" concepts proposed first by Hooke then expanded upon by others. First, the cell theory recognizes that compartmentalization is a universal phenomenon of all or nearly all parts of both plants and animals, and that therefore the cell is a universal structural unit of all living creatures. Second, the cell theory recognizes that 2 the ubiquitous structural units are also distinct physiological and developmental units, capable of a separate and individual existence under certain conditions. The level of resolution employed by cell biologists is the organ, tissue or cell. Organ or tissue culture involves the explanting of a piece of tissue from an animal directly into an in vitro environment. The tissue may be a thin slice, a larger chunk, or a complete organ. Cell culture involves the culturing of dissociated cells obtained from tissue explants. The dissociated cells may be primary cultures derived directly from tissue, they may be secondary or subsequent passages of cells from primary cultures, or they may be continuous established cell lines. Studies at in vitro levels form the basis for the construction of models which may be used to design and implement experiments in vivo on whole organs or organisms to test the validity of a model. Similarly, models based on results obtained from in vivo studies may be tested and expanded by using the tools of molecular and cellular biology. The combined use of in vivo and in vitro studies provides a powerful approach to study mechanisms of growth control, replication, and differentiation. The research presented in my thesis describes two in vitro cell culture systems. My thesis research focusses on (1) the derivation and long-term culture of SFME cells, (2) defining the growth response of SFME cells to medium supplements, (3) investigating the relationship between the nonsenescent nature of SFME cells and their responses to serum and EGF, and (4) applying the serum-free cell culture methods that allowed the extended culture of SFME cells to the multipassage culture of human embryonic astrocytes. 3 REVIEW OF LITERATURE Animal cell culture. In order for cell culture to be carried out successfully several criteria need to be met. The populations of cells have to be grown from single cells, the cell population has to grow continuously for an extended period of time, the culture has to consist of normal cells, and finally the cells must represent and display the properties of cells from their tissue of origin. Ross Grenville Harrison has been credited with the founding of modern cell culture. In 1907 he performed an experiment designed to elucidate the process that takes place in vivo resulting in extension of nerve fibers between nerve cell bodies during embryonic development (Harrison, 1907). He isolated fragments from frog embryos known to give rise to nerve fibers and cultured them in vitro in a lymph clot on a cover slip. The cover slip was then inverted over a hollow slide and sealed with paraffin. Using this approach Harrison was able to culture the tissue for a period of several weeks and continually observe its development under a light microscope. He observed that in a large number of cases fibers extended from the mass of nerve tissue and extended out into the surrounding lymph clot. The most remarkable feature of the fiber was its enlarged, ameboid end, from which numerous filaments extended. These projections have since been named growth cones. This experiment showed that nerve fibers develop by elongation from the nerve cell body rather than from protoplasmic bridges formed between cells. More important, this work demonstrated that cells could survive and develop in vitro in a manner closely resembling their normal embryonic development in vivo. Having found that tissues from chick embryos grow readily in artificial media, nutrient agar and bouillon, Lewis and Lewis (1911) proposed the great advantages of culturing such tissues in media composed entirely of defined components. In their work they studied the growth promoting effects of sugars and amino acids on chick embryo 4 explants in solutions of various simple salts. However, long-term growth was not achieved. Carrel (1912) proposed that death of cultures may result from preventable occurrences; such as accumulation of catabolic substances and exhaustion of the medium. By passaging chick embryo tissue explants into fresh medium and including plasma and embryo extract in the medium, Carrel found the tissue could be passaged more than 20 times over a period of many months. Carrel's view was generally accepted that the entire growth-promoting activity of his medium for fibroblasts resided in the embryo extract. Plasma was regarded as an almost inert substratum. Because of his success, over the next several decades most investigators adopted modifications of his approach, and little attempt was made to develop more defined media. Based on his earlier work developing media for plant tissue culture, White (1946) developed the first defined medium able to support the prolonged survival but not active growth of heart muscle and skeletal muscle from chick embryos. This medium was based on the current knowledge of the nutrient requirements of animals and included glucose, salts, amino acids, and vitamins. While this medium did not allow extended culture, it demonstrated the potential of fully synthetic media and its use in defining the nutritional requirement of cells. Soon after, Morgan et al. (1950) developed a defined medium designated Mixture 199. This medium contained 68 defined compounds and supported survival of chick embryo tissues for peroids averaging 4-6 weeks; however, no substantial proliferation of cells was obtained. Around this time, Sanford et al. (1948) developed the first permanent cell line. Sanford cloned a strain of transformed, continuously passaged mouse fibroblast cells (L strain) isolated originally by Earle et al. (1943). This development led to an era of advances in the cultivation of animal cells in chemically defined media. Work by several investigators, based on the earlier medium formulations of White (1946) and Morgan (1950), led to the development of the first defined media that would support the active 5 growth of L cells in the absence of serum (Healy et al., 1954; Evans et al., 1955). Healy's medium formulation, which contained 58 defined components, yielded 5- to 6-fold increases of L cells per week. Evan's medium formulation, designated NCTC 109, contained 71 defined components including 25 amino acids, 18 vitamins, 6 coenzymes, 5 nucleic acid precursors, 3 fatty acids, glucose and glutathione. This medium yielded 10- to 13-fold increases of L cells per week. However, the inclusion of horse serum gross globulin greatly increased the mitogenic activity of NCTC 109, indicating the need to identify additional medium supplements. Nearly all the early successes in animal cell cultivation achieved in chemically defined media were obtained with strain L cells. Although investigators developed numerous basal nutrient media and began to optimize the concentration of sugars, salts, amino acids, nucleic acid precursors, vitamins and other components for cells in culture, investigators over the next 20 years still relied on undefined medium supplements in order to achieve acceptable growth of animal cells in culture. The supplement most often used was bovine, calf or fetal calf serum. This practice of supplementation of media with serum is still widely used today. Using dialyzed serum as a medium supplement, Eagle systematically studied the nutritional requirements of mammalian cells in an effort to establish their minimal essential nutritional requirement (1955). These studies were conducted with He La cells, a human cervical carcinoma cell line isolated by Gey et al. (1953), and with the transformed mouse fibroblast L cell line (Sanford et al., 1948). Eagle showed that in addition to a small amount of dialyzed serum, 13 amino acids, 7 vitamins, 6 inorganic ions and glucose or another carbohydrate were required for cell growth. While limited by the inclusion of serum, Eagle's work contributed greatly to the understanding of the nutritional requirement of animal cells and resulted in the formulation of Eagle's minimal essential medium (MEM) (Eagle et al., 1959). Important advances in enzymology also resulted from this work. For instance, it was observed that while glutamine is essential for most serially propagated cell lines, high concentrations of glutamic acid can substitute for glutamine (Eagle et al., 1956). 6 De Mars (1958) observed that at high concentrations of glutamic acid, cells formed greatly elevated levels of glutamine synthetase. This increased enzyme formation was productinhibited; the presence of glutamine inhibited the formation of glutamine synthetase. This was the first reported case of enzyme induction in serially propagated mammalian cell cultures. Hayflick and Moorhead (1961, 1965) described and characterized the development of numerous cell strains of human origin (WI series), derived from fetal tissue and cultured in Eagle's minimal essential medium supplemented with 10% calf serum. One of the goals of this work was to create from normal tissue human cell strains which could be used as models to develop viral vaccines. Cells were passaged twice weekly (2:1 split ratio) for as many as 55 passages over a period of 8 months and retained a diploid karyotype. Without exception, all of the strains were fibroblasts, and all cultures eventually entered a period of growth crisis or senescence and stopped growing. Based on their work Hayjlick and Moorhead advanced the hypothesis that the finite lifetime of diploid cell strains in vitro may be an expression of aging or senescence at the cellular level. Todaro and Green (1963) used a quantitative approach to study the growth of mouse embryo cells in culture. They examined in detail the growth properties of mouse embryo cells from the time they were placed in culture until an established cell line was developed. Particular attention was paid to the cells as they entered growth crisis, or senescence, and as they "converted" to become an immortalized cell line. Using various passaging schemes based on time between passaging and plating density, they observed that all cultures showed at first a progressively declining growth rate, followed by a period of little or no growth, then the emergence, in most cultures, of an established cell line within 3 months of culture. The established cell lines acquired several growth properties distinct from their cells of origin. The established cells had a much greater ability to grow at low cell density and in most cases a greater ability to form high density multilayered cultures. However 3T3 cells (3 day passage, 3 x 105/plate), which unlike most cultures 7 did not reach confluent densities during establishment, remained contact inhibited under normal culture conditions. These cells also remained nontumorigenic, while cells which were not contact inhibited were tumorigenic. Most important, all of the established cells were found to have near-tetraploid karyotypes and other gross chromosomal abnormalities. These chromosomal alterations were observed to occur very rapidly following the emergence from growth crisis, suggesting a chromosomal shift in a large fraction of the diploid or quasidiploid population. Swiss, BALB, and NIH lines (3T3, 3T6, and 3T12), which were derived using this approach, continue to be used extensively in studies of growth regulation, differentiation, and carcinogenesis (Todaro and Green, 1963; Aaronson and Todaro, 1968; Jainchill et al., 1969; Reznikoff et al., 1973). Studies concerning the nutritional requirements of mammalian cells in vitro were continued by Ham and coworkers. They developed media which were optimized for the clonal growth of particular cell types using a systematic approach in which the components of the defined medium were titrated against one another, in the presence of decreasing amounts of serum, until little or no supplementation with dialysed serum was required to obtain clonal cell growth (Ham,1963,1965; McKeehan and Ham,1978; Bettger and Ham, 1981). Using this approach they developed the well-known media F10 and F12 for the clonal growth of Chinese hamster ovary (CHO) cells in the absence of serum, MCDB 105 for the growth of normal diploid fibroblasts in 1% dialyzed serum, and hormonesupplemented MCDB 108 and 110 for the clonal growth of normal diploid fibroblasts in the absence of serum. These medium formulations are still widely used today. Serum-free mouse embryo (SFME) cells were derived in and are routinely cultured in a basal nutrient medium consisting of a one-to-one mixture of Ham's F12 and Dolbecco's modified Eagle's medium (Loo et al., 1987). Another approach taken to understand the role of serum in cell culture was to isolate and characterize growth promoting activities from serum (Temin et al., 1972; Brooks, 1975). In general, these studies were unsuccessful. Purification of growth stimulatory 8 factors from serum has proved to be a very difficult task for numerous reasons. Large amounts of starting material are required, many of the active factors found in serum are bound to carrier proteins, active factors are often synergistically associated with other factors, and finally many of the protein components of serum are physically similar (Barnes and Sato, 1980). Sato and coworkers, recognizing that a primary role of serum was to provide hormones, developed a unique approach which has eliminated the requirement of serum in cell culture medium for many cell types (Sato, 1975, Hayashi and Sato, 1976; Rizzino et al., 1979; Barnes and Sato, 1980a,b; Barnes, 1987). These studies were based on the following reasoning. Cells in vivo exist in and react to a complex set of nutritional, hormonal, and stromal influences. If a cell is to survive or proliferate when removed from an animal and placed in culture, the medium must carry out the functions previously served by the complicated external in vivo environment of the cell. The approach Sato and coworkers took was to begin with culture medium whose serum concentration had been reduced and could not support significant cell proliferation. Purified hormones and serum factors were then added to this medium individually or in various combinations. Factors which restore cell growth were included in the medium and the serum concentration was reduced until cell growth ceased. The process of hormone addition and serum reduction was repeated until the serum was completely eliminated or no further improvements could be made by addition of known factors (Rizzino et al., 1979). The serum factors identified using this approach fall into several categories: hormones or growth factors such as insulin and epidermal growth factor (EGF), transport proteins such as transferrin and albumin, attachment factors such as fibronectin and serum spreading factor, and nutrients such as fatty acids, polyamines and trace elements. Using this serum-free approach, serum-free media have been developed which will support the growth of many cell types, including cells of endocrine origin, epithelial origin, neural and related origins, and mesodermal origin (Barnes, 1987). While most of the cells 9 capable of long-term proliferation in serum free medium are either immortalized cell lines, transformed cell lines or cell lines derived from tumors, several normal cell lines have been derived and maintained long-term in culture under serum-free conditions. Orly et al. (1980) cultured primary rat granulosa cells in a 1:1 mixture of Ham's F12 and Dulbecco's modified Eagle's medium (F12:DME) supplemented with insulin, hydrocortisone, transferrin and fibronectin. The cells remained healthy and responsive to follitropin (FSH) for at least 60 days. Cell response to FSH was suppressed by serum. Using Eagle's MEM supplemented with nonessential amino acids, various trace elements, insulin, transferrin, EGF, and 3, 5, 3'-tri-iodothyronine (T3), Kan and Yamane (1982) cultured human diploid fibroblasts for up to 80 population doublings over a peroid of more than 80 days before growth ceased. The addition of bovine serum albumin (BSA) to the serum-free medium enhanced growth while delipidated BSA was ineffective. Reconstitution of delipidated BSA with certain lipids restored the ability of BSA to enhance growth, suggesting that BSA provided nutritional lipids. Using Ham's F12 supplemented with 0.1-0.5% calf serum and six hormones, Ambesi-Impiombato et al. (1980) cultured primary rat thyroid cells (FRTL strain) for more than three years, with maintenance of highly differentiated functions. Hammond et al. (1984) reported the long-term growth of normal human mammary epithelial cells for more than 70 population doublings by using basal nutrient medium MCDB 170 supplemented with insulin, hydrocortisone, EGF, ethanolamine and bovine pituitary extract. Brandi et al. (1986), using Coon's modified Ham's F12 supplemented with bovine hypothalamic extract, bovine pituitary extract, EGF, insulin, transferrin, selenous acid, hydrocortisone, T3, retinoic acid, and galactose, cultured bovine parathyroid cells for more than 140 population doublings before the culture began to deteriorate. The cells were epithelioid and maintained highly differentiated functions. Finally, in collaboration with my coworkers, I succeeded in the long-term culture of mouse and human astrocyte precursor cells. Serum-free mouse embryo (SFME) cells, 10 cultured in a nutrient medium (F12:DME) supplemented with insulin, transferrin, EGF, high-density lipoprotein (HDL), and fibronectin, have maintained a diploid karyotype with no detectable chromosomal abnormalities for more than 200 generations. The cells did not undergo growth crisis and remain in culture today. SFME cells are absolutely dependent on EGF for survival, growth inhibited by serum or plasma, and nontumorigenic in syngeneic hosts (Loo et al., 1987, 1989a,b, 1990; Rawson et al., 1990, 1991a,b). Treatment of SFME cells with serum or transforming growth factor beta (TGF beta) leads to the appearance of glial fibrillary acidic protein, a specific marker for astrocytes (Sakai et al., 1990; Lazarides, 1982). Human embryonic brain cells, cultured in a nutrient medium (F12:DME) supplemented with insulin, transferrin, EGF, fibroblast growth factor (FGF), heparin, HDL, and fibronectin, were maintained for a maximum of 70 generations before entry into growth crisis (Loo et al., 1991). Human embryonic brain cells expressed glial fibrillary acidic protein (GFAP) in response to serum or TGF beta similar to SFME cells. However, unlike SFME cells the human embryonic brain cells were more responsive to FGF than EGF, they did not acutely require EGF or FGF for survival, and they were not growth inhibited by serum. Most important, the human embryonic brain cells, unlike SFME cells, could not be maintained in vitro for periods beyond that observed with other human cell types in conventional serum-containing medium. Brain cell diversity and mammalian cell culture. In the earliest stages of mammalian development an invasion of mesodermal cells triggers the induction of overlying ectodermal cells to become neuroectoderm (Spemann, 1938). The neuroectoderm then rolls up along the length of the embryo and pinches off to form the neural tube. The cells of the neural tube are precursors for the major differentiated cell types in the central nervous system (CNS): astrocytes, oligodendrocytes, neurons and ependymal cells (Sauer, 1935). Recently, much research has focussed on identifying mechanisms which regulate the production of differentiated cells in the CNS (McKay, 1989). Specifically, investigators 11 are looking at the lineal relationship between neurons, type-1 and type-2 astrocytes, and oligodendrocytes. Neurobiologists are using cell culture methods to study the development of the CNS. Both cell lines and primary culture of brain cells are used. Many cell lines have been established from brain-derived tumors, including the mouse C1300 neuroblastoma cell line (Sato et al., 1969; Schubert et al., 1969), the rat C6 glioma cell line (Benda, et al., 1969), the rat RN22 schwannoma cell line (Pfeifer, 1973), and the rat pheochromocytoma cell line PC12 (Greene, 1976). The cell lines retain differentiated functions which are associated with their cells of origin in vivo. They serve as valuable models for many research interests, including membrane electrophysiology and ion transport, neurotransmitter metabolism, cellular endocrinology, neurotoxicity and regulation of growth and differentiation. The isolation of stem cell lines are of particular interest. One clonal stem cell line, RT4-AC, derived from a rat peripheral neurotumor, spontaneously differentiated in culture into two morphologically distinct cell types: a glial-type cell, which expressed the glial marker S-100 protein, and a neuronal-type cell, which possessed veratridine-dependent sodium influx (Tomozawa and Sueoka, 1978). Established cell lines have also been developed by transformation of embryonic cells with various oncogenes, including c-myc and SV40 large T antigen. Ten-day embryonic mouse neuroepithelial cells transformed with c-myc differentiate in response to FGF into astrocytes, containing GFAP, and neurons, expressing the A2B5 marker and neurofilaments (Bartlett et al., 1988). This finding indicated that some cells in the neuroepithelium at embryonic day 10 are multipotent and are not restricted to either the glial or neuronal cell lineage. Recently, Anderson and coworkers used a retrovirallytransformed adrenal chromaffin cell line to study neuronal development. The cell line they used differentiates to contribute to neurons of the sympathetic nervous system and secretory cells in the adrenal gland (Stemple et al., 1988; Barinaga et al., 1990). Anderson 12 found that FGF induced the cells to synthesize NGF (nerve growth factor) receptors. Following priming by FGF the cells became responsive to NGF. Exposure to NGF caused the cells to differentiate and become sympathetic neurons. In vivo, cells which escape priming by FGF are thought to move on to the adrenal gland where they become secretory cells under the influence of cortisol. A recent approach to develop established cell lines was to derive cultures from the cerebral cortex of transgenic mice that carried the polyoma virus large T antigen gene. Cell lines established by this approach synthesize laminin and neural cell adhesion molecules and induce primary neurons to develop neuritic processes. Upon reaching confluence the cells expressed GFAP, and expression of GFAP is reversibly correlated with cell division arrest (Galiana, et al., 1990). Tumor-derived and oncogene transformed cell-lines have the advantage of providing large quantities of homogeneous populations of cells. It must be remembered however, that these cell lines have acquired properties associated with transformed and malignant cells, and should not be considered to represent normal cells. They may be tumorigenic in vivo and often have reduced requirements for growth factors (Scher et al., 1978; Paul et al., 1978; Cherington et al., 1979; Chiang et al., 1985). Studies using transformed cell lines can be extremely valuable when coupled with complementary in vivo and primary culture studies. Primary cultures have been used extensively to study the in vitro differentiation and interactions of glial and nerve cells. Refinements in chemically defined media have facilitated the growth of purified astrocytes, oligodendrocytes and neurons in culture (Brunner et al., 1982; Bottenstein, 1985, 1986; Morrison and de V ellis, 1984; Conn, 1990). Raff, Noble and coworkers have used primary culture of the rat optic nerve as a model to study glial cell development. The rat optic nerve contains the axons of retinal ganglion neurons that project from the eye to the brain. Oligodendrocytes and astrocytes structurally and functionally support the axons in the nerve. Raff et al. (1983) showed that 13 cultured rat optic nerve contained two types of astrocytes which could be distinguished by morphology, cell-surface markers, and response to growth factors. They have been called type-1 and type-2 astrocytes. Type-1 astrocytes were first detected at embryonic day 16, oligodendrocytes on the day of birth, and type-2 astrocytes at the beginning of the second week following birth (Miller et al., 1985). Using this system, Raff and coworkers showed the rat optic nerve contains a bipotential glial progenitor cell (O -2A progenitor cell) found in the rat CNS whose development was dependent on cell-cell interactions. The 0-2A progenitor cell developed into a type-2 astrocyte or an oligodendrocyte in vitro. Raff suggests that a collaboration between ciliary neurotrophic factor (CNTF) and the extracellular matrix (ECM) influenced the differentiation of 0-2A progenitor cells. CNTF, a protein produced by type-1 astrocytes in culture, induced 0-2A cells to express GFAP in serum-free cultures of perinatal optic nerve cells (Hughes et al., 1988; Lillien et al., 1988, 1990; Raff, 1989). GFAP was expressed only transiently, however, and later the culture reverted to become differentiated oligodendrocytes. The addition of ECM was required to achieve stable differentiation to type-2 astrocytes. Type-1 astrocytes derive from a different precursor cell (Raff et al., 1984). The mechanisms regulating the differentiation pattern of 0-2A progenitor cells are actively being studied. Cell death. The concept of cell death evolved early in the history of cellular pathology and has long been regarded as a degenerative phenomenon resulting from injury. Only recently has cell death been investigated as a mechanism necessary for normal growth and development of multicellular organisms. Based on morphological and biochemical criteria, cell death has been divided into two classifications: necrosis and apoptosis (Wyllie et al., 1980). The occurrence of necrosis has been associated with nonphysiological conditions such as severe hypoxia and ischemia, major changes in environmental temperature, complement-mediated cell membrane disruption, and exposure to toxins. Necrotic insults 14 affect cells in groups rather than singly. No evidence has shown a connection between necrosis and normal embryonic development. Morphological features of necrosis include gross swelling of the matrix of mitochondria, swelling and eventural rupture of nuclear, organelle, and plasma membranes and dissolution of ribosomes and lysosomes. Chromatin initially appears as marginated, loose clumps, but with swelling of the nucleus and rupture of its membrane, the marginated chromatin masses become dispersed and often disappear (Wyllie et al., 1980). In contrast to necrosis, apoptosis or programmed cell death appears to be intrinsically programmed and mediates several important physiological and pathological processes (Wyllie et al., 1980; Arends et al., 1990). It has been shown to occur in developmentally regulated cell death in the embryo (Wyllie, 1981; Searle et al., 1982; Hinchliffe, 1981) and require protein and RNA synthesis (Oppenheim et al., 1990; Schwartz et al., 1990). It also has been observed with the deletion of autoreactive T-cells during thymus development (Smith et al., 1989; S hi et al., 1989) and following the addition of specific regulatory hormones (Cohen and Duke, 1984) or the deprivation of specific growth factors (Kerr et al., 1973; Koury and Bondurant, 1990; Martin et al., 1988; Rodriguez-Tarduchy and Lopez-Rivas, 1989; Kyprianou and Isaacs, 1989). Apoptosis affects single cells rather than groups and has been observed to occur in three morphologically distinct stage (Wyllie et al., 1980; Arends et al., 1990). During the first stage chromatin condenses into a crescent-shaped cap that abuts the nuclear membrane and the nucleus becomes convoluted and shrinks, forming a pyknotic nucleus. The total cell volume decreases, the cytoplasm condenses, and the endoplasmic reticulum swells. Mitochondria and other organelles remain normal. In the second stage, which may overlap with the first stage, the nuclear and cellular membranes bud and separate into multiple membrane-bounded apoptotic bodies containing condensed cytoplasm and in some cases nuclear material. The apoptotic bodies are dispersed in the intercellular spaces and are either transported to an adjacent lumen or phagocytosed by neighboring cells or 15 macrophages. In the final stage degeneration of the nucleus and cytoplasmic structures proceeds, while the cell body remains small. In addition to the morphological uniqueness of apoptosis, a biochemical hallmark of apoptosis has been cleavage of chromatin into oligonucleosome- sized fragments (Arends et al., 1990). Evidence suggests that cleavage results from activation of a calcium- and magnesium-dependent endonuclease activity capable of cleaving chromatin at internucleosomal sites. In many cases, apoptosis has been prevented by inhibitors of RNA and protein synthesis, suggesting that macromolecular synthesis may be required for programmed cell death. Apoptosis has been studied both in vivo and in vitro by using several model systems. In the intact adult male, the supply of androgen regulates a balance between cell death and proliferation of the prostate gland. Castration-induced androgen deprivation in the adult male rat led to loss of nuclear androgen receptors and involution of the ventral prostate. Calcium- and magnesium-dependent DNA fragmentation and accumulation of apoptotic bodies was observed (Kyprianou and Isaacs, 1988). Actinomycin D and cycloheximide, inhibitors of cellular RNA and protein synthesis, partially inhibited regression of the ventral prostate. Primary sympathetic neurons in culture require nerve growth factor (NGF) for survival. Neurons deprived of NGF for 48 hours died, and exhibited ultrastructural and physiological properties consistent with apoptosis (Martin et al., 1988). Agents capable of elevating cAMP prevented neuronal cell death, while activators of protein kinase C did not. As with the rat ventral prostate, inhibitors of RNA and protein synthesis prevented neuronal cell death following NGF withdrawal. Erythropoietin, a glycoprotein produced in the kidney and liver, controls erythrocyte production by regulation of erythroid progenitor cells. In the presence of erythropoietin, progenitor cells survived and differentiated into reticulocytes. In the absence of erythropoietin however, DNA fragmentation and death of erythroid progenitor 16 cells occurred within 16 hours. Cycloheximide inhibits DNA fragmentation and cell death (Koury and Bondurant, 1990). In the interleukin -2 (IL-2) dependent T-lymphocyte cell line CTLL -2, deprivation of IL-2 led to DNA fragmentation and cell death within 10 hours (Rodriguez-Tarduchy and Lopez-Rivas, 1989). Phorbol 12,13-dibutyrate, an activator of protein kinase C, inhibited DNA fragmentation and blocked cell death resulting from IL-2 deprivation. In another system, glucocorticoid hormones have been shown to kill immature thymocytes through activation of programmed cell death (McConkey et al., 1989; Arends et al., 1990). Glucocorticoid treatment of immature thymocytes resulted in an increase in cytosolic calcium, calcium-dependent DNA fragmentation and cell death. Quin-2, which buffers cytosolic calcium, and inhibitors of RNA and protein synthesis limited the increase in cytosolic calcium and prevented DNA fragmentation and cell death. Inhibitors of calmodulin also prevented cell death. Studies which showed that inhibitors of RNA and protein synthesis blocked apoptosis suggests that synthesis of new RNA and protein may be required for activation of the cell death program. In the case of growth factor-deprivation induced apoptosis, presence of the growth factor may suppress the expression of proteins which directly or indirectly participate in programmed cell death. Much attention has focussed on isolating and characterizing genes and proteins whose expression is regulated during programmed cell death. Using the technique of differential cloning, several labs have cloned genes whose expression increased. By using the in vivo rat ventral prostate system, several androgen-repressed genes have been cloned and identified. Expression If sulfated glycoprotein-2 (SGP-2) and the related protein testosterone-repressed prostate message-2 (TRPM-2) were highly increased in the ventral prostate following castration (Montpetit et al., 1986; Leger et al., 1987; Bettuzzi et al., 1989). SGP-2 is a major glycoprotein secreted by Sertoli cells. Other androgen repressed genes include glutathione S-transferase (Bettuzzi et al., 1989), c-myc, 17 c-fos, heat shock protein 70K and alpha tubulin (Ruttyan et al., 1988), as well as other genes yet to be identified. The complexity of this in vivo system has made it difficult to determine the relationship between androgen deprivation and expression of the identified genes. What role any of these genes may play in apoptosis remains to be determined. Genes regulated during programmed cell death in the in vitro systems have not been identified. Orientation to format. The following four chapters and two appendicies constitute results of my thesis research. Each chapter and appendix represents work related to the long-term growth and characterization of mouse and human astrocyte precursor cell lines. Each of the chapters and appendices has been submitted and published in a peer review journal. They are presented in publication format, including a brief SUMMARY, an INTRODUCTION to provide the reader with background information and an introduction to the scientific approach taken, a METHODS section, a RESULTS section, an ILLUSTRATIONS section containing the data of the manuscript, and a DISCUSSION section. I was the principal investigator of the research presented in the first four manuscripts, thererore they represent chapters of my thesis. I was co-principal investigator of the last two manuscripts. Since I was a major contributor to these manuscripts, and the work contributes significantly to my thesis research, they are included as appendices. All references are compiled at the end of the thesis. 18 CHAPTER 2 Extended Culture of Mouse Embryo Cells Without Senescence: Inhibition by Serum Deryk Loo, Janice Fuquay, Cathleen Rawson and David Barnes Published in: Science. Volume 236, pp. 200-202 (1987). Coauthor contribution: D.L., primary investigator; J.F., technical assistant; C.R., post-doctoral scientist; D.B., research director 19 SUMMARY Mouse embryo cells cultured in vitro in serum-supplemented media undergo growth crisis, resulting in the loss of genomically normal cells prior to the appearance of established, aneuploid cell lines. Mouse embryo cells established and maintained for multiple passages in the absence of serum did not exhibit growth crisis or gross chromosomal aberration. Cells cultured under these conditions were dependent on epidermal growth factor for survival. Proliferation was reversibly inhibited by serum or platelet-free plasma, suggesting that mouse embryo cultures maintained by conventional procedures are under the influence of inhibitory factors. 20 INTRODUCTION Mouse embryo cells cultured in conventional serum-supplemented media rapidly lose proliferative potential, leading to growth crisis or senescence followed by the appearance of genomically altered immortalized (established) cell lines. Swiss, Balb, and NIH lines (3T3, 3T6, and 3T12) as well as C3H 10T1/2, which are commonly used in studies of carcinogenesis and growth control, were derived in this way and exhibit near- tetraploid karyotypes with major chromosomal abnormalities (Todaro and Green, 1963; Aaronson and Todaro, 1968; Jainchill et al., 1969; Reznikoff et al., 1973). Because extended culture of some cell types has been achieved by replacing serum in the culture medium (Orly et al., 1980; Barnes and Sato, 1980b; Ambesi-Impiombato et al., 1980; Brandi et al., 1986; Hammond et al., 1984; Masui et al., 1986; Giguere et al., 1982), we developed a serum-free approach to the culture of mouse embryo cells. These procedures allowed extended cell proliferation in the absence of detectable crisis or gross genomic alterations. Growth of cultures derived under these conditions was markedly inhibited by both serum and plasma, suggesting the presence of circulating inhibitors distinct from platelet-associated factors. 21 METHODS AND RESULTS Cells from 16-day-old mouse embryos were cultured in nutrient medium supplemented with 10% calf serum (Todaro and Green, 1963; Aaronson and Todaro, 1968; Jainchill et al., 1969; Reznikoff et al., 1973) or in medium in which serum was replaced with insulin, transferrin, epidermal growth factor (EGF), high-density lipoprotein (HDL), and fibronectin. This medium was developed originally for the growth of the serum-derived, established C3H 10T1/2 and Swiss NIH 3T3 mouse embryo cell lines (Pipas et al., 1983; Chiang et al., 1985) and will also support the growth, at least in the short term, of early passage mouse embryo cells cultured initially in serum. Growth of serum-derived cultures in this medium is enhanced by the addition of platelet-derived growth factor (PDGF). The medium formulation is related in nutritional and hormonal composition to other media developed for the growth of established lines of mouse embryo cells (Serrero et al., 1979; Shipley and Ham, 1981). Cultures carried in serum-containing media, in the presence or absence of additional supplementation with the above factors, underwent a well-defined crisis (Fig. 2.1). A single batch of unheated serum was used for the serum-containing cultures for the duration of the experiment. Similar results were obtained when the experiment was repeated with a different batch of serum or with heat-inactivated serum (56°C, 30 minutes). Cultures maintained in serum-free media grew exponentially without a significant time lag and represented a major portion of the population of embryo cells initially plated. Cell number in primary cultures 6 days after plating was three times higher in serum-free medium than in serum-containing medium. Cells of serum -free, cultures appeared morphologically homogeneous upon light microscopic examination by the second or third passage and remained so upon long-term culture. Cultures were derived in serum-free medium from both Balb/c and Swiss embryos. Initiation and long-term growth in the absence of serum was carried out with three 22 independent Swiss mouse embryo cultures; similar results were observed in all cases. Cells spread poorly in low-density serum-free cultures, occasionally growing as detached clumps. At high density the cells exhibited elongated, bipolar, spindle-shaped morphology and organized into characteristic fibroblastic swirls. Swiss mouse embryo cells cultured for 200 generations in serum-free medium were nontumorigenic as measured by injection of 107 cells subcutaneously into 4- and 7-week-old male and female athymic Swiss mice. Mice were observed for 6 months. Post-crisis mouse embryo cells derived in serumcontaining medium exhibited aneuploid karyotypes as expected, but cells cultured under serum-free conditions retained chromosome numbers in the diploid range (Fig. 2.2). Karyotype analysis by Giemsa banding was carried out at the same time as chromosome counts. All of the metaphases from Swiss mouse embryo cultures in serum- containing medium contained one or more translocated marker chromosomes after 20 population doublings had occurred. Giemsa banding indicated that one marker was the result of a Robertsonian fusion involving chromosome 5 and an unidentifiable chromosome; the other consistently observed marker was a minute chromosome. No chromosomal abnormalities were identified in the serum-free derived cultures. Trans locations were detected in some clones derived from the serum-free Swiss mouse cultures, suggesting that cloning or freezing procedures may affect karyotype. One clone containing a translocation was examined further. This clone remained nontumorigenic and had a modal chromosome number of 40. Cultures derived in serumfree medium contained a small fraction of cells (4 to 7%) with karyotypes in the tetraploid range, similar to that reported for pre-crisis Swiss mouse embryo cells maintained in serum-containing media (Todaro and Green, 1963). Mouse embryo cells derived in serum-free medium required EGF for survival (Fig. 2.3). The EGF requirement was also observed if MCDB 402, a formulation developed for Swiss 3T3 cells (Shipley and Ham, 1981), was used as the basal nutrient medium. Individual omission of the other supplements resulted in decreased cell growth, but did not 23 result in immediate cell death. Flow cytometric analysis indicated that cells maintained in the absence of EGF accumulated in the G1 phase of the cell cycle prior to loss of viability (Rawson and Barnes, unpublished data). The EGF requirement of mouse embryo cells in vitro suggests that this molecule or EGF-like factors (Marquardt et al., 1983) may play a critical role in fetal development. A comparison of biochemical properties suggest that EGF is not the survival factor identified previously for 3T3 cells (Lipton et al., 1972). Mouse embryo cells derived in serum-free medium were capable of growing to very high densities (Fig. 2.2 ). Under these conditions the cells were smaller than at lower densities, forming multilayered piles and aggregates. It has been shown previously that fibroblastic cultures can be grown to high cell densities if adequate nutritional conditions are maintained (Dulbecco and Elkington, 1973). The apparent decrease in growth rate of serum-free mouse embryo cultures at high density was the result of both increased cell loss due to depletion of nutrients and accumulation of cells in G1 due to rapid depletion of EGF. Survival of the cultures at very high cell densities was precarious because of rapid depletion of EGF. Addition of serum to the culture medium led to inhibition of growth; supplementation of serum-containing medium with the serum-free medium marginally improved cell growth (Fig. 2.3). Inhibition of cell proliferation was reversible upon replacement of serum-containing with the serum-free medium. Flow cytometric analysis indicated that cells in serum-containing medium accumulated in the G1 phase of the cell cycle. Both calf serum and platelet-free plasma were effective at inhibiting cell growth (Fig. 2.4). In addition to the inhibitory activity of plasma factors, platelet-derived growth factors (Masui et al., 1986; Tucker et al., 1984) also may be inhibitory for serum-freederived mouse embryo cells, since the inhibitory activity per milligram of protein was somewhat higher for serum than for plasma. Inhibitory activity was detected in six 24 commercial lots of calf serum, all of which were capable of supporting the growth of 3T3 mouse embryo cell lines established by conventional procedures. 25 DISCUSSION Our work indicates that mouse embryo cells exhibiting the characteristics of precrisis cultures can be maintained on a long-term basis if serum is replaced by hormonal and nutritional supplements to the basal nutrient medium. Although the precise relationship of the cell type in the cultures derived in serum-free medium to those derived in serumcontaining media remains to be resolved, several lines of evidence suggest that the serum- free-derived cells may be of mesodermal origin, like serum-derived lines. Under appropriate conditions the cells derived in serum-free medium were capable of expressing classical fibroblastic morphology. In addition, malignant cells derived from serum-free cultures by calcium phosphate-mediated oncogene transfection formed undifferentiated sarcomas in athymic mice (Fuquay et al., unpublished data). The phenomena of crisis (or senescence) of diploid rodent embryo cells that have been maintained in serum-supplemented media and the transfection-induced immortalization or establishment of genomically altered lines are being actively studied. Inhibition of growth by serum or plasma factors and selection of cells unresponsive to these factors may contribute to the processes of crisis and immortalization under conventional culture conditions. Although we found it possible in serum-free medium to carry out extended culture of mouse embryo cells without an obvious crisis, human fibroblasts have been reported to undergo senescence in serum-free medium at approximately the same number of population doublings as serum-grown cultures (Kan and Yamane, 1972). These results may reflect differences in species or in serum-free medium formulations. In any case, mouse embryo cells in serum-free media may provide a stable model system for the identification and study of growth inhibitors and examination of the mechanisms by which cells become hyperploid, malignantly transformed, or otherwise genomically aberrant. 26 ACKNOWLEDGMENTS We thank A. Rizzino for calf serum and plasma and information on TGF beta concentrations, K. Nusser and A. Helmrich for assistance and suggestions, T. Ernst and W. Peterson for karyotype analysis, and J. Willard for flow cytometry assistance. D.B. is a recipient of a Faculty Research Award from the American Cancer Society. Janice Fuquay prepared mouse embryo cells for karyotyping. Cathleen Rawson prepared culture medium and assisted with cell culture. David Barnes provided financial and scientific support. 27 Fig. 2.1. Growth of Balb/c mouse embryo cells upon successive transfer in serum-containing or serum-free media. Cultures were grown in (0), medium supplemented with 10% calf serum (Gibco); (0), serum-free medium supplemented with bovine insulin (10 µg/ml; Sigma), human transferrin (20 pg/m1; Sigma), EGF (50 ng/ml; Collaborative Research), human HDL (20 µg/ml; Meloy) on flasks precoated with human fibronectin (20 µg/m1; Meloy) as described (Barnes et al., 1983), except without the use of bovine serum albumin; (II), cultures containing both the serum-free supplements and 10% calf serum. Mouse embryo cells were prepared as described (Todaro and Green, 1963). Cultures were initiated at 105 cells per 25-cm2 flask, medium was changed 3 or 4 days after plating, and cells were passaged 6 or 7 days after plating. Cells remained subconfluent under these circumstances. Serum-free cultures were passaged with the use of trypsin and trypsin inhibitor (Barnes and Sato, 1980b). Cell number per flask was determined on the first day after plating and at passage by counting suspensions of trypsinized cells in a Coulter particle counter and by hemocytometer. Three or more replicate flasks were initiated at each passage; cell number per flask for replicates varied less than 10% from the average. Nutrient medium was a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 (Gibco) containing 10 nM sodium selenite, sodium bicarbonate (1.2 g/liter), 15 mM Hepes buffer, penicillin (200 IU/ml), streptomycin (200 gg/m1), and ampicillin (25 p.g/ml) (Barnes and Sato, 1980b). 28 Figure 2.1 1024 1020 1016 1012 108 104 100 Days 29 Fig. 2.2. Karyotype of mouse embryo cell cultures. (A) Swiss mouse embryo cells cultured in medium containing 10% calf serum for two population doublings in vitro; 25-hour doubling time (primary culture). (B) Swiss mouse embryo cells cultured in medium containing 10% calf serum for 20 population doublings; 23-hour doubling time (post-crisis culture). (C) Swiss mouse embryo cells cultured in serum-free medium for 200 population doublings; 20-hour doubling time. (D) Balb/c mouse embryo cells cultured in medium containing 10% calf serum, seven population doublings; 30-hour doubling time. (E) Balb/c mouse embryo cells cultured in serum-free medium for 70 population doublings; 24-hour doubling time. Balb/c cultures and Swiss cultures in serum-free medium were initiated and passaged as described previously for the derivation of Swiss 3T3 lines (Todaro and Green, 1963). Swiss cells in serum-free medium were maintained initially in the presence of human PDGF (1 unit/m1; Collaborative Research). PDGF stimulation of cell growth in the presence of the other supplements was minimal, and addition of PDGF was discontinued at 60 population doublings. 30 Figure 2.2 60 A 1 20 15 5 40 0 5 D 1 , 50 I I, 1111a . 10 20 60 100 Chromosomes per metaphase 31 Fig. 2.3. Effect of medium supplements on growth of mouse embryo cells that were derived in serum-free medium. Cultures were grown in (0), serum-free medium with supplements as described in Fig. 2.1; (), medium supplemented with 10% calf serum; (), medium containing both the serum-free supplements and 10% calf serum; (0), serum-free medium with all supplements as described in Fig 2.1 except EGF; (A), medium supplemented with 10% calf serum for the first 3 days followed by a change to serum-free medium with the supplements. Swiss mouse embryo cells initiated and grown for approximately 200 generations in serum-free medium were used for the experiment. Similar results were obtained with Balb/c mouse embryo cells that had been derived in serum-free medium. Cells were plated at 105 per 35-mm dish. Cell number was determined at the indicated times after plating by counting cell suspensions in a Coulter particle counter, individual values varied less than 10% from the average. All cells in the serum-free plates from which EGF was omitted were dead by day 5. 32 Figure 2.3 107 3 x 106 .c i 0) 106 a. ,A N 15 c 3 x 105 a 105 a 0-1...sa_.- .4`-=-M.."--.0-. 1- WEI '0 3 x 104 1 1 3 % i I I I 5 7 9 11 Days after plating 33 Fig. 2.4. Growth inhibition of serum-free-derived mouse embryo cells by plasma or serum. Cells were plated in serum-free medium supplemented as described in Fig. 2.1 in the presence of the indicated concentrations of calf serum (I) or plasma (0). Cell number was determined 6 days later by counting cell suspensions in a Coulter particle counter, individual values varied less than 10% from the average. Plasma was defibrinated before use as described (Pledger et al., 1978) and serum was similarly treated. Cells were plated at 105 per 35-mm dish. Swiss mouse embryo cells initiated and grown for approximately 200 generations in serum-free medium were used for the experiment. Similar results were obtained with serum-free-derived Balb/c mouse embryo cells. Assay of calf plasma indicated less than 2 pg of transforming growth factor beta per milligram of plasma protein. 34 Figure 2.4 4 0 31 omm, 0 0 0.1 1.0 Plasma or serum protein (mg/ml) 10 35 CHAPTER 3 Serum-Free Mouse Embryo Cells: Growth Responses In Vitro Deryk Loo, Cathleen Rawson, Angela Helmrich, and David Barnes Published in: Journal of Cellular Physiology. Volume 139, pp. 484-491 (1989). Coauthor contribution: D.L., primary investigator; C.R., post-doctoral scientist; A.H., technical assistant; D.B., research director 36 SUMMARY We have derived serum-free mouse embryo (SFME) cultures in a basal nutrient medium supplemented with insulin, transferrin, epidermal growth factor (EGF), highdensity lipoprotein (HDL), and fibronectin. These cells are nontumorigenic, lack gross chromosomal aberrations, and exhibit several other unique properties, including dependence on EGF for survival and growth inhibition by serum. We have examined the concentration dependence of the growth stimulatory effects of protein supplements used in the SFME medium formulation and surveyed other supplements that might act as alternative or complementary additions to the culture medium. Insulin could be replaced by insulinlike growth factor I and EGF could be replaced by transforming growth factor alpha in the same concentration range. Transferrin could be replaced by higher concentrations of lactoferrin. Deterioration of cultures in the absence of EGF began within 8 hours of the removal of the growth factor, and could be prevented by the addition of fibroblast growth factor/heparin-binding growth factor. Attachment proteins other than fibronectin were effective on SFME cells, but limited success was obtained when substituting other lipid preparations for HDL. These data introduce a precise system for exploring the unusual characteristics of SFME cells and contribute additional information that may be useful in the extension of these approaches to other cell types and species. 37 INTRODUCTION Mouse embryo cells cultured multipassage in basal nutrient medium supplemented with serum eventially undergo growth crisis or senescence, often followed by the appearance of genomically altered, immortalized and sometimes tumorigenic cell lines (Todaro and Green, 1963). Replacing serum in the culture medium by growth factors and other supplements allows extended culture of some cell types that cannot be maintained on a long-term basis in conventional serum-containing media (Ambesi-Impiombato et al., 1980; Orly et al., 1984; Gospodarowicz et al., 1981; Hammond et al., 1984; Brandi et al., 1986). We have applied a similar serum-free approach to the culture of mouse embryo cells. We have reported that mouse embryo cultures can be initiated and maintained on a multi-passage basis in a rich basal nutrient medium supplemented with insulin, transferrin, epidermal growth factor (EGF), high-density lipoprotein (HDL), and fibronectin (Loo et al., 1987). Serum-free mouse embryo (SFME) cells derived in this manner display several unique properties. SFME cells do not exhibit growth crisis or gross chromosomal aberration and are nontumorigenic in syngeneic or athymic mice; these cells are dependent on EGF for survival and are growth inhibited by serum or platelet-free plasma (Loo et al., 1987). Because an appreciation of the behavior of SFME cells in serum-free culture is necessary to further pursue an understanding of the unusual properties of the cells, as well as to allow the further application and modification of this approach to cells derived from embryos, neonates, or adults of mice and other species, we have examined in detail the behavior of SFME cells in serum-free culture. In this report we characterize the effects on SFME cells of growth stimulatory protein supplements to the basal serum-free medium. 38 MATERIALS AND METHODS Materials. Bovine insulin, human transferrin, human lactoferrin, trypsin, bovine serum albumin (BSA), heparin, polylysine, and soybean trypsin inhibitor were obtained from Sigma Chemical Corp. (St. Louis, MO). Nerve growth factor (NGF) was obtained from Bioproducts for Science, Inc. (Indianapolis, IN). Recombinant human insulin-like growth factor (IGF I) was obtained from Amgen (Thousand Oaks, CA). Synthetic human transforming growth factor alpha (TGF alpha) was obtained from Peninsula Laboratories (Palo Alto, CA). Mouse laminin and type IV collagen was obtained from BRL (Bethesda, MD). Type I collagen was obtained from Collagen Corp. (Palo Alto, CA). Bovine acidic fibroblast growth factor/heparin binding growth factor-1 (endothelial cell growth factor; Schreiber et al., 1985) and basic fibroblast growth factor/heparin binding growth factor-2 (FGF/HBGF), human transforming growth factor beta (TGF beta), and human platelet- derived growth factor (PDGF) was obtained from R and D Inc. (Minneapolis, MN). Mouse EGF was obtained from Upstate Biotechnologies (Lake Placid, NY). Powdered medium formulations and antibiotics were obtained from Grand Island Biological Company (Grand Island, NY). Sodium selenite and ethylenediaminetetraacetate (EDTA) were obtained from Fisher Scientific (Pittsburgh, PA). 4-(2-hydroxy- ethyl)-1-piperazine-ethanesulfonic acid (HEPES) was obtained from Research Organics, Inc. (Cleveland, OH). Gelatin-Sepharose was obtained from Pharmacia Chemicals (Piscataway, NJ). Plasticware for cell culture was obtained from Falcon. Human serum spreading factor was prepared as described (Barnes and Silnutzer, 1983). Human plasma was obtained from volunteers. Serum-free cell culture procedures. Cultures of Balb/c SFME cells giving rise to the continuous cultures used for the experiments described in this report were initiated from 16-day-old mouse embryos (Loo et al., 1987). The embryos were minced and dispersed in 0.25% crude trypsin with 1 mM EDTA in phosphate-buffered saline without calcium or 39 magnesium (PBS) and incubated for 15 minutes at 37°C. Large chunks of tissue were discarded, and the cell suspension was centrifuged at low speed from the trypsin solution and resuspended in 10 ml of serum-free basal nutrient medium containing 1 mg/ml soybean trypsin inhibitor. Cells were then recentrifuged, resuspended in fresh serum-free basal nutrient medium, and plated in serum-free basal nutrient medium with growth stimulatory supplements as described below. Passaging of SFME cells was accomplished by trypsinization followed by dilution of the cells into an equal volume of soybean trypsin inhibitor solution and centrifugation from solution. Cells were routinely grown in a 5% CO2-95% air atmosphere at 37°C and passaged weekly, plating at a density of 105 viable cells/25 cm2 flask. SFME cells are extremely sensitive to trypsin; only a few seconds of exposure to the trypsin solution at room temperature was necessary for removal from the flask and small volumes of trypsin and trypsin inhibitor solution are needed (0.5 ml per 25 cm2 flask). Loss of cells sometimes occurred when passaging under serum-free conditions if dilute cell suspensions were manipulated. This is presumably due to adherence of the cells to pipets and tubes. The problem could be reduced somewhat by maintaining high cell densities in the suspensions during passaging and by the addition of 1 mg/ml BSA (Cohn Fraction V) to the trypsin inhibitor solution. SFME cultures have been passaged continuously without evidence of growth crisis or senescence for 200 doublings (Loo et al., 1987). Serum-free culture medium. The basal nutrient medium was a one-to-one mixture of Dulbecco-modified Eagle's medium containing 4.5 g/L glucose and Ham's F12 (Ham and McKeehan, 1979) supplemented with 15 mM HEPES, pH 7.4, 1.2 g/L sodium bicarbonate, sodium selenite (10 nM), penicillin (200 U/ml), streptomycin (200 µg/ml), and ampicillin (25 tg/m1) (F12:DME) (Mather and Sato, 1979). Details and general approaches of serum-free culture techniques and medium preparations are published elsewhere (Ham and McKeehan, 1979; Sato et al., 1982; Bettger and Ham, 1982; Barnes, 1987). Cells were cultured in F12:DME supplemented with insulin (10 gg/m1), transferrin 40 (25 14/m1), human high-density lipoprotein (HDL) (10 p.g/ml), and EGF (50 ng/ml), in dishes or flasks precoated with bovine fibronectin (10 gg/m1). Plating was accomplished by adding the appropriate cell number in 1 ml of prewarmed medium to culture vessels previously precoated with fibronectin and preincubated in the incubator for approximately 15 minutes with the remaining portion of medium (e.g., 1 ml of cell suspension added to 1 ml of medium in a 35 mm-diameter plated or 4 ml of medium in a 25 cm2 flask). Insulin, transferrin, EGF, and HDL were then added directly to the individual plates or flasks as small aliquots from concentrated stocks. Insulin was prepared at 1 mg/ml in 20 mM HC1 and transferrin at 5 mg/ml in F12:DME; both were filter sterilized. EGF was obtained as sterile, lyophilized powder and reconstituted at 50 1.4/m1 with sterile PBS. FGF/HBGF was obtained as sterile, lyophilized powder and reconstituted at 10 pg/ml with sterile PBS containing 1 mg/ml BSA. Stocks of insulin, transferrin, EGF and FGF/HBGF were stored at -86°C in aliquots until needed, then stored at 4°C after thawing. HDL (density=1.068-1.121 g/cc) was prepared by KBr ultracentrifugation as described (Gospodarowicz, 1984) and filter sterilized. Protein concentration of HDL stocks were 10-20 mg/mi. Fatty-acid-free BSA was added to the HDL solutions at a final concentration of 1 mg/ml to act as a trap for toxic lipid liberated by decomposition of the HDL with time. Stocks of HDL were stored at 4°C. Fibronectin was prepared by gelatin-affinity chromatography (Ruoslahti et al., 1982) and filter sterilized. Stocks were maintained at approximately 1 mg/ml in 50 mM Tris, pH 7.5, containing 4 M urea. Fibronectin was provided to cells by precoating plates with a solution of fibronectin at the indicated concentration made by adding a small volume of concentrated stock to F12:DME (1 ml per 35 min-diameter plate) and incubating for 30 minutes at 37°C in a CO2 incubator, followed by removal of the precoating solution and one wash with 2 or 5 ml of F12:DME. Stocks of fibronectin were stored at -86°C in aliquots until needed, then stored at 4°C after thawing. 41 Experimental procedures. Cells cultured continuously in the complete serum-free medium were detached from stock flasks by trypsinization, diluted into trypsin inhibitor solution, centrifuged, resuspended, counted, and plated at 105/35 mm-diameter dish (or, otherwise, at the density indicated) in media of the indicated composition. Cell number was determined 5 days after plating, or otherwise at the indicated times after plating, by counting trypsinized cell suspensions in a Coulter particle counter. Data shown are the average of values from duplicate plates. Individual values among replicate plates varied less than twelve percent from the mean. Stock SFME cultures used for the experiments described in this report had undergone between 70 and 100 population doublings. 42 RESULTS Effects of insulin and transferrin on SFME cells. Virtually all cell types in serum- free culture are responsive to insulin (Sato et al., 1982; Barnes, 1987). The lowest concentration of insulin to which SFME cells responded maximally was 3 .tg/m1 (Fig. 3.1). Response of cells to nonphysiological concentrations of insulin often is taken to indicate that insulin is mimicking the action of IGF's, and these peptide growth factors frequently are found to stimulate growth at concentrations that are lower than the effective concentrations of insulin (Rosenfeld and Hintz, 1986). However, we found for SFME cells that IGF I was effective at stimulating cell growth in approximately the same concentration range as insulin (Fig. 3.1). No additional effect of IGF I (1 tg/ml) was observed if it was added to SFME cells in the presence of a maximally effective concentration of insulin (10 1.4/m1). The IGF used in these experiments was maximally active at 1-5 ng/M1 in the stimulation of thymidine incorporation in chick embryo fibroblasts, a standard assay of insulin-like growth factor activity, and similar results on SFME cells were obtained with two different IGF lots (not shown). As with insulin, almost all cell types in serum-free culture are responsive to transferrin (Sato et al., 1982; Barnes, 1987). The lowest concentration of transferrin to which SFME cells responded maximally was 10 p.g/m1 (Fig. 3.2). Lactoferrin has been reported to stimulate cell growth and replace transferrin for some cell systems (Hashizume et al., 1987), and we found that lactoferrin also would stimulate SFME cell growth, although effective concentrations of lactoferrin were approximately tenfold higher than those of transferrin (Fig. 3.2). No additional effect of lactoferrin (30 pg/m1) was observed if it was added to SFME cells in the presence of a maximally effective concentration of transferrin (30 µg/ml). Effects of EGF and FGF /HBGF on SFME cells. Many cell types in serum-free culture are mitogenically responsive to nanomolar concentrations of EGF (Sato et al., 1982; 43 Barnes, 1987). Maximal stimulation of SFME cell growth by EGF was observed at 30 ng/ml (Fig. 3.3). TGF alpha, which binds to the EGF receptor and mimics the action of EGF (Marquardt et al., 1984), was also effective in the same concentration range as EGF, although EGF was of somewhat higher specific activity than TGF alpha. Addition of both EGF and TGF alpha together at 100 ng/ml produced an effect no larger than the addition of either separately. SFME cells were strikingly dependent on EGF for survival in serum-free medium; when EGF was omitted from the medium, greater than 90% of the cells were dead within 24-48 hours (Fig. 3.4). This result was qualitatively different from those obtained when any of the other medium supplements were singly omitted from the medium; SFME cells in the presence of EGF and the individual absence of any of the other components were capable of survival and growth, although cell number under these conditions was lower than that of control plates in the complete serum-free medium (e.g., Figs. 3.1, 3.2, 3.4). In experiments in which EGF was omitted and then restored to the medium and the cultures allowed to recover for 4 days in the complete serum-free medium, we found that incubations without EGF for as little as 8 hours led to significant reduction in the ability of the cultures to recover, suggesting that cell death begins in the cultures very soon after the omission of EGF (Fig. 3.5). By comparison, little time-dependent effect of the omission and readdition of insulin was observed in this protocol. Neither PDGF (1-10 ng/ml), NGF (1-10 ng/ml), nor TGF beta (0.1-10 ng/ml) could replace EGF for survival or growth of SFME cells, but cells did survive and proliferate slowly if EGF was replaced with FGF/HBGF (Figs. 3.4, 3.6). TGF beta added in addition to EGF resulted in no effect or variable, small growth inhibition, although this factor markedly improved cell adhesion and spreading. PDGF added in addition to EGF was marginally stimulatory: about a 10% increase in cell number over controls without PDGF was observed. 44 Both acidic FGF/HBGF-1 (endothelial cell growth factor) and basic FGF/HBGF-2 were effective and proliferation-promoting effects of FGF also were observed when the growth factor was added to the complete serum-free medium containing EGF (Figs. 3.6, 3.7). Heparin potentiated the effect of both basic and acidic FGF/HBGF (Figs. 3.6, 3.7). Potentiation of the FGF effect is thought to occur because the growth factor is more stable when bound to heparin (Schreiber et al., 1985; Gospodarowicz and Cheng, 1986; Klagsbrun et al., 1987). Effects of attachment proteins and HDL on SFME cells. In addition to hormones like insulin, IGF I, EGF, FGF, or TGF alpha and binding proteins like transferrin or lactoferrin, attachment factors are also required for the proper anchorage of cells in serum- free culture (Barnes, 1987). SFME cells were derived from primary culture using fibronectin as an attachment factor, and responded maximally to about 10 14/m1 of this protein (Table 3.1). Laminin and serum spreading factor, other attachment factors used in serum-free medium formulations for various cell types (Grotendorst et al., 1982; Barnes and Silnutzer, 1983; Barnes, 1987), also supported attachment and stimulated growth of SFME cells (Table 3.1). Combinations of these glycoproteins with fibronectin did not produce effects on cell growth larger than the effects of each alone. Polylysine precoating of culture dishes as described by McKeehan and Ham (McKeehan and Ham, 1976) was effective at promoting attachment, but was inhibitory for cell growth unless fibronectin was also present. Type I and type IV collagen and gelatin were ineffective at supporting SFME cell attachment or stimulating cell growth (not shown). Interestingly, SFME cells in the absence of fibronectin were capable of extended growth as unattached multi-cellular aggregates. Cells in plates without fibronectin underwent about five population doublings in 6 days (Table 3.1). In many cases, additional nutritional lipid supplements beyond those provided by the basal medium are required for maximal growth of cells in serum-free culture (Ham and McKeehan, 1979; Sato et al., 1982; Bettger and Ham, 1982; Barnes, 1987). HDL is 45 growth stimulatory for many cell types in serum-free medium (Gospodarowicz, 1984; Rizzino, 1984; Hoshi and McKeehan, 1984), and is also effective on SFME cells (Fig. 3.8). Delipidated (apo) HDL did not exhibit the stimulatory activities of HDL on SFME cells. Low-density lipoprotein (LDL) at similar concentrations was growth stimulatory, but considerable variability in activity was observed among LDL preparations, and some preparations were toxic at doses higher than 1 pg/ml. Very low-density lipoprotein (VLDL) was inhibitory even at 1 µg/ml (not shown). These effects are presumably due to the toxic nature of the particular lipid components or lipid decomposition products of these lipoprotein fractions. The lipid composition of HDL is primarily phospholipid and fatty acid cholesterol esters (Gospodarowicz, 1984). Replacement of HDL with linoleic acid, oleic acid, arachadonic acid, dioleoyl phosphatidyl choline, dilinoleoyl phosphatidyl choline, diarachadonyl phosphatidyl choline, or cholesterol, with or without fatty-acid-free BSA present as a lipid carrier, produced modest stimulatory effects in some cases, but the effect of HDL could not be entirely replaced by these supplements and toxicity was observed at higher doses of some lipids (e.g., greater than 500 nM linoleic acid) due to detergent effects (not shown). 46 DISCUSSION SFME cells exhibit a number of unusual characteristics for cells that can be grown indefinitely in vitro. These cells are growth inhibited by serum or platelet-free plasma and dependent on EGF (or FGF) for survival (Loo, et al., 1987). Here we have described the responses of SFME cells to growth stimulatory supplements included in the serum-free medium formulation in which these cells were derived, and also characterized the effects of FGF on these cells. The formulation used for SFME cells was developed originally for serum-free culture of C3H 10T1/2 and Swiss NIH 3T3 mouse embryo cell lines (Pipas et al., 1984; Chaing et al., 1985) and is related in nutritional and hormonal composition to other media developed for the growth of established lines of mouse embryo cells and related cell types (Serrero et al., 1979; Darmon et al., 1981; Shipley and Ham, 1983). The serum-free medium employed embodies a highly specific formulation that imparts an intense selective pressure for the SFME cell type and the distinctive survival and growth characteristics described. Not only are these cultures selected in the absence of serum proteins such as albumin and platelet factors such as platelet-derived growth factor and transforming growth factor beta, but also are cultured in lower concentrations of transferrin, HDL, and fibronectin than one might expect to find in conventional serumsupplemented medium. Thus, the extracellular environment governing the survival and growth of our serum-free cultures is significantly different than that of cells cultured in medium supplemented with 10% serum. Although it is not surprising that SFME cells respond to insulin and transferrin, it is unusual to find that insulin and IGF I are effective at similar concentrations. Although the optimal concentration of insulin or IGF I for growth stimulation of SFME cells is higher than might be expected to exist under physiological conditions, suboptimal growth- promoting effects of concentrations in the nanomolar range were observed for both 47 peptides. The relevant receptor presumably is of low affinity for these ligands; perhaps both insulin and IGF I are interacting with the IGF II receptor. The ubiquitous requirement of serum-free cultures for transferrin (Sato et al., 1982; Barnes, 1987) is presumed to be due to the function of this protein as a carrier of iron, and the iron-binding lactoferrin molecule could replace transferrin for SFME cells, as has been observed for some other cell types (Hashizume et al., 1987). The optimal concentration of transferrin for growth stimulation of SFME cells was within the physiological range; the concentration of transferrin in serum is greater than 1 mg/nil (Beck, 1982). Both EGF and TGF alpha at physiologically relevant concentrations were capable of supporting survival and growth of SFME cells. These peptides in the range of 10-100 ng/ml were strongly growth stimulatory for SFME cells, but even 1 ng/ml was capable of preventing cell death and supporting some proliferation beyond the initial plating density (Fig. 3.3). TGF alpha levels can reach 1 ng/ml in culture medium from cells producing this peptide and are present at about 1 ng/mg protein in mouse embryos (Twardzik et al., 1982; Salomon et al., 1987). The level of circulating EGF in the mouse is about 1 ng/ml, but can be boosted to over 100 ng/ml by appropriate stimulation of the submaxillary gland (Carpenter and Cohen, 1979). Circulating levels of EGF in vivo should be sufficient to allow survival of SFME cells injected into mice in tumorigenicity studies, especially when one considers that the concentration in vivo is constant, while EGF levels in the cell culture experiments described were diminished over the course of the experiment. Replacement of EGF with FGF allowed survival and slow growth of the cells, but the rate of proliferation was reduced compared to that in the presence of EGF, and the gross morphology of cells was different in medium containing EGF compared with medium in which EGF was replaced with FGF. The stimulation of SFME cell growth by FGF in medium containing EGF suggests that serum-free medium containing FGF in addition to the other components might be useful in the initiation and multipassage culture of rodent embryo cells. We have utilized medium containing 10 ng/ml FGF in addition to 48 the other supplements used routinely with SFME cells to initiate cultures from C3H mouse and Fischer rat embryos, as well as newborn Balb/c mice, and have obtained cells with properties similar to those of Balb/c SFME cells. Cell types derived in serum-containing medium, including mouse embryo lines such as the various 3T3 and 10T1/2 strains as well as human fibroblasts, are generally mitogenically responsive to EGF or FGF (Serrero et al., 1979; Darmon et al., 1981; Shipley and Ham, 1983; Pipas at al., 1984; Chiang et al., 1985; Phillips and Cristofalo, 1988), but are not dependent on these peptides for survival. The cellular function required for survival of SFME cells that is presumably met by activation of the EGF/TGF alpha receptor is unknown, as is the mechanism by which FGF supplants the requirement. The dramatic requirement of the peptide growth factors for survival of these mouse embryo cells in culture suggests that an EGF-like or FGF-like factor may be critical for proper fetal development. SFME cells responded to fibronectin and other glycoprotein attachment factors in serum-free culture. Even in the absence of any attachment factor, SFME cells were capable of sustained growth. The ability to grow under anchorage-independent conditions is a phenomenon not usually observed in nontumorigenic cells that also are capable of adherent growth. SFME cells grow to very high densities in adherent culture (Fig. 3.6), also an unusual property for nontumorigenic cells. Extended maintenance of high-density cultures leads to an apparent plateau in cell number per plate that is the result of increased cell loss due to medium depletion of nutrients and growth arrest prior to cell death due to depletion of growth factors (Loo et al., 1987). SFME cells in high-density culture grow in multilayered arrangements throughout the dishes, and no formation of foci is observed. Nutritional lipids were supplied to the basal nutrient medium by the addition of HDL. The concentration of human HDL used in serum-free medium for SFME cells (10 14/m1) is considerably less than that found in human plasma (about 1 mg/ml). Limited 49 success was obtained when substituting other lipoprotein fractions or lipid preparations for HDL. Although most of the growth stimulatory supplements to the basal nutrient medium used for SFME cells are present in commercial sera, SFME cells do not grow in conventional serum-containing media (Loo et al., 1987). The serum-free cell culture techniques described here allow the elimination of undefined inhibitory serum factors from the medium and also provide an assay system for the identification and study of these inhibitory activities. In addition to usefulness in the study of growth-inhibitory factors, SFME cells also provide a novel model for the study of other phenomena related to the regulation of proliferation of animal cells, including the exceptional activities of peptide growth factors and actions of oncogenes to block serum inhibition of growth or mimic peptide growth factor action (Barnes et al., 1989). The explicit control of the culture environment afforded by the SFME system confers advantages in approaching these topics experimentally, as well as providing the only means by which cells exhibiting these properties have been isolated. Application of these techniques to the direct serum-free culture of untransformed cells from other species, including humans, should provide interesting results in the future. 50 ACKNOWLEDGMENTS We thank Kevin Nusser and Wayne Englander for help with this work. Support was provided by NM-NCI 40475 and NIH-AG 07560. D.B. is the recipient of Research Career Development Award 01226 from the National Cancer Institute. Cathleen Rawson prepared HDL and performed insulin/IGF I dose response experiment. Angela Heinrich prepared medium and performed experiments involving attachment factors. David Barnes provided financial and scientific support. 51 Table 3.1. Effect of attachment factors on SFME cell growth. Cells were plated at 105/dish in F12:DME medium supplemented with insulin (10 gg/m1), transferrin (2514/m1), HDL (10 µg/ml), and EGF (50 ng/ml). Dishes were precoated with attachment factors as indicated (1 ml precoating solution/plate) and washed prior to plating as described in Materials and Methods. Cell number was determined 5 days after plating. Attachment factor Experiment 1 None Fibronectin (1 µg /ml) Fibronectin (2.5 µg /ml) Fibronectin (5 14/m1) Fibronectin (10 µg /ml) Fibronectin (25 µg /ml) Experiment 2 None Fibronectin (10 µg /ml) Serum spreading factor (10 µg /ml) Laminin (10 µg /ml) Polylysine (100 µg /ml) Fibronectin (10 p.g/m1) + Serum spreading factor (10 i.g /ml) Cells/dish 4.1 4.7 5.1 5.3 5.9 6.1 (±.13) x 106 (±.19) x 106 (-±.17)x 106 (±.17) x 106 (-±.36)x 106 (±.72) x 106 4.1 6.6 5.7 5.3 2.2 (±.03) x 106 (±.11)x 106 (±.36) x 106 (±.08) x 106 (±.19)x 106 6.4 ( ±.44) x 106 Fibronectin (10 1.1.g/m1) + Laminin (10 µg /ml) Fibronectin (10 p.g /ml) + Polylysine (100 µg /ml) 6.0 (±.04) x 106 6.0 (±.47) x 106 52 Fig. 3.1. Growth response of SFME cells to insulin and IGF I. Cells were plated at 105/dish in F12:DMA basal nutrient medium supplemented with transferrin (25 p.g/ml), HDL (10 µg/ml), and the indicated concentration of insulin or IGF I. Dishes were precoated with fibronectin (10 j.t.g/m1). Cell number was determined 5 days after plating. 0, insulin; S. IGF 1. 6 5 4 GI 3 2 1 fr.-4r 0 0 // 0.001 61- 0.01 0.1 Insulin or IGF (µg/m1) I.0 10.0 53 Fig. 3.2. Growth response of SFME cells to transferrin and lactoferrin. Cells were plated at 105/dish in F12:DME basal nutrient medium supplemented with insulin (10 mg/m1), EGF (50 ng/ml), and the indicated concentration of transferrin or lactoferrin. Dishes were precoated with fibronectin (10 µg/ml). Cell number was determined 5 days after plating. 0, transferrin; 0, lactoferrin. 2 o 1 kJ ...If' 0 . 0' ,w . .. . o' , 0 - y ,i -. 0 ' 0 II L 0.1 1.0 10.0 Transferrin or Lactoferrin (µg/m1) 100.0 54 Fig. 3.3. Growth response of SFME cells to EGF and TGF alpha. Cells were plated at 105/dish in F12:DME medium supplemented with insulin (1014/m1), transferrin (2514/m1), HDL (10 µg/ml), and the indicated concentration of EGF or TGF alpha. Dishes were precoated with fibronectin (10 µg/m1). Cell number was determined 5 days after plating. 0, EGF; , TGF alpha. 6 5 3 2 1 0 1 I0 EGF or TGF alpha (ng/ml) 100 55 Fig. 3.4. Photomicrographs of SFME cells. A: Complete serum-free medium. B: Medium from which insulin was omitted at the time of plating. C: Medium from which EGF was omitted at the time of plating. D: Medium in which EGF was replaced by basic FGF (10 ng/ml) and heparin (10 mg/nil). Photographs were taken 2 days after plating. Composition of complete serum-free medium was as indicated in Materials and Methods. B 6? 0 a 0a a 2, O 0 a J 3 O 3 r, 56 Figure 3.5. Recovery of SFME cells from plating in growth-factor-deficient medium. Cells were plated at 5 x 104/dish in F12:DME basal nutrient medium supplemented with insulin (10 µg/m1), transferrin (25 tg/m1), and HDL (10 .Lg/m1) (without EGF) or in medium supplemented with EGF (50 ng/ml), transferrin (25 1./g/m1), and HDL (10 .tg/m1). At the indicated times, insulin (0) or EGF () was added to the medium in plates deficient in that factor and cell number was determined four days later. Medium was changed 48 hours before cell counts. Cells in plates maintained without EGF i for more than 24 hours were dead (see Fig. 3.4) 5 0 A O O .. 0 I 0 I 48 24 Hours Without EGF or Insulin 72 57 Fig. 3.6. Growth response of SFME cells to acidic and basic FGF in the absence of EGF. Cells were plated at 105/dish in F12:DME basal nutrient medium supplemented with insulin (10 µg/ml), transferrin (25 tg/ml), and HDL (10 µg/ml) (0), and 10 ng/ml basic FGF with 10 µg/ml heparin (0), 10 ng/ml acidic FGF (A), or 10 ng/ml acidic FGF with 10 µg/m1 heparin (A). Control cultures (I) contained insulin, transferrin, HDL, and EGF (50 ng/ml). Dishes were precoated with fibronectin (10 .tg/ml). Cell number was determined on the indicated days after plating. Medium was changed 4 days after plating. IC 3 x 106 106 3 x I05 105 0 2 4 Days After Plating 6 8 58 Fig. 3.7. Growth response of SFME cells to FGF in the presence and absence of EGF and heparin. Cells were plated at 5 x 104/dish in F12:DME basal nutrient medium supplemented with insulin (10.tg/ml), transferrin (25 1.4/m1), HDL (10 14/m1), and the indicated concentrations of basic FGF, with or without heparin (10 p.g/ml) and with or without EGF (50 ng/ml). Dishes were precoated with fibronectin (10 p.g/ml). Cell number was determined 5 days after plating. 0, without EGF, without heparin; I, without EGF, with heparin; 0, with EGF, without heparin; I, with EGF, with heparin. 3 x 106 106 I oif ( -- ---' 0 T - --a - - ... 0 0 0 oil.° ., . 0 111,,,........../ 105 lil -sr ------7". 111 3 x 104 ... . ...-,0 / T/ 0 0.1 1.0 FGF ( ng/ml) 10.0 59 Fig. 3.8. Growth response of SFME cells to HDL. Cells were plated at 105/dish in F12:DME basal nutrient medium supplemented with insulin (1014/m1), transferrin (25 µg/ml), EGF (50 ng/ml), and the indicated concentration of HDL (A) or delipidated (apo) HDL (). Dishes were precoated with fibronectin (10 µg/m1). Cell number was determined 5 days after plating. 8 0 7 6 6. 0 x 5 4E11E0 .111. Mln. Mln. 4 3 2 1 0 5 10 15 0 20 High Density Lipoprotein (i.s.g/m1) 60 CHAPTER 4 Glucocorticoid and Thyroid Hormones Inhibit Proliferation of Serum-Free Mouse Embryo (SFME) Cells Deryk Loo, Cathleen Rawson, Molly Schmitt, Katherine Lindburg, and David Barnes Published in: Journal of Cellular Physiology (1990). Volume 142, pp. 210-217. Coauthor contribution: D.L., primary investigator, C.R., post-doctoral scientist; M.S. and K.L., technical assistants; D.B., research director 61 SUMMARY Mouse embryo cells derived in a serum-free medium formulation (SFME cells) do not exhibit growth crisis or chromosomal abnormalities and are nontumorigenic in vivo; these cells are also reversibly growth inhibited by serum or platelet-free plasma (Loo et al., 1987). A portion of the inhibitory activity of serum could be extracted by charcoal, a procedure that removes steroid and thyroid hormones. Both L-3,5,3'-triiodothyronine (T3) and hydrocortisone inhibited growth of SFME cells in a reversible manner. The inhibitory activity of serum also was partially removed by treatment with anion exchange resin in a procedure designed to deplete serum of thyroid hormone. However, the effect of serum on untransformed SFME cells could not be prevented by addition of the antiglucocorticoid RU38486, and ras-transformed clones of SFME cells, which are capable of growing in serum-containing medium, retained inhibitory responses to glucocorticoid and, with some clonal variability, to T3. These results suggest that glucocorticoid or thyroid hormones may contribute to the inhibitory activity of serum on SFME cells, but additional factors are also involved. 62 INTRODUCTION Mouse embryo cells derived in serum-free basal nutrient medium supplemented with insulin, transferrin, epidermal growth factor (EGF), high-density lipoprotein (HDL), and fibronectin exhibit a number of unusual properties. These serum-free mouse embryo (SFME) cells do not exhibit an obvious growth crisis or chromosomal abnormalities and are nontumorigenic in vivo (Loo et al., 1987,1989a). These cells are also dependent on EGF for survival and are markedly inhibited by serum added to the medium at levels that are stimulatory for cell lines derived in serum-containing medium (Loo et al., 1987). Sera from a variety of species, including calf, adult, and fetal bovine sera, human serum and mouse serum, as well as platelet-free plasma, exert similar inhibitory activity on SFME cells. Fibroblast growth factor (FGF) can replace EGF in the serum-free formulation, although growth with FGF in place of EGF is suboptimal (Loo et al., 1989b). The influence of serum on untransformed SFME cells is likely to represent a meaningful physiological response because the effect occurs at low serum concentrations, is a reversible growth inhibition, rather than a toxicity, and results in the accumulation of cells in the G1 phase of the cell cycle, rather than random inhibition at all points in the cycle (Loo et al., 1987). Furthermore, SFME cells transformed by the human Harvey ras oncogene no longer require EGF for survival, are capable of sustained proliferation in serum-containing medium, and are tumorigenic in syngeneic or athymic mice (Barnes et al., 1989). The ability of oncogene-transformed SFME cells to overcome growth inhibition by serum raises the possibility that the loss of response to the inhibitory activity may reflect a component of the malignant phenotype. In this paper we report experiments designed to examine the nature of the inhibitory activity of serum on SFME cells. We found that a portion of the activity was extracted by charcoal, a classical procedure for removal of hydrophobic hormones (Westphal et al., 1975). Both L-3,5,3'-triiodothyronine (T3) and glucocorticoid at concentrations 63 comparable to those found in serum were capable of inhibiting growth of SFME cells. These inhibitory activities were specific, reversible, and synergistic with charcoal-extracted serum. The combination of T3 and hydrocortisone at optimal concentrations produced an inhibitory effect greater than the effect of each separately, and this effect was of the same magnitude as that seen with an optimal amount of serum. In addition, the inhibitory activity of serum was partially removed by treatment with anion exchange resin, which removes thyroid hormone (Samuels et al., 1979), as well as other molecules, from serum. However, the effect of serum on untransformed SFME cells could not be prevented by addition of the antiglucocorticoid RU38486 (Moguilewsky and Philibert, 1984); rastransformed clones of SFME cells retained inhibitory responses to glucocorticoid; and some clones retained inhibitory responses to T3. 64 MATERIALS AND METHODS Materials. Bovine insulin, human transferrin, steroids and thyroid hormone and derivatives, Norit A charcoal, trypsin, and soybean trypsin inhibitor were obtained from Sigma Chemical Corp., St. Louis, MO. AG 1-X8 resin was obtained from Bio-Rad, Richmond, CA. Mouse EGF was obtained from Upstate Biotechnologies, Lake Placid NY. HDL (density = 1.068-1.121 g/cc) was prepared by KBr ultracentrifugation as described (Gospodarowicz, 1984) and filter sterilized after dialysis. Powdered medium formulations and antibiotics were obtained from Grand Island Biological Company, Grand Island, NY. Sodium selenite and ethylenediaminetetraacetate (EDTA) were obtained from Fisher Scientific, Pittsburgh, PA; 4-(2-hydroxy-ethyl)-1-piperazine-ethanesulfonic acid (HEPES) was obtained from Research Organics, Inc., Cleveland, OH. Plasticware for cell culture was obtained from Falcon. RU38486 was obtained from Roussel-Uclaf, 111 Route de Noisy, 93230 Romainville, France. SFME cell culture. Details of derivation and behavior of SFME cells and serumfree culture methods have been published (Barnes, 1987; Loo et al., 1987, 1989a,b). The basal nutrient medium was a one-to-one mixture of Dulbecco-modified Eagle's medium containing 4.5 g/L glucose and Ham's F12 (Ham and McKeehan, 1979; Mather and Sato, 1979) supplemented with 15 mM HEPES, pH 7.4, 1.2 g/L sodium bicarbonate, sodium selenite (10 nM), penicillin (200 U/ml), streptomycin (200 p.g/m1), and ampicillin (25 .tg/ml) (F12:DME). Cells were cultured in F12:DME supplemented with insulin (10 .tg/ml), transferrin (25 .tg/ml), human high-density lipoprotein (HDL) (10 pg/m1), and EGF (50 ng/ml), in dishes or flasks precoated with bovine fibronectin (10 .tg/ml). Passaging of SFME cells was accomplished by rapid trypsinization at room temperature (0.25% crude trypsin with 1 mM EDTA in phosphate-buffered saline without calcium or magnesium) followed by 65 dilution of the cells into an equal volume of soybean trypsin inhibitor solution (1 mg/ml in F12:DME) and centrifugation from suspension. Plating was accomplished by adding the appropriate cell number in 1 ml of prewarmed medium to culture vessels previously precoated with fibronectin and preincubated in the incubator (5% CO2-95% air atmosphere at 37°C) for approximately 15 minutes with the remaining portion of medium. Insulin, transferrin, EGF, and HDL were added as small aliquots from concentrated stocks directly to the individual culture vessels immediately after plating cells. Fibronectin was provided to cells by precoating plates with a solution of fibronectin made by adding 5 gl of a 1 mg/ml stock to F12:DME (1 mg per 35 mm-diameter plate) and incubating for 60 minutes at 37°C in a CO2 incubator, followed by removal of the precoating solution and one wash with 1 ml of F12:DME. Insulin was prepared at 1 mg/ml in 20 mM HC1 and transferrin at 5 mg/ml in F12:DME; both were filter sterilized. EGF was obtained as sterile, lyophilized powder and reconstituted at 50 µg/ml with sterile PBS. Fibronectin stocks were filter sterilized and adjusted to approximately 1 mg/ml in 50 mM Tris, pH 7.5 containing 4 M urea. Stocks of insulin, transferrin, EGF, and fibronectin were stored at -86°C in aliquots until needed and then stored at 4°C after thawing. Fatty-acid-free bovine serum albumin was added to HDL solutions to a final concentration of 1 mg/nil and aliquots were stored at 4°C. Steroids were prepared at 10-3 M in absolute ethanol and stored at -20°C. Thyroid hormone and derivatives were prepared at 10-5 M in 0.2 N NaOH and stored at -20°C. Human Ha-ras-transformed SFME cells were derived by calcium phosphate- mediated transfection (Loo et al., 1989a) with a plasmid containing the oncogene (pUCEJ6.6) (Shih and Weinberg, 1982; Land et al., 1983) obtained from the American Type Culture Collection, Rockville, MD. Transformed cells were obtained by selection in medium without EGF and isolated with cloning cylinders. Assay of cell growth. Cells cultured continuously in the complete serum-free medium were detached from stock flasks by trypsinization, diluted into trypsin inhibitor 66 solution, centrifuged, resuspended, counted, and plated at 105/35 mm-diameter dish in media of the indicated composition. In experiments employing antiglucocorticoid, RU38486 was added as small volumes of dilutions in F12:DME medium from a stock (1 mM) in absolute ethanol. Cell number was determined at the indicated times after plating by counting trypsinized cell suspensions in a Coulter particle counter. Data shown are the average of values from duplicate plates. Stock SFME cultures used for the experiments described in this report had undergone between 60 and 120 population doublings. Transformed cells had undergone approximately 30 to 60 doublings after cloning. Measurements of T3 receptor levels in untransformed and transformed SFME cells were carried out according to the procedure of Samuels et al. (Samuels et al., 1975). Serum treatments. Serum was extracted twice with 50 mg/ml charcoal (Norit A) for 30 minutes at 56°C, followed by centrifugation to remove charcoal particles and filtration through a 0.2 1.t.m filter (Westphal et al., 1975). Charcoal treatment reduced the radiolabeled tracer dexamethasone and T3 added prior to treatment by 99.8%. Control serum was incubated without charcoal for 30 minutes at 56°C, followed by centrifugation. Charcoal treatment reduced the protein concentration of the serum by 11%. For resin treatment, serum was incubated with AG 1-X8 (chloride) resin at room temperature (50 mg resin/ml serum, three changes at intervals of 5, 18, and 24 hours (Samuels et al., 1976). Resin was autoclaved before use and sterility of serum was confirmed after resin treatment. Control serum was incubated in the same protocol without resin. Resin treatment reduced radiolabeled tracer T3 and dexamethasone added prior to treatment by greater than 99.9%. Resin treatment reduced protein concentration of the serum by 6%. 67 RESULTS Inhibition of SFME cells by glucocorticoid and thyroid hormone. Serum is strongly inhibitory for SFME cell growth, even when added to medium supplemented with all of the growth - stimulatory supplements used in the serum-free medium formulation (Loo et al., 1987). In attempts to characterize the inhibitory activity of calf serum on SFME cells, we found that incubation of serum with activated charcoal at 56°C diminished or eliminated the inhibitory activity of calf serum at low concentrations, although residual inhibitory activity remained at higher serum concentration (Fig. 4.1). Serum was extracted twice with charcoal; further extractions did not lead to additional reduction in inhibitory activity. Incubation of serum at 56°C in the absence of charcoal did not affect the inhibitory activity (Fig. 4.1). The inhibitory activity of calf plasma and human serum and plasma also was partially removed by charcoal treatment. Lipoproteins are inhibitory for some cell lines (Leffert and Weinstein, 1976; Ito et al., 1982; Barnes and Reed, 1989), and might be expected to be removed to some extent by charcoal treatment, but delipidation of calf serum or human plasma did not remove the inhibitory activity on SFME cells (not shown). Because charcoal treatment is an established procedure for removal of steroids from serum, we tested steroid hormones for inhibitory activity on SFME cells in medium containing 2.5% charcoal-treated serum. Glucocorticoids were inhibitory at concentrations that may be obtained in serum (Figs. 4.2,4.3). Other classes of steroids had no effect on SFME cell proliferation. Similarly, charcoal treatment removes thyroid hormones, and we found that T3 also was inhibitory for SFME cell growth at physiological concentrations (Fig. 4.4). Thyroxine (T4) was also active in this assay, while related compounds that do not possess thyroid hormone activity in vivo did not inhibit SFME cell growth (Fig. 4.5). Both hydrocortisone and T3 acted synergistically with charcoal-treated serum (Fig. 4.6). The synergistic effect of charcoal-treated serum with thyroid hormone was not mimicked by T4 binding protein 68 added with T4 under serum-free conditions. The synergistic effect of charcoal-treated serum with either hydrocortisone or T3 also was not mimicked with bovine serum albumin at 1 mg/m1 (not shown). Contribution of glucocorticoid and thyroid hormone to the inhibitory activity of serum on SFME cells. SFME cells plated in F12:DME medium with growth-stimulatory supplements, charcoal-treated calf serum, and either hydrocortisone or T3 grew more slowly than controls throughout the culture period, and growth rate was further reduced if both hydrocortisone and T3 were present (Fig. 4.7A). This inhibition was of similar magnitude to that observed with 10% calf serum, and the effect, like that of calf serum, was reversible upon removal of the inhibitory supplement (Fig. 4.7B). Treatment of serum with AG 1-X8 anion exchange resin, which removed thyroid hormone, significantly reduced the growth inhibitory activity of the serum, although resin-treated serum was still strongly inhibitory at a concentration of 10% (Fig. 4.8). This treatment, like charcoal treatment, also removed glucocorticoid from the serum. However, addition of antiglucocorticoid RU38486 at 1 tM did not reduce the inhibitory activity of serum, although this compound was capable of reversing the inhibitory effect of added hydrocortisone on SFME cells (Table 4.1). RU38486 at 11.1M also was ineffective at further reducing the residual inhibitory activity of resin-treated sreum at a concentration of 10% (Fig. 4.8), and resin treatment of charcoal-treated serum did not reduce inhibitory activity beyond that of treatment with resin or charcoal only. SFME cells cultured for 5 days in medium supplemented with the growth-stimulatory factors of the serum-free formulation and additionally with 10% resun-treated, charcoal-treated serum were growth inhibited by 85%, compared with controls containing no serum. SFME cells transformed with the human Ha-ras oncogene grow in the presence of serum concentrations that are strongly inhibitory for untransformed SFME cells (Barnes et al., 1989). We tested several clones of ras-transformed SFME cells for inhibitory activity of triiodothyronine and hydrocortisone; results with two clones am shown in Table 4.2. 69 Both clones were inhibited by hydrocortisone or hydrocortisone and T3 together to a degree similar to that observed with untransformed SFME cells. Inhibition of rastransformed cells by T3 was reduced in one clone, but was not reduced in another (Table 4.2). The concentration dependence of the inhibitory effect of T3 on the latter clone was similar to that of untransformed SFME cells. T3 receptor level in the clone that was less inhibited by T3 was not reduced, relative to untransformed SFME cells (not shown). 70 DISCUSSION The unusual inhibitory response of SFME cells to serum and the ability of tumorigenic derivatives to overcome the serum inhibition prompted an examination of the molecules in serum that might be responsible. Although the platelet-derived peptide transforming growth factor (TGF) beta is inhibitory for some cell types in culture (Tucker et al., 1984; Sporn and Roberts, 1985), platelet-free plasma containing no detectable TGF beta is strongly inhibitory for SFME cell growth (Loo et al., 1987), and addition of purified TGF beta over a wide range of concentrations produced little or no SFME cell growth inhibition under a variety of conditions. Other potentially inhibitory peptides-tumor necrosis factor, interleukins, and interferon (Clemens and McNurlan, 1985; Tsai and Gaffney, 1986; Saneto et al., 1986; Kohase et al., 1987; Barnes and Reed, 1989) are probably not responsible for the inhibitory effect of serum on SFME cells because tumor necrosis factor and interleukins I and II had no effect when tested on SFME cells, and serum containing no detectable interferon was strongly inhibitory for SFME cell growth (unpublished results). We have presented evidence in this report that nonpeptide hormones are capable of inhibiting SFME cell growth. Removal of steroid and thyroid hormones by treatment with charcoal reduced the inhibitory activity of serum on SFME cells. Of the steroids tested, only glucocorticoids were inhibitory for SFME cell growth, and thyroid hormones (T3 or T4) also were inhibitory. The effect of T3 and hydrocortisone together in the presence of 2.5% charcoal- treated calf serum was greater than either separately, and was similar in magnitude to that observed with 10% calf serum. It is possible that hydrocortisone or thyroid hormones inhibit cell growth coordinately with the induction of a differentiated state in SFME cells, but if this is the case, the differentiation is not terminal, because the inhibitory effect of these hormones, like that of serum, was reversible. 71 Steroid and thyroid hormones induce multiple effects on cells, depending on the cell type, but display similarities in receptors and mechanisms of action at the molecular level (Evans, 1988; Beato, 1989). The action of hydrocortisone on SFME cells led to reduced growth, but did not cause cell death, and thus differs from the irreversible toxicity of this steroid on thymocytes (Kaiser and Edelman, 1977; Bourgeois and Gasson, 1985). The effect of glucocorticoid on SFME cells resembles the inhibitory activity of steroids on chicken fibroblasts and human mammary cells in culture (Lippman et al., 1976; Rubin, 1977). Both hydrocortisone and T3 were active at concentrations found in serum, and both were maximally effective when added in the presence of a low concentration of charcoal- treated calf serum. This effect may be due to binding proteins of serum that modulate the action of steroid and thyroid hormones. These proteins may not be extracted with charcoal, or may be present in excess of the ligands. We were not able to mimic the effect of charcoal-treated serum with T4-binding protein or albumin, another steroid and thyroid hormone-binding serum protein, and the molecular nature of the factor or factors responsible for the synergism of hydrocortisone and T3 with charcoal-treated serum is unknown. We were unable to alter the inhibitory activity of serum by addition of antiglucocorticoid, although the antihormone clearly was capable of preventing the effect of added hydrocortisone in the presence of charcoal-treated serum. Furthermore, rastransformed cells capable of growing in serum-containing medium remained inhibited by hydrocortisone. Although the degree to which endogenous glucocorticoid in serum may contribute to the inhibitory activity of serum on SFTAE cell growth is unclear, these cells may provide a useful model for the study of glucocorticoid inhibition of proliferation under well-controlled culture conditions. We obtained a partial removal of the inhibitory activity of serum by treatment with anion exchange resin that was designed to remove thyroid hormone, suggesting that 72 endogenous thyroid hormone may play a role in serum inhibition of SFME cells. However, the resin treatment also removed glucocorticoid; partial removal of steroid from serum by this treatment has been reported previously, and has been attributed to adsorption to the hydrophobic support material of the resin (Samuels et al., 1979). In addition, we found that some clones of ras-transformed SFME cells capable of growing in serumcontaining medium remained inhibited by T3, indicating that escape from serum inhibition of SFME cell growth is not solely the result of loss of inhibitory response to thyroid hormone. Recent evidence that the erbA oncogene is related to the T3 receptor (Weinberger et al., 1986; Sap et a., 1986) has focused speculation that this oncogene may represent a mutationally activated receptor capable of functioning in the absence of ligand in a growthpromoting manner analogous to the mutationally activated EGF receptor, or erbB oncogene (Downward et al., 19484; Samuels et al., 1988; Evans, 1988). An alternative possibility raised by observations that thyroid hormone can be growth inhibitory is that a thyroid hormone receptor might become altered in a manner that leads to interference with the inhibitory function, thus releasing a cell from T3-mediated negative growth control. Recently a non-hormone-binding erbA protein capable of inhibiting thyroid hormone action has been reported (Koinig et al., 1989). Although the phenomena we have reported here may explain partially the effect of serum on SFME cell growth, significant growth-inhibitory activity existed in serum that was exhaustively extracted with charcoal or anion exchange resin, or with a combination of both treatments. Charcoal-treated or resin-treated serum at a concentration of 10% remained strongly inhibitory for SFME cells. In addition, both treatments are capable of extracting from serum factors other than steroid and thyroid hormones, such as peptides (Titani et al., 1978). It appears that the effect of serum on SFME cells involves at least one additional factor that is not removed by charcoal extraction. The nature of this factor remains to be explored. 73 ACKNOWLEDGMENTS We thank Carrie Smith for technical assistance. Supported by NIH -NAI- 07560. D. Barnes is the recipient of Research Career Development Award NEI-NCI-01226. Cathleen Rawson assisted with all growth curve experiments. Molly Schmitt prepared charcoal-treated calf serum and assisted with SFME stock culturing. Katherine Lindburg prepared medium and assisted with experiments using transformed-SFME cells. David Barnes provided financial and scientific support. 74 Table 4.1. Calf serum inhibition of SFME cells in the presence of antiglucocorticoid RU38486. Experiment 1 (control): SFME cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL, fibronectin, and 2.5% charcoal-treated calf serum, with or without hydrocortisone (100 nM) and RU38486 (1 gM). Cell number was determined 5 days after seeding. Experiment 2: Plates were supplemented with insulin, transferrin, EGF, HDL, fibronectin, and the indicated concentrations of serum, with or without RU38486 (1 pM). Cell number was determined 6 days after seeding. Cells x 10-6 Experiment 1 hydrocortisone + hydrocortisone Experiment 2 0% calf serum 1.0% calf serum 2.5% calf serum 5.0% calf serum 10% calf serum RU38386 + RU38386 1.56 ± .19 0.43 ± .07 1.91 ± .07 1.58 ± .13 4.66 2.19 .288 .145 .159 ± .32 ± .47 ± .04 ± .01 ± .01 4.39 1.98 .258 .144 .152 ± .11 ± .48 .02 ± .01 ± .02 75 Table 4.2. Glucocorticoid and triiodothyronine inhibition of ras-transformed SFME cells. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL, fibronectin, and 2.5% charcoal-treated calf serum, with or without hydrocortisone (100 nM) and, T3 (10 nM), or both. Plates containing calf serum (10%) also received insulin, transferrin, EGF, HDL, and fibronectin. Cell number was determined 6 days after plating. SFME Control Hydrocortisone Triiodothyronine Hydrocortisone + triiodothyronine Calf serum 2.38 0.73 0.91 0.28 ± 0.20 ± 0.01 ± 0.01 ± 0.02 0.21 ± 0.03 Cells x 10m86 ras-SFME clone A 2.75 ± 0.19 0.92 ± 0.05 1.00 ± 0.04 0.40 ± 0.03 ras-SFME clone B 3.65 ± 0.37 0.93 ± 0.01 2.41 ± 0.09 0.47 ± 0.05 1.19 ± 0.01 2.06 ± 0.18 76 Fig. 4.1. Partial removal of inhibitory activity of calf serum for SFME cells by charcoal treatment. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented with insulin (10 1.4/m1), transferrin (25 1.4/m1), EGF (50 ng/ml), and HDL (1014/m1) and the indicated concentrations of calf serum. Dishes were precoated with bovine fibronectin (10 p.g/m1). Serum was extracted with charcoal at 56°C as described. Control serum was incubated at 56°C without charcoal. Cell number was determined 6 days after plating by counting trypsinized cell suspensions in a Coulter particle counter. (II), untreated serum; (0), 56°C-treated serum; (0), charcoal-treated serum. Error bars indicate variation of individual samples from the mean. 7 b6 45- x .c 5 60 4 , 3 a° N T 2 U -) I 0 0.0 2.5 5.0 Serum ( %) 7.5 10.0 77 Fig. 4.2. Steroid inhibition of SFME cell growth. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL and fibronectin as described. Dishes also contained 2.5% charcoal- treated calf serum and the indicated steroids (10 nM). Cell number was determined 6 days after plating. Error bars indicate variation of individual samples from the mean. 7 6 5 4 3 2 0 0 V 0 SI SI C 0 CU it UJ O 78 Fig. 4.3. Concentration dependence of hydrocortisone inhibition of SFME cell growth. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL and fibronectin as described. Dishes also contained 2.5% charcoal-treated calf serum and the indicated concentration of hydrocortisone. Cell number was determined 6 days after plating. Error bars indicate variation of individual samples from the mean. I 4- i /iNr---I EN I 0 I 0 // I 10-10 I i 1 10-9 10-8 10-7 Hydrocortisone ( M ) 79 Fig. 4.4. Concentration dependence of triiodothyronine inhibition of SFME cell growth. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL and fibronectin as described. Dishes also contained 2.5% charcoal-treated calf serum and the indicated concentration of T3. Cell number was determined 6 days after plating. Error bars indicate variation of individual samples from the mean. 0 , 1J-711 ' 0 10-12 I 4 3 o 2 I 0 IO-II i0-10 10-9 Triiodothyronine (M) 10-8 80 Fig. 4.5. Specificity of thyroid hormone inhibition of SFME cells. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL and fibronectin as described. Dishes also contained 2.5% charcoal-treated calf serum and 1 nM triiodothyronine (T3), thyroxine (T4), diiodothyronine (T2), iodotyrosine (I-tyr) or diiodotyrosine (I2-tyr). Cell number was determined 6 days after plating. Error bars indicate variation of individual samples from the mean. a 7 6 5 4 3 2 1 0 Control T3 T4 T2 I-Tyr I2-Tyr 81 Fig. 4.6. Concentration dependence of charcoal-treated calf serum for glucocorticoid and T3 inhibition of SFME cell growth. Cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL and fibronectin as described. Dishes also contained 10 nM triiodothyronine (0) or 100 nM hydrocortisone (0) and the indicated concentrations of charcoal-treated serum. Data are expressed as percent of cell number in control plates containing the indicated concentration of charcoal-treated serum but without triiodothyronine or hydrocortisone. Cell number was determined 6 days after plating. Error bars indicate variation of individual samples from the mean. 2.5 5.0 Serum (%) 7.5 10.0 82 Fig. 4.7. Time course and reversibility of triiodothyronine and hydrocortisone inhibition of SFME cell growth. Cells were plated at 105/35 mmdiameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL, fibronectin, and 2.5% charcoal-treated calf serum. Some dishes also were supplemented with insulin, transferrin, EGF, HDL, fibronectin, and 10% calf serum. A: (0), serum-free medium without T3 or hydrocortisone; (A), with hydrocortisone; (V), with T3. B: (0) serum-free medium without T3 or hydrocortisone, as in A; (), with both T3 and hydrocortisone; (), 10% calf serum; (15) T3 and hydrocortisone added at plating, changed on day 3 to medium without T3 and hydrocortisone; (0), 10% calf serum added at plating, changed on day 3 to medium without calf serum. Error bars indicate variation of individual samples from the mean. I07 10 6 0 103 3 5 Days After Plating 7 9 3 5 7 Days After Plating 9 11 83 Fig. 4.8. Partial removal of inhibitory activity of calf serum by anion exchange resin. Serum was extracted with resin as described. SFME cells were plated at 105/35 mm-diameter dish in F12:DME medium supplemented as described with insulin, transferrin, EGF, HDL, fibronectin, and the indicated concentration of serum. (0), unextracted calf serum; (0), resin-extracted serum; (0), resin-extracted serum plus antiglucocorticoid RU38486 (1 iiM). Error bars indicate variation of individual samples from the mean. Serum ( %) 84 CHAPTER 5 Serial Passage of Embryonic Human Astrocytes in Serum-Free, Hormone-Supplemented Medium Deryk Loo, Yoshio Sakai, Cathleen Rawson, and David Barnes Published in: Journal of Neuroscience Research. Volume 28, pp. 101-109 (1991). Coauthor contribution: D.L., primary investigator, Y.S., visiting professor; C.R., research associate; D.B., research director 85 SUMMARY We applied serum-free cell culture methods that allow extended proliferation of mouse astrocyte precursor cells to the multipassage culture of embryonic human brain cells. Cells were cultured in nutrient medium supplemented with insulin, transferrin, epidermal growth factor, fibroblast growth factor, heparin, high-density lipoprotein, and fibronectin. Cultures were maintained for a maximum of 70 population doublings before proliferation ceased. The cells synthesized glial fibrillary acidic protein, an astrocyte marker, and expression of this protein was increased by incubation of the cells with transforming growth factor beta or serum. These results identify extracellular factors important for proliferation and differentiation of embryonic human astrocytes and provide a controlled system for multipassage culture. 86 INTRODUCTION Our laboratory has derived and characterized cell cultures from 16 day mouse embryos in basal nutrient medium supplemented with insulin, transferrin, epidermal growth factor (EGF), high-density lipoprotein (HDL), and fibronectin (Loo et al., 1987, 1989a, b, 1990; Rawson et al., 1990; Shirahata et al., 1990; Sakai et al., 1990). These serum-free mouse embryo (SFME) cells exhibit a number of unusual properties. These cells do not lose proliferation potential or develop gross chromosomal aberration when cultured for more than ten times the number of population doublings that can be achieved with mouse embryo cells in conventional, serum-containing medium (Loo et al., 1987), and proliferation of SFME cells is reversibly inhibited by serum (Loo et al., 1987, 1990; Shirahata et al., 1990). Recently we reported that treatment of SFME cells with serum or transforming growth factor beta (TGF beta) leads to the appearance of glial fibrillary acidic protein (GFAP), an intermediate filament protein of about 50,000 daltons that is a specific marker for astrocytes (Sakai et al., 1990; Lazarides, 1982; De Armond et al., 1983; de Vellis et al., 1986; Morrison et al., 1985). TGF beta is a platelet-derived growth factor that influences growth and differentiation of a number of cell types (Sporn and Roberts, 1988; Massague, 1987; Rizzino, 1988). Cells with properties like those of SFME cells can be isolated directly from mouse brain (Sakai et al., 1990), and formulations of serum-free media developed specifically for astrocytes (Morrison and de Vellis, 1981; Raff et al., 1983; Morrison et al., 1985; Raff et al., 1984) are similar to the medium formulation used for SFME cells. Fibroblast growth factor (FGF), pituitary extract (a source of FGF), or EGF are used in these astrocyte media, and FGF can partially replace EGF for SFME cell survival and growth (Loo et al., 1989b). 87 Results with mouse cells led us to ask if similar cultures could be developed from embryonic human brain. Here we report the multipassage growth of human astrocytes from 20- to 24-week-old embryos in media similar to that used for SFME cells. Several of these cultures were maintained for 60 to 70 population doublings using this approach, but, unlike SFME cells, the human cells eventually ceased proliferation. These cultures were different in several other ways from SFME cultures. For instance, human cells were dependent on FGF for proliferation and were less responsive to EGF than to FGF, while SFME cells were dependent on EGF for survival and less responsive to FGF (Loo et al., 1989b; Rawson et al., 1990). Response of the human cells to mitogens, in general, decreased with population doubling level. Medium supplemented with 10% calf serum did not support growth of the human cells well unless supplemented additionally with the growth factors of the serum-free medium. GFAP expression was increased in the cells by TGF beta or serum, but human cells, unlike SFME cells, expressed GFAP at easily detectable levels in the absence of TGF beta or serum. These results show that a cell type related to SFME cells is present in embryonic human brain, identify extracellular factors important for proliferation and differentiation of these cells, and provide a well-controlled system of multipassage culture for further study. 88 MATERIALS AND METHODS Cell culture. Embryonic tissue (brains from 20- to 24-week-old embryos from saline-induced abortion) was obtained from the International Institute for the Advancement of Medicine, Essington, Pennsylvania. For initiation of cultures, brains were dissected under sterile conditions and dissociated by the procedure of Brunner at al. (Brunner et al., 1982). Brains were minced and fragments were washed twice in 10 ml sterile basal nutrient medium, then transferred to a sterile 10 cm-diameter tissue culture dish and cut into 1-3 mm-sized pieces. Trypsin was added (10 ml of 0.5 mg/ml crude porcine trypsin in medium) and incubated at 37°C for 10 minutes. Tissue fragments were resuspended by pipetting and 3 ml of a RNase/DNase solution (1 mg/ml each in phosphate-buffered saline, PBS) was added and incubated at 37°C for 5 minutes. Five milliliters of a collagenase/hyaluronidase solution (4 mg/ml each in PBS) was added and incubated at 37°C for 30 minutes, resuspending tissue every 5 minutes. This was followed by the addition of 3 ml RNase/DNase, 5 ml trypsin inhibitor (1 mg/ml in medium), and an additional 3 ml of RNase/DNase with pipetting between each addition. The tissue digest was filtered through a 30 gm pore nylon filter into a conical polypropylene tissue culture tube and centrifuged at 600g for 5 minutes. The cell pellet was washed once with medium and cells were plated in tissue culture flasks and cultured in serum-free medium with the supplements as described below. Detailed procedures for the culture of Baib /c SFME cells have been published (Loo et al., 1989a,b), and similar procedures were employed for embryonic human brain cultures The basal nutrient medium was a 1:1 mixture of Dulbecco-modified Eagle's medium containing 4.5 g/L glucose and Ham's F12 (Ham and McKeehan, 1979; Mather and Sato, 1979) supplemented with 15 mM HEPES, pH 7.4, 1.2 g/L sodium bicarbonate, sodium selenite (10 nM), penicillin (200 U/ml), streptomycin (200 p.g/m1), and ampicillin (2514/m1) (F12:DME). SFME cells were cultured in F12:DME supplemented with insulin 89 (lOgg/m1), transferrin (10 gg/m1), HDL (10 µg/ml), basic FGF (10 ng/ml), heparin (1 p.g/ml), and EGF (50 ng/ml) in dishes or flasks precoated with bovine fibronectin (10 gg/1111) Bovine insulin and human transferrin were obtained from Sigma Chemical Corp. (St. Louis, MO). Bovine basic FGF and human TGF beta were obtained from R and D Inc. (Minneapolis, MN). Mouse EGF was obtained from Upstate Biotechnologies (Lake Placid, NY). HDL and fibronectin were prepared as described (Loo et al., 1989a). Passaging of human embryonic brain cells was accomplished by trypsinization followed by dilution of the cells into an equal volume of soybean trypsin inhibitor solution and centrifugation from solution. Cells were routinely grown in a 5% CO2-95% air atmosphere at 37°C and passaged at a 1:10 dilution. Medium was changed or additional growth factors were added 4 days after plating, and cultures were passaged or medium was changed 7 days after plating. Population doublings were calculated from passage dilution at each trypsinization. Western blots. For the preparation of extracts, cells were scraped from 10 cm- diameter plates into 1 ml ice-cold PBS, pH 7.2, with 0.02% PMSF. Cell suspensions were centrifuged and pellets were stored at -86°C until extracts were prepared. To prepare extracts, one to two volumes of extract buffer (0.1% Triton X-100, 0.02% PMSF, 0.125 M Trizma base, pH 6.8, 250 units/ml DNase) was added to cell pellets and thoroughly mixed using constant pipetting. Then 0.5% SDS and 0.5% 2-mercaptoethanol were added to each lysate followed by extensive mixing. Protein assays were performed on the lysates using the method of Bradford (Bradford et al., 1976). For preparation of tissue extract from uncultured brain, minced tissue fragments were treated as described for cell suspensions. Cell extracts (40 .tg protein per lane) were reduced with 2-mercaptoethanol and fractionated by electrophoresis on a 10% polyacrylamide slab gel (80 V/29 mA for 5 minutes followed by 100 V/35 mA for 15 minutes, then 25 V/6 mA for 13.5 hours). The 90 polyacrylamide gel was removed from between glass plates and encased in an electroblotting apparatus (Transblot, BioRad). Proteins were electrophoretically transferred from the gel to nitrocellulose filters (0.45 gm, Schleicher and Schuell) in transfer buffer (150 mM glycine, 20 mM Trizma base, pH 8.3, 20% methanol, at 4°C) at 75 V for 3 hours. The nitrocellulose filters were air dried at room temperature for 16 hours. The nitrocellulose blots were immersed in TBS (20 mM Tris, pH 7.5, 500 mM NaC1) and incubated with gentle agitation at room temperature for 2 minutes. Then filters were incubated for 1 hour in blocking solution (3% gelatin (w/v) in TBS) and washed for 10 minutes in TTBS (20 mM Tris, pH 7.5, 500 mM NaC1, 0.05% Tween-20). In order to detect GFAP, filters were incubated for 1.5 hours in a 1:1,000 dilution of anti-GFAP (rabbit anti-bovine GFAP antisera, Biomedical Technologies, Inc.) in antibody buffer (1.0% gelatin (W/V) in TTBS) then washed twice, 5 minutes each in TTBS. Filters were then incubated for 1.5 hours in a 1:3,000 dilution of affinity-purified goat anti-rabbit IgG conjugated with horseradish peroxidase (BioRad, blotting grade) in antibody buffer, washed twice, 5 minutes each, in TTBS and once for 5 minutes in TBS. Filters were incubated with substrate (.067 mg/ml 3-amino-9-ethylcarbazole, 50 mM Tris- HC1, pH 7.3) for 10 minutes to 1 hour with gentle agitation in the dark at room temperature. When bands were visible development was stopped by decanting substrate solution and washing the filter twice with water, l0 minutes each time, at room temperature in the dark. Experiments with rabbit antibody were confirmed with mouse monoclonal anti-GFAP (Boehringer-Mannheim) and anti-mouse IgG conjugated with horseradish peroxidase (BioRad). GFAP culture immunoassay. Cells were plated at 5 x 105 cells per 35 mmdiameter dish. At the indicated time, cells were fixed by aspirating medium and slowly adding 1 ml 70% ethanol followed by incubation for 5 minutes at room temperature, followed by an identical treatment with 95% ethanol. The ethanol was then aspirated and plates were air dried. Fixed cells were incubated with a solution containing 15 mM 91 NaPO4, pH 7.1, 1% Triton X-100, 0.6 M KC1, 10 mM MgC12, 2 mM EDTA, and 1 mM EGTA for 5 minutes at room temperature and then washed with 1 ml PBS for 5 minutes at room temperature. One milliliter of PBS with 10% calf serum (CS) was added and incubated for 1 hour at room temperature. The 10% CS solution was aspirated and 1 ml of anti-GFAP antiserum (1:1,000 in PBS with 10% CS) was added and incubated at room temperature for 1 hour followed by two washes with 2 ml PBS, each 5 minutes at room temperature. Controls received an equivalent amount of normal rabbit serum in place of anti-GFAP. Affinity-purified goat anti-rabbit IgG (H & L) conjugated with horseradish peroxidase (BioRad, blotting grade) (1 ml of a 1:3,000 dilution in PBS with 10% CS) was added and incubated for 1 hour at room temperature. This was aspirated and plates were washed twice, 5 minutes each, in 2 ml PBS at room temperature. One milliliter of 2 mM 2,2'-azino-di-(3-ethyl-benzothiazolin-sulfonate) in 0.1 M sodium acetate, 0.05 M Naphosphate and 2.5 mM H202 was added and the plates were incubated for 30 minutes in the dark at room temperature. Absorbance at 405 nm was determined for each sample. Cell number per dish for each condition was determined by trypsinizing replicate dishes cultured identically to those used for the GFAP assay and counting in a Coulter particle counter. Control values from plates receiving normal rabbit serum in place of anti-GFAP were less than 20% of those with anti-GFAP and were subtracted from the corresponding value with anti-GFAP in each case. Variation in individual plates in GFAP per cell, expressed as (A405)/106 cells, was less than 10% from the average of duplicate dishes. Immunocytochemical assay was carried out in the same manner except substrate was 3amino-9-ethylcarbazole (.067 mg/ml) in 50 mM Tris-HC1, pH 7.3, and incubation was 3 to 24 hours in the dark at room temperature. 92 RESULTS Serial passage culture of embryonic human astrocytes. Human brain cultures (22 week-old embryo) initiated in the serum-free medium developed for SFME cells and further supplemented with FGF and heparin initially grew slowly (Fig. 5.1). Ultimately a population of homogeneous appearance emerged (Fig. 5.2) and, in the example shown in Figure 5.1, proliferation rate increased. Similar results were obtained with two cultures initiated independently from the same specimen and carried independently (Fig. 5.1). Human foreskin fibroblasts initiated and carried in the serum-free medium initially proliferated rapidly, but growth halted abruptly after about 30 population doublings (Fig. 5.1). Companion cultures from the same preparations initiated in medium supplemented only with 10% serum could be maintained for only a few generations in this medium before proliferation ceased (not shown). Ultimately proliferation rate of the brain-derived cells declined and the cultures reached a point at which cell proliferation ceased. Figure 5.3 is an example of data from a culture (24 week-old embryo) that did not recover from the initial slow growth rate in serum-free medium, but exhibited a behavior similar to that of the cells of Figure 5.1 when 0.5% serum was added at the time of passaging in addition to the other medium supplements. The medium was changed to serum-free medium the following day, so the cells were exposed to medium containing the small amount of serum for less than 24 hours at each passage. Continuous supplementation of cultures with 0.5% or 1.0% calf serum did not prevent the ultimate loss of proliferative potential after 60 to 70 population doublings. Similarly, addition of 2% or 10% serum to the medium of cells carried serum-free to population doubling levels of 60 to 70 as shown in Figure 5.1 did not restore proliferative potential. Another independent culture (23 week-old embryo) derived in serum-free medium underwent the initial growth and loss of proliferative potential, but did not resume growth when 1% serum was added to the medium (not shown). Attempts were made to culture 93 cells from three other embryos (7 to 11 weeks old), and all were successfully maintained in primary culture and passaged, but long-term culture was not undertaken. Clonal growth of all of the cultures was poor in serum-free medium but was substantially improved by supplementation of the medium with serum in addition to the other factors. Growth characteristics of embryonic human astrocytes in vitro. Cells passaged in serum-free medium supplemented with insulin, transferrin, EGF, FGF, heparin, HDL, and fibronectin initially underwent a lengthy lag period after plating before proliferation was observed. Additional hormonal supplements such as steroids and thyroid hormones and other peptide growth factors did not improve growth. Little proliferation was observed in plates containing medium supplemented only with 10% calf serum, while cells proliferated with no lag in medium supplemented with both the factors of the serum-free medium formulation and 10% calf serum. The pattern of cell growth in these media was the same in both low-passage and high-passage cultures, but the overall level of growth was reduced in the high-passage cultures (Fig. 5.4). Individual omission of each of the peptide growth factors from the serum-free medium reduced the cell number per plate after 7 days, and similar results were seen in both low- and high-passage cells, although growth was generally reduced in the highpassage culture. Cell number was reduced more by omission of FGF/heparin or insulin than by omission of EGF (Fig. 5.5). The response of the human cells to removal of both FGF and EGF from the medium was qualitatively different from the response of SFME cells. Greater than 80% of the human cells remained alive in the absence of these growth factors through the 7-day period of the experiment, while all SFMF. cells under identical conditions die within 48 hours (Loo et al., 1989a; Rawson et a1., 1990). Results shown in Figures 5.4 and 5.5 were obtained with the culture for which multipassage growth characteristics are shown in Figure 5.1. Similar results were obtained with the culture for which growth characteristics are shown in Figure 5.3. 94 Expression of GFAP by embryonic human astrocytes in vitro. GFAP was detected in a quantitative assay using both early and late passage cultures of human brain cells cultured in serum-free medium (Fig. 5.6). Although the human brain cells, like SFME cells, increased expression of GFAP in response to TGF beta and calf serum, a significant amount of GFAP was evident in these cultures in the immunoassay and in Western blots (Fig. 5.7) in the absence of serum or TGF beta. SFME cells do not produce easily detectable amounts of GFAP in the absence of the inducers, but markedly express GFAP in response to serum or TGF beta in immunoassays and Western blots identical with those described here for human cells, while mouse 3T3 cells are negative in these assays (Sakai et al., 1990). Western blots of extracts of the serum-free human brain cells probed with antiserum to neurofilaments or neuron-specific enolase were negative and no induction of these markers was observed when the cells were incubated with 10% calf serum (not shown). Western blot controls with normal rabbit serum in place of anti-GFAP were negative. Results identical to those shown in Figure 5.7 were obtained with monoclonal antibody to GFAP in place of rabbit anti-GFAP and negative results were obtained with an equivalent concentration of nonspecific mouse IgG in place of monoclonal anti-GFAP. Immunohistochemical staining detected expression of GFAP in greater than 80% of the cells of cultures incubated 48 hours in medium with 10% calf serum, with over half of the cells staining very strongly. Results were similar with cultures at 20 population doublings and 60 population doublings, indicating that the medium is supportive of astrocyte precursors over multipassage culture. Controls with normal rabbit serum in place of antiGFAP showed no stained cells. 95 DISCUSSION We have applied the serum-free approach developed for mouse embryo cells to human embryonic brain cells and achieved extended multipassage culture of human astrocytes. Although rodent astrocytes are routinely cultured short-term under conventional, serum-containing or serum-free conditions (de Vellis et al., 1986; Morrison and de Vellis, 1981; Morrison et al., 1985; Raff et al., 1983, 1984), extended culture of these cells in serum containing medium results in spontaneous transformation and alterations in expression of differentiated properties (Bressler and de Vellis, 1985). Previously cell culture of human embryonic astrocytes under conventional conditions has been limited to a few passages (Grenier et al., 1989; Rutka et al., 1987; Milsted et al., 1990). Several cultures were maintained for 60 to 70 population doublings before proliferation ceased, providing a useful new culture model. This is sufficient proliferative potential to allow long-term experiments such as transfection and selection of cells expressing exogenous genes integrated into the genome or chronic exposure to chemical agents and evaluation of toxic or carcinogenic effects. The population doubling levels observed in this system are similar to those obtained under conventional conditions for human fibroblasts and in serum-free medium for mammary epithelia: cells (Hayflick, 1965; Martin et al., 1970; Hammond et al., 1984; Cristofalo and Stanuli.;-Praeger, 1982). For long-term growth one of the cultures required at each passage the addition of a low concentration of serum for a short period. The serum may provide undefined growth factors, but may also contribute nutritional factors, adhesion proteins, or other factors that may help the cells recover from the trypsinization (Barnes, 1987; Ham and McKeehan, 1979). The medium employed for serum-free culture of human astrocytes was a modification of the medium used for SFME cells; FGF was added in addition to the other 96 components and heparin also was added to stabilize FGF in the culture medium (Gospodarowicz and Cheng, 1986). FGF is growth stimulatory for SFME cells in the presence of the other factors (Loo et al., 1989b), and is a component of a serum-free medium formulated specifically for astrocytes (Morrison et al., 1985; Morrison and de Vellis, 1982). Of the three peptide growth factors in the serum-free medium--FGF, EGF, and insulin--the human astrocyte cultures were most strongly stimulated to proliferate by FGF. SFME cells are most responsive to EGF (Loo et al., 1987, 1989b). SFME cells are markedly inhibited by serum in the culture medium (Loo et al., 1987; Shirahata et al., 1990); multiple factors in serum may be responsible for inhibition of SFME cell proliferation, including glucocorticoid and thyroid hormones (Loo et al., 1990). Human embryonic astrocytes derived in the serum-free medium were not inhibited by serum when it was added to medium containing the appropriate supplements used in the serum-free formulation. We were not able to maintain multipassage cultures of human embryonic astrocytes in medium supplemented only with serum, and it is unlikely that initiation and continuous maintenance of the cultures in medium containing both calf serum and the supplements of the serum-free medium would be successful. because this might be expected to lead to growth of multiple cell types and possibly to overgrowth of fibroblasts. Multiple serum factors have been shown to regulate expression of GFAP in rodent astrocyte cultures (Bologa et al., 1988). Based on results with SFME cells (Sakai et al., 1990), we anticipated that serum and TGF beta could influence expression of differentiation on the human cells derived in the serum-free medium. Immunoassay of fixed cells demonstrated an increase in GFAP in the human cells after treatment with TGF beta or serum, and immunoblots with the same antiserum confirmed that the protein recognized was of the appropriate molecular weight for GFAP. TGF beta may play a role in control of differentiation of astrocytes in the developing human brain, and the growth factor released from platelets at the site of a wound or other lesion also may contribute to GFAP expression in the brain in response to trauma. The quantitatively greater response of 97 the cells to calf serum than to TGF beta in this assay is consistent with the view that other factors in serum contribute to increased GFAP expression (Bologa et al., 1988). Serum-free cultures from human embryonic brain are similar to serum-free mouse astrocyte precursor cultures in TGF beta response and GFAP expression, but the human cells differ from the mouse cells in ability to survive for longer periods in the absence of EGF and FGF, in growth factor requirements for optimal proliferation, and in growth response to serum. Most important, the serum-free human cultures, unlike SFME cells, could not be maintained in vitro for periods beyond that observed with other human cell types in conventional serum-containing medium. High-passage cells did not show a loss of response to a specific mitogen, but instead were generally less responsive under all growth conditions tested. One hypothesis regarding "senescence" in vitro is that loss of proliferative potential is related to increased differentiation of the cells with increased population doubling level (Cristofalo and Stanulis-Praeger, 1982; Martin et al., 1970). In our cultures low- and high-passage cells expressed GFAP at approximately equal levels and remained responsive to TGF beta and serum in the expression of the differentiation marker, indicating that the loss of proliferative potential with increasing population doublings was not correlated with increased expression of differentiated function. The higher-passage cells were somewhat less responsive to serum and TGF beta for expression of the differentiation marker than were the lower-passage cells. Several explanations exist for the difference in potential culture lifetime between human and mouse serum-free cultures. Possibly the mouse cells have undergone an undetected genomic alteration leading to "immortalization" and the human cells did not undergo this alteration. It is well established that mouse cells in culture are much more likely to experience spontaneous genetic changes than are human cells (Cristofalo and Stanulis-Praeger, 1982; Martin et al., 1970; Todaro and Green, 1963). However, such a change must be considerably more subtle than the change accompanying "immortalization" 98 in mouse embryo cells in conventional culture, because the conventional alterations lead to gross karyotypic abnormalities (Todaro and Green, 1963; Loo et al., 1989a). In addition, if this explanation is correct, the spontaneous genetic change leading to immortalization occurs reproducibly in multiple initiations of cultures from several mouse strains (Loo et al., 1987,1989a). Another explanation, suggested by requirement of the human cells for additional medium supplementation beyond that required for mouse cells, is that the medium used for the human cells remains suboptimal, and additional medium formulations would allow further growth of the human cultures. Finally, the differences in the behavior of the human and mouse cultures with regard to serum inhibition of growth and growth factor responses suggest that the cell type represented by the human cultures is related to, but not identical with, SFME cells. It is possible that a human homologue of SFME cells remains to be cultured and this cell type is capable of population doublings exceeding that reported in this paper. It is also possible that the precise SFME cell type simply does not exist in humans; significant differences exist in the developmental neurobiology of glial cells in these two species (Choi, 1981; Levitt et al., 1983). 99 ACKNOWLEDGMENTS This work was supported by NM-NAT-07560. D. Barnes is the recipient of Research Career Development Award NIH-NCI-01226. Yoshio Sakai performed the GFAP western immunoblot. Cathleen Rawson assayed the response of embryonic human astrocytes to peptide growth factors in serumfree medium. David Barnes provided financial and scientific suppon 100 Fig. 5.1. Long-term culture of embryonic human astrocytes in serum-free medium. Procedures for initiation and serial culture are described in the text. Population doublings were calculated from passage dilution at each trypsinization. x x x and xx , replicate cultures from human brain (22 week-old embryo) initiated and carried independently; x x -x , culture of human foreskin fibroblasts initiated and carried by identical procedures. 60 40 POP DBL 20 0 0 100 300 200 Days 400 500 101 Fig. 5.2. Photomicrograph of embryonic human astrocytes in serum-free medium. Multipassage growth characteristics of the cells are shown in Figure 5.1. Photograph was taken at 40 population doublings. 102 Fig. 5.3. Long-term culture of embryonic human astrocytes in medium supplemented with low concentrations of calf serum. The culture was initiated from human brain (24 week-old embryo) and carried for 20 population doublings in serum- free medium identical with that used for the experiment of Figure 5.1 xxx . At this point cultures were divided into three groups and serially passaged with the supplements of the serum-free medium and the following: 0.5% calf serum added at the time of plating and removed upon medium change the following day, continuously in the medium, medium, . 1E51 . ++. 0.5% calf serum present 1.0% calf serum present continuously in the Population doublings were calculated from passage dilution at each trypsinization. 80 60 POP 40 D131. 20 0 0 100 200 Days 300 400 103 Fig. 5.4. Growth of embryonic human astrocytes in serum-free and serumsupplemented media. Cells were plated in complete serum-free medium with all supplementary factors (SF), basal nutrient medium (F12:DME) supplemented only with 10% calf serum (10%CS), or complete serum-free medium supplemented additionally with 10% calf serum (CS + factors). Cells were plated at 105/35 mm-diameter plate and cell number was determined by counting trypsinized cell suspensions 7 days after plating. Medium was exchanged for fresh medium of the same composition 4 days after plating. Open bars are date from cultures at 10 population doublings. Solid bars are data from same cultures at 50 population doublings. Multipassage growth characteristics of the cells are shown in Figure 5.1. Data are the average of cell counts from two replicate dishes. Individual values for replicates did not vary more than 5% from the average. 30 SF 10%CS CS+f actors 104 Fig. 5.5. Response of embryonic human astrocytes to peptide growth factors in serum-free medium. Cells were plated in complete serum-free medium containing all supplementary factors (SF) or in medium individually lacking EGF, FGF, or insulin (I) or lacking both EGF and FGF. Cells were plated at 105/35 mm-diameter plate and cell number was determined by counting trypsinized cell suspensions 7 days after plating. Medium was exchanged for fresh medium of the same composition 4 days after plating. Open bars are data from cultures at 10 population doublings. Solid bars are data from cultures at 50 population doublings. Multipassage growth characteristics of the cells are shown in Figure 5.1. Data are the average of cell counts from two replicate dishes. Individual values for replicates did not vary more than 5% from the average. SF -EGF -FGF -EGF&FGF -I 105 Fig. 5.6. Expression of GFAP in embryonic human astrocytes. Serum-free human astrocytes at 10 and 40 population doublings were incubated in the complete serum- free medium as described (solid bars), in this medium supplemented with 10 ng/ml TGF beta (crosshatched bars) or in basal nutrient medium (F12:DME) supplemented with 10% calf serum (open bars). Cells were fixed after 2 days and assayed for GFAP as described in the text. Control values from plates receiving normal rabbit serum in place of antiGFAP were less than 20% of that obtained with anti-GFAP and were subtracted from the corresponding value with anti-GFAP in each case. Multipassage growth characteristics of the cells are shown in Figure 5.1. GFAP (A405) per 1 6 10 cells 10 POP DBLS 40 POP DBLS 106 Fig 5.7. Western immunoblot of GFAP in extracts of embryonic human astrocytes in serum-free medium. Extracts were made from cultures after 10 population doublings in vitro. A, B: Extracts from two independent cultures probed with anti-GFAP (multipassage growth characteristics of the cells are shown in Figs. 5.1, 5.3). C: Blank. D: Human brain extract probed with anti-GFAP. E: Cell culture extract as in A, probed with normal rabbit serum in place of anti-GFAP. F: Human brain extract as in D, probed with normal rabbit serum in place of anti-GFAP. GFAP is sensitive to degradation by intracellular proteinases, resulting in multiple bands detected in Western blots for GFAP in control brain extracts (De Armond et al., 1983) and occasionally in cell extracts. 68-_ ' ........., 4.1111or 43- A B C D E F 107 CHAPTER 6 Conclusion and Future Perspectives CONCLUSION The common thread binding this thesis together is the use of a serum-free approach to the long-term culture of mammalian embryonic astrocytes. A serum-free approach to the long-term culture of mouse embryo cells was undertaken based in part on the following reasoning. Serum is not a physiologically relevent substance for most cell types since most cells in vivo are not continually exposed to blood serum. Furthermore, serum is a very complex material, and contains substances which may effect both cell function and cell growth in vitro. The entry into and subsequent exit from growth crisis of mouse embryo fibroblasts cultured in serum-containing medium may reflect in part an adaptation of the cells to long-term culture in the presence of antagonistic serum factors. We reasoned that by replacing serum with a set of def..:ed growth supplements we could avoid some of the drawbacks of serum, and perhaps culture cells that were genetically and physiologically more normal than cell lines previously derived using serum-containing media. The derivation of SFME cells, detailed in chapter 2, forms the cornerstone of my thesis. This research demonstrated for the first time that karyotypically normal mouse 108 embryo cells could be cultured long-term without growth crisis using a serum-free approach. SFME cells derived using this approach displayed several unique properties. The cells were dependent on EGF for survival and were reversibly growth inhibited by serum. Later, research by Yoshio Sakai (1990) showed that SFME cells expressed glial fibrillary acidic protein (GFAP), an intermediate filament protein expressed uniquely by astrocytes, when grown in the presence of calf serum or transforming growth factor beta (TGF beta). GFAP expression was reversible following removal of calf serum or TGF beta. Sakai's work suggests SFME cells represent astrocyte precursor cells or proastroblasts. SFME cells may provide a unique cell culture model to study the biology of astrocyte precursor cells. In addition, the long-term culture of SFME cells provides evidence that normal cells can be cultured long-term under the proper environmental conditions. Following the derivation of SFME cells my thesis research branched into several projects based on observations made during the long-term culture of SFME cells. The experiments I conducted were aimed at answering the following questions. First, what type of cell do SFME cells represent in vivo, and what are the optimal growth conditions for the cells? Second, can the serum-free approach used for the long-term culture of SFME cells be extended to the long-term culture of human embryonic astrocytes? Third, by what mechanism are SFME cells growth inhibited by serum? Fourth, by what mechanism does EGF deprivation cause SFME cell death? Chapter 3 detailed the growth responses of SFME cells in vitro. This work examined precisely the growth requirements of SFME cells in vitro and detailed the cell culture methods used to derive SFME cells. The concentration dependence of the growth stimulatory effects of protein supplements used in the serum-free medium formulation was determined. A survey of other supplements that might act as alternative or complementary additions to the culture medium was also done. Three main conclusions of this work were (1) transforming growth factor alpha (TGF alpha), at equivalent concentrations, replaced 109 EGF for SFME cell survival and growth, (2) fibroblast growth factor partially replaced EGF and allowed for survival and slow growth of SFME cells, and (3) the viability of SFME cells was reduced when cells were deprived of EGF for as little as 8 hours. In chapter 4 and appendix 1 the inhibition of SFME cell growth by calf serum was explored. Research detailed in chapter 4 showed that SFME cells were reversibly growth inhibited by low concentrations of calf serum or plasma, and cells arrested in the G1 phase of the cell cycle. Additionally, charcoal extraction, a procedure that removes hydrophobic molecules, removed a portion of the inhibitory activity from serum. Both glucocorticoid hormones and thyroid hormones (T3 and T4), molecules that would be removed by charcoal, were growth inhibitory for SFME cells. The inhibitory activity was specific, reversible, and synergistic with charcoal-extracted serum. The primary conclusion from these results was that glucocorticoid or thyroid hormones may contribute to the inhibitory activity of serum on SFME cells, but additional unidentified inhibitory factors were also involved and remain to be identified. Appendix 1 reported that SFME cells cultured in serum-containing medium arrested in the G1 phase of the cell cycle, with a greatly reduced rate of incorporation of precursors into DNA and thymidine kinase activity. A reduction in rate of incorporation of amino acids into protein was not observed. These observations are associated with physiologically relevant inhibition of cell growth, and are consistent with growth inhibition associated with induction of cell differentiation. In addition, appendix 1 revealed that variant SFME cells capable of growing in serum-containing medium were selected in medium containing 10% calf serum using a 3T3 protocol. Proliferation of variant cells was observed with a frequency of about 1 in 106 SFME cells following a one-month lag of no cell growth, similar to the growth crisis period observed with serum-derived mouse fibroblasts. Karyotypic analysis revealed that variant SFME cells capable of growing in serum displayed abnormal karyotypes with a strong tendency toward hyperploidy, suggesting that serum-containing medium selected for, or induced, an abnormal karyotype 110 in SFME cells. While gene dosage may play a role in release from serum inhibition, SFME cells made polyploid by cell fusion remained inhibited by serum. In addition, close examination of the relationship between exit from growth crisis and karyotype shift from a diploid to a tetraploid karyotype of the population of serum-derived mouse embryo cells suggested that cells exited from growth crisis one or two generations before the karyotype switch occurred. These observations suggest that exit from growth crisis of serum-grown mouse embryo cells resulted from one or more changes within the cell, either genetic or epigenetic, which allow the cells to be refractile to inhibition by serum. In addition, the change appeared to reduce the cells ability to properly maintain a normal karyotype. Chapter 5 detailed research addressing whether SFME-like cells could be cultured from human embryonic brain. This chapter described the long-term growth of astrocyte cells from human embryonic brain with properties common to SFME cells. The human astrocytes displayed a morphology similar to SFME cells in culture and expressed GFAP when grown in the presence of calf serum or TGF beta. However, the human astrocytes differed from SFME cells in ability to survive for longer periods in the absence of EGF and FGF, in growth factor requirements for optimal proliferation, and in growth response to serum. The human astrocytes also expressed detectable amounts of GFAP when grown serum-free. Most important, the serum-free human cultures, unlike SFME cells, could not be maintained in vitro for periods beyond that observed with other human cell types in conventional serum-containing medium. Several explanations were proposed in chapter 5 for the difference in potential culture lifetime between the human and mouse serum-free cultures. The mouse cells may have undergone an undetectable genetic change which resulted in the ability of the cells to avoid growth crisis. The genome of human cells is more stable than rodent cells in vitro, so genetic changes required for human cells to escape growth crisis may occur less frequently. The medium formulation used to culture the human astrocytes may have been suboptimal, and additional formulations might have allowed for further growth. The 111 growth stimulatory effect of calf serum added to cells growing in the serum-free medium suggests other growth stimulatory factors exist. The human astrocytes may be a related but not identical cell type to SFME cells. It is possible that SFME-type cells remain to be cultured from embryonic human brain, or perhaps SFME-type cells do not exist in embryonic human brain. Finally, appendix 2 described research directed at understanding the mechanism by which EGF deprivation led to death of SFME cells. Electron microscopic examination of SFME cells showed that cells became small and severely degenerate. growth arrested in the G1 -phase of the cell cycle, and displayed characteristic pyknotic nuclei following removal of EGF. Chromatin isolated from cultures undergoing EGF deprivation-dependent cell death exhibited a pattern of fragmentation suggesting that DNA was cleaved by activated nucleases at regions unprotected by histones. At the same time mRNA from these cells remained intact. Many of these characteristics have been associated with cell death in several growth factor-dependent systems, both in vitro and in vivo (refer to introduction). It is well established that apoptosis, or programmed cell death, is required for the normal development and survival of all multicellular organisms. It has been suggested that cell death, following growth factor withdrawal in growth factor-dependent systems, may occur by apoptosis. It has been further suggested, based on results from experiments using RNA and protein synthesis inhibitors, that apoptosis, or programmed cell death in these growth factor-dependent systems, required the synthesis of RNA and protein. SFME cell death following EGF withdrawal is blocked by inhibitors of RNA and protein synthesis. The general conclusion of appendix 2 is that SFME cells, when deprived of EGF, rapidly entered a cell death program, termed apoptosis, that is morphologically, physiologically, and biochemically distinct from necrosis. 112 FUTURE PERSPECTIVES Research using SFME cells falls into two broad categories. One, experiments may be designed to answer questions about SFME cells, in order to expand our knowledge of the biology of SFME cells. Two, experiments may be designed where SFME cells are used as an in vitro model to study a particular phenomenon. These two categories of experiments often overlap. I believe it is important that experiments involving SFME cells continue to pursue both lines of research. In the next several paragraphs I will briefly pose some broad questions that I feel are important to follow. Some fall into category one, others into category two, and still others somewhere in between. I will then briefly propose some ways in which these questions might be pursued. First, how do SFME contribute to the adult mouse brain? It would be valuable to be able to show that SFME cells contribute to the normal development of the mouse brain, and would open up numerous research avenues. One direct approach would be to create SFME cells that express a specific, detectable markers, such as beta galactosidase or luciferase. Once available, the cells could be transplanted into the brains of mice at various stages of development. The mouse brain is not fully developmed at birth, and newborn mice may prove to be the best candidate. Progress of the transplanted SFME cells may be followed by sacrificing mice later in development and assaying thin tissue slices for the specific marker expressed by the SFME cells. At the same time, geneticin (G -418)resistant SFME cells could be transplanted into similar mice, and later in development the brain could be recovered and the presence of G-418-resistant cells assayed for in culture using G-418-containing serum-free medium. If transplanted SFME cells contribute to normal brain development then the role of SFME cells in vivo may be addressed. For instance, how do SFME cells contribute to reactive gliosis? Do SFME cells participate in the repair of brain lesions? Evidence suggests that lesioning of the rat brain results in an increase in membrane EGF receptor 113 content of astrocytes adjacent to the lesion, and a decrease in an EGF receptor-like astrocyte mitogen inhibitor (Nieto-Sampedro, 1988). In addition, macrophages and microglia are attracted to lesioned areas of the brain, and tissue plasminogen activator (TPA) protein has been found associated with lesioned areas of the brain. Both low density lipoprotein (LDL) receptors and TPA contain EGF-like domains and may promote SFME cell growth. Finally, TGF beta may be released from platelets at the site of the wound and promote astrocyte differentiation. An interplay between growth inhibitory and growth stimulatory agents may modulate, perhaps through the EGF receptor, the response of astrocytes to injuries to the brain. Second, what are the additional growth inhibitory factors in serum, and by what mechanism do they cause growth inhibition of SFME cells? Purifying the inhibitory components from serum using biochemical techniques has not been very successful. One approach that may yield information about the mechanism of serum inhibition is to use the technique of differential cloning to identify genes that are regulated by serum. This approach may lead to the identification of genes that regulate SFME cell growth and may also identify cell-specific markers of astrocyte precursor cells. Another approach might be to examine the signal transduction pathway of the EGF receptor in SFME cells following exposure to serum. Although the level of membrane-associated EGF receptors is similar between SFME cells growth serum-free or in serum-containing medium, the phosphorylation state of the receptor may be altered, or the EGF receptor complex as a whole may be altered. Finally, gene expression between normal SFME cells and serum- selected SFME cells may be compared in order to examine whether gene expression of serum-selected SFME cells differs from normal SFME cells. Third, do genes repressed by EGF exist in SFME cells, and do they participate in the EGF-deprivation dependent cell death program? One approach would be to continue using differential cloning techniques to identify genes that are specifically expressed following EGF withdrawal. Although I was unable to identify EGF-repressed genes 15 114 hours following EGF withdrawal, examining gene expression at earlier time points following growth factor removal may prove more successful. Earlier time points may avoid artifacts, such as the differential accumulation of fragmented DNA, resulting from cellular degradation as the cells undergo apoptosis. Another approach might be to examine the EGF receptor signal transduction pathway of SFME cells grown in the presence and absence of EGF. For instance, blocking points in the signal transduction may lead to SFME cell death and identify points of the pathway important for EGF-repression of programmed cell death. Finally, the serum-free approach used for the long-term culture of both mouse and human embryonic astrocytes may be extended to the long-term culture of astrocytes recovered from individuals afflicted with degenerative brain disorders, including Alzheimer's disease and Parkinson's disease. Comparisons between cultures of astrocyte cells from normal and diseased tissues may provide important information about the role astrocytes may play in the disease state. 115 BIBLIOGRAPHY Aaronson, S.A. and Todaro, G.J. 1968. Development of 3T3-like lines from Balb/c mouse embryo cultures: transformation susceptibility to SV40. J. Cell Phys. 72:141-148. Ambesi-Impiombato,F.S., Parks, L.A.M. and Coon H.G. 1980. Culture of hormonedependent functional epithelial cells from rat thyroids. Proc. Natl. Acad. Sci. USA. 77:3455-3459. Arends, M.J., Morris, R.G. and Wyllie, A.H. 1990. Apoptosis: the role of the endonuclease. Amer. J. Path. 136:593-608. Augusti-Tocco, G. and Sato, G. 1969. Establishment of functional clonal lines of neurons from mouse neuroblastoma. Proc. Natl. Acad. Sci. USA. 64:311-315. Barinaga, M. 1990. Technical advances power neuroscience. Science. 250:908-909. Barnes, D. 1987. Serum-free animal cell culture. BioTechniques. 5:1-9. Barnes, D. and Sato, G. 1980a. Methods for growth of cultured cells in serum-free medium. Anal. Biochem. 102:255-270. Barnes, D. and Sato, G. 1980b. Serum-free cell culture: a unifying approach. Cell. 22:649-655. Barnes, D. and Silnutzer, J. 1983. Isolation of human serum spreading factor. J. Biol. Chem. 258:12548-12552. 1989. Effects of oncogenes on EGF requirement and serum inhibition of serum-free derived mouse embryo (SFME) cells. J. Cell Biol. 107:4452 (abstract). Barnes, D., Loo, D., Shirahata, S., Rawson, C. and Chang, Y. Barnes, D.W. and Reed, D.J. 1989. Cytotoxicity and cell culture conditions. In: Milo, G.E. (ed): "Transformation of Human Diploid Fibroblasts." Barnes, D.W., Silnutzer, J., See. C. and Shaffer, M. 1983. Characterization of human serum spreading factor with monoclonal antibodies. Proc. Natl. Acad. Sci. USA. 80:1362-1366. Bartlett, P.F., Reid, H.H., Bailey, K.A. and Bernard, 0. 1988. Immortalization of mouse neural precursor cells by the c-myc oncogene. Proc. Natl. Acad. Sci. USA. 85:3255-3259. Baserga, R., Waechter, D.E., Soprano, K.J. and Galanti, N. 1982. Molecular biology of cell division. Ann. N.Y. Acad. Sci. 397:110-120. Beato, M. 1989. Gene regulation by steroid hormones. Cell. 56:335-344. Beck, W.S. 1982. Hypochromic anemias I. Iron deficiency and excess. In: Beck, W.S. (ed): "Hematology." MIT Press, Cambridge, MA. 102-103. 116 Benda, P., Lightbody, J., Sato, G., Levine. L. and Sweet, W. 1969. Differentiated rat glial cell strain in tissue culture. Science. 161:370-371. Bettger, W.J. and Ham, R.G. 1982. The nutrient requirements of mammalian cells grown in culture. Adv. Nutr. Res. 4:249-286. Bettger, W.J., Boyce, S.T., Walthall, B.J. and Ham, R.G. 1981. Rapid clonal growth and serial passage of human diploid fibroblasts in a lipid-enriched synthetic medium supplemented with epidermal growth factor, insulin, and dexamethasone. Proc. Natl. Acad. Sci. USA. 78:5588-5592. Bettuzzi, S., Hiipakka, R.A., Gilna, P. and Liao, S. 1989. Identification of an androgenrepressed mRNA in rat ventral prostate as coding for sulphated glycoprotein 2 by cDNA cloning and sequence analysis. Biochem. J. 257:293-296. Bishop, J.M. 1987. The molecular genetics of cancer. Science. 235:305-311. Bologa, L., Cole, R., Chiappelli, F., Saneto, R.P. and de Vellis, J. 1988. Expressoin of glial fibrillary acidic protein by differentiated astrocytes is regulated by serum antagonistic factors. Brain Res. 457:295-302. Bottenstein, J.E. 1985. Growth and differentiation of neural cells in defined media. In: Bottenstein, J.E. and Sato, G. (eds): "Cell Culture in the Neurosciences." Plenum Press, NY. Bottenstein, J.E. 1986. Growth requirements in vitro of oligodendrocyte cell lines and neonatal rat brain oligodendrocytes. Proc. Natl. Acad. Sci. USA. 83:1955-1959. Bourgeois, S. and Gasson, J.C. 1985. Genetic and epigenetic bases of glucocorticoid resistance in lymphoid cell lines. Biochem. Actions Horm. 12:310-351. Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248254. Brandi, M.L., Fitzpatrick, L.A., Coon, H.G. and Aurbach, G.D. 1986. Bovine parathyroid cells: cultures maintained for more than 140 population doublings. Proc. Natl. Acad. Sci. USA. 83:1709-1713. Bressler, J.P. and de Vellis, J. 1985. Neoplastic transformation of newborn rat astrocytes in culture. Brain Res. 348:21-27. Brooks, R.F. 1975. Growth regulation in vitro and the role of serum. In: Allison, A.C. (ed): "Structure and Function of Plasma Proteins." Plenum, NY, 1. 112. Brunner, G., Lang, K., Wolfe, R.A., McClure, D.B. and Sato, G.H. 1982. Selective cell culture of brain cells by serum-free, hormone-supplemented media: a comparative morphological study. Dev. Brain Res. 2:563-575. Buttyan, R., Zakeri, Z., Lockshin, R. and Wolgemuth, D. 1988. Cascade induction of cfos, c-myc, and heat shock 70K transcripts during regression of the rat ventral prostate gland. Mol. Endocrin. 2:650-657. 117 Carpenter, G. and Cohen, S. 1979. Epidermal growth factor. Annu. Rev. Biochem. 48:193-216. Carrel, A. 1912. On the permanent life of tissues outside of the organism. J. Exp. Med. 15:516-528. Cherington, P.V., Smith, B.L. and Pardee, A.B. 1979. Loss of epidermal growth factor requirement and malignant transformation. Proc. Natl. Acad. Sci. USA. 76:3937-3941. Chiang, L.-C., Silnutzer, J., Pipas, J.M. and Barnes, D.B. 1985. Selection of transformed cells in serum-free media. In Vitro Cell. Dev. Biol. 21:707-712. 1981. Radial glia of developing human fetal spinal cord: golgi, immunohistochemical and electron microscope study. Dev. Brain Res. 1:249-267. Choi, B.H. Clemens, M.J. and mcNurlan, M.A. 1985. Regulation of cell proliferation and differentiation by interferons. Biochem. J. 226:345-350. Cohen, J.J. and Duke, R.C. 1984. Glucocorticoid activation of a calcium-depenent endonuclease in thymocyte nuclei leads to cell death. J. Immunology. 132:38-42. Conn, P.M. 1990. Methods in neurosciences. Volume 2. Cristofalo, V.J. and Stanulis-Praeger, B.M. 1982. Cellular senescence in vitro. Adv. Cell Culture. 2:1-53. Darmon, M., Serrero, G., Rizzino, A. and Sato, G. 1981. Isolation of myoblastic, fibroadipogenic and fibroblastic clonal cell lines from a common precursor and study of their requirement for growth and differentiation. Exp. Cell Res. 132:313-327. de Vellis, Wu, D. and Kumar, S. 1986. Enzyme induction and regulation of protein synthesis. In: Fedoroff, S. and Vernadakis, A. (eds): "Biochemistry, Physiology, and Pharmacology of Astrocytes." Academic Press, NY. 2:209-237. DeArmond, S.J., Fajardo, M. and Eng, L.F. 1983. Degradation of glial fibrillary acidic protein by a calciium dependent proteinase: an electoblot study. Brain Res. 262:275-282. Demars, R. 1958. The inhibition by glutamine of glutamyl transferase formation in cultures of human cells. Biochim. et Biophys. Acta. 27:435-436. Downward, J., Yarden, Y., Mayer., E., Scrace, G., Totty, N., Stockwell, P., Ullrich, A., Schlessinger, J. and Waterfield, M.D. 1984. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 307:521-527. Duke, R.C. and Cohen, J.J. 1986. IL-2 addiction: withdrawl of growth factor activates a suicide program in dependent T cells. Lymphokim Res. 5:289-299. Dulbecco, R. and Elkington, J. 1973. Conditions limiting multiplication of fibroblastic and epithelial cells in dense cultures. Nature. 246:197-199. Eagle, H. 1955a. Nutrition needs of mammalian cells in tissue culture. Science. 122:501504. 118 Eagle, H. 1955b. The minimum vitamin requirements of the L and He La cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J. Exp. Med. 102:595-600. Eagle, H. 1955c. The specific amino acid requirements of a human carcinoma cell (strain He La) in tissue culture. J. Exp. Med. 102:37-48. Eagle, H. 1955d. The specific amino acid requirements of a mammalian cell (strain L) in tissue culure. J. Biol. Chem. 214:839-853. Eagle, H. 1959. Amino acid metabolism in mammalian cell cultures. Science. 130:432437. Eagle, H., Oyama, V.I., Levy, M., Horton, C.L. and Fleischman, R. 1956. The growth response of mammalian cells in tissue culture to L-glutamate and L-glutamic acid. J. Biol. Chem. 218:607-617. Earle, W.R., Schilling, E.L., Stark, T.H., Straus, N.P., Brown, M.F. and Shelton, E. 1943. Production of malignancy in vitro. IV. the mouse fibroblast cultures and changes seen in the living cells. J. Natl. Cancer Inst. 4:165-212. Ernst, T., Jackson, C. and Barnes, D. 1990. Factors influencing karyotypic stability of serum-free mouse embryo (SFME) cells. Cytotech. in press. Evans, R.M. 1988. The steroid and thyroid hormone receptor superfamily. Science. 240:889-895. Evans, V.J., Bryant, J.C., Fioramonti, M.C., McQuilkin, W.T., Sanford, K.K., and Earle, W.R. 1955. Studies of nutrient media for tissue cells in vitro. Cancer Research. 16:77-86. Farber, R.A. and Liskay, R.M. 1974. Karyotypic analysis of a near-diploid established mouse cell line. Cytogenet. Cell Genet. 13:384-396. Galiana, E., Borde, I., Marin, P., Rassoulzadegan, M., Cuzin, F., Gros, F., Rouget, P. and Evrard, C. 1990. Establishment of permanent astroglial cell lines, able to differentiate in vitro, from transgenic mice carrying the polyoma virus large T gene: an alternative approach to brain cell immortalization. J. Neurosci. Res. 26:269-277. Gey, G.O., Coffman, W.D. and Kubicek, M.T. 1952. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 12:264265. Giguere, L., Cheng, J. and Gospodarowicz, D. Factors involved in the control of proliferation of bovine corneal endothelial cells maintained in serum-free medium. J. Cell Phys. 110:72-80. Gospodarowicz, D. 1984. Preparation and uses of lipoproteins to culture nortnal diploid and tumor cells under serum-free conditions. In: Barnes, D.W., Sirbasku, D.A. and Sato, G.H. (eds): "Cell Culture Methods for Molecular and Cell Biology." Alan R. Liss, NY. 1:69-88. Gospodarowicz, D. and Cheng, J. 1986. Heparin protects basic and acidic FGF from inactivation. J. Cell Phys. 28:475-484. 119 1981. Factors controlling the proliferative rate, final cell density and life span of bovine vascular smooth muscle cells in culture. J. Cell Biol. 89:568-578. Gospodarowicz, D., Hirabayashi, K., Giguere, L. and Tauber, H. Gray, J.W. and Coffin, P. 1979. Flow cytometry. Methods Enzymol. 58:233-238. Greene, L.A. and Tisch ler, A.S. 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA. 73:2424-2428. Grenier, Y., Ruijs, T.C.J., Olivier, A., Robitaille, Y. and Antel, J.P. 1989. DNA synthesis by young adult human-derived astrocytes in vitro. Brain Res. 480:87-91. 1989. Thymidine kinase synthesis is repressed in nonreplicating muscle cells by a translational mechanism that does not affect the polysome distribution of thymidine kinase mRNA. Proc. Natl. Acad. Sci. USA. 86:4987-4991. Gross, M.K. and Merrill, G.F. Grotendorst, G., Kleinman, H., Rohrbach, D., Hewitt, A., Varner, H., Horigan, E., Hassel, J., Terranova, V. and Martin, G. 1982. Role of attachment factors in mediating the attachment, distribution, and differentiation of cells. In: Sato,G. and Sirbasku, D. (eds): "Cold Spring Harbor Conference on Cell Proliferation." Cold Spring Harbor Press, Cold Spring Harbor, NY, 9:403-413. Ham, R.G. 1963. An improved nutrient solution for diploid chinese hamster and human cell lines. Exp. Cell Research. 29:515-526. Ham, R.G. 1965. Clonal growth of mammalian cells in a chemically defined, synthetic medium. Proc. Natl. Acad. Sci. USA. 53:288-293. Ham, R.G. and McKeehan, W.L. 1979. Media and growth requirements. Methods Enzymol. 58:44-93. Hamburger, V., Brunso-Bechtold, J.K. and Yip, J.W. 1981. Neuronal death in the spinal ganglia of the chick embryo and its reduction by nerve growth factor. J. Neurosci. 1:60-71. Hammond, S.L., Ham, R.G. and Stampfer, M.R. 1984. Serum-free growth of human mammary epithelial cells: rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc. Natl. Acad. Sci. USA. 81:5435-5439. Harrison, R.G. 1907. Observations on the living developing nerve fiber. Proc. Soc. Exp. Biol. Med. 4:140-143. Hashizume, S., Kuroda, K. and Murakami, H. 1984. Cell culture assay of biological activity of lactoferrin and transferrin. Methods Enzymol. 147:302-314. Hayashi, I. and Sato, G. 1976. Replacement of serum by hormones permits growth of cells in defined medium. Nature. 259:132-134. Hayat, M.A. 1986. Basic techniques for transmission electron microscopy. Academic Press, Orlando, FL. 226. 120 Hayflick, L. 1965. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Rsearch. 37:614-636. Hayflick, L. and Moorhead P.S. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Research. 25:585-621. Healy, G.M., Fisher, D.C. and Parker, R.C. 1954. Nutrition of animal cells in tissue culture. IX. synthetic medium No. 703. Can. J. Biochem. Physiol. 32:327-337. Hinchliffe, J.R. 1981. Cell death in embryogenesis. In: Bowen, I.D. and Lockshin, R.A. (eds): "Cell Death in Biology and Pathology." Chapman and Hall Press, London, NY, 35-78. Hoshi, H. and McKeehan, W.L. 1984. Brain cell derived and liver cell derived factors are required for growth of human endothelial cells in serum-free culture. Proc. Natl. Acad. Sci. USA. 81:6413-6417. Hughes, S.M., Lillien, L.E., Raff, M.C., Rohrer, H. and Sendtner, M. 1988. Ciliary neurotrophic factor induces type-2 astrocyte differentiation in culture. Nature 335:70-73. Isofort, R.J. 1990. Frequency and mechanisms of factor independence in IL-3-dependent cell lines. Somatic Cell Mol. Genet. 16:109-121. Ito, F., Takii, Y., Sizuki, J. and Masamune, Y. 1982. Reversible inhibition by human serum lipoproteins of cell proliferation. J. Cell Phys. 13:1-13. Jainchill, J.L., Aaronson, S.A. and Todaro, G.J. 1969. Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. J. Virol. 4:549-553. Kaiser, N. and Edelman, I.S. 1977. Calcium dependence of glucocorticoid-induced lymphocytolysis. Proc. Natl. Acad. Sci. USA. 74:638-642. Kan, M. and Yamane, I. 1982. In vitro proliferation and lifespan of human diploid fibroblasts in serum-free BSA-containing medium. J. Cell. Phys. 111:155-162. Kerr, J.F.R. and Searle, J. 1973. Deletion of cells by apoptosis during castration-induced involution of the rat prostate. Virchows Arch. 13:87-102. Klagsbrun, M., Sullivan, R., Smith, S., Rybka, R. and Shing, Y. 1987. Purification of endothelial cell growth factors by heparin affinity chromatography. Methods Enzymol. 147:95-106. Koenig, R.J., Lazar, M.A., Hodin, R.A., Brent, G.A., Larsen, P.R., Chin, W.W. and Moore, D.D. 1989. Inhibition of thyroid hormone action by a non-hormone binding cerbA protein generated by alternative mRNA splicing. Nature. 337:659-661. Kohase, M., May, L., Tamm, I., Vilcek, J. and Sehgal, P. 1987. A cytokine network in human diploid fibroblasts: interactions of beta interferons, tumor necrosis fator, plateletderived growth factor and interleukin 1. Mol. Cell. Biol. 7:273-280 Koury, M.J. and Bondurant, M.C. 1990. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science. 248:378-381. 121 Kyprianou, N. and Isaacs, J.T. 1988. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology. 122:552-562. Land, H., Parada, L.F. and Weinberg, R.A. 1983. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 304:596-602. Lazarides, E. 1982. Intermediate filaments: a chemically heterogeneous, developmentally regulated class of proteins. Annu. Rev. Biochem. 51:219-250. Leffert, H.L. and Weinstein, D.B. 1976. Growth control of differentiated fetal rat hepatocytes in primary monolayer culture IX. Specific inhibition of DNA synthesis initiation by very low density lipoprotein and possible significance to the problem of liver regeneration. J. Cell. Biol. 70:20-27. Leger, J.G., Montpetit, M.L. and Tenniswood, M.P. 1987. Characterization and cloning of androgen-repressed mRNAs from the rat ventral prostate. Biochem. Biophys. Res. Comm. 147:196-203. Levitt, P., Cooper, M.L. and Rakic, P. 1983. Early divergence and changing proportions of neuronal and glial precursor cells in primate cerebral ventricular zone. Dev. Biol. 96:472-484. Lewis, R.L. and Lewis W.H. 1911. The cultivation of tissues from chick embryos in solutions of NaCl, CaC12, KCl and NaHCO3. The Anatomical Record. 5:277-285. Lil lien, L.E. and Raff, M.C. 1990a. Analysis of the cell-cell interactions that control type2 astrocyte development in vitro. Neuron. 4:525-534. Li llien, L.E. and Raff, M.C. 1990b. Differentiation signals in the CNS: type-2 astrocyte development in vitro as a model system. Neuron. 5:111-119. Lillien, L.E., Sendtner, M., Rohrer, H., Hughes, S.M. and Raff, M.C. 1988. Type-2 astrocyte development in rat brain cultures is initiated by a CNTF-like protein produced by type-1 astrocytes. Neuron. 1:485-494. Lippman, M., Bolan, G. and Huff, K. 1976. The effects of glucocorticoids and progesterone on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res. 36:4602-4609. Loo, D., Rawson, C., Helmrich, A. and Barnes, D. 1989b. Serum-fee mouse embryo (SFME) cells: growth responses in vitro. J. Cell Phys. 139:484-491. Loo, D., Rawson, C., Schmitt, M., Lindburg, K. and Barnes, D. 1990. Glucocorticoid and thyroid hormones inhibit proliferation of serum-free mouse embryo (SFME) cells. J. Cell Phys. 142:210-217 Rawson, C.L. and Barnes, D.W. 1987. Extended culture of Loo, D.T., Fuquay, mouse embryo cells without senescence: inhibition by serum. Science. 236:200-202. Loo, D.T., Rawson, C.L., Ernst, T., Shirahata, S. and Barnes, D.W. 1989a. Serum-free primary culture of mouse embryo cells. In Baserga, R. (ed): "Cell Growth and Cell Division: A Practical Approach." London: IRL Press, pp17-34. 122 Loo, D.T., Sakai, Y., Rawson, C.L. and Barnes, D.W. 1991. Serial passage of embryonic human astrocytes in serum-free, hormone-supplemented medium. J. Neurosci. Res. 28:101-109. Maniatis, T., Fritsch, E.F. and Sambrook, J. 1982. Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. Marquardt, H., Hunkapiller, M.W., Hood, L.E. and Todaro, G. 1984. Rat transforming growth factor type 1: structure and relation to epidermal growth factor. Science. 223:1079-1082. Martin, D.P., Schmidt, R.E., Di Stefano, P.S., Lowry, 0.H., Carter, J.G. and Johnson, E.M. 1988. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106:829-844. Martin, G.M., Sprague, C.A. and Epstein, C.J. 1970. Replicative life-span of cultivated human cells. Lab. Invest. 23:86-92. Massague, J. 1987. The TGF-beta family of growth and differentiation factors. Cell. 49:437-438. Masui, T., Wakefield, L.M., Lechner, J.F., LaVeck, M.A., Sporn, M.B. and Harris, C.C. 1986. Type beta transforming growth factor is the primary differentiation-inducing serum factor for normal human bronchial epithelial cells. Proc. Natl. Acad. Sci. USA. 83:2438-2442. Mather, J.P. and Sato, G.H. 1979. The use of hormone-supplemented serum-free media in primary cultures. Exp. Cell Res. 124:215-221. McConkey, D.J., Nicotera, P., Hartzell, P., Bellomo, G., Wyllie, A.H. and Orrenius, S. 1989. Glucocorticoids activate a suicide process in thymocytes through an elevation of cytosolic calcium concentration. Arch. Biochem. Biophys. 269:365-370. McKay, R.D.G. 1989. The origins of cellular diversity in the mammalian central nervous system. Cell. 58:815-821. McKeehan, W. and Ham, R. 1976. Stimulation of clonal growth of normal fibroblasts with substrata coated with basic polymers. J. Cell Biol. 71:727-734. McKeehan, W.L., Genereux, D.P. and Ham, R.G. 1978. Assay and partial purification of factors from serum that control multiplication of human diploid fibroblasts. Biochem. Biophys. Res. Comm. 80:1013-1021. Merrill, G.F., Hauschka, S.D. and McKnight, S.L. 1984. tk enzyme expression in differentiating muscle cells is regulated through an int,mal segment of the cellular tic gene. Mol. Cell. Biol. 4:1777-1784. Miller, R.H., David, S., Patel., R., Abney, E.R. and Raff, M.C. 1985. A quantitative immunohistochemical study of macroglial cell development in the rat optic nerve: in vivo evidence for two distinct astrocyte lineages. Dev. Biol. 111:35-41. Milsted, A., Barna, B.P. Ronsohoff, R.M., Brosnihan, K.B. and Ferrario, C.M. 1990. Astrocyte cultures derived from human brain tissue express angoitensinogen mRNA. Proc. Natl. Acad. Sci. USA. 87:5720-5723. 123 Moguilewsky, M. and Philibert, D. 1984. RU38486: potent antiglucocorticoid activity correlated with strong binding to the cytosolic glucocorticoid receptor followed by an impaired activation. J. Steroid Biochem. 20:271-276. Montpetit, M.L., Lawless, K.R. and Tenniswood, M. 1986. Androgen-repressed messages in the rat ventral prostate. The Prostate. 8:25-36. Morgan. J.F., Morton, H.J. and Parker, R.C. 1950. Nutrition of animal cells in tissue culture. I. inital studies on a synthetic medium. Proc. Soc. Exp. Biol. Med. 73:1-8. Morrison, R.S. and de Vellis, J. 1981. Growth of purified astrocytes in a chemically defined medium. Proc. Natl. Acad. Sci. USA. 78:7205-7209. Morrison, R.S. and de Vellis, J. 1984. Preparation of a chemically defined medium for purified astrocytes. In: Barnes, D.W., Sirbasku, D.A. and Sato, G.H. (eds): "Cell Culture Methods for Molecular and Cell Biology." Alan R. Liss, NY. 4:15-22. Morrison, R.S., de Vellis, J., Lee, Y.L., Bradshaw, R.A. and Eng, L.F. 1985. Hormones and growth factors induce the synthesis of glial fibrillary acidic protein in rat brain astrocytes. J. Neurosci. Res. 14:167-176. Nieto-Sampedro, M. 1988. Astrocyte mitogen inhibitor related to epidermal growth factor receptor. Science. 240:1784-1786. Nicola, N.A. 1989. Hemopoietic cell growth factors and their receptors. Annu. Rev. Biochem. 58:45-77. Olivier, A.R., Ballou, L.M. and Thomas, G. 1988. Differential regulation of S6 phosphorylation by insulin and epidermal growth factor in Swiss mouse 3T3 cells: insulin activation of type 1 phosphatase. Proc. Natl. Acad. Sci. USA. 85:4720-4724. Oppenheim, R.W., Prevette, D., Tytell, M. and Homma, S. 1990. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA sytthesis: evidence for the role of cell death genes. Dev. Biol. 138:104-113. Orly, J., Sato, G. and Erickson, G. 1980. Serum suppresses the expression of hormonally-induced functions in cultured granulosa cells. Cell. 20:817-827. Pardee, A.B., Dubrow, R., Hamlin, J.L. and Kletzien, R.F. 1978. Animal cell cycle. Annu. Rev. Biochem. 47:715-750. Paul, D., Brown, K.H., Rupniak, H.T. and Ristow, H.J. 1978. Cell cycle regulation by growth factors and nutrients in normal and transformed cells. In Vitro Cell. Dev. Biol. 14:76-84. Pfeiffer, S.E. 1973. In: Sato, G. (ed): "Tissue Culture of the Nervous System." Plenum Press, NY. Phillips, P.D. and Cristofalo, V.J. 1988. Classification system based on the functional equivalency of mitogens that regulate WI-38 cell proliferation. Exp. Cell Res. 175:396403. 124 Pipas, J.M., Chaing, L.C. and Barnes, D.W. 1984. Effect of cell type, hormonal conditions and viral genetic background on transforming infections of S V40. Cancer Cells. 2:355-363. Pledger, W.J., Stiles, C.D., Antoniades, H.N. and Scher, C.D. 1978. An ordered sequence of events is required before BALB/c-3T3 cells become committed to DNA synthesis. Proc. Natl. Acad. Sci. USA. 75:2839-2843. Raff, M.C. 1989. Glial cell diversification in the rat optic nerve. Science. 243:14501455. Raff, M.C., Abney, E.R. and Miller, R.H. 1984. Two glial cell lineages diverge prenatally in rat optic nerve. Dev. Biol. 106:53-60. Raff, M.C., Abney, E.R., Cohen, J., Lindsay, R. and Noble, M. 1983. Two types of astrocytes in cultures of developing rat white matter: differences in morphology, surface gangliosides, and growth characteristics. J. Neurosci. 3:1289-1300. Raff, M.C., Miller, R.H. and Noble, M. 1983. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 303:390-396. Raff, M.C., Williams, B.P. and Miller, R.H. 1984. The in vitro differentiation of a bipotential glial progenitor cell. EMBO J. 3:1857-1864. Rawson, C., Cosola-Smith, C. and Barnes, D. 1990. Death of serum-free mouse embryo cells caused by epidermal growth factor deprivation is prevented by cycloheximide, 12-0tetradecanoylphorbol-13-acetate, or vanadate. Exp. Cell Res. 186;177-181. Rawson, C., Loo, D., Helmrich, A., Ernst, T., Natsuno, T., Merrill, G. and Barnes, D. 1991b. Serum inhibition of proliferation of serum-free mouse embryo cells. Exp. Cell Res. 192:271-277. Rawson, C.L., Loo, D.T., Duimstra, J.R., Hedstrom, O.R., Schmidt, E.E. and Barnes, D. 1991a. Death of serum-free mouse embryo cells caused by epidermal growth factor deprivation. J. Cell Biol. 113:671-680. Reznikoff, C.A., Brankow, D.W. and Heidelberger, C. 1973. Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Research. 33:3231-3238. Rizzino, A. 1984. Behavior of transforming growth factors in serum-free media--an improved assay for gransforming growth factors. In Vitro Cell Dev. Biol. 20:815-822. Rizzino, A. 1988. Transforming growth factor beta: multiple effects on cell differentiation and extracellular matrices. Dev. Biol. 130:411-422. Rizzino, A., Rizzino, H. and Sato, G. 1979. Defined media and the determination of nutritional and hormonal requirements of mammalian cells in culture. Nutrition Reviews. 37:369-378. Rodriguez-Tarduchy, G. and Lopez-Rivas, A. 1989. Phorbol esters inhibit apoptosis in IL-2 dependent T lymphocytes. Biochem. Biophys. Res. Comm. 164:1069-1075. 125 Rosenfeld, R.G. and Hintz, R.L. 1986. Somatomedin receptors: structure, function and regulation. In: Conn, P.M. (ed): "Receptors." Academic Press, Orlando, FL. 3:281329. Rubin, H. 1977. Antagonistic effects of insulin and cortisol on coordinate control of metabolism and growth in culture fibroblasts. J. Cell. Phys. 91:249-260. Ruoslahti, E., Hayman, E., Peirschbacher, M. and Engvall, E. 1982. Fibronectin; purification, immunochemical properties and biological activities. Methods Enzymol. 82:803-830. Rutka, J.T., Gib lin, J.R. Balkissoon, R., Wen, D., Myatt, C.A., McCulloch, J.R. and Rosenblum, M.L. 1987. Characterization of fetal human brain cultures. Dev. Neurosci. 5:154-173. Sakai, Y., Rawson, C., Lindburg, K. and Barnes, D. 1990. Serum and transforming growth factor beta regulate glial fibrillary acidic protein in serum-free-derived mouse embryo cells. Proc. Natl. Acad. Sci. USA. 87:8378-8382. Salomon, D.S., Perroteau, I., Kidwell, W.R., Tam, J. and Derynck, R. 1987. Loss of growth responsiveness to epidermal growth factor and enhanced production of alphatransforming growth factors in ras-transformed mouse mammary epithelial cells. J. Cell Phys. 130:397-409. Samuals, H.H., Stanley, F. and Shapiro, L.E. 1976. Dose-dependent depletion of nuclear receptors by L-triiodothyronine: evidence for a role in induction of growth hormone synthesis in cultured GH1 cells. Proc. Natl. Acad. Sci. USA. 73:3877-3881. Samuels, H.H., Forman, B.M., Horowitz, Z.D. and Ye,Z.S. 1988. Regulation of gene expression by thyroid hormone. J. Clin. Invest. 84:957-967. Samuels, H.H., Stanley, F. and Casanova, J. 1979. Depletion of L-3,5,3'triiodothyronine and L-thyroxine in euthyroid calf serum for use in cell culture studies of the action of thyroid hormone. Endocrinology. 105:80-85. Saneto, R.P., Altman, A., Knob ler, R.L. Johnson, H.M. and de Vellis, J. 1986. Interleukin -2 mediates the inhibition of oligodendrocyte progenitor cell proliferation in vitro. Proc. Natl. Acad. Sci. USA. 83:9221-9225. Sanford, K.K., Earle, W.R., and Likely, G.D. 1948. The growth in vitro of single isolated tissue cells. J. Natl. Cancer Inst. 9:229-246. Sap, J., Munoz, A., Damm, k., Goldberg, Y., Ghysdael, J., Leutz, A., Beug, H. and Vennstrom, B. 1986. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 324:635-640. Sasaki, M.S. and Kodama, S. 1987. Establishment and some mutational characteristics of 3T3-like dear-diploid mouse cell line. J. Cell. Physiol. 131:114-122. Sato, G., Pardee, A. and Sirbasku, D (eds). 1982. In: "Growth of Cells in Hormonally Defined Media." Cold Spring Harbor Press, Cold Spring Harbor, NY. Sato, G.H. 1975. The role of serum in cell culture. In: Litwack, G. (ed): "Biochemical Action of Hormones." Academic Press, 391-396. 126 Scher, C.D., Pledger, W.J., Martin, P., Antoniades, H. and Stiles, C.D. 1978. Transforming viruses directly reduce the cellular growth requirement for a platelet derived growth factor. J. Cell Phys. 97:371-380. Scherer, W.F., Syverton, J.T. and Gey, G.O. 1953. Studies on the propagation in vitro of poliomyelitis viruses. J. Exp. Med. 97:695-709. Schreiber, A., Kenney, J,. Lowalski, W., Firesel, B., Mehlman, T. and Maciag, T. 1985. The interaction of endothelial cell growth factor with heparin: characterization by receptor and antibody recognition. Proc. Natl. Acad. Sci. USA. 82:6138-6142. Schubert, D., Humphreys, S., Baroni, C. and Cohn, M. 1969. In vitro differentiation of a mouse meuroblastoma. Proc. Natl. Acad. Sci. USA. 64:316-323. Schwartz, L.M., Kosz, L. and Kay, B.K. 1990. Gene activation is required for developmentally programmed cell death. Proc. Natl. Acad. Sci. USA. 87:6594-6598. Searle, J., Kerr, J.F.R. and Bishop, C.J. 1982. Necrosis and apoptosis: distinct modes of cell death with fundamentally different significance. Pathol. Annu. 17:229-259. Serrero, G., McClure, D. and Sato, G. 1979. Growth of mouse 3T3 fibroblasts in serum-free, hormone-supplemented media. Cold Spring Harbor Conf. Cell Prolif. 6:523530. Shi, Y., Sahai, B.M. and Green, D.R. 1989. Cyclosporin A inhibits activation-induced cell death in T-cell hybridomas and thymocytes. Nature. 339:625-626. Shih, C. and Weinberg, R.A. 1982. Isolation of a transforming sequence from a human bladder carcinoma cell line. Cell. 29:161-169. Shipley, G.D. and Ham, R.G. 1983. Multiplication of swiss 3T3 cells in a serum-free medium. Exp. Cell Res. 146:249-260. Shirahata, S., Rawson, C., Loo, D., Chang, Y. and Barnes, D. 1990. Ras and neu oncogenes reverse serum inhibition and epidermal growth factor dependence of serum-free mouse embryo (SFME) cells. J. Cell. Physiol. 144:69-76. Smith, C.A., Williams, G.T., Kingston,R., Jenkinson, E.J. and Owen, J.T. 1989. Antibodies to CD3a-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature. 337: 181-184. Spemann, H. 1938. In "Embryonic Development and Induction." New Haven, CT, Yale Univ. Press. Sporn, M.B. and Roberts, A.B. 1985. Autocrine growth factors and cancer. Nature. 313:745-747. Sporn, M.B. and Roberts, A.B. 1988. Peptide growth factors are multifunctional. Nature. 332:217-219. Stemple, D.L., Mahanthappa, N.K. and Anderson, D.J. 1988. Basic FGF induces neuronal differentiation, cell division, and NGF dependence in chromaffin cells: a sequence of events in sympathetic development. Neuron. 1:517-525. 127 Temin, H.M., Pierson, R.W. and Dulak, N.C. 1972. The role of serum in the control of multiplication of avian and mammalian cells in culture. In: Rothblat, G. and Cristofalo, V.J. (eds): "Growth, Nutrition and Metabolism of Cells in Culture." Academic Press, NY, 1:50-81. Thoenen, H. and Barde, Y. -A. 1980. Physiology of nerve growth factor. Physiol. Rev. 60:1284-1335. Titani, K., Koide, A., Ericsson, L., Kumar, S., Hermann, J., Wade, R.D., Walsh, K.A., Neurath, H. and Fischer, E.H. 1978. Sequence of the carboxyl-terminal 492 residues of rabbit muscle glycogen phosphorylase including the pyrodoxal 5'-phosphate binding site. Biochemistry. 17:5680-5695. Todaro, G.J. and Green, H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established cell lines. J. Cell Biol. 17:299-313. Tomozawa, Y. and Sueoka, N. 1978. In vitro segregation of different cell lines with neuronal and glial properties from a stem cell line of rat neurotumor RT4. Proc. Natl. Acad. Sci. USA. 75:6305-6309. Tsai, S. and Gaffney, E.V. 1986. Inhibition of cell proliferation by interleukin 1 derived from monocytic leukemia cells. Cancer Res. 46:1271-1278. Tucker, R., Shipley, G., Moses, H. and Holley, R. 1984. Growth inhibitor for BSC-1 cells closely related to platelet type beta transforming growth factor. Science. 226:705707. Twardzik, D.R., Ranchalis, J.E. and Todaro, G.J. 1982. Mouse embryonic transforming growth factors related to those isolated from tumor cells. Cancer Res. 42:590-593. Weinberger, C., Thompson, C.C., Ong, E.S., Lebo, R. Gruol, D.J. and Evans, R.M. 1986. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 374:641-646. Westphal, U., Burton, R.M. and Harding, G.B. 1975. Characterization of steroidbinding glycoproteins: methodological comments. Methods Enzymol. 36:91-104. White, P.R. 1946. Cultivation of animal tissues in vitro in nutrients of precisely known constitution. Growth. 10:231-289. Worton, R.G. and Duff, C. 1979. Karyotyping. Methods Enzymol. 58:322-344. Wyllie, A.H. 1981. Cell death: a new classification separating apoptosis from necrosis. In: Bowen, I.D. and Lockshin, R.A. (eds): "Cell Death in Biology and Pathology." Chapman and Hall Press, London, NY, 9-34. Wyllie, A.H., Kerr, J.F.R. and Currie, A.R. 1980. Cell death: the significance of apoptosis. In: "International Review of Cytology." Academic Press, NY. 68;251-306. Yanishevsky, R.M. and Stein, G.H. 1981. Regulation of the cell cycle in eukaryotic cells. Int. Rev. Cytol. 69:223-259. Yanker, B.A. and Shooter, E.M. 1982. The biology and mechanism of action of nerve growth factor. Annu. Rev. Biochem. 51:845-868. 128 Yip, H.K. and Johnson, E.M. 1984. Developing dorsal root ganglion neurons require trophic support from their central processes: evidence for a role of retrogradely transported nerve growth factor from the central nervous system to the periphery. Proc. Natl. Acad. Sci. USA. 81:6245-6249. APPENDICES 129 APPENDIX 1 Serum Inhibition of Proliferation of Serum-Free Mouse Embryo Cells Cathleen Rawson, Deryk Loo, Angela Helmrich, Ted Ernst, Tetsuya Natsuno, Gary Merrill, and David Barnes Published in: Experimental Cell Research. Volume 192, pp. 271-277 (1991). Coauthor contribution: C.R., primary investigator, D.L., co-primary investigator, A.H., contributing investigator, T.E., technical assistant; T.N., visiting scientist; G.M., collaborating scientist; D.B., research director 130 SUMMARY Serum-free mouse embryo (SFME) cells, derived in medium supplemented with insulin, transferrin, high density lipoprotein, epidermal growth factor, and fibronectin, do not undergo crisis, maintain a predominately diploid karyotype with no detectable chromosomal abnormalities for well over 100 population doublings in vitro, and are growth inhibited by concentrations of serum that are growth-stimulatory for most cell lines in culture. Serum inhibition of SFME cell proliferation was reversible and was not prevented by addition of the supplements of the serum-free medium, even when added repeatedly during the culture period. The serum effect on SFME cell proliferation could be detected after incubation in serum-containing medium for as little as 8 hours. SFME cells in serum-containing medium were arrested in the G1 phase of the cell cycle with a greatly reduced rate of incorporation of precursors into DNA and thymidine kinase activity, while a reduction in rate of incorporation of amino acids into protein was not observed. SFME cultures maintained for extended periods in serum-containing medium underwent a crisis- like period followed by the appearance of variant cells capable of growing in serumsupplemented medium. These cells exhibited abnormal karyotype and were resistant to several inhibitors of proliferation active on the parent SFME cell type. 131 INTRODUCTION Mouse embryo cells initiated in the absence of serum in a rich basal nutrient medium supplemented with insulin, transferrin, epidermal growth factor (EGF), high density lipoprotein, and fibronectin display several unusual properties (Loo et al., 1987,1989a, 1989b, 1990; Rawson et al., 1990; Shirahata et al., 1990) that distinguish them from mouse embryo cells established in conventional protocols utilizing serum as a source of growth factors (e.g., 3T3 cells) (Todaro and Green, 1963; Aaronson and Todaro, 1968; Jainchill et al., 1969; Reznikoff et al., 1973). Serum-free mouse embryo (SFME) cells do not exhibit growth crisis or chromosomal aberration when carried in vitro for more than 10 times the number of population doublings that can be obtained before crisis with mouse embryo cells in serum-containing medium (Loo et al., 1987, 1989b; Rawson et al., 1990). In addition, SFME cells are dependent on EGF for survival and are growth inhibited by serum. Transformation of SFME cells with the ras or neu oncogenes releases SFME cells from the growth inhibitory effect of serum as well as the EGF requirement for survival (Loo et al., 1987; Shirahata et al., 1990). Part of the growth inhibitory activity of serum may be due to glucocorticoids and thyroid hormones, both of which can inhibit growth of SFME cells. However, additional factors that contribute to the inhibitory activity on SFME cells are also present in serum (Loo et al., 1990). In order to better understand mechanisms by which serum inhibits proliferation of SFME cells and oncogenic activity abrogates this response, we examined the characteristics of SFME cells under the influence of serum inhibition and also isolated and characterized rare SFME variants that did not undergo growth inhibition in response to serum. Exposure of SFME cells to medium with 10% calf serum caused a reversible inhibition of cell growth with accumulation of cells in the G1 phase of the cell cycle and depletion of S phase cells. Serum caused an inhibition of incorporation of thymidine into SFME cells which was 132 associated with a decrease in thymidine kinase activity but was not accompanied by a decrease in amino acid incorporation. SFME cells cultured in serum-containing medium underwent a crisis period of approximately 1 month similar to that observed with mouse embryo cells derived in serum- containing media (Todaro and Green, 1963; Aaronson and Todaro, 1968; Jainchill et al., 1969; Reznikoff et al., 1973), followed by the appearance of variant cells capable of growing in serum-containing medium. The variant cells exhibited an abnormal, hyperploid karyotype and were resistant to growth inhibition by hydrocortisone, thyroid hormone or gamma-interferon, suggesting an alteration had occurred in a basic mechanism by which SFME cells are growth inhibited. 133 MATERIALS AND METHODS Cell culture. Detailed procedures for the initiation and continuous culture of Balb/c SFME cells have been published (Loo et al., 1989a, 1989b). The basal nutrient medium was a one-to-one mixture of Dulbecco's modified Eagle's medium containing 4.5 g/liter glucose and Ham's F12 (Mather and Sato, 1979; Ham and McKeehan, 1979) supplemented with 15 mM Hepes, pH 7.4, 1.2 g/liter sodium bicarbonate, sodium selenite (10 nM), penicillin (200 U/ml), streptomycin (200 µg/ml), and ampicillin (25 .tg/ml) (F12:DME). SFME cells were cultured in F12:DME supplemented with insulin (10 µg/ml), transferrin (10 p.g/ml), human high density lipoprotein (HDL) (10 µg/m1), and EGF (50 ng/ml) in dishes or flasks precoated with bovine fibronectin (1014/m1). Bovine insulin and human transferrin were obtained from Sigma Chemical Co. (St. Louis, MO). Mouse EGF was obtained from Upstate Biotechnologies (Lake Placid, NY). Fibronectin and HDL were prepared as described (Loo et al., 1989a; Gospodarowicz, 1984). Calf serum (CS) was obtained from Grand Island Biological Co. (Grand Island, NY). Gamma- interferon was obtained from Amgen (Thousand Oaks, CA). Stock SFME cultures used for the experiments described in this report had undergone between 60 and 120 population doublings. Flow cytometry. Cells cultured as indicated were harvested by trypsinizing in 0.25% crude trypsin with 1 mM EDTA in phosphate-buffered saline without calcium or magnesium (PBS) and diluted into an equal volume of F12:DME containing 1 mg/m1 soybean trypsin inhibitor. Cells were washed in F12:DME and resuspended in cold PBS. Cold 70% ethanol was added dropwise with gentle mixing. Fixed cells were centrifuged and resuspended in a solution containing chromomycin A3 (20 ii.g/m1) and 15 mM MgC12 to give a concentration of 1 to 5 x 106 cells/ml. Suspensions were incubated 30 minutes at room temperature in the dark. Cells were centrifuged from suspension and resuspended in the same volume of PBS. 134 Basic procedures for flow cytometry were described by Gray and Coffin (1979). Flow cytometry was carried out with an Epics flow cytometer-cell sorter (Coulter Epics Division, Hialeah, FL). Chromomycin A3-stained cells were excited with an output of 200 mW of 457 nm light emitted from an argon laser. The resulting cell-associated fluorescence was captured through a 495-nm longpass absorbance filter (Coulter) and a blocking interference filter (Corion). Assays. Cells were plated in F12:DME with serum-free supplements or with 10% dialyzed CS. After 24 hours medium was changed to fresh medium of the same composition and isotopes (4 liCi/m1 [3H]-thymidine, New England Nuclear, NET042H or 14C-amino acid mixture, New England Nuclear, NEC445) were added three hours after the medium change. Four hours later medium was removed, cells were washed three times with cold PBS, and macromolecules were precipitated with 10% trichioroacetic acid and counted in a liquid scintillation counter. Cell number per plate was determined by counting suspensions of trypsinized cells from duplicate plates in a Coulter particle counter. Thymidine kinase assay was carried out as described (Merrill, et al., 1984). Karyotyping. Procedures for the analysis of mitotic chromosomes from SFME cell cultures have been previously published (Loo et al., 1989a; Worton and Duff, 1979). Fifty to two-hundred metaphases from each culture were stained with a 3% Giemsa stain and analyzed at 1000X with an oil immersion objective. Cells containing fewer than 35 chromosomes were considered artifacts of the procedure and excluded from the analyses. 135 RESULTS Effects of serum on SFME cells. SFME cells plated in basal nutrient medium supplemented with 10% CS do not proliferate and addition of the supplements of the serum-free medium with 10% CS allows approximately one population doubling over a period of about 48 hours before growth arrest (Loo et al., 1987) (Fig. A1.1). No further growth is observed if the supplements are added repeatedly over the course of the experiment, and initial exposure of SFME cells to medium containing 10% CS for 2 days followed by addition of the supplements to the serum-containing medium resulted in no growth response to the supplements (Fig. A1.1). In experiments in which SFME cells were incubated in medium containing 10% CS for various times and then switched to serum-free medium with the growth-stimulatory supplements, exposure of cells to 10% CS for as little as 8 hours produced a measurable reduction in cell number, and the effect of serum did not increase markedly upon exposure of SFME cells to serum-containing medium for longer periods (Fig. A1.2). Because EGF is critical for the growth of SFME cells (Loo et al., 1987, 1989b; Rawson et al., 1990), we examined levels of the EGF receptor on SFME cells in serum-free and serum-containing media. No differences were observed, and SFME cells in serum-containing medium also exhibited the ability to reexpress EGF receptors on the cell surface after down regulation by a preincubation with the ligand (not shown). Flow cytometric analysis indicated that SFME cells cultured in serum-containing medium for 2 days accumulated in the G1 phase of the cell cycle, with S phase cells disappearing from the cultures (Fig. A1.3). The cytometric pattern was similar to that observed in serum-derived Balb/c 3T3 cells growth arrested by incubation on 0.5% CS, except that the amount of DNA per SFME cell was less than that observed with 3T3 cells because SFME cells remain predominately diploid while 3T3 cells are approximately tetraploid (Todaro and Green, 1963; Aaronson and Todaro, 1969; Jainchill et al., 1969). 136 The serum-induced arrest of SFME cell proliferation was reflected in a reduced incorporation of thymidine into DNA and reduced levels of thymidine kinase, an S phase- dependent enzyme (Table A1.1). A decrease in thymidine kinase activity was associated with a reduction in mRNA for this enzyme detectable by Northern hybridization analysis (not shown). Incorporation of amino acid into protein was not reduced under conditions in which DNA synthesis was inhibited by CS. Variant SFME cells capable of growing in serum-containing medium. SFME cells cultured for 96 population doublings in serum-free medium were carried for an extended period in a 3T3 protocol (plating at 3 x 105 cells/25-cm2 flask, passaging every 3 days (Todaro and Green, 1963)) in medium containing 10% CS. Cell proliferation was observed in serum-containing medium after a period of about 1 month (Fig. A1.4). Karyotyping of cells capable of growth in serum-containing medium showed a strong tendency toward hyperploidy; 44 of 50 metaphases examined contained greater than the diploid number of 40 chromosomes (Fig. A1.5). Chromosomal abnormalities were also evident in these metaphases. The SFME culture selected in serum-containing medium in the 3T3 protocol may have been derived from several variant cells present in the initial population or generated during the selection in serum-containing medium. Each variant might confer a characteristic diploid, hyperploid, or polyploid contribution to the total karyotype of the population. Because of this we selected directly from SFME cultures clonally derived variants capable of growing in serum-containing medium to determine if all cells capable of growing in serum-containing medium exhibited abnormal karyotype and to determine if a common structural abnormality could be detected in all clones. SFME cells were plated in medium containing 10% CS at low density (105 cells/80-cm2 dish) and cultured with weekly medium changes for 5 weeks. Colonies capable of growing in serum-containing medium developed with a frequency of about 1 in 106 SFME cells. Five of these were propagated and karyotyped (Fig. A1.6). All clones 137 exhibited a degree of hyperploidy fivefold or greater than that of the parent cells cultured in serum-free medium (Table A1.2). Three clones showed two subpopulations: one with chromosome numbers hyperploid but near-diploid and one with chromosome numbers approximately tetraploid. One clone was largely near-tetraploid and one clone was moderately hyperploid with less polyploidy (Fig. A1.6, Table A1.2). Structural alterations in chromosomes were also evident as metacentric and submetacentric chromosomes (all normal mouse chromosomes are acrocentric) and as double minute chromosomes (Table A1.2), but no structural alteration was common to all five clones. Although hyperploidy was a common feature of all clones, it was not sufficient for release of SFME cells from serum inhibition of proliferation, because SFME cells made hyperploid by polyethylene glycol-induced fusion did not grow in serum-containing medium. The proliferative behavior of one of the clones was examined further. This clone grew slowly in medium supplemented only with 10% CS, and grew well in serum-free medium or in medium containing both the supplements of the serum-free medium and 10% CS, indicating that the inhibitory proliferative response was not elicited by CS on these cells (Fig. A1.7). We have reported previously that glucocorticoid and thyroid hormones are inhibitory for growth of SFME cells in the presence of low concentrations of charcoal- treated CS (Loo et al., 1990). Gamma-interferon was also markedly inhibitory for SFME cells (Fig. A1.8), although none of these factors are solely responsible for the effect of serum on SFME cells (Loo et al., 1990). The variant clone that was not inhibited by serum also showed reduced inhibitory response to all three of these factors (Fig. A1.8). 138 DISCUSSION SFME cells are inhibited by CS, but continue to grow for about one population doubling if plated in 10% CS with insulin, transferrin, EGF, HDL, and fibronectin. Further addition of these factors upon extended culture did not lead to any additional proliferation in the presence of CS and cells plated for 48 hours in 10% CS before the addition of the factors did not undergo cell division. The growth inhibitory effect is reversible upon removal of serum from the culture medium, suggesting that the phenomenon is not simply due to toxicity of serum for SFME cells. Furthermore, serum inhibition of proliferation was associated with buildup of the cells in the 01 phase of the cell cycle and reduction of the fraction of cells in S phase, a decrease in thymidine kinase activity, and a reduction in DNA synthesis. All of these phenomena are associated with physiologically relevant inhibition of cell division such as that observed when cells become growth arrested upon growth factor depletion or upon induction of differentiation (Pardee et al., 1978; Yanishevsky and Stein, 1981; Baserga et al., 1982). Growth factor depletion is not applicable in this circumstance, but coordinate inhibition of proliferation and induction of differentiation of SFME cells by serum is consistent with the observed lack of inhibition of incorporation of amino acid by CS treatment of SFME cells. Almost without exception mouse embryo cell lines derived in serum-containing media are hyperploid with one or more chromosomal abnormality; even lines in which karyotypic abnormalities are minimal are still aneuploid or karyotypically unstable (Todaro and Green, 1963; Aaronson and Todaro, 1968; Jainchill et al., 1969; Reznikoff et al., 1973; Farber and Liskay, 1974; Sasaki and Kodama, 1987). Exposure of SFME cells that had been grown long-term in serum-free medium to a 3T3 passage regimen in serum- containing medium led to a peroid of no proliferation, followed by the appearance of variant cells capable of growing in serum-containing medium and expressing abnormal, 139 hyperploid karyotype with structural chromosomal abnormalities. This behavior is similar to the phenomena of crisis and immortalization observed in mouse embryo cells derived initially in serum-containing medium and suggests that serum-containing medium selected for, or induced, an abnormal karyotype in SFME cells. Variant SFME cells selected for the ability to grow in serum no longer were inhibited by serum factors and exhibited reduced responses to several hormones that were growth inhibitory for SFME cells. An alteration apparently occurred in these cells that led to reduced inhibitory growth responses in general, unrelated to a specific response to a particular factor that might be present in serum. Although no consistent chromosomal abnormality was detected in the clones capable of growing in serum, the tendency toward hyperploidy in these cells suggests that gene dosage effects may provide selective growth advantage for variant clones under the selective conditions employed, as is probably the case with postcrisis, hyperploid mouse embryo cell lines arising from cultures initiated in serum-containing medium. This explanation is not completely simple, however, because SFME cells made polyploid by cell fusion remained inhibited by serum. Further work is necessary to determine the relationship between abnormal karyotype, immortalization or escape from crisis, and escape from serum inhibition observed in variant SFME cells. 140 ACKNOWLEDGMENTS This work was supported by NIH-NAI-07560. D. Barnes is the recipient of RCDA NIH-NCI01226. G. Merrill is the recipient of RCDA NIH-AG00334. Flow cytometry was carried out through the Oregon State University Environmental Health Sciences Center Flow Cytometry Core Unit (NIEHS ES00210). Cathleen Rawson assisted with cell culture experiments and performed flow cytometry. Angela Heinrich provided cell culture support. Ted Ernst prepared karyotypes and statistical data. Tetsuya Natsuno performed the amino acid incorporation experiment. Gary Merrill performed the thymidine incorporation and thymidine kinase activity experiments. David Barnes provided financial and scientific support. 141 Table A1.1. Synthetic and Enzymatic Activities of SFME Cells in Serum- Free and Serum-Containing Media. SFME cells were plated in medium supplemented with insulin, transferrin, EGF, HDL, and fibronectin (serum-free) or 10% dialyzed CS. 24 hours later, plates were assayed for DNA synthesis (thymidine incorporation, 4 hour pulse), protein synthesis (amino acid incorporation, 4 hour pulse), and thymidine kinase activity as described under Materials and Methods. Serum-free Calf serum 56.6 ± 4.5 4.05 ± 0.40 (cpm X 10-4/106 cells) 13.3 ± 0.40 17.7 ± 2.0 Thymidine kinase activity (pmol/min/106 cells) 91.0 ± 5.55 1.30 ± 0.18 Thymidine incorporation (cpm X 10-4/106 cells) Amino acid incorporation 142 Table A1.2. Karyotypic Abnormalities of Variant SFME Clones Capable of Growing in Serum-Containing Media. Parent cultures were initiated in serum-free medium and had undergone 96 population doublings in this medium at the time of the experiment. The normal mouse contains 40 acrocentric chromosomes; metacentric and submetacentric chromosomes are indications of structural abnormalities. Hyperploid cells were those with more than 40 chromosomes. Polyploid cells were considered to be cells containing more than 70 chromosomes. Parent % Hyperdiploid % Polyploid % with metacentric chromosome % with submetacentric chromosome % with double minute chromosomes 16 6 1 0 1 Clone A Clone B Clone C Clone D Clone F 78 64 56 0 6 82 30 49 96 52 4 98 82 80 23 0 100 100 18 81 25 0 0 60 5 143 Fig. A1.1. Culture of SFME cells in serum-containing medium. Cells were seeded at 105/35 mm-diameter plate in the media listed below and cell number was determined on the indicated days after plating. (0), medium containing 10% CS; (I), medium containing 10% CS with addition of 10 µg/ml insulin, 10 µg/ml transferrin, 10 p.g/m1HDL, and 50 ng/ml EGF on Day 3 after plating; (), medium containing 10% CS, insulin, transferrin, HDL, and EGF; (0), medium containing 10% CS, insulin, transferrin, HDL, and EGF with additional insulin, transferrin, HDL, and EGF added on Days 2, 4, and 6 after plating. All plates were precoated with fibronectin. m t 0 ..-1 X 2 .1mm, i 0 1 2 3 4 5 Days After Plating 6 7 144 Recovery of SFME cells from plating in serum-containing Fig. A1.2. medium. Cells were plated at 5 x 104/35 mm-diameter dish in medium containing 10% CS and precoated with fibronectin. At the indicated times, the medium was removed, plates were washed once with serum-free medium, and medium supplemented with insulin, transferrin, FIDL, and EGF was added. Cell number was determined 4 days later. 5 O in 1 0 -4 x 4 3 2 0 0---°"-----...........0 O 1 0 0 24 hrs 48 hrs 72 hrs Preincubation With 10% Calf Serum 145 Fig. A1.3. Flow cytometric analysis of SFME cells in serum-containing medium. Tracings show relative cell number (Y axis) versus relative amount of DNA per cell (X axis). (A), 3T3 cells growing in medium containing 10% CS; (B), 3T3 cells growth arrested by incubation in medium containing 0.5% CS for 2 days; (C), SFME cells growing in serum-free medium supplemented with insulin, transferrin, EGF, HDL, and fibronectin; (D), SFME cells growth arrested in medium containing 10% CS for 2 days. A B .k- C Jar D 146 Fig. A1.4. Selection of variant SFME cells capable of growing in serumcontaining medium. SFME cells initiated and cultured for 96 population doublings in serum-free medium as described were cultured in medium containing 10% CS, plating at 3 x 105/25-cm2 flask and passaging every 3 days (3T3 protocol). Cell number was determined at each passage. Total cell number decreased during the first 5 pasages due to loss of cells upon transfer. An increase in cell number was first detected 27 days after beginning the experiment. 7 102 6 5 4 101 3 2 1 100 0 10 20 30 Days 40 50 147 Fig. A1.5. Karyotype of variant SFME cells selected by culture in serumcontaining medium. SFME cells initiated and cultured for 96 population doublings in serum-free medium as described were cultured in medium containing 10% CS, plating at 3 x 105 cells/25-cm2 flask and passaging every 3 days. Karyotyping was carried out as described in the text. (A), parent culture at 96 population doublings; (B), parent culture after an additional 6 population doublings in serum-free medium; (C), variants after approximately seven population doublings in serum-containing medium. A 80 80 70 70 60 6 50 80 70 In 50 N a, a, co g .c 40 50 co Col 40 40 -a; co c8 ;0 2 O 30 o 30 30 a, a, cn a pl ; 20 ccci C ,_,J 20 20 a, a- 10 0 10 10 _.A11L 35 38 41 I 44 >80 Number of Chromosomes 35 38 41 44 >80 Number of Chromosomes 35 38 41 44 66 69 72 75 78 81 84 87 90,160 Number of Chromosomes 148 Fig. A1.6. Karyotype of variant SFME clones capable of growing in serumcontaining medium. Clones were derived and karyotyped as described in the text. C B A 50 30 50 e t0 a a .0 in 04. e a .= a 230 a a I; 30 m 30 2 2 o 2 "O 2. 0 e 0 so o a a Z .zi 10 m a. 0. iii Iii I Hill I I 35 36 41 44 611 69 72 75 75 SI 0 0 33 36 41 44 86 55 72 75 75 et 64 87 90.136 Number of Chromosomes 36 36 54 41 44 68 , E 50 40 a a z 30 00 S 2 0 0 1i 20 o a 20 0 u 10 10 oll 35 ss III 4. 50 0 0. 7 SO 5.1 a .5 r. II 7. $35 II Cl so Number of Chromosomes 59 72 75 75 65 64 87 Number o Chromosomes Number of Chromosomes 0 I1 35 36 44 I . 76 11111 1 71 52 65 Number of Chromosomes 149 Fig. A1.7. Growth of variant SFME cells in serum-containing and serumfree medium. Variant SFME cells (clone shown in Fig. A1.6E) were plated at 105/35 mm-diameter plate and cell number was determined on the indicated days after plating. (), medium containing 10% CS; (0), medium containing 10% CS with addition of 10 p.g/ml HDL and 50 ng/ml EGF; (0), medium containing insulin, transferrin, HDL, and EGF. All plates were precoated with fibronectin. 3 X 106 U) .1.) .-4 0. 10 6 C_ a) 0 al -) 3 X 105 v-1 a) U 105 0 2 4 6 Days After Plating a 150 Fig. A1.8. Reduced inhibition of variant SFME cells by thyroid hormone, glucocorticoid, and gamma-interferon. Parent or variant SFME cells capable of growing in serum-containing medium (clone shown in Figs. A1.6E and A1.7). were plated at 105/35 mm-diameter dish in medium supplemented with insulin, transferrin, EGF, frDL, fibronectin, and 2.5% charcoal-treated CS (Shirahata, et al., 1990), with or without hydrocortisone (100 nM), T3 (10 nM), gamma-interferon (3 ng/ml) or CS (10%). Cell number was determined 6 days after plating. Data for parent SFME cultures are on the top; data for variant clone are on the bottom. rn Cu 0 .-4 0 L 41 C 0 U Cr) F- + U 1 + UI-1 + U) L) + r-i 0 C- 4.) C 0 0 I uI u_ cr) + + + rn + u 151 APPENDIX 2 Death of Serum-Free Mouse Embryo Cells Caused by Epidermal Growth Factor Deprivation Cathleen Rawson, Deryk Loo, Julie Duimstra, Olaf Hedstrom, Edward Schmidt, and David Barnes Published in; The Journal of Cell Biology. Volume 113, Number 3, pp. 671-680 (1991). Coauthor contributions: C.R., primary investigator; D.L., co-primary investigator; J.D., technical assistant; O.H. and E.S., collaborating scientists; D.B., research director 152 SUMMARY Serum-free mouse embryo (SFME) cells, derived in medium in which serum is replaced with growth factors and other supplements, are proastroblasts that are acutely dependent on epidermal growth factor (EGF) for survival. Ultrastructurally, an early change found in SFME cells deprived of EGF was a loss of polysomes which sedimentation analysis confirmed to be a shift from polysomes to monosomes. The ribosomal shift was not accompanied by decreased steady-state level of cytoplasmic actin mRNA examined as an indicator of cellular mRNA level. With time the cells became small and severely degenerate and exhibited nuclear morphology characteristic of apoptosis. Genomic DNA isolated from cultures undergoing EGF deprivation-dependent cell death exhibited a pattern of fragmentation resulting from endonuclease activation characteristic of cells undergoing apoptosis or programmed cell death. Flow cytometric analysis indicated that cultures in the absence of EGF contained almost exclusively G1 -phase cells. Some of the phenomema associated with EGF deprivation of SFME cells are similar to those observed upon NGF deprivation of nerve cells in culture, suggesting that these neuroectodermal-derived cell types share common mechanisms of proliferation control involving peptide growth factor-dependent survival. 153 INTRODUCTION We have described the long-term culture of mouse embryo cells derived and passaged under conditions in which the serum supplement to the basal nutrient medium is replaced by growth factors and other supplements: insulin, transferrin, EGF, high-density lipoprotein, and fibronectin (Loo et al., 1987, 1989a, b). These serum-free mouse embryo (SFME) cells exhibit a number of unusual properties. Unlike mouse embryo cells derived in conventional, serum-supplemented media which undergo growth crisis and immortalization, SFME cells do not lose proliferative potential or develop gross chromosomal aberration when cultured for more than 10 times the number of population doublings that can be achieved with mouse embryo cells in conventional, serum-containing medium (Todaro and Green, 1963; Loo et al., 1987, 1989a; Ernst et al., 1990). Proliferation of SFME cells is reversibly inhibited by serum (Loo et al., 1987, 1990; Rawson et al., 1991), and treatment of SFME cells with serum or transforming growth factor beta leads to the appearance of glial fibrillary acidic protein (GFAP), an intermediate filament protein that is a specific marker for astrocytes (Sakai et al., 1990). Cells with properties like those of SFME cells also can be isolated directly from mouse brain (Sakai et al., 1990), and a modification of the SFME medium formulation allows the multipassage serum-free culture of human brain cells that express GFAP (Loo et al., 1991). These results suggest that SFME cells are proastroblasts. SFME cells are acutely dependent on EGF for survival, and cycloheximide or actinomycin D prevents death caused by EGF deprivation (Raawson et al., 1990), suggesting that EGF-dependent survival may depend on suppression of synthesis of proteins that cause cell death, a phenomenon similar to that reported for neuronal "programmed cell death" in the absence of nerve growth factor (NGF) (Martin et al., 1988). Thus, multiple cell types of neuroectodermal origin may share common mechanisms of proliferative control involving peptide growth factor-dependent survival. 154 SFME cell death in the absence of EGF also is delayed by orthovanadate, an inhibitor of phosphotyrosine phosphatases, and 12- O- tetradecanoylphorbol 13-acetate, an activator of protein kinase C (Rawson et al., 1990). Greater than 90% of SFME cells are dead within 48 hours after removal of EGF from the culture medium, and decreased cell survival is observed after EGF deprivation for as little as 8 hours (Loo et al., 1987, 1989b). Individual omission of any of the other medium supplements leads to reduced SFME cell growth, but does not induce cell death (Loo et al., 1989b). NGF, platelet-derived growth factor, and transforming growth factor beta will not substitute for EGF in this system, while fibroblast growth factor will allow survival and slow growth of SFME cells in the absence of EGF (Loo et al., 1989b). We have suggested that at least two alternative mechanisms for the control of cell proliferation may exist: one related to intrinsic limitation of proliferative potential observed in conventional cell cultures, and the other governed by growth factor-dependent survival observed in SFME cell cultures (Sakai et al., 1990). We examined ultrastructural, biochemical, and physiological responses of SFME cells to EGF deprivation as a step toward defining the primary mechanisms leading to death of these cells. Early changes included a shift of ribosomes from a polysomal organization to monosomes. The ribosomal shift was not accompanied by decreased steady-state level of actin messenger RNA in the cytoplasm. With time the cells became small and degenerate, and pyknotic nuclei appeared. Flow cytometric analysis indicated that cultures in the absence of EGF contrained almost exclusively a diploid amount of DNA (G1- phase). Genomic DNA isolated from cultures undergoing EGF deprivation-dependent cell death exhibited a pattern of fragmentation associated with programmed cell death in other systems (Arends et al., 1990). 155 MATERIALS AND METHODS Cell culture. Detailed procedures for the initiation and continuous culture of Balb/c SFME cells have been published (Loo et al., 1989a,b). The basal nutrient medium was a one-to-one mixture of DME containing 4.5 g/1 glucose and Ham's F12 (Mather and Sato, 1979; Ham and McKeehan, 1979) supplemented with 15 mM Hepes, pH 7.4, 1.2 g/1 sodium bicarbonate,sodium selenite (10 nM), penicillin (200 U/ml), streptomycin (200 tg/m1) and ampicillin (25 p.g/ml) (F12:DME). Stock cultures of SFME cells were cultured in F12:DME supplemented with insulin (10 µg/ml), transferrin (1014/m1), human high density lipoprotein (HDL) (10 µg/ml) and EGF (50 ng/ml) in dishes or flasks precoated with bovine fibronectin (10 µg/ml). Methods for preparation, storage, and use of medium supplements and fibronectin precoating have been published (Loo et al., 1989b). Plating was accomplished by adding cells in prewarmed medium to plates or flasks previously precoated with fibronectin and preincubated in the incubator (5% CO2-95% air atmosphere at 37°C) for 15 minutes with the remaining portion of medium. Insulin, transferrin, EGF, and HDL were added directly to the individual culture vessels as small aliquots from concentrated stocks immediately after plating the cells. Bovine insulin, human transferrin, trypsin, and soybean trypsin inhibitor were obtained from Sigma Chemical Co. (St. Louis, MO). Mouse EGF was obtained from Upstate Biotechnologies (Lake Placid, NY). HDL (density = 1.068-1.121 g/cc) was prepared by KBr ultracentrifugation as described (Gospodarowicz, 1984; Loo et al., 1989b) and filter sterilized after dialysis. Preparation of fibronectin has been described (Loo et al., 1989b). Electron microscopy. SFME cells were plated in 10-cm-diameter plates (2 x 107 cells/plate) in serum-free medium with 5 ng/ml EGF. The following day some plates were changed to serum-free medium without EGF (10m1/plate). Control plates were changed to serum-free medium with 50 ng/ml EGF. At the indicated times 8 ml of the culture medium were carefully removed from a plate, 3 ml of fixative (1.75% glutaraldehyde in 0.1 M 156 sodium cacodylate buffer, pH 7.40) were added, and plates were then incubated at room temperature for 30 minutes. After fixation, the cells were removed from the dishes with a rubber policeman and were resuspended in fixative, pelleted, and the pellet was minced into pieces 1 mm3. The samples were washed through two changes of 0.2 M sodium cacodylate buffer, pH 7.4, and were postfixed for 1 hour in 1.0% 0s04 in 0.15 M sodium cacydylate buffer. Samples were washed through two changes of 0.2 M sodium cacodylate buffer and dehydrated through a graded acetone series (X3), infiltrated through several mixtures of acetone/resin, and embedded in freshly prepared Medcast-Araldite 502 resin (Ted Pella, Inc., Irvine, CA) and polymerized at 60°C for 24 hours (hayat, 1986). Ultrathin sections were cut on an ultramicrotome (model MT-500; Sorvall Instruments Div., DuPont Co., Newton, CT) with a sapphire knife. Sections 60-70 nm in thickness were collected on 300 hexagonal mesh copper grids and stained with saturated ethanolic uranyl acetate followed by bismuth subnitrate. The sections were examined with a transmission electron microscope (EM 10/A; Zeiss, Oberkochen, Germany) at an accelerating voltage of 60 kV. DNA fragmentation analysis. SFME cells (2 x 107/plate) were seeded in 10-cm- diam. plates in serum-free medium with 5 ng/ml EGF. The following day some plates were changed to serum-free medium without EGF (10 ml/plate). Control plates were changed to serum-free medium with 50 ng/ml EGF. At the indicated times duplicate cells were harvested by scraping into cold phosphate-buffered saline with calcium or magnesium (PBS) followed by centrifugation at 4°C in 1,000 g for 5 minutes. The supernatant was aspirated and the cell pellets were immediately stored at -86°C. Cell pellets were thawed on ice and incubated at 37°C for 4 hours in 4 ml of lysis buffer (200 mM Tris, pH 8.5, 100 mM EDTA, 50 j.ig/m1 proteinase K, 1% SDS). The DNA solution was extracted twice with phenol. The aqueous phase was dialyzed overnight against 10 mM Tris HC1, pH 7.5, 1 mM EDTA. After dialysis the DNA solution was incubated at 37°C for 5 hours with 50 p.g/ml of DNase-free RNase A. Proteinase K (50 157 µg/ml) was added and the DNA solution was further incubated at 37°C for 5 hours. The DNA solution was extracted once with phenol and once with phenol:chloroform:isoamyl alcohol (25/24/1). DNA was precipitated in ethanol and resuspended in distilled water. DNA concentration was determined from the absorbance at 260 nm. For analysis of fragmentation, DNA (20 µg/lane) was fractionated by electrophoresis for 16 hours at 40 V on a 1.0% agarose gel with 90 mM Tris HC1, 90 mM boric acid, 2 mN EDTA, pH 8.0 as running buffer. DNA was visualized by shortwave UV illumination after ethidium bromide staining (Maniatis et al., 1982). A 123 by DNA ladder (Bethesda Research Laboratories, Gaithersburg, MD) was used as a standard. Enzyme assay. Culture medium (0.5 ml) from Balb SFME or Balb 3T3 cultures maintained for the indicated times under serum-free conditions in the presence or absence of EGF was added to an equal volume of lactate dehydrogenase assay reagent (100 mM lactate, 14 mM nicotinamide adenine dinucleotide, pH 8.9; #228-10; Sigma Diagnostics, St. Louis, MO). Enzyme units were quantified by determining the rate of absorbance change at 340 nm over a 60 second time interval. Flow cytometry. SFME cells were seeded at 2 x 107 cells per 10-cm-diam. dish and cultured overnight in serum-free medium with 5 ng/ml EGF. The following day some plates were changed to serum-free medium without EGF (10 ml/plate). Control plates were changed to serum-free medium with 50 ng/ml EGF. At the indicated times cells were analyzed for cell size by measuring forward angle light scatter (FALS) or cell cycle by measurement of DNA staining with chromomycin A3 (IGFL). For size analysis cells were removed from plates with 0.25% crude trypsin/1 mM EDTA in PBS and diluted into an equal volume of F12:DME containing 1 mg/ml soybean trypsin inhibitor. Cells were washed with ice-cold PBS by centrifugation and resuspension in 5 ml PBS. Forma lin (550 gl, 37%) was added while vortexing and fixed cells were stored at 4°C. 158 For chromomycin staining, cells were removed from plates, centrifuged, resuspended in 5.0 ml ice-cold PBS as described above, and then pelleted by centrifugation. The supernatant was aspirated, leaving 0.5 ml in the bottom of the tube, and 5.0 ml of ice-cold 70% ethanol was added dropwise while vortexing the cell pellet. Cells were stored in ethanol at 4°C until all samples could be processed together. Fixed cells were centrifuged, resuspended in chromomycin A3 (20 p.g/m1) with 15 mM MgC12, and incubated for 0.5 hours at room temperature in the dark. Cells were centrifuged from solution, resuspended (5 x 105 to 5 x 106 cells/m1) in ice-cold PBS, and filtered through a 40 pm nylon filter. Basic procedures for flow cytometry were as described by Gray and Coffino (1979). Flow cytometry was carried out with an Epics flow cytometer-cell sorter (Coulter Electronics Inc, Hialeah, FL). Chromomycin A3-stained cells were excited with an output of 100 mW of 457-nm light emitted from an argon laser. The resulting cell-asociated fluorescence was captured through a 495-nm-long pass absorbance filter (Coulter Electronics Inc.) and blocking interference filter (Corion Corp., Holliston, MA). Cell sizing was analyzed with a ND-1 filter for FALS. Polysome profiles. SFME cells were plated at 2 x 10 cells/10-cm-diam. dish and cultured for 24 hours in serum-free medium with 5 ng/ml EGF. The medium was changed on one plate to serum-free conditions with 50 ng/ml EGF and on the other plate to serumfree conditions without EGF. After 6 hours the medium was aspirated, plates were washed twice with PBS containing 10 gg/m1 cycloheximide, and then stored frozen at -86°C. All subsequent manipulations were performed on ice or at 4°C. Plates were scraped in 2x lysis buffer (500 mM NaCl, 50 mM MgC12, 100 mM Tris, pH 7.5, 1% Triton X-100, 400 U/ml RNasin, and 40 gg/m1cyclohexirnide) and if necessary sterile water was added to achieve a lx lysis buffer concentration. Lysates were centrifuged for 10 minutes at 13,000 rpm in a precooled (model SS34; Sorvall Instruments Div., DuPont Co.) rotor. The supernatant was carefully 159 transferred to the top of a freshly prepared 15-50% sucrose gradient (250 mM NaC1, 25 mM MgC12, 50 mM Tris, pH 7.5, 20 µg/ml cycloheximide, and 15-50% sucrose). Gradients were centrifuged for 130 minutes in a precooled rotor (model SW40; Beckman Instruments Inc., Palo Alto, CA) at 32,000 rpm and scanned from top to bottom at 254 nm using a model 185 density gradient fractionator (ISCO, Lincoln, NE) at a flow rate of 0.375 ml/minute (Gross and Merrill, 1989). Absorbance was monitored by a UA-5 absorbance/fluorescence detector and type 6 optimal unit (ISCO), which was calibrated to an internal standard immediately before use. Northern blot hybridization analysis. For isolation of cytoplasmic RNA, plates containing SFME cells were washed with ice-cold PBS, cells were scraped into cold PBS and centrifuged (4°C, 1,000 g, 5 minutes). Cell pellets were resuspended in 10 mM Tris HC1, pH 7.6, 140 mM NaC1, 1.5 mM MgC12, 200 p.g/ml heparin, lysed by added 0.5% NP-40, and vortexed for 20 seconds, and then centrifuged at 4,000 g for 10 minutes to pellet nuclei. 1% SDS and 10 mM EDTA were added to the supernatant followed by extraction twice each with phenol, phenol:chloroform:isoamyl alcohol (25/24/1), and chloroform:isoamyl alcohol (24/1). RNA was precipitated in ethanol and stored in diethylpyrocarbonate-treated water at -86°C. Poly(A)+ RNA was prepared from cytoplasmic RNA using standard methods (Maniatis et al., 1982). Poly(A)+ RNA (1.0 lig/lane) was fractionated by electrophoresis on a 1.2% agarose gel containing 6.6% formaldehyde and transferred for 24 hours to a nitrocellulose membrane (Maniatis et al., 1982). The plasmid pCD-beta actin was linearized with HindIII and labeled (5 x 108 cpm/p.g DNA) by oligonucleotide-primed extension (kit from Boehringer Mannheim Diagnostics, Indianapolis, IN). The membrane was prehybridized for 4 hours, and then hybridized with probe (2 x 106 cpm/ml) for 20 hours in 50% formamide, 50 mM sodium phosphate, pH 6.5, 0.8 M NaC1, 1 mM EDTA, 0.1% SDS, 0.05% BSA, 0.5% Ficoll, 0.05% polyvinylpyrrolidone, 250 Mg/ml sonicated salmon sperm DNA, and 500 µg/ml yeast RNA at 57°C. Filters were washed twice with lx SSC 160 and 0.1% SDS at room temperature for 30 minutes each, and then twice with 0.5x SSC and 0.1% SDS at 65°C for 30 minutes each. Autoradiography was carried out on film (XOMAT AR; Kodak Co., Rochester, NY) for 12-36 hours at -86°C. 161 RESULTS Ultrastructural changes associated with EGF deprivation. SFME cells were cultured in the absence of EGF and fixed for EM 4, 8, 16, and 24 hours later. Control cells grown in the presence of EGF were fixed at the beginning and end of the experiment. Cells grown in the presence of EGF had a high nuclear to cytoplasmic ratio relative to most cultured cells and contained abundant aggregates of polyribosomes that were generally uniformly distributed (Fig A2.la and b). An additional prominent cytoplasmic feature of control cells was frequent large vacuoles and multivesicular bodies (MVB) that tended to be regionally located. A well-developed Golgi complex and abundant coated vesicles were present and intermediate filaments and microtubules were prominent. Cytoplasmic lipid droplets were rarely seen. Nuclear chromatin appeared as a thin peripheral rim of heterochromatin with small aggregates of heterochromatin also randomly dispersed throughout the nuclear matrix. In most nuclei, single or multiple compact nucleoli were seen either centrally or marginally located adjacent to the nuclear envelope. SFME cells cultured in the absence of EGF for 4 hours exhibited a loss of polyribosomes (Fig A2. 1c) and predominance of monosomes. When SFME cells were cultured for 8 hours without EGF, cellular degeneration, cell death, and other ultrastructural pathologic alterations were visible. Cellular morphology was pleomorphic; some cells were small and round, while other cells were larger and elongated with distinct ameboid-like cytoplasmic projections (Fig A2.2a and b). Some of the small cells were severely degenerate and contained electron-dense nuclei with a thick band of marginated chromatin in a circumscribed or crescent form with nucleoli condensed or absent. Cytoplasm in these degenerate cells was condensed and cytoplasmic organelles were scant but recognizable; occasional cytoplasmic blebs or projections were found. A portion of cytoplasmic and nuclear matrix of most degenerate cells displayed a light-grey granular matrix. The few mitochondria found in degenerate 162 cells with light grey cytoplasmic matrix were normal, whereas the few profiles of rough ER observed were slightly dilated or vesicular with a loss of polyribosomes. Dead cells were distinguished by prominent electron-dense cytoplasm containing lucent cytoplasmic vacuoles and pyknotic nuclei. The nuclei had either a wide, electrondense marginated band of chromatin with the remaining nuclear matrix of similar electron density to that of the cytoplasm, or the nucleus was round and filled with electron-dense chromatin. Nucleoli were not found in these cells. The predominant population of cells in the absence of EGF appeared smaller than control cells cultured in the presence of EGF because of a loss of cytoplasm. Cytoplasmic projections with the remaining portion of the cell out of the plane of section and cytoplasmic debris were scattered throughout the fields, suggesting that cells may have been in an early phase of disintegration. Frequently, nuclei were found that contained large nucleoli composed primarily of a light granular component (Fig. A2.2c). At 16 hours, a greater proportion of SFME cells grown without EGF were ultrastructurally altered than at 8 hours; however, the extent of the ultrastructural changes, except for minor differences, were similar to those changes described at 8 hours. In the remaining cells the nuclear envelope was frequently deeply indented or invaginated resulting in segmented nuclei. Nucleoli, if found, were condensed and small. Cytoplasmic vacuoles appeared expanded and occupied a larger proportion of the cytoplasm. Lipid droplets were commonly found, Golgi were atrophied, intermediate filaments, and coated vesicles were difficult to find, and aggregates of polyribosomes were sparse. Nuclear karyorrhexis, and apparent segmental loss of the nuclear envelope was occasionally seen in some early degenerate cells, but cytoplasmic organelles in these cells were recognizable. This finding suggests that nuclear condensation and eventual pyknosis occurred before cytoplasmic condensation and increased electron density. The majority of SFME cells cultured without EGF for 24 hours were either dead or degenerate (Fig. A2.3a). Dead cells, severely degenerate cells, cytoplasmic projections, 163 and cellular debris were similar in appearance to that described above, but were a greater fraction of the total population relative to 8 and 16 hour samples. The remaining cells appeared to be degenerate because the cytoplasm was filled with large vacuoles and abundant lipid droplets. Vacuoles often contained abundant membranous material in circular or concentric myelin figure profiles (Fig. A2.3b). Vacuoles also contained variable amounts of electron-dense amorphous debris and vesicles. Biochemical and physiological changes associated with EGF deprivation. Fragmentation of DNA isolated from SFME cells cultured without EGF was detected 8 hours after removal of the growth factor (Fig. A2.4). The pattern of degradation into oligonucleosome-length fragments, generating a regularly spaced "ladder," is characteristic of programmed cell death (Arends et al., 1990) resulting from activation of endonuclease cleaving chromatin into polynucleosomes. Lactate dehydrogenase released by cells into the culture medium was assayed as an indication of cell lysis. A large increase in enzyme activity was only observed in SFME cell medium at relatively late times after removal of EGF from the cultures (Fig. A2.5). No major release of lactate dehydrogenase over the same time course was detected in identical experiments with 3T3 mouse embryo cells, which are not acutely dependent on EGF for survival (Shipley and Ham, 1983). Nuclease assay of medium from SFME cells cultured in the absence of EGF did not detect increased activity before the time in which a general release of cellular enzymes was detected by assay of lactate dehydrogenase, indicating that nuclease causing the cleavage pattern of Fig. A2.4 probably was not released as cells underwent the progressive changes in the early phases leading to cell death resulting from EGF deprivation. Incubation of SFME cells in the presence of EGF with extracts of SFME cells cultured without EGF for 24 hours did not lead to cell death. Light and electron microscopic examination suggested that SFME cells became smaller after removal of EGF. This was demonstrated quantitatively by flow cytometric analysis of forward angle light scatter. The average size of the population of cells became 164 smaller with time after removal of EGF, while SFME cells from parallel cultures incubated with EGF did not show this change (Fig. A2.6). Analysis of DNA content of SFME cells incubated without EGF showed that the percentage of cells in G2 and S phase of the cell cycle markedly decreased with time after the removal of EGF (Fig. A2.7). Most of the cells 24 hours after removal of EGF were in the 01 stage of the cell cycle. No timedependent change in the relative percentages of cells in cell cycle stages was seen over the course of the experiment when EGF was included in the medium. To confirm the electron microscopic observations that polysomal aggregation decreased in SFME cells upon removal of EGF, we generated polysome profiles by gradient centrifugation and spectrophotometrically quantified the relative polysome and monosome content of SFME cells cultured for 6 hours with or without EGF (Fig. A2.8). Integration of the spectrographic tracings revealed that polysome-associated ribosomes were reduced 50% in EGF-deprived cells, relative to controls, with a corresponding increase in monosomes and ribosomal subunits. It is unlikely that the shift from polysomes to monosomes was because of an overall reduction in steady state level of mRNA in the cultures incubated without EGF, because Northern blot hybridization analysis did not detect a change in actin mRNA in SFME cells after 16 hours without EGF(Fig. A2.9). 165 DISCUSSION NGF-dependent neurons in vitro represent a well-studied growth factor-dependent cell culture system. NGF is necessary to establish cultures of these cells and neurons die within 48 hours in vitro if the growth factor is omitted from the medium (Yanker and Shooter, 1982; Thoenen and Barde, 1980; Martin et al., 1988). The behavior of these cells in vitro has been compared to programmed neuronal cell death in vivo, and the ability of NGF to promote neuronal survival in vivo has been demonstrated (Hamburger et al., 1981; Yip and Johnson, 1984). EGF-dependent SFME cells represent another growth factor- dependent culture system of neural tissue origin (Sakai et al., 1990). Growth factordependent systems also exist among hematopoietic stem cells: interleukin-dependent lymphocytes and erythropoietin-dependent reticulocyte progenitors (Koury and Bondurant, 1990; Rodriguez-Tarduchy and Lopez-Rivas, 1989; Nicola, 1989). Identification of SFME cells as astrocyte precursors suggests mechanistic and developmental relationships may exist between the NGF dependence of neurons and the EGF dependence of SFME cells. In one respect, however, the SFME culture system more closely resembles the hematopoietic cells. Like lymphocytes and reticulocyte precursors, and unlike neurons, the cells are capable of proliferation in vitro, and the growth factor promotes both proliferation and survival. A common feature among all the growth factor- dependent cell types is the loss of the growth factor requirement after oncogenic transformation; in some instances malignant transformation is accompanied by autocrine production of growth factor or activation of a receptor (Shirahata et al., 1990; Isfort, 1990; Bishop, 1987). Analysis of DNA from SFME cells cultured without EGF revealed a pattern consistent with the notion that the cells underwent degradation of chromatin in the regions unprotected by nucleosomes. This pattern is characteristic of cells undergoing apoptosis or programmed cell death, through which an endogenous endonuclease is activated (Arends, 166 1990). The time course of appearance of fragmented DNA in cultures incubated without EGF indicates that increased levels of fragmented DNA occurred coordinately with increased frequency of appearance of dead cells with pyknotic nuclei. Degenerate cells in our cultures show ultrastructural similarities to "phase 1" apoptotic cells, identified by Arends et al. (1990) as undergoing early phases of programmed cell death, and may not yet have undergone extensive DNA fragmentation associated with pyknotic nuclei. Cells identified as degenerate appeared before the large scale manifestation of dead cells with pyknotic nuclei, also suggesting that degenerate cells are precursors of apoptotic cells. In experiments in which EGF was removed for 8 to 24 hours and then added back, we found that incubation for only 8 hours in the absence of EGF reduced by 50% the number of cells that ultimately were recovered after EGF deprivation (Loo et al., 1989b), suggesting that the degenerate cells traverse an irreversible pathway leading to death. Incubation for 24 hours in the absence of EGF reduced by 80% the number of cells that recovered after EGF deprivation (Loo et al., 1989b; Rawson et al., 1990). At this time most of the cells in the cultures appeared either dead or degenerate by ultrastructural analysis and significant leakage of cytoplasmic enzyme into the culture medium was detected. Assay of cytoplasmic enzyme release into the medium did not detect a large release at earlier time points when ultrastructural and some biochemical changes were obvious, indicating that plasma membrane integrity was maintained through this early period and implying that degenerate cells were not highly permeable. Nuclease activity was not released in a specific manner before the general breakdown of membrane integrity and extracts of dying cells did not kill SFME cells in the presence of EGF, suggesting that cell death resulting from EGF deprivation is not recruited in some cells by release of enzyme from other cells. Flow cytometry showed that the cultures in the absence of EGF for 24 hours were entirely in the G1 phase of the cell cycle. This probably reflects an inability of 167 cells to progress into S phase in the absence of EGF, and may also indicate an initial preferential loss of cells in S or G2 in the absence of EGF. Ultrastructural analysis of SFME cells cultured in the absence of EGF identified a time-dependent reduction in polysomes, alterations in nuclear morphology, a reduction in cell size and cytoplasmic volume and the appearance of lipid or membrane-containing vacuoles and apoptotic nuclei. The appearance of lipid-containing vacuoles and multivesicular bodies and a progression to small cell size has been noted in neurons deprived of NGF (Martin et al., 1988). In both culture systems apoptotic nuclei appear with little evidence of a transition state, suggesting that this change occurs rapidly (Martin et al., 1988). Primary disturbances in ER are observed in the neuronal system, but were not prominent in SFME cells without EGF, and the shift from polysomes that we observed with SFME cells is not seen in neurons cultured without NGF (Martin et al., 1988). The reduction in SFME cell size perceived microscopically was confirmed by flow cytometric analysis. This effect appears to be the result of fragmentation and loss of cytoplasm from the cells; the small cells are essentially nuclei surrounded by a minimal amount of cytoplasm and plasma membrane. A loss of polysomes was the earliest change in SFME cells observed in the electron micrographs after removal of EGF, and the EGFdependent polysome shift was confirmed in sedimentation profiles. mRNA remained intact in these cells, suggesting that an EGF-dependent regulation of translation may be responsible. EGF and other peptide-growth factors stimulate phosphorylation of ribosomal protein S6 in other cell culture systems (Oliver et al., 1988), but the functional significance of this effect has not been established. The relationship of the ultrastructural, biochemical, and physiological alterations we have observed in EGF-deprived SFME cells to the primary mechanism of cell death in this system, as well as the means by which inhibitors of protein and RNA synthesis prevent cell death, remains to be determined. 168 ACKNOWLEDGMENTS Supported by National Institutes of Health National Aging Institute-07560 and Council for Tobacco Research 1813. D. Barnes is the recipient of Research Career Development Award, NIH-National Cancer Institute-01226. Flow cytometry was carried out through the Oregon State University Environmental Health Sciences Center Flow Cytometry Core Unit (NIEHS ES00210). Cathleen Rawson performed flow cytometry experiments and assisted with polysome profiles. Julie Duimstra and Olaf Hedstrom provided electron microscopy data. Edward Schmidt performed polysome profile experiments. David Barnes provided financial and scientific support. 169 Fig. A2.1. SFME cells cultured with EGF or without EGF for 4 hours. (A), with EGF, low power. Polygonal or rounded cells, found in sheets, contain large, round to oblong, slightly indented nuclei. Nuclei usually have a thin peripheral rim of heterochromatin, abundant finely dispersed chromatin, and prominent nucleoli. (B), with EGF, at higher magnification, the cytoplasm is rich in organelles: abundant polyribosomes (P), prominent clear vacuoles (V) containing delicate membranous-like material, multivesicular bodies (X), well-defined Golgi complex (G), coated vesicles (arrows), short, slightly dilated profiles of RER (arrowhead), and mitochondria (M). (C), without EGF for 4 hours. The first significant alteration detected was decreased polyribosomic aggregates giving the cytoplasm an increased electron lucency. A few cytoplasmic lipid droplets (L) were also found. A well-developed Golgi complex (G), clear vacuoles (V), multivesicular bodies (X), intermediate filaments (F), and mitochondria (M) were ultrastructurally similar to corresponding control organelles. Bars: (A) 10 gm; (B) 1 p.m; (C) 1 pm. 170 Figure A2.1. A, B 14+ ' :.41. Ira . f,... 4 171 Figure A2.1. C im:aith14.-r }arv , 172 Fig. A2.2. Cell death and degeneration in SFME cells cultured for 8 hours in the absence of EGF. (A and B), low power magnification of SFME cells depicting pleomorphic cells, severely degenerate cells (arrows), dead cells (D), cellular debris (arrowhead), and elongated cytoplasmic projections with the remaining portion of the cell out of the plane of section (P). (C), higher magnification of SFME cells showed elongated cytoplasmic projections (P), cytoplasmic blebs (B), atrophied Golgi (G), and lack of aggregates of poyribosomes. peripherally located. Note the prominent light granular nucleolus (Nu) 173 Figure A2.2. A, B 174 Figure A2.2. C 175 Fig. A2.3. SFME cell death and severe degeneration after 24 hours of EGF deprivation. (A), dead (D) or severely degenerate cells (arrows), and dispersed cellular debris (arrowheads) were a major feature at this time period. The cytoplasm of remaining cells contained abundant large vacuoles, some of which compressed adjacent structures (i.e.., nucleus). Nuclei of these cells were usually indented or partially segmented. (B), the presence of abundant cytoplasmic lipid droplets was an additional feature observed during this time period. Note the numerous lipid droplets (L), peripheral collapsed vacuoles, and condensed granular cytoplasm of a severely degenerate cell. In adjacent cells, cytoplasmic vacuoles contained a predominance or a mixture of the following structures: circular membranous material, vesicles, or cellular debris. Multivesicular bodies (X) or in some vacuoles, membranous whorls (myelin figures) (M) were also seen. Bars: (A) 1011m; (B) 1 p.m. 176 Figure A2.3. ff 7, 0 , . -.:4 %,,e .', .- " Z 4 4 e'' ; ',. 4 -%, .... ..--:., :4.: ,?1' v. -.!:.71.--1.44c:. , ;, , :. /5 . II, ° .> , ... ,; 4.111 -5'''.- .44471:11,s ;,i. '.) -L.., ,,, :, " ..1,...,...-. . ,", tit:* r ..' .2.:4;',..--r, , T.- : , to) . , .1. - 177 Fig A2.4. DNA fragmentation in EGF-deprived SFME cells. Cultures were changed to medium without EGF and incubated for up to 24 hours. DNA was isolated from samples during this time course, fractionated by electrophoresis, and visualized by ethidium bromide staining. (A), 0 h without EGF; (B), 4 h without EGF; (C), 8 h without EGF; (D) 16 h without EGF; (E), 24 h without EGF; (F), 24 h with EGF; (G), 123-bp DNA ladder standard. Arrowheads mark bands comprising oligonucleosome ladders indicative of DNA fragmentation by endonuclease (Arends et al., 1990). ABCDEFG 178 Fig. A2.5. Release of cytoplasmic enzyme in culture medium by SFME cells in the absence of EGF. SFME cells or 3T3 cells were incubated the indicated times in serum-free medium with or without EGF and culture medium assayed for lactate dehydrogenase as described. 3T3 cells are mouse embryo control cells that are not acutely dependent on EGF for survival. (), with EGF; (), without EGF. 50 40 LDH 30 Iml. IND 20 10 0 Fil 1 4 HR 8 HR SFME i 24 HR now f-M 4 HR 8 HR 3T3 1 24 HR 179 Fig. A2.6. Flow cytometric size analysis of SFME cells. Cultures were incubated with or without EGF and processed as described for flow cytometry size analysis by measurement of forward angle light scattering. Tracings show relative cell number (Y axis) versus relative volume per cell (X axis). (A), no EGF, 0 h; (B), no EGF, 8 h; (C), no EGF, 24 h; (D), no EGF, 48 h; (E-H), EGF present, 0, 8, 24, 48 h. 180 Fig. A2.7. Flow cytometric DNA analysis of SFME cells. Cultures were incubated with or without EGF and processed as described for analysis of DNA content per cell. Tracings show relative cell number (Y axis) versus relative amount of DNA per cell (X axis). (A), no EGF, 0 h; (B), no EGF, 8 h; (C), no EGF, 24 h; (D-F), EGF present, 0, 8, 24 h. A 181 Fig. A2.8 Polysome profiles of SFME cells. Cells were incubated with or without EGF, lysed in the presence of cycloheximide, and polysome distributions were determined by sedimentation in a sucrose gradient and spectroscopic measurement as described. (A), without EGF, 6 h; (B), control, with EGF. Arrows show direction of sedimentation. (T), absorptive material at the top of the gradient; (R), monosomes and ribosomal subunits; (P), polysomes. 182 Fig. A2.9. Northern blot hybridization analysis of actin mRNA from SFME cells. SFME cells were cultured as described in serum-free medium with (A) or without (B) EGF for 16 hours followed by isolation of poly-A-containing RNA and blot hybridization as described. The blot was probed with labeled plasmid containing the gene for beta actin. -4 28 S 110 le AB -418s