X-ray photoelectron spectroscopy investigation of eTieH thin films magnetron sputtered Mg I.J.T. Jensen
advertisement

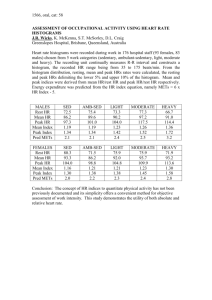
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 Available online at www.sciencedirect.com journal homepage: www.elsevier.com/locate/he X-ray photoelectron spectroscopy investigation of magnetron sputtered MgeTieH thin films I.J.T. Jensen a,*, A. Thøgersen b, O.M. Løvvik b, H. Schreuders d, B. Dam d, S. Diplas b,c a Department of Physics, University of Oslo, P/O Box 1048 Blindern, 0316 Oslo, Norway SINTEF Materials and Chemistry, P/O Box 124 Blindern, 0314 Oslo, Norway c Center for Materials Science and Nanotechnology, P/O Box 1126 Blindern, 0318 Oslo, Norway d Materials for Energy Conversion or Storage (MECS), DelftChemTech, Faculty of Applied Science, Technical University Delft, P/O Box 5045, NL-2600 GA Delft, The Netherlands b article info abstract Article history: Thin film samples of Mg80Ti20 (MgeTi) and Mg, both with and without H, were investigated Received 22 January 2013 in a series of X-ray photoelectron spectroscopy (XPS) measurements. The samples were Received in revised form covered with a thin protective layer of Pd, which was removed by Arþ sputtering prior to 22 May 2013 data acquisition. This sputtering was found to reduce both oxides and hydrides. A distinct, Accepted 26 May 2013 previously unknown peak was revealed in the Mg KLL spectrum of the MgeTieH samples, Available online 8 July 2013 located between the metallic and the MgO component. This peak was attributed to trapping of H in very stable interstitial sites at the interface between Ti nano-clusters and the Keywords: Mg matrix, based on earlier density functional theory calculations and supported by Thin films so-called Bader analysis. The latter was performed in order to study the theoretical charge Magnesium distribution between Mg, Ti and H, establishing a link between the position of the previ- Titanium ously unknown peak and the effect of H on the valence state of Mg. The composition of the Immiscible elements samples was studied both by energy dispersive spectroscopy using transmission electron Interface microscopy and by quantitative XPS analysis. Final state Auger parameters (AP) were obtained for metallic Mg, MgO and MgH2, as well as Mg affected by trapped H. No difference XPS between the AP values from the metallic components was found between the Mg and the MgeTi samples. The AP values for MgO and MgH2 were consistent with previous reports in literature; several eV lower than the metallic value. Mg in the vicinity of trapped hydrogen, on the other hand, showed a more metallic character, with its corresponding AP value less than 1 eV below the AP for pure Mg. Copyright ª 2013, Hydrogen Energy Publications, LLC. Published by Elsevier Ltd. All rights reserved. 1. Introduction The discovery of so-called switchable mirrors of Y and La hydrides [1] in 1996 sparked an interest in thin film hydrides, leading to the discoveries of several hydride systems with tunable electrical and optical properties [2e6]. MgeTi thin films stand out among them, as they appear black rather than transparent in the fully hydrogenated state [6]. Possible applications for MgeTieH and other switchable hydride films range from coatings on solar collectors [6] and smart windows * Corresponding author. Present address: SINTEF Materials and Chemistry, P/O Box 124 Blindern, 0314 Oslo, Norway. Tel.: þ47 98230513. E-mail address: IngvildThue.Jensen@sintef.no (I.J.T. Jensen). 0360-3199/$ e see front matter Copyright ª 2013, Hydrogen Energy Publications, LLC. Published by Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.ijhydene.2013.05.142 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 [7] to optical hydrogen sensors [8] and semiconductor devices [9]. In addition to their exotic optical behavior MgeTi films are interesting from a scientific point of view for a number of reasons: Mg and Ti are immiscible, which means that the structure of samples produced by non-equilibrium techniques such as magnetron sputtering [6] is difficult to predict. They show excellent hydrogen storage capacity and kinetics, even at room temperature [10], and what is more: they are stable over repeated cycles of hydrogen loading and unloading [11]. The latter is really quite remarkable considering the inherent meta-stability of MgeTi alloys. Rather, one would expect segregation into pure Mg and Ti upon cycling, as is the case for several other Mg containing alloys [5,12e14]. As already mentioned the limited solubility of Ti in Mg makes the crystal structure of MgeTi thin film samples difficult to predict, either with or without hydrogen. No phase separation has been found with X-ray diffraction (XRD) or transmission electron microscopy (TEM); instead the samples were found to be single phase crystalline all the way from before hydrogen absorption to after hydrogen desorption [15]. However, evidence for chemical short range order was obtained from simulation of optical isotherms from hydrogenography by Gremaud et al. [16] in 2008 and further verified by a combination of X-ray diffraction and extended X-ray absorbtion fine structure (EXAFS) spectroscopy [17]. Density functional theory (DFT) calculations later showed that the formation enthalpy of the system was in fact lowered significantly when the Ti atoms were arranged in small nanoclusters of sizes well below the X-ray detectable range [18]. In this configuration the electron interaction between Mg and Ti was reduced and the Ti atoms in the center of the clusters could obtain a state close to elemental Ti. In a further DFT investigation hydrogen was introduced to the MgeTi nanocluster model, revealing very stable H sites at the interface between Mg and Ti, with hydrogenation energies down to 115 kJ/(mol H2). It was found that with respect to the mixing enthalpy, which is the cohesive energy relative to standard state elements, the MgeTieH system changes from metastable to stable for hydrogen contents above 0.07 H per metal [19]. Further it was found that the DFT results were in good correspondence with the experimental work of Vermeulen et al. [20], who found increased solid solubility of H in MgeTi. In their work it was also observed that the amount of H being retrieved from a MgeTi sample was less than the amount initially absorbed, pointing to the presence of sites where H was very strongly bound. Upon hydrogenation Mg100yTiy thin films form a MgH2like rutile structure (body centered tetragonal) or a TiH2-like fluorite structure (face centered cubic) depending on the amount of Ti [21]. The turning point between formation of a MgH2-like and a TiH2-like hydride phase was reported to be at a composition 10 < y < 13 [15]. However, the reasons why such relatively small amounts of Ti would cause Mg to form a cubic hydride remains a riddle. What is known, from extensive optical and electrical investigations [15], is that regardless of the Ti content, there must be a phase with a similar band gap as the conventional MgH2 rutile structure present in the hydrogenated samples. Thus the electrical properties of fluorite MgH2 appear to be similar to those of the conventional rutile MgH2. 10705 Limited published work was found on the MgeTieH system using spectroscopic techniques like X-ray photoelectron spectroscopy (XPS). The close overlap of the hydride, oxide, and hydroxide components of the Mg peaks makes data interpretation a challenge. Friederichs et al. [22] studied different powders of Mg and Mg hydride under controlled exposure to oxygen and air. In their analysis, the MgH2, MgO and Mg(OH)2 components were not separated in the Mg 2p peak, while in the Mg KLL peak the Mg(OH)2 component was shifted to slightly lower kinetic energy EK compared to MgO. Nevertheless they did detect distinct Mg 2p-KLL Auger parameter (AP) values for MgH2 and MgO, 1231.8 eV and 1231.2 eV, respectively. Vapor deposited Mg100yTiy samples with y up to 25.3 were investigated by high energy XPS by Mitchell et al. [23] From AP and charge-transfer calculations, small indications of charge-transfer from Mg to Ti were found. In this work thin films of Mg80Ti20 with and without H were studied by XPS and compared to their Mg counterparts. The results were interpreted in view of recent DFT calculations, which were extended to include a study of the charge distribution between Mg and Ti in the theoretical model using socalled Bader analysis. 2. Experimental Mg80Ti20 (MgeTi) and Mg films covered with a thin protective layer of Pd were deposited in a UHV system (base pressure ¼ 106 Pa) by DC/RF magnetron sputtering. The former was made by co-sputtering of Mg (99.95%) and Ti (99.999%) targets in 0.3 Pa of Ar, on single-crystal Si(100) substrates. The substrate was kept at room temperature. In order to increase the homogeneity and ensure an even thickness of the film the substrate was continuously rotated during sputtering. The typical deposition rate was 2.2 A/s at 150 W RF for Mg and 0.42 A/s at 131 W DC for Ti. A Pd (99.98%) caplayer was sputtered with a rate of 0.58 A/s at 25 W DC to prevent oxidation. The Mg films were deposited at similar conditions. The investigated samples were produced in two batches. In batch 1 the investigated film was 100 nm thick with a 5 nm Pd layer on top and in batch 2 the film was 200 nm thick with a 1.5 nm Pd layer. The first batch was hydrogenated ex situ in 10 bar H2 at 80 C for about a day. The second batch was hydrogenated at room temperature in situ directly after sputtering with 100% hydrogen at 1 bar for about a day. The thin Pd layer of 1.5 nm is just enough to load the sample with H, but not enough to stop oxidation completely. Formation of oxide is believed to prevent unloading of H. The samples were investigated by XPS using a KRATOS AXIS ULTRADLD instrument with monochromatic Al Ka radiation (hv ¼ 1486.6 eV) operated at 15 kV and 15 mA. Typical vacuum level during measurement was in the order of 107 Pa. The Pd layer was removed by sputtering an area typically about 2 mm 2 mm with an Arþ ion beam of 0.5 or 2 kV delivering 100 mA of current. To minimize cratering effects and avoid detection of Pd the measurements were done using a “small spot” aperture with a diameter of 110 mm. High resolution spectra were acquired with pass energy 20 eV and step size 0.1 eV/s. Two different series of experiments were performed. 10706 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 In the over-night exposure (ON) experiments the measured sample was left in position in the XPS analysis chamber for about 24 h before the same measurement was repeated. Magnesium is highly reactive to oxygen, which means that it will oxidize even in UHV. Thus the results from the ON experiment show the changes in spectra upon gentle oxidation, comprising data from Mg, Mg with H and MgeTi with H. A comparative depth profile (DP) experiment was performed for MgeTi with and without H. For each sample measurements where done at intervals of 5 min of Arþ sputtering at 0.5 kV, starting at 5 min and ending at 30 min total sputtering time. The sputtering time was not converted to sample depth as the craters were too shallow to be measured by available methods, e.g., stylus profilometry or atomic force microscopy. Details of the measurements are listed in Table 1. The Mg 2p, Ti 2p and O 1s photoelectron peaks and the Mg KLL Auger peak were acquired. In some of the measurements the Mg 1s peak was also obtained. Data processing was done using CasaXPS [24]. All spectra were fitted with a standard Shirley background [25] and calibrated setting the Mg 2p metal component to 49.4 0.05 eV [26], due to the absence of adventitious carbon. In quantifications Scofield cross-sections were used for the relative sensitivity factors [27]. Cross-sectional transmission electron microscopy (TEM) samples were prepared by ion-milling using a Gatan precision Table 1 e List of measurements, labeled by measurement numbers (M no.). The experiments are categorized as initial, depth profile (DP) or over-night exposure (ON). The compositions refer to Mg (Mg) and Mg80Ti20 (MgeTi) with and without H. Information about sample batch number and ArD sputtering parameters are also given. See text for details. M no. Experiment Composition Batch no. Arþ sputtering Volt. [kV] Time [min] M1 M2 Initial Initial Mg MgeTi 1 1 2 2 3.5 3.5 M3 M4 M5 M6 M7 M8 DP DP DP DP DP DP MgeTi MgeTi MgeTi MgeTi MgeTi MgeTi 2 2 2 2 2 2 0.5 0.5 0.5 0.5 0.5 0.5 5 10 15 20 25 30 M9 M10 M11 M12 M13 M14 DP DP DP DP DP DP MgeTieH MgeTieH MgeTieH MgeTieH MgeTieH MgeTieH 2 2 2 2 2 2 0.5 0.5 0.5 0.5 0.5 0.5 5 10 15 20 25 30 M15 M16 M17 M18 M19 ON ON ON ON ON MgeTieH MgeTieH MgeTieH MgeH MgeH 1 1 1 1 1 2 2 2 2 2 10 M20 M21 ON ON Mg Mg 2 2 2 2 a Same as above, measured w2 h later. b Same as above, next day measurement. ion polishing system with 5 kV gun voltage. The sample was analyzed in a 200 keV JEOL 2010F microscope with a Gatan imaging filter and detector, and a NORAN Vantage DIþ energy dispersive spectroscopy (EDS) system. A low intensity beam and minimal exposure were used in order to reduce the knock-on damage by the electron beam. DFT calculations at the PBE-GGA level [28] were performed for a Mg81.25Ti18.75 composition using the Vienna ab-initio simulation package (VASP) [29,30]. A 4 4 2 Mg unit cell (64 atoms) was used as a starting point, with Ti arranged in nano-clusters. Hydrogen was introduced to the system gradually. In this structure the sites available for hydrogen will have the amount of Ti in the first coordination sphere (nTi) varying from 6 to 0. The sites where filled according to their stability, in the following order: nTi ¼ 3, 6, 2, 1 and 0. A detailed description of the calculations can be found in Ref. [20]. In the present work Bader analysis [31] was employed to investigate the charge transfer upon hydrogenation. This is a method to determine which electrons belong to which atoms in the DFT unit cell, where a surface is drawn around each atom perpendicular to minima in the charge density. 3. Results and discussion 3.1. Microstructure and composition The TEM image in Fig. 1 shows a 200 nm thick MgeTieH film from sample batch 2, on a Si substrate. A 3 nm thick SiOx film is visible at the MgeTieH/Si interface, and the Pd capping layer at the surface is estimated to be about 10 nm thick. However, the surface is rough and the thickness of the Pd layer may therefore be overestimated. The electron diffraction pattern presented in Fig. 1 shows the Mg [100] and Si [110] zone axis. The Mg grains grow perpendicular to the substrate surface, in the [002] direction. The lattice parameters measured on the diffraction pattern of Mg (P63/mmc) was a ¼ b ¼ 3.11 A a b 5 b 40 b Fig. 1 e Cross-sectional TEM image of the film, with an electron diffraction pattern as an inset for the Mg [100] and the Si [110] zone axis. i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 10707 and c ¼ 5.10 A. The composition of bulk film was measured by EDS to be 84 at.% Mg and 16 at.% Ti within an uncertainty of 1 at.%. No Ti clusters could be observed with TEM. Fig. 2 shows compositional quantification obtained by XPS, based on the Mg 2p, Ti 2p and O 1s spectra. The amount of Ti detected is generally higher than the expected 20 at.%, especially for the unhydrogenated samples. However, the accuracy of such quantifications rely on many factors, especially for inhomogeneous samples, and can typically be anywhere from 1 to 10 at.%. Nevertheless, there appears to be a relative difference in Ti content between the DP measurements of MgeTi with and without H (M9-14 and M3-8, respectively). An option to consider is whether this is related to differences in volume per metal atom before and after hydrogenation. The apparent drop in Ti content upon attempted hydrogenation (M9-14) is accompanied by a slight increase in oxygen content. As will be shown later, the H to metal ratio is also found to be higher in the Ti rich regions of the samples compared to the Mg rich regions. For the limited analysis depth of the XPS technique such reductions of Ti atoms per volume in the hydrogenated sample could possibly be sufficient to cause the apparent drop in the Ti content of the sample. Fig. 2 also shows that the oxygen content was somewhat increased in the first DP measurements (M3 and M9) closest to the surface and the initial ON measurements (M15 and M18) that were hydrogenated ex situ. The final ON measurements (M16, M17 and M19), which were left in UHV in the XPS after removal of the protective Pd layer, show a substantial increase of oxygen. 3.2. Mg KLL spectra Fig. 3 shows high resolution Mg KLL spectra from MgeTi and Mg before and after hydrogenation. In the unhydrogenated state (Fig. 3a and b) the spectra are dominated by the metallic peak at about 1186 eV [32]. Also prominent is the peak at about 1180.6 eV, which is due to MgO [33]. Traces of Mg(OH)2 is hidden in the shoulder on the low EK side of MgO [22]. The bulk plasmon loss peak is clearly seen at 1175.2 eV for Mg [34] and is less intense for MgeTi at 1174.2 eV (Fig. 3a and b, respectively.). Between the bulk plasmon and the oxygen related Auger peak the surface plasmon loss peak can be vaguely Fig. 2 e Quantification data obtained by XPS. The measurement numbers refer to Table 1. Fig. 3 e Mg KLL spectra from Mg (M1), MgeTi (M2), MgeH (M18) and MgeTieH (M13) (bottom upwards). The components are labeled as follows: M [ metallic, O [ MgO, OH [ Mg(OH)2, H [ MgH2, PL [ bulk plasmon. For roman numbered components see text. discerned [34]. One or two weak peaks (I and II) are found between the metallic and the oxygen related Auger peaks. After hydrogenation the Mg sample (Fig. 3c) shows a drastic increase in intensity in the region of MgO and Mg(OH)2. This corresponds well with previous work [22] where MgH2 was found to overlap with MgO. The metallic peak is still clearly visible, meaning that either the sample was not fully loaded from the start, or the sample has lost H during the sputtering and/or measurement. The hydrogenated MgeTi spectrum on the other hand (Fig. 3d), is more surprising. Rather than an increase in intensity of the region of MgO consistent with MgH2, hydrogenated MgeTi shows a strong peak much closer to the metallic one, at about 1183.9 eV (peak IH). Along with this comes a broader, less distinct feature just above 1182 eV (peak IIH). The spectrum for the hydrogenated Mg sample also exhibits increased intensity in the region between metallic Mg and MgH2, which can be fitted with one or more components. For simplicity the region was fitted with just one component (IIIH). Peaks I, II and IIIH can be attributed to understoichiometric oxide, hydroxide and hydride, respectively, as will be discussed below. The extra peaks IH and IIH in the hydrogenated MgeTi sample, however, appear to be previously unknown. No contaminating species other than traces of Pd have been found in the survey spectra of these samples. 10708 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 The positions of peaks I and IH appear to be quite close to each other. One option to consider is whether these peaks have similar origins, for instance from interactions between Mg and the Pd caplayer. The formation of MgePd alloys (Mg6Pd, Mg5Pd2) is often observed in hydrogen cycling of Pdcapped Mg and Mg2Ni thin films [35,36]. Mg KLL spectra from such MgePd alloys are not readily accessible in literature, so a direct comparison has not been possible. As shown in Fig. 4, comparisons of the DP spectra for MgeTi with and without H do indeed show increased levels of Pd consistent with MgePd alloy formation upon hydrogenation, as described in literature [35,36]. Fig. 5 shows the corresponding Mg KLL spectra from the unhydrogenated sample. From this it is evident that while the Pd 3d peak vanishes with sample depth, the I peak in the Mg KLL spectrum only decreases slightly. Electron attenuation lengths l were calculated according to Tanuma et al. [37] assuming a Mg matrix, in order to compare the maximum sample depth d ¼ 3l where the signal arise from for Mg KLL and Pd 3d. The values were l ¼ 2.96 nm and 2.90 nm, respectively. Thus the sample volume analyzed in Figs. 4 and 5 should be practically the same. In general the relative Pd levels in the samples after Arþ sputtering do not correlate with the relative intensities of the unknown peaks. For instance, the Pd levels in the deepest MgeTieH DP measurements are similar to that of the Mg sample, see Fig. 6. From Fig. 3 on the other hand, it is obvious that the corresponding intensities of the I and IH peaks are very different. Furthermore, if the IH peak was caused by enhanced alloying of Mg and Pd and subsequent MgePd hydride formation it should also be present in similar amounts in the MgeH spectra, which is not the case. More likely the I peak can be understood after a close look at the corresponding O 1s spectra. These spectra, both in MgeTi and Mg, exhibit a fairly strong shoulder at the higher binding energy (EB) side of the MgO component. Fig. 7 compares the O 1s spectrum of unhydrogenated MgeTi fitted with one and two oxide peaks. According to Göpel et al. [38] the Fig. 4 e Pd 3d spectra from the DP measurements of MgeTi (left) and MgeTieH (right). The Pd content at the first depth is about 40% higher in the hydrogenated sample. Sample depth increases from top downwards. Fig. 5 e Mg KLL spectra from the DP measurements of MgeTi without H. Sample depth increases from top downwards. The components are labeled as follows: M [ metallic, O [ MgO, OH [ Mg(OH)2, I: see text. presence of under-stoichiometric oxide should give an O 1s peak shifted to higher EB accompanied by a shift to lower EB in the corresponding cation peak. In an investigation of the stoichiometry and morphology of MgO films Wollenschläger et al. [39] assigned a peak at 1183.5 eV in the Mg KLL spectrum Fig. 6 e Pd 3d spectra from the Mg measurement M1 (a) and the MgeTieH measurements M13 (b) and M14 (c) showing Pd levels of about 1 at.%. i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 10709 and IIH peaks in the Mg KLL spectrum from MgeTieH will be discussed further in separate sections. 3.3. Mg 1s, Mg 2p and Ti 2p spectra Figs. 8 and 9 show the Ti 2p and Mg 2p spectra corresponding to Fig. 3. In the Ti 2p spectra the key challenge is to be able to distinguish correctly between the metallic, hydride and oxide components. For instance, the intensity of the TiO component will depend strongly on the peak shape of the metallic component. For the unhydrogenated samples we have chosen to follow the fitting procedure proposed by Biesinger et al. [43]. For the metallic peak an asymmetric peak-shape La(1.1,5,7) was used, where the first two parameters define the spread on either side of a Lorentzian component. The Lorentzian curve is convoluted by a Gaussian with the width specified by the last parameter. For more details about the LA peak shape see Ref. [43]. The oxide peaks were fitted with a mixed GaussianeLorentzian component. This resulted in a fit consisting almost entirely of metallic Ti with only about 5% of the intensity attributed to TiO and TiO2. For comparison purposes and lack of better suggestions from literature the hydride component was fitted with the same peak shape as the metallic one. Unlike MgH2, TiH2 is conducting, thus the use of the metallic peak shape seems reasonable. This choice of fitting parameters resulted in increased intensity in the regions of the TiO, Ti2O3 and TiO2 components in the hydrogenated sample compared to in the unhydrogenated state. The Fig. 7 e O 1s spectra from MgeTi (M2), fitted with one (bottom) or two (top) Mg oxide peaks. to under-stoichiometric magnesium oxide. Arþ sputter reduction of oxides is a well known phenomenon [40]. In the work of Kurth et al. [41] the initial stages of oxidation of Mg was studied at oxygen partial pressures between 1.3 108 and 1.3 105 Pa, revealing a clear oxygen deficiency with respect to MgO for the early oxide. Thus we find the presence of under-stoichiometric magnesium oxide to be a plausible explanation for peak I in the Mg KLL spectrum of unhydrogenated samples. Along the same lines peak II can be thought of as under-stoichiometric Mg hydroxide. This is supported by a tendency of peak II to disappear with increasing sample depth (not shown). Compositional changes due to Arþ ion sputtering have also been reported for Ti hydrides by Lamartine et al. [42] Similar to the oxide case we believe that the broad feature in the MgeH spectrum (peak IIIH) is due to under-stoichiometric Mg hydride, MgHx<2, caused by the Arþ sputtering. This would mean that in reality the IIIH peak is made up of several components arising from a gradual decrease of hydrogen content. H is generally thought to be more easily desorbed from MgeTieH than from MgH2 [20]. Thus the MgeTieH samples should also be affected by Arþ sputtering, yet the region between the metal and the MgH2 peak positions in the Mg KLL spectrum is not smooth as in the spectrum from MgeH. On the contrary, the IH peak is a very distinct feature, likely to have an equally distinct origin. The IH Fig. 8 e Ti 2p spectra from MgeTi (M2, bottom) and MgeTieH (M13, top). 10710 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 Fig. 9 e Mg 2p spectra from Mg (M1), MgeTi (M2), MgeH (M18) and MgeTieH (M13) (bottom upwards). The spectra are normalized using the metallic peak, but in reality this peak will have lost intensity in the spectra from hydrogenated samples compared to the unhydrogenated ones. The components are labeled as follows: M [ metallic, O [ MgO, OH [ Mg(OH)2, H [ MgH2. hydride component was shifted about 0.3 eV to higher EB as compared to the metallic component. The position of the titanium hydride peak at 454.3 eV corresponds fairly well with literature: 454.4 eV was reported (without Ti metal reference) by Lesiak et al. [44], 454.6 eV, and a 0.6 eV shift compared to Ti metal was reported by Lisowski et al. [45] and by Kang et al. [46]. However, the shift observed in Fig. 8 is smaller than what would be expected from pure TiH2. Lamartine et al. [42] found a linear relationship between the H:Ti ratio and the shift in Ti 2p, and according to this the Ti in our samples appears not to be fully hydrogenated. Rather it seems that the H:Ti ratio was around 1, half of what is the case in fully loaded TiH2. All the Mg 2p spectra show a clear metallic peak, which was set to 49.4 eV in order to calibrate the energy scale [26]. On the high EB side of the metallic peak Mg oxide, hydroxide and hydride components partly overlap to form a broader continuous peak [22]. For the unhydrogenated samples this region is weak in intensity, reflecting the low level of oxygen contamination in the samples. For the hydrogenated samples, on the other hand, the intensity in this region is strongly increased due to hydride overlapping with the existing oxide. A certain shift in the peak position is observed between MgeTieH and MgeH, consistent with the differences seen also in the Mg KLL spectra. Fig. 10 shows the Mg 1s peak for Mg (M1), MgeTi (M2), MgeH (M18) and MgeTieH (M15). In the first two the metallic component at 1302.9 eV is clearly seen, while in the hydrogenated samples it is present as a shoulder on the low EB side. In the MgeTi spectrum the oxide peak is increased quite a lot compared to in the Mg spectrum. The quantification in Fig. 12 also shows increased oxygen levels in the MgeTi measurement compared to Mg. This difference will be enhanced further in the Mg 1s peak, which is very surface sensitive. Due to low EK it is collected from shallow sample depths only. For the MgeH sample the hydride/oxide component is dominant, Fig. 10 e Mg 1s spectra from Mg (M1), MgeTi (M2), MgeH (M18) and MgeTieH (M15) (bottom upwards). The components are labeled as follows: M [ metallic, O [ MgO, OH [ Mg(OH)2, H [ MgH2. with the MgeTieH peak shifted to slightly lower EB (1304.35 eV) compared to MgeH (1304.71 eV), as seen also in the case of Mg 2p. 3.4. Auger parameters Final state Auger parameters (AP) obtained in this work are listed in Table 2. The Mg 1s-KLL final state AP for the metallic components was 2488.8 0.1 eV for both Mg (M1) and MgeTi (M2), while the Mg 2p-KLL final state AP for the metallic components was 1235.3 0.1 eV for all samples. No significant differences between Mg and MgeTi were found in either of the cases and the AP values were in correspondence with literature values for pure Mg [47]. The final state AP is defined as: a ¼ EB ðphotoelectronÞ þ EK ðAuger electronÞ; (1) where EB and EK are the binding energy and kinetic energy, respectively. The final state Auger parameter a is essentially the energy distance between a photoelectron peak and a related Auger-peak in the spectra, making it unaffected by energy referencing and charging problems. Differences in the Auger parameter reflect differences in the system’s ability to screen the core hole created upon photoemission. It can be divided into the relaxation of the photoionized atom itself, e.g., shrinkage of the valence band, and the response of the surroundings through either charge transfer or polarization. Table 2 e Final state Auger parameters for Mg. Composition Mg MgeTi MgeH MgeTieH MgeTieH Phase 1s-KLL AP [eV] 2p-KLL AP [eV] Metallic Metallic MgH2 IH MgO 2488.8 2488.8 2485.7 2488.4 e 1235.3 1235.3 1232.1 1234.6 1231.2 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 The fact that there was no difference between the AP values of Mg and MgeTi means that no strong effect of interactions between Mg and Ti could be detected. Fig. 11 shows Mg 2p-Mg KLL final state Auger parameters for the metallic and the dominant non-metallic phases in the hydrogenated samples with and without Ti. The AP is unaffected by energy referencing problems provided that the photoelectron and Auger peaks being compared originate from the same phase. In the Mg 2p spectra the oxide, hydroxide and hydride phase overlap to form a broad peak (the non-metallic peak), which represent a problem in this context. Rather than trying to deconvolute the individual components this peak was fitted with just one component, based on the assumption that the dominant peak in the Mg KLL spectrum would also dominate the position of the peak maximum in the Mg 2p spectrum. For the MgeTieH samples two APs were measured, combining the non-metallic Mg 2p peak with the Mg KLL MgO and IH component, respectively. MgO is not a dominant phase in this case, which means that the previous argument regarding the fitting of the Mg 2p component is not valid. It is however a well known phase with AP values for comparison readily available from literature. The AP for MgO shows good consistency across the data set for all but two measurements (M9 and M17). M9 is the first of the DP measurements, suggesting that the analyzed area was still affected by the surface of the film at this sputtering depth, e.g., possible alloying with Pd. M17 is an ON next day measurement, which showed extensive formation of hydroxide possibly affecting the position of the non-metallic Mg 2p peak. Disregarding these two anomalies, the AP was found to be 1231.25 eV with a standard deviation of 0.02 eV. This is consistent with AP ¼ 1231.2 eV found for MgO by Diplas et al. [48] and Friederichs et al. [22] The AP for the IH peak was found to be 1234.60 eV with a standard deviation of 0.07 eV, still disregarding M9 and M17. For the MgeH samples (M18 and M19) only one AP was calculated, relating the non-metallic Mg 2p peak to the Mg KLL peak with expected overlapping contribution from MgH2 and MgO. This AP shows a significant deviation from the AP of MgO found above. The AP for M18 was 1232.1 eV, while the AP for the next day measurement (M19) with hydroxide formation had decreased slightly to Fig. 11 e Mg 2p-KLL Auger parameter values (AP) of the hydrogenated samples with standard deviations. The measurement numbers refer to Table 1. 10711 1231.9 eV. Variations in O 1s positions in the data set (e.g., Figs. 7 and 14) indicate a certain effect of sample charging, making it difficult to reliably determine exact positions for the individual oxide and hydride peaks. The AP values however, are not affected by charging and thus the present result indicates that it should be possible to separate the oxide and hydride phase of Mg in a dedicated XPS experiment. The unhydrogenated samples have AP values for MgO similar to the MgeTieH samples, but the low oxygen content makes it hard to determine the exact position of the non-metallic Mg 2p peak in each spectrum. The Mg 1s-KLL AP values corresponding to the dominant peak in the Mg 1s spectra were found to be 2488.4 eV and 2485.7 eV for MgeTieH (M15) and MgeH (M18), respectively. The overall trend in shifts between MgO, MgH2 and the phase responsible for the IH peak corresponds very well with the Mg 2p-KLL AP results. 3.5. The origin of the IH peak If Mg and Ti do not interact to any large extent one would expect MgH2 and TiH2 to be formed more or less independently, but given the difference between the spectrum c and d in Fig. 3 this is clearly not what we observe in our sample. As mentioned in Section 1, MgH2 forms a TiH2-like structure (face centered cubic rather than body centered tetragonal) for the composition of our sample. Could the difference in crystal structure account for the difference observed in the Mg KLL spectra? Probably not, as it is known from optical and electrical investigations that the band gap must be similar to that of conventional rutile MgH2 [15]. Thus one would not expect a shift of several eV in the Auger spectrum and it seems that fluorite MgH2 can be ruled out as a cause of the IH peak. Another option to consider is that the hydrogenated MgeTi samples undergo H desorption in the XPS instrument, for instance due to the initial Arþ sputtering, as mentioned also for MgeH in Section 3.2. From the Ti 2p spectrum a H:Ti ratio of about 1 was estimated, half of what is expected for fully loaded TiH2. Whether there is any hydrogen connected to Mg is more of an open question, since TiH2 is significantly more stable than MgH2 in bulk form [15]. As the Mg KLL peaks of MgO Fig. 12 e Ratio of the peak with contribution from MgO and/ or MgH2 relative to total amount of Mg in the Mg KLL spectrum. The measurement numbers refer to Table 1. 10712 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 Fig. 13 e Mg KLL spectra from MgeTieH taken immediately after Pd removal (M15, bottom)and after 24 h in UHV (M17, top). The components are labeled as follows: M [ metallic, O [ MgO, OH [ Mg(OH)2, IH and IIH [ see text. and MgH2 are not easily separated, there is no direct way to determine whether the latter is present in the hydrogenated MgeTi samples. Instead we look at the ratio of the peak with contribution from MgO and/or MgH2 relative to total amount of Mg in the Mg KLL spectrum, Fig. 12. We see that for the two MgeH measurements (M18 and M19) the ratio is dramatically increased, giving clear indications of the presence of conventional MgH2. In the MgeTieH case, however, nothing like that happens. In fact, if one compares the depth profile data with and without H (M3-M8 and M9-M14), which should be directly comparable, the MgO level is nearly the same. Thus, the content of conventional MgH2 in the MgeTieH samples seems to be very low, if any at all. We will return to this issue in Section 3.6. Fig. 13 shows what happens when a hydrogenated MgeTi sample is left without the Pd protection in UHV for 24 h (the ON experiments). The corresponding fitting parameters are given in Table 3. The metallic peak suffers a reduction of more than 50% in intensity while the Mg(OH)2 peak intensity increases drastically. The latter is supported also by a corresponding change in the O 1s spectrum, see Fig. 14. The decrease of the metallic and increase of the hydroxide peak is similar for the hydrogenated Mg sample (not shown). For comparison Fig. 14 also shows oxidation of unhydrogenated Mg after 24 h in UHV, where both the oxide and hydroxide components in the O 1s peak increase evenly. Thus the rapid growth of Mg(OH)2 in M17 gives strong indications of the presence of H in both the MgeH and the MgeTieH samples. Fig. 13 shows that the relative area of the IH peak decreases while IIH increases in vacuum. This leads us to conclude that IH must be related primarily to hydrogen while IIH is somehow related to the oxidation process. A possible explanation of the IH peak can be found in our previously published DFT work, where we investigated the introduction of H into a model with Ti nano-clusters in an Mg matrix [19]. It was found that a certain amount of H was likely to remain trapped in especially stable sites on the interface between Mg and the Ti clusters, even after dehydrogenation had been attempted. As already mentioned it could be that the hydrogenated MgeTi sample was actually desorbing H due to the Arþ sputtering and what was measured was the metallic rather than the hydride phase. In this case the unknown peak IH in Figs. 3 and 13 could be due to H trapped on the very stable interstitial sites on the interface between Mg and the Ti nanocluster. To match the experimental deviation from maximum M:H ratio, it was proposed from our calculation that sites with 6, 3 and 2 nearest neighbor Ti atoms would take part in the trapping, i.e., the sites within the Ti nano-clusters as well as the ones on the interface. Such a picture would fit well with our current experimental findings: In the metallic hcp structure H prefers to occupy octahedral voids, leading to a H:M ratio of 1. This is the ratio of H:Ti estimated from the Ti 2p spectrum. In the present work the theoretical charge distribution between Mg, Ti and H was investigated by performing Bader Table 3 e Fitting parameters for the spectra in Fig. 13. Peak Mg met. IH IIH MgOc Mg(OH)2 Directly after Pd removal (M15) a After 24 h in UHV (M17) Position EK [eV] FWHM Rel. area [%] Position EK [eV] FWHMa Rel. area [%] 1185.9 1184.0 1182.1 1180.6 1179.3 0.8 2.1 2.0 1.5 2.0 31.7 36.9 16.3 10.3 4.8 1186.0 1183.9 1181.8 1180.7 1179.5 0.8 2.3 2.2 1.6b 2.4b 11.1 18.1 24.5 8.6 37.8 a The metallic peak was fitted with peak shape GL(99)T(1.2) and the non-metallic peaks with GL(50). Please refer to CasaXPS [24] for definition of peak shapes. b Upwards restricted. c Position restricted to 5.3 eV below metallic peak, based on data from non-hydrogenated samples. i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 10713 the Mg core hole screening efficiency caused by hydrogen trapped on especially stable interstitial sites at the interface between Mg and the Ti nano-clusters. 3.6. Fig. 14 e O 1s spectra from MgeTieH (left, M15 and M17) and unhydrogenated Mg (right, M20 and M21) taken immediately after Pd removal (bottom) and after 24 h in UHV (top). analysis [31] on the DFT results obtained in our previous work [19]. Fig. 15 shows the calculated average number of valence electrons per atom as function of hydrogen content x, as obtained by Bader analysis. Here it is clearly seen how Mg loose electrons compared to the elemental state, regardless of the H content. The average number of remaining valence electrons per Mg is found to be w1.5, which is less than the elemental 2 electrons, but more than the ionic MgO state without remaining valence electrons on the Mg sites. Thus the Bader analysis supports that introduction of H to MgeTi would result in a peak situated between the metal and the oxide component in the Mg KLL spectrum. It is also consistent with the AP values presented in Section 3.4. The AP of metallic Mg and MgO are separated by several eV, reflecting the substantially reduced core hole screening on Mg sites in the ionic oxide. For MgH2 the Mg AP is close to that of MgO, while the AP calculated for the IH phase is much closer to the metallic value. In view of our theoretical findings we think this is due to a small reduction of The origin of the IIH peak The IIH peak remains a topic for discussion. The region between IH and MgO is very broad, without distinct features, not unlike the IIIH peak. For simplicity it has been fitted with just one component (IIH), but there might be more than one, with under-stoichiometric MgHx<2 as a likely candidate. From the ON experiment, however, we found IIH to increase upon oxygen exposure, thus pointing to the involvement of oxygen. One possibility is that oxygen connects to understoichiometric MgHx<2 to produce under-stoichiometric Mg(OH)2. Additionally, the strong affinity of H for the sites on the interface between the Mg matrix and the Ti nanoclusters could perhaps be extended to O as well. In the hydrogenation process O could move to the interface between Mg and Ti and interact with H to result in a hydroxide-like MgeTieOeH constellation. This would also explain the increased oxygen levels seen in the Ti 2p spectra of the MgeTi samples after hydrogenation, giving different peak positions depending on the site occupied by O. However, such claims would have to be investigated further both experimentally and by DFT calculations. 4. Conclusions In this work thin films of Mg80Ti20 were studied by XPS before and after hydrogen loading. No significant trace of conventional MgH2 was found. Instead new, previously unknown peaks were revealed between the position of the metal and the oxide in the Mg KLL spectrum. From the shift in the Ti 2p spectrum the H:Ti ratio appeared to be 1 rather than 2, which would have been the fully loaded state. It was concluded that most probably the sample had undergone hydrogen desorption due to the Arþ sputtering prior to XPS measurement. However, the presence of remaining H was evident from over night exposure-experiments in the analysis chamber, where oxygen increase was seen primarily in the form of Mg(OH)2. The most prominent new peak in the Mg KLL spectrum (IH) was proposed to be due to H trapped in especially stable interstitial sites at the interface between Ti nano-clusters and the Mg matrix. Such stable H sites have previously been predicted both experimentally [20] and theoretically [19]. An additional peak (IIH) in the Mg KLL spectrum was found to increase upon oxygen exposure. This peak was suggested to be due to MgeTi hydroxide formation. references Fig. 15 e Results from Bader analysis of Mg81.25Ti18.75Hx. Average number of valence electrons per atom as function of hydrogen content x. Horizontal lines give values for elemental Ti, Mg and H; 4, 2 end 1 valence electrons respectively. [1] Huiberts JN, Griessen R, Rector JH, Wijngaarden RJ, Dekker JP, de Groot DG, et al. Yttrium and lanthanum hydride films with switchable optical properties. Nature 1996;380:231e4. [2] van der Sluis P, Ouwerkerk M, Duine PA. Optical switches based on magnesium lanthanide alloy hydrides. Appl Phys Lett 1997;70(25):3356e8. 10714 i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 [3] Richardson TJ, Slack JL, Armitage RD, Kostecki R, Farangis B, Rubin MD. Switchable mirrors based on nickel-magnesium films. Appl Phys Lett 2001;78(20):3047e9. [4] Lohstroh W, Westerwaal RJ, Noheda B, Enache S, Giebels IAME, Dam B, et al. Self-organized layered hydrogenation in black Mg2NiHx switchable mirrors. Phys Rev Lett 2004;93(19):197404. [5] Giebels IAME, Isidorsson J, Griessen R. Highly absorbing black Mg and rare-earth-Mg switchable mirrors. Phys Rev B 2004;69:205111. [6] Borsa DM, Baldi A, Pasturel M, Schreuders H, Dam B, Griessen R, et al. Mg-Ti-H thin films for smart solar collectors. Appl Phys Lett 2006;88:241910. [7] Anders A, Slack JL, Richardson TJ. Electrochromically switched, gas-reservoir metal hydride devices with application to energy-efficient windows. Thin Solid Films 2008;517:1021e6. [8] Pasturel M, Slaman M, Borsa DM, Schreuders H, Dam B, Griessen R. Stabilized switchable “black state” in Mg2NiH4/Ti/ Pd thin films for optical hydrogen sensing. Appl Phys Lett 2006;89:021913. [9] Karazhanov SZ, Ulyashin AG, Vajeeston P, Ravindran P. Hydrides as materials for semiconductor electronics. Philos Mag 2008;88(16):2461e76. [10] Niessen RAH, Notten PHL. Electrochemical hydrogen storage characteristics of thin film Mg (X, X ¼ Sc, Ti, V, Cr) compounds. Solid-State Lett 2005;8:A534e8. [11] Vermeulen P, Niessen RAH, Notten PHL. Hydrogen storage in metastable MgyTi1y thin films. Electrochem Commun 2006;8:27e32. [12] Gonzalez-Silveira M, Gremaud R, Baldi A, Schreuders H, Dam B, Griessen R. Effect of H-induced microcstructural changes on pressure-optical transmission isotherms for Mg-V thin films. Int J Hydrogen Energy 2010;35:6959e70. [13] Di Vece M, Zevenhuizen SJM, Kelly JJ. Optical switching properties from isotherms of Gd and GdMg hydride mirrors. Appl Phys Lett 2002;81:1213. [14] Nagengast DG, van Gogh ATM, Kooij ES, Dam B, Griessen R. Contrast enhancement of rare-earth switchable mirrors through microscopic shutter effect. Appl Phys Lett 1999;75:2050e2. [15] Borsa DM, Gremaud R, Baldi A, Schreuders H, Rector JH, Kooi B, et al. Structural, optical and electrical properties of MgyTi1yHx thin films. Phys Rev B 2007;75:205408. [16] Gremaud R, Baldi A, Gonzalez-Silveira M, Dam B, Griessen R. Chemical short-range order and lattice deformations in MgyTi1yHx thin films probed by hydrogenography. Phys Rev B 2008;77:144204. [17] Baldi A, Gremaud R, Borsa DM, Baldè CP, van der Eerden AMJ, Kruijtzer GL, et al. Nanoscale composition modulations in MgyTi1yHx thin film alloys for hydrogen storage. Int J Hydrogen Energy 2009;34:1450e7. [18] Jensen IJT, Diplas S, Løvvik OM. Density functional calculations of Ti nanoclusters in the metastable Mg-Ti system. Phys Rev B 2010;82:174121. [19] Jensen IJT, Diplas S, Løvvik OM. Hydrogen induced stabilization of meta-stable Mg-Ti. Appl Phys Lett 2012;100:111902. [20] Vermeulen P, Wondergem HJ, Graat PCJ, Borsa DM, Schreuders H, Dam B, et al. In situ electrochemical XRD study of (de)hydrogenation of MgyTi100y thin films. J Mater Chem 2008;18:3680e7. [21] Vermeulen P, Graat PCJ, Wondergem HJ, Notten PHL. Crystal structures of MgyTi100y thin film alloys in the as-deposited and hydrogenated state. Int J Hydrogen Energy 2008;33:5646e50. [22] Friedrichs O, Sánchez-López JC, López-Cartes C, Dornheim M, Klassen T, Fernández A. Chemical and [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] microstructural study of the oxygen passivation behaviour of nanocrystalline Mg and MgH2. Appl Surf Sci 2006;252:2334e45. Mitchell T, Diplas S, Tsakiropoulos P, Watts JF, Matthew JAD. Study of alloying behaviour in metastable Mg-Ti solid solutions using Auger parameter measurements and chargetransfer calculations. Philos Mag A 2002;82(4):841e55. http://www.casaxps.com; 2012. Shirley DA. High-resolution x-ray photoemission spectrum of the valence bands of gold. Phys Rev B 1972;5(12):4709e14. Ley L, McFeely FR, Kowalczyk SP, Jenkin JG, Shirley DA. Many-body effects in x-ray photoemission from magnesium. Phys Rev B 1975;11(2):600e12. Scofield JH. Hartree-Slater subshell photoionization crosssections at 1254 and 1487 eV. J Electron Spectrosc Relat Phenom 1976;8(2):129e37. Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B 1992;46:6671e87. Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 1996;54(16):11169e86. Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a planewave basis set. Comp Mater Sci 1996;6(1):15e50. Henkelman G, Arnaldsson A, Jónsson H. A fast and robust algorithm for Bader decomposition of charge density. Comput Mater Sci 2006;36:254e360. Van Attekum PMTM, Trooster JM. A plasmon gain satellite in the KLL Auger spectrum of Mg and A1 metal. J Phys F Met Phys 1978;8(7):169e73. Carlson TA, Dres WB, Nyberg GL. Study of KLL Auger processes for light elements above Z ¼ 10. Phys Scr 1977;16:211e6. Steiner P, Reitner FJ, Höchst H, Hüfner S. The KLL Auger spectra of Na and Mg metal and their plasmon structure. Phys Stat Sol B 1978;90:45e51. Slack JL, Locke JC, Song SW, Ona J, Richardson TJ. Metal hydride switchable mirrors: factors influencing dynamic range and stability. Sol Energy Mater Sol Cells 2006;90:485e90. Eijt SWH, Kind R, Singh S, Schut H, Legerstee WJ, Hendrikx RWA, et al. Positron depth profiling of the structural and electronic structure transformations of hydrogenated Mg-based thin films. J Appl Phys 2009;105:043514. Tanuma S, Powell CJ, Penn DR. IMFP values determined from optical data and a theoretical model. Surf Interface Anal 1991;17:911. Göpel W, Anderson JA, Frankel D, Jaehnig M, Phillips K, Schäfer JA, et al. Surface defects of TiO2(110): a combined XPS, XAES and ELS study. Surf Sci 1984;139:333e46. Wollschläger J, Viernow J, Tegenkamp C, Erdös D, Schröder KM, Pfnür H. Stoichiometry and morphology of MgO films grown reactively on Ag(100). Appl Surf Sci 1999;142:129e34. González-Elipe AR, Munuera G, Espinos JP. Compositional changes induced by 3.5 keV Arþ ion bombardment in Ni-Ti oxide systems. A comparative study. Surf Sci 1989;220:368e80. Kurth M, Graat PCJ, Carstanjen HD, Mittemeijer EJ. The initial oxidation of magnesium: an in situ study with XPS, HERDA and ellipsometry. Surf Interface Anal 2006;38:931e40. Lamartine BC, Haas TW, Solomon JS. Characterization of TiHx and TiD0.9 surfaces: AES, ELS, SIMS and XPS studies. Appl Surf Sci 1980;4:537e55. Biesinger MC, Lau LWM, Gerson AR, Smart RSC. Resolving surface chemical states in XPS analysis of first row transition i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 8 ( 2 0 1 3 ) 1 0 7 0 4 e1 0 7 1 5 metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl Surf Sci 2010;257:887e98. n anský M. Elastic electron [44] Lesiak B, Zemek J, Jiricek P, Cer backscattering from Ti: grain size effect. Surf Interface Anal 2004;36:816e9. [45] Lisowski W, Keim EG, van der Berg AHJ, Smithers MA. Structural and chemical characterisation of titanium deuteride films covered by nanoscale evaporated palladium layers. Anal Bioanal Chem 2006;385:700e7. 10715 [46] Kang XD, Wang P, Cheng HM. Improving hydrogen storage performance of NaAlH4 by novel two-step milling method. J Phys Chem C 2007;111:4879e84. [47] Fuggle JC. XPS, UPS and XAES studies of oxygen adsorption on polycrystalline Mg at 100 and 300 K. Surf Sci 1977;69:581e608. [48] Diplas S, Tsakiropoulos P, Brydson RMD, Watts JF. Development of Mg-V alloys by physical vapour deposition. Part 2 e characterisation of corrosion products formed in 3 wt% NaCl. Mater Sci Technol 1998;14:699e711.