This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike License. Your use of this
material constitutes acceptance of that license and the conditions of use of materials on this site.
Copyright 2006, The Johns Hopkins University and Rafael A. Irizarry. All rights reserved. Use of these materials
permitted only in accordance with license rights granted. Materials provided “AS IS”; no representations or
warranties provided. User assumes all responsibility for use, and all liability related thereto, and must independently
review all materials for accuracy and efficacy. May contain materials owned by others. User is responsible for
obtaining permissions for use from third parties as needed.
BIOINFORMATICS AND COMPUTATIONAL
BIOLOGY SOLUTIONS USING R AND
BIOCONDUCTOR
Biostatistics 140.688
Rafael A. Irizarry
Preprocessing Affymetrix
GeneChip Data
Credit for some of today’s materials:
Ben Bolstad, Leslie Cope, Laurent
Gautier, Terry Speed and Zhijin Wu
Expression
1
Affymetrix GeneChip Design
5’
3’
Reference sequence
…TGTGATGGTGGGGAATGGGTCAGAAGGCCTCCGATGCGCCGATTGAGAAT…
CCCTTACCCAGTCTTCCGGAGGCTA Perfectmatch
CCCTTACCCAGTGTTCCGGAGGCTA Mismatch
NSB & SB
NSB
Terminology
•
Each gene or portion of a gene is represented by 1q to 20
oligonucleotides of 25 base-pairs.
•
Reporter/Feautre/Probe: an oligonucleotide of 25 base-pairs,
i.e., a 25-mer.
Perfect match (PM): A 25-mer complementary to a reference
sequence of interest (e.g., part of a gene).
Mismatch (MM): same as PM but with a single homomeric base
change for the middle (13th) base (transversion purine <->
pyrimidine, G <->C, A <->T) .
Probe-pair: a (PM,MM) pair.
Probe-pair set: a collection of probe-pairs (1q to 20) related to
a common gene or fraction of a gene.
Affy ID: an identifier for a probe-pair set.
The purpose of the MM probe design is to measure nonspecific binding and background noise.
•
•
•
•
•
•
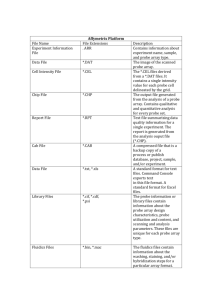
Affymetrix files
• Main software from Affymetrix company
MicroArray Suite - MAS, now version 5.
• DAT file: Image file, ~10^7 pixels, ~50 MB.
• CEL file: Cell intensity file, probe level PM and
MM values.
• CDF file: Chip Description File. Describes
which probes go in which probe sets and the
location of probe-pair sets (genes, gene
fragments, ESTs).
2
Expression Measures
• 10-20K genes represented by 11-20
pairs of probe intensities (PM & MM)
• Obtain expression measure for each
gene on each array by summarizing
these pairs
• We already discussed background
adjustment and normalization. We
assume this has been done.
• There are many methods
Data and notation
• PMijg , MMijg = Intensity for perfect match and
mismatch probe in cell j for gene g in chip i.
– i = 1,…, n -- from one to hundreds of chips;
– j = 1,…, J -- usually 11 or 20 probe pairs;
– g = 1,…, G -- between 8,000 and 20,000 probe sets.
• Task: summarize for each probe set the probe level
data, i.e., PM and MM pairs, into a single expression
measure.
• Expression measures may then be compared within
or between chips for detecting differential expression.
MAS 4.0
• GeneChip® MAS 4.0 software used AvDiff up
until 2001
AvDiff =
1
"
% (PM
j
# MM j )
j $"
where A is a set of “suitable” pairs, e.g., pairs with d j = PMj -MMj
within 3 SDs of the average of d(2) , …, d(J-1)
• Obvious
problems:
!
– Negative values
– No log scale
3
Why use log?
Original scale
Log scale
Li and Wong’
Wong’s observations
• There is a large probe effect
• There are outliers that are only noticed
when looking across arrays
• Non- linear normalization needed
(discussed in previous lecture)
PNAS vol. 98. no. 1, 31-36
Probe effect
4
Probe effect makes correlation deceiving
Correlation for absolute expression of replicates
looks great! But…
Probe effect makes correlation deceiving
•It is better to look at relative expression because
probe effect is somewhat cancelled out.
•Later we will see that we can take advantage of
probe effect to find outlier probes.
Li & Wong
• Li & Wong (2001) fit a model for each probe set, i.e.,
gene
PM ij " MM ij = # i$ j + %ij , %ij & N(0,' 2 )
where
– θi: model based expression index (MBEI),
– φj: probe sensitivity index.
•! Maximun likelihood estimate of MBEI is used as
expression measure for the gene in chip i .
• Non-linear normalization used
• Ad-hoc procedure used to remove outliers
• Need at least 10 or 20 chips
5
There is one more reason why
PM-MM is undesirable
Especially for large PM
log2PM-log2MM
We see bimodality
(log2PM+log2MM)/2
6
Two more problems with MM
• MM detect signal
• MM cost $$$
MAS 5.0
• Current version, MAS 5.0, uses Signal
signal = Tukey Biweight{log(PM j " MM *j )}
• Notice now log is used
• But what about negative PM-MM ?
!
•
•
•
•
MM* is a new version of MM that is never larger than PM.
If MM < PM, MM* = MM.
If MM >= PM,
– SB = Tukey Biweight (log(PM)-log(MM))
(log-ratio).
– log(MM*) = log(PM)-log(max(SB, +ve)).
Tukey Biweight: B(x) = (1 – (x/c)^2)^2 if |x|<c, 0 ow.
Can this be improved?
Rank of
Spikeins
(out of
12626)
We will discuss P/M/A calls later
141
250
364
368
480
586
686
838
945
1153
1567
NA
NA
NA
NA
7
MBEI not much better
RMA
•
•
•
Robust regression method to estimate expression measure and
SE from PM* (background adjusted normalized PM)
Use quantile normalization
Assume additive model
log 2 (PM ij* ) = ai + b j + "ij
•
•
•
Estimate RMA = ai for chip i using robust method, such as
median polish (fit iteratively, successively removing row and
column medians, and accumulating the terms, until the process
stabilizes).
!
Works with n=2 or more chips
This is a robust multi-array analysis (RMA)
Can this be improved?
Rank of
Spikeins
(out of
12626)
141
250
364
368
480
586
686
838
945
1153
1567
NA
NA
NA
NA
8
RMA
Rank of
Spikeins
(out of
12626)
1
2
3
4
7
11
15
21
35
122
1182
230
450
1380
11700
Irizarry et al. (2003) NAR 31:e15
QC from probe level models
•
•
•
•
•
RMA fits a probe level model
From these fits we can obtain residuals
We can also get weights if we use formal robust regression
procedures instead of median polish
These probe-level residuals and summaries of their size can be
used for quality control
Software available: affyPLM Bioconductor package (Ben
Bolstad)
Images of probe level data
This is the raw data
9
Images of probe level data
Log scale version much more informative
Images of probe level data
Residuals (or weights) from probe level
model fits show problem clearly
Images of probe level data
Here is a more subtle artifact. Can you see it?
The strong probe effect does not let you.
10
Images of probe level data
Probe level fit residuals really show it
Other pseudo-chip images
Weights
Residuals
Positive
Residuals
Negative
Residuals
NUSE
Normalized
Unscaled
Standard
Errors
11
Can RMA be improved?
RMA attenuates
signal slightly to
achieve gains in
precision
method slope
MAS 5.0 0.69
RMA
0.61
More on this later (if time permits)
Detection
Detection
• The detection problem:
“Given the probe-level data, which mRNA
transcripts are present in the sample?”
• Biologists are mostly interested in expression levels,
and so detection has received less attention
• To date only Affymetrix has tackled this, with
– Rank-based tests
– Implemented in MAS5.0
12
MAS Rank-based Detection
The test used in MAS 5.0 compares the following two hypotheses
H0 : median (PMj - MMj)/(PM j + MMj) = τ ;
H1 : median (PMj - MMj)/(PM j + MMj) > τ.
Significance levels: 0 < α1 < α 2 < 0.5. If p is the p-value for the (rank)
test, MAS 5.0 calls a transcript
absent: if p > α 2 ,
marginal: if α 1 ≤ p ≤ α 2 , and
present: if p < α 1.
Typically tests are carried out with τ = 0.15, α 1 = .04 and α 2 = .06.
Expression Detection
MAS 5.0
Remember uncertainty
• Some data analysts remove probesets
called absent from further analysis
• This creates false negatives:
HG95
Present
Absent
P
M
82% 1%
0%
0%
A
17%
100%
HGU133 P
M
Present 77% 3%
Absent 0%
0%
A
20%
100%
From spike-in experiments
13
Consistency across reps
Consistency across reps
Current work
• We need better estimates of means and
variances of bivariate normal
background noise
• Use observed MM intensities along with
sequence information
• We also have a solution that does not
use the MM
14
Predict NSB with sequence
• Fit simple linear
model to yeast on
human data to
obtain base/position
effects
Predict NSB with sequence
• Fit simple linear
model to yeast on
human data to
obtain base/position
effects
• Call these affinities
and use them to
obtain parameters
for background
model
Also explains MM thing
15
Also explains MM thing
Does it help?
• We can predict
empirical results
with model
• Accuracy of
expression
measures
improves…
Does it help?
•
•
•
We can predict
empirical results with
model
Accuracy of expression
measures improves…
Without adding too
much variance
16
Alternative background adjustment
•
•
Use this stochastic model
Minimize the MSE:
.( " s˜ %+ 2
1
E 0)log$ ', S > 0,PM, MM 3
0/* # s &32
•
•
!
To do this we need to specify distributions for the different
components
Notice this is probe-specific so we need to borrow strength
!
*These
parametric distributions were chosen to provide a closed form solution
Good example
Not always pretty
17
Problems similar to
expression arrays?
• Background
• Normalization
• Probe effect
• Outliers
Probe effect for SNPs
Background problem
18
Need for normalization
Need for Normalization
19
Length effect (PCR?)
Sequence effect
Sequence effect
20
Target Related Tasks
• Genotyping: AA, AB, BB?
– Current approaches use likelihood models,
clustering, and classification (if we know
truth for some)
• Copy number estimation
• LOH
21