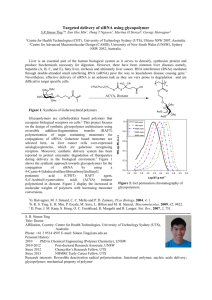
Document 11206699
advertisement

Strategies for Combinatorial Development of siRNA Conjugate Delivery Systems 0z C\J Q LL = -U By Rosemary L. Kanasty Submitted to the Department of Chemical Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering at the Massachusetts Institute of Technology June 2015 2015 Massachusetts Institute of Technology All rights reserved. Signature of Au thor...... Sicinature redacted ..Rosemary L. Kanasty Department of Chemical Engineering &Certified by...... Signature red acted...Rbr .Lne .......................... Robert S. Langer David H4 Koch (1962?) Instituten Professor S. SSignature redacted Certified by......... ............ v........ ......... .... Thesis Supervisor T ..................... Daniel G. Anderson Samuel A. Goldblith Professor of Applied Biology, Chemical Engineering and Health Sciences & Technology Thesis Supervisor Signature redacted ..................... Accepted by ....... ,V......... . ., ............................... Richard D. Braatz <ofessor of Chemical Engineering Chairman, Committee for Graduate Students 02 z Strategies for Combinatorial Development of siRNA Conjugate Delivery Systems By Rosemary Lynn Kanasty Abstract RNA interference (RNAi), which can reversibly silence the expression of any gene, has vast potential as a therapeutic to treat many diseases. A wide variety of small interfering RNA (siRNA) delivery systems has been studied, including lipid nanoparticles (LNPs) and small molecule conjugates. LNPs are among the most potent and diverse nonviral delivery systems, and they have enjoyed continued improvement in efficacy as new generations of lipids have been explored. This steady progress in lipid performance has relied heavily on studies using combinatorial synthesis and high-throughput screening of large libraries to discover novel delivery materials. Conjugate delivery systems are attractive for their purity, well-defined structures, low ratios of delivery material to siRNA, and potentially broad therapeutic windows, yet relatively few efficacious conjugates are reported in the literature. As synthetic challenges prevent the study of large conjugate libraries, the development of conjugate systems has relied on individual synthesis of new materials. There is currently a need for new directions in the development of conjugate delivery systems, which demands methods to enable high-throughput screening of conjugate libraries. Here we present a viable synthetic strategy for creating combinatorial libraries of hundreds of siRNA conjugate delivery materials. We first demonstrate the synthesis of novel sequence-defined polymers that can incorporate a broad diversity of chemical properties relevant to delivery into oligomers with high purity. We develop methods of synthesis, purification, and siRNA conjugation that are translatable to high-throughput synthesis in multiwell plates. We then demonstrate the high-throughput capability of this method by synthesizing a library of over 500 novel siRNA conjugate materials. We explore applications of these novel conjugates as potential delivery materials in cell-based screens. We also develop strategies for conjugating materials to siRNA multivalently, which may improve delivery. This work enables the synthesis of hundreds of novel conjugate delivery materials in parallel. The study of these combinatorial libraries has the potential to open vast new avenues in the development of therapeutic siRNA conjugates. 2 Table of Contents A B ST R A CT ......................................................................................................................................... 2 A CK N O W LED G EM EN T S ................................................................................................................. 6 CH A PT ER 1:BA CK G R O U N D ......................................................................................................... 7 1.1 M OTIVATION FOR siRN A THERAPEUTICS ............................................................................................. 7 1.2 CHALLENGES OF IN VIVO siRN A DELIVERY ........................................................................................... 7 1.2.1 Pharmacodynamic considerations............................................................................................. 8 1.2.1.1 RNA structure and Gene Silencing ...................................................................................................................... 8 1.2.1.2 Im m unostim ulation ............................................................................................................................................... 10 1.2.1.3 Off-target gene silencing ...................................................................................................................................... 14 1.2.1.4 Saturation of the RNAi m achinery ................................................................................................................... 16 1.2.1.5 Delivery m aterial toxicity .................................................................................................................................... 17 1.2.2 Pharmacokinetic considerations.............................................................................................. 19 1.2.2.1 siRNA in circulation ................................................................................................................................................20 1.2.2.2 Extravasation ............................................................................................................................................................21 1.2.2.3 Cell uptake ..................................................................................................................................................................23 1.2.2.4 Elim ination .................................................................................................................................................................26 1.2.3 Conclusions.........................................................................................................................................27 1.3 STRATEGIES FOR siRN A DELIVERY ...................................................................................................... 28 1.3.1 Cy clodextrin polym er nanoparticles....................................................................................... 30 1.3.2 Lipid nanoparticles.........................................................................................................................31 1.3.2.1 Cationic and ionizable lipids .............................................................................................................................. 32 1.3.2.2 Shielding lipid ........................................................................................................................................................... 34 1.3.2.3 Cholesterol ................................................................................................................................................................. 35 1.3.2.4 Targeting Ligands ................................................................................................................................................... 35 1.3.3 Conjugate delivery system s ......................................................................................................... 36 1.3.3.1 Dynam ic PolyConjugates ..................................................................................................................................... 36 3 1.3.3.2 T rianten nary G al N A c-siR N A ............................................................................................................................... 39 1.3.3.3 O ligon ucleotid e N an op articles .......................................................................................................................... 40 1 .3.4 Co n clusio ns ......................................................................................................................................... 1 .4 T H E SIS G OA LS............................................................................................................................................ 41 43 CHAPTER 2: SEQUENCE-DEFINED OLIGOMERIC MATERIALS FOR SYNTHESIS OF NUCLEIC ACID CONJUGATE LIBRARIES ................................................................................... 44 2 .1 IN TRO D U C TIO N ........................................................................................................................................ 44 2.2: POTENTIAL CHEMICAL STRATEGIES FOR SYNTHESIZING CONJUGATE LIBRARIES .................. 45 2 .2.1 In trod u ction ....................................................................................................................................... 45 2.2.2 DegradableEster-linked Oligomers................................................................................. 46 2.2.3 Oligocarbamatesbased on 2-amino 4-hydroxybutyric acid.................................... 48 2.2.4 Oligocarbamatesbased on 4-amino 3-hydroxybutyric acid..................................... 49 2.2.5 Oligocarbamatesbased on 4-amino 2-hydroxybutyric acid..................................... 51 2.2.6 Oligocarbamatesbased on hydroxyproline................................................................... 54 2.3 HYDROXYPROLINE-BASED OLIGOCARBAMATES PROVIDE A ROBUST PLATFORM FOR THE SYNTHESIS OF SIRNA CONJUGATE LIBRARIES ............................................................................................... 57 CHAPTER 3. HIGH-THROUGHPUT SYNTHESIS OF A HYDROXYPROLINE OLIGOMERSIRNA CONJUGATE LIBRARY............................................................................................................65 3 .1 IN T R O D U CTIO N ........................................................................................................................................ 65 3.2 HIGIH-THROIJGHPUT SYNTIESIS OF IYDROXYPROLINE-BASED OLIGOCARBAMATES............ 66 3.3 CONJUGATION OF OLIGOCARBAMATES TO SIRNA AND CONJUGATE PURIFICATION................ 70 3.4 CONCLUSIONS AND RECOMMENDATIONS....................................................................................... 71 CHAPTER 4: POTENTIAL APPLICATIONS OF HYDROXYPROLINE-BASED OLIGOCARBAMATES ............................................................................................................................73 4 .1 IN T R O D U C T IO N ........................................................................................................................................ 73 4 4.2 LIBRARY SCREENING IN DUAL HELA CELLS .................................................................................... 74 4.2 LIBRARY SCREENING IN K B CELLS .................................................................................................... 76 4.3 81 HYDROXYPROLINE OLIGOMERS AS LIPID-LIKE NANOPARTICLE DELIVERY SYSTEMS............. 4.4. CONJUGATE MODIFICATIONS AS ENHANCERS IN OTHER DELIVERY SYSTEMS.........................85 4.5. RECOMMENDATIONS FOR FUTURE IN VITRO SCREENS ............................................................... 88 CHAPTER 5. STRATEGIES FOR MULTIVALENT CONJUGATION TO SIRNA.............90 CHAPTER 6. CONCLUSIONS....................................................................................................94 APPENDIX 1. ABBREVIATIONS AND SYMBOLS ............................................................... 96 APPENDIX 2. MATERIALS AND METHODS.......................................................................97 A PPEN DIX 2.1 LIST OF M ATERIALS ............................................................................................................. 97 A PPEN DIX 2 .2 M ETH OD S .............................................................................................................................. 97 APPENDIX 3. SUPPLEMENTARY DATA ............................................................................... 104 APPENDIX 3.1 'H NM R AND 13C N M R SHIFTS ...................................................................................... 104 APPENDIX 3.2 LCMS ANALYSIS OF TRIMER AND HEXAMER SYNTHESIS ............................................ 110 APPENDIX 3.3 HIGH-THROUGHPUT PURIFICATION OF SIRNA CONJUGATES ..................................... 122 APPENDIX 3.4 HPLC ANALYSIS OF SIRNA CONJUGATES ...................................................................... 123 APPENDIX 3.5 SYNTHESIS OF MULTIVALENT DBCO AND BCN LINKERS .......................................... 125 APPENDIX 4. REFERENCES ..................................................................................................... 128 5 Acknowledgements I would like to thank my thesis advisors, Prof. Bob Langer and Prof. Dan Anderson, for their unwavering support, positivity, and their fostering of a laboratory culture that inspires openness and imagination. Bob is both a considerate mentor and an inspiring role model who is very deserving of the widespread respect and admiration he has garnered. Dan's guidance, along with his creativity, ambition, and penchant for big ideas has motivated me and made me a better scientist. I am grateful to have enjoyed the support of a thesis committee that offers unparalleled scientific expertise, consistent encouragement, and exemplary personal mentorship. For this I would like to thank Prof. Sangeeta Bhatia and Prof. Dane Wittrup. I am extremely fortunate to have had the daily guidance of Dr. Arturo Vegas, who has generously contributed his time to training me throughout my graduate career. I have him to thank not only for my laboratory skill set, but for years of scientific guidance, experimental troubleshooting, professional mentorship, and emotional support through all of challenges of graduate school. Among the many brilliant scientists I've met in the Langer and Anderson labs, I would like to especially thank Luke Ceo, Owen Fenton, Omar Khan, Ben Tang, Karsten Olejnik, Omid Veiseh, Kevin Kauffman, Hok Hei Tam, Abel Cortinas, Robert Dorkin, Abigail LyttonJean, Katie Whitehead, Kevin Love, Ana Jaklenec, Josh Doloff, Chris Alabi, Leon Bellan, H6loise Ragelle, Eunha Kim, Minglin Ma, Chris Levins, Eddie Eltoukhy, David Nguyen, Pam Basto, Eric Pridgen, Aleks Radovic-Moreno. Yizhou Dong, Weiheng Wang, Guarav Sahay, Hao Yin, Patrick Fenton, Beata Chertok, and Avi Schroeder. I would also like to thank our collaborators at Alnylam: Klaus Charisse, Martin Maier, Don Foster, and Stuart Milstein. For funding I owe my gratitude to the National Science Foundation, Alnylam Pharmaceuticals, and the MIT Skoltech Initiative. I must also thank my parents for enthusiasm for science and engineering and for encouraging me as I pursued my goals. Finally, I would like to thank my friends and family for their support, confidence, patience, and comfort over the last several years. 6 Chapter 1: Background Reproduced with permission from: Kanasty et al., Molecular Therapy, 20121; Kanasty et al., Nature Materials, 20132; and Yin et al., Nature Reviews Genetics, 20143. 1.1 Motivation for siRNA therapeutics Since the discovery of RNA interference (RNAi) in mammalian cells, there has been great interest in harnessing this pathway for the treatment of disease. RNAi is an endogenous pathway for post-transcriptional silencing of gene expression that is triggered by double-stranded RNA, including endogenous microRNA (miRNA) and synthetic short interfering RNA (siRNA). By activating this pathway, siRNAs can silence the expression of virtually any gene with high efficiency and specificity, including targets traditionally considered to be "undruggable." The therapeutic potential of this method is far-reaching, and siRNA-based therapeutics are under development for the treatment of diseases ranging from viral infections 4s to hereditary disorders 6 to cancers 7,8. Large amounts of effort and capital have been invested in bringing siRNA therapeutics to the market. At least 22 RNAibased drugs have entered clinical trials (Table 1), and many more are in the developmental pipeline. 1.2 Challenges of in vivo siRNA delivery siRNA molecules must be delivered intracellularly in order to trigger RNAi, but their large, anionic, and hydrophilic structure prevents them from diffusing across cell membranes to reach their site of action. A broad array of delivery materials has been developed to mediate delivery and to improve the overall pharmacokinetics of siRNA when administered systemically (reviewed elsewhere, see reference 9). While the design of siRNA delivery systems is usually focused on the immediate goal of delivering enough of the injected dose into enough target cells to generate a particular therapeutic effect, a broader awareness of the fate of the entire injected dose and its many other interactions with the body is essential in the development of safe and effective systems. siRNAs and their delivery vehicles can induce immunogenicity, toxicity, and off-target effects in addition to target gene silencing. Various factors can mediate the degradation, clearance, and cellular uptake of siRNA delivery systems, affecting their potency. This chapter presents a brief summary of important pharmacodynamic and pharmacokinetic considerations in siRNA delivery and intends to introduce the reader to the numerous interactions between the body and siRNA, but is not a comprehensive review of PD and PK data for various delivery systems. For more detailed information on particular topics, the reader is referred to reviews of more limited scope and of greater depth. 7 1.2.1 Pharmacodynamic considerations siRNA delivery systems can elicit both intended effects (e.g. target gene silencing) as well as unintended consequences, including immune stimulation, toxicity, and off-target silencing. The pharmacodynamics of siRNA delivery systems are dependent not only upon the siRNA therapeutic, but also upon the biomaterials included in the delivery vehicle. .- rxjfriva.j Pd III - sR A /0nmotm *\ av .e -BE=~A *- 'if andii / ci XeA KS oalang K jrd Perfect hybricabt*on Partia i,? rizANA to 3-1t-Rg I Vaarni .ciivmin fr ot1ple hyt1 up% rreRNA I tutin T,ge rtiOiNA siRNA pathway m~RNA pathway Figure 1-1. sIRNA and miRNA pathways. Exogenous double-stranded RNA dsRNA) introduced into the cytoplasm is cleaved by Dicer into 21-nt fragments with 2-nt overhangs on the 3' ends. The antisense strand, or guide strand, is loaded into the RNA-tnduced silencing complex and the sense or passenger strand is cleaved by Argonaute-2 Ago-2). The activated RISC-siRNA complex seeks out messenger RNA (mRNA) that hybridizes with the guide strand and cleaves this mRNA. The same silenang machinery particpates in gene silencing by endogenous miRNAs MiRNAs usually hybridize imperfectly with target mRNA. which leads to translational repression rather than cleavage of target mRNA. Off-target gene silencing by synthetic siRNA is thought to occur when the siRNA seed region is partialy complementary to 3'UTR sequences, causing siRNAs to enter the miRNA pathway 1.2.1.1 RNA structure and Gene Silencing 8 The central component of any siRNA formulation is the siRNA itself. The silencing of specific genes through RNA interference machinery (Figure 1.1) is the intended pharmacodynamic effect of siRNA. Over the past few years, a number of design criteria have been developed to improve the specificity and potency of RNA, some of which has been driven by an increased understanding of the biochemistry driving RNA-interference 10,11 When an siRNA molecule is delivered inside of a cell, it can be loaded into the RNAinduced silencing complex (RISC) (Figure 1.1). Argonaute 2 (Ago 2, Figure 1.1), a protein within RISC, unwinds the siRNA. The two strands of siRNA are referred to the passenger strand and the guide strand. The passenger strand is cleaved while the activated RISC-guide strand complex seeks out and cleaves mRNA that is complementary to the guide strand. The nucleotide sequence of the siRNA affects the efficiency of each of these steps and therefore affects the potency of gene silencing. A number of guidelines have emerged for selection of the most active siRNA sequences, and these principles have been incorporated into various siRNA design algorithms 12. The enhancement of siRNA asymmetry and minimization of secondary structure in mRNA target sites are both important considerations in sequence selection. When siRNA enters the RNAi pathway (Figure 1.1), one strand is loaded into the RISC complex to become the guide strand and its complementary strand is cleaved. If the incorrect strand is selected for RISC loading, the strand intended to be the guide strand is cleaved, leading to reduced potency of target gene silencing. Incorrect strand selection can also trigger the silencing of off-target genes complementary to the intended passenger strand. siRNA asymmetry refers to the preference for one strand to be loaded into RISC over the other. Increasing the asymmetry of the duplex helps to ensure that the desired strand is loaded into RISC to direct gene silencing. Strand selection by RISC is determined by the thermodynamic stability of the ends of the duplex: the 5' end with the lowest hybridization stability is loaded into RISC to become the guide strand 13-15. Thermodynamic asymmetry is increased by enriching the A-U base pair content in the 5' end of the antisense strand. Unlocked nucleic acids (UNA, Figure 1.4) destabilize duplexes and can enhance asymmetry when incorporated into the 5' end of the guide strand 16-18. Asymmetry can also be enhanced by modification of passenger strand ends. For siRNAs to be functional, the 5' end of the antisense strand must have a terminal phosphate 15. 5'-OH -terminated siRNAs are rapidly phosphorylated in the cytoplasm 19, but modification of the terminal hydroxyl can prevent phosphorylation and prevent RISC loading 20. Methylation of 9 the 5' end of the passenger strand is a common strategy to ensure proper strand selection by RISC 20. The presence of delivery materials can affect RISC loading. Delivery materials that complex with siRNA to form nanoparticles must dissociate from siRNA once inside the cell to allow RISC loading. For example, the cationic lipids of many lipoplex delivery systems undergo a phase transition in acidic environments such as endosomes. The conversion from a lamellar phase to hexagonal packing promotes both endosomal release and release of siRNA from the delivery material 21. In other systems, delivery materials are conjugated directly to the ends of siRNA strands. As the 5' end of the guide strand is essential for RISC loading, conjugation of delivery materials is usually avoided in this position. Delivery materials are usually conjugated to the 3' or 5' ends of the passenger strand, though conjugation to the 3' end of the guide strand can be effective as well 13,22,23. The level of accessibility of the mRNA target site also affects the potency of siRNA 14,24,25. Folding of mRNA can block access to the target site, as activated RISC cannot unfold secondary structures in mRNA 24. Secondary structure prediction algorithms can help to choose mRNA target sites with high accessibility 12,14. Self-folding of the siRNA guide strand can also impede target site recognition, and inverted repeats are often avoided in siRNA sequences 26,27. An example of a siRNA design algorithm that considers RNA secondary structures is the OligoWalk web server, which selects active siRNA sequences based on their free energy changes upon hybridization with target mRNA 12. The internal stability of the length of the duplex can also affect activity. A systematic analysis of a siRNA library determined that the most active sequences had a GC content of 36% - 52% 11. It has been proposed that higher GC content may inhibit RISC loading and release of the cleaved sense strand, while lower GC content may inhibit activity by weakening hybridization between the guide strand and mRNA 10. Several studies of siRNA sequences have confirmed a preference for certain nucleotides in specific positions along siRNA strands 11,28. Several of these positional preferences are consistent with the idea of thermodynamic asymmetry of siRNA ends: A or U is preferred at position 1 of the antisense strand, G or C is preferred at position 19, and AU-richness in positions 1 - 7 is favored. In addition, A or U is favored at position 10, the site of substrate cleavage by Ago2. 1.2.1.2 Immunostimulation 10 TLR-Indspondent Pathways TLR-Oependmnt Pathways WVAYO% SARNA 'I V cytos A A ME h I Figure 1-2 sMA invokes Ihmunmostmulation via pathogen recognitIn. receptor. TLR3 (red' exists on both the cell surface and in subcellular compartments of select cells. TLR7 (orange) and TLR8 (yellow) exist solely in the endosomes and lysosomes of specialized immune cells and recogruze siRNA in a sequencedependent manner. PKR, and RIG-1 are present in the cytoplasm of certain cell types and can detect and react to siRNA in a sequence-independent fashion. When activated, receptors trigger an immune signaling cascade that causes an increased transcription of mRNA that encodes Type I interferons and inflammatory cytokines. The cascade and resulting inflammatory response are unique for each receptor. siRNA can be recognizedby the innate immune system In addition to mediating RNA interference, siRNA molecules have the potential to induce the innate immune system 29. The interactions between siRNA and the immune system are complex and only an overview is provided here. For a detailed review, see reference 30. The process of immunological recognition and response to non-self nucleic acids such as siRNA is governed by the innate immune system. Innate immune responses are 11 characterized by an induction of small signaling molecules called cytokines, including interleukins 31, Type I interferons 32 and tumor necrosis factor alpha (TNF-a) 33. The stimulation of pattern recognition receptors (PRRs) triggers the innate immune response. PRRs recognize distinct pathogenic patterns that are not present on self-cells 34,35 Importantly for siRNA delivery, the innate immune system has evolved to include multiple ZPRRs that recognize different aspects of RNA structure, providing a redundancy that makes immunostimulation difficult to escape (Figure 1.2). Two general classes of siRNArecognizing PRRs include Toll-like receptors and cytoplasmic receptors 36. Toll-like receptors (TLRs) are a class of PRRs that recognize structurally conserved regions of foreign pathogens, and each member of the TLR family is responsible for the detection of varying pathogen-associated molecules. Of the 10 human TLRs and 12 murine TLRs that have been discovered to date 37, at least three have relevance to siRNA delivery. These include TLR3, which recognizes dsRNA, and TLR7 and TLR8, which recognize singlestranded RNA (ssRNA). TLR3 responds to dsRNA, which is typically a characteristic of viral replication found in lysed or apoptotic virally-infected cells 38. In humans, TLR3 is expressed in endosomes and on the cell surface of select cell populations. TLRs 7 and 8 are important PRRs that respond to ssRNA in a sequence-specific manner. They are located exclusively in the intracellular vesicles, including endosomes, lysosomes, and the endoplasmic reticulum of plasmacytoid dendritic cells, B cells, and myeloid cells 35. More specifically, TLR 7 is located primarily in the endosomes of plasmacytoid dendritic cells and TLR 8 in endosomes of myeloid cells 39. In addition to endosomal TLRs, several cytoplasmic receptors mediate immune responses to siRNA (Figure 1.2). PKR, a protein kinase that responds to dsRNA 40 and siRNA 35, can cause the inhibition of protein translation and an interferon response when activated 4 0. RIG-I, another cytoplasmic PRR expressed in fibroblasts and dendritic cells 41, can provoke a strong interferon response in the presence of various forms of siRNA 35. Several features of siRNA delivery systems influence the nature and degree of immunostimulation. These include the siRNA sequence, the siRNA structure and chemistry, and the materials used to construct the delivery vehicle 29. The sequence of siRNA influences immune stimulation through TLRs 7 and 8, which recognize ssRNA in a sequence-specific manner 42,43. Generally, siRNAs rich in GU-rich motifs tended to provoke more immunostimulatory activity while a decrease in the presence of uridine residues had the opposite effect. The substitution of certain residues within an RNA molecule has been 12 demonstrated to diminish pro-inflammatory response. In particular, substituting guanosine or uridine residues with adenosine resulted in decreased cytokine and interferon activity 43. The chemical structure of the siRNA duplex can also influence the degree of innate immune response. RIG-1, for example, has been shown to bind to single-stranded or doublestranded RNA containing uncapped 5'-triphosphate groups resulting in an interferonmediated immune response 44-46. Uncapped RNA is a sign of viral infection, and subsequently induces inflammatory action. Additionally, it has been reported that bluntended dsRNA (no overhangs) is also capable of provoking immunostimulatory activity through recognition by RIG-I 35. Nucleotide structure also has a profound effect on innate immune system activation. For example, it has been established that modifying the 2' group on the ribose ring of the RNA backbone can reduce or even eliminate the innate immune response. Locked nucleic acids 47, unlocked nucleic acids 48, as well as 2'-F, 2'-H (i.e. 2'-deoxy) and 2'-O-methyl modifications (2'-O-Me) (Figure 1.4) 4,22 have all been shown inhibit immune activity to some extent. Relative to other types of nucleotide modification, 2'-O-Me modifications successfully inhibit TLR7/8-mediated recognition of siRNA without diminishing RNAi . potency 4 2 siRNA Delivery Vehicles Influence Immunostimulation Although single doses of naked siRNA do not necessarily cause innate immune activation 49, siRNA delivery materials can have a substantial effect on siRNA-mediated immune activity. Delivery vehicles can account for immune responses varying up to two orders of magnitude 39,50,51 and can act through a variety of mechanisms. Different delivery materials escort siRNA molecules across cell membranes in unique ways and into differing subcellular compartments, exposing the siRNA to differing numbers and types of PRRs. For example, cationic materials are frequently employed for siRNA delivery applications, as they readily condense RNA due to electrostatic interactions. In general, cationic and lipid materials are believed to deliver siRNA through the mediation of endosomal uptake into the cell and subsequent endosomal escape into the cytoplasm of the cell 52. When formulated with a cationic delivery material, siRNA is trafficked through several subcellular locations, allowing it to interact with Toll-like receptors in the 13 endosomal compartments of immune cells as well as with RIG-I and PKR (Figure 1.2), which are present in the cytoplasm of most cell types. Therefore, this type of delivery material would expose its cargo to many pattern recognition receptors, rendering the siRNA more prone to innate immune recognition than a delivery material that avoided endosomal and lysosomal delivery routes. The size, charge, and biodistribution of a delivery vehicle may also alter the intensity and nature of a siRNA-induced immune response. Judge and colleagues found that SNALPencapsulated and polyethylenimine-complexed siRNA caused a different immune response than siRNA incorporated with polylysine, which forms a larger delivery particle. It has also been shown that the immunostimulatory properties of the lipid-like delivery vehicle ND985 (Figure 1.3) depend significantly on formulation approach 53. 1.2.1.3 Off-target gene silencing Early reports described siRNA-induced silencing as extremely specific: a single mismatch in the target site completely abolished silencing 54. However, it was soon demonstrated that siRNAs with partial sequence homology to off-target mRNAs could affect the expression of many genes 55,56. Off-target silencing both complicates experimental results and can lead to toxicity 57. One cause of off-target silencing is improper strand selection by RISC. Passenger-strand silencing can be avoided by selecting siRNA sequences with high thermodynamic asymmetry or by chemically modifying the sense strand, as described above. Guide strand off-targeting commonly occurs through complementarity of the siRNA 5' end to the 3' untranslated regions (UTRs) of mRNA 29. Positions 1-8 of the 5' end of the guide strand are referred to as the "seed region," a sequence that plays a key role in target recognition by endogenous miRNAs 58. miRNAs regulate gene expression through incomplete hybridization of the seed region with 3'UTRs, which results in gene suppression by translational inhibition and deadenylation (Figure 1.1) 59,60. As 3' UTR sequences are shared by many mRNAs, a single miRNA can regulate hundreds of genes 61-65. siRNAs and miRNAs share the same silencing machinery, and siRNA off-targeting is thought to result from siRNAs entering the miRNA pathway (Figure 1.1) 29. These miRNA-like effects can result in downregulation of scores of off-target genes 18,66,67. 14 Though miRNA-like effects are responsible for most off-target gene regulation 10,68, partial complementarity of siRNAs to coding regions of mRNA can also cause unintended silencing. Significant gene silencing has been observed with as little as 11 nt of continuous sequence homology in coding regions 56. To avoid miRNA-like off-targeting, siRNA design algorithms attempt to select sequences with minimal seed-region complementarity to 3' UTRs 69. However, partial homology with some UTRs is unavoidable: an analysis of the human 3'UTR database revealed that even the most infrequent 7-nt seed sequence still had complementarity with 17 different 3' UTR sequences 70. Alternatively, chemical modification can be used to minimize miRNA-like effects. Efforts have focused on modifications that destabilize hybridization between the seed sequence and the 3'UTR. The most common modification for minimizing off-target effects is 2'-OMe in the guide strand seed region, specifically at position 2. In a microarray study, this modification mitigated effects on 80% of off-target transcripts 67. It has been suggested that the bulkiness of the 2'-OMe affects the conformation of the complex formed from Ago, the guide strand, and the target mRNA in a way that inhibits binding to partially-complementary targets more than to fully-complementary targets. Incorporation of UNA in the seed region, particularly in position 7, also destabilizes siRNA-3'UTR hybridization 48, and was able to reduce the number of off-target transcripts by >90% 16,17. Both methylation and UNA were able to reduce off-targeting without reducing the potency of on-target silencing 16,17,67. While destabilization of the seed region duplex has been shown to mitigate offtargeting, stabilization of the length of the duplex may improve specificity by other mechanisms. Of the four human Argonaute proteins, Ago-2 is the only one with In target cleavage 24. for siRNA-mediated activity required endonuclease coimmunoprecipitation studies, siRNA duplexes interacted with all four Ago proteins, however, strands of duplexes with increased hybridization stability were only efficiently separated by Ago2. In the same study, duplexes stabilized by LNA demonstrated both reduced seed-region off-targeting and improved on-target potency 71. Modifications such as methylation, UNA, and LNA can provide the additional benefit of reducing the immunostimulatory effects of siRNA without reducing potency 4,22,42,48. Optimization of siRNA sequence along with combinations of chemical modifications enables 15 the design of siRNAs that potently silence target genes with minimal off-target silencing or immunogenicity. 1.2.1.4 Saturation of the RNAi machinery Exogenous siRNAs share components of the RNAi pathway with endogenous miRNAs, and competition for RNAi machinery may inhibit normal gene regulation by miRNAs. Several studies have demonstrated the saturability of the RNAi pathway and the toxic effects that can result from saturation 72-76. RNAi pathway saturation was first observed in experiments using high expression of short hairpin RNAs (shRNAs). ShRNAs are transcribed from virus or plasmid vectors and have a stem-loop structure similar to that of pre-miRNAs that enters the RNAi pathway. In cultured cells, overexpression of shRNAs from plasmid vectors delivered by transfection reagents inhibited miRNA activity by saturating Exportin-5 77, a protein required for transport of shRNAs and miRNA from the nucleus to the cytoplasm. In adult mouse livers, sustained high expression of shRNAs, achieved using viral delivery vectors, resulted in dose-dependent liver injury and fatality that was associated with decreased expression of liver miRNAs 72. Subsequent studies demonstrated that competition for RNAi machinery could also occur downstream from Exportin-5, in the pathway relevant to silencing by synthetic siRNAs. When multiple siRNAs were co-transfected into cells, efficacies of individual sequences were reduced, suggesting competition for downstream silencing machinery 73,78. siRNAs were also observed to affect the activity of endogenous miRNAs and of co-transfected shRNAs 73. Overexpression of Expo-5 only partially relieved this competition, suggesting saturation of downstream components of RISC 73. Competition between transfected siRNAs and endogenous miRNAs was also suggested by an examination of genome-wide transcript levels from a set of 150 published experiments. The analysis found that transfection of siRNA, while silencing genes complementary to siRNA sequences, also led to increased expression of common miRNA targets 76. While Xpo-5 was previously identified as one saturable component of the RNAi pathway, recent overexpression studies with shRNA have implicated the Ago proteins as key factors in the saturation of RNAi. In adult mice, overexpression of Xpo-5 enhanced shRNA efficiency but increased hepatotoxicity. Ago-2/Xpo-5 coexpression enhanced shRNA silencing, reduced hepatotoxicity, and increased RNAi stability 79. Of the four human Argonautes, Ago-2 was identified as the primary determinant of RNAi efficacy, toxicity, and persistence both in cell culture and in mouse liver 79. 16 The harmful effects of RNAi saturation may be avoidable by limiting doses of exogenous RNA 79. Hepatotoxic effects in mice were abrogated when shRNAs were expressed under milder promoters 80. Therapeutic levels of gene silencing in vivo have been observed without perturbing miRNA function. In mice and hamsters, LNP-delivered siRNAs achieved 80% silencing of clinically-relevant gene targets without affecting endogenous miRNA activity 80. 1.2.1.5 Delivery material toxicity Systemic or cellular toxicity can also be caused by delivery reagents themselves. In many of the most efficient in vivo delivery systems, the therapeutic dose of siRNA is accompanied by more than 7 times its weight in delivery materials 81-83, making vehiclerelated toxicity an important consideration. Delivery material toxicity can occur through various mechanisms including immunostimulation (see above), membrane destabilization, and alterations in cellular signaling or gene expression. One element of the toxicity of lipoplexes and polyplexes is often attributed to their positive charge, which is exacerbated in low pH environment of the endosomes and lysosomes 84. Mice treated with certain cationic lipid nanoparticles showed signs of hepatotoxicity and a systemic interferon type I response, attributed in part to activation of TLR 4 85. This effect was not observed in mice treated with neutral or negatively-charged particles 85. Cellular toxicity of cationic lipids has been linked to increased production of reactive oxygen species 86,87 and subsequent rise in cellular calcium levels 86, which were not Cholesterol-derived cationic observed with neutral or negatively-charged lipids. amphiphiles are known to inhibit protein kinase c (PKC) 84,88, which can cause cytotoxicity. Cationic lipid toxicity varies greatly with structure and formulation. Cholesterolderived lipids with quaternary ammonium head groups can be potent PKC inhibitors and are more toxic than those with tertiary amines 84,88. Introduction of heterocyclic rings into head groups can delocalize charge and in some cases reduce this toxicity 84,89,90. Lipids that contain biodegradable linkages between their polar head groups and hydrophobic tails are also associated with lower toxicity 84,91. Use of ionizable cationic lipids, which are neutral in circulation but become positively charged in the acidic endosomal environment, may reduce nonspecific disruption of cell membranes 92. Formulation of cationic lipids with 17 nucleic acids partially neutralizes their positive charge, and the toxicity profiles of lipids alone can be distinct from those of lipids in formulation with nucleic acid 84,93. Unmodified PEI has been shown to induce systemic and/or cellular toxicity depending on structure and formulation. Both linear and branched PEI can cause cytotoxicity by damage to cell membranes and by depolarization of mitochondrial membranes, leading to apoptosis 94. PEI formulations with high ratios of delivery material to siRNA can also cause systemic toxicity due to their high positive charges. Particles with high surface charges interact with the negatively charged membranes of blood cells, which can lead to erythrocyte aggregation. Large aggregates can have difficulty passing through capillaries, leading to serious systemic effects 95. Grafting of PEG to PEI can mitigate the problem of erythrocyte aggregation 9s and reduce overall cytotoxicity 95,96. PEG-PEI also is not inert and can affect cellular processes differently than PEI alone. PEG-PE has been shown to activate apoptotic pathways in a cell-line and concentration-dependent manner 97. c. Cholesterol-siRNA conjugate a. Stable nucleic acid lipid particle (SNALP) 0 H d. Lipidoid 98N12-5(1) H N H N N H H 0 H Cationic lipid Artf\j Poly(ethylene glycol) Neutral lipid Cholesterol e. Lipidoid C12-200 HO b. Cyclodextrin OH OH 0 HO 0 \fOH 0 HO HO H H -H9H O N f. Poly(ethylene imine) H OH NH, H OH 00 NH NH-, Figure 1-3. Examples of siRNA delivery systems. (a) Stable nucleic acid lipid particles (SNALPs) encapsulate siRNA as cargo. They are formulated with cationic and neutral lipids, cholesterol, and poly(ethylene glycol) (PEG). (b) Cyclodextrin polymers form nanoparticles when complexed with siRNA. (c) Delivery materials such as cholesterol can be directly conjugated to siRNA. (d,e) Lipid-like delivery agents are often formulated with cholesterol and PEG when administered in vivo. (f) Poly(ethylene imine) can be linear or branched and is often grafted with PEG. 18 Microarray studies have begun to reveal that delivery materials can induce gene expression changes, which can potentially cause toxicity and other off-target effects. Commonly-used transfection reagents have been shown to induce their own gene expression signatures even in the absence of siRNA 96-98. The cationic lipids Oligofectamine and Lipofectin altered the expression of dozens of genes involved in various cellular processes including proliferation, differentiation, and apoptosis pathways 98. PEI (25kDa) and PEI-PEG altered the expression of several genes involved in apoptosis and inflammatory signaling 96,97. In some stably-transfected cell lines, signaling triggered by PEG-PEI increased CMV promoter activity, causing an increase in reporter gene expression rather than siRNA-mediated knockdown 97. Gene signatures of these materials can change when the material is formulated with nucleic acid 99 and can vary greatly for different Structure of the delivery material delivery materials, cell types, and treatment times. affects gene expression signatures as well. When injected intratumorally, branched PEI induced more gene expression changes that linear PEI 99, consistent with the higher reported toxicity of branched PEI 100. An understanding of the alterations in gene expression due to delivery materials is necessary to properly interpret experimental results and to develop delivery formulations with minimal off-target effects. While delivery materials alone can induce gene expression changes and cause toxicity, delivery materials in formulation with siRNA may cause toxicity and off-target effects Delivering siRNAs into simply introducing siRNA into different cellular environments. endosomes increases their potential to stimulate endosomal TLRs to elicit an immune response. Delivering siRNAs into the cytoplasm increases their potential to saturate the RNAi machinery or to enter the miRNA pathway, causing off-target silencing. As such, delivery materials alone and in formulation with siRNA (even siRNA sequences that lack targets) can have distinct gene signatures 99. 1.2.2 Pharmacokinetic considerations Pharmacokinetic considerations determine the ability of delivery systems to reach the cytoplasm of target cells and therefore to induce a therapeutic effect. The fate of intravenously administered siRNA depends heavily upon its structure, chemical modification, and delivery formulation. These factors affect a delivery system's stability in circulation, its tendency to extravasate, or exit blood vessels, its ability to enter cells, and its likelihood of being cleared from circulation without being delivered. 19 Pharmacokinetic parameters such as circulation half-lives, area under the concentration-time curve, elimination rates, and bioavailability are functions of these various physiological processes. For example, a short circulation half-life could be explained by rapid excretion of the formulation in urine, or it may result from the rapid uptake of the formulation by tissues. This section aims to describe the mechanisms and processes that shape pharmacokinetic behavior of siRNA delivery systems. Detailed pharmacologic data for various delivery systems are beyond the scope of this review. 1.2.2.1 siRNA in circulation Intravenously-administered siRNA is exposed to serum proteins, nucleases, and cells of the innate immune system. The base base base resulting processing and /5 0 biodistribution of injected siRNA 0 0 varies with both the structure of the H CH 3 siRNA and the delivery system in which it is carried. Successful siRNA RNA 2'-F-RNA 2'-OMe-RNA delivery has often required the use of a base base delivery vehicle because naked, unmodified siRNA is degraded by 0 serum RNAses and cleared from the 5/ bloodstream within minutes 0 LNA base 0 0 nanoparticles H 04__0 In contrast, many delivery formulations, such as lipid and polymer systems, protect siRNA from nucleases by encapsulating it as cargo inside UNA base 4101102. 103,104. H base 0 o4s _ base 0 H Phosphodiester H Phosphorothioate Figure 1-4. Common chemical modifications to the siRNA backbone. The 2'-OH of the ribose ring can be replaced with methoxy (2'-OMe) or fluorine (2'-F) moieties. Locked nucleic acids (LNA) contain a methylene bridge connecting the 2' oxygen to the 4' carbon. Unlocked nucleic acids (UNA) lack a bond between the 2' and 3'carbons. The nuclease stability of unformulated siRNA can also be dramatically improved by making chemical modifications to the RNA backbone 105. A common strategy involves modification of the ribose 2'OH group, as this functional group is critical to the mechanism of many serum RNAses 106,107. Among the most effective backbone modifications for serum stability improvement are the 20 substitution of the ribose 2' hydroxyl with 2'-Fluorine or 2'-Methoxy groups (Figure 1.4). If placed judiciously within the duplex, such modifications can add stability without affecting RNAi activity 22,102. A similar effect is achieved by incorporation of locked nucleic acid (LNA) nucleotides, which contain a methylene bridge connecting the 2' oxygen to the 4' carbon 47,108 (Figure 1.4). Additionally, modifications to the linkages between sugars can be incorporated into the 3' overhangs of siRNA to confer resistance to exonucleases. The most common of these is the replacement of certain phosphodiester linkages with phosphorothioate linkages (Figure 4) 109. Combinations of chemical modifications have been shown to improve siRNA stability in plasma 22,110 as well as increase its circulation time 4,22,102. The mechanism by which chemical modification improves circulation time in vivo is unknown. As renal filtration is expected to be slower for large molecules (such as intact siRNA) than for small molecules (such as fragments of degraded siRNA), an increase in serum stability may partly explain the increase in circulation time for chemically stabilized siRNA. In addition to enzymes, circulating siRNAs also interact with or bind to various components of the blood, such as red blood cells and serum proteins, which can influence their pharmacokinetics. In some cases, interaction between serum proteins and siRNA can improve delivery outcomes by increasing circulation time and enhancing uptake into target tissues 111,112. For example, the incorporation of cholesterol-conjugated siRNA into serum lipoproteins has been shown to improve its circulation time, reduce renal clearance, and facilitate uptake into hepatocytes via lipoprotein receptors 111. It has also been found that certain ionizable lipid nanoparticles are subject to apolipoprotein E adsorption, which can mediate their uptake by hepatocytes via the low-density lipoprotein receptor 112. In other cases, interaction with serum proteins or red blood cells can be detrimental, leading to formation of aggregates that are readily opsonized or worse. Large aggregates can have difficulty passing through capillaries, which can lead to accumulation in lung capillary beds and even lung hemorrhage 9s. More commonly, adsorption of serum proteins, especially components of the complement system, leads to rapid clearance of particles by the cells of the mononuclear phagocyte system (MPS) 113,114. Phagocytosis of particles is stimulated by the interaction of adsorbed proteins with receptors on phagocytes 113. Adsorption of these proteins can be minimized by functionalizing the surface of particles with hydrophilic polymers such as poly(ethylene glycol) (PEG) 113,11s,116. PEGylation of siRNA delivery systems is a widely-used strategy for minimizing protein adsorption and reducing nonspecific uptake to create long-circulating delivery systems 113,114. 1.2.2.2 Extravasation 21 Circulating siRNA must leave the bloodstream by crossing the vascular endothelium in order to reach many target tissues. The structure and permeability of the endothelium vary across different tissue types, making some tissues more accessible to macromolecular therapeutics than others 117. In healthy tissues, endothelia are described as continuous or discontinuous and fenestrated or non-fenestrated (Figure 1.5). In continuous endothelium, cells are connected by tight junctions and adherens junctions that * allow the passage of water and small solutes but prevent the passage of molecules larger than 3 Endothel Let nm Macromolecules such as addr-fer1 iuntiuit' !Urdeut albumin Continuous, nonfenestrated capillary hetirt. lunys .Mi %Lk'trit I spae uf D'e -f 117 Continuous, fenestrated capillary iterti 1'.1d Iy I ~and rtneKtae traverse endothelia continuous by transcytosis 118. In fenestrated endothelia, contain cells channels, transcellular or fenestrae, that allow direct passage of water small solutes across endothelial cells. Fenestrae in continuous 5irus .dstEC - uendothelia Liver sinusoidal capillary $N-isitated endothel idi' ..'t , are typically 40 - 60 nm in diameter 119 and are spanned by a glycoprotein thin diaphragm that prevents of passage the macromolecules Figure 1-5. Endoth"elia heterogeneity. Capillary endothelta can be continuous or discontinuous. fenestrated or nonfenestrated. The structure of the endothelium affects the permeability of the vasculature to the passage of siRNA therapeutic formulations. 117,120. Discontinuous found endothelium, liver in primarily is sinusoids, The diaphragm. lack a that nm 117,121) 200 (100 fenestrae by large characterized sinusoidal endothelium also lacks an organized basement membrane 117 and therefore provides minimal resistance to the passive transport of macromolecules and nanoparticles from the bloodstream to the liver interstitium. This, along with the well-perfused nature of the liver, may explain why the liver is one of the primary sites of accumulation of many systemically-administered siRNA formulations 4,81,92,122-124. Though the structure of the liver endothelium allows access to hepatocytes, therapeutics can also be absorbed by resident 22 macrophages in the liver, called Kupffer cells 125. In some cases, accumulation of therapeutic particles the liver is due mainly to phagocytosis by Kupffer cells rather than uptake by hepatocytes 125. Another site that can have increased vascular permeability is malignant tissue. In tumors, blood vessels can be heterogeneous in size, spacing, tortuosity, and permeability 119,126-128. Tumor endothelia can have large fenestrae 129, unusually thick or thin basement membranes 119, and intercellular gaps that can be as wide as a few microns 130. As a result, tumor endothelia are sometimes more permeable than those of healthy tissues 131,132, allowing delivery systems to extravasate relatively easily. Certain tumors also have poorlyfunctioning lymphatic vessels 133,134, interstitial fluid is not efficiently drained from tumor tissue and nanoparticles tend to accumulate in the interstitium. This accumulation, due mostly to high vascular permeability in addition to poor lymphatic drainage, is known as the enhanced permeation and retention (EPR) effect, which is exploited by many nanoparticle systems for passive tumor targeting 119,135,136. The EPR effect aids larger particles (-100 nm) to accumulate in regions of leaky vasculature in tumors; however, smaller particles (< 60 nm) diffuse more readily through the interstitium and more deeply penetrate tissues 119,137. Phagocytosis can also play a major role in the clearance of siRNA delivery systems from the blood. As a result, accumulation of siRNA in immune cells is also observed 138. In one study, fluorescence molecular tomography imaging showed that siRNA delivered by lipidoid nanoparticles exited the blood pool primarily into the spleen, bone marrow, and liver, with a blood half-life of 8.1 minutes. siRNA taken up by the liver was excreted by the hepatobiliary tract, while siRNA in the spleen and bone marrow localized to phagocytic cells including neutrophils, macrophages, dendritic cells, and monocytes. This biodistribution pattern was exploited to use siRNA to reduce the accumulation of monocytes in sites of inflammation by silencing of the chemokine receptor CCR2. Knockdown of this target in inflammatory monocytes attenuated the damaging effects caused by inflammation in multiple disease models, including atherosclerotic plaques, myocardial infarction, diabetes, and cancer 138. 1.2.2.3 Cell uptake Delivery systems that extravasate and diffuse into tissues must cross cell membranes into the cytoplasm to trigger RNAi. Most siRNA delivery formulations traverse cell membranes through membrane-derived transport vesicles in the tightly regulated process 23 of endocytosis. Endocytosis encompasses two broad categories: phagocytosis and pinocytosis. While phagocytosis is only associated with specific cell subtypes, pinocytosis is common to all cells. Phagocytosis is used by specialized immune cells including macrophages, neutrophils, and monocytes to clear the body of large particles such as pathogens or dead cells 139-141. Prior to involution by phagocytosis, foreign materials are opsonized, which involves their binding to opsonin proteins such as immunoglobulins, complement proteins, and blood serum proteins. Complexation with these proteins marks foreign materials for phagocytosis 140-143. Rho-family GTPases together with specific cell-surface receptors and signaling cascades determine the mode of phagocytosis 139,144. Macrophages, for example, utilize Fc receptors to recognize antibody-bound targets. Resulting activation of Cdc42 and Rac triggers actin assembly and creates finger-like protrusions that engulf antibody-bound targets 140,141,145. The formation of the resulting vesicle, called a phagosome, stimulates the cell's inflammatory responses and the entrapped target is bombarded with hydrolases, oxygen radicals, and a low pH environment in an attempt to destroy the foreign material 146. Macropinocytosis is a non-specific endocytic process similar to phagocytosis that occurs in most cells. Rho-GTPase mediates actin-driven membrane protrusions that extend out and collapse over large volumes of extracellular milieu, entrapping particles in large vesicles called macropinosomes 147-149. These large vesicles can be up to 1-5 tm in diameter, and their intracellular fate is uncertain, although there is evidence that they shrink and fuse with lysosomal compartments for degradation of entrapped materials. Clathrin-mediated endocytosis (CME), the most prolific internalization mechanism, occurs constitutively in all tissue types and participates in the uptake of vital nutrients and in intercellular signaling through receptor-dependent and receptor-independent pathways 148,150-153. Iron-bound transferrin protein, for example, is internalized after association with transferrin receptors initiates invagination of the plasma membrane. Pits that form in the membrane around protein-bound receptors are coated by clathrin, a triskelion-structured protein that assembles into polygonal cages on the cytoplasmic face of the membrane 150,151,153. Receptor-independent CME is able to induce these internalization events simply by non-specific ionic or hydrophobic interactions with the plasma membrane. Fission of the coated pit from the membrane is mediated by the GTPase dynamin, forming a clathrincoated vesicle roughly 100-120 nm in size. Internalization occurs on the order of minutes, and resulting vesicles undergo endosomal maturation. Early endosomes have a pH around 6 and later fuse with acid hydrolase-containing prelysosomal vesicles to form late 24 endosomes (pH -5). The acidic and enzymatic environment of the lysosome is aimed at degradation of the vesicular cargo 154. Caveolae-mediated endocytosis (CvME) is used by cells to internalize serum proteins such as albumin and essential metabolites such as cholesterol and folic acid. CvME involves flask-shaped invaginations, on the order of 50-100 nm, of the plasma membrane called The dimeric protein caveolin is responsible for the shape and caveolae 150,1S2,155,156. structure of caveolae, whose membranes are enriched with cholesterol and sphingolipids. CvME is a tightly regulated process that is believed to be driven by receptor-ligand interactions, with vesicle formation being mediated by the GTPase dynamin. Caveolinassociated vesicles do not contain any enzymatic mixtures and do not enter a degradative pathway by maturing into lysosomes like CME. The internalization kinetics of CvME are much slower than those of CME, with an uptake half-time on the order of 20 minutes. Recently, endocytic pathways have been uncovered that do not fall into any of the previously-described categories 1s2,155-157. These processes are often referred to as clathrinand caveolae-independent pathways, and occur at cholesterol-rich microdomains of the plasma membrane that are similar to caveolae. These lipid "rafts" are small structures approximately 40-50 nm in size, and have unique membrane protein compositions that may play a role in vesicle formation. The mechanisms that stimulate this endocytic pathway are still poorly understood, and currently the only way to determine the contribution of this pathway to a cargo's internalization is by elimination of the other pathways. Some systems In general, siRNA delivery particles enter cells via endocytosis. incorporate targeting ligands designed to stimulate receptor-mediated endocytosis 158. Other systems, such as lipophilic conjugates and some ionizable lipid nanoparticles, exploit endogenous targeting ligands by associating with serum lipoproteins, which are taken up by hepatocytes by receptor-mediated endocytosis 111. Untargeted siRNA lipoplexes are also taken up mostly by endocytosis, but a small number of complexes enter cells through another pathway, possibly by direct fusion of the lipoplex with the cell membrane. Interestingly, this minor pathway was shown to be responsible for functional siRNA delivery in certain systems, as blocking endocytosis did not abolish gene silencing 159. Uptake of siRNA delivery systems by endocytosis leads to sequestration of the delivery system inside intracellular vesicles. Escape from these vesicles may represent a major barrier to triggering RNAi and thereby affect the efficacy of delivery systems. While the mechanisms of vesicular escape of synthetic materials are still poorly-understood, polymers 25 with many protonatable groups are thought to induce lysis of endosomes by pH buffering 52,160. Protons are transported into endosomes accompanied by counter ions, and absorption of protons by polymers leads to increased concentrations of counter ions, osmotic swelling, and endosomolysis. Certain ionizable lipid systems undergo a phase transition at low pH, resulting in membrane destabilization that promotes release of material from endosomes 92,161. By converting from a lamellar to an inverted hexagonal phase, these lipids are thought to increase their interactions with the endosomal membrane, leading membrane destabilization and inducing endosomal release. Still other systems employ acid-labile materials 162 or modified cell-penetrating peptides 163. Dominska and Dykxhoorn provide a more detailed review of how various delivery systems overcome the problem of endosomal release 164. A recent report showed that for a leading lipid delivery system, approximately 70% of the siRNA taken up by cells underwent 00 . endocytic recycling and exocytosis 1.2.2.4 Elimination siRNA that is not degraded or endocytosed is removed from circulation by excretion in urine or bile. Renal filtration occurs in the specialized capillaries of the kidney glomerulus (Figure 1.6). Water and small molecules are able to pass through capillary walls into the urinary space, while Gamerulmr macromolecules remain in circulation. The E6 %e:,ent glomerular filtration barrier is composed of Mem~rbrane the the endothelium, three layers: glomerular basement membrane (GBM), and a layer of epithelial cells called podocytes 165. Fe-e-,t ated endotheiuf Fgure 1-6. The glomrular filtration barrier. The capillaries of the kidney glomerutus participate in the filtration of blood to form urine. The glomerular filtration barrier consists of three layers endothelial cells, the glomnerular basement membrane, and podocytes. Molecules smaller than -8 nm in diameter are able to pass through this barer into the urinary space. The glomerular endothelium is continuous and populated with abundant 60-100 nm 166. Endothelial cells are fenestrations supported by the glomerular basement membrane, which provides an additional Podocytes play an diffusion barrier 167. important role in the size selectivity of the Slit 168. barrier filtration glomerular diaphragms, composed of proteins that bridge the space between podocytes, create -8 nm pores 169-171 that filter waste prior to excretion in the urine 172,173. 26 The liver, which is also part of the body's clearance system, mediates the secretion of compounds into bile ducts, and are eliminated from the body via the intestine 174-176. Hepatocytes secrete bile into hepatic ducts, where it can be emptied directly into the small intestine or stored in the gallbladder. Various studies have shown that naked, unstabilized siRNA is rapidly eliminated from the bloodstream via renal filtration, with serum half-lives of less than 5 min 4,101,110. Chemically-stabilized siRNAs have been reported to circulate for 30 - 50 min 4,110, and also to undergo renal clearance 4,110. While kidney filtration has often been assumed to be the major route of siRNA elimination, a recent study demonstrated the importance of the hepatobiliary pathway in the clearance of chemically-stabilized siRNAs, both naked and in formulation 177. 1.2.3 Conclusions The many interactions between siRNA and the body continue to be elucidated as more delivery systems are studied. In the design of delivery vehicles, it is natural to focus on the aspects of pharmacokinetics and pharmacodynamics that contribute directly to the desired therapeutic effect, yet many other interactions exist that affect the overall potency and tolerability of a formulation. The information contained in this review provides a brief overview of these interactions, which can serve as a guide in design of new systems and in the optimization of existing systems. 27 M Table 1. Selected RNAi drugs in clinical trials (continued on next page) Delivery System Drug ALN-RSV01 TD101 Sponsor Alnylam Pharmaceuticals Pachyonychia Congenita Project Status ClinicalTrials.gov Identifier Disease Phase RSV nucleocapsid Respiratory Syncytial Virus Infections 2 Completed NCT00658086 K6a (N171K mutation) Pachyonychia congenita 1 Completed NCT00716014 2 Terminated NCT00363714 1 Active NCT01064505 Acute Primary AngleClosure Glaucoma 2 Recruiting NCT01965106 Kidney injury, acute renal failure Delayed Graft 1 Completed NCT00554359 1,2 Active NCT00802347 2 Active NCT01445899 2 Completed NCT00713518 2 Completed NCT00306904 2 Completed NCT00259753 Ocular Pain, Dry eye 1, 2 Recruiting NCT01776658 ADR32 Ocular Hypertension, Open Angle Glaucoma 2 Completed NCT01739244 CTGF Cicatrix, Scar prevention 1 Active NCT01780077 Target gene Age-Related Macular AGN211745 Allergan VEGFR1 Degeneration, Choroidal Neovascularization Optic Atrophy, Nonarteritic Anterior Quark QPI-1007 Naked siRNA PF-655 (PF- 04523655) bevasiranib Pharmaceuticals CASP2 Quark PharmaceuticQuNP als p53 Quark Pharmaceutica Is RTP8O1 (Propri etary target) Opko Inc. Health' VEGF Neuropathyic Function, Complications of kidney transplant Choroidal Neovascularization, Diabetic Retinopathy, Diabetic Macular Edema Age Related Macular Degeneration Diabetic Macular Edema Macular Degeneration SYL1001 SYL040012 RXi-109 I Sylentis, S.A. ___________ Sylentis, S.A. RMi PharmaceuticI als TRPV1 _____________syndrome__________ 1.3 Strategies for siRNA delivery A key challenge to realizing the broad potential of siRNA-based therapeutics is the need for safe and effective delivery methods. Unmodified siRNA is unstable in the bloodstream, can be immunogenic, and does not readily cross membranes to enter cells 9. Therefore, chemical modifications and/or delivery materials are required to bring siRNA to its site of 28 action without adverse effects. A broad diversity of materials is under exploration to address the challenges of in vivo delivery. These include polymers 178, lipids 81,92, peptides 179, antibodies 180, aptamers 181,182, or small molecules 101,183 as delivery materials. Successful systems have been developed by rational design or discovered utilizing high-throughput screens 81,92. Table 1 (continued). Selected RNAi drugs in Delivery System Drug Sponsor clinical trials Target gene Disease Phase ClinicalTrials.gov Identifier Status ALN-VSPO2 Alnylam Pharmaceuticals KSP and VEGF Solid tumors 1 Completed NCT01158079 siRNA- MD Anderson EphA2 Advanced 1 Cancer Center Active NCT01591356 EphA2-DOPC 1, 2 Recruiting NCT01808638 1, 2 Recruiting NCT01262235 1 Recruiting NCT01518881 Atu027 ence cancers PKN3 Therapeutics AG TKM-080301 Tekmira Pharmaceutical Company Pancreatic Ductal Carcinoma PLK1 Cancer VP24, Lipid nanoparticle s (LNPs) TKM-100201 s (LNs) Tekmira Pharmaceutical CopanyEbola Company VP35, Zaire Lpolymera se Tekmira PRO-040201 Pharmaceutical Ebola Virus Infection Hypercholestero ApoB lemia 1 Terminated NCT00927459 Hypercholestero lemia Transthyretin mediated amyloidosis 1 Completed NCT01437059 3 Active NCT01960348 Fibrosis 1 Recruiting Transthyretin mediated amyloidosis 1 Recruiting NCT01814839 Hepatitis B 1 Recruiting NCT01872065 Company siRNAGaINAc conjugate Dynamic Poly- ALN-PCSO2 Alnylam Pharmaceuticals PCSK9 ALN-TTR02 Alnylam Pharmaceuticals TTR ND-L02s0201 Nitto Denko Corporation ALN-TTRsc Alnylam Pharmaceuticals TTR Arrowhead Research conserved regions of Corporation HBV Calando Pharmaceuticals RRM2 Solid tumors 1 Active Silenseed Ltd KRAS Pancreatic Cancer 2 Active ARC-520 Conjugate Cyclodextrin NP LODER polymer CALAA-01 siG12D LODER HSP47 NCT01858935 I NCT00689065 NCT01676259 Here we review a selection of promising systems for systemic siRNA delivery, with a focus on the design of materials and the approach to the delivery problem. These systems, all with reported in vivo efficacy, are diverse in size, shape, structure, chemistry, and mechanism of action. This diversity reflects our still-developing understanding of the mechanisms that underlie much of the delivery process and highlights the still vast space for innovation and creativity in the field of siRNA delivery. 29 1.3.1 Cyclodextrinpolymer nanoparticles . Cyclodextrin polymer (CDP)-based nanoparticles (Figure 1.7) entered clinical trials for siRNA delivery less than a decade after their introduction, 184. Davis and coworkers first introduced the cyclodextrin delivery system for plasmid DNA in 1999, with re-optimization of the system for siRNA delivery years later 185-192. This was the first targeted nanoparticle siRNA delivery system to enter clinical trials for cancer 184 PrEGMW=5SO AD-PEG AD-PEG-Tf siRNA nn Fgm 1-7. Cyludwdln podmmv .mnapwllms. Composition ofthe cyclodextrin delivery system developed by the Davis group. CDPs are polycationic oligomers (n-5) synthesized by a step-growth polymerization between diamine-bearing cyclodextrin monomers and dimethyl suberimidate, yielding oligomers with amidine functional groups 185. The strong basicity of these amidine groups mediates efficient condensation of nucleic acids with the CDPs at N/P ratios as low as 3. End-capping of the polymer termini with imidazole functional groups was shown to facilitate endosomal escape 193, resulting in improved delivery efficacy of both plasmid DNA and siRNA 192,194. 30 Though nanocomplexes composed of only CDP and siRNA were able to mediate efficient delivery in vitro, these complexes required additional formulation components for Both adamantane-PEG (AD-PEG) and stabilization and efficacy in vivo 188,194-196. (AD-PEG-TO were incorporated to improve particle adamantane-PEG-transferrin properties in vivo 192,196,197. Adamantane (AD) is a hydrophobic molecule that forms a stable inclusion complex with the cyclic core of the cyclodextrin structure. This noncovalent interaction allowed for surface modification of CDP-siRNA nanoparticles with AD-modified excipients by a chemical interaction that is orthogonal to the ionic forces that facilitate siRNA binding. PEG shielding was necessary to prevent protein-induced aggregation in serum, but PEGylation also reduced cellular uptake and silencing efficacy. To recover efficacy, the protein transferrin was conjugated to the free end of AD-PEG as a targeting agent. Inclusion of this AD-PEG-Tf conjugate enabled multivalent binding to the CD71 transferrin receptor, improving efficacy 198. The CDP-siRNA delivery system has been evaluated in several therapeutically relevant animal models. In a xenograft model for Ewing's Sarcoma 192, CDP nanoparticles formulated with siRNA targeting the oncogenic EWS-FLl1 fusion gene induced gene knockdown and when had antiproliferative effects with no measured innate immune responses administered intravenously 192. In a syngeneic subcutaneous mouse tumor model, the targeted CDP delivery system showed potent silencing against the validated cancer target ribonucleotide reductase subunit 2 (RRM2) 199. The clinical translatability of the delivery system was evaluated in cynomolgus monkeys, which indicated that the nanoparticles can be tolerated up to 27 mg siRNA/kg with translated efficacy in the range of 0.6-1.2 mg siRNA/kg 200. Finally, clinical potential was established when Davis and co-workers showed RNAi-specific gene inhibition in human melanoma patients (phase I clinical trial) by monitoring siRNA-mediated cleavage of RRM2 mRNA 8. Several factors contributed to the translation of this delivery system: 1) the low toxicity of the cationic polymer, 2) its facile condensation with nucleic acids, 3) the steric stabilization by PEGylation of the CDP-siRNA nanoparticles in a stable and non-covalent fashion, and 4) the inclusion of a ligand to enhance in vivo uptake and efficacy. These lessons provide useful guidelines for designing future polycationic delivery vehicles. 1.3.2 Lipid nanoparticles The activity of liposomal siRNA formulations was first reported in non-human primates in 200682. Since then a number of lipid nanoparticle (LNP) RNAi drugs have entered clinical trials (Table 1), including treatments targeting hypercholesterolemia, transthyretin mediated amyloidosis, and cancer 2 01 20 2 . Prior to their use with siRNA, liposomes were studied for decades as delivery vectors for DNA-based drugs, due to their 31 . ability to protect entrapped oligonucleotides from nuclease degradation and renal clearance, and for their ability to promote cellular uptake and endosomal escape 203. To date, a number of different lipid and lipid-like structures and formulation methods have been developed, generating a wide variety of LNPs2O4,20s. In the study of these diverse systems, several features have emerged as particularly effective for siRNA delivery. They include the use of cationic or ionizable lipids, shielding lipids, cholesterol, and targeting moieties, 206 1.3.2.1 Cationic and ionizable lipids Many liposomes used for siRNA delivery include a cationic or ionizable lipid. Positively charged lipids serve several functions: improving the entrapment of the negatively charged siRNA, increasing cellular uptake, and facilitating endosomal escape. Several studies have determined that cationic lipids, which have a constitutive net positive charge, have been shown to be less efficacious 20 7 and more toxic 88 than ionizable lipids, whose charge is dependent upon the pH of the surrounding environment. As a result, recent research has focused on the development of novel ionizable lipids. The composition of these lipids is generally divided into three parts: an amine head group, a linker group, and hydrophobic tails (Figure 1.8a). Many of these lipids have been synthesized in a combinatorial manner 83 208 by altering these three sections in a systematic fashion in attempt to develop structure-function correlations. To minimize toxicity without sacrificing efficacy, the pKa of an ionizable lipid should be low enough to remain unprotonated during circulation, but high enough to become protonated in either the early or late endosome. Protonation is necessary to promote membrane fusion and lipid mixing with the anionic lipids in the endosomal membrane61 2 09 z ,2 1 0 . In a study of the effect of lipid pKa on in vivo gene silencing using 53 ionizable lipids, an efficacious range of pKa was found to occur between 5.4 and 7.6. Within this range, efficacy increased as pKa approached an optimum value of 6.44211. Overall liposomal pKa appears to be more important for efficacy than the pKa of individual lipid components. When two lipids of pKas 5.64 and 6.93 were co-formulated, the mixed liposomes had an overall pKa 6.93 and exhibited four-fold greater in vivo silencing than liposomes formulated with either lipid alone 211. It should be noted that though there is strong correlation between pKa and efficacy, particles with comparable pKas can have disparate levels of efficacy in ViVo92,209. Lipid transition temperature refers to the temperature at which lipid membranes shift from the more stable lamellar phase to the less-stable hexagonal phase configuration (Figure 1.8d) 2 12. This transition promotes destabilization of the endosomal membrane and release of siRNA from both nanoparticles and endosomes 213. Lipids with lower transition temperatures, which more readily shift from lamellar to hexagonal phase 214 to promote 32 endosomal release, have a small polar head groups and large, unsaturated hydrophobic tails (Figure 1.8b). Lipids with large polar head groups and fully saturated hydrophobic tails are more likely to adopt the stable lamellar phase configuration. Successful lipid formulations are engineered to remain in the lamellar phase during circulation and to transition to the hexagonal phase within endosomal compartments. Amine Head Group a 0 0 Linker Group b a 0 c Conical Hydrophobic Tails I I I I II I Cylindrical DLinDMA Liposomal d membrane DLin-KC2-DMA En dosomal m embrane Lamellar Phase Hexagonal Phase Figure 1-8. Lipid structures and shapes. a) Ionizable lipids are composed of three sections, the amine head group, the linker group, and the hydrophobic tails. b) Lipids with a large head groups and tails composed of saturated hydrocarbons tend to adopt a cylindrical structure, while lipids with small head groups and unsaturated tails tend to adopt a conical structure. c) The structures of siRNA delivery lipids DLinDMA and DLin-KC2-DMA. d) The mixing of cationic (orange) and anionic (blue) lipids promotes the transition from the more stable lamellar phase to the less stable hexagonal phase, facilitating fusion of the liposomal and endosomal membranes. The structure of a lipid's hydrophobic tail affects its pKa, transition temperature, and potency. In a study of lipids with identical head groups, linkers, and tail lengths, 33 . increased unsaturation of hydrophobic tails resulted in decreased pKa, decreased transition temperature, and increased in in vivo efficacy. The most efficacious of the lipids in this study, DLinDMA (Figure 1.8c) was subsequently tested in cynomolgus monkeys where it was shown to be capable of silencing 90% of mRNA in hepatocytes 8 2 Variations on the amine head group and linker portions of lipids also affect in vivo efficacy. For the lipid DLinDMA (Figure 1.8c), replacing the ether linker with an ester group reduced efficacy, while a ketal linker improved efficacy 9 2. The amine head group was modified by altering the length of the carbon chain connecting the amine to the ketal group. As the chain length increased, the pKa of the formulation increased and the transition temperature remained constant. Maximal efficacy was obtained using DLin-KC2-DMA (Figure 1.8c), which had a pKa of 6.792. . While many of the ionizable structures examined are two-tailed species with a single amine, it has also been demonstrated that multi-tailed species with numerous amines can be highly efficacious. Several combinatorial libraries have been generated using multi83 2 08 amine head groups bound to numerous hydrophobic tails of various lengths 8 1. 1.3.2.2 Shielding lipid Lipid-anchored PEG is a common component in liposomes. PEG groups serve many purposes: reducing particle size 21 s.2 1 6, preventing aggregation during storage, increasing circulation time, and reducing uptake by unintended targets such as red blood cells and macrophages 2 16,217. Shielding lipids can also reduce cellular uptake by target cells, and have been shown to reduce efficacy in vitro and in vivo 2 1 6. After endocytosis, PEG can sterically and electrostatically block the interaction between the liposome and endosomal membrane, hindering membrane fusion and preventing endosomal release. One strategy for improving the efficacy of PEGylated nanoparticles involves incorporation of acid-sensitive bonds connecting PEG to the liposome. In a comparison of stable carbamate linkers and oxime linkers designed to degrade at pH 5.5, liposomes with oxime-linked PEG were stable at pH 7.4 but showed improved release of siRNA at pH 5.5 in vitro. Oxime-linked PEGylated liposomes also demonstrated improved gene silencing in 2 17 . ViV0 Another method to reduce the negative effects of shielding components involves the use of a pH-sensitive modified PEG that binds to liposomes through ionic interactions. The liposomal core consists of a HEMA-lysine-modified cholesterol as well as DOPE, and the PEG is covalently modified with HEMA-histidine-methacrylic acid. At neutral pH the PEG copolymer has a net negative charge while the liposomal core has a net positive charge. In the endosome, imidazole and methacrylic acid residues become protonated, and the net charge of the PEG becomes positive. This results in the release of PEG from the lipid core, 34 . exposing the positively charged liposomal membrane and allowing it to fuse with the endosome 21 8 1.3.2.3 Cholesterol Many liposomal formulations include cholesterol, which can associate with lipid bilayers 219 . Up to 25% cholesterol, increasing cholesterol content lowers the transition temperature of liposomal membranes containing conical-shaped lipids, facilitating their conversion from lamellar to hexagonal phase 220. Cationic liposomes with less than 10% cholesterol released less than 5% of entrapped drug within two hours when mixed with anionic liposomes, but liposomes with 17% cholesterol released more than 90% of 2 13 . entrapped drug within 5 minutes 1.3.2.4 Targeting Ligands To improve the biodistribution of liposomes, many formulations employ endogenous or exogenous targeting ligands. An endogenous targeting ligand is a molecule, often a serum protein, which binds to the liposome during circulation and directs the particle to the ligand's natural cellular target. Exogenous targeting ligands are added to liposomal formulations before injection to bind desired surface proteins on target cells. Advancements have been made in developing and understanding both forms of delivery, thereby improving control of the biodistribution of liposomes in vivo. . The lipoprotein ApoE has been utilized as an endogenous targeting ligand by DLinKC2-DMA based ionizable liposomes. In wild type mice, the formulation silenced greater than 90% of factor VII serum protein with a dose of 0.2 mg siRNA/kg. However, in ApoE knockout mice the same formulation achieved less than 20% silencing at the same dose. Silencing efficacy was restored by incubation of liposomes with recombinant ApoE prior to injection into ApoE knockout mice' 1 2 Retinol binding protein (RBP) is also used as an endogenous targeting ligand. This serum protein binds vitamin A and transports it to cells expressing the RBP receptor, including hepatic stellate and pancreatic stellate cells as well as stellate cells that have become proliferative, fibrogenic, and contractile myofibroblasts 22 ,222. Delivery of RBPmodified nanoparticles to stellate cells has been reported to have efficacy that is five-fold higher in cirrhotic rats as compared to normal rats. These liposomes have been reported to reduce collagen production, thereby reducing liver and pancreatic fibrosis and improving 222 . survival rates in rats 2 2' The use of exogenous ligands has also been examined as a means of targeting distribution and improving efficacy in vivo. The small molecule N-acetylgalactosamine (GalNAc) has been studied as a ligand to target liposomes to the liver, due to its ability to 35 . bind to the asialoglycoprotein receptor on the surface of hepatocytes11 2, while folate has been used to target delivery to rapidly dividing cancer cells 2 2 3 - 2 2 5 . Exogenous ligands are generally attached to the distal end of the lipid anchored PEG groups 12 ,2 2 3 - 2 2 5 . - The LNP drug ALN-VSP, under development at Alnylam Pharmaceuticals, is an example of a lipid delivery system that achieves success by careful incorporation of each of these components. The particles contain the ionizable lipids DLin-DMA and DPPC, the PEGlipid MPEG200-C-DMA, cholesterol, and two siRNAs targeting two genes - vascular endothelial growth factor and kinesin spindle protein 226,227. The resulting particles are 80 100 nm in diameter, have essentially no surface charge at physiological pH, and accumulate in the liver and spleen in preclinical models 226. ALN-VSP was recently evaluated in Phase I clinical trials for the treatment of advanced solid tumors with liver involvement. In this dose-escalation study the drug was generally well tolerated up to 1.0 mg/kg and demonstrated antitumor activity, including complete response in one patient with endometrial cancer. RNAi mechanism of action was confirmed by the detection of mRNA cleavage products in tissue biopsies 226 1.3.3 Conjugate delivery systems A number of promising systems have been developed by directly conjugating delivery material to the siRNA cargo. This approach leads to well-defined, singlecomponent systems that employ only equimolar amounts of delivery material and siRNA. The first conjugate delivery systems to show efficacy in vivo consisted of siRNA conjugated to cholesterol 101 and other lipophilic molecules 111. Other conjugate delivery systems have been developed by attaching siRNA to polymers, peptides, antibodies, aptamers, and small molecules 228. This review will discuss in detail two of the most clinically advanced conjugate platforms, Dynamic Polyconjugates and N-acetylgalactosamine (GalNAc) conjugates, as well as one newly developed system based on oligonucleotide nanoparticles. The first two systems are among the most developed conjugate delivery systems and are both drug candidates. However this work is not well represented in the scientific literature because they are under development by private companies as proprietary technologies. Though not all widely discussed, these systems provide useful lessons on the design of conjugate materials that may benefit the design of future systems. 1.3.3.1 Dynamic PolyConjugates In 2007, Rozema and coworkers reported the development of siRNA-polymer conjugate delivery systems designed to respond to intracellular environments, which they termed Dynamic PolyConjugates 229. These conjugates incorporate several components, each intended to play a particular role in the delivery process (Figure 1.9). The siRNA cargo 36 is attached to a membrane-disrupting polymer via a hydrolysable disulfide linker. The activity of the polymer is masked by PEG side chains while the system is in circulation. Ligands are incorporated to induce uptake by target cells via receptor-mediated endocytosis. The PEG is designed to be shed in the acidic environment inside the endosome, exposing the membrane-active polymer and triggering endosomal release. The disulfide linkage is cleaved in the reducing environment of the cytosol, releasing the siRNA from the delivery polymer. The siRNA itself is chemically modified to improve nuclease stability and to reduce off-target effects. ?M_ BF h rrrl 0 011N H'NH H3 H 1 01 **' 'NH3 +H3 Nll 'NHI + 0 o IQ + 3 _0 0 Is VW4 0 SH wlAV AQM0 Cytoplasm 1X II1 52 1 If Endosome HLHNINH I + 0 0 0 + + 00"" 'S )OWMllr' Figure 1-9. Dynamic PolyConjugates. Dynamic PolyConjugate materials are designed to respond to the acidic environment of the endosome and to the reducing environment of the cytoplasm. In circulation, the membrane-disrupting PBAVE polymer (black) is shielded by polyethylene glycol (PEG). After cell uptake, the PEG chains are shed as the pH of the endosome lowers, exposing the polymer and causing endosomal release. In the cytoplasm, the disulfide bond linking the siRNA to the polymer is reduced, freeing siRNA to trigger RNAi. N-Acetylgalactosamine (GaINAc) is included in the formulation as a targeting ligand to facilitate uptake by hepatocytes. The membrane-active polymer, poly(butyl amino vinyl ether) or PBAVE (Figure 19), has amphipathic side chains that include alkyl groups interspersed with amines that are reversibly linked to the PEG shielding agent and N-acetylgalactosamine (GalNAc) targeting ligands. The alkyl chains are important for membrane activity, as longer alkyl chains (propyl or butyl) were shown to improve the polymer's ability to lyse liposomes in solution 230. PEG and targeting ligands are reversibly linked to the polymer backbone using carboxylated dimethyl maleic acid (CDM) chemistry (Figure 1-9), which allows for the release of the PEG shielding agent in the acidic environment of the endosome. 37 The Dynamic PolyConjugate system was effective at silencing two different genes in the liver when administered intravenously, apolipoprotein B (Apo-B) and peroxisome proliferator-activated receptor alpha (ppara). PolyConjugates were designed to target the liver by incorporation of GalNAc ligands, which bind to the asialoglycoprotein receptor (ASGPR) on hepatocytes. Silencing of Apo-B was dose-dependent and produced the expected phenotypic effects, including reduction in serum cholesterol 229. The importance of individual components of the PolyConjugate was explored, revealing some structure-function insight. The attached GaINAc ligand was essential for both uptake by hepatocytes and in vivo silencing activity. Replacement of GalNAc with glucose greatly reduced hepatocellular uptake, and replacement with mannose directed uptake to nonparenchymal liver cells that express mannose receptors. In primary hepatocytes, it was reported that the PEG shielding moiety must be linked to the polymer via a reversible linkage, as attachment with a nonhydrolyzable linkage abolished silencing activity 229. Newer generations of Dynamic PolyConjugates are under development at Arrowhead Research Corporation 231. The original PBAVE polymers were synthesized by uncontrolled polymerization, resulting in heterogeneity in size and composition. Newer generation DPC polymers are synthesized using controlled radical polymerizations, including atom transfer radical polymerization (ATRP) and reversible additionfragmentation chain transfer (RAFT) to produce homogenous polymers that are more amenable to optimization 231. Hydrolyzable bonds are incorporated into different positions, including the polymer backbone and side chains, a strategy aimed at reducing toxicity. The company also reports the development of longer-circulating DPCs by employing more stable bonds between the membrane-active polymer and the PEG shielding agent 231. The longer circulation time is intended to facilitate the targeting of tissues other than the liver. To this end, the use of other classes of targeting ligands have been explored, including peptides, antibodies, small molecules, glycans, lectins, and nucleic acids. Latest generation DPCs have been reported to induce 99% knockdown of liver genes after a single 0.2 mg/kg dose in nonhuman primates, with the effect lasting nearly 7 weeks 231. A 2012 report on DPC showed that the masked PBAVE polymer could be co-injected with cholesterol-siRNA to induce gene silencing in the liver 232. Though the polymer was not covalently attached to the siRNA in this case, both were targeted to the hepatocytes and colocalized in endosomes, though they did not interact with each other in solution. It was demonstrated that the GalNAc ligand, ASGPR, and the cholesterol moiety were all essential for gene silencing. Interestingly, LDL and LDLR were not required for silencing, though they are required for silencing by cholesterol-siRNA alone 111. This co-injection strategy is reported to be used by the Arrowhead clinical candidate for the treatment of Hepatitis B (HBV), ARC-520 233. This drug contains two cholesterolsiRNAs targeting conserved regions of HBV transcripts. The original PBAVE polymer has 38 been replaced with a melittin-like peptide with similar reversibly-masked endosomolytic properties 234,235. The drug is currently in Phase I clinical trials (Table 1). 1.3.3.2 Triantennary GaINAc-siRNA A liver-targeted siRNA conjugate composed simply of chemically stabilized siRNA with a trivalent targeting ligand has shown promise in the treatment of several diseases. Under development at Alnylam Pharmaceuticals, the conjugates ALN-TTRsc, ALN-PCS, and ALN-AT3 are being studied for the treatment of transthyretin amyloidosis, hypercholesterolemia, and hemophilia, respectively. In this system the 3' terminus of the siRNA sense strand is attached to three GalNAc molecules via a triantennary spacer (Figure 1-10). HO H H OH 0 X FF fNOH OH HAc 00 0'N N 0 *- NA_ 0 OH 0 NHAC Figure 1-10. N-Acetylgalactosamine (GaINAc)-siRNA conjugates. Structure of the triantennary GaINAcsiRNA conjugate used in several drug candidates from Alnylam Pharmaceuticals. The structure of this delivery material is designed for high-affinity binding to its target, ASGPR on hepatocytes. Variations of this triantennary ligand have previously been studied for the purposes of targeting drugs or liposomes to the liver 236,237, and certain trends relating geometry to binding affinity and cell uptake are documented. Multivalency of the sugar ligand greatly improves cell uptake 238,239 and spacing of the sugar moieties also plays a role. In a study of triantennary galactose ligands, binding affinity increased with spacer length over a range of 4 - 20 angstroms 236. Alnylam's GalNAc conjugate similarly employs a triantennary GalNAc ligand with 20-Angstrom spacing that binds to the ASGPR with high affinity (Kd = 2.48 nM 240). A comparison of subcutaneous and intravenous administration of this conjugate revealed both greater accumulation of siRNA in the liver and improved knockdown of the target gene by subcutaneous administration 240. The three drug candidates based on this conjugate are therefore administered subcutaneously. ALN-TTRsc, designed to silence transthyretin (TTR) for the treatment of TTRmediated amyloidosis, is the most clinically advanced of Alnylam's GalNAc conjugates. In nonhuman primates, ALN-TTRsc administered subcutaneously reduced circulating TTR protein by 70% after one week of daily dosing at 2.5 mg/kg 6. This level of TTR expression 39 was maintained by weekly administration of the same dose, and serum TTR gradually returned to pre-dosing levels upon cessation of treatment. Circulating TTR mRNA levels decreased concomitantly. No evidence of cytokine induction, complement activation, or other adverse effects was observed at a dose of 300 mg/kg, indicating a wide therapeutic window. The expected therapeutic phenotype, the reduction of TTR deposits in peripheral tissues, was confirmed in mice by histology 6. A Phase IlIl clinical trial of ALN-TTRsc (Revusiran) was initiated in late 2014 (Tablel). Two other drugs are under investigation using the same GaINAc targeting ligand to deliver siRNA to hepatocytes. By changing the siRNA sequence, this conjugate has been used to silence the expression of two circulating proteins: PCSK9, which plays a role in hepatocellular uptake of low-density lipoprotein, and antithrombin, which regulates thrombin and plays a role in blood clotting. ALN-PCSsc, a conjugate targeting PCSK9 for the treatment of hypercholesterolemia, showed dose-dependent silencing of the target gene in humanized mice, with an EC5O of 0.3 mg/kg 6. ALN-AT3 targets antithrombin (AT) for the treatment of hemophilia and rare bleeding disorders. A single dose of 1.0 mg/kg ALN-AT3 reduced serum AT protein levels in nonhuman primates by 50% and weekly doses as low as 0.5 mg/kg stably reduced serum AT levels by 75-80%. The expected phenotype, increased in serum thrombin levels, was observed to occur in a dose-dependent manner 6. 1.3.3.3 OligonucleotideNanoparticles The construction of 3D nanoparticles of defined composition from nucleic acids has generated significant interests, due to its unique ability to produce a population of molecularly identical nanoparticles with strictly-defined characteristics 241. This approach has been adapted to deliver siRNA molecules 2 4 2 ,2 4 3 . Oligonucleotide nanoparticles (ONPs) were composed of complementary DNA fragments designed to hybridize into predefined three-dimensional structures. Lee and coworkers 242 modified a previously-described method 244 of constructing DNA tetrahedra by incorporating single-stranded overhangs on each edge. SiRNAs were modified by extension of the 3' sense strands with DNA overhangs that enabled hybridization to the edges of the tetrahedra (Figure 1.11). By using unique overhang sequences, six siRNA strands could be attached to each particle, each in a specified position. The resulting particles had a hydrodynamic diameter of about 29 nanometers. ONPs modified with folate ligands were used to study the minimum number of targeting ligands required for delivery and to probe the optimal arrangement of these ligands, questions that are difficult or impossible to address using many other nanoparticle systems. As the position of each siRNA on the tetrahedron could be controlled, siRNAs with or without targeting ligand were assembled into the particles to achieve the desired number and position of folate ligands. A minimum of three folate ligands was required to achieve significant gene silencing. Incorporation of more than three ligands did not drastically improve silencing efficiency. In addition, the positioning of the three ligands was critical. ONPs with three ligands arranged to maximize local density (all three ligands arranged 40 . around one side or one vertex) showed efficient silencing. Arranging ligands to be distant from one another abolished silencing activity. Interestingly, ligand positioning did not affect the rate of cellular uptake - it was hypothesized that the arrangement of these ligands may affect intracellular trafficking of particles 242 Six single-stranded oligonucleotides One-step self-assembly N/v\a AW _i ' tP l Six folate-modified siRNAs with 3'overhangs Figure 1-11. Self-assembly of oligonucleotide nanopartictes. DNA tetrahedra carrying six siRNAs were synthesized in a single step through hybridization of complementary strands. Positioning of each siRNA on the surface of the tetrahedron could be controlled by using unique sequences in each of the siRNA 3'overhangs. This precise synthesis method enabled the study of structure-function relationships such as number and spatial orientation of folate targeting ligands. Folate-ONPs were evaluated for both biodistribution and gene silencing ability in After intravenous injection, mice with folate receptor-expressing tumor xenografts. particles accumulated in both the tumor and kidneys, a pattern consistent with the expression profile of the high affinity folate receptor 245. At a dose of 2.5 mg/kg, folate-ONPs without significant silenced luciferase expression in the tumor by -60% immunostimulation. The DNA particle was required for this efficacy, as free folate-siRNA demonstrated no significant silencing in this model. ONPs are set apart from other nanoparticle systems by the exceptional control over particle structure that is afforded by DNA self-assembly. Particle size, shape, and surface chemistry are known to influence many aspects of performance 246, but the heterogeneity of ONPs many systems precludes the study of specific structure-function relationships. provide a platform to gain structure-function information that can benefit many delivery technologies. 1.3.4 Conclusions 41 The delivery systems that have shown efficacy in vivo exhibit great diversity in structure, size, chemistry, and overall approach to the delivery problem. The delivery systems discussed here are all highly efficacious, promising drug candidates, yet they are each unique in many aspects of their designs. Some have precisely-defined structures while others are heterogeneous. Their sizes range from hundreds of nanometers to about the size of a single siRNA. Some are held together firmly by covalent bonds or precise hydrogen bonding; others are associated by hydrophobic or ionic interactions. Nanoparticles composed of synthetic lipids are among the most potent formulations in the developmental pipeline. Conjugate systems enjoy precisely defined molecular structures with minimal amounts of delivery material, apparently broad therapeutic windows, and their efficacy continues to improve. Though a variety of delivery systems has been developed in the laboratory, challenges remain in bringing the full potential of RNAi to the clinic. The most advanced systems are nanoparticles formed by mixing and self-assembly of various components. This formulation method presents additional challenges in the scale-up of the manufacturing process. Tightly-controlled mixing processes are required to achieve consistent quality of the drug product 2 47 . Microfluidic methods are already in use to improve the quality and reproducibility of LNP formulations 21s, 248. Currently, the most clinically advanced systems target siRNA to well-perfused tissues such as the liver where fenestrated or discontinuous endothelium allows the passage of macromolecules to target tissues. Delivery to less accessible tissues remains a major challenge. Safe and effective delivery of siRNA to the many tissues where it has therapeutic potential will likely require development of a variety of delivery systems, each optimized to address the specific challenges of delivery to a particular tissue. As our mechanistic understanding of the delivery process remains incomplete, we have only some guidelines about optimal delivery material characteristics. Nanoparticulate delivery systems aim for particle sizes of 20 - 200 nm, large enough to avoid renal filtration but small enough to evade phagocytic clearance. Shielding agents such as PEG have proven valuable in preventing nonspecific interactions and avoiding immune recognition in circulation. Chemical modification of siRNA is usually employed to improve nuclease stability and reduce immunostimulation. Exploitation of endogenous or exogenous targeting ligands is often beneficial to improve uptake by target cells. Membrane-disrupting materials that are shielded or neutralized in circulation but become active in the endosome have proven efficacious. Yet even these established guidelines are not set in stone. For example, it is assumed that siRNA must escape from endosomes in order to trigger RNAi, yet we observe efficient silencing from delivery vehicles that incorporate no moiety aimed at achieving this, such as GalNAc conjugates. Such delivery systems reach the RNAi machinery via unknown avenues - they may follow undiscovered intracellular trafficking pathways, they may escape from endosomes by an unidentified mechanism, or they may enter cells in an unknown non-endocytic manner. Continued research into diverse delivery platforms will help to elucidate the biological phenomena that are currently unclear and more guidelines will emerge to aid in the design of delivery materials. Today, important 42 pieces of the delivery process remain unexplained, and there is much room for creativity and innovation in the design of siRNA delivery materials. 1.4 Thesis goals The goal of this thesis is to build the capacity to study siRNA conjugate delivery systems using combinatorial methods. With the recent advancement of certain conjugate platforms to clinical trials, there is great interest in developing novel conjugate delivery systems to However, compared to an target various tissues and treat various disease states. materials, relatively few new abundance of lipid and polymer nanoparticle delivery conjugate delivery materials are reported in the literature. The vast diversity of effective lipid and polymer nanoparticle systems has been developed by the synergy of two approaches: rational design and combinatorial screening. By contrast, the development conjugate delivery systems has relied on rational design alone. Combinatorial library screens have the potential to open new avenues in siRNA conjugate delivery, but conjugate libraries have not been studied due to the many technical hurdles to their synthesis. This thesis aims to overcome these technical hurdles to develop a comprehensive strategy for creating siRNA conjugate libraries that can incorporate vast diversity in chemical structures. In Chapter 2 of this thesis, chemical strategies are developed for library synthesis and siRNA conjugation that are shown to be robust and amenable to high-throughput synthesis. In Chapter 3 this chemical strategy is used to synthesize a library of over 500 siRNA conjugates using a semi-automated process. In Chapter 4 various applications of these materials are explored. Chapter 5 presents strategies for conjugating multiple copies of these materials to each siRNA duplex. 43 Chapter 2: Sequence-defined oligomeric materials for synthesis of nucleic acid conjugate libraries 2.1 Introduction RNA interference-based drugs represent a promising new class of therapeutics with the potential to treat a wide range of human diseases. The major barrier to clinical translation of these drugs is the safe and efficient delivery of nucleic acid to target cells. A wide variety of delivery materials has been studied for more than a decade. Some of these materials have advanced to clinical trials and begun to fulfill the promise of RNAi therapeutics. Yet there is still a need for the discovery of novel materials to target different tissues, increase efficacy, and provide new mechanistic insight into the delivery process. Some of the most potent RNA delivery systems have been developed using a combinatorial chemistry approach, in which large libraries of novel materials are synthesized rapidly and then are screened for their ability to complex with and functionally deliver RNA. This high-throughput approach has led to the discovery of a number of efficacious lipid-like materials that would not likely have been developed by relying on rational design alone. Furthermore, the structure-function information gained from studying libraries can help to guide the rational design of new generations of materials. Conjugate delivery systems, in which delivery material is covalently attached to RNA strands, have been of interest recently. These single-component systems require only an equimolar ratio of delivery material to RNA, and the most advanced clinical candidates are reported to have very broad therapeutic windows 240 . A major limitation to the discovery of new conjugate delivery materials is that they are less amenable to the combinatorial chemistry and high-throughput screening approach that has led to the discovery of many potent nanoparticle formulations. Conjugate systems are usually designed and synthesized individually, making the exploration novel materials laborious. Here we report a synthetic strategy for the high-throughput synthesis of siRNA conjugate libraries. We develop a general method of synthesizing sequence-defined oligomeric materials using a hydroxyproline backbone and a fluorous purification tag. By modifying the carboxylic acid of hydroxyproline, we create a diverse set of monomer units that incorporate various functionalities of interest for siRNA delivery. The hydroxyprolinebased monomers are linked together sequentially to form oligocarbamates. The same chemistry is used to incorporate functionalities for siRNA attachment. Our method enables 44 the exploration of vast chemical space in well-defined materials with high conversion and high purity. Mild reaction conditions and simple purification methods make this strategy readily scalable to multiwell plate format for high-throughput synthesis. In this chapter, we first describe several alternative chemical strategies for the synthesis of synthetic oligomers that were explored and found to be insufficiently robust. The insight gained from studying these potential strategies led us to develop the very This method is then thoroughly robust hydroxyproline-based synthetic method. characterized to demonstrate its applicability to high-throughput synthesis of siRNA conjugates. 2.2: Potential chemical strategies for synthesizingconjugate libraries. 2.2.1 Introduction The aim of this project is to develop a systematic method of making libraries of siRNA conjugate delivery materials. These materials should be able to incorporate a wide variety chemical structures and properties, including particular functionalities that have been observed to promote delivery in other systems. For example, in lipid delivery systems protonatable nitrogen groups, such as dimethylamino groups, are associated with several effective delivery systems 92,249. Alkyl groups are also a major component of these systems. PEG chains are regularly incorporated to improve in vivo performance11 3 114 . A useful synthetic strategy would enable the researcher to incorporate such structures at will and combine these structures to yield predictable products with high purity. Our general strategy for creating such libraries is modeled on peptide chains. Peptides, formed from combinations of amino acid building blocks, can have enormous structural diversity created from a limited number of starting monomers. Each monomer contains the same functionalities necessary for linking monomers together into peptide chains, along with a variable side chain that introduces structural diversity. We envisioned a library of synthetic peptide-like molecules, where monomer side chains are chosen by the researcher. 45 In creating a set of peptide-inspired synthetic oligomers, there are many potential avenues ranging from naturally occurring peptides to completely synthetic materials. Naturally occurring cell penetrating peptides have been studied in various delivery applications 250 . These have the advantages of being non-immunogenic, endogenous sequences, but they are limited in number and allow only the incorporation of natural amino acids. Attachment of natural peptides to siRNA can be challenging. An alternative strategy could pursue the combination of synthetic amino acids, forming a peptide backbone with synthetic side chains. This allows more room for the incorporation of structures of choice, including functionalities for siRNA attachment. Still, peptide synthesis requires multiple protection and deprotection steps, which adds complexity to highthroughput synthesis. We hypothesized that we could create a set of entirely synthetic monomer units that would allow the incorporation of non-natural side chains and that could be linked together without the need for protection and deprotection steps. A synthetic strategy that is scalable and amenable to high-throughput synthesis must meet several requirements. First, the chemical reactions for linking monomers together in to dimers, trimers, and longer chains must proceed smoothly, with high conversion and minimal side reactions, regardless of the properties of the individual monomers being incorporated. Though monomer side chains may be very hydrophobic, very hydrophilic, charged, neutral, large, or small, coupling chemistry must be robust for any combination. Second, a general purification strategy is needed to separate oligomer chains from excess reagents. Purification must also be effective regardless of the properties of each oligomer. Third, functionalities for siRNA attachment must be incorporated, such as azide or cyclooctyne groups. Fourth, a general strategy for conjugating the materials to siRNA and purifying the conjugates is needed. Finally, all of theses steps must be amenable to 96-well plate format, and should be mild and robust enough to be largely carried out by a liquid-handling robot. In this section, we discuss various strategies that were explored for achieving this aim. We discuss the limitations of various chemistries and the observations that led us to develop a strategy that is robust and reliable. 2.2.2 Degradable Ester-linked Oligomers 46 a. 0 3 OH HO HN NH 2 0ZNH 0 R1 b. 0 R ,0OH HN, Rl O Cl H TMS 0 0 Cl HO 0 DMF HN, R R2 2 NH 0 0 R 0 O OH R HN, Rl O Cl 0 0 Cl HO 0 DMF HN, R 0 HN' R.. TMS 0 O 3 0 0 HN R OH 0 HN RR 3 Figure 2.1 Degradable ester-linked oligomers. a.) Monomers were synthesized from amine-substituted butyrolactone. b.) Monomers were coupled together using oxalyl chloride. Recyclization of the lactone monomers reduced the efficiency of monomer coupling. Biodegradability of delivery materials is often desirable as it is observed to reduce the toxicity of some 251 One . systems for strategy synthesizing degradable materials is by linking a hydroxyl group to a carboxylic acid to form ester linkages. Esters are hydrolyzed over in aqueous time environments. To create degradable oligomers, we synthesized model monomers from amino butyrolactone (Figure 2.1). The amine could be readily modified with the desired side chain by amidation. Opening the lactone ring made acid and hydroxyl groups available for monomers linking together into chains. These lactonebased monomers were linked together using oxalyl chloride (Figure 2.1). In pilot experiments, formation from dimer of monomers was detectable by LC-MS analysis, but was inefficient. Reclosure of the lactone ring was highly favored over formation of dimer, even when protecting groups 47 were added to the free hydroxyl (Figure 2.1). The stability of the five-membered lactone ring relative to the linear ester may make these oligomers too unstable to be usable. For this reason, we moved to a synthetic strategy that prevented ring closure, as described in the next section. 2.2.3 Oligocarbamates based on 2-amino 4-hydroxybutyricacid To improve the stability of oligomer chains, we explored replacing the ester linkages with more stable carbamates. Using the same monomer backbone structure, we modified the acid moiety with our side chain of choice, leaving the amine free for linking monomers together (Figure 2.2). Using a starting monomer with a protected amine, the free hydroxyl was activated with excess 1,1-carbonyldiimidazole (CDI). After quenching of excess reagent and thorough drying of the intermediate, the second monomer was added in excess, yielding the carbamate dimer. In pilot experiments, the formation of dimer from monomer proceeded with about 40% conversion when left overnight. Adjusting reaction conditions including temperature, solvent, and amount of excess amine, and addition of 4-(dimethylamino)pyridine (DMAP) catalyst did not reliably improve the yield of product. The reaction of the starting monomer with CDI to form the imidazole-N-carboxylic ester intermediate proceeded smoothly, suggesting that the amine of the second monomer was limiting the progress of the reaction. We hypothesized that a less hindered amine might react more efficiently with the imidazolide intermediate. To test this, we coupled a starting monomer to a model amine compound (amine-PEG, Figure 2.2c). This reaction proceeded smoothly for a variety of reaction conditions and consistently gave complete conversion of starting monomer to carbamate product, according to LC-MS analysis. Based on these results, we moved to a new monomer backbone structure with a less hindered amine. 48 a. PG NH2 PGNH NH PG NH OH NH O 0 CO 0= NH 2 ROH 0 ROH 0 b. c. R1 NH 0 Ri PG OH H PG OH H R2 0 0 N#N AN' OH H2NX R NH 0 N'N KN I H 2N in R2 NH 0 0 PG OH ON N PGH H H flO n N, 40% 100% 0 N'N C) NH OH H-jN I. RI PG N H N from amine-substituted butyrolactone. b.) Monomers FR?2 Ri NH 0 Figure 2.2 Oligocarbamates based on 2-amino 4-hydroxybutyric acid. a.) Monomers were synthesized N 0 N H O N H oH were coupled together using carbonyldiimidazole (CDI) with limited efficiency. c.) The same reaction proceeded efficiently when a model amine was used, suggesting that changing the structure of the monomer backbone may improve coupling efficiency. 2.2.4 Oligocarbamates based on 4-amino 3-hydroxybutyricacid We next explored 4-amino 3-hydroxybutyric acid as a monomer backbone structure (Figure 2.3) because the amine in this molecule is less hindered than in the previous backbone structure and our previous results suggested that this change could improve the reactivity of the amine. As before, we modified the acid moiety of the backbone with the desired side chain by amidation. In the first monomer of the chain, the amine was protected leaving the 49 alcohol to react with CDI. In pilot studies, formation of the imidazole-N-carboxylic ester intermediate proceeded smoothly, and carbamate was formed efficiently upon addition of the second monomer. Conversion of monomer to dimer was consistently over 90% based on LC-MS analysis. These pilot studies were done using monomers whose side chains were uncharged and hydrophobic, such as benzyl groups. OH 'NH 0 H 2N lip H O Figure 2.3 Oligocarbamates based on 4-amino 3-hydroxybutyric acid. Monomer coupling proceeded smoothly with some monomer side chains, but with others coupling efficiency was reduced by elimination. 0 OH PG'N 0 N N' HN'Ri 0 H 0 0 PGIN R N N IN R = dimethylarnino, morpholino R 2 R*NH 1 *NH benzyl, 0 H 2N dodecyl 0 H OH PG'N HNRi 0 H G 0 OH itN H 0'N HNR1 0 NN 0 H N \zJ PG ,N 0 O N H R2N I~1 HN NH 0 PG' 0 O N H 0H' Y 0 HN, R 2 50 To study the versatility and robustness of this method, we synthesized a set of monomers with varying side chains, including aliphatic (dodecyl), aromatic (benzyl), hydrophilic (dioxolane), and positively charged (dimethylamino, morpholino). We then linked these monomers into dimers and trimers in various combinations and observed the efficiency of each reaction by LC-MS. Monomer coupling proceeded smoothly for hydrophobic and uncharged side chains, but was less efficient when side chains were positively charged. With positively charged side chains we observed elimination of the hydroxyl group upon activation with CDI (Figure 2.3). The elimination byproduct was observed in significant amounts (up to 50%), preventing elongation of the chain. These results indicated that in order to incorporate monomer side chains that are positively charged, we would need to change the monomer backbone to a structure that was less likely to undergo elimination. 2.2.5 Oligocarbamates based on 4-amino 2-hydroxybutyricacid Based on prior results, we next explored 4-amino 2-hydroxybutyric acid as a monomer backbone structure. As before, the carboxylic acid group was modified to incorporate variable side chains, and the alcohol of the first monomer was linked to the amine of the second monomer using CDI to form oligocarbamates (Figure 2.4). In pilot experiments, monomer coupling proceeded smoothly and with high yield based on LC-MS analysis. Pilot reactions were carried out using monomers with hydrophobic side chains, usually benzyl. As before, we tested the versatility of this method by synthesizing a set of monomers with varying side chains, including aliphatic (dodecyl), aromatic (benzyl), hydrophilic (dioxolane), and positively charged (dimethylamino, morpholino). We then linked these monomers into dimers and trimers in various combinations and observed the efficiency of each reaction by LC-MS. As in our previous strategy, monomer coupling proceeded smoothly for hydrophobic and uncharged side chains, but was less efficient when side chains were positively charged. In this case, the major side reaction was the formation of an oxazolidinedione ring (Figure 2.4) upon addition of CDI. This byproduct was present in significant amounts (up to 40%) for certain monomer side chains. 51 0 OH J H 2N Figure 2.4 Oligocarbamates based on 4-amino 2-hydroxybutyric acid. Monomer coupling proceeded smoothly with some monomer side chains, but with others coupling efficiency was reduced by cyclization of the imidazoleN-carboxylic ester intermediate. OH 0 NH PG% N OH H 0 IN IIN N R1 O NH PG N H JO IN *N R2 O OH H 2N H 2N 1 NH O PG 1N Q H O O N H N \:zj 0OH ) N GN PGN H NH 0 NH HI N-i R2 Ri 0 0 1 NH 0 N R1 1 N 0 N JO HH NH NH H 52 H2 OH Figure 2.5 Oligocarbamates based on 4-amino 2-hydroxybutyric acid. Monomer side chains were modified to afford a fully substituted amide nitrogen. This change was designed to prevent the cyclization reaction shown in Figure 2.4. OH H 2N 1 R O N PG OH H o N'N I N-' a WN R1 N PGO H R2 O N. OH H 2N 1 R2 O O NPGN O H & 0 HN NOH N H OH R 0 NN N N' (OH HN O 0 ' H O PGN N O-4 R2 IN1 PG N N H N N H H 53 53 To avoid the inadvertent formation of oxazolidinediones, we synthesized a new set of monomers in which all of the side chains were formed by amidation using secondary amines, yielding tertiary amides unlikely to cyclize (Figure 2.5). We observed a similar trend in monomer coupling - reactions proceeded smoothly with hydrophobic and uncharged side chains, but yield of desired product was lower with positively charged side chains. Two side reactions contributed to this decreased efficiency. First, we observed that at least one monomer (dimethylamino) was unstable over time and tended to undergo cyclization to form a 5-membered amide ring (Figure 2.5). Though this problem could be mitigated by using freshly prepared monomer in excess, it would be preferable to be able to Second, formation of a different store monomers for some time before use. oxazolidinedione ring structure was observed after monomers had been linked together into dimers (Figure 2.5). This rearrangement resulted in loss of a monomer side chain. These results demonstrated that the formation of stable 5-membered ring structures is favored when certain side chains are present, and would likely limit the utility of this synthetic strategy for the synthesis of libraries. For this reason, we moved to a new monomer backbone to avoid this ring formation. 2.2.6 Oligocarbamates based on hydroxyproline In our previous synthetic strategies for making oligocarbamates, the efficiency of coupling monomers together into chains was limited by several side reactions that involved the cyclization of monomers or monomer chains to form five-membered rings. As five- and six-membered rings are generally quite stable, it may be difficult to avoid the intramolecular cyclization of a linear oligomer backbone over time. Based on our observations, we hypothesized that a rigid monomer backbone that intentionally includes five-membered rings may be more stable. For this reason we chose hydroxyproline as a monomer backbone structure. 54 HN Figure 2.6 Oligocarbamates based on hydroxyproline. This strategy affords highly efficient monomer coupling as long as the imidazole-N-carboxylic ester intermediate is kept dry. -H OH N PG oOH 0)2 NRT 0 N N H 20 0 PG, N N'\N NZ/ 0 N R1 00H Q HN 0 NR2 0 PG, N 0 N ON 00 N 0 N N"' 0 N-Rj oOH R2 Ri N 0FI N^N )-N 55 NR2 As before, monomer side chains are added to the backbone by amidation of the carboxylic acid. As in our previous strategy, we used tertiary amides to avoid unwanted reaction of the amide nitrogen during oligomer synthesis. The hydroxyl and amine on hydroxyproline were then coupled together using CDI to form carbamates. In this case, monomer coupling proceeded smoothly for all side chains tested, including aliphatic (methyldodecyl), aromatic (methylbenzyl), hydrophilic uncharged (dioxolane), and Complete positively charged (dimethylamino, morpholino, imidazole) (Figure 2.6). conversion of monomer to dimer was consistently observed when a 2- to 6-fold molar excess of amine (second monomer) was used. The only side reaction observed using this synthetic strategy was the gradual hydrolysis of the imidazole-N-carboxylic ester intermediate. This side reaction is easily avoided by limiting its exposure to moisture. Simple procedural adjustments were sufficient to avoid this degradation. First, reactions were carried out in a dry nitrogen environment using dry solvents and thoroughly dried monomers. Second, quenching of excess CDI reagent was done with an equimolar amount of water to CDI, rather than excess water. Finally, an extra drying step was added to the workup of the imidazole carbonyl ester intermediate - benzene was added to the intermediate, which was dried under vacuum to remove residual water in the form of the benzene-water azeotrope. Using this method, we were able to consistently achieve high conversion of monomer to dimer, trimer, and longer chains. These chains were highly stable over time, even when exposed to heat and acidic environments. When coupled with a fluorous purification strategy (discussed in the next section), we were able to achieve good recovery of product with excellent purity. This method is a robust candidate for scale-up to a high-throughput strategy. Reactions proceed reliably for a variety of different side chains, and the only observed side reaction (hydrolysis of the intermediate), results in the loss of a monomer from a chain. While this side reaction is undesirable, it is both avoidable and predictable. In highthroughput synthesis, some impurities will invariably be present in some wells. Preliminary screening of materials is usually carried out on crude mixtures, the composition of which is deconvoluted after hits are identified. As this process can be difficult and timeconsuming, it is extremely helpful to know what impurities to expect. In the synthetic strategy developed here, it is advantageous that the side reactions that do occasionally occur are very predictable, which will facilitate the detailed analysis of results in the future. 56 2.3 Hydroxyproline-basedoligocarbamates provide a robust platform for the synthesis of siRNA conjugate libraries Pilot studies of various chemical strategies indicated that hydroxyproline-based oligocarbamates could provide an effective method for synthesizing siRNA conjugate libraries. To test the robustness and versatility of this method we first synthesized a set of 6 diverse monomers and then combined these into 14 different trimers. Monomers were synthesized by amidation of the carboxylic acid on CbZ-protected hydroxyproline (Sigma), followed by deprotection of the amine (Figure 2.7a). Fluorous-CbZ-protected starting monomers were synthesized by coupling of fluorous-CbZ-NHS ester (Fluorous Technologies) to hydroxyproline, followed by amidation of the carboxylic acid. Coupling of monomers was carried out by adding excess carbonyldiimidazole (CDI, Sigma) to starting monomer in dimethylformamide (DMF) (Figure 2.7b). After 1 h, excess CDI was quenched with water and the activated imidazole-N-carboxylic ester was dried thoroughly. The second monomer was then added in excess and reacted overnight to yield the carbamate product. After the addition of each monomer, solid phase extraction (SPE) on fluorous silica (Fluorous Technologies) provided a simple and general method for purifying fluoroustagged oligomer chains. For a general and scalable purification strategy, we take advantage of a fluorous Molecules tagged with this protecting group that serves as a purification handle. perfluorocarbon chain can be rapidly recovered from a reaction mixture by solid phase extraction through fluorous silica (FSPE, Figure 2.7c). The reaction mixture in DMF is loaded onto the silica matrix and washed with an aqueous solvent, removing excess monomer and any impurities that lack a fluorous tag. The fluorous product is then eluted with acetone. This method afforded efficient separation of oligomers from excess reagents for a wide range of monomer and oligomer properties. Fluorous tags provide the benefit of facile purification afforded by solid-phase synthesis, but with the advantage of solutionphase reaction kinetics and direct analysis of materials without the need for cleavage of materials from the solid support. This method can be readily scaled for high-throughput purification, as fluorous columns are available in 96-well plate format. 57 a. 0 O OH N 0 0 - HN OH O RH0 R NHRNH (: OH b. -OH 0 0 C. OH N O C8 F 1 7 Reaction mixture 10 Ri N.) N N''N 2.) HN 80% MeOH/H 20 Acetone -OH NAN Fluorous R2 silica Silica 0 0 0 C8 F 17 N 0 oR - -OH o C8 F17 RiR2 1.) N 2 0 o 0 0_ S0 coupling using CDI c.) Purification by fluorous solid phase extraction (FSPE) 3 0 O o O N *~ON 0O N o- R1 R2 R3 C0F17 product oligocarbamates. a.) Monomer synthesis b.) Monomer OH N R Fluorous Figure 2.7 Synthesis and purification of hydroxyproline .) HN K 1 Impurities OH H Reactions were monitored by LCMS (Figures 2.8, 2.10, 2.11, Appendix 2.2). Monomer coupling consistently proceeded smoothly and with high conversion, based on the shift in elution time and the detected product mass. Minimal impurities were observed, with the most common impurity being unreacted starting material. This was most likely due to hydrolysis of the imidazole-N-carboxylic ester intermediate, as formation of the intermediate proceeded reliably with complete conversion. Hydrolysis was easily avoided by thorough drying of reagents and by minimizing exposure of the intermediate to moisture. Purity of products was calculated by integration of the LC trace at 214 nm. Product structure was confirmed by 1H NMR and 13 C NMR (Appendix 2.1). 58 811.2047 [M+H]+ AJL C.F, 00 C0, O O '-0H 1120,2130 [M+HI+ A I CF,. .. '''.'j..i'....''i'',..'..'. [M+HI+ 02 0.50 1.00 1.60 2.06 ... 2.50 3.00 min 500 100 1380.5671 1 m/Z '00 Figure 2.8 LCMS analysis of trimer synthesis. LC traces of monomer, dimer, and trimer show UV absorbance at 214 nm. Mass spectra show ions contributing to the corresponding peak in the MS trace. For the 14 trimers synthesized, product purity was consistently high (averaging 94% for dimers and 92% for timers), and recovered yield was variable, averaging 72% per step. We observed reduced yield for the most hydrophilic oligomers, which was due to partial elution of product in the aqueous wash. Reducing the amount of washing improved recovery in subsequent steps. This may also be avoided by using longer perfluorocarbon tags. 59 1 a. 2 3 4 6 N, I N 5 N N R= N N N 0 Rf'~O N b. N R0,' 0 OH R4 0 R1 R2 Purity (%) / Yield (%) R1\R2 1 1 2 90/55 3 4 2 100/48 3 6 96/80 90/94 100/90 100/48 1_ 5 97/76 5 6 4 95/54 80/68 87/86 97/85 1_1100/68 C. 0 Rf^'O fl NH 0O K Rl.0 RI Ri R2 R3 R2 -OH Purity (%) / Yield (%) R1R2\R3 1 2 3 4 6 71/23 12 14 100/48 21 73/83 89/14 78/34 1100/67 25 34 5 100/64 90/54 36 100/82 42 100/87 44 91/58 46 100/97 53 54 99/86 63 95/60 Figure 2.9 Purity and Yield of various monomer combinations. A set of 14 unique trimers was synthesized from six starting monomers. a.) Monomer side chains for monomers 1- 6. b.) Purity and yield are reported for each dimerization reaction. c.) Purity and yield are reported for each trimerization reaction. 60 To evaluate the utility of this method for synthesizing longer chains, we selected a subset of the trimers, which exhibited varying hydrophilicity and charge, and extended these chains to hexamers (Figure 2.10, Appendix 2.2). As before, monomer coupling proceeded smoothly, with essentially complete conversion. Recovered yield averaged 80% per step, an improvement over the average yield observed during trimer synthesis. This improvement is most likely due to adjustments made to the purification process - the amount of aqueous washing was reduced by half, which improved yields without sacrificing purity. ZNOH Z Z [M+H]+ N- N- N- N- 1728.9980 + [M+2H] 2 864.967 6 1020.0685 [M+2H] 2 . 43, yO -OH 0 N- - QO ~N OZ1 OZ DOH O C8F17 0 0 783.0773 0 [M+3H] 3 . O N 1174.6709 [M+2H]24 0.5 1.0 1.5 2.0 2.5 3.0 mn 500 1000 1500 m/z Figure 2.10 LCMS analysis of hexamer synthesis. LC traces of tetramer, pentamer, and hexamer show UV absorbance at 214 nm. Mass spectra show ions contributing to the corresponding peak in the MS trace. We next explored whether hydroxyproline-based oligomers could be conjugated to biomacromolecules. Using the same CDI chemistry, we attached a short polyethylene glycol with an azide functionality to a subset of five selected trimers (Figure 2.11). After purification by FSPE, we removed the fluorous protecting group by heating in trifluoroacetic acid (Figure 2.11). 61 815.2253 cg7^ o C.F N .<)r N [M+H]+ [M+H]+ ~ 1153.3219 N N- NH [M+H]+ 1394.6890 N' N N N ~5N 3 Ok< ... 0 [M+H]+ . . . . . . . . . . . . . . . . . [M+2H] 2 + . 1638.8097 N . . . N N 0 . O 0 < . 0 CsFI?-- 529.8145 [M+H]+ 1058.7411 0.50 0.75 1.00 1.25 1.50 1.75 2.00 2.25 '2.50 2.75 3.00 min 500 1000 1500 m/z Figure 2.11 LCMS analysis of trimer synthesis, coupling to azido PEG, and removal of purification tag. LC traces show UV absorbance at 214 nm. Mass spectra show ions contributing to the product peak in the MS trace. The final LC trace shows peaks representing deprotected product (1.2 min), fluorous-tagged starting material (2.35 min), and free fluorous tag (2.8 min). The azide-modified trimers were conjugated to dibenzocyclooctyne (DBCO)modified siRNA using a 10-fold molar excess of the azide to DBCO. Both fluorous-protected trimers and deprotected trimers conjugated readily to the siRNA using a solvent system of 30% acetonitrile in phosphate-buffered saline (PBS). Deprotected trimers were conjugated without purification, and the presence of the free fluorous protecting group did not appear to affect conjugation efficiency. Excess azide and free fluorous tag were removed from the mixture by ethanol precipitation. Conjugation efficiency was analyzed by HPLC (Figure 2.12, Appendix 2.3). Conjugation consistently proceeded essentially to completion, based on the shift in elution time by H PLC. 62 siRNA Conjugates by HPLC 14 0 12:0 0 C 80 .0 0 - Unmodified siRNA -- 341-siRNA -F341-siRNA 6 0 0 2 0 5 10 15 20 25 30 35 40 45 50 55 60 Time (min) Conjugation of fluorous-protected trimers to siRNA -Unmodified E60 o 120 siRNA s13-F214 ci10 N120 100 s13-F341 80 M 60 0 40 -- s13-F365 40 $ 0 ..--- __ 5 10 15 20 25 30 35 40 45 50 55 60 Time (min) Conjugation of deprotected trimers to siRNA 140 -341 E 120 8 -443 100 -215 @ 80 -Unmodified C60 0 siRNA 40 20 0' 5 10 15 20 25 30 35 40 45 50 55 60 Time (min) Figure 2.12 HPLC analysis of oligocarbamate conjugation to siRNA. Top trace: Azide-modified trimer 341 (structure shown in Figure 2.11) was conjugated to DBCO-modified siRNA. Unmodified siRNA = siRNA-DBCO alone, F341 -siRNA = fluorous protected trimer 341 conjugated to siRNA-DBCO, 341 -siRNA = deprotected trimer 341 conjugated to siRNA-DBCO. Middle trace: Samples of other fluorous-tagged trimers conjugated to siRNA. Side chains corresponding to each number shown in Figure 2.9a. Bottom trace: Samples of other deprotected trimers conjugated to siRNA. Our results indicate that hydroxyproline-based oligocarbamates offer a viable strategy for the high-throughput synthesis of siRNA conjugate libraries. Conversion of monomer to dimer, trimer, and longer chains proceeded smoothly with this chemistry regardless of the properties of the individual monomers. Oligocarbamate chains were all purified by one general method, solid phase extraction through fluorous silica, and were isolated with good yields and outstanding purity. We were able to easily incorporate a functional group for conjugation to biomolecules using the same CDI chemistry and the Deprotection was achieved by simple heating in same FSPE purification method. 63 trifluoroacetic acid, and deprotected oligomers could be purified by FSPE or conjugated directly to siRNA with out purification. Conjugation of various trimers to siRNA was demonstrated using a general method. Finally, a simple and scalable strategy for purification of conjugates, ethanol precipitation, was also demonstrated. Importantly, all of the steps of the synthesis of these carbamate materials, their purification, and their conjugation to biomacromolecules can be carried out in 96-well plate format. 64 Chapter 3. High-Throughput synthesis of a hydroxyproline oligomer-siRNA conjugate library 3.1 Introduction In the previous chapter, we showed that hydroxyproline-based oligomers are wellsuited to high-throughput library synthesis. The chemistry of linking monomers together proceeds efficiently and the FSPE purification method provides excellent purity, regardless of monomer properties. Functional groups for siRNA attachment are easily incorporated using the same chemistry and purification method. Purification tags are easily removed, and deprotected products can be conjugated to biomolecules with or without purification. In this chapter, we apply this strategy to the synthesis of hundreds of diverse materials in parallel. The process is semi-automated, making use of liquid-handling robots to mix reactants in various combinations. Purification is carried out in 96-well plate format using Fluorous SPE plates. We use LCMS analysis to characterize a subset of the materials. We also develop methods of high-throughput conjugation to siRNA as well as highthroughput purification of siRNA conjugates. -N N/ OH OH N0 HN HNO OH HN D-OH No ow /N HN NH "*0H H 04 wHN D 4H Figure 3.1 Monomers for library synthesis. A set of seven unique starting mono- mers was synthesized from hydroxyproline. 65 Using this method we are able to synthesize 504 unique siRNA conjugates. We are able to incorporate a variety of material properties that are relevant to siRNA delivery in a manner that allows for the study of structure-function relationships. The strategy developed here is a robust method for synthesizing libraries of diverse materials conjugated to biomolecules, which can enable the discovery new therapeutically relevant materials, including siRNA conjugate delivery materials. 3.2 High-throughputsynthesis of hydroxyproline-basedoligocarbamates Beginning with seven unique monomers (Figure 3.1), we synthesized a set of 252 unique trimers in multiwell plates. Individual monomers were synthesized conventionally and loaded onto a liquid-handling robot for library synthesis (Figure 3.2). To reduce the exposure of the reactions to moisture, all reaction vessels were dried overnight in a 150'C oven and the space inside the robot was purged with dry nitrogen. First, solutions of starting, fluorous-protected monomers were distributed into wells of a 96-well plate, followed by a solution of CDI. After one hour, plates were cooled to 00 C excess CDI was quenched with 3:1 acetonitrile: water. After quenching, plates were removed from the robot and transferred to a Genevac EZ-2 Evaporator to remove solvent. Once concentrated, the imidazole-N-carboxylic ester intermediates were further dried by redissolving them in benzene and again removing solvents under vacuum in the Genevac. Once thoroughly dried, the plates were transferred back to the liquid-handling robot, which distributed solutions of (deprotected) monomers into each of the wells. The reactions were then stirred overnight at room temperature. 66 Conventional synthesis of 7 starting monomers Purification was achieved by FSPE in 96-well plates. Silica was preconditioned with 15% water in DMF, the reaction mixture was loaded, and 15% water in DMF was used to wash away excess monomer and other impurities. Fluorous product was eluted with THF, and the silica was cleaned with a 0.5% solution of TFA in 1:1 methanol:THF. 0 o HN H N 1N 0N) 0-P HNOH HN o 0_ OH HN 'OH HN OH Automated trimer synthesis --> 252 compounds LCMS analysis showed that synthesis proceeded largely as expected (Figures 3.3, 3.5). Monomer coupling proceeded smoothly for various combinations of monomers. The most common impurity observed was unconverted starting material, most likely due to the hydrolysis of the imidazole-N- High-throughput purification in 96-we 11 Fluorous SPE plates (Figure 3.4). carboxylic ester Hydrolysis was observed more frequently than in pilot studies. This is most likely due to the longer time required for the workup after CDI quenching. In individual benchtop reactions, solvents could be evaporated quickly due to their LCMS Characterization Upon relatively small volumes. scale-up to hundreds of reactions, the larger solvent volume required greater time for evaporation, allowing more time for exposure to moisture and hydrolysis of the intermediate. This problem could be mitigated by using a more Figure 3.2 High-throughput synthesis workflow volatile solvent for this step, such as Overall, tetrahydrofuran (THF). purity of trimers averaged 68% based on integration of LC traces at 214 nm. 67 0 893.3083 0K [M+H]+ OH 0 CNF! ,N 1785.6145 1149.5431 [M+H]+ 1409.7048 0 [M+H]+ 2.0 1.5 min 3.0 2.5 1000 500 1500 m/z Figure 3.3 LCMS analysis of high-throughput trimer synthesis. LC traces of monomer, dimer, and trimer show UV absorbance at 214 nm. Mass spectra show ions contributing to the corresponding peak in the MS trace. 0o ~ OO 0 3 K No..O 0 COF1D 1.0 1.5 Figure 3.4 Impurities. The most common impurity detected in the library was unreacted starting material. In this case, LCMS analysis shows the presence of unreacted dimer. N _-OH _ H 2.5 2.0 0 O 0 Cs CgF17 3.0 1395.6764 [M+H]+ -).oj -OH O min 1153.5270 [M+H]+ t 500 1000 1 500 1000 1500 m/z An azide functionality was then added to the trimer library to enable siRNA attachment. Additional washing steps were added to the purification protocol to remove any traces of excess azide-PEG amine, which, if not completely removed, could reduce the 68 efficiency siRNA conjugation. This extra purification may have reduced recovered yield, but this was acceptable because very small amounts of material are required for siRNA conjugation. A selection of the trimer azides was weighed, and the average recovery was 9.6 mg, roughly a 51% theoretical yield. 811.2047 [M+H]+ H COF17 OLO "I''"I''"..''"." ". [M+H]+ - N 1120.3473 .... ............. "I"M'" I"' " I" !"""'l "if " "- O C&IF OO O SN\ N 4 N 1380.5056 OH [M+H]+ [M+2H] + 2 N 690.7457 - N _-0 S 7 C F1 O O N 1.0 1.5 2.0 N N O ) , 0 1624.7058 'N0 [M+H]+ N 500 1000 1500 M/z Figure 3.5 LCMS analysis of high-throughput trimer synthesis and attachement of azido-PEG. LC traces show UV absorbance at 214 nm. Mass spectra show ions contributing to the corresponding peak in the MS trace. Each of the 252 trimer-azides was split into two batches, one of which underwent deprotection to remove the fluorous purification tag. This doubled the total number of compounds in the library to 504 (252 trimer-azides with fluorous tags and 252 without Deprotection was carried out using iodotrimethylsilane (TMSI) in fluorous tags). dichloromethane at 0*C. After quenching with methanol, the reaction mixture was incubated with sodium thiosulfate beads to remove the iodine byproduct before purification by FSPE. Pilot studies of this reaction showed complete deprotection of the CbZ group without detectable degradation of the trimer backbone (Appendix 3.2). 69 3.3 Conjugation of oligocarbamates to siRNA and conjugate purification We next developed methods of conjugating the carbamate-azide library to siRNA and purifying the siRNA conjugates. While conjugation could easily be carried out in 96well plates, the purification strategy required adjustments to accommodate this format. The standard method of purifying nucleic acid by ethanol precipitation involves briefly freezing samples followed by centrifugation at very high forces (10,000 - 20,000 x g). Adapting this procedure to 96-well plates required adjustments because standard centrifuges designed to run plates reach only forces of 3,000 x g. We compensated for this difference by freezing the samples longer (overnight) and centrifuging longer (90 min). We tested whether purification by ethanol precipitation in 96-well plates afforded consistent recovery of siRNA conjugates. As conjugation of a hydrophobic material to siRNA may alter the solubility of the nucleic acid in ethanol, we were concerned that we may experience a loss of conjugated siRNA when purifying by this method. Also, the slower centrifugation speeds may be insufficient to pellet out precipitated nucleic acid. To test this, we conjugated a hydrophobic trimer (fluorous-protected benzyl-benzylbenzyl, F333) to two different siRNA sequences (siLuc and GaINAc-modified siTTR) across the diagonal of a 96-well PCR plate. The samples were purified by ethanol precipitation as described above, dried, and redissolved in PBS. The concentration of siRNA was measured using a NanoDrop spectrophotometer. Based on the absorbance of the samples at 260 and 280 nm, the average recovery of siRNA was 96%, with all eight samples over 89%. To test whether the added carbamate material contributed significantly to absorbance at 260 (and therefore could skew the measurement of siRNA concentration) we also measured the absorbance spectrum of a concentrated sample of the carbamate alone, which showed peak absorbance at 230 nm and negligible absorbance at 260 nm (Appendix 3.3). We concluded that the standard spectrophotometry method for measuring siRNA concentration (using the absorbance of the sample at 260 and 280 nm) is a reliable method for measuring the concentration of siRNA conjugated to the carbamate materials, and that ethanol precipitation in 96-well plate format affords consistently high recovery of siRNA. We then used this method to conjugate the library of 504 carbamate azide materials HPLC analysis of selected wells showed consistently efficient to siRNA sequences. conjugation and good recovery (Figure 3.6). We observed the same for several siRNA sequences (siLuc, Figure 3.6; siGFP, siAHA1) as well as for siRNA sequences that were also conjugated to a targeting ligand (Folate-siGFP, Figure 2.12, GalNAc-siTTR, Figure 3.6). 70 siLuc conjugates by HPLC 140 E 120 100 80 U C siRNA -Unmodified siLuc-F126 60 -siLuc-111 o 40 20 0 3 7 5 9 11 13 15 17 19 23 21 25 27 29 31 35 33 37 39 Time (min) E GaINAc-siTTR conjugates 80 unmodified 0- - GaINAc-siTTR-72 o- - GaNAc-siTTR-F12 6 0 20 7 9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39 41 43 45 47 49 51 53 Time (min) Figure 3.6 HPLC analysis of oligocarbamate library conjugation to siRNA. Top trace: Azide-modified trimers were conjugated to DBCO-modified luciferase siRNA (top trace) and to an siRNA targeting transthyretin that was modified with both the DBCO moiety and a trivalent N-acetylgalactosamine ligand (GaINAc-siTTR, bottom trace). 3.4 Conclusionsand recommendations Our results show that this method of making hydroxyproline-based oligocarbamates is a viable strategy for the synthesis of siRNA conjugate libraries. In synthesizing over 500 compounds in parallel, we generally observed the results expected based on pilot studies. Monomer coupling and oligomer purification proceeded smoothly, azide functionalities were easily added to chains, and conjugation to siRNA was efficient. 71 We did observe some reduction in efficiency and purity compared to pilot studies, and we believe these issues are easily addressable. For example, reduced efficiency of the CDI coupling reaction was likely due to the increased handling time of the imidazole-Ncarboxylic ester intermediate. This problem can be avoided by using a more volatile solvent for the addition of CDI (THF rather than DMF). Removal of fluorous purification tags can also be made simpler. While pilot studies of this reaction using TMSI showed efficient deprotection without degradation of the trimers, later data suggested that in some cases a small amount of degradation could occur. If a small amount of unwanted cleavage occurs, free azide-PEG-amine may be present in the deprotected product. Even a small amount of this free azide could greatly reduce the efficiency of siRNA conjugation and therefore delivery efficiency. We found that deprotection by heating in trifluoroacetic acid avoided this degradation. In addition, this obviates the need for incubation with thiosulfate beads, leaving a cleaner final product free of thiosulfate salts. We also found that TFA-deprotected trimers could be conjugated to siRNA without FSPE purification, as free fluorous tag does not affect the conjugation reaction. The free fluorous protecting group is then removed during ethanol precipitation of siRNA conjugates. The semi-automated nature of this method places some limitations on the possible throughput. FSPE purifications were not automated and were carried out using a multichannel pipette. Automation of this process is possible but may require some effort. When plate-based FSPE was done by hand, the FSPE plates were fitted to a vacuum manifold to draw solvents through the silica quickly. This plate-compatible vacuum system is not available on most liquid handling robots. FSPE purification can also be done by gravity alone; the plates are loaded with coarse-grained silica for this reason. Using our semi-automated method, the largest number of compounds one could practically synthesize in parallel would be 384, equivalent to four 96-well plates. Splitting each into two batches before deprotection, as we have done here, can double this number to 768. Our method of hydroxyproline oligomer synthesis enabled us to synthesize 504 unique siRNA conjugates in parallel. While purity and yield of materials were acceptable in this pilot study, we also found some simple ways to improve the efficiency and simplicity of the process. Our data demonstrate the robustness of this method and its utility for the synthesis and study of libraries of siRNA conjugate delivery materials. 72 Chapter 4: Potential applications of hydroxyproline-based oligocarbamates 4.1 Introduction In our efforts to develop the capacity to synthesize combinatorial libraries of siRNA conjugates, we have also developed a novel strategy for the synthesis of sequence-defined polymers. These polymers, which can incorporate a wide variety of side chains, could find applications in many fields, including molecular recognition, molecular self-assembly, peptidomimetics, novel enzyme inhibitors, and various other applications that can benefit from library screening. As our original purpose for developing this methodology was for the study of siRNA delivery materials, we chose to focus first on this application, though we hope that other researchers may find these materials useful in other fields. There are a number of strategies available to assess the ability of these materials to deliver RNA to cells. For in vitro assays, cell types, gene targets, siRNA sequences, and Delivery materials can be conjugated to siRNA siRNA modifications can all vary. monovalently or multivalently. They can be the only material conjugated to the siRNA, or they can be conjugated to an siRNA strand that is also modified with a targeting ligand. They can be administered as purified conjugates or with an added excess of delivery material, as a colloidal nanoparticle system. Conjugates can be combined with other delivery materials in an effort to improve efficacy of other systems. Our initial screening efforts focused on assessing the ability of the materials to mediate delivery 1) on their own as monovalent conjugates, 2) as monovalent conjugates alongside a targeting ligand, 3) as conjugates in synergy with delivery lipids, and 4) as delivery lipids themselves, forming nanoparticles. We chose cell types and gene targets for ease of analysis high-throughput, primarily using reporter genes in common cell lines. For the 504 materials screened, we did not observe functional delivery that proved to be reproducible in the assays chosen. Though no outstanding hits were discovered in this first attempt at a library screen, our data provide a number of lessons to guide future screens. We discuss possible explanations for our data as well as our recommendations for future researchers who may be interested in screening this library or other conjugate libraries for functional delivery. Our methodology and recommendations provide a viable 73 platform for researchers to study conjugate libraries with a higher probability of discovering efficacious delivery materials. 4.2 Library screening in dual HeLa cells The first in vitro assay that we chose to screen our conjugate library was in dual HeLa cells, using an siRNA sequence targeted to the reporter gene firefly luciferase. The dual HeLa cell line expresses two luciferase reporter genes (Firefly luciferase and Renilla luciferase) whose expression levels can be measured by a simple luminescence reading. Renilla luciferase serves as an internal control for cell number and cytotoxicity, while firefly luciferase is the reporter gene targeted for knockdown by siRNA. We chose this assay because it is a well-established method for studying functional siRNA delivery in vitro and because it is well suited to high-throughput screening. Reagents are commercially available (Dual-Glo Assay, Promega), the assay protocol is easily carried out in multiwell plates, and luminescence measurements can be taken quickly using a plate reader. We screened the library in these cells at several doses and with varying cell culture conditions. Our initial screens included doses of 500 nM and 200 nM with and without serum for all 504 compounds. We noticed significant well-to-well variation, even among untreated wells, resulting in large error bars. We nonetheless identified several lead compounds that appeared to show knockdown of firefly luciferase without knockdown of Renilla. - The selected compounds were rescreened at multiple doses ranging from 500 nM 5 nM (Figure 4.1). We did not observe the dose-dependent silencing that would be expected for true siRNA delivery. Furthermore, performance at the 500 nM dose did not replicate the findings from the original screen at this dose for any of the 20 compounds. Most showed no silencing at all at the highest dose. For further verification, we conducted an additional repeat screen at 500 nM on 40 of the top-performing compounds from our original screen. None of these compounds showed significant silencing. Overall, results of our initial library screens showed a lack of reproducibility, a lack of dose-responsiveness, and consistently large error bars (Figures 4.1). Silencing by positive control formulations was evident, however, suggesting that compounds with strong 74 enough silencing ability should be detectable despite the noisy readout. Our data thus far suggested that none of the compounds screened was potent enough to be detected by this assay at the current dosing levels. In attempt to overcome this issue, we screened the compounds again at higher doses (2 ptM, repeated twice, and 5p.M). Results were similarly noisy, with a coefficient of variation averaging 0.3 for each treatment, even for non-treated wells. - - - -- - -- - - --- - Rescreen of top-performing compounds: 500 nM - -- 2 .5- E Firefly 0 Renilla 2- - - - - - - - - - - -T- 1 .5 1 0.5 0 Z ~00 I- 0 < ri(4 m n << LA <c -1 C- n ao n oU i 4 M -T UUU n 1r M -I n r JL_- i -i n V LL 00U i 00(f 0 XT X , m q , m -t M Random variation? 12 Firefly ---- - Re fl-a ___1.01 10 -- - .T -T ___ M Increased gene expression: Cell stress? -;o 1.2 - 088 - Firefly 0.44 . Renilla 0.2 2 0.0 0 . 06 , V1N Dose response: Conjugate 331 -- 1.4 0.8 Z 00000WU.WLULUL Figure 4.1 Conjugate screens in dual HeLa cells. This assay produced very noisy data, obscuring results. Compounds that appeared to be demonstrating silencing of firefly luciferase did not show the same effect upon repeat screening and did not demonstrate dose-responsive silencing. In some cases increased gene expression was observed, which could be due to cell stress or random noise. We explored whether various aspects of our methods could be causing the high well-to-well variability in this assay. Using 96-well plates of non-treated cells, we carried out the assay according to the manufacturer's protocol and examined well-to-well variation. We adjusted various aspects of our methods of cell culture, plating, assay timing, and robotics procedures. More specifically we explored plating cells at different densities (20,000 - 100,000k cells/well), adjusting robotics methods and carrying out the assay by hand, changing time points, plating cells at different levels of confluency, thawing new batches of cells, plating cells at various passage numbers, using fresh reagents and reagents that had been previously prepared and frozen, adjusting the ratio of media to reagent, and having different researchers plate cells. None of these changes reliably reduced the The coefficient of variation in luciferase readings observed well-to-well variability. averaged 0.2 or higher (Figure 4.2). This level of error would make it difficult to identify 75 any compounds that silence relatively weakly; only highly potent delivery systems could be identified over the background noise. As our data indicated that none of our conjugates silenced strongly enough to be detected by this assay, we decided to move to a different cell line that exhibited less background noise in reporter gene readout. Variation in non-treated cells Renilla Firefly 300000 MH 30000 H 250000 MG 25000 SG 200000 SF 20000 MF 150000 mE 15000 EE 100000 MD 10000 MD 50000 C 5000 SC 0 EB 0 KB MA 8 9 10 11 12 Firefly/Renilla 'H 20 SG 15 SF 10 KE MD 5B 0' 1 2 3 4 5 6 MB B 6 78 9 10 11 12 MA Figure 4.2 Random variation in non-treated dual HeLa cells. 96-well plates of non-treated dual HeLa cells showed significant levels of background variation in luciferase expression. 4.2 Library screening in KB cells We next screened the library in a KB cell line, which expresses the reporter gene green fluorescent protein (GFP). Expression level of GFP was measured by quantifying total fluorescence of cell lysates using a multiwell plate reader 48 h after transfection. Control experiments showed relatively low well-to-well variation in non-treated cells (CV=0.07), making this cell line an attractive alternative to the dual HeLas discussed in the previous section. 76 We conjugated the 504 library materials to siGFP and screened them for activity at a dose of 1 piM in KB cells (Figure 4.3). Background noise levels in this assay were much improved over the previous assay. However, none of the materials showed significant improvement in silencing ability over siRNA alone. KB cell screen: 1 uM, no targeting ligand - 1.6 ------- -.--.. 1.4 - 1.8 - ~--~ 0. 6 - - 1.2 0- Figure 4.3 siGFP conjugates in KB cells. Selected results of high-throughput screen of 504 siRNA conjugates targeting GFP in KB cells. We also conjugated the library materials to an siGFP sequence bearing a folate targeting ligand. KB cells are known to express folate receptors and are used to study folate-targeted delivery 223,24 2. Folate targeting of drug delivery systems is thought to aid in 245 , but folate-targeted siRNA alone does cellular uptake via receptor-mediated endocytosis not induce RNAi (Figure 4.4). Presumably folate-targeting is sufficient to aid cellular uptake but not endosomal escape' 83. By combining our library materials with folate targeting, we hoped to identify compounds that may aid in endosomal release or trafficking to the RNAi machinery. Such compounds could potentially benefit a number of ligand-targeted delivery systems. In our first screen using the folate-targeted sequence, we identified five compounds that showed modest silencing at a 500 nM dose (Figure 4.4a). We repeated the transfection with these five lead compounds at doses of 500 nM and 1 lM (Figure 4.4b). Of the five, two showed silencing that replicated the data from the initial screen at 500 nM, and both showed dose-dependent silencing. A parallel cytotoxicity assay (Cell Titer-Glo, Promega) showed no detectable toxicity at either dose (Figure 4.4c). The two lead compounds were and FluorousF214 and F215 (Fluorous-protected-dioxolane-dimethylamino-C12 carbamates two these Delivery by protected-dioxolane-dimethylamino-morpholino). appeared to be folate-dependent, as they did not induce silencing in the original screen without folate targeting (Figure 4.4d). 77 1.4 -t------ - 1.6 w 0.8 0-2 I-< 0: 0 r- r- r rN r- r- rN N- N- oo o 0 opo oo oo co oo o o o-a)q)a o-) 0, a)~ q0) o'l a) o- c U Repeat screen: Folate-siGFP-carbamates b. 1.4 T_ 1.2 C 0 la #A LU Cell Viability ATP concentration by Cell Titer-Glo MluM 1000000 M 500 nM 1 800000 0.8 600000 0.6 ti 1 uM 400000 0.4 500 nM 200000 0.2 0 '- 0 ., q iN 4 Control siRNA IF D4 1F D5 14 KB Cell screen Folate-siGFP-carbamates d. of performance of compounds F214 and F215 when conjugated to siRNA sequences with and without folate targeting ligand seems to indicate folate-dependent silencing. a X W a 1.2 - 0.8 0.6 0 .4 0.2 * 500 nM -- - 1 - - - -- - - - C 0 *; - compounds that demonstrated reproducible, dose-dependent silencing. d.) The comparison 1.4 - Figure 4.4 siGFP conjugates with folate targeting ligand in KB cells, a.) Selected results of high-throughput screen of 504 folate-siRNA conjugates targeting GFP in KB cells. Arrows indicate compounds that were selected for rescreening. b.) Repeated screen of selected compounds. c.) Cell viability assay results for . ------ KB cell screen: 500 nM, folate-targeted siRNA a. 0 Z - << < Encouraged by these results, we synthesized larger batches of the two lead compounds by conjugating more of the trimers to the folate-modified siRNA. The same batch of carbamate material was used as had been used previously, it was conjugated to the same siRNA sequence, and conjugation was confirmed by HPLC. Upon transfection of KB cells with the new batch of conjugate, we did not observe the same gene silencing effect. No significant reduction in GFP expression was observed (Figure 4.5). 78 We took several approaches to attempt to replicate the original silencing data with the two lead compounds. The original batch of conjugate, which had repeatably showed silencing, had been used completely in previous experiments, leaving none of the original efficacious batch available for comparison to new batches. As the only difference between the newest batch and the previous batch was scale at conjugation, we first repeated the siRNA conjugation step at the original scale, following exactly the same protocol used originally. We also made additional batches with adjusted conjugation conditions, varying concentration, time, and ratios of reactants. Upon transfection, these new batches also did not show significant silencing, even at micromolar doses (Figure 4.5). F214 and F215 conjugates with folate, different batches 1.2 __ T i.T T 1 T I I 0 I I- 1 4-- III 0.8 CL W 0.6 t ~ 250 nM 0. 500nM 0.4 0.2 0 I 1 uM I_____ kF o (~V ~~ \0 ((f t>1- <V N Different batches of lead conjugates Figure 4.5 Loss of activity in new batches of lead compounds. We also explored the possibility that the differences in results may have resulted from variation in cell culture or plating conditions. We repeated the transfection experiments at different cell plating densities, with freshly thawed cells, and with cells at various passage numbers. These adjustments did not recover silencing activity. We also explored delivery with these lead compounds in another GFP-expressing cell line, 4T1 cells. We did not observe significant silencing by either F214 or F215 at several plating densities. 79 We next explored whether degradation of the carbamate material or the siRNA sequence may have resulted in the loss of activity. The carbamates were purified by HPLC and fractions of the purified, intact trimers were isolated and confirmed by LCMS. We also isolated fractions of some impurities from these materials (which appeared from LCMS analysis to be dimer-azides, F21-azide). These materials were conjugated to the freshest available batches of the siRNA sequence. At doses of 500 nM and 1 pM, these conjugates did not induce silencing (Figure 4.6). These experiments were repeated again later, with newly synthesized carbamate materials, and the results were analyzed by flow cytometry. Again, significant silencing again was not observed, indicating that these particular materials as monovalent purified conjugates most likely do not effectively deliver siRNA in this system, and that the effects observed in the original data sets must have been caused by something unpredicted. HPLC-purified carbamate conjugates (folate-targeted) 1.2 0.8 0.6 0.4 0 nM EM50 El uM 0.2 0 Figure 4.6 HPLC-purified carbamate materials. HPLC purification of carbamate materials did not recover silencing activity in KB cells. We also explored whether excess carbamate material present in the conjugate solution could have contributed to the original silencing effect. Here, we did see some evidence of silencing with a large excess of carbamate (roughly a 2:1 weight ratio of carbamate: siRNA, Figure 4.7). Though it is unclear whether this could explain the silencing observed in the first set of experiments, it does suggest that increasing the amount of delivery material relative to siRNA may improve delivery. Adding excess delivery material also improved the efficacy of some of another material from the library (Figure 4.7). This led us to explore methods of multivalent conjugation and of formulation of these materials as nanoparticles, discussed in other sections. 80 Conjugates with excess carbamate 1.4 0 0. -; 1.2 M 1 Conjugate W +2 uL carbamate 0.8 0.6 +10 uL - X- C:L 0.4 k +30 uL 0.2 0 ~ .O P~~ D <N Figure 4.7 Conjugates formulated with excess carbamate material. Addition of excess carbamate material to purified conjugtes showed some degree of recovered silencing, suggesting that the additional material may improve delivery. 4.3 Hydroxyprolineoligomers as lipid-likenanoparticledelivery systems In the process of characterizing conjugate materials, we observed evidence that some of the carbamate materials may form complexes with siRNA. For example, excess carbamate material was regularly added to siRNA in conjugation reactions, and this excess material had to be removed by ethanol precipitation before HPLC analysis. If the excess carbamate material was not removed, the siRNA product did not elute from an HPLC column (Figure 4.8). HPLC analysis of purified conjugates vs. siRNA with excess carbamate 50:1 mol carbamate F126: mol Luc siRNA 100 E C 0 M 0i U unmodified siRNA 80 siRNA + excess carbamate purified siRNA conjugate 60 40 C 20 0 5 7 9 11 13 15 17 19 21 23 25 27 29 31 33 35 Time (min) Figure 4.8 HPLC studies suggest the formation of complexes when excess carbamate is added to siRNA. Conjugates were usually prepared by the addition of a large excess of carbamate-azide material to DBcO-modified siRNA, followed by removal of excess carbamate by ethanol precipitation and HPLC analysis. If excess conjugate was not removed by ethanol precipitation prior to HPLC analysis, no siRNA was observed to elute from the HPLC column. To test whether the carbamate materials could form particles when mixed with siRNA, a subset of the library materials was formulated with siRNA and particle size was measured by dynamic light scattering (DLS, Malavern ZetaSizer). Formulation was done by 81 ethanol injection, a method that is typically used for making nanoparticles from nucleic acid and cationic lipids. The materials selected were the ones most similar to delivery lipids: those with at least two C12 chains and some cationic component (dimethylamino, morpholino, or imidazole). Of eighteen library materials tested, five formed nanoparticles after simple formulation, with average diameters ranging from 260 - 540 nm. The same materials were also formulated with 1% PEG lipid, C14-PEG2000, a lipid commonly used in lipid nanoparticle formulations. With 1% PEG lipid add, all 18 materials tested formed nanoparticles (Figure 4.9), with average diameters ranging from 100 - 500 nm. Some of these particles showed stability over time, with particle size remaining constant over a 24-h period (Figure 4.9). Carbamate Nanoparticle Size 25 - Particle stability over time Formulated with 1% PEG lipid F414 + 1% PEG lipid I -20 - -i30 -F414 -F441 15 - 10 -1 5 446 0X 464 IA C '544 "- min 24 h 0 M 5 1 10 100 Diameter (nm) 1000 10000 1 10 100 1000 Diameter (nm) 10000 Figure 4.9 Carbamate-siRNA particle sizing by dynamic light scattering. Lipid-like carbamates formed nanoparticles when formulated with siRNA and PEG-lipid. Some formulations showed long-term stability. We next explored whether the particles could deliver siRNA to KB cells. Particles were prepared by the same ethanol injection method but with varying amounts of PEG lipid (none, 0.25%, and 1.25% PEG lipid) and at a higher concentration (10X). The particles were The purpose of formulating at a higher then diluted in PBS and added to cells. concentration was to reduce the final concentration of ethanol added to cells, as toxicity was observed when particles were not diluted. At a dose of 65 nM in KB cells, some formulations stood out as showing silencing of GFP (Figure 4.11). The transfection was repeated with these formulations at two doses (65 nM and 130 nM). One particular carbamate (F144), which showed the strongest silencing of any of the materials, also showed silencing in the repeated experiment. Silencing activity for this material was dosedependent and relatively independent of the amount of PEG lipid present (Figure 4.12). We also explored different ratios of carbamate material to siRNA and found increasing silencing with increasing ratio of carbamate to siRNA (Figure 4.12). A cytotoxicity assay indicated 82 good cell viability for all formulations (Figure 4.13). However, one repeated transfection showed weaker silencing for the same formulations (those with F144) that had showed silencing in previous experiments (not shown). Cationic Lipid complexes 0 1.5 0. 5:1 wt:wt siRNA ------ 1 T- No PEG L aJ 0.5 Lo PEG Hi PEG 0 F_ 0 4~ 0 -4 1-T -J I~T rq : t r- ZT R LI, -* - LI, -t -1 .014 TT k.0 1: .- 1 Iz L U I, '-4 .-4 r- j 'Z3 LnLICIC o'0 Ir '0 -T -1 ui W. -4 .-4 LI mNY1 U Figure 4.11 Carbamate-siRNA complexes in KB cells. Lipid-like carbamates chosen from the library were formulated as nanoparticle complexes with varying amounts of PEG lipid for delivery to KB cells. Hi PEG = 1.25 mol% PEG-lipid, Lo PEG = 0.25 mol% PEG-lipid. Nanoparticle complexes: repeated F144 at different wt:wt siRNA -- - - -- - - No PEG - 1 .2 1 _TTI C .0 0.8 1.2 T--- 1 Wt:wt 1A 0. 6 -~ 0.4 0.2 U 0.8 a No PEG *2:1 e0.60.6 * lo PEG 0.4 hi PEG 0.2 10:1 0 0 -- <<<<N <)t Figure 4.12 Carbamate-siRNA complexes in KB cells, varying in dose and formulation composition. Only one compound showed repeated, dose-dependent silencing when formulated as particles. Increasing the ratio of carbamate to siRNA appeared to improve silencing. Hi PEG = 1.25 mol% PEG-lipid, Lo PEG = 0.25 mol% PEG-lipid. 83 Cell viability ATP levels measured by Cell Titer-Glo 2 1.8 1.6 1.4 1.2- - ------* no PEG M low PEG 0.4 high PEG 0.2 0 Figure 4.13 Carbamate-siRNA complexes in KB cells, cell viability. To address the discrepancy in results from experiment to experiment, we repeated the transfection and analyzed the results by flow cytometry. Previous experiments relied on average measurements of fluorescence in each well, and we hoped that data points on individual cells could provide clearer information as to whether the observed changes in GFP levels were truly due to efficacious siRNA delivery. Several of the carbamates were formulated with siRNA and different ratios of PEG lipid (no PEG, 0.25%, and 1% PEG, 8:1 wt A propidium iodide stain was included in the flow cytometry carbamate: wt siRNA). analysis to gate out dead cells. The data revealed very few GFP-negative cells, even for formulations that had showed silencing of GFP in previous experiments. Two of the formulations showed a very modest population of GFP-negative cells (Figure 4.14), amounting to of cells 4-5% transfected. Carbamate NP transfection in KB cells, FACS analysis ~No PEG m 12 10 "0 10 8 10 Lo PEG _H 40F C M. 100- 1 1 Z 4 2 0 102 F CA 0 PBS F144 F146 F341 F414 F141 Positive controls: Lipo (95% transfection), C12-200 (98% transfection) Figure 4.14 Carbamate-siRNA complexes in KB cells, flow cytometry analysis. Formulations that previously appeared to show silencing did not show activity when GFP expression was analyzed by flow cytometry. a.) Percent transfected cells by formulation b.) Histograms of GFP expression for non treated cells (red) and positive controls (blue = C12-200, green = Lipofectamine RNAiMax) c.) Histograms of GFP expression for non-treated cells (red) and F144 Lo-PEG formulation (blue). Hi PEG = 1.25 mol% PEG-lipid, Lo PEG = 0.25 mol% PEG-lipid. 84 There are several possible reasons for the variation in performance of these formulations across experiments. Formulation procedures, concentration, and ratios of components were adjusted for different experiments and particle size and stability were not characterized for all formulations. These changes in formulation may affect particle formation and stability, and this should be explored more thoroughly if such nanoparticle formulations are to be pursued as delivery agents. The data do suggest that higher ratios of delivery material to siRNA may improve delivery efficiency, suggesting that multivalent conjugation may be helpful to the performance of conjugates. 4.4. Conjugate modificationsas enhancers in other delivery systems Another application of the carbamate library could be to enhance the efficacy of other delivery systems. One efficacious delivery system, a trivalent N-acetylgalactosamine (GalNAc) conjugate targeting hepatocytes, is currently in clinical trials for the treatment of transthyretin amyloidosis. The drug consists of a chemically stabilized siRNA sequence targeting transthyretin (TTR) conjugated to a trivalent linker and three GaINAc ligands. Our collaborators at Alnylam Pharmaceuticals generously provided a version of this molecule that also contained a DBCO group for conjugation to our library of azides. We conjugated the 504 materials to this sequence and screened the conjugates for delivery to primary hepatocytes. TTR mRNA levels were measured by a branched DNA (bDNA) assay. The library of conjugates was added to plated primary hepatocytes at doses ranging from 500 nM - 0.1 nM. Most of the materials showed no improvement over the parent sequence, which achieved 50% silencing at the lowest dose. A small number of these materials (four trimers) showed some modest improvement at the lowest dose (Figure 4.15), but the improvement was not large enough to merit extensive follow up. 85 GaINAc-siTTR conjugates in primary hepatocytes 70.0 50.0 - -I M 500nM - 60.0 40.0 0 1OnM 30.0 M 1nM 20.0 * 0.lnM 10.0 0.0 Parent 766 611 351 F766 726 646 331 666 334 335 411 F333 Figure 4.15. Carbamates conjugated to GaINAc-modified siTTR in primary hepatocytes. Topperforming compounds. As a control experiment to verify that the siRNA sequences could still induce RNAi when modified with the carbamates, the library of conjugates was delivered to primary hepatocytes using Lipofectamine. This confirmed that the sequence was still effective when conjugated to the carbamate trimers. Interestingly, when combined with this transfection reagent a number of the materials showed improved silencing over the parent (Figure 4.16). Analysis of the top 25 most effective materials showed that this group of materials was enriched for monomer 1 (dimethylamino) and monomer 3 (benzyl). In particular, these groups appeared twice in the same monomer about four times as frequently as they did on average in the library. Primary hepatocyte transfection: Synergy with Lipofectamine 70.0 60.0 50.0 40.0 30.0 TT T 20.0 J.U C.0a T i iI I I II L u ii1ti 0 1OnM 4i-i - M0.1nM --- -- ~~-i U11iEEi~ ii I ii ILdI iUiUJJIIhI - 10.0 I I 0~ Figure 4.16 Carbamate-siTTR-GaINAc conjugtes transfected with Lipofectamine. Top -performing compounds. 86 We further explored the possibility that our carbamate materials might synergize with delivery lipids in other systems. We first explored transfection with Lipofectamine in dual HeLa cells using the top-performing compounds from the previous screen (Figure 4.17). Again the modification did not hinder silencing activity, but we did not observe the same synergistic improvement in silencing either. We next explored whether we could replicate the original results in primary hepatocytes. Using the top four carbamates from the original screen combined with Lipofectamine, we did not observe any improvement in silencing over the parent sequence (Figure 4.18). We also explored possible synergy with the lipid C12-200, but did not observe significant improvement in efficacy with the addition of the carbamate materials (Figure 4.18). Transfection with Lipofectamine in Dual HeLas 1.6 0 1.4 1.2 T T * 60 nM ::I 0.1 >. 0.8 JI E I E 30nM S0.6 U. a ElO nM 0.4 M1 0.2 nM 0 NT Parent F111 F251 345 336 Figure 4.17 Carbamate-siLuc conjugates transfected with Lipofectamine in Dual HeLa cells Transfection with C12-200 in primary hepatocytes Transfection with Lipofectamine in primary hepatocytes C 1.6 .0 1.4 0. 1.2 11 400.8 0.26 .0 C 40InM 2 2 1.5 T C1 X 1 40 nM 0.5 & 20 nM 0 Figure 4.18 Carbamate-siTTR-GaINAc conjugates transfected with Lipofectamine and C12-200 in primary hepatocytes. The synergy between the carbamates and Lipofectamine observed in Figure 4.16 was not observed upon repetition of the experiment. 87 It is unclear why synergistic silencing observed in the library screen was not replicated in subsequent experiments, even in the same cell type with the same siRNA sequence. The original observation could be an artifact of noise in the assay readout: in a screen of hundreds of wells there is inevitably a distribution in readout due experimental error, and the lower tail of the distribution may appear to be silencing. As silencing was not observed upon replication of the experiment, we were not convinced of an actual effect of enhanced delivery in this system. 4.5. Recommendationsfor future in vitro screens As a first attempt to screen our conjugate library for functional delivery, we explored various cell-based assays that had previously been useful for screening libraries of lipid nanoparticles. These assays are useful for high-throughput screening because they are simple to execute and they allow for quick measurement of gene expression levels using a microplate reader. However, our results indicate plate reader measurements, which report a total level of gene expression for a well of cells, may not be sensitive enough for conjugate library applications. Interpretation of screening data was complicated by signal-to-noise issues, lack of reproducibility, and even inconsistent controls. Though these cell-based assays were sufficiently sensitive for LNP screens in the past, our data suggest that improvements are needed in order to study libraries of conjugates. None of the conjugate materials in our library appear to silence strongly enough to be consistently detected over the background variation that is unavoidably present in the assays used. There are several reasons why our conjugate library cannot be expected to induce in vitro silencing with the same potency as previous lipid libraries. First, past lipid libraries have been based around a particular delivery lipid that is already known to be efficacious. As we lack a comparable starting point for our conjugate library, we have developed a random library whose range of potency is unknown. Second, LNPs are able to deliver hundreds of siRNA molecules per particle, so a single internalization event can have a significant impact on gene expression, leading to strong signals in cellular assays. Conjugates must achieve hundreds of internalization events to achieve the same level of silencing. Third, the dose of delivery material in conjugate assays is significantly lower than that used in LNP screens. A typical LNP screen (100 ng siRNA in 100 uL, 10:1 wt lipid: wt siRNA) uses only -70 nM siRNA, but this is delivered with roughly 1 ptg delivery lipid per well. A comparable conjugate screen at 1 ptM siRNA includes only -150 ng delivery material per well. Overall, it is not surprising that the high-throughput assays studied may not be sensitive enough to detect the modest levels of silencing we might reasonably expect out of our first attempt at a conjugate library. 88 There are two ways to address this issue: one is to improve assay sensitivity and the other is to improve the potency of the delivery conjugates. To improve assay sensitivity, our results suggest that measuring gene expression levels by flow cytometry is superior to microplate reader methods. By flow cytometry, we observe consistent performance of positive controls and we are able to reproducibly detect very modest levels of silencing (Figure 4.14). Another strategy may be to fluorescently label the siRNA conjugates and observe their internalization or association with cells by flow cytometry, rather than measuring functional delivery. To improve the potency of delivery materials, one method may be to increase the representation of delivery material per siRNA strand via multivalent conjugation (Chapter 5). We observe some evidence that addition of excess delivery carbamate may improve performance (Figure 4.7). Also, we have performed our assays using sequences developed several years ago, and since then significant progress has been made in sequence design for improved potency. Use of improved stabilization chemistry on the siRNA backbone may also improve results. Our work on cell-based screening methods has provided important information to guide future assay development for in vitro screening of conjugate libraries. We have demonstrated the need for more sensitive measurements of gene expression levels and have provided several recommendations for improving upon current assays. In the next chapter, we provide methods for multivalent conjugation of delivery materials, which may improve the potency of conjugates in future screens. 89 Chapter 5. Strategies for multivalentconjugationto siRNA The ability to conjugate multiple copies of a material to siRNA would be beneficial for several applications. In the case of the delivery materials developed here, increasing the representation of the carbamate library materials on each siRNA strand may increase the chances of observing delivery in cell-based screens. Multivalent conjugation would also be useful in the study of novel targeting ligands, which often benefit from multiple ligandreceptor interactions 23 ,240. We developed methods for making siRNA with trivalent clickable groups (azides and cyclooctynes) in order to allow facile attachment of multiple groups to siRNA. This simple method employs commercially available multiarm PEGs (Creative PEGWorks) and can be used to convert any DBCO-modified sequence into trivalent azide, trivalent DBCO, or trivalent bicyclononyne (BCN). a. + / N3-\-(H2H2,nO 3 oCHCH Oln,-N3 2H2)~ \ CH CH2O NaN n N3 9'C (CH 2CH 2 O)n-N3 b. N3 N3 excess 70 Trivalent azide siRNA - E 60 50 -siRNA-(N3)3 40 --- siRNA C30 (0 201 10 0 0 10 20 30 40 50 60 Time (min) Figure 5.1 Synthesis of trivalent azide-siRNA. a.) Reaction scheme b.) HPLC analysis of DBCO-modified siRNA alone and the same siRNA after conjugation to 4-arm PEG azide We first conjugated DBCO-modified siRNA to 4-arm PEG azide (Figure 5.1). Under dilute conditions, the siRNA was added slowly to a large excess (50-100 eq) of the multiarm PEG to afford one-to-one coupling of the two components. HPLC analysis indicated efficient coupling of the two components in a single product peak (Figure 5.1). 90 We also synthesized 4-arm PEG DBCO and 4-arm PEG BCN (Figure 5.2) from purchased 4-arm PEG- NHS ester (Creative PEGWorks) coupled to excess DBCO amine and BCN amine (Sigma). Completely substituted products were isolated by reversed phase flash chromatography. Azide-modified siRNA was made by adding a large excess of diazido PEG (Sigma) to the same DBCO-modified siRNA (Figures 5.3, 5.4). The 4-arm PEG-cyclooctynes were then conjugated to the azide- siRNA (Figures 5.3, 5.4). After each conjugation step, siRNA was thoroughly purified by ethanol precipitation to remove excess reagents. 0 H2N-- N (CH 2CH 2O)n, 0 \\ O (CHH2O)n 2 CH2 0 'kCCH ,(CH 2 CH 2 H)fCH (CH 2 CH2O)n 9 (CH2 CH 20)n (CH 2CH 2O) I - O)n YO 0 (CH2CH20)n 0 H H H2N 'O' 0 O O-, 0 H H (CH 2CH 2O)n,0 H 0 II '(CH 2CH 20)n OxH : (CH 2CH 20)n 9 H H Figure 5.2 Synthesis of 4-arm PEG-DBCO and 4-arm PEG BCN. 91 \- (CH2CHz0)n, .(CHYc-l CH (-JCHCH2(CH 0) excess excess N 3 b. Trivalent DBCO siRNA E 80 C 70 60 j 50 a 40 m 30 .0 _-S13 _S13 azide f1~S13 azide 113-3DBCO o 20 .010 0 15 10 20 30 25 35 40 45 50 6 55 0 Time (min) Figure 5.3 Synthesis of trivalent DBCO-siRNA. a.) Reaction scheme b.) HPLC analysis of DBCO-modified siRNA alone ("S13"), the same siRNA after conjugation to diazido PEG ("S13 azide"), and after conjugation to 4-arm PEG DBCO ("S13-3DBCO") dCH20J.. -:D \_CH2C2~ a. n \CHCHOJ) N3 eoes excess JC CH H1 HCpH~ _N3 excess HH HH H b. Trivalent BCN siRNA 513-3BCN E 70 60 - 50 _ 40 c 513 azide S13 30 - 0 20 10 10 15 20 25 30 35 Time (min) 40 45 50 55 60 Figure 5.4 Synthesis of trivalent BCN-siRNA, a.) Reaction scheme b.) HPLC analysis of DBCO-modified siRNA alone ("S13"), the same siRNA after conjugation to diazido PEG ("S13 azide"), and after conjugation to 4-arm PEG BCN ("S13-3BCN") We next explored whether the library materials developed in Chapter 2 could be conjugated to the trivalent DBCO-siRNA and trivalent BCN-siRNA. Roughly 3 eq of a sample oligocarbamate azide (trimer 341, Figure 2.11) were added to the trivalent DBCO and BCN sequences. By HPLC we observed three shifted peaks in each case (Figures 5.5 and 5.6). We expect that these peaks correspond to mono-, di-, and tri-substituted products, but additional analysis is needed. 92 N -N -N -N ' a. H >N AA p -,p C. b. N Trivalent DBCO conjugates E50 J 30 S13-3DBCO - 40 S13-3DBCO-341 - o 20 .0 IO 10 .0 35 30 40 45 50 55 60 Time (min) Figure 5.5 Conjugation of carbamate trimers to trivalent DBCO-siRNA. a.) Reaction scheme b.) HPLC analysis of trivalent DBCO-siRNA ("S13-3DBCO") and the same conjugated to trimer 341 ("S13-3DBCO-341") N,-'O-O--oO- NY 0O-ko N IH -N 0,; N a. ] H H H JOOOQ HH H b. Trivalent BCN conjugates 45 E 40 S13-3BCN U 335 -S13-3BCN-341 0 o 10 5 0 30 35 40 45 50 55 60 Time (min) Figure 5.6 Conjugation of carbamate trimers to trivalent BCN-siRNA. a.) Reaction scheme b.) HPLC analysis of trivalent BCN-siRNA ("S13-3BCN") and the same conjugated to trimer 341 ("S13-3BCN-341") 93 Chapter 6. Conclusions The field of RNAi therapeutics is coming of age as dozens of RNAi drugs are now in clinical trials (Table 1). The clinical success of certain conjugate platforms has engendered renewed interest in the development of conjugate delivery systems. However, when compared to the vast diversity LNP delivery materials, there are relatively few efficacious conjugate systems reported in the literature. The diversity of successful LNP systems derives from the cooperation of two development approaches: rational design and combinatorial screening. In a rational design approach, individual materials are synthesized with the aim of testing a particular hypothesis. In a combinatorial approach, large libraries of diverse molecules are synthesized and screened to identify hits whose structures may not have been predictable. Hypothesis-driven, rational design methods allow us to systematically test our understanding of the delivery mechanism. However, there are important gaps in our understanding of the delivery process. In particular the intracellular mechanism of transport from the endosome to the RNAi machinery in the cytosol is poorly understood. For many systems, including successful conjugate systems, we have little to no information about how this process happens or how to make it more efficient. When our understanding of mechanism is lacking to this degree, it can be difficult to develop hypotheses to test. Combinatorial screens play the essential role of providing new leads, informing us of what hypotheses to make. Data gathered from screening results can provide structure-activity information to guide the rational design of new materials, and mechanistic understanding gained from rational design experiments can guide the choice of chemical space to explore in new combinatorial screens. The synergy of these two methods has propelled the field of nanoparticle-based delivery systems forward at an impressive rate. Conjugate delivery materials have thus far been developed exclusively by rational design, as the molecularly defined nature of conjugate systems makes a combinatorial chemistry approach challenging. The chemical methods used to make lipid libraries are poorly suited to making conjugate libraries as they do not allow for facile incorporation of functionalities for siRNA attachment and they rarely provide the high purity that is required when materials are attached to siRNA in just a 1:1 molar ratio. Without combinatorial methods for developing new conjugate delivery materials, the discovery of novel conjugate systems has been much slower than that of nanoparticle systems. 94 In this thesis, we have developed a reliable synthetic strategy for creating libraries of conjugate delivery materials. In the process we have demonstrated a novel method for synthesizing sequence-defined polymers that may find applications in a number of fields. We showed that our methodology is scalable to high-throughput automated synthesis and we were able to synthesize a library of over 500 novel siRNA conjugates in parallel. We have done significant work on the development of cell-based assays for the screening of conjugate libraries. We have also developed platforms for multivalent conjugation of library materials to siRNA. Recent clinical success has reinvigorated interest in conjugates, and there is a need for the discovery of novel conjugate delivery materials. Combinatorial library screening is essential to providing new leads in the development of these systems, but synthetic challenges have thus far precluded the use of this method. The synthetic approach developed in this thesis enables the study of conjugate libraries of vast structural diversity, including hundreds of conjugate materials in parallel. The study of conjugate libraries, enabled by this work, has the potential to open vast new directions in the field of siRNA delivery. 95 Appendix 1. Abbreviationsand Symbols Oligomer naming system: "F253" refers to fluorous-tagged ("F") trimer with side chains 2, 5, and 3, with 2 being nearest the fluorous tag. The structure of F253 is shown in Figure 2.8. Structures of monomer side chains corresponding to each number are shown in Figure 2.9a. "253" refers to the same trimer after removal of the fluorous tag. Addition of an azide (Figure 2.11) is indicated by "F341-azide" or "341-azide," or is implied if, for example the trimer is attached to DBCO-modified siRNA. ASGPR - Asialoglycoprotein receptor BCN - Bicyclononyne, (1R,8S,9s)-Bicyclo[6.1.0]non-4-yn C12-200 - Lipid formulation from Anderson et al. (Figure 1.5e) used as positive control for siRNA delivery DBCO - Dibenzocyclooctyne DCM - Dichloromethane DMF - Dimethylformamide FSPE - Fluorous solid phase extraction GalNAc - N-acetylgalactosamine HPLC- High performance liquid chromatography LCMS - Liquid chromatography with tandem Mass Spectrometry Lipo - Lipofectamine 2000 or Lipofectamine RNAiMax MeCN - Acetonitrile MeOH - Methanol NMR - Nuclear magnetic resonance spectroscopy PBS - Phosphate-buffered saline siRNA - Short interfering RNA SPE - Solid phase extraction TFA - Trifluoroacetic acid THF - Tetrahydrofuran TMSI - lodotrimethylsilane 96 Appendix 2. Materials and Methods Appendix 2.1 List of Materials Materials All Fluorous materials (Fluorous CbZ-OSu, Fluorous silica, Fluorous 96-well SPE plates): Fluorous Technologies Multiarm PEGs (4 arm PEG NHS ester, 4 arm PEG azide): Creative PEGWorks RNA sequences were generously provided by Alnylam Pharmaceuticals Cellular assays: Promega Dual Glo, Promega CellTiter-Glo Transfection reagents: Lipofectamine 2000, Lipofectamine RNAiMax (Life Technologies) Cell culture materials: Life Technologies All other reagents were purchased from Sigma Aldrich Instrumentation Flash chromatography: Teledyne ISCO CombiFlash LCMS : Waters Acquity UPLC with a Xevo QToF LC/MS NMR - VARIAN Inova-500 NMR Spectrometer with an Oxford Instruments Ltd. superconducting magnet H PLC: Agilent HPLC with a Waters OST C18 column Dynamic Light Scattering: Malavern ZetaSizer Liquid Handling robot, chemical synthesis: Freeslate CM3 Liquid Handling robot, cellular assays: Tecan Freedom Evo Microplate reader: Tecan Infinite Flow cytometry: BD LSR II HTS, BD FACSDIVA Appendix 2.2 Methods I. Synthesis and Purification of monomers Materials a. Solid phase extraction through fluorous silica (FSPE). containing the Fluorous purification tag were routinely purified by solid In a typical bench scale phase extraction through Fluorous silica. purification for 40 g silica (or 2-3 g reaction mixture), the silica was first washed with 50 mL DMF and then preconditioned with 120 mL 85:15 methanol:water. The reaction mixture was loaded into the silica in up to 10 mL DMF. Non-Fluorous impurities were removed by washing with 150-200 mL 85:15 methanol :water. The fluorous product was then collected by washing the silica with 200-250 mL THF or acetone. The columns were then washed with 200 - 300 mL of 1:1:0.01 methanol:THF:TFA; this fraction often 97 b. contained additional product that could be recovered. This general procedure was scaled to accommodate the amount of material to be purified. Fluorous-CbZ-protected monomers i. Fluorous-CbZ-protected hydroxyproline. N-[4-(1H, 1H, 2H, 2Hperfluorodecyl) benzyloxy-carbonyloxy] succinimide (16 mmol, 11.12 g) was combined with 1.5 eq. (24 mmol, 3.14 g) trans-4hydroxy-L-proline in 65 mL anhydrous DMF. Triethylamine (2 eq, 3.9 mL) was added and the mixture was stirred at room temperature for 2 days or until LCMS analysis of the mixture showed that the NHS ester starting material was completely reacted. The product was purified by solid phase extraction on Fluorous silica. The reaction mixture was split into two batches and loaded on to 60 g of fluorous silica as described. Batches that required further purification after solid phase extraction were re-purified by normal phase flash chromatography on a silica column using a gradient of hexane to ethyl acetate. 90-95% yield. ii. Fluorous-CbZ-protected monomer 1. Fluorous-CbZ-protected hydroxyproline (2 mmol, 1.42 g) was dissolved in 4 mL DMF. Amine side chain (N,N,N' trimethylethylenediamine, 1.5 eq, 3 mmol, 389 microliters) and DIPEA (2 eq, 4 mmol, 695 pL) were added to the mixture. PyBOP (1.2 eq, 2.4 mmol, 1.25 g) was dissolved in 4 mL DMF and added to the reaction. The mixture was stirred at 500 C overnight. Product was purified by solid phase extraction through 40g of fluorous silica. If further purification was required, the product was purified by normal phase flash chromatography on a 25-gram silica column using a gradient of dichloromethane to Ultra solvent (88% dichloromethane, 20% methanol, 2% ammonium hydroxide). 60% yield. iii. Fluorous-CbZ-protected monomer 2. The same procedure was followed as for Fluorous-CbZ-protected monomer 1, using 2methylaminomethyl-1,3-dioxolane (1.5 eq, 3 mmol, 341 ptL) as the amine side chain. Batches that required further purification after solid phase extraction were re-purified by reverse-phase HPLC (Agilent column info here) using a gradient of 5-95% acetonitrile. 60% yield. iv. Fluorous-CbZ-protected monomer 3. The same procedure was followed as for Fluorous-CbZ-protected monomer 1, using Nbenzylmethylamine (1.5 eq, 3 mmol, 385 iiL) as the amine side chain. Batches that required further purification after solid phase extraction were re-purified by normal phase flash chromatography on a 25-gram silica column using a gradient of dichloromethane to 80:20 dichloromethane:methanol. 80% yield. 98 v. Fluorous-CbZ-protected monomer 4. The same procedure was followed as for Fluorous-CbZ-protected monomer 1, using Nmethyldodecylamine (1.5 eq, 3 mmol, 751 ptL) as the amine side chain. Batches that required further purification after solid phase extraction were re-purified by normal phase flash chromatography on a 25-gram silica column using a gradient of hexane to ethyl acetate. 80% yield. vi. Fluorous-CbZ-protected monomer S. The same procedure was followed as for Fluorous-CbZ-protected monomer 1, using 4morpholinopiperidine (1.5 eq, 3 mmol, 504 mg) as the amine side chain. Batches that required further purification after solid phase extraction were re-purified by normal phase flash chromatography on a 25-gram silica column using a gradient of dichloromethane to Ultra solvent (88% dichloromethane, 20% methanol, 2% ammonium hydroxide). 60% yield. vii. Fluorous-CbZ-protected monomer 6. The same procedure was followed as for Fluorous-CbZ-protected monomer 1, using 2-[2-(1HImidazol-1-yl) ethyl] piperidine dihydrochloride (1.2 eq, 2.4 mmol, 605 mg) as the amine side chain, with an additional 2.4 eq of DIPEA to solubilize the amine in a portion of the DMF prior to addition to the reaction. This reaction was run at room temperature overnight. Batches that required further purification after solid phase extraction were re-purified by reverse phase HPLC using a gradient of 20 - 95% acetonitrile. 60% yield. viii. Fluorous-CbZ-protected monomer 7. The same procedure was followed as for Fluorous-CbZ-protected monomer 1, using PEG500amine (1.5 eq, 3 mmol, 1500 mg) as the amine side chain. Batches that required further purification after solid phase extraction were re-purified by reversed phase flash chromatography. 60% yield. c. CbZ-protected monomers i. CbZ-protected monomer 1. Z-Hyp-OH (Aldrich) (13.2 mmol, 3.5 g) Amine side chain (N,N,N' was dissolved in 20 mL DMF. trimethylethylenediamine, 1.5 eq, 19.8 mmol, 2.57 mL) and DIPEA (2 eq, 26.4 mmol, 4.59 mL) were added to the mixture. PyBOP (1.2 eq, 15.8 mmol, 8.24 g) was dissolved in 30 mL DMF and added to the reaction. The mixture was stirred at 50*C overnight. Product was purified by reverse phase flash chromatography on a C18 column using a slow gradient from 100% water to 95% acetonitrile. 90% yield. ii. CbZ-protected monomer 2. The same procedure was followed as for CbZ-protected monomer 1, using 2-methylaminomethyl-1,3dioxolane (1.5 eq, 19.8 mmol, 2.25 mL) as the amine side chain. Product was purified by reverse phase flash chromatography on a 99 d. C18 column using a slow gradient from 100% water to 95% acetonitrile. 88% yield. iii. CbZ-protected monomer 3. The same procedure was followed as for CbZ-protected monomer 1, using N-benzylmethylamine (1.5 eq, 19.8 mmol, 4.96 mL) as the amine side chain. Product was purified by normal phase flash chromatography on a silca column using a gradient of hexane to ethyl acetate (5-95%). 95% yield. iv. CbZ-protected monomer 4. The same procedure was followed as for CbZ-protected monomer 1, using N-methyldodecylamine (1.5 eq, 19.8 mmol, 2.56 mL) as the amine side chain. Product was purified by normal phase flash chromatography on a silca column using a gradient of hexane to ethyl acetate (5-95%). 95% yield. v. CbZ-protected monomer 5. The same procedure was followed as for CbZ-protected monomer 1, using 4-morpholinopiperidine (1.5 eq, 19.8 mmol, 3.33 g) as the amine side chain. Product was purified by normal phase flash chromatography on a silica column using a gradient dichloromethane to Ultra solvent (88% dichloromethane, 20% methanol, 2% ammonium hydroxide). 80% yield. vi. CbZ-protected monomer 6. Z-Hyp-OH (Aldrich) (15.1 mmol, 4 g) was dissolved in 10 mL DMF. Amine side chain (2-[2-(1H-Imidazol1-yl) ethyl] piperidine dihydrochloride, 1.2 eq, 18.1 mmol, 4.56 g) was combined with DIPEA (3.5 eq, 52.8 mmol,9.2 mL) in 15 mL DMF and added to the reaction mixture. PyBOP (1.2 eq, 18.1 mmol, 9.42 g) was dissolved in 35 mL DMF and added to the reaction. The mixture was stirred at room temperature overnight. Product was purified by reverse phase flash chromatography on a C18 column using a slow gradient from 100% water to 95% acetonitrile. 55% yield. Removal of CbZ protecting group to yield monomers 1-6. i. Monomer 1. CbZ-protected monomer 1 was dissolved in DMF to a concentration of 0.25 M and added to palladium catalyst (30 wt% on activated carbon) at a ratio of roughly 3:1 CbZ-protected monomer: palladium on carbon. A hydrogen balloon was attached to the reaction flask, which was stirred for at least 3 h at room temperature. After reaction, the hydrogen balloon was removed and -5 mL of triethylamine was added to the mixture, which was allowed to stir for another 10 min. The catalyst was removed by filtration and rinsed thoroughly with methanol and water. The eluent was concentrated and purified by reverse phase flash chromatography on a C18 column using several column volumes of 100% water followed by a slow gradient up to 20% acetonitrile. 53% yield. 100 II. ii. Monomer 2. The same reaction and purification methods were used as for monomer 1. 74% yield. iii. Monomer 3. The same reaction procedure used as for monomer 1. Product was purified by reverse phase flash chromatography on a C18 column using a gradient of 5-95% acetonitrile. 90% yield. iv. Monomer 4. The same reaction procedure used as for monomer 1. Product was purified by reverse phase flash chromatography on a C18 column using a gradient of 5-95% acetonitrile. 80% yield v. Monomer 5. The same reaction procedure used as for monomer 1. Product was purified by reverse phase flash chromatography on a C18 column using several column volumes of 100% water followed by a slow gradient up to 50% acetonitrile. 70% yield. vi. Monomer 6. The same reaction procedure used as for monomer 1. Product was purified by reverse phase flash chromatography on a C18 column using several column volumes of 100% water with 1% triethylamine, followed by a slow gradient up to 20% acetonitrile with 1% triethylamine. 65% yield. Oligomer synthesis and purification a. Bench top synthesis and purification for individual batches. Fluorous CbZ-protected monomer was weighed in an oven-dried glass vial which was fitted with a rubber septum and fitted with a line flowing dry nitrogen. Ten to twelve molar equivalents of carbonyldiimidazole were added in DMF and the mixture was stirred to dissolved the starting monomer to a final concentration of 0.1 M. After stirring for one hour at room temperature, the mixture was cooled to 0' C before quenching. Excess CDI was quenched with a 1:1 mixture of water and acetonitrile such that a minimum amount of water was added to quench excess CDI (1 molar equivalent of water: excess CDI). The mixture was then evaporated to dryness, then resuspended in benzene and evaporated twice to remove residual water. The material was placed back under nitrogen flow and the second monomer (6 equivalents, deprotected) was added in DMF to a final concentration of 0.1 M starting monomer. The mixture was stirred at room temperature overnight, then purified by solid phase extraction through fluorous silica, as described previously. Yields ranged from 85 - 100% Synthesis steps were b. High-throughput synthesis and purification. carried out by a Freeslate CM3 robot, using 1-mL glass vials in 96-well plate format. All vials, stir bars, and reaction blocks were dried overnight at 1500 C before use. The interior of the robot was purged with nitrogen such that moisture levels were below 150 ppm for the duration of the synthesis. Reagent addition scripts were written in Automation Studio and executed using the Library Studio software. i. Stock solutions. Monomers were dissolved in anhydrous DMF to a concentration of 0.24M for fluorous CbZ-protected monomers and 101 0.58M for deprotected monomers, with the exception of Monomer 3, which was dissolved in 1:1 DMF:THF to 0.12M, and Monomer 5, which was dissolved in 1:1 DMF: MeCN to 0.12M. CDI was dissolved in anhydrous DMF until the solution was saturated. Solutions were stored in the dry environment of the robot. ii. Monomer coupling. Using the single tip lookahead feature of the Freeslate CM3, 12 umol of starting (fluorous CbZ-protected) monomer was added to each well (100 tL of stock solution for monomers 3 and 5, 50 iL for other monomers). Wells were stirred at room temperature while 125 ptL of saturated CDI solution was added to each well using the 6-channel tip or single tip lookahead feature. After addition, the wells were stirred for 1 h. Plates were then cooled to 0 0 C and 5 pL of a 1:1 mixture of acetonitrile: water was added to each well. Alternatively, quenching of CDI could be done outside the robot by placing the plates on ice and adding MeCN: water using a multichannel pipette. After quenching, plates were removed from the robot and dried on a Genevac EZ-2 for -1h. Benzene (50-100 ptL) was added to each well using a multichannel pipette and the plates were stirred briefly before being dried again for -45 min. Benzene was added one more time and the plates were dried thoroughly before being replaced in the robot's glove box. Deprotected monomer stock solution (125 iiL) was added to each well using the robot's 6-tip pipette or single-tip lookahead feature. The wells were stirred overnight at 22'C. iii. Oligomer purification. Oligomers were purified by solid phase extraction through fluorous silica in 96-well plate format. Before use, fluorous silica plates were washed with 1 mL THF per well, followed by 3 mL DMF, using a multichannel pipette to transfer solvents and vacuum to pull solvents through the silica. Each well was then conditioned with 3 mL of 85:15 DMF:water before sample loading. The reaction mixture was then loaded onto the silica in 125 iL DMF. The silica washed with 3 mL 85:15 DMF:water to remove excess reagents, and then the product was eluted with 4 mL of THF. Wells were then washed with 5 mL of 1:1:0.01 THF: MeOH:TFA. This fraction was collected and a selection of wells was analyzed by LCMS as it often contained additional product that could be combined with the previous fraction. Wells were then washed with an additional 2 mL of DMF before their next use. Product fractions were concentrated and transferred to glass 1-mL vials in 96-well plates for the next synthesis step. Yields ranged from 70 - 99%. iv. Coupling to Azido-PEG-amine. CDI coupling was carried out in the same manner as for oligomer synthesis, with the exception that only 102 v. vi. 2 eq of amine (11-Azido-3,6,9-trioxaundecan-1-amine ) were added to the mixed imidazolide intermediate. The same purification Purification of Oligomer-PEG-azides. method was used as for oligomer purification, but with additional washing: after loading the crude Removal of fluorous purification tag. Protected oligomers were dissolved in 80 [tL of 1:1 acetonitrile:benzene using anhydrous solvents. In a dry nitrogen environment, the reaction vessels were cooled to 0"C before the addition of 25 piL of a 1M solution of iodotrimethylsilane (TMSI) in dichloromethane. After stirring for 45 min, the reactions were quenched by the addition of 50 iL of methanol. Thiosulfate bound to polystyrene beads (Sigma) was added to each well and the solutions were incubated overnight to remove residual iodine. The solution of deprotected product was then removed from the well with a pipette, concentrated, and purified by FSPE. Deprotected product was collected in the aqueous fraction. trifluoroacetic acid for 3h. siRNA conjugation, purification, and characterization Purified oligomer-PEG-azides were a. siRNA conjugation conditions. dissolved in 500 ptL MeCN. The DBCO-modified siRNA sequence was dissolved in PBS and stored at a concentration of 1 mg/mL. Oligomer-PEGazide stock solution was added to siRNA stock solution, PBS, and acetonitrile such that the final concentration of siRNA was 0.25 mg/mL in 30% MeCN. For high-throughput synthesis, reactions were prepared in PCR plates and allowed to stir on an orbital shaker overnight. Plates were then frozen at - III. Deprotection was also achieved by heating to 50'C in b. c. 20'C for 1-3h and allowed to thaw in a refrigerator. Purification of siRNA conjugates. The reaction mixture was dried on a Genevac EZ-2 and then redissolved in 30% MeCN/PBS to a concentration of 1 mg/mL. One tenth volume of 3M sodium acetate buffer solution and then 5 volumes of 200 proof ethanol were added to this. The plates were covered and frozen at 20'C overnight, then moved to a -80'C freezer for 1-3 h. For high-throughput synthesis, the plates were then centrifuged at 3,200 rcf for 90 minutes, carefully decanted, and dried. For single-batch synthesis in microcentrifuge tubes, tubes were centrifuged at 15,000 rcf for 20 min before decanting. HPLC characterization of conjugates. Samples were diluted in 0.1M triethylamino acetate (TEAA) buffer to a concentration of 1 pg/50 [tL. Five hundred ng - 1 sg of siRNA was injected for each run on a Waters XBridge OST C18 (2.5 m, 2.1 x 50 mm) column and run on a gradient of 10-95% B (A = 0.1M TEAA in water, B = 0.1M TEAA in MeCN) in 60 minutes at a flow rate of 0.2 mL/min. 103 Appendix 3. Supplementary Data Appendix 3.1 H NMR and "C NMR shifts Monomer 1 Fluorous protected: 0 IH NMR (500 MHz, Chloroform-d) 6 7.37 - 7.27 (m, 2H), 7.19 (m, 2H), 5.18 - 5.01 (m, 2H), 4.88 - 4.75 (m, I H), 4.56 (d, J= 17.5 Hz, 1 H), 3.80 3.71 (m, 1H), 3.70 -- 3.52 (m, 2H), 3.47 - 3.23 (m, IH), 3.15 (d, J= 0.9 Hz, 1H), 2.99 (d, J= 0.9 Hz, 1H), 2.90 (m, 3H), 2.50 - 2.30 (m, 2H), 2.29 - 2.16 (m, 7H), 2.16 - 2.03 (m, 3H). OA - CBF17 N fO NMR (126 MH z, CDC13) 6 173.09, 155.47, 139.58, t35.64, 129.70, 128.97, 120.66, 118.77, 116.76, 113.84, 111.52, 109.11, 70.20, 67.49, 57.79, 56.15, 50.85, 48.66, 46.94, 46.04, 39.38, 35.82, 33.51, 26.75. 13C Deprotected: HN ""'OH N 'H NMR (500 MHz, Methanol-d 4 ) 6 4.41 (m, I H), 4.19 (m, 1H), 3.60 - 3.51 (m, I H), 3.55 - 3.41 (m, 1H), 3.32 - 3.21 (m, 1H), 3.09 (s, 2H), 2.97 (s, IH), 2.78 (m, 1H), 2.64 - 2.41 (m, 2H), 2.30 (d, J= 10.6 Hz, 6H), 2.20 - 2.06 (m, IH), 1.82 (m, IH). N 3 C NMR (126 MHz, Methanol-d 4) 6 173.89, 72.65, 57.22, 56.15, 55.08, 46.13, 45.17, 44.96, 40.01, 34.40. Monomer 2 Fluorous protected: / -/ N 0 N 'OH 'H NMR (500 MHz, 2H), 5.19 - 4.76 (m, (m, 211), 3.51 - 3.26 2.86 (m, 3H), 2.44 - Chloroform-d) 6 7.35 - 7.24 (m, 2H), 7.24 - 7.17 (m, 4H), 4.67 - 4.55 (m, 1H), 4.03 - 3.72 (m, 4H), 3.71 - 3.53 (m, 1H), 3.24 (s, IH), 3.07 (s, IH), 3.02 (s, 1H), 2.96 2.34 (m, 2H), 2.34 - 2.00 (m, 2H). - C8F- 7 - NMR. (126 M Hz, CDC13) 6 173.41,1,54.87, 139.00,1.35.05, 128.97, 128.36, 120.22, 11.8.10, 116.01, 113.19, 110.77, 108.57, 102.26, 70.18, 69.41, 66.89, 65.03, 55.36, 51.89, 50.79, 39.01, 36.58, 32.88, 26.16. 13C Deprotected: 104 'H NMR (500 MHz, Methanol-d 4 ) 6 5.08 - 4.93 (m, 1H), 4.49 - 4.40 (m, 1H), 4.31 (m, 1H), 4.03 - 3.91 (m, 2H), 3.91 - 3.80 (m, 2H), 3.71 - 3.46 (m, 2H), 3.33 - 3.23 (m, 1H), 3.16 (s, 1H), 3.03 (s, 1H), 2.80 (m, 1H), 2.22 - 2.06 (m, 1H), 1.80 (m, 1H). 0 - N o 13 HN C NMR (126 MHz, cd 3od) 6 175.27, 103.14, 73.25, 66.23, 65.89, 57.61, 55.76, 51.90, 40.61, 36.70. 'OH Monomer 3 Fluorous protected: / C 8F1 7 0 S H NMR (500 MHz, Chloroform-d) 6 7.54 - 6.91 (m, 9H), 5.24 - 4.99 (m, 2H), 4.99 - 4.70 (m, 2H), 4.68 - 4.38 (m, 2H), 3.86 - 3.53 (m, 2H), 3.04 (m, 2H), 2.91 (m, 2H), 2.85 (d, J = 19.5 Hz, 1H), 2.45 - 2.21 (m, 3H), 2.16 (m, I1H). N "C NMR (126 MHz, CDC1 3) 6 172.71, 155.04, 139.05, 136.71, 135.18, 129.07, 128.49, 128.18, 127.91, 126.54, 119.86, 118.35, 116.38, 113.19, 110.86, 108.71, 70.19, 66.91, 55.48, 53.21, 51.55, 38.93, 34.67, 31.87, 26.06. Deprotected: HN 'H NMR (500 MHz, Methanol-d4 ) 6 7.35 - 7.13 (m, 5H), 4.77 (m, I H), 4.66 - 4.41 (m, 3H), 3.39 (m, 1H), 3.35 - 3.22 (m, IH), 2.88 (d, J = 4.1 Hz, 3H), 2.49 (m, IH), 1.99 (m, 0 N- I H). 13 - C NMR (126 MHz, cd 3od) 6 174.95, 137.99, 129.95, 129.68, 128.75, 128.49, 127.67, 73.41, 57.80, 55.82, 52.78, 40.90, 34.65. Monomer 4 Fluorous protected: / \ 0 = o=K 1H NMR (500 MHz, Chloroform-d) 6 7.35 - 7.28 (m, 1H), 7.27 - 7.23 (m, 1H), 7.23 - 7.14 (m, 2H), 5.28 - 4.93 (m, 2H), 4.91 - 4.67 (m, 1H), 4.59 (d, J= 18.2 Hz, 1H), 3.92 - 3.74 (m, 1H), 3.74 - 3.57 (m, 1H), 3.57 - 3.42 (m, 1H), 3.36 - 3.19 (m, 1H), 3.12 (s, 1H), 3.04 - C8 F 17 S..OH C 12H 2 5 -N 105 2.79 (m, 4H), 2.47 - 2.30 (m, 2H), 2.28 - 2.06 (m, 211), 1.63 - 1.49 (m, 2H), 1.44 - 1.03 (m, 18H), 0.89 (t, J= 6.9, 2.6 Hz, 3H). ' 3 C NMR (126 MHz, CDC1 3) 6 172.32, 154.89, 138.91, 135.05, 128.91, 128.31, 120.08, 118.12, 116.01, 113.17, 110.80, 108.51, 69.68, 66.90, 55.46, 50.03, 48.55, 38.79, 35.03, 34.15, 32.87, 31.94, 29.66, 29.37, 28.79, 28.45, 26.92, 26.17, 22.68, 13.97. Deprotected: 'H NMR (500 MHz, Methanol-d4) 6 4.81 - 4.70 (m, 1 H), 4.63 - 4.57 (m, I H), 3.52 - 3.25 (m, 4H), 3.06 (s, 2H), 2.98 (s, 1H), 2.59 - 2.43 (m, 1H), 2.05 - 1.90 (m, 1H), 1.72 - 1.52 (m, 2H), 1.36 - 1.27 (m, 18H), 0.94 - 0.86 (m, 3H). OH HN "C NMR (126 MHz, cd 3od) 6 168.97, 71.30, 58.65, 54.95, 50.24, 38.99, 34.47, 32.89, 30.63, 30.60, 30.55, 30.53, 30.50, 30.31, 28.89, 27.57, 23.56, 14.40. N 0 k2H"25 Monomer 5 Fluorous protected: 'H NMR (500 MHz, Chloroform-d) 6 7.37 - 7.25 (m, 2H), 7.25 - 7.15 (m, 2H), - 5.30 - 4.96 (m, 2H), 1H), 3.82 - 3.64 (m, (m, 2H), 2.75 - 2.48 1.69 (m, 2H), 1.62 - 0 O=< N 0 "OH N- 4.95 - 4.78 (m, 1H), 4.70 - 4.42 (m, 2H), 4.18 - 3.83 (in, 5H), 3.61 - 3.45 (m, 1H), 3.31 - 2.96 (m, 1H), 2.95 - 2.76 (m, 4H), 2.47 - 2.30 (m, 3H), 2.29 - 1.99 (m, 3H), 1.97 1.05 (m, 2H). - C 8F1 3 C NMR (126 MHz, cd3od) 6 171.29, 155.67, 139.55, 135.66, 128.96, 128.51, 121.17, 118.75, 116.61, 113.33, 111.55, 108.84, 69.86, 67.59, 62.33, 55.78, 50.11, 45.20, 42.29, 39.17, 33.51, 29.68, 28.88, 26.77. NN 0 Deprotected: HN N N 00 OH 'H NMR (500 MHz, - 4.20 (n, IH), 4.06 - 3.05 (m, IH), 2.88 - 2.42 (m, I H), 2.18 - 1.25 (m, 211). Methanol-d 4) 6 - 3.96 (m, IH), - 2.78 (m, 1H), - 2.07 (m, 1 H), 4.57 3.72 2.77 2.08 - 4.48 (m, IH), 4.46 3.66 (m, 4H), 3.33 2.64 (m, I H), 2.65 1.92 (m, I H), 1.92 - 4.36 3.24 2.55 1.72 (m, (m, (m, (m, 1H), 4.29 2H), 3.18 4H), 2.54 I H), 1.53 3 1C NMR (126 MHz, cd 3od) 6 172.51, 73.23, 67.88, 62.95, 62.88, 57.56, 55.66, 50.97, 50.85, 45.24, 42.55, 40.80, 29.75, 28.97. Monomer 6 106 Fluorous protected: 'H NMR (500 MHz, Chloroform-d) 6 7.98 (d, J= 33.5 Hz, 1H), 7.39 - 7.27 (m, 2H), 7.20-7.13 (m, 2H), 7.10 (s, 1H), 7.05 (d,J= 1.7 Hz, 1H), 5.11 5.02 (m, 2H), 4.92 - 4.79 (m, 2H), 4.58 (dq, J= 4.9, 2.3 Hz, 1H), 4.11 3.91 (m, 2H), 3.84 (d, J= 14.0 Hz, 1H), 3.81 - 3.73 (m, 1H), 3.63 (dt, J 11.5, 1.8 Hz, IH), 3.21 - 3.10 (m, IH), 2.95 - 2.80 (m, 2H), 2.42 - 2.25 (m, 4H), 2.25 - 2.18 (m, 1H), 2.17 - 2.03 (m, 1H), 1.81 - 1.69 (m, 2H), 1.69 1.50 (m, 3H), 1.49 - 1.27 (m, 1H). - C 8F 17 - N *"OH - N ( N/ 3 1C NMR (126 MHz, CDCI3) 6 171.50, 155.07, 139.04, 136.89, 135.14, 128.95, 128.49, 128.25, 125.55, 119.62, 118.23, 116.07, 113.26, 110.85, 108.63, 69.91, 66.92, 60.53, 55.63, 50.12, 45.48, 40.96, 37.86, 32.89, 31.47, 29.33, 26.07, 21.16, 19.18. Deprotected: 'OH - N 'H NMR (500 MHz, Methanol-d4 ) 6 7.79 - 7.65 (m, IH), 7.27 - 7.11 (m, 1H), 7.01 6.89 (m, 1H), 4.41 (ddq, J= 11.0, 5.5, 2.7 Hz, 2H), 4.25 - 4.01 (m, 2H), 3.95 (dt, J= 12.6, 7.3 Hz, 2H), 3.80 - 3.73 (m, 1H), 3.33 - 3.17 (m, 2H), 2.85 - 2.72 (m, 1H), 2.43 - 2.27 (m, 1H), 2.25 - 2.08 (m, LH), 2.07 - 1.87 (m, 1H), 1.87 - 1.68 (m, 2H), 1.67 1.37 (m, 4H). - HN CN 3 1C NMR (126 MHz, CD30D) 6 172.80, 138.56, 128.96, 120.66, 72.97, 58.02, 55.58, 49.85, 48.08, 45.16, 41.15, 32.15, 29.81, 27.12, 19.96. Trimers F122 - - Chloroform-d) 6 7.37 - 7.27 (m, 2H), 7.23 - 7.13 (m, 2H), 5.41 - 4.94 (m, 5H), 4.94 4.37 (m, IH), 4.20 - 4.00 (m, lH), 4.00 - 3.82 (m, 7H), 3.82 - 3.57 (m, 7H), 3.57 3.19 (m, 2H), 3.19 - 3.09 (m, 3 H), 3.09 - 2.77 (m, IOH), 2.57 - 2.42 (m, 2H), 2.42 1.99 (m, 4H). - 'H NMR (500 MHz, 4.55 (m, 4H), 4.56 3.36 (m, 3H), 3.36 2.22 (m, 8H), 2.22 - F144 - - Chloroform-d) 6 7.35 - 7.24 (m, 2H), 7.22 - 7.15 (m, 2H), 5.25 - 4.93 (m, 4H), 4.92 3.57 (m, 811), 3.54 - 3.35 (m, 2H), 3.30 - 3.04 (m, 5H), 3.00 - 2.95 (m, 2H), 2.94 2.68 (m, 2H), 2.55 - 2.42 (m., 2H), 2.40 - 2.19 (m, 9H), 2.14 - 1.94 (m, 3H), 1.75 1.00 (m, 36H), 0.86 (td, J= 7.0, 2.3 Hz, 611). - 'H NMR (500 MHz, 4.42 (m, 6H), 3.88 2.86 (m, 4H), 2.85 1.44 (m, 4H), 1.44 - 3 1C NMR (126 MHz, CDC13) 6 172.35, 154.24, 139.69, 135.50, 129.09, 120.88, 118.79, 116.51, 113.65, 111.45, 108.94, 78.00, 77.94, 77.74, 77.49, 74.32, 73.03, 70.17, 67.59, 57.60, 56.62, 55.68, 53.28, 51.33, 50.50, 48.90, 46.38, 40.79, 40.15, 39.61, 37.79, 36.31, 35.24, 34.49, 33.54, 32.60, 30.32, 30.04, 29.46, 29.12, 27.75, 27.47, 26.86, 23.36, 14.78. F146 107 'H NMR (500 MHz, Chloroform-d) 6 7.50 2H), 7.07 - 7.01 (m, I H), 6.95 (d, J= 11.4 (m, 2H), 3.94 - 3.44 (m, 1OH), 3.22 - 3.10 (m, IOH), 2.15 - 1.83 (m, 6H), 1.75 - 1.61 3H). (d, J= 16.9 Hz, IH), 7.37 - 7.28 (m, 2H), 7.19 (d, J= 7.6 Hz, Hz, I H), 5.26 - 4.94 (m, 4H), 4.87 - 4.35 (m, 5 H), 4.17 - 4.00 (m, 3H), 3.02 - 2.82 (m, 6H), 2.76 - 2.45 (m, 5H), 2.38 - 2.20 (m, 4H), 1.46 - 1.37 (m, 2H), 1.25 (s, 18H), 0.91 - 0.83 (m, 3 C NMR (126 MHz, CDC1 3) 6 172.32, 154.92, 139.80, 137.99, 135.14, 129.10, 129.05, 119.51, 116.45, 113.23, 110.97, 108.87, 106.88, 103.84, 70.94, 69.75, 67.56, 64.46, 56.77, 55.80, 54.71, 53.26, 46.35, 46.21, 43.82, 41.56, 36.96, 32.61, 31.09, 30.35, 30.06, 26.88, 24.55, 23.89, 23.39, 23.37, 19.49, 14.81. F214 - 'H NMR (500 MHz, Chloroforin-d) 6 7.31 - 7.22 (m, 2H), 7.19 - 7.14 (m, 2H), 5.26 - 5.00 (m, 4H), 4.98 4.60 (m, 4H), 4.42 (s, IH), 3.94 - 3.55 (m, 1 H), 3.39 (s, 4H), 3.25 - 3.15 (m, 2H), 3.11 - 3.00 (m, 3 H), 2.98 - 2.78 (m, 9H), 2.48 - 2.39 (m, 1H), 2.38 - 2.07 (m, 12H), 2.08 - 1.85 (m, IH), 1.47 (s, 2H), 1.24 (d, J= 31.8 Hz, 18H), 0.90 - 0.78 (m, 3 H). 3 C NMR (126 MHz, CDC 3) 6 173.04, 154.70, 139.67, 135.41, 129.13, 120.59, 118.78, 116.63, 113.69, 111.38, 109.18, 102.86, 74.48, 70.59, 69.81, 67.65, 65.68, 57.82, 56.93, 55.83, 53.06, 51.02, 49.34, 46.31, 39.84, 38.42, 37.08, 35.66, 34.67, 33.51, 32.56, 30.16, 27.34, 26.83, 23.33, 14.74. F215 Chloroform-d) 6 7.35 - 7.26 (m, 2H), 7.22 - 7.18 (m, 2H), 5.41 - 5.17 (m, 2H), 5.16 4.61 (m, 4H), 4.60 - 4.37 (i, 2H), 4.28 - 4.02 (m, IH), 4.01 - 3.55 (m, 15 H), 3.55 (s, 3H), 3.11 - 2.77 (m, 1OH), 2.76 - 2.62 (in, I H), 2.54 (s, 4H), 2.48 - 2.14 (m, I1 H), 1.77 - 1.58 (m, 1H). - H NMR (500 MHz, 5.02 (m, 2H), 5.01 3H), 3.32 - 3.11 (m, 2.13 - 1.78 (m, 5H), 13 C NMR (126 MHz, CDC 3) 6 173.01, 155.41, 139.63, 135.90, 129.10, 120.98, 118.80, 116.66, 114.02, 111.45, 109.27, 102.88, 74.91, 74.12, 70.36, 67.77, 65.70, 62.36, 57.62, 55.79, 53.46, 52.51, 51.44, 50.51, 45.91, 42.37, 38.45, 37.16, 33.52, 30.39, 29.99, 29.39, 28.84, 28.52, 26.87, 23.39, 22.15. F341 - 'H NMR (500 MHz, Chloroform-d) 6 7.38 - 7.28 (m, 3H), 7.26 - 6.95 (m, 6H), 5.32 - 4.96 (m, 4H), 4.92 4.60 (m, 3H), 4.58 - 4.39 (m, 3H), 3.83 - 3.51 (m, 6H), 3.41 (s, 4H), 3.28 - 3.17 (m, IH), 3.10 - 2.58 (m, 12H), 2.49 - 2.20 (m, 11 H), 2.16 - 2.03 (m, 2H), 1.99 - 1.87 (m, 1H), 1.70 - 1.38 (m, 2H), 1.38 - 0.94 (m, 18H), 0.86 (t, J= 8.3, 4.7, 1.4 Hz, 3H). 3 C NMR (126 MHz, CDC1 3) 6 172.47, 154.63, 139.65, 137.35, 135.49, 129.65, 129.31, 129.07, 128.47, 127.42, 120.81, 118.71, 116.50, 113.91, 111.55, 109.10, 74.40, 70.81, 69.77, 67.52, 57.94, 56.69, 55.84, 53.36, 52.00, 51.19, 50.23, 48.59, 46.29, 40.19, 39.53, 38.99, 37.67, 36.61, 34.74, 33.59, 32.58, 30.31, 30.03, 29.47, 27.51, 26.95, 23.34, 14.75. F365 108 - H NMR (500 MHz, Chloroform-d) 6 7.49 - 6.63 (m, 12H), 5.39 - 5.03 (m, 4H), 5.01 - 4.64 (m, 3H), 4.62 - 4.37 (m, 4H), 4.23 - 3.88 (m, 3H), 3.88 - 3.46 (m, I1 H), 3.45 - 3.38 (m, 3H), 3.19 - 2.95 (m, 3H), 2.95 2.73 (m, 4H), 2.71 - 2.20 (m, 11H), 2.18 - 1.76 (m, 6H), 1.74 - 1.42 (m, 4H), 1.43 - 1.16 (m, 3H). 13C NMR (126 MHz, CDC13) 6 171.78, 154.63, 140.19, 137.44, 136.86, 135.46, 130.05, 129.59, 129.32, 129.08, 128.33, 127.22, 120.45, 120.04, 118.92, 116.63, 113.80, 111.43, 109.22, 74.35, 70.67, 67.81, 62.50, 55.99, 53.35, 52.11, 51.23, 50.42, 45.29, 44.30, 42.51, 42.14, 39.37, 37.87, 34.90, 33.40, 32.30, 30.09, 28.87, 26.96, 25.31, 19.26. F425 - 'H NMR (500 MHz, Chloroform-d) 6 7.33 - 7.20 (m, 2H), 7.20 - 7.12 (m, 2H), 5.29 - 5.00 (m., 4H), 4.97 4.65 (m, 311), 4.63 - 4.35 (m, 3H), 4.06 - 3.79 (m, 5H), 3.77 - 3.50 (m, IOH), 3.39 (d, J= 1.0 H1z, 5H), 3.17 - 2.94 (m, 4H), 2.93 - 2.85 (m, 4H), 2.84 - 2.73 (m, 2H), 2.61 - 2.12 (m, 11 H), 2.11 - 1.71 (m, 5H), 1.67 - 1.38 (m, 2H), 1.36 - 1.03 (m, 18H), 0.83 (t, J= 6.3, 1.8 Hz, 3H). "C NMR (126 MHz, CDC1 3) 6 171.92, 154.47, 139.80, 135.48, 129.68, 129.07, 120.87, 118.74, 116.60, 113.40, 111.29, 109.19, 102.50, 74.12, 70.66, 69.80, 67.65, 65.69, 62.48, 55.90, 53.03, 51.30, 50.45, 49.05, 44.52, 42.18, 39.92, 37.55, 36.21, 34.57, 33.47, 32.55, 30.25, 30.16, 30.00, 29.02, 27.48, 26.82, 23.31, 14.72. F443 - 'H NMR (500 MHz, Chloroform-d) 6 7.40 - 7.02 (m, 9H), 5.26 - 4.96 (m, 4H), 4.95 - 4.72 (m, 2H), 4.70 4.35 (m, 4H), 3.87 - 3.57 (m, 6H), 3.40 (s, 6H), 3.13 - 2.97 (m, 3H), 2.93 - 2.82 (m, 6H), 2.82 - 2.68 (m, 2H), 2.39 - 2.13 (m, 6H), 1.75 - 1.34 (m, 4H), 1.34 - 0.89 (m, 36H), 0.85 (t, J= 6.9, 2.1 Hz, 6H). 1 3C NMR (126 MHz, CDC1 3) 6 172.61, 154.63, 139.61, 136.74, 135.41, 129.90, 129.47, 129.02, 128.28, 127.00, 120.44, 118.75, 116.64, 114.01, 111.56, 109.45, 74.51, 70.21, 67.52, 55.95, 53.39, 50.98, 48.83, 39.75, 37.98, 34.71, 33.51, 32.57, 30.30, 30.14, 30.11, 30.02, 29.45, 27.53, 26.83, 23.33, 14.73. F465 - - 'H NMR (500 MHz, Chloroform-d) 6 7.42 - 7.32 (m, IH), 7.33 - 7.22 (m, 2H), 7.23 - 7.13 (m, 2H), 7.12 6.90 (m, 2H), 5.41 - 4.96 (m, 5H), 4.97 - 4.68 (m, 2H), 4.68 - 4.36 (m, 3H), 4.27 - 3.89 (m, 3H), 3.89 3.38 (, i5H), 3.37 - 2.98 (m, 4H), 2.98 - 2.75 (m, 5H), 2.53 (d, J= 7.8 Hz, 6H), 2.43 - 2.15 (m, 6H), 2.14 - 1.78 (m, 5H), 1.78 - 1.44 (m, 5H), 1.46 - 1.26 (m, 2H), 1.27 - 1.07 (m, 18H), 0.89 - 0.76 (m, 3H). 13 C NMR (126 MHz, CDC1 3) 6 172.11, 170.75, 154.66, 139.95, 136.94, 135.29, 129.97, 129.07, 120.46, 120.07, 118.92, 116.50, 113.55, 111.43, 109.20, 74.55, 70.43, 67.80, 62.54, 55.91, 53.20, 51.21, 50.45, 48.89, 45.28, 44.30, 42.32, 39.40, 37.77, 35.47, 33.55, 32.57, 30.21, 29.00, 27.55, 26.85, 25.72, 23.34, 19.39, 14.75. F534 109 - 'H NMR (500 MHz, Chloroform-d) 6 7.55 - 6.78 (m, 9H), 5.41 - 4.93 (m, 4H), 4.93 - 4.30 (m, 6H), 4.24 3.91 (m, I H), 3.91 - 3.51 (m, 9H), 3.42 (d, J= 0.6 Hz, 4H), 3.14 - 2.76 (m, 8H), 2.74 - 2.45 (m, 7H), 2.25 (s, 6H), 2.15 - 1.59 (m, 6H), 1.28 (s, 20H), 0.91 - 0.76 (m, 311). 13 C NMR (126 MHz, CDC1 3) 6 172.66, 170.53, 154.52, 139.77, 137.29, 135.67, 130.09, 129.39, 129.05, 128.24, 127.18, 120.63, 118.77, 116.63, 113.58, 111.27, 109.19, 74.71, 71.43, 69.96, 67.67, 62.28, 55.90, 53.28, 51.36, 50.33, 45.07, 42.30, 39.77, 37.24, 35.37, 34.37, 33.48, 32.59, 30.32, 30.03, 29.05, 27.17, 23.62, 14.78. F542 Chloroform-d) 6 7.37 - 7.22 (m, 2H), 7.20 - 7.11 (m, 214), 5.23 - 5.05 (m, 311), 5.04 4.34 (m, 4H), 4.18 - 3.81 (m, 5H), 3.81 - 3.49 (m, 1IH), 3.38 (s, 6H), 3.19 - 2.92 (m, 4H), 2.77 - 2.53 (m, 3H), 2.52 - 2.22 (m, 9H), 2.05 - 1.80 (m, 4H), 1.66 - 1.37 (m, 18H), 0.83 (t, J= 7.1, 3.6, 1.7 Hz, 3H). - 'H NMR (500 MHz, 4.71 (m, 3H), 4.68 4H), 2.91 - 2.80 (m, 2H), 1.37 - 0.94 (m, 13 C NMR (126 MHz, CDCI 3) 6 172.93, 170.53, 154.93, 154.19, 139.57, 135.54, 128.98, 120.65, 118.70, 116.61, 113.66, 111.32, 109.17, 102.81, 74.34, 70.17, 67.68, 65.72, 62.35, 55.86, 52.88, 51.17, 50.42, 48.45, 45.12, 42.24, 39.77, 37.37, 34.84, 33.59, 32.55, 30.05, 28.92, 27.19, 23.31, 14.71. F632 - H NMR (500 MHz, Chloroform-d) 6 7.60 - 7.39 (m, IH), 7.38 - 6.67 (m, 1IH), 5.37 - 4.88 (m, 5H), 4.86 - 4.59 (m, 4H), 4.58 - 4.48 (m, IH), 4.47 - 4.31 (m, IH), 3.98 - 3.65 (m, 9H), 3.65 - 3.52 (m, 3H), 3.41 (d, J= 0.5 Hz, 8H), 3.23 - 3.12 (m, IH), 3.10 - 2.73 (m, 8H), 2.73 - 2.47 (m, 3H), 2.42 - 2.17 (m, 5H), 2.15 2.10 (m, 1H), 2.06 - 1.89 (m, 2H), 1.77 (s, 111). 13 C NMR (126 MHz, CDC1 3) 6 171.54, 154.31, 139.73, 137.67, 137.23, 135.53, 129.63, 129.33, 128.95, 128.34, 127.09, 119.70, 118.90, 116.63, 114.00, 111.38, 109.47, 107.03, 102.71, 74.55, 69.60, 67.55, 65.71, 56.04, 52.76, 51.31, 47.16, 44.83, 41.40, 40.17, 37.79, 35.16, 33.05, 30.14, 27.02, 19.84. Appendix 3.2 LCMS analysis of trimer and hexamer synthesis Trimers synthesized in Chapter 2: LC traces @ 214 nm. Mass spectra show ions contributing to peak in MS trace of trimer. 110 F122 I 0.50 1: 1KF122_fresh_1CuL 176 (1 596) Crm (172 181) 1308.3196 100-i 2.50 225 200 1.2575 1.00 75 L1309.3483 2175 3.00 p Time 3.25 TOF MIS ES+ 1 14e6 654.6742 250 ........ .. ,. 500 . . . . . . . .1. 1 000 . . H- 1000 750 1 1250 1500 1500 0.75 &SO 1M 125 150 1 1 75 . 200 .... F144 , .2.2 . . 225 RKF144_dil 284 (2.547) Cm (22:288) 1472.6122 100- m/z .1 F14 Fl F144 11750 2.50 2.7 2.75 Time 3.00 32 1: TOF MS ES+ 9.28e .1473.6342 1474.6632 736.8069 0250 500 750 1000 1250 1500 1750 111 Fl F146 F146 F14 ----------------0 050 1 125 175 150 2.00 2.25 250 2.75 3.00 3.25 1: TOF MS ES+ 1.96e6 RK_F146fresh_3uL 194 (1.741) Cm (189:200) 726.7626 100 Iol -727.7617 1452.5959 I I1-1 UL 750 500 250 II m/Z 1000 1250 1500 1750 F214 1.00 0 50 2.00 1 0 F21 3.00 2.50 F214 F2 PKF214_fresh-dil_2 242 (2.175) Cm (240:245) 1390.5074 100- 1: TOF MS ES+ 4.92e5 1 390.4775-J1 392.5259 695.7391 0- 250 -%- 'I 500 750 100I 1250 -pP-FR-; m/Z 1500 112 F215 0,50 I1.00 200 1 .so F215 250 F2 F21 RKF215_fresh_dil 152 (1,368) Cm (150 157) 1 TOF MS ES+ 8 07eD 681.1881 100,1 ,,681.6757 L1362.4178 1361.4189 .,682.1821 0- 500 250 I7 750 F253 ,....,I.. 1250 1000 m/Z 250 300 1760 F2 F25 F253 050 ,. .. . , .. , Timem 075 100 150 125 175 200 225 275 PKF253 _dil_5uL 195 (1 .749) Cm (195:198) 1380.5576 100, 325 1: TOF MS ES+ 5.25e5 1381.5792 ,1382.5966 690.7449 0 m/Z 260 500 750 1000 1250 1500 1750 113 F341 F341 F3 F34 [341 [341 F3 F34 0.O PR 05 10 2A1.''2 12 .5 0 ,5 200 2.50 2.25 2.75 F341 BuL 259 (2.317) Cm (256:261) . 1: TCF MS ES+ 1 03e6 1394.6890 100- 1Tlime 3.25 1 3,00 l1395.7103 ,1396.7494 I 0 250O 5 0 750 1000 1250 1750 1500 ,I M/Z F365 L; 1.5k F365 1.00 0.50 2.00 2.50 F36 3.00 F3 RK_F365_5uL 161 (1.454) Cm (159:164) 721.7708 100] 1: TOF MS ES+ 5 92e5 L722.2648 1442.6532 Li 443.6576 ,722.7689 I 0 -I 250 500 L 750 1 m/Z 1000 1250 1500 1750 114 F425 0 50 0.75 1.00 1.50 15 1.75 200 2 2 25 . 275 3.00 8.56e5 1458.7059 100 ,1459.7343 j+ 1460.7714 I I i-i 500 750 1500 1250 1750 F4 1.50 1.00 M/Z _________________I 1000 F443 0.50 3.2f 1: TOF MS ES+ RK_F425_8uL 252 (2.258) Cm (250:254) 250 F4 F42 F425 2.00 2.50 3.50 3.00 RK F443 long_dil 380 (3.392) Cm (378:386) 1491.8455 100- F44, F443 Time 4.00 1: TOF MS ES+ 4.40e5 ,1492.8517 02.0 A m/z A 600 750 1000 1250 1500 1750 115 F465 a 1 00 . 0.50 1.50 I F4 F46 F465 RkF465 3.00 2.50 2.00 1 TOF M-AS ES+ 7 39e5 uL 205 (1 .844) Cm (202 207) 760.8513 L761 3452 1520.8215 ------- 0 250 500 750 1500 1250 1000 F5 F53 F534 . .. ., 1.00 ,0* ... 1',,......,. , ,...0.75 0.50 1.25 1.50 m/z 1760 F534 1.75 2.00 2.25 2.50 2.75 3.00 .2i Time 3.25 1: TOF MS ES+ RKF534_5uL 251 (2.250) Cm (248:253) 8.18e5 1462.5201 100- !146 4.5765 731.7589 0 500 750 1000 1250 1500 1760 I m/z 116 F54/F542 F5 FS 42 Time 0.75 0.50 1.50 1.25 1.00 200 1,75 225 250 100- .4 F6 F632 0.75 0.50 rri/ . .I 1060 750 500 1.25 1.00 325 579e5 1458.7223 250 300 1; TOF MS ES+ RKF542_6uL 241 (2.166) Cm (240:243) 0 2,75 1250 1500 1750 2. .0 F63/F632 1.5 .75 20 RKF632_fresh 201 (1.810) Cm (194:203) 389.3875 100-, 27 .0 .71 Tim2 3.25 1: TOF MS ES+ 7 .21e6 1390.3912 695.1772 0 250 500 750 1000 1250 1500 1750 117 F25326 F253262 F2532,F253262 214 0,50Da Range 3 1440-1 3.0e-1 2.001.0e-1-- 0.60 0.00 1.00 1.20 1.40 160 1.80 200 220 2.40 2.60 2.80 3.00 3.20 RKF2532_Ac_6uL 192 (1.724) Cm (189:195) 1636.7245 6.93e5 100- RKF26326_Ac _1uL 150 1.416) Cm (156 1162) 3 24e5 978.4323 100 PKF253264_Ac 201 (1 810) Cm (199:203) 4 33e5 1147.5789 100- 0 500 M/Z 1000 1500 118 F4433 F443341, F44334 F443341 214 O.5ODa 342e-1 6.0e-14.0e-1 7 2 50 3009 2.0e-1- 0.00.50 i 1.00 ,.5 ,2.00 i , ' ' 1.50' 1.00 Time 1 '2.0 Ib I 2.50 3.00 3.50 4.00 4.50Ti e RK_F4433_Ac 384 (3.437) Cm (379:387) 1: TOF MS ES+ 3,29e5 1751.9911, 1773.9697 10 -1775.9764 875.3495 1302.7145 I - [1- " . . RK F44334_Ac 417 (3.724) Cm (408:417)1 TOF MS ES+ 4.29e4 875.3499 100- K876.3543 .1046.0966 -Pp-L k . A.A RKF443341_Ac_dil 322 (2.887) Cm (319:327) 3 44e5 1166.6953 100- 1166.1857, ,1167.1918 ,1167.6902 0 600 L1000 .,. 1500 i m/z 119 F54236, F542363 F542363 F5423 2'14 U.bUUa Range: 3 12 7e-1 3.0 e-1 2.0 e-1 1 .0 e-1 0.0 0. 0.50 0.75 . 1.00 1.25 1.50 1.75 2.00 2.26 2.50 2.75 3.00 T me 3.25 RKF5423_Ac_diI3 246 (2 208) Cm (244:248) 6.83e5 1718.8962, ,1720.9270 859.8965 0 ;,. ...-. ., .- ..y 4v, 1 . ' 11 0- 1 RKF54236_Acdil 202 (1 818) Cm (201 205) 1019.5101 5.986e5 10U-U ,1020.0222 ,1020.5206 i ------,I; -----nIIII' ' II I4 I - OL I RK F542363_Ac dil 206 (1 652) Cm (202:210) 1149.5846 5 08e 1001149.0811, ,1150.0858 11 I.. - II ,1150.5851 50 I ll ' 100101 15 0 m/z 120 High throughput synthesis (Chapter 3): F122 RKFMmerl ] 26-Nov-2014 1: TOF MS ES+ RKFMmerl 175 (1.571) Cm (171:140) 2.3996 796.0820 4 Diode Array 214 100 Range 1 320 1.0 [.1139 nfl 0 40 1 . 120 1.0 1.40 2.0 220 2.60 2.40 2.80 3.40400 ri-q 3.20 RKan. 1.8. Pmmc_0iay Range. :2 RK L2_1AB dimer di2 182 (1.641) Cm (178:186) 1212 3157-e 1052.3145 100 1054.4021 9.230e-1 7 04 F.6- 50.0e-1 00 1. TOF MS ES+ a0 :796.2473 .~__________ 0.80 1.20 1.0 141 40 2.0 14 2.20 240 2.40 4: Diode Array 214 Range: 5,8999-1 RK L2_1AB _nimer 5.0e-1 040 0. 1.4 140 0-14 140 0 3.20 3.0 2.40 1 TOF MS ES+ 183 (1 649) Cm (179.188) 1 12e6 1052.3486 1308.4878 RKL21A8_utriumer 100 654.7222 2.40 220 2.40 2.0 2.60 300 RK_L2_1A0_tinmerPEG-1OuL 214 Range '1310.5492 3.20 5 292e-1 4: Diode Array R) L2 1A8 trimerPEG 10--- tOuL 194 (1.741) Cm (109 201) 1296.5067 1552,6423 0.0 .0 040O . . .2. 1.40 120 100 8 27e0 1553.0747 O~e-I 140 1460 ... 2400 220O 240 240O ..... 3.00 2.40 7673 645.7134 Time 320 20 4 1554.3144 112343 76.793 1123 4324 700 103 1250 1750 1500 mnz 27,jun,2013 HT synthesis: Removal of purification tag using TMSI STOF MS 1, an s 1579.45591 DenprTeV 4: Dod1 May 8%0+1 SM Ix 6,0- 50-U 10 6 1 R.ng, 151048 Mass: 1578.5 1 23021 797.1402 8".1704 302 100+1 1 4. IO 1,9 2w 211) 2,A i RE~pprohtA34 2 4 3 DA-)M 3 20 Z 2w 7 3,I0 "14.3 637.43330","1 "u'4N28w e .Eer 1084.3104 M + PEG + Na [,..3i. 2.5-1 Mass: 998.5 7T 21 27' 57 76 21 23 170 2 2110 30 3.10 187,058 low 3104 30.^''^+^20..1 13 35230 32 ol 369 1071370 2Q23 'I.....6W560264 260 ' 4Do ' VA421Q sOJ 107314 %, 34116 144416 60 ' 1653 011727 21301 , RK 10bD 121 120D ' nb 1600 Appendix 3.3 High-throughputpurificationof siRNA conjugates HT purification of siRNA conjugates: Yield Large, hydrophobic carbamate (F333-azide) + siLuc-DBCO GalNAc-siTTR-DBCO Concdntration 7.00 I 0 1.00 2.00 "k 1.00i - 2211 - 0.00-230 2 250 260 270 280 20 3Ui Wavelength mn 310 320 330 340 Sample (% of control) Al (siLuc-F333) 96.2 B2 (siTTR-F333) 92.6 C3 (siLuc-F333) 103.7 D4 (siTTR-F333) 89.1 E5 (siLuc-F333) 93.4 F6 (siTTR-F333) 100.1 G7 (siLuc-F333) 96.5 H8 (siTTR-F333) 92.1 Average % yield 95.5 3g0 Carbamate alone (5-10x the concentration in the conjugation reaction) Absorbance spectra taken by NanoDrop spectrophotometry of recovered siRNA conjugates after ethanol precipitation. Eight test samples and three controls, which were made up to a concentration corresponding to 100% theoretical yield. Carbamate alone refers to F333 azide modification. 122 Appendix 3.4 HPLC analysis of siRNA conjugates Conjugation of Fluorous-protected carbamates to siRNA 140 E 120 W 100 80 e 60 -513 S13-F425 S13-F542 40 0 20 03 13 23 43 33 53 Time (min) Conjugation of deprotected carbamates to siRNA 90 -Unmodified E 80 siRNA 70 60 50 C 40 . 30 020 -- 425 -- 542 S10 0 5 10 15 35 30 Time (min) 25 20 40 45 50 55 60 Conjugation of deprotected carbamates to siRNA 140 -Unmodified siRNA -341 E 120 2100 N % -443 80 C 60 o 40 S20 0 5 10 15 20 25 35 30 45 40 50 60 55 Time (min Comparison of Fluorous-protected and deprotected conjugates for the same trimer: siRNA conjugation by HPLC 160 E 140 N @J W 0U .0 0 120 -Unmodified siRNA 100 -214-siRNA 80 60 40 20 0 -S13-F214 5 10 15 20 25 30 35 40 45 50 55 60 Time (min) 123 siRNA conjugation by HPLC 180 E 160 140 Unmodified siRNA - 120 -- 365-siRNA S100 80 m F365-siRNA - 060 40 20 0 5 10 15 20 25 35 40 45 50 55 60 40 45 50 55 60 40 45 50 55 60 Time (min) siRNA conjugation by HPLC 100 E 80 - Unmodified siRNA % 60 40 -- 215-siRNA -- F215-siRNA 0 0 10 5 15 20 25 30 35 Time (min) siRNA conjugation by HPLC 100 E 80 60 -Unmodified siRNA i 40 - 542-siRNA -- F542-siRNA .0 4 20 0 5 10 15 20 25 30 35 Time (min) 124 Appendix 3.5 Synthesis of multivalentDBCO and BCN linkers Starting material 4-arm PEG2000-NHS, analyzed by LCMS, was observed to separate into two fractions of observed ionized masses 1255 and 1326. Assuming this is (m+2)/2, the average mass of these two fractions would be 2508 and 2650, respectively. Mass spectra are shown below: 1: TOF MS ES+ 4.64e3 RK_4PEGNHS-std 202 (1.819) Cm (199:204) 100- 526.2375 614.2972 482.2097,2 ,746.3916 ,943.1784 1326.2477 436.8330 OL b-#NNYFq -lwl1-I 1: TOF MiS ES+ 4.45e3 - K_4PEG_NHS_std 193 (1.732) C:rri (189:194) 852.1204 100896.1545 793.0830 1255.7 155 482.2162,70.2737 ,1 343.7802 1 409.8275 1111. 0 The expected mass of fully substituted 4-arm PEG BCN (Figure 5.2, bottom) is then 3344 and 3486 for these two fractions, respectively. (m+2)/2 = 1673 and 1744. We isolated two fractions with approximately these masses. Mass spectra are shown below: 125 1: TOF MS ES+ 1 48e4 RKN4165 _BCN_t15 259 (2.316) Cm (251:26;). 409.2156 100- 541.3085 I I 629.3728 673.4063 691.4463 ,,746.4561 .779.5088 1612.5868 824.5240 Ii. 0" I Ii IMJ11M~I 1 TOF MS ES+ 1 2Fe4 RkN4165_BCN_t14 259 (2 316) Crm (250-262) 409.2083 541.3003 1u62.34 629.3648 673.3978 717,4310 ..735.4689 779.5023 ,823.5262 824.51 76 I24. 911.5886 1148.7468 1634.5918 1722.6543 2 .1810.7227 AJ911-588 200 400 ,181 600 800 1000 1200 1400 1600 /.7227 1800 The expected mass of fully substituted 4-arm PEG DBCO (Figure 5.2, top) is then 3152 and 3294 for these two fractions, respectively. (m+2)/2 = 1577 and 1648. We isolated three fractions with approximately these masses. Mass spectra are shown below. The middle fraction (ionized mass = 1648) was used in siRNA conjugation experiments. 126 1: TOF MS ES+ 1.25e4 RK_N4165_DBCOt13 297 (2.656) Cm (291305) 981.8841 100-1 111 1040.6005 952.5297 0 1494. 3894 ,1560.4440 1604.4799 -I . "I Imyv- 1 TOF MS 1. 53e4 P1N4165 DBCO_t12 296 (2.639) Crri (285:308) 835.7128 1084.6349 1001128.6672 813.6952 , .1143.3467 802.6871,, '1158.0253 791.6752, 1648.5165 1 692.5463 1715.0632 1759.1033 S1173.0353 ,1781.115 4 769.6570, I- L RKN4165_['BCOt1l1 290 (259 7) Cm (282:298) 100- 857 .7286 846. 7208 835. 7080 820.44 400 600 946.0546 1172.7037 \/ .957.0656 1 92 i U 200 1: TOF MS ES+ &52e3 901.7662 879 .7455, ,923.7844 - 8- ,1055.2777 69 I 1 0 .9573 ,A084.6360 ,1099.6469 689.4061 583.3304 800 .1 Ii . . . . . ,1. .. , ,.. .1. , . . 1000 1781.1196 1200 m/z 1400 1600 1800 1H NMR comparisons of the starting 4-arm PEG -NHS and the two cyclooctyne products confirmed that these fractions were fully substituted, based on the peak integral ratio of cyclooctyne signatures to PEG protons. 4 arm PEG NHS: 'H NMR (500 MHz, Methanol-d 4) 6 4.6 (s, 2H), 3.6 - 3.7 (in, 58H), 3.45 (s, 2H), 2.8 - 2.9 (m, 4H). 4 arm PEG DBCO: 'H NMR (500 MHz, Methanol-d4) Phenyl protons: 6 7.2 - 7.7 (m, 9H), PEG protons: 3.55 - 3.7 (m, 58H), 3.45 (s, 2H) 4 arm PEG BCN: 'H NMR (500 MHz, Methanol-cd 4) PEG protons: 3.55 - 3.8 (in, 68H), 3.4-3.45 (i, 4H), BCN protons: 2.1 - 2.4 (m, 7H), 1.56 - 1.7 (m, 2H), 1.3 - 1.5 (m, I H), 0.9 - 1.05 (m, 2H). 127 Appendix 4. References 1. Kanasty, R. L., Whitehead, K. a, Vegas, A. J. & Anderson, D. G. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol. Ther. 20, 513-24 (2012). 2. Kanasty, R., Dorkin, J. R., Vegas, A. & Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 12, 967-77 (2013). 3. Yin, H. et al. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15, 541-555 (2014). 4. Morrissey, D. V et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 23, 1002-7 (2005). 5. Okumura, A., Pitha, P. M. & Harty, R. N. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. U. S. A. 105, 3974-9 (2008). 6. Alnylam RNAi Roundtable: Conjugate Delivery. (2012). at <http://www.alnylam.com/capella/wp-content/uploads/2012/12/ALNYRNAiRoundtable-ConjugateDelivery-Dec-14-20 12.pdf> 7. Shen, H., Sun, T. & Ferrari, M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 19, 367-73 (2012). 8. Davis, M. E. et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067-70 (2010). 9. Whitehead, K. a, Langer, R. & Anderson, D. G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129-38 (2009). 10. Pei, Y. & Tuschl, T. On the art of identifying effective and specific siRNAs. Nat. Methods 3, 670-676 (2006). 11. Reynolds, A. et al. Rational siRNA design for RNA interference. Nat. Biotechnol. 22, 326-30 (2004). 12. Lu, Z. J. & Mathews, D. H. OligoWalk: an online siRNA design tool utilizing hybridization thermodynamics. Nucleic Acids Res. 36, W104-8 (2008). 13. Schwarz, D. S. et al. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199-208 (2003). 14. Tafer, H. et al. The impact of target site accessibility on the design of effective siRNAs. Nat. Biotechnol. 26, 578-83 (2008). 15. Nykanen, a, Haley, B. & Zamore, P. D. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell 107, 309-21 (2001). 128 16. Vaish, N. et al. Improved specificity of gene silencing by siRNAs containing unlocked nucleobase analogs. Nucleic Acids Res. 39, 1823-1832 (2010). 17. Bramsen, J. B. et al. A screen of chemical modifications identifies position-specific modification by UNA to most potently reduce siRNA off-target effects. Nucleic Acids Res. 38, 5761-73 (2010). 18. Laursen, M. B. et al. Utilization of unlocked nucleic acid (UNA) to enhance siRNA performance in vitro and in vivo. MoL. Biosyst. 6, 862 (2010). 19. Weitzer, S. & Martinez, J. The human RNA kinase hClp1 is active on 3' transfer RNA exons and short interfering RNAs. Nature 447, 222-6 (2007). 20. Chen, P. Y. et al. Strand-specific 5'-O-methylation of siRNA duplexes controls guide strand selection and targeting specificity. RNA 14, 263-74 (2008). 21. Zuhorn, I. S., Engberts, J. B. F. N. & Hoekstra, D. Gene delivery by cationic lipid vectors: overcoming cellular barriers. Eur. Biophys. J. 36, 349-62 (2007). 22. Chiu, Y.-L. siRNA function in RNAi: A chemical modification analysis. Rna 9, 10341048 (2003). 23. Khvorova, A., Reynolds, A. & Jayasena, S. D. Functional siRNAs and miRNAs exhibit strand bias. Cell 115, 209-16 (2003). 24. Ameres, S. L., Martinez, J. & Schroeder, R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell 130, 101-12 (2007). 25. Brown, K. M., Chu, C.-Y. & Rana, T. M. Target accessibility dictates the potency of human RISC. Nat. Struct. Mol. Biol. 12, 469-70 (2005). 26. Koberle, C., Kaufmann, S. H. E. & Patzel, V. Selecting effective siRNAs based on guide RNA structure. Nat. Protoc. 1, 1832-9 (2006). 27. Patzel, V. et al. Design of siRNAs producing unstructured guide-RNAs results in improved RNA interference efficiency. Nat. Biotechnol. 23, 1440-4 (2005). 28. Huesken, D. et aL. Design of a genome-wide siRNA library using an artificial neural network. Nat. Biotechnol. 23, 995-1001 (2005). 29. Jackson, A. L. & Linsley, P. S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57-67 (2010). 30. Whitehead, K. A., Dahlman, J. E., Langer, R. S. & Anderson, D. G. Silencing or Stimulation? siRNA Delivery and the Immune System. Annu. Rev. Chem. Biomol. Eng. 2, 77-96 (2011). 129 31. Feve, B. & Bastard, J.-P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 5, 305-11 (2009). 32. Garcia-Sastre, A. & Biron, C. a. Type 1 interferons and the virus-host relationship: a lesson in detente . Science 312, 879-82 (2006). 33. Locksley, R. M., Killeen, N. & Lenardo, M. J. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487-501 (2001). 34. Akira, S., Uematsu, S. & Takeuchi, 0. Pathogen recognition and innate immunity. Cell 124, 783-801 (2006). 35. Marques, J. T. et al. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 24, 559-65 (2006). 36. Takeuchi, 0. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805-20 (2010). 37. Akira, S., Takeda, K. & Kaisho, T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2, 675-80 (2001). 38. Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373-84 (2010). 39. Robbins, M., Judge, A. & MacLachlan, I. siRNA and innate immunity. Oligonucleotides 19, 89-102 (2009). 40. Meurs, E. et al. Molecular Cloning and Characterization Human Double-Stranded Kinase Induced by Interferon of the Protein. Cell 62, 379-390 (1990). 41. Kato, H. et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19-28 (2005). 42. Judge, A. D. et al. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 23, 457-62 (2005). 43. Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526-9 (2004). 44. Hornung, V. et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science 314, 994-7 (2006). 45. Pichlmair, A. et al. RIG-1-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 314, 997-1001 (2006). 46. Kim, D.-H. et al. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat. Biotechnol. 22, 321-5 (2004). 130 47. Elm6n, J. et al. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 33, 439-47 (2005). 48. Langkjaer, N., Pasternak, A. & Wengel, J. UNA (unlocked nucleic acid): a flexible RNA mimic that allows engineering of nucleic acid duplex stability. Bioorg. Med. Chem. 17, 5420-5 (2009). 49. Heidel, J. D., Hu, S., Liu, X. F., Triche, T. J. & Davis, M. E. Lack of interferon response in animals to naked siRNAs. Nat. Biotechnol. 22, 1579-82 (2004). 50. Kornek, M. et al. 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP)-formulated, immune-stimulatory vascular endothelial growth factor a small interfering RNA (siRNA) increases antitumoral efficacy in murine orthotopic hepatocellular carcinoma with liver fibrosis. Mol. Med. 14, 365-73 (2008). 51. Cubillos-ruiz, J. R. et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. 119, (2009). 52. Akinc, A., Thomas, M., Klibanov, A. M. & Langer, R. Exploring polyethyleniminemediated DNA transfection and the proton sponge hypothesis.J. Gene Med. 7, 657-63 (2005). 53. Nguyen, D. N. et al. Drug delivery-mediated control of RNA immunostimulation. Mol. Ther. 17, 1555-62 (2009). 54. Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494-8 (2001). 55. Holen, T. et al. Tolerated wobble mutations in siRNAs decrease specificity, but can enhance activity in vivo. Nucleic Acids Res. 33, 4704-10 (2005). 56. Jackson, A. L. et al. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635-637 (2003). 57. Fedorov, Y. Off-target effects by siRNA can induce toxic phenotype. Rna 12, 11881196 (2006). 58. Lai, E. C. Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 30, 363-4 (2002). 59. Doench, J. G. & Sharp, P. a. Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504-11 (2004). 60. Wu, L., Fan, J. & Belasco, J. G. MicroRNAs direct rapid deadenylation of mRNA. Proc. Nat. Acad. Sci. U. S. A. 103, 4034-9 (2006). 131 61. Farh, K. K.-H. et al. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science 310, 1817-21 (2005). 62. Krek, A. et al. Combinatorial microRNA target predictions. Nat. Genet. 37, 495-500 (2005). 63. Lewis, B. P., Shih, I., Jones-Rhoades, M. W., Bartel, D. P. & Burge, C. B. Prediction of mammalian microRNA targets. Cell 115, 787-98 (2003). 64. Lim, L. P. et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769-73 (2005). 65. Xie, X. et al. Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals. Nature 434, 338-45 (2005). 66. Semizarov, D. et al. Specificity of short interfering RNA determined through gene expression signatures. Proc. NatL. Acad Sci. U. S. A. 100, 6347-52 (2003). 67. Jackson, A. L. et al. Position-specific chemical modification of siRNAs reduces 'offtarget' transcript silencing. RNA 12, 1197-205 (2006). 68. Birmingham, A. et al. 3' UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3, 199-204 (2006). 69. Boese, Q. et al. Mechanistic insights aid computational short interfering RNA design. Methods Enzymol. 392, 73-96 (2005). 70. Naito, Y., Yoshimura, J., Morishita, S. & Ui-Tei, K. siDirect 2.0: updated software for designing functional siRNA with reduced seed-dependent off-target effect. BMC Bioinformatics10, 392 (2009). 71. Petri, S. et al. Increased siRNA duplex stability correlates with reduced off-target and elevated on-target effects. Rna 737-749 (2011). doi:10.1261/rna.2348111 72. Grimm, D. et al. Fatality in mice due to oversaturation of cellular microRNA / short hairpin RNA pathways. Nature 441, 537-541 (2006). 73. Castanotto, D. et al. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 35, 5154-64 (2007). 74. Vickers, T. a, Lima, W. F., Nichols, J. G. & Crooke, S. T. Reduced levels of Ago2 expression result in increased siRNA competition in mammalian cells. Nucleic Acids Res. 35, 6598-610 (2007). 75. Koller, E. et al. Competition for RISC binding predicts in vitro potency of siRNA. Nucleic Acids Res. 34, 4467-76 (2006). 132 76. Khan, A. A. et al. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 27, 549-55 (2009). 77. Yi, R., Doehle, B. P., Qin, Y., Macara, I. G. & Cullen, B. R. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 11, 220-6 (2005). 78. Hutvaigner, G., Simard, M. J., Mello, C. C. & Zamore, P. D. Sequence-specific inhibition of small RNA function. PLoS Biol. 2, E98 (2004). 79. Grimm, D. et al. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver.J. Clin. Invest. 120, 3106-19 (2010). 80. Giering, J. C., Grimm, D., Storm, T. A. & Kay, M. A. Expression of shRNA from a tissuespecific pol 11 promoter is an effective and safe RNAi therapeutic. MoL. Ther. 16, 1630-6 (2008). 81. Akinc, A. et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 26, 561-9 (2008). 82. Zimmermann, T. S. et al. RNAi-mediated gene silencing in non-human primates. Nature 441, 111-4 (2006). 83. Love, K. T. et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Nat/. Acad. Sc. U. S. A. 107, 1864-9 (2010). 84. Lv, H., Zhang, S., Wang, B., Cui, S. & Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery.j. Control. Release 114, 100-9 (2006). 85. Kedmi, R., Ben-Arie, N. & Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 31, 6867-75 (2010). 86. Soenen, S. J. H., Brisson, A. R. & De Cuyper, M. Addressing the problem of cationic lipid-mediated toxicity: the magnetoliposome model. Biomaterials30, 3691-701 (2009). 87. Dokka, S., Toledo, D., Shi, X., Castranova, V. & Rojanasakul, Y. Oxygen radicalmediated pulmonary toxicity induced by some cationic liposomes. Pharm. Res. 17, 521-5 (2000). 88. Bottega, R. & Epand, R. M. Inhibition of protein kinase C by cationic amphiphiles. Biochemistry 31, 9025-30 (1992). 89. Solodin, I. et al. A novel series of amphiphilic imidazolinium compounds for in vitro and in vivo gene delivery. Biochemistry 34, 13537-44 (1995). 133 90. Van der Woude, I. et al. Novel pyridinium surfactants for efficient, nontoxic in vitro gene delivery. Proc. NatL. Acad. Sci. U. S. A. 94, 1160-5 (1997). 91. Leventis, R. & Silvius, J. R. Interactions of mammalian cells with lipid dispersions containing novel metabolizable cationic amphiphiles. Biochim. Biophys. Acta 1023, 124-32 (1990). 92. Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172-6 (2010). 93. Xu, Y., Hui, S. W., Frederik, P. & Szoka, F. C. Physicochemical characterization and purification of cationic lipoplexes. Biophys. J. 77, 341-53 (1999). 94. Moghimi, S. M. et aL. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol. Ther. 11, 990-5 (2005). 95. Malek, A. et al. In vivo pharmacokinetics, tissue distribution and underlying mechanisms of various PEI(-PEG)/siRNA complexes. Toxicol. Appl. Pharmacol. 236, 97-108 (2009). 96. Beyerle, A., Irmler, M., Beckers, J., Kissel, T. & Stoeger, T. Toxicity pathway focused gene expression profiling of PEI-based polymers for pulmonary applications. Mol. Pharm. 7, 727-37 (2010). 97. Merkel, 0. M. et aL. Polymer-related off-target effects in non-viral siRNA delivery. Biomaterials32, 2388-98 (2011). 98. Omidi, Y. et al. Toxicogenomics of non-viral vectors for gene therapy: a microarray study of lipofectin- and oligofectamine-induced gene expression changes in human epithelial cells.J. Drug Target. 11, 311-23 (2003). 99. Akhtar, S. & Benter, I. Toxicogenomics of non-viral drug delivery systems for RNAi: potential impact on siRNA-mediated gene silencing activity and specificity. Adv. Drug Deliv. Rev. 59, 164-82 (2007). 100. Kircheis, R., Wightman, L. & Wagner, E. Design and gene delivery activity of modified polyethylenimines. Adv. Drug Deliv. Rev. 53, 341-58 (2001). 101. Soutschek, J. et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432, 173-8 (2004). 102. LAYZER, J. M. In vivo activity of nuclease-resistant siRNAs. RNA 10, 766-771 (2004). 103. Buyens, K., Demeester, J., De Smedt, S. S. & Sanders, N. N. Elucidating the encapsulation of short interfering RNA in PEGylated cationic liposomes. Langmuir 25, 4886-91 (2009). 134 104. Akinc, A. et al. Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol. Ther. 17, 872-9 (2009). 105. Deleavey, G. F., Watts, J. K. & Damha, M. J. in Currentprotocols in nucleic acid chemistry / edited by Serge L. Beaucage ... [et al.] Chapter 16, Unit 16.3 (2009). 106. Turner, J. J., Jones, S. W., Moschos, S. a, Lindsay, M. a & Gait, M. J. MALDI-TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A-like activity. Mol. Biosyst. 3, 43-50 (2007). 107. Raines, R. T. Ribonuclease A. Chem. Rev. 98, 1045-1066 (1998). 108. Mook, 0. R., Baas, F., de Wissel, M. B. & Fluiter, K. Evaluation of locked nucleic acidmodified small interfering RNA in vitro and in vivo. Mol. Cancer Ther. 6, 833-43 (2007). 109. Braasch, D. a et al. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg. Med. Chem. Lett. 14, 1139-43 (2004). 110. Gao, S. et al. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol. Ther. 17, 1225-33 (2009). 111. Wolfrum, C. et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 25, 1149-57 (2007). 112. Akinc, A. et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 18, 1357-64 (2010). 113. Alexis, F., Pridgen, E., Molnar, L. K. & Farokhzad, 0. C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 5, 505-15 (2008). 114. Romberg, B., Hennink, W. E. & Storm, G. Sheddable coatings for long-circulating nanoparticles. Pharm. Res. 25, 55-71 (2008). 115. Bazile, D. et al. Stealth Me.PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system.J. Pharm. Sci. 84, 493-8 (1995). 116. Peracchia, M. T. et al. Visualization of in vitro protein-rejecting properties of PEGylated stealth polycyanoacrylate nanoparticles. Biomaterials20, 1269-75 (1999). 117. Aird, W. C. Phenotypic heterogeneity of the endothelium: 1. Structure, function, and mechanisms. Circ. Res. 100, 158-73 (2007). 118. Minshall, R. D., Tiruppathi, C., Vogel, S. M. & Malik, A. B. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem. Cell Biol. 117, 105-12 (2002). 135 119. Jain, R. K. & Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 7, 653-64 (2010). 120. Aird, W. C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 100, 174-90 (2007). 121. Wisse, E., Jacobs, F., Topal, B., Frederik, P. & De Geest, B. The size of endothelial fenestrae in human liver sinusoids: implications for hepatocyte-directed gene transfer. Gene Ther. 15, 1193-9 (2008). 122. Merkel, 0. M. et al. Stability of siRNA polyplexes from poly(ethylenimine) and poly(ethylenimine)-g-poly(ethylene glycol) under in vivo conditions: effects on pharmacokinetics and biodistribution measured by Fluorescence Fluctuation Spectroscopy and Single Photon Emission Com.J. Control. Release 138, 148-59 (2009). 123. Frank-Kamenetsky, M. et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 105, 11915-20 (2008). 124. Shi, B. et al. Biodistribution of Small Interfering RNA at the Organ and Cellular Levels after Lipid Nanoparticle-mediated Delivery.j. Histochem. Cytochem. 59, 727-40 (2011). 125. Schroeder, A., Levins, C. G., Cortez, C., Langer, R. & Anderson, D. G. Lipid-based nanotherapeutics for siRNA delivery.J. Intern. Med. 267, 9-21 (2010). 126. Fukumura, D., Duda, D. G., Munn, L. L. & Jain, R. K. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation 17, 206-25 (2010). 127. Jain, R. K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58-62 (2005). 128. Vakoc, B. J. et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat. Med. 15, 1219-23 (2009). 129. Roberts, W. G. & Palade, G. E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 57, 765 (1997). 130. Hashizume, H. et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 156, 1363-80 (2000). 131. Hobbs, S. K. et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc. Natl. Acad. Sci. U. S. A. 95, 4607-12 (1998). 136 132. Yuan, F. et al. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. CancerRes. 54, 4564-8 (1994). 133. Padera, T. P. et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 296, 1883-6 (2002). 134. Leu, A. J., Berk, D. A., Lymboussaki, A., Alitalo, K. & Jain, R. K. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 60, 4324-7 (2000). 135. Maeda, H., Wu, J., Sawa, T., Matsumura, Y. & Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review.j. Control. Release 65, 27184 (2000). 136. Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug. Chem. 21, 797-802 (2010). 137. Wong, C. et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. U. S. A. 108, (2011). 138. Leuschner, F. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 29, 1005-1010 (2011). 139. Caron, E. & Hall, A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 282, 1717-21 (1998). 140. Groves, E., Dart, A. E., Covarelli, V. & Caron, E. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell. Mol. Life Sci. 65, 1957-76 (2008). 141. Swanson, J. A. & Baer, S. C. Phagocytosis by zippers and triggers. Trends Cell Biol. 5, 89-93 (1995). 142. Owens, D. E. & Peppas, N. a. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int.J. Pharm. 307, 93-102 (2006). 143. Vonarbourg, A., Passirani, C., Saulnier, P. & Benoit, J.-P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials27, 4356-73 (2006). 144. Vachon, E. et al. CD44 is a phagocytic receptor. Blood 107, 4149-58 (2006). 145. Gold, E. S. et al. Dynamin 2 is required for phagocytosis in macrophages.J. Exp. Med. 190, 1849-56 (1999). 146. Claus, V. et al. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes.j. Biol. Chem. 273, 9842-51 (1998). 137 147. Racoosin, E. L. & Swanson, J. a. M-CSF-induced macropinocytosis increases solute endocytosis but not receptor-mediated endocytosis in mouse macrophages.J. Cell Sci. 102 (Pt 4, 867-80 (1992). 148. Stromhaug, P. E., Berg, T. 0., Gjoen, T. & Seglen, P. 0. Differences between fluid-phase endocytosis (pinocytosis) and receptor-mediated endocytosis in isolated rat hepatocytes. Eur. J. Cell Biol. 73, 28-39 (1997). 149. Swanson, 150. Conner, S. D. & Schmid, S. L. Regulated portals of entry into the cell. Nature 422, 3744 (2003). 151. Mukherjee, S., Ghosh, R. N. & Maxfield, F. R. Endocytosis. Physiol. Rev. 77, 759-803 (1997). 152. Hillaireau, H. & Couvreur, P. Nanocarriers' entry into the cell: relevance to drug delivery. Cell. Mol. Life Sci. 66, 2873-96 (2009). 153. Jones, A. R. & Shusta, E. V. Blood-brain barrier transport of therapeutics via receptormediation. Pharm. Res. 24, 1759-71 (2007). 154. Bareford, L. M. & Swaan, P. W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 59, 748-58 (2007). 155. Medina-Kauwe, L. K. 'Alternative' endocytic mechanisms exploited by pathogens: new avenues for therapeutic delivery?Adv. Drug Deliv. Rev. 59, 798-809 (2007). 156. Mayor, S. & Pagano, R. E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8, 603-12 (2007). 157. Marsh, M. & Helenius, A. Virus entry: open sesame. Cell 124, 729-40 (2006). 158. Yu, B., Zhao, X., Lee, L. J. & Lee, R. J. Targeted delivery systems for oligonucleotide therapeutics. AAPSJ. 11, 195-203 (2009). 159. Lu, J. J., Langer, R. & Chen, J. A novel mechanism is involved in cationic lipid-mediated functional siRNA delivery. Mol. Pharm. 6, 763-71 (2009). 160. Boussif, 0. et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 92, 7297-301 (1995). 161. Hafez, I. M., Maurer, N. & Cullis, P. R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 8, 1188-96 (2001). J. A. & Watts, C. Macropinocytosis. Trends Cell Biol. 5, 424-8 (1995). 138 162. Guo, X., MacKay, J. A. & Szoka, F. C. Mechanism of pH-triggered collapse of phosphatidylethanolamine liposomes stabilized by an ortho ester polyethyleneglycol lipid. Biophys.J. 84, 1784-95 (2003). 163. Endoh, T. & Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug Deliv. Rev. 61, 704-9 (2009). 164. Dominska, M. & Dykxhoorn, D. M. Breaking down the barriers: siRNA delivery and endosome escape.J. Cell Sci. 123, 1183-9 (2010). 165. Jarad, G. & Miner, J. H. Update on the glomerular filtration barrier. Curr. Opin. Nephrol. Hypertens. 18, 226 (2009). 166. Fogo, A. B. & Kon, V. The glomerulus - a view from the inside - the endothelial cell. Int. J. Biochem. Cell Biol. 42, 1388-1397 (2010). 167. Miner, J. H. Glomerular basement membrane composition and the filtration barrier. Pediatr. Nephrol. (2011). doi:10.1007/s00467-011-1785-1 168. Patrakka, J. & Tryggvason, K. New insights into the role of podocytes in proteinuria. Nat. Rev. Nephrol. 5, 463-8 (2009). 169. Wartiovaara, J. et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. Filtration114, 1475-1483 (2004). 170. Rodewald, R. & Karnovsky, M. J. Porous substructure of the glomerular slit diaphragm in the rat and mouse.j. Cell Biol. 60, 423-33 (1974). 171. He, X. M. & Carter, D. C. Atomic structure and chemistry of human serum albumin. Nature 358, 209 - 215 (1992). 172. Patrakka, J. & Tryggvason, K. Molecular make-up of the glomerular filtration barrier. Biochem. Biophys. Res. Commun. 396, 164-9 (2010). 173. Kawachi, H. et al. Role of podocyte slit diaphragm as a filtration barrier. Nephrology (Carlton). 11, 274-81 (2006). 174. Ghibellini, G., Leslie, E. M. & Brouwer, K. L. R. Methods to evaluate biliary excretion of drugs in humans: an updated review. Mol. Pharm. 3, 198-211 (2006). 175. Fleck, C. & Brdunlich, H. Factors determining the relationship between renal and hepatic excretion of xenobiotics. Arzneimittelforschung. 40, 942-6 (1990). 176. Fagerholm, U. Prediction of human pharmacokinetics-biliary and intestinal clearance and enterohepatic circulation.j. Pharm. Pharmacol.60, 535-42 (2008). 177. Huang, Y. et al. Elimination pathways of systemically delivered siRNA. Mol. Ther. 19, 381-5 (2011). 139 178. Singha, K., Namgung, R. & Kim, W. J. Polymers in small-interfering RNA delivery. Nucleic Acid Ther. 21, 133-47 (2011). 179. Mo, R. H., Zaro, J. L. & Shen, W.-C. Comparison of cationic and amphipathic cell penetrating peptides for siRNA delivery and efficacy. MoL. Pharm. 9, 299-309 (2012). 180. Yao, Y. et al. Targeted delivery of PLK1-siRNA by ScFv suppresses Her2+ breast cancer growth and metastasis. Sci. TransL. Med. 4, 130ra48 (2012). 181. Dassie, J. P. et aL. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 27, 839-49 (2009). 182. Neff, C. P. et aL. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci. Transi. Med. 3, 66ra6 (2011). 183. Thomas, M. et aL. Ligand-targeted delivery of small interfering RNAs to malignant cells and tissues. Ann. N. Y. Acad. Sci. 1175, 32-9 (2009). 184. Davis, M. E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol. Pharm. 6, 65968 (2009). 185. Gonzalez, H., Hwang, S. J. & Davis, M. E. New class of polymers for the delivery of macromolecular therapeutics. Bioconjug. Chem. 10, 1068-74 (1999). 186. Hwang, S. J., Bellocq, N. C. & Davis, M. E. Effects of structure of beta-cyclodextrincontaining polymers on gene delivery. Bioconjug. Chem. 12, 280-90 (2001). 187. Pun, S. H. et aL. Cyclodextrin-modified polyethylenimine polymers for gene delivery. Bioconjug. Chem. 15, 831-40 (2004). 188. Pun, S. H. & Davis, M. E. Development of a nonviral gene delivery vehicle for systemic application. Bioconjug. Chem. 13, 630-9 (2002). 189. Reineke, T. M. & Davis, M. E. Structural effects of carbohydrate-containing polycations on gene delivery. 1. Carbohydrate size and its distance from charge centers. Bioconjug. Chem. 14, 247-54 (2003). 190. Popielarski, S. R., Mishra, S. & Davis, M. E. Structural effects of carbohydratecontaining polycations on gene delivery. 3. Cyclodextrin type and functionalization. Bioconjug. Chem. 14, 672-8 (2003). 191. Davis, M. et aL. Self-Assembling Nucleic Acid Delivery Vehicles via Linear, WaterSoluble, Cyclodextrin-Containing Polymers. Curr. Med. Chem. 11, 179-197 (2004). 140 192. Hu-Lieskovan, S., Heidel, J. D., Bartlett, D. W., Davis, M. E. & Triche, T. J. Sequencespecific knockdown of EWS-FLII by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res. 65, 8984-92 (2005). 193. Mishra, S., Heidel, J. D., Webster, P. & Davis, M. E. Imidazole groups on a linear, cyclodextrin-containing polycation produce enhanced gene delivery via multiple processes. j. Control. Release 116, 179-91 (2006). 194. Bartlett, D. W. & Davis, M. E. Physicochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjug. Chem. 18, 456-68 (2007). 195. Mishra, S., Webster, P. & Davis, M. E. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur. J. Cell Biol. 83, 97-111 (2004). 196. Bellocq, N. C., Pun, S. H., Jensen, G. S. & Davis, M. E. Transferrin-containing, cyclodextrin polymer-based particles for tumor-targeted gene delivery. Bioconjug. Chem. 14, 1122-32 (2003). 197. Bartlett, D. W. & Davis, M. E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 34, 322-33 (2006). 198. Bartlett, D. W., Su, H., Hildebrandt, I. J., Weber, W. a & Davis, M. E. Impact of tumorspecific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc. Natl. Acad. Sci. U. S. A. 104, 15 54954 (2007). 199. Bartlett, D. W. & Davis, M. E. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol. Bioeng. 99, 975-85 (2008). 200. Heidel, J. D. et al. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc. Natl. Acad. Sci. U. S.A. 104, 5715-21 (2007). 201. Alabi, C., Vegas, A. & Anderson, D. Attacking the genome: emerging siRNA nanocarriers from concept to clinic. Curr. Opin. Pharmacol. 12, 427-33 (2012). 202. Burnett, J. C., Rossi, J. J. & Tiemann, K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol. J. 6, 1130-46 (2011). 203. Xu, Y. & Szoka, F. C. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 35, 5616-23 (1996). 204. Zhang, S., Zhi, D. & Huang, L. Lipid-based vectors for siRNA delivery.j. Drug Target. 20, 724-35 (2012). 141 205. Kesharwani, P., Gajbhiye, V. & Jain, N. K. A review of nanocarriers for the delivery of small interfering RNA. Biomaterials33, 7138-50 (2012). 206. Huang, L. & Liu, Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu. Rev. Biomed. Eng. 13, 507-30 (2011). 207. Sato, Y. et al. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo.J. Control. Release 163, 267-76 (2012). 208. Mahon, K. P. et al. Combinatorial approach to determine functional group effects on lipidoid-mediated siRNA delivery. Bioconjug. Chem. 21, 1448-54 (2010). 209. Zhang, J., Fan, H., Levorse, D. a & Crocker, L. S. Ionization behavior of amino lipids for siRNA delivery: determination of ionization constants, SAR, and the impact of lipid pKa on cationic lipid-biomembrane interactions. Langmuir 27, 1907-14 (2011). 210. Hafez, I. M., Ansell, S. & Cullis, P. R. Tunable pH-sensitive liposomes composed of mixtures of cationic and anionic lipids. Biophys.J. 79, 1438-46 (2000). 211. Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Enyl. 51, 8529-33 (2012). 212. Zuhorn, I. S. et al. Nonbilayer phase of lipoplex-membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol. Ther. 11, 801-10 (2005). 213. Zhigaltsev, I. V, Maurer, N., Wong, K. F. & Cullis, P. R. Triggered release of doxorubicin following mixing of cationic and anionic liposomes. Biochim. Biophys. Acta 1565, 129-35 (2002). 214. Koltover, I. An Inverted Hexagonal Phase of Cationic Liposome-DNA Complexes Related to DNA Release and Delivery. Science (80-.). 281, 78-81 (1998). 215. Belliveau, N. M. et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 1, e37 (2012). 216. Bao, Y. et al. Effect of PEGylation on Biodistribution and Gene Silencing of siRNA/Lipid Nanoparticle Complexes. Pharm. Res. 30, 342-51 (2013). 217. Kolli, S. et al. pH-Triggered nanoparticle mediated delivery of siRNA to liver cells in vitro and in vivo. Bioconjug. Chem. (2013). doi:10.1021/bc3004099 218. Lin, S.-Y. et al. Sterically polymer-based liposomal complexes with dual-shell structure for enhancing the siRNA delivery. Biomacromolecules13, 664-75 (2012). 219. Virtanen, J. A., Ruonala, M., Vauhkonen, M. & Somerharju, P. Lateral organization of liquid-crystalline cholesterol-dimyristoylphosphatidylcholine bilayers. Evidence for 142 domains with hexagonal and centered rectangular cholesterol superlattices. Biochemistry 34, 11568-81 (1995). 220. Takahashi, H., Sinoda, K. & Hatta, I. Effects of cholesterol on the lamellar and the inverted hexagonal phases of dielaidoylphosphatidylethanolamine. Biochim. Biophys. Acta 1289, 209-16 (1996). 221. Sato, Y. et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 26, 431-42 (2008). 222. Ishiwatari, H. et al. Treatment of pancreatic fibrosis with siRNA against a collagenspecific chaperone in vitamin A-coupled liposomes. Gut 1-12 (2012). doi:10.1136/gutjnl-2011-301746 223. Yoshizawa, T., Hattori, Y., Hakoshima, M., Koga, K. & Maitani, Y. Folate-linked lipidbased nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur. J. Pharm. Biopharm. 70, 718-25 (2008). 224. Feng, C. et al. Silencing of the MYCN gene by siRNA delivered by folate receptortargeted liposomes in LA-N-5 cells. Pediatr. Surg. Int. 26, 1185-91 (2010). 225. Wan, K. et al. In vivo tumor imaging using a novel RNAi-based detection mechanism. Nanomedicine 8, 393-8 (2012). 226. Tabernero, J. et al. First-in-Man Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 406-417 (2013). doi:10.1158/2159-8290.CD-12-0429 227. Alsina, M. et al. Open-Label Extension Study of the RNAi Therapeutic ALN-VSPO2 in Cancer Patients Responding to Therapy. American Society of Clinical Oncology Meeting (2012). at <http://www.alnylam.com/capella/wpcontent/uploads/2012/06/ALN-VSP-ExtensionStudyPoster-ASCO-June2012panel.pdf> 228. Jeong, J. H., Mok, H., Oh, Y.-K. & Park, T. G. siRNA conjugate delivery systems. Bioconjug. Chem. 20, 5-14 (2009). 229. Rozema, D. B. et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 104, 12982-7 (2007). 230. Wakefield, D. H., Klein, J. J., Wolff, J. a & Rozema, D. B. Membrane activity and transfection ability of amphipathic polycations as a function of alkyl group size. Bioconjug. Chem. 16, 1204-8 (2005). 231. Lewis, D. Dynamic Polyconjugates ( DPC ) Technology: An Elegant Solution to the siRNA Delivery Problem. (2006). at <http://www.arrowheadresearch.com/sites/default/files/udocs/ArrowheadResear chCorporation-DPCTechnologyWhitePaper.pdf> 143 232. Wong, S. C. et al. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther. 22, 380-90 (2012). 233. Macron, D. Arrowhead Presents Preclinical Data on HBV Candidate, Subcutaneous Delivery Tech. Gene Silencing News (2012). at <http://www.arrowheadresearch.com/sites/default/files/news/GeneSilenNews_1 11612_reprint.pdf> 234. Rozema, D. B., Ekena, K., Lewis, D. L., Loomis, A. G. & Wolff, J. A. Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjug. Chem. 14, 51-7 (2003). 235. Wooddell, C. I. et al. Hepatocyte-targeted RNAi Therapeutics for the Treatment of Chronic Hepatitis B Virus Infection. Mol. Ther. (2013). doi:10.1038/mt.2013.31 236. Biessen, E. a et al. Synthesis of cluster galactosides with high affinity for the hepatic asialoglycoprotein receptor.]. Med. Chem. 38, 1538-46 (1995). 237. Rensen, P. C. N., van Leeuwen, S. H., Sliedregt, L. a J. M., van Berkel, T. J. C. & Biessen, E. a L. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J. Med. Chem. 47, 5798-808 (2004). 238. Baenziger, J. U. & Fiete, D. Galactose and N-acetylgalactosamine-specific endocytosis of glycopeptides by isolated rat hepatocytes. Cell 22, 611-20 (1980). 239. Rensen, P. C. et al. Determination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepatocytes in vitro and in vivo.J. Biol. Chem. 276,37577-84 (2001). 240. Kallanthottathil, R. Conjugation Strategies for In Vivo siRNA Delivery. (2012). at <http://www.alnylam.com/capella/wp-content/uploads/2012/11/ALNY-OTSConjugate-Oct2012.pdf> 241. Smith, D., Schtller, V., Engst, C., Raidler, J. & Liedl, T. Nucleic acid nanostructures for biomedical applications. Nanomedicine (Lond). 8, 105-21 (2013). 242. Lee, H. et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 7, 389-93 (2012). 243. Shu, D., Shu, Y., Haque, F., Abdelmawla, S. & Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 6, 658-67 (2011). 244. Goodman, R. P. et al. Rapid chiral assembly of rigid DNA building blocks for molecular nanofabrication. Science 310, 1661-5 (2005). 144 245. Xia, W. & Low, P. S. Folate-targeted therapies for cancer. J. Med. Chem. 53, 6811-24 (2010). 246. Petros, R. a & DeSimone, J. M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 9, 615-27 (2010). 247. Gindy, M. E., Leone, A. M. & Cunningham, J. J. Challenges in the pharmaceutical development of lipid-based short interfering ribonucleic acid therapeutics. Expert Opin. Drug Defiv. 9, 171-82 (2012). 248. Chen, D. et al. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc. 134, 6948-51 (2012). 249. Alabi, C. a et al. Multiparametric approach for the evaluation of lipid nanoparticles for siRNA delivery. Proc. NatL. Acad. Sci. U. S. A. 110, 12881-6 (2013). 250. Boisgu6rin, P. et al. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. (2015). doi:10.1016/j.addr.2015.02.008 251. Whitehead, K. a et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun. 5, 4277 (2014). 145