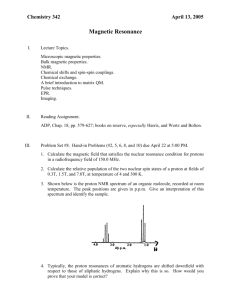
Three Dimensional Molecular Architectures for the Synthesis
advertisement

Three Dimensional Molecular Architectures for the Synthesis and Improved Properties of High Performance Polymers by MASSACHUSMpS INSTITTE OF TEcHNOLOGY Brett VanVeller JUN 0 7 2011 B.Sc. (First Class Honors) Chemistry, McMaster University, 2006 LIBRARIES Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of ARCHIVES Doctor of Philosophy at the Massachusetts Institute of Technology June 2011 © Massachusetts Institute of Technology, 2011. All rights reserved. Signature of Author: Department of Chemistry May 18, 2010 Certified by: Accepted by: Timothy M. Swager Thesis Supervisor Robert W. Field Studies Graduate on Committee Departmental Chairman, 2 This doctoral thesis has been examined by a Committee of the Department of Chemistrv as folinw- Professor Stephen L. Buchwald: Thesis Committee Chair Professor Timothy M. Swager: T#is Supervisor Professor Barbara Imperiali: Department of Chemistry 4 Three Dimensional Molecular Architectures for the Synthesis and Improved Properties of High Performance Polymers by Brett VanVeller Submitted to the Department of Chemistry on May 18, 2011 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemistry. ABSTRACT Conjugated polymers have found various uses in optoelectronic applications, including chemical sensors, light-emitting diodes, and photovoltaic materials. In this thesis, we investigate the effect of having a molecular architecture that is both rigid and three-dimensional might play in the synthesis and performance of conjugated polymers. We discuss the efficient synthesis of a hydrophilic monomer bearing a threedimensional noncompliant array of hydroxyl groups that prevents water-driven excimer features of hydrophobic poly(p-phenylene ethynylene) backbones. We also use the detection of 3-nitrotyrosine as a probe to learn more about its physical state in solution. We further utilize the monomer above in a biocompatible post-polymerization functionalization reaction, taking advantage of the polymer's structural motif for the controllable attachment of biotin. The utility of this method is demonstrated for a model biosensor that responds to streptavidin. Finally, we discuss how rigid molecular architectures can be harnessed to bring two reacting groups together for the annulation of various rr-systems. The optical effects of the transformation are both notable and predictable, and the molecules have potential as monomers for conjugated polymer application in high performance organic light emitting diodes and photovoltaic devices. Thesis Supervisor: Timothy M. Swager, Title: John D. MacArthur Professor of Chemistry Dedicatedto Courtney, my love, whose support made this possible Table of Contents Title Page Signature Page Abstract Dedication Table of Contents List of Abbreviations List of Figures List of Schemes List of Tables 1 3 5 6 7 9 10 17 19 Chapter 1. Fluorescent Conjugated Polymer Sensors: A Primer 20 1.1 Introduction 1.2 Electronic Structure 1.3 Exciton Migration and Fluorescence Quenching Amplification 1.3.1 Energy Transfer to an Emissive Quencher 1.4 Higher Order Exciton Migration and Amplification 1.5 Conclusion 1.6 References 21 21 23 25 26 27 28 Chapter 2. Rigid Hydrophilic Structures for Improved Properties of Conjugated Polymers in Water 29 2.1 Introduction and Design Considerations 2.2 Monomer Synthesis 2.3 Polymer Synthesis 2.4 Monomer Effects on Spectral Purity 2.5 Polymer Solution Properties 2.6 Conclusion 2.7 Experimental Section 30 32 35 37 39 41 42 2.7.1 Optimization of 4 -+ 6-anti/syn: 2.7.2 Optimization of 6-anti/syn -- 8-anti and 8-syn: 2.8 References Chapter 2 Appendix. 1H-NMR, 47 48 50 C-NMR, UV-vis and 13 53 Fluorescence data, and X-ray Structures Chapter 3. Biocompatible Post-Polymerization Functionalization 69 3.1 3.2 3.3 3.4 70 72 73 75 Introduction Post Polymerization Functionalization and Linkage Structure Effect of Functionalization on Polymer Properties Response to Streptavidin as a Model Biosensor 3.5 Alternative Sensore Designs for Detection of Native Analytes 3.5.1 Results and Discussion 3.6 Conclusion 3.7 Experimental Section 3.8 References 78 78 82 82 85 Chapter 3 Appendix. 'H-NMR, "C-NMR, UV-vis and Fluorescence data 87 Chapter 4. Trypticene Diol Scaffolds for rr-System Elaboration 91 4.1 Introduction 4.2 Dihalogen Substituted Trypticene Diols 4.3 Cyclization with Alkynes 4.3.1 Synthesis of Model Compounds for Cyclization 4.3.2 Polymer Synthesis and Cyclization 4.3.3 Photophysical Properties 4.3.4 Electronic Properties 4.3.5 Conclusions: Alkyne Cyclization 4.4 Cyclization with Phenyl Groups 4.4.1 Future Applications of Phenyl Cyclized Trypticene Diols 4.5 Cyclization with Thiophenes 4.5.1 Future Applications of Thiophene Cyclized Trypticene Diols 4.6 Conclusions 4.7 Experimental Section 4.8 References Chapter 4 Appendix. 'H-NMR, Curriculum Vitae Acknowledgements 13C-NMR, IR Data, and X-ray Structures 92 94 95 96 99 100 103 105 106 107 108 111 113 113 123 126 146 148 List of Abbreviations ATR-IR attenuated total reflection-infrared spectroscopy CPE conjugated polyelectrolyte DCE 1,2-dichloroethane DMF NN-dimethyl formamide DMSO dimethyl sulfoxide ESI electrospray ionization EtOH ethanol GPC gel permeation chromatography HOMO highest occupied molecular orbital HRMS high-resolution mass spectrometry h photon LED light-emitting diode lowest unoccupied molecular orbital LUMO MeCN acetonitrile MeOH methanol Mn number-average molecular weight MS mass spectrometry NMR nuclear magnetic resonance OD optical density PDI polydispersity index PhMe toluene PLED polymer light-emitting diode PPE poly(para-phenylene ethynylene) PPV poly(para-phenylene vinylene) PPP poly(para-phenylene) PT polythiophene fluorescence quencher Q rt room temperature Sn singlet electronic state TBAF tetrabutylammonium fluoride THF tetrahydrofuran UV-vis ultraviolet-visible List of Figures Figure 1.1: Examples of conjugated polymers 21 Figure 1.2: Schematic of the energy gap (Eg) between the HOMO and LUMO levels and the interaction of light with (a) small molecule MOs and (b) a conjugated polymer energy band system. 23 Figure 1.3: Schematic of the band structure of a generic conjugated polymer showing (a) exciton generation, migration, and relaxation (b) exciton generation, migration, and deacitivation by electron transfer with quencher (Q). 24 Figure 1.4: Unconjugated system of single molecule fluorophores. Binding of a quenching analyte (Q) affects the florescence of only one molecule. 25 Figure 1.5: Schematic of the band structure of a generic conjugated polymer showing exciton generation, migration, and energy tranfer to an emissive dye quencher. 26 Figure 1.6: Representation of exciton migration in conjugated polymers in a) dilute solution (isolated chains), b) supramolecular aggregate in solution, c) solid film. Purple star represents the site of exciton generation. 27 Figure 2.1: Effect of interpolymer p-p interactions on spectral purity and signal intensity 30 Figure 2.2: Supramolecular mitigation of excimer formation. Physical separation prevents interchain exciton migration 31 Figure 2.3: Penticene (2). Its rigid structure prevents excimer formation yet is compact, allowing for productive interchain exciton migration 31 32 Figure 2.4: Three dimensional, rigid monomer scaffolds. Goal - apply the design principles of pentiptycene to aqueous media Figure 2.5: UV-vis and fluorescence spectra of P1-anti and P-peg in both water and 1 X PBS solution 38 Figure 2.6: Solubility enhancement of quantum yield of P1-anti . UV-vis absorption trace (left) of P1-anti in water (black), 1:1 water/glycerol (red), 1:1 water/methanol (green) (units of e = L ' mol-1 cm'). Fluorescence trace (right) of P1-anti in water (black, <F = 19%), 1:1 water/glycerol (red, (DF 49%), 1:1 water/methanol(red, <DF= 50%). 40 Figure 2.7: Fluroescence spectra for the addition of micromolar quantities of 3nitrotyrosine to P1-anti in IX PBS (plot) and associated Stern-Volmer plot (inset). Schematic representation of soluble polymer aggregates for each measured Ksv. Figure 2.A.1: 1H NMR spectrum of 4 in CDCl 3 (400 MHz) Figure 2.A.2: 13C NMR spectrum of 4 in CDCl 3 (125 MHz) Figure 2.A.3: 1H NMR spectrum of 6-anti/syn in CDCl 3 (400 MHz) Figure 2.A.4: 13 C NMR spectrum of 6-anti/syn in CDCl 3 (125 MHz) Figure 2.A.5: 'H NMR spectrum of 6-anti/syn enriched in 6-anti in CDCl 3 (400 MHz) Figure 2.A.6: (125 MHz) 13C NMR spectrum of 6-anti/syn enriched in 6-anti in CDCl 3 Figure 2.A.7: 'H NMR spectrum of 7 in CDCl 3 (400 MHz) Figure 2.A.8: 'H NMR spectrum of 8-anti in CDCl 3 (400 MHz) Figure 2.A.9: 13 C NMR spectrum of 8-anti in CDCl 3 (125 MHz) Figure 2.A.10 : 1H NMR spectrum of 8-syn in CDCl 3 (400 MHz) Figure 2.A.11 : 13C NMR spectrum of 8-syn in CDCl 3 (125 MHz) Figure 2.A.12 'H NMR spectrum of 9-anti in DMF-d 7 (400 MHz) Figure 2.A.13 13C NMR spectrum of 9-anti in DMF-d 7 (125 MHz) Figure 2.A.14 'H NMR spectrum of 9-syn in DMF-d7 (400 MHz) Figure 2.A.15 13C NMR spectrum of 9-syn in DMF-d 7 (125 MHz) Figure 2.A.16 'H NMR spectrum of 1-anti in DMF-d 7 (400 MHz) Figure 2.A17: 13C NMR spectrum of 1-anti in DMF-d 7 (125 MHz) Figure 2.A.18 'H NMR spectrum of 1-syn in DMF-d 7 (400 MHz) Figure 2.A.19 13C NMR spectrum of 1-syn in DMF-d 7 (125 MHz) Figure 2.A.20: 1H NMR spectrum of P1-anti in D20 (600 MHz) Figure 2.A.21: Stem-Volmer quenching plots of P1-anti with micromolar quantities of 3-nitrotyrosine under different solvent conditions. (a) water, (b) IX PBS, (c) 1:1 water/methanol. (d) shows the minimal fluorescence quenching response of P-peg to 3-nitrotyrosine (Chapter 2.# for explanation). 64 Figure 2.A.22: Crystal Structure of 6-anti. 65 Figure 2.A.23: Crystal Structure of 7. 66 Figure 2.A.24: Crystal Structure of 8-anti. 67 Figure 2.A.25: Crystal Structure of 8-syn. 68 Figure 3.1: (a) Absorbance spectra of P1 and 3a in water and PBS solution. (b) Fluorescence spectra of P1 and 3a in water and PBS solution. 74 Figure 3.2: Effect of quantum yield of 3a at different pH. Measurements performed in PBS where pH was adjusted with HCl or NaOH. 74 Figure 3.3: (a) Energy transfer schematic showing how intra- and interchain exciton migration and enery transfer to TRXS can lead to amplification. (b) Addition of 9.15 pmol aliquots of TRXS to 3.46 nmol (based on repeat unit of 3a). (c) Emission of P1 in the presence of 100 pmol of TRXS (black) and direct excitation of 100 pmol of TRXS (red). (d) Stern-Volmer quenching analysis of data in (b). 76 Figure 3.4: Comparison of quenching response (Ksv) to TRXS between two polymers (a) one containing a rigid structural monomer and (b) one bearing only linear PEG side groups. 77 Figure 3.5: Proposed sensing scheme for native, non-dye streptavidin labelled 80 Figure 3.6: (a) Absorbance spectra of P1 and 13 in water and PBS solution. (b) Fluorescence spectra of P1 and 13 in water and PBS solution. (c) and (d) Addition of 9.15 pmol aliquots of streptavidin to 3.46 nmol 13 and 14 respectively (based on repeat unit). (e) Emission of 13 (black) and 14 (cyan) at 390 nm excitation and emission of their respective appended Texas Red X T (13, red and 14, green) at 580 nm excitation. (f) Stern-Volmer quenching analysis of data in (e). (f) Addition of 9.15 pmol aliquots of streptavidin to 3.46 nmol (based on repeat unit of 13). Inset: Stern-Volmer quenching analysis of data in (f). 81 Figure 3.A.1: 1H NMR spectrum of compound 5 in CDCl 3 (400 MHz) 88 Figure 3.A.2: 3 C NMR spectrum of compound 5 in CDC 3 (125 MHz) 88 Figure 3.A.3: 'H NMR spectrum of compound 6 in CDC13 (400 MHz) 89 Figure 3.A.4: 13C NMR spectrum of compound 6 in CDC 3 (125 MHz) 89 Figure 3.A.5: 'H NMR spectrum of compound 3b in D2 0 (600 MHz) 90 Figure 3.A.6: 'H NMR spectrum of compound 3a in D20 (600 MHz) 90 Figure 4.1: Rational for the potential of trypticene diols for cyclization and extended planar -systems. 93 Figure 4.2: Absorbance traces for 28 (black) and 29 (green) and fluorescence traces for 28 (blue) and 29 (red) in CHCl 3. 101 Figure 4.3: Absorbance traces for 30 (black) and 31 (green) and fluorescence traces for 30 (blue) and 31 (red) in CHC13. 101 Figure 4.4: Absorbance traces for 30 (black) and 31 (green) and fluorescence traces for 30 (blue) and 31 (red) in thin film. 102 Figure 4.5: Distyrylbenzene 32 and potential nucleophilic site in 31 103 Figure 4.6: Cyclic voltammograms for 29 (a) and 31 (b) in solution, and thin films of 31 (c), and 30 (d). Measurements performed under nitrogen in degassed solvents 104 Figure 4.7: General transformation for a PPE to a chain rigidified PPV via gold(I) catalysis 105 Figure 4.8: Absorbance and fluorescence data for compound 33 107 Figure 4.9: Absorbance and fluorescence data for 39 (a) and 41 (b). 109 Figure 4.10: Comparison of the 'H NMR spectra of 3 (top) and 3 (bottom). 111 Figure 4.11: (a) Planarized monomer for improved charge mobility. (b) Favorable interactions between fullerene and trypticenes to prevent phase segregation. 112 Figure 4.A.1: 'H NMR spectrum of 10 in CDCl 3 (400 MHz) 127 Figure 4.A.2: 127 13C NMR spectrum of 10 in CDCl 3 (125 MHz) Figure 4.A.3: 'H NMR spectrum of 11 in CDC13 (400 MHz) Figure 4.A.4: NMR spectrum of 11 in CDC13 (125 MHz) 13C 127 128 Figure 4.A.5: H NMR spectrum of 12 in CDC13 (400 MHz) 128 NMR spectrum of 12 in CDC13 (125 MHz) 129 Figure 4.A.7: 'H NMR spectrum of 13 in CDC13 (400 MHz) 129 Figure 4.A.6: 13C Figure 4.A.8: 13C NMR spectrum of 13 in CDC13 (125 MHz) Figure 4.A.9: 'H NMR spectrum of 14 in CDC13 (400 MHz) Figure 4.A.10: 13C NMR spectrum of 14 in CDC13 (125 MHz) 129 130 130 Figure 4.A.11: 'H NMR spectrum of 15 in CDC13 (400 MHz) 130 NMR spectrum of 15 in CDC13 (125 MHz) 131 Figure 4.A.13: 'H NMR spectrum of 16 in CDCl 3 (400 MHz) 131 Figure 4.A.14: "C NMR spectrum of 16 in CDC13 (125 MHz) 131 Figure 4.A.15: 'H NMR spectrum of 22 in CDC13 (400 MHz) 132 NMR spectrum of 22 in CDC13 (125 MHz) 132 Figure 4.A.17: 1H NMR spectrum of 23 in CDC13 (400 MHz) 132 C NMR spectrum of 23 in CDC13 (125 MHz) 133 Figure 4.A.19: 'H NMR spectrum of 24 in CDCl 3 (400 MHz) 133 C NMR spectrum of 24 in CDC13 (125 MHz) 133 Figure 4.A.21: 'H NMR spectrum of 25 in CDCl 3 (400 MHz) 134 Figure 4.A.22: 13 C NMR spectrum of 25 in CDC13 (125 MHz) 134 Figure 4.A.23: 'H NMR spectrum of 26 in CDC13 (400 MHz and 600 MHz) 134 Figure 4.A.24: gCOSY spectrum of 26 in CDC13 (600 MHz) 135 Figure 4.A.25: 13C NMR spectrum of 26 in CDC13 (125 MHz) 135 Figure 4.A.12: Figure 4.A.16: Figure 4.A.18: Figure 4.A.20: 13C 13C Figure 4.A.26: 'H NMR spectrum of 27 in C12CD-CDC12 (400 MHz) Figure 4.A.27: 13 C NMR spectrum of 27 in C12CD-CDC12 (125 MHz) 135 136 Figure 4.A.28: 1H NMR spectrum of 28 in CDC13 (400 MHz) 136 Figure 4.A.29: 13C NMR spectrum of 28 in CDCl3 (125 MHz) 136 Figure 4.A.30: H NMR spectrum of 29 in CDCl 3 (400 MHz) 136 C NMR spectrum of 29 in CDC13 (125 MHz) 137 Figure 4.A.32: 'H NMR spectrum of 30 in CDC13 (400 MHz) 137 Figure 4.A.33: 'H NMR spectrum of 31 in CDC13 (400 MHz) 137 Figure 4.A.31: Figure 4.A.34: C NMR spectrum of 31 in CDCl 3 (125 MHz) Figure 4.A.35: 1H NMR spectrum of 32 in CDCl 3 (400 MHz) 138 138 C NMR spectrum of 32 in CDC13 (125 MHz) 138 Figure 4.A.37: 'H NMR spectrum of 33 in CDCL3 (400 MHz) 139 NMR spectrum of 33 in CDC13 (125 MHz) 139 Figure 4.A.36: Figure 4.A.38: 13C Figure 4.A.39: 1H NMR spectrum of 36 in CDC3 (400 MHz) 139 Figure 4.A.40: 13C NMR spectrum of 36 in CDCl 3 (125 MHz) 140 Figure 4.A.41: 'H NMR spectrum of 37 in CDC13 (400 MHz) 140 NMR spectrum of 37 in CDCl 3 (125 MHz) 140 Figure 4.A.43: 'H NMR spectrum of 40 in CDC13 (400 MHz) 140 Figure 4.A.42: 13C 13 C NMR spectrum of 40 in CDC13 (125 MHz) 141 Figure 4.A.45: 'H NMR spectrum of 41 in CDC13 (400 MHz) 141 Figure 4.A.46: 13C NMR spectrum of 41 in CDCl 3 (125 MHz) 141 Figure 4.A.47: FT-AT-IR spectrum of 39. 142 Figure 4.A.48: Crystal Structure of 23. 143 Figure 4.A.44: Figure 4.A.49: Crystal Structure of 27. 144 Figure 4.A.50: Crystal Structure of 33. 144 Figure 4.A.51: Crystal Structure of 38. 145 List of Schemes Scheme 2.1. Synthesis of the carbon framework Scheme 2.2: Osmium tetroxide catalyst turnover prevented by dioxo-diosmium species 34 Scheme 2.3: Rationale for why Upjohn conditions do not result in catalyst turnover 34 Scheme 2.4: Dihydroxylation 35 Scheme 2.5: Deprotection to reveal 1-anti 35 Scheme 2.6: Oxidative Palladium Acetylene Homocoupling 36 Scheme 2.7: Homopolymerization and Copolymerization 37 Scheme 3.1: Molecules of interest discussed in Chapter 2. 70 Scheme 3.2: Post-polymerization functionalization reaction 72 Scheme 3.3: Model studies and proposed mechanism for azepane formation 73 Scheme 3.4: Synthesis of fractionally dye conjugated polymer sample 80 Scheme 4.1: Synthesis of trypticene diols through [2 + 2 + 2] cylcoaddtion 93 Scheme 4.2: Synthesis of 1,4-dibromo substituted trypticene diols 95 Scheme 4.3: Proposed cyclization of TDs onto alkynes 95 Scheme 4.4: PPE main chain reduction 96 Scheme 4.5: Spontaneous metal-free 5-exo-dig cyclization 97 Scheme 4.6: Electrophilic activation of 6-endo-dig cyclization 98 Scheme 4.7: Electrophilic activation of 6-endo-dig cyclization of more soluble variants 98 Scheme 4.8: Polymerization and post- cyclization 99 Scheme 4.9: Synthesis of planarized phenylene model compound 106 Scheme 4.10: Future application for ladder phenylene polymers 108 Scheme 4.11: Oxidative cyclization of trypticene diols with thiophenes 109 Scheme 4.12: Cyclization by acid catalyzed trans-etherification 110 List of Tables Table 2.1: Peak Analysis data for Figure 2.5 38 Table 2.2: Screening of conditions for the di-aryne cycloaddition of 4 and 5. 48 Table 2.3: Screen of reaction conditions for the dihydroxylation of 6-antilsyn 49 Table 2.A.1: Summary of photophysical data of P1-anti 64 Table 1.A.1: X-ray Crystallographic Data 65 Table 3.A.1: Summary of photophysical data of 3a 90 Table 4.1: Summary of photophysical data 103 Chapter 1 Fluorescent Conjugated Polymer Sensors: A Primer 1.1 Introduction Conjugated polymers are, most typically, organic polymers whose n-bonding structure allows for electronic delocalization (orbital conjugation) along the backbone. This extensive orbital delocalization is responsible for the interesting photophysical and electronic properties that have allowed these materials to find use in a variety of applications including light emitting diodes, field-effect transistors, photovoltaics and chemical sensors. 1 Further, organic conjugated polymers-in addition to the mechanical and processing advantages inherent to polymers-offer unique opportunities to easily tailor their structure and properties through organic synthesis (as an alternative to other inorganic materials used for these purposes). 2 Several examples of different conjugated polymer structures relavent to the following chapters are shown in Figure 1.1. This chapter does not seek to encompass the science behind the vast application of conjugated polymers listed above, but instead seeks some explanation and unpacking of the basic principles behind fluorescent conjugated polymer sensors. Thus, we will begin with a brief explanation of the transduction mechanisms. n Polyacetylene PA Poly(p-phenylene vinylene) PPV n Polythiophene PT Poly(p-phenylene ethynylene) S Poly(p-phenylene) PPP -n __ -nPP n n n PPE Poly(p-phenylene butadiynylene PPB Figure 1.1: Examples of conjugated polymers 1.2 Electronic Structure The fluorescence sensing ability of conjugated polymers follows directly from their electronic structure. Consider a small molecule containing an isolated double bond (Figure 2.2a). Upon absorption of a photon with energy greater than the energy gap (Eg), a y-electron can be promoted from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) to form a singlet excited state. Once in this excited state the electron may relax down to the ground state by non-radiative vibrational relaxation (called internal conversion) or emission of a photon (fluorescence from a singlet excited state or phosphorescence from a triplet excited state). In the case of conjugated polymers, fluorescence is the dominant form of emission due to negligible transition rates from singlet to triplet states. Orbital interactions-double bond conjugation-causes a decrease in the value of Eg by elevating the HOMO and depressing the LUMO and allowing for lower energy photon absorption (Figure 1.2b). As the extent of conjugation continues, the density of molecuar orbital states are more often thought of as a band structure, analogous to schemes presented in the solid-state physics for inorganic semiconductors like silicon. The mixing of HOMO levels produces a broadened electron filled band called the valence band, and the corresponding LUMO levels produce the empty conductance band. In an analogous manner to small molecule excitation, a suitably energetic photon can promote an electron from the valence band to the conduction band. The resultant excited state is typically refered to as an "exciton". An exciton is a quasiparticle consisting of an electrostatically bound electron-hole pair. 3 Because of the band nature of the conjugated polymer back bone, the exciton is permited to migrate to another location within the band until it relaxes back to the ground state (lifetime <1 ns). Finally, as an aside, the relative HOMO and LUMO levels can be greatly affected by substituents on the polymer backbone. Electron donating substituents act to raise the HOMO level relative to the LUMO making the polymer more prone to oxidation, while electron-withdrawing groups lower the LUMO relative to the HOMO making reduction more facile. The result in both situations is a decrease in the band gap energy and generally a red-shift in absorbance and fluorescence. a) b) L- LUMO - -.-- Conduction Band LUMO--- AL --V Eg HOMO E HOMO tVLy : Valence .. ... ........... Band Isolated double bond Isolated double bond * Conjugated polymer Figure 1.2: Schematic of the energy gap (Eg) between the HOMO and LUMO levels and the interaction of light with (a) small molecule MOs and (b) a conjugated polymer energy band system. 1.3 Exciton Migration and Fluorescence Quenching Amplification Modulation of how excitons return to the ground state is the basis for the fluorescent conjugated polymer's sensing response. Figure 1.3a shows the band structure of a generalized conjugated polymer. As discussed above, because all of the repeating units in the polymer are electrically linked through conjugation, the exciton is permitted to migrate along the polymer chain until it relaxes back to the ground state by the competing processes of fluorescence or non-radiative relaxation. If an analyte with LUMO levels suitably matched to the polymer's Eg comes in contact with the polymer-the exciton can relax to the ground state through a nonradiative electron transfer mechanism with the LUMO of the analyte-quenching the excited state (Figure 1.3b). Further, the efficient transport of excitons along the polymer allows for excitons generated at various locations on the backbone to migrate to the site of quenching. As a result, one quencher can lead to an amplified decrease in the fluorescence process through enhanced non-radiative relaxation (greater sensitivity to quencher). 4 a) Conduction Band - --- * Valence - Band n Figure 1.3: Schematic of the band structure of a generic conjugated polymer showing (a) exciton generation, migration, and relaxation (b) exciton generation, migration, and deacitivation by electron transfer with quencher (Q). 24 The above process stands in stark contrast to a sensing scheme involving unconjugated single molecule fluorescence quenching (Figure 1.4). Binding of a quencher results in fluorescence "turn-off' of only one molecule, leaving emission from unbound fluorophores intact (linear quenching response). hXx LO fkET W / Figure 1.4: Unconjugated system of single molecule fluorophores. Binding of a quenching analyte (Q) affects the florescence of only one molecule. 1.3.1 Energy Transfer to an Emissive Quencher The energy transferred to the quenching analyte need not be fated to non-radiative vibrational relaxation. The analyte that quenches the polymer fluorescence may itself be a fluorescent dye. If the energy levels of the dye are suitably matched to the polymer's Eg then energy transfer can occur following either the Dexter or the dipole-dipole (Fdrster) mechanism to yield the emmissive dye excited state (Figure 1.5). In this case emission from the dye is expected to be greater than direct excitation of itself due to the funneling of numerous polymer excitons to the dye quencher. Conduction -- ' Band ---- * Energy Transfer Eg V - HOMO Valence -©---,- ----- Band Figure 1.5: Schematic of the band structure of a generic conjugated polymer showing exciton generation, migration, and energy tranfer to an emissive dye quencher. 1.4 Higher Order Exciton Migration and Amplification Critical to the sensitivity of the quenching response is the efficiency with which the exciton can migrate to the site of quenching before it relaxes to the ground state. The model of exciton migration discussed in Section 1.3 best approximates a conjugated polymer in dilute solution (Figure 1.6a). That is, once the exciton is generated, its movement can be thought as a one-dimensional random walk back and forth along the isolated chain. However, the nature of this singular pathway for migration is somewhat inefficient. In the back and forth walk, there is a high probability of sampling the same "unquenched" polymer segment numerous times and returning to the ground state while never encountering the bound quencher. Thus, migration efficiency can be enhanced by decreasing the number of "visits" to unbound polymer segments. If the polymers are brought within close proximity of eachother-within a supramolecular aggregate (Figure 1.6b) for instance-then the exciton can hop to an adjacent chain (through a dipole-dipole coupling energy transfermechanism). This effect introduces interchain migration in addition to intrachain migration. The result is a higher dimensional random walk that increases the number of migration pathways, there by reducing the number of times an exciton revisits an unbound binding site, and providing a greater realized exciton migration efficiency.5 Further, it allows for excimers on adjacent chains to contribute to the amplified quenching response. When spin into a film the polymers are aggregated very closely and the excitons can migrate even more freely (Figure 1.6c). Revisitation of empty binding sights is at a minimum and polymer films generally provide the most sensitive chemical sensors. Our group has reported a highly fluorescent conjugated polymer film that undergoes amplified fluorescence quenching in response to only trace amounts of trinitrotoluene (TNT) vapor. 6 hv\ h a) b) c) Figure 1.6: Representation of exciton migration in conjugated polymers in a) dilute solution (isolated chains), b) supramolecular aggregate in solution, c) solid film. Purple star represents the site of exciton generation. 1.5 Conclusion With this basic understanding of the operation of fluorescent conjugated polymer sensors in mind, we can now examine other factors affecting their performance. Chapter 2 will discuss low energy emissive defects that are the result of the close association of polymers within an aggregate (Figure 1.6b). These defects appear as broad ill-defined bands in the emission spectrum and can behave like energy pot-holes that slow exciton migration. Chapter 3 will touch on how their removal can lead to a greater sensing response. 1.6 References (1) (a) ConjugatedPolymers: The Novel Science and Technology of Highly Conducting and Nonlinear Optically Active Materials; Bredas, J.-L.; Silbey, R. J., Eds.; Kluwer Academic Publishers: Boston, 1991. (b) Conjugated Conducting Polymers; Kiess, H. G.; Baeriswyl, D., Eds.; Springer-Verlag: New York, 1992. (c) Barashkov, N. N.; Gunder, 0. A. FluorescentPolymers; Ellis Horwood: New York, 1993. (d) Conjugated Polymers and Related Materials: The Interconnection of Chemical and Electronic Structure; Salaneck, W. R.; Lundstr6m, I.; Rainby, B. G., Eds.; Oxford University Press: New York, 1993. (e) Advances in Synthetic Metals: Twenty Years of Progress in Science and Technology; Bernier, P.; Lefrant, S.; Bidan, G., Eds.; Elsevier: New York, 1999. (f) Roth, S.; Carroll, D. One-Dimensional Metals: Conjugated Polymers, Organic Crystals, CarbonNanotubes, 2nd ed.; Wiley-VCH: Weinheim, 2004. (g) Handbook of Conducting Polymers, 3rd ed.; Skotheim, T. A.; Elsenbaumer, R. L.; Reynolds, J. R., Eds.; CRC Press: New York, 2007. (2) Moliton, A.; Hiorns, R. C. Polym. Int. 2004, 53, 1397-1412. (3) (a) Turro, N. J. Modern Molecular Photochemistry; University Science Books: Sausalito, CA, 1991. (b) Yan, M.; Rothberg, L.; Hsieh, B. R.; Alfano, R. R.Phys. Rev. B 1994, 49, 9419-9422. (4) (a) Zhou, Q.; Swager, T. M. J Am. Chem. Soc. 1995, 117, 7017-7018. (b) Zhou, Q.; Swager, T. M. J. Am. Chem. Soc. 1995, 117, 12593-12602. (5) (a) Levitsky, I. A.; Kim, J.; Swager, T. M. J. Am. Chem. Soc. 1999, 121, 1466-1472. (b) Hennebicq, E.; Pourtois, G.; Scholes, G. D.; Herz, L. M.; Russell, D. M.; Silva, C.; Setayesh, S.; Grimsdale, A. C.; Millen, K.; Bredas, J.-L.; Beljonne, D. J. Am. Chem. Soc. 2005, 127, 4744-4762. (6) (a) Yang, J. S.; Swager, T. M. J Am. Chem. Soc. 1998, 120, 5321-5322. (b) Yang, J. S.; Swager, T. M. J. Am. Chem. Soc. 1998, 120, 11864-11873. Chapter 2 Rigid Hydrophilic Structures for Improved Properties of Conjugated Polymers in Water Adapted and reproduced in part with permission from: VanVeller, B.; Miki, K.; Swager, T. M. Org. Lett. 2010, 12, 1292-1295 2.1 Introduction and Design Considerations Fluorescent conjugated polyelectrolytes (CPEs) are conjugated polymers made water soluble by pendant ionic functionalities along the polymer backbone. CPEs have seen application in a variety of chemical and biological sensing schemes' because the intra- and interchain migration of excitons along the polymer backbone results in the amplification of fluorescence signals relative to small-molecule fluorophores.'a However, the rigid hydrophobic main chains of CPEs are prone to interpolymer itit interactions leading to broad, ill-defined emission features and reduced luminescence (Figure 2.1).' "Film induced" or Solvent induced aggregation "excimer" formation rigid hydrophobic main chain E. broad, ill-defined emission features W Wavelength Figure 2.1: Effect of interpolymer x-it interactions on spectral purity and signal intensity Supramolecular and macromolecular approaches to mitigate these effects have been developed that effectively isolate polymer chains from each other.4 However, these strategies can result in large interpolymer separation that can reduce energy transfer between chains (Figure 2.2), which is the basis of some transduction schemes need that aggregation figure to elaborate this point (see Section 1.4 ). isolated chains, therefore no excimer formation large side chains reduced interchain energy transfer efficiency C I reduced amplification response Figure 2.2: Supramolecular mitigation of excimer formation. Physical separation prevents interchain exciton migration / * Rigid, three-dimensional structure * Prevents strong quenching interactions in thin film * "Minimalist" structure maintains energy transfer efficiency H H 2 high efficiency interchain energy transfer maintained Figure 2.3: Penticene (2). Its rigid structure prevents excimer formation yet is compact, allowing for productive interchain exciton migration The three-dimensional, noncompliant structure of iptycene-type monomers (2, Figure 2.3 and 2.4) has been shown by our laboratory to prevent strong quenching interactions between polymer chains while still promoting productive interpolymer energy transfer necessary for amplification. We hypothesized that monomer 1, with its three dimensional hydrophilic architecture, might impart the same properties to water soluble polymers as 2 does in non-polar media (Figure 2.4). Organic H H 2 / Figure 2.4: Three dimensional, rigid monomer scaffolds. Goal - apply the design principles of pentiptycene to aqueous media 2.2 Monomer Synthesis The synthesis of 1 began with p-bromanil (3); double addition of TIPS acetylide to 3 followed by SnCl 2 reductive aromatization provided 4 (Scheme 2.1).7 We envisioned that the three dimensional carbon framework might be established in one step via a double aryne-cycloaddition protocol.8 In the event, addition of n-BuLi to dienophile precursor 4 at -45 *C produces a benzyne intermediate after initial lithium-halogen exchange. This putative dienophile underwent [4+2] cycloaddition with 59 to provide a mixture of 6-anti and 6-syn cycloadditon products in a ratio of 1.7:1. The reaction was evaluated under a variety of solvent and temperature conditions (a more detailed discussion can be found in the Experimental Section 2.7.1), but the ratio of isomers varied only slightly. The isomers were not easily separated so the mixture was carried on to the next step; however, sufficient quantities of 6-anti could be isolated to confirm the stereochemistry by X-ray crystallography (Scheme 2.1). The structure of 6-syn was determined from a dioxo-diosmium bridged species isolated in a subsequent step (vide infra Scheme 2.2). Despite a modest yield, this procedure creates considerable structural complexity in a single step. Further, this transformation can be performed on multigram scales (>5 g). Scheme 2.1. Synthesis of the carbon framework TIPS OC 0X 0 Br 1TIPS Br Br 2. SnC -- 5 (3 eq.) n-BuLi (2.4 eq.) Li 2 90% overall Br Tol/Hex -45 0C to rt 0 67% TIPS 0 0 (1.7:1) / / o + / 7/ TIPS TIPS inseparable mixture anti: syn / o0 / 00 0 0 6-anti ntl TIPS 6-syn Modeling studies suggested that the double dihydroxylation of 6 would occur on the less hindered face of the alkene in 6-anti/syn to produce the corresponding isomers 8anti and 8-syn (Scheme 2). Unfortuantely, all attempts with a variety of NMO mediated osmium tetroxide dihydroxylation conditions (Upjohn process)' 0 ' showed no conversion of starting material. However, 'H NMR analyses of the crude reaction mixtures revealed the amount of 6-syn diminished relative to 6-anti with increasing Os04 loadings. To investigate this observation further, a reaction using a stoichiometric amount of Os04 was performed and 7 was obtained. While dioxo-diosmium type species are known,' 2 the cisdioxo variant 7 demonstrates remarkable stability in a variety of solvents and can be purified by silica gel chromatography. The steric bulk surrounding the osmium in this system likely prevents re-oxidation by NMO, thus trapping the osmium catalyst and halting the catalytic cycle (Scheme 2.3). Scheme 2.2: Osmium tetroxide catalyst turnover prevented by dioxo-diosmium species TIPS 7 o stoich. 0O Os0 4 0 6-antiVsyn TIPS (1.7:1) NMO Os0 4 V (Upjohn X process) TIPS TIPS OH H HO O OH OH 7 TIPS + HO HO . 0 0 7 0 TIPS 8-anti 8-syn Scheme 2.3: Rationale for why Upjohn conditions do not result in catalyst turnover TIPS O 0 ' 0 H H _'O I Hydrolysis Os0 TIPS These observations led us to examine the Sharpless asymmetric dihydroxylation (SAD), where conditions allow for hydrolysis of the osmate ester (7) prior to reoxidation in free solution.' 3 After some modification of the SAD conditions (see Experimental Section 2.7.2 for further details), separable isomers of 8-anti and 8-syn were isolated in excellent yield and the geometry of dihydroxylation was confirmed by X-ray crystallography (Scheme 2.4). Finally, global deprotection of 8-anti (see Experimental Section for 8-syn) with BC13 to remove the acetonide groups followed by TBAF to remove the TIPS groups provided 1-anti (Scheme 2.5). 4 Scheme 2.4: Dihydroxylation TIPS o 0 ---- / / 0 O 0 t-BuOH/H 2 0 (1:1), rt OH 6-antl/syn TIPS 90% overall (1.7:1) TIPS TIPS K2 Os 2(OH) 4 (3 mol%) DABCO (15 mol%) K3 Fe(CN) 6 (6 eq) K2 CO3 (6 eq) MeSO 2NH 2 (6 eq) / - TIPS // S + OH 0 HO OH HO HO HO .- 0 C /7 TIPS 8-anti (57%) 0 0 (33%) 8-Syn X-ray of 8-syn Scheme 2.5: Deprotection to reveal 1-anti TIPS HO HO O 8n OH // OH TIPS O BC3 DCM 88% TBAF OH 1H THF 92% 'OH -OH 8-anti 2.3 Polymer Synthesis Initial attempts at polymerization of 1-anti focused on homopolymerization using palladium mediated oxidative homocoupling of acetylenes (Scheme 2.6 and 2.7).'1 The reaction makes use of the facile way in which butadiynes (K) can be formed through reductive elimination from Pd" (a common reaction to activate Pd" precatalysts to Pd). Scheme 2.6 shows the mechanism of how these butadiynes can be applied to polymer synthesis. As stated, the Pd" (J), serves as the active catalyst begins with a double transmetallation step to form G followed by reductive elimination to produce A and K. A stoichiometric oxidant is required to convert A back to the Pd" species J, and the cycle repeats. In contrast to the Sonogashira reaction where the amine base is required in stoichiometric quantities, here the amine acts as a proton transfer catalyst. Scheme 2.6: Oxidative Palladium Acetylene Homocoupling OH R3NH+X CuX 2 R3 N + | OH LnPd" H O 2 R3NH+ + + :NR 3 | Oxidant L O CuF oxidation LnPd0 A double transmetallation reductive elimination R2 R2 DE /R2 R2 LnPd" G GR 2 Product K Unfortunately, the reaction proved unsuccessful in all attempts at direct homopolymerization of 1-anti and only insoluble material could be isolated. Attempts to polymerize diol protected versions of 1-anti and then deprotect after polymerization also failed to yield water soluble homopolymers (Scheme 2.7). These observations suggest the rigid structure of 1-anti may not provide enough favorable entropic interactions with water to produce a soluble homopolymer. Copolymerization however, with the sulfonated diiodide 103a,1s by Sonogashira polymerization produced high molecular weight (M" = 14,500, PDI = 1.7, DP = 18) and highly water-soluble (>8 mg/mL) polymers (P1-anti) in good yield. Scheme 2.7: Homopolymerization and Copolymerization n HO HO HO HO OH OH OH OH Short oligomers and insoluble material ui H(i-P r)2 benzoquinone DMF/ H20 Deprotection R=H H RO OR OR OR // R = protecting groups: OR n RO RO Pd(PPh 3)4, Cul, benzoquinone HN(i-Pr)2, solvent /0RO // RO RO RO esters, orthoesters, acetals - O RO OR OR OR OR H Na 3S O NaO 3 S,- O HO HO ,O n /O 10 A~- SO3 Na Pd(PPh 3)4, Cui DMF, H20, HN(i-Pr) 2 88% R=H 6H OH SO 3 Na HO HO // OH OH P1-anti 2.4 Monomer Effects on Spectral Purity The immediate effects of incorporating a monomer such as 1-anti with its noncompliant three dimensional architecture can be seen in Figure 2.5. The emission characteristics of P1-anti in comparison to an analogous control polymer (P-peg)5 a lacking a deaggregating structure' 6 is evident by the sharper, well-defined emission features. Further, sensing of biological analytes will likely not take place in pure water but in higher ionic strength solutions, such as phosphate buffered saline (PBS). Indeed, P1-anti shows the same sharp emission features in 1X PBS with a quantum yield within the error of measurement (Table 2.1). The control polymer however, shows lower energy emission features, consistent with the formation of low-energy emissive traps arising from interpolymer n-n interactions. 0.08 - 0.070.060.050.040.030.02 0.01 0.00300 350 NaO 3 S,- 400 450 500 Wavelength (nm) O n / HO NaO O 450 500 550 600 650 700 Wavelength (nm) 3 S-,O n /O SO 3 Na " SO3Na HO HO O)H O 400 550 600 HO3 HO ... / OH* OH P1-anti MeO water OF19% o F O Z 0 Ooe P-peg N PBSF=7% M water, 4)F GF s PBS, 19 F=12% Figure 2.5: UV-vis and fluorescence spectra of P1-anti and P-peg in both water and I X PBS solution Table 2.1: Peak Analysis data for Figure 2.5 Entry 1 2 3 4 5 6 <DF P1-anti, water P1-anti, X PBS P-peg, water P-peg, 1X PBS P1-anti, water:glycerol (1:1) P1-anti, water:methanol (1:1) a Arbitrary 1 17% 1 9% 1 4 5 peak heighta 41.81 36.99 22.10 10.86 143.7 149.9 fwhma, ratioc 32.50 24.82 92.47 100.29 28.48 29.51 1.30 1.50 0.24 0.11 5.04 5.08 units. b fwhm = full width at half maximum. ' Ratio = (peak height):(fwhm). Peak analysis data in Table 2.1 shows numerically what is visually represented in Figure 2.5. By examining the ratio between the peak height and the peak's full width at half maximum, it is clear that the presence of 1-anti (P1-anti) produces distinctly sharper fluorescence signals with visible vibrational features (Table 2.1, entries 1-4). The blue shifted absorbance and emission of P1-anti relative to the control is due to a lower HOMO level resulting from the absence conjugated oxygen lone pairs in 1-anti (See Section 1.2 for further explanation). 2.5 Polymer Solution Properties Interestingly, despite the sharper emission peaks of P1-anti, there is not an observed increase in quantum yield often seen when bulky side chains are used. This is possibly due to close proximity of the the hydroxyl groups of 1-anti to the polymer chain. The hydrogen bonding of the aqueous media with the hydroxyl group array might encourage non-radiative vibration relaxation of the excited state. This theory is supported by the increase in quantum yield for entries 5 and 6 in Table 2.1 (also see Figure 2.6); the presence of alcohol solvent does decreases the overall hydrogen bonding of the media. However, we believe that this increase in quantum yield may be due to water:alcohol solvent mixtures being more solubilizing for P1-anti. This would imply that the P1-anti, while soluble in water and PBS, is infact in an aggregated form and undergoing selfquenching while not experiencing excimer formation. 50 , 4 - 03 Ii 0 0- Cu 300 I 350 400 450 Wavelength (nm) 500 400 I i I 450 500 550 Wavelength (nm) 600 Figure 2.6: Solubility enhancement of quantum yield of P1-anti . UV-vis absorption trace (left) of P1-anti in water (black), 1:1 water/glycerol (red), 1:1 water/methanol (green) (units of E= L 'mol-* cm'1). Fluorescence trace (right) of P1-anti in water (black, DF = 19%), 1:1 water/glycerol (red, F= 49%), 1:1 water/methanol(red, <DF = 50%). To further understand the nature of P1-anti in solution, Stern-Volmer quenching experiments were performed with 3-nitrotyrosine, a modified tyrosine residue associated with over 50 disease states (Figure 2.7).17 A greater quenching constant can be expected for more aggregated polymers because of increased interchain exciton migration and transfer to the electron-deficient nitroaromatic quencher (see Section 1.4 for details). Indeed, a Ksv of 27,737 was found in PBS and the Ksv decreased for better solvents (4,774 in water and 1,232 in water:methanol), indicating a decrease in aggreagation with better solvents. Quenching experiments performed with P-peg as control showed no response, presumably because the aggregated emissive states of the polymer provide lower energy states than the LUMO of 3-nitrotyrosine. 4x10 1.17 - 3x10 - - U 1.14 1.11 " O2N . 3-nitrotyrosine /CO 2 1.511 21 NH 3+ 1.02 096 0 NaO 2X4(6 /O 11HHO HO SONa 3 " 7/HO HO 0 [ SA,-O 3 6 0 - 450 Ksv 500 550 Wavelength (nm) OH OOH 600 PBS 27,737 Water Water/MeOH 4,774 1,232 Figure 2.7: Fluroescence spectra for the addition of micromolar quantities of 3nitrotyrosine to P1-anti in 1X PBS (plot) and associated Stern-Volmer plot (inset). Schematic representation of soluble polymer aggregates for each measured Ksv. 2.6 Conclusion In summary, we have developed an expedient synthesis of monomer 1-anti (and 1-syn, see Experimental Section). The hydrophilic three-dimensional structure of 1-anti was shown to promote spectral purity with decreased lower energy excimer emission in both water and PBS despite an aggreagted state. Further interchain energy migration was shown through Stem-Volmer analysis and work towards applying interpolymer energy transfer for further sensing applications is underway. 2.7 Experimental Section Materials: Silica gel (40 prm) was purchased from SiliCycle. All solvents used for photophysical experiments were spectral grade. Pd(PPh 3)4 was purchased from Strem Chemicals, Inc. All other reagent grade materials were purchased from Aldrich, TCI America, and Alfa Aesar, and used without further purification. Experimental: NMR Spectroscopy: 'H and 13C NMR spectra for all compounds were acquired in CDCl 3, D2 0 and DMF-d 7 on a Bruker Avance Spectrometer operating at 400 and 100 MHz, respectively. The chemical shift data are reported in units of 6 (ppm) relative to residual solvent. Gel Permeation Chromatography (GPC): Polymer molecular weights were determined using a triple detection method for calibration with poly(acrylic acid) standards on a Viscotek TDA 305-040 instrument equipped with two Viscotek A-MBHMW-3078 columns. and analyzed with light scattering and refractive index detectors. Samples were dissolved in 5% NH4 0H. Absorption and Emission Spectroscopy: Fluorescence spectra were measured on a SPEX Fluorolog-T3 fluorometer (model FL-321, 450 W Xenon lamp) using right-angle detection. Ultraviolet-visible absorption spectra were measured with an Agilent 8453 diode array spectrophotometer and corrected for background signal with a solvent filled cuvette. Fluorescence quantum yields of P1-anti in both water and 1X PBS were determined relative to perylene and are corrected for solvent refractive index and absorption differences at the excitation wavelength. The quantum yield of P-peg in IX PBS was determined relative to P-peg in pure water. Lifetime measurements: Time resolved fluorescence measurements were performed by exciting the samples with 160 femtosecond pulses at 390 nm from the double output of a Coherent RegA Ti:Sapphire amplifier. The resulting fluorescence was spectrally and temporally resolved with a Hamamatsu C4780 Streak Camera system. Stern-Volmer Quenching Experiments: A 4 mL solution of P1-anti, at an appropriate concentration for UV-vis and fluorescence measurements, was prepared and 1 mL this solution was used to dissolve 3-nitrotyrosine (0.045-0.055 mg). 20 [tL additions of the 1 mL were made to the original P1-anti solution and measurements taken after addition and mixing (5 additions). The 3-nitrotyrosine absorbance overlaps with P1-anti so a corrected Stern-Volmer protocol was applied to the data following reference 8. Synthetic Procedures Diaryne precursor 4. The procedure of Bowles et. al.' was used with little modification. TIPS acetylene (6.93 g, 54.3 mmol) was dissolved in 190 mL of dry THF under argon. Following, n-BuLi (52 mmol, 1.6 M in Hexane) was added dropwise at room temperature and the mixture was stirred for 30 min. Compound 3 (10g, 23.6 mmol) was added in one portion and the reaction immediately turned dark. The solution was stirred at 60 C for 30 min and cooled to room temperature. The reaction was quenched with 200 mL of sat. NH 4C1 and extracted with one portion Hexanes, then DCM. The combined organic layers were concentrated in vacuo and the residue was dissolved in 500 mL of MeCN. SnCl 2 (18 g, 95 mmol) and 2 mL of water was added and the reaction refluxed for 12 hrs. After cooling to room temperature, the suspension was filtered through a thick pad of alumina, eluting with hexanes until UV-active material no longer eluted from the bottom of the column (checked with TLC at 254nm). The solvent was removed under reduced pressure and the residue recrystallized from EtOAc to give 4 (95%). 1H NMR (400 MHz, CDCl 3 ): d 1.16 (m, 42H). 13 C NMR (125 MHz, CDC13) d 129.2, 128.36, 105.8, 104.8, 18.9, 11.5. HRMS (EI) calcd. for C2 8H42 Br 4 Si2 [M] 753.9556, found 753.9524. Dicycloaddition products 6-anti/6-syn. To a suspension of 4 (5 g, 6.63mmol) and 59 (3.03 g, 11.94 mmol) in dry toluene at -45 0C under argon was added dropwise 10 mL nBuLi (1.6 M in hexane) that had been further diluted to 40 mL of hexanes by syringe pump over 1.5 h. Once addition was complete the reaction was allowed to warm to rt and stirred for 2 hr. The reaction was quenched with methanol and water and the volatiles were removed in vacuo. The residue was taken up and partitioned between water and DCM (50 mL each). The aqueous phase was washed with DCM (2 x 50 mL) and the combined organic layers were dried over Na2 SO 4 and the solvent removed in vacuo. The residue was purified by flash chromatography (silica gel, 6:4 Hexanes:DCM, Rf= 0.4) to yield 4 as an inseparable mixture of 6-anti/6-syn mixture (67%, 1.7:1 by NMR). 6-anti: 'H NMR (400 MHz, CDCl 3): d 6.49 (m, 4H), 4.65 (m, 4H), 4.20 (m, 4H), 1.40 (s, 6H), 1.25 (s, 6H), 1.17 (m, 42H). 13 C NMR (125 MHz, CDCl 3): d 140.6, 132.8, 116.8, 112.3, 101.8, 99.6, 78.6, 44.0, 26.0, 25.5, 19.0, 11.5. HRMS (ESI) caled. for C4 6H6 6 0 4 Si2 [M+Na*] 761.4392, found 761.4415. 6-syn: 1H NMR (400 MHz, CDC13): d 6.44 (m, 4H), 4.65 (m, 4H), 4.27 (m, 4H), 1.41 (s, 6H), 1.28 (s, 6H), 1.17 (m, 42H). 13 C NMR (125 MHz, CDCl 3): d 140.7, 132.8, 116.7, 112.3, 101.9, 99.5, 78.6, 44.0, 26.0, 25.6, 19.0, 11.5. HRMS (ESI) calcd. for C4 6 H6 60 4 Si2 [M+Na*] 761.4392, found 761.4415. Diosmium bridged species 7: To a solution of 6-anti/6-syn (10 mg, 0.013 mmol) in 2 mL of acetone was added solid Os04 (0.158 mmol). The solution turned dark and was stirred for 15 h. The reaction was partitioned betweened water and DCM and the organic layer was collected, dried with Na 2 SO 4 and evaporated to dryness. Silica gel chromatography (8:2) Hexanes:EtOAc provided 7 as a purple solid. (80%). 1H NMR (400 MHz, CDCl 3): d 5.83 (m, 4H), 4.53 (in, 4H), 4.36 (in, 4H), 1.56 (s, 6H), 1.36 (s, 6H), 1.17 (in, 42H). HRMS (ESI) calcd. for C4 6H 66 0 12 0s 2 Si 2 Na [M+Na*] 1273.3220, found 1095.4094, 1363.3888, 1430.5004, parent ion not found. X-ray crystal structure, see Figure S2. Dihydroxylation to give 8-anti and 8-syn. A round bottom flask was charged with K3Fe(CN)6 (5.34 g, 16.23 mmol), K2 C0 3 (2.24 g, 16.23 mmol), MeSO 2NH 2 (1.54 g, 16.23 mmol), K2 0sO2 (OH)4 (60 mg, 0.163 mmol) and DABCO (46 mg, 0.407 mmol), 27 mL of t-BuOH and 27 mL H20. After 10 min of stirring, 6-anti/syn was added and the reaction suspension stirred at room temperature for 24 h. The reaction was quenched with 8 g of sodium sulfite, stirred for 10 min, and finally diluted with 2M KOH and stirred for 2 h. The mixture was extracted with EtOAc (2 x 80 mL) and the combined organic layers were combined, washed with 2M KOH (2 x 80 mL), dried over Na 2 SO 4 and the solvent removed in vacuo. The combine organic layers were further washed with 2M KOH, dried over NaSO 4 and concentrated in vacuo. The residue was purified by flash chromatography (silica gel, DCM) to remove unreacted starting material, then elution with (8:4 hexanes:EtOAc, Rf = 0.3) to yield 8-anti (57%), finally elution with (1:1 hexanes:EtOAc, Rf= 0.4) to yield 8-syn (33%) as separable isomers (90% overall). 8-anti: 'H NMR (400 MHz, CDCl 3): d 4.60 (m, 4H), 4.20 (m, 4H), 4.13 (m, 4H), 1.91 (br s, 40H), 1.53 (s, 6H), 1.31 (s, 6H), 1.16 (m, 42H). 13 C NMR (125 MHz, CDCl 3): d 137.7, 122.2, 111.6, 101.3, 100.9, 74.9, 65.2, 45.1, 26.0, 24.0, 19.0, 11.5. HRMS (ESI) calcd. for C4 6H7 00sSi 2 [M+Na*] 829.4507, found 829.4434. 8-syn: 'H NMR (400 MHz, CDCl 3): d 4.41 (m, 4H), 4.05 (m, 4H), 4.02 (m, 4H), 2.54 (br s, 40H), 1.50 (s, 6H), 1.26 (s, 6H), 1.15 (m, 42H). 13C NMR (125 MHz, CDCl 3): d 137.7, 122.0, 111.8, 101.3, 101.1, 75.2, 65.4, 45.1, 26.1, 24.0, 19.0, 11.5. HRMS (ESI) calcd. for C4 6H7 00sSi 2 [M+Na*] 829.4507, found 829.4448. Acetonide deprotection to give 9-anti. Under argon, 8-anti (1 g, 1.24 mmol) was dissolved in 20 mL of dry DCM and cooled to -78 C. Boron trichloride (6.19 mmol, 1M in DCM) was added dropwise and the reaction was allowed to warm to room temperature and stirred for 2 h. The reaction was quenched with ice (100 g) and the mixture was stirred at 80'C where upon the DCM was distilled off. After 1 h the reaction was cooled in an ice bath, filtered and washed with cold water to give 9-anti as a white powder (88%). 1H NMR (400 MHz, DMF-d 7 ): d 5.18 (m, 4H), 4.52 (m, 4H), 3.41 (m, 4H), 3.96 (in, 4H), 3.62 (in, 4H), 1.18 (in, 42H). 13 C NMR (125 MHz, DMF-d 7): d 138.7, 120.4, 104.1, 97.42, 65.5, 64.4, 48.6, 18.7, 11.6. HRMS (DART FT-MS) calcd. for C4 0H62 0 8Si 2 [M+H+] 727.4056, found 727.4049. Acetonide deprotection to give 9-syn. Under argon, 8-syn (1 g, 1.24 mmol) was dissolved in 20 mL of dry DCM and cooled to -78 C. Boron trichloride (6.19 mmol, 1M in DCM) was added dropwise and the reaction was allowed to warm to room temperature and stirred for 2 h. The reaction was quenched with ice (100 g) and the mixture was stirred at 800C where upon the DCM was distilled off. After 1 h the reaction was cooled in an ice bath and filtered and dried to give crude 9-syn as a powder. The syn isomer required a further reflux in IM HCl for 2 h after which the suspension was cooled in an ice bath, filtered and dried to give pure 9-syn as a white powder (75%). 1H NMR (400 MHz, DMF-d7 ): d 5.19 (m, 4H), 4.52 (in, 4H), 4.27 (m, 4H), 3.97 (m, 4H), 3.58 (m, 4H), 1.18 (m, 42H). '3 C NMR (125 MHz, DMF-d 7): d 138.8, 120.4, 104.0, 97.5, 65.6, 64.4, 48.7, 18.7, 11.6. HRMS (DART FT-MS) caled. for C40 H62 0 8 Si 2 [M+NH 4+] 744.4321, found 744.4330. TIPS removal to give 1-anti. Tetrabutylammonium fluoride (2.41 mmol, IM in THF) was added to 9-anti in 5 mL DMF and 15 mL THF and stirred for 3 h. The THF was removed in vacuo, and acetic acid (2 mL) and chloroform (50 mL) were added. The product precipitated with cooling in an ice bath. Filtration yielded 1 as a white solid (92%). 'H NMR (400 MHz, DMF-d7 ): d 5.10 (in, 4H), 4.53 (m, 4H), 4.49 (s, 2H), 4.35 (m, 4H), 3.87 (m, 4H), 3.61 (in, 4H). 13C NMR (125 MHz, DMF-d): d 138.7, 119.7, 86.5, 80.2, 65.4, 64.3, 48.4. HRMS (DART FT-MS) calcd. for C2 2 H22 0 8 [M+H*] 415.1387, found 415.1377. TIPS removal to give 1-syn. The same procedure was used as for 1-anti. (95%). 'H NMR (400 MHz, DMF-d 7 ): d 5.10 (in, 4H), 4.53 (m, 4H), 4.50 (s, 2H), 4.31 (m, 4H), 3.87 (in, 4H), 3.58 (m, 4H). "3 C NMR (125 MHz, DMF-d 7): d 138.7, 119.7, 86.5, 80.1, 65.5, 64.2, 48.4. HRMS (DART FT-MS) calcd. for C22 H22 0 8 [M+NH 4 *] 432.1653, found 432.1654. Calcd. for C22 H22 0 8 [M+H t ] 415.1393, found 415.1393. Polymer Synthesis: A suspension of 1-anti (38.2 mg, mmol) and 10 (60 mg, mmol) in 2.4 mL of DMF:H 2 0 (1:1) was degassed with three cycles of freeze-pump-thaw. A mixture of Pd(PPh 3)4 (4 mg, 3.5 tmol) and CuL (8 mg, 42 tmol) in 1.2 mL DMF:HN(iPr)2 was added by cannula and the reaction stirred at 55 C.After 12 h the mixture was diluted to 50 mL with water and stirred for several hours to completely dissolve the polymer. The solution was centrifuged and particulate removed by decanting the yellow supernatant. The volume of the solution was reduced in vacuo (~15 mL) and added dropwise to 60 mL of acetone with stirring. The mixture was strongly basified with aqueous NaOH and the polymer precipitated as an orange gel. The supernatant was decanted off and discarded; 60 mL of fresh acetone was added to the gel residue and stirred, then left to stand. The supematant was discarded and this process was repeated twice more with EtOAc, then THF. The gel residue was then suspended in 50 mL of water and dialyzed against de-ionized water with 6 changes of water. Lyophilization yielded P1-anti as a yellow solid (88%). GPC gave M, = 14,500 Mw = 24,700 PDI = 1.7 DP = 18. 'H NMR (400 MHz, D2 0): d7.42 (s, 2H), 4.61 (broad, 4H), 4.35 (broad t, 4H), 4.03 (broad, 4H), 3.88 (braod, 4H), 3.16 (broad t, 4H), 2.33 (broad t, 4H). Synthetic Optimizations - Extended Discussions 2.7.1 Optimization of 4 -> 6-anti/syn: Upon addition of n-BuLi, 4 undergoes lithium halogen exchange to give an anion, which then eliminates the adjacent bromide to produce a benzyne-like intermediate. This putative dienophile undergoes [4+2] cycloaddtion with 5. The reaction was evaluated with slow addition of n-BuLi in hexanes to a chilled suspension of 4 and 5 in toluene. Initial temperature screens revealed that -45 C gave the highest yields (Table S2, entries 1-4). Additional equivalents of diene did not greatly improve the yield and complicated product purification (entry 6). A smaller silyl group resulted in an improved yield (entry 7), but product solubility was greatly reduced with this variant. Tetrahydrofuran was also tested as the reaction solvent but appears to be inferior to toluene (entry 8). Interestingly, the ratio of product isomers varied only slightly (6-anti : 6-syn, 1.7:1) across all conditions. A slower rate of addition (2 hr.) of n-BuLi gave cleaner reaction products but did not greatly affect the yield. Table 2.2: Screening of conditions for the di-aryne cycloaddition of 4 and 5. R R n-BuLi (2.5 eq) Toluene Hexane Br Br + n 0 Br Br 4 5 Syringe pump addition 2h Entry T [0C] n 3 1 -78 2 3 -45 3 3 -15 4 rt 3 2 5 -45 6 6 -45 3 7 -45 3 -45 8 a THF used in place of tolu ene 2.7.2 Optimization of 6-anti/syn -> R 0 0 /7 6-anti R isomers not separable R TIPS TIPS TIPS TIPS TIPS TIPS TMS TIPS 0//0 /7 0 (1.7:1 6-syn Yield [%] 51 67 55 44 60 65 75 28a 8-anti and 8-syn: Modeling studies suggested dihydroxylation would occur on the less hindered face of the alkene and the modified NMO conditions of Sharpless et. al. were initially tried. Unfortunately, all attempts with a variety of NMO conditions proved unsuccessful (Table S3, entries 1-5). Increased osmium loading results in a reduction of the amount of 6-syn observed in the crude product mixture. (entries 2-5). To investigate this observation further, a stoichiometric reaction was performed (entry 6) and the product 7 was obtained. While dioxo-diosmium type species are known, this particular example demonstrates remarkable stability to a variety of solvents and even silica gel. The steric bulk surrounding the osmium in this system likely prevents re-oxidation by NMO, thus trapping the osmium catalyst and halting the catalytic cycle (Figure refer to above figure). These observations lead us to examine the Sharpless Asymmetric Dihydroxylation (SAD), where conditions allow for hydrolysis of the osmate ester before reoxidation. It was hoped that attack of a smaller hydroxide anion would be faster than a bulky NMO molecule, resulting in more efficient catalytic turnover. Indeed, initial attempts using ADmix-c and the hydrolysis accelerant methane-sulfonamide looked promising (entry 7). Switching to a smaller tertiary amine, 1,4-diazobicyclo[2.2.2]octane (DABCO), and increasing the catalyst loading resulted in higher conversions (entries 9-10). Upon purification, 6-anti still contained small amounts of osmium that were observed to precipitate as osmium(0) in the subsequent step (acetonide deprotection). Thus, after reductively quenching the reaction with Na 2 SO 3 , the reaction was stirred with 2M KOH for several hours to completely hydrolyze the residual osmate ester (entry 11). The result was a near quantitative conversion. Table 2.3: Screen of reaction conditions for the dihydroxylation of 6-antilsyn TIPS TIPS OH // O 0 Conditions / N. / HOH 0nHO / T 6-anti/syn mix TIPS (1.7:1) C 8-anti Entry 1 2 3 4 5 6 7 8a Os catalyst 0.1% K20sO 2(OH) 4 0.5% K20sO 2(OH) 4 2% OS04 5% Os0 4 10% OsO4 Stoich. OS04 AD-mix-a (2 eq) 0.2% K20sO2(OH) 4 6 K3Fe(CN)6 Solvent H20/t-BuOH (1:1) Pyridine Acetone/H 20(9:1) Acetone/H20(9:1) Acetone/H 2 0(9:1) Acetone H20/t-BuOH (1:1) H20/t-BuOH (1:1) 9 3% K2 0sO2(OH) 4 6 K3Fe(CN)6 H20/t-BuOH (1:1) 10 6% K2 0sO2(OH) 4 6 K3Fe(CN)6 H20/t-BuOH (1:1) 6% K2 0sO 2(OH) 4 6 K3Fe(CN) 6 H20/t-BuOH (1:1) 11 b Additives 4 Citric acid 8-syn Conv. Yield [%] 1%] 1%] [%] rt rt rt rt rt rt [h] 15 15 15 15 15 15 48 48 -anti 0 0 0 0 0 0 44 53 -syn 0 0 2 5 8 86 90 85 -anti 0 0 0 0 0 0 14 20 -syn 0 0 0 0 0 0 80 75 50 48 85 >95 80 90 rt 24 95 >95 84 93 rt 24 95 >95 95 93 [0C] 120 AD-mix-a: (DHQ) 2PHAL (1 mol%), K20sO2(OH) 4 (0.4 mol%), K 3Fe(CN)6 (3 eq.), K CO (3 eq.) 2 3 b After reductive quenching with Na 2 SO3 , 2M KOH was added and the mixture stirred for several hours a Yield Conv. rt 3 MeSO 2NH 2 0.1 DABCO 3 MeSO 2NH 2 6 K2C03 0.1 DABCO 6 MeSO 2NH 2 6 K2 CO3 0.15 DABCO 6 MeSO 2NH 2 6 K2 CO3 0.15 DABCO 6 MeSO2NH 2 6 K2 C0 3 TI PS Time Temp Reoxidant 2.2 NMO 2.2 NMO 2.2 NMO 2.2 NMO 2.2 NMO HO OH 0 TIPS TIPS separable isomers 2.8 References (1) (a) Thomas, S. W.; Joly, G. D.; Swager, T. M. Chem. Rev. 2007, 107, 1339. (b) Satrijo, A.; Swager, T. M. J. Am. Chem. Soc. 2007, 129, 16020. (c) Wosnick, J. H.; Mello, C. M.; Swager, T. M. J. Am. Chem. Soc. 2005, 127, 3400. (d) Gaylord, B. S.; Heeger, A. J.; Bazan, G. C. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10954. (e) Wang, D.; Gong, X.; Heeger, P. S.; Rininsland, F.; Bazan, G. C.; Heeger, A. J. Proc. Natl. Acad Sci. U.S.A. 2002, 99, 49. (f) Wang, S.; Gaylord, B. S.; Bazan, G. C. J Am. Chem. Soc. 2004, 126, 5446. (g) Kumaraswamy, S.; Bergstedt, T.; Shi, X.; Rininsland, F.; Kushon, S.; Xia, W.; Ley, K.; Achyuthan, K.; McBranch, D.; Whitten, D. Proc. Natl. Acad Sci. U.S.A. 2004, 101, 7511. (h) Phillips, R. L.; Miranda, 0. R.; You, C. C.; Rotello, V. M.; Bunz, U. H. F. Angew. Chem. Int. Ed. 2008, 47, 2590. (i) You, C. C.; Miranda, 0. R.; Gider, B.; Ghosh, P. S.; Kim, I. B.; Erdogan, B.; Krovi, S. A.; Bunz, U. H. F.; Rotello, V. M. Nat. Nanotechnol. 2007, 2, 318. (j) Yang, C. Y.; Pinto, M.; Schanze, K.; Tan, W. Angew. Chem. Int. Ed. 2005, 44, 2572. (k) Pinto, M. R.; Schanze, K. S. Proc.Natl. Acad. Sci. USA 2004, 101, 7505. (1)Xu, H.; Wu, H.; Huang, F.; Song, S.; Li, W.; Cao, Y.; Fan, C. Nucleic Acids Res. 2005, 33, e83. (m) Ho, H. A.; Najari, A.; Leclerc, M. Acc. Chem. Res. 2008, 41, 168. (n) Ho, H. A.; Boissinot, M.; Bergeron, M. G.; Corbeil, G.; Dore, K.; Boudreau, D.; Leclerc, M. Angew. Chem. Int. Ed. 2002, 41, 1548. (o) Nilsson, K. P. R.; Inganas, 0. Nat. Mater. 2003, 2, 419. (p) Sigurdson, C. J.; Nilsson, K. P. R.; Homemdnn, S.; Manco, G.; Polymenidou, M.; Schwarz, P.; Leclerc, M.; Hammarstrom, P.; Wiithrich, K.; Aguzzi, A. Nat. Methods 2007, 4, 1023. (q) Li, C.; Numata, M.; Takeuchi, M.; Shinkai, S. Angew. Chem. Int. Ed. 2005, 44, 6371. (r) Feng, F.; Wang, H.; Han, L.; Wang, S. J. Am. Chem. Soc. 2008, 130, 11338. (s) Feng, F.; Tang, Y.; Wang, S.; Li, Y.; Zhu, D. Angew. Chem. Int. Ed. 2007, 46, 7882. (t) Tang, Y.; Feng, F.; He, F.; Wang, S.; Li, Y.; Zhu, D. J. Am. Chem. Soc. 2006, 128, 14972. (u) An, L.; Liu, L.; Wang, S.; Bazan, G. C. Angew. Chem. Int. Ed. 2009, 48, 4372. (2) Swager, T. M. Acc. Chem. Res. 1998, 31, 201. (3) (a) Tan, C.; Pinto, M. R.; Schanze, K. S. Chem. Commun. 2002, 446. (b) Tan, C.; Alas, E.; MUller, J. G.; Pinto, M. R.; Kleiman, V. D.; Schanze, K. S. J. Am. Chem. Soc. 2004, 126, 13685. (c) Chen, L.; McBranch, D. W.; Wang, H. L.; Helgeson, R.; Wudl, F.; Whitten, D. G. Proc. Natl. Acad Sci. U.S.A. 1999, 96, 12287. (4) For a review see: Frampton, M. J.; Anderson, H. L. Angew. Chem. Int. Ed. 2007, 46, 1028. (5) (a) Lee, K.; Cho, J. C.; DeHeck, J.; Kim, J. Chem. Commun. 2006, 1983. (b) Khan, A.; Miller, S.; Hecht, S. Chem. Commun. 2005, 584. (c) Jiang, D. L.; Choi, C. K.; Honda, K.; Li, W. S.; Yuzawa, T.; Aida, T. J Am. Chem. Soc. 2004, 126, 12084. (d) Kuroda, K.; Swager, T. M. Chem. Commun. 2003, 26. (e) Terao, J.; Tsuda, S.; Tanaka, Y.; Okoshi, K.; Fujihara, T.; Tsuji, Y.; Kambe, N. J. Am. Chem. Soc. 2009, 131, 16004. (f) Terao, J.; Tanaka, Y.; Tsuda, S.; Kambe, N.; Taniguchi, M.; Kawai, T.; Saeki, A.; Seki, ShuJ Am. Chem. Soc. 2009, 131, 18046. (6) Swager, T. M. Acc. Chem. Res. 2008, 41, 1181. (7) Bowles, D. M.; Palmer, G. J.; Landis, C. A.; Scott, J. L.; Anthony, J. E. Tetrahedron 2001,57,3753. (8) (a) Hart, H.; Lai, C.; Nwokogu, G. C.; Shamouilian, S. Tetrahedron 1987, 43, 5203. (b) Chen, C. L.; Sparks, S. M.; Martin, S. F. J. Am. Chem. Soc. 2006, 128, 13696. (9) (a) Baran, A.; Segen, H.; Balci, M. Synthesis 2003, 1500. (b) Yang, N. C. C.; Chen, M. J.; Chen, P. J. Am. Chem. Soc. 1984, 106, 7310. (10) VanRheenen, V.; Kelly, R. C.; Cha, D. Y. TetrahedronLett. 1976, 1973. (11) Dupau, P.; Epple, R.; Thomas, A. A.; Fokin, V. V.; Sharpless, K. B. Adv. Synth. Catal. 2002, 344, 421. (12) (a) Phillips, F. L.; Skapski, A. C. J. Chem. Soc., Dalton Trans. 1975, 2586. (b) Casey, C. P. J. Chem. Soc., Chem. Commun. 1983, 126. (c) Cartwright, B. A.; Griffith, W. P.; Schr6der, M.; Skapski, A. C. J. Chem. Soc., Chem. Commun. 1978, 853. (d) Cleare, M. J.; Hydes, P. C.; Griffith, W. P.; Wright, M. J. J. Chem. Soc,. Dalton Trans. 1977, 941. (e) Marzilli, L. G.; Hanson, B. E.; Kistenmacher, T. J.; Epps, L. A.; Stewart, R. C. Inorg. Chem. 1976, 15, 1661. (13) Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483. (14) Williams, V. E.; Swager, T. M. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 4669. (15) Chen, W.; Joly, A. G.; Malm, J. 0.; Bovin, J. 0.; Wang, S. J. Phys. Chem. B. 2003, 107, 6544. (16) CPE copolymers with 2 are not readily soluble in water, see Zheng, J.; Swager, T. M. Chem. Commun. 2004, 2798. (17) Neumann, H.; Hazen, J. L.; Weinstein, J.; Mehl, R. A.; Chin, J. W. J. Am. Chem. Soc. 2008, 130, 4028 and references there in. Chapter 2 Appendix 1H-NMR, "C-NMR, UV-vis and Fluorescence data, and X-ray Structures TIPS B B Br Br 4 TIPS 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 2.A.1: 'H NMR spectrum of 4 in CDCl 3 (400 MHz) TIPS || Br Br Br Br 4 TIPS 210 200 Figure 2.A.2: 190 13 180 170 160 150 140 130 120 110 100 90 80 70 C NMR spectrum of 4 in CDC13 (125 MHz) 60 50 40 30 20 ppm TIPS // o / / TIPS TIPS inseparable mixture anti : syn (I. 7 1) TIPS / / /i 6-anti / N/ TIPS /P o6 6-syn ~i1 7.0 6.5 6.0 5.5 5.0 4.5 i~i 4.0 3.5 3.0 2.5 2.0 1.5 L1.0 0.5 ppm 0 ppm Figure 2.A.3: H NMR spectrum of 6-anti/syn in CDC13 (400 MHz) TIPS inseparable mixture anti :syn (1.7 :1) TIPS 0 / TIP G-1/ i TIPS 200 Figure 2.A.4: 6-anti 180 13 TIPS 160 140 o 6-syn 120 100 80 60 40 C NMR spectrum of 6-anti/syn in CDC13 (125 MHz) 20 TIPS 0 T - TIPS I A 7.0 6.5 Figure 2.A.5: 6.0 5.5 5.0 6-anti 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm NMR spectrum of 6-anti/syn enriched in 6-anti in CDC13 (400 MHz) TIPS T , O TIPS 200 Figure 2.A.6: 6-anti 180 13 160 140 120 100 80 60 40 20 0 ppm C NMR spectrum of 6-anti/syn enriched in 6-anti in CDC13 (125 MHz) 56 0 0 TI PS N 7 fIPS I 7.5 7.0 I I I I 6.5 6.0 5.5 I~ 5.0 I I I 4.5 4.0 3.5 TIPS 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 2.A.7: H NMR spectrum of 7 in CDC13 (400 MHz) TIPS A-O O OH OH TIPS 7.0 6.5 HO -HO 6.0 / 0 8-anti 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 Figure 2.A.8: H NMR spectrum of 8-anti in CDC13 (400 MHz) 1.5 1.0 0.5 ppm TIPS O O- / HO HO OH OH O TIPS 200 Figure 2.A.9: $-anti 180 13 C 160 140 120 100 80 60 40 20 0 ppm NMR spectrum of 8-and in CDC13 (125 MHz) TIPS OH OH ' 0 HO HO - AO n TIPS 8-syn JL__ t 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 Figure 2.A.10: 'H NMR spectrum of 8-syn in CDC13 (400 MHz) 1.5 1.0 0.5 ppm TIPS OH HO 0 // O 8-syn TIPS I .. 200 Figure 2.A.11: 180 13 C 160 140 120 I. --. 100 - L 80 - . 60 - - -- 40 NMR spectrum of 8-syn in CDC13 (125 MHz) ll 20 0 ppm TIPS/ 8.0 7.5 7.0 6.5 9-anti 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 1.5 2.0 1.0 0.5 ppm Figure 2.A.12: 'H NMR spectrum of 9-anti in DMF-d 7 (400 MHz) TIPS HO HO /HO HO OH OH TIPS 200 Figure 2.A.13: 180 13C 160 140 120 OH / OH 9-anti 100 80 60 40 NMR spectrum of 9-anti in DMF-d 7 (125 MHz) 20 0 ppm TIPS OH HO OH HO HO HO OH OH 9-syn TIPS k--LJA-j, 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 ki 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 2.A.14: 'H NMR spectrum of 9-syn in DMF-d 7 (400 MHz) TIPS OH OH OH // TIPS 200 Figure 2.A.15: 180 13 160 140 120 HO HO HO HO OH 9-syn 100 80 60 40 C NMR spectrum of 9-syn in DMF-d 7 (125 MHz) 20 0 ppm H HO HO " HO HO OH O OH OOH 1-anti H LJL" 8.0 7.0 7.5 6.5 6.0 5.5 5.0 4.0 4.5 3.5 2.5 3.0 2.0 1.5 I 1.0 0.5 ppm Figure 2.A.16: H NMR spectrum of 1-anti in DMF-d 7 (400 MHz) L ---. A& 200 160 180 Figure 2.A17: 13C 140 - 120 I-- lj"_&L6AL 80 100 40 60 0 20 ppm NMR spectrum of 1-anti in DMF-d 7 (125 MHz) H / HO HO HO OH HO HO OH OH H 1-syn Lii ------------ 8.0 7.5 Figure 2.A.18: 7.0 1H 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 NMR spectrum of 1-syn in DMF-d 7 (400 MHz) 1.5 1.0 0.5 ppm H OH OH / HO HO HO HO* OH // H 200 180 Figure 2.A.19: 13 160 140 120 OH 1-syn 100 80 60 40 0 20 ppm C NMR spectrum of 1-syn in DMF-d 7 (125 MHz) NaO 3 S S 0O n 10-O HO HO -- SO Na 3 HO HO - OH OH OH OH P1-anti 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 Figure 2.A.20: 1H NMR spectrum of P1-anti in D20 (600 MHz) 1.5 1.0 ppm Table 2.A.1: Summary of photophysical data of P1-anti Em Xmax (nm) Abs max (nm) 451 435 P1-anti,water 462 451 P1-anti, 1X PBS 1.07 (ns) 0.61 Tsoln 1.17 (b) 1.06 (a) 1.051 1.04 1.1 U u- 1.03 .02 1.08 1.01 1.00 - 1.05 1.02 - U U U 0.990.98 <DF 19% 17% 1.20 , , , log E 4.68 4.74 0.99 , , , , 2 0 , 4 , , 6 8 0.96 - 0 - 2 4 6 [Q] 1 mol/L [Q] 1 mol/L 0.990 - 0.985 (c) (d) 0.980 0.975 U U 0.970- 0.9601 2 6 4 jmol/L [Q] 8 450 500 550 600 650 Wavelength (nm) 700 Figure 2.A.21: Stern-Volmer quenching plots of P1-anti with micromolar quantities of 3-nitrotyrosine under different solvent conditions. (a) water, (b) 1X PBS, (c) 1:1 water/methanol. (d) shows the minimal fluorescence quenching response of P-peg to 3nitrotyrosine (Chapter 2.# for explanation). Table 1.A.1: X-ray Crystallographic Data Empirical formula FW Space group a (A) b (A) c (A) a (deg) #6(deg) y (de ) V(A) z dcic (g cm-3) p (mm-') Omax (deg) Reflections collected Unique data GOF RI [I>2o(1)], wR2 (all data) Apmaxmin (e-A -3) 6-anti C46H66O4Si 2 739.17 P2(1)/c 13.6286 10.5494 16.1848 90.00 104.6470 90.00 2251.32 2 1.090 0.117 29.13 36249 C46H660 Os 12 2Si2 1290.65 P2(1)/c 16.6965 11.8460 26.3463 90.00 98.013 90.00 5160.1 4 1.661 8-anti C46H70O8Si 2 806.4609 P-1 9.0065 12.0657 12.5677 93.4460 94.1180 105.0570 1311.03 1 1.205 8-syn C 46H700 8Si2 806.4609 P-1 12.7591 13.1605 14.7690 70.182 76.378 74.595 2220.1 2 1.207 1.131 68.89 7754 5.025 29.57 0.124 29.57 143487 35043 5364 1.021 0.0443, 0.1222 14476 1.089 0.0207, 0.0468 7347 1.029 0.0377, 0.1053 1.050 0.0457, 0.1201 0.852, -0.459 1.318, -0.455 0.521, -0.211 0.722, -0.568 7754 Figure 2.A.22: Crystal Structure of 6-anti. Suitable crystals were grown using t-BuOH-DCM solution followed by slow solvent evaporation. Anisotropic thermal ellipsoids set at 50% probability. Figure 2.A.23: Crystal Structure of 7. Suitable crystals were grown using DCM-Hexane solution followed by slow solvent evaporation. One of the TIPS groups showed disorder in the structure (top). These features are omitted for clarity (bottom). Anisotropic thermal ellipsoids set at 50% probability. 66 Figure 2.A.24: Crystal Structure of 8-anti. Suitable crystals were grown using (9:1:90) DCM-THF-Hexane solution followed by slow solvent evaporation. Anisotropic thermal ellipsoids set at 50% probability. THF found with structure not shown. Figure 2.A.25: Crystal Structure of 8-syn. Suitable crystals were grown (9:1:90) DCM-THF-Hexane solution followed by slow solvent evaporation. The TIPS groups and other parts of the structure showed disorder (top, anisotropic thermal ellipsoids set at 50% probability). These features are omitted for clarity (bottom, anisotropic thermal ellipsoids set at 30% probability). Chapter 3 Biocompatible Post-Polymerization Functionalization Adapted and reproduced in part with permission from: VanVeller, B.; Swager, T. M. Chem. Commun. 2010,46,5761-5763 3.1 Introduction (PPF) Strategies for the post-polymerization functionalization of polymers are advantageous in that they allow for tuning of a polymer's properties without synthetically retreating to the monomer stage. In effect, a variety of different polymer properties and/or architectures can be realized late in the synthetic process from a common precursor. Further, PPF permits the incorporation of functional groups that may be incompatible with polymerization conditions. This chapter will focus specifically on side chain modification of PPE polymers, while Chapter 4 will discuss PPF strategies targeted at alterations in the conjugated polymer backbone. Several strategies for the PPF of conjugated polymers have been reported. A number of designs involve substitution reactions with pendant halogen,I alcohol, 2 or carboxylic acid moieties, 3 and application of high yielding click chemistries 4 like the 1,3dipolar cycloaddtion of alkynes and azides 5 or thiol-conjugate addition 6 have also been reported. Two potential drawbacks are characteristic of the above strategies: (i) an appropriately functionalized monomer specific for the intended PPF must be incorporated into the polymer synthesis-often in protected form and (ii) it can be difficult to control the extent of functionalization. Scheme 3.1: Molecules of interest discussed in Chapter 2. NaO 3 S, H .O n /HO HO HO O HO O0 OH OH// H HO OH OH OH H 1OH// SONa - HO HO HO OH OH OH p1 Chapter 2 discussed the synthesis of a rigid hydrophilic monomer (1) that-when incorporated into poly(p-phenylene ethynylenes) (PPEs) 7 (P1)-lead to increased spectral purity by preventing hydrophobically induced aggregate emission (Scheme 3.1).8 We envisioned that the three dimensional array of vicinal hydroxyl groups might be further elaborated through periodate oxidation and reductive amination (P1-+2->3, Scheme 3.2).9 Similar processes have been widely applied for bioconjugation through periodate oxidation of carbohydrate residues, making this process compatible with existing bioconjugation schemes. Herein we report a biocompatible post-polymerization biotinylation of P1, where (i) the need for a PPF specific monomer is negated by activation of an existing structural motif (already present in the polymer for purposes outlined above, also see Chapter 2) and (ii) the extent of functionalization can be controlled by the equivalents of the NaIO 4 reagent. Further, the improved spectral purity imparted by the presence of 1 in 3a is not lost. In turn, this results in a more sensitive signal amplified biosensorlo response to fluorophore-labeled streptavidin, a tetrameric protein with high biotin affinity (4 x 10-14 M)1 1 that has been applied to a variety of conjugated polymer affinitychromic3, 12 and agglutination 2b biosensor designs. 3.2 Post Polymerization Functionalization and Linkage Structure Treatment of P1 with 0.2 equivalents of NaIO 4 in water generated 1,6dialdehyde moieties at random positions along the backbone (2, Scheme 3.2).13 Subsequent incubation with an excess of amine-containing compound (a or b) in aqueous alkaline solution generated the putative Schiff base, which was reduced in situ to the tertiary amine azepane 3 with NaCNBH 3 (vide infra, Scheme 3 Scheme 3.2: Post-polymerization functionalization reaction NaO 3 S-O n-0.2n - O-' HO HO NaIO 4 P1 (0.2 eq.) P1 0 H2 /HO HO OH OH // 0 NaO 3 O / O''SONa SONa HO HO 1. a orb 2. NaCNBH 3 H20 O OH OH OH OH (40 mM) SO3 Na O' HO HO OH Na2HP04 -0.2n n-O.2n NaO 3 S,-,O OH /, .2n 0'O HO OH SONa HO HO HO OH OH OH OH OH 2 OH OH OH 3a R = Biotin-PEG -N OH 3b R = Piv-Lys-N 0 HN 0 NH H S ,)N 0 H '^ NH2 B a, Bioin-PEG3-NH2 O HN t-Bu 0 NH2 b, Piv-Lys-NH 2 The proposed azepane linkage in 3 is based on two model studies. Firstly, the broad nature of the 'H NMR signals of 3a overlapped with the weaker biotin signals making determination of the extent of functionalization difficult. Thus, 3bexhibiting a strong, unobstructed pivalamide signal-was prepared under identical conditions for 3a. Integration analysis revealed a 18-20% incorporation of b (Chapter 3 Appendix, Figure 3.A.5). Therefore, while 0.2 equivalents of NaIO 4 oxidant should generate 0.4 aldehyde equivalents, there appears to only be 0.2 equivalents of the incorporated amine. Secondly, acetonide protected 4-a synthetic intermediate in the synthesis of 18-was treated with periodate anion and produced the tetraaldehyde 5 (Scheme 3.3a). Addition of an excess of butyl amine and NaCNBH 3 to 4 in methanol gave 5 as the major product. Such products have been observed for bridging 1,6dialdehydes1 5 and likely form via a 7-exo-trig reductive cyclization to install one amine for every dialdehyde present (Scheme 3.3b). Thus, we propose the PPF in Scheme 3.2 proceeds in an analogous manner, allowing for the extent of functionalization to be controlled by the molar equivalents of NaIO 4. Scheme 3.3: Model studies and proposed mechanism for azepane formation (a) TIPS // HO O HO OH OH ~ 0 0// NaIO 4 Os cat. TBAIO THF/H 20 reflux TIPS (b) . .. 00 // 0 TIPS BuNH 2 NaCNBH 3 5 ' 7/ 7-exo-tri ,NBu 0 MeOH, reflux N / Bu' 7-exo-trig TIPS H .NHBu N 0 1. BuNH 2 2. NaCNBH 3 O .- NBu O ONBu TIPS TIPS 7/ 0 0 6 Bu ig I Bu NaCNBH 3 10 10 Bu -- . 1 11 3.3 Effect of Functionalization on Polymer Properties The effect of the described PPF method on the photophysical properties of the polymer can be seen in Figure 3. la and 3.1b. The absorbance and fluorescence maxima of 3a show excellent overlap with the parent polymer P1 in both water and PBS solution, indicating that the oxidation and reductive amination reactions leave the conjugated polymer backbone intact. The origin of the reduced quantum yield of 3a is unclear. It is possible that the newly installed amine lone pairs-held closely to the polymer backbone-might quench the polymer excited state through excited state photo-electron transfer (PET) from the electron rich amine to the polymer. To test this hypothesis the pH of the solution was varied (pH = 1-12, Figure 3.2). In theory, at low pH, protonated quaternary ammonium groups would be unable to participate in PET and the quantum yield should increase sharply relative to higher pH. As seen in Figure 3.2, no dramatic changes were observed over the pH range; the minor deviations in quantum yield are likely due to solvent effects. The reduced quantum yield may be attributed to replacing diol moieties with the relatively insoluble biotin, leading to a more aggregated state of the polymer and diminished quantum yield. In any event, the effect of incorporating 1 in 3a is still present as no lower energy excimer emission is observed and spectral purity is maintained. (a) 1.0j.8 0.6 0.40.2 0.0300 350 500 450 400 Wavelength (nm) 400 550 450 500 550 600 Wavelength (nm) 650 Figure 3.1: (a) Absorbance spectra of P1 and 3a in water and PBS solution. (b) Fluorescence spectra of P1 and 3a in water and PBS solution. 1 3 9 7.4 12 pH Figure 3.2: Effect of quantum yield of 3a at different pH. Measurements performed in PBS where pH was adjusted with HCl or NaOH. 3.4 Response to Streptavidin as a Model Biosensor The response to streptavidin in the presence of 3a is represented schematically in Fig. 3.3a, where Texas Red XTM-labeled streptavidin (TRXS) is able to gather the biotinylated polymers (3a) into a supramolecular aggregate. Amplification is achieved through the funneling of polymer excitons to the lower energy Texas Red XTM dyes through intra- and interchain energy migration within the aggregate. The results of serial additions of TRXS to 3a at room temperature in PBS solution are shown in Figure 3.3b. As anticipated, a decrease in the 3a emission and a corresponding increase in dye emission was observed (see Section 1.3.1 for details). The amplifying effect of the polymer sensor can be seen through direct excitation of the dye (Figure 3.3c, red). Finally, incubation of TRXS with P1 showed no response (Figure 3.3c, black). To better understand the nature of the interaction between TRXS and 3a and to determine the overall quenching response, we determined the Stern-Volmer quenching constant for the polymer emission (460 nm) in Figure 3.3b. The SternVolmer plot showed positive curvature (Figure 3.3d), which is likely due to additional energy migration pathways within the polymer assembly' 6 produced by the strong biotin-streptavidin association. Further, no detectable excited state lifetime change was observed with increasing TRXS concentration, indicating that static quenching is the dominant mechanism of energy transfer. A comparison of the response of 3a to a previous system (12),1o which lacks the rigid hydrophilic monomer 1, showed a 100 fold greater quenching response based on a Ksv values (2x10 7 mol-I vs 1 x 105 mol- 1, Figure 3.4). This higher sensitivity is likely due to enhanced energy transfer-more excitons reaching the dye-through avoidance of lower energy excimers (broad featureless emission in Figure 3.4b). These states-negated by the presence of 1 in P1-are localized and perhaps too low in energy to undergo transfer to the dye. (a) Energy Transfer (ET) Schematic dye-labeled streptavidin 3a (b) (c) CO C I 450 500 550 600 650 Wavelength (nm) (d) 3.5 700 *1 450 500 550 600 650 Wavelength (nm) 700 U -U 3.0 Ksv =2 x10 7 u.. 2.5 - U- = U 2.0 U 1.5 U U 1.0 0 20 80 40 60 [Q] nmol/L 100 Figure 3.3: (a) Energy transfer schematic showing how intra- and interchain exciton migration and enery transfer to TRXS can lead to amplification. (b) Addition of 9.15 pmol aliquots of TRXS to 3.46 nmol (based on repeat unit of 3a). (c) Emission of P1 in the presence of 100 pmol of TRXS (black) and direct excitation of 100 pmol of TRXS (red). (d) Stern-Volmer quenching analysis of data in (b). (b) (a) C C C 450 500 550 600 650 Wavelength (nm) 700 500 450 600 650 550 Wavelength (nm) (Ksv=2x107 700 Ksv= 1 X 105 MeO 'OX ln-0.2n HI NaO 3S, MeOk MeO O Figure 3.4: Comparison of quenching response (Ksv) to TRXS between two polymers (a) one containing a rigid structural monomer and (b) one bearing only linear PEG side groups. 3.5 Alternative Sensore Designs for Detection of Native Analytes In principle, the sensing scheme described above (Section 3.4 and Figure 3.3a) can be extended to any analyte that exhibits multivalent binding of a specific ligand bound to P1. Indeed, we hypothesize that the higher the binding valency of the analyte, the greater the response because the larger the aggregate the greater opportunity for absorption, exciton formation, and amplification. However, the scheme outlined in Figure 3.3a is contingent on the streptavidin protein (or general multivalent analyte of interest) bearing a dye suitable for energy transfer. It would be advantages to sense native analytes that do not require modification with a specific dye, and we hoped to extend the PPF procedure even further to allow sensing of native streptavidin (i.e. no dye labelling). To this end we synthesized polymers 13 (Scheme3.4) using the same PPF strategy as above (reductive amination following periodate oxidation). Biotin along with a small amount of Texas Red XTM (an amount of dye calculated to match the dye content in Figure 3.3b) were attached in a one-step procedure. We hoped the result would be a polymer sample rich in biotinylated polymer (3a) with a fraction of the polymers bearing both dye and biotin (Figure 3.5). A schematic of the proposed sensor's operation can be seen in Figure 3.5. In an analogous manner to Figure 3.3a, the biotinylated polymers form the supramolecular aggregate induced by the presence of streptavidin. Once polymers bearing the dye are incorporated into the aggregate, enhanced migration pathways allow for the funneling of excitons from the polymer aggregate into the dye for a turn on response. 3.5.1 Results and Discussion The absorbance and fluorescence speactra of 13 in comparison to P1 are shown in Figure 3.6a and b. Initial results were encouraging as the small signal at -620 nm of 13 (Figure 3.6b) was attributed to emission from Texas Red XTM following energy transfer from the polymer (polymer excitation at 400 nm). Further, enhanced dye fluorescence upon excitation of the polymer (Figure 3.6e, black) compared to direct excitation of the dye (Figure 3.6e, red) confirmed amplified energy transfer. The result of serial additions of streptavidin are shown in Figure 3.6c. Unfortunately, the sensing scheme discussed above was not realized in practice as there was no observed increase in dye emission with added streptavidin (as with the analogous TRXS expeiment in Figure 3.3b). To confirm the presence of biotin attached to the polymer, 13 was subjected to the same experiment with TRXS as in Figure 3.3b and turn-on energy transfer to the dye was observed in an analogous way (Figure 3.6f), albeit with reduced Ksv likely the result of a lower biotin content relative to P1. The failure of the proposed sensing scheme is somewhat unclear. It seems apparent that energy transfer to the dye is occuring (Figure 3.6c and e). However, it appears that aggregate formation with streptavidin is not generating additional, fruitful exciton migration pathways to allow for greater amplification of the dye (see Chapter 1.4). Polymer 14 was then synthesized with a calculated dye content approaching one dye per polymer. Polymer 14 can be considered to be approaching the theoretical limit of amplification, as every polymer in the sample is capable of energy transfer to a pendant dye even without the occurrence of aggreagation. From Figure 3.6e it is clear that there is energy transfer to the dye occuring (signal at 620 nm with polymer excitation at 400 nm) and amplification of the dye's fluorescence is occuring following the same arguments for 13 as above for Figure 3.6e. Despite the fact that every polymer theoretically possess a pendant dye, the effective energy transfer to the dye is quite small (see the relative peak heights in Figure 3.6d). The failure of the sensor design may lie in the fact that initial energy transfer to the dye is quite poor and outweighs any exciton migration enhancement. Scheme 3.4: Synthesis of fractionally dye conjugated polymer sample NaO 38 ,O n 'O HO HO - SO 3 Na HHO HO 1. NalO 4 (0.2 eq.) 2. Binary Mixture 3. NaCNBH 3 H OH H OH Binary Mixture N 0 H2 N - HN o NH SONH(CH2)5 NH2 0 3S T 0 NH 0 13 =1:24 a Texas Red X~~ 14 =1:4 q_ streptavidin excess Figure 3.5: Proposed sensing scheme for native, non-dye labelled streptavidin ( b) 1 (a) 1-0 0.8 08 - - 13 in H20 - PI in PBS - 13 in PBS 1 -P1 -13 -P1 13 , 1.0- - P1 in H 0 2 0- 0.6- S0.6- 0.4- .04 10.2z 0.0- . inH20, DF =19% inH20, <DF = 6% inPBS, <F =17% in PBS, <DF = 5% 0.2 0.0 300 350 400 450 500 Wavelength (nm) 550 400 450 500 550 600 650 700 Wavelength (nm) (d) (c) 400 450 500 550 600 Wavelength (nm) 650 400 450 500 550 600 Wavelength (nm) 650 (f) 1.6 15 1.4 Mu Ksv=6x10 6 * 10 0 400 450 500 550 600 650 700 Wavelength (nm) 400 450 20 40 60 80 [Q]nmo/L 10( - 500 550 600 650 700 Wavelength (nm) Figure 3.6: (a) Absorbance spectra of P1 and 13 in water and PBS solution. (b) Fluorescence spectra of P1 and 13 in water and PBS solution. (c) and (d) Addition of 9.15 pmol aliquots of streptavidin to 3.46 nmol 13 and 14 respectively (based on repeat unit). (e) Emission of 13 (black) and 14 (cyan) at 390 nm excitation and emission of their respective appended Texas Red XTM (13, red and 14, green) at 580 nm excitation. (f) Stern-Volmer quenching analysis of data in (e). (f) Addition of 9.15 pmol aliquots of streptavidin to 3.46 nmol (based on repeat unit of 13). Inset: Stern-Volmer quenching analysis of data in (f). 3.6 Conclusion In summary, a biocompatible PPF strategy has been developed, which takes advantage of existing monomer functionality and design. Further, the extent of functionalization can be controlled through the equivalents of NaIO 4 . Finally, a highly sensitive (Ksv = 2x10 7 mol-1) turn-on model biosensor based on ET between 3a and TRXS was demonstrated where the presence of 1 lead to dramatically increased sensitivity. 3.7 Experimental Section Materials: Silica gel (40 pim) was purchased from SiliCycle. All solvents used for photophysical experiments were spectral grade. Pd(PPh 3)4 was purchased from Strem Chemicals, Inc. All other reagent grade materials were purchased from Aldrich, TCI America, and Alfa Aesar, and used without further purification. Experimental: NMR Spectroscopy: 1H and 13C NMR spectra for all compounds were acquired in CDCl 3, D2 0 and DMF-d 7 on a Bruker Avance Spectrometer operating at 400 and 100 MHz, respectively. The chemical shift data are reported in units of 6 (ppm) relative to residual solvent. Gel Permeation Chromatography (GPC): Polymer molecular weights were determined using a triple detection method for calibration with poly(acrylic acid) standards on a Viscotek TDA 305-040 instrument equipped with two Viscotek A-MBHMW-3078 columns and analyzed with light scattering and refractive index detectors. Samples were dissolved in 5% NH4 0H. Absorption and Emission Spectroscopy: Fluorescence spectra were measured on a SPEX Fluorolog-t3 fluorometer (model FL-321, 450 W Xenon lamp) using right-angle detection. Ultraviolet-visible absorption spectra were measured with an Agilent 8453 diode array spectrophotometer and corrected for background signal with a solvent filled cuvette. Fluorescence quantum yields of 3a in both water and 1X PBS were determined relative to perylene and are corrected for solvent refractive index and absorption differences at the excitation wavelength. General protocol for energy transfer assays in PBS: 50 p.L of a stock polymer solution (0.056 mg/mL in PBS) was diluted with PBS to a total volume of 3 mL in a fluorescence cuvette. To this was added aliquots of Texas Red-XTM labeled streptavidin (0.5 piL of a 1 mg/mL solution) and fluorescence emission was taken at each addition. Excitation wavelength was 426 nm. Synthetic Procedures Biotin functionalization, synthesis of 3a: Polymer 1 (11.8 mg, 14.6 iimol based on repeat unit) was dissolved in 4 mL of H20 and NaIO 4 (2.92 tmol in 0.2 mL) was added dropwise under vigorous stirring. After 30 min, a (Biotin-PEG 3-NH 2, 3 mg, 7 Rmol in 1 mL of 0.2M Na2HPO 4) was added and the reaction was stirred for 20 min. A solution of NaCNBH 3 (15 mg, 239 [mol in 1 mL of 40 mM Na 2HPO 4) was added and the reaction stirred for 3 hours. The reaction was dialyzed against water with 5 changes of water and lyophilized to yield 3a. GPC gave Mn = 38,474, PDI = 3.4. 'H NMR (600 MHz, D20): 67.44 (s, 2H), 4.64 (broad, 4H), 4.37 (broad, 4H), 4.05 (broad, 4H), 3.89 (broad, 4H), 3.80-3.30 (biotin, PEG), 3.18 (broad, 4H) 2.35 (broad, 4H), 1.30-0.90 (biotin). Piv-Lysine functionalization, synthesis of 3b: Prepared using identical conditions as above for 3a except that b (Piv-Lys-NH 2) was used in place of a. GPC gave Mn = 49,073, PDI = 4.9. 1H NMR (600 MHz, D20): 67.42 (s, 2H), 4.61 (broad, 4H), 4.35 (broad, 4H), 4.03 (broad, 4H), 3.88 (broad, 4H), 3.16 (broad, 4H), 2.33 (broad, 4H), 1.05 (broad, tBu, 1.6-1.8*). *Based on three experiments and corresponds to 18-20%. Synthesis of tetraaldehyde 5: A solution NaIO 4 (0.200 g, 0.935 mmol) in 10 mL of water was added to a solution of 4 (0.200 g, 0.248 mmol) in 10 mL of THF. Solid TBAIO 4 (54 mg, 0.124 mmol) was added directly and the solution was refluxed for 30 min. After cooling, the reaction was partitioned between EtOAc and brine and the organic phase collected. The aqueous layer was washed with fresh EtOAc and the combined organic layers were dried over Na 2 S0 4 and concentrated in vacuo. The residue was eluted through a silica gel plug using EtOAc to give 5 (95%). 1H NMR (400 MHz, CDCl 3): d 9.65 (d, J=2, 4H), 4.98 (nfo, actual ddd, J=6.6, 2.8, 2, 4H), 4.89 (dd, J=6.6, 2.8, 4H), 1.48 (s, 6H), 1.46 (s, 6H), 1.12 (s, 42H). 13C NMR (125 MHz, CDCl 3) 8 197.6, 134.1, 125.9, 110.7, 105.7, 101.2, 73.9, 54.5, 25.9, 24.2, 18.8, 11.4. HRMS (EI) calcd. for C4 6H66 O8 Si 2 [M+H] 803.4369, found 803.4344. Synthesis of amine 6: To a solution of 5 (0.150 g, 0.186 mmol) in 10 mL of MeOH was added butyl amine (82 mg, 1.12 mmol). After stirring for 10 min at room temperature, NaCNBH 3 (0.250 g, 3.98 mmol) was added and the mixture was refluxed for 3 hours. Once cool, 1 mL of Sat. NaHCO 3 was added and the solvent was removed in vacuo. The residue was partitioned between DCM and sat. NaHCO 3 . The organic layer was dried over Na 2 SO 4 and evaporated to dryness. Silica gel chromatography (EtOAc:Hex, 8:2) provided 6 (65%) as a white solid. 1H NMR (400 MHz, CDCl 3): d 4.33 (dd, J=4.0, 2.4, 4H), 4.00 (m, 4H), 2.84 (d, J=12, 4H), 2.58 (dd, J=1 1.8, 7.5, 4H), 2.29 (br t, 4H), 1.64 (s, 6H), 1.40 (s, 6H), 1.28 (in, 4H), 1.12 (br s, 42H), 1.06 (m, 4H), 0.74 (t, J=7.4, 6H). 13 C NMR (125 MHz, CDCl 3) 6 139.5, 119.8, 110.8, 103.2, 98.3, 76.5, 57.4, 48.7, 41.5, 28.7, 25.9, 24.7, 20.7, 19.0, 14.1, 11.6. HRMS (El) calcd. for C54H 88N 2O 4 Si2 [M+H] 885.6355, found 885.6357. 3.8 References 1 (a) C. Xue, F. T. Luo, H. Y. Liu, Macromolecules, 2007, 40, 6863; (b) C. Xue, V. R. R. Donuru, H. Y. Liu, Macromolecules, 2006, 39, 5747; (c) Y. Li, G. Vamvounis, J. Yu, S. Holdcroft, Macromolecules, 2001, 34, 3130; (d) J. Tolosa, C. Kub, U. H. F. Bunz, Angew. Chem. Int. Ed., 2009, 48, 4610. 2 (a) C. A. Breen, T. Deng, T. Breiner, E. L. Thomas, T. M. Swager, J. Am. Chem. Soc., 2003, 125, 9942; (b) J. N. Wilson, Y. Wang, J. J. Lavigne, U. H. F. Bunz, Chem. Commun., 2003, 1626. 3 S. Bernier, S. Garreau, M. Bera-Aberem, C. Gravel, M. Leclerc, J. Am. Chem. Soc., 2002, 124, 12463. 4 H. C. Kolb, M. G. Finn, K. B. Sharpless, Angew. Chem. Int. Ed., 2001, 40, 2004. 5 (a) B. C. Englert, S. Bakbak, U. H. F. Bunz, Macromolecules, 2005, 38, 5868; (b) T. L. Benanti, A. Kalaydjian, D. Venkataraman, Macromolecules, 2008, 41, 8312; (c) H. B. Bu, G. Gbtz, E. Reinold, A. Vogt, S. Schmid, R. Blanco, J. L. Segura, P. Bauerle, Chem. Commun., 2008, 1320; (d) Q. Chen, B. H. Han, J. Polym. Sci., PartA: Polym. Chem., 2009, 47, 2948. 6 G. C. Bailey, T. M. Swager, Macromolecules, 2006, 39, 2815. 7 B. VanVeller, T. M. Swager, Poly(aryleneethynylene)s. In Design and Synthesis of ConjugatedPolymers, M. Leclerc, J. Morin, Eds. Wiley-VCH: Weinheim, 2010; pp 175-200. 8 B. VanVeller, K. Miki, T. M. Swager, Org. Lett., 2010, 12, 1292. 9 G. T. Hermanson, Bioconjugation Techniques. Elsevier Science: San Diego, 1996. 10 J. Zheng, T. M. Swager, Chem. Commun., 2004, 2798. 11 N. M. Green, Methods Enzymol., 1990, 184, 51. 12 E. Geiger, P. Hug, B. A. Keller, Macromol. Chem. Phys., 2002, 203, 2422. 13 IH NMR analysis of 2 showed little bias regarding which diol moiety (endo or exo) was oxidized. 14 Based on three experiments. 15 (a) G. F. Painter, A. Falshaw, H. Wong, Org. Biomol. Chem., 2004, 2, 1007; (b) V. Bonnet, R. Duval, V. Tran, C. Rabiller, Eur. J. Org. Chem., 2003, 4810; (c) A. Robinson, G. L. Thomas, R. J. Spandl, M. Welch, D. R. Spring, Org. Biomol. Chem., 2008, 6, 2978; (d) P. R. Brooks, S. Caron, J. W. Coe, K. K. Ng, R. A. Singer, E. Vazquez, M. G. Vetelino, H. H. Watson, D. C. Whritenour, M. C. Wirtz, Synthesis, 2004, 1755; (e) A. H. Fray, D. J. Augeri, E. F. Kleinman, J. Org. Chem., 1988, 53, 896; (/) P. Stoy, J. Rush, W. H. Pearson, Synth. Commun., 2004, 34, 3481. 16 (a) I. A. Levitsky, J. S. Kim, T. M. Swager, J. Am. Chem. Soc., 1999, 121, 1466; (b) Satrijo, A.; Swager, T. M., J. Am. Chem. Soc., 2007, 129, 16020. Chapter 3 Appendix 'H-NMR, 3 C-NMR, UV-vis and Fluorescence data 0 0~0 TIPS J -L 10 9 8 7 6 5 4 3 2 1 ppm 20 ppm Figure 3.A.1: H NMR spectrum of compound 5 in CDC13 (400 MHz) TIPS 0~ o 0< TIPS 210 200 Figure 3.A.2: 190 13 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 C NMR spectrum of compound 5 in CDCl 3 (125 MHz) 30 TIPS XO 0 // N O N O'? N'// ii AA 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 LI~ . 2.0 1.5 1.0 0.5 ppm Figure 3.A.3: H NMR spectrum of compound 6 in CDC13 (400 MHz) TIPS O N N O TIPS ii I 4114 No 190 180 Figure 3.A.4: 170 13 160 150 140 130 11- IN I'mah ill, 120 110 100 I Ld-W-1. .. .. I I 90 80 70 60 50 40 C NMR spectrum of compound 6 in CDCl 3 (125 MHz) 30 20 ppm NaO 3 S-.. O n-O 2n . O HO HO O OH NaO3 S O O /O HO OH OH / OH 0.2n ..- HO 'SO 3 Na N 0 - / 6H OH f-8U NH OH OH -'SONa "HO HO . OH 0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 3.A.5: 'H NMR spectrum of compound 3b in D20 (600 MHz) NaO3 ... .. ... n-02n S0 HO HO OH OH OH OH H OH H 0OH SONa HO HO HN NHf NH S 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 Figure 3.A.6: 1H NMR spectrum of compound 3a in D20 (600 MHz) Table 3.A.1: Summary of photophysical data of 3a Abs Xmax (nm) Em Xmax log E (nm) 4.52 451 436 3a, water 461 4.58 3a, 1X PBS 450 O)F 8% 7% 1.0 0.5 ppm Chapter 4 Trypticene Diol Scaffolds for n-System Elaboration 4.1 Introduction Critical to the performance of many polymer electronics is the presence of chain planarity along the polymer backbone. Coplanarity of the adjacent monomers (often aromatic moieties of some structural type, see Chapter 1, Figure 1.1) allows for more effective overlap of adjacent it-orbitals, resulting in less energetic disorder along the backbone. The result is more efficient exciton delocalization and charge transport (along the a-way). Strategies for the synthesis and introduction of highly planarized, a-extended, monomers have been reported for a variety of different classes of conjugated polymers. However, the synthetic strategy is often only applicable within one structural class. Iptycenes are molecules built upon [2,2,2]-ring systems in which the bridges are aromatic rings 1, and their three dimensional shape creates interstitial free volume around the molecule. A growing number of research efforts have been reported using the rigid three-dimensional structure of iptycenes to introduce new supramolecular interactions and achieve new material properties.1' 2 Free Volume\\ OH 2 3 1 The simplest member of the iptycene family is the trypticene 2. Recently, Taylor and Swager reported an efficient synthesis of a trypticene diol (TD, 3), bearing alcohols at the bridgehead carbons (Scheme 4. 1).3 Starting from anthraquinone (4), a moderately selective syn-addition of lithium acetylide provided 5. Once deprotected (5-+>6) the newly installed syn-alkynes were shown to be geometrically disposed to Rh catalyzed [2 + 2 + 2] cycloaddition with either terminal alkynes or norbornadiene (an acetylene equivalent, vide infra) to give the TDs (7 and 8). Scheme 4.1: Synthesis of trypticene diols through [2+ 2 +2] cylcoaddtion Li = KOH R' Toluene 2.1:1 selectivity MeOH, THF R1 = TMS R1 = Ph, TMS R1 ~~~ 5or6 H' 5 6 R2 Rh(PPh 3)3CI 5 - Toluene, 100 OC [Rh(cod)C] 2 Toluene, 100 OC () R1= Ph, H R' =Ph We hoped to extend this method into 1,4-dihalogen variants suitable for Pdcatalyzed cross-coupling with a variety of a-systems (Figure 4.1). We theorized the rigid scaffold enforced by the trypticene would hold the alcohol and newly coupled it-system in close proximity to promote cyclization under appropriate conditions. The result would be a higher order planarized trypticene it-system. When incorporated into conjugated polymers, this should allow for an increase in conjugation length. Additionally, this strategy would allow us to evaluate cyclization with x-systems of different structural classes. Rigid scaffold enforces close proximity of alcohol and reacting partner X Pd-catalyzed cross-coupling Cyclization HO X 1,4-dihalogen subsituted trypticene diol Extended planar n-system Figure 4.1: Rational for the potential of trypticene diols for cyclization and extended planar i-systems. 4.2 Dihalogen Substituted Trypticene Diols Synthesis of TDs-bearing the 1,4-halogen substitution pattern proposedfollowed the same strategy of the initial TDO synthesis outlined in Scheme 4.1. However, for reasons unclear, the presence of bromine substituents did not permit direct transfer of previous synthetic conditions. Thus, a modified synthesis is presented herein (Scheme 4.2). Starting from commercially available 9, a Sandmeyer reaction afforded dibromoanthroquinone 10 in excellent yield. Double addition of TIPS acetylide to 10 in toluene provided the syn-addition product (11) with modest selectivity (2.3:1, the analogous TMS acetylide reagent gave a 1:1 mixture of syn and anti addition products). Finally, fluoride deprotection revealed the cyclization substrate 12. procedure showed Wilkinson's catalyst [Rh(PPh 3)Cl] The previous to be superior for [2 + 2 + 2] cycloaddtion, but this protocol was completely ineffective for 12-again, the failure of these conditions for the dibromo substrate is unclear. The ruthenium catalyst [CpRu(PPh 3)2C] in dioxane was found to be superior for the [2 + 2 + 2] cycloaddtion with terminal alkynes to yield enantiomeric products (13-15). Finally, the formal cycloaddtion with acetylene was obtained (16) by reaction of norbornadiene in the presence of [Rh(cod)C12], a reaction postulated to proceed by [2 + 2 + 2] cycloaddtion followed by retro-Diels-Alder reaction and expulsion of cyclopentadiene.4 With the 1,4-dibromo trypticene diol scaffold in hand, we sought to investigate the proposed cyclization pathway outlined in Figure 4.1 with three different 7-systems: alkynes (Section 4.3), phenyls (Section 4.4), and thiophenes (Section 4.5). 94 Scheme 4.2: Synthesis of 1,4-dibromo substituted trypticene diols TIPS o CuBr 2 t-BuONO O MeCN 65 OC 94% NH2 9 o Br Br Li-=---TIPS TBAF Toluene 65 OC, 70% THF, rt 91% 10 H/ 12 R OH HO Br /\ (*) Br CpRu(PPh 3)2C 12 Dioxane 105 OC [Rh(cod)CI] 2 / W. OH Br -- Toluene 105 OC, 87% HO Br 16 13 R = n-Bu, 89% 14 R = n-Oct, 85% 15 R = n-Dec, 86% 4.3 Cyclization with Alkynes Our initial investigation of substituent-cyclized TDs focused on alkynes due to the large literature surrounding alcohol cyclization onto alkynes using Lewis acids (Scheme 4.3).5 Further, in the context of polymer chemistry, we envisioned such a transformation might also provide an uncommon form of post-polymerization modification. Scheme 4.3: Proposed cyclization of TDs onto alkynes H ~- R Sonogashira cross-coupling / Lewis acid catalyzed cyclization R /O 6-endo-dig 0- R 18 Chapter 3 discussed post-polymerization functionalization as an efficient strategy for late stage polymer diversification in the context of side chain modification or elaboration. A far more rare type of post-polymerization modification is alteration of the polymer's main chain or backbone. This type of transformation is inherently different as the polymer's identity as a conjugated polymer is fundamentally changed to a different class of conjugated polymer. Scheme 4.4 outlines some examples that have been applied to main chain alteration of poly(p-phenylene ethynylene)s (PPEs) through reduction of the triple bonds. The full reduction of PPEs to the unconjugated poly(p-xylene)s can be achieved under a variety of hydrogenation conditions (19-+->20),6-8 and the partial reduction to cis-poly(p-phenylene vinylene)s (PPVs) was recently reported by Moslin and Swager (19--21).9 Scheme 4.4: PPE main chain reduction R 2. i-PrMgCI or R R 20 TosNHNH 2 NPr 3, 110 0 C -n n 1 1. Ti(Oi-Pr)4 R RhCI(PPh 3)3/H 500 bar, 350 0C2 - R 3. H20 19 -n - 21 R In the context of the transformation outlined in Scheme 4.3, such a reaction of a PPE possessing the TD moiety would result in a trans-PPV.Further, it would provide for a rarely seen substitution of the vinyl segment of a PPV with an electron donating oxygen group,10 11 all while creating greater planarity in the PPV backbone. 4.3.1 Synthesis of Model Compounds for Cyclization Compound 16 was used to investigate the cyclization reaction with alkyne substituents. Compound 22 was initially synthesized by Sonogashira cross coupling for investigation as a polymerizable monomer (Scheme 4.5). Interestingly, upon deprotection, spontaneous metal-free cyclization occurred on one side of the molecule to give 23. NMR analysis indicated the product to be the 5-exo-dig cyclization (based on vinyl coupling constants), and X-ray crystallography revealed how closure of the fivemembered ring prevents cyclization on the other side by pulling the alkyne and alcohol apart. Investigation of molecular models suggested that cyclization of the alcohol oxygen onto the terminal carbon of the alkyne (6-endo-dig) would lead to a less strained 6- membered ring with less structural distortion, and might allow for double cyclization to take place. Scheme 4.5: Spontaneous metal-free 5-exo-dig cyclization \ HO OH PdCl2 (PPh 3) 2 Cul _ Toluene, NEt 3 Br /''''/HO 85 16 Br / SiMe 3 / SiMe 3 OH .~~~ // 0C, 22 Me3 Si TBAF, THF or KOH, MeOH, THF 87% .d 5-exo-dig 0 X-ray of 23 -- / HO //23 H Phenyl acetylene analogue 24 was found to not undergo spontaneous cyclization, which allowed for exploration of the 6-endo-dig cyclization through electrophilic activation of the alkyne (Scheme 4.6). Gratifyingly, treatment with electrophilic iodine sources5h provided the double cyclization product (25)-consistent with the 6-endo-dig hypothesis above. Further, electrophilic gold(III) proved moderately effective in promoting the 6-endo-dig isomerization of 24 to the mono- and doubly cyclized compounds 26 and 27. Compound 27 displayed poor solubility making analysis and further method development difficult. To this end, compound 14, bearing a solubilizing alkyl side chain was elaborated to the diphenyl acetylene 28 (Scheme 4.7). Subsequent 6-endo-dig isomerization of the alkyne to the oxygen substituted alkene (28--+29) using a more active gold(I) catalyst system provided 29 in good yield (spot to spot conversion by TLC). We now felt confident that we could extend these methods to reactions on polymers for the post polymerization alteration of PPEs into PPVs. Finally, as an aside, the 1H NMR chemical shift for the bridgehead alcohol in the alkyne precursors 22, 24, and 28 was found further downfield (6 5.6-5.9) relative to more typical benzyl alcohols (6 4.5) indicating some through space deshielding effects from the phenylene ethynylene Rsystem. Scheme 4.6: Electrophilic activation of 6-endo-dig cyclization ICI, DCM - / OH HO 16 Br ~780C to rt Ph 86% PdCl 2(PPh 3)2 Br Cul OR 12,NaHCO 3 MeCN, rt 24% Toluene, NEt 3 85 OC, 93% AuCl3 6-endo-dig THF:MeCN (4:1), rt X-ray of 27 H 0 - 27 Yo - 40% Scheme 4.7: Electrophilic activation of 6-endo-dig cyclization of more soluble variants n-Oct /I' / 110 10 14 Br / -Ph OH /Io__ Ph3PAuCI (6 mol%) AgSbF6 (6 mol%) B PdCl 2(PPh 3)2 Br Cul _ - /O _ THF:DCE (1:1) rt, 4 h, 85% Toluene, NEt 3 85 OC, 93% 98 0 - 29 4.3.2 Polymer Synthesis and Cyclization To examine the effectiveness of the electrophilic gold(l) conditions in the context of a polymer, compound 14 was polymerized with 1,4-diethynylbenzene under Sonogashira conditions (Scheme 4.8). Compound 14, in addition to the n-octyl side chain, is also racemic. This leads to a region-random orientation of the n-octyl side chain along the polymer that assists in solubility (i.e. interpolymer crystallization is impeded by the random orientation of side chains). Unfortunately, while reasonably high molecular weight polymer was isolated 30 (M, = 12,500, PDI = 1.44, DP = 24), the yield was low due to large quantities of insoluble polymer presumably too long in length to be soluble. In any case, 30 was subjected to a two stage addition of gold(I) catalyst to produce cyclized polymer 31 (Mn = 14,700, PDI = 1.16 DP = 28) as expected. The increase in molecular weight of 31 relative to 30 is likely due to the increased rigidity along the chain. The rigidity increases the persistence length of the polymer making it appear larger under size exclusion measurements. Scheme 4.8: Polymerization and post- cyclization n-Oct n-Oct PdCl 2(PPh 3 )2 OH / HO 14 Br + uiOH \ -85 Toluene, NEt3 OC H Br 30 n-Oct n // Ph3PAuCI (20 mol%) AgSbF6 (20 mol%) THF:DCE (1:1) rt, 24 h / 0 O 0 / - 99 4.3.3 Photophysical Properties The effect of the transformation on the x-system of 29 can be seen in Figure 4.2. Both 28 and 29 show very small stokes shifts when considering the band edges of absorption (ignoring the (0,0) absorption transition is not the Xmax). However both the absorbance and fluorescence maximums of 29 red-shift by almost 100 nm (Figure 4.2b and d)(Table 1). This closing of the band gap energy is ascribed to both the increased planarity of 29 and the presence of electron rich oxygen donor atoms on the alkene, which raise the HOMO level. Indeed, the band gap of 29 appears to be among the smallest for any phenylene vinylene trimer bearing either electron withdrawing or donating substituents. 12-18 The increased planarity was also evidenced by the distinctly sharper emission features of 29 which reveal the vibrational transitions. Further, an associated increase in fluorescence quantum yield was observed for 29 compared to 28 (Table 1), likely due to removal of vibrational relaxation through planarization and rigidification. Interestingly, the (0,0) transition of both 28 and 29 are not the most intense bands in the absorption spectrum (Figure 4.2a and c), but they are the most intense in the emission spectrum. This indicates the terminal phenyl rings for both 28 and 29 may not be coplanar in the ground state but are so in the excited state. Similar effects observed between the model small molecule compounds 28 and 29 were also seen for polymers 30 and 31. Both polymer band gaps are diminished relative to their small molecule analogues due to greater delocalization along the conjugated polymer backbone (Figure 4.3). Again, small Stokes shifts were observed for both polymers and polymer 31 showed approximately a 100 nm red shift in absorbance and emission upon cyclization (Figure 4.3b and d) (Table 1). Further, the planarity imparted to 31 preserves the vibrational fine structure even in a less structurally defined polydisperse polymer. In contrast to 29, polymer 31 did not have an associated increase in fluorescence quantum yield relative to precursor polymer 30. This is likely due to the fluorescence lifetime (TF) of 31 being more than double that of 30 (Table 1) allowing more time for non-radiative deactivation of the excited state. It is indeed ironic that the cyclization, originally designed to impart better spectral features and properties through planarization, prolongs the lifetime of the excited state such that polymer 31, hoisted by its own petard, has a lower observable quantum yield. 100 0) 0 - C 300 400 500 600 Wavelength (nm) Figure 4.2: Absorbance traces for 28 (black) and 29 (green) and fluorescence traces for 28 (blue) and 29 (red) in CHCl 3. C 0) %%00 CD 300 400 500 600 Wavelength (nm) 700 Figure 4.3: Absorbance traces for 30 (black) and 31 (green) and fluorescence traces for 30 (blue) and 31 (red) in CHCl3 . 101 The thin film spectra of both polymers are shown in Figure 4.4. Both polymers remain visibly fluorescent in thin film, and 31 still maintains a significant red shift relative to 30 (Figure 4.4b and d). It would appear that the trypticene is not sufficient to prevent n-t interactions between polymer chains as both polymers display significant broadening of their absorbance and fluorescence spectra; however, polymer 31 appears to retain some vibrational structure in its fluorescence spectrum (Figure 4.4c). C: (0 C 300 400 500 600 700 Wavelength (nm) Figure 4.4: Absorbance traces for 30 (black) and 31 (green) and fluorescence traces for 30 (blue) and 31 (red) in thin film. 102 Table 4.1: Summary of photophysical data Compound abs Amax em 2Amaxa log , DF TF Eg,opticaib [ns] [eV] [nm] [nm] 28 322 349 4.74 0.65 1.11 3.5 29 395 427 4.63 0.89 1.77 2.8 30 372 408 4.62 0.74 0.53 2.8 31 460 507 5.23 0.46 1.28 2.4 30, film 384 495 0.15 1.20c 2.8 31, film 435 523 0.03 1.10d 2.3 a Measured in CHCl 3, b Band gap, estimated by onset of absorption spectrum c Bi-exponential lifetime 82.9%. Second exponential 3.5 ns, 17.1% d Bi-exponential lifetime 91.7%. Second exponential 3.0 ns, 8.3% 4.3.4 Electronic Properties Cyclic voltammetry was used to investigate the redox behavior of the cyclized model 29 and the two polymers discussed above (Figure 4.6). In the case of the molecules discussed, the newly introduced electron donating oxygen atoms should result in a more electron rich system that is more readily oxidized as opposed to reduced (however, the rigid structure may limit the extent of donation relative to an alkoxy side chain because the oxygen must an energy penalty to assume sp2 hybridization for donation). This was indeed the case as 29 showed reversible single electron oxidation at +1.05 and +1.39 V in solution (Figure 4.6a). It is interesting to note that the completely unsubstituted distyrylbenzene 32 (Figure 4.5) is more readily reduced (single electron reductions at -2.48 V and -2.74 V) further demonstrating the power of substitution to tune the HOMO-LUMO levels of conjugated polymers. n-Oct n - 0 enol ether, potential nucleophile t__ 1'0 32 31 Figure 4.5: Distyrylbenzene 32 and potential nucleophilic site in 31 103 8.0 (a)-Scan 1 -Scan 2 - Scan 3 - Figure 4.6: Cyclic voltammograms for 29 (a) and 31 (b) in solution, and thin films of 31 (c), and 30 (d). Measurements performed under nitrogen in degassed solvents, DCM for (a) and (b) and MeCN for (c) and 4.0- 0.0 -4.0 2.0 - (b) -Scan (d). 1 Scan 2 -Scan 3 n-Oct 1.0 k -Oc" 0 - 0.0 L -1.0 10 (c) -Scan 8.0 - 1 -Scan2 - Scan 3 n-Oct 6.0 ZNn 4.0 \ 0 2.0 0.0 -2.0 4.0 - 3.0 (d) - Scan 1 - Scan 2 Scan 3 2.01.0 0.0 -1.0 L. 0.0 1.5 1.0 0.5 Potential (Vvs. SCE) 2.0 104 Electrolyte = 0.1 AgNO 3/0.1 TBAPF 6. 1.6 mm diameter Pt button electrode. Scan rate = 50 mV/sec / ZN - In contrast to 29, polymer 31 undergoes irreversible oxidation at a similar potential in solution (Figure 4.6b). This irreversibility is even more evident in the thin film (Figure 4.6c) where solid-state effects such as counter ion penetration into the film may prevent reversibility. The difference in behavior of 29 and 31 is unclear. Another possibility regarding the thin film of 31 is the nucleophilic nature of the enol ether present (Figure 4.5). Perhaps, cations generated on adjacent chains are attacked by the enol ether and irreversibility crosslinked. PPEs typically display poor electrochemical behavior so it is not surprising that 30 also displays irreversible oxidation. The polymer shows a first wave oxidation. The propensity to oxidize might stem from through space donation of the nonbonding electrons of the alcohol to the 7-system.19 4.3.5 Conclusions: Alkyne Cyclization In summary, we have developed a highly efficient strategy to introduce planarity along a conjugated polymer backbone using a catalytic main chain alteration of a PPE into a different class of conjugated polymer, a PPV (Figure 4.7). As predicted, the PPV demonstrates red shifted photophysics, and the established planarity provides it with well defined vibrational features. The effect of the trypticene, the simplest member of the iptycene family, does not appear sufficient to suppress unfavorable a-7U interactions between polymer chains in thin film. Finally, while the small molecule model compounds displayed reversible redox behavior, the polymer versions suffered from irreversible oxidation. n-Oct n-Oct \n Au(I) OH HO MO -- Figure 4.7: General transformation for a PPE to a chain rigidified PPV via gold(l) catalysis 105 4.4 Cyclization with Phenyl Groups We next turned out attention to cylcization of TDs onto other ir-systems. Phenyl groups were a promising target as the cyclization would result in a highly planarized penylene moiety. Phenylenes have received wide attention as blue emitting materials for organic light emitting diodes (OLEDs). 20 Thus, the highly planarized structure might lead to a higher performing phenylene through greater charge delocalization and mobility. Scheme 4.9: Synthesis of planarized phenylene model compound (3 eq.) &B(OH)2 ,Br PdCI 2 (PPh 3 ) 2 ) THF K3 PO4 (0.5M) 60 0C, 98% I- / - OH y / / HO K2 S 2 0 8 (4 eq.) Cu(OAc) 2 (8 eq.) KOAc (12 eq.) AcOH, 113 0C - 77% \ / 32 /0 0 - \ / 33 X-ray of 33 To investigate the feasibility of TD cyclization with benzene, 32 was synthesized in a straight forward manner from 16 under Suzuki conditions (Scheme 4.9). The cyclization was achieved under oxidative conditions using Cu(II) and persulfate. Presumably, the reaction occurs by oxidation of the phenyl rings followed by attack by the proximal alcohol and subsequent rearomatization. Oxygen radical attack by the alcohol may also be playing a role as oxygen radical generation has been associated with these conditions.n-2 4 106 = 81%8 4abs = 3 5 nm (IDF Xem = 359 nm 0 -0 3- 0300 400 350 Wavelength (nm) 450 Figure 4.8: Absorbance and fluorescence data for compound 33 The effect of the cyclization and planarization of the terphenylene moiety in 33 is evident from sharp absorbance and emission spectrum (Figure 4.8) similar to 29 above. The rigidity of the system preserves the vibronic structure of the electronic transitions and endows it with a fairly high quantum yield. 4.4.1 Future Applications of Phenyl Cyclized Trypticene Diols Current synthetic efforts are focused on the synthesis of polymers based on 33 (Scheme 4.10). Suzuki poly-condensation polymerization of 14 with benzene-1,4bis(pinacol boronic ester) should lead to the alternating copolymer 34. Following, oxidative cyclization would lead to a polymer with monomers held in what is called a ladder polymer structure (the polymer is composed of fused ring systems). Further, the alternating trypticene motif would result in a polymer possessing a repeating structure that more closely resembles a pentypticene architecture, a molecular shape more effective at decreasing interpolymer x-7i interactions (Chapter 2, Figure 2.3). The racemic nature of 14 would also be helpful for solubility as discussed above for 31. Unfortunately, the presence of benzylic positions in 35 might require a more mild oxidation protocol. 107 Scheme 4.10: Future application for ladder phenylene polymers n-Oct (3 eq.) O /B / OH Br Br n-Oct O0 & B /O Pd cat. HO 34 14 Br n-Oct Oxidation --------------- n-uct / /-- / 0/ ~-35 4.5 Cyclization with Thiophenes Finally, thiophenes were investigated in the cyclization with TDs (Scheme 4.11 and 4.12). The Suzuki cross-coupling between TD and thiophene boronic acid was found to be most efficient using a newly developed palladium precatalyst (16-+36 and 37). Unfortunately, the oxidative conditions applied to the phenylene series failed to yield any cyclized product (36--38). This unsurprising result is likely due to the electron rich nature of thiophene and its propensity for oxidative polymerization. To investigate this result further, a methyl-capped analogue was prepared (36) and subjected to the oxidative conditions. A highly polar molecule was isolated from the reaction mixture which, based on preliminary characterization (IR, 1H NMR), was found to possess sulfonic and carboxylic residues (39). Further, based on its absorbance and fluorescence spectrum (Figure 4.9a) appears to be a cyclized product. From these results is appears the thiophene moiety is too sensitive to undergo cyclization under such strongly oxidizing conditions. 108 Scheme 4.11: Oxidative cyclization of trypticene diols with thiophenes (3 eq.) R s B(OH) 2 -R Pd cat. K2S208 (4 eq.) Cu(OAc) 2 (8 eq.) KOAc (12 eq.) 0 Ac H R =H THF K3PO4 (0.5M) -/O 38 Ss 40 0C, 98% K2 S20 8 (4 eq.) Cu(OAc) 2 (8 eq.) KOAc (12 eq.) AcOH, 113 OC R = Me Pd cat. C02H XPhos-Pd-NH 2 CI 0 - \ -:0 HO2C 39 (tentative structural assignment) O 350 400 450 500 550 600650 Wavelength (nm) 300 350 400 450 Wavelength (nm) Figure 4.9: Absorbance and fluorescence data for 39 (a) and 41 (b). 109 500 Scheme 4.12: Cyclization by acid catalyzed trans-etherification OMe (3 eq.) \\MeO OH C/s Br - HO 16 Br / B(pinacol) OH 3 Pd cat. /- THF K3PO4 (0.5M) rt, 82% H HOH TsOH eH 2 0 Toluene - 85 0C S \13 S 0 S 40 75% 0 - 38 OMe X-ray of 38 In general, 3-methoxythiophene derivatives are known to undergo substitution of the methyl ether group under acid catalyzed conditions. In the context of 40 the alcohol of the TD is held in close proximity to the carbon in the 3-position of the thiophene (Scheme 4.12). Thus, upon protonation of the methoxy group, a 6-membered transition state attack of the TD alcohol on at the thiophene 3-position resulted in the desired cyclized product 41. As expected, the photophysical characteristics of 41 are in alignment with all of the cyclized TD molecules presented thus far, sharp vibrational transitions and minimal stokes shift (Figure 4.9b). An interesting result is also observed in the 1H NMR spectra of the 40 and 41. The methoxy signal along with the TD alcohol and even the protons on the "wings" of the TD are broadened, perhaps due to rotamers and intermolecular hydrogen bonding (Figure 4.10 top). Upon cyclization, removal of the rotational degrees of freedom produces a sharply defined spectrum (Figure 4.10 bottom). 110 OMe H OH - H H 0 H 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 Figure 4.10: Comparison of the 'H NMR spectra of 40 (top) and 38 (bottom). 4.5.1 Future Applications of Thiophene Cyclized Trypticene Diols Compound 41 might show promise as a monomer for polymer solar cells based on the bulk heterojunction (BHJ) design. The BHJ structure is usually formed by blending a p-type conjugated polymer with an n-type fullerene to form a bicontinuous, interpenetrating network. Power conversion is achieved through absorption of a photon by the conjugated polymer and charge separation of the excited electron onto the n-type fullerene, leaving the hole on the p-type polymer. Critical to the success of this process is the interaction between polymer and fullerene. Crystallization and phase separation between the two materials can often lead to inefficient charge transfer and lower current output. Finally, also critical to device efficiency is the charge mobility within the polymer; as once charge separation occurs, the hole must rapidly diffuse to the electrode or charge recombination might result (i.e. a waste of a photon). 111 It is our hope that the structural characteristics of 38 might lend some improvement to both the phase separation and charge mobility issues mentioned above. Monomers similar to 38, possessing the same planarized thiophene-phenylene-thiophene structural motif, have already been shown to improve hole mobility in solar cells with efficiencies greater than 6% (Figure 4.11 a).26 Further, pendant alkyl phenyl groups in 41 are stereochemically perpendicular to the planar a-system. The phenyl "wings" of the trypticene in 38 are similarly disposed, indicating the three-dimensional effect might not be detrimental to charge mobility within the polymer. Regarding the charge separation interface (heterojunction), trypticenes have long been known to form intercalating networks with fullerenes (Figure 4.11b). 27 -30 It is conceivable that the polymers incorporating 38 would be less prone to phase segregation and reduced charge separation efficiency. (a) (b) C6H13C6H13 41 C6H13C6H13 Figure 4.11: (a) Planarized monomer for improved charge mobility. (b) Visualization of favorable interactions between fullerene and trypticenes to prevent phase segregation (based upon X-ray crystal structures 27 30) 112 4.6 Conclusions Critical to the design of conjugated polymers is the effective conjugation length. We have shown that the rigid structure of TDs promotes cyclization of the TD alcohol onto alkynes, phenyls and thiophenes to form relatively unstrained 6-membered rings. Several applications of these newly formed planar a-systems have been discussed from OLEDs to photo-voltaic materials. 4.7 Experimental Section Materials: Silica gel (40 pIm) was purchased from SiliCycle. All solvents used for photophysical experiments were spectral grade. All reagent grade materials were purchased from Aldrich, TCI America, Strem Chemicals Inc., and Alfa Aesar, and used without further purification. Experimental: NMR Spectroscopy: 1H and 13C NMR spectra for all compounds were acquired in CDCl 3, CDCl 2- CDCl 2 and THF-d8 on a Bruker Avance Spectrometer operating at 400 and 125 MHz, respectively. The chemical shift data are reported in units of 6 (ppm) relative to residual solvent. Gel Permeation Chromatography (GPC): Polymer molecular weights were determined using a triple detection method for calibration with poly(acrylic acid) standards on a Viscotek TDA 305-040 instrument equipped with two Viscotek A-MBHMW-3078 columns. and analyzed with light scattering and refractive index detectors. Samples were dissolved in 5%NH 40H. Absorption andEmission Spectroscopy: Fluorescence spectra were measured on a SPEX Fluorolog-t3 fluorometer (model FL-321, 450 W Xenon lamp) using right-angle detection. Ultraviolet-visible absorption spectra were measured with an Agilent 8453 diode array spectrophotometer and corrected for background signal with a solvent filled 113 cuvette. Fluorescence quantum yields of P1-anti in both water and IX PBS were determined relative to perylene and are corrected for solvent refractive index and absorption differences at the excitation wavelength. The quantum yield of P-peg in IX PBS was determined relative to P-peg in pure water. Lifetime measurements: Time resolved fluorescence measurements were performed by exciting the samples with 160 femtosecond pulses at 390 nm from the double output of a Coherent RegA Ti:Sapphire amplifier. The resulting fluorescence was spectrally and temporally resolved with a Hamamatsu C4780 Streak Camera system. Electrochemistry: Electrochemical measurements were carried out using an Autolab PGSTAT 10 or PGSTAT 20 potentiostat (Eco Chemie) in a three-electrode cell configuration consisting of a quasi- internal Ag wire reference electrode (BioAnalytical Systems) submerged in 0.01 AgNO3 / 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in anhydrous CH3CN, a Pt button (1.6 mm in diameter), 5-pm interdigitated micro-, or ITO-coated glass electrodes as the working electrode, and a Pt coil or Pt gauze as the counter electrode. The ferrocene/ferrocenium (Fc/Fc+) redox couple was used as an external reference. Half-wave potentials of Fc/Fc+ were observed between 88-95 mV in CH3CN and 210-245 mV (all vs. Ag/Ag+) in CH2Cl2. Synthetic Procedures Synthesis of 10: 1,4-diaminoanthraquinone (2g, 8.39 mmol) was added to 16 mL of acetonitrile and CuBr 2 (4.22 g, 18.89 mmol, 2.25 equiv) and t-butylnitrite (t-BuONO) (1.95 g, 18.89 mmol, 2.25mmol) were subsequently added. The mixture was stirred at 650 C for 5 h. Volatiles were removed in vacuo and the residue was partitioned between DCM and IM HCl and washed with 1M HCl twice more. The organic layer was dried with Na 2SO 4 and concentrated in vacuo. The residue was purified via column chromatography (4:1, DCM:Hexanes) to give 10 (94%). 1H NMR (400 MHz, CDC3): 6 8.21 (dd, J=6, 3.6, 2H), 7.81 (s, 2H), 7.80 (dd, J=6, 3.6, 2H). 13 C NMR (125 MHz, CDCl3) 6 181.7, 140.7, 134.3, 133.6, 133.5, 127.0, 122.1. HRMS (ESI) calcd. for C14 H6 Br 2 O 2 [M+Na] 366.8627, found 386.8614. 114 Synthesis of 11: TIPS acetylene (2.63 g, 3.24 mL 14.44 mmol) was dissolved in 28 mL of dry toluene under argon. Following, n-BuLi (13.9 mmol, 1.6 M in Hexane) was added dropwise at room temperature and the mixture was stirred for 10 min. Compound 10 (1 g, 2.78 mmol) was added in one portion. The solution was stirred at 60 C for 2 h and cooled to room temperature. The reaction was quenched with sat. NH 4 C1 and the volatiles were removed in vacuo. The residue was partitioned between DCM and water and the organic layer was dried over Na2 SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (4:1, DCM:Hexanes) to give 11 (70%). 1H NMR (400 MHz, CDCl3): 6 7.92 (dd, J=6, 3.2, 2H), 7.57 (s, 2H), 7.43 (dd, J=6, 3.2, 2H), 4.53 (s, 2H), 0.96 (m, 42H). 13 C NMR (125 MHz, CDCl3) 6 136.7, 136.2, 134.5, 128.9, 128.4, 123.2, 108.54, 89.3, 66.7, 18.7, 11.4. HRMS (ESI) calcd. for C36H5 oBr 20 2 Si 2 [M+Na] 751.1608, found 751.1592. NOTE: The trans-diolisomer elutes faster than the syn-diol isomer. Synthesis of 12: Compound 11 (1 g, 1.37 mmol) was dissolved in 13 mL of THF and TBAF (3.0 mmol, IM in THF) was added. After stirring for 15 h at 23 'C, the reaction was partitioned between EtOAc and Brine. The organic layer was washed with Brine twice more and then dried with Na2 SO 4 and concentrated in vacuo. The residue was purified by silica gel chromatography (DCM to 1:1 DCM:EtOAc) to give 12 (91%). 1H NMR (400 MHz, CDCl3): 6 7.95 (dd, J=6, 3.6, 2H), 7.62 (s, 2H), 7.51 (dd, J=6, 3.6, 2H), 4.60 (s, 2H), 2.69 (s, 2H). 13 C NMR (125 MHz, CDCl3) 6 136.7, 136.4, 134.1, 129.6, 128.1, 123.2, 84.9, 75.6, 66.1. HRMS (ESI) calcd. for Ci 8H10Br 20 2 [M+H] 438.8940, found 438.8924. Synthesis of 13: Compound 12 (50 mg, 0.12 mmol) and CpRu(PPh 3)2Cl (8.71 mg, 0.012 mmol) were suspended in 1.2 mL dry dioxane under argon. 1-Hexyne (0.1mL, 5.98 mmol) was added and the flask was sealed and heated at 105 0C for 12 h. The reaction was cooled to 23" C and the volatiles removed in vacuo. The residue was purified by silica gel chromatography (9:1, hexanes:EtOAc) to give 13 (89%). 1H NMR (400 MHz, CDCl3): 6 7.68 (m, 2H), 7.57 (d, J=7.6, 1H), 7.51 (d, J=1.3, lH), 7.19 (dd, J=5.6, 3.2, 2H), 7.00 (s, 2H), 6.99 (dd, J=7.6, 1.6, lH), 4.55 (s, 1H), 4.51 (s, 1H), 2.60 (pseudo t, 115 J=8, 2H), 1.57 (m, 2H), 1.33 (m, 2H), 0.91 (t, J=7.4, 3H). 13C NMR (125 MHz, CDC13) 6 144.8, 144.7, 143.7, 143.6, 143.4, 141.4, 140.9, 133.6, 133.5, 126.3, 126.2, 126.1, 120.3, 120.03, 120.01, 119.9, 114.4, 114.3, 80.8, 80.7, 35.9, 33.9, 22.7, 14.2. HRMS (ESI) calcd. for C24H20Br 2O2 [M+H] 498.9903, found 498.9911. Synthesis of 14: Compound 12 (0.4 g, 0.96 mmol) and CpRu(PPh 3)2 C1 (69 mg, 0.096 mmol) were suspended in 9 mL of dry Dioxane under argon. 1-Decyne (0.68 g, 0.8 mL, 4.98 mmol) was added and the flask was sealed and heated at 105 0C for 12 h. The volatiles were removed in vacuo and the residue was purified by column chromatography (7:3, Hexanes:DCM) to give 14 (85%). 'H NMR (400 MHz, CDC13): 6 7.68 (m, 2H), 7.57 (d, J=7.6, 11H), 7.51 (d, J=1.2, 1H), 7.19 (dd,. J=5.2, 3.2, 2H), 7.00 (s, 2H), 6.99 (dd, J=7.6, 1.6, 1H), 4.55 (s, 1H), 4.51 (s, 1H), 2.59 (pseudo t, J=8, 2H), 1.58 (m, 2H), 1.25 (m, 10H), 0.88 (t, J=7.2, 3H). 13C NMR (125 MHz, CDCl3) 6 144.8, 144.7, 143.7, 143.6, 143.4, 141.5, 140.8, 133.6, 133.5, 126.3, 126.2, 126.1, 120.2, 120.03, 120.0, 119.94, 114.4, 114.3, 80.82, 80.77, 36.2, 32.1, 31.8, 29.7, 29.6, 29.4, 22.9, 14.3. HRMS (ESI) calcd. for C28H28Br 2 O2 [M+H] 555.0529, found 555.0535. Synthesis of 15: Compound 12 (0.4 g, 0.96 mmol) and CpRu(PPh 3)2C1 (69 mg, 0.096 mmol) were suspended in 9 mL of dry Dioxane under argon. 1-Dodecyne (0.68 g, 4.98 mmol) was added and the flask was sealed and heated at 105 0 C for 12 h. The volatiles were removed in vacuo and the residue was purified by column chromatography (7:3, Hexanes:DCM) to give 15 (86%). 1H NMR (400 MHz, CDCl3): 6 7.70 (m, 2H), 7.59 (d, J=7.6, 1H), 7.54 (d, J=1.2, 1H), 7.20 (dd, J=5.6, 3.2, 2H), 7.01 (dd. J=7.6, 1.6, 1H), 6.98 (s, 2H), 4.58 (s, 1H), 4.54 (s, 1H), 2.60 (pseudo t, J=7.8, 2H), 1.59 (m, 2H), 1.27 (m, 14H), 0.91 (t, J=6.8, 3H). 13C NMR (125 MHz, CDCl3) 6 144.8, 144.7, 143.7, 143.6, 143.4, 141.5, 140.9, 133.5, 133.4, 126.3, 126.2, 126.1, 120.2, 120.03, 120.0, 119.9, 114.4, 114.3, 80.8, 80.7, 36.2, 32.1, 31.8, 29.8, 29.7, 29.6, 29.5, 22.9, 14.4. HRMS (ESI) calcd. for C30 H 32 Br 2 O 2 [M+H] 583.0842, found 583.0832. Synthesis of 16: Compound 12 (0.4 g, 0.96 mmol) and [Rh(cod)Cl] 2 (12 mg, 0.024 mmol) were suspended in 9 mL of dry toluene under argon. Norbornadiene (0.88 g, 1.03 116 mL, 9.57 mmol) was added and the flask was sealed and heated at 105 0 C for 15 h. The volatiles were removed in vacuo and the residue was purified by column chromatography (3:2, Hexanes:DCM) to give 16 (87%).'H NMR (400 MHz, CDCl3): 6 7.69 (dd, J=5.6, 3.2, 4H), 7.19 (dd, J=5.6, 3.2, 4H), 7.02 (s, 2H), 4.56 (s, 2H). 13C NMR (125 MHz, CDCl3) 6 144.6, 143.5, 133.6, 126.3, 120.1, 114.4, 80.8. HRMS (ESI) caled. for C2 0H1 2 Br 2 O 2 [M+H] 442.9277, found 442.9292. Synthesis of 22: Compound 16 (0.2 g, 0.45 mmol), Pd(PPh3)2Cl 2 (20 mg, 0.027 mmol), Cul (10 mg, 0.054 mmol) was suspended in 2.25 mL dry Toluene under argon. TMS acetylene (0.124 g, 1.28 mmol) was added by syringe along with 2.25 mL NEt 3 . The reaction was sealed and heated at 85 C for 12 h. The volatiles were removed in vacuo and the residue was purified by silica gel chromatography (1:3, DCM:Hexanes) to give 22 (94%). 1H NMR (400 MHz, CDCl3): 6 7.73 (dd, J=5.6, 3.2, 4H), 7.16 (dd, J=5.6, 3.2, 4H), 5.93 (s, 2H), 0.38 (s, 18H). 13 C NMR (125 MHz, CDC13) 6 147.1, 144.2, 130.9, 125.9, 120.2, 115.5, 104.8, 101.9, 80.8, -0.11. HRMS (ESI) calcd. for C30 H30 O2 Si 2 [M+H] 479.1857, found 479.1869. Synthesis of 23: Compound 22 (0.150 g, 0.31 mmol) was dissolved in 4 mL THF, TBAF (0.69 mmol, IM in THF) was added and the reaction was left to stir for 2 h. The reaction mixture was diluted with brine and washed with DCM twice. The combined organic fractions were dried over Na 2SO 4 and the volatiles were removed in vacuo. The residue was purified by silica gel chromatography (6:4, Hexanes:DCM) to give 23 (87%). 1H NMR (400 MHz, CDCl3): 6 7.73 (i, 2H), 7.58 (in, 2H), 7.17 (d, J=8, 1H), 7.14 (m, 2H), 7.12 (m, 2H), 7.08 (d, J=8, 1H), 5.06 (d, J=2.8, 1H), 4.78 (s, 1H), 4.75 (d, J=2.8, 1H), 3.56 (s, 1H). 13C NMR (125 MHz, CDCl3) 6 162.8, 150.4, 145.0, 141.8, 140.4, 132.8, 127.5, 126.0, 125.8, 120.7, 119.5, 118.4, 114.0, 89.8, 85.4, 83.7, 82.6, 80.6. HRMS (ESI) calcd. for C24H1 40 2 [M+H] 335.1067, found 335.1059. Synthesis of 24: Compound 16 (0.2 g, 0.45 mmol), Pd(PPh3)2 Cl 2 (20 mg, 0.027 mmol), Cul (10 mg, 0.054 mmol) was suspended in 2.25 mL dry Toluene under argon. Phenylacetylene (0.138 g, 0.148 mL, 1.35 mmol) was added by syringe followed by 2.25 117 mL NEt 3. The reaction was sealed and heated at 85 C for 12 h. The volatiles were removed in vacuo and the residue was purified by silica gel chromatography (1:4, DCM:Hexanes) to give 24 (93%). 1H NMR (400 MHz, CDCl3): 6 7.76 (dd, J=5.6, 3.2, 4H), 7.66 (m, 4H), 7.45 (m, 6H), 7.19 (s, 2H), 7.17 (dd, J=5.6, 3.2, 4H), 5.68 (s, 2H). 13 C NMR (125 MHz, CDCl3) 6 146.5, 144.2, 131.8, 130.9, 129.6, 128.9, 126.0, 122.1, 120.2, 115.5, 98.3, 86.1, 81.0. HRMS (ESI) calcd. for C3 6H2 2 0 2 [M+H] 485.1547, found 485.1556. Synthesis of 25 using 12: Compound 24 (0.05 g, 0.1 mmol) and NaHCO 3 (0.052 g, 0.6 mmol) were suspended in 2 mL MeCN. Iodine (0.16 g, 6 mmol) was added and the reaction was stirred at rt for 15 h. Saturated Na 2S2O3 was added and left for 10 min. The reaction was diluted with water and washed with DCM (2x). The volatiles were removed in vacuo and the. residue was purified by silica gel chromatography (6:4, Hexanes: DCM) to give 25 (24%). -using IC: Compound 24 (0.02 g, 0.4 mmol) dissolved in 3.2 mL dry DCM and cooled to -78 0C. ICl (0.014 g, 0.086 mmol) was added dropwise and the reaction was allowed to warm to rt. After 10 min saturated Na 2S2O3 was added and left for a further 10 min. The reaction was diluted with water and washed with DCM (2x). The volatiles were removed in vacuo and the residue was purified by silica gel chromatography (6:4, Hexanes: DCM) to give 25 (86%). 1H NMR (400 MHz, CDCl3): 6 7.84 (in, 4H), 7.60 (in, 10H), 7.15 (dd, J=5.6, 3.2, 4H), 6.92 (s, 2H). 13 C NMR (125 MHz, CDCl3) 6 153.3, 144.1, 138.4, 131.2, 130.0, 129.8, 128.5, 127.1, 126.9, 126.0, 120.2, 83.1, 70.1. HRMS (ESI) calcd. for C3 6H20 12 0 2 [M+H] 737.9547, found 737.9565. Synthesis of 26 and 27: Compound 24 (72 mg, 0.15 mmol) and AuCl 3 (6 mg, 0.02 mmol) were dissolved in 1.6 mL THF and 0.4 mL MeCN. The solution was initially clear but a yellow solid precipitated after a few minutes and stirring was continued at rt for 12 h. The volatiles were removed in vacuo and the residue was purified by silica gel chromatography (7:3, Hexanes:DCM) to give 27 (40%) and 26 (29%). 27: 1H NMR (400 MHz, Cl 2CD-CDCl2): 6 8.06 (dd, J=7.6, 1.2 4H), 7.71 (dd, J=5.6, 3.2, 4H), 7.56 (m, 4H), 7.49 (in, 2H), 7.19 (dd, J=5.6, 3.2, 4H), 6.70 (s, 2H), 6.30 (s, 2H). 118 13C NMR (125 MHz, Cl 2 CD-CDCl2) 6 150.8, 144.2, 134.5, 131.9, 129.0, 128.6, 125.5, 124.7, 124.3, 120.9, 120.0, 96.1, 82.5. HRMS (ESI) calcd. for C3 6H2 2 0 2 [M+H] 487.1693, found 487.1701. 26: 1H NMR (600 MHz, CDC3): 6 8.08 (dd, J=8.4, 1.2, 2H), 7.80 (dd, J=7.8, 1.2, 2H), 7.64 (dd, J=7.5, 2.1, 2H), 7.61 (dd, J=6.9, 0.6, 2H), 7.55 (in, 2H), 7.49 (in, 1H), 7.43 (in, 3H), 7.17 (ddd, J=7.8, 7.2, 1.2, 2H), 7.13 (ddd, J=7.2, 7.2, 1.2, 2H), 7.10 (d, J=7.8, 1H), 6.72 (d, J=7.8, 1H), 6.26 (s, 1H), 5.67 (s, 1H). 13 C NMR (125 MHz, CDCl3) 6 151.9, 144.3, 143.8, 142.8, 135.0, 134.4, 131.9, 131.7, 129.6, 129.2, 128.9, 128.8, 126.9, 125.9, 125.8, 125.2, 122.5, 120.9, 120.7, 119.8, 112.6, 96.3, 95.8, 86.6, 83.1, 81.5. HRMS (ESI) caled. for C3H22O2 [M+H] 487.1693, found 487.1713. Synthesis of 28: Compound 14 (0.2 g, 0.36 mmol), Pd(PPh 3)2Cl 2 (15 mg, 0.022 mmol), Cul (10 mg, 0.054 mmol) was suspended in 2.25 mL dry Toluene under argon. Phenylacetylene (0.11 g, 0.118 mL, 1.08 mmol) was added by syringe followed by 2.25 mL NEt 3. The reaction was sealed and heated at 85 C for 18 h. The volatiles were removed in vacuo and the residue was purified by silica gel chromatography (1:4, DCM:Hexanes) to give 28 (93%). 1H NMR (400 MHz, CDCl3): 6 7.56 (in, 2H), 7.687.63 (in, 5H), 7.59 (d, J=1.2, 1H), 7.46-7.44 (in, 6H), 7.19-7.15 (m, 4H), 6.98 (dd, J=7.6, 1.2, 1H), 5.68 (s, 1H), 5.63 (s, 1H), 2.59 (pseudo t, J=8, 2H), 1.59 (in, 2H), 1.25 (in, 10H), 0.86 (t, J=6.8, 3H). 13 C NMR (125 MHz, CDCl3) 6 146.8, 146.7, 144.4, 144.3, 144.1, 141.55, 141.1, 131.8, 130.9, 130.8, 129.5, 128.9, 125.9, 125.8, 125.7, 122.2, 120.3, 120.1, 120.0, 115.4, 98.24, 98.22, 86.3, 86.2, 81.1, 81.0, 36.3, 32.1, 31.9, 29.8, 29.7, 29.5, 22.9, 14.3. HRMS (ESI) calcd. for C44 H3 8 0 2 [M+H] 599.2945, found 599.2935. Synthesis of 29: Compound 28 (0.1 g, 0.167 mmol), Ph 3PAuCl (5 mg, 0.01 mmol) and AgSbF 6 (3.5 mg, 0.01 mmol) were dissolved in 3 mL of THF:1,2-dichloroethane (1:1) and stirred at rt for 4 h. The volatiles were removed in vacuo and the residue was purified by silica gel chromatography (9:1, Hexanes:DCM) to give 29 (85%).1H NMR (400 MHz, CDC13): 6 8.06 (m, 4H), 7.66 (in, 2H), 7.57-7.43 (in, 8H), 7.12 (dd, J=5.4, 3, 2H), 6.92 (dd, J=7.6, 1.2, 1H), 6.62 (s, 2H), 6.26 (s, 1H), 6.25 (s, 1H), 2.55 (pseudo t, J=8, 2H), 1.54 (m, 2H), 1.26-1.23 (in, 10H), 0.85 (t, J=6.8, 3H). 13C NMR (125 MHz, CDC13) 6 119 151.1, 151.0, 145.0, 144.9, 144.6, 140.8, 135.2, 129.1, 128.9, 128.8, 125.6, 125.5, 125.3, 125.1, 125.0, 124.5, 121.1, 120.3, 120.1, 120.0, 119.9, 96.6, 96.5, 83.0, 36.2, 32.1, 32.0, 29.7, 29.6, 29.5, 22.9, 14.3. HRMS (ESI) caled. for C44 H38 0 2 [M+H] 599.2945, found 599.2963. Synthesis of 30: Compound 14 (50 mg, 0.113 mmol), 1,4-diethynylbenzene (14.3 mg, 0.113 mmol), Pd(PPh3)Cl 2 (2.38 mg, 3.4 gmol), Cul (15 mg, 79 ptmol) were dissolved in 1 mL dry toluene and 0.7 mL NEt 3 under argon. The reaction was stirred at 85 *C for 3 d and partitioned between DCM and sat. NH 4Cl. The reaction was extracted with sat. NH 4 C1 twice and the organic portion was dried over Na 2 SO 4 and evaporated to dryness in vacuo. The residue was dissolved in a minimum amount of DCM, filtered to remove insoluble material, and precipitated from MeOH. The precipitation procedure was repeated twice more to give 30 (32%). 1H NMR (400 MHz, THF-d 8): 6 7.75-7.55 (m, 6H), 7.23 (m, 2H), 7.09 (m, 2H), 6.93 (d, J=7.6, 1H), 6.21 (br, 2H), 5.98 (s, 1H), 5.96 (s, 1H), 2.58 (br, 2H), 1.57 (br, 2H), 1.28 (br, 1OH), 0.88 (br t, 3H). GPC: Ma = 12,500, PDI = 1.44, DP = 24. Synthesis of 31: Polymer 30 (50 mg, 0.123 mmol) was dissolved in 1 mL THF:1,2dichloroethane (1:1). A solution of Ph 3PAuCl (6.5 mg, 13 [tmol) and AgSbF 6 (4.4 mg, 13 tmol) was premixed in 1 mL THF: 1,2-dichloroethane (1:1) and filtered through celite into the reaction mixture. After 12 h of stirring at room temperature, a second preparation of catalyst solution was added and left to stir for an additional 12 h. The reaction mixture was diluted with IM HCl and extracted with fresh DCM twice. The combined organic fractions were dried over Na 2 SO4 and evaporated to dryness in vacuo. The residue was dissolved in a minimum amount of DCM and precipitated from MeOH. The precipitation procedure was repeated twice more to give 31 (74%). 1H NMR (400 MHz, CDCl3): 6 8.174 (br, 2H) 7.77-7,48 (m, 4H), 7.19 (br, 2H), 7.01 (br, 1H), 6.59 (br, 1H), 6.32 (br, 1H), 4.82 (s, 1H) 4.79 (s, 1H), 2.61 (br, 2H), 1.59 (br, 2H), 1.26 (br, 1OH), 0.85 (br, 6H). 3 C NMR (125 MHz, CDCl3, partial) 6 125.8, 125.0, 124.4, 120.2, 32.0, 29.6, 29.4, 22.8. GPC: Ma = 14,700, PDI = 1.16, DP = 28. 120 Synthesis of 32: Compound 16 (0.15 g, 0.34 mmol), phenylboronic acid (0.84 g, 1.01 mmol), Pd(PPh3 )2 Cl 2 (14 mg, 0.02 mmol) were dissolved in 2 mL degassed THF and 2 mL K3 PO 4 (0.5 M, degassed) under argon. The reaction was sealed and heated at 60 C for 6 h. The reaction was diluted with sat. NH4 Cl and washed with DCM twice. The combined organic layers were dried over Na2 SO 4 and volatiles were removed in vacuo. The residue was purified by silica gel chromatography (6:4, Hexanes:DCM) to give 32 (98%). 1H NMR (400 MHz, CDC13): 6 7.52 (m, 10H), 7.38 (m, 4H), 7.13 (dd, J=5.4, 3, 4H), 6.81 (s, 2H), 3.04 (s, 2H). 13C NMR (125 MHz, CDCl3) 6 145.0, 142.7, 141.0, 135.2, 129.4, 128.8, 128.4, 128.3, 125.7, 119.6, 81.0. HRMS (ESI) calcd. for C3 2 H2 2 0 2 [M+H] 439.1693, found 439.1687. Synthesis of 33: Compound 32 (85 mg, 0.19 mmol), K2S208 (0.21 g, 0.78 mmol), CuOAc 2 (0.31 g, 1.56 mmol) and KOAc (0.30 g, 3.1 mmol) was suspended in 2 mL of AcOH and heated at 113 0C for 6 h. Once cool the reaction was diluted with 10% NaOH. The mixture was extracted with DCM (3x) and the combined organic fractions were dried over Na2 SO 4 and volatiles were removed in vacuo. The residue was triturated with EtOAc (2 mL) and filtered to give 33 (77%). 1H NMR (400 MHz, CDCl3): 6 7.67 (dd, J=5.6, 3.2, 4H), 7.64 (dd, J=8, 1.6, 2H), 7.40 (s, 2H), 7.39 (dd, J=8, 1.2, 2H), 7.35 (ddd, J=8, 7.2, 1.6, 2H), 7.12 (dd, J=5.6, 3.2, 4H), 7.03 (ddd, J=8, 7.2, 1.2, 2H). 13 C NMR (125 MHz, CDCl3) 6 153.1, 143.9, 138.6, 134.3, 130.2, 125.8, 124.1, 122.7, 122.2, 120.3, 118.5, 117.8, 81.8. HRMS (ESI) caled. for C32Hi 8 0 2 [M+H] 435.1380, found 435.1360. Synthesis of 36: Compound 16 (0.05 g, 0.113 mmol), Pd cat. (4.5 mg, 5.7 mmol) and 2thiophene boronic acid (0.043 g, 0.338 mmol) were dissolved in 1 mL degassed THF under argon. Following, 2 mL of degassed 0.5 M K3 PO 4 was added by syringe and the reaction was stirred at 40 "C for 12 h. The reaction was diluted with water and extracted with DCM (3x). The combined organic extracts were dried over Na2 SO 4 and the volatiles were removed in vacuo. The residue was purified by silica gel chromatography (6:4, Hexanes:DCM) to give 36 (98%). 1H NMR (400 MHz, CDCl3): 6 7.58 (dd, J=5.6, 3.2, 4H), 7.53 (dd, J=5.2, 1.2, 2H), 7.19 (dd, J=5.2, 3.2, 2H), 7.15 (dd, J=5.6, 3.2, 4H), 7.09 (dd, J=3.2, 1.2, 2H), 3.74 (s, 2H). 13 C NMR (125 MHz, CDCl3) 6 144.6, 141.1, 129.6, 121 128.3, 128.1, 127.6, 127.4, 125.9, 119.8, 80.9. HRMS (ESI) caled. for C2 8 Hi 8 0 2 S2 [M+H] 449.0675, found 449.0657 Synthesis of 37: Compound 16 (0.1 g, 0.226 mmol), Pd cat. (4.5234 mg, 5.7 mmol) and 5-methyl-2-thienylboronic acid (0.123 g, 0.338 mmol) were dissolved in 1 mL degassed THF under argon. Following, 2 mL of degassed 0.5 M K3 PO 4 was added by syringe and the reaction was stirred at 40 C for 12 h. The reaction was diluted with water and extracted with DCM (3x). The combined organic extracts were dried over Na2 SO 4 and the volatiles were removed in vacuo. The residue was purified by silica gel chromatography (6:4, Hexanes:DCM) to give 37 (70% plus unreacted 16) 1H NMR (400 MHz, CDCl3): 6 7.62 (dd, J=5.6, 3.2, 4H), 7.15 (dd, J=5.6, 3.2, 4H), 6.89 (s, 2H), 6.87 (d, J=3.2, 2H), 6.84 (m, 2H), 4.10 (s, 2H), 2.60 (s, 6H). 3 C NMR (125 MHz, CDCl3) 6 144.8, 144.4, 142.4, 138.5, 129.6, 128.5, 128.1, 125.8, 125.5, 119.8, 80.9, 15.6. HRMS (ESI) calcd. for C3 0H22 0 2 S2 [M+H] 477.0988, found 477.0995. Synthesis of 40: Compound 16 (0.1 g, 0.225 mmol), Pd cat. (8.8 mg, 11.2 pmol) and thiophene (0.162 g, 0.675 mmol) were dissolved in 2 mL degassed THF under argon. Following, 4 mL of degassed 0.5 M K3 PO 4 was added by syringe and the reaction was stirred at rt for 12 h. The reaction was diluted with water and extracted with DCM (3x). The combined organic extracts were dried over Na 2 SO4 and the volatiles were removed in vacuo. The residue was purified by silica gel chromatography (6:4->2:8, Hexanes:DCM) to give 40 (good yield). 1H NMR (400 MHz, CDC3): 6 7.58 (br m, 4H), 7.37 (d, J=5.6, 2H), 7.13 (br m, 4H), 6.98 (d, J= 5.6, 2H), 6.90 (s, 2H), 4.69 (br m, 2H), 3.82 (br s, 6H). 13 C NMR (125 MHz, CDCl)(Partial) 6 145.6, 144.6, 130.8, 125.6 (broad), 125.0, 120.2-119.5 (broad), 116.6, 80.8, 59.1. HRMS (ESI) calcd. for C30 H2 2 0 4 S2 [M+H] 511.1032, found 511.1035. Synthesis of 38: Compound 40 (38.5 mg, 75.4 pmol) and a catalytic amount of ptoluenesulfonic acid monohydrate were dissolved in 1 mL toluene and heated at 85 0C overnight. The reaction was diluted with DCM and washed with sat. NaHCO 3 twice. The organic fraction was dried over Na 2SO4 and volatiles removed in vacuo. The residue was 122 purified by silica gel chromatography (8:2, Hexanes:DCM) to give 41 (71%). 1H NMR (400 MHz, CDCl3): 6 7.67 (dd, J=5.6, 3.2, 4H), 7.18 (d, J=5.2, 2H), 7.14 (dd, J=5.6, 3.2, 4H), 7.08 (d, J=5.2, 2H), 6.78 (s, 2H). 13 C NMR (125 MHz, CDCl3) 6 151.6, 143.7, 132.7, 125.9, 122.9, 122.5, 120.4, 119.4, 117.7, 112.0, 84.3. HRMS (ESI) caled. for C 30 H22 0 4 S2 [M+H] 447.0508, found 447.0525. 4.8 References 1. Swager, T. M. Acc. Chem. Res. 2008, 41, 1181-1189. 2. Chong, J. H.; MacLachlan, M. J. Chem. Soc. Rev. 2009, 38, 3301-3315. 3. Taylor, M. S.; Swager, T. M. Org. Lett. 2007, 9, 3695-3697. 4. Wu, Y.; Hayama, T.; Baldridge, K. K.; Linden, A.; Siegel, J. S. J. Am. Chem. Soc. 2006, 128, 6870-6884. 5. Seo, S.; Marks, T. J. Chem. Eur. J. 2010, 16, 5148-5162. (b) Trost, B. M.; Gutierrez, A. C.; Livingston, R. C. Org. Lett. 2009, 11, 2539-2542. (c) McDonald, F. E.; Reddy, K. S.;Diaz, Y. J. Am. Chem. Soc. 2000, 122, 4304-4309. (d) McDonald, F. E. Chem. Eur.J. 1999, 5, 3103-3106. (e) Trost, B. M.; Rhee, Y. H. J Am. Chem. Soc. 1999, 121, 1168011683. (f) Compain, P.; Gord, J.; Vatele, J.-M. Tetrahedron 1996, 52, 10405-10416. (g) Compain, P.; Gore, J.; Vatele, J.-M. TetrahedronLett. 1995, 36, 4059-4062. (h) Mancuso, R.; Mehta, S.; Gabriele, B.; Salerno, G.; Jenks, W. S.; Larock, R. C. J. Org. Chem. 2010, 75, 897-901. 6. Marshall, A. R.; Bunz, U. H. F. Macromolecules 2001, 34, 4688-4690. 7. Ricks, H. L.; Choudry, U. H.; Marshall, A. R.; Bunz, U. H. F. Macromolecules 200, 36, 1424-1425. 8. Beck, J. B.; Kokil, A.; Ray, D.; Rowan, S. J.; Weder, C. Macromolecules 2002, 35, 590-593. 9. Moslin, R. M.; Espino, C. G.; Swager, T. M. Macromolecules 2008, 42, 452-454. 10. Moslin, R. M.; Andrew, T. L.; Kooi, S. E.; Swager, T. M. J. Am. Chem. Soc. 2009, 131, 20-21. 11. Greenham, N. C.; Moratti, S. C.; Bradley, D. D. C.; Friend, R. H.; Holmes, A. B. Nature 1999, 365, 628-630. 12. Renak, M. L.; Bartholomew, G. P.; Wang, S.; Ricatto, P. J.; Lachicotte, R. J.; Bazan, G. C. J. Am. Chem. Soc. 1999, 121, 7787-7799. 123 13. Sandros, K.; Sundahl, M.; Wennerstrom, 0.; Norinder, U. J. Am. Chem. Soc. 1990, 112, 3082-3086. 14. Marri, E.; Pannacci, D.; Galiazzo, G.; Mazzucato, U.; Spalletti, A. J. Phys. Chem. A. 2003,107, 11231-11238. 15. Sancho-Garcia, J.C.; Bredas, J.; Beljonne, D.; Comil, J.; Martinez-Alvarez, R.; Hanack, M.; Poulsen, L.; Gierschner, J.; Mack, H.; EgelhaafH.; Oelkrug, D. J. Phys. Chem. B. 2005, 109, 4872-4880. 16. Meier, H.; Gerold, J.; Kolshorn, H.; Mhfiling, B. Chem. Eur. J. 2004, 10, 360-370. 17. Lin, H.; Tsai, C.; Huang, G.; Lin, J. J. Poly. Sci. PartA 2006, 44, 783-800. 18. Wong, M. S.; Li, Z. L.; Shek, M. F.; Chow, K. H.; Tao, Y.; D'Iorio, M. J. Mater. Chem. 2000, 10, 1805-1810. 19. Taylor, M. S.; Swager, T. M. Angew. Chem. Int. Ed. Engl. 2007, 46, 8480-8483. 20. Grimsdale, A. C.; Mullen, K. Phenylene-BasedLadder Polymers, in Design and Synthesis of ConjugatedPolymers, ed. Leclerc, M. and Morin, J. Wiley-VCH, Weinheim, 2010, pp. 227-245. 21. Giordano, C.; Belli, A. J Org. Chem. 1979, 44, 2314-2315. 22. Decorzant, R.; Vial, C.; Naf, F.; Whitesides, G. M. Tetrahedron 1987, 43, 1871-1879. 23. Camaioni, D. M.; Alnajjar, M. S. J. Org. Chem. 1985, 50, 4456-446 1. 24. Minisci, F.; Citterio, A.; Giordano, C. Acc. Chem. Res. 1983, 16, 27-32. 25. Kieseritzky, F.; Allared, F.; Dahlstedt, E.; Hellberg, J. TetrahedronLett. 2004, 45, 6049-6050. 26. Chen, Y.; Yu, C.; Fan, Y.; Hung, L.; Chen, C.; Ting, C. Chem. Commun. 2010, 46, 6503-6505. 27. Veen, E. M.; Postma, P. M.; Jonkman, H. T.; Spek, A. L.; Feringa, B. L. Chem. Commun. 1999, 1709-1710. 28. Konarev, D. V.; Drichko, N. V.; Lyubovskaya, R. N.; Shul'ga, Y. M.; Litvinov, A. L.; Semkin, V. N.; Dubitsky, Y. A.; Zaopo, A. J. Mol. Struct. 2000, 526, 25-29. 29. Konarev, D. V.; Zubavichus, Y. V.; Valeev, E. F.; Slovokhotov, Y. L.; Shul'ga, Y. M.; Lyubovskaya, R. N. Synth. Met. 1999, 103, 2364-2365. 30. Konarev, D. V.; Khasanov, S. S.; Otsuka, A.; Maesato, M.; Saito, G.; Lyubovskaya, R. N. Angew. Chem. Int. Ed. Engl. 2010, 49, 4829-4832. 124 125 Chapter 4 Appendix H-NMR, "C-NMR, IR Data, and X-ray Structures 126 o Br O Br I 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 ppm Figure 4.A.1: H NMR spectrum of 10 in CDC13 (400 MHz) O Br O Br vwTr- R-111"r-ri Fly-mr 200 180 Figure 4.A.2: 13C 160 140 120 100 80 60 40 20 0 ppm NMR spectrum of 10 in CDC13 (125 MHz) TIPS OH Br I~ N OH Br TIPS I 8.0 iII 7.5 I 7.0 6.5 6.0 5.5 Figure 4.A.3: lH NMR spectrum of 4.5 4.0 3.5 3.0 in CDC 3 (400 MHz) 127 2.5 2.0 1.5 . 1.0 0.5 ppm TIPS OH Br OH Br TIPS 11 I --i -l.I---J.---A200 180 Figure 4.A.4: 13 160 140 120 100 80 ,A. -L-~.iAj - L .v 60 40 20 0 . ppm C NMR spectrum of 11 in CDC13 (125 MHz) H OH Br OH Br H -1 . 8.0 7.5 7.0 ... .... 6.5 .... 6.0 .... 5.5 ... 5.0 4.5 4.0 3.5 3.0 Figure 4.A.5: 'H NMR spectrum of 12 in CDC13 (400 MHz) 128 2.5 2.0 1.5 1.0 0.5 ppm H OH Br OH Br H II 210 200 190 13 Figure 4.A.6: 180 170 160 150 140 130 120 110 100 90 80 60 50 40 30 20 ppm C NMR spectrum of 12 in CDCl 3 (125 MHz) jUL 7.5 70 I 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Al 2.0 1.5 1.0 0.5 ppm Figure 4.A.7: H NMR spectrum of 13 in CDC13 (400 MHz) il~[ Br 200 Figure 4.A.8: 180 13 C 160 140 i I~ 120 100 80 60 NMR spectrum of 13 in CDC13 (125 MHz) 129 40 20 0 ppm ilJ~iCi 7.5 7.0 6.5 6.0 Z 5.5 5.0 4.5 4.0 Ld 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 4.A.9: 1H NMR spectrum of 14 in CDC13 (400 MHz) OH HO 200 Br 180 Figure 4.A.10: Br _ 13C 160 140 120 100 80 40 60 20 0 ppm NMR spectrum of 14 in CDC13 (125 MHz) C0OH 21 OH Br HO Br 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Figure 4.A.11: 1H NMR spectrum of 15 in CDC13 (400 MHz) 130 2.0 1.5 1.0 0.5 ppm C1 0H2 OH HO 1 Br - Br 200 180 Figure 4.A.12: 13 160 140 120 100 80 60 40 20 0 ppm C NMR spectrum of 15 in CDC13 (125 MHz) OH Br HO Br 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 1.5 2.0 1.0 0.5 ppm Figure 4.A.13: H NMR spectrum of 16 in CDC13 (400 MHz) OH Br HO Br 200 Figure 4.A.14: 180 3 160 140 120 100 80 60 C NMR spectrum of 16 in CDC13 (125 MHz) 131 40 20 0 ppm SiMe3 / OH /0 s'HO Me3 Si IIII I 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 4.A.15: H NMR spectrum of 22 in CDC13 (400 MHz) OH SiMe3 / HO Me3Si" 150 140 130 Figure 4.A.16: 7.5 7.0 3 120 110 100 90 80 70 60 50 40 30 20 10 ppm C NMR spectrum of 22 in CDC13 (125 MHz) 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Figure 4.A.17: 'H NMR spectrum of 23 in CDC13 (400 MHz) 132 2.0 1.5 1.0 0.5 ppm 0 HO+ H I I 200 180 Figure 4.A.18: 160 140 120 100 80 60 40 Ii1~ 0 ppm I I 7.5 7.0 Figure 4.A.19: ",0164 20 C NMR spectrum of 23 in CDCl 3 (125 MHz) 13 6.5 1H NMR LL."" 200 Figure 4.A.20: 6.0 LJA 180 5.5 5.0 4.5 I I 4.0 3.5 I'' I' 3.0 2.5 2.0 1.5 1.0 0.5 ppm spectrum of 24 in CDC13 (400 MHz) AALkjlkgjiw 160 -A'" 140 120 100 13 80 60 C NMR spectrum of 24 in CDC13 (125 MHz) 133 40 20 0 ppm Ai 8.0 7.5 I 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 A -,L. 1.5 1.0 0.5 ppm Figure 4.A.21: 'H NMR spectrum of 25 in CDC13 (400 MHz) L. 200 2 Figure 4.A.22: 180 13C 160 160 , si LA"AALAAwmJL 140 1 , 120 1 100 8 60 6 40 4 220 0 ppm NMR spectrum of 25 in CDC13 (125 MHz) @400MHz 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 @600MHz H HO 8.1 8.0 7.9 7.8 7.7 7.6 7.5 7.4 7.3 7.2 7.1 0.5 ppm - 7.0 6.9 6.8 6.7 6.6 6.5 6.4 6.3 6.2 6.1 6.0 5.9 5.8 5.7 ppm Figure 4.A.23: 'H NMR spectrum of 26 in CDC13 (400 MHz and 600 MHz) 134 I I I ppm -5.5 -6.0 -6.5 -7.0 -7.5 -8.0 -8.5 8.5 8.0 7.5 6.5 7.0 6.0 ppm 5.5 Figure 4.A.24: gCOSY spectrum of 26 in CDCl 3 (600 MHz) A ,.I 200 180 Figure 4.A.25: 13C I j .- 140 160 120 i- 100 40 60 80 20 0 NMR spectrum of 26 in CDC13 (125 MHz) H 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 Figure 4.A.26: lH NMR spectru m of 27 in CL2CD-CDC12 (400 MHz) 135 1.5 1.0 0.5 ppm 200 Figure 4.A.27: 8.0 7.5 180 13 160 140 120 100 80 - AH ,. L60 40 20 0 ppm C NMR spectrum of 27 in CL2CD-CDC12 (125 MHz) 7.0 6.5 6.0 5.5 4.5 5.0 4.0 3.5 3.0 2.5 2.0 1.5 0.5 ppm 1.0 Figure 4.A.28: 'H NMR spectrum of 28 in CDC13 (400 MHz) n-Oct 200 180 Figure 4.A.29: 160 13C NMR 140 120 100 40 60 20 0 spectrum of 28 in CDCl 3 (125 MHz) 0 - H -ii 8.5 80 it 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 Figure 4.A.30: 'H NMR spectrum of 29 in CDC13 (400 MHz) 136 2.5 2.0 1.5 1.0 ppm j HI KI5H I L.. 200 180 Figure 4.A.31: 8.5 8.0 I I~ 13 160 hi 140 120 .1. I 11,1 100 a 0, 1. 1..- -AA. 80 . .1. . 1 1. 40 .1 p 20 C NMR spectrum of 29 in CDC13 (125 MHz) I I I 7.5 7.0 6.5 ~ 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.0 1.5 1.0 ppm 2.0 1.5 1.0 ppm Figure 4.A.32: lH NMR spectrum of 30 in CDC13 (400 MHz) 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 Figure 4.A.33: IH NMR spectrum of 31 in CDC13 (400 MHz) 137 2.5 n-Oct n \ /0 0 200 13 Figure 4.A.34: - 150 100 / [ppm] OH HO - 1\ -J(t I I 7.5 0 50 C NMR spectrum of 31 in CDC13 (125 MHz) / I 7.0 6.5 6.0 5.5 5.0 I 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 4.A.35: 1H NMR spectrum of 32 in CDC13 (400 MHz) / HO 200 Figure 4.A.36: 180 13 C 160 140 120 100 NMR spectrum of 32 in CDC 138 80 3 60 (125 MHz) 40 OH - 20 0 ppm /O 0 0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 - 3.5 3.0 2.5 2.0 1.5 0.5 ppm 1.0 Figure 4.A.37: H NMR spectrum of 33 in CDCl 3 (400 MHz) /0 0 jjAjA".A -7 200 180 Figure 4.A.38: 13 160 140 L I-- - - l TTF"VrW--T-I'F'W- 120 100 80 60 40 20 0 ppm C NMR spectrum of 33 in CDC13 (125 MHz) h i Ii 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Figure 4.A.39: 'H NMR spectrum of 36 in CDC 3 (400 MHz) 139 2.0 1.5 1.0 0.5 ppm 200 160 Figure 4.A.40: 13 160 140 120 100 80 60 40 20 0 ppm C NMR spectrum of 36 in CDC13 (125 MHz) Me V, I 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 0.5 ppm 1.0 Figure 4.A.41: 1H NMR spectrum of 37 in CDCl 3 (400 MHz) HO - S Me T - 200 180 Figure 4.A.42: 1 3 I 1 160 1 140 120 IO 100 I 80 60 40 20 0 ppm C NMR spectrum of 37 in CDC13 (125 MHz) os | S oue 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Figure 4.A.43: H NMR spectrum of 40 in CDC13 (400 MHz) 140 2.0 1.5 1.0 0.5 ppm \ MeO / OH| SHO S S OMe 200 180 Figure 4.A.44: 13 160 140 120 7.5 80 60 40 20 0 ppm C NMR spectrum of 40 in CDCl 3 (125 MHz) JL1 8.0 100 0 7.0 6.5 6.0 5.5 5.0 - I I 4.5 4.0 I 3.5 I 3.0 2.5 2.0 1.5 1.0 0.5 ppm Figure 4.A.45: 1H NMR spectrum of 41 in CDC13 (400 MHz) S 0 2 200 Figure 4.A.46: 6 180 13C ' 160 -p ' 140 2 '120 0 100 8 6 80 60 NMR spectrum of 41 in CDC13 (125 MHz) 141 2 40 20 0 ppm 84 - 82- 80- 78- 76- 400 3500 3000 25;0 Wavenumbers(cm-1) Figure 4.A.47: FT-AT-IR spectrum of 39. 142 2000150 100 X-ray Crystal Structures: a) b) c) d) Figure 4.A.48: Crystal Structure of 23. (a) Suitable crystals were grown by vial-in-vial vapor diffusion using CHCl 3:Pentane as the solvent:antisolvent mixture. b) Anisotropic thermal ellipsoids set at 50% probability. c) and d) Crystal twins separated for clarity. 143 A Figure 4.A.49: Crystal Structure of 27. (a) Suitable crystals were grown by vial-in-vial vapor diffusion using (DCM and 1,1,2,2tetrachloroethane):MeOH as the (solvent): antisolvent mixture (solvent molecules removed for clarity. b) Anisotropic thermal ellipsoids set at 50% probability. Figure 4.A.50: Crystal Structure of 33. Suitable crystals were grown by slow evaporation of CHCl3. Anisotropic thermal ellipsoids set at 50% probability. 144 Figure 4.A.51: Crystal Structure of 38. Suitable crystals were grown by vial-in-vial vapor diffusion using CHCl 3 :Pentane as the solvent:antisolvent mixture. 145 BRETT VANVELLER Department of Chemistry - Massachusetts Institute of Technology 77 Massachusetts Avenue, Bldg 18-031 Cambridge, MA 02139-4307 USA (617) 452-2569 - bvv@mit.edu EXPERIENCE 2006-2011 Graduate Student with Professor Timothy M. Swager Massachusetts Institute of Technology 2005-2006 Researcher with Professor Alex Adronov McMaster University 2004 (summer) Researcher with Professor M6nica Barra University of Waterloo EDUCATION 2011 Ph.D., Chemistry, Massachusetts Institute of Technology 2006 B.Sc., Chemistry, McMaster University ACADEMIC HONORS & AWARDS 2010 2008 2007 2007 2007 2006 2006 2006 2006 2005 2005 2005 2004 2004 2004 2004 2002 David A. Johnson Summer Graduate Fellowship National Science and Engineering Research Council Postgraduate Scholarship Massachusetts Institute of Technology Department of Chemistry Award for Outstanding Teaching David A. Johnson Award Macromolecular Science and Engineering Division of the Chemical Institute of Canada Undergraduate Research Thesis Award National Science and Engineering Research Council Postgraduate Scholarship The Society of Chemical Industry Merit Award The Provost's Honour Roll Medal The Robert S. Haines Memorial Scholarship The Canadian Society for Chemistry Prize The J. L. W. Gill Prize The Donald G. McNabb Scholarship National Science and Engineering Research Council Undergraduate Student Research Award Reactive Intermediates Student Exchange Award The Chemical Institute of Canada Prize The University Senate Scholarship The McMaster Honour Award 146 PUBLICATIONS (5) (4) (3) (2) (1) Polat, B. E.; Lin, S.; Mendenhall, J. D.; VanVeller, B.; Langer, R.; Blankschtein, D. An Experimental and Molecular Dynamics Investigation into the Amphiphilic Nature of Sulforhodamine B, J. Phys. Chem. B 2011, 115, 1394-1402. Cover Article Andrew, T. L.; VanVeller, B.; Swager, T. M. Synthesis of Azaperylene-9,10-dicarboximides, Synlett 2010, 3045-3048. VanVeller, B.; Swager, T. M. Biocompatible Post-Polymerization Functionalization of a Water Soluble Poly(p-Phenylene Ethynylene), Chem. Comm un. 2010, 46, 5761-5763. VanVeller, B.; Swager, T. M., Poly(aryleneethynylene)s. In Design and Synthesis of Conjugated Polymers, Leclerc, M.; Morin, J., Eds. Wiley-VCH: Weinheim, 2010; pp 175-200. VanVeller, B.; Miki, K.; Swager, T. M. Rigid Hydrophilic Structures for Improved Properties of Conjugated Polymers and Nitrotyrosine Sensing in Water, Org. Lett. 2010, 12, 1292-1295. PRESENTATIONS 2010 2010 2010 2004 VanVeller, B.; Swager, T. M. Design and synthesis of rigid 3D hydrophilic structures for improved properties of conjugated polymers in water and post-polymerization strategies for sensing of proteins and influenza. Presented at the 240t ACS National Meeting, Boston, Massachusetts, USA. VanVeller, B.; Swager, T. M. Rigid Hydrophilic Structures for Improved Properties of Conjugated Polymers in Water and Applications in Biosensing. Presented at the 9 3 rd Canadian Chemistry Conference and Exhibition, Toronto, Ontario, CAN. VanVeller, B.; Swager, T. M. Rigid Hydrophilic Structures for Improved Properties of Conjugated Polymers in Water and Applications in Biosensing. Presented at the 9 th International Symposium on Functional nt-Electron Systems, Georgia Institute of Technology, Atlanta, Georgia, USA. VanVeller, B.; Barra, M. Synthesis of the Elusive 5-Deazariboflavin: a Probe for the Study of Electron Transfer Mechanisms. Presented at Reactive Intermediate Student Exchange Symposium, University of Saskatchewan, Emma Lake, Saskatchewan, CAN. TEACHING EXPERIENCE 2006-2007 Massachusetts Institute of Technology, Department of Chemistry: Classroom Teaching Assistant for Organic Chemistry and General Chemistry Prepared lessons for two one hour tutorials and held office hours each week. Received The MIT Department of Chemistry Award for Outstanding Teaching 2005-2006 McMaster University, Department of Chemistry: Classroom Teaching Assistant for General Chemistry Prepared lessons for two one hour tutorials and held office hours each week. 2004 (fall) McMaster University, Department of Chemisty: Laboratory Teaching Assistant for General Chemistry Instruction for one three hour laboratory session each week. OTHER RELEVENT EXPERIENCE 2009-2010 Executive planning committee, "Chemistry and Policy: Solving Problems at the Interface" symposium. 2010 Fall ACS National Meeting, Boston, MA. http: / /web.mit.edu / gsspc /. Press: Chemical & Engineering News 2011, 89, 54-55. 2007-2010 Assistant Editor of the journal Synfacts 147 Acknowledgements The past five years in graduate school have been both challenging and enriching in no small part thanks to the many wonderful people I had the pleasure of working with here at MIT. First and foremost, I must thank my advisor Tim Swager. He has been an inspiring and imaginative mentor who always granted me freedom to explore whatever chemical curiosity I could cook up. His wonderful creativity and sense of humor made it a pleasure to work with him these past five years. I would also like to thank my thesis committee, Profs. Stephen Buchwald and Barbara Imperiali, for guiding my academic progress. I thank Prof. Buchwald in particular for his advice and recommendation when selecting my future postdoctoral position. I am also indebted to my many coworkers in the Swager lab. There are far too many people to name who made my experience at MIT so memorable. Jeewoo Lim was my year mate and a great colleague to discuss chemistry with over a beer at the Muddy Charles. Trisha Andrew taught me everything I know about photophysics and using the instruments. Jason Cox, for the many conversations we shared, I will miss them once I leave. I wish to thank my office mates Barney Walker, Joel Batson, and Scott Meek, for all the wonderful discussions both chemistry and non-chemistry related. There are far too many to thank and this document was supposed to be submitted to the registrar last week. I gratefully acknowledge the organization s that financially supported my research: the Institute for Soldier Nanotechnologies (ISN) and the Natural Science and Engineering Research Council (NSERC) of Canada. Finally, I thank my wife Courtney for filling my life with happiness. Without her my life would not be as full. The last five years have been such a fun adventure and I can't wait for the next chapter together as our family has expanded by one-our new daughter Helena is now only 10 weeks old. 148