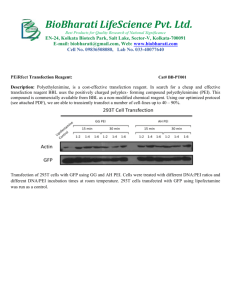
SPECIFIC AND EFFICIENT SEP 2 22010 LIBRARIES
advertisement

SPECIFIC AND EFFICIENT IN VIVO DELIVERY OF DNA AND siRNA BY POLYETHYLENIMINE AND ITS DERIVATIVES by MASSACHUSETTS INSTITUTE OF TECHNOLOGY JENNIFER A. FORTUNE SEP 2 22010 B.A. Chemistry Wheaton College, 2003 LIBRARIES Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of ARCHVES Doctor of Philosophy in Biological Chemistry at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September 2010 @ 2010 Massachusetts Institute of Technology All rights reserved Signature of Author:. Department of Chemistry August 3, 2010 Certified by: Alexander M. Klibanov Novartis ChairProfessorof Chemistry and Bioengineering Thesis Supervisor Accepted by:. Robert W. Field Haslam and Dewey Professor of Chemistry Chairman,DepartmentalCommittee on GraduateStudents This Doctoral Thesis has been examined by a committee of the Department of Chemistry as follows: JoAnne Stubbe Novartis Professorof Chemistry and Professor of Biology Thesis Chair Alexander M. Klibanov Novartis Chair Professorof Chemistry andBioengineering Thesis Supervisor John M.Essigmann William R. and Betsy P. Leitch Professor of Chemistry andiologicalEngineering SPECIFIC AND EFFICIENT IN VIVO DELIVERY OF DNA AND siRNA BY POLYETHYLENIMINE AND ITS DERIVATIVES by JENNIFER A. FORTUNE Submitted to the Department of Chemistry on August 3, 2010 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biological Chemistry ABSTRACT Linear polyethylenimine (PEI) is the "gold standard" of polycationic gene delivery vectors. However, little focus has been placed on enhancing or understanding the specificity of PEImediated gene delivery. Herein we evaluated the effect of chemical modification on the specificity of PEI-mediated nucleic acid delivery. We found that low molecular weight PEI (2 kDa) does not mediate efficient gene expression while high molecular weight (> 87 kDa) leads to toxicity. However, linear PEI of 25 kDa is an efficient gene delivery vector for both DNA and siRNA. Therefore, this PEI was chemically modified to explore the relationship between structure and specificity. First, PEI was covalently attached to a monoclonal anti-angiotensin I-converting enzyme (ACE) antibody (PEI-9B9) and evaluated for its ability to target PEI-9B9 polyplexes following intravenous delivery in a rat. Although mAb 9B9 retains affinity for its substrate ACE, PEI-9B9 does not enhance delivery to its intended target, the lung. Clearance of PEI-9B9 from circulation likely occurs before antibody binding to the surface expressed antigen. Next, we evaluated the ability of hydrophobic modification to modulate specificity of PEIbased gene delivery. Linear PEI was alkylated with variable length hydrocarbon chains at varying percent modification and evaluated for effective and specific gene delivery following intravenous delivery in mice. Modest alkylation (11% modification with ethyl chains to produce N-ethyl-PEI) enhances gene delivery in the lung 26-fold while quadrupling the ratio of gene product expressed in the lung relative to other organs. Interestingly, specificity profiles of the various alkyl chain derivatives vary among the organs examined. Additionally, a topical approach to gene delivery was investigated. Small branched PEI was cross-linked to gold to create PEI-gold nanoparticles (PEI-GNPs). These polycations were complexed with DNA and delivered topically to scratched rabbit cornea. PEI-GNPs effectively transfected corneal endothelium and evoked expression of the plasmid DNA without causing significant immunogenicity or toxicity. Finally, the effect of radiation on biologics was evaluated using a rigorously controlled experimental design with extreme conditions to unequivocally determine if radiofrequency radiation (RFR) has a non-thermal effect on biologics. Neither enzymes nor living cells (both bacterial and mammalian) were affect non-thermally by RFR. Thesis Supervisor: Alexander M. Klibanov Title: Novartis Chair Professor of Chemistry and Bioengineering ACKNOWLEDGMENTS I wish to express my gratitude to my thesis advisor, Alex Klibanov. Thank you for providing an environment where I was granted tremendous independence but also much needed guidance and advice. I take with me many lessons that will help me far beyond science and a surplus of antidotal stories to keep me smiling along the way. The bird is in your hands. I would also like to acknowledge committee members and faculty who played a critical role during my graduate work; JoAnne Stubbe, my thesis chair, John Essigmann, Stuart Licht, Liz Nolan, and Rajiv Mohan. I am exceedingly grateful to both past and present colleagues, especially Ken Hamill, Hector Hernandez, C. Ainsley Davis, Chia H. Wu, Mathew Tantama, Nebojsa Milovic, Alisha Weight, and Alyssa Larson, for helpful discussions and for their friendship. Without you, graduate school would have been a far more frustrating and far less enjoyable place. I am thankful to my parents, Domenic and Karen, my sisters and brothers, Jamie, Chris, Jess, Jill, and Leif, and my in-laws, Roseann and Chris, for constant support and enjoyable diversions. Thank you for listening to my rants and sharing joy in my successes. And most importantly, to my husband Bill, I can't put into words how integral a part of this thesis process you played. Thank you for making these years memories to be looked upon with a smile. I love you. TABLE OF CONTENTS Abstract Acknowledgements Table of Contents List of Figures List of Tables Abbreviations 3 4 5 7 8 9 I. Gene Therapy and Vectors for In Vivo Nucleic Acid Delivery A. Introduction B. References 10 31 II. Fully Hydrolyzed Linear Polyethylenimine Effects Functional In Vivo Delivery of Plasmid DNA and siRNA A. Introduction B. Results and Discussion C. Materials and Methods D. References 42 48 57 61 III. Specificity of Gene Delivery In Vivo Mediated By Polyethylenimine Conjugated to an Anti-ACE Antibody A. Introduction B. Results and Discussion C. Materials and Methods D. References 65 66 74 79 IV. On the Mechanism of Highly Effictive Gene Transfection In Vivo by Alkylated Polyethylenimine A. Introduction B. Results and Discussion C. Materials and Methods D. References 83 84 95 96 V. Polyethylenimine Mediates Specific In Vivo Gene Delivery Upon Topical Application A. Introduction B. Results and Discussion C. Materials and Methods D. References 100 101 109 113 VI. Radio Frequency Radiation (RFR) Causes No Non-Thermal Damage in Enzymes and Livng Cells A. Introduction B. Results and Discussion C. Materials and Methods D. References 116 117 124 127 Curriculum Vitae 131 LIST OF FIGURES Figure 1.1 Figure 1.2 Figure 1.3 Figure 1.4 Figure 1.5 Figure 2.1 Figure 2.2 Figure 2.3 Figure 2.4 Figure 2.5 Figure 2.6 Figure 3.1 Figure 3.2 Figure 3.3 Figure 3.4 Figure 3.5 Figure 4.1 Figure 4.2 Figure 4.3 Figure Figure Figure Figure 4.4 4.5 5.1 5.2 Figure 5.3 Figure 5.4 Figure 6.1 Figure 6.2 DNA transfection of a cell Diversity of viruses Cationic lipid delivery vectors PAMAM dendrimers: structure and characteristics Structure of polyethylenimine Efficient in vivo gene delivery by linear PEI Cytotoxicity of high molecular weight linear PEI Schematic of synthesis route for preparation of linear PEI Gene delivery by low molecular weight linear PEI in mice Biodistribution profile of pDNA delivered by low molecular weight linear PEI in mice In vivo siRNA knockdown of caveolin- 1 by linear PEI and its effects In vivo gene delivery by linear PEI in rats Schematic of synthesis of PEI-9B9 conjugates Binding affinity of PEI-conjugated anti-ACE antibody 9B9 In vivo gene delivery by PEI conjugated to 9B9 In vivo gene delivery by PEI conjugated to 9B9 at low doses Schematic of synthetic route for N-alkylated linear PEI derivatives Characterization of N-alkylated linear PEI by buffering capacity and DNA exclusion Specificity and efficacy of gene delivery of N-alkylated linear PEI derivatives in mice Effect of %modification by N-alkylation on gene delivery in vivo Biodistribution profile of N-alkyl PEI derivatives in mice Schematic of synthetic route for preparation of PEI-GNPs In vivo detection of PEI-GNP/GFP plasmid polyplexes by silver staining and detection of expressed GFP by fluorescence microscopy Immunogenicity of topical delivery of PEI-GNP polyplexes in the cornea Toxicity of topical delivery of PEI-GNP polyplexes in the cornea Effect of RFR on enzymatic activity of p-galactosidase and HRP Effect of RFR on bacteria and mammalian cells 11 16 20 24 28 45 47 50 51 52 55 67 69 70 71 73 85 87 89 90 93 102 103 105 108 120 122 LIST OF TABLES Table 1.1 Table 2.1 Table 4.1 Characteristics of viral delivery vectors Silencing of influenza infection by PEI-mediated siRNA delivery Biodistribution of plasmid delivered by N-alkylated linear PEI derivatives 17 46 92 ABBREVIATIONS Ab-SPDP ACE ANOVA s-gal BSA BCA DAPI DNA DTT Epi GFP HCl HIV HRP kb mAb 9B9 mRNA NMR N/P ratio NP-siRNA OxPAPC PAMAM PBS pDNA PEI PEI-9B9 PEI-GNP PEI-SPDP PEOZ PLL RFID RFR RFID RLU RNA RNAi SAR SCID siRNA TUNEL VILI Antibody conjugated to SPDP angiotensin I-converting enzyme analysis of variance enzyme P-galactosidase bovine serum albumin bicinchoninic acid 4',6-diamidino-2-phenylindole deoxyribonucleic acid dithiothreitol epithelial scrape green fluorescent protein hydrochloric acid human immunodeficiency virus enzyme horseradish peroxidase kilobases (kilonucleotides) mouse anti rat ACE monoclonal antibody (mAb) messenger RNA nuclear magnetic resonance ratio of nitrogen in PEI to phosphate in DNA influenza nucleopotein siRNA oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine polyamidoamine phosphate buffered saline plasmid DNA polyethylenimine PEI-mAb 9B9 conjugates PEI-gold nanoparticle PEI conjugated to SPDP poly(2-ethyl-2-oxazoline) poly-(L)-lysine radiofrequency identification radiofrequency radiation radiofrequency identification relative light unit ribonucleic acid RNA interference specific absorption rate severe combined immunodeficiency short interfering RNA terminal deoxyribonucleotidyl transferase dUTP nick end labeling ventilator induced lung injury I. GENE THERAPY AND VECTORS FOR IN VIVO NUCLEIC ACID DELIVERY A. Introduction Gene therapy is the treatment of a genetic or acquired disease by administration of a therapeutic nucleic acid'. Originally, the term was restricted to the delivery of a gene encoding a functional copy of a protein 2' 3 . However, with the advancement of nucleic acid technology, the term gene therapy has expanded to include delivery of therapeutic, non-gene nucleic acids such as siRNA and mRNA 4 . Although the promise of gene therapy has been a reality for decades, delivery of therapeutic nucleic acids has proven to be a formidable obstacle. There are several barriers that must be overcome for exogenous, therapeutic nucleic acids to function within a cell (Figure 1.1)5-8. First, the nucleic acid must be taken up by a clinically relevant cell type. For some therapies, any cell is capable of mediating the desired response, whereas in other instances, transfection of a specific cell type is required. The strong negative charge of nucleic acids and the proteoglycans expressed on the exterior surface of cells discourages interaction of delivered nucleic acids with the cell surface 9 ' . Additionally, there are varied cell types within an organism and nucleic acids do not inherently prefer interaction with one type over another. For this reason, successful in vivo delivery of nucleic acids is often achieved through complexation with a vector' ". Nucleic Acid Vector + + + ~ r"-.j + Electrostatic Interaction Complex Extracellular Matrix Cellular Interaction and Endocytosis + + * +rP' Endosomal . + + Unpackaging Escape + RNA Nuclear Transport and Uptake Unoackaaina + + + Cytoplasm Figure 1.1. Critical steps for transfection of a cell by either DNA or siRNA. 11 Second, the nucleic acid must circumvent degradation in the late endosome or lysosome and escape from the organelle . Uptake of nucleic acids by cells occurs via an endocytic mechanism1 2,13. The endocytosed nucleic acids are then subjected to low pH and the presence of nucleases in the late endosome and lysosome 4 . These conditions are designed to retain and degrade endocytosed particles. For DNA or RNA to mediate a therapeutic effect, the nucleic acid must remain intact and escape from the vesicle into the cytoplasm of the cell. Following escape from the endosome, the nucleic acid must localize to the region of the cell where it is capable of effecting a therapeutic outcome. For RNA, this is the cytoplasm. However, for plasmid DNA (pDNA), translocation to the nucleus through an exceptionally viscous cytoplasm 1'16 and subsequent nuclear uptake are required (see Figure 1.1). pDNA is generally considered too large to be actively transported through nuclear pores and thus requires a different method for uptake into the nucleus17 . The exact methods of translocation and uptake remain unknown, but are agreed to be a major limiting factor in successful gene delivery'. Finally, once in the compartment where it can effect a clinically relevant event, the nucleic acid must be unpackaged from the vector utilized in its delivery 8 . It is only following dissociation from the vector that the nucleic acid can provide a therapeutic benefit. Types of Gene Therapy There are two main approaches to gene therapy, ex vivo and in vivo. In ex vivo gene therapy, cells are removed from the host organism, injected with the therapeutic nucleic acid, and then reintroduced into the organism 9. Transfer of nucleic acids into the cells occurs in a laboratory setting where high tech equipment and tools are readily available. With the recent advances in this field, DNA can be easily injected directly into the nucleus while siRNA is transferred to the cytoplasm20 . The ability to specifically deliver nucleic acids to the desired cellular compartment is clearly advantageous. However, this method is not without limitations. Only cells that can be removed and effectively reintroduced in a functional way can be treated with ex vivo gene therapy. To date, nearly all ex vivo gene therapy studies addresses diseases of the circulatory system2 1 . Due to the nature of ex vivo gene therapy, most diseases cannot be treated in this manner. Additionally, this type of gene therapy is not commercially favorable and exists as a medical service rather than a pharmaceutical formulation. In contrast, in vivo gene therapy involves treatment of cells in their natural environment, a living organism, and is applicable to nearly all diseases. Nucleic acids (most frequently in complex with a vector) are administered either topically, by direct injection into a tissue, or by systemic intravenous injection. The nucleic acids must then effectively reach the desired cells, cross the cell membrane, reach the necessary cellular compartment, and effectively unpack from the vector before they can exhibit a therapeutic effect. Although this method of gene therapy is broadly applicable and therapeutic nucleic acids have been developed and tested, the lack of a safe and efficient delivery vector has limited in vivo gene therapy successes. Therapeutic Effects Previously, two distinct methods of administering nucleic acids for gene therapy have been discussed. It is equally important to understand the various ways in which nucleic acids can mediate a therapeutic effect. pDNA delivery leads to expression of a therapeutic protein while siRNA delivery mediates silencing of aberrant cellular proteins or foreign viral proteins. Delivery of pDNA that is subsequently transcribed and translated into a functional, therapeutic protein is the classical description of gene therapy. It was first proposed for treatment of genetic diseases characterized by lack of expression of a functional copy of a single, essential protein 2. The first clinical trials for pDNA gene therapy addressed cystic fibrosis (CF)22 '2 and X-linked sever combined immune deficiency (SCID)24 , specifically adenosine deaminase deficiency related SCID2 1 2 5 . SCID and CF are recessive genetic diseases characterized by mutation of a single protein without which the individual will eventually die. In contrast, silencing of aberrant cellular proteins or viral proteins is a more recent expansion of the definition of gene therapy. With the advent of RNAi, the idea of in vivo gene silencing became a clinical possibility. Diseases characterized by over expression of a protein that is harmless in low levels (or even essential) but which manifests as a diseased state when over expressed could be treated by this method; cancer is the classical example of such a disease2 6 Finally, silencing of viral proteins is another example of gene therapy albeit furthest from the original concept. The goal of anti-viral siRNA delivery is not to repair or treat a genetic abnormality but rather to treat a viral infection 26 . For efficient viral infection, proteins essential to the production of new virions must be synthesized2 7 . Silencing the expression of any of these proteins would effectively halt a viral infection. Although this approach is applicable to any viral infection, it is most frequently discussed in the context of a therapy for HIV or hepatitis, as there are currently no successful treatments for these conditions27,28 Vectors for Gene Therapy Myriad nucleic acid constructs capable of mediating a therapeutic effect in vivo are in existence and the number is growing 2 9 , 30 . However, as Verma said, three problems remain for successful gene therapy, "delivery, delivery, delivery!"3 1 In an effort to translate the theory of gene therapy into a clinical reality, much work in recent years has focused on vectors for gene delivery. Though many compounds and approaches have been examined, the key players include viruses, cationic lipids, and cationic polymers. Viral Vectors Viruses have evolved to encompass a wide array of properties as demonstrated in Figure 1.2. Viral genomic nucleic acid can be in the form of DNA or RNA and may be enveloped or exist as a naked capsid. In addition to these major classification differences, viral tropisms vary greatly. Nucleic acid packaging capacity, cell or tissue specificity, and expression and immunogenicity profiles differ significantly as seen in Table 1.11. Similarly, routes of entry and cellular targeting, cellular uptake, and nuclear entry mechanisms are diverse. For example, adenovirus infects through mucosal exposure while Dengue virus is directly injected into the host by mosquito transfer32 ; additionally, cytomegalovirus uses the epidermal growth factor receptor to gain entry into cells while retrovirus are far more promiscuous . Viruses currently being investigated for their potential application in gene delivery include adenoviruses, adenoassociated viruses, retroviruses, the herpes simplex virus (HSV), the papilloma virus, and more 34 Non-enveloped; Picomaviridae, Caliciviridae Positive Strand Enveloped; Togaviridae, Flaviviridae, Corovaviridae RNAe Wuses NeaieStranEd( Enveloped; Paramyxoviridae, Bunyaviridae, Rhabdoviridae, Orthomyxoviridae Positive / Negative Strands Double Capsid; Reoviridae (Rotavirus, Colorado tick virus) Positive Strand via DNA Enveloped; Retroviridae (HIV- 1,T cell leukemia virus Figure 1.2. General classification of viruses with potential use as delivery vectors for gene therapy. Adapted from Polymeric Gene Delivery'. . Virus Packaging Capacity poxviridae 25 kb adenoviridae < 7.5 kb .. retrovirus 8 kb parvovirus <4 kb Cell / Tissue Application Outcome of Gene Therapy epidermal transient expression respiratory highly immunogenic epithelia broad: hepatocytes endothelial cells transient expression smooth muscle cells immunogenic airwayairwaycellshighly cells ocular tissues neurons broad. durable expression stem cells toxicity concerns neurons muscles mostly dividing cells: genome integration ocells expression tdurable lymphocytes safety concerns hepatocytes bone marrow cells Broad: slow expression onset integrating or nonintegrating dividing cells non-dividing cells durable expression Table 1.1. Key characteristics of viral families commonly employed for gene delivery, adapted from Polymeric Gene Delivery'. Note that the average size of a human gene is 10-15 kb. Abbreviation: kb = kilobases (kilonucleotides) Though many properties differ among viral strains, viral life cycles share several strict commonalities. All viruses have evolved to efficiently bind to cell surfaces with specificity, infect host cells, escape from endosomes (when relevant), traverse the cytoplasm to the nucleus, cross the nuclear membrane, and mediate expression of their genomic material to produce the proteins essential for continued infection35. These steps exactly mirror the desired pathway of gene delivery. Unfortunately, there are caveats to employing viruses as gene delivery vectors that greatly curb enthusiasm about their clinically relevant transfection efficacies. Technologically, viruses have two major limitations. They demonstrate limited cargo capacity (Table 1.1) and are challenging from a production and quality control standpoint'. Additionally, viruses evolve very rapidly leading to unpredictable mutations and are recognized by the host's defenses initiating a significant immune response 36. These two caveats present major safety concerns for the use of viruses as gene delivery vectors in humans. In fact, early clinical trials have only magnified these concerns with outcomes of cancer and even death3 7' 3 Given the early safety failures of virus mediated gene delivery, recent years have seen a significant increase in development of alternative vectors. Two major classes of compounds have emerged as leaders in the field of non-viral nucleic acid delivery vectors, specifically, cationic lipids and polycations. Although the gene delivery efficiency of these agents is currently inferior to that of viruses, the ability to design for and control safety in such vectors makes them very attractive targets as gene delivery vectors3 9' 40 Non- Viral Cationic Lipids Cationic lipids have been employed as gene delivery agents for over a decade41 42 . A cationic lipid consists of three major components: a hydrophilic head group, a hydrophobic tail that will self assemble to form micelles or bilayer liposomes in aqueous media, and a linker to join the two11 . Figure 1.3 depicts the most commonly employed cationic lipids and demonstrates how varied these structures can be. Though all cationic lipids shown in Figure 1.3 are used for transfection, a set of critical features for efficient in vivo transfection has been identified8 '4 3 . They include: (1) a head group consisting of a tertiary or quaternary amine; (2) sufficient membrane-destabilizing/fusion characteristic (mediated by unsaturation and/or acyl chain length in the tail group); (3) a cholesterol component; and (4) a surplus of positive charge. For cationic lipid mediated delivery, the first step is mixing of lipids to create selfassembled liposomes, basically a lipid bilayer4 4 . Upon addition of nucleic acids, the high cationic charge density on the surface of liposomes interacts electrostaticly with the negatively charged phosphate groups of the nucleic acids forming complexes, referred to as lipoplexes, that range from 80-400 nm in size8 . The ratio of cationic lipid to nucleic acid determines the overall charge of the resultant lipoplex; an excess of positive charge is desirable. Following administration, lipoplexes encounter cell surfaces that are highly negatively charged due to an abundance of sulfated proteoglycans. Interaction between the positively charged lipoplex and the cell surface leads to endocytosis and release of the nucleic acid into the cytoplasm45-49. Subsequent translocation to the nucleus and expression of pDNA produces the desired gene product. DOTMA O Ns DC-Chol DOGS NH2 NH HN H2N GL-67 H N H2N 0 N O NH2 DMRIE HOO Figure 1.3. Cationic lipids commonly employed for gene delivery. Liposome mediated gene delivery is not without limitations, low transfection efficiency being the most critical 46 . Release of nucleic acid from the endosome is very poor as liposomes are not inherently fusogenic. Recent progress has demonstrated enhanced efficiency following the incorporation of fusogenic lipids, however, improvements have been minimal at best5 . Additionally, liposomes can be toxic to cells. Although this toxicity is concentration dependent and can be modulated in a predictable manner, it remains an obstacle that must be overcome as low concentrations of lipoplexes correlate with low transfection efficiencies51 52 . Non- Viral Polycationic Vectors Polycations are a second major class of non-viral vectors for nucleic acid delivery and have been studied for decades. The diversity among gene delivery agents classified as polycations is vast and ranges from polypeptides to synthetic amino polymers and dendrimers to even glucosamine-based polysaccharides. These polycations can be linear or branched, low or high molecular weight, and biodegradable or not. The one physical property shared among vectors in this class is a high positive charge density at neutral pH, almost always due to the presence of primary amines. To function as an efficient gene delivery vector, a polycation must interact with the negatively charged phosphate group of a nucleic acid, condense it into a compact particle (referred to as a polyplex), protect the nucleic acid from degradation by nucleases, and enhance both the uptake of the nucleic acid into the cell and the resultant gene expression or silencing. The three most extensively investigated polycationic vectors include poly-(L)-lysine, polyamidoamine dendrimers (PAMAM Starburst dendrimer), and polyethylenimine (PEI). Poly-(L)-lysine Poly-(L)-lysine (PLL) was the first polycation demonstrated to enhance nucleic acid delivery in vitro. It was shown to condense DNA into toroid and rod shaped structures of 25-50 nm and 40-80 nm in size, respectively 53 . Additionally, formation of such polyplexes has been shown to be critically dependent upon the size of the polymer3,54 , salt concentration of the medium5 5 , and most importantly, the charge ratio between the cationic polymer and anionic DNA 53 . For PLL, a 2:1 weight ratio of PLL to DNA generates ideal polyplexes56 . These polyplexes are of an appropriate size (<200 nm) and charge (overall positive charge) to interact 57 with and be taken up by a cell, the first step in successful in vitro gene delivery Unfortunately, following endocytic uptake, PLL exhibits poor gene delivery properties 58' 59 . Without the addition of endosomolytic6"'a6 or fusogenic agents, most PLL polyplexes are unable to escape from endosomes and ultimately localize in lysosomes where the low pH of the vesicle degrades the polyplex and prevents expression of the desired gene product. In addition to poor escape from endosomes, PLL demonstrates other properties that prevent its success as an in vivo gene delivery vector. PLL is composed of repeating units of lysine, a common amino acid, and is therefore not biologically inert. It has been shown to enhance cellular processes during transfection such as endocytosis 62, 63 and cell division 64 and to activate phospholipases and proteins alike65 66 . Upregulation of proteins and cellular processes can mediate unpredictable effects and is therefore an undesirable side effect. Moreover, the cytotoxicity of PLL is high at nearly 50 % cell death following incubation with PLL-DNA polyplexes at concentrations necessary for transfection' 56. Finally, PLL demonstrates no inherent specificity for a particular cell or tissue type. As previously described, specificity is a critical component of gene delivery systems. However, it should be noted that targeting of PLL through the attachment of various ligands has demonstrated some success, but only in cell 57,67,68 culture systems7, Polyamidoamine Dendrimers Polyamidoamine (PAMAM) dendrimers, though technically polymers, possess a classic dendrimer core-shell structure composed of three elements; a core, an interior generation of repetitive branching units, and finally, terminal functional groups as seen in Figure 1.41,69,70 For PAMAM, the terminal groups are amines that are highly charged at neutral pH. By design, these dendrimers assume a spherical shape from generation 5 and above (where generation corresponds to the number of branching steps introduced during synthesis of these polymers) 71' 72. The spherical shape and surface charge of PAMAM dendrimers makes them impeccable synthetic mimics of histones, the cellular proteins responsible for binding to and winding DNA into an ordered structure within the nucleus7 3 ,74 Upon interaction with nucleic acids, PAMAM dendrimers form electrostatic interactions with the charged phosphate groups of the nucleic acid, effectively condensing nucleic acids into a small polyplex and protecting them from nuclease degradation7 5 . The size of the dendrimer (determined by the generation number) and also the ratio of dendrimer to nucleic acid are critical factors for successful polyplex formation 76. As with PLL, the medium in which the interaction occurs affects the characteristics of the resultant polyplex. Similarly, the ability of PAMAM dendrimers to bind to and be taken up by cells is dependent upon the size and charge of the polyplexes. NH2 G= 4 G =3 0 . H NH2 N" G= 2 G=1 NH2 G=O NH2 CORE INTERIOR SURFACE Determines size, shape directionality and multiplicity Branch cell amplification region Cationic terminal groups Generation 0 1 2 3 4 5 Surface Groups 4 8 16 32 64 128 Molecular Weight 517 1,430 3,256 6,909 14,215 28,826 Diameter (nm) 1.4 1.9 2.6 3.6 4.4 5.7 Figure 1.4. Structure of a G4 PAMAM dendrimer, adapted from Polymeric Gene Delivery', and a table which describe the growth and characteristics of these dendrimers. Note that with each generation, the number of surface groups doubles while the diameter grows only 1-2 nm. As described above, PAMAM dendrimers share many commonalities with PLL. However, these dendrimers mediate significantly greater in vivo pDNA expression than PLL due to their ability to escape from endosomes without the assistance of lysosomal disruption agents. Higher generation PAMAM dendrimers (i.e. G7 or G9) induce appreciable membrane leakage. This behavior is described by a membrane-bending model whereby the anionic membranes are grossly distorted by the fixed, extended radius of curvature associated with large, spherical dendrimers 77'78. This distortion induces lipid mixing, which allows for escape of polyplexes. Additionally, it should be noted that there is some evidence for the ability of the surface amines of the dendrimer to buffer the pH of the late endosome, which may play a role in endosomal release79 . Following escape from the endosome, PAMAM dendrimers again show dissimilarity to PLL. Polyplexes formulated with this dendrimer do not fully dissociate from nucleic acids within the cytoplasm. There is evidence to suggest that the polyplexes are taken up by the nucleus as a complex and separate once inside the nucleus80 . Given the ability of PAMAM dendrimers to mediate siRNA silencing, clearly some of the polyplexes must dissociate in the cytoplasm. However, very little is known about the processing of polyplexes following endosomal escape. Although PAMAM dendrimers are superior to PLL as a gene delivery agent, they too have limitations. PAMAM dendrimers are more efficient at endosomal escape but addition of lysotropic agents has been demonstrated to enhance delivery significantly8 1 , suggesting incomplete release from endosomes. Additionally, optimal conditions require a high dendrimer to DNA charge ratio (6:1), often causing toxicity to cells82 . Moreover dendrimers by design have a very rigid structure that does not allow for significant chemical modification, greatly limiting the versatility of this polymer for gene delivery. Polyethylenimine Polyethylenimine (PEI) was first identified as an efficient gene delivery agent in the lab of Jean-Paul Behr in 199583. He and colleagues demonstrated the ability of PEI to condense DNA into polyplexes capable of mediating gene expression both in vitro and in vivo. The structures of both branched and linear PEI are shown in Figure 1.5. The starting material and synthesis employed dictate both the structure (linear vs. branched) and size (molecular weight) of the PEI polymer produced. A catalyzed ring opening of aziridine creates a branched structure8 4 while both polymerization of oxazoline followed by hydrolysis of side chain amides and aziridine polymerization at low temperatures result in a linear polymer8 5. Every third atom of the ethylenimine polymer backbone is an amine, 90% of which can be protonated at neutral pH8 6. As such, PEI possesses the greatest charge density of any cationic polymer in use for gene delivery to date. Linear PEI possesses only secondary amines while most commercially available branched PEI have a ratio of 1:2:1 of primary, secondary, and tertiary amines 4 . These amines are essential to the ability of PEI to mediate gene expression. Like other polycations, the extensive positive charge of PEI enables condensation of nucleic acids into small polyplexes that can be endocytosed by cells8 7 . However, once inside the cell, PEI is adept at mediating endosomal escape via a so-called proton sponge effect8 3 . As the endosome is acidified, nitrogen atoms in the PEI backbone accept the free protons, preventing appreciable lowering of endosomal pH. As such, a substantial influx of protons (and the corresponding Cl- counterions) is required if the endosome is to mature into a late endosome with a pH of between 5 and 6. The significant salt gradient created between the interior of the endosome and the cytoplasm by the influx of HCl leads to diffusion of water into the organelle. The increased internal pressure causes rupture of the endosomal membrane allowing for release of the polyplexes into the cytoplasm"3' 88 . Addition of lysotropic and/or fusogenic agents does not enhance release of polyplexes89' 90. Although little is known about subsequent processing of polyplexes, several studies have suggested that PEI mediates transport through the cytoplasm and into the nucleus, all the while protecting DNA from digestion by nucleases 91-93 . Recent work in our lab and others has shown linear PEI to be an exceptional delivery agent for nucleic acids both in vitro and in vivo"' ". Linear PEI effects significant levels of gene expression from pDNA as well as knockdown by siRNA while exhibiting minimal to no toxicity. Additionally, the simple chemical structure of PEI makes it readily amenable to chemical modifications. The versatility of linear PEI can be exploited to overcome the current shortfalls of polycation mediated gene delivery, efficacy and specificity. NH2 A HN NH H2N HN NH H NH N HN HN N-sNH 2 H N? NH?N 2NH N- H2N H2N B H H H H H H H H Figure 1.5. Structures of branched (A) and linear (B) polyethylenimines (PEIs) Chapter Preview Our lab has devoted significant effort to both understanding and improving gene delivery mediated by polyethylenimine. For PEI to effect clinically relevant levels of gene expression or RNA silencing, there are two obstacles that must be overcome, enhanced efficacy and specificity. This is the focus of our lab and specifically my thesis work. Recent work in our lab demonstrated PEI mediated in vitro gene delivery does not directly translate to in vivo work. The added complexity of in vivo systems (blood component interactions, pharmacokinetic concerns, clearance systems, etc.) cannot be replicated in cell culture and thus use of such systems is exceptionally limited. As a result, all work in our lab is conducted in vivo in animal models. Our lab recently characterized the ability of linear PEI to mediate gene delivery in vivo. We explored the ability of linear PEI to effect pDNA expression. Additionally, we investigated the ability of linear PEI to silence expressed pDNA and viral genetic material9 5. However, the ability of linear PEI to mediate silencing of endogenous proteins is not known. In Chapter 2, we demonstrate the in vivo utility of linear PEI for delivery of siRNA against an endogenous target. Additionally, we evaluate the ability of low molecular weight linear PEI (2.5 kDa) to deliver nucleic acids with reduced toxicity. This work allows for complete characterization of linear PEI mediated gene delivery in mice. In subsequent chapters, we investigate whether chemical modification of PEI can enhance the efficacy or specificity of gene delivery. In Chapter 3, we address the specificity of linear PEI delivery. To date PEI has been employed to successfully deliver nucleic acids to the lungs of mice but has failed to mediate high expression in other organs. Herein we investigate if covalent attachment of an antibody to PEI can alter the in vivo specificity of gene delivery. The use of antibodies for targeted drug delivery has been demonstrated in other systems. However, the in vivo properties of gene delivery agents vary greatly and targeting of PEI with antibodies remains unstudied. Although antibodies demonstrate great specificity for cell surface receptors and ligands, they are also capable of initiating an immune reaction in vivo. Therefore in Chapter 4 we use systematic chemical modification by introducing short alkyl groups of varying length to investigate how minor changes in polymer structure alter the specificity of gene expression. Additionally, we mechanistically explore the observed changes in specificity so that we can design more specific and efficacious polymers. Although our focus has been predominantly chemical modification of linear PEI for intravenous in vivo gene delivery, several diseases lend themselves to treatment by topical applications of PEI, a method that allows for strict control over specificity. In Chapter 5 we address the potential of chemically modified PEI for topical treatment of corneal disease of the eye. Here we cross-link small branched PEIs (which are non-toxic but which demonstrate poor transfection efficiency) to gold to make nanoparticles (designated PEI-GNPs). These gold-crosslinked PEIs are then combined with nucleic acid to form polyplexes and administered topically to rabbit cornea to investigate their ability to mediate successful in vivo gene delivery. The uptake of PEI-GNPs and the expression of their cargo DNA were evaluated. Additionally, activation of immune response and apoptosis were examined to determine the toxicity of these topically delivered gene delivery vectors. With continued success in this field, gene therapy will hopefully one day be a clinical reality. At that time, one can imagine vials of freeze-dried polyplexes being prepared and shipped to hospitals and treatments centers around the world. Currently, radio frequency identification technology (RFID) is employed to track, monitor, and provide quality control for shipments of pharmaceuticals. Unfortunately, the non-thermal effect of such radiation on sensitive biological pharmaceuticals continues to be hotly debated. In Chapter 6, we investigate the existence of such effects through rigorous experimental control and protocols. B. References 1. Amiji, M. M., Polymeric Gene Delivery. PrinciplesandApplications. CRC Press LLC: 2005. 2. Friedmann, T.; Roblin, R., Gene therapy for human genetic disease? Science 1972, 175, (25), 949-55. 3. Fischer, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M., 20 years of gene therapy for SCID. Nat Immunol 11, (6), 457-60. 4. Kim, S. S.; Garg, H.; Joshi, A.; Manjunath, N., Strategies for targeted nonviral delivery of siRNAs in vivo. Trends Mol Med 2009, 15, (11), 491-500. 5. Bally, M. B.; Harvie, P.; Wong, F. M.; Kong, S.; Wasan, E. K.; Reimer, D. L., Biological barriers to cellular delivery of lipid-based DNA carriers. Adv Drug Deliv Rev 1999, 38, (3), 291315. 6. Wiethoff, C. M.; Middaugh, C. R., Barriers to nonviral gene delivery. JPharmSci 2003, 92, (2), 203-17. 7. Nishikawa, M.; Huang, L., Nonviral vectors in the new millennium: delivery barriers in gene transfer. Hum Gene Ther 2001, 12, (8), 861-70. 8. Rolland, A.; Sullivan, S. M., PharmaceuticalGene Delivery Systems. 2003; Vol. 131. 9. Belting, M.; Sandgren, S.; Wittrup, A., Nuclear delivery of macromolecules: barriers and carriers. Adv Drug Deliv Rev 2005, 57, (4), 505-27. 10. Wiethoff, C. M.; Smith, J. G.; Koe, G. S.; Middaugh, C. R., The potential role of proteoglycans in cationic lipid-mediated gene delivery. Studies of the interaction of cationic lipid-DNA complexes with model glycosaminoglycans. JBiol Chem 2001, 276, (35), 32806-13. 11. Hall, J. C.; Dunlap, J. C.; Friedmann, T.; Gianelli, F., Advances in Genetics. 1999; Vol. 41. 12. Apodaca, G., Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic 2001, 2, (3), 149-59. 13. Guy, J.; Drabek, D.; Antoniou, M., Delivery of DNA into mammalian cells by receptor- mediated endocytosis and gene therapy. Mol Biotechnol 1995, 3, (3), 237-48. 14. Lechardeur, D.; Verkman, A. S.; Lukacs, G. L., Intracellular routing of plasmid DNA during non-viral gene transfer. Adv Drug Deliv Rev 2005, 57, (5), 755-67. 15. Luby-Phelps, K.; Taylor, D. L.; Lanni, F., Probing the structure of cytoplasm. JCell Biol 1986, 102, (6), 2015-22. 16. Luby-Phelps, K.; Castle, P. E.; Taylor, D. L.; Lanni, F., Hindered diffusion of inert tracer particles in the cytoplasm of mouse 3T3 cells. Proc Natl Acad Sci USA 1987, 84, (14), 4910-3. 17. Lukacs, G. L.; Haggie, P.; Seksek, 0.; Lechardeur, D.; Freedman, N.; Verkman, A. S., Size-dependent DNA mobility in cytoplasm and nucleus. JBiol Chem 2000, 275, (3), 1625-9. 18. Kang, H. C.; Lee, M.; Bae, Y. H., Polymeric gene carriers. CritRev Eukaryot Gene Expr 2005, 15, (4), 317-42. 19. Goldspiel, B. R.; Green, L.; Calis, K. A., Human gene therapy. Clin Pharm 1993, 12, (7), 488-505. 20. Boggs, S. S., Targeted gene modification for gene therapy of stem cells. Int J Cell Cloning 1990, 8, (2), 80-96. 21. Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J. L.; Bousso, P.; Deist, F. L.; Fischer, A., Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, (5466), 669-72. 22. Rosenfeld, M. A.; Collins, F. S., Gene therapy for cystic fibrosis. Chest 1996, 109, (1), 241-52. 23. Crystal, R. G.; McElvaney, N. G.; Rosenfeld, M. A.; Chu, C. S.; Mastrangeli, A.; Hay, J. G.; Brody, S. L.; Jaffe, H. A.; Eissa, N. T.; Danel, C., Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat Genet 1994, 8, (1), 42-5 1. 24. Buckley, R. H., Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol 2004, 22, 625-55. 25. Aiuti, A.; Slavin, S.; Aker, M.; Ficara, F.; Deola, S.; Mortellaro, A.; Morecki, S.; Andolfi, G.; Tabucchi, A.; Carlucci, F.; Marinello, E.; Cattaneo, F.; Vai, S.; Servida, P.; Miniero, R.; Roncarolo, M. G.; Bordignon, C., Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 2002, 296, (5577), 2410-3. 26. White, P. J., Barriers to successful delivery of short interfering RNA after systemic administration. Clin Exp PharmacolPhysiol 2008, 35, (11), 1371-6. 27. von Laer, D.; Baum, C.; Protzer, U., Antiviral gene therapy. Handb Exp Pharmacol2009, (189), 265-97. 28. Pawlotsky, J. M.; Chevaliez, S.; McHutchison, J. G., The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology2007, 132, (5), 1979-98. 29. Hackett, P. B.; Ekker, S. C.; Largaespada, D. A.; Mclvor, R. S., Sleeping beauty transposon-mediated gene therapy for prolonged expression. Adv Genet 2005, 54, 189-232. 30. Papadakis, E. D.; Nicklin, S. A.; Baker, A. H.; White, S. J., Promoters and control elements: designing expression cassettes for gene therapy. Curr Gene Ther 2004, 4, (1), 89-113. 31. Jaroff, L., Fixing the genes. Time 1999, 153, (1), 68-70, 73. 32. Modis, Y.; Ogata, S.; Clements, D.; Harrison, S. C., A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA 2003, 100, (12), 6986-91. 33. Wang, X.; Huong, S. M.; Chiu, M. L.; Raab-Traub, N.; Huang, E. S., Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, (6947), 45661. 34. Kootstra, N. A.; Verma, I. M., Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol 2003, 43, 413-39. 35. El-Aneed, A., An overview of current delivery systems in cancer gene therapy. J Control Release 2004, 94, (1), 1-14. 36. Roth, J. A.; Cristiano, R. J., Gene therapy for cancer: what have we done and where are we going? JNatl Cancer Inst 1997, 89, (1), 21-39. 37. Marshall, E., Gene therapy death prompts review of adenovirus vector. Science 1999, 286, (5448), 2244-5. 38. Kaiser, J., Gene therapy. Seeking the cause of induced leukemias in X-SCID trial. Science 2003, 299, (5606), 495. 39. Ledley, F. D., Nonviral gene therapy: the promise of genes as pharmaceutical products. Hum Gene Ther 1995, 6, (9), 1129-44. 40. Lollo, C. P.; Banaszczyk, M. G.; Chiou, H. C., Obstacles and advances in non-viral gene delivery. Curr Opin Mol Ther 2000, 2, (2), 13 6-42. 41. Felgner, P. L.; Gadek, T. R.; Holm, M.; Roman, R.; Chan, H. W.; Wenz, M.; Northrop, J. P.; Ringold, G. M.; Danielsen, M., Lipofection: a highly efficient, lipid-mediated DNAtransfection procedure. Proc Natl A cad Sci U SA 1987, 84, (21), 7413-7. 42. Brigham, K. L.; Meyrick, B.; Christman, B.; Magnuson, M.; King, G.; Berry, L. C., Jr., In vivo transfection of murine lungs with a functioning prokaryotic gene using a liposome vehicle. Am JMed Sci 1989, 298, (4), 278-81. 43. Barteau, B.; Chevre, R.; Letrou-Bonneval, E.; Labas, R.; Lambert, 0.; Pitard, B., Physicochemical parameters of non-viral vectors that govern transfection efficiency. Curr Gene Ther 2008, 8, (5), 313-23. 44. Safinya, C. R., Structures of lipid-DNA complexes: supramolecular assembly and gene delivery. Curr Opin Struct Biol 2001, 11, (4), 440-8. 45. Legendre, J. Y.; Szoka, F. C., Jr., Delivery of plasmid DNA into mammalian cell lines using pH-sensitive liposomes: comparison with cationic liposomes. Pharm Res 1992, 9, (10), 1235-42. 46. Zabner, J.; Fasbender, A. J.; Moninger, T.; Poellinger, K. A.; Welsh, M. J., Cellular and molecular barriers to gene transfer by a cationic lipid. JBiol Chem 1995, 270, (32), 18997-9007. 47. Labat-Moleur, F.; Steffan, A. M.; Brisson, C.; Perron, H.; Feugeas, 0.; Furstenberger, P.; Oberling, F.; Brambilla, E.; Behr, J. P., An electron microscopy study into the mechanism of gene transfer with lipopolyamines. Gene Ther 1996, 3, (11), 1010-7. 48. Wrobel, I.; Collins, D., Fusion of cationic liposomes with mammalian cells occurs after endocytosis. Biochim Biophys Acta 1995, 1235, (2), 296-3 04. 49. McLean, J. W.; Fox, E. A.; Baluk, P.; Bolton, P. B.; Haskell, A.; Pearlman, R.; Thurston, G.; Umemoto, E. Y.; McDonald, D. M., Organ-specific endothelial cell uptake of cationic liposome-DNA complexes in mice. Am JPhysiol 1997, 273, (1 Pt 2), H387-404. 50. Nakanishi, M.; Mizuguchia, H.; Ashihara, K.; Senda, T.; Akuta, T.; Okabe, J.; Nagoshi, E.; Masago, A.; Eguchi, A.; Suzuki, Y.; Inokuchi, H.; Watabe, A.; Ueda, S.; Hayakawa, T.; Mayumi, T., Gene transfer vectors based on Sendai virus. J Control Release 1998, 54, (1), 61-8. 51. San, H.; Yang, Z. Y.; Pompili, V. J.; Jaffe, M. L.; Plautz, G. E.; Xu, L.; Feigner, J. H.; Wheeler, C. J.; Feigner, P. L.; Gao, X.; et al., Safety and short-term toxicity of a novel cationic lipid formulation for human gene therapy. Hum Gene Ther 1993, 4, (6), 781-8. 52. Stewart, M. J.; Plautz, G. E.; Del Buono, L.; Yang, Z. Y.; Xu, L.; Gao, X.; Huang, L.; Nabel, E. G.; Nabel, G. J., Gene transfer in vivo with DNA-liposome complexes: safety and acute toxicity in mice. Hum Gene Ther 1992, 3, (3), 267-75. 53. Kwoh, D. Y.; Coffin, C. C.; Lollo, C. P.; Jovenal, J.; Banaszczyk, M. G.; Mullen, P.; Phillips, A.; Amini, A.; Fabrycki, J.; Bartholomew, R. M.; Brostoff, S. W.; Carlo, D. J., Stabilization of poly-L-lysine/DNA polyplexes for in vivo gene delivery to the liver. Biochim Biophys Acta 1999, 1444, (2), 171-90. 54. Ward, C. M.; Read, M. L.; Seymour, L. W., Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood 2001, 97, (8), 2221-9. 55. Liu, G.; Molas, M.; Grossmann, G. A.; Pasumarthy, M.; Perales, J. C.; Cooper, M. J.; Hanson, R. W., Biological properties of poly-L-lysine-DNA complexes generated by cooperative binding of the polycation. JBiol Chem 2001, 276, (37), 34379-87. 56. Lee, M.; Nah, J. W.; Kwon, Y.; Koh, J. J.; Ko, K. S.; Kim, S. W., Water-soluble and low molecular weight chitosan-based plasmid DNA delivery. Pharm Res 2001, 18, (4), 427-31. 57. Wagner, E.; Cotten, M.; Foisner, R.; Birnstiel, M. L., Transferrin-polycation-DNA complexes: the effect of polycations on the structure of the complex and DNA delivery to cells. Proc Natl Acad Sci USA 1991, 88, (10), 4255-9. 58. Erbacher, P.; Roche, A. C.; Monsigny, M.; Midoux, P., Putative role of chloroquine in gene transfer into a human hepatoma cell line by DNA/lactosylated polylysine complexes. Exp Cell Res 1996, 225, (1), 186-94. 59. Zauner, W.; Kichler, A.; Schmidt, W.; Mechtler, K.; Wagner, E., Glycerol and polylysine synergize in their ability to rupture vesicular membranes: a mechanism for increased transferrinpolylysine-mediated gene transfer. Exp Cell Res 1997, 232, (1), 137-45. 60. Choi, Y. H.; Liu, F.; Kim, J. S.; Choi, Y. K.; Park, J. S.; Kim, S. W., Polyethylene glycol- grafted poly-L-lysine as polymeric gene carrier. J Control Release 1998, 54, (1), 39-48. 61. Mislick, K. A.; Baldeschwieler, J. D.; Kayyem, J. F.; Meade, T. J., Transfection of folate- polylysine DNA complexes: evidence for lysosomal delivery. Bioconjug Chem 1995, 6, (5), 5125. 62. Shen, W. C.; Ryser, H. J., Conjugation of poly-L-lysine to albumin and horseradish peroxidase: a novel method of enhancing the cellular uptake of proteins. Proc Natl Acad Sci US A 1978, 75, (4), 1872-6. 63. Pruzanski, W.; Saito, S., The influence of natural and synthetic cationic substances on phagocytic activity of human polymorphonuclear cells. An alternative pathway of phagocytic enhancement. Exp Cell Res 1978, 117, (1), 1-13. 64. Kundahl, E.; Richman, R.; Flickinger, R. A., The effect of added HI histone and polylysine on DNA synthesis and cell division of cultured mammalian cells. J Cell Physiol 1981, 108, (3), 291-8. 65. Shier, W. T.; Dubourdieu, D. J.; Durkin, J. P., Polycations as prostaglandin synthesis inducers. Stimulation of arachidonic acid release and prostaglandin synthesis in cultured fibroblasts by poly(L-lysine) and other synthetic polycations. Biochim Biophys Acta 1984, 793, (2), 238-50. 66. Kyriakis, J. M.; Avruch, J., pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J Biol Chem 1990, 265, (28), 17355-63. 67. Wu, G. Y.; Wu, C. H., Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. JBiol Chem 1987, 262, (10), 4429-32. 68. Cotten, M.; Langle-Rouault, F.; Kirlappos, H.; Wagner, E.; Mechtler, K.; Zenke, M.; Beug, H.; Birnstiel, M. L., Transferrin-polycation-mediated introduction of DNA into human leukemic cells: stimulation by agents that affect the survival of transfected DNA or modulate transferrin receptor levels. Proc NatlAcad Sci USA 1990, 87, (11), 4033-7. 69. Tomalia, D. A.; Frechet, J. M. J., Discovery of dendrimers and dendritic polymers: A brief historial perspective. JPolymer Sci 2002, 40, 2719-2728. 70. Tomalia, D. A.; Baker, H.; Dewald, J. R.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P., A new class of polymers: Starburst-dendritic macromolecules. Polym. J 1985, 17, 117-132. 71. Caminati, G.; Turro, J.; Tomalia, D. A., Photophysical investigation of starburst dendrimers and their interactions with anionic and cationic surfactants. JAm Chem Soc 1990, 112, 8515-8522. 72. Potschke, D.; Ballauff, M.; Lindner, P.; Fischer, M.; Vogtle, F., Analysis of the structure of dendrimers in solution by small angle neutron scattering including contrast variation. Macromolecules 1999, 32, 4079-4087. 73. Chen, W. Q.; Turro, N. J.; Tomalia, D. A., Using ethidium bromide to probe the interactions between DNA and dendrimers. Langmuir 2000, 16, 15-19. 74. Bielinska, A. U.; Kukowska-Latallo, J. F.; Baker, J. R., Jr., The interaction of plasmid DNA with polyamidoamine dendrimers: mechanism of complex formation and analysis of alterations induced in nuclease sensitivity and transcriptional activity of the complexed DNA. Biochim Biophys Acta 1997, 1353, (2), 180-90. 75. Bielinska, A.; Kukowska-Latallo, J. F.; Johnson, J.; Tomalia, D. A.; Baker, J. R., Jr., Regulation of in vitro gene expression using antisense oligonucleotides or antisense expression plasmids transfected using starburst PAMAM dendrimers. Nucleic Acids Res 1996, 24, (11), 2176-82. 76. Bielinska, A. U.; Chen, C.; Johnson, J.; Baker, J. R., Jr., DNA complexing with polyamidoamine dendrimers: implications for transfection. Bioconjug Chem 1999, 10, (5), 84350. 77. Zhang, Z. Y.; Smith, B. D., High-generation polycationic dendrimers are unusually effective at disrupting anionic vesicles: membrane bending model. Bioconjug Chem 2000, 11, (6), 805-14. 78. Wattiaux, R.; Laurent, N.; Wattiaux-De Coninck, S.; Jadot, M., Endosomes, lysosomes: their implication in gene transfer. Adv Drug Deliv Rev 2000, 41, (2), 201-8. 79. Haensler, J.; Szoka, F. C., Jr., Polyamidoamine cascade polymers mediate efficient transfection of cells in culture. Bioconjug Chem 1993, 4, (5), 372-9. 80. Yoo, H.; Juliano, R. L., Enhanced delivery of antisense oligonucleotides with fluorophore-conjugated PAMAM dendrimers. Nucleic Acids Res 2000, 28, (21), 4225-3 1. 81. Kukowska-Latallo, J. F.; Chen, C.; Eichman, J.; Bielinska, A. U.; Baker, J. R., Jr., Enhancement of dendrimer-mediated transfection using synthetic lung surfactant exosurf neonatal in vitro. Biochem Biophys Res Commun 1999, 264, (1), 253-261. 82. Brazeau, G. A.; Attia, S.; Poxon, S.; Hughes, J. A., In vitro myotoxicity of selected cationic macromolecules used in non-viral gene delivery. Pharm Res 1998, 15, (5), 680-4. 83. Boussif, 0.; Lezoualc'h, F.; Zanta, M. A.; Mergny, M. D.; Scherman, D.; Demeneix, B.; Behr, J. P., A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc NatlAcad Sci USA 1995, 92, (16), 7297-301. 84. Kichler, A.; Behr, J. P.; Erbacher, P., Nonviral Vectorsfor Gene Therapy. Academic Press: San Diego, 1999. 85. Brissault, B.; Kichler, A.; Guis, C.; Leborgne, C.; Danos, 0.; Cheradame, H., Synthesis of linear polyethylenimine derivatives for DNA transfection. Bioconjug Chem 2003, 14, (3), 5817. 86. Suh, J.; Paik, H. J.; Hwang, P. K., Ionization of polyethylenimine and polyellylamine at various pHs. Bioorg Chem 1994, 22, 318. 87. Bieber, T.; Meissner, W.; Kostin, S.; Niemann, A.; Elsasser, H. P., Intracellular route and transcriptional competence of polyethylenimine-DNA complexes. J Control Release 2002, 82, (2-3), 441-54. 88. Klemm, A. R.; Young, D.; Lloyd, J. B., Effects of polyethyleneimine on endocytosis and lysosome stability. Biochem Pharmacol 1998, 56, (1), 41-6. 89. Boussif, 0.; Zanta, M. A.; Behr, J. P., Optimized galenics improve in vitro gene transfer with cationic molecules up to 1000-fold. Gene Ther 1996, 3, (12), 1074-80. 90. Kircheis, R.; Kichler, A.; Wallner, G.; Kursa, M.; Ogris, M.; Felzmann, T.; Buchberger, M.; Wagner, E., Coupling of cell-binding ligands to polyethylenimine for targeted gene delivery. Gene Ther 1997, 4, (5), 409-18. 91. Godbey, W. T.; Wu, K. K.; Mikos, A. G., Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci US A 1999, 96, (9), 5177-81. 92. Pollard, H.; Remy, J. S.; Loussouarn, G.; Demolombe, S.; Behr, J. P.; Escande, D., Polyethylenimine but not cationic lipids promotes transgene delivery to the nucleus in mammalian cells. JBiol Chem 1998, 273, (13), 7507-11. 93. Suh, J.; Wirtz, D.; Hanes, J., Efficient active transport of gene nanocarriers to the cell nucleus. Proc Natl Acad Sci USA 2003, 100, (7), 3878-82. 94. Thomas, M.; Lu, J. J.; Chen, J.; Klibanov, A. M., Non-viral siRNA delivery to the lung. Adv Drug Deliv Rev 2007, 59, (2-3), 124-33. 95. Thomas, M.; Lu, J. J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A. M., Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci U S A 2005, 102, (16), 5679-84. II. FULLY HYDROLYZED LINEAR POLYETHYLENIMINE EFFECTS FUNCTIONAL IN VIVO DELIVERY OF PLASMID DNA AND siRNA A. Introduction Gene therapy has yet to become a clinical reality due to the lack of a vector capable of delivering therapeutic levels of nucleic acid to cells in vivo'-. Virus mediated delivery has achieved clinically useful levels of DNA/siRNA delivery but has been plagued by significant 6-9 safety concerns - . In contrast, cationic polymers and lipids are safe but evoke lower levels of nucleic acid delivery than their viral counterparts2,1, 1. In an effort to achieve virus-like levels of gene delivery safely, our lab has devoted significant effort to understanding and improving transfection mediated by the cationic polymer polyethylenimine (PEI). Branched PEI first emerged as a successful gene delivery agent in 1995. Since then many labs have worked to enhance delivery by this polymer 13' 7 . Much work has been completed using mammalian cells grown in culture as it is a convenient and inexpensive way to 18 screen gene delivery vectors' . Unfortunately, results in our lab have demonstrated that in vitro and in vivo results fail to correlate directly with one another. This is presumably due to the stark differences between cell culture and in vivo systems. In cell culture, gravity and diffusion are responsible for positioning polyplexes, nucleic acid/polymer complexes, in contact with the cell surface whereby they are endocytosed. In vivo, systemic circulation and pharmacokinetics play key roles in dictating the interactions of polyplexes with the various cells types' ~21. For this reason, our lab has limited the use of cell culture to initial toxicity screens of polymers and all transfection studies are conducted in vivo. Initial transfection studies in our lab with commercially available linear PEI revealed specific accumulation of PEI-based polyplexes (nucleic acid/PEI complexes) in the lung. Although this phenomenon is not well understood, there are a few factors likely to contribute to this observed phenomenon. The lung is the first pass organ following intravenous delivery. This means that polyplexes injected in the tail vein first travel to the lung where they can be taken up by lung cells. It is only after passage through the lung that the polyplexes reach the heart and are distributed to the rest of the organs. Additionally, as the lung is designed for rapid exchange of gases, the capillaries in the lung are very small. To better gauge the size, a single red blood cell must distort its shape to pass through the capillaries. This translates into a long retention time in the lung and close proximity to the endothelial cells of the blood vessel, encouraging interaction and the potential for uptake. Recently our lab described the synthesis of a fully deacylated linear PEI for in vivo gene delivery whose efficiency far surpasses that of commercially available polymer2 2 . Commercially available linear PEI (25 kDa) retains 11 % of the propionyl groups from the original polymerization reaction (See Fig 2.3). Complete acid hydrolysis of these groups enhances gene delivery in the lung 10,000 fold (Figure 2.1)22. Additionally, we found that co-delivery of the luciferase gene and anti-luciferase siRNA with the fully hydrolyzed linear PEI of 25 kDa silences greater than 75 % of luciferase expression at N/P ratios of 5 and 7.5. Moreover, we synthesized a fully hydrolyzed linear PEI of 87 kDa and demonstrated greater than 90% suppression of luciferase activity with anti-luciferase siRNA delivered at N/P ratios as low as 3.75 and 522 Finally, we evaluated the ability of fully hydrolyzed linear PEI to reduce viral titer through delivery of anti-viral siRNA. Anti-viral siRNA in complex with fully hydrolyzed linear PEI of 87 kDa reduced in vivo influenza viral titer greater than 40% when administered to mice before challenge with the virus (Table 2.1). The siRNA employed was directed toward the influenza nucleoprotein (siRNA-NP), a protein essential to continued viral infection. Although 40% is a tremendous reduction in viral titer, it is less than the 90% reduction in luciferase observed 22 . This is not surprising as co-delivery of the gene of interest and the siRNA to silence that gene should allow for transfection of the same population of cells. In contrast, silencing of viral infection requires delivery to all infected cells, some of which may not be accessible to delivered polyplexes. Additionally, viruses are adept at surviving challenges and it takes longer to eliminate a viral infection than protein expression. It is important to note that a 40% reduction in viral titer is significant for treatment of influenza. Even minor reductions in viral titer facilitate elimination of the virus by the host's immune system. In this chapter, we investigate the ability of fully hydrolyzed linear PEI to silence expression of an endogenous gene through delivery of siRNA. This is a critical step in siRNA mediated gene therapy and has not been demonstrated with fully hydrolyzed linear PEI to date. Additionally, through careful selection of the protein target (caveolin- 1), we demonstrate the ability of this vector to mediate delivery to endothelial cells. Although uptake of polyplexes by endothelial cells has been hypothesized, it has not been experimentally demonstrated in an in vivo model with linear PEI. Moreover, we demonstrate a functional response to caveolin-1 knockdown through assay of endothelial cell barrier function, which is believed to occur in a caveolin- 1 dependent manner. - -"44 - - - - .. 0 I.M.IIII . ........- - - . ....... 109 105 - S110 at .. . .... 100- .2 106 1. 10 3 $ 100 7.5 . 1031 . 10 10- $ \ne5.0 rn5.0 Figure 2.1. Comparison of the delivery efficiencies to different organs in mice of a plasmid containing the luciferase gene mediated by linear PEIs, commercial linear (A) and hydrolytically pure linear PE125 (B). Only the mean values are shown. Challenge Mean virus titer in the lung Decrease in viral titer P value 5% glucose (control) 3.08 ± 0.4 ---- ---- PEI/GFP-siRNA 3.00 ± 0.0 2.6 % 0.64 PEI Alone 3.00 ±0.0 2.6% 0.64 PEI/NP-siRNA 1.86 ± 0.5 40% 0.002 Table 2.1. Inhibition of virus production in the lungs of influenza-infected mice by delivering influenza nucleoprotein siRNA mediated by linear, hydrolytically pure PEI of 87 kDa. Adapted from Thomas, 2005 22 -- M '--n ...... ..... ........ ... ... ........ . .... ... .. . ......................... 0 40 20 N/P 60 100 80 120 100 80 0 60 40 20 0 3 6 [PEI], pig/mi 9 12 Figure 2.2. Cytotoxicities induced by linear polyethylenimines as determined by the MTT cellular assay: commercial (red circles) and fully deacylated (green triangles) PEIs of 25 kDa, as well as hydrolytically pure PEIs of 22 kDa (yellow circles), 87 kDa (blue diamonds), and 217 kDa (pink squares) in A549 cells. Error bars are present for all data points but some may be too small to be seen. Additionally, although linear PEI of ~ 22-25 kDa has been shown to be a successful gene delivery agent, interest in lower molecular weight linear PEI abounds. Higher molecular weight 1 22 23 , , . This trend extends to linear polymers correlate with increased toxicity for branched PE71 PEI as high molecular weight fully hydrolyzed linear PEIs of 87 kDa and 217 kDa demonstrate elevated toxicity (relative to linear PEI of 22 kDa) in cell culture (Figure 2.2)22. As such, it would be preferable to employ a low molecular weight linear PEI for in vivo gene delivery studies if it is capable of mediating substantial gene expression or siRNA knockdown. In this chapter, we examine the in vivo dependence of the molecular weight of linear PEI as it pertains to nucleic acid delivery and mechanistically explore differences observed in an effort to identify the most efficient, non-toxic fully hydrolyzed linear PEI vector. B. Results and Discussion Low Molecular Weight Linear PEI Studies with branched PEI have demonstrated molecular weight to be a critical factor in the success of PEI mediated gene delivery both in vitro and in vivo. Polymers greater than 25 kDa exhibit toxicity while smaller polymers of 2 kDa fail to mediate gene expression. Herein we investigate what role polymer size plays for in vivo transfection with linear PEI and investigate the mechanism of said effect. Low molecular weight linear PEI of 2 kDa was synthesized from low molecular weight poly(2-ethyl-2-oxazoline) (PEOZ), as shown in Figure 2.3. The ability of low molecular weight linear PEI to deliver pDNA in vivo was examined in a mouse model. As seen in Figure 2.4, low molecular weight PEI does not mediate significant gene expression. Delivery in the lung is over 300-fold lower than with linear PEI of 22 kDa. To better understand the observed phenomenon, the ability of low molecular weight PEI to reach the desired target, the lung, was investigated. Using radiolabeled plasmid DNA, the localization of polyplexes (PEI/DNA complexes) following intravenous tail vein injection was determined. The tissue biodistribution was assayed at 15 minutes as PEI polyplexes are rapidly cleared from circulation (greater than 90% clearance in 15 min). As seen in Figure 2.5, the biodistribution profile of low molecular weight linear PEI is nearly identical to that of the higher molecular weight linear PEI of 22 kDa molecular weight. Total plasmid accumulation in the lung at 15 min for both 22 kDa and 2.5 kDa linear PEI are statistically indistinguishable. Given the similar biodistribution profiles, it is clear that uptake by the lung is not altered through the use of low molecular weight PEI. Therefore, the reduced efficiency of low molecular weight linear PEI is a result of a post-uptake step in the transfection process. Lower molecular weight linear PEI possesses fewer amines per polymer and therefore should not chelate to DNA with the same affinity as the higher molecular weight PEI. This could cause dissociation of the polymer and DNA in the cytoplasm leading to degradation of the plasmid and reduced expression and warrants additional investigation. Unfortunately, the low levels of expression observed with transfection by low molecular weight linear PEI prohibits use of this polymer for in vivo delivery. Therefore, the ability of linear PEI to mediate knockdown of an endogenous target was examined with linear PEI of 22 kDa. O0 n 24 % (wt/vol) HCI N 110 0C, 96 h Figure 2.3. Synthetic route for the preparation of linear PEI from poly(2-ethyl-2-oxazoline) (PEOZ). PEOZ is refluxed in HCl to cleave off propionyl groups via a standard acid catalyzed amide cleavage. n 120 0 4W CL 80 MINN 40 0Spleen Kidney Liver Organ Lung Heart Figure 2.4. Gene delivery of luciferase encoding pDNA with linear PEI of 22 kDa (grey) and 2.5 kDa (black) in mice as measured by luciferase expression. 70 tg of pDNA was delivered and animals were assayed at 24 h. Values shown are averages +/- standard deviation. RLU/10Os in yaxis label stands for relative light units of luciferase emission collected over a period of 10 seconds. 4 3 1 , 0 U 2 C 0 1 ! 0 - Spleen Kidney Liver Lung Heart Organ Figure 2.5. Biodistribution profile of 22 kDa (grey) and 2.5 kDa (black) linear PEI polyplexes in a mouse model. 70 tg of pDNA was delivered and animals were assayed at 24h. Values are presented as averages +/- standard deviation. In Vivo Knockdown of an Endogenous Target With the knowledge that linear PEI of 22-25 kDa is the most effective gene delivery agent in vivo in mice, we investigated its ability to effectively reduce endogenous protein levels by delivery of siRNA. To study this, we employed a ventilator induced lung injury (VILI) model. High tidal volume (HTV) ventilation (20 ml/kg) is known to cause damage to lungs. Specifically it results in acute inflammation and barrier regulation dysfunction. In short, the endothelial cells lining the blood vessels in the lung fail to regulate passage of proteins and cells from the blood into the lung tissue. This results in an increase in levels of protein and also total cell count. Recently, it was found that OxPAPC (oxidized 1-palmitoyl-2-arachidonoyl-snglycero-3-phosphocholine) exhibits potent barrier-protective effects for lung endothelial cells 24 , 26. Lungs injured by HTV ventilation poorly regulate transfer across the endothelium while injured lungs treated with OxPAPC demonstrate normal barrier properties. It has been suggested that these effects are mediated via caveolin enriched microdomains24 There exist a specialized subset of lipid rafts known as caveolin enriched microdomains that exist only in endothelial cells. These microdomains exhibit high levels of caveolin- 1 expression relative to other lipid rafts and have been implicated in endothelial cell barrier function and in interactions with the actin cytoskeleton24,25. As such, they play a critical role in determining what can and cannot cross the endothelial cell barrier in the lung, and in other highly vascularized tissues such as the heart and liver. It has been suggested that these caveolin- 1 enriched microdomains play a critical role in OxPAPC mediated lung protective effects. Herein we demonstrate in vivo knockdown of caveolin- 1 and assess the effect of this knockdown on OxPAPC mediated barrier protection. Anti-caveolin- 1 siRNA was delivered to the lungs of mice in complex with fully hydrolyzed linear PEI of 22 kDa via jugular vein injection in a dose response manner. As seen in Figure 2.5A, when 10 mg/kg of si-caveolin1 was delivered, caveolin-1 expression was nearly undetectable while a significant reduction in concentration was visible even at a dose of 6 mg/kg. Importantly, at a dose of 6 mg/kg, non-specific reduction of caveolin- 1 in the heart and liver were not observed. This demonstrates the first tissue specific in vivo knockdown of an endogenous target by fully hydrolyzed linear PEI. This is a critical requirement of any gene delivery vector that might ultimately be employed for gene therapy. Additionally, these results provide mechanistic information about gene delivery mediated by fully hydrolyzed linear PEI. To affect endothelial barrier protection in a caveolin-1 dependent manner, silencing of caveolin- 1 in endothelial cells must occur. This represents the first conclusive evidence of transfection of endothelial cells by fully hydrolyzed linear PEI. However, it is important to note that these results indicate only that endothelial cells are transfected and do not negate the possibility of transfection of additional cells types within the lung. This consideration warrants additional investigation. 6 mglkg 4 mglkg 4 1O mtlg 0\ 6mglkg 6mglkg Heart Uver o~ Caveoli Actin Lung B si-Cavolini nsRNA VILI OxPAPC + VIU 100% . VILI OxPAPC + VILI 103.1% 91.2% Figure 2.6. Depletion of pulmonary caveolin-1 expression impairs the protective effects of OxPAPC on ventilator induced lung injury (VILI). A) Linear PEI mediated lung specific transfection and depletion of caveolin-1 in dose-dependent manner as verified by Western blot2 7 . B) Effects of caveolin-1 depletion on the attenuation of lung vascular leak by OxPAPC. Evans blue dye (30 ml/kg) was injected into the external jugular vein 2 h before termination of ventilation to assess vascular leak. Lungs were harvested and imaged against a white background. Insets depict the quantitative measurement of Evans blue-labeled albumin extravasation in the shown lung preparation. Evans blue accumulation in the lungs from small nuclear RNA VILI animals was taken as 100% (n=4 per condition). Above we demonstrated siRNA mediated silencing of in vivo caveolin- 1 expression. However, for this knockdown to be relevant for gene therapy, it must also mediate a change in cellular function. In healthy lungs, jugular vein injection of dye-conjugated albumin does not result in accumulation of the dye in the lungs of the animal as barrier function prevents transfer across the endothelial cell barrier. However, VILI damages the ability of the lungs to effectively modulate the endothelial cell barrier and injected dye-conjugated albumin passively extravasates into the tissue. Upon visual inspection, healthy lungs appear mostly white while injured lungs are blue. This damage is repaired by administration of OxPAPC, in a caveolin- 1 specific manner. Therefore, to investigate the effect of knockdown of caveolin- 1, mice were treated with anti-caveolin- 1 siRNA and the ability of OxPAPC to restore barrier function in these animals was assessed. As seen in Figure 2.6B, caveolin-1 knockdown eliminates OxPAPC mediated barrier protection. When lung injury was induced by ventilation, OxPAPC repaired the barrier function in mice treated with non-specific (ns) siRNA whereas those treated with anti-caveolin- 1 siRNA failed to recover from lung injury. The observed differences in barrier function between the control and treatment groups represent a clear change in tissue function following knockdown of an endogenous target. The inset of Figure 2.6B shows the quantitative values for accumulation of dye. In conclusion, fully hydrolyzed linear PEI of ~22 kDa is the premier PEI vector for lung targeted gene delivery. In addition to mediating lung specific expression of plasmid DNA and reduction in viral titer, it safely silences endogenous protein expression in vivo in lung endothelial cells without altering protein expression in other highly vascularized tissues. C. Materials and Methods Materials. Nal 1 (sodium iodide) was purchased from Perkin Elmer. PEOZ of 5kDa was purchased from Polysciences. All other chemicals were from Sigma-Aldrich (St. Louis, MO) and were of the highest purity available and were used as received. 1H NMR spectra were recorded using a Bruker 400-MHz NMR spectrometer with chemical shifts expressed with reference to the chloroform peak in CDCl 3 (7.24 ppm). Plasmid and its iodination. gWiz Luc encoding the firefly luciferase gene was purchased from Aldevron (Fargo, ND). This ready-to-use plasmid, containing the luciferase gene under the control of a modified promoter from the cytomegalovirus immediate early gene, was obtained as a 5.0 mg/ml stock solution in water. Iodination of the plasmid was achieved using a modified version of previously described methods 28,29. Briefly, two iodobeads from Thermo Scientific (Rockford, IL) were incubated with 30 1dof 0.35 M sodium acetate buffer, pH 4.0, and 400 pmol of NaI12 5 (containing 1 mCi of radiation) at 50 *C for 15 min. To that, 100 tg of gWiz Luc (in 20 tl) was added and incubated for an additional 30 min. The iodinated plasmid was purified by sequential desalting on Minitrap and Midi-trap desalting columns (GE Healthcare). Linear PEI synthesis. Fully deacylated linear PEI was synthesized from commercial 50 kDa and 5 kDa poly(2-ethyl-2-oxazolines) (PEOZs) as previously described 2 2 . Briefly, 3.0 g of the PEOZ was added to 120 ml of 24% (w/v) HCl, followed by refluxing for 96 h. The PEOZ crystals dissolved completely in 2 h, and a white precipitate appeared 3 h later. The solution was adjusted to pH 10 with 10 M NaOH and the precipitate, isolated by vacuum filtration, was washed with cold water and lyophilized to obtain the desired product as the free base. The resultant white powder was confirmed by NMR at the MIT DCIF to be pure PEI base through the disappearance of the -CH 3 and -CH 2 peaks. Gene delivery in mice via tail vein injection. All animal experiments conducted in this study adhered to the Principles of Laboratory Animal Care (National Institutes of Health publication no. 85-23, revised in 1985). To obtain the desired N/P ratios (those of PEI nitrogen to DNA phosphate), appropriate volumes of PEI stock solutions were diluted to 500 [d in 5% aqueous glucose and added to an equal volume of the glucose solutions containing 350 [tg of the plasmid DNA (gWiz Luc), followed by pipette mixing. The resulting polyplexes were incubated at room temperature for 10 min. Then 6- to 8-week-old Swiss Webster female mice (Taconic Farms) were injected intravenously via tail vein with 200 1dof the polyplexes containing 70 tg of DNA. After 24 h, the mice were euthanized by CO 2 inhalation; their lungs, kidneys, livers, hearts, and spleens were collected, washed with PBS, and suspended in lysis buffer prepared by mixing 4 ml of 5x passive lysis buffer (Promega), 800 tl of 8.7 mg/ml phenylmethylsulfonyl fluoride (PMSF) in methanol, 400 [l of protease inhibitor mixture, and 14.8 ml of water. The samples were freeze-thawed, homogenized by probe-sonication for 40 sec in 20 sec increments at 8W, and centrifuged 22 . Then 10 tl of the supernatants was mixed with 100 d of the luciferase assay reagent (Promega), and the luminescence was measured using an Optocomp I luminometer (MGM Instruments, Hamden, CT). Protein concentrations were determined using the bicinchoninic acid (BCA) assay with a BSA standard, and the results were expressed as mean SD (n = 4). Mouse perfusion and radiation measurements. Polyplexes were prepared as described in the previous section. 1 [tCi of plasmid DNA was included with the unlabeled DNA to obtain the desired dose of nucleic acid. At 5, 10, or 15 min or 24 h, mice were anesthetized with a lethal dose of Avertin, the vena cava was cut, a blood sample was collected, and the animals were perfused with PBS using a peristaltic pump at a flow rate of about 20 ml/min for 5-10 min. Once the perfusion ran clear, the organs were dissected and assayed for gamma counts using a 5min read time. The results were expressed as mean - SD (n = 4). Jugular vein delivery of small interfering RNA in mice. Adult male C57BL/6J mice, 8-10 weeks old, with average weight 20-25 grams (Jackson Laboratories, Bar Harbor, ME) were bred at the University of Chicago animal care center. siRNAs from Dharmacon (Lafayette, CO) had the following sequences - siCaveolinl: 5'-ACGUAGACUCCGAGGGACAUU-3'; control siRNA (Luciferase): 5'- UAAGGCUAUGAAGAGAUA-3'. Fully hydrolyzed linear PEI of 22- kDa was used as a vector for siRNA-induced caveolin knockdown. PEI/siRNA were complexed at an N/P ratio of 10 (ratio of monomers of PEI to monomers of siRNA). Required amounts of PEI were brought to 200 1din a final concentration of 5% aqueous glucose and added to the equal volume of the glucose solution containing amounts of siRNA to reach 4 mg/kg, 6 mg/kg and 10 mg/kg. The resulting polyplexes were incubated at room temperature for 10 min. Obtained PEI-siRNA polyplexes (400 pl) were injected into jugular vein of 8- to 10-week-old C57BL/6 male mice under anesthesia. After 72 h, the mice were sacrificed; their lungs, livers and hearts were collected and homogenized in 1 ml of SDS lysis buffer containing protease inhibitor cocktail set III (Calbiochem, NJ). Western blot. Following treatment of animals as described above, lung were collected, washed in cold PBS and frozen. Lungs were then homogenized in buffer containing T-PER tissue protein extraction reagent (Prod #78510, Thermo Scientific), IX protease inhibitor cocktail (P8340, Sigma), and IX phosphatase inhibitor cocktails 1 & 2 (P2850/P5726, Sigma) according to manufacturer's instructions. Per left lung, 1 ml of buffer was used. Following homogenization, samples were centrifuged at 10,000 rpm for 20 min at 4 *C and supernatants were collected. Protein concentration was determined by BCA Protein Assay (#23223, Thermo Scientific) according to manufacturer's instruction with a BSA standard. All samples were then adjusted to a protein concentration of 2 mg/ml. Each protein sample was diluted 2X in sample buffer to a volume of 100 1tl, boiled for 3 min, and electrophoresed via SDS PAGE. Per well, 20 tg of protein was loaded. Following electrophoresis, blotting was performed by standard methods using 5% milk in TBST (50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 0.1% Tween-20), with a mouse or rabbit anti-caveolin-1 primary antibody and HRP-linked anti-mouse or anti-rabbit secondary antibody from Cell Signaling. HRP was visualized by standard methods following blotting. Actin levels in all samples were quantified as a control in an identical manner but with anti-actin primary antibody. Mechanical ventilation of mice. After 72 h of siRNA delivery in mice with siCaveolin or nsRNA at dose of 6 mg/kg, mice were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and acepromazine (1.5 mg/kg). A tracheotomy was performed and the trachea was cannulated with a 20-gauge one-inch catheter (Penn-Century Inc., Philadelphia, PA), which was tied into place to prevent air leak. The animals were placed on mechanical ventilator (Harvard Apparatus, Boston, MA) for 4 hours with high tidal volume (30 ml/kg, 75 breaths per minute and 0 PEEP, HTV) ventilation. Mice were randomized to concurrently receive sterile saline solution or OxPAPC (1.5 mg/kg, i.v. via jugular vein) to yield 4 groups: nsRNA VILI, nsRNA VILI + OxPAPC, siRNA VILI, and siRNA VILI + OxPAPC. After the experiment, animals were sacrificed by exsanguination under anesthesia. Assessment of pulmonary vascular leakage by Evans blue. Two hours prior to the termination of HTV, Evans blue was injected intravenously at a dose of 30 mg/kg. At the end of ventilation, thoracotomy was performed, and the lungs were perfused free of blood with PBS containing 5 mM EDTA. Both left lung and right lung were excised and imaged by Kodak digital camera. After imaging, lungs were blotted dry, weighed and homogenized in PBS (1 ml/1 00 pg tissue). Homogenized tissue was incubated with 2 volumes of formamide (18 h, 60 0 C), centrifuged at 12,000 g for 20 min. Optical density of the supernatant was determined by spectrophotometry at 620 nm and 740 nm. The concentration of extravasated Evans blue dye (EBD) (micrograms of EBD per gram lung) in lung homogenates was calculated using a standard curve of EBD. The mean value of nsRNA VILI group was considered as 100% injury; injury to the other three groups were calculated as the ratio of the mean value to the mean value of nsRNA VILI group. D. References 1. Aigner, A., Gene silencing through RNA interference (RNAi) in vivo: strategies based on the direct application of siRNAs. JBiotechnol 2006, 124, (1), 12-25. 2. Davis, M. E., Non-viral gene delivery systems. Curr Opin Biotechnol 2002, 13, (2), 128- 31. 3. Kim, S. S.; Garg, H.; Joshi, A.; Manjunath, N., Strategies for targeted nonviral delivery of siRNAs in vivo. Trends Mol Med 2009, 15, (11), 491-500. 4. Ledley, F. D., Nonviral gene therapy: the promise of genes as pharmaceutical products. Hum Gene Ther 1995, 6, (9), 1129-44. 5. Jaroff, L., Fixing the genes. Time 1999, 153, (1), 68-70, 73. 6. Flotte, T. R., Gene therapy: the first two decades and the current state-of-the-art. J Cell Physiol 2007, 213, (2), 301-5. 7. Kaiser, J., Gene therapy. Seeking the cause of induced leukemias in X-SCID trial. Science 2003, 299, (5606), 495. 8. Marshall, E., Gene therapy death prompts review of adenovirus vector. Science 1999, 286, (5448), 2244-5. 9. Roth, J. A.; Cristiano, R. J., Gene therapy for cancer: what have we done and where are we going? JNatl CancerInst 1997, 89, (1), 21-39. 10. Eliyahu, H.; Barenholz, Y.; Domb, A. J., Polymers for DNA delivery. Molecules 2005, 10, (1), 34-64. 11. Lollo, C. P.; Banaszczyk, M. G.; Chiou, H. C., Obstacles and advances in non-viral gene delivery. Curr Opin Mol Ther 2000, 2, (2), 136-42. 12. Boussif, 0.; Lezoualc'h, F.; Zanta, M. A.; Mergny, M. D.; Scherman, D.; Demeneix, B.; Behr, J. P., A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 1995, 92, (16), 7297-301. 13. Bologna, J. C.; Dom, G.; Natt, F.; Weiler, J., Linear polyethylenimine as a tool for comparative studies of antisense and short double-stranded RNA oligonucleotides. Nucleosides Nucleotides Nucleic Acids 2003, 22, (5-8), 1729-31. 14. Breunig, M.; Lungwitz, U.; Liebl, R.; Fontanari, C.; Klar, J.; Kurtz, A.; Blunk, T.; Goepferich, A., Gene delivery with low molecular weight linear polyethylenimines. J Gene Med 2005, 7, (10), 1287-98. 15. Godbey, W. T.; Barry, M. A.; Saggau, P.; Wu, K. K.; Mikos, A. G., Poly(ethylenimine)- mediated transfection: a new paradigm for gene delivery. J Biomed Mater Res 2000, 51, (3), 321-8. 16. Kircheis, R.; Schuller, S.; Brunner, S.; Ogris, M.; Heider, K. H.; Zauner, W.; Wagner, E., Polycation-based DNA complexes for tumor-targeted gene delivery in vivo. J Gene Med 1999, 1, (2), 111-20. 17. Thomas, M.; Ge, Q.; Lu, J. J.; Chen, J.; Klibanov, A. M., Cross-linked small polyethylenimines: while still nontoxic, deliver DNA efficiently to mammalian cells in vitro and in vivo. PharmRes 2005, 22, (3), 373-80. 18. Goetz, A. S.; Andrews, J. L.; Littleton, T. R.; Ignar, D. M., Development of a facile method for high throughput screening with reporter gene assays. JBiomol Screen 2000, 5, (5), 377-84. 19. Aigner, A., Cellular delivery in vivo of siRNA-based therapeutics. Curr Pharm Des 2008, 14, (34), 3603-19. 20. Breunig, M.; Lungwitz, U.; Liebl, R.; Klar, J.; Obermayer, B.; Blunk, T.; Goepferich, A., Mechanistic insights into linear polyethylenimine-mediated gene transfer. Biochim Biophys A cta 2007, 1770, (2), 196-205. 21. Jeong, G. J.; Byun, H. M.; Kim, J. M.; Yoon, H.; Choi, H. G.; Kim, W. K.; Kim, S. J.; Oh, Y. K., Biodistribution and tissue expression kinetics of plasmid DNA complexed with polyethylenimines of different molecular weight and structure. J Control Release 2007, 118, (1), 118-25. 22. Thomas, M.; Lu, J. J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A. M., Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci U S A 2005, 102, (16), 5679-84. 23. Thomas, M.; Klibanov, A. M., Enhancing polyethylenimine's delivery of plasmid DNA into mammalian cells. Proc Nad Acad Sci USA 2002, 99, (23), 14640-5. 24. Birukova, A. A.; Malyukova, I.; Mikaelyan, A.; Fu, P.; Birukov, K. G., TiamI and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol 2007, 211, (3), 608-17. 25. Birukova, A. A.; Malyukova, I.; Poroyko, V.; Birukov, K. G., Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am JPhysiolLung Cell Mol Physiol 2007, 293, (1), L199-211. 26. Nonas, S.; Birukova, A. A.; Fu, P.; Xing, J.; Chatchavalvanich, S.; Bochkov, V. N.; Leitinger, N.; Garcia, J. G.; Birukov, K. G., Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care 2008, 12, (1), R27. 27. Singleton, P. A.; Chatchavalvanich, S.; Fu, P.; Xing, J.; Birukova, A. A.; Fortune, J. A.; Klibanov, A. M.; Garcia, J. G. N.; Birukov, K. G., Akt-Mediated Transactivation of the SIPI Receptor in Caveolin-Enriched Microdomains Regulates Endothelial Barrier Enhancement by Oxidized Phospholipids. Circ Res 2009, 104, (8), 978-986. 28. Markwell, M. A., A new solid-state reagent to iodinate proteins. I. Conditions for the efficient labeling of antiserum. Anal Biochem 1982, 125, (2), 427-32. 29. Piatyszek, M. A.; Jarmolowski, A.; Augustyniak, J., Iodo-Gen-mediated radioiodination of nucleic acids. Anal Biochem 1988, 172, (2), 356-9. III. SPECIFICITY OF GENE DELIVERY IN VIVO MEDIATED BY POLYETHYLENIMINE CONJUGATED TO AN ANTI-ACE ANTIBODY A. Introduction Linear polyethylenimine (PEI) is one of the premier polycationic nucleic acid delivery agents. Studies in our lab and others have demonstrated successful gene and siRNA delivery mediated by linear PEI in vivo in mice and to a diverse array of cells in vitro 6 . Specificity of in vivo delivery is highly desirable as it allows for reduced dosages of polyplexes and eliminates toxicity associated with high dosing. Many labs have worked to enhance the efficacy of PEImediated gene delivery3 , 4,7-3 but none have demonstrated modulation of the specificity of fully hydrolyzed linear PEI in an in vivo model. The use of antibodies to mediate interaction between a compound and a cell with which it would not otherwise interact is well documented 14 . As early as the 1980s, antibodies were used to mediate binding of red blood cells to collagen-coated surfaces. More recently, antibodies against cancer cell markers have been employed in directed therapies in an effort to enhance drug efficacy while reducing dose related toxicity16 ' 17. Additionally, conjugation of antibodies to gene therapy vectors for targeting of nucleic acids has been reported 8 '19. Myriad studies have demonstrated enhanced in vitro gene delivery to a diverse set of cells using appropriate antibody conjugates 20 ,21. Cell specific enhancement of up to 3-fold has been shown by attachment of mAb G250 to branched PEI of 25 kDa while similar results have been demonstrated by others using antibodies targeted to different cell lines. However, the in vivo use of antibody targeting to alter the specificity of unmodified linear PEI-based nucleic acid delivery remains unstudied and is of great interest. Angiotensin-converting enzyme (ACE) is a membrane-anchored glycoprotein expressed on the luminal surface of endothelial cells with preferential expression in the lung 23, 24 . As such, ACE is an optimal target for directed lung therapies. Recently a monoclonal antibody (mAb) (designated 9B9) that is cross reactive to human and rat ACE was developed. Following intravenous injection in rats, all lung endothelial cells demonstrate binding of 9B9 while only 20% of endothelial cells in the spleen interact substantially with the anti-ACE mAb and binding by all other organs occurs in less than 10% of endothelial cells 25 . Herein we examine if conjugation of 9B9 to fully hydrolyzed, unmodified linear PEI enhances delivery of polyplexes to the lung following intravenous tail vein injection in rats. B. Results and Discussion Use of linear PEI in rats has been limited to intratracheal instillations for specific lung delivery. Therefore, we first investigated the ability of linear PEI to effect gene expression following intravenous delivery in Sprague Dawley rats. The tissue expression profile and transfection efficiency of linear PEI in rats is shown in Figure 3.1. Expression across most organs is low with greatest transgene expression observed in the spleen. Polyplexes formulated with linear PEI enhance pDNA expression in the spleen 10-fold over DNA alone while maintaining at least a 5-fold greater expression than in other organs. 6000 0 4000 0 0, E 0n 2000 1 OJ SL- Spleen Kidney Liver Lung Heart Tissue Figure 3.1. Transfection efficiency and specificity of linear PEI as measured by luciferase gene expression in vivo in rats for no treatment (grey), DNA only (black), and polyplexes with linear PEI 22 kDa (white). A dose of 1.7 tg pDNA / g rat was delivered intravenously via tail vein injection at an N/P ratio of 8 and expression was assessed at 24 h. Values presented as averages with standard deviation (n=4). To investigate the ability of 9B9 to mediate enhanced lung expression and specificity of PEI-delivered pDNA, antibody-PEI conjugates were synthesized. Linear PEI contains only secondary amines that cannot be selectively modified. Therefore, to prevent conjugation of more than one antibody to an individual polymer chain, polymers were first modified with a single functional group through which antibody conjugation would occur. An excess of polymer was reacted with antibody as described in the methods section and as depicted in Figure 3.2. Conjugates were formulated at a 1.3:1 polymer to antibody ratio as determined by BCA antibody quantification and TNBS polymer quantification (data not shown). This ratio was selected to prevent interference of the polymer with the ability of the antibody to bind its target, ACE. Following complexation, PEI conjugated 9B9 was evaluated for retention of ACE binding functionality as described in the Methods. As seen in Figure 3.3, 9B9 retains significant affinity for ACE even in conjugation with PEI. Conjugates of 9B9 and PEI, designated PEI-9B9, were examined for the ability to enhance gene expression in the lung. A non-specific mouse IgGiK isotype antibody was separately linked to PEI as a control (PEI-Ab). As shown in Figure 3.4, attachment of 9B9 to linear PEI did not affect PEI-mediated gene delivery. Enhanced lung expression and specificity were not observed when PEI-9B9 was employed as the gene delivery vector. Expression in the spleen remained at least 5-fold greater than that observed in the lung and PEI-9B9 mediated lung expression was not statistically distinguishable from that of PEI-Ab or PEI alone. () H Antibody P n A H B (ii) -m OH SH OH a-N H 0 0 H N N C 0 A + C NS PEI B + HS 0 0 NN (i) N- H N0 H N SS O D SH S 0 O H NN + N O Figure 3.2. Schematic representation of the synthetic route for attachment of an antibody to PEI. Both antibody and polymer were modified by SPDP and then a disulfide exchange was used to link the antibody to the polymer. (i) Sulfo-LC-SPDP, PBS pH 7.4, 60 min at RT; (ii) DTT, PBS pH 7.4, 30 min, RT; (iii) PBS pH 7.4, 24 h at RT; A PEI-SPDP; D = PEI-Ab conjugate at ratio of 1.3 to 1. = PEI-SPDP; B = Ab-SPDP; C = reduced ~50 >40 30 m 20 00 1 0 i10 a. 0 0 8 6 4 2 Concentration of Monoclonal Antibody, ug/mi 10 Figure 3.3. Binding affinity of 9B9 to angiotensin I-converting enzyme (ACE) both as a free antibody (triangles) and when conjugated to linear PEI (diamonds). standard deviation and are present but may be too small to see. Error bars represent 10000 L. 7500 T 0. 0 0) 5000 E -. 2500 0 - Spleen --- - Kidney Liver Lung Heart Tissue Figure 3.4. Transfection efficiency and specificity of linear PEI as measured by luciferase gene expression in vivo in rats for delivery mediated by PEI alone (grey), PEI-Ab (black), and PEI-9B9 (white). A dose of 1.7 tg pDNA / g rat (30 [tg of antibody) was delivered intravenously via tail vein injection at an N/P ratio of 8 and expression was assessed at 24 h. Values presented as average with standard deviation (n=4). Given the ability of PEI-conjugated 9B9 to bind ACE, the formulation of the polyplexes or the pharmacokinetics of gene delivery by PEI prevent antibody targeting of linear PEI polyplexes in vivo. It is possible that during formulation, condensation of DNA by 9B9-PEI leads to burying of 9B9 so it is not surface exposed. This would prevent 9B9 from binding to ACE following injection. To reduce the likelihood of such burying, the amount of free PEI (no 9B9 conjugation) in the formulation was reduced by decreasing the amount of DNA delivered 8.5-fold and the increasing the amount of 9B9 included in the formulation from 30 tg to 75 rig. As seen in Figure 3.5, lower concentrations of free PEI and greater amounts of 9B9 did not lead to enhancement of lung delivery by polyplexes formulated with PEI-9B9 relative to PEI alone. Overall gene expression is dramatically reduced in the spleen due to the lower dose of pDNA, but conjugation to 9B9 does not have an effect on the efficacy or specificity of delivery. These data suggest that it is likely pharmacokinetics that prevent 9B9 from mediating enhanced expression. Interaction of the polyplexes with components of the circulatory system might shield 9B9, preventing binding to ACE. Alternatively (or possibly additionally) polyplexes formulated with PEI are cleared from circulation within 5-10 min while binding of 9B9 to ACE is optimal at 30-40 min. This reduced exposure time might limit the binding of 9B9 to ACE and thus the enhancement in specificity. Although the use of covalent attachment of antibodies for targeting of gene delivery vectors has demonstrated in vitro success, its applicability to in vivo delivery by linear PEI is greatly limited by the physiology and pharmacokinetics of in vivo systems. Alternative chemical modifications should be explored for their ability to alter the specificity and enhance the efficacy of gene delivery by linear PEI. 400 C '300 . 0 1200 0 0 ft.ft100 -J 0 Spleen Lung Tissue Figure 3.5. Transfection efficiency and specificity of linear PEI as measured by luciferase gene expression in vivo in rats for delivery mediated by PEI alone (black) and PEI-9B9 (grey). A doses of 0.2 tg pDNA / g rat containing 75 tg 9B9 were delivered intravenously via tail vein injection at an N/P ratio of approximately 7.5 and transgene expression was assessed at 24 h. Values presented as average with standard deviation (n=4). C. Materials and Methods Materials. IgGiK isotype control antibody was purchased from Biolegend (San Diego, CA). mAb 9B9 antibody was generously provided by Dr. Sergei Danilov (University of Illinois, Chicago). LC-Sulfo-SPDP was purchased from Thermo Scientific (Rockford, IL). All other reagents were purchased from Sigma Aldrich and were of the highest purity possible and were used as received. Synthesis of linear PEI. Fully deacylated linear PEI was synthesized as described in Chapter 2 of this thesis. Synthesis of SPDP modified PEI (PEI-SPDP). Linear PEI was modified with SPDP according to the manufacturers instructions. Briefly, 6.1 mg of 22 kDa linear PEI (0.278 tmol) was buffer exchanged into PBS-EDTA buffer (100 mM sodium phosphate, 150 mM NaCl, 1 mM EDTA, 0.02% sodium azide) with a final volume of 971 l. Sulfo-LC-SPDP was prepared at 20 mM in water. Immediately following preparation, 0.58 tmol was added to the PEI solution and mixed well. The reaction was allowed to proceed at room temperature for 60 min. PEI-SPDP was then purified on a pre-equilibrated desalting column (GE Healthcare, PD 10 desalting column #17-0851-01). The entire 1 ml sample was loaded on the column followed by 1.5 ml of PBS-EDTA buffer. The sample was eluted with 3 ml of PBS-EDTA and concentrated to approximately 400 [d using a Millipore concentrator (Amicon Ultra 3kDa MWCO membrane #UFC900324). Characterization of PEI-SPDP. Purified PEI-SPDP was assayed for both SPDP and PEI content. SPDP content was assessed according the manufacturers protocol. Briefly, an appropriate dilution of the sample was prepared in 250 tl and reduced with 10 tl of 15 mg/ml dithiothreitol (DTT) at room temperature for 15 minutes. The sample was then read against an appropriate blank at 343 nm to determine the amount of liberated pyridine-2-thione. The amount of incorporated SPDP was back calculated using the extinction coefficient of pyridine-2-thione (8080 M-1 cm-1) assuming 100% conversion. The concentration of PEI was determined using a TNBS assay as previously described2 6 Briefly, the sample containing PEI was diluted appropriately in 100 mM sodium borate buffer, pH 9.3. Then 100 [d of each sample and a standard curve of PEI polymer (from stocks of known concentration) were loaded into a 96 well plate. To each sample was added 0.31 [tmol of TNBS in water to a final volume of 102.5 1d.Samples were incubated at room temperature for 30 min and the A420nm was read. The concentration of PEI in the samples was determined from the standard curve in the range of 0 to 56 [tg/ml. Synthesis of SPDP modified antibody (Ab-SPDP). Anti-ACE monoclonal antibody 9B9 and IgGiK isotype antibody were modified with SPDP according to the manufacturers instructions. Briefly, 1.5 mg of antibody (10 nmol) was buffer exchanged in PBS-EDTA, pH 7.5, with a final volume of 975 [d. Sulfo-LC-SPDP was prepared at 20 mM in water. Immediately following preparation, 0.5 [tmol of SPDP was added to the antibody solution and mixed well. The reaction was allowed to proceed at room temperature for 60 min. Ab-SPDP was purified like PEI-SPDP above. It was concentrated to approximately 400 [d using a Millipore concentrator (Amicon Ultra 50kDa MWCO membrane #UFC905024). Characterization of Ab-SPDP. The SPDP content of Ab-SPDP was determined according to the manufacturer's protocol as described above for PEI-SPDP. Concentration of antibody was determined from the extinction coefficient of the antibody at 280 nm (1.46 mg-1 cm-I ml). Conjugation of antibody to PEI (PEI-Ab). To 0.278 [mol of SPDP modified PEI was added 23 mg/ml DTT at a v/v ratio of 2:1. The sample was allowed to react for 30 min at room temperature before free DTT was removed on a pre-equilibrated desalting column (GE Healthcare, PD10 Desalting Column). Specifically, the sample was loaded on the column in a volume of less than 1.25 ml and PBS-EDTA was added to a final volume of 2.5 ml. Reduced PEI was then eluted with 3 ml of PBS-EDTA. The sample was concentrated to less than 1 ml and purified on a second pre-equilibrated desalting column. Eluted reduced PEI-SPDP was then added immediately to 10 nmol of Ab-SPDP and allowed to react at room temperature for 24h. After incubation, free PEI-SPDP was removed by extensive washings through an Amicon Ultra concentrator with a 50 kDa MWCO membrane (Millipore, Billerica, MA). Characterization of PEI-Ab. PEI-Ab was characterized by FPLC to confirm that all antibody was modified by PEI (recall that all free polymer was removed in prior purification steps). PEI-Ab was loaded on to a weak cation exchange column (Pall CM Ceramic Hyper D F AcroSep Chromatography Column) in PBS buffer, pH 7.5 at a flow rate of 0.2 ml/min in a total volume of 2 ml. The column was then washed with 5 ml of the same buffer and subsequently washed with 10 ml of PBS pH 7.5 with 1 M NaCl to remove free antibody bound to the column. Finally the column was washed with 10 ml of 1 M HCl to elute any remaining compounds bound to the column. Under these conditions, PEI binds nearly irreversibly to the column and cannot be eluted with anything less than 1 M HCl. On the contrary, free antibody, with a pI of 9-10, binds to the column at pH 7.5 but is easily eluted with 1 M NaCl. Finally, 1 M HCl is required to elute PEI-Ab conjugates. This protocol was used to confirm purity of PEI-Ab conjugates following each synthesis. A pure batch resulted in no elution with 1 M NaCl with all the bound sample eluting in the final acid wash. Recall, this method was used only to confirm purity, not to purify the conjugates as an acid wash destroys the conjugates. Conditions were optimized to create complexes that did not require purification. Following confirmation of purity, PEI-Ab was characterized to determine the ratio of polymer to antibody in the conjugate using the methods described above. Briefly, the antibody concentration was assessed using its absorbance at 280 nm and its extinction coefficient as PEI does not absorb at 280 nm. PEI concentration was assessed using the TNBS assay with a free antibody control. In all cases the ratio of PEI to antibody was nearly 1.3:1. Antigen-binding activity of mAb 9B9. (Plate immunoprecipitation assay). 96-well plates (Coming, Coming, NY) were coated with 50 pl of 10 ptg/ml affinity-purified goat antimouse IgG (Pierce, Rockford, IL) and stored overnight at 4'C. After washing with PBS/0.05% Tween 20, the wells were incubated with anti-ACE mAb 9B9 or it is conjugate with PEI (2 jig/ml) in PBS/BSA (0.1 mg/ml) for 2 h at RT and washed. Wells were then incubated with ACE. After washing of unbound ACE, plate-bound ACE activity was measured by adding a substrate for ACE (Hip-His-Leu or Z-Phe-His-Leu) directly into wells as described previously2 7 . Briefly, cleavage of the His-Leu peptide bond creates a primary amine that reacts with fluorescamine or ortho-phthalaldehyde (OPA) to produce a fluorescent product. The fluorescence is measured at ex340/em455 for OPA and ex390/em465 for fluorescamine. Gene delivery in rats. All animal experiments conducted in this study adhered to the Principles of Laboratory Animal Care (National Institutes of Health publication no. 85-23, revised in 1985). First, the desired amount of antibody was taken as an aliquot of the PEI-Ab solution. The amount of PEI in this sample was calculated and to obtain the desired N/P ratio, (that of PEI nitrogen to DNA phosphate), an appropriate volume of linear PEI stock solution was added to the PEI-Ab aliquot and diluted to 480 pl in 5% aqueous glucose. The PEI sample was then added to an equal volume of the glucose solution containing the desired amount of the plasmid DNA (gWiz Luc), followed by pipette mixing. The resulting polyplexes were incubated at room temperature for 10 min. Then 6- to 10-week-old Sprague Dawley female rats (Taconic Farms) were injected intravenously via tail vein with 800 tl of the polyplexes while anesthetized with isoflorane. After 24 h, the rats were euthanized by CO 2 inhalation; their lungs, kidneys, livers, hearts, and spleens were collected, washed with PBS, and suspended in lysis buffer prepared by mixing 4. ml of 5X passive lysis buffer (Promega), 800 tl of 8.7 mg/ml phenylmethylsulfonyl fluoride (PMSF) in methanol, 400 tl of protease inhibitor mixture (sigma), and 14.8 ml of water. The samples were freeze-thawed, homogenized by probe-sonication at 8 W for 30 sec, and centrifuged at 4000 RPM. Then 10 [d of the supernatants was mixed with 100 1dof the luciferase assay reagent (Promega), and the luminescence was measured using an Optocomp I luminometer (MGM Instruments, Hamden, CT). Protein concentrations were determined using the bicinchoninic acid (BCA) assay with a BSA standard, and the results were expressed as mean - SD (n = 4). D. References 1. Brissault, B.; Leborgne, C.; Guis, C.; Danos, 0.; Cheradame, H.; Kichler, A., Linear topology confers in vivo gene transfer activity to polyethylenimines. Bioconjug Chem 2006, 17, (3), 759-65. 2. Reed, S. E.; Staley, E. M.; Mayginnes, J. P.; Pintel, D. J.; Tullis, G. E., Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. J Virol Methods 2006, 138, (1-2), 85-98. 3. Thomas, M.; Lu, J. J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A. M., Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. ProcNatl Acad Sci U S A 2005, 102, (16), 5679-84. 4. Thomas, M.; Lu, J. J.; Zhang, C.; Chen, J.; Klibanov, A. M., Identification of novel superior polycationic vectors for gene delivery by high-throughput synthesis and screening of a combinatorial library. Pharm Res 2007, 24, (8), 1564-71. 5. Wightman, L.; Kircheis, R.; Rossler, V.; Carotta, S.; Ruzicka, R.; Kursa, M.; Wagner, E., Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo. J Gene Med 2001, 3, (4), 362-72. 6. Zaric, V.; Weltin, D.; Erbacher, P.; Remy, J. S.; Behr, J. P.; Stephan, D., Effective polyethylenimine-mediated gene transfer into human endothelial cells. J Gene Med 2004, 6, (2), 176-84. 7. Gebhart, C. L.; Kabanov, A. V., Evaluation of polyplexes as gene transfer agents. J Control Release 2001, 73, (2-3), 401-16. 8. Kleemann, E.; Dailey, L. A.; Abdelhady, H. G.; Gessler, T.; Schmehl, T.; Roberts, C. J.; Davies, M. C.; Seeger, W.; Kissel, T., Modified polyethylenimines as non-viral gene delivery systems for aerosol gene therapy: investigations of the complex structure and stability during airjet and ultrasonic nebulization. J Control Release 2004, 100, (3), 437-50. 9. Kunath, K.; von Harpe, A.; Fischer, D.; Petersen, H.; Bickel, U.; Voigt, K.; Kissel, T., Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecularweight polyethylenimine. J Control Release 2003, 89, (1), 113-25. 10. Lungwitz, U.; Breunig, M.; Blunk, T.; Gopferich, A., Polyethylenimine-based non-viral gene delivery systems. Eur JPharmBiopharm 2005, 60, (2), 247-66. 11. Mennesson, E.; Erbacher, P.; Piller, V.; Kieda, C.; Midoux, P.; Pichon, C., Transfection efficiency and uptake process of polyplexes in human lung endothelial cells: a comparative study in non-polarized and polarized cells. J Gene Med 2005, 7, (6), 729-38. 12. Neu, M.; Fischer, D.; Kissel, T., Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med 2005, 7, (8), 992-1009. 13. Neu, M.; Germershaus, 0.; Mao, S.; Voigt, K. H.; Behe, M.; Kissel, T., Crosslinked nanocarriers based upon poly(ethylene imine) for systemic plasmid delivery: in vitro characterization and in vivo studies in mice. J Control Release 2007, 118, (3), 370-80. 14. Abeloff, M. D.; Armitage, J. 0.; Niederhuber, J. E.; Kastan, M. B.; McKenna, W. G., Clinical Oncology. Churchill Livingstone: Philadelphia, PA, 2008. 15. Samokhin, G. P.; Smirnov, M. D.; Muzykantov, V. R.; Domogatsky, S. P.; Smirnov, V. N., Effect of flow rate and blood cellular elements on the efficiency of red blood cell targeting to collagen-coated surfaces. JAppi Biochem 1984, 6, (1-2), 70-5. 16. Barth, B. M.; Sharma, R.; Altinoglu, E. I.; Morgan, T. T.; Shanmugavelandy, S. S.; Kaiser, J. M.; McGovern, C.; Matters, G. L.; Smith, J. P.; Kester, M.; Adair, J. H., Bioconjugation of calcium phosphosilicate composite nanoparticles for selective targeting of human breast and pancreatic cancers in vivo. ACS Nano 2010, 4, (3), 1279-87. 17. Schliemann, C.; Neri, D., Antibody-based vascular tumor targeting. Recent Results Cancer Res 2010, 180, 201-16. 18. Froelich, S.; Ziegler, L.; Stroup, K.; Wang, P., Targeted gene delivery to CD 117- expressing cells in vivo with lentiviral vectors co-displaying stem cell factor and a fusogenic molecule. BiotechnolBioeng 2009, 104, (1), 206-15. 19. Rivest, V.; Phivilay, A.; Julien, C.; Belanger, S.; Tremblay, C.; Emond, V.; Calon, F., Novel liposomal formulation for targeted gene delivery. Pharm Res 2007, 24, (5), 981-90. 20. Trubetskoy, V. S.; Torchilin, V. P.; Kennel, S. J.; Huang, L., Use of N-terminal modified poly(L-lysine)-antibody conjugate as a carrier for targeted gene delivery in mouse lung endothelial cells. Bioconjug Chem 1992, 3, (4), 323-7. 21. Suh, W.; Chung, J. K.; Park, S. H.; Kim, S. W., Anti-JL1 antibody-conjugated poly (L- lysine) for targeted gene delivery to leukemia T cells. J Control Release 2001, 72, (1-3), 171-8. 22. Duan, Y.; Zheng, J.; Han, S.; Wu, Y.; Wang, Y.; Li, D.; Kong, D.; Yu, Y., A tumor targeted gene vector modified with G250 monoclonal antibody for gene therapy. J Control Release 2008, 127, (2), 173-9. 23. Muzykantov, V. R.; Martynov, A. V.; Puchnina, E. A.; Danilov, S. M., In vivo administration of glucose oxidase conjugated with monoclonal antibodies to angiotensinconverting enzyme. The tissue distribution, blood clearance, and targeting into rat lungs. Am Rev Respir Dis 1989, 139, (6), 1464-73. 24. Muzykantov, V. R.; Atochina, E. N.; Kuo, A.; Barnathan, E. S.; Notarfrancesco, K.; Shuman, H.; Dodia, C.; Fisher, A. B., Endothelial cells internalize monoclonal antibody to angiotensin-converting enzyme. Am JPhysiol 1996, 270, (5 Pt 1), L704-13. 25. Danilov, S. M.; Gavrilyuk, V. D.; Franke, F. E.; Pauls, K.; Harshaw, D. W.; McDonald, T. D.; Miletich, D. J.; Muzykantov, V. R., Lung uptake of antibodies to endothelial antigens: key determinants of vascular immunotargeting. Am JPhysiol Lung Cell Mol Physiol 2001, 280, (6), L1335-47. 26. Snyder, S. L.; Sobocinski, P. Z., An improved 2,4,6-trinitrobenzenesulfonic acid method for the determination of amines. Anal Biochem 1975, 64, (1), 284-8. 27. Danilov, S. M.; Chumachenko, P. V.; Andreeva, Y. V.; Printseva, 0. Y.; Lacis, R. V., Angiotensin converting enzyme expression in the inflammation zone of human myocardial infarction and rat model of skin injury. JHypertensSuppl 1993, 11, (5), S232-3. IV. ON THE MECHANISM OF HIGHLY EFFECTIVE GENE TRANSFECTION IN VIVO BY ALKYLATED POLYETHYLENIMINE A. Introduction The promise of gene therapy has yet to be realized for lack of a safe, efficacious, and specific delivery vector 1. Viral vectors are naturally equipped to evoke maximal gene expression but have failed to prove safety in clinical trials 3. In contrast, cationic polymers and lipids demonstrate superior safety but do not produce clinically relevant levels of gene expression 4 . Polyethylenimine (PEI) is considered a leading polycationic vector for gene delivery 8. Work in our lab and others have demonstrated successful in vivo nucleic acid delivery in mice with various substituted PEIs -1O. Branched PEI first emerged as a "gold standard" of non-viral gene delivery in terms of efficacy but was plagued by toxicity concerns ". Less toxic lower molecular weight branched PEIs, specifically those of 2-kDa molecular weight, failed to afford sufficient gene expression 15. 12 However, N-dodecylation of this PEI dramatically enhanced the protein expression mediated by it to levels at least comparable to those the 25-kDa polycation 16. More recently, linear 22kDa PEI has emerged as a premier PEI for gene delivery 10 17; it exhibits a 21-fold enhancement in protein expression over leading branched PEIs and with minimal toxicity 10. Unfortunately, in vivo nucleic acid delivery by linear PEI is limited to the lung as it has a natural specificity for this organ. Attempts to alter or enhance the specificity of linear PEI through covalent attachment of a targeting antibody have failed to effect the desired outcome (Chapter 3, unpublished results). In the present study, we synthesized and mechanistically explored N-alkylated linear PEls for their ability to enhance and direct the specificity of gene delivery in vivo in mice. We found that covalent derivatization of a small fraction of PEI's amino groups with short-chained alkyls enabled a 26-fold enhancement of gene expression in the mouse lung, while also nearly quadrupling the amount of expression in this organ relative to others. Interestingly, the effect of N-alkylation varied among tissues and did not alter uptake of polyplexes into cells; rather, it seemed to affect an intracellular transfection step. B. Results and Discussion While much has been done to improve its transfection efficiency, linear PEI, currently a "gold standard" of polycationic gene delivery, still does not measure up to characteristics required for clinical utility. In this work, we prepared a series of N-alkylated linear PEI derivatives with the goal of developing a more efficient and specific vector for in vivo transfection. In particular, methyl-, ethyl-, propyl-, butyl-, and octyl- PEIs were synthesized from the corresponding linear iodoalkanes and fully deacylated linear PEI as shown in Figure 4.1. Alkylation conditions were optimized to derivatize approximately 11% of the backbone amines for all polycations (quantified by 'H NMR spectroscopy). The ability to buffer endosomes/lysozomes and condense DNA is a necessary requirement for efficient gene delivery by polycationic vectors. To assess the effect of Nalkylation on the ability of PEI to mediate these steps, the PEIs were characterized by acid titration and ethidium bromide (EtdBr) displacement from plasmid DNA. As shown in Figure 4.2A, all N-alkylated PEIs retain significant buffering capacity. Likewise, all of them condense plasmid DNA to an appreciable degree (Figure 4.2B). CH2 R N H m + ICH2 R ( R = H, CH 3 , CH2CH3, (CH 2)2 CH3 , (CH2)6 CH3 o) H N H LN Xy Figure 4.1. Schematic of the synthetic route for alkylated linear PEI derivatives. (i) iodoalkanes (methyl iodide, ethyl iodide, propyl iodide, butyl iodide, or octyl iodide) were individually reacted in ethanol at 60 C for 6 h to produce the resulting alkyl-PEIs (N-methyl-PEI, N-ethylPEI, N-propyl-PEI, N-butyl-PEI, or N-octyl-PEI respectively) with 11 % alkylation. m = 512; x = 456; and y = 56 It is noteworthy that N-ethyl-PEI demonstrates reduced DNA condensation and buffering capacity, suggesting that the fluid phase dynamics and DNA interactions of alkylated polyamines are complicated and that further research is required to fully understand the relationship between the chemical structure of N-alkylated linear PEI and their biophysical properties. This unique 20 behavior is observed following ethylation of other polycations as wellis Both condensation/decondensation of DNA and endosomal/lysosomal buffering are critical steps in cell transfection 7,21; although ideal conditions for these steps have not been established, it is known that effective DNA condensation to form polyplexes and subsequent decondensation inside the cell are in direct competition 22. These observations alone are insufficient to predict the transfection properties of ethyl-PEI or any of the other N-alkylated PEI derivatives 23 Since our unpublished work suggested a lack of correlation between in vitro (i.e., in cell culture) and in vivo (i.e., in animal models) PEI-mediated gene expression, the N-alkylated linear PEI derivatives were investigated for their ability to efficiently and specifically transfect cells in vivo. Figure 4.3 depicts the gene expression profiles in mice of the parent linear PEI and by its N-alkylated derivatives. One can see that in the lung, the tissue which demonstrates over 96% of all luciferase expression for the parent polycation, methyl-, ethyl-, and propyl- PEIs exhibited enhanced luciferase expression relative to the parent by 8-, 26-, and 7-fold, respectively (Fig. 4.3A). In contrast, longer alkyl chains negatively affected the transfection efficiency in the lung: while pulmonary luciferase expression mediated by butyl-PEI is marginally reduced, octyl-PEI demonstrates 200-fold lower expression in the lung than the unmodified PEI (Fig. 4.3A). 12 100 AC 7--- I o 50-- 200 4) 0 125 Volume of HCI, u I 250 0 2 4 N /P Ratio Figure 4.2. The effect of N-alkylation on the buffer capacity and DNA binding efficiency of linear PEI. (A) Acid titration profiles of aqueous solutions of the underivatized PEI (solid squares), methyl-PEI (open squares), ethyl-PEI (solid triangles), propyl-PEI (open triangles), butyl-PEI (solid circles), octyl-PEI (open circles), and NaCl as a control (X symbols). The corresponding 113 mM solutions were adjusted to pH 11.5 at room temperature and then titrated with 0.5 M HCl. In the case of NaCl, 0.04 M HCl was used as 0.5M HCl does not allow for visualization of the titration curve. (B) Displacement of the intercalated fluorophore EtdBr from plasmid DNA by the underivatized PEI (solid squares), methyl-PEI (open squares), ethyl-PEI (solid triangles), propyl-PEI (open triangles), butyl-PEI (solid circles), and octyl-PEI (open circles). N/P ratio is the ratio between the nitrogen atoms in the polycation and the phosphate groups of the bases in the plasmid. Interestingly, the other mouse tissues examined each demonstrate a unique expression profile with respect to the alkyl chain length (Fig. 4.3). In the heart and liver, propyl-PEI performs the best by 3.5- and 581-fold, respectively, over the parent polycation (Figs. 4.3B and 4.3D), while in the kidneys longer alkyl chain mediates enhanced gene expression with octyl-PEI providing 221-fold greater expression than unmodified PEI (Fig. 4.3E). In the spleen, ethyl- and octyl- PEIs produce similar levels of luciferase expression that are twice those of the parent's (Fig. 4.3C). The changes in the luciferase expression profile can be summarized by the ratio of expression in the lungs relative to all other organs combined. While for the parent PEI that ratio is 28:1, it jumps to 119:1 and 117:1, respectively, for the methyl and ethyl derivatives. For the three remaining derivatives (propyl-, butyl-, and octyl- PEIs), however, the ratio drops to 15:1, 2:1, and 0.06:1, respectively. Although alkylation with small alkyl chains does not alter specificity of delivery from the lung, octyl-PEI-mediated delivery preferentially localizes to the spleen. Moreover, significant accumulation is visible in the kidney where transfection by octyl-PEI dominates as well. This suggests that for appropriate applications N-alkylation can be used to modulate the organ specificity of gene expression. This is especially true for delivery of siRNA as it is catalytic in nature. 3- g Lung 2- 0- 0) (I) 0 50 - 25 - -J 0U) 1.~ a, 8 4- Spleen C., 4- 0 ~ .be C.) C.) 2 Liver C.) 4, 0. (I) 0 oE26E 0.141 Non Me Et P B 0 Figure 4.3. Comparison of the gene expression in the lungs, heart, spleen, liver, and kidney of a plasmid containing the luciferase gene mediated by the following N-alkylated linear PEI derivatives: Non=unmodified, Me=methyl, Et=ethyl, P=propyl, B=butyl, and O=octyl. 0) E . 0 5 11 14 20 N-Alkylation, % Figure 4.4. Gene expression in the lungs of a plasmid containing the luciferase gene mediated by N-ethyl-PEI as a function of the degree of the polycation's alkylation. Given the dramatic differences in protein expressions across mouse organs with 11% alkylation, we investigated the effect of varying degrees of N-alkylation with the best performing ethyl-PEI. As shown in Figure 4.4, the 11% alkylation happens to produce the most efficient gene expression in the lungs as compared with the 5%, 14%, and 20% derivatization. Higher degrees of N-alkylation of linear PEI were not tested due to drastically diminished solubility of the resultant polycations. Since protein expression is ultimately required for gene therapy to become a clinical reality, it is an appropriate endpoint measurement. However, it is also of mechanistic interest to establish in what organ the delivered plasmid ends up. To this end, we treated mice with I125_ labeled DNA in complexes with the N-alkylated linear PEI derivatives to follow the localization of the delivered plasmid. The biodistribution of the polyplexes was determined in vivo in a time dependent manner with time points taken in five minute increments following administration and also at 24 h, the time point at which protein concentration was determined above. At 24 h, most of the plasmid had been excreted; what remained resided predominantly in the clearance organs, specifically the kidneys and liver (0.1-0.5 % of injected dose for the best transfecting ethyl-PEI) as observed in Table 4.1. Less than 0.01 % of the injected dose remained in the lung at 24 h regardless of the delivery vector. This is not surprising as nearly 80% of the free plasmid was cleared from circulation in just 5 min and 90% at 15 min (Figure 4.5A). As demonstrated in Fig. 4.5B, the tissue distribution of naked plasmid DNA, linear PEI, and the most competent N-alkylated derivative (ethyl-PEI) at 15 min is very similar to that at 24 h. The kidneys and liver retained the greatest percentages of delivered radiation (about 2% of the injected dose each for ethyl-PEI), while the lung retained much less (only 0.35% of the injected dose). Polycation Spleen Kidney Liver Lung Heart Blood unmodified PEI 0.03 0.11 0.49 0.01 0.00 0.02 N-methyl-PEI 0.02 0.08 0.30 0.01 0.00 0.03 N-ethyl-PEI 0.02 0.12 0.30 0.02 0.01 0.06 N-propyl-PEI 0.02 0.11 0.30 0.01 0.00 0.03 N-butyl-PEI 0.02 0.09 0.29 0.01 0.00 0.03 N-octyl-PEI 0.03 0.05 0.27 0.01 0.00 0.01 Table 4.1. Biodistribution of 1 2 5-labeled gWiz Luc plasmid delivered with linear PEI and its Nalkylated derivatives. Values are the percentages of injected dose still remaining in select tissues at 24 h. 100 12 A 0) 08 B 0 40 - 0 5 10 15 Spleen Kidney Liver Lung Heart Blood Time, min Figure 4.5. (A) Pharmacokinetic profile of 112 5-labeled gWiz Luc plasmid DNA with no delivery vector with an initial dose of 70 tg of the plasmid containing approximately 1 [Ci of 1 2 5 radiation. (B) Comparison of the organ biodistribution of 1 2 5 -labeled plasmid containing the luciferase gene complexed with no vector (black), linear PEI (light grey), and N-ethylated linear PEI (dark grey) at 15 min. Interestingly, delivery of plasmid via a polycationic vector does not change the biodistribution of the plasmid at either 15 min or 24 h as compared to naked plasmid. We previously hypothesized that enhanced interactions with cell membranes due to increased hydrophobicity stemming from N-alkylation of branched PEI could enhance polyplex uptake and subsequent gene expression 16. However, studies described herein revealed no appreciable change in the biodistribution of free plasmid DNA relative to that delivered with a vector, either linear PEI itself or its N-ethylated derivative. The localization of 1 2 5-labeled plasmid DNA is similar across all tissues and all polycationic vectors at 24 h and across all those investigated at 15 min (Fig. 4.5B). This suggests that at least in the case of linear PEI, its N-alkylation does not enhance uptake of the polyplexes and instead plays some other role. The expression of protein from delivered plasmid DNA requires the successful completion of several steps: (i) uptake by endocytosis; (ii) endosomal/lysosomal escape; (iii) transport to, and uptake by, the nucleus; (iv) decondensation of DNA from the polycation; and (v) transcription and translation of delivered plasmid. Alkylation of PEI may confer an enhanced ability to complete any one of these steps on the polyplexes formulated with this polymer. Biodistribution studies suggest that uptake is not the step that is perturbed by N-alkylation. Given the changes to buffering capacity and DNA condensation seen with N-ethylated PEI and the greatly enhanced protein expression observed as a result, it is likely that either endosomal escape (which requires significant buffering of the endosome) or the decondensation of DNA are responsible for the enhanced efficacy seen in the lung with N-ethyl PEI. C. Materials and Methods Materials and NMR. Nal was purchased from Perkin Elmer. All other chemicals were from Sigma-Aldrich (St. Louis, MO) and were of the highest purity available and used as received. NMR spectra were recorded in the MIT DCIF using a Bruker 400-MHz NMR spectrometer. Plasmid and its iodination. Plasmid was handled and iodinated as described in Chapter 2 of this thesis. Linear PEI synthesis and its N-alkylation. Fully deacylated linear PEI was synthesized as described in Chapter 2 of this thesis. The linear PEI obtained was reacted with 10 mol % of iodomethane, iodoethane, iodopropane, iodobutane, or iodooctane in ethanol as previously described 16. The N-alkylated crude products, obtained upon rotary evaporation, were dissolved in water, adjusted to pH 10 with 10 M NaOH, isolated by vacuum filtration, washed with cold water, and lyophilized. Pure products (~11% alkyl group substitution) were obtained as white hygroscopic solids on lyophilization. As such, they were stored in a drying chamber at - 20 *C. 1H NMR spectroscopy (with CDCl 3 as an internal standard) was used to determine the percent of N-alkylation through comparison of the integration of the 0.5 - 1 ppm triplet from the protons of the terminal methyl group of the alkane with the 2.5 - 3 ppm peak of the polymer's ethylene protons. pH titration of N-alkyl-PEIs to determine buffering capacity. Acid titrations were carried out using a 1 ml solution of each N-alkyl-PEI (113 mM in -CH 2CH 2NH- units) adjusted to pH 11.5 with NaOH. Sequential additions of 20-r1 volumes of 0.5 M HCl were performed, and the pH after each addition was measured; 113 mM aqueous NaCl was titrated similarly as a control using 0.04 M HCl. Ethidium bromide (EtdBr) displacement assay to determine DNA binding of Nalkyl-PEls. To a 200 tl solution of gWiz Luc DNA (4.1 tg) and EtdBr (0.63 tg) in 10 mM PBS in a black 96-well plate, 5-rd aliquots of 0.63 mM N-alkyl-PEI stock solutions were added sequentially. Fluorescence spectra of free EtdBr (F1), of EtdBr/DNA (FD), and of EtdBr/DNA after each addition of PEI (Fc) were recorded (x = 523 nm and Xm = 587 nm). Relative fluorescence values were calculated using the formula [(Fc - F1) / (FD - F1)] x 100%. Gene delivery in mice. Gene delivery in mice was conducted in Chapter 2 of this thesis for tail vein injection. Mouse perfusion and radiation measurements. Biodistribution studies were conducted as described in Chapter 2 of this thesis. D. References 1. El-Aneed, A., An overview of current delivery systems in cancer gene therapy. J Control Release 2004, 94, (1), 1-14. 2. Marshall, E., Gene therapy death prompts review of adenovirus vector. Science 1999, 286, (5448), 2244-5. 3. Cavazzana-Calvo, M.; Thrasher, A.; Mavilio,, F., The future of gene therapy. Nature 2004, 427, (6977), 779-81. 4. Verma, I. M.; Somia, N., Gene therapy -- promises, problems and prospects. Nature 1997, 389, (6648), 239-42. 5. Nimesh, S.; Aggarwal, A.; Kumar, P.; Singh, Y.; Gupta, K. C.; Chandra, R., Influence of acyl chain length on transfection mediated by acylated PEI nanoparticles. Int JPharm2007, 337, (1-2), 265-74. 6. Godbey, W. T.; Wu, K. K.; Mikos, A. G., Poly(ethylenimine) and its role in gene delivery. J Control Release 1999, 60, (2-3), 149-60. 7. Aigner, A., Gene silencing through RNA interference (RNAi) in vivo: strategies based on the direct application of siRNAs. JBiotechnol2006, 124, (1), 12-25. 8. Thomas, M.; Lu, J. J.; Zhang, C.; Chen, J.; Klibanov, A. M., Identification of novel superior polycationic vectors for gene delivery by high-throughput synthesis and screening of a combinatorial library. Pharm Res 2007, 24, (8), 1564-71. 9. Singleton, P. A.; Chatchavalvanich, S.; Fu, P.; Xing, J.; Birukova, A. A.; Fortune, J. A.; Klibanov, A. M.; Garcia, J. G. N.; Birukov, K. G., Akt-Mediated Transactivation of the SIPI Receptor in Caveolin-Enriched Microdomains Regulates Endothelial Barrier Enhancement by Oxidized Phospholipids. Circ Res 2009, 104, (8), 978-986. 10. Thomas, M.; Lu, J. J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A. M., Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci U S A 2005, 102, (16), 5679-84. 11. Boussif, 0.; Lezoualc'h, F.; Zanta, M. A.; Mergny, M. D.; Scherman, D.; Demeneix, B.; Behr, J. P., A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A 1995, 92, (16), 7297-301. 12. Choosakoonkriang, S.; Lobo, B. A.; Koe, G. S.; Koe, J. G.; Middaugh, C. R., Biophysical characterization of PEI/DNA complexes. J Pharm Sci 2003, 92, (8), 1710-22. 13. Kunath, K.; von Harpe, A.; Fischer, D.; Petersen, H.; Bickel, U.; Voigt, K.; Kissel, T., Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecularweight polyethylenimine. J Control Release 2003, 89, (1), 113-25. 14. Thomas, M.; Klibanov, A. M., Conjugation to gold nanoparticles enhances polyethylenimine's transfer of plasmid DNA into mammalian cells. Proc Natl A cad Sci USA 2003, 100, (16), 9138-43. 15. Breunig, M.; Lungwitz, U.; Liebl, R.; Fontanari, C.; Klar, J.; Kurtz, A.; Blunk, T.; Goepferich, A., Gene delivery with low molecular weight linear polyethylenimines. J Gene Med 2005, 7, (10), 1287-98. 16. Thomas, M.; Klibanov, A. M., Enhancing polyethylenimine's delivery of plasmid DNA into mammalian cells. Proc Natl Acad Sci USA 2002, 99, (23), 14640-5. 17. Wightman, L.; Kircheis, R.; Rossler, V.; Carotta, S.; Ruzicka, R.; Kursa, M.; Wagner, E., Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo. JGene Med 2001, 3, (4), 362-72. 18. San Juan, A.; Letourneur, D.; Izumrudov, V. A., Quaternized poly(4-vinylpyridine)s as model gene delivery polycations: structure-function study by modification of side chain hydrophobicity and degree of alkylation. Bioconjug Chem 2007, 18, (3), 922-8. 19. Zhang, C.; Ding, Y.; Ping, Q.; Yu, L., Novel Chitosan-Derived Nanomaterials and Their Micelle-Forming Properties. JournalofAgriculturaland Food Chemistry 2006, 54, (22), 84098416. 20. Chen, C. C.; Hsu, C. H.; Kuo, P. L., Effects of alkylated polyethylenimines on the formation of gold nanoplates. Langmuir 2007, 23, (12), 6801-6. 21. Davis, M. E., Non-viral gene delivery systems. Curr Opin Biotechnol 2002, 13, (2), 128- 31. 22. Neu, M.; Fischer, D.; Kissel, T., Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med 2005, 7, (8), 992-1009. 23. von Harpe, A.; Petersen, H.; Li, Y.; Kissel, T., Characterization of commercially available and synthesized polyethylenimines for gene delivery. J ControlRelease 2000, 69, (2), 309-22. 24. Markwell, M. A., A new solid-state reagent to iodinate proteins. I. Conditions for the efficient labeling of antiserum. Anal Biochem 1982, 125, (2), 427-32. 25. Piatyszek, M. A.; Jarmolowski, A.; Augustyniak, J., Iodo-Gen-mediated radioiodination of nucleic acids. Anal Biochem 1988, 172, (2), 356-9. V. POLYETHYLENIMINE MEDIATES SPECIFIC IN VIVO GENE DELIVERY UPON TOPICAL APPLICATION A. Introduction Polyethylenimine (PEI) has been employed as a gene delivery agent since 19951. PEI is a particularly versatile gene delivery vector as it is readily amenable to chemical modification. Our lab has devoted much effort to employing chemical modification to enhance the gene 2 delivery efficacy and/or specificity of PEI in vivo following systemic intravenous delivery . However, intravenous delivery faces pharmacokinetic factors that have proven to be a significant obstacle to altered and enhanced specificity. Fortunately, gene therapy targets that can be reached by topical application abound. Topical application of PEI varies greatly from its use in intravenous delivery. Topical delivery more closely resembles in vitro cell culture gene delivery as polyplexes are administered directly to cells. The size of the particle is responsible for positioning the polyplex in contact with the cell surface through gravity and diffusion. Polyplexes do not encounter components of the circulatory system and do not face biodistribution or clearance concerns. Additionally, systemic toxicity is not a concern as gene delivery agents are applied directly to the target site and cannot accumulate in other organs. To date, the use of PEI for topical treatment has been limited to applications in the lung as it is a highly desirable target for gene therapy for treatment of cystic fibrosis and may other diseases 6'7. However, there are many topical targets that are of great interest. For example, there are over 2 dozen diseases that affect the cornea, the clear outer covering of the eye, a tissue 100 exceptionally amenable to direct application of gene therapy agents8 . Herein we investigate the use of chemically modified PEI for topical gene delivery in the eye. B. Results and Discussion Specificity of gene delivery in vivo is an obstacle for PEI-mediated gene delivery. Previous work in our lab and others has focused on enhancing specificity following systemic intravascular delivery and the associated complications as discussed in Chapters 2-4. Herein, we explore the direct application of PEI-based polyplexes for specific gene delivery in the eye. For this study, we employed a gold-conjugated PEI as the gene delivery vector. A gold conjugated vector was chosen instead of linear PEI as topical delivery more closely mirrors in vitro cell culture delivery. Gravity and diffusion are responsible for positioning polyplexes in contact with the cell surface and therefore larger complexes are more readily taken up by cells. Gold is a convenient way to crosslink polymer chains together and also allows for determination of the amount of the complex that is taken up by cells via silver staining. This PEI-gold vector, designated PEI-GNP where GNP stands for gold nanoparticle, was synthesized by linking PEI to gold nanoparticles via the bis-succinimide ester of 4,4'dithiodibutyric acid according to the reaction mechanism shown in Figure 5.1. A Au/PEI molar ratio of 15 was employed in these studies as confirmed by elemental analysis. Although PEIGNPs are larger than vectors typically employed for gene delivery, their use for topical application negates concerns over their size. In intravenous delivery, large polyplexes may obstruct blood flow through the capillaries and are too large to extravasate between cells that line the blood vessel. Clearly these are not concerns for topical delivery. 101 0 0 HoSu, DCC/DMF OH HO ' 0 0 N-O,,-7 O-N 0 00 (i) () H H N HSCH 2CH2OH/H 20 HN HN HN H H N \ .N 0 0 0 SH PE12/THF/CH 2CI2 SH H HAuCl 4 , NaBH 4/H20 H N HN HN (iv) A O HN WNH HN H N S -S S s O S S NN 0 /NH N H Figure 5.1. Schematic representation of the synthesis of PEI-GNPs. i: 4,4'-dithiobutyric acid is converted to the corresponding succinimidyl ester; ii: crosslinking of PEI with Bis-(Nhydroxysuccinimido)-4'4-dithiodibutyrate; mercaptoethanol; iv: hydroxysuccinimide; formation DCC, iii: of PEI-GNP reduction conjugates. dicyclohexylcarbodiimide; tetrahydrofuran. 102 of DMF, the disulfide Abbreviations: with s- HOSu, N- dimethylformamide; THF, .... 81h EpiScr No EpiScr Figure 5.2. Transfection of cornea by PEI-GNP in complex with GFP plasmid DNA. Light microscopy images following silver staining (a) and fluorescent microscopy images (b) of rabbit corneal sections at 8 h post 5 min application of PEI-GNP/plasmid GFP at an N/P ratio of 180, both with and without epithelial scraping. Polyplexes are visible as dark spots (a) and GFP expression is seen as green (b). 103 . i ..... ....... -............. To investigate the ability of PEI-GNPs to specifically deliver genes in vivo following direct administration, the eye was selected. The outer most layer of the eye that is exposed to air is called the cornea. The cornea is responsible for protecting the eye in the same way that skin protects muscles below it' 0 . When protecting an individual from infection and particulate matter, the cornea is good. However, it also makes treatment of cells deeper in the corneal tissue a challenging endeavor. Fortunately, advances in medicine have helped to alleviate this problem. Scraping of the outer cells of the cornea (about 8-10 cells deep) is now a commonly employed procedure for treatment of the eye. It allows for removal of the protective layers and access to the lower layers. In two weeks, the outer layer of the cornea is able to regenerate itself and continue offering a protective effect 0 . In our work, we exploited this technique to determine if we could transfect cells deep within the cornea. PEI-GNPs were complexed with plasmid DNA encoding GFP at an optimal N/P ratio of 180. Previous studies in tissue culture determined that this high ratio is successful for in vitro gene delivery9 . Polyplexes were then applied to the surface of the cornea for 5 min either directly or after epithelial scraping, a common clinical procedure in ocular therapies used to gain greater access to the stroma layer below the corneal epithelium"' . We then evaluated uptake of PEI-GNP polyplexes by the stroma at 8 h post incubation (Figure 5.2A). Unscraped cornea are also shown as a reference. 104 m .. . . ......... 8 h' No Epi Scr Epi Scr Figure 5.3. Detection of neutrophils (inflammatory cells) in rabbit cornea sections at 8 h post 5 min application of PEI-GNP vectors in complex with GFP plasmid DNA at an N/P ratio of 180 via the CD1 lb marker and fluorescent microscopy. Cells possessing CD11b marker appear pink and are indicated by white arrows on the image with epithelial scraping. 105 Although uptake of polyplexes is interesting, if that uptake does not lead to expression of the delivered plasmid DNA, it is not particularly useful. Therefore we then investigated the ability of PEI-GNPs to successfully effect expression of GFP following topical application to epithelium scraped cornea. As shown in Figure 5.2B, GFP expression was detected in the cornea. Nuclei were stained blue with DAPI while GFP expression appears as a bright green spots. Nearly every nuclei demonstrates co-fluorescence with GFP indicating substantial uptake and expression of PEI-GNP/GFP polyplexes. This represents the first demonstration of topical gene delivery to any organ other than the lung with PEI. Unfortunately, successful in vivo delivery is often accompanied by toxicity and/or immunogenicity. We have worked with intravenous delivery of linear PEI long enough to know what doses will cause toxicity in mice and rats. However, as we had never applied PEI in vivo in a topical manner, the toxicity associated with such delivery remained a concern. Therefore, we investigated toxicity associated with topical delivery of PEI-GNPs to the cornea with two methods, detection of inflammatory cells and the initiation of the apoptotic pathway. Inflammation is a common occurrence in cells that have been subjected to non-normal stimuli. However, significant inflammation due to acute toxicity is rapid and indicative of a major problem 13 . To evaluate provocation of an immune response by delivery of PEI-based polyplexes, we directly detected the presence of inflammatory cells via the CD 11b marker, an antigen only and always expressed on inflammatory cells. An antibody to CD11 b, which was conjugated to the fluorophore Alexa Flour 594, was incubated with tissue sections to identify those where CD11b was expressed. As seen in Figure 5.3, inflammatory cells were detected throughout the corneal section. It is important to note that although inflammation was apparent, 106 it was only minimal in nature. It is the same inflammation observed following only epithelial scraping (data not shown). Next, we looked at induction of an apoptotic effect due to transfection with these polyplexes. Apoptotic pathways lead to digestion of DNA to produce 3'-OH groups which otherwise do not exist. As such, the most widely used assay to detect apoptosis in living animals is a fluorescence based TUNEL assay. In this assay, tissue sections are treated with terminal deoxynucleotidyl transferase (a specialized DNA polymerase) and modified DNA bases (containing digitoxin) to add additional bases to the 3'-OH created by the apoptotic pathway. These bases are then visualized by addition of an anti-digitoxin antibody that is conjugated to a fluorescent dye (in this case, rhodamine). As shown in Figure 5.4, apoptosis is visible in our tissue sections. However, it is important to note that all toxicity is localized to the region of the cornea that was subjected to epithelial scraping. This manual removal of surface cells is known to induce apoptosis that resides with repopulation of the area with new cells within two weeks. It is important to note that there is no apoptosis in the deeper layer of the stroma. This work represents the first topical application of PEI-based polyplexes for gene delivery in the eye. PEI-GNPs are able to successful mediate expression of the cargo DNA without exhibiting appreciable toxicity. Both inflammatory and apoptotic markers of toxicity were normal. These results indicate the future work should include evaluation of PEI-based agents for topical delivery of nucleic acids to achieve efficient and specific delivery. 107 ................... ............. . 8 .................. ... .......... h Epi Scr No EpiScr Figure 5.4. Detection of apoptosis (via terminal 3'-OH DNA cleavage) in rabbit cornea sections at 8 h post 5 min application of PEI-GNP vectors in complex with GFP plasmid DNA at an N/P ratio of 180 via a TUNEL assay and fluorescent microscopy. 108 ..... ............ ..... C. Materials and Methods Materials. The 2 kDa PEI, HAuCl 4 trihydrate, NaBH4 , 4,4'-dithiodibutyric acid, 2mercaptoethanol, dicyclohexylcarbodiimide, and N-hydroxysuccinimide were purchased from Sigma. All solvents used, also from Sigma, were of the highest purity available. Spectra Por CE dialysis tubing with a molecular mass cutoff of 500 Da was from Spectrum Laboratories (Houston), and cellulose dialysis tubing with a molecular mass cutoff of 12 kDa was from Sigma. Elemental analyses were performed by Columbia Analytical Services (Tucson). Synthesis of PEI-GNP. Bis-(N-hydroxysuccinimido)-4,4'-dithiodibutyrate was first synthesized following a modified literature procedure 9 . 25 mmol of 4,4'-dithiodibutyric acid and 65 mmol of hydroxysuccinimide were dissolved at room temperature in 25 ml of DMF. To this solution was added with stirring, 55 mmol of dicyclohexylcarbodiimide in 20 ml of DMF. Following precipitation of dicyclohexylurea, the reaction was cooled in an ice bath with stirring for 12 h. It was diluted with ethyl acetate and the dicyclohexylurea was removed by filtration. The ethyl acetate layer was washed with brine and dried over anhydrous sodium sulfate. The solvent was evaporated and the sample was dissolved in methylene chloride and filtered to remove additional dicyclohexyl urea. Saturated potassium carbonate was used to wash the filtrate 3X before a brine wash, drying over sodium sulfate and solvent evaporation. The Bis-(Nhydroxysuccinimido)-4,4'-dithiodibutyrate was then solubilized in minimal hot methanol and allowed to crystallize while cooling to room temperature. The structure of the pure crystals was confirmed by NMR at the MIT DCIF and mass spectral analysis. PEI was modified with bis-(N-hydroxysuccinimido)-4,4'-dithiodibutyrate following a literature protocol 9 . Briefly, 2.5 mmol of branched PEI of 2 kDa was dissolved in 40 ml of a 5:3 ratio of methylene chloride/THF solution. To this solution was added 2.31 mmol of bis-(N- 109 hydroxysuccinimido)-4,4'-dithiodibutyrate in 15 ml of THF drop wise with stirring over 3 min followed by a 15 h stirring and evaporation of the solvent. The material obtained was suspended in 50 ml of water, treated with 2.24 ml of @-mercaptoethanol, and stirred for an additional 2 h. The resulting solution was filtered and dialyzed extensively against water in a 500 Da cutoff membrane. This thiol-modified PEI was then conjugated to gold nanoparticles. An aqueous solution (25 ml) containing 0.48 mmol of the thiol-modified PEI was combined with 1.43 mmol of HAuCl4*3H 2 0 and stirred for 10 min before drop wise addition of aqueous NaB4 solution (71.4 mmol in 36 ml) over 90 sec followed by stirring for 24 h. The conjugate obtained was dialyzed extensively against water with a 12 kDa cutoff membrane. A measured portion of the obtained solution was lyophilized and analyzed for elemental content (57.8 % gold, Au/PEI molar ratio = 15). The amount of PEI in the stock solution was calculated based on the mass of the dried solid and the ratio of gold to PEI. For example, if 1 ml of the solution produced 0.2 g of solid, then 42.2 % of that (or 0.084 g) is polymer. To calculate the concentration of PEI in the stock solution, 0.084 g was divided by the original volume of 1 ml to give a concentration of 84 mg/ml. Transfection of rabbit cornea. New Zealand White rabbits 6-8 wks old were obtained from Myrtle's Rabbitary (TN). All procedures involving these animals adhered to current regulations for use of animals for research purposes. Rabbits were anesthetized with a mixture of zylene and ketamine at a dose of 10 and 50 mg/kg respectively. Once anesthetized, one half of the rabbit cornea were epithelial scraped by standard procedures. Then polyplexes were prepared as follows. 10 tg of GFP encoding plasmid DNA was complexed with PEI-GNP at an N/P ratio of 180 in PBS pH 7.4 and 10% glucose buffer and allowed to incubate for 10 min at 37 110 C. Then, 100 [d of polyplexes were applied to the cornea of each rabbit eye and allowed to incubate for 5 min. Eyes were then copiously washed with balanced salt solution eye drops (Alcon, Ft. Worth, TX). At 8 h post incubation, rabbits were sacrificed by an overdose of intravenous pentobarbitone and the eyes were removed. Corneal sections were cut from sections frozen in OCT medium and used for further assays. Silver staining to detect delivered polyplexes. Sections of rabbit corneal tissues 8 [m thick were prepared at 8 h post 5 min topical application of PEI-GNP polyplexes at an N/P ratio of 180 for both epithelial scraped and untreated cornea. These sections were counterstained with silver following the manufacturer's protocol (Invitrogen LI Silver Enhancement Kit #L24919, Carlsbad, CA) and visualized using light microscopy (Leica with spot software). Briefly, tissue sections were rinsed well with deionized water to remove all ions. Samples were then treated with an initiator and silver enhancer for 20 minutes to cause deposition of silver on the GNPs. The reaction was quenched by copious washing with deionized water for 5 minutes. Silver stained GNPS were then visualized with light microscopy. GFP detection following transfection. Sections of rabbit corneal tissues 8 [tm thick were prepared at 8 h post topical application of PEI-GNPs in complex with GFP encoding plasmid DNA at an N/P ratio of 180 for both epithelial scraped and untreated cornea. Vectashield mounting medium containing DAPI (Vector Laboratories, Inc. Burlingame, CA) was used to visualize nuclei in the tissue sections. The sections were viewed and photographed with a fluorescent microscope with spot software equipped with a digital camera (Leica, Wetzlar, Germany and SpotCam RT KE, Diagnostic Instruments Inc., Sterling Heights, MI, USA). Samples were excited at 488 nm with emission measured at 519 nm and a FITC cutoff. GFP appears as a green dot while nuclear staining appears blue. 111 CD11b immunohistochemistry for detection of inflammation. In response to trauma, inflammatory cells (neutrophils) will locate to the site of damage. These cells express a unique marker designated CD 11b. To detect inflammation caused by gene delivery, corneal sections were assayed for the presence of CD 11b following topical application of PEI-GNP polyplexes. Tissue sections (7 im) were incubated with a primary antibody (rat anti-mouse antibodies (BD Pharmingen, San Jose, CA)) at 1:50 dilution in 1xHEPES containing 5% BSA at room temperature for 90 minutes. Note, mouse CD1 lb has high homology to rabbit and anti-mouse CDl lb is commonly used for detection of rabbit CD 11b. Incubation with an Alex Flour 594 conjugated goat anti-rat IgG secondary antibody (Alex Flour 594, Molecular Probes, Eugene, OR) at a dilution of 1:500 for 60 minutes at room temperature was employed for visualization of neutrophils. Vectashield mounting medium containing DAPI (Vector Laboratories, Inc. Burlingame, CA) was used to visualize nuclei in the tissue sections. The sections were viewed and photographed under a Leica fluorescent microscope (Leica, Wetzlar, Germany) equipped with a digital camera (SpotCam RT KE, Diagnostic Instruments Inc., Sterling Heights, MI) with an excitation at 590 nm, emission at 617 nm, and a Texas red cutoff. TUNEL assay for detection of apoptosis. Apoptosis leads to generation of 3'-OH DNA termini. These are considered markers of toxicity and can be assessed by a fluorescence-based TUNEL assay according to vendor's instructions. Corneal sections were fixed in acetone at 20'C for 2 min, dried at room temperature for 5 min, and then placed in PBS balanced salt solution. A fluorescence-based TUNEL assay was used according to the manufacturer's instructions using Apop Tag Red in situ apoptosis detection kit (Chemicon International #7165, Temecula CA, USA). Briefly, following fixation, sections were incubated in equilibration buffer at 15 1d/cm 2 for 10 seconds at room temperature. Excess liquid was removed by blotting and 112 TdT (terminal deoxynucleotidyl transferase) enzyme was applied at 11 tl/cm 2 and incubated for 1 h at 37'C in a humidified chamber. The reaction was stopped by agitation for 15 sec followed by incubation for 10 min at room temperature in stop/wash buffer. Sections were washed with three changes of PBS for 1 min each and then dried by careful blotting. digoxigenin conjugate (rhodamine) was applied to the slide at 13 1d/cm 2 Warmed anti- and incubated in a dark, humidified chamber for 30 min at room temperature. Slides were then washed in 4 changes of PBS for 2 min per wash at room temperature. Before visualization, samples were counterstained by addition of a mounting medium containing 0.5-1 [g/ml of DAPI and mounted under a glass coverslip. Tissue sections were visualized by fluorescence microscopy using standard rhodamine and DAPI excitation and emission filters (ex. 540 nm / em. 550 nm and ex. 365 nm / em. 480 nm respectively). D. References 1. Boussif, 0.; Lezoualc'h, F.; Zanta, M. A.; Mergny, M. D.; Scherman, D.; Demeneix, B.; Behr, J. P., A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 1995, 92, (16), 7297-301. 2. Thomas, M.; Ge, Q.; Lu, J. J.; Chen, J.; Klibanov, A. M., Cross-linked small polyethylenimines: while still nontoxic, deliver DNA efficiently to mammalian cells in vitro and in vivo. Pharm Res 2005, 22, (3), 373-80. 3. Thomas, M.; Klibanov, A. M., Enhancing polyethylenimine's delivery of plasmid DNA into mammalian cells. Proc NatlAcad Sci USA 2002, 99, (23), 14640-5. 113 4. Thomas, M.; Lu, J. J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A. M., Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci U S A 2005, 102, (16), 5679-84. 5. Thomas, M.; Lu, J. J.; Zhang, C.; Chen, J.; Klibanov, A. M., Identification of novel superior polycationic vectors for gene delivery by high-throughput synthesis and screening of a combinatorial library. Pharm Res 2007, 24, (8), 1564-71. 6. Gautam, A.; Waldrep, J. C.; Orson, F. M.; Kinsey, B. M.; Xu, B.; Densmore, C. L., Topical gene therapy for pulmonary diseases with PEI-DNA aerosol complexes. Methods Mol Med 2003, 75, 561-72. 7. Rudolph, C.; Schillinger, U.; Plank, C.; Gessner, A.; Nicklaus, P.; Muller, R.; Rosenecker, J., Nonviral gene delivery to the lung with copolymer-protected and transferrinmodified polyethylenimine. Biochim Biophys Acta 2002, 1573, (1), 75-83. 8. National Eye Institute (2010). Facts About the Cornea and Corneal Disease. www.nei.nih.gov/health/cornealdisease. 9. Thomas, M.; Klibanov, A. M., Conjugation to gold nanoparticles enhances polyethylenimine's transfer of plasmid DNA into mammalian cells. Proc Natl Acad Sci USA 2003, 100, (16), 9138-43. 10. Smolin and Thoft's The Cornea:Scientific Foundationsand ClinicalPractice.Lippincott Williams & Wilkins: 2004; Vol. Fourth Edition. 11. Barbosa, F. L.; Goes, R. M.; de Faria, E. S. S. J.; Haddad, A., Regeneration of the corneal epithelium after debridement of its central region: an autoradiographic study on rabbits. Curr Eye Res 2009, 34, (8), 636-45. 114 12. Choi, C. Y.; Kim, J. Y.; Kim, M. J.; Tchah, H., Transmission electron microscopy study of corneal epithelial flaps following removal using mechanical scraping, alcohol, and epikeratome techniques. JRefract Surg 2008, 24, (7), 667-70. 13. Colella, P.; Cotugno, G.; Auricchio, A., Ocular gene therapy: current progress and future prospects. Trends Mol Med 2009, 15, (1), 23-31. 115 VI. RADIO FREQUENCY RADIATION (RFR) CAUSES NO NON-THERMAL DAMAGE IN ENZYMES AND LIVING CELLS A. Introduction The U.S. Food and Drug Administration (FDA) has stressed RFID technology for the combating of drug counterfeiting. This technology relies on the use of RFR to identify and detect electronic tags included by the manufacturer in product packaging. RFR is defined as 2 electromagnetic radiation with frequencies in the range from 3 KHz to 300 GHz , i.e., similar to those used in such common electronic devices as radios, microwave ovens, cellular phones, and wireless data networks. There is no consensus, however, as to the effect of RFR on pharmaceutical products in general and on biologics (considered particularly vulnerable due to their complex structures) specifically, thus leading to safety and utility concerns. In the last two decades, much research has been devoted to studying the effect of RFR on biologics 3 -6. To best understand the consequences of RFR exposure, the putative effects can be classified into two distinct categories: thermal and non-thermal. The former occur due to a significant rise in sample temperature, as measured by a temperature probe, because RFR can generate heat under conditions of high power and/or prolonged exposure 7'8 . The stand-alone, heat-induced deterioration of biological, e.g., protein-based products is well documented and understood 9"0 ; thermal effects of RFR on such systems can be predicted and thus require no further study. In contrast, non-thermal effects, i.e., those that arise despite no appreciable increase in sample temperature, are both controversial and ill defined. No consensus has been reached even about the existence of such effects, let alone their mechanisms. 116 The lack of agreement on the existence of non-thermal effects of RFR can be attributed to the following factors: (i) the corresponding studies are often not reproducible and report effects only slightly above the noise level"; (ii) critical testing parameters are frequently missing from publications12,13; (iii) accurate sample temperature maintenance, a critical component for uncovering non-thermal effects, is often lacking 6 1, ,14, 5 ; (iv) reported studies lack sufficient breadth to make generalizations 516 ' ; (v) no plausible mechanism is evident for said effects; and (vi) about half of the published literature documents the inability of RFR to cause non-thermal effects on biologics, while the rest arrives at the opposite conclusion 6 . In this work, we have undertaken to definitively investigate the existence of non-thermal RFR effects on diverse biological agents. To this end, we have adhered to the following experimental strategy: (i) a RFR device which reliably maintains global sample temperature to within ±1C has been employed; (ii) a contained system which provides accurate determination of RFR specific absorption rate (SAR) values has been utilized; (iii) a great excess of RFR has been employed with respect to its intensity and the length of exposure; (iv) the effect of RFR on both non-living (enzymes) and living (bacterial and mammalian cells) biologics has been investigated under the same conditions; and (v) to increase the sensitivity of the experiments, samples were exposed to RFR under such conditions where slight degradation was detectable even without RFR so that any extra degradation due to the latter would be readily measurable. B. Results and Discussion Many published studies aimed at determining whether RFR induces non-thermal effects on biologics fail to prove accurate maintenance of sample temperature ''14'1 , thus preventing differentiation between thermal and non-thermal effects. Additionally, SAR values are often 117 either not provided at all or there is insufficient information as to how they were determined; this makes it difficult to attribute the observed effects to a particular cause because it is unclear how much of the incident radiation the sample actually absorbed. Finally, investigations in this field are often inadequately designed and/or are insufficiently rigorous leading to irreproducibility 1',. In the present study, we have designed and utilized what we believe to be a superior experimental approach to definitively elucidate whether diverse biologics are indeed affected, in a non-thermal way, by RFR of the most commonly used commercial frequencies, i.e., 2.45 GHz, 915MHz, and 13.56MHz. The effect of RFR exposure on two unrelated enzymatic proteins, on Gram-positive and Gram-negative bacteria, and on human spleen-derived lymphoblast cells was quantitatively examined under well-defined and rigorously controlled conditions. We eliminated the thermal effects through the use of a previously characterized apparatus 7 capable of maintaining sample temperature upon RFR exposure to within +1C of the desired value, as measured by an accurate fiber optic temperature probe. We also determined the SAR values for the experimental conditions employed (as described in Methods). Finally, we conducted our study under stressed conditions to increase the likelihood of observing a putative non-thermal effect by deliberately reducing the stability of the biologics to enhance experimental sensitivity. Specifically, we employed elevated sample temperatures, grossly extended exposure times, and greatly exaggerated radiation power relative to lifetime exposure under standard RFID technology use. Such extreme irradiation conditions, combined with the broadness of the frequencies employed, should provide sound data to evaluate the effect of RFR on biologics. The two enzymes studied, horseradish peroxidase (HRP) and p-galactosidase (3-gal), represent model, unrelated, commercially important (e.g., in clinical assays) enzymes with 118 distinct functions: HRP is a heme-containing oxidoreductase, while P-gal is a hydrolase. They were subjected to RFR in aqueous buffers at a nearly neutral pH and 50'C for up to 24 h (these conditions were selected to afford a noticeable but not excessive rate of inactivation in the absence of radiation); in a control experiment, each enzyme was incubated under the same conditions but without RFR. The remaining enzymatic activities were measured as a function of time of exposure/incubation (see Methods). The loss in activity of HRP and $-gal over a 24-h RFR exposure is shown in Figure 6.1 A and 6.1B with the time point data represented relative to those of the control sample. Samples with RFR-unaffected enzymatic activities would thus yield horizontal lines at a 100% level. One can see that the irradiated samples indeed do not appreciably deviate from control values over the course of the exposure and remain at nearly 100%. Statistical analysis (ANOVA) of the results obtained at all three RFR frequencies employed, performed as outlined in Methods, has revealed that the rates of enzyme inactivation with and without radiation are indistinguishable from the control within a 95% confidence interval (DFd; n = 4). The same is also true for all of the irradiated samples compared with one another (data not shown). Since the loss in enzymatic activity is not detectably amplified by exposure to RFR, we conclude that there is no observable non-thermal effect of RFR on either HRP or p-gal. 119 ]T 100 IT__ I ---L T 50 A 0 100 50 - B 0 I 1 12 D I 2 Time, h Figure 6.1. Time courses of the change in enzymatic activity (relative to the non-irradiated control) of horseradish peroxidase (A) and p-galactosidase (B) at 50"C with RFR of 2.45-GHz (squares), 915-MHz (triangles), and 13.56-MHz (circles) frequencies. For experimental conditions, see Methods. Error bars are present for all data points but may be too small to see. 120 Next, two distinct bacterial strains were examined, namely S. aureus (Gram-positive) and E. coli (Gram-negative). Both were irradiated at each of the three frequencies tested for 4 h at 300 C in 0.85% aqueous NaCl. As seen in Figures 6.2A and 6.2B, there is no detectable change in the number of viable cells over time relative to the control (i.e., without RFR) for both bacteria. Statistical analysis has confirmed that the changes were indistinguishable at a 95% confidence interval for irradiated and control samples (DFd; n = 4). The same is also true for all of the irradiated samples compared with one another (data not shown). We conclude, therefore, that none of the RFR frequencies investigated appreciably affects the viability of either bacterium. Finally, the putative non-thermal effect of RFR on human cells, namely TK6 spleenderived lymphoblast suspension cell line, has been examined. The cells were exposed to radiation for up to 4 h at 37'C in Dulbecco's PBS medium at pH 7.4. As can be seen in Figure 6.2C, cellular vitalities (relative to the control) with and without irradiation are comparable for all the frequencies studied: they all have been determined to be statistically indistinguishable at a 95% confidence interval (DFd; n = 5). The same is also true for all irradiated samples compared with one another (data not shown). 121 100 50- A "$ C o 100 - S 50- B 0 1I LO E 100 50 C 0 2 4 Time, h Figure 6.2. Time courses of the change in the number of live cells (relative to the non-irradiated control) of E. coli (A) and S. aureus (B) at 30'C and of human TK6 spleen-derived lymphoblast cells at 37'C (C) with RFR of 2.45-GHz (squares), 915-MHz (triangles), and 13.56-MHz (circles) frequencies. For experimental conditions, see Methods. 122 Thus neither enzymatic activities nor cell viability/vitality are affected by exposure to RFR at any of the most commonly employed frequencies for RFID technology (Figs. 1 and 2). Enzymatic activity is arguably among the most sensitive measures of the intactness of protein structure' 0 . Therefore, a further study of the effect of RFR on the enzymes appears unwarranted because no effect on catalytic activity has been observed. Likewise, since growth is one of the pivotal cellular processes altered when a living cell is stressed, we have chosen this variable as our endpoint measurement and have observed no radiation-induced changes. Our conclusion that RFR induces no non-thermal effects in biochemical or biological systems agrees with the majority of rigorous studies on the subject, including very recent ones2 3' 24 In closing, the results of this study point to the safety of the RFID technology for use with pharmaceutical protein formulations and live cells alike. Radiation at 13.56 MHz, 915 MHz, or 2.45 GHz frequency induces no detectable non-thermal effect on biologics even under conditions far harsher than those to be encountered in real-life application of RFID technology in terms of irradiation exposure and power; nor is the use of this technology under real-life conditions likely to lead to a noticeable temperature rise. It is worth noting, however, that some studies have suggested that pulsed-wave (PW) RFR can mediate non-thermal effects through harmful permeabilization of cellular membranes of living organisms 2- . Since the present work deals exclusively with the effects of continuous RFR, those of PW RFR may warrant further investigation. Finally, it is important to note that uses of RFR unrelated to RFID technology, such as with cellular phones, should be evaluated independently as such uses have the potential to cause thermal effects. 123 C. Materials and Methods Materials. Dulbecco's PBS and heat-inactivated fetal bovine serum (FBS) were from Gibco (Grand Island, NY). Enzymes and chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and were of the highest purity commercially available. S. aureus (strain 33807), E. coli (strain CGSC4401), and human TK6 cells (spleenocyte-derived lymphoblasts; CRL-8015) were purchased from the American Type Culture Collection (ATCC) (Rockville, MD). Bacteria and human cell culture. A 500- or 250-pl suspension of E. coli or S. aureus (10" cells/ml), respectively, was added to 10 ml of sterilized pH 7 Luria-Bertani (LB) Broth (10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl per liter) in a sterile 50-ml centrifuge tube and shaken at 200 rpm and 370 C for 2-4 h (Innova 4200 Incubator Shaker; New Brunswick Scientifica, Edison, NJ). At an OD 600 of about 0.7, cells were harvested by centrifugation at 6,000 rpm for 10 min (Sorvall RC-5B; DuPont Instruments, Wilmington, DE), washed twice 7 with 0.85% NaCl, and diluted to 1 x 108 cells/ml for E. coli and 1 x 10 cells/ml for S. aureus in 0.85% aqueous NaCl. Human TK6 cells were cultured in an RPMI-1640 medium (ATCC) containing 10% FBS following the ATCC protocol' 7 . Briefly, TK6 non-adherent cells were maintained at a density of 3 x 105 cells/ml at 370 C and 5% CO 2 in a humidified environment with daily medium renewal. Cells were harvested by centrifugation at 2,000 rpm for 5 min (GS-6 Centrifuge, Beckman, Fullerton, CA), washed with Dulbecco's PBS, and diluted to 1 x 106 cells/ml in Dulbecco's PBS. Instrumentation. The RFR-generating instrument used was previously described by Cox et al. 7 ; in this work, no external incubator was used to maintain the sample temperature as the instrument regulated temperature better without it. The RFR frequencies were 13.56 MHz, 915 MHz, or 2.45 GHz at an incident power of- 21 W. SAR values were determined as described 124 by Felicia C.A.I. Cox in her Master of Engineering thesis' 8 . Briefly, the reflected and transmitted powers over the course of a 2-h exposure to RFR were subtracted from the incident power for a blank control. The same calculation was performed for an exposure with the sample to determine the amount of power absorbed. SAR values were then calculated and found to be at least 20 W/kg at 915 MHz and 200 W/kg at 2.45 GHz; at 13.56 MHz they could not be determined as the amount of radiation absorbed at this wavelength was within the noise of the instrument. RFR exposure. Aliquots (2 ml each) of (i) 1 mg/ml enzymes in PBS, pH 7.4; (ii) E. coli and S. aureus bacteria (1 x 108 and 1 x 107 cells/ml, respectively) in 0.85 % NaCl; or (iii) human TK6 cells at 1 x 106 cells/ml in Dulbecco's PBS were transferred to 2-ml vials (Agilent #51820714) for RFR exposure. Incubation temperatures were 50 30 ± 1*C for bacteria, and 37 1VC for the enzymes, 25 ± VC or l 0o C for the human cells. Aliquots were removed at time intervals of up to 24 h for the enzymes and up to 4 h for the cells and stored at 4'C until assayed. Enzymatic assays. HRP enzymatic activity was assessed using the Sigma-Aldrich protocol with the chromogenic substrate 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS)1 9 . Briefly, 1 mg/ml HRP samples were diluted to 25 nM in PBS at pH 7.4. The reducing substrate was prepared at a concentration of 9.1 mM in 100 mM Na acetate buffer (pH 5) and hydrogen peroxide at 0.5% in water. Diluted 10-gl HRP samples were mixed with 480 gl of ABTS and 10 gl of H2 0 2 . The rate of the reaction was monitored by measuring absorbance at 405 nm for 3 min (U-3010 spectrophotometer, Hitachi, Japan). f-Galactosidase (p-gal) enzymatic activity was assessed with o-nitrophenyl galactoside (ONPG) as a substrate using the Promega assay system 20 . Briefly, 50-pI aliquots of p-gal samples in PBS, pH 7.4, were mixed with 50 pl of Promega2X Assay Buffer (200 mM Na 125 phosphate, pH 7.3, 2 mM MgCl 2 , 100 mM p-mercaptoethanol, and 1.33 mg/ml ONPG), and the rate of the reaction was monitored by measuring absorbance at 420 nm for 3 min on a plate reader (SpectraMAX 190, Molecular Devices, Sunnyvale, CA). Cell viability and vitality assays. Following the RFR exposure, bacterial cell aliquots were analyzed for viability using the Molecular Probes Live/Dead BacLight Bacterial Viability Kit (L7012)21 . Briefly, control cells were grown as described above, washed with 0.85% aqueous NaCl, and resuspended in 2 ml of that salt solution. One milliliter of cell suspension was added to 20 ml of either 0.85% NaCl or 70% isopropyl alcohol, followed by incubation at room temperature for 1 h with brief shaking every 15 min. Negative and positive control cells (all-dead and all-live, respectively) were harvested by centrifugation at 6,000 rpm and adjusted to 1 x 108 cell/ml for E. coli and 1 x 107 cells/ml for S. aureus. Live and dead cells were mixed at different ratios to produce a standard curve from 100% to 0% live cells. Aliquots (50 pl) of controls and all irradiated samples were analyzed in triplicate according to the Molecular Probes plate reader protocol (SpectraMAX Gemini XS, Molecular Devices, Sunnyvale, CA). Human cell aliquots were analyzed for cell vitality using the Cell Vitality Assay Kit (Molecular Probes L3495 1) according to the manufacturer's protocol. Briefly, negative control cells were prepared by incubation of TK6 cells with 2 mM H2 0 2 under standard cell growth conditions for 4 h. Positive controls were freshly harvested cells. Live and dead cells were mixed at different ratios to produce a standard curve from 100% to 0% vitality. Aliquots (90 pl) of controls and all irradiated samples were analyzed in triplicate according to the manufacturer's fluorescent plate reader protocol. Statistical analysis. The data analyzed were in the form of changes of enzymatic activity or cellular viability/vitality (as % of control) as a function of time of the RFR exposure for 126 enzymes and cells, respectively. Unaffected samples would thus appear as horizontal lines. To ascertain whether the rates of change (slopes of the linear regressions) were in fact zero (null hypothesis Ho: P=O), a standard Analysis of Variance (ANOVA) was employed22 . Calculated F values were compared with critical tabulated F values from Zar to evaluate the null hypothesis at a 95% confidence interval with the appropriate degrees of freedom2. D. REFERENCES 1. FDA, Combatting Counterfeit Drugs; February 18, 2004. 2. Habash, R. W. Y., Bioeffects and TherapeuticApplications of Electromagnetic Energy. CRC Press: Boca Raton, FL, 2008. 3. Erwin, D. N., Mechanisms of biological effects of radiofrequency electromagnetic fields: an overview. Aviat Space Environ Med 1988, 59, A21 -A3 1. 4. Foster, K. R., Mechanisms of interaction of extremely low frequency electric fields and biological systems. Radiat Prot Dosimetry 2003, 106, 301-3 10. 5. Hardell, L.; Sage, C., Biological effects from electromagnetic field exposure and public exposure standards. Biomed Pharmacother2008, 62, 104-109. 6. Nittby, H.; Grafstrom, G.; Eberhardt, J. L.; Malmgren, L.; Brun, A.; Persson, B. R.; Salford, L. G., Radiofrequency and extremely low-frequency electromagnetic field effects on the blood-brain barrier. Electromagn Biol Med 2008, 27, 103-126. 7. Cox, F. I.; Sharma, V. K.; Klibanov, A. M.; Wu, B. I.; Jin, A.; Engels, D. W., A method to investigate non-thermal effects of radio frequency radiation on pharmaceuticals with relevance to RFID technology. ConfProc IEEE Eng Med Biol Soc 2006, 1, 4340-4343. 127 8. Foster, K. R.; Glaser, R., Thermal mechanisms of interaction of radiofrequency energy with biological systems with relevance to exposure guidelines. Health Phys 2007, 92, 609-620. 9. Mozhaev, V. V.; Berezin, I. V.; Martinek, K., Structure-stability relationship in proteins: fundamental tasks and strategy for the development of stabilized enzyme catalysts for biotechnology. CR C CritRev Biochem 1988, 23, 235-281. 10. Ahern, T. J.; Klibanov, A. M., Analysis of processes causing thermal inactivation of enzymes. Methods Biochem Anal 1988, 33, 91-127. 11. Hoyto, A.; Juutilainen, J.; Naarala, J., Ornithine decarboxylase activity of L929 cells after exposure to continuous wave or 50 Hz modulated radiofrequency radiation--a replication study. Bioelectromagnetics2007, 28, 501-508. 12. Cotgreave, I. A., Biological stress responses to radio frequency electromagnetic radiation: are mobile phones really so (heat) shocking? Arch Biochem Biophys 2005, 435, 227-240. 13. Guisasola, C.; Desco, M.; Millan, 0.; Villanueva, F. J.; Garcia-Barreno, P., Biological dosimetry of magnetic resonance imaging. JMagn Reson Imaging 2002, 15, 584-590. 14. Porcelli, M.; Cacciapuoti, G.; Fusco, S.; Massa, R.; d'Ambrosio, G.; Bertoldo, C.; De Rosa, M.; Zappia, V., Non-thermal effects of microwaves on proteins: thermophilic enzymes as model system. FEBS Lett 1997, 402, 102-106. 15. Schwarz, C.; Kratochvil, E.; Pilger, A.; Kuster, N.; Adlkofer, F.; Rudiger, H. W., Radiofrequency electromagnetic fields (UMTS, 1,950 MHz) induce genotoxic effects in vitro in human fibroblasts but not in lymphocytes. Int Arch Occup Environ Health 2008, 81, 75 5-767. 16. Vijayalaxmi; Obe, G., Controversial cytogenetic observations in mammalian somatic cells exposed to radiofrequency radiation. Radiat Res 2004, 162, 481-496. 128 17. Skopek, T. R.; Liber, H. L.; Penman, B. W.; Thilly, W. G., Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: Possibility for a rapid human cell mutation assay. Biochem Biophys Res Commun 1978, 84, 411-416. 18. Cox, F. C. A. I., MIT, Cambridge, MA, 2006. 19. Porstmann, B.; Porstmann, T.; Nugel, E., Comparison of chromogens for the determination of horseradish peroxidase as a marker in enzyme immunoassay. J Clin Chem Clin Biochem 1981, 19, 435-439. 20. Rosenthal, N., Identification of regulatory elements of cloned genes with functional assays. Methods Enzymol 1987, 152, 704-720. 21. Berney, M.; Hammes, F.; Bosshard, F.; Weilenmann, H. U.; Egli, T., Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight Kit in combination with flow cytometry. Appl Environ Microbiol 2007, 73, 3283-3290. 22. Zar, J. H., BiostatisticalAnalysis. 4th edn.; Prentice Hall: Upper Saddle River, N.J., 1999. 23. Obermayer, D.; Gutmann, B.; Kappe, C. 0., Microwave chemistry in silicon carbide reaction vials: separating thermal from nonthermal effects. Angew Chem Int Ed Engl 2009, 48, 8321-8324. 24. Microwave chemistry's thermal effect. Chem Engn News 2009, 87 (41), 43. 25. d'Ambrosio, G.; Massa, R.; Scarfi, M. R.; Zeni, 0., Cytogenetic damage in human lymphocytes following GMSK phase modulated microwave exposure. Bioelectromagnetics 2002, 23, 7-13. 26. Lai, H.; Singh, N. P., Acute low-intensity microwave exposure increases DNA single- strand breaks in rat brain cells. Bioelectromagnetics 1995, 16, 207-210. 129 27. Lee, S.; Johnson, D.; Dunbar, K.; Dong, H.; Ge, X.; Kim, Y. C.; Wing, C.; Jayathilaka, N.; Emmanuel, N.; Zhou, C. Q.; Gerber, H. L.; Tseng, C. C.; Wang, S. M., 2.45 GHz radiofrequency fields alter gene expression in cultured human cells. FEBS Lett 2005, 579, 48294836. 130 CURRICULUM VITAE EDUCATION Ph.D. in Biological Chemistry, Massachusetts Institute of Technology 2010 Thesis title: "Specific and Efficient In Vivo Delivery of DNA and siRNA by Polyethylenimine and Its Derivatives" Professor Alexander M. Klibanov Advisor: 2003 B.A. in Chemistry, Wheaton College EXPERIENCE Graduate Research Assistant, Massachusetts Institute of Technology 2003-2010 - Synthesized and chemically modified various polyethylenimines e Investigated the effect of chemical structure on the gene delivery efficacy and specificity of polyethylenimine in vivo in animal models - Investigated the effect of radio frequency radiation on biologics, both enzymes and living cells e Participated in the writing of NIH/NSF grant proposals and progress reports 2003-2008 Teaching Assistant, Massachusetts Institute of Technology Aided in the development of new laboratory protocol for undergraduate biochemistry module. Involved formulating new laboratory goals and experiments and subsequent optimization of the experiments and instruction of undergraduate students and their TAs. 2003-2008 Laboratory Safety Officer, Massachusetts Institute of Technology Ensured that the laboratory was in compliance with federal and state laws covering academic labs. Summer 2005 Research Intern, Alnylam Pharmaceuticals, Cambridge, MA Worked with company scientists to evaluate use of polyethylenimine for in vivo delivery of novel siRNA therapeutics against various targets. AWARDS/AFFILIATIONS BTP Training Grant Fellowship 2004-2007 2003-present Member of Phi Beta Kappa PUBLICATIONS Klibanov AM, Schefiliti JA (2004). Biotechnol Lett 26, 1103-6. Singleton PA et. al. (2009) Circ Res 104, 978-86. Fortune JA, Wu BI, Klibanov AM (2010). Biotechnol Prog Epublished ahead of print. Fortune JA, Danilov SM, Klibanov AM (2010). Biotechnol Lett (Submitted). 131