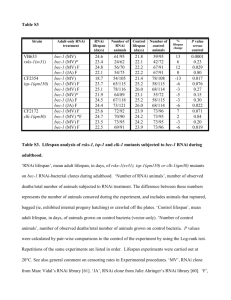
Identification of New Genes and Pathways C. elegans
advertisement

Identification of New Genes and Pathways that Act to Delay C. elegans Aging by Alina Berdichevsky B. Sc. Tel-Aviv University, 1999 M. Sc. Weitzmann Institute of Science, 2001 Submitted to the Department of Biology in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY Septemb r 2007 @ 2007 Alina Berdichevsky. All rights reserved. The author hereby grants MIT permission to reproduce and to distribute paper and electronic copies of this thesis document in whole or in part. Signature of Author: eep6artment of Biology, eptembf 2007 Certified by: ) H. Robert Horvi) Professor of Biology Thesis Supervisor Leonard Guarinte Professor of Biology Thesis supervisor Accepted by: Steven Bell Steven Bell Cnair of the Giraduate Committee Department of Biology i OF TEOHNOLOGY ARCH-VE•S FEB 2 2008 t i8zm LIBRARIES I 1 Identification of New Genes and Pathways that Act to Delay C. elegans Aging Alina Berdichevsky Abstract Aging of an organism is determined by both stochastic and genetic components. The importance of genes is illustrated by the discovery of single gene mutations that alter lifespans of species ranging from invertebrates C. elegans and D. melanogasterto mice. To better understand the mechanisms underlying genetic regulation of the rate of aging, we have studied aging and lifespan determination in C. elegans. We have investigated the mechanism of action of the sir-2.1 gene, which extends C. elegans lifespan when overexpressed. We discovered that sir-2.1 acts to promote longevity in parallel to low insulin signaling, in a stress-dependent pathway that converges with the insulin-like pathway on a forkhead transcription factor DAF-16. We discovered that 14-3-3 proteins play a role in C. elegans lifespan determination. 14-3-3 proteins interact with the SIR-2.1 protein, and the 14-3-3 genes par-5 andftt-2 are required for the longevity mediated by sir-2.I1overexpression, indicating that 14-3-3 proteins act to delay C. elegans aging in the sir-2.I1-dependent longevity pathway. To identify new genes that act to delay C. elegans aging we developed a genetic screen for mutants that prematurely accumulate the age-related fluorescent pigment lipofuscin. In this screen, we isolated lossof-function mutations in the gene kat-1. These mutations confer increased lipofuscin accumulation, short lifespan, and other abnormalities characteristic of premature aging. kat-1 encodes a ketoacyl thiolase involved in fatty acid beta-oxidation, suggesting that defects in fat metabolism can affect the regulation of aging. Thesis Supervisors: H. Robert Horvitz Professor of Biology Leonard Guarente Professor of Biology Acknowledgments I would like to thank my advisors Lenny Guarente and Bob Horvitz for their guidance, their constant encouragement and support, and for giving me the freedom of choice. Thanks for all members of my graduate committee during these years: Marcia Haigis, Dennis Kim, Terry OrrWeaver, and especially Chris Kaiser and Richard Hynes for their input and ideas and for reading this thesis. I'd like to thank many members of the Guarente and the Horvitz labs for their advice. Special thanks to Nick Bishop, Gil Blander, Brendan Galvin, Niels Ringstad and Hillel Schwartz for ideas, arguments, discussions, and critical reading of my manuscripts. Thanks to Beth Castor and Na An for making the lab feel more like home, and for all the help with strains, sequencing, and deletion hunt. I would like to thank Peter Reddien for the great time during my rotation and for recruiting me to the Horvitz lab. I'd like to thank all friends of mine both in US and abroad who shared a lot of happy and distressing moments. Brendan Galvin was my sarcastic roommate all these years, and I would like to thank him for always being in the right place at the right time for me and for all the balloons blown on weekends for Ron. Takashi Hirose was a great bay-mate for the last two years, and a good friend. I would like to thank my parents who managed to survive these six years so far from their daughter and grandchildren, and who have always encouraged me to go on. Igor, Ron, and Dana filled my life and I would not be able to carry on without them. Table of contents A bstract.......................................................................... 2 Acknowledgments................................................................. 3 Table of C ontents .................................................................. .4 Chapter 1. Introduction: Genetic and Molecular Pathways that Control A ging................... ................................................ 11 O verview .......................................................................... 12 The conserved features of aging ........................................... 13 Isolation of first mutants with abnormal aging rates... C. elegans Aging ...................... .............. 15 ..................................... 17 An insulin-like pathway controls the rate of aging in C. elegans And other organisms......................................................... 18 Dauer pathway .................................................. 18 Some but not all dauer pathway mutants age abnormally....19 Molecular cloning of dauer pathway genes reveals a genetic pathway that controls aging.........................20 The role of insulin-like pathway in aging isconserved........21 Aging, DNA damage, and the stress response ............................. 22 Sirtuins and lifespan determination......................................23 Mitochondria and the metabolic control of aging......................25 The "rate of living" hypothesis.... ............... ....... 25 C. elegans mitochondrial mutants are long-lived ............ 25 Metabolic activity during aging and calorie restriction.......26 Other genes and pathways that control C. elegans aging .............. 27 Mammalian genes that regulate aging .................................... 28 Conclusions .................................................................... 30 R eferences......................... .........................................31 Figure 1. Current theories proposed to explain the cause of aging .... 45 Figure 2. Genes from one of the two arms of the dauer pathway regulate C. elegans longevity......................................... 47 Figure 3. Insulin/IGF pathway regulates C. elegans longevity and stress-resistance....................... ...................... 49 Chapter 2. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend lifespan.......................................... Summary ................................................... Introduction .............................. R esults............................. ....... 51 ............. 52 ..................................... 53 ............................................ 56 C. elegans SIR-2.1 interacts with 14-3-3 proteins ......................... 56 C. elegans 14-3-3 proteins can interact with DAF-16 and act to retain DAF-16::GFP in the cytoplasm.............................57 sir-2.1 overexpression slows DAF-16::GFP exit from the nucleus during recovery from stress .................................. .............. 60 The 14-3-3 genesftt-2 and par-5 are required for sir-2.I1-mediated lifespan extension....................................61 sir-2.1, par-5 andftt-2 are not required for lifespan extension in animals with reduced insulin/IGF signaling ........................... 63 sir-2.I1can promote resistance to stress in anftt-2-dependentway.... 63 sir-2.1 can promote daf-16-dependent transcription in a manner dependent onftt-2.........................................................64 Following stress, SIR-2.1 interacts with DAF-16 in a 14-3-3-dependent fashion.................................. .................. 65 Discussion........................... ............................................ 67 C. elegans SIR-2.1 and 14-3-3 proteins can function in a stress-response pathway of longevity...............................67 SIR-2.1 and 14-3-3 may affect longevity by regulating DAF-16 Forkhead in parallel to the insulin-like pathway.....................69 A model for the roles of SIR-2.1 and 14-3-3 proteins in DAF-16 regulation and longevity .................................................... 70 Experimental procedures....................................................71 Antibody production, immunofluorescence, and cell fractionations ..71 Immunoprecipitation and western blots................................72 Lifespan, stress-resistance and RNAi analyses ......................... 73 Strains...........................................................................74 Molecular biology..............................................................77 Acknowledgments .............................................................. 78 R eferences ........................................................................... 79 T ables............................................................................... 86 Table 1. Reducing par-5 andftt-2 by RNAi results in nuclear accumulation of DAF-16::GFP........................................................ 86 Table 2. Analyses of lifespan experiments ............................... 87 Table 3. daf-16 targets upregulated in a sir-2.I1overexpresser ........ Figures ......................... ........................................ 88 89 Figure 1. C. elegans SIR-2.1 interacts with 14-3-3 proteins........89 Figure 2. C. elegans 14-3-3 proteins interact specifically with SIR-2.1 and DAF-16 ............................................. 92 Figure 3. 14-3-3 proteins affect DAF-16 localization..... ......... 94 Figure 4. Overexpression of sir-2.1 delays DAF-16 exit from the nucleus during recovery from stress .......................96 Figure 5. Overexpression of sir-2.1 is required for longevity and stress resistance of the pklsl642 transgenic animals.............98 Figure 6. ftt-2 and par-5 are required for sir-2.I1-mediated lifespan extension..................... ................................. 100 Figure 7. Overexpression of sir-2.1 leads to stress-resistance and activation of the daf-16 target gene sod-3 in an ftt-2-dependent manner.............................. 102 Figure 8. DAF-16 and SIR-2.1 interact in worms following stress, and this interaction depends on 14-3-3....................104 Figure 9. daf-2 RNAi results in low insulin-like signaling........ 106 Figure 10. A model for the roles of SIR-2.1 and 14-3-3 in DAF-16 regulation of stress resistance and lifespan ............108 Chapter 3. 3-Ketoacyl thiolase controls the rate of aging in C. elegans.. 110 Abstract ...................................................................... 111 Introduction ......................................................... R esults.......................... ...... 112 ......................................... 115 Aging pigment that accumulates in old worms fluoresces with a spectrum similar to that of mammalian lipofuscin ....... 115 Mutants with abnormal aging rates show abnormal fluorescence accumulation .............................. 116 Screen for mutants with increased lipofuscin..................116 Mutants isolated in the screen correspond to at least three complementation groups ........................................ 117 Three isolates premature display multiple attributes of aging... 118 Mapping and cloning of the gene identified by n4430........... 120 T02G5.8 encodes a conserved thiolase involved in fatty acid metabolism............................................. 121 kat-1 delays aging independently of most known longevity pathways ........................................................ 122 kat-1 mutants are sensitive to starvation .................................. 123 Other fatty-acid beta-oxidation genes affect aging ........................ 123 Discussion.......................................................................... 126 KAT-1 thiolase regulates C. elegans aging..............................126 Fatty acid oxidation in aging and disease ................................. 127 Does impaired fatty acid beta-oxidation cause aging? ...................... 128 Experimental procedures..........................................................130 Strains ........................................................................ 130 Isolation of mutants with high lipofuscin-like fluorescence ........ 130 Linkage group assessment ........................................ 131 Polymorphism mapping ................................. ................ 131 Transgenic animals ........................................ .......131 RNAi analyses ............................................ .......132 Allele sequences .......................................... ...... 132 Analyses of lifespan and stress-resistance ......................... 132 Behavioral assays ........................................ Electron Microscopy ......................................... 133 ...... 134 Acknowledgments .................................................... 135 References .......................................................... 135 Figure and tables ................................................................... 143 Table 1. Characterization of screen isolates............................ 143 Table 2. Epistasis Analyses of kat-1 with other aging pathways..... 144 Table 3. kat-1 mutants tissue aging...................................... 145 Table 4. Analyses of lipofuscin accumulation and lifespan in animals subjected to RNAi knock down of fatty acid beta- oxidation genes....................... ..................................... 146 Figure 1. Old worms accumulate autofluorescent pigment, and amount of the pigment correlates with lifespan in aging mutants.......147 Figure 2. A screen for mutants with high accumulation of lipofuscin-like fluorescent pigment....................................149 Figure 3. Mutations in gene specified by n4430, n4434, and n4435 result in increased fluorescence and short lifespan........ ............. 151 Figure 4. Behavior abnormalities of n4430, n4434, and n4435 mutants ......................................................... 153 Figure 5. kat-1 tissue aging...............................................155 Figure 6. Mapping and cloning of kat-1 ... ......... ......... .. . 157 Figure 7. kat-1 controls fluorescence accumulation and lifespan.......159 Figure 8. kat-1 encodes a conserved ketoacyl thiolase................161 Figure 9. kat-1 stress resistance ........................................... 163 Figure 10. 3-ketoacyl thiolase catalyzes the last step in fatty acid beta-oxidation pathway ..................................................... 165 Figure 11. Reducing fatty acid oxidation genes causes increased lipofuscin accumulation in C. elegans....................... .. 167 Addendum to Chapter 3. FLU-1 kynurenine monooxigenase controls intestinal fluorescence accumulation in C. elegans..............................169 A bstract... .......................... ............................................. 170 Results ............................................................................. 171 Caracterization of LGV isolates......................................171 Mapping and cloning offlu-1 ........................................ 171 flu-i encodes a conserved metabolic enzyme, kynurenine 3monooxygenase ..................................................... 173 D iscussion......................... .......................... .................. 175 Conclusion and future directions............................................177 R eferences.......................... ............................................ 178 Tables .................... ............................. .............. 179 Table 1. Three of the seven screen isolates with mutations that map to chromosome V have coding mutations in the gene encoding FLU-1 R07B7.5 kinurenine 3-monooxygenase............179 Figures............................. ............................................. 180 Figure 1. Chromosome V mutants accumulate high amounts of autofluorescence..................................................... . 180 Figure 2. Mapping and cloning offlu-1 ... ... ... ... .................. 182 Figure 3. flu-1 encodes a 3-kynurenine mooxygenase, a conserved metabolic enzyme.................................... 184 Figure 4. Kynurenine metabolic pathway in tryptophan m etabolism ..................................................... ....... 186 Appendix 1. A Stress Response Pathway Involving Sirtuins, Forkheads, and 14-3-3 Proteins ..................................................................... 188 Introduction: Sir2 and mechanisms of life span determination .... 189 14-3-3 proteins: new players in life span determination ........... 190 Mechanism of SIR-2.1 effect on life span..........................192 SIR-2.1 pathway for life span extension..............................193 Emerging group of DAF-16 co-factors ..... References............................... Figures.... ....................... ............................... 193 196 ............. .............. . .............. 200 Figure 1. SIR-2.1 mechanism of life span extension. ............. 200 Figure 2. DAF-16 can be activated by different cofactors..........202 Chapter 1 Introduction Genetic and Molecular Pathways that Control Aging Overview Aging of an organism is generally accompanied by a progressive dysfunction of organs and tissues causing physiological decline and eventually death. Several theories have been proposed to explain the causes of aging, including oxidative stress leading to accumulation of damage to proteins and DNA, mitochondrial dysfunction, and changes in metabolic rate (Figure 1). Free radicals, or reactive oxygen species, are by-products of cellular metabolism and accumulate during aging, causing oxidative stress and posing a threat to cells and tissues. A "rate of living" hypothesis suggests that the rate of aging is inversely proportional to metabolic rate, i.e. higher metabolic rate accelerates aging and limits lifespan. Mitochondria provide energy to cells, and mitochondrial dysfunction caused by an accumulation of oxidative damage has also been suggested as a cause of aging. Over the last three decades, experiments involving C. elegans and other organisms have provided evidence supporting many of these theories and have demonstrated that in addition to the stochastic component of the aging process certain genes regulate the timing of aging. Interfering with the function of such genes and the pathways they regulate can significantly change the aging rate of an organism, resulting in delayed aging and increased longevity or premature aging (progeria). At least some of the mechanisms that control lifespan are conserved from nematodes to mammals, as are certain metabolic, cellular and behavioral features of aging organisms. Studies of short-lived and long-lived mutants of simple organisms might advance our understanding of the genetic regulation of human aging and identify potential therapeutic targets that can be manipulated to delay aging and aging-related disease. The conserved features of aging Aging can be described as an increased probability of death with time, increased susceptibility to various diseases and decreased resistance to injury and infection (Knight 1995; Kirkwood and Austad 2000). While lifespans of individuals of the same species might differ substantially, a population shows a characteristic lifespan distribution, with a predictable mean and maximum lifespan (Piantanelli et al. 1992). During the first two thirds of an organism's lifespan, the mortality is very low, and it increases exponentially during the third phase of the lifespan (Witten and Eakin 1997). This mortality pattern is conserved in evolution, with species as different as nematodes and humans showing similar shapes of lifespan curves, although the absolute values for mean and maximum lifespan vary greatly (Finch 1990; Piantanelli et al. 1992). Historically, longevity has often been used as the ultimate measure of aging and the terms longevity and aging have been used interchangeably. However, a distinction has to be made between longevity, which is an interval of time between the birth and the death of an organism, and the rate of aging, which reflects the physiological decline of an organism during the second half of its lifespan, which precedes death. As aging is a complex phenomenon and lifespan can be limited by different factors, alteration of the rate of aging does not necessarily imply an effect on longevity (Kirkwood 1997). Also, numerous diseases limit longevity without affecting the rate of aging, complicating the distinction between short lifespans caused by premature aging and unrelated disease. Comprehensive studies of the rate of aging in simple organisms were limited because of the lack of reliable markers of aging in species other than mammals. Therefore, descriptive studies of aging in simple organisms are valuable, because they facilitate the distinction between old and sick animals, and allow one to follow the rate of aging. Aging-related diseases, i.e. diseases with time of onset in the late adulthood and exponential increase in prevalence with age, might differ in diverse species, but some basal aspects of organismal changes with age seem to be conserved (see below). Damage to cells, both from external sources (UV light, toxic chemicals) and internal metabolism products (reactive oxygen species) contributes to aging (Finch 1990; Shigenaga et al. 1994; Kirkwood and Austad 2000; Stevnsner et al. 2002; Kujoth et al. 2005). At the cellular level, many cell types stop dividing after a limited number of replications (Hayflick 1979). Such postreplicative cells accumulate damage that eventually causes malfunction and/or death of the cell. Old cells from different tissues show structural defects, such as the appearance of protein aggregates, necrosis-like membrane structures, and accumulation of fluorescent substances, often called lipofuscin ("fluorescent lipid") as oxidized lipids are major components of this material (Chen and Hillman 1999; Hinault et al. 2006; Widmer et al. 2006). These features are observed in many multicellular organisms, including worms, flies, and mammals (Miquel et al. 1974; Klass 1977; Clokey and Jacobson 1986; Hosokawa et al. 1994; Brunk and Terman 2002; Herndon et al. 2002; Gerstbrein et al. 2005). On a tissue level, reproductive organs, digestive organs and muscles seem to be universally affected in old age (Thomson and Keelan 1986; Herndon et al. 2002; Di Iorio et al. 2006). The nervous system, on the other hand, greatly suffers from aging in mammals, but might be relatively unaffected in nematodes (Garigan et al. 2002; Herndon et al. 2002; Miller and O'Callaghan 2003; Well et al. 2007). Conserved behavioral aspects of old age include progressive slowing of locomotion (which might be attributed to muscle failure), decrease in food intake, and cessation of reproduction (Klass 1977; Glenn et al. 2004; Huang et al. 2004). Finally, old age is associated with metabolic changes, which might result from defects in mitochondrial metabolic pathways controlling energy production. Mitochondrial DNA accumulates numerous mutations with age, leading to mitochondrial dysfunction, which results in reduction in oxidative phosphorylation efficiency, fat metabolism abnormalities, and promotes hyperglycemia and insulin resistance (Shigenaga et al. 1994; Wallace 2005; Reddy and Rao 2006). Isolation of first mutants with abnormal aging rates Until recently, it was broadly thought that aging stems from multiple stochastic events leading to multi-system failure, and therefore represents a passive deterioration process that is not controlled by individual genes or cellular functions. During the last three decades, scientists began questioning the idea that aging is entirely stochastic, because similar organisms with very comparable metabolic rates often had lifespans that differed two to five fold. The examples include mice and bats, rats and chipmunks, and even inbred and outbred mice (Austad and Fischer 1991). Several groups began searching for mutants in simple organisms with abnormal aging rates, and such mutants were indeed found. In 1983 Michael Klass screened for aging genes in C. elegans and identified several mutants with long lifespans (Klass 1983). Mutants in age-i, one of the genes isolated in Klass's screen, have long lifespans, increased resistance to stress, and impaired reproduction (Friedman and Johnson 1988). Later, age-i and other C. elegans mutants with abnormal aging were key for the identification of conserved genetic pathways that control aging and longevity. These pathways will be described below. Another exciting story was the discovery of methuselah gene in Drosophila,mutations in which resulted in 25% lifespan extension (Lin et al. 1998). Mutations in several Drosophilagenes were isolated in longevity screens and other genetic experiments. Many of these mutants exhibited increased resistance to stress in addition to extended lifespan (Seong et al. 2001; Helfand and Rogina 2003). Several groups have studied the genetic control of aging in budding yeast. In S. cerevisiae,a yeast mother cells can give rise to a limited number of daughter cells and subsequently stops dividing. This replicative aging can be followed and modulated genetically (Kaeberlein et al. 2001). Studies of yeast replicative aging identified a new class of aging genes: silent information regulators, or sirs (Kaeberlein et al. 1999). One gene from this group, sir2, was shown to also regulate longevity of fruit flies and nematodes (Tissenbaum and Guarente 2001; Rogina and Helfand 2004), suggesting that the sir2 role in the regulation of aging is conserved. In the last decade, several mouse mutants with abnormal aging rates were also identified, and targeted genetic manipulations were shown to cause longer lifespans or progeria in mice (Hekimi 2006). In particular, mitochondrial function and DNA repair seem to be key processes that determine the rate of aging in mice. Studies of aging and longevity of rodents have always been hampered by the fact that their lifespans are relatively long, ranging from three to five years, which makes genetic analysis of mutants very time-consuming. This difficulty might explain in part why the majority of current knowledge about the genetic control of aging and longevity came from the study of simple organisms, primarily the nematode C. elegans. C. elegans Aging The first experiments describing aging of the nematode C. elegans were conducted in the 1970s by Michael Klass (Klass 1977). C. elegans has a short lifespan, on average around three weeks, and, as noted above, shows a sharp increase in mortality rate in the last third of its lifespan, similar to that observed in mammals. Behavioral analysis of aging C. elegans nematodes showed that worms gradually slow the rates of locomotion, feeding, and defecation with age (Klass 1977; Huang et al. 2004). Other groups studied accumulation of the fluorescent pigment lipofuscin, which is known to accumulate with age in postreplicative cells in many organisms, including humans. The autofluorescent pigment accumulation in old nematodes was shown to localize to the secondary lysosomes, much like human lipofuscin, and the rate of lipofuscin accumulation was altered in aging mutants (Clokey and Jacobson 1986; Gerstbrein et al. 2005; Chapter 3 of this thesis). Excellent genetic tools developed for studies of C. elegans biology helped isolate numerous mutants with abnormal lifespans and significantly promoted our understanding of genetic and molecular pathways involved in aging. An insulin-like pathway controls the rate of aging in C. elegans and other organisms Dauer pathway Studies of C. elegans development identified mutants that subsequently defined the first genetic pathway involved in lifespan regulation in multiple species. Nematodes develop from a fertilized egg to adulthood through four larval stages L1 through L4, separated by moltings. Both the timing of development and developmental decisions in C. elegans are tightly regulated by temperature and food availability. When the environment becomes unfavorable, either because the temperature is too high, food is scarce, or the worms are too crowded, an alternative developmental pathway is activated, which is called the dauer pathway. Dauer is an alternative third larval stage, and the decision to become dauer has to be made before the transition between the L2 and L3 larval states. Dauers are structurally different from L3 larvae. They are extremely resistant to stress, have low metabolic rate, do not require feeding, and therefore are able to survive for extended periods of time in harsh environmental conditions (Riddle 1997). Genetic screens for mutants with abnormal dauer formation resulted in the identification of mutants that entered the dauer state even when conditions were favorable (Daf-c, dauerconstitutive mutants), and mutants that failed to initiate the dauer program even when conditions were unfavorable (Daf-d, dauer-defective mutants) (Albert and Riddle 1988). Epistasis experiments helped order the mutants into a dauer pathway, which consisted of two groups of genes acting in parallel to promote dauer formation (Larsen et al. 1995). Some but not all dauer pathway mutants age abnormally In 1993, Cynthia Kenyon made a historic observation that one of the dauer pathway mutants, a constitutive-dauer mutant daf-2 has an extremely long lifespan, more than twice as long as than that of the wild type (Kenyon et al. 1993). Both longevity and constitutive dauer formation of daf-2 mutants require a functional daf-16 gene, mutations in which result in a dauerdefective phenotype. Later it was shown that the longevity of age-i mutants also depends on daf-16, and that age-i animals are dauer-constitutive, similar to the daf-2 mutants, suggesting that the dauer pathway controls longevity in C. elegans (Larsen et al. 1995). Moreover, the dauerconstitutive mutants age-1 and daf-2 not only had longer lifespans but also maintained the appearance and behavior of young animals much longer than the wild type, arguing that these mutations indeed change the aging rate of mutant organisms (Kenyon et al. 1993; Herndon et al. 2002). Importantly, not all the mutants of the dauer pathway had lifespan abnormalities. In fact, only mutants in one of the two arms of the dauer pathway had abnormal lifespans (Figure 2), suggesting that aging and entry into dauer state are differentially controlled (Larsen et al. 1995). The timing of aging regulation was also different from that of dauer entry: even though the same genes were shown to control dauer formation and lifespan, dauer formation depended on signaling during embryogenesis and early larval development, whereas longevity effects required signaling during adulthood (Dillin et al. 2002a). 19 Molecular cloning of dauer pathway genes reveals a genetic pathway that controls aging It took several years before the molecular identities of the above longevity genes were uncovered. Studies primarily in the Ruvkun laboratory led to cloning of genes responsible for mutant phenotypes in daf-2, age-i, and daf-16 animals (Kimura et al. 1997; Lin et al. 1997; Paradis and Ruvkun 1998; Wolkow et al. 2002). It was shown that daf-2 encodes the C. elegans homolog of insulin/IGF receptor, age-1 encodes a PI3 kinase, and daf-16 encodes a forkhead transcription factor homologous to mammalian Foxo. These proteins are members of the insulin signaling pathway that has been studied extensively in mammalian cells (Cantley 2002). Subsequent genetic and molecular studies identified multiple additional members of the insulin/IGF pathway in C. elegans and in mammals, including akt-1 and akt2 genes encoding kinases homologous to the mammalian AKT/PKB, sgk-1 that encodes SGK kinase, PTEN phosphatase gene daf-18, and others (Braeckman et al. 2001). The biochemical events involved in signal transduction along the kinase cascade upon activation of the insulin pathway were elucidated primarily in mammalian cells (Saltiel and Kahn 2001), but experiments in C. elegans provided important information on the genetic interactions among insulin-like pathway genes, and studies in nematodes and mammals were complementary. The insulin/IGF-1 signaling (Figure 2) originates by binding of an insulin-like molecule to the DAF-2 transmembrane receptor, which activates a cascade of intracellular kinases including PI3 kinase AGE-1, AKT/PKB and the DAF-16 forkhead family transcription factor (Guarente and Kenyon 2000; Finch and Ruvkun 2001; Kenyon 2005). Mutations that decrease signaling by the DAF-2 receptor pathway cause an extension of the lifespan 20 of adults (and more frequent entry into the dauer developmental state by larvae) by triggering the nuclear localization of DAF-16 (Henderson and Johnson 2001; Lee et al. 2001; Lin et al. 2001). Such mutations also increase the resistance of animals to oxidative and genotoxic stress. The longevity and stress resistance engendered by a reduction in insulin/IGF signaling is abolished in mutants defective in daf-16 (Murakami and Johnson 1996; Lin et al. 1997; Paradis and Ruvkun 1998; Honda and Honda 1999). The forkhead transcription factor DAF- 16 can activate a plethora of target genes that promote longevity and stress-resistance (Cahill et al., 2001; Honda and Honda, 1999; Lee et al., 2003; McElwee et al., 2003). The role of the insulin-like pathway in aging is conserved A major impact of the studies of the C. elegans insulin-like pathway in worm lifespan determination was that the role of this pathway in aging regulation is evolutionary conserved. Studies of Drosophilashowed that mutants in the Drosophilainsulin receptor and insulin-receptor substrate were extremely long-lived and stress-resistant, much like C. elegans mutants (Helfand and Rogina 2003). Mutations in the insulin pathway in mammals cause insulin resistance, hyperglycemia and diabetes mellitus, and mutations in the IGF somatotrophic axis cause dwarfism. Recent findings from studies of mice, however, indicate that dwarf mice with mutations in growth hormone (GH), that have low levels of circulating IGF, and IGFR (IGF receptor) mutants have 25% to 60% longer lifespans than the wild type (Flurkey et al. 2001). Also, tissue specific mutations in the insulin receptor (IR) gene cause an increase in mean and maximum lifespan (Bluher et al. 2002). An important observation is that effects of insulin/IGF mutations on murine lifespan are often gender-specific. This phenomenon does not have an explanation at this point but suggests that hormonal signal circuitries and other gender-specific biological processes have to be further studied to understand the differential effects on lifespan of insulin pathway mutations in males and females. Aging, DNA damage, and the stress response Accumulation of damage to DNA and proteins resulting from oxidative stress as a consequence of normal metabolism is now a widely accepted explanation of the aging process. This hypothesis emerged from the ideas of Harman (1956) in the middle of the last century and was supported by a clear association between aging and the presence of oxidative stress and oxidative damage. When the level of damage reaches a certain threshold, cellular dysfunction occurs, eventually leading to organ dysfunction characteristic to aging. Therefore, the ability of the organism to control the rate of damage accumulation and to repair the damage is likely important for longevity and the rate of aging. In accordance with this idea, many human disorders caused by mutations in genes that control DNA maintenance and repair of DNA damage result in a phenotype that resembles premature aging and are referred to as progeroid disorders. Examples include Xeroderma Pigmentosum, which results from Nucleotide Excision Repair defects; Werner Syndrome, caused by mutations in a DNA helicase; and Ataxia Telangiectasia, caused by mutations in the ATM kinase required for repair of double-stranded DNA breaks (Shiloh and Rotman 1996; Lehmann 2003; Niedernhofer et al. 2006; Orren 2006). Importantly, patients with any of these disorders show many but not all attributes of aging and hence they are referred to as "segmental" progeroid syndromes. One common characteristic of these progeroid diseases is greatly increased cellular sensitivity to stress. In simple organisms, mutations that cause stress sensitivity often also cause short lifespan and other attributes of premature aging. Conversely, increased resistance to stress is a characteristic of long-lived mutants of the insulin-like pathway in nematodes and fruit flies (Honda and Honda 1999; Guarente and Kenyon 2000; Braeckman et al. 2001; Honda and Honda 2002). However, resistance to stress does not always parallel longevity. In fact, some long-lived mutants are less resistant to certain stressors, while increased resistance to stress is not always associated with longevity (Munoz and Riddle 2003). These results imply that variables other than stress response contribute to the determination of longevity and aging rate. Sirtuins and lifespan determination sir2 (silent information regulator 2) encodes a conserved protein deacetylase, which controls stress-resistance and lifespan determination in different organisms. The role of sir2 in aging was first discovered in S. cerevisiae,where it was identified in a screen for genes that control yeast replicative aging. In S. cerevisiae, extra copies of sir2 cause significant extension of replicative lifespan, while deletion of sir2 results in lifespan shortening (Kaeberlein et al. 1999). Furthermore, sir2 is required for the lifespan extension caused by calorie restriction in S. cerevisiae (Lin et al. 2002). Many organisms, including C. elegans, Drosophila,mice and 23 humans, have multiple sir2 homologs (sirtuins). The nematode genome encodes four sirtuins, sir-2.I1- sir-2.4, and humans have seven sirtuins, SIRT1-SIRT7, many of which have important roles in metabolism or genomic integrity (Frye 2000; Haigis and Guarente 2006). Some sirtuins, including the yeast ortholog Sir2 and the worm SIR2.1 possess NAD-dependent deacetylase activity (Imai et al. 2000; Viswanathan M., personal communication). Studies of the yeast and mammalian sirtuins indicated that they can bind and deacetylate histones as well as several other proteins with important roles in metabolism and stress response. Some examples of sirtuin-binding proteins are p53, forkhead transcription factors, and PGC-1 (Luo et al. 2001; Brunet et al. 2004; Daitoku et al. 2004; Rodgers et al. 2005; Gerhart-Hines et al. 2007). Enzymatic activity of Sir2 is tightly controlled by the NAD/NADH ratio, and it was proposed that Sir2 might act as a sensor of the metabolic state of the cell and transfer this information to downstream effectors (Guarente and Kenyon 2000). Introducing extra copies of the closest C. elegans homologue of the yeast sir2, sir-2.I1into C. elegans genome results in increased resistance to stress and 30-50% extension of the worm lifespan (Tissenbaum and Guarente 2001; Berdichevsky et al. 2006). Furthermore, deleting sir-2.I1 causes 15% shortening of lifespan (Viswanathan et al. 2005). Experiments by us and others indicate that C. elegans sir-2.I1acts to promote longevity via a stress-dependent pathway that converges with the insulin-like pathway at the level of transcription factor DAF-16 (Berdichevsky et al. 2006; Wang et al. 2006). Overexpressing the fly sir2 homologue also results in lifespan extension, and the mechanism underlying this effect in flies may be similar to calorie restriction (Rogina and Helfand 2004). Mitochondria and the metabolic control of aging The "rate of living" hypothesis Mitochondrial DNA is especially prone to the accumulation of damage and mutations caused by reactive oxygen species (ROS), byproducts of oxidative phosphorylation. Since ROS production is proportional to metabolic rate, it has been suggested that metabolic rate is coupled to lifespan (Pearl 1928). This "rate of living" hypothesis proposes that duration of lifespan inversely depends on the metabolic rate of an organism. C. elegans mitochondrial mutants are long-lived Recent studies of nematodes support the idea of an inverse relationship between metabolic rate and aging: mutants in many C. elegans mitochondrial genes that affect components of the oxidative phosphorylation chain and therefore inhibit energy production develop slowly, show decreased rates of locomotion, decreased brood sizes, and have up to 2-fold longer lifespans (Felkai et al. 1999; Feng et al. 2001; Dillin et al. 2002b). The mitochondrial gene clk-i encodes a member of the ubiquinone pathway, a component of ETC complex IV, and clk-i mutants are long-lived (Felkai et al. 1999). This effect is additive to that of loss-of-function of daf-2 insulinlike receptor gene, and double mutants between daf-2 and clk-i live much longer than any single mutant, indicating that mitochondrial function affects lifespan in parallel to the insulin-like pathway. Other C. elegans mitochondrial genes that when mutated cause long lifespan include isp-1, a complex IV protein; ctb-1, another complex IV protein encoded by mitochondrial genome (Feng et al. 2001); and lrs-2, a mitochondrial leucyl tRNA-synthetase (Lee et al. 2003). RNAi knockdown of several genes encoding mitochondrial ETC subunits extend the worm's lifespan. In a genome-wide RNAi screen for genes causing lifespan extension of C. elegans, approximately 20% of genes the knockdown of which promotes longevity encode proteins involved in mitochondrial electron transport chain (ETC), suggesting a pivotal role for the mitochondria and therefore metabolic rate in the regulation of longevity (Lee et al. 2003; Aguilaniu et al. 2005). Moreover, mice heterozygous for the mClkl, the homolog of C. elegans clk-I gene were reported to have extended lifespans (Liu et al. 2005). Metabolic activity during aging and calorie restriction While in some mutants reduced metabolic activity is associated with increased longevity, aging organisms also show reduced metabolism. A comprehensive study of C. elegans metabolic activity showed a linear reduction in oxygen consumption and ATP content with age (Braeckman et al. 2002). Similar results were obtained from studies of mouse, rat, and human tissues (Meacci et al. 1993; Imre et al. 2000; Martin et al. 2007). Metabolic defects associated with mammalian aging lead to several disorders, such as diabetes mellitus and metabolic syndrome (Shigenaga et al. 1994; Toth and Tchernof 2000; Saltiel and Kahn 2001). The most well-known means to extend lifespan is to limit food consumption. Calorie restriction (CR) promotes longevity and delays agingrelated disorders in all organisms tested, including worms, flies, mice and primates (Koubova and Guarente 2003). It had been suggested in the past that CR causes lifespan extension by reducing metabolic rate, much like mitochondrial mutants, therefore limiting damage to cells and delaying tissue and organismal aging. Recent results, however, do not support this hypothesis. It appears that at least in some species, calorie-restriction does not lead to reduced metabolism. Studies of yeast and worms show that CR actually causes increased respiration and oxygen consumption (Houthoofd et al. 2002a; Houthoofd et al. 2002b; Lin et al. 2002; Bishop and Guarente 2007). Additionally, mammals subjected to CR are more active than agematched controls and do not display differences in body temperature or ATP production (McCarter RJM 1997; Weed et al. 1997). These findings argue that reduced metabolic rate is not always associated with delayed aging and longer lifespan. The molecular mechanisms underlying the CR effect on aging are largely unknown. CR extends lifespan independently of the insulin-like signaling, as insulin-like pathway mutants respond normally to CR (Houthoofd et al. 2003). sir2 homologs are required for CR-induced increase in respiration and lifespan in yeast, and possibly in flies (Lin et al. 2000; Rogina and Helfand 2004). Studies are underway to identify genes and pathways that play a role in the beneficial effects of calorie restriction on aging and aging-related disease (Bishop and Guarente 2007; Panowski et al. 2007). Other genes and pathways that control C. elegans aging Directly reducing reproduction can also have a positive effect on C. elegans lifespan, as ablation of worm germline cell precursors by laser or mutation results in lifespan extension (Hsin and Kenyon 1999), suggesting that signals from the germline limit lifespan in worms. The germline signaling effect on lifespan requires functional DAF-16 Foxo, but is likely independent from insulin/IGF receptor DAF-2, since reducing insulin-like pathway activity in the germline signaling mutants causes synergistic effect on lifespan (Arantes-Oliveira et al. 2003). Nematodes can sense aspects of their environment, such as temperature and food availability, and modulate their behavior accordingly. C. elegans mutants with defects in sensory perception are often long-lived (Apfeld and Kenyon 1999; Alcedo and Kenyon 2004). This effect is unlikely to be caused by food restriction, because CR can extend lifespans of the C. elegans sensory mutants to the same extent as of the wild type (Bishop N., personal communication). Recently, several reports indicated that inhibiting translation in adult nematodes results in increased lifespan (Hansen et al. 2007; Syntichaki et al. 2007). Also, autophagy has been shown to play a role in lifespan determination, as longevity of insulin-like pathway mutants requires the becI gene, which is involved in autophagy, and overexressing autophagy genes extends worm lifespan (Melendez et al. 2003). Taken together, these findings suggest that deregulation of protein synthesis and/or accumulation of misfolded proteins in aging nematodes contribute to aging, and preventing protein accumulation by either inducing autophagy or attenuating protein synthesis can delay aging. Mammalian genes that regulate aging. The relatively long lifespans of mice and rats have limited the genetic analysis of aging pathways in mammals, and only a small number of genes have been shown so far to affect mammalian aging. On the other hand, the interpretation of progeroid mutants is easier in rodents than in invertebrates, because old rodents display more markers characteristic of human aging. Many human progeroid disorders caused primarily by defects in DNA repair have been recapitulated in mice, including Werner syndrome, Ataxia Telangiectasia, Xeroderma Pigmentosum (XP), and Cockayne syndrome (van de Ven et al. 2007). For XP mice, analysis of the progeroid phenotype of mutants was extended beyond anatomical descriptions, and transcriptional profiling of mutant animals showed remarkable similarity between profiles of progeroid mutants and old animals (Niedernhofer et al. 2006; van der Pluijm et al. 2006). A gain-of-function mutation in the p53 protein, which is involved in maintaining genomic stability, also causes a progeroid phenotype in mice, possibly because of increased programmed cell death leading to degeneration of various organs and tissues (Tyner et al. 2002). A mitochondrial link to aging was also found in mice: overexpression of a mutator DNA polymerase in mitochondria leads to an increased number of mutations in mitochondrial DNA, and premature aging (Kujoth et al. 2005). By contrast, superoxide dismutase mimetics and overexpression of peroxisomal catalase in mitochondria extend murine lifespan, presumably by reducing the rate of reactive oxygen species accumulation (Melov et al. 2000; Schriner et al. 2005). For some human and mouse progeroid syndromes, mutations responsible for the phenotype may be identified, but the mechanisms by which they act or pathways that they regulate are unknown. Examples include human Hutchinson-Gilford progeroid syndrome resulting from mutations in the LaminA gene (Eriksson et al. 2003), and the apparently causal role of the Klotho protein in mouse aging (Kurosu et al. 2005; Masuda et al. 2005). 29 Conclusions This is a very exciting time for studies of the genetic regulation of aging. Numerous pathways that affect longevity have recently been identified. It is clear that genes involved in responses to DNA damage, mitochondrial function and metabolic pathways contribute to the determination of lifespan. Studies of simple organisms like the nematode C. elegans are extremely valuable for the identification of new pathways that regulate longevity and for understanding the genetic interactions among different longevity pathways. It is tempting to speculate that one or more of these pathways act in humans and can be manipulated to delay aging or aging-related disease. One limitation of the field is its focus on genes that limit lifespan, mutations in which cause increased longevity. There are few comprehensive studies of genes that act to slow the rate of aging, primarily caused by the complication of distinguishing progeria from sickness in simple organisms. Development of reliable biomarkers of aging should solve this problem and facilitate studies of genes that promote longevity. This thesis describes three such genes, C. elegans sir-2.1,ftt-2, and kat-1, which act in new pathways of aging regulation. References Aguilaniu, H., Durieux, J., and Dillin, A. 2005. Metabolism, ubiquinone synthesis, and longevity. Genes Dev 19(20): 2399-2406. Albert, P.S. and Riddle, D.L. 1988. Mutants of Caenorhabditiselegans that form dauer-like larvae. Dev Biol 126(2): 270-293. Alcedo, J. and Kenyon, C. 2004. Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron 41(1): 45-55. Apfeld, J. and Kenyon, C. 1999. Regulation of lifespan by sensory perception in Caenorhabditiselegans. Nature 402(6763): 804-809. Arantes-Oliveira, N., Berman, J.R., and Kenyon, C. 2003. Healthy animals with extreme longevity. Science 302(5645): 611. Austad, S.N. and Fischer, K.E. 1991. Mammalian aging, metabolism, and ecology: evidence from the bats and marsupials. J Gerontol 46(2): B47-53. Berdichevsky, A., Viswanathan, M., Horvitz, H.R., and Guarente, L. 2006. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125(6): 1165-1177. Bishop, N.A. and Guarente, L. 2007. Two neurons mediate diet-restrictioninduced longevity in C. elegans. Nature 447(7144): 545-549. Bluher, M., Michael, M.D., Peroni, O.D., Ueki, K., Carter, N., Kahn, B.B., and Kahn, C.R. 2002. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell 3(1): 25-38. Braeckman, B.P., Houthoofd, K., and Vanfleteren, J.R. 2001. Insulin-like signaling, metabolism, stress resistance and aging in Caenorhabditis elegans. Mech Ageing Dev 122(7): 673-693. -. 2002. Assessing metabolic activity in aging Caenorhabditiselegans: concepts and controversies. Aging Cell 1(2): 82-88; discussion 102103. Brunet, A., Sweeney, L.B., Sturgill, J.F., Chua, K.F., Greer, P.L., Lin, Y., Tran, H., Ross, S.E., Mostoslavsky, R., Cohen, H.Y., Hu, L.S., Cheng, H.L., Jedrychowski, M.P., Gygi, S.P., Sinclair, D.A., Alt, F.W., and Greenberg, M.E. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303(5666): 2011-2015. Brunk, U.T. and Terman, A. 2002. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. FreeRadic Biol Med 33(5): 611-619. Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science 296(5573): 1655-1657. Chen, S. and Hillman, D.E. 1999. Dying-back of Purkinje cell dendrites with synapse loss in aging rats. JNeurocytol 28(3): 187-196. Clokey, G.V. and Jacobson, L.A. 1986. The autofluorescent "lipofuscin granules" in the intestinal cells of Caenorhabditiselegans are secondary lysosomes. Mech Ageing Dev 35(1): 79-94. Daitoku, H., Hatta, M., Matsuzaki, H., Aratani, S., Ohshima, T., Miyagishi, M., Nakajima, T., and Fukamizu, A. 2004. Silent information regulator 2 potentiates Foxo 1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA 101(27): 1004210047. Di lorio, A., Abate, M., Di Renzo, D., Russolillo, A., Battaglini, C., Ripari, P., Saggini, R., Paganelli, R., and Abate, G. 2006. Sarcopenia: age- 32 related skeletal muscle changes from determinants to physical disability. Int JlmmunopatholPharmacol19(4): 703-719. Dillin, A., Crawford, D.K., and Kenyon, C. 2002a. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298(5594): 830-834. Dillin, A., Hsu, A.L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A.G., Kamath, R.S., Ahringer, J., and Kenyon, C. 2002b. Rates of behavior and aging specified by mitochondrial function during development. Science 298(5602): 2398-2401. Eriksson, M., Brown, W.T., Gordon, L.B., Glynn, M.W., Singer, J., Scott, L., Erdos, M.R., Robbins, C.M., Moses, T.Y., Berglund, P., Dutra, A., Pak, E., Durkin, S., Csoka, A.B., Boehnke, M., Glover, T.W., and Collins, F.S. 2003. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423(6937): 293-298. Felkai, S., Ewbank, J.J., Lemieux, J., Labbe, J.C., Brown, G.G., and Hekimi, S. 1999. CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditiselegans. Embo J 18(7): 1783-1792. Feng, J., Bussiere, F., and Hekimi, S. 2001. Mitochondrial electron transport is a key determinant of life span in Caenorhabditiselegans. Dev Cell 1(5): 633-644. Finch, C. 1990. Longevity, Senescence, and the Genome. . Chicago,IL, USA: Chicago UniversityPress. Finch, C.E. and Ruvkun, G. 2001. The genetics of aging. Annu Rev Genomics Hum Genet 2: 435-462. Flurkey, K., Papaconstantinou, J., Miller, R.A., and Harrison, D.E. 2001. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci USA 98(12): 6736-6741. Friedman, D.B. and Johnson, T.E. 1988. A mutation in the age-i gene in Caenorhabditiselegans lengthens life and reduces hermaphrodite fertility. Genetics 118(1): 75-86. Frye, R.A. 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273(2): 793-798. Garigan, D., Hsu, A.L., Fraser, A.G., Kamath, R.S., Ahringer, J., and Kenyon, C. 2002. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161(3): 1101-1112. Gerhart-Hines, Z., Rodgers, J.T., Bare, O., Lerin, C., Kim, S.H., Mostoslavsky, R., Alt, F.W., Wu, Z., and Puigserver, P. 2007. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-lalpha. Embo J26(7): 1913-1923. Gerstbrein, B., Stamatas, G., Kollias, N., and Driscoll, M. 2005. In vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditiselegans. Aging Cell 4(3): 127-137. Glenn, C.F., Chow, D.K., David, L., Cooke, C.A., Gami, M.S., Iser, W.B., Hanselman, K.B., Goldberg, I.G., and Wolkow, C.A. 2004. Behavioral deficits during early stages of aging in Caenorhabditis elegans result from locomotory deficits possibly linked to muscle frailty. J GerontolA Biol Sci Med Sci 59(12): 1251-1260. Guarente, L. and Kenyon, C. 2000. Genetic pathways that regulate ageing in model organisms. Nature 408(6809): 255-262. 34 Haigis, M.C. and Guarente, L.P. 2006. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev 20(21): 2913-2921. Hansen, M., Taubert, S., Crawford, D., Libina, N., Lee, S.J., and Kenyon, C. 2007. Lifespan extension by conditions that inhibit translation in Caenorhabditiselegans. Aging Cell 6(1): 95-110. Harman, D. 1956. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11(3): 298-300. Hayflick, L. 1979. Progress in cytogerontology. Mech Ageing Dev 9(5-6): 393-408. Hekimi, S. 2006. How genetic analysis tests theories of animal aging. Nat Genet 38(9): 985-991. Helfand, S.L. and Rogina, B. 2003. Genetics of aging in the fruit fly, Drosophilamelanogaster.Annu Rev Genet 37: 329-348. Henderson, S.T. and Johnson, T.E. 2001. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditiselegans. Curr Biol 11(24): 1975-1980. Hemdon, L.A., Schmeissner, P.J., Dudaronek, J.M., Brown, P.A., Listner, K.M., Sakano, Y., Paupard, M.C., Hall, D.H., and Driscoll, M. 2002. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419(6909): 808-814. Hinault, M.P., Ben-Zvi, A., and Goloubinoff, P. 2006. Chaperones and proteases: cellular fold-controlling factors of proteins in neurodegenerative diseases and aging. JMol Neurosci 30(3): 249265. Honda, Y. and Honda, S. 1999. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditiselegans. FasebJ 13(11): 13851393. -. 2002. Oxidative stress and life span determination in the nematode Caenorhabditiselegans. Ann N YAcad Sci 959: 466-474. Hosokawa, H., Ishii, N., Ishida, H., Ichimori, K., Nakazawa, H., and Suzuki, K. 1994. Rapid accumulation of fluorescent material with aging in an oxygen-sensitive mutant mev-1 of Caenorhabditis elegans. Mech Ageing Dev 74(3): 161-170. Houthoofd, K., Braeckman, B.P., Johnson, T.E., and Vanfleteren, J.R. 2003. Life extension via dietary restriction is independent of the Ins/IGF-1 signalling pathway in Caenorhabditis elegans. Exp Gerontol 38(9): 947-954. Houthoofd, K., Braeckman, B.P., Lenaerts, I., Brys, K., De Vreese, A., Van Eygen, S., and Vanfleteren, J.R. 2002a. Axenic growth up-regulates mass-specific metabolic rate, stress resistance, and extends life span in Caenorhabditiselegans. Exp Gerontol 37(12): 1371-1378. -. 2002b. No reduction of metabolic rate in food restricted Caenorhabditis elegans. Exp Gerontol 37(12): 1359-1369. Hsin, H. and Kenyon, C. 1999. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399(6734): 362-366. Huang, C., Xiong, C., and Kornfeld, K. 2004. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditiselegans. Proc Natl Acad Sci USA 101(21): 80848089. Imai, S., Armstrong, C.M., Kaeberlein, M., and Guarente, L. 2000. Transcriptional silencing and longevity protein Sir2 is an NADdependent histone deacetylase. Nature 403(6771): 795-800. 36 Imre, S., Firbas, J.H., and Noble, R.C. 2000. Reduced lipid peroxidation capacity and desaturation as biochemical markers of aging. Arch Gerontol Geriatr31(1): 5-12. Kaeberlein, M., McVey, M., and Guarente, L. 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13(19): 2570-2580. -. 2001. Using yeast to discover the fountain of youth. Sci Aging Knowledge Environ 2001(1): pel. Kenyon, C. 2005. The plasticity of aging: insights from long-lived mutants. Cell 120(4): 449-460. Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. 1993. A C. elegans mutant that lives twice as long as wild type. Nature 366(6454): 461-464. Kimura, K.D., Tissenbaum, H.A., Liu, Y., and Ruvkun, G. 1997. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditiselegans. Science 277(5328): 942-946. Kirkwood, T.B. 1997. The origins of human ageing. Philos Trans R Soc Lond B Biol Sci 352(1363): 1765-1772. Kirkwood, T.B. and Austad, S.N. 2000. Why do we age? Nature 408(6809): 233-238. Klass, M.R. 1977. Aging in the nematode Caenorhabditiselegans: major biological and environmental factors influencing life span. Mech Ageing Dev 6(6): 413-429. -. 1983. A method for the isolation of longevity mutants in the nematode Caenorhabditiselegans and initial results. Mech Ageing Dev 22(3-4): 279-286. 37 Knight, J.A. 1995. The process and theories of aging. Ann Clin Lab Sci 25(1): 1-12. Koubova, J. and Guarente, L. 2003. How does calorie restriction work? Genes Dev 17(3): 313-321. Kujoth, G.C., Hiona, A., Pugh, T.D., Someya, S., Panzer, K., Wohlgemuth, S.E., Hofer, T., Seo, A.Y., Sullivan, R., Jobling, W.A., Morrow, J.D., Van Remmen, H., Sedivy, J.M., Yamasoba, T., Tanokura, M., Weindruch, R., Leeuwenburgh, C., and Prolla, T.A. 2005. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309(5733): 481-484. Kurosu, H., Yamamoto, M., Clark, J.D., Pastor, J.V., Nandi, A., Gurnani, P., McGuinness, O.P., Chikuda, H., Yamaguchi, M., Kawaguchi, H., Shimomura, I., Takayama, Y., Herz, J., Kahn, C.R., Rosenblatt, K.P., and Kuro-o, M. 2005. Suppression of aging in mice by the hormone Klotho. Science 309(5742): 1829-1833. Larsen, P.L., Albert, P.S., and Riddle, D.L. 1995. Genes that regulate both development and longevity in Caenorhabditiselegans. Genetics 139(4): 1567-1583. Lee, R.Y., Hench, J., and Ruvkun, G. 2001. Regulation of C. elegans DAF16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol 11(24): 1950-1957. Lee, S.S., Lee, R.Y., Fraser, A.G., Kamath, R.S., Ahringer, J., and Ruvkun, G. 2003. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33(1): 40-48. Lehmann, A.R. 2003. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85(11): 1101-1111. 38 Lin, K., Dorman, J.B., Rodan, A., and Kenyon, C. 1997. daf-16: An HNF3/forkhead family member that can function to double the life-span of Caenorhabditiselegans. Science 278(5341): 1319-1322. Lin, K., Hsin, H., Libina, N., and Kenyon, C. 2001. Regulation of the Caenorhabditiselegans longevity protein DAF- 16 by insulin/IGF- 1 and germline signaling. Nat Genet 28(2): 139-145. Lin, S.J., Kaeberlein, M., Andalis, A.A., Sturtz, L.A., Defossez, P.A., Culotta, V.C., Fink, G.R., and Guarente, L. 2002. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418(6895): 344-348. Lin, Y.J., Seroude, L., and Benzer, S. 1998. Extended life-span and stress resistance in the Drosophilamutant methuselah. Science 282(5390): 943-946. Liu, X., Jiang, N., Hughes, B., Bigras, E., Shoubridge, E., and Hekimi, S. 2005. Evolutionary conservation of the clk-l-dependent mechanism of longevity: loss of mclkl increases cellular fitness and lifespan in mice. Genes Dev 19(20): 2424-2434. Luo, J., Nikolaev, A.Y., Imai, S., Chen, D., Su, F., Shiloh, A., Guarente, L., and Gu, W. 2001. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107(2): 137-148. Martin, C., Dubouchaud, H., Mosoni, L., Chardigny, J.M., Oudot, A., Fontaine, E., Vergely, C., Keriel, C., Rochette, L., Leverve, X., and Demaison, L. 2007. Abnormalities of mitochondrial functioning can partly explain the metabolic disorders encountered in sarcopenic gastrocnemius. Aging Cell 6(2): 165-177. Masuda, H., Chikuda, H., Suga, T., Kawaguchi, H., and Kuro-o, M. 2005. Regulation of multiple ageing-like phenotypes by inducible klotho 39 gene expression in klotho mutant mice. Mech Ageing Dev 126(12): 1274-1283. McCarter RJM, S.I., Ikeno Y, Higami Y, Hubbard GB, Yu BP, McMahan CA. 1997. Physical activity as a factor in the action of dietary restriction on aging: Effects in Fischer 344 rats. AGING-CLINICAL AND EXPERIMENTAL RESEARCH 9((1-2)): 73-79. Meacci, E., Vannini, F., Vasta, V., Farnararo, M., and Bruni, P. 1993. Effect of aging on insulin regulation of fructose 2,6-bisphosphate metabolism in human fibroblasts. Biochem Mol Biol Int 30(1): 91-98. Melendez, A., Talloczy, Z., Seaman, M., Eskelinen, E.L., Hall, D.H., and Levine, B. 2003. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301(5638): 1387-1391. Melov, S., Ravenscroft, J., Malik, S., Gill, M.S., Walker, D.W., Clayton, P.E., Wallace, D.C., Malfroy, B., Doctrow, S.R., and Lithgow, G.J. 2000. Extension of life-span with superoxide dismutase/catalase mimetics. Science 289(5484): 1567-1569. Miller, D.B. and O'Callaghan, J.P. 2003. Effects of aging and stress on hippocampal structure and function. Metabolism 52(10 Suppl 2): 1721. Miquel, J., Tappel, A.L., Dillard, C.J., Herman, M.M., and Bensch, K.G. 1974. Fluorescent products and lysosomal components in aging Drosophilamelanogaster.J Gerontol 29(6): 622-637. Munoz, M.J. and Riddle, D.L. 2003. Positive selection of Caenorhabditis elegans mutants with increased stress resistance and longevity. Genetics 163(1): 171-180. Murakami, S. and Johnson, T.E. 1996. A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditiselegans. Genetics 143(3): 1207-1218. Niedernhofer, L.J., Garinis, G.A., Raams, A., Lalai, A.S., Robinson, A.R., Appeldoorn, E., Odijk, H., Oostendorp, R., Ahmad, A., van Leeuwen, W., Theil, A.F., Vermeulen, W., van der Horst, G.T., Meinecke, P., Kleijer, W.J., Vijg, J., Jaspers, N.G., and Hoeijmakers, J.H. 2006. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444(7122): 1038-1043. Orren, D.K. 2006. Werner syndrome: molecular insights into the relationships between defective DNA metabolism, genomic instability, cancer and aging. FrontBiosci 11: 2657-2671. Paradis, S. and Ruvkun, G. 1998. Caenorhabditiselegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 12(16): 2488-2498. Pearl, R. 1928. The rate of living: Being an account of some experimental studies on the biology of life duration. . AA Knopf New York. Piantanelli, L., Rossolini, G., and Nisbet, R. 1992. Modelling survivorship kinetics: a two-parameter model. Gerontology 38(1-2): 30-40. Reddy, J.K. and Rao, M.S. 2006. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am JPhysiol GastrointestLiver Physiol 290(5): G852-858. Riddle, D., Blumenthal, T, Meyer, BJ, Priess, J. 1997. C. elegans II. CSHL Press, Cold Spring Harbor,New York. Rodgers, J.T., Lerin, C., Haas, W., Gygi, S.P., Spiegelman, B.M., and Puigserver, P. 2005. Nutrient control of glucose homeostasis through a complex of PGC-lalpha and SIRT1. Nature 434(7029): 113-118. Rogina, B. and Helfand, S.L. 2004. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U SA 101(45): 15998-16003. Saltiel, A.R. and Kahn, C.R. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414(6865): 799-806. Schriner, S.E., Linford, N.J., Martin, G.M., Treuting, P., Ogburn, C.E., Emond, M., Coskun, P.E., Ladiges, W., Wolf, N., Van Remmen, H., Wallace, D.C., and Rabinovitch, P.S. 2005. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308(5730): 1909-1911. Seong, K.H., Ogashiwa, T., Matsuo, T., Fuyama, Y., and Aigaki, T. 2001. Application of the gene search system to screen for longevity genes in Drosophila.Biogerontology2(3): 209-217. Shigenaga, M.K., Hagen, T.M., and Ames, B.N. 1994. Oxidative damage and mitochondrial decay in aging. ProcNatl Acad Sci US A 91(23): 10771-10778. Shiloh, Y. and Rotman, G. 1996. Ataxia-telangiectasia and the ATM gene: linking neurodegeneration, immunodeficiency, and cancer to cell cycle checkpoints. J Clin Immunol 16(5): 254-260. Stevnsner, T., Thorslund, T., de Souza-Pinto, N.C., and Bohr, V.A. 2002. Mitochondrial repair of 8-oxoguanine and changes with aging. Exp Gerontol 37(10-11): 1189-1196. Syntichaki, P., Troulinaki, K., and Tavernarakis, N. 2007. eIF4E function in somatic cells modulates ageing in Caenorhabditiselegans. Nature 445(7130): 922-926. Thomson, A.B. and Keelan, M. 1986. The aging gut. Can JPhysiol Pharmacol64(1): 30-38. Tissenbaum, H.A. and Guarente, L. 2001. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditiselegans. Nature 410(6825): 227230. Toth, M.J. and Tchernof, A. 2000. Lipid metabolism in the elderly. Eur J Clin Nutr 54 Suppl 3: S121-125. Tyner, S.D., Venkatachalam, S., Choi, J., Jones, S., Ghebranious, N., Igelmann, H., Lu, X., Soron, G., Cooper, B., Brayton, C., Hee Park, S., Thompson, T., Karsenty, G., Bradley, A., and Donehower, L.A. 2002. p53 mutant mice that display early ageing-associated phenotypes. Nature 415(6867): 45-53. van de Ven, M., Andressoo, J.O., Holcomb, V.B., Hasty, P., Suh, Y., van Steeg, H., Garinis, G.A., Hoeijmakers, J.H., and Mitchell, J.R. 2007. Extended longevity mechanisms in short-lived progeroid mice: identification of a preservative stress response associated with successful aging. Mech Ageing Dev 128(1): 58-63. van der Pluijm, I., Garinis, G.A., Brandt, R.M., Gorgels, T.G., Wijnhoven, S.W., Diderich, K.E., de Wit, J., Mitchell, J.R., van Oostrom, C., Beems, R., Niedernhofer, L.J., Velasco, S., Friedberg, E.C., Tanaka, K., van Steeg, H., Hoeijmakers, J.H., and van der Horst, G.T. 2006. Impaired genome maintenance suppresses the growth hormone-insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol 5(1): e2. Viswanathan, M., Kim, S.K., Berdichevsky, A., and Guarente, L. 2005. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell 9(5): 605-615. Wallace, D.C. 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359-407. Wang, Y., Oh, S.W., Deplancke, B., Luo, J., Walhout, A.J., and Tissenbaum, H.A. 2006. C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech Ageing Dev 127(9): 741-747. Weed, J.L., Lane, M.A., Roth, G.S., Speer, D.L., and Ingram, D.K. 1997. Activity measures in rhesus monkeys on long-term calorie restriction. Physiol Behav 62(1): 97-103. Well, D., Yang, H., Houseni, M., Iruvuri, S., Alzeair, S., Sansovini, M., Wintering, N., Alavi, A., and Torigian, D.A. 2007. Age-related structural and metabolic changes in the pelvic reproductive end organs. Semin Nucl Med 37(3): 173-184. Widmer, R., Ziaja, I., and Grune, T. 2006. Protein oxidation and degradation during aging: role in skin aging and neurodegeneration. Free Radic Res 40(12): 1259-1268. Witten, T.M. and Eakin, T. 1997. Multiphasic models of survival: analysis of mortality rate change regions and the issue of finite species lifespan. Exp Gerontol 32(3): 259-285. Wolkow, C.A., Munoz, M.J., Riddle, D.L., and Ruvkun, G. 2002. Insulin receptor substrate and p55 orthologous adaptor proteins function in the Caenorhabditiselegans daf-2/insulin-like signaling pathway. J Biol Chem 277(51): 49591-49597. Figures Figure 1. Current theories proposed to explain the cause of aging. It is generally accepted that oxidative stress plays a central role in aging. Free radicals, or reactive oxygen species, are by-products of cellular metabolism and accumulate during aging, causing oxidative stress. The result is damage to proteins and DNA, which in turn can lead to mitochondrial dysfunction, energy deficit and metabolic abnormalities associated with aging. A "rate of living" hypothesis suggests that the rate of aging is inversely proportional to metabolic rate, i.e. higher metabolic rate accelerates aging and limits lifespan. 45 Figure 1 Oxidative stress Damage to DNA and proteins I AGING Mitochondrial dysfunction Metabolic Metabolic abnormalities 46 Figure 2. Genes from one of the two arms of the dauer pathway regulate C. elegans longevity (edited from Larsen 1995). daf-2 and daf-7 mutants are Daf-c (form dauers constitutively), indicating that these genes have a role in promoting continuous, non-Dauer development. Four other Daf-c genes, daf-1, daf-4, daf-8, and daf-14, have genetic interactions similar to those of mutations in daf-7, suggesting that they function with daf-7. Genetic epistasis analysis indicated that Daf-d (dauer-defective) genes daf-3 and daf-5 act downstream of daf-7 pathway, because mutations in any of these genes suppress the effects of mutations in daf-7, daf-1, daf4, daf-8 or daf-14. daf-3 and daf-5 seem to function specifically in this pathway since they do not strongly suppress mutations in Daf-c genes daf-2 and age-1/daf-23. By contrast, dauer-defective gene daf-16 suppresses the effects of mutations in daf-2 and age-1/daf-23, but not of mutations in daf7. Mutations in daf-2 and age-1/daf-23 genes confers longevity to adults, while mutations in daf-7, daf-1, daf-4, daf-8 or daf-14 do not. 47 Figure 2. daf-7 d Sdaf-4 daf-8 " daf-14 daf-3 daf-5 DAUER ENTRY daf-2 -* age-1/daf-23 Hdaf-16 ADULT LONGEVITY Figure 3. Insulin/IGF pathway regulates C. elegans longevity and stress-resistance. The signaling cascade is initiated by binding of an insulin-like substrate molecule to the receptor encoded by the daf-2 gene. This binding event leads to activation of age-i-encoded PI3-kinase, which generates PtdIns-3,4,5-P3, whereas the DAF-18 PTEN phosphatase homolog degrades this second messenger. PtdIns-3,4,5-P 3 and its derivative PtdIns-3,4P2 lead to the activation of PKB-family kinases, encoded by akt-1, akt-2, and sgk-1 . Phosphorylation by PKB blocks the activity of the daf-16 gene product (forkhead / winged helix transcription factor) by phosphorylation, which leads to DAF-16 exclusion from the nucleus. This translocation event prevents activation of the daf-16 target genes, some of which function in stress-resistance and lifespan determination. 49 Figure 3. INSULIN / IGF DAF-2 Insulin-like Receptor AGE-1 PI3K I PI(3,4,5)P 3 AKT- 1/AKT-2 /SGK- 1 PKB/SGK Longevity Stress Resistance 50 DAF-18 PTEN Chapter 2. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend lifespan Ala Berdichevsky, Mohan Viswanathan, H. Robert Horvitz and Leonard Guarente This chapter is modified from the following manuscript: Berdichevsky, A., Viswanathan, M., Horvitz, H.R., and Guarente, L. 2006. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125(6): 1165-1177 Mohan Viswanathan contributed anti-SIR-2.1 polyclonal serum that I affinity purified, and made and initially characterized the sir-2.1overexpressing strain NL3909. We analyzed the NL3909 transcriptional profiles together. SUMMARY The longevity of Caenorhabditiselegans is promoted by extra copies of the sir-2.1 gene in a manner dependent on the forkhead transcription factor DAF-16. We identify two C. elegans 14-3-3 proteins as SIR-2.1 binding partners and show that 14-3-3 genes are required for the lifespan extension conferred by extra copies of sir-2.1. 14-3-3 proteins also are required for SIR-2.1-induced transcriptional activation of DAF-16 and stress resistance. Following heat stress, SIR-2.1 can bind DAF-16 in a 14-3-3-dependent manner. By contrast, low insulin-like signaling does not promote SIR-2.1 / DAF-16 interaction, and sir-2.1 and the 14-3-3 genes are not required for the regulation of lifespan by the insulin-like signaling pathway. We propose the existence of a stressdependent pathway in which SIR-2.1 and 14-3-3 act in parallel to the insulin-like pathway to activate DAF-16 and extend lifespan. 52 INTRODUCTION C. elegans lifespan is controlled by an insulin/IGF signaling pathway, which includes the DAF-2 transmembrane receptor, a series of intracellular kinases, and the DAF-16 forkhead family transcription factor (Guarente and Kenyon, 2000; Finch and Ruvkun, 2001; Kenyon, 2005). This insulin/IGF pathway regulates the generation of dauer larvae, which are long-lived developmental variants that arise from second-stage (L2) larvae when conditions are harsh (Larsen et al., 1995; Vanfleteren and Braeckman, 1999). Mutations that decrease signaling by the DAF-2 receptor pathway cause an extension of the lifespan of adults (and more frequent entry into the dauer developmental state by larvae) by triggering the nuclear localization of DAF-16 (Henderson and Johnson, 2001; Lee et al., 2001; Lin et al., 2001). Such mutations also increase the resistance of animals to oxidative and genotoxic stress. The longevity and stress resistance engendered by a reduction in insulin/IGF signaling is abolished in mutants defective in daf-16 (Murakami and Johnson, 1996; Lin et al., 1997; Paradis and Ruvkun, 1998; Honda and Honda, 1999). The forkhead family of transcription factors includes mammalian FOXO 1, 3, and 4. In mammals, activation of the insulin/IGF pathway causes phosphorylation of forkhead proteins and their retention in the cytoplasm (Brunet et al., 1999; Nakae et al., 1999; Tang et al., 1999). Nuclear localization of forkhead proteins can be elicited by depletion of insulin from serum or by stress (Tran et al., 2003; Brunet et al., 2004; Essers et al., 2004; Van Der Heide et al., 2004; van der Horst et al., 2004). C. elegans sir-2.1 and its orthologs in Sacharomyces cerevisiae and Drosophilamelanogastercan regulate lifespan (Kaeberlein et al., 1999; Tissenbaum and Guarente, 2001; Rogina and Helfand, 2004). Transgenic worms with extra copies of sir-2.1 live longer than the wild type. Because this longevity requires daf-16, it was proposed that sir-2.1 works by downregulating insulin signaling (Tissenbaum and Guarente, 2001). This genetic inference was bolstered by the finding that transgenes overexpressing sir2.1 conferred no further extension in lifespan to daf-2 mutants (Tissenbaum and Guarente, 2001). Nevertheless, the mechanistic link between sir-2.I1 and the insulin/IGF pathway and its target daf-16 is unknown. The proteins encoded by yeast SIR2 and its orthologs are NADdependent deacetylases (Blander and Guarente, 2004). The mammalian homolog Sirtl interacts with many proteins, including transcription factors, such as p53, PPARO, and NF- OB, and transcriptional cofactors, such as p300 and CBP (Luo et al., 2001; Langley et al., 2002; Takata and Ishikawa, 2003; Picard et al., 2004; van der Horst et al., 2004; Vaquero et al., 2004; Yeung et al., 2004; Bouras et al., 2005). In mammals, Sirtl can bind to and deacetylate forkhead proteins. Deacetylation of forkhead transcription factors by Sirt 1 can result in either repression or activation of the transcription of their target genes (Brunet et al., 2004; Motta et al., 2004; Bouras et al., 2005) and appears to increase the resistance of mammalian cells to DNA damage-induced apoptosis (Luo et al., 2001; Brunet et al., 2004; Cohen et al., 2004; Motta et al., 2004). 14-3-3 proteins are highly conserved small acidic proteins that bind phosphoserine and phosphothreonine residues in a context-specific manner 54 (Durocher et al., 2000). Through interactions with their partners, 14-3-3 proteins regulate key biological processes, such as the cell cycle, apoptosis, and transcription (Tzivion et al., 2001). 14-3-3 proteins affect their targets by multiple mechanisms, including by activating or inhibiting intrinsic protein activities, by altering subcellular protein localization, and by scaffolding interactions between two binding partners. In mammalian cells, 14-3-3 proteins bind to phosphorylated forkhead (Brunet et al., 1999; Durocher et al., 2000; Obsil et al., 2003), and this binding leads to retention of forkhead in the cytoplasm, rendering it inactive. Mammalian 14-3-3 proteins can also bind the C. elegans forkhead protein DAF-16 when it is phosphorylated (Cahill et al., 2001). In C. elegans, two genes, par-5 andftt2, encode 14-3-3-like proteins, both of which are enriched in the embryo and in the gonad (Wang and Shakes, 1997; Morton et al., 2002). Mutations in par-5 affect the asymmetry of early embryonic cell divisions and cause maternal-effect lethality (Morton et al., 2002). ftt-2 is expressed in the soma (Wang and Shakes, 1997); the effect of the loss offtt-2 function has not been described. In this report, we show that SIR-2.1 acts with 14-3-3 proteins to affect DAF-16 activity and lifespan. Our findings suggest that SIR-2.1 and 14-3-3 may act in parallel to the insulin-like pathway to regulate DAF-16 and extend lifespan. RESULTS C. elegans SIR-2.1 interacts with 14-3-3 proteins. To identify SIR-2.1-interacting proteins in C. elegans, we raised a polyclonal antibody that recognizes worm SIR-2. 1. Immunoflurescence studies using this antibody showed nuclear staining in wild-type animals (Figure 1A, left), consistent with the reported nuclear localization of the mammalian homolog Sirtl (Langley et al., 2002). No staining was seen in sir-2.1(ok434) mutants, which are deleted for the sir-2.I1gene (Figure 1A, right), indicating that this antibody is specific to SIR-2.1. We isolated SIR2.1 protein complexes by co-precipitation with the anti-SIR-2.1 antibody and analyzed the complexes by mass spectrometry. We assayed lysates of three different strains: the wild type (N2), sir-2.I (ok434A) (which does not produce full length SIR-2. 1 protein), and a transgenic sir-2.I1overexpresser (O/E). We detected SIR-2.1 and one additional protein in the sir-2.I (O/E) lysate (Figure 1B). This protein was subjected to mass spectrometry, which revealed that it contained both C. elegans 14-3-3-like proteins, PAR-5 and FTT-2, and no other proteins (Figure IC). PAR-5 and FTT-2 share 88% sequence identity and are 78% (PAR-5) and 82% (FTT-2) identical with human 14-3-3ý. To address whether SIR-2.1/14-3-3 interaction also occurs in wildtype animals with endogenous levels of SIR-2. 1, we repeated the anti-SIR2.1 immuno-precipitations and analyzed the precipitates by western blotting using antibody raised against mouse 14-3-3 recognizing both C. elegans 143-3 proteins (Figure ID). We detected 14-3-3 immunoreactivity in SIR-2.1 56 precipitates isolated from both the wild type and the SIR-2.1overexpressing strain. The precipitation of another C. elegans deacetylase protein, HDA-1, from wild-type worm lysates did not contain 14-3-3, indicating that the interaction is specific to SIR-2.1 (see Figure 2A). 14-3-3 proteins in C. elegans are mostly localized to the cytoplasm (Wang and Shakes, 1997). In mammals, 14-3-3 proteins are known to shuttle between the cytoplasm and the nucleus (Brunet et al., 2002; Van Der Heide et al., 2004). To see whether 14-3-3 proteins are present in C. elegans nuclei, we performed cell fractionation experiments. While we detected SIR-2.1 protein in the nuclear fraction but not in the cytosolic fraction, 143-3 proteins were present in both nuclear and cytosolic fractions (Figure lE). These observations are consistent with the hypothesis that the interaction between SIR-2.1 and 14-3-3 proteins occurs in the nucleus. C. elegans 14-3-3 proteins can interact with DAF-16 and act to retain DAF-16::GFP in the cytoplasm. Since mammalian 14-3-3 proteins bind phosphorylated FOXO proteins (the homologs of DAF-16) and control their subcellular localization (Brunet et al., 1999; Brunet et al., 2002; Cahill et al., 2001; Obsil et al., 2003), we investigated whether C. elegans 14-3-3 proteins can bind DAF- 16 and affect the localization of DAF-16. We immunoprecipitated a functional DAF-16::GFP translational fusion protein (Lee et al., 2001) from worm lysates using an anti-GFP antibody and observed that 14-3-3 proteins co-immunoprecipitated with DAF-16::GFP. The interaction between 14-3-3 proteins and DAF-16::GFP 57 was likely specific to DAF-16 since 14-3-3 proteins did not co-precipitate with a LIN-53::GFP translational fusion protein in a similar experiment (Figure 2B). Thus 14-3-3 proteins can physically interact with DAF- 16, similar to the interaction between mammalian 14-3-3 and the DAF- 16 homologue FOXO. We next investigated whether low 14-3-3 levels will affect the subcellular localization of DAF-16. In this experiment, we used RNAmediated gene interference (RNAi) to reduce the expression of the 14-3-3 genes. We constructed four non-overlapping RNAi clones, two targeting each of the two 14-3-3 genes, par-5 andftt-2 (see Experimental Procedures). Progeny of animals fed with bacteria expressing par-5 RNAi expressed a maternal-effect lethal phenotype similar to that previously described forpar-5 loss-of-function (If) mutants (Morton et al., 2002). The progeny of animals fed withftt-2 RNAi frequently retained eggs and died of internally hatched progeny, a phenotype similar to that offtt-2(lf) mutants (see below). Feedingpar-5 orftt-2 RNAi did not cause any apparent abnormalities in animals that were transferred to RNAi plates as L4 larvae or as adults. At the protein level, par-5(RNAi) had a minimal effect on the level of total 14-3-3 proteins (data not shown), whereas ftt-2(RNAi) caused substantial reduction in 14-3-3 protein level (see Figure 5B below). It is important to note that the two C. elegans 14-3-3 genes are very similar, so it is possible that RNAi of one gene would result in reduction of function of the other. Nevertheless, we believe that at least some of the RNAi effects are gene-specific, since par-5(RNAi) but notftt-2(RNAi) resulted in a maternal-effect lethal phenotype similar to that caused by mutation in par5, andftt-2(RNAi) but not par-5(RNAi) resulted in a lower level of 14-3-3 proteins. 58 Under normal conditions, DAF-16::GFP has a diffuse mostly cytoplasmic localization. Reducing the expression ofpar-5 orftt-2 or both by RNAi caused a pronounced nuclear accumulation of DAF-16::GFP (Figure 3A). Nuclear DAF-16::GFP localization was most prominent in the intestinal cells but was also apparent in muscle and hypodermis (we scored animals as positive for nuclear DAF-16::GFP if most or all intestinal cells showed nuclear GFP). We did not see GFP accumulation in neuronal nuclei, perhaps because of the previously described insensitivity of this tissue to RNAi (Timmons et al., 2001). RNAi targeting offtt-2 led to nuclear DAF-16::GFP in 68% or 100% of animals, depending on the RNAi clone used, while par-5 RNAi caused nuclear DAF-16::GFP in about 50% of animals (Table 1). The par-5 RNAi result suggests that this biological assay of DAF-16::GFP localization is likely more sensitive than western blots, in which we could not detect a significant reduction in 14-3-3 level following par-5(RNAi) feeding. In addition to altering the localization of DAF-16::GFP, ftt-2(RNAi) but not par-5(RNAi) caused arrest at the L2-L3 larval stages of the progeny of daf-16::gfp animals. Larval arrest was not observed when wild-type animals were subjected toftt-2(RNAi) and may have been a consequence of the increased nuclear localization of overexpressed DAF-16::GFP. To confirm that loss offtt-2 function can alter DAF-16 localization, we generated anftt-2 mutant strain by screening a library of chemically mutagenized animals using PCR (Jansen et al., 1997; Liu et al., 1999). ftt-2(n4426A) worms bear a 668 bp deletion that removes part of the promoter and the start codon of the predictedftt-2 gene and therefore is a good candidate for being a null allele offtt-2. These mutants develop normally but die in early adulthood, because of bursting at the vulva or internal hatching of progeny (data not shown). We introduced the daf16::gfp transgene into theftt-2(n4426A) strain. Larvae homozygous forftt2(A) had nuclear DAF- 16::GFP, similar to animals treated withftt-2(RNAi) (Figure 3B), indicating thatftt-2 function may be required for the proper subcellular localization of DAF-16. These experiments suggest that, as proposed for the homologous proteins in mammals, C. elegans 14-3-3 proteins can act to retain the forkhead protein DAF-16 in the cytoplasm. sir-2.1 overexpression slows DAF-16::GFP exit from the nucleus during recovery from stress. We next asked whether sir-2.1 levels could also affect the cellular localization of DAF- 16. We crossed the daf-1 6::gfp transgene into two different long-lived strains over-expressing sir-2.1, geln3, and pkIsl 642. The DAF-16::GFP localization in these strains was cytoplasmic, as in the control strain with wild-type levels of sir-2.1 (data not shown). Therefore we concluded that under normal conditions SIR-2.1 overexpression does not affect DAF-16 localization. DAF-16 is known to translocate into the nucleus after heat shock. To investigate a possible more subtle effect of SIR-2.1 on DAF-16 localization, we studied the kinetics of stress-induced nuclear localization in the strain with extra copies of sir-2.1. We found that in a wild type background and in a strain over-expressing SIR-2. 1, DAF-16::GFP entered the nucleus with similar kinetics, such that within 30 min. of a shift to 370 C over 80% of animals in both strains had nuclear DAF-16::GFP (see Figure 4). The kinetics of relocalization of DAF-16::GFP to the cytoplasm, however, differed: loss of nuclear localization occurred significantly more slowly in 60 the strain with the higher level of SIR-2. 1. For example, after 30 min. of recovery, the over-expressing strain showed more than double the number of worms that had cells with nuclear DAF- 16 (46% as compared to 18%; Figure 4B). These experiments suggest that following stress increased SIR2.1 function can cause DAF-16 to remain in the nucleus, and therefore promote the function of DAF-16 to mediate a cellular stress response. The 14-3-3 genesftt-2 and par-5 are required for sir-2.1-mediated lifespan extension. As FTT-2 and PAR-5 interact with SIR-2.1 and affect DAF-16 localization, we asked whether 14-3-3 genes regulate C. elegans lifespan. We used RNAi to reduce the levels of par-5 andftt-2 in adults and assayed their lifespans. As controls, we measured lifespans of animals fed bacteria expressing a control RNAi vector and animals fed daf-16 RNAi. RNAi reduction ofpar-5,ftt-2 and daf-16 had similar effects on the lifespans of worms with wild-type levels of sir-2.1: they resulted in a shortening of lifespan by approximately 20% compared to the vector control (see Figure 6A). Next we asked whether downregulation of 14-3-3 genes would affect lifespan in long-lived animals with extra copies of sir-2.1. The sir-2.I1gene R1 1A8.4 is predicted to be a downstream gene of a two-gene operon (CEOP4372). To overexpress sir-2.1 with its regulatory regions we made a new transgenic strain NL3909 pkIs1642 by microparticle bombardment. The construct in the NL3909 strain contains the entire operon, including the sir-2.1 coding sequence plus 2.5 kb of sequence upstream of sir-2.1 containing the gene R11A8.5, predicted to encode a glutathione S- transferase-related protein, as well as 600 bases further upstream. Animals carrying pkls 1642 transgene showed marked extension of lifespan relative to the wild type (Figure 5A). Control experiments indicated that overexpression of sir-2.1 but not of R11A8.5 is required for lifespan extension ofpklsl642 transgenic animals (see Experimental Procedures and Figure 5A). Overexpression of sir-2.1 inpkls1642 strain also promoted increased resistance to heat shock (Figure 5B). Reduction of 14-3-3 levels in animals with extra copies of sir-2.1 fully suppressed the lifespan extension observed in this strain (Figure 6B). Whileftt-2 RNAi resulted in a shortened lifespan indistinguishable from the effect of daf-16 RNAi, par-5 RNAi suppressed the lifespan of the sir-2.1 overexpresser to a wild-type duration. Table 2 details the data for these and all other lifespan experiments presented in Figure 6. To address the possibility that the worms fed with the par-5 orftt-2 RNAi bacteria have non-specifically shortened lifespans because they are sick, we tested the effects ofpar-5 andftt-2 RNAi on the lifespans of sir2.1 (ok434A) and daf-1 6(lf) mutants, both of which have slightly shorter lifespans than the wild type. In these strains reducing 14-3-3 gene expression caused little or no reduction in lifespan (see Figures 6C and 6D). More strikingly, reducing 14-3-3 gene expression in a daf-2 mutant, which like the sir-2.I1-overexpressing strain displays an increased lifespan, did not shorten lifespan (see below). Thus, the effects ofpar-5 and ftt-2 RNAi on lifespan probably are not caused by non-specific sickness. Rather, par-5 andftt-2 appear to be specifically required for the sir-2.1-dependent lifespan extension. Our findings indicate that reduction of eitherpar-5 orftt-2 triggered nuclear localization of DAF-16 but did not promote longevity. This 62 observation suggests that nuclear localization of DAF- 16 may not be sufficient to activate target genes and extend lifespan. sir-2.1, par-5 and ftt-2 are not required for lifespan extension in animals with reduced insulin/IGF signaling. C. elegans sir-2.1 has been suggested to affect lifespan via the insulin-like pathway (Tissenbaum and Guarente, 2001). To analyze the interactions of the 14-3-3 genes with the insulin-like pathway, we tested the effects of reducing par-5 andftt-2 in a daf-2 mutant, which has extended lifespan. In this experiment, the lifespan extension of daf-2(el370) mutants was fully suppressed by daf-16 RNAi, as expected. By contrast, par-5 RNAi andftt-2 RNAi had no effect (Figure 6E). Likewise, the loss of sir2.1 did not reduce the long lifespan of the daf-2 mutant and may have lengthened it slightly (Figure 6F). These findings indicate that sir-2.1, par-5 andftt-2 do not function in the insulin-like pathway downstream of daf-2. Instead, sir-2.1 and the 14-3-3 genes may act upstream of the DAF-2 insulin-like receptor (for instance, by controlling production of its ligands) or in a pathway of lifespan determination parallel to that of insulin signaling. Since daf-16 is required for the lifespan extension by extra copies of sir-2.1, any parallel pathway must converge on the DAF- 16 transcription factor. sir-2.1 can promote resistance to stress in anftt-2-dependent way. DAF- 16 function is important for the stress response (Hsu et al., 2003; Lamitina and Strange, 2005; Lin et al., 1997). As SIR-2.1 63 overexpression led to extension of lifespan in a daf-16- andftt-2-dependent manner, we asked whetherftt-2 might also be involved in stress resistance caused by extra copies of sir-2.1. We examined the effects of sir-2.I1andftt2 on resistance to heat stress, by determining the survival of animals subjected to 320 C heat shock (Figure 7A), and to oxidative stress, by assaying development in 0.25 mM paraquat (Figure 7B). In both cases, the sir-2.1 overexpressing strain was stress-resistant compared to the wild type, and the sir-2.1 deletion strain was stress-sensitive (Figure 7A and B). Moreover, anftt-2 loss-of-function mutation also caused sensitivity to stress and completely abolished the stress resistance of the sir-2.1-overexpressing strain (Figure 7A and B). These findings show that sir-2.1 and 14-3-3 function together in determining stress resistance. sir-2.1 can promote daf-16-dependent transcription in anftt-2dependent fashion. The genetic interactions between sir-2.I1and daf-16 led us to speculate that sir-2.I1may activate daf-16. To test this hypothesis we used a GFP reporter for a known daf-16 target, sod-3 (Cahill et al., 2001; Honda and Honda, 1999; Lee et al., 2003; McElwee et al., 2003). This sod-3::gfp reporter, which has been used to assay daf-16 activity (Libina et al., 2003), was crossed into strains with high-copy-number (geln3) or low-copynumber (pkls1642) transgenes carrying extra copies of sir-2.I1,and the sod3::gfp signal in these strains was visualized by fluorescence microscopy (Figure 7C-E). In both SIR-2.1 overexpressing strains, the GFP signal was much stronger than that in the wild type, indicating that transcription from the sod-3 promoter was upregulated. A loss-of-function mutation in daf-16 64 eliminated this upregulation of sod-3 (Figure 7G, H), suggesting that it results from the activation of DAF- 16 by the elevated levels of SIR-2. 1. Transcriptional array data also indicated an elevated expression of sod-3 and other DAF-16 targets in animals overexpressing SIR-2.1 (Viswanathan et al., 2005 and Table 3). Next we tested whether this activation of DAF-16 by SIR-2. 1 would be influenced by reducing the levels of 14-3-3. Animals bearing a deletion inftt-2 did not show upregulation of sod-3::gfp expression (Figure 7J), although these mutants displayed a nuclear localization of DAF-16::GFP (Figure 3B). However, the upregulation of sod-3 by the SIR-2.1overexpressing transgene was abolished by deletion offtt-2 (Figure 7K). Moreover, the requirement forftt-2 was specific for the activation of DAF16 by SIR-2.1 overexpression, as the ftt-2 deletion did not reduce the high sod-3.:.gfp expression seen in the daf-2(e3 70) mutant (see Figure 7F and I). In summary, overexpression of SIR-2.1 appears to promote DAF-16 transcriptional activity and requires 14-3-3 to do so. Following stress, SIR-2.1 interacts with DAF-16 in a 14-3-3-dependent manner. In mammals, both 14-3-3 proteins and a SIR-2.1 homolog Sirt1 can physically interact with mammalian FOXO proteins (Brunet et al., 1999; Brunet et al., 2002; Obsil et al., 2003; Brunet et al., 2004; Motta et al., 2004; van der Horst et al., 2004). To ask whether comparable interactions occur among the related proteins of C. elegans, we used a functional DAF16::GFP (Lee et al., 2001) and antisera to GFP and SIR-2.1 for coimmunoprecipitation experiments and analyzed the precipitates using western blots (Figure 8A). Since SIR-2.1 is a nuclear protein and DAF-16 is normally cytoplasmic (Henderson and Johnson, 2001; Lee et al., 2001; Lin et al., 2001), we reasoned that any interaction would require nuclear entry of DAF-16. Heat stress causes nuclear accumulation of DAF-16 (Henderson and Johnson, 2001; Lin et al., 2001). Thus, we immunoprecipitated DAF-16::GFP or SIR-2.1 from untreated animals and from animals that had been heat-shocked and assayed for coimmunoprecipitation of SIR-2.1 and DAF-16. As expected, no SIR2. 1/DAF- 16 complex was observed in the absence of heat shock. After heat shock, SIR-2.1 co-immunoprecipitated with DAF- 16 (Figure 8A). Thus, following stress DAF-16 can physically interact with SIR-2.1 proteins in C. elegans. Notably, 14-3-3 was found in the DAF-16::GFP immunoprecipitate before and after heat shock and not in that of the control animals lacking DAF-16::GFP (Figure 8A and 2B). One of the known biological roles of 14-3-3 proteins is to act as a scaffold, promoting interactions between two different proteins (Tzivion et al., 2001). We asked whether 14-3-3 proteins were important for the interaction between SIR-2.1 and DAF-16. We immunoprecipitated DAF- 16::GFP protein from lysates of animals grown onftt-2(RNAi) or vector RNAi and subjected to heat shock and assessed the levels of SIR-2.1 in the precipitates. ftt-2(RNAi) markedly reduced the amount of SIR-2.1 that coprecipitated with DAF-16 (Figure 8B). par-5(RNAi) had no effect on the level of 14-3-3 proteins in worms or the co-immunoprecipitation of SIR-2.1 with DAF-16 (data not shown). We conclude that following heat stress SIR2.1 interacts with DAF- 16 in a 14-3-3-dependent manner. We tested whether reducing insulin-like signaling, which should cause accumulation of dephosphorylated nuclear DAF-16, will affect interactions between DAF-16, 14-3-3 and SIR-2.1 proteins. RNAi reduction of daf-2 results in decreased insulin-like signaling leading to lifespan extension and accumulation of DAF-16::GFP in the nucleus ((Henderson and Johnson, 2001; Murphy et al., 2003) and Figure 9). However, unlike heat shock, daf-2(RNAi) did not promote the interaction between SIR-2.1 and DAF-16 (Fig. 8B). After heat shock of the daf-2 RNAi treated worms, a low level of DAF-16/SIR-2.1 interaction was observed, which may be due to a fraction of DAF-16 that was not affected by daf-2 RNAi. These findings suggest that DAF-16 phosphorylation and interaction with 14-3-3 may be important for the formation of SIR-2.1/ DAF-16 complex. DISCUSSION C. elegans SIR-2.1 and 14-3-3 proteins can function in a stress-response pathway of longevity. The longevity of C. elegans is increased by extra copies of the sir-2.I1 gene, and this effect is dependent on daf-16 (Tissenbaum and Guarente, 2001). Here we provide evidence for a mechanistic basis of this genetic interaction: SIR-2. 1 and DAF-16 proteins can physically interact. We found that SIR-2.1 functions in stress responses, as worms overexpressing SIR-2.1 were resistant to heat and oxidative stresses and sir-2.I1loss of function mutants were stress-sensitive. We also found that SIR-2.1 can activate DAF-16-dependent transcription, as overexpression of SIR-2.1 increased transcription of the DAF-16 target gene sod-3 in a daf-16-dependent way. Moreover, we found that 14-3-3 proteins are required for the lifespan extension, stress resistance and sod-3 activation conferred by extra copies 67 of sir-2.1. Deletion offtt-2 prevented stress resistance and sod-3 activation by SIR-2.1 overexpression, and RNAi of either par-5 orftt-2 suppressed the SIR-2.1-mediated lifespan extension. We are not certain that par-5 functions in the lifespan extension caused by SIR-2.1 overexpression, because RNAi ofpar-5 may also targetftt-2. Mammalian 14-3-3 proteins can interact with phosphorylated FOXO proteins and cause their retention in the cytoplasm (Brunet et al., 1999; Durocher et al., 2000; Obsil et al., 2003). Insulin signaling promotes this retention by causing phosphorylation of FOXO proteins. Here we demonstrate that 14-3-3 proteins appear to be important for the cytoplasmic localization of DAF-16, since reducing the expression levels of par-5 orftt2 results in nuclear accumulation of DAF-16::GFP. Thus, C. elegans 14-3-3 proteins can act to retain the C. elegans forkhead protein DAF-16 in the cytoplasm, similar to their mammalian homologs. Our data thus suggest that two different 14-3-3 protein complexes are present in C. elegans: a DAF-16/14-3-3 complex in the cytoplasm and a SIR-2.1/14-3-3 complex in the nucleus. Consistent with this hypothesis, we could not detect an interaction between DAF-16 and SIR-2. 1 under normal conditions. However, heat shock, which is known to promote DAF-16 localization to the nucleus (Henderson and Johnson, 2001; Lin et al., 2001), triggered the formation of a SIR-2. 1/DAF-16 complex. Both phosphorylation of DAF- 16 at the AKT sites and FTT-2 appear to be required for this interaction between DAF- 16 and SIR-2. 1, since RNAi of daf-2 orftt-2 reduced their association. In summary, 14-3-3 proteins are required for the daf-16-dependent biological effects of SIR-2.1 as well as for the physical association of SIR2.1 and DAF-16. Our results suggest that 14-3-3 genes and sir-2.1 act together in a stress-mediated pathway of longevity. Even under normal conditions animals may experience a basal level of stress, which may explain the effects of SIR-2.1 and 14-3-3 in an absence of a specific stress treatment. Our data indicate that 14-3-3 proteins play two antagonistic roles in DAF-16 regulation. First, as previously described in mammals, 14-3-3 proteins bind to phosphorylated DAF-16 and promote its retention in the cytoplasm. Second, 14-3-3 proteins function in a previously unknown pathway to facilitate the association of DAF- 16 with SIR-2.1 in the nucleus to elicit activation of sod-3, stress resistance and extension of lifespan. A recent report suggests that the C. elegans P3-Catenin ortholog bar-i can activate DAF-16 following stress (Essers et al., 2005). It will be interesting to see if bar-i participates in the 14-3-3/SIR-2. 1-mediated mechanism of DAF- 16 regulation. SIR-2.1 and 14-3-3 may affect longevity by regulating DAF-16 Forkhead in parallel to the insulin-like pathway. It has been suggested that sir-2.I1promotes longevity by downregulating the insulin-like signaling pathway (Tissenbaum and Guarente, 2001). We observed that sir-2.1, par-5 andftt-2 were not required for lifespan extension in a daf-2 mutant. Furthermore,ftt-2 was not required for the activation of sod-3 by a reduction in insulin-like signaling, even thoughftt-2 was required for activation of sod-3 by SIR-2.1 overexpression. Our findings therefore indicate that sir-2.1 andftt-2 do not act in the insulin/IGF pathway downstream of DAF-2. We cannot rule out the possibility that SIR-2.1 functions upstream of the DAF-2 receptor, for example, by regulating insulin production. However, the stress-inducible 69 14-3-3-dependent physical interaction between SIR-2.1 and DAF-16 suggests that SIR-2.1 and 14-3-3 act in a pathway parallel to insulin-like signaling that like the insulin-like pathway converges on the DAF-16 transcription factor. A model for the roles of SIR-2.1 and 14-3-3 in DAF-16 activation and longevity. Our results suggest the following model for the role of SIR-2.1 in DAF-16 activation (Figure 10). Under normal conditions, DAF-16 is mostly inactive, present in the cytoplasm and bound by 14-3-3 proteins. A reduction in insulin/IGF signaling, for example in daf-2 mutants, renders DAF-16 nuclear by reducing its state of phosphorylation by AKT kinase, thereby dissociating it from 14-3-3. Nuclear localization of DAF-16 is not sufficient for activation of its target genes, since reducing expression offtt2 rendered DAF-16 nuclear but did not activate the DAF-16 target sod-3 or extend lifespan. That nuclear DAF-16 is not necessarily active is consistent with the finding that overexpression of DAF-16 mutated at all known AKT sites leads to constitutively nuclear DAF-16 localization but does not extend lifespan (Lin et al., 2001). It is possible that DAF-16 requires an additional cofactor that acts with DAF- 16 to regulate transcription when insulin/IGF signaling is reduced. Alternatively, the nuclear DAF-16 that accumulates in worms with lowftt-2 levels may be non-functional because DAF-16 is insufficiently dephosphorylated. In response to stress, forkhead proteins translocate to the nucleus (Brunet et al., 2004; Henderson and Johnson, 2001; Lin et al., 2001; Tran et al., 2003) in a process that may involve the JNK kinase signaling pathway (Oh et al., 2005). We propose that SIR-2.1 binds the nuclear DAF-16 produced by stress, but not the nuclear DAF-16 produced by low insulinlike signaling. The resulting SIR-2.1/DAF-16 complex promotes DAF-16dependent transcription, stress resistance and longevity, in a manner dependent on 14-3-3 proteins. One possibility is that 14-3-3 scaffolds the interaction between SIR-2.1 and DAF-16 and a ternary complex among SIR-2.1, DAF-16 and 14-3-3 participates in the transcriptional activation of DAF-16 target genes. Another possibility is that 14-3-3 mediates a modification of DAF-16 and/or SIR-2.1 following stress, triggering the association of a binary complex of DAF-16 and SIR-2.1. This alternative model requires two additional steps involving an unknown 14-3-3-mediated modification of either SIR-2.1 or DAF-16 and dissociation of 14-3-3 from both SIR-2.1 and DAF- 16. Our proposed activation of FOXO proteins by a pathway involving SIR-2.1 and 14-3-3 may be a general molecular mechanism for the regulation of longevity by Sir2 orthologs in metazoa. EXPERIMENTAL PROCEDURES Antibody production, immunofluorescence, and cell fractionations Rabbit polyclonal anti-SIR-2.1 antiserum was raised against purified fulllength His-tagged SIR-2.1 protein and affinity-purified using His-tagged SIR-2.1 bound to nitrocellulose membranes. The mouse monoclonal antibody to GFP was anti-AFP mAb 3E6 (QbioGene). The anti-DAF-16 rabbit polyclonal antibody was a gift from G. Ruvkun. The mouse monoclonal anti-14-3-3 1(H8) antibody was purchased from Santa Cruz Biotechnology. To control for total protein content in loading we used the anti-aTubulin mouse monoclonal antibody Dmla (Sigma). HDA-1 rabbit polyclonal antibody was purchased from Santa Cruz. Whole-mount immunofluorescence of larvae and adult animals was performed as described (Finney and Ruvkun, 1990) with purified anti-SIR2.1 antibody and visualized by Nomarski fluorescence microscopy. Cell fractionations were performed as described (Chen et al., 2000). Celamin antibody was from Y. Gruenbaum (Liu et al., 2000). SQV-4 antibody was obtained from Ho-Yon Hwang (Hwang and Horvitz, 2002). Immunoprecipitation and western blots Worm lysates for immunoprecipitation (IP) were made by sonication of pelleted worms in 5 volumes of IP buffer (10 mM Hepes-KOH, 250 mM NaC1, 0.5% NP-40, 10% glycerol, 5 mM EDTA, 1 mM DTT, Protease Inhibitors: one Complete Mini tablet (Roche Biochemical) per 10 ml IP buffer), for three 10 sec bursts on ice, and the lysates were centrifuged at 10,000 rpm for 20 min at 40 C. The supernatant was saved, and total protein concentration was determined using a BioRad Dc Protein Assay kit. For mass spectrometry analysis, 10 mg of total protein was used per IP. For western blot analysis, 1-3 mg of total protein was used per IP. Input lanes contained 20-50 [tg of total protein. Immunoprecipitations were performed by first binding the antibody to Amersham fast-flow Protein A beads for 2 hr at 40 C, followed by washes with IP buffer, and incubation with worm lysates at 40 C. Precipitated proteins were eluted by boiling in SDS protein sample buffer. The samples were then centrifuged 5 min at 13,000 rpm to separate Protein A beads, and the supernatants were loaded onto 4-15% gradient polyacrylamide gels 72 (BioRad). For mass spectrometry analysis, gels were stained with Coomassie blue, and bands were excised and sent to the Taplin Biological Mass Spectrometry Unit at Harvard Medical School for analysis. For western blot analysis, proteins were transferred to nitrocellulose membranes. Membranes were blocked with 5% milk in PBST (20 mM sodium phosphate pH 7.2, 150 mM NaCl, 0.01% Tween-20) for 30 min at room temperature, and then incubated with the indicated primary antibodies in PBST for 60 min at room temperature or overnight at 4VC. The membranes were then incubated with anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies that were purchased from Bio-Rad, and the signal was detected using Western Lightning Chemiluminescence reagent (Perkin Elmer). Lifespan, stress-resistance and RNAi analyses Unless stated otherwise, lifespan assays were performed at 200 C and initiated by transferring L4 larvae of the indicated genotypes to plates containing 10 [tM fluorodeoxyuridine (FUDR). Unless indicated otherwise, all RNAi and lifespan experiments were performed using N-terminal par-5 andftt-2 RNAi clones. In many cases, the plates contained Carbenicillin, Tetracyclin, and IPTG, which are required for the standard RNAi feeding protocol (Kamath et al., 2003). We used Prism 4 software to perform statistical analyses of survival curves. Animals that crawled off the plate, burst at the vulva or died from internal hatching of progeny were excluded from the analyses. In all lifespan assays reported here, between 90% and 95% of animals died naturally and were scored. For RNAi analysis of DAF-16::GFP localization, L4 animals were transferred to RNAi plates containing bacteria induced to express an RNAi clone or carrying empty RNAi feeding vector as a control (Kamath et al., 2003). The phenotype of progeny was scored after 48-72 hr at 20 0 C. Heat-shock assays were performed at 320 C using one-day-old adults. For each genotype, 50-70 animals were transferred to 5 cm plates, and viability was scored every two to eight hrs after shift to 32TC. Statistical analysis was performed using Prism 4 software, as for the lifespan assays. Assays of paraquat sensitivity were performed at 200 C by transferring arrested Li larvae to plates containing 0.25 mM paraquat. Development was assessed 2, 3, and 4 days later, and the percent of animals that developed to adulthood was scored. All strains assayed contained muIs84[sod-3::gfp]in the background; expression of this transcriptional reporter did not alter stress resistance (data not shown). The student t-test was used to evaluate the significance of differences in the means. Strains Standard nematode growth medium (NGM) (Brenner, 1974) was used for C. elegans growth and maintenance at 20 0 C. Unless otherwise stated, plates were seeded with E. coli OP50 bacteria (Brenner, 1974). NL3909 pkIsl642 [unc-119 sir-2.1] is a low-copy sir-2.I1transgenic overexpresser strain produced by microparticle bombardment. The transgene inpkIs1642 includes sir-2.1 coding sequence plus 2.5 kb of upstream sequence, including the predicted gene R11A8.5, subcloned into a pRP2510 vector (a gift of E. Ivanov). The construct was introduced into unc-119(ed3) worms, and transgenic animals were selected based on the unc-119(+) phenotype (coordinated locomotion and ability to form dauer larvae). The presence of the transgene was verified by PCR. As a control, the vector pRP2510, which contains the unc-119 gene alone, was transformed into unc-119(ed3) animals, generating strain NL3908 pkls1641 [unc-119]. To test whether the effects on stress resistance and lifespan observed in the NL3909 strain are indeed attributable to sir-2.1, we used RNAi to reduce expression of each of sir-2.1 and R11A8.5 genes in the pkls1642 background. Northern blot analysis confirmed that RNAi of both genes is specific: that is, sir-2.I1RNAi reduced the level of the sir-2.I1 transcript but did not affect the level of R11A8.5 transcript; similarly, R11A8.5 RNAi reduced the level of the R11A8.5 transcript but did not affect sir-2.1 level. Then we performed lifespan and heat-stress assays of pkls1642 transgenic animals fed control RNAi, sir-2.1 RNAi or R 11A8.5 RNAi bacteria. Both lifespan extension and stress resistance ofpkIsl642 animals were suppressed by sir-2.1 RNAi but were not significantly affected by R11A8.5 RNAi (See Figure 4). Strain VC199 sir-2.1(ok434) was obtained from the C. elegans Gene Knockout Consortium via the Caenorhabditiselegans Genetics Center and was outcrossed five times to the wild type prior to use. ftt-2(n4426) was obtained by screening a deletion library. This mutant has a 668 bp deletion of the promoter and most of the first exon of the predictedftt-2 gene, from 22920 to 23598 on cosmid F52D10. The strain was outcrossed six times to the wild type. Presence of the deletion was verified by PCR. The muls84[sod-3::gfp]transgene from strain CF 1553 was from the CGC. The muls84[sod-3::gfp]transgene was crossed into the following genetic backgrounds by standard genetic methods: geln3[sir-2.1 rol75 6(sul 006)], pkls1642[sir-2.1 unc-119], daf-16(mgDf50); pks1l642, daf-16(mgDf50); geln3,ftt-2(n4426), pklsl642; ftt-2(n4426), daf-2(e1370); ftt-2(n4426). The daf-16::gfp transgene xrls87 [daf-16a::GFP::DAF-I6brol- 6(su1006)] was a gift from R. Lee and G. Ruvkun (Lee et al., 2001). The daf-16::gfp transgene xrls87 [daf-]6a::GFP::DAF-16b rol-6(sul 006)] was crossed into N2, geln3, and pkls1642 backgrounds by standard genetic methods, and the presence of geln3 was verified by anti-SIR-2.1 western blot. To generate daf-16::gfp;ftt-2(n4426) animals we crossed daf-16::gfp transgenic males withftt-2(n4426) hermaphrodites, generatingftt-2 (+/A) heterozygotes, and examined the progeny of theftt-2 (+/A) worms for DAF16::GFP localization. Two classes of GFP-expressing progeny were observed: the first, constituting more than 85% of GFP-expressing progeny, had cytoplasmic DAF-16::GFP and at least one wild-type copy offtt-2; the second class had nuclear DAF-16::GFP in all cells. Worms of this second class arrested as L1 or L2 larvae and were shown by single-animal PCR to be homozygous for the ftt-2 deletion. lin-53(n833) unc-76(e911); lin-15A(n767); nIs119[LIN-53::GFP] was a gift from M. Harrison (personal communication). Molecular biology To constructpar-5 andftt-2 RNAi clones, the cDNAs of these genes were amplified by RT-PCR from N2 RNA purified using Trizol (Invitrogen). To construct the N-terminal RNAi clones for par-5 andftt-2, we used the primers AAAATGGAAGATGGGGGTTC and GGAATACCGCGTCAAGGTAA for par-5 and AGAGTCAAGTTGTCACGGAGAAG and 76 CGATGTTTTCGTCCTATTTTCTG primers forftt-2. The cDNA fragments were then digested with SacI endonuclease (New England Biolabs), resulting in a 168-611 fragment for par-5 and a 275-614 fragment forftt-2, and cloned into the SacI site of the pPD 129.36 RNAi feeding vector. In addition, AGA GCT CGG ACT CGA CCT TGA TCA TGC and AGA GCT CTT AGT TTC CAG CTT CCT GG, and AGA GCT CTG ACG CCA TCG CTG AGC TCG and AGA GCT CAG TTG CCT CCC TCT GTC TCG TTG GC primers were used to amplify a C-terminal portion of the par-5 andftt-2 genes, respectively, from N2 genomic DNA and to introduce a SacI site at the 5' and 3' ends of the amplified fragment. SacI cleavage resulted in new non-overlapping fragments (639-747 for par5 and 614-745 forftt-2) that were cloned into a SacI site of pPD 129.36 to obtain non-overlapping C-terminal RNAi clones for par-5 andftt-2. The feeding RNAi strain for daf-16 was a gift of S. Lee and G. Ruvkun. The RNAi clone for daf-2 was made by subcloning cDNA corresponding to exon 9 of daf-2 into the pPD 129.36 RNAi feeding vector. Acknowledgments We thank the Caenorhabditiselegans Genetics Center for providing strains, Gary Ruvkun for the daf-16:.:gfp strain, daf-16 RNAi clone and DAF-16 antiserum, Ronald Plasterk for help with constructing the low-copy sir-2.I1 overexpresser, Andrew Hellman for PCR-screening the deletion library to identify theftt-2(n4426A) strain, Na An and Beth Castor for strain maintenance and determining DNA sequences, respectively, members of the Guarente and Horvitz laboratories for suggestions, and Nick Bishop, Gil Blander, Brendan Galvin, Mike Hurwitz, Niels Ringstad and Hillel Schwartz for critically reading the manuscript. A.B. was supported by a graduate fellowship from the American Federation for Aging Research. A.B. and H.R.H. were supported by the Ellison Medical Foundation. H.R.H. is an Investigator of the Howard Hughes Medical Institute. M.V. was supported by a fellowship from the Jane Coffin Childs Memorial Fund for Medical Research. L.G. was supported by grants from the NIH. References Blander, G., and Guarente, L. (2004). The Sir2 family of protein deacetylases. Annu Rev Biochem 73, 417-435. Bouras, T., Fu, M., Sauve, A. A., Wang, F., Quong, A. A., Perkins, N. D., Hay, R. T., Gu, W., and Pestell, R. G. (2005). SIRT1 deacetylation and repression of P300 involves lysine residues 1020/1024 within the cell-cycle regulatory domain 1. J Biol Chem. 280, 10264-10276 Brenner, S. (1974). The genetics of Caenorhabditiselegans. Genetics 77, 71-94. Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and Greenberg, M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857-868. Brunet, A., Kanai, F., Stehn, J., Xu, J., Sarbassova, D., Frangioni, J. V., Dalal, S. N., DeCaprio, J. A., Greenberg, M. E., and Yaffe, M. B. (2002). 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol 156, 817-828. Brunet, A., Sweeney, L. B., Sturgill, J. F., Chua, K. F., Greer, P. L., Lin, Y., Tran, H., Ross, S. E., Mostoslavsky, R., Cohen, H. Y., et al. (2004). Stressdependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011-2015. Cahill, C. M., Tzivion, G., Nasrin, N., Ogg, S., Dore, J., Ruvkun, G., and Alexander-Bridges, M. (2001). Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-33-independent pathways. J Biol Chem 276, 13402-13410. Chen, F., Hersh, B. M., Conradt, B., Zhou, Z., Riemer, D., Gruenbaum, Y., and Horvitz, H. R. (2000). Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science 287, 1485-1489. Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., Howitz, K. T., Gorospe, M., de Cabo, R., and Sinclair, D. A. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390-392. Durocher, D., Taylor, I. A., Sarbassova, D., Haire, L. F., Westcott, S. L., Jackson, S. P., Smerdon, S. J., and Yaffe, M. B. (2000). The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol Cell 6, 1169-1182. Essers, M. A., de Vries-Smits, L. M., Barker, N., Polderman, P. E., Burgering, B. M., and Korswagen, H. C. (2005). Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308, 1181-1184. Essers, M. A., Weijzen, S., de Vries-Smits, A. M., Saarloos, I., de Ruiter, N. D., Bos, J. L., and Burgering, B. M. (2004). FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. Embo J 23, 4802-4812. Finch, C. E., and Ruvkun, G. (2001). The genetics of aging. Annu Rev Genomics Hum Genet 2, 435-462. Finney, M., and Ruvkun, G. (1990). The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell 63, 895-905. Guarente, L., and Kenyon, C. (2000). Genetic pathways that regulate ageing in model organisms. Nature 408, 255-262. Henderson, S. T., and Johnson, T. E. (2001). daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditiselegans. Curr Biol 11, 1975-1980. Honda, Y., and Honda, S. (1999). The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditiselegans. Faseb J 13, 1385-1393. Hsu, A. L., Murphy, C. T., and Kenyon, C. (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 11421145. 80 Hwang, H. Y., and Horvitz, H. R. (2002). The Caenorhabditiselegans vulval morphogenesis gene sqv-4 encodes a UDP-glucose dehydrogenase that is temporally and spatially regulated. Proc Natl Acad Sci U S A 99, 14224-14229. Jansen, G., Hazendonk, E., Thijssen, K. L., and Plasterk, R. H. (1997). Reverse genetics by chemical mutagenesis in Caenorhabditiselegans. Nat Genet 17, 119-121. Kaeberlein, M., McVey, M., and Guarente, L. (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13, 2570-2580. Kamath, R. S., Fraser, A. G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., et al. (2003). Systematic functional analysis of the Caenorhabditiselegans genome using RNAi. Nature 421, 231-237. Kenyon, C. (2005). The plasticity of aging: insights from long-lived mutants. Cell 120, 449-460. Lamitina, S. T., and Strange, K. (2005). Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am J Physiol Cell Physiol 288, C467-474. Langley, E., Pearson, M., Faretta, M., Bauer, U. M., Frye, R. A., Minucci, S., Pelicci, P. G., and Kouzarides, T. (2002). Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J 21, 23832396. Larsen, P. L., Albert, P. S., and Riddle, D. L. (1995). Genes that regulate both development and longevity in Caenorhabditiselegans. Genetics 139, 1567-1583. Lee, R. Y., Hench, J., and Ruvkun, G. (2001). Regulation of C. elegans DAF-16 and its human ortholog FKHRL 1 by the daf-2 insulin-like signaling pathway. Curr Biol 11, 1950-1957. Lee, S. S., Kennedy, S., Tolonen, A. C., and Ruvkun, G. (2003). DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644-647. Libina, N., Berman, J. R., and Kenyon, C. (2003). Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489-502. Lin, K., Dorman, J. B., Rodan, A., and Kenyon, C. (1997). daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditiselegans. Science 278, 1319-1322. Lin, K., Hsin, H., Libina, N., and Kenyon, C. (2001). Regulation of the Caenorhabditiselegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28, 139-145. Liu, J., Rolef Ben-Shahar, T., Riemer, D., Treinin, M., Spann, P., Weber, K., Fire, A., and Gruenbaum, Y. (2000). Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol Biol Cell 11, 39373947. Liu, L. X., Spoerke, J. M., Mulligan, E. L., Chen, J., Reardon, B., Westlund, B., Sun, L., Abel, K., Armstrong, B., Hardiman, G., et al. (1999). High-throughput isolation of Caenorhabditiselegans deletion mutants. Genome Res 9, 859-867. Luo, J., Nikolaev, A. Y., Imai, S., Chen, D., Su, F., Shiloh, A., Guarente, L., and Gu, W. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107, 137-148. McElwee, J., Bubb, K., and Thomas, J. H. (2003). Transcriptional outputs of the Caenorhabditiselegans forkhead protein DAF-16. Aging Cell 2, 111-121. Morton, D. G., Shakes, D. C., Nugent, S., Dichoso, D., Wang, W., Golden, A., and Kemphues, K. J. (2002). The Caenorhabditiseleganspar-5 gene encodes a 14-3-3 protein required for cellular asymmetry in the early embryo. Dev Biol 241, 47-58. Motta, M. C., Divecha, N., Lemieux, M., Kamel, C., Chen, D., Gu, W., Bultsma, Y., McBumey, M., and Guarente, L. (2004). Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551-563. 82 Murakami, S., and Johnson, T. E. (1996). A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditiselegans. Genetics 143, 1207-1218. Murphy, C. T., McCarroll, S. A., Bargmann, C. I., Fraser, A., Kamath, R. S., Ahringer, J., Li, H., and Kenyon, C. (2003). Genes that act downstream of DAF- 16 to influence the lifespan of Caenorhabditiselegans. Nature 424, 277-283. Nakae, J., Park, B. C., and Accili, D. (1999). Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 274, 15982-15985. Obsil, T., Ghirlando, R., Anderson, D. E., Hickman, A. B., and Dyda, F. (2003). Two 14-3-3 binding motifs are required for stable association of Forkhead transcription factor FOXO4 with 14-3-3 proteins and inhibition of DNA binding. Biochemistry 42, 15264-15272. Oh, S. W., Mukhopadhyay, A., Svrzikapa, N., Jiang, F., Davis, R. J., and Tissenbaum, H. A. (2005). JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A 102, 4494-4499. Paradis, S., and Ruvkun, G. (1998). Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-i PI3 kinase to the DAF16 transcription factor. Genes Dev 12, 2488-2498. Picard, F., Kurtev, M., Chung, N., Topark-Ngarm, A., Senawong, T., Machado De Oliveira, R., Leid, M., McBurney, M. W., and Guarente, L. (2004). Sirtl promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429, 771-776. Rogina, B., and Helfand, S. L. (2004). Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 101, 15998-16003. Takata, T., and Ishikawa, F. (2003). Human Sir2-related protein SIRTI associates with the bHLH repressors HES 1 and HEY2 and is involved in HES 1- and HEY2-mediated transcriptional repression. Biochem Biophys Res Commun 301, 250-257. Tang, E. D., Nunez, G., Barr, F. G., and Guan, K. L. (1999). Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 274, 16741-16746. Timmons, L., Court, D. L., and Fire, A. (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditiselegans. Gene 263, 103-112. Tissenbaum, H. A., and Guarente, L. (2001). Increased dosage of a sir-2 gene extends lifespan in Caenorhabditiselegans. Nature 410, 227-230. Tran, H., Brunet, A., Griffith, E. C., and Greenberg, M. E. (2003). The many forks in FOXO's road. Sci STKE 2003, RE5. Tzivion, G., Shen, Y. H., and Zhu, J. (2001). 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 20, 6331-6338. Van Der Heide, L. P., Hoekman, M. F., and Smidt, M. P. (2004). The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J 380, 297-309. van der Horst, A., Tertoolen, L. G., de Vries-Smits, L. M., Frye, R. A., Medema, R. H., and Burgering, B. M. (2004). FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J Biol Chem 279, 28873-28879. Vanfleteren, J. R., and Braeckman, B. P. (1999). Mechanisms of life span determination in Caenorhabditiselegans. Neurobiol Aging 20, 487-502. Vaquero, A., Scher, M., Lee, D., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2004). Human SirTI interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16, 93-105. Viswanathan, M., Kim, S. K., Berdichevsky A., and Guarente L. (2005). A role for sir-2.I1regulation of ER stress response genes in determining C. elegans lifespan. Dev Cell 9, 605-15. Wang, W., and Shakes, D. C. (1997). Expression patterns and transcript processing offtt-1 andftt-2, two C. elegans 14-3-3 homologues. J Mol Biol 268, 619-630. Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., Frye, R. A., and Mayo, M. W. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J 23, 23692380. TABLES Table 1: Reducingpar-5 andftt-2 by RNAi results in nuclear accumulation of DAF-16::GFP % nuclear DAF-16::GFPa % arrested L3 larvae (n) Adults (n) larvae RNAi vector 0 (123) 0 (>200) 0 par-5 RNAi (N-terminal) 18 (82) 50 (80) 0 ftt-2 RNAi (N-terminal) 100 (91) NA 100 par-5+ftt-2 RNAi 100 (66) NA 96 ND 48 (219) 0 68 (520) NA 80 (N-terminal) par-5 RNAi (C-terminal) ftt-2 RNAi (C-terminal) GFP localization was scored using fluorescence microscopy to observe the progeny of young adults fed the indicated RNAi bacteria. a All animals with any cells containing nuclear GFP were scored as positive for nuclear DAF-16::GFP. n = total number of animals examined, ND = not determined, NA = non-applicable because of larval arrest. Table 2: Analyses of lifespan experiments sir-2.1 genotype RNAi WT WT WT WT WT WT (+) (+) (+) (+) (+) (+) Vector par-5 ftt-2 Vector par-5 ftt-2 20 17 17 14 14 14 70(1) 56(1) 61(1) 69(1) 65(1) 71(1) P=0.0407 P=0.0007 P<0.0001b P=0.0840 P=0.0844 pkIsl641 WT (+) Vector 20 101(2) P= 0 .4 14 9 b [unc-119] WT (+) WT (+) WT (+) par-5 ftt-2 daf-16 16 16 14 104(2) 100(2) 35(1) P<0.0001 P<0.0001 P<0.0001 sir-2.1(ok434) If(-) If(-) Vector par-5 16 15 94(2) 75(2) P< 0 .0 00 1b P=0.0412 If(-) If(-) ftt-2 daf-16 14 14 68(2) 43(1) P=0.0006 P<0.0001 O/E (++) Vector 30 122(2) P< 0 .0 0 01 b [sir-2.1, unc-119] O/E (++) par-5 19 138(2) P<0.0001 daf-2(e1370) O/E (++) O/E (++) WT (+) ftt-2 daf-16 Vector 15 15 41 169(2) 94(2) 100(2) P<0.0001 P<0.0001 P< 0 .0 00 1b par-5 ftt-2 daf-16 None 38 38 18 39 54(1) 106(2) 111(2) 122(2) P=0.7011 P=0.1219 P<0.0001 daf-2(e1370) WT WT WT WT sir-2.1(ok434) If (-) None 10 91(2) daf-2(e13 70); sir-2.1(ok434) If (-) None 43 143(2) Genotype N2 daf-16(mgDf50) pkIsl642 (+) (+) (+) (+) Median # animals P value vs. Shown lifespan, that died controla in figure: (# trials) days 6A--D 6D 6A 6C 6B 6E 6F P<0.0001 c All statistical analyses were performed with Prism4 software, which uses KaplanMeier survival analysis, and the log-rank test to analyze survival curves. If, loss-of-function; O/E, overexpression. aUnless stated otherwise, p-values compare the lifespans of worms grown on bacteria expressing a particular RNAi clone to the lifespans of worms of the same genotype grown on bacteria expressing the RNAi vector control. bp-value vs. the lifespans of wild-type (N2) worms grown on an RNAi vector control. Cp-value compares the lifespans of sir-2.1(ok434) animals to daf-2(e 13 70); sir2(ok434) double mutants. 87 Table 3: daf-16 targets upregulated in a sir-2.1 overexpresser. Reported as daf-16 target (ref) (3) Elevation fold in sir-2.1 o/e# Description 2.8 lys-7 protein involved in the response to pathogenic bacteria (3) 2.3 C05E4.9 gei-7 member of the malate synthase and the isocitrate lyase family (1, 2, 3) 2.7 sod-3 superoxide dismutase C08A9.1 (3) 2.3 dod-12 acyl-coA dehydrogenase C55B7.4 (3) 2.2 Protein of unknown function E01G4.3 (3) 3.5 Protein of unknown function F15E6.4 (3) 2.4 D-aspartate oxidase F18E3.7 (3) 2.5 F21C10.10 Protein of unknown function (3) 2.3 Member of a protease protein family F28A12.4 (3) 6.3 acs-2 fatty-acid acyl-coA ligase F28F8.2 (3) 2.2 Protein of unknown function F43H9.4 (3) 3.1 F45D3.4 Protein of unknown function (3) 2.0 gst-4 glutathione S-transferase K08F4.7 (3) 2.3 Member of a transporter/facilitator family K09C4.5 (3) 2.1 Protein similar to asparagine synthase M02D8.4 (3) 2.3 R12A1.4 ges-1 carboxylesterase expressed in gut cells (3) 2.8 Member of the carboxylesterase protein family T02B5.1 (3) 2.5 T10B9.1 cyp-13A4/dod-1 cytochrome P450 family 3.9 (3) hsp-16.1 heat shock protein T27E4.8 (3) 2.0 W01B11.6 Thioredoxin 4.0 (3) W06D12.3 fat-5 delta-9 fatty acid desaturase ZC410.5 Protein of unknown function 2.0 (3) "For a full list of genes that change expression levels in animals with extra copies of sir-2.1, see ref.4: Viswanathan et al, 2005. Gene C02A12.4 References 1. Libina, N., Berman, J. R., and Kenyon, C. (2003). Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489-502. 2. McElwee, J., Bubb, K., and Thomas, J. H. (2003). Transcriptional outputs of the Caenorhabditiselegans forkhead protein DAF-16. Aging Cell 2, 111-121. 3. Murphy, C. T., McCarroll, S. A., Bargmann, C.I., Fraser, A., Kamath, R. S., Ahringer, J., Li, H., and Kenyon, C. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditiselegans. Nature 424, 277-283. 4. Viswanathan, M., Kim, S. K., Berdichevsky A., and Guarente L. (2005). A role for sir-2.1 regulation of ER stress response genes in determining C. elegans lifespan. Dev Cell 9, 605-15 88 FIGURES Figure 1: C. elegans SIR-2.1 interacts with 14-3-3 proteins. (A) SIR-2.1 is a nuclear protein. Whole-mount larvae and adults were incubated with anti-SIR-2.1 antibody followed by washes and incubation with FITC-conjugated secondary anti-rabbit antibody. Fluorescence microscopy revealed nuclear staining in wild-type adult hermaphrodites (WT, left) but not in a sir-2.I1deletion strain sir-2.1(ok434) mutants (sir2.1(A), right). Black arrows point at intestinal nuclei, and white arrow points at germline nuclei. (*) marks non-specific staining of the pharynx muscle. (B) 14-3-3 proteins coimmunoprecipitate with SIR-2.1. Sonicates were prepared from synchronized 1-day-old adults of three strains with different levels of sir-2.1: sir-2.1(ok434), (sir-2.I (A)), the N2 wild-type strain (WT), and the sir-2.1 high-copy transgenic overexpresser geln3 (sir2.1(0/E)). All samples contained 10 mg of total protein. The SIR-2.1 band and a 30 kDa co-precipitating protein band are indicated by arrows. H, heavy chain; L, light chain. (C) Peptides identified by mass spectrometry of the 30 kDa band. The numbers represent the first and the last amino acids of each recovered peptide. (D) SIR-2.1 and 14-3-3 interact in wild-type animals. SIR-2.1 was precipitated from lysates of wild type, sir-2.1(A) and SIR-2.1 (O/E) animals, and precipitates were analyzed using western blots with the indicated antibodies. Input lanes contained 50 [tg of lysate proteins. Mock IP, to control for non-specific antibody interactions we substituted the pre-immune serum for the SIR-2.1 antibody. (E) Both SIR2.1 and 14-3-3 are present in worm nuclear fractions. Western blot of subcellular fractionations of wild-type embryos, separated into nuclear and 89 cytosolic fractions. First lane contains crude extract prior to separation. N, nuclear extract, C, cytosolic extract. Ce-lamin, a nuclear fraction marker, SQV-4, C. elegans UDP glucose dehydrogenase, a cytosolic marker. 90 Figure 1. /ý(p -- kDa 90- -SIR-2.1 60" 403020- WT. DAPI --ý6 si-.£ & -H -14 -3-3] A C. Protein (molecular mass, Da) FFT-2 WT, SIR-2,1 IF (28,067) iOK___ Input MOCK Input IP SIR-2.1 IP SIR-2.1: WT A OIE OIE WT A OlE PAR-6 (28,163) Tubulin - I 14-3-3 NICN C SQV-4 [1Ce-lamin I SIR-2.1 14-3-3 VOEDUIM KHELGACELSN VTEAEL•sNE• ,LAEVASGD YLWMEA ODRNSV•E SQ1QSYBFIAIK AtiE SIR-2.1 i- -I E. Peptides identified Pw 1 21-25 3031-43 130-140 131-146 14-148 225-24 3-435 AnQamuwsnum ONKWISROAba• 13013 1--1M 22D~2. Figure 2. C. elegans 14-3-3 proteins interact specifically with SIR-2.1 and DAF-16. (A): 14-3-3 co-precipitates with SIR-2.1 but not with HDA-1 proteins. Immunoprecipitations were performed from wild-type animal lysates with indicated antibodies, and analyzed by western blot with the same antibodies and with 14-3-3 antibodies. (B): C. elegans 14-3-3 proteins can bind DAF-16. Anti-GFP precipitations were performed from strains expressing DAF-16::GFP and LIN-53::GFP fusion proteins, respectively. 14-3-3 co-precipitated with DAF-16::GFP translational fusion protein but not with a LIN-53::GFP translational fusion protein. LC, light chain. Input lanes contained 50 Rg of lysate proteins. See Figure 1 and Experimental Procedures for details. Figure 2. A. antiHDA-1 IP antiSIR-2.1 In IP In Western blot with precipitating Ab I-- I !!! € Western blot with anti-14-3-3 Ab B. Strain: WB: LIN-53::GFP DAF-16::GFP anti-GFP anti-DAF-16 IP In TP TuI IP with iti-GFP Ab 14-3-3 LC estern blot with mnti-14-3-3 Ab Figure 3: 14-3-3 proteins affect DAF-16 localization. (A) Reducing par5,ftt-2, or both by RNAi results in nuclear accumulation of DAF-16::GFP. Top left panel shows control animals fed bacteria carrying the RNAi vector. Top right panel shows DAF-16::GFP accumulation in the nuclei of intestinal cells in worms treated with par-5 RNAi. Bottom panels show nuclear accumulation of DAF- 16::GFP in the intestinal and hypodermal cells offtt-2 RNAi (left) or double par-5 + ftt-2 RNAi animals (right). White arrows mark GFP accumulation in hypodermal nuclei, and white arrowheads mark nuclei of intestinal cells. Images of L3 progeny of animals fed with bacteria expressing the RNAi clones indicated were taken at 400X magnification using Nomarski fluorescence microscopy, 2,000 ms exposure time. (B)ftt-2 deletion results in nuclear DAF-16::GFP. Left, DAF-16::GFP in animals with wild-typeftt-2. Right, nuclear accumulation of DAF-16::GFP inftt-2(n4426A) animals. Images of L1 larvae were taken at 1000X magnification using fluorescence microscopy, 1,000 ms exposure time. 94 Figure 3. A. vecto RNi B. par- RNm Figure 4. Overexpression of sir-2.1 delays DAF-16 exit from the nucleus during recovery from stress. (A and B) DAF-16::GFP localization in animals with endogenous SIR-2.1 levels and in animals overexpressing SIR-2.1 30 minutes after heat pulse. Worms from indicated genotypes were subjected to heat pulse of 30 min at 370 C. DAF-16::GFP localizations was monitored before the heat pulse, and every 10 min thereafter. For recovery worms were transferred to 20 0 C, and DAF-16::GFP localization was again monitored every 10 min. (B) Kinetics of DAF-16::GFP subcellular relocalization during recovery from heat stress. 96 Figure 4. sir-d(I B. 100 90 7O 30 20 10 0 I Ti mfn f5 20 mihm 3 mE sW Eu k Time of recovery after heat shock 97 121 min Figure 5. Overexpression of sir-2.1 is required for lifespan extension and stress resistance of the pklsl642 transgenic animals. (A): sir-2.1 RNAi but not R11A8.5 RNAi suppresses lifespan extension ofpklsl642 animals. Median lifespans are: pkls]641 + vector RNAi: 24 days (n=l 12); pkIs1642 + vector RNAi: 32 days (n=l 13); pkls]642 + R11A8.5 RNAi: 34 days (n= 116); pkls1642 + sir-2.1 RNAi: 24 days (n=115). (B): pkIs1642 animals fed sir-2.1 RNAi show reduced resistance to heat stress relative to animals fed control RNAi vector or R11A8.5 RNAi bacteria. Median survivals at 32TC are: N2, 25 hours (n=64); pkls]642 + vector RNAi: 48 hours (n=68); R11A8.5 RNAi: 40 hours (n=78), p>0.05, ns; sir-2.1 RNAi: 25 hours (n=66), p<0.001. Figure 5. A. !641 + vector RNAi 642 + vector RNAi '642 + R11A8.5 RNAi 642 + sir-2.1 RNAi 0 days i i •'l r-2.1 RNAi dtor RNAi 1A8.5 RNAi hours Figure 6:ftt-2 and par-5 are required for sir-2.1-mediated lifespan extension. Lifespan analyses were performed using RNAi plates as described in Experimental Procedures. A-E: The black dashed line represents the lifespans of wild-type (N2) control animals fed bacteria carrying control RNAi vector, and solid lines represent lifespans of animals of indicated genotypes fed with bacteria carrying control vector (black), par-5 RNAi (magenta),ftt-2 RNAi (blue), or daf-16 RNAi (red). The sir2.1(wt) animals in (A) and pk1642[sir-2.10/EJanimals carried the unc-119 coinjection marker. Experiments were performed at 20 0 C. (A) par-5 andftt2 RNAi shorten the lifespans of animals with endogenous sir-2.I1levels by -25%. (B) RNAi targeting par-5 orftt-2 prevents the lifespan extension by extra copies of sir-2.1. Worms fedftt-2 RNAi had lifespans similar to those of animals fed daf-16 RNAi (-50% reduction compared to the vector control), and par-5 RNAi had a weaker effect, as lifespans were shortened to the level of the wild type (-30% reduction relative to the vector control). (C) Effects ofpar-5,ftt-2, and daf-16 RNAi on the short lifespan of sir-2.1 loss-of-function animals. Reductions in the expression levels of any of the three genes resulted in a similar slight (<10%) shortening of the life span of sir-2.1(ok434) animals. (D) par-5 andftt-2 RNAi do not significantly shorten the life span of daf-16(mgDf50) animals. (E)ftt-2 RNAi do not significantly shorten the long life span of daf-2(e1370) animals. (F) Loss of sir-2.1 function does not shorten the life span of daf-2(e1370) animals. Epistasis analysis of sir-2.1 and daf-2 genes showed that double mutants bearing loss-of-function mutations in both genes had long life spans, similar to those of daf-2(l) animals. The experiment in (F) was performed at 250 C. 100 Figure 6. . -- -- -- .-- oW rNAi 0lbA IA. fI5u j , edaer Niw r-5 R"4NA -2 RNAi Q1(OE)+par-5 RNAi U1(cO+#I-2 RNMi 1(EeW+daf-s RNAi Led vekr RNAi D. n 4 0).VK RNA O*)par-5RNA O+it-2RNAi ul rjWr-5 RN4Ai lftt-2 RNAi H+da-16 RNAi LAi or RNAi ! dWys E. --- -a i 12WalVMayrr;SSRW apsect Jfeg 1370); sir-2.1(A) 4e.137r0W R*M 1370) -2*137)%peRNMi #Vf3r+R RNM I duy do, 101 Figure 7. Overexpression of sir-2.1 leads to stress-resistance and activation of the daf-16 target gene sod-3 in anftt-2-dependent manner. (A-B): sir-2.1 andftt-2 promote stress resistance. WT, wild type; sir-2.1(A), sir-2.1(ok434); ftt-2(A), ftt-2(n4426); sir-2.I (o/e), pkIs1642. A. Young adults of the indicated genotypes were subjected to heat shock at 320 C, and their viability was scored over the next 40 hr at the indicated time points. B. Synchronized L larvae were transferred to plates containing 0.25 mM paraquat. The number of animals that developed past the L4 stage was assessed 96 hr later. Error bars represent standard deviations from triplicate experiments. The p values vs. the wild type are: sir-2.1(A), p=0.012 ; sir- 2.1(o/e), p=0.001;ftt-2(A), p=0.004;sir-2.1(o/e);ftt-2(A) relative to sir2.1(o/e), p=0.001. (C-K) SIR-2.1 overexpression promotes activation of sod-3 transcription, in daf-16 andftt-2-dependent manner. The psod3::gfp(muls84) reporter was crossed into the indicated genetic backgrounds, and the expression of the reporter was observed using fluorescence microscopy. Note higher levels of sod-3::gfp expression in both strains overexpressing sir-2.1, the high-copy overexpresser geln3 (D), and the lowcopy overexpresser pkIsl642 (E), relative to that in the wild type (C). In daf-16(mgDf5O) animals sod-3::gfp expression was not increased by transgenes carrying extra copies of sir-2.I1(compare D to G and E to H). Theftt-2 deletion did not have a pronounced effect on sod-3::gfP expression in the wild-type background (J) but totally prevented induction of sod3::gfp expression in worms with extra copies of sir-2.1 (compare K to E). This effect offitt-2(A) was specific for sir-2.1, since anftt-2 deletion did not reduce elevated sod-3::gfp expression in the daf-2(lf) strain (compare I to 102 F). All images were taken at 50X magnification using Nomarski fluorescence microscopy, with an exposure time of 300 ms. Figure 7. --- *-21 L. PI "/,,--,•| ' + ( tit) 2- 2.f +);ft-2(& }( .2f(OQE);9f-2(+) 2.f(oE);fft-2(2Q 0 4 8 i S 12 1i 20 24 28 32 3X 40 sir-2.1 + OIE A + O/E Mft-2+ + + A A hr D E C=, si-.IOE pi wl typ e- Gl s ir,O rt.H++ F 103 62 Figure 8. DAF-16 and SIR-2.1 interact in worms following stress, and this interaction depends on 14-3-3. (A) Coimmunoprecipitation of SIR2.1 with DAF-16::GFP and 14-3-3 proteins following heat shock. Worm lysates were prepared from geln3 worms (lacking DAF-16::GFP) or animals overexpressing DAF-16::GFP fusion protein. SIR-2.1 or DAF16::GFP were precipitated from worm lysates using SIR-2.1 antibody or an anti-GFP monoclonal antibody. Precipitates were separated by SDS PAGE and analyzed using western blots with antibodies to DAF-16 (polyclonal serum), SIR-2.1, and 14-3-3. +, animals were heat-shocked for 45 min at 37TC prior to lysis (B) Coimmunoprecipitation of SIR-2.1 and DAF16::GFP in animals with low insulin signaling and reduced levels of 14-3-3 proteins. Lysates were made as in (A). Worms were grown on RNAi plates containing L4440 vector control bacteria (vector), daf-2 RNAi bacteria, or ftt-2 RNAi bacteria. The lysates were analyzed as in (A). (C) A summary of immunoprecipitation data above: associations between SIR-2.1, 14-3-3, and DAF-16 under normal conditions, following stress, and upon low insulinlike signaling. 104 Figure 8. input IP antibody Pre SIR-2.1 DAF-16::GFP - + + Heatshock +4 + DAF-16 GFP 4-444- --- - - SIR-2.1 N i I --- ....... - - ... *LC -- Tubulin IP anti-GFP vector def-2 ftt-2 -I- - + input vector daf-2 fW-2 -.Heat shock -+ RNAi - + DAF-16 DAF-16 L SIR-2.1I i SIR-2.1 _ 4 Tubulin RNAi 114-3-3 :___..__._.._.______....__.__.~_~~_~,_, rl~~~~~l~~~--~~---~----1 input vector def-2 Heatshock IPanti-SIR-2.1 ftt-2 vector def-2 Wf-2 - + - DAF-16 DAF-16 I ---- ... SIR-2.1 .i 4 14-3-3 Complexes I -- ------ ----- m 1 Normal i Stress conditions SIR-2.1 eee 2 14-3-3 Low insulin signaling Low insulin + stress SIR-2.1/14-3-3 + + + + DAF-16/14-3-3 + + -/- +1- SIR-2_1/DAF-16 . + - 105 +1- Figure 9. daf-2 RNAi results in low insulin-like signaling. (A): DAF16::GFP accumulates in nuclei of animals subjected to daf-2 RNAi. daf16::gfp animals were fed daf-2 RNAi bacteria or control RNAi vector bacteria for two generations. The picture shows young adult progeny of animals transferred to RNAi plates at L1 larval stage. Arrows point at intestinal nuclei, arrowheads point at hypodermal nuclei. (B): Animals fed daf-2 RNAi show significant lifespan extension relative to animals fed control RNAi vector. Median lifespans are: vector RNAi: 20 days (n=64), daf-2 RNAi: 32 days (n=52), p<0.001. 106 Figure 9. A. veto U - B. - vedor RNA -- daf-2 RNAi I I' ._ 0 4 8 12 16 20 24 28 32 36 40 44 48 days 107 Figure 10. A model for the roles of SIR-2.1 and 14-3-3 in DAF-16 regulation of stress resistance and lifespan. We propose that following stress SIR-2.1 binds DAF-16 in the nucleus in a 14-3-3-dependent manner and the resulting complex participates in transcriptional activation of DAF16 target genes. 14-3-3 may promote the interaction between SIR-2.1 and DAF- 16 either by scaffolding the complex or through a modification of DAF-16 or SIR-2.1 following stress. Under low insulin-like signaling conditions DAF-16 is not phosphorylated at the Akt sites, becomes dissociated from 14-3-3 and accumulates in the nucleus. Nuclear DAF-16 produced by low insulin-like signaling does not bind SIR-2.1 and does not require sir-2.1 and 14-3-3 function for activation. It is possible that another nuclear cofactor promotes DAF-16 activity under low insulin conditions. 108 Figure 10. Iss eir I (d so -ff LITO RPSfl T "~> fie span J StresAR / f Reduced irisulin/IGF signaling cytoplasm nucleus 109 Chapter 3 3-Ketoacyl thiolase controls the rate of aging in C. elegans Ala Berdichevsky, Erika Hartweig, Leonard Guarente, and H. Robert Horvitz Erika Hartweig performed the electron microscopy experiments in this chapter. 110 ABSTRACT Studies of long-lived C. elegans mutants identified several genes that function to limit the nematode lifespan, i.e. mutate to extend lifespan. By contrast, studies of genes that normally function to delay aging were limited. We identified a keto-acyl thiolase gene kat-1 in a genetic screen for C. elegans mutants that age prematurely. Loss of kat-1 function results in marked accumulation of aging pigment lipofuscin, short lifespan, early behavioral decline, and other phenotypes characteristic of premature aging. kat-1 encodes a conserved metabolic enzyme involved in fatty acid oxidation. Our findings indicate that kat-1 acts to delay C. elegans aging independently of known pathways for longevity determination, and suggest that fatty acid oxidation may be important for successful aging of C. elegans. 111 INTRODUCTION Over the last few decades, several lines of experiments using C. elegans and other organisms have resulted in progress towards understanding molecular mechanisms of aging (Russell and Seppa 1987; Guarente and Kenyon 2000; Braeckman et al. 2001; Gershon and Gershon 2002). C. elegans aging, like aging of many organisms, has both predetermined and stochastic components: animals of identical genotype will not have identical lifespans, but a population under controlled conditions will show a predictable distribution (Klass 1977). Loss-of-function mutations in single genes in C. elegans can substantially increase lifespan (Kenyon et al. 1993; Tissenbaum and Ruvkun 1998; Hsin and Kenyon 1999; Arantes-Oliveira et al. 2003), indicating that lifespan determination in nematodes has a major genetic component. Research in the field has largely focused on genes that normally function to reduce longevity, that is, genes that mutate to extend lifespan. This focus has been primarily a consequence of the difficulties in distinguishing accelerated aging from sickness in short-lived mutants. While longevity remains the most commonly used measure of the nematode aging rate, additional biomarkers of C. elegans aging have been characterized (Klass 1977; Garigan et al. 2002; Herndon et al. 2002; Gerstbrein et al. 2005). These biomarkers include behavioral decline and tissue aging. Strikingly, old nematodes display several features characteristic of mammalian aging. First, aging of C. elegans is characterized by a progressive decline in locomotion, both in velocity and in coordination: aging worms move more slowly, become uncoordinated, and eventually cease movement and become paralyzed (Klass 1977; Herndon et al. 2002; Huang et al. 2004). Aging animals also show decreased feeding and 112 defecation rates (Klass 1977; Huang et al. 2004). Food consumption by C. elegans is facilitated by contractions of the pharyngeal muscle that grinds the bacteria and pumps it into the intestine (pharyngeal pumping). Old worms show a gradual decrease of pharyngeal pumping rate, followed by a cessation of pumping around day 16 of adulthood. Lack of visible pumping does not lead to immediate death, and animals can live for more than a week without feeding (Huang et al. 2004). Old nematodes, like old humans, show progressive tissue and organ deterioration. Muscles degenerate in aged worms, and gonad, intestine, and connective tissues deteriorate (Herndon et al. 2002). Herndon et al. (2002) observed that senescence in C. elegans is tissue-specific. While the nervous system of C. elegans maintains normal anatomy and expresses appropriate markers until a very old age, the muscle degenerates extensively: body-wall muscle nuclei change shape and disintegrate, and sarcomeres become disrupted. Old worms also seem to undergo misregulation of biosynthetic processes and display thickening of their cuticles (normally, the cuticle is synthesised during the larval stages prior to molting and not in adulthood) and a production and diffusion throughout the body cavity of yolk proteins, even though the oocytes that normally take up yolk proteins are no longer produced (Garigan et al. 2002; Herndon et al. 2002). The intestinal cells of C. elegans accumulate autofluorescent material in the cytoplasm, just as post-mitotic mammalian cells accumulate the aging pigment lipofuscin (Clokey and Jacobson 1986; Gerstbrein et al. 2005). Lipofuscin consists of variable lipid peroxidation products, oxidized proteins, and iron, and resists proteolytic degradation (Yin 1996). This material accumulates in the secondary lysosomes, the cell's waste compartment (Koenig 1964; Clokey and Jacobson 1986; Brunk and Terman 113 2002). The amount of lipofuscin gradually increases throughout adulthood, and C. elegans autofluorescent granules can be detected by fluorescence microscopy. Tissue degeneration, the accumulation of lipofuscin, and behavioral decline provide criteria to distinguish between aged and sick worms and therefore can be used to facilitate the study of the aging process in C. elegans. Specifically, these features provide biomarkers to identify genes involved in the regulation of aging rate and for characterizing mutants that show premature aging, or progeria, and may reveal a new class of aging genes in C. elegans and other organisms. Here we report the identification of a gene, kat-1, that controls the rate of aging in C. elegans. We isolated kat-1 in a genetic screen for mutants with increased accumulation of the aging pigment lipofuscin, and animals bearing loss-of-function mutations in kat-1 age and die prematurely. kat-1 encodes a conserved enzyme involved in fatty acid metabolism and was previously implicated in the control of fat accumulation in nematodes. Our results indicate that C. elegans kat-1 acts in a new pathway that controls longevity and the rate of aging. 114 RESULTS Aging pigment that accumulates in old worms fluoresces with a spectrum similar to that of mammalian lipofuscin Of the biomarkers of aging we could use to screen for C. elegans mutants that age prematurely, we chose to focus on the age-related autofluorescence accumulation. This autofluorescence was readily detectable in living old animals under fluorescence-equipped dissecting microscope, whereas young animals accumulated negligible amounts of autofluorescence (Figure lA). We determined the spectrum of the autofluorescent pigment that accumulates in old C. elegans hermaphrodites and compared it with the spectrum of mammalian lipofuscin. We prepared lysates of wild-type worms of different ages and measured the fluorescence in these extracts by fluorimeter using different excitation/emission combinations (data not shown). The best signal-to-noise ratio was observed using the excitation wavelength of 350 nm. At this excitation, the emitted fluorescence peaked at approximately 430 nm, and the magnitude of the peak fluorescence correlated with the age of the animals (Figure IB). Notably, mammalian lipofuscin fluoresces with a very similar spectrum: its excitation and emission maxima are 345 nm and 430 nm, respectively (Csallany and Ayaz 1976; Brunk and Terman 2002). These experiments show that worm aging pigment is similar to that accumulating in old mammalian cells. The characterization of the fluorescence signal by fluorimetry helped us define a custom filter set for monitoring intestinal autofluorescence (excitation 365 nm, emission 420-440 nm). Subsequent experiments were performed with this new filter set, which allowed detection of increased 115 fluorescence signal in younger worms, facilitating screening and recovery of the isolates. Mutants with abnormal aging rates show abnormal fluorescence accumulation Next we wanted to determine whether mutants with abnormal lifespans will show differences in accumulation of the lipofuscin-like aging pigment. Mutations in genes involved in the insulin-like pathway significantly alter the C. elegans lifespan and rate of aging (Finch and Ruvkun 2001; Kenyon 2005). We examined a long-lived mutant with a loss-of-function mutation in the insulin-like receptor daf-2 gene and a short-lived mutant with a lossof-function mutation in the forkhead transcription factor gene daf-16. We also tested autofluorescence accumulation in another short-lived mutant defective in the gene sir-2.1, which acts in a different aging pathway (Berdichevsky et al. 2006). Both short-lived mutants accumulated more lipofuscin-like fluorescence than the wild-type animals, at all ages tested. The long-lived daf-2 mutants accumulated less autofluorescence than did the wild type (Figure IC). Similar observations have been reported by Gerstbrein et al. (2005). These results indicate that lipofuscin accumulation is a reliable marker of aging, and that this biomarker is suitable for screening for mutants that age prematurely. Screen for mutants with increased lipofuscin To identify genes that when disrupted cause premature aging, we performed a screen for mutants that show premature accumulation of lipofuscin autofluorescence (Figure 2). We mutagenized L4 hermaphrodites with EMS and isolated synchronized F2 progeny. We then 116 treated the L4 F2 worms with 5' fluoro-deoxyuridine (FUdR), which blocks DNA replication and prevents worm eggs from hatching. This treatment allows observation of aged F2 worms without mixing these animals with the next generation. The F2 animals were screened for increased lipofuscin accumulation when they reached the second or third day of adulthood. Wild-type animals of this age accumulate a negligible amount of intestinal autofluorescense (Figure 1). Isolates with high levels of lipofuscin were moved to plates without FUdR for recovery. The phenotype of high autofluorescence was retested for in the progeny of the original isolates. We screened approximately 20,000 haploid genomes and picked 51 candidate mutants, 16 of which did not produce viable progeny. Of the 35 fertile isolates, 24 failed to retest. We obtained 11 mutant strains that reliably exhibited the premature accumulation of intestinal autofluorescence. Mutants isolated in the screen correspond to at least three complementation groups We outcrossed the isolates from the screen six times to the wild type before characterization. Three of the isolates, n4430, n4434, and n4435, exhibited a weakly semidominant phenotype: heterozygous animals showed weak accumulation of autofluorescence and were distinguishable from both the wild type and the mutant. Among the progeny of these heterozygous animals approximately one quarter showed strong fluorescence accumulation, one quarter had no detectable fluorescence and one half showed weak accumulation of autofluorescence. The phenotype of the n4501 isolate was recessive, whereas the phenotypes of the other seven 117 mutants, n4427, n4428, n4429, n4432, n4433, and n4436, were strongly semidominant: heterozygous animals showed high levels of autofluorescence but were still distinguishable from the homozygous mutants. We mapped n4430, n4434, n4435, and n4501 to linkage group II (LGII), and n4427, n4428, n4429, n4432, n4433, and n4436 mapped to LGV (data not shown). n4430, n4434, and n4435 failed to complement, while n4501 complemented n4430 for the increased lipofuscin accumulation, and therefore represents another complementation group. The progeny of crosses between different chromosome V mutants all showed the mutant phenotype. However the interpretation of these complementation tests is complicated by the semidominant phenotype caused by these mutations. In short, the 11 isolates with high autofluorescence represent at least three complementation groups, two on LGII and one or more on LGV (Table 1), suggesting that at least three genes control autofluorescence accumulation in C. elegans. Three isolates premature display multiple attributes of aging To assess whether our mutants age prematurely, we tested them for other aging-related characteristics. Progeroid mutants should have shorter lifespans than wild-type animals. Therefore, we performed lifespan analysis of our mutants (see Table 1 and Figure 3). Seven had 25-40% shorter lifespans than the wild type, whereas four had normal or almost normal lifespans (Table 1). Notably, all three allelic LGII mutants had short lifespans (Figure 3). The other LGII mutant, n4501, had only a slight (15%) shortening of lifespan, and of LGV mutants, four had shorter lifespans, while others had nearly wild-type lifespans. 118 We also assessed the behavioral declines with age of our mutants. The most obvious behavioral phenotype of aged worms is a progressive slowing of the rates of locomotion and pharyngeal pumping with time. Young worms produce robust sinusoidal movements with about 50 bodybends per minute. After about 9 days of adulthood worms slow down their locomotory rate by 50%, and by day 18 most have stopped crawling completely (Hemdon et al. 2002; Huang et al. 2004). A similar slowing of the pharyngeal pumping occurs (Huang et al. 2004). We have characterized the behavioral decline with age of our screen isolates. The three allelic LGII mutants n4430, n4434, and n4435 initially had wild-type rates of locomotion and pumping, but from the mid-point in their lives (day 9) showed a premature decline in both locomotion and pumping (Figure 4A, B). The LGII mutants n4430, n4434, and n4435 also showed premature onset of uncoordinated locomotion, another biomarker of aging in worms (Herndon et al. 2002), similar to that observed in daf-16(lf) mutants that age prematurely (Figure 4C). None of the other mutants, including those with short lifespans, developed behavioral abnormalities with age (see Table 1 and Figure 4A, B). To further analyze the phenotype of allelic LGII mutants we performed electron microscopy analysis to look at tissue integrity in aging wild type and in n4430 mutant animals. Old wild-type animals show profound decline in the integrity of many organs, including intestine, gonad, and muscle, and accumulation of electron-dense material (Herndon et al. 2002). The intestinal lumen of old worms is often bloated with bacteria, and the intestinal cells show membrane swirls typical of necrosis (Garigan et al. 2002). n4430 animals showed early decline of intestine and gonad 119 compared to the wild type, greatly increased accumulation of electrondense material, and early appearance of necrosis-like membrane swirls (Table 3 and Figure 5). Based on these results, we conclude that the gene mutated in n4430, n4434, and n4435 is likely to control normal onset and progression of aging in C. elegans. Mapping and cloning of the gene identified by n4430 We cloned the gene mutated in n4430 animals using a combination of positional cloning, polymorphism mapping and RNAi. We genetically mapped n4430 on LGII to the region between dpy-10 and unc-4 and close to unc-104 (Figure 6A). We then used polymorphism mapping and bli-2 n4430 unc-4/CB4856 recombinants to narrow the region to approximately 155 kb, spanned by three cosmids, C15F1, C52E12 and C06B7. We injected of a pool of these three cosmids and observed rescue of the premature autofluorescence observed in n4430 animals (see Figure 6B). When injected individually, only cosmid C06B7, which contains 16 predicted genes, rescued the fluorescence accumulation of n4430 animals. By contrast, cosmid ZK1127, which contains 12 of the 16 C06B7 ORFs, did not rescue (Figure 6). In parallel, we knocked down each of the genes contained in C06B7 by feeding dsRNAs, and found that reducing one of the 16 genes on C06B7, T02G5.8, results in high autofluorescence. We injected a PCR product containing the T02G5.8 gene into n4430 animals and observed that TO2G5.8 can rescue fluorescence accumulation abnormalities in the n4430 mutants (Figure 7A). Importantly, the same fragment also restored normal lifespan to the short-lived n4430 mutants (Figure 7B). We determined the 120 sequence of TO2G5.8 in n4430 mutants and found a missense mutation (G1550A) in the last exon of this gene (Figure 6C). We determined the sequences of TO2G5.8 in the two allelic mutants, n4434 and n4435, and found lesions in both (n4434: G504A, n4435: G1402A) (Figure 6). C. elegans mutants with increased intestinal autofluorescence were previously isolated by Babu (1974). Mutations in one of the genes he described, flu-3, resulted in an increase in gut autofluorescence similar to that observed in the n4430 mutants, and mapped close to dpy-10 on LGII (Babu 1974).flu3(el001) failed to complement n4430 for intestinal fluorescence accumulation, suggesting that they are allelic. We sequenced T02G5.8 influ3(el001) and found that T02G5.8 is mutated in this strain (Ala271Thr, Ala279Val), confirming that T02G5.8 isflu-3. T02G5.8 encodes a conserved thiolase involved in fatty acid metabolism. The gene T02G5.8 encodes a protein with 52% identity and 70% similarity to the mammalian enzyme acetoacetyl CoA thiolase (EC 2.3.1.9), which is involved in the beta-oxidation of fatty acids in mitochondria and peroxisomes (Kegg pathway database, http://www.genome.ad.jp/kegg/pathway.html). T02G5.8 was named kat-1 (for keto-acyl thiolase-1) by Mak et al (2006), who isolated this gene in a screen for C. elegans mutants with abnormal fat metabolism (see Discussion). We will use this name, instead offlu-3, below. kat-1 is well conserved from worms to humans, and mutations identified in our screen result in substitutions of conserved amino acids (Figure 8). To analyze the null phenotype of kat-1, we obtained a deletion allele of kat-1, tm1037, from the Japanese knock-out consortium (National Bioresource Project for the 121 Experimental Animal Nematode C. elegans). kat-1(tml037A) showed increased lipofuscin fluorescence, a shortened lifespan, and early behavioral decline comparable to those observed for the kat-1 mutants n4430, n4434, and n4435 (Figure 4C and data not shown). kat-1 delays aging independently of most known longevity pathways Several pathways are known that control C. elegans aging (Kenyon 2005). To see whether kat-1 acts to delay aging via one of these known longevity pathways, we crossed the kat-1 mutation into backgrounds of long-lived C. elegans mutants and tested whether kat-1 is required for the lifespan extension of these mutants. In particular we made kat-1 double mutants with mutants defective in the insulin-like receptor gene daf-2 (Kenyon et al. 1993), mitochondrial mutant isp-1 (Feng et al. 2001), germline specification mutant mes-1 (Berkowitz and Strome 2000), and chemosensory mutants osm-5 and che-3 (Apfeld and Kenyon 1999). In most cases we saw that kat-1 is not required for the longevity induced by mutations in these genes (see Table 2). The only exception was the germline signaling gene mes-1. Lack of a germline significantly increases the lifespan of C. elegans (Hsin and Kenyon 1999), and mes-1 temperature sensitive mutants that lacked a germline had a 63% longer lifespan than mes-1 animals that had a germline (Table 2). Although kat-1; mes-1 double mutants showed a lifespan extension relative to kat-1 alone, this extension was only 26% (Table 2). These results suggest that kat-1 acts to delay aging via an unknown pathway, which may be partially required for lifespan regulation by germline signaling. 122 kat-1 mutants are sensitive to starvation As many aging mutants show an altered resistance to stress, we tested stress responses of kat-1 mutants. We assayed three types of stresses: heat stress, oxidative stress, and starvation. In contrast to daf-1 6 progeroid animals, which are more sensitive to both heat and oxidative stress, kat-1 mutants showed wild-type sensitivity to these stressors (Figure 9A and B). kat-1 was not needed for the increased resistance to heat shock of longlived animals bearing mutations in the daf-2 insulin-like receptor gene (Figure 9A), suggesting that it is not involved in cellular responses to heat and oxidative stress. By contrast, kat-1 was important for normal response to starvation stress, because kat-1 mutants show increased sensitivity to starvation compared to wild type (Figure 8C). For example, after nine days without food, over 90% of kat-1 mutants had died, while two thirds of the wild-type animals were still alive. Notably, kat-1 mutants also were less responsive to calorie restriction (CR) relative to the wild type, as they showed a less significant lifespan extension upon CR (35% extension for the wild type vs. 24% extension for the kat-1 mutants, see Table 2), consistent with the idea that kat-1 function is required for coping with energy deficit. Other fatty-acid beta-oxidation genes affect aging Mutations in the gene kat-1 result in abnormalities in fat metabolism (Mak et al. 2006). The enzyme encoded by kat-1 likely catalyzes the last step of fatty acid breakdown, and kat-1 loss-of-function might result in a decreased activity of the entire lipid beta-oxidation pathway. Beta-oxidation of fatty acids involves binding of the camitine molecule, which is required for the transport of fatty acids into the mitochondria. This reaction is 123 catalyzed by carnitine transferase and it is a rate-limiting step of fatty acid beta-oxidation (Shepherd et al. 1966). Other enzymatic steps include CoA binding and subsequent oxidation reactions that lead to the production of 3ketoacyl CoA, which is cleaved into acetyl CoA and acyl CoA chain, now shortened by two carbons (Figure 10). C. elegans genome contains multiple genes encoding homologs of enzymes that catalyze different steps of betaoxidation pathway (see Table 4). We hypothesized that the progeroid phenotype of kat-1 mutants is the result of an impaired fat oxidation by these animals. We therefore asked whether other genes involved in fatty acid beta-oxidation also affect the rate of worm aging. Specifically, we used RNAi to reduce expression of the C. elegans enzymes predicted to catalyze different steps of the fatty acid oxidation pathway and assessed lifespan and fluorescence accumulation in these animals. The results of these experiments are summarized in Table 4. While RNAi of many of the genes tested did not cause aging abnormalities, reducing several fatty-acid oxidation genes, including cpt-1 carnitine palmitoyl transferase 1, ech-2 enoyl dehydrogenase 2, ard-1 dehydrogenase 1, and another ketoacyl thiolase T02G5.4, resulted in a significant shortening of lifespan and/or premature accumulation of lipofuscin fluorescence (see Figure 11 and Table 4). RNAi reduction of other putative beta-oxidation enzymes did not lead to any visible abnormalities, suggesting that they are either not involved in the regulation of aging, or that they act redundantly with other genes encoding members of the same category of enzymes. Alternatively, the RNAi may not have caused knock down of these genes to the extent required to see the abnormalities. 124 These results are consistent with the hypothesis that the premature aging phenotype of kat-1 mutants is not kat-l-specific but rather results from down-regulation of the fatty acid beta-oxidation pathway. 125 DISCUSSION KAT-1 thiolase regulates C. elegans aging. We report the isolation of C. elegans loss-of-function mutations in the ketoacyl thiolase gene kat-1 in a genetic screen for animals that age prematurely. Here we show that kat-1 mutants have multiple defects characteristic of accelerated aging: they precociously accumulate high levels of aging pigment lipofuscin, show premature tissue aging, and have short lifespans. These animals also show early behavioral decline typical of old age. This finding indicates that kat-1 functions to promote longevity. The phenotype of kat-1 loss-of-function mutants is unlikely to be caused by non-specific sickness, because these mutants are not more sensitive to heat and oxidative stress than the wild type. In addition, young kat-l(lf) animals do not show any behavioral or tissue abnormalities apart from increased lipofuscin. Therefore, we believe that kat-1 generally functions to maintain normal rate of aging in worms, and its loss-offunction accelerates the aging process. Mutations in kat-1 were previously identified in a screen for mutants displaying synthetic fat accumulation with tub-1 loss-of-function. tub-i encodes a homologue of mammalian Tubby, which is involved in glucose and fat metabolism (Wang et al. 2006). kat-1 acts in parallel to tub-i in the mitochondria of intestinal and muscle cells to control triglyceride accumulation in C. elegans (Mak et al. 2006). Defects in the mitochondrial ketoacyl-CoA thiolase activity are found in patients with a rare metabolic disease characterized by impaired ketone body metabolism, lactic acid build-up, and inability to sustain starvation (Fukao et al. 2001). We found 126 that C. elegans KAT-1 also results in sensitivity to starvation, suggesting that the two proteins may play similar roles in energy homeostasis. Fatty acid oxidation in aging and disease Fatty acid beta-oxidation is a crucial process for acquiring energy from dietary fats and fat stores (Ockner 2004). Fat is the main energy store in the cell, and fatty acid oxidation is pivotal at times of food deprivation, such as starvation or fasting (Finn and Dice 2006), and in organs that heavily rely on energy produced from fat, such as liver, skeletal muscle, white adipose tissue, and the heart (Dhalla et al. 1992; Wolfe 1998; Ockner 2004). Fatty acid oxidation is carried out in mitochondria and peroxisomes, and integrity of these organelles is crucial for effective energy production from fat fuels (Reddy and Mannaerts 1994; Shigenaga et al. 1994; McGarry 1998; Reddy and Rao 2006). Consistent with the crucial role of beta-oxidation in energy homeostasis, mutations in genes encoding metabolic enzymes and upstream regulators of fatty acid oxidation have been implicated in a variety of human disorders. For example, mutations in the mitochondrial trifunctional protein that possesses fatty acid dehydrogenase, enoyl-coA hydratase and hydroxyacyl-coA dehydrogenase activities result in severe metabolic disease characterized by inability to oxidize fats, liver abnormalities, and inability to sustain starvation (Angdisen et al. 2005). Accordingly, mutations in several fatty acid metabolism enzymes in rodents also cause metabolic defects and early death (Ibdah et al. 2005; Tolwani et al. 2005; Huyghe et al. 2006; Kao et al. 2006). Normal mitochondrial energy production via oxidative phosphorylation results in an accumulation of reactive oxygen species that ultimately cause damage to mitochondria and mitochondrial dysfunction (Singh 2006). 127 Mitochondrial DNA mutations and non-functional mitochondria accumulate during aging (Wallace 2005; Kujoth et al. 2006). Numerous studies over the past 30 years showed an association between aging and decreased fatty acid oxidation. Fatty acid oxidation rates are low in a variety of tissues in old animals, such as skeletal muscles, liver, adipose tissue and myocardial muscle (Mori et al. 1988; Paradies et al. 1995; Guo et al. 2007). Tissues from old animals show decreased activities of multiple enzymes implicated in fatty acid metabolism (a few examples include Acetyl-coA dehydrogenase, AMPK, PGC-lbeta, and camitine acetyltransferase (Mori et al. 1988; Nogalska et al. 2003; Gonzalez et al. 2004)). Accumulation of triglycerides and various products of incomplete lipid oxidation also occurs at old age, leading to so-called lipotoxicity, and is associated with age-related disorders like type 2 diabetes, metabolic syndrome and Alzheimer's disease (Montine and Morrow 2005; Reddy and Rao 2006; Weinberg 2006). Does impaired fatty acid beta-oxidation cause aging? Despite the apparent association between decline in fatty acid oxidation and aging, it is unclear whether aging causes metabolic abnormalities, or metabolic abnormalities contribute to the aging process, and both ideas are discussed in the field (Toth and Tchernof 2000; Slawik and Vidal-Puig 2006). That loss of function of KAT-1, an enzyme involved in fatty acid breakdown, causes progeria raises the possibility that fatty acid beta-oxidation pathway is important for the control of aging and lifespan. Reducing expression of several beta-oxidation genes caused effects similar to that of kat-1, namely increased lipofuscin and short lifespan. These results are consistent with the hypothesis that beta-oxidation pathway is 128 involved in the regulation of C. elegans aging. Notably, overexpressing katI did not result in lifespan extension, perhaps due to the fact that other steps of the pathway were limiting. Indeed, the reaction catalyzed by kat-1 is not the rate-limiting reaction in fatty acid oxidation (Shepherd et al. 1966). What causes the aging abnormalities of kat-1 mutants and of the animals with a presumptive reduction of fatty acid oxidation? One possibility is that inability to efficiently process fatty acids may result in insufficient cellular energy, leading to cellular and organismal death. Alternatively, defects in fatty acid oxidation might lead to the accumulation of toxic substances originating from intermediates of lipid peroxidation; such toxic substances might promote cellular damage and aging. Further experiments to identify compounds that accumulate in kat-1 mutants as well as studies of the energy utilization throughout the C. elegans lifespan might shed light on the mechanistic basis of the effect of kat-1 and of fatty acid oxidation on aging. 129 EXPERIMENTAL PROCEDURES Strains Strains were cultured as described by Brenner (Brenner 1974) and maintained at 20 0 C unless specified otherwise. The wild-type strain described in this study was the Bristol strain N2 (Brenner 1974). The following mutations were used (described by Riddle (1997) unless otherwise noted): LGI: dpy-5(e61), daf-16(mgDf50) (Ogg et al. 1997), che-3(e1124) LGII: unc-85(n319), bli-2(e768), dpy-10(e128), unc-104(e1265), unc4(e120), bli-1(e769), kat-1(tm1037, n4430, n4434, m4435, e1001) (this study). LGIII: unc-32(e189), daf-2(e1370) LGIV: unc-5(e53), sir-2.1(ok434), isp-l(qm150) LGV: dpy-11(e224), unc-76(e911), n4427, n4428, n4429, n4431, n4432, n4433, n4436 (this study) LGX: lon-2(e678), osm-5(p813), mes-l(bn74ts) Isolation of mutants with high lipofuscin-like fluorescence We mutagenized N2 hermaphrodites at late L4 stage with ethyl methanesulphonate (EMS) as described by Brenner (1974). After recovery on food for 1 hour, individual Po animals were transferred to 50 mm Petri plates. After 5 to 6 days the Fl animals were dissolved by bleaching in NaOH and bleach solution. Bleaching dissolves larvae and adults, but not embryos, and this procedure results in a synchronized culture, which is important when studying aging. The F2 animals were transferred to 50 mm Petri plates, 100 animals per plate. At the L4 stage, fluorodeoxyuridine 130 (FUdR) was added to plates to final concentration of 25 uM. Animals were screened for high intestinal autofluorescence at day 2 or 3 of adulthood. We screened animals equivalent to approximately 20,000 mutagenized haploid genomes using this procedure. Linkage group assessment We used the following markers to determine the chromosomal linkage of newly isolated mutations that caused high autofluorescence: dpy-5 I, bli2 II, unc-32 III, unc-5 IV, dpy-11 V lon-2 X We generated animals heterozygous for the new mutation and for three of these markers. We picked progeny with high autofluorescence and determined if these progeny segregated the above markers. Polymorphism mapping We used CB4856 and bli-2 n4430 unc-4 strains for single nucleotide polymorphisms (SNP) mapping, similarly to the method described by (Wicks et al. 2001). We crossed CB4856 males with bli-2 n4430 unc-4 hermaphrodites, picked F2 animals recombinant between bli-2 and unc-4 (showing only one marker) and obtained homozygotes for the recombinant chromosome. We tested animals in next generation for the presence of the n4430 mutation and identified the recombination site by following sequence polymorphisms specific to N2 or CB4856 genome. Transgenic animals Germline transformation was performed as described by Mello et al. (1991) by injecting cosmid DNA (5-20 ng/ul) or PCR products (5-50 ng/ul) into kat-1 (n4430); unc- 76(e911) mutants. An unc-76 rescuing construct 131 p76-16B (Bloom and Horvitz 1997) was used as co-injection marker. NonUnc transgenic lines were established and scored. RNAi analyses We used the whole-genome C. elegans RNAi library (Kamath et al. 2003) as a source for dsRNA-expressing bacterial strains we used in our study. Feeding RNAi was performed as described by Fire et al. (1998), and progeny of animals subjected to RNAi were scored at day 3 of adulthood for autofluorescence accumulation. As RNAi negative controls, animals were fed HT115 bacteria containing pL4440, a backbone vector in which library constructs were subcloned. Allele sequences We used PCR-amplified regions of genomic DNA as templates in determining gene sequences. For each gene investigated we determined the sequences of the entire gene plus 250 bases of the upstream promoter sequence. At least two independent PCR products were used to confirm an observed mutation. All sequences were determined using an automated ABI 373 DNA sequencer (Applied Biosystems). Analyses of lifespan and stress-resistance Unless stated otherwise, lifespan assays were performed at 20 0 C and initiated by transferring L4 larvae of the indicated genotypes to plates containing 10 [tM fluorodeoxyuridine (FUdR). We used Prism 4 software to perform statistical analyses of survival curves. Animals that crawled off the plate, burst at the vulva or died from internal hatching of progeny were 132 excluded from the analyses. In all lifespan assays reported here, between 90% and 95% of animals died naturally and were scored. Heat-shock assays were performed at 320C using one-day-old adults. For each genotype, 50-70 animals were transferred to 5 cm plates, and viability was scored every 2 - 8 hrs after shift to 320 C. Statistical analysis was performed using Prism 4 software, as for the lifespan assays. Assays of paraquat sensitivity were performed at 20 0 C by transferring arrested L1 larvae to plates containing 0.25 mM paraquat. Development was assessed 2, 3, and 4 days later, and the percentage of animals that developed to adulthood was scored. Starvation survival analyses were performed with synchronized L larvae in M9 with or without food. Survival was assayed every 2 - 4 days by movement of larvae in liquid droplets. The student t-test was used to evaluate the significance of differences in the means. Behavioral assays Animals were grown on regular NGM plates without FUdR and transferred every other day to separate them from their progeny. For behavioral assays, animals were transferred to fresh NGM plates (seeded with bacteria the night before) and maintained for 30 min. before locomotion and/or pumping rates were counted. Locomotion rate was determined by counting body bends per minute of moving animals in the center of a fresh bacterial loan. Pumping rate was assayed by counting the number of movements of the pharyngeal muscle per 30 seconds in animals present in the bacterial loan. At least 15 animals per genotype per time point were assayed. Classification of aging animals into behavioral classes 133 was done as described by Hemdon et al. (2002). At least 80 animals per genotype were assayed. Electron Microscopy Adult hermaphrodites of various ages were fixed in a mixture of 0.8% gluteraldehyde and 0.8% OsO4 in 0.1 M cacodylate buffer (pH 7.2) for one hr at 4°C in the dark. Worms were cut in this fixative with a razor blade, the head regions discarded, and postfixed in 2% OsO4 in 0.1 M cacodylate buffer overnight at 40 C. Approximately four posterior regions were then aligned in an agar block, and the agar blocks were dehydrated and infiltrated into an Epon-Araldite mixture. Sections of the midbody region (-30 nm) were collected on Pioloform-coated slotted grids. Sections were post-stained with 1% uranyl acetate and Reynolds lead citrate. Sections were viewed using a JEOL 1200EX electrone microscope at 80 KV. Sections from at least ten animals per genotype were examined and analyzed. 134 Acknowledgments We thank the Caenorhabditiselegans Genetics Center for providing strains, Dr. Shohei Mitani and the Japanese knock-out consortium for the kat1(tm1037) strain, Na An and Beth Castor for strain maintenance and determining DNA sequences, respectively, Brendan Galvin, Niels Ringstad, Hillel Schwartz, and other members of the Guarente and Horvitz laboratories for suggestions. References Angdisen, J., Moore, V.D., Cline, J.M., Payne, R.M., and Ibdah, J.A. 2005. Mitochondrial trifunctional protein defects: molecular basis and novel therapeutic approaches. Curr Drug Targets Immune Endocr Metabol Disord5(1): 27-40. Apfeld, J. and Kenyon, C. 1999. Regulation of lifespan by sensory perception in Caenorhabditiselegans. Nature 402(6763): 804-809. Arantes-Oliveira, N., Berman, J.R., and Kenyon, C. 2003. Healthy animals with extreme longevity. Science 302(5645): 611. Babu, P. 1974. Biochemical genetics of Caenorhabditiselegans. Molec Gen Genet 135: : 39-44. Berdichevsky, A., Viswanathan, M., Horvitz, H.R., and Guarente, L. 2006. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125(6): 1165-1177. Berkowitz, L.A. and Strome, S. 2000. MES-1, a protein required for unequal divisions of the germline in early C. elegans embryos, resembles receptor tyrosine kinases and is localized to the boundary 135 between the germline and gut cells. Development 127(20): 44194431. Bloom, L. and Horvitz, H.R. 1997. The Caenorhabditiselegans gene unc76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc Natl Acad Sci US A 94(7): 3414-3419. Braeckman, B.P., Houthoofd, K., and Vanfleteren, J.R. 2001. Insulin-like signaling, metabolism, stress resistance and aging in Caenorhabditis elegans. Mech Ageing Dev 122(7): 673-693. Brenner, S. 1974. The genetics of Caenorhabditiselegans. Genetics 77(1): 71-94. Brunk, U.T. and Terman, A. 2002. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med 33(5): 611-619. Clokey, G.V. and Jacobson, L.A. 1986. The autofluorescent "lipofuscin granules" in the intestinal cells of Caenorhabditiselegans are secondary lysosomes. Mech Ageing Dev 35(1): 79-94. Csallany, A.S. and Ayaz, K.L. 1976. Quantitative determination of organic solvent soluble lipofuscin pigments in tissues. Lipids 11(5): 412-417. Dhalla, N.S., Elimban, V., and Rupp, H. 1992. Paradoxical role of lipid metabolism in heart function and dysfunction. Mol Cell Biochem 116(1-2): 3-9. Feng, J., Bussiere, F., and Hekimi, S. 2001. Mitochondrial electron transport is a key determinant of life span in Caenorhabditiselegans. Dev Cell 1(5): 633-644. Finch, C.E. and Ruvkun, G. 2001. The genetics of aging. Annu Rev Genomics Hum Genet 2: 435-462. 136 Finn, P.F. and Dice, J.F. 2006. Proteolytic and lipolytic responses to starvation. Nutrition 22(7-8): 830-844. Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. 1998. Potent and specific genetic interference by doublestranded RNA in Caenorhabditiselegans. Nature 391(6669): 806811. Fukao, T., Scriver, C.R., and Kondo, N. 2001. The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency (betaketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol Genet Metab 72(2): 109-114. Garigan, D., Hsu, A.L., Fraser, A.G., Kamath, R.S., Ahringer, J., and Kenyon, C. 2002. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161(3): 1101-1112. Gershon, H. and Gershon, D. 2002. Caenorhabditiselegans--a paradigm for aging research: advantages and limitations. Mech Ageing Dev 123(4): 261-274. Gerstbrein, B., Stamatas, G., Kollias, N., and Driscoll, M. 2005. In vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditiselegans. Aging Cell 4(3): 127-137. Gonzalez, A.A., Kumar, R., Mulligan, J.D., Davis, A.J., and Saupe, K.W. 2004. Effects of aging on cardiac and skeletal muscle AMPK activity: basal activity, allosteric activation, and response to in vivo hypoxemia in mice. Am JPhysiolRegul Integr Comp Physiol 287(5): R1270-1275. 137 Guarente, L. and Kenyon, C. 2000. Genetic pathways that regulate ageing in model organisms. Nature 408(6809): 255-262. Guo, W., Pirtskhalava, T., Tchkonia, T., Xie, W., Thomou, T., Han, J., Wang, T., Wong, S., Cartwright, A., Hegardt, F.G., Corkey, B.E., and Kirkland, J.L. 2007. Aging results in paradoxical susceptibility of fat cell progenitors to lipotoxicity. Am JPhysiolEndocrinolMetab 292(4): E1041-1051. Herndon, L.A., Schmeissner, P.J., Dudaronek, J.M., Brown, P.A., Listner, K.M., Sakano, Y., Paupard, M.C., Hall, D.H., and Driscoll, M. 2002. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419(6909): 808-814. Hsin, H. and Kenyon, C. 1999. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399(6734): 362-366. Huang, C., Xiong, C., and Kornfeld, K. 2004. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditiselegans. ProcNatl Acad Sci USA 101(21): 80848089. Huyghe, S., Mannaerts, G.P., Baes, M., and Van Veldhoven, P.P. 2006. Peroxisomal multifunctional protein-2: the enzyme, the patients and the knockout mouse model. Biochim Biophys Acta 1761(9): 973-994. Ibdah, J.A., Perlegas, P., Zhao, Y., Angdisen, J., Borgerink, H., Shadoan, M.K., Wagner, J.D., Matem, D., Rinaldo, P., and Cline, J.M. 2005. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 128(5): 1381-1390. Kamath, R.S., Fraser, A.G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., Welchman, 138 D.P., Zipperlen, P., and Ahringer, J. 2003. Systematic functional analysis of the Caenorhabditiselegans genome using RNAi. Nature 421(6920): 231-237. Kao, H.J., Cheng, C.F., Chen, Y.H., Hung, S.I., Huang, C.C., Millington, D., Kikuchi, T., Wu, J.Y., and Chen, Y.T. 2006. ENU mutagenesis identifies mice with cardiac fibrosis and hepatic steatosis caused by a mutation in the mitochondrial trifunctional protein beta-subunit. Hum Mol Genet 15(24): 3569-3577. Kenyon, C. 2005. The plasticity of aging: insights from long-lived mutants. Cell 120(4): 449-460. Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. 1993. A C. elegans mutant that lives twice as long as wild type. Nature 366(6454): 461-464. Klass, M.R. 1977. Aging in the nematode Caenorhabditiselegans: major biological and environmental factors influencing life span. Mech Ageing Dev 6(6): 413-429. Koenig, H. 1964. Neuronal lipofuscin in disease. Its relation to lysosomes. Trans Am Neurol Assoc 89: 212-213. Kujoth, G.C., Leeuwenburgh, C., and Prolla, T.A. 2006. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res 66(15): 7386-7389. Mak, H.Y., Nelson, L.S., Basson, M., Johnson, C.D., and Ruvkun, G. 2006. Polygenic control of Caenorhabditiselegans fat storage. Nat Genet 38(3): 363-368. McGarry, J.D. 1998. Glucose-fatty acid interactions in health and disease. Am J Clin Nutr 67(3 Suppl): 500S-504S. 139 Mello, C.C., Kramer, J.M., Stinchcomb, D., and Ambros, V. 1991. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J 10(12): 3959-3970. Montine, T.J. and Morrow, J.D. 2005. Fatty acid oxidation in the pathogenesis of Alzheimer's disease. Am JPathol166(5): 1283-1289. Mori, S., Morisaki, N., Saito, Y., and Yoshida, S. 1988. Age-related changes of long-chain fatty acid metabolism in rat liver. Mech Ageing Dev 44(2): 175-183. Nogalska, A., Pankiewicz, A., Goyke, E., and Swierczynski, J. 2003. The age-related inverse relationship between ob and lipogenic enzymes genes expression in rat white adipose tissue. Exp Gerontol 38(4): 415-422. Ockner, R.K. 2004. Integration of Metabolism, Energetics, and Signal Transduction Ogg, S., Paradis, S., Gottlieb, S., Patterson, G.I., Lee, L., Tissenbaum, H.A., and Ruvkun, G. 1997. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389(6654): 994-999. Paradies, G., Ruggiero, F.M., Petrosillo, G., Gadaleta, M.N., and Quagliariello, E. 1995. Carnitine-acylcarnitine translocase activity in cardiac mitochondria from aged rats: the effect of acetyl-L-camitine. Mech Ageing Dev 84(2): 103-112. Reddy, J.K. and Mannaerts, G.P. 1994. Peroxisomal lipid metabolism. Annu Rev Nutr 14: 343-370. Reddy, J.K. and Rao, M.S. 2006. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am JPhysiol GastrointestLiver Physiol 290(5): G852-858. 140 Russell, R.L. and Seppa, R.I. 1987. Genetic and environmental manipulation of aging in Caenorhabditiselegans. Basic Life Sci 42: 35-48. Shepherd, D., Yates, D.W., and Garland, P.B. 1966. The rate-limiting step in the oxidation of palmitate or palmitoyl-coenzyme A by rat-liver mitochondria. Biochem J 98(1): 3C-4C. Shigenaga, M.K., Hagen, T.M., and Ames, B.N. 1994. Oxidative damage and mitochondrial decay in aging. ProcNatl Acad Sci US A 91(23): 10771-10778. Singh, K.K. 2006. Mitochondria damage checkpoint, aging, and cancer. Ann N YAcad Sci 1067: 182-190. Slawik, M. and Vidal-Puig, A.J. 2006. Lipotoxicity, ovemutrition and energy metabolism in aging. Ageing Res Rev 5(2): 144-164. Tissenbaum, H.A. and Ruvkun, G. 1998. An insulin-like signaling pathway affects both longevity and reproduction in Caenorhabditiselegans. Genetics 148(2): 703-717. Tolwani, R.J., Hamm, D.A., Tian, L., Sharer, J.D., Vockley, J., Rinaldo, P., Matern, D., Schoeb, T.R., and Wood, P.A. 2005. Medium-chain acylCoA dehydrogenase deficiency in gene-targeted mice. PLoS Genet 1(2): e23. Toth, M.J. and Tchernof, A. 2000. Lipid metabolism in the elderly. Eur J Clin Nutr 54 Suppl 3: S121-125. Wallace, D.C. 2005. The mitochondrial genome in human adaptive radiation and disease: on the road to therapeutics and performance enhancement. Gene 354: 169-180. Wang, Y., Sebum, K., Bechtel, L., Lee, B.Y., Szatkiewicz, J.P., Nishina, P.M., and Naggert, J.K. 2006. Defective carbohydrate metabolism in 141 mice homozygous for the tubby mutation. Physiol Genomics 27(2): 131-140. Weinberg, J.M. 2006. Lipotoxicity. Kidney Int 70(9): 1560-1566. Wicks, S.R., Yeh, R.T., Gish, W.R., Waterston, R.H., and Plasterk, R.H. 2001. Rapid gene mapping in Caenorhabditiselegans using a high density polymorphism map. Nat Genet 28(2): 160-164. Wolfe, R.R. 1998. Metabolic interactions between glucose and fatty acids in humans. Am J Clin Nutr 67(3 Suppl): 519S-526S. Yin, D. 1996. Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radic Biol Med 21(6): 871-888. 142 FIGURES AND TABLES Table 1. Characterization of screen isolates Mutation Phenotype wild-type Chromosome Complementation Locomotory decline group Pumping decline Mean lifespan, da3 200C(250 C) - - normal normal 21(12) n4430 Weakly semidominant II 1 premature premature 14(11) n4434 Weakly semidominant II 1 premature premature 14(9) n4435 Weakly semidominant II 1 premature premature 14(10) n4501 Recessive II 2 normal normal 18 n4427 Semidominant V 3* normal normal 16(11) n4428 Semidominant V 3* normal normal 19(11) n4429 Semidominant V 3* normal normal 19(11) n4431 Semidominant V 3* normal normal 19(11) n4432 Semidominant V 3* normal normal 12(10) n4433 Semidominant V 3* normal normal 19(11) n4436 Semidominant V 3* normal normal 16(11) * these mutants might or might not be allelic, as they cause a semidominant phenotype. 143 Table 2. Epistasis studies of kat-1 with genes in other aging pathways Pathway Insulin-like pathway Mitochondrial respiration Sensation Gemiline signaling Genotype Median life span N/ # of trials wild-type (20C125c*) 20/12 >500/>10 kat-1(n4430) (20c25c*) 17/10 >500/>10 daf-2(e1370)* 35 76/2 kat-1(n4430);daf-2(e 370) * 32 92/2 daf-16(mgDf50) 12 157/2 daf-16(m gDf50);kat-1(n4430) 11 261/3 isp-I(qml50)* 14 42/1 isp-1(qml50);)kat-1(n4430)* 12 44/1 osm-5(pr813) 41 50/2 kat-l (n4430);osm-5(pr813) 38 43/2 che-3(el 24) 26 50/2 che-3(e1124); kat-1 (n4430) 25 47/2 mes-l(bn74) gonad + 19 44/2 gonad - 31 54/2 gonad + 18 53/2 gonad - 24 36/2 kat-1(n4430); mes-1(bn74) CR wild-type, CR 49/1 kat-1(n4430), CR 44/1 *, experiments performed at 25 0 C; CR, calorie restriction. 144 Table 3. n4430 mutants display premature tissue aging Gonad Electron-dense deposits I 1 1 wild type day 8 (15) 1.5C 1.6 2.1 wild type day 13 (5) 1.8 3.2 2.8 type day 18 (5) Iwild wild type day 23 (4) 2.4 3.4 3.2 3.25 4 3.75 n4430 day 2 (5) 1i 1 1.8 n4430 day 8 (14) 2.7 3.6 3.2 Worms analyzed (n) Gut wild type day 2 (5) Key: 1 = no decline, i.e. youthful-looking tissue/ no deposits 2 = tissues showing early markers of degeneration/ few deposits 3 = marked degeneration of tissues/ deposits cover up to 50% of section 4 = total degeneration, i.e. organ is indistinguishable/deposits cover over 50% of section Electron microscopy images were analyzed for the accumulation of electrondense deposits and abnormalities in gonad and intestine. Each animal was given a score for gonad, intestine, and electron-dense deposits. The numbers represent mean scores of all animals in the group. For example, the "Gut" score of 3.25 for 23-day-old wild-type adults indicates that 3 out of 4 animals had completely degenerated intestines scored as 4, and 1 animal had markedly degenerated yet distinguishable intestine, scored as 3. 145 Table 4. Analyses of lipofuscin accumulation and lifespan in animals subjected to RNAi knock down of fatty acid beta-oxidation genes. cpt-1 Y46G5A.17 nd Lifespan (%control) nd cpt-2 R07H5.2 ++ short (75) F37C12.7 ++ dead R09E10.3 ++ short (75) acs-1 7 C46F4.2, F47G6.2, R07C3.4, R09E10.4 n n Y76A2B.3 C48B4.1, F25C8.1 n n short (72) short (70-85) F08A8.3, F08A8.4, T10E9.9, F58F9.7, F28A10.6, K05F1.3, n n ech-2 F38H4.8 ++ short (75) ech-9 FOIG10.3 ++ short (85) Lipofuscin Step in beta-oxidation (Enzyme) FA transport into mitochondria (Carnitine O-palmytoyl transferase) Gene(s) CoA binding Fatty Acid acyl-coA ligase/ Acyl coA synthetase FA breakdown Fatty Acid acyl-coA oxidase/ Fatty Acid acyl co-A dehydrogenase/ Enoyl CoA hydratase K06A5.6, T25G12.5 ard-1 FOIG4.2 Iva (na) ech-1 C29F3.1, ech-8 FOIG10.2, ech-3 F43H9.1 n n ech-7 Y105E8A.4, B0272.3, hacd-1 R09B5.6 n short (75-85) TO2G5. 7 n short (85) n kat-1 T02G5.8 +++- short (85) Acetyl coA release Acetoacetyl co-A thiolase T02G5.4 nd, not done; n, normal; Iva, larval arrest; na, not applicable; ++/+++ indicate increased lipofuscin accumulation. 146 Figure 1. Old worms accumulate autofluorescent pigment, and amount of the pigment correlates with lifespan in aging mutants. A. Intestinal autofluorescence of 1-day-old (top) and 12-days-old (bottom) wild-type adult hermaphrodites was captured using a DAPI filter (excitation 365-380 nm, emission 450 nm), with an exposure time of 1000 ms., magnification 50x. Here and elsewhere ages of animals are indicated as days post-development, i.e. day 1 is first day of adulthood. B. Emission spectrum of fluorescence of worm extracts prepared from wild-type animals of indicated ages, at 350 nm excitation. The fluorescence was measured using by spectroflurimetry. At least 100 animals were used for each extract (see Experimental Procedures for details). C, D. Emission spectrum of fluorescence of worm extracts prepared from 2-days-old (C) and 8-days-old (D) wild-type adults (wt, blue curves), long-lived daf-2(e1370) mutants animals (purple curves), and short-lived daf-16(mgDf50) (cyan curves) and sir2.1 (ok434) (yellow curves). 147 Figure 1. B. A. 14000 12000 I-day-old 10000 wild type 8000 OOF,- W - -- -- - -- 6000 4000 2000 12-day-old wild type 77T7- I 0 ;50 390 430 470 510 550 Emissio, -day C- 2 -day 5 -day 8 -day 590 nam 12 D. I0R00 12000 10000 10000 BA~ill 4000 6000 4000 4000 2000 2000 S 350 t 350 390 430 470 510 550 o90 3 430 470 510 550 590 Emissbi , sm ir-21 d1iy Am daf4My Emissiona m - wtday2 -duf-2di2 wir-2•dry2 d-16day2 I 148 L- wtday8 -d-•lqAv Figure 2. A screen for mutants with high accumulation of lipofuscin-like fluorescent pigment. We screened synchronized F2 progeny of EMS-mutagenized hermaphrodites at day 3 of adulthood for high lipofuscin-like autofluorescence. Initial isolates were retested in the next generation. 149 Figure 2. EMS/ Fl adults I Bleach to synchronize progeny Screen young adult progeny for high autofluorescence -" Retest candidates for high autofluorescence of young adults 150 Figure 3. Mutations in gene specified by n4430, n4434, and n4435 result in increased fluorescence and short lifespan A. Intestinal lipofuscin of 3-day-old wild-type and n4430 adults. B. n4430, n4434, and n4435 animals have shortened lifespans. Median lifespans were as follows: N2, 18 days, n=79; n4430, 13 days, n=105; n4434, 16 days, n=95; n4435, 13 days, n=122. 151 Figure 3. B. A. n4430 wild type Lipofuscin accumulation • . L, 80 ~~Ctl;~7 60 40 = 20 wild type C. days - wild type T kat-1 (n4430) Skat-1(n4434) - kat-1(n4435) 152 n4430 7- Figure 4. Behavior abnormalities of n4430, n4434, and n4435 mutants. A, B. Decline in locomotion rate (A) and in pharyngeal pumping rate (B) with age was assessed in wild-type animals (grey bars), three LGII mutants n4430, n4434, n4435 (blue and light blue bars), and two chromosome V mutants with high intestinal fluorescence, n4427 (scarlet bars) and n4433 (pink bars). Three LGII mutants behave normally as young adults, but show premature decline in locomotion and pumping evident from day 9 of their lifespan. C. Aging animals were classified into four groups based on their locomotion: A (green bars), animals with robust coordinated sinusoidal locomotion; B (yellow bars), uncoordinated and/or sluggish animals; C (red bars), animals that do not move forward or backward (but do move their heads or shudder upon prodding); D (black bars), paralyzed/dead animals. Four allelic mutants were tested: n4430 (1), n4434 (2), n4435 (3), and tm1037 (see below) (4). Wild-type animals (N2) and progeroid daf16(mgDf50) mutants were used as controls. 153 Figure 4. A. ·.E 40 SrN2 35 I,1, I w4427 Ili I il IT1435 II 0 day 1 day 5 day 9 day 13 day 16 SN2 B. .1 In• ,••.t Pi a II , PI III b6 a E3 I k class FB U I1 -" day 1 Cc 100% llI day 5 day 5 day 9 day 9 -- day 13 IN - 20% 1234 z day 21 day 27 11 -1111111111 111111 IF 40% 11234 day 16 day 13 60% 0% day 16 1234 Iz 1234 MS --F 154 1 1234 1234 ze4 t ýZzu it~ Figure 5. kat-1 tissue aging. Electron microscopy analyses were performed to look at tissue aging of kat-] mutants. All animals of a given age were grown identically and fixed on the same day. Figures show midbody cross-sections of wild-type worms and n4430 mutants of indicated ages. Eight days old n4430 mutants show degeneration of the gonad and the intestine, with evident packing of bacteria in the lumen and increased necrotic membrane swirls (red arrows), which are present in the wild type only at later time points, and accumulation of electron-dense material (red arrowheads). i, intestine; g, gonad. 155 Figure 5. I I I . I wt, dayI 2 wt day 8 Ibo Sc. wt. day 131 =me.,-,.q ma r L j, •. vt --- day 13 e. f. n4430. davy 8 1 J. wk 156 day 18 re R. 4430, day 8 n4430 dav8 I Figure 6. Mapping and cloning of kat-1. A. Three-point mapping placed the n4430 mutation right of or close to unc-104 on chromosome II. B. The 155 kb n4430-containingregion identified by polymorphism mapping is covered by three cosmids, one of which (C06B7) rescued the fluorescence accumulation by n4430 animals. C. Mutation sites and amino acid changes of the three alleles of T02G5.8 identified in our screen and the e1001 allele. 157 Figure 6. A. -3.22 -0.98 I--I_ 1.75 0 0.21 2.85 m.u. B. C15F1 T02G5 C52E12 ZK1127 C06B7.• ... ,, i Lines rescued/total: Cosmid: / /· .• C15F1] / C52E12 .............................. 11/11 C06B7 i C SF1 ................................... 0/3 C52E12 .................................. 0/4 C06B7..................................7/7 T02G5.9 TO2GS.Sl ZK1127 ................................... 0/3 '-, ,,_ T02GS.6 msh-4 hat-1 krs-1 TO2GS.7 A A T02G5.8 II A4434 G134E 1 II I I n4435 4430 elOOl4 C393Y G368E A279T A273V, tm103 7 deletion 0.1 kb 158 Figure 7. kat-1 controls fluorescence accumulation and lifespan. A. Intestinal fluorescence accumulation in young (1-day-old), middle-aged (5days-old) and old (12-days-old) n4430 animals compared with wild-type nematodes (N2), progeroid daf-16(l animals, and n4430 animals injected with genomic fragment containing T02G5.8 ORF and 2.5 kb of upstream promoter sequence. Images were taken as in Figure 1. B. Lifespan of n4430 animals compared with wild-type (N2) lifespan and n4430 animals injected with the rescuing genomic fragment (as in A). Median lifespans were as follows: N2, 18 days, n=109, kat-](n4430), 15 days, n=95, kat-](n4430); unc-76(e911); nls]234 [unc-76, T02G5.8), 18 days, n=83. 159 Figure 7. A. Wild type kat1(n4430) day I day 5 day 12 B. days -l- Wild type * n4430 n4430 + T02G5.8 160 kat-1(n4430); alerrnAr; RI Figure 8. kat-1 encodes a conserved ketoacyl thiolase. Alignments of KAT-1 amino acid sequence with sequences of human, mouse, and drosophila homologous proteins. Dark blue, amino acids identical in all four sequences. Light blue, sequences identical in 2-3 of the four sequences. Arrows indicate amino acids mutated in the screen isolates. 161 Figure & IML-SSGHRI --------------- FlyACA IEV CLERSVRSKPTL -- MRALVYALHOVVRRPLLRO H]• Aca1 MIVLRALLRSGARSRSPLLRR - ki:: I YAT-1T fly ACAT1 iS RRITTSRRLSH I YVERSYVSKPTL SQ Liv TNY 10 I T UI, AA K TA I PL 10 U SV E S GSLL j L- ssv BID 0 IS-+ KI, KAT-I RyACAIf MBm Acail Hýý Acai MG TK KAT-1 Fly ACATI Mamse ACaml I~m •Acal '"- -- I 7 411P LE Q I OME S WON VIKS A K T Aý' T .IENI TISl I I S Mous Acatl A=ctI Hmusu HaT allfl] A271T-- 0vDV 0I 1 MSeI A279V EQSL KAGI RLN% I R LM \ I Fy ACATI Huma Acld Marn Acatl [man Arui D AnVTV Lu Fly ACATi EAT-I V - A H oo a KIQ00Q i L ( SM anM LTOF I P LI I. V · Y Sil V OVv I 1 v 3 U _ ,&r.c f 7i im IVi -••"S A"ILLSM L JE nm - U.- niI Figure 9. kat-1 stress resistance. A. kat-1 mutants do not show increased sensitivity to heat shock. Animals were subjected to 32C heat shock at first day of adulthood, and survival was measured during the next 64 hours. At least 60 animals are presented in each curve. B. kat-1 mutants do not show increased sensitivity to oxidative stress. Wild-type and kat1 (n4430) animals were allowed to lay eggs on plates containing 25 mM and 50 mM paraquat. Fraction of animals developing to adulthood was measured after 4 days and 6 days respectively. Development of least 300 eggs per PQ concentration per genotype was monitored. A representative experiment of three independent experiments is shown. C. Survival of arrested Ll larvae from wild type and kat-1 mutants in M9 buffer lacking bacteria was monitored every 2 to 3 days. At every time point, samples containing approximately 100 animals were observed in a drop of M9, and percent of swimming animals was determined. The numbers shown are the means of three samples per time point per genotype. 163 Figure 9. A.i It- - 9.. 777 so . * kat-f (n4430) rr dF-2(el370) --& At-I (n4430); da-2(ef370) a Be74 am ,, I 0 Pua B. estanc Paraquat resistance 10t0 so 70 60 50 Swildtyp-qe g W-l·n~f~q 40 30 20 10 0 0.5s at P&,IULM 149 starvation toQle C nce 60 mwi type pa k4sii4 40 . a-·tmes 20 I 9 13 days 3 Figure 10. 3-ketoacyl thiolase catalyzes the last step in fatty acid beta-oxidation pathway. Fatty acid oxidation starts by binding of a CoA group to fatty acids, catalyzed by acyl-CoA synthases; the resulting acyl-CoAs bind camitine (in the reaction catalyzed by camitine palmitoyltransferase), which facilitates entry into mitochondrial matrix. Inside mitochondria, acyl-coAs are converted to enoyl-CoAs by acyl-CoA oxidases and acyl-CoA dehydrogenases, enoyl CoA is reduced by enoylCoA hydratase into 3-hydroxyacyl-CoA. 3-hydroxyacyl-CoA dehydrogenase then catalyzes production of 3-Ketoacyl-CoA. Finally, the last step of the fatty acid oxidation is catalyzed by a 3-ketoacyl thiolase (e.g. KAT-1 in C. elegans) and it involves binding of another CoA group and cleavage of a thiol bond that produces Acetyl-CoA and Acyl-CoA chain shortened by two carbons (see box). This Acyl-CoA molecule can enter the cycle again to be processed further. 165 Figure 10. Fatty acid + CoA Acyl-CoA synthetase Carntine palmitoyltransferase Acyl-CoA - *I.-o Acyl-CoA oxidase Acyl-CoA dehydrogenase Enoyl-CoA Enoyl-CoA hydratase 3-Hydroxyacyl-CoA 3-Hydroxyacyl-CoA dehydrogenase 3-Ketoacyl-CoA 3-Ketoacyl-CoA thiolase CoA 0 0 R-CH -CH -C-C-CoA 2 52 COA-SH o R-CH 2 -C -CoA ~2t Acetyl-CoA 0 + CHl-C-CoA 3 166 Acyl-CoA Figure 11. Reducing fatty acid oxidation genes causes increased lipofuscin accumulation in C. elegans. Intestinal autofluorescence of 3days-old wild-type animals grown on bacteria expressing indicated RNAi clones was photographed as described in Figure 1. Note that RNAi knockdown of ard-1 (acyl-CoA dehydrogenase) resulted in larval arrest at L2-L4 stages. Rare escapers that reached adulthood were small and sterile. The image below shows ard-1 (RNAi) adult escapers. 167 Figure 11. kat-1 (RNAi) Vector (RNAi) TO2GS.4 thiolase (RNAi) 168 ard-1 (RNAi) Addendum to Chapter 3. FLU-1 kynurenine monooxigenase controls intestinal fluorescence accumulation in C. elegans. 169 ABSTRACT Characterization of mutants isolated in genetic screens for increased accumulation of autofluorescence led to identification offlu-1 gene, which controls intestinal fluorescence levels in C. elegans. In our screen, we isolated three alleles offlu-1, all of which confer high levels of intestinal autofluorescence. Some but not all mutations influ-1 were associated with reduced longevity, and none resulted in premature behavioral decline, indicating that whileflu-1 may control some aspects of aging, it is unlikely to be involved in the general regulation of aging process. We cloned the gene mutated influ-1 and found thatflu-1 encodes a kynurenine monooxygenase, an enzyme involved in tryptophan metabolism. 170 RESULTS Characterization of LGV isolates. In our screen for C. elegans mutants with premature accumulation of intestinal autofluorescence, we isolated seven mutants that mapped to chromosome V (see Chapter 3 Table 1). Mutations in these isolates semidominantly cause increased accumulation of autofluorescence. Lifespan analysis of the seven isolates showed that four mutants had normal lifespans, whereas three mutants, n4427, n4432, and n4436 had shorter lifespans than the wild type (Table 1). Quantitation of the autofluorescence in n4427 and n4432 showed that these mutations resulted in strong autofluorescence accumulation, much stronger than that seen in a daf-16 progeroid mutant used as a control (Figure 1). The semi-dominant phenotype of these mutants complicated the interpretation of complementation analysis. Genetic mapping indicated that all seven mutations were close to each other on chromosome V: all showed similar linkage to dpy-11 on LGV (data not shown), raising the possibility that they were alleles of the same gene. We set to map and clone the gene mutated in one of the shorter-lived mutants, n4427. Mapping and cloning offlu-1. We used a combination of positional cloning, polymorphism cloning and RNAi to map and clone the gene mutated in n4427. We first located it to the right of or close to dpy-11 on V (see Figure 2A): in the cross between n4427 and unc-46 dpy-11, 20/20 Unc-non-Dpy recombinants showed high autofluorescence, and 0/7 Dpy-non-Unc recombinants showed high autofluorescence. We then mapped it to the right of or close to unc-23: in 171 the cross between n4427 and dpy-11 unc-23, 0/16 Unc-non-Dpy recombinants showed high autofluorescence, and 3/3 Dpy-non-Unc recombinants showed high autofluorescence. Next- we mapped the mutation to the left of unc-76: in the cross between n4427 and dpy-11 unc76, 18/34 Unc-non-Dpy recombinants showed high autofluorescence, and 13/27 Dpy-non-Unc recombinants showed high autofluorescence. Finally, we mapped the mutation to the right of or close to lon-3 and left of or close to rol-4-(Figure2A): in the cross between n4427 and unc-42 lon-3, 5/5 Unc-non-Lon recombinants showed high autofluorescence, and 0/15 Lonnon-Unc recombinants showed high autofluorescence. Similarly, in the cross between n4427 and rol-4 dpy-21, 0/26 Rol-non-Dpy recombinants showed high autofluorescence, and 4/4 Dpy-non-Rol recombinants showed high autofluorescence. We then double-cis-marked n4427 with dpy-11 and unc-76 to use the resulting strain in single nucleotide polymorphism (SNP) mapping. We performed SNP mapping as described (Wicks et al. 2001), and were able to narrow down the region to approximately 450 kbp interval on chromosome V. The above region was covered by 11 cosmids (Figure 2B). Eight of these eleven cosmids, when injected in pooled combinations, did not rescue fluorescence accumulation in n4427. We then used RNAi to knock down the candidate genes in the region determined by SNP mapping, including genes not covered by the injected cosmids (see Figure 2C). We performed the RNAi experiments in an eri-I genetic background, which is hypersensitive to RNAi (see Chapter 3 Experimental Procedures for details). Of the 48 RNAi clones tested, one RNAi clone, RO7B7.5, resulted in a striking accumulation of autofluorescence in eri-1 animals. We sequenced the coding regions of RO7B7.5 in n4427 and found a mutation in 172 the fourth exon of this gene. DNA sequencing of RO7B7.5 in all other LGV isolates revealed that two other isolates n4432 and n4433 also carried mutations in this gene. Notably, n4433 animals, which carried a nonsense mutation in RO7B7.5 did not show lifespan abnormalities despite high accumulation of fluorescence (see Discussion). In the other chromosome Vlinked isolates, n4428, n4429, n4431, and n4436, we did not find mutations in the coding regions of RO7B7.5 (Table 1). These isolates may represent alleles of other gene(s), or they can carry mutations in the non-coding regulatory regions of RO7B7.5. Sequencing of introns and promoter regions of RO7B7.5 in these mutants should answer this question. Babu (1974) performed a genetic screen for mutants with abnormal autofluorescence. One of the genes he described,flu-1, mapped to chromosome V and exhibited similar effects on gut autofluorescence to the RO7B7.5 mutants. The molecular identity of theflu-1 gene was unknown, but extracts offlu-1 mutant animals showed decreased kynurenine monooxygenase activity (Siddiqui and Babu 1980). We sequenced RO7B7.5 in theflu-1(e1002) allele and identified a mutation in the coding region of the gene. Interestingly, the mutation in e1002 changed the same amino acid as the mutation in n4427 (see Figure 2D). Therefore, RO7B7.5 isflu-1, and we will use this name below. flu-i encodes a conserved metabolic enzyme, kynurenine 3monooxygenase. R07B7.5 flu-i encodes a protein with high similarity to metabolic enzyme involved in the oxidative metabolism of tryptophan, kynurenine 3monooxygenase (EC1.14.13.9). The C. elegans FLU-1 protein is 45% identical and 68% similar to human Kmo-1 (Figure 3). This enzyme uses 173 oxygen, NADPH, and FAD for the catalytic oxidation of L-kynurenine to 3hydroxykynurenine and water (Figure 4A). Kynurenine is a key compound in the kynurenine pathway, a major pathway for tryptophan metabolism in mammals, which is involved in synthesis of nicotinic acid (Fig 4B). Previous studies by Babu et al showed thatflu-1 (el 002) animals accumulate high amounts of anthranilic acid and have low concentrations of 3-hydroxy-kynurenine caused by the block in this step of the pathway (Siddiqui and Babu 1980 and Figure 4B). That the nonsense mutation in n4433 and RNAi knock down of RO7B7.5 result in high accumulation of gut autofluorescence suggests that this phenotype is caused by loss of function of kynurenine monooxygenase. In accordance with this, extracts fromflu-i(e1002) mutants had less than 10% of the wild-type kynurenine hydroxylase specific activity (Siddiqui and Babu 1980). The semidominant nature of theflu-1 mutations implies haplo-insufficiency, suggesting that the kynurenine pathway in C. elegans is dose-sensitive. 174 DISCUSSION The essential amino acid tryptophan is metabolized via several metabolic pathways, which result in the generation of several biologically important molecules. Tryptophan metabolites include serotonin, the 'sleep' hormone melatonin, an odor molecule indole, and the neurotransmitter tryptamine (Moroni 1999). The main pathway of the tryptophan metabolism is the kynurenine pathway involved in the biosynthesis of quinolinic acid, the precursor for nicotinic acid and NAD. Tryptophan metabolism and kynurenine pathway in particular are important for normal brain function, because different intermediates of the kynurenine pathway can act as agonists or antagonists of certain glutamate receptors (Moroni 1999). Imbalances of the kynurenine pathway metabolism are implicated in numerous neurodegeneration diseases, such as Huntington's disease, Alzheimer's disease, ALS, and AIDS-dementia (Stone et al. 2001; Stone et al. 2003). We isolated mutants in a kynurenine monooxygenase geneflu-1 in a screen for C. elegans mutants with elevated intestinal autofluorescence. We obtained at least three alleles of theflu-1 gene, all of which have mutations in the coding region offlu-1 and likely result in loss-of-function of this gene. flu-1 was first characterized by Babu (1974), who isolated aflu-1 mutant in a similar screen. We have now determined the molecular lesion in the original mutant strain and shown thatflu-1 encodes a conserved metabolic enzyme kynurenine monooxygenase. Kynurenine monooxygenase catalyzes the conversion of kynurenine to 3hydroxykynurenine. Accordingly, biochemical studies by Babu and colleagues showed thatflu-1 mutants have decreased levels of 3175 hydroxykynurenine and increased levels of anthranilic acid, another product of kynurenine. The idea behind our screen was to identify mutants that show increased accumulation of aging pigment lipofuscin and are aging prematurely. However, several observations suggest thatflu-1 is not involved in the control of aging rate in C. elegans. First, the lifespan of a flu-1 (n4433) mutant, which has an early stop mutation in the flu-1 gene, is not shorter than that of wild type. Theflu-i (el 002) mutant also has wildtype lifespan. Two otherflu-1 alleles, n4427 and n4432 confer shorter lifespans than the wild type; however, both mutants have missense mutations influ-1. The amino acid mutated in n4427 is identical to the amino acid mutated in e1002, even though the resulting substitutions are different. These data suggest that short lifespan observed in n4427 and n4432 may not be specific to theflu-i loss-of-function. However, it is still possible that mutations in n4432 and n4427 result in a dominant-negative effect and cause stronger lifespan phenotype than n4433 loss-of-function. Rescue experiments reintroducing wild-typeflu-1 gene into n4432 and n4427 strains should clarify this issue. Animals mutated influ-1 show no gross abnormalities apart from high fluorescence accumulation. None of the flu-1 mutants showed behavioral abnormalities typical of premature aging mutants, such as progressive decline in locomotion and pharyngeal pumping with age (see Chapter 3 Figure 6), suggesting thatflu-1 does not generally control the rate of aging in C. elegans, and at most is involved in regulation of some aspects of the nematode aging. 176 CONCLUSIONS AND FUTURE DIRECTIONS To conclude, flu-] mutants may not be involved in aging regulation, but they may be useful in the studies of the kynurenine pathway and tryptophan biology. In particular, additional genetic screens for animals with abnormal fluorescence accumulation may result in identification of mutants in other steps of these pathways. Screens for suppressors of high fluorescence accumulation may identify upstream and downstream regulators of the pathway and shed light on the structural aspects of the FLU-1 kynurenine monooxygenase. It would be interesting to determine whetherflu-1 mutants have any defects in signal transduction in the nervous system, as young adults or as aging adults, that would parallel the abnormalities caused by deregulation of tryptophan metabolism in mammals. Finally, efforts are underway to develop therapeutic agents that target the kynurenine pathway (Schwarcz 2004). The profound increase in autofluorescence in worm kynurenine pathway mutants may make C. elegans nematodes useful for screening of potential drug candidates that target enzymes of the kynurenine pathway. 177 References Babu, P. 1974. Biochemical genetics of Caenorhabditiselegans. Molec Gen Genet 135: 39-44. Moroni, F. 1999. Tryptophan metabolism and brain function: focus on kynurenine and other indole metabolites. Eur JPharmacol375(1-3): 87-100. Schwarcz, R. 2004. The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol4(1): 12-17. Siddiqui, S.S. and Babu, P. 1980. Kynurenine hydroxylase mutants of the nematode Caenorhabditiselegans. Mol Gen Genet 179(1): 21-24. Stone, T.W., Behan, W.M., Jones, P.A., Darlington, L.G., and Smith, R.A. 2001. The role of kynurenines in the production of neuronal death, and the neuroprotective effect of purines. JAlzheimers Dis 3(4): 355366. Stone, T.W., Mackay, G.M., Forrest, C.M., Clark, C.J., and Darlington, L.G. 2003. Tryptophan metabolites and brain disorders. Clin Chem Lab Med 41(7): 852-859. Wicks, S.R., Yeh, R.T., Gish, W.R., Waterston, R.H., and Plasterk, R.H. 2001. Rapid gene mapping in Caenorhabditiselegans using a high density polymorphism map. Nat Genet 28(2): 160-164. 178 TABLES Table 1. Three of the seven screen isolates with mutations that map to chromosome V have coding mutations in the gene encoding FLU-1 R07B7.5 kinurenine 3-monooxygenase. Allele Mapped to R07B7.5 Mean lifespan, Decline in Dominance chromosome genotype days 200 C / 250 C locomotion, pumping wild type na na WT 21/12 normal e1002 strong SD V A137T 19/11, ** n4427 strong SD V A137V 16/11, p<0.05 normal n4428 strong SD V WT* 19/11, ** normal n4429 SD V WT* 19/11, ** normal n4431 SD V WT* 19/11, ** normal n4432 strong SD V P407L 12/10, p<0.05 normal n4433 strong SD V Q 115ochre 19/11, ** normal n4436 strong SD V WT* 16/11, p<0.05 normal nd SD, semidominant; na, not applicable; WT, wild type; *, only sequences of coding regions were determined; ** , not significant (p>0.05). 179 FIGURES Figure 1. Chromosome V mutants accumulate high amounts of autofluorescence. Autofluorescence was quantitated in extracts of 3-dayold and 7-day-old animals of above genotypes. For each genotype/age, 100 or more animals were used to make the extract, and the amounts were normalized by protein content. Fluorescence was measured using spectrofluorimeter, with excitation of 350nm. The values in the table represent the emission peak at 420 nm. 180 Figure 1. 12000 ' 10000 r 8000 I 6000 C 'a 4M0o ] day 3 l day 7 C )00 2] 0 I N2 N2 I d•-la• daf-16lf) I n4427 V 181 n4432 V Figure 2. Mapping and cloning offlu-1. A. 3-point positional mapping was used to mapflu-1 to a region between lon-3 and rol-4. B. SNP mapping allowed to narrow down the region to the one shown, covered by 11 cosmids. Injections of 8 of these cosmids (shown in black) did not result in rescue of the fluorescence accumulation phenotype. C. RNAi was used to knock down genes present in the area defined by SNP mapping. RNAi of R07B7.5 (red) resulted in strong accumulation of autofluorescence. D. Structure of R07B7.5 flu-] as predicted in Wormbase with sites of mutations and amino acid changes in screen isolates. 182 Figure 2. tlr46 -. 6 w23 dPYI tLc.6 4 ,v1i 0-2 I Or I) --2 -2.36 K12G1 1 7.29 3.74 4.27 13.18 F58H1 C13C4 D2023 R31 R07B7 C52E4 C08F12 F10C2 C 02C8 3 recombinants 5 recombinants RNAi: -R311 R313 - --.3 .4 .5 - .2 A A r R07B7.5 I 0.1 kb R07B7 F1OC2 R31.2 ' A .14-4 - .4 .5 .6 -- * 4-.3 .7 A I n4433, Q15ochre n4427, A237V e1002, A237T 183 A I A In4432, P407L Figure 3.flu-1 encodes a 3-kynurenine mooxygenase, a conserved metabolic enzyme. Alignment of the C. elegans FLU-i R07B7.5 protein with its closest C. briggsae homolog, and with the human 3-kinurenine monooxygenase 1 Kmo-1. C. elegans FLU-1 is 48% identical and 69% similar to Kmo- 1. 184 Fig•ure 3. oeFLU-lp obFLU-1p human kmo-1 MDSSVIQ ceFLU-1lp cbFLU-1p human kio-1 --n4433, Q>ochre obFLU-lp E to---UIIII human kmo-1 Illllllli~Plrm*rP41s~ oeFLU-lp obFLU.lp human kmo-1 S-"f 4 4 27,A>V; e1002, A>T oeFLU-lp obFLU-lp human kmo-1 B~lldCrlClr ceFLU-1p obFLU-lp humnm krno-1 (S H C oeFLU-lp obFLU-p human kmo-1 s 77- L ~rm - FA - D nUDL Q LCLP U ~rp( ~I PslPrrrr~ IllrsrrrslPs~rssrro~ f-- n4432, P>L ceFLU-lp obFLU-lp human kinmo-i - IM 1 0 FF I: MS IceFLU-1lp obFLU-lp human kmo-1 -UI -P F P- RPWMiWIRHFRNTTCFPRKRVDSLEQISLISRLLSHSEILOT Figure 4. Kynurenine metabolic pathway in tryptophan metabolism. A. Enzymatic reaction catalyzed by FLU-1. Kinurenine 3-monooxygenase catalyzes the convertion of L-kinurenine into 3-hydroxykinurenine. B. Kinurenine 3-monooxygenase in Tryptophan metabolism. In addition to serving as a building block for proteins, tryptophan can be metabolized by a variety of pathways leading to generation of biologically active molecules such as hormones, neurotransmitters and enzymatic cofactors. Kinurenine 3-monooxygenase catalyzes one of the reactions of the Kinurenine pathway that leads to the production of Quinolinic acid, a precursor of NAD. 186 Figure 4. A. kynurenine 3monooxygenase L-kyurankm "M"ydroxykynmrenkin B. PROTEINS A Tryptamine 5-Hydroxytryptopha kynureine 3-monooxygenase Kynarenine 3-HydroxyI Acetyl-c oA1 Anthranili acij[dE Quinolinic Acid NAD + NADP Nicatinamide ADPR 187 Melatonin 188 Appendix 1. A Stress Response Pathway involving Sirtuins, Forkheads, and 14-3-3 proteins. Ala Berdichevsky and Leonard Guarente This chapter was published: Berdichevsky, A. and Guarente, L. 2006. Cell Cycle 5(22): 1165-1177. Review. Acknowledgments: We thank Nicholas Bishop and Robert H. Horvitz for critical comments on this perspective. 189 Introduction. Sir2 and mechanisms of life span determination The conserved protein deacetylase Sir2 can affect life span of yeast, flies, and worms: introducing extra copies of the sir2 gene results in life span extension, while deletion of sir2 shortens lifespan (Blander and Guarente 2004). It is likely that revealing how sir2 affects longevity in simple organisms can improve our understanding of mechanisms of human aging. What do we know about mechanisms of life span extension? From studies of worms many mitochondrial genes have emerged as important for life span determination: worms with loss-of-function mutations in genes encoding components of the mitochondrial electron transport chain have much longer life spans. However, the development and behavior of these mutants are slowed down, resulting in a "slow-living" phenotype (Lee et al. 2003; Aguilaniu et al. 2005). Another conserved mechanism of life span extension involves the insulin-like pathway, attenuation of which can extend life span of worms, flies, and even mice (Kenyon 2005). In this pathway, a signaling cascade originates at the insulin-like receptor and eventually results in phosphorylation and exclusion from the nucleus of transcription factor DAF-16, the activation of which is required for the very long life span and marked stress-resistance of insulin-like signaling C. elegans mutants (Lin et al. 1997). The best-known means of extending life span, which works in every species tested, including primates, is calorie restriction (CR). Reducing calorie intake has been also shown to alter metabolism and delay aging-related disease (Koubova and Guarente 2003). Which, if any, of the above mechanisms might sir2 affect, and how is it acting? In S. cerevisiae,sir2 acts to prevent recombination at the rDNA loci, reducing the number of extrachromosomal DNA circles that cause 190 aging in yeast (Guarente 2000). This mechanism, however, is specific to yeast cells, implying that in higher eukaryotes sir2 functions in a different way to affect life span. In flies, sir2 may act as part of a calorie restriction longevity pathway and respond to the histone deacetylase protein encoded by Rpd3 (Rogina and Helfand 2004). There is some controversy about whether C. elegans sir-2.1 (the closest homologue of yeast sir2) is required for longevity induced by calorie restriction in worms. A recent report (Wang and Tissenbaum 2006) showed genetic analysis with a sir-2.1 mutant that suggested that sir-2.1 may be essential for CR-induced life span extension, while others did not see the same effect in similar and different experimental settings (N. Bishop, and M. Viswanathan, unpublished observations). It has been proposed in the past that sir-2.1 acts through the insulin-like pathway (Tissenbaum and Guarente 2001; Tissenbaum and Guarente 2002). This hypothesis stemmed from the fact that the forkhead transcription factor DAF-16, encoded by a downstream gene in the insulin-like pathway that is required for life span extension by low insulin-like signaling, is also required for life span extension by sir-2.1 overexpression in worms (Tissenbaum and Guarente 2001). However, the mechanism of how sir-2.1 extends worm life span was not known until a short time ago. 14-3-3 proteins: new players in life span determination Recently, we reported that C. elegans SIR-2.1 forms a complex with two proteins of a conserved 14-3-3 family, PAR-5 and FTT-2 (Berdichevsky et al. 2006). Studies of mammalian cells suggested that 143-3 proteins, which frequently serve as adaptors modulating activity and/or localization of their binding partners (Tzivion et al. 2001), interact with the 191 mammalian DAF-16 homologue FOXO, and that this interaction retains FOXO in the cytoplasm, inactive (Brunet et al. 1999). Is this also true for worms? Indeed, when we down-regulate 14-3-3 proteins in C. elegans, DAF-16 becomes constitutively nuclear, implying that worm 14-3-3 proteins can act similarly to mammalian 14-3-3 to sequester DAF-16 in the cytoplasm (Berdichevsky et al. 2006). One model that could explain the requirement for 14-3-3 proteins for the long life span granted by SIR-2.1 overexpression is that SIR-2.1 acts by preventing 14-3-3 from sequestering DAF-16. This appears not to be the case, however, because overexpression of SIR-2. 1 does not result in relocalization of DAF- 16 into the nucleus. Also, despite the evident role of 14-3-3 in DAF-16 subcellular localization, low levels of 14-3-3 proteins are not beneficial for longevity. Instead, low 14-3-3 levels cause shortening of life span in wild-type worms, and actually eliminate the extension of life span by sir-2.1 overexpression, indicating that 14-3-3 genes act in the same life span determination pathway as sir-2.1, downstream of sir-2.1 (Berdichevsky et al. 2006). Therefore, like SIR-2.1 and DAF-16, 14-3-3 proteins act positively on lifespan. More recently, Wang et al (2006) also reported SIR-2.1/14-3-3 physical interaction, as well as requirement for 14-3-3 proteins for the life span extension mediated by SIR-2.1 overexpression. Notably, they demonstrate that overexpression of 14-3-3 proteins causes extension of life span in a daf-16-dependent manner, much like SIR-2.1 overexpression (Wang et al. 2006). This result further supports the overall positive role of 14-3-3 proteins in C. elegans longevity. 192 Mechanism of SIR-2.1 effect on lifespan We found that C. elegans SIR-2.1 protein can interact with DAF- 16. This interaction is readily detectable when the worms are stressed by heat shock, but in the absence of heat shock (under normal conditions) a SIR2. 1/DAF- 16 complex was not detected. 14-3-3 proteins, which interact with both SIR-2.1 and DAF-16 independent of heat shock, play a vital role in the formation of the SIR-2.1/DAF-16 complex, as reducing 14-3-3 levels greatly decreases the stress-induced interaction between DAF-16 and SIR2.1. It seems likely, therefore, that SIR-2.1 overexpression results in increased levels of complex formation between SIR-2.1 and DAF-16, leading to DAF- 16 activation (Figure 1). This hypothesis is supported by the fact that expression of a canonical DAF- 16 target gene, sod-3, as well as other DAF-16 targets, is elevated in worms overexpressing sir-2.1, and 143-3 proteins are again required for this effect (Berdichevsky et al. 2006). Therefore, in C. elegans, SIR-2.1 and 14-3-3 can act as DAF-16 coactivators, binding to DAF- 16 and triggering transcription of its targets upon stress. 14-3-3 proteins either physically bridge SIR-2.1 and DAF-16, or promote their interaction by modifying one or both proteins in response to stress. In mammalian cells, a similar complex between SirTI (a homologue of SIR-2. 1) and FOXO (a homologue of DAF-16) has been reported, and stress is also important for this interaction (Brunet et al. 2004; Motta et al. 2004). However, it is unknown whether mammalian 14-3-3 is required for complex formation between SirTi and FOXO. Also, in mammals SirTi can directly deacetylate FOXO, and this deacetylation can modulate FOXO activity by repressing some of FOXO's target genes while activating others (Brunet et al. 2004). So far, there is no evidence for SIR-2.1-mediated 193 repression of DAF- 16 targets in worms, but the physical interaction between the two proteins is conserved from worms to mammals. It is tempting to speculate that human SirT1 may also modulate response to stress and organismal aging through interaction with DAF-16/FOXO. SIR-2.1 pathway for life span extension. Thus, sir-2.1, 14-3-3 genes and daf-16 act together to determine longevity, but do they act within the insulin-like pathway? If that were the case, one would expect 14-3-3 and sir-2.1 to be required for the life span extension by reduction in insulin-like signaling. However, the long life span of mutants defective in the insulin-like receptor DAF-2 does not depend on sir-2.1 or 14-3-3 (Berdichevsky et al. 2006; Wang et al. 2006; Wang and Tissenbaum 2006). Thus sir-2.1 and 14-3-3 do not act downstream of the insulin-like receptor to extend life span. Like stress, low insulin-like signaling results in accumulation of DAF-16 in the nucleus. However, unlike stress, low insulin-like pathway activity does not promote the formation of SIR-2. I1/DAF- 16 complex (Berdichevsky et al. 2006). Taken together, these results indicate that sir-2.1 and 14-3-3 likely act to promote longevity in parallel to the insulin-like pathway, in a separate stressdependent pathway that also converges on daf-16. Is stress then good for the worm? It may be: a mild heat shock early in life can produce life span extension of up to 30% (Apfeld et al. 2004). It will be interesting to determine whether this effect requires 14-3-3 and/or SIR-2.1. Emerging group of DAF-16 co-factors Several studies suggest that nuclear localization of DAF- 16 is not sufficient for life span extension and stress resistance: as noted above, 194 worms with low 14-3-3 levels have constitutively nuclear DAF-16 yet they are short-lived and hypersensitive to stress. This implies that either phosphorylated DAF-16 that accumulates in the nucleus upon reduction in 14-3-3 levels is inactive, or that another cofactor is required for DAF- 16 activity. Overexpression of a constitutively nuclear form of DAF- 16 lacking all the Akt phosphorylation sites also does not lead to extended longevity and stress resistance (Lin et al. 2001), supporting the notion that other cofactors play a vital role in DAF-16 activation. Binding of such cofactors to DAF-16 may depend on DAF-16 localization and possibly posttranscriptional modifications of DAF-16. For 14-3-3/DAF-16 interaction, phosphorylation of DAF-16 at the Akt sites, which requires activity of the insulin-like signaling cascade, is essential (Figure 1) (Cahill et al. 2001). Stress may lead to additional modifications of DAF-16 and/or SIR-2.1, promoting their interaction. Several recent reports bring to light new factors in the stress-induced relocalization of DAF- 16 into the nucleus. Jun N-terminal kinase (JNK) can directly phosphorylate DAF-16 and facilitate its nuclear localization following heat shock (Oh et al. 2005). It would be interesting to see whether phosphorylation by JNK is required for SIR-2. 1/DAF-16 interaction and life span extension by SIR-2.1 or 14-3-3 overexpression. Upon oxidative stress, MST1 kinase (CST-1 in C. elegans) phosphorylates DAF-16/FOXO and promotes its nuclear translocation (Lehtinen et al. 2006). MST1 phosphorylation disrupts FOXO's interaction with 14-3-3 proteins, suggesting that 14-3-3 proteins may not act as DAF16 co-activators following oxidative stress. It is plausible that different stressors will induce distinct modifications that determine the specificity of the cofactor (Figure 2). A recent report suggests that bar-], a worm 195 homologue of beta-catenin, may act as a DAF-16 co-activator upon oxidative stress: like SIR-2.1 and 14-3-3 proteins during heat stress, betacatenin/BAR-1 can bind FOXO/DAF-16 and activate transcription of its targets when worms are subjected to oxidative stress (Essers et al. 2005). Under low insulin-like signaling, an unphosphorylated DAF-16 accumulates in the nucleus. In this case, 14-3-3 and SIR-2.1 do not bind DAF-16, but another cofactor, SMK-1 (a homologue of mammalian Smekl), does. This nuclear protein is required for life span extension by low insulin-like signaling (Wolff et al. 2006). Another pathway for life span determination that converges on DAF16 involves signaling through germline cells and the somatic gonad: worms lacking the germline live 40% longer than their fertile counterparts, and this effect depends on DAF- 16 as well as another nuclear co-factor, KRI- 1 (Hsin and Kenyon 1999; Berman and Kenyon 2006). So it seems that DAF-16 does not act alone, but rather selects its allies depending on the situation, an interesting fact considering the number of longevity pathways that converge on DAF-16. So far, the search for DAF-16 transcriptional targets has been limited to genes with expression that is altered when DAF-16 is activated by reducing insulin-like signaling. It is possible that under different conditions, specific cofactor binding determines which of the variety of DAF- 16 targets will be induced. Further experiments should clarify this issue. 196 References Aguilaniu, H., Durieux, J., and Dillin, A. 2005. Metabolism, ubiquinone synthesis, and longevity. Genes Dev 19(20): 2399-2406. Apfeld, J., O'Connor, G., McDonagh, T., Distefano, P.S., and Curtis, R. 2004. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. Berdichevsky, A., Viswanathan, M., Horvitz, H.R., and Guarente, L. 2006. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125(6): 1165-1177. Berman, J.R. and Kenyon, C. 2006. Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124(5): 1055-1068. Blander, G. and Guarente, L. 2004. The Sir2 family of protein deacetylases. Annu Rev Biochem 73: 417-435. Brunet, A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S., Anderson, M.J., Arden, K.C., Blenis, J., and Greenberg, M.E. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96(6): 857-868. Brunet, A., Sweeney, L.B., Sturgill, J.F., Chua, K.F., Greer, P.L., Lin, Y., Tran, H., Ross, S.E., Mostoslavsky, R., Cohen, H.Y., Hu, L.S., Cheng, H.L., Jedrychowski, M.P., Gygi, S.P., Sinclair, D.A., Alt, F.W., and Greenberg, M.E. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303(5666): 2011-2015. Cahill, C.M., Tzivion, G., Nasrin, N., Ogg, S., Dore, J., Ruvkun, G., and Alexander-Bridges, M. 2001. Phosphatidylinositol 3-kinase signaling 197 inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem 276(16): 1340213410. Essers, M.A., de Vries-Smits, L.M., Barker, N., Polderman, P.E., Burgering, B.M., and Korswagen, H.C. 2005. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308(5725): 1181-1184. Guarente, L. 2000. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev 14(9): 1021-1026. Hsin, H. and Kenyon, C. 1999. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399(6734): 362-366. Kenyon, C. 2005. The plasticity of aging: insights from long-lived mutants. Cell 120(4): 449-460. Koubova, J. and Guarente, L. 2003. How does calorie restriction work? Genes Dev 17(3): 313-321. Lee, S.S., Lee, R.Y., Fraser, A.G., Kamath, R.S., Ahringer, J., and Ruvkun, G. 2003. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33(1): 40-48. Lehtinen, M.K., Yuan, Z., Boag, P.R., Yang, Y., Villen, J., Becker, E.B., DiBacco, S., de la Iglesia, N., Gygi, S., Blackwell, T.K., and Bonni, A. 2006. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125(5): 9871001. Lin, K., Dorman, J.B., Rodan, A., and Kenyon, C. 1997. daf-16: An HNF3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278(5341): 1319-1322. 198 Lin, K., Hsin, H., Libina, N., and Kenyon, C. 2001. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28(2): 139-145. Motta, M.C., Divecha, N., Lemieux, M., Kamel, C., Chen, D., Gu, W., Bultsma, Y., McBurney, M., and Guarente, L. 2004. Mammalian SIRT1 represses forkhead transcription factors. Cell 116(4): 551-563. Oh, S.W., Mukhopadhyay, A., Svrzikapa, N., Jiang, F., Davis, R.J., and Tissenbaum, H.A. 2005. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A 102(12): 4494-4499. Rogina, B. and Helfand, S.L. 2004. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 101(45): 15998-16003. Tissenbaum, H.A. and Guarente, L. 2001. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410(6825): 227230. -. 2002. Model organisms as a guide to mammalian aging. Dev Cell 2(1): 9- 19. Tzivion, G., Shen, Y.H., and Zhu, J. 2001. 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 20(44): 6331-6338. Wang, Y., Oh, S.W., Deplancke, B., Luo, J., Walhout, A.J., and Tissenbaum, H.A. 2006. C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF- 16/FOXO. Mech Ageing Dev. Wang, Y. and Tissenbaum, H.A. 2006. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev 127(1): 48-56. 199 Wolff, S., Ma, H., Burch, D., Maciel, G.A., Hunter, T., and Dillin, A. 2006. SMK- 1, an essential regulator of DAF- 16-mediated longevity. Cell 124(5): 1039-1053. 200 Figures: Figure 1: SIR-2.1 mechanism of life span extension. (a) SIR-2.1 binds DAF-16 in the 14-3-3-dependent manner. We propose that SIR-2.1 can bind and activate DAF-16 in the nucleus. 14-3-3 likely bridges between SIR-2.1 and DAF- 16, or modifies one or both of them in response to stress to promote their interaction. (b) In the absence of 14-3-3 proteins, SIR-2.1 cannot bind DAF-16, and DAF-16 remains inactive despite nuclear localization, resulting in shorter life span and sensitivity to stress. (c) When SIR-2.1 is overexpressed, more DAF-16/SIR-2.1 complexes are formed, leading to DAF-16 activation and life span extension. 201 Figure 1. a. Wild type + stress SIR-2. 14-3-3 ... DAF-16 active r b. 14-3-3A ~IP..9 I Life span S•tressRI c. SIR-2.1 ole SIR-2.1 Life spar Stress R 202 4"14-3-3 DAF-16 active · ·--- --·- --- Figure 2: DAF-16 can be activated by different cofactors. Under normal conditions, the majority of DAF-16 molecules are present in the cytoplasm, inactive, bound and sequestered by 14-3-3 proteins. When insulin-like signaling is low, unphosphorylated DAF-16 is unable to bind 14-3-3 and accumulates in the nucleus, where it can interact with, and get activated by SMK-1. In worms that lack a germline, DAF-16 also accumulates in the nucleus, and is activated by KRI-1 protein. It is unknown whether such nuclear DAF-16 is still phosphorylated at the Akt sites and bound by 14-3-3. It is also not known what other modifications may be involved in DAF-16 activation and/or nuclear localization in germlineless worms. Stress also results in accumulation of DAF-16 in the nucleus. Heat stress-induced relocalization of DAF-16 likely involves phosphorylation by JNK. Since the insulin-like pathway is still active in this case, DAF-16 is phosphorylated at the Akt sites and can interact with 14-33 and SIR-2. 1, which activate DAF-16 in this case. Finally, activation of DAF-16 by oxidative stress may involve phosphorylation by MSTl/CST-1 kinase and interaction with BAR- 1. In every case, lack of a cofactor abolishes DAF-16-dependent life span extension and resistance to stress. 203 ~-C4DAF-I6'j SITR-2.·'3?.,· DAF-16 IBAR-1t DA~LF-16e active r.. Stress I Not DAF-16 active SMK-I 1443-3 DAF-16 active nucleus cytoplasm Figure 2 204