Transport of Phonons and Electrons Sangyeop Lee
advertisement

Transport of Phonons and Electrons
in Thermoelectric Materials and Graphene
ARCHVES
MiASSAC"E-1TSTS 'N7TI)TE
by
J
JUL 302015
Sangyeop Lee
, LIBR A RIEF-S
Submitted to the Department of Mechanical Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITTUE OF TECHNOLOGY
June 2015
0 Massachusetts Institute of Technology 2015. All rights reserved
Signature redacted
.DIartment of Mechanical Engineering..
Signature of Author ............... ............... ......
May 26, 2015
Signature redacted
C e rtifi e d by .......................................... ..... .............................
......
*
Gang Chen
Carl Richard Soderberg Professor of Power Engineering
Thesis Supervisor
Signature redacted
A ccepted by ..........................................
.........................................
David E. Hardt
Chairman, Department Committee on Graduate Students
2
Transport of Phonons and Electrons in Thermoelectric Materials and Graphene
by
Sangyeop Lee
Submitted to the Department of Mechanical Engineering on May 26, 2015,
in partial fulfillment of the requirements for the degree of Doctor of Philosophy
Abstract
Understanding transport of phonons and electrons plays a critical role in developing energy
conversion and information devices. Thermoelectric materials, which directly convert heat to electricity
or vice versa, require both extremely low thermal conductivity and high thermoelectric power factor.
However, a good understanding of low thermal conductivity is still lacking even for several good
thermoelectric materials that have been studied over several decades. For the information devices,
graphene has recently drawn much attention for various applications including high speed transistors due
to its high electron mobility and high thermal conductivity. However, the graphene's high thermal
conductivity has yet to be fully understood. There have been many studies based on diffusive-ballistic
phonon transport, but no conclusive explanation for the graphene's high thermal conductivity has been
drawn.
In this thesis, we investigate the transport of phonons and electrons in thermoelectric materials
and graphene using both first principles calculations and experimental characterizations. We start by
studying phonon transport in Bi and Bi-Sb alloys using first principles calculations. A notable observation
from this calculation is that a strong long-range interaction exists in Bi and Sb along a specific
crystallographic direction. We further show that this long-range interaction is also found in other good
thermoelectric materials, and is a key to understanding their low thermal conductivity. The long-range
interaction is explained with resonant bonding which many good thermoelectric materials commonly
share. The particularly strong resonant bonding in group IV-VI materials leads to the low thermal
conductivity through the long-range interaction and resulting softening of optical phonons that strongly
scatter acoustic phonons.
We study electron transport in thermoelectric materials with two-dimensional discontinuities,
such as grain boundaries. We set up an experimental system to measure thermo- and galvano-magnetic
electron transport coefficients of a Bi2 Te 27 SeO.3 nanocomposite sample to examine the electron filtering
effect by many grain boundaries in the nanocomposite. The experimental results indicate that the
nanocomposite sample exhibits the electron filtering effect and it would be possible to increase the
thermoelectric power factor by engineering the potential barrier of grain boundaries.
While thermoelectric applications require materials with low thermal conductivity, electronic and
optoelectronic devices often require high thermal conductivity. Graphene is attractive for these
applications because of its unique electrical, optical, and thermal properties. We use first-principles
calculations to reveal that the phonon transport in graphene is not diffusive unlike many threedimensional materials, but is hydrodynamic due to graphene's two-dimensional features. The
hydrodynamic phonon transport is demonstrated through a drift motion of phonons, phonon Poiseuille
flow, and second sound, all of which are not possible in both diffusive and ballistic phonon transport.
Thesis Supervisor: Gang Chen
Title: Carl Richard Soderberg Professor of Power Engineering
3
4
Acknowledgements
This thesis could not be completed without the help from many people. Here
I would like to thank
several people whom I am much indebted to.
First, I would like to thank my advisor, Prof. Gang Chen. I am very fortunate to have studied
under his guidance. He gave me almost complete freedom in choosing my research topic and making
progress so that I could be trained as an independent researcher. He also emphasized the importance of
having a big picture and asking an important question. All these inspiring comments and guidance will be
an invaluable asset for my future research career.
I also thank my thesis committee members. Prof. Mildred Dresselhaus spent a tremendous
amount of time for me. She kindly suggested me for several times to come to her office and to discuss
about my research progress and future directions. She also carefully revised my thesis and journal papers,
and gave me back many detailed comments. Prof. Nicolas Hadjiconstantinou asked me several important
questions regarding my hydrodynamic phonon transport work in Chapter 5. While I was trying to answer
those questions, I could develop a better understanding of the hydrodynamic phonon transport. I also
thank Prof. Alexie Kolpak for her encouragements and valuable inputs to my research.
I have to thank Prof. Keivan Esfarjani now at Rutgers University and Prof. David Broido at
Boston College. The first principles calculation of phonon transport that I mainly used in this thesis was
developed by these two people. Collaboration with them was an exceptional chance for me, and without
their help, I could not learn so quickly the first principles calculations of phonon transport.
I also thank
Prof. Joseph Heremans at Ohio State University. He kindly allowed me to spend a week in his laboratory
and to learn the method of four coefficients which is presented in Chapter 4.
Working with my lab mates was another great source of learning. In particular, I would like to
thank several people here. Bolin Liao and Maria Luckyanova were very helpful in revising my papers. I
thank them for their comments on my writing. I also enjoyed discussion with them, and the discussion
often gave me good insights. I also thank several people for their help in my experimental studies:
Kimberlee Collins, Daniel Kraemer, Kenneth McEnaney, Austin Minnich, Qing Hao, and Andy Muto.
Finally, I would like to thank my family and friends. I thank my parents and parents in law for
their love and prayer. I also thank my wife, Jac, and my 7 year old son, Junwoo. My wife, Jae, also
pursued her professional career and in fact she was busier than
I, but she supported me more than she
could do. Along with my wife, my son, Junwoo, was a constant source of happiness for me. I also thank
many friends, particularly John Hong, Dong-Hoon Yi, and Gyuwon Hwang, for their friendship and
encouragements.
5
6
Table of Contents
1. Introduction ........................................................................................................
17
1.1. Heat and Charge Transport in Thermoelectric and Information Processing Devices........ 17
1.2. Thesis Outline ....................................................................................................................
2. Phonon Transport in Bi, Sb, and Bi-Sb Alloys ............................................
2 . 1. B ackground ........................................................................................................................
20
23
23
2.2. First Principles Calculations of Phonon Transport and Thermal Conductivity .............. 25
2.2.1. Second- and Third-order Force Constants..............................................................
25
2.2.2. Scattering Rates and Peierls-Boltzmann Transport Equation .................................
33
2.3. Results and Discussions ................................................................................................
39
2.3.1. Phonon thermal conductivity....................................................................................
39
2.3.2. Phonon Mean Free Path Distributions....................................................................
47
2 .4 . C on clu sion ..........................................................................................................................
50
3. Low Thermal Conductivity of IV-VI Materials from Resonant Bonding....51
3 . 1. B ackground ........................................................................................................................
51
3.2. Resonant Bonding in IV-VI, V2-VI3, and Element V Materials ....................................
53
3.3. Long-range Interaction due to the Resonant Bonding ..................................................
60
3.4. Strong Three-Phonon Scattering in IV-VI Materials .....................................................
67
3.4.1. Large Anharmonicity of Ferroelectric Soft Phonon Modes ....................................
67
3.4.2. Large Phase Space for Three-Phonon Scattering .....................................................
73
7
3 .5 . Co n clu sion ..........................................................................................................................
75
4. Experimental Characterization of Electron Filtering Effect in
Nanocomposite Bi 2 Te 2 .7Seo.3 . -- . .. .
. ..
.
. ..
. ..
. . ..... .
. .. . .
. .. ... ... 77
4 . 1. B ack gro und ........................................................................................................................
77
4.2. The Method of Four Coefficients....................................................................................
79
4.3. Experimental Setup ............................................................................................................
86
4.4. Results and Discussions ................................................................................................
89
5. Hydrodynamic Phonon Transport in Suspended Graphene......................95
5.1. B ack ground ........................................................................................................................
95
5.2. Drift Motion of Phonons ................................................................................................
98
5.2.1. Details of First Principles Calculations ..................................................................
5.2.2. Displaced Phonon Distribution..................................................................................
5.3. Phonon Poiscuille Flow....................................................................................................
99
101
105
5.3.1. Criteria for Phonon Poiseuille Flow ..........................................................................
106
5.3.2. Characteristics of Phonon Poiseuille Flow................................................................
11I
5.3.3. Possible Experiments for Observing Phonon Poiseuille Flow ..................................
117
5.4 . Second Sound ...................................................................................................................
119
5.4.1. Criteria for Second Sound .........................................................................................
119
5.4.2. Possible Experiments for Observing Second Sound .................................................
122
5.5. Origin of the Hydrodynamic Phonon Transport in Graphene..........................................
128
5 .6 . C onclu sion ........................................................................................................................
13 1
8
6. Summary and Future Directions
...............................
6 .1. Sum mary ..........................................................................................................................
6.2. Future Directions .....................................................
9
135
133
133
List of Figures
Figure 2-1 Crystal structure of Bi and Sb. The void and filled atoms represent two basis
atoms. RI, R 2, and R 3 are primitive lattice vectors and a is a rhombohedral angle between
two primitive lattice vectors. The values of a are 57030 for Bi and 57084 for Sb, which are
27
close to 600 of the simple cubic structure.........................................................................
Figure 2-2 Force constants of Bi and Sb versus interatomic distance (a) Trace values of
second-order force constant tensors and (b) two-body third-order force constants .......
29
Figure 2-3 Phonon dispersion of Bi and Sb. (a) and (b) represent Bi and Sb cases, respectively.
Dots are experimental values from Refs. [48] for Bi and [49] for Sb. The location of high
symmetry points in the Brillouin zone are plotted in (c) for Bi, Sb, and Bi-Sb alloys. ........ 30
Figure 2-4 Acoustic mode Gruneisen parameters of (a) Bi and (b) Sb comparing inclusion
up to the fourth- and tenth-neighbors, to the references. The reference Grineisen
parameters are calculated using the difference of phonon frequencies of two different crystal
vo lumes..................................................................................................................................
33
Figure 2-5 Comparison of Normal, Umklapp and mass disorder scatterings. The squares
represent the first Brillouin zone ........................................................................................
35
Figure 2-6. Thermal conductivity of Bi (a) in the binary direction and (b) in comparison
between the binary and the trigonal directions. Kph in (b) is calculated with the single mode
relaxation time approximation and using third-order force constants up to the tenthneighbors. The solid lines and dots represent our first principles calculation results and the
experimental data from Ref. [32], respectively. The Full and SMRT in the legend represent
solution of the Peierls-Boltzmann equation using the full iterative method and the single
mode relaxation time approximation, respectively. ..........................................................
40
Figure 2-7. The thermal conductivity of Sb (a) in the binary direction and (b) in comparison
between the binary and the trigonal directions. The solid lines and dots in (b) represent our
first principles calculation results and the experimental data from Ref. [26], respectively.
The Full and SMRT in the legend represent solution of the Peierls-Boltzmann equation
using the full iterative method and the single mode relaxation time approximation,
resp ectively ............................................................................................................................
44
10
Figure 2-8. Thermal conductivity of the Bi-Sb alloys. (a) The effect of Sb content on the
phonon thermal conductivity, showing that inclusion of even small amount of Sb
significantly reduce phonon thermal conductivity. (b) Comparison between the total and
phonon thermal conductivity of Bi, Sb, and Big8 Sb] 2 , and (c) an enlarged plot for the
Bi88 Sb1 2 data. The experimentally measured total thermal conductivity values are from Ref.
[2 6 , 32 ]. .................................................................................................................................
46
Figure 2-9. Phonon mean free path distribution (a) Bi, BiwSbi, Bi8 8Sb 2 , and Sb at 100 K, (b)
Bi and Bi8gSb 12 at lOOK, and (c) Bi at 50, 100, 200, and 300 K for the binary and the
trigonal directions. In (b) and (c), the accumulated thermal conductivity is normalized by the
49
phonon therm al conductivity value ...................................................................................
Figure 3-1 Normalized thermal conductivity of binary III-V and IV-VI compounds at 300
K. The solid lines are for a guide to the eyes. ...................................................................
53
Figure 3-2 Rocksalt-like crystal structures of PbTe, Bi 2Te3 , and Bi. The number on each
atom indicates the shell number. Bi 2Te 3 , Bi and Sb have distorted rocksalt structures and
have different numbers for shells than the exact rocksalt case. The numbers on the Bi 2Te 3
and Bi atoms indicate the equivalent shell numbers as for a rocksalt structure in the absence
o f lattice distortion .................................................................................................................
55
Figure 3-3 Electronic band structure and projected density-of-states of PbTe showing weak
sp -hybridization ...................................................................................................................
56
Figure 3-4 Electronic band structure and projected density-of-states of PbSe showing weak
sp -hybridization ...................................................................................................................
57
Figure 3-5 Electronic band structure and projected density-of-states of PbS showing weak
sp -hyb ridization ...................................................................................................................
57
Figure 3-6 Electronic band structure and projected density-of-states of SnTe showing weak
sp -hyb ridization ...................................................................................................................
58
Figure 3-7 Electronic band structure and projected density-of-states of Bi2 Te 3 showing
w eak sp -hybridization .........................................................................................................
59
Figure 3-8 Electronic band structure and projected density-of-states of Bi showing weak sp-
hy brid ization ........................................................................................................................
11
59
Figure 3-9 Electronic band structure and projected density-of-states of Sb showing weak
sp -hybridization ...................................................................................................................
60
Figure 3-10 Normalized trace of interatomic force constant tensors versus atomic distances.
(a) lead chalcogenides and SnTe (group IV-VI), (b) NaCl and InSb, (c) Bi 2Te3 (group V2V1 3), and (d) Bi and Sb (group V). The element in the parenthesis indicates interaction
between the corresponding atom and other atoms. For example, 'PbTe(Pb)' means
63
interaction between Pb and other atoms in PbTe. ............................................................
Figure 3-11 Electron density distribution and polarization in NaCl and PbTe. (a-d) the
electron density distribution at the ground state. (e-h) the electron density distribution
change by a displacement of the center atom. The plot is on the (100) plane and each black
. . . . . . . . . .. . . . . . . . . . . . . . . . .. . .
65
dot represents an atom . The unit is A ...............................................
Figure 3-12 Diatomic 1D chain. The numbers on the atoms indicate the shell number, with an
increasing neighbor distance with increasing number. The black circles denote A atoms and
w hite circles denote B atom s............................................................................................
69
Figure 3-13 Near ferroelectric behavior due to resonant bonding. (a) Optical phonon
dispersion in a model 1D atomic chain, showing the softening of the optical phonons due to
the long-range interactions. Three numbers in the legend represent relative interaction
strength of first, second and third-nearest neighbors in the ID chain. (b-d) Soft TO phonon
modes along the trigonal direction for lead chalcogenides, Bi 2Te3 , and Bi and Sb,
respectively, calculated based on first-principles. Lines and circles are calculation and
experimental data, respectively. The experimental data are from Ref. [48, 49, 73, 77, 78].
The red dotted line in b is after removing the fourth, eighth, fourteenth-nearest neighbor
interactions in PbTe, which do not show the soft TO mode. (b-d) are plotted on the same
scale for the y-axis. (e) Calculated Griineisen parameters of TO mode, showing strongly
anharmonic behavior of the TO phonons of lead chalcogenides. The dotted line denotes the
Grdneisen parameters of the LA mode in PbTe for comparison........................................
70
Figure 3-14 Analysis of phonon transport in IV-VI and III-V materials by first principles
calculation. (a) Calculated and experimental phonon thermal conductivity. Lines and
squares are results by experiments and calculations, respectively. (b-c) Phonon mean free
path distributions and phonon lifetime, showing significant three-phonon scattering in IV-VI
materials. The accumulated thermal conductivity in (b) is normalized by the thermal
conductivity value of the corresponding material. The data in (b-c) are for the 300 K case.
The experimental thermal conductivity values in (a) are from Ref. [62, 90, 91] and other
calculation results for PbTe, PbSe and GaAs are from Ref. [36, 88]...............................
72
12
Figure 3-15 Lower thermal conductivity of PbTe compared to Bi due to more significant
resonant bonding. (a) Comparison of phonon dispersions showing the smaller group
velocity of acoustic phonons in Bi (b) Comparison of thermal conductivity showing the
lower therm al conductivity of PbTe .................................................................................
72
Figure 3-16 Phase space volume for three-phonon scattering. (a) Phase space volumes for
three phonon scattering of tV-VI and III-V, showing a large scattering phase space for PbSe
and PbS. The solid line is for a guide to the eyes. (b) Comparison of the phonon dispersion
of PbS and AISb, showing significantly dispersed optical phonons of PbS. (c) Contribution
of each scattering process to total scattering phase space volume. The scattering phase space
and phonon dispersion data are normalized by the inverse of the largest optical phonon
frequency of each material for comparison. .....................................................................
74
Figure 4-1. A schematic picture of a potential barrier at a grain boundary in an n-type
semiconductor. Ec, EF, and Ev represent a conduction band edge, Fermi level, and a
valence band edge, respectively ........................................................................................
78
Figure 4-2. A schematic picture of the Seebeck effect........................................................
83
Figure 4-3. A schematic picture of the Hall effect................................................................
84
Figure 4-4. Schematic pictures of Nernst effect depending on the energy dependence of
electron scattering rates. Note that there is no transverse electric field when r = 0; the hot
electrons are preferentially deflected upward when r > 0 and the cold electrons tend to go
dow nw ard when r < 0....................................................................................................
86
Figure 4-5. A sample with various probe wires and the configuration of the measurement
setu p ......................................................................................................................................
88
Figure 4-6. A prepared sample assembly on the cold finger, showing the heater location and
the ceram ic p late..................................................................................................................
89
Figure 4-7. Measurement data of the four transport coefficients. (a) electrical resistivity, (b)
Seebeck coefficient, (c) Hall coefficient, and (d) Nernst coefficient ................................
90
Figure 4-8. Fermi level from the method of four coefficients. The black points are from Ref.
[103] for com parison. ............................................................................................................
91
Figure 4-9. Density-of-states effective mass from the method of four coefficients. The blue
line is for eye-guide. The inset schematically shows the first light carrier pocket and the
13
second heavy carrier pocket. The density-of-states effective mass is in unit of mo, physical
92
mass of a free electron (mo=9. I x 10-1 kg).......................................................................
Figure 4-10. Electron mobility from the method of four coefficients. The black line is from
93
Ref. [107] for comparison. ...............................................................................................
Figure 4-11. Scattering exponent representing the energy dependence of the electron
scattering rates from the method of four coefficients. The black line and the circles are
from Refs. [103, 107] for comparison. The brown line represents the case where the
scattering by phonons is predominant over other scattering mechanisms. .......................
94
Figure 5-1. Different macroscopic transport phenomena in the hydrodynamic and diffusive
regimes. (a-b) The steady state heat flux profiles in hydrodynamic and diffusive regimes,
respectively, under a temperature gradient. (c-d) The propagation of a heat pulse in the
hydrodynamic and diffusive regimes, respectively. The width and length of the sample are
assumed to be much larger than the phonon mean free path.............................................
97
Figure 5-2. The mode Grineisen parameters of graphene. The circles are calculated from the
finite difference of phonon frequencies with different crystal volumes by 1% and the lines
100
are calculated using both second- and third-order force constants......................................
Figure 5-3. The displaced distribution of phonons at 100 K in the reciprocal space of a
graphene sheet. The '3C isotope concentration is 0.1%. (a) A contour plot of the normalized
deviation of the distribution of flexural acoustic (ZA) phonons in graphene. The hexagon
represents the first Brillouin zone and a temperature gradient is applied along the xdirection. (b) The normalized deviation of the distribution of the three acoustic branches in
graphene along the x-direction at qy=0 (M-F-M). The linear dependence on q, indicates drift
motion of acoustic modes, according to Eq. (5.3). The same slope for all three acoustic
branches means that acoustic phonons have the same drift velocity, regardless of
polarization and w avevector................................................................................................
102
Figure 5-4. The phonon mode thermal conductivity of graphene with 0.1 % 13 C at 100 K.
(a) A contour plot of the mode thermal conductivity of ZA phonon modes in graphene. (b)
The mode thermal conductivity of the three acoustic modes in graphene along the xdirection at qy= 0 ...................................................................................................................
102
Figure 5-5. The normalized deviation of the distribution of the acoustic branches in pure
bismuth at 300 K along the trigonal-direction (T-r-T). A temperature gradient is applied
in the same direction. Only one TA branch is included in the plot since the two TA branches
14
are degenerate along the line, T-F-T. The inset shows the first Brillouin zone of bismuth
103
w ith the high sym metry points............................................................................................
%
Figure 5-6. Comparison of N-scattering and R-scattering rates in graphene with a 0.1
concentration of isotope 13 C at 100 K. The Matthiessen's rule is used to combine U104
scattering and isotope scattering rates into the R-scattering rate. .......................................
Figure 5-7. A schematic picture describing the random walk of phonons ..........................
109
Figure 5-8. The wide window of sample widths for phonon Poiseuille flow in graphene as
1 10
compared to diam ond........................................................................................................
Figure 5-9. Comparison between N- and R-scattering rates in graphene and diamond at
100 K, showing extremely strong N-scattering in graphene. The condition of isotope
content is specified in the plots. The isotope content of 1.1% 13 C in (b,d) represents the
naturally occurring case. The figure (a) is duplicated from Fig. 5-6 for comparison. ........ 111
Figure 5-10 Geometry of two-dimensional duct. The width and depth of the duct are
represented as L and d, respectively....................................................................................
112
Figure 5-11 Effects of sample width on the heat flux profile and thermal conductivity (a)
The shape of the heat flux profiles when transport is close to the hydrodynamic limit
(L/A=0. 1) and close to the diffusive limit (L/A=20) (b) Dependence of the thermal
conductivity on sample width, L. The vertical axis represents the exponent value (a) in the
simple power law relation, K~La..........................................................................................
116
Figure 5-12 The possible frequency ranges of second sound in graphene and diamond. (a)
The content of isotope 13C is fixed at 0.01 %. (b) The sample size is fixed at 1000 pm. (c-d)
Contour plots of second sound frequency range in graphene with respect to sample size and
isotope content for 50 and 100 K, respectively. The second sound frequency range is defined
as Q,,ppe/Qiower, where
,,pper and ieower are the upper and lower bounds of second sound
frequency, respectively. The frequency range is plotted on a log scale in the contour plots.
Second sound in diamond is not possible in the given range of temperature, sample size, and
isotope conten t.....................................................................................................................
122
Figure 5-13 The speed of second sound in graphene with respect to temperature............. 126
Figure 5-14 Comparison between the delay time by ballistic transport and second sound
propagation in the heat pulse experiment. The delay time is per distance between the
15
source and the sensor, W, in [m]. The inset illustrates the configuration of the sample with a
point heat source and a point heat sensor for observing second sound............................... 128
Figure 5-15 Cumulative weighting factors for averaging scattering rates for quadratic
dispersion in two-dimensional materials and for linear dispersion in three-dimensional
materia ls.............................................................................................................................
13 1
16
1. Introduction
I .1Heat and Charge Transport in Thermoelectric and Information
Processing Devices
Thermoelectric energy conversion has drawn much attention for waste heat recovery and
solid-state cooling applications due to its advantages over conventional thermo-mechanical
energy conversion. Thermoelectric energy conversion devices have no moving parts, high
reliability, and easy scalability. These advantages have led to some noteworthy applications
including automotive climate control seats, diode lasers temperature stabilization, power for
deep-space mission spacecraft, remote terrestrial areas, and potential applications in power
generation from solar irradiation and waste heat recovery [1-3]. However, the low efficiency
compared to conventional thermo-mechanical cycles has limited thermoelectric devices to niche
applications where the conventional cycles cannot be easily applied [4].
The maximum efficiency of thermoelectric energy conversion devices is determined by
the thermoelectric figure-of-merit, ZT:
S2 U
ZT = -
where S,
O-, K,
K
T
(1.1)
and T are the Seebeck coefficient, electrical conductivity, thermal conductivity,
and temperature, respectively. The numerator, Sea, is called the thermoelectric power factor.
The above expression, Eq. (1.1), implies three important requirements for a material to exhibit
17
high thermoelectric figure-of-merit: a large thermoelectric effect, small Ohmic losses, and small
heat leakage through thermoelectric material.
Recently, the thermoelectric figure-of-merit was significantly improved by reducing
thermal conductivity. The reduction of thermal conductivity was achieved by introducing
nanostructures into conventional thermoelectric materials [5]. One example is nanocomposite
thermoelectric materials which consist of many nanograins and grain boundaries [6, 7]. These
grain boundaries were demonstrated to strongly scatter phonons and significantly reduce thermal
conductivity. Further reduction of thermal conductivity by the use of nanostructures would
require the detailed information about phonon dynamics, such as spectral contribution of
phonons to thermal transport and spectral distribution of phonon mean free paths. This detailed
information can help for the rational design of nanostructures in terms of characteristic size and
shape. The recently developed first principles calculation can provide such detailed information
about phonon dynamics [8, 9]. In Chapter 2, we use this first principles calculation for Bi, Sb,
and Bi-Sb alloys to quantify phonon mean free paths as well as intrinsic phonon thermal
conductivity values.
In parallel to applying nanostructures to the conventional thermoelectric materials, there
also have been many efforts to find new thermoelectric materials [10, 11]. Searching for new
thermoelectric materials had been very tedious for a long time since the synthesis and the
experimental
characterization of new materials are extremely time-consuming processes.
Recently developed high-throughput first principles calculations have a potential to make this
process much faster [12, 13]. Once a specific material group, such as half-Heusler alloy, is
identified, the high-throughput calculation roughly estimates thermal conductivity values of all
possible compounds in the periodic
table and finds good candidate
compounds.
This
combinatorial search can be even more powerful if we have good physical insights into the
fundamental relation between the thermal conductivity and chemical bonding. However,
establishing such a relation between the thermal conductivity and chemical bonding has not been
actively pursued so far, to our best knowledge. There have been only few past studies to find
such a relation [14, 15]. These previous studies compare the thermal conductivity values of a
wide range of materials to seek any correlation of or general trends in the thermal conductivity
values. However, in these previous studies, the thermal conductivity values were only correlated
18
with basic properties such as atomic mass, Debye temperature, lattice constant, and thermal
expansion coefficient, through several simple empirical formulas. This motivates us to pursue
establishing a link between the low thermal conductivity of several conventional thermoelectric
materials and their chemical bonding in Chapter 3.
In addition to reducing the thermal conductivity, increasing the thermoelectric power
factor is another pathway to achieving a higher thermoelectric figure-of-merit from Eq. (1.1).
However, this pathway has been less successful compared to reducing the thermal conductivity,
partially due to the lack of understanding of electron transport in thermoelectric materials. For
example, nanocomposite materials with many grain boundaries were very successful in
increasing the scattering rates of phonons [6, 7], but only few studies characterized the details of
the electron transport in nanocomposite materials [16]. If properly understood and engineered,
many discontinuities such as grain boundaries or interfaces that have been successfully used to
reduce thermal
conductivity can also provide an engineering platform to increase the
thermoelectric power factor. As a step towards this goal, in Chapter 4, we experimentally
characterize electron transport in thermoelectric materials.
For the information processing devices such as integrated transistors, thermal transport
plays an important role in improving the performance of devices. The charge flow in these
devices necessarily causes Joule heating and appropriate cooling is required to maintain
temperature in an operational range.
However, cooling of those devices is challenging. The
volumetric heat generation is exceedingly large because so many transistors are integrated into
very small volume. In addition, each transistor in the devices impedes thermal transport just as
nanostructures significantly reduce thermal conductivity in the nanostructured thermoelectric
materials. As a result, hot spots, in which the generated heat is not discharged but accumulated,
often occur, and severely degrade the performance and reliability of those devices. There have
been many engineering attempts to avoid hot spots. Numerical and experimental studies were
carried out to predict and detect local hot spots [17, 18] and microscale thermoelectric coolers
were developed to remove hot spots [19]. However, the clock frequency of transistor is still
limited by those thermal issues [18].
19
A recently discovered two-dimensional material, graphene, has the potential to solve
those thermal issues owing to its extremely high thermal conductivity [20]. However, the
extremely high thermal conductivity of graphene remains poorly understood. In particular, the
role of flexural phonon modes in thermal transport should be understood better. The flexural
phonon modes are vibrational eigenmodes in which atoms vibrate in the out of plane direction,
and thus are a distinguishable feature of two-dimensional materials relative to typical threedimensional materials. In the early days, the flexural acoustic phonons were considered to
negligibly contribute to thermal transport due to their small group velocity and extremely large
anharmonicity [21, 22]. However, in fact, the flexural acoustic phonon branch has turned out to
be the largest contributor among the three acoustic phonon branches in graphene [23, 24]. The
remaining question is: why do the flexural acoustic phonons carry much despite of their
extremely large anharmonicity? This question leads to our discussion in Chapter 5 about a new
regime of phonon transport, hydrodynamic phonon transport, in graphene.
1.2.Thesis Outline
The purpose of this thesis is to promote our fundamental understanding of phonons and
electrons and thereby solve the aforementioned challenges. We devote the first three chapters
(Chapter 2, 3, and 4) to the transport of phonons and electrons in thermoelectric materials, and
then Chapter 5 is devoted to the transport of phonons in graphene. These chapters are also
arranged in such a way that we discuss the transport phenomena with reducing dimensionality:
from bulk three-dimensional thermoelectric materials (Chapter 2 and 3), to three-dimensional
materials with two-dimensional interfaces such as grain boundaries (Chapter 4), to a twodimensional atomically thin material, which is graphene (Chapter 5).
Chapter 2 discusses the phonon transport in Bi, Sb, and Bi-Sb alloys from first principles
calculations. Those materials have been the best thermoelectric materials for cryogenic
applications, but their phonon thermal conductivity values were not well known previously. We
use the first principles calculation to quantify and detail the phonon transport in those materials.
20
One interesting observation from this calculation is that those materials exhibit strong long-range
interaction along a specific crystallographic direction. This long-range interaction will be more
extensively discussed in Chapter 3. Chapter 2 also briefly summarizes the first principles
calculation method that we use to study phonon transport in Chapter 3 and Chapter 5.
Chapter 3 shows that the strong long-range interaction in Bi and Sb is also observed in
other good thermoelectric materials such as group IV-VI and V2-VI 3 materials. These materials,
group IV-VI (e.g., PbTe), V 2 -VI 3 (e.g., Bi2 Te 3), and element V (Bi-Sb alloys), are the current
best thermoelectric materials at high temperature (above 500 K), intermediate temperature (300
to 500 K), and cryogenic temperature (100 to 300 K), respectively. We introduce resonant
bonding to explain this common feature of the long-range interaction in those seemingly
different materials. Then, we use first principles calculations to find a link between the resonant
bonding and the low thermal conductivity of group TV-VT materials in which resonant bonding is
particularly significant.
Chapter 4 experimentally studies electron transport in thermoelectric materials with a
focus on the electron transport across a two-dimensional interface, which is a grain boundary.
We experimentally measure galvano- and thermo-magnetic properties of a Bi 2Te 2.7SeO.3
nanocomposite sample. From these measured coefficients, we estimate several characteristics of
electron transport including the energy dependence of electron scattering rates. The estimation
indicates that the potential barrier at grain boundaries largely affect electron transport and cause
the electron filtering effect, which can potentially lead to the improvement of the thermoelectric
power factor.
Chapter 5 uncovers a fundamental reason for the extremely high thermal conductivity of
graphene using the first principles calculations. In graphene, unlike many three-dimensional
materials we study in Chapter 2 and 3, most of the phonon scattering processes conserve crystal
momentum and do not directly cause resistance in thermal transport. The momentum-conserving
nature of phonon scattering in graphene is similar to that of a molecule scattering in a fluid. From
this feature, we show that the phonon transport in graphene is not diffusive unlike many threedimensional materials, but is hydrodynamic. We associate this hydrodynamic phonon transport
with graphene's two dimensional features.
21
Finally, Chapter 6 presents possible future directions and concludes this thesis.
22
2. Phonon Transport in Bi, Sb, and Bi-Sb Alloys
Bi and Bi-Sb alloys have been the best thermoelectric materials at cryogenic temperatures
for several decades [25]. However, their phonon thermal conductivity values, which are basic
information to further enhance thermoelectric figure-of-merit, was not well known. This is
because the electron contribution to the total thermal conductivity is considerably large and
comparable to the phonon contribution. Separating the electron and phonon contributions to the
total thermal conductivity in experiments is challenging. However, the quantitative accuracy and
predictive power of the first principles calculation enable us to quantify the phonon thermal
conductivity values. In this chapter, we present the calculated phonon thermal conductivity
values of and phonon mean free paths in Bi, Sb, and Bi-Sb alloys from the first principles
calculation.
In
addition,
we observe a strong
long-range
interaction
along a specific
crystallographic direction in Bi and Sb, which will lead to our discussion in Chapter 3.
2.1.Background
Bi and Bi-Sb alloys have long been studied for their promising low temperature
thermoelectric applications. Bi and Sb have a rhombohedral crystal structure, which is a Peierl's
distortion of the simple cubic crystal. The small structural distortion results in Brillouin zone
folding and a small overlap between conduction and valence bands, thereby causing semimetallic
behavior and conduction by the both electrons and holes. Since the semimetallic behavior causes
cancellation of the hole and electron contributions to the power factor, bulk Bi is not a good
thermoelectric
material.
However,
Bi has
a large
thermomagnetic
effect and a large
thermomagnetic figure-of-merit [26]. The thermomagnetic effect is particularly pronounced
below 10 K due to the extremely long mean free path of the electrons in Bi [27]. Additionally, Bi
23
nanowires become semiconducting as their diameters approach several nanometers, thereby
exhibiting a large thermoelectric power factor [28, 29]. As a conventional bulk thermoelectric
material, Bi1 .xSb, has drawn more attention than Bi, since alloying with a small amount of Sb
causes Bi,Sb, to become a narrow gap semiconductor, which is advantageous for high
thermoelectric
efficiency.
Currently,
Bit-.,Sb,
(x
~ 0.12)
is the
best
available
n-type
thermoelectric material below 200 K [25].
Before discussing the thermal transport by phonons, we would like to emphasize that
electrons, in addition to phonons, carry a considerable amount of heat in Bi, Sb, and Bi-Sb alloys.
Therefore, both phonons and electrons contribute to the total thermal conductivity, which can be
expressed as
Ktot =
where Ktat and
Kph
Kph +
Ke
(2.1)
are the total thermal conductivity and the phonon thermal conductivity,
respectively. The term
Ke
includes the thermal conductivity of electrons, holes, and the bipolar
contribution (hereafter electron thermal conductivity). The electron thermal conductivity of Bi,
Sb, and Bi-Sb alloys is expected to contribute substantially to the total thermal conductivity since
these materials are either semimetals or semiconductors with a very narrow band gap.
Accurate methods to separate the phonon and electron contributions to the total thermal
conductivity are crucial to developing better thermoelectric materials. However, separating the
phonon and electron contributions is experimentally nontrivial. The phonon thermal conductivity
can be directly measured under a high magnetic field, because such fields largely suppress
electron transport. Previous measurements [30, 31] in practical temperature ranges (100 to 300 K)
utilized this method, but the prior measurements are mainly limited to transport along the binary
crystallographic direction. We could not find any reports on the phonon thermal conductivity
along the trigonal direction, which are expected to have a greater thermoelectric figure-of-merit
than for the binary direction and thus is of more interest. Another way to separate the phonon and
electron contributions to the total thermal conductivity is to estimate the electron thermal
24
conductivity using either the Wiedemann-Franz law or other electron transport properties, such
as the electrical conductivity and Seebeck coefficient [32]. Such an approach provides a
reasonable qualitative analysis, but validity of the Wiedemann-Franz law and the simple electron
transport models used in the estimation of the electron thermal conductivity is sometimes
questionable for quantitative purposes [33].
In this chapter, we study the lattice dynamics and quantify the phonon thermal
conductivity values for Bi, Sb, and Bi-Sb alloys from first principles and the Peierls-Boltzmann
transport equation. As shown in recent works [8, 9, 34-38], this approach provides an excellent
agreement with experimental data for many pair-bonded materials, such as Si, GaAs, and Si-Ge
alloys. We follow the same approach, but pay special attention to the range of interatomic
interactions. This is because Bi, unlike other pair-bonded materials, has significant interaction
strength out to large number neighbors, such as the ninth-nearest neighbor [39, 40].
2.2. First Principles Calculations of Phonon Transport and Thermal
Conductivity
2.2.1. Second- and Third-order Force Constants
We calculated the second- and third-order force constants using the density functional
theory. The calculation of the second-order force constants of Bi and Sb is based on the real
space approach [41]. We calculated the force exerted on each atom when we displace one or
multiple atoms in a 4x4x4 supercell which consists of 128 atoms. For the supercell calculation,
we used 30 Ry for the cut-off energy of the plane wave basis and a 4x4x4 k-mesh for Brillouin
zone sampling, both of which were carefully checked for the convergence of the calculation
results.
The
calculation
was
performed
with the
ABINIT
package
[42]
and
HGH
pseudopotentials [43]. The valence electrons in these pseudopotentials are 6s 2 6p 3 and 5s 2 5p 3 for
Bi and Sb, respectively. The spin-orbit interaction is included in all calculations, because of the
25
strong spin-orbit interaction in Bi and Sb [441. The second-order force constants are then fitted to
the calculated
displacement-force
data set, while enforcing translational
and rotational
invariances. In the fitting process, we considered up to the fourteenth-neighbors to include the
previously reported long-range interaction occurring at the ninth-neighbor [39, 40]. The ninthneighbors are shown by the atom labeled C in Fig. 2-1 where the origin atom is described by
atom A. Bi and Sb both have a slightly distorted simple cubic crystal structure. Due to this small
crystallographic distortion, the six first-neighbors in the cubic structure become three firstneighbors and three second-neighbors. In Fig. 2-1, the atom B is the first-neighbor to the atom A
and the second-neighbor to the atom C. The almost collinear chain consisting of AB and BC
forms the ninth-neighbor relation and the atom C is the ninth-neighbor to the atom A. In the
following discussions, the fourth- and the ninth-neighbors are frequently mentioned to discuss
the range of the force constants for Bi and Sb. The fourth- and the ninth-neighbors in the
rhombohedral crystal structure of Bi correspond to the second-neighbor (separated by Via) and
the fourth-neighbors (separated by 2a), respectively, in the undistorted cubic structure, where a
is the lattice constant of the simple cubic structure.
26
a
0
Origin 0
*atom-
F'
eigftpr
inth
neighbor
RI
Figure 2-1 Crystal structure of Bi and Sb. The void and filled atoms represent two basis atonrs. RI, R2,
and R3 are primitive lattice vectors and a is a rhombohedral angle between two primitive lattice vectors.
The values of a are 57'30 for Bi and 57*84 for Sb, which are close to 60* of the simple cubic structure.
The third-order force constants were calculated by taking finite differences of the secondorder force constants [45]. We built a 3x3x3 supercell consisting of 54 atoms and we displaced
one of the two basis atoms along the +R1 direction in Fig. 2-1 by 0.04 A. The displacement value
of 0.04 A was chosen after carefully checking the convergence of third-order force constants
with respect to the displacement values. The size of the supercell was large enough to include the
significant ninth-neighbor interaction. In addition, the large size of the supercell minimizes the
effect from the periodic images of the displaced atom due to periodic boundary conditions. For
the calculation, a cut-off energy of 30 Ry and a 3x3x3 k-mesh are used. We then calculate the
27
second-order force constants using density functional perturbation theory [46, 471. All of the
procedures are repeated for another supercell with the displacement along the -R, direction. By
taking the finite differences of the second-order force constants of the two different supercells,
the third-order force constants with respect to the R1 direction are calculated. Rotational
invariance with respect to the trigonal direction is then applied to calculate the third-order force
constants with respect to the R 2 and R3 directions. Translational invariance is applied to the thirdorder force constants by modifying the self-interaction terms.
We calculated the phonon dispersions and mode Groneisen parameters to validate the
calculated second- and third-order force constants. In Fig. 2-2a, we plot the trace of the secondorder force constant tensors versus distance. Both Bi and Sb have the interactions of significant
magnitude occurring at the ninth-neighbors, which agree well with the previous reports [39, 40].
In Fig. 2-3, the calculated phonon dispersions for Bi and Sb are compared with the experimental
values. Both calculated phonon dispersions are similar to the experimental data, confirming the
accuracy of the calculated second-order force constants.
28
1
(a)
1)
0
e*
-
ca.
oo
0
-1
9th neighbors
U-
0
-2
0 -3
LL
-4
-5
*0
I
2
3
*
I
4
5
Bi
6
0
Sb
7
8
distance (A)
CO
10
(b)
8
C
6
0
-
eB
O Sb
0
0
-8
4
40
9th neighbors
S2
0
'2
M
I
2
0)
3
4
5
distance (A)
6
7
8
Figure 2-2 Force constants of Bi and Sb versus interatomic distance (a) Trace values of second-order
force constant tensors and (b) two-body third-order force constants
29
(a)
3
I0
N
1
2
C
01
LL
000
N
I
43-
.
0
C.r
(c)
respectively. Dots are
Figur'e 2-3 Phonon dispersion of Bi and Sb. (a) and (b) represent Bi and Sb cases,
symmetry points in the
experimental values from Refs. [48] for Bi and [49] for Sb. The location of high
Brillouin zone are plotted in (c) for Bi, Sb, and Bi-Sb alloys.
30
Since the ninth-neighbors in Bi and Sb have significant second-order force constants, the
third-order force constants at the ninth-neighbors should also be of interest. In Fig. 2-2b, we plot
the two-body third-order force constants as a function of distance. Each dot represents a thirdorder force constant. As seen in Fig. 2-2b, the third-order force constants have substantial values
The importance
at the ninth-neighbors.
of the
ninth-neighbor interaction
on
crystal
anharmonicity can be checked with the mode Gruneisen parameters. The Gruneisen parameter, y,
of a phonon mode with wavevector (q) and polarization (s) is calculated with the calculated
third-order force constants using the following expression[9]:
y(qs) =
-
6u
2
(qs)
DbRRb1,Rzb 2
2
'MMb
Xoabe(-qs, b1 /)e(qs, b2 y)
(2.2)
where o, T, and M represent the phonon frequency, third-order force constant, and atomic mass.
Here, afly, R 1 , and b, denote the polarization, translational vector, and basis atom. Also, X and
e are the atomic position and eigenvector, respectively. In order to investigate the effects of the
ninth-neighbor interaction on the crystal anharmonicity, we used two different sets of the thirdorder force constants: one includes up to the fourth-neighbors and the other includes up to the
tenth-neighbors.
To evaluate the reliability of the third-order force constants, the reference mode
Grlneisen parameters are also calculated. For the reference mode Gruneisen parameters, we used
density functional perturbation theory to calculate the phonon frequencies for two different
crystal volumes: a crystal at equilibrium and one with the volume increased by 1%. We then take
the finite differences of the two different phonon frequencies and calculate the mode Grineisen
parameters from the definition of the Grineisen parameter:
Inw(qs)
d nV
y(qs) =
31
(2.3)
where V is the crystal volume. Shown in Fig. 2-4 are the calculated acoustic mode Gruneisen
parameters.
In
Fig. 2-4, we show
that the
acoustic mode Gruneisen
parameters
are
underestimated over a wide range of wave-vectors when the third-order force constants are
considered only up to the fourth-neighbors. Even after considering up to the eighth-neighbors,
the mode Grnneisen parameters are relatively unchanged. This is consistent with the negligible
third-order force constants at the fifth-, sixth-, seventh-, and eighth-neighbors as shown in Fig. 22b. However, when extending the range up to the tenth-neighbors, the calculated acoustic mode
Griineisen parameters agree reasonably well with the reference Gruneisen parameters. This
confirms that the ninth-neighbor interaction is playing a significant role in the anharmonic
properties. The optical mode Gruneisen parameter was also determined from third-order force
constants that included up to the fourth-neighbor and the tenth-neighbor interaction terms. Both
cases yielded similar values for the optical Griineisen parameter.
32
(a)
10
8-
E
CL
C:
C
Reference
upto 4th neighbor
upto 10th neighbor
-
L..
-
6
4
2
0
__
_
-2
-4
(b)
-
X K
I-
T
W
1L
X
6
---
i)
-
E
1
4
Reference
upto 4th neighbor
upto 10th neighbor
cc
L..
C
0
C
2
C
0
-91
X K
U
T
XW
L
I
X
Figure 2-4 Acoustic mode Grflneisen parameters of (a) Bi and (b) Sb comparing inclusion up to the
fourth- and tenth-neighbors, to the references. The reference Grineisen parameters are calculated
using the difference of phonon frequencies of two different crystal volumes.
2.2.2. Scattering Rates and Peierls-Boltzmann Transport Equation
The phonon thermal conductivity can be calculated from the distribution function of the
phonon modes. We calculate the distribution function by solving the linearized Boltzmann
equation with the scattering rates due to the three-phonon process and mass disorder. The
scattering rate of the three-phonon process is given by [50]
33
W, = 21r|V3 (-q1 s1 ,-q 2s 2 ,q 3s 3 ) 2 N N2(N3' + 1)8(-W - W2 +
W2, 3 = 2iV3 (-q1 s1 , q 2 S 2 , q 3 s 3 )I 2N 1 (N 2 + 1)(N3 + 1)6(-o
1
(3)
+ W2 + W3)
(2.4)
(2.5)
where 1, 2, and 3 denotes phonon modes in the three phonon process. Here, 1 indicate a phonon
mode with a wavevector (q 1 ) and polarization (sl). In addition, N' indicates the Bose-Einstein
equilibrium distribution function. Eqs. (2.4) and (2.5) represent a coalescence and a decay
process, respectively. The delta-function in Eqs. (2.4) and (2.5) represents energy conservation
of the three-phonon scattering process. In addition to the energy conservation, the three phonon
modes should meet the conservation of crystal momentum with the reciprocal primitive vector, G.
This requirement can be expressed with the expressions given below for the coalescence process
and decay process, respectively:
q1 + q 2 = q 3 + G
q
+
2 q
3+G
(2.6)
(2.7)
where G is a reciprocal primitive vector including a null vector. When G is a null vector, the
three-phonon scattering is called Normal scattering (hereafter N-scattering), while it is called
Umklapp scattering (hereafter U-scattering) when G is a non-zero vector. The N-scattering and
U-scattering is schematically compared in Fig. 2-5. The three-phonon scattering matrix element,
V3 , in Eqs. (2.4) and (2.5) is given by [50]
)
V3 (q 1 s1 , 4 2 s 2 , q 3 s 3
8No 1 j
20 3
12
Ofy (Obl, R2b2,
.mb
Reb3)eiq2-R ?
i3-R3
cabeflb2 eyb 3
1
blb 2 b 3 afly R 2 R 3
34
mbmb
3
2
(2.8)
where IDfgy(Obi, R2 b2 , R 3 b 3 ) is a third-order force constant with Cartesian coordinates ay and
Rb representing the lattice vector and basis atom. Here, eab 1 denotes the phonon eigenvector
component of the basis atom b, along direction a while N is the total number of wave-vectors in
the first Brillouin zone.
(a)
(b)
(c)
q,
q3
q/2
N le
q2G7
Normal scattering
Umnklapp scattering
mass disorder
scattering
Figure 2-5 Comparison of Normal, Umklapp and mass disorder scatterings. The squares represent
the first Brillouin zone.
To study the effects of alloying on phonon thermal conductivity, the virtual crystal
approximation is used [51]. The atomic mass and the force constants of the virtual crystal were
linearly interpolated between Bi and Sb, weighted by the composition ratio of each constituents.
The lattice constant of the virtual crystal is also averaged according to the composition ratio,
which is well justified by the fact that the Bi-Sb alloy follows Vegard's law [52]. Three-phonon
scattering is calculated using the virtual crystal approximation while the atomic mass disorder is
treated as an additional elastic scattering mechanism. This approach was very successful in
predicting the Si-Ge alloy thermal conductivity[38]. The mass disorder scattering rate is
e,* - eb
mx203N10(N20 + 1)
b
35
2
6(6
1
- 62
)
=
(2.9)
with the mass variance factor g, defined by g =
ijfi (1 - Mi/Mavg
2
where
fg
is the fraction of
element i.
One of the numerical uncertainties in the scattering rate calculation is from dealing with
the energy conservation. Due to the computational limitations, the Brillouin zone is sampled with
a relatively coarse mesh. To find sets of three phonons satisfying the energy and momentum
conservation, each point in the coarse mesh is usually broadened by a Gaussian function.
However, numerical uncertainties arise from the tuning of two adjustable parameters, mesh size
and Gaussian width. To avoid this artifact, a tetrahedron method is utilized for the Brillioun zone
integrations of 5-functions when calculating the scattering rates according to Eqs. (2.4) to (2.9)
[53]. With this method, the mesh size is the only adjustable parameter; consequently, the
calculation should converge as the mesh size is increased. For our calculation of Bi, Sb, and BiSb alloys, the mesh size of 16x16x 16 was enough for convergence.
Using the scattering rates that are calculated with the given expressions above, the
Peierls-Boltzmann transport equation can be solved. The original form of the Peierls-Boltzmann
transport equation is
(q
tNqs
)scatt
T
(2.10)
=
k
Nqs
In most cases, the non-equilibrium distribution function, Nqs, deviates only slightly from the
equilibrium distribution function, N' 5 . Based on this, the non-equilibrium distribution function
can be linearized as follows:
dN 0
Nq = No
s+gqo
(2.11)
where I is a linearized deviation of the distribution function from equilibrium, defined as
'P
=
(N 0
-
N)/(dN0 /3). Here, p is the phonon frequency normalized by temperature, defined
as hw/kBT. Putting the scattering rates of the three-phonon process and mass disorder shown in
36
Eqs. (2.4) to (2.9) with the linearized distribution function in Eq. (2.11) into the PeierlsBoltzmann equation, we obtain:
-Vqls,
(aBN
- VT I
)T
W32(T1 + T 2 - J3 ) +2W3(Pl-W
2
-l
3
)
(2.12)
2,3
2
)
+ IW2(Tj1 2
The linearized Boltzmann equation above is basically a large set of linear equations. The
size of the matrix is around 25000
x
25000 if we sample the Brillouin zone with 16x 16x 16
points and there are 6 phonon branches. We solve this equation iteratively to find the deviation
of the phonon distribution function, P [35, 54]. In this iterative method, the deviation of phonon
distribution can be separated into a zeroth-order solution, qif, given by the single mode
relaxation time approximation, and a remaining part, AT.
P -P0
(2.13)
+ AT
We start from the zeroth-order solution, To , given by the single mode relaxation time
approximation. The single mode relaxation time approximation assumes only one phonon mode
is ever out of equilibrium and the time for the non-equilibrium mode to relax to equilibrium,
represented by the relaxation time, which is then calculated. In this case, P 2 and
T 3
in Eq. (2.12)
can be set to zero, meaning that only phonon mode I is out of equilibrium while phonon modes 2
and 3 stay at equilibrium. This assumption will lead to a simple solution of Eq. (2.12) as follows:
37
(2.14)
a = hw 1 N,(Nl + 1)via
TQI
where a is the direction in the Cartesian coordinate. Above, Q1 represents the total scattering rate
of phonon mode 1, defined as:
Q1 =
W,2 + 1Wi'+LW
(2.15)
2
2,3
Then, the remaining part of the solution, A'P, in Eq. (2.13) can be updated at each iteration step
according to the modified form of Eq. (2.12) which is given below.
1+T)+1
3
3
[W
2(-%
2 P2 )+ 1 P
2 +' ) + W
AT =
(2.16)
W2,3
~
~
W12~l
W
Q12,3
1
2
1
(.6
The above expression can be easily derived by putting Eqs. (2.13) and (2.14) into Eq. (2.12).
When calculating AIP according to Eq. (2.16), values from the previous iteration step can be
2
and P 3
.
used for
Once we calculate the non-equilibrium phonon distribution function from the PeierlsBoltzmann
equation, the phonon thermal conductivity
can be calculated.
The thermal
conductivity tensor, icp, can be defined by the Fourier's heat conduction law:
Kap
VpT
(qa
2.17)
where qa represent the heat flux along the direction of a. The heat flux, qa, can be expressed
with the non-equilibrium phonon distribution as follows,
38
q
=Z
Therefore, the thermal conductivity tensor,
Kap
=
hwvaN0 (N 0 + 1)Y
icg,
(2.18)
is simply
1hvNO(No + 1)
We used both the full iterative method
and the
(2.19)
single mode relaxation
time
approximation to calculate the phonon thermal conductivity from the Peierls-Boltzmann equation
and we compare the results from the two methods.
2.3.Results and Discussions
2.3.1. Phonon thermal conductivity
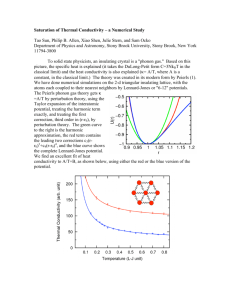
In Fig. 2-6a, we show that the ninth-neighbor interaction has a significant effect on the
lattice thermal conductivity. Here, we compare the phonon thermal conductivity in the binary
direction calculated with the two different force constant sets: one set includes up to the tenthneighbor and the other includes up to the fourth-neighbor for the third-order force constants. In
both cases, the second-order force constants include up to the fourteenth-neighbor, otherwise, the
phonon dispersion is not stable and the phonon frequencies of some modes have imaginary
values. As shown in the mode Gruneisen parameter plot (Fig. 2-4), when the ninth-neighbor
interaction is not included for the third-order force constants, the crystal anharmonicity is largely
underestimated.
Figure 2-6a explicitly
shows that the phonon thermal conductivity
is
significantly overestimated when the ninth-neighbor interaction is not included for the thirdorder force constants. However, when the third-order force constants include up to the tenth-
39
neighbor interactions, the calculated phonon thermal conductivity is half of the value obtained
when including only third-order force constants up to the fourth-neighbor.
(a)
30
--- - p UPtO
E
--
Kph
Kph
-
10th (Full)
upto 10th (SMRT)
upto 4th (Full)
.. KPh upto 4th
0 Kph (Gallo)
o
20
0
0
E
-
Kph
(SMRT)
(Uher)
10
0
C
50
100
150
250
200
300
Temperature (K)
binary
(b)Kph
-
3
o
Z'20
U
trigonal
Kph binary (Gallo)
Kph trigonal (Gallo)
tot binary (Gallo)
Ktot trigonal (Gallo)
U
010
C 0
50
100
150
200
250
300
Temperature (K)
Figure 2-6. Thermal conductivity of Bi (a) in the binary direction and (b) in comparison between the
binary and the trigonal directions. Kph in (b) is calculated with the single mode relaxation time
approximation and using third-order force constants up to the tenth-neighbors. The solid lines and dots
represent our first principles calculation results and the experimental data from Ref. [32], respectively.
The Full and SMRT in the legend represent solution of the Peierls-Boltzmann equation using the full
iterative method and the single mode relaxation time approximation, respectively.
40
The calculated results with the ninth-neighbor interaction are validated by comparing
these results to the previously reported experimental data [30, 31]. Figure 2-6a shows that our
calculation results with the ninth-neighbor interaction agree well with the experimental data by
Uher [30]. Our calculation is further confirmed by comparing to another measurement by Kagan
[31], showing that the phonon thermal conductivity value is around 5 W/m-K at 250 K. In
contrast, another reported value for the phonon thermal conductivity by Gallo [32], which is
calculated from the difference between the measured total thermal conductivity and the
calculated electron thermal conductivity as briefly discussed later, shows disagreement with our
calculation near room temperature. Our calculated phonon thermal conductivity is twice the
reported value[32] at room temperature. This disagreement could stem from the simple electron
transport model used in the referenced work [32]. Instead of directly measuring the phonon
thermal conductivity, Gallo obtained the electron thermal conductivity from an electron transport
model using a parabolic band structure and an electron scattering rate that obeys a simple power
law. The measured Seebeck coefficient and electrical resistivity determines the electron
contribution to the thermal conductivity, and then the phonon thermal conductivity is calculated
by subtracting the deduced electronic thermal conductivity from the measured total thermal
conductivity. To reiterate, our calculation near room temperature is well validated by Kagan's
direct measurement[3 I].
We also see in Fig. 2-6a that the results from the single mode relaxation time
approximation are similar to the calculations from the full iterative solution of the PeierlsBoltzmann equation. This is because the temperatures in our calculations are high compared to
the Debye temperature of Bi (120 K). When the temperature is not significantly smaller than the
Debye temperature, Umklapp scattering is dominant over Normal scattering. In this case, the
single mode relaxation time approximation is usually a good approximation.
In Fig. 2-6b, we compare the binary and the trigonal directions of Bi in terms of their
phonon thermal conductivity values. The previous work based on obtaining the electron thermal
conductivity[32], mentioned above, estimates that the phonon thermal conductivity along the
trigonal direction is half of the value of that along the binary direction in Bi at room temperature.
Our calculation shows that the phonon thermal conductivity value along the trigonal direction is
smaller than that along binary direction, but the difference is less than 10 %.
41
The relatively
similar value of the phonon thermal conductivity along the trigonal direction compared to that
along binary direction can be explained by the fact that the rhombohedral structure of Bi is close
to the simple cubic structure. The structure of Bi is only slightly stretched along the trigonal
direction from the simple cubic structure. Therefore, the atomic bonding is slightly softer in the
trigonal direction than in the binary direction, resulting in the lower phonon thermal conductivity
in the trigonal direction. However, the distortion from the exact cubic structure is very small: the
rhombohedral angle of Bi (a in Fig. 2-1) is 57030, very similar to 600 for the exact cubic
structure [52]. This very small distortion explains the almost isotropic phonon thermal
conductivity of Bi shown in Fig. 2-6b. The almost isotropic phonon thermal conductivity of Bi is
in contrast with its well-known highly anisotropic electron transport properties[25]. This shows
that the small distortion of crystal structure of Bi affects the electron and the phonon transport to
a very different extent. Even though the distortion of the Bi crystal structure is very small from
the exact cubic structure, this small distortion causes highly anisotropic shapes to occur in the
very small electron and hole pockets responsible for its electronic transport properties, giving
rise to largely anisotropic electron transport behavior. However, the small distortion does not
much affect the lattice vibrational properties, and thus the phonon thermal conductivity is
observed to be almost isotropic.
We also compare the calculated phonon thermal conductivity and the experimentally
measured total thermal conductivity of Bi in Fig. 2-6b in order to estimate the relative
contributions from phonons and electrons to the total thermal transport. In the binary direction,
the phonon thermal conductivity value is around 60 % of the measured total thermal conductivity
at 100 K, and its contribution decreases with temperature. In the trigonal direction, the phonon
%
contribution is more significant than in the binary direction, with a contribution of around 75
at 100 K. Based on this large contribution from phonons, we have large room in the trigonal
direction to reduce the thermal conductivity effectively by enhancing phonon scattering, as was
recently demonstrated in Bi1ASbO 6 Te 3 and PbTe [7, 55]. In particular, the large lattice
contribution in the trigonal direction would be interesting because the electron transport in this
direction of Bi has a favorable feature for a high thermoelectric power factor. It is known that the
electrons in the trigonal direction of Bi have an extremely large value for the product of the
42
mobility and the density-of-states effective mass, p(m*/m)3/2, due to the high anisotropy in its
electronic structure, which is directly related to the thermoelectric power factor [25].
Many features of the phonon thermal conductivity in Sb, presented in Fig. 2-7, show
strong similarities to the thermal conductivity of Bi. The ninth-neighbor interaction in Sb is also
significant, and the phonon thermal conductivity is significantly overestimated without including
this contribution in the calculation. The single mode relaxation time approximation is valid for
Sb since its Debye temperature is also small (- 200 K). The distortion from the exact cubic
structure is also small for Sb, as it is in Bi, resulting in an almost isotropic phonon thermal
conductivity. The most noticeable difference between Bi and Sb is the contribution of the
phonon thermal conductivity to total thermal conductivity, comparing Fig. 2-6b and Fig. 2-7b.
The phonon contribution is comparable to the electron contribution in Bi, but phonons contribute
only a small portion of the total thermal conductivity in Sb. In other words, the electron
contribution is very significant in Sb, because the carrier density in Sb is two orders of
magnitude larger than that of Bi [56].
43
(a)
60
upto 10th (Full)
Kph
---
ph upto 10th (SMRT)
-K - ph upto 4th (Full)
16 CSM r
... ph Uptu 4 I
C
-
' 40
20
0
E
I- I
0
100
250
200
150
(b)60
--
p
- - -
Kph
0
*
*
*
40
300
Temperature (K)
a
binary
trigonal
tot binary (Yim)
Ktot trigonal (Yim)
C
0 20
0 "Z
--
L
0
100
150
200
250
300
Temperature (K)
Figure 2-7. The thermal conductivity of Sb (a) in the binary direction and (b) in comparison between
the binary and the trigonal directions. The solid lines and dots in (b) represent our first principles
calculation results and the experimental data from Ref. [26], respectively. The Full and SMRT in the
legend represent solution of the Peierls-Boltzmann equation using the full iterative method and the single
mode relaxation time approximation, respectively.
The phonon thermal conductivity of Bi, Sb, and Bi-Sb alloys is presented in Fig. 2-8. Our
calculation for Bi88 Sb12 agrees well with the experimental data by Kagan[3 1], showing ~ 3 W/m-
K at around 100 K and ~ 2 W/m-K at around 250 K for Bi87Sb 3 . Figure 2-8a shows that the
44
phonon thermal conductivity of Bi can be significantly reduced by alloying with small
concentrations of Sb. The composition Bi 88 Sb1 2 , which has the highest thermoelectric figure-ofmerit among the Bi-Sb alloys [25], has four-times smaller phonon thermal conductivity value
than Bi at 100 K. In order to study the anisotropy of phonon transport, we compare the phonon
thermal conductivity values in the binary and the trigonal direction. The Bi-Sb alloy, like its Bi
and Sb constituents, has smaller phonon thermal conductivity values in the trigonal direction, but
the difference between the trigonal and the binary directions is very small, indicating a
predominantly isotopic phonon thermal conductivity.
45
(a)
--
20
binary
trigonal
-
T=100 K
15
=3
-)
10
C
8
5
E
0
20
0
100
80
60
40
Sb content (%)
(b)
pure Bi binary
BMSb. binary
A
40
>1
(Yim)
A
B
pure Sb binary
A
30 0
.)
0
U)
-c
E
-
....
A
Kph *
Kp
Ktot (Gallo)
Ktot (Yim)
*
Kph A Ktot (Yim)
A
A-
20
10-
100
50
300
250
200
150
Temperature (K)
(C)
6
binary
BisSbe
C-)
Kph 0
-
Lib trigonal -
0
.0
C
8
(Y
- - -Kpn 0 Ktot (y~m
4
e
Ktot
)
BSb,. tngonal - - Kph 0Ktot
-
-
0
0
e
00
0
2
E
0'
0
100
150
200
250
300
Temperature (K)
Figure 2-8. Thermal conductivity of the Bi-Sb alloys. (a) The effect of Sb content on the
thermal conductivity, showing that inclusion of even small amount of Sb significantly reduce
thermal conductivity. (b) Comparison between the total and phonon thermal conductivity of Bi,
Bi8 lSb12, and (c) an enlarged plot for the Bi88Sb12 data. The experimentally measured total
conductivity values are from Ref. [26, 32].
46
phonon
phonon
Sb, and
thermal
The comparison between the experimentally measured total thermal conductivity and the
calculated phonon thermal conductivity in Figs. 2-8(b) and 2-8 (c) indicates whether electrons or
phonons are the predominant heat carrier in Bi, Sb, and BiRgSb12. Unlike Bi and Sb, the total
thermal conductivity of Bi88Sb]2 comes predominantly from phonon contributions at low
temperature. Around 75 K, the calculated phonon thermal conductivity value of Bi8 8 Sb]2 is
comparable to the measured total thermal conductivity value for either the trigonal or the binary
direction. The electron thermal conductivity, in this case, is expected to be small due to the
positive electronic band gap (-30 meV) of Bi88 Sb 2 [56]. The number of charge carrier in
BiggSb12 is much lower than that in Bi and Sb, resulting in the smaller electron thermal
conductivity. However, from comparing the measured total thermal conductivity and the
calculated phonon thermal conductivity, the electron thermal conductivity increases with
temperature. This can be explained by the increasing charge carrier density and also increasing
bipolar thermal transport as temperature increases. From Fig. 2-8c, the electron contribution
becomes comparable to the phonon contribution near room temperature. Another noticeable
feature in the phonon thermal conductivity value of Bi88 Sb12 is that its insensitivity to
temperature variation. This is because mass disorder scattering, a temperature independent
process, is the dominant phonon scattering mechanism in this alloy.
2.3.2. Phonon Mean Free Path Distributions
Finally, we show in Fig. 2-9 the accumulated thermal conductivity, Kac, versus phonon
mean free path. The accumulated thermal conductivity we show in Fig. 2-9 is defined as [57, 58]
Kacc(A) =
KqsX
qs
47
(A)
(2.20)
where Kqs represents the thermal conductivity of the phonon mode with wave vector q and
polarization 2. Here, A is a phonon mean free path and X(A) is a step function: X(A) = 1 when
Aqs < A, and X(A) = 0 otherwise. The accumulated thermal conductivity shows the range of
mean free paths of the phonon modes that significantly contribute to thermal transport[57, 58].
From Fig. 2-9a, we see that most of heat is carried by phonons with mean free paths ranging
from 10 nm to 100 nm at 100 K. However, the phonon mean free path range of the BigsSb12 alloy
is slightly different from that of Bi in the 50 to 100 nm region in Fig. 2-9b: the mean free path
range of the alloy is extended to longer mean free paths compared to Bi. This is because the alloy
scattering is very effective for scattering high frequency phonons, but not as effective for low
frequency phonons. If the alloy scattering is approximated by a point defect scattering
mechanism, the Rayleigh scattering model shows that the scattering rate is proportional to the
fourth power of phonon frequency.
Fig. 2-9a shows that the nanostructures in the 10 to 100 nm range scale can significantly
contribute to phonon scattering, ultimately resulting in a greatly reduced thermal conductivity in
both Bi and the Bi-Sb alloy. In addition to the reduction in the phonon thermal conductivity, it is
known that Bi nanowires becomes semiconducting and exhibit a high power factor when the
diameter is on the order of 10 nm [28, 29]. If harmonic and anharmonic force constants of Bi
nanowires are not drastically different from those of bulk phase Bi, the phonon mean free path
distribution from bulk Bi calculations can guide the design of Bi nanowires for high ZT. To
provide a strategy for reducing phonon thermal conductivity through nanostructuring, we present
the phonon mean free path distributions of Bi at various temperatures in Fig. 2-9c. From Fig. 29c, we see that nanostructures having characteristic sizes of around 10 nm would be effective for
suppressing phonon thermal conductivity in the temperature range of 100 K to 300 K as they are
expected to reduce the phonon thermal conductivity by a factor of 10 at 100 K to 3 at 300 K if
boundary scatterings are assumed to be completely diffuse.
48
(a)
-
Bi
20 .pure
Bi Slb, --
pure Sb
-
E
15
binary - -
..-
-
T=100 K
trigonal
- binary - - - trigonal
..-
- binary - - - trigonal
binary- - - trigonal
ure
Sb
M
pure Bi
cc =10
-
0
E
Bi.S
5
0
(b)
1
10
- - -- -
-
(b)
100
Phonon mean free path (nm)
7:
=3
1.2
-0
"a
T=100 K
pure Bi binary
-
Bi- Sb- 2 binary
.0
0 .8
C
E
go .6
0
E a .4
E (-D
CU
Z
U.2
0.0
10
100
Phonon mean free path (nm)
(C)
CU
50K 100K 200K 300K ---
1.2
1.0
binary
- - -trigonal
binary - - -trigonal
binary - - - trigonal
binary - - - trigonal
pure Bi-
E : 0.8
300K
C, 80.6
N
0
---1
Z
50K
E 0.4
-4S0.2
0.0 L
1
10
---
100
Phonon mean free path (nm)
Figure 2-9. Phonon mean free path distribution (a) Bi, Bi4>Sb, BiggSb, and Sb at 100 K, (b) Bi and
Bi88 Sb12 at 100K, and (c) Bi at 50, 100, 200, and 300 K for the binary and the trigonal directions. In (b)
and (c), the accumulated thermal conductivity is normalized by the phonon thermal conductivity value.
49
2.4. Conclusion
In conclusion, we calculate the phonon thermal conductivity values of Bi, Sb and Bi-Sb
alloys from first principles. We explicitly show that the significant ninth-neighbor interaction is
important for anharmonic interatomic force constants, phonon scattering, and phonon thermal
conductivity. Our calculation agrees well with the experimental phonon thermal conductivity
values for the binary direction. We also provide the phonon thermal conductivity values for the
trigonal direction, which has not been directly measured. From our calculation, the phonon
thermal conductivity values are almost isotropic in these materials, showing a significant contrast
with the highly anisotropic electron transport in Bi. This implies that the small distortion in the
crystal structure can affect the electron and the phonon transport to a much different extent. By
comparing our calculated phonon thermal conductivity to the measured total thermal
conductivity, we compare the relative thermal conductivity contributions from phonons and
electrons. The phonon thermal conductivity is comparable in magnitude to the electron thermal
conductivity in Bi. In Sb, however, the electron contribution to the total thermal conductivity is
much more dominant because of the larger charge carrier concentration. In Bi88 Sb, 2 , the phonon
thermal conductivity is the dominant contributor below 75 K, but becomes less significant as the
temperature increases. Finally, we calculate the phonon mean free path distributions at various
temperatures, which provide a useful guide in determining appropriate nanostructure sizes for
achieving significant phonon thermal conductivity reduction.
50
3. Low Thermal Conductivity of IV-VI Materials
from Resonant Bonding
The long-range interaction that has been reported in Chapter 2 for Bi and Sb is also
observed in other thermoelectric materials, such as group IV-VI and V 2-VI 3 materials. These
group IV-VI, V2 -VI3, and V materials are the currently available best thermoelectric and phase
change materials. In this chapter, we discuss a link between the low thermal conductivity and
resonant bonding. Our first-principles calculations reveal that the long-range interaction along
the <100> direction of the rocksalt structure exists in lead chalcogenides, SnTe, Bi 2Te 3, Bi and
Sb due to the resonant bonding that is common to all of them. This long-range interaction in lead
chalcogenides and SnTe causes optical phonon softening, strong anharmonic scattering, and a
large phase space for three-phonon scattering processes, which explain why rocksalt IV-VI
compounds have much lower thermal conductivity than zincblende III-V compounds.
3.1. Background
,
Most good thermoelectric and phase change materials are found in group IV-VI, V 2 -VI 3
and V materials. For example, PbTe, Bi 2Te3 , and Bi1 ,Sb, have been the best thermoelectric
materials in the intermediate, room, and low temperature ranges, respectively [7, 25, 55, 59].
Alloys of GeTe and Sb 2Te 3 (GST) have been the most popular materials for optical storage
technologies, such as compact disc and phase change random access memory [60, 61]. These
51
materials all have low thermal conductivity, which is crucial for thermoelectric and phase change
memory applications.
Usually, group IV-VI, V2-VI 3, and V materials have low thermal conductivity. This
becomes particularly obvious when IV-VI materials are compared to III-V compounds. For
example, the thermal conductivity of SnTe is only 4 W/m-K (Ref. [62]) while that of InSb,
adjacent to SnTe in the periodic table, is 16 W/m-K at room temperature. The low thermal
conductivity of rocksalt IV-VI materials (hereafter IV-VI materials) compared to those of
zincblende Ill-V materials (hereafter Ill-V materials) have been attributed to their differences in
the crystal structure. While 111-V materials have tetrahedral bonding, many IV-VI materials have
octahedral bonding. The bond length is usually longer in octahedral structures than in tetrahedral
structures, resulting in weaker bonding and lower thermal conductivity [15].
Our first-principles calculations show that there are more reasons for the low thermal
conductivity of IV-VI materials than that discussed above. To compare the thermal conductivity
of many different III-V and IV-VI materials, we normalize their thermal conductivity by their
harmonic properties using the formula suggested by Slack [63]. The formula gives reasonable
predictions for many materials with zineblende and rocksalt structures [14]. According to the
formula, thermal conductivity,
K,
is roughly correlated to several parameters through the formula:
B = n 1/5D
3
(3.1)
Y2
where B is a numerical coefficient, M is the average mass of the basis atoms, n is the number of
phonon branches, and y is the Gru-neisen parameter. The average volume per atom is denoted by
63, and OD is the acoustic Debyc temperature. Here, the harmonic properties include M, OD, and
6; these three properties determine the average group velocities of acoustic phonons and they
reflect the bonding stiffness. In Fig. 3-1 we plot the thermal conductivity,
K,
of IV-VI and Ill-V
materials normalized by the harmonic properties, Rn'/38SD 3 , as a function of the mass ratio of
the basis atoms. There are two distinct differences between the thermal conductivity of IV-VI
52
and III-V materials: 1) overall, IV-VI compounds still have much lower thermal conductivity
than Ill-V compounds even after normalization, and 2) the thermal conductivity difference
between IV-VI and III-V materials is amplified when the mass ratio is small.
C
0
0
16
N
2
PA'
GaPbO
AISbO
GaPG
~
-
InAsO
-
3
V
GaAs
A0
InSbo
11
Pb~s
s
SnTe
PbTe*
0
z
0.2
0.4
0.6
0.8
1.0
Mass ratio
Figure 3-1 Normalized thermal conductivity of binary I1-V and IV-VI compounds at 300 K. The
solid lines are for a guide to the eyes.
In this chapter, we show that those seemingly different IV-VI, V2-V 3 , and V materials
commonly have long-range interactions along the <100> direction of rocksalt as a result of the
resonant bonding. Then, we infer that the significant long-range interaction in IV-VI materials
play a key role in their low thermal conductivity.
3.2. Resonant Bonding in IV-VI, V2-VI 3, and Element V Materials
Resonant bonding can be understood as resonance or hybridization between different
electronic configurations: three valence p-electrons alternate their occupancy of six available
covalent bonds that exist between a given atom and its octahedral neighbors [64]. For example,
53
in PbTe, the sp-hybridization is small and the s-band is lower than the p-band by 1.5 eV [65].
Therefore, we can consider only p-electrons for valence states and each atom has three valence
electrons on average. Given PbTe's octahedral structure and its three valence electrons per atom,
the choice of bond occupation is not unique. This leads to a hybridization between all the
possible choices of states for the three electrons forming the six bonds. This description for
resonant bonding that is presented here is based on
IV-VI compounds and their isoelectronic V
elements for simplicity, but the resonant bonding exists in even more complicated materials such
as V 2 -V
3
and many alloys of IV-VI and V2-V1
3
materials [66]. In general, the unsaturated
covalent bonding by p-electrons with rocksalt-like crystal structures can be regarded as resonant
bonding [67].
Materials
with
resonant bonding have several
features.
First,
because of their
,
coordination number of six, they have rocksalt-like crystal structures. Many group IV-VI, V 2 -VI 3
and V compounds have rocksalt-like crystal structures, as shown in Fig. 3-2.
54
PbTe
Bi
.05
3 .49A
1b
Bi Te2
(U
23
>:
>4
Figure 3-2 Rocksalt-like crystal structures of PbTe, Bi 2Te3, and Bi. The number on each atom
indicates the shell number. Bi 2Te3 , Bi and Sb have distorted rocksalt structures and have different
numbers for shells than the exact rocksalt case. The numbers on the Bi Te and Bi atoms indicate the
2
3
equivalent shell numbers as for a rocksalt structure in the absence of lattice distortion.
In addition, these materials have very weak sp-hybridization and the s-bands are well
below the p-bands. Several past studies show that PbTe, Bi Te , and Bi satisfy this
condition [65,
2
3
68, 69]. In order to confirm resonant bonding in those materials, we also calculated the electronic
band structure of rocksalt IV-VI, Bi 2Te 3, Bi, and Sb using density functional theory calculation
packages (Quantum Espresso and Abinit) [42, 70] with norm conserving Perdew-Zunger[71] and
Hartwigsen-Goedecker-Hutter[43] pseudopotentials. In particular, the spin-orbit interaction
is
included in the calculation because the heavy elements, such as Bi, Pb, Te, Sb, and Se, exhibit
strong spin-orbit interaction. The calculated electronic band structure and wavevector resolved
density-of-states are presented in Figs.3-3 to 3-9. From these figures, it can be clearly seen
that
55
the sp-hybridization in all of these materials is very weak, supporting the concept of resonant
bonding.
PbKe
Projected density-of-states
ib
4
2
0
-2
..-...-....-.
-.
P
band
-4
LU
-e
-8
band
-
-10
-12
14
Total Pb s Te s
Pb p Te p
k-resolved projected densit -of-states
C
5i
s-baid
r
xw
t.
r
KX
r
p-band
x w
t.
r
KX
Figure 3-3 Electronic band structure and projected density-of-states of PbTe showing weak sphybridization
56
Projected density-of-states
PbSe
4
4
band
-12
band
r
x
I
W
K X Total
r
s Ses Pbp Sep'
k-resolved projected density-of-states
C
C
p-band
r
x W
r
KX
r
x w
i
r
KX
Figure 3-4 Electronic band structure and projected density-of-states of PbSe showing weak sphybridization
Projected density-of-states
PbS
4
2'
0..................
..... J,
-2
}band
C
-10
}12band
-14
x
w
K
Total Pbs
Ss
Pbp
Sp
k-resolved projected densit -of-states
C
C
io10
nd
r
x w
p-band
i
r
KX
r
x w
L
r
Kx
Figure 3-5 Electronic band structure and projected density-of-states of PbS showing weak
sphybridization
57
Projected density-of-states
SnTe
b
4
2
0
a -2
band
.4
C
S
-10
.12
AAi
band
I
X W
L
K X Total Sns
V
Tes Snp Tep
k-resolved projected densiV r-of-states
v0
.5
C
0
U.J IC
p-band
V
x
W I
I
f,
x W
L
I-
K
X
Figure 3-6 Electronic band structure and projected density-of-states of SnTe showing weak sphybridization
58
rojected density-of-states
Bi 2Te3
.....
.......
b}
.......
-2
p
band
54:
w- 10-
S
band
-14
r z
UI
L total
I
F
Bi
Tel
Te2
S
P
k-resolved projected density-of-states
2C
-24
'A
U
r-
F
z
p-banc
I-Z
U
L
F
F
L
Figure 3-7 Electronic band structure and projected density-of-states of Bi2Te3 showing weak sphybridization
Projected density-of-states
Bi
4
2
0
-2
-4
band
8
u-
-10
band
-12
-14
W
T
L
Total Bus Bi2s Biip Si2p
k-resolved projected density-of-states
>
s-ba
K
p-band
I
T
W
L
K
F
W
Figure 3-8 Electronic band structure and projected density-of-states of Bi showing weak sphybridization
59
Projected density-of-states
Sb
4
b
-2p
band
-4
.6
band
r
K
L Total Sb1 s Sb2 s Sb1 p Sb2 p
W
T
k-resolved projecte densit -of-states
C
00
p-band
s-_
U
K
W
A
L
K
I
T
W
L
Figure 3-9 Electronic band structure and projected density-of-states of Sb showing weak sphybridization
One very important feature of resonant bonding is that the electron density distribution is
highly delocalized. As a result, the materials in this group have a large electronic polarizability,
large dielectric constants, and large Born effective charges [72, 73]. For example, the dielectric
constants of PbTe and Bi2Te 3(l) are 33 and 50, respectively, while for Si this value is 11.76 [7476]. The large electronic polarizability from resonant bonding could explain certain electronic
properties of thermoelectric and phase change materials [64, 67].
3.3. Long-range Interaction due to the Resonant Bonding
Along with the structural, electronic, and optical characteristics of resonant bonding
discussed in the previous section, here we discuss the lattice dynamic characteristics in the
resonant bonding materials.
60
We calculated the harmonic force constants of IV-VI, V2 -VI3, and V materials using firstprinciples density functional theory. We found that a common feature among these compounds is
the presence of long-range interactions along the <100> direction of the rocksalt structure. In
order to compare the long-range interactions of these different materials regardless of their
detailed crystal structures and bonding stiffness, we normalized the traces of their interatomic
force constant (IFC) tensors by the trace values of their self-interaction IFC tensors. The IFC
tensor is
i2E
a 2E
dRxdRx
aRxdRy
aRxaRz
RYORx
dRyORy
a 2E
dRyaRz
a2 E
(2E
a 2E
aRzaRX
3RZORy
a 2E
a2E
tR
a2E
1R j
=
(3.2)
aRzOiRzi
where E and Ra represent energy and atomic displacements along the a direction, respectively.
By taking the trace of their IFC tensors, we can assess the bonding stiffness regardless of their
crystal structure or coordinate system. Finally, we need to normalize the trace values of many
materials to compare their interaction ranges since different materials have slightly different
bonding stiffness ranges. The normalization is done by taking the trace value of their self-
interaction force constants:
a
normalized trace of IFC =
2
E
OxOR
(12E
a2_E
+
3RO,xaROx +
In the above expression,
a2
a2E
RO,yaRO y
a
+
2
E
2E
(3.3)
ORO,zaRO,7
represents the second-order force constant along the x-
direction between the origin atom (described as "0" in RO,x) and the n-th neighbor atom
(described as "n" in Rn,x). The
a2E
represents the self-interaction force constant along the x-
direction, which means the force constant of one specific atom when the atom itself is displaced.
61
By taking the trace values and normalizing them, we could compare the interatomic interaction
ranges of many materials with different crystal structures and different bonding stiffness.
The long-range interaction is particularly very significant in IV-VI materials. From Fig.
3-10a, fourth-nearest neighbors, separated by 6
A
(e.g. Pb-Pb and Te-Te), have interactions
which are comparable to those of first-nearest neighbors, spaced 3
A
apart, and are much
stronger than second- and third-nearest neighbor interactions. In addition, eighth-nearest
neighbors, separated by 9
A,
have even positive force constants, giving them the behavior of
"anti-springs". Fourteenth-nearest neighbors, separated by 12
A,
also have non-negligible force
constants. The force constants at the fourteenth-nearest neighbors are clearly distinguished from
other force constants nearby when using finer q mesh in the electron response calculation to
capture the long-range interaction more accurately. These first-, fourth-, eighth-, and fourteenthnearest neighbors form a chain along the <100> direction in rocksalt structures, as shown in Fig.
3-2. Other rocksalt IV-VI materials, such as PbSe, PbS, and SnTe, exhibit very similar behaviors.
These behaviors were not captured by earlier works on the lattice dynamics of PbTe and PbSe
within the shell model [77, 78]. To compare lead chalcogenides with other materials, in Fig. 310b we show that the IFCs of NaCl and InSb decrease with distance. NaCl and InSb are chosen
as prototypes of ionic and sp-hybridized covalent bonding materials, respectively. The longrange interactions along the <100> direction in NaCl are much smaller compared to those in
PbTe. In the case of InSb, interactions besides for first-nearest neighbor interactions are
negligible.
62
Q
0.0
a
b
a)
4-)
-0.1 -14t
P PbTe(Pb)
4hg0PbTe(Te)
I St
a)
IN
h bor
0P~(b
o
PbSe(Se)
0 PbS (Pb)
-02
o
o
NaCl (CI)
0 InSb (in)
o InSb (Sb)
* SnTe(Sn)
o SnTe(Te)
-0l3
0
0 NaCI (Na)
PbS(S)
---0
5
10
0
%I0
1
0.0
8th neighbor
8
-0.1
equivalent
4th nelghbor
equivalent
4th neighbor
equivalent
IN
-02
0
z
0 BI 2Te 3(rel)
o B 2Te3 (Te2)
o B 2Te, (Bi)
10
O Bi
0 Sb
0
5
10
distance (A)
Figure 3-10 Normalized trace of interatomic force constant tensors versus atomic distances. (a) lead
chalcogenides and SnTe (group IV-VI), (b) NaCl and InSb, (c) Bi 2 Te 3 (group V 2-VI), and (d) Bi and Sb
(group V). The element in the parenthesis indicates interaction between the corresponding atom and other
atoms. For example, 'PbTe(Pb)' means interaction between Pb and other atoms in PbTe.
We need to point out that the long-range interactions we observe are different from the
long-range Coulomb interaction, which cannot explain why fourth-nearest neighbor interactions
are stronger than second- or third-nearest neighbor interactions. The long-range and nonmonotonically decreasing interaction is due to the long-range electronic polarizability. The
second-order force constant can be expressed by using the Hellman-Feynman theorem [79, 80].
63
J
dn
dr+ nzdEnrn
n dVe-n
cIRIaRJJORJ dR1 f
fR1R
a2_E2
d
E=
-drV+
_
(3.4)
_
where, R and n are respectively the atomic position and electron density distribution as a
function of distance, r. Here Ven and E,_,
refer to potentials of the electron-nucleus and
nucleus-nucleus interactions, respectively. In the above expression, the second term on the right
hand is non-zero only for the self-interaction terms (I = j) and the third term decreases
monotonically with distance. Hence, the long-range and non-monotonically decreasing IFC with
distance along <100> cannot be attributed to the second and third terms. Since
V,V,/dR in the
first term is also decreasing with distance, the an/dR, electron distribution change due to the
atomic displacement, must be the only reason for the long-range interatomic interaction effect.
We confirm that the electron polarization is long-range in PbTe, but short-ranged in NaCl,
by calculating the electron density distributions in PbTe and NaCl. We carried out density
functional theory calculations for two different cases: without any displacement and with a small
displacement of one atom. The calculation results are shown in Fig. 3-11. We displaced a Pb
atom and a Na atom for PbTe and NaCl, respectively. The displacement is 2% of the fourthnearest neighbor distance in the -x direction in Fig. 3-11. Since a periodic boundary condition is
used in the electron distribution calculation, there is an effect from the periodic images of the
displaced atom. Therefore, we calculated large enough superclls (24 atoms) to minimize this
effect. After calculating the electron density of the two cases, we took a finite difference of these
two cases to calculate the change in the electron density distribution by the displacement.
In Figs. 3-1la and 3-11 b, we compare electron density distributions at the ground state of
PbTe and NaCI. From the ground state electron density distribution, it is clear that PbTe has a
largely delocalized electron density distribution due to resonant bonding, but electrons in NaCl
are highly localized due to its ionic bonding. In Fig. 3-11 e, the electron polarization in NaCl is
short-ranged and the electrons surrounding the fourth-nearest neighbors are not perturbed much.
However, electron polarization in PbTe in Fig. 3-11 f is long-range and reach fourth-nearest
neighbor. The electron density distribution surrounding the fourth-nearest neighbors is largely
disturbed by the displacement of the center atom.
64
NaCI
Electron density at ground state
a
e
density change
due to displacement
1,-
total
4 bneighbor
3
b
0
2
.
PbTe
total
02
0.5
023
*0.2
04
.03
C
0.2
s-band
0.1
0.0
d
I
Ii
0-
i
025
0.2
p-band
0.1'
GA
0.05
.
Figure 3-11 Electron density distribution and polarization in NaCI and PbTe. (a-d) the electron
density distribution at the ground state. (e-h) the electron density distribution change by a displacement of
the center atom. The plot is on the (100) plane and each black dot represents an atom. The unit is A- 3
The long-range polarization in PbTe can be explained by the resonant bonding. In
resonant bonding, if one atom is displaced along the +x direction, it perturbs the px orbital of the
adjacent atom. In other words, the bonding electrons on the -x side of the adjacent atom easily
move to the +x side since both sides are in the same px orbital[8 1]. This perturbation can persist
over long ranges due to the large electronic polarizability and the collinear bonding in resonant
bonding materials. The band-by-band electron polarization analysis in Fig. 3-11 confirms that the
long-range polarization is due to the resonantly occupied p-electrons. PbTe has very weak sphybridization and we plot the electronic polarization for s-band and p-band electrons separately
in Fig. 3-11 f-h. From Fig. 3-11g, the polarization of the s-electron is short-range and does not
reach the fourth-nearest neighbor. However, the p-electron distribution in Fig. 3-11 h exhibits the
long-range polarization. This analysis shows that the easily polarized electrons in PbTe are
65
resonantly occupied p-electrons, rather than the lone pair Pb s electrons suggested in recent work
[82].
The resonant bonding picture discussed above also applies to V 2-VI 3 (Bi 2Te 3) and V (Bi
and Sb) materials. Bi 2Te 3 has a rhombohedral structure which can be understood as a deformed
rocksalt structure with a layer spacing as shown in Fig. 3-2. This rocksalt-like structure contains
five resonantly bonded layers of atoms (Tel-Bi-Te2-Bi-Tel), and is separated from the next
quintuple layer by weak Tel-Tel van der Waals interaction. The structural deviation from the
exact rocksalt structure within the quintuple layer is small. The lengths of the strongest bond
(Tel-Bi), 3.03
A,
and that of the second-strongest bond (Bi-Te2), 3.22
A,
are similar. In addition,
the angles of Tel-Bi-Te2 and Bi-Te2-Bi are 174.60 and 1800, making them similar to rocksalt in
structure, since the rocksalt has an angle of 1800 exactly. Due to this small structural distortion,
the resonant bonding exists in a weakened form. As shown in Fig. 3-10c, the IFCs of Bi 2Te 3
show a behavior similar to that of lead chalcogenides, but the long-range interactions are
weakened due to the structural distortions, resulting in a weakened resonant bonding. Bi-Bi and
Tel-Te2, both spaced about 6
A
apart, have interactions which are equivalent to the fourth-
nearest neighbor interactions in an exact rocksalt structure, and are less significant compared to
the PbTe case. The interactions of Bi-Tel (at a distance of 9
A)
separated by Te2-Bi, which are
equivalent to the eighth-nearest neighbor interactions in PbTe, also have positive force constants,
but their magnitudes are smaller than those of the PbTe case. However, it is noticeable that the
force constants of Te2 atoms are very similar to those of the Te atom in PbTe, as predicted in
previous work[73]. This is because the resonant bonding around the Te2 atom is well maintained:
the Bi-Te2 and Te2-Bi bond lengths are same and they make an angle of 1800, as shown above.
Another noticeable observation in Bi 2Te 3 is that there is no long-range interaction between atoms
separated by Tel-Tel. It is well known that Tel-Tel has van der Waals type bonding due to the
induced dipole-dipole interaction[68], prohibiting the long-range interaction caused by the longrange electron polarization shown in Fig. 3-11.
Pure Bi and Sb have rhombohedral crystal structures which are Peierls distortions of the
simple cubic structure [83]. With the two basis atoms, the structure can be understood as a
rocksalt structure stretched along the <111> direction. Because of this distortion, the six first-
66
nearest neighbors in the rocksalt structure become the three first-nearest neighbors and the three
second-nearest neighbors. The distances between the first- and the second-nearest neighbors are
3.05 A and 3.49 A, respectively (See Fig. 3-2). Considering that the distances between the firstand the second-nearest neighbors are 3.03
A and
3.22
A,
respectively, in Bi 2Te3 , the distortion
from the rocksalt structure is much larger in Bi and Sb than in Bi 2Te 3. Therefore, the resonant
bonding is further weakened in Bi and Sb. This structural distortion further weakens the longrange interactions, as shown in Fig. 3-10d. However, the ninth-nearest neighbors, separated by 6
A,
have interactions which correspond to the fourth-nearest neighbor interactions in rocksalt and
which are thus expected to be significant, and are found to be significant in the calculation.
3.4. Strong Three-Phonon Scattering in IV-VI Materials
3.4.1. Large Anharmonicity of Ferroelectric Soft Phonon Modes
The long-range interaction along the <100> direction is related to the existence of a soft
transverse optical (TO) mode. This directional long-range interaction was also predicted and
considered as a main reason for the ferroelectric behavior in perovskite BaTiO 3 and PbTiO 3 [84].
We use a simple lattice dynamics theory to show that the long-range interactions along
<100> lead to the soft TO mode. The dynamical matrix in a diatomic crystal with basis atoms A
and B is
D(q)
=
OORe iq-R
pOR iq.R
#ieiqwhere each sub-matrix X
4OAeq-R
eiq-R
JpOR
1
eq-Rj
R
(3.5)
q-
is 3x3 matrix including x, y, and z direction. Here, the 0AA
denotes a second-order force constants between the basis atoms A with the distance of R. As q
67
approaches zone center, the phase term, eiq-R, becomes unity. Therefore, in the limit of q -> 0,
the dynamical matrix becomes a simple sum of force constants without the phase term:
D(q -> 0) =
(3.6)
O
In the above expression, the off-diagonal terms in each sub-matrix (OA",
and X
#/2),
#
0
,
OA,
which show dynamical coupling between the transverse directions (i.e., x-y,
y-z,
and x-z directions), become exactly zero due to the cubic symmetry. Then, the non-zero terms in
the dynamical matrix are only the diagonal terms in each sub-matrix and the most significant
contribution to the diagonal terms is from interatomic interactions along the <100> direction as
we show in Fig. 3-10.
As a result, the lattice dynamics in the resonant bonding materials can be approximated
with the lattice dynamics of a 1 D diatomic chain that contains the long-range interaction. Figure
3-12 shows a simple diatomic 1 D chain. The chain consists of two basis atoms with the equal
spacing. One atom is assumed to be twice heavier than the other atom. We cut-off the
interatomic interaction at the third-nearest neighbors and assume the interactions with atoms
beyond this range are negligible. Also we assume that the basis atoms A and B are identical in
terms of the force constants. Therefore, the force constants can be expressed as
First-nearest neighbor force constant: q1 = -a#
Second-nearest neighbor force constant:
02 =
-#0
Third-nearest neighbor force constant: 03 = -yo
(3.7)
(3.8)
(3.9)
where 0 is a constant and a, fl, and y represent the relative strength of each force constant. The
self-interaction term is decided by the acoustic sum rule,
Self-interaction force constant:
68
45
= 2(a +
fl + y)#
(3.10)
Basis atom A
Basis atom B
Figure 3-12 Diatomic ID chain. The numbers on the atoms indicate the shell number, with an increasing
neighbor distance with increasing number. The black circles denote A atoms and white circles denote B
atoms.
Using the force constants above, in the limit of zero wavevector, a dynamical matrix in Eq. (3.5)
can be written as
D(q -+ 0) =
201 +203
(3.11)
00 +2 2 2
In Fig. 3-13a, we plot the phonon dispersion of the ID diatomic chain with varying values for a,
fl, and y in Eqs. (3.7) to (3.10) to show the effects of the long-range interaction on optical
phonon dispersion. The self-interaction force constant is kept constant. The numbers in the
legend of Fig. 3-13a denote values of a, P, and y in Eqs. (3.7) to (3.10). In Fig. 3-13a, as the
second- and third-nearest neighbor interactions in the I D chain (equivalent to fourth- and eighthnearest neighbor interactions in rocksalt) increased, the zone center TO phonon frequency
decreased. When the long-range interaction is significant as represented by the case of '6/6/-2' in
the figure, the long-range interaction spring constants, q2 and
0
3
,
in Eq. (3.11) reduce the
magnitude of each term of the dynamical matrix and thereby softening TO phonon mode.
69
a
20
613
4
.... PbTe (LA)
~
E 15
o00O
~
e--SnTe
PbTe
-
d
C
b
Bi2Tel
13
C
Sb
:10
ci)
ID
14)
L
bse
-- PbS
chain model
2
1st/2nd/3rd
- 8/2/0
*
---
PbTe
5/5/0 1--5-PbSi
- /6/-2
0.5 0.0
0.0
C5-
---- PbSe
--
SnTej
-
Bi2Te
0
0.5 0.0
0.5 0.0
0.5
1
0
0.5
Figure 3-13 Near ferroelectric behavior due to resonant bonding. (a) Optical phonon dispersion in a
model 1 D atomic chain, showing the softening of the optical phonons due to the long-range interactions.
Three numbers in the legend represent relative interaction strength of first, second and third-nearest
neighbors in the ID chain. (b-d) Soft TO phonon modes along the trigonal direction for lead
chalcogenides, Bi2Te 3, and Bi and Sb, respectively, calculated based on first-principles. Lines and circles
are calculation and experimental data, respectively. The experimental data are from Ref. [48, 49, 73, 77,
78]. The red dotted line in b is after removing the fourth, eighth, fourteenth-nearest neighbor interactions
in PbTe, which do not show the soft TO mode. (b-d) are plotted on the same scale for the y-axis. (e)
Calculated GrUneisen parameters of TO mode, showing strongly anharmonic behavior of the TO phonons
of lead chalcogenides. The dotted line denotes the Gruneisen parameters of the LA mode in PbTe for
comparison.
The pronounced softening of the actual TO modes in lead chalcogenides, shown in Fig.
3-13b, is consistent with this ID model. To further confirm that the long-range interactions along
<100> are the main reason for the near ferroelectric behavior, the phonon dispersion is calculated
for a fictitious PbTe without the fourth- and eighth-nearest neighbor interactions. The TO mode
in the fictitious PbTe is not softened as shown in Fig. 3-13b. By comparison, Bi 2Te3, Bi, and Sb
have weakened long-range interactions due to distortion of the structure, and their TO modes are
not as soft as those of IV-VI materials (Figs. 3-13c and 3-13d).
The TO phonon softening leads to strong anharmonicity and phonon scattering by the TO
modes [85]. To show the anharmonicity of the modes, we plot in Fig. 3-13e the calculated mode
Griineisen parameters of TO modes in resonant bonding materials. The TO modes in lead
chalcogenides and SnTe have remarkably large mode Gruneisen parameters. Bi 2Te 3, Bi, and Sb
70
also have increasing Grflneisen parameters as the zone center is approached, but the magnitude is
not as large as in lead chalcogenides. The strongly anharmonic TO modes in IV-VI materials
lead to their low phonon thermal conductivity and this was predicted and experimentally
observed [85-89].
We further confirm using first principles calculations that the low thermal conductivity of
IV-VI is due to the strongly anharmonic TO modes. In Fig. 3-14, we show detailed phonon
transport characteristics in several Ill-V and IV-VI materials, calculated from first principles. We
calculate the phonon transport of SnTe and InSb in addition to PbTe, PbSe and GaAs from the
literature [36, 881. It is interesting to directly compare SnTe and InSb since these materials are
close to each other in the periodic table, and therefore they have similar Debye temperature and
mass ratio. The thermal conductivity calculation of InSb using first principles was reported by
other group [37]. The calculated and experimental thermal conductivity values in Fig. 3-14a
show large contrast between Ill-V and IV-VI materials. We further contrast the two different
material groups by analyzing phonon mean free path and phonon life time. For the phonon mean
free path, we present accumulated thermal conductivity as a function of phonon mean free path
at 300 K, defined in Eq. (2.20). The accumulated thermal conductivity function shows the mean
free path ranges of phonon modes that significantly contribute to thermal conductivity. From
Figs. 3-14b and 3-14c, it is clearly seen that the IV-VI materials exhibit much shorter mean free
path ranges and phonon lifetime compared to the Ill-V materials. The significant effect of soft
TO mode on phonon thermal conductivity is also substantiated by the comparison between PbTe
and Bi in Fig. 3-15. The thermal conductivity of PbTe is smaller than that of Bi by factor of two
as shown in Fig. 3-15b, even though the group velocity of acoustic phonons of PbTe is smaller
than that of Bi [30, 90] (Fig. 3-15a).
71
-
a
-PbTe
--
-O
100
10
1 SnTe
m
*PbSe
OInSb
*
*
*
0
0
C
b
GAs
100.
SnTe
PbTe
PbSe
InSb
GaAs
0A
10
0
10
0.2
200
(
300
0
o
0
1
-GaAs
0.0
10
-STe
*-.-PbTe
-PbS.
-
-
0
-
C 10-
5
400
61~
1000
100
10
1
Phonon frequency (THz)
Phonon mean free path(nm)
Temperature (K)
by first principles
Figure 3-14 Analysis of phonon transport in IV-VI and 111-V materials
and squares are results
calculation. (a) Calculated and experimental phonon thermal conductivity. Lines
and phonon
distributions
path
free
by experiments and calculations, respectively. (b-c) Phonon mean
The accumulated thermal
lifetime, showing significant three-phonon scattering in IV-VI materials.
corresponding material. The
conductivity in (b) is normalized by the thermal conductivity value of the
[62,
data in (b-c) are for the 300 K case. The experimental thermal conductivity values in (a) are from Ref.
88].
[36,
Ref.
from
are
90, 91] and other calculation results for PbTe, PbSe and GaAs
a
b
-PbTe
-B
Z 25
E
U
20
13
3
PbTe (exp)
PbTe (cal)
aBi (exp)
Bi (cal)
15
N
C
2
0
E
U-
(D
-5
0
L.
F~
X
50
100
150
200
250
300
Temperature (K)
resonant
Figure 3-15 Lower thermal conductivity of PbTe compared to Bi due to more significant
phonons
acoustic
of
velocity
group
smaller
bonding. (a) Comparison of phonon dispersions showing the
in Bi (b) Comparison of thermal conductivity showing the lower thermal conductivity of PbTe
72
3.4.2. Large Phase Space for Three-Phonon Scattering
Along with the phonon anharmonicity, the phonon lifetimes also depend on the threephonon scattering phase space available that meets energy and momentum conservation
requirements. The difference in the volume of the scattering phase space explains the second
difference between the
I1-V and IV-VI materials in Fig. 3-1 (i.e., the thermal conductivity
difference between III-V and IV-VI materials is much increased when the mass difference
between the two atoms is large). The phase space integral for three phonon scattering is the
volume satisfying energy and momentum conservation in the three phonon process. Therefore, it
can be defined as [92]
Phase space volume =
f(Ol +
dq
dq'
2
-
(03)
1
(3.12)
where 1, 2, and 3 represent phonon states defined in Chapter 2.2. The two 6-functions are for
energy conservation of the phonon coalescent and decay processes.
Fig. 3-16a compares the inverse of the three-phonon scattering phase space volume of
IV-VI and III-V materials. Assuming constant phonon mode anharmonicity, the inverse of the
three-phonon scattering phase space volume relates linearly to phonon lifetime and hence to the
thermal conductivity. We calculate the phase space volume as defined in Eq. (3.12). For the
integration of the 6-functions, we used a q-grid of 20x20x20 with the tetrahedron method which
is introduced in Chapter 2. As seen in the expression for the phase space volume in Eq. (3.12),
the calculation of phase space volume involves the integration of 6 -functions of phonon
frequencies. Therefore, the phase space volume is inversely proportional to phonon frequency
scale. To compare many materials with different phonon frequency scale, we normalized the
phase space volume by the inverse of the maximum optical phonon frequency.
We can see from Fig. 3-16a that III-V materials with dissimilar atomic masses such as
AlSb and InP have much smaller phase spaces than other III-V materials. This is a common
73
behavior for many materials which have large atomic mass differences[931. The large mass
difference causes a large gap to appear between the acoustic and optical phonon bands as shown
in Fig. 3-16b. With such a large gap, low frequency acoustic phonons cannot couple to optical
phonons and thus have fewer chances to be scattered. Only high frequency acoustic phonons,
which are limited in a small region of reciprocal space, can be coupled with optical phonons. As
the mass ratio is reduced from unity, the acoustic-optical phonon gap becomes larger and the
scattering phase space is further reduced, leading to longer lifetimes and a larger thermal
conductivity. As can be seen from Fig. 3-16c, the phase space in AISb is mostly due to (a,a,a)
and the phase spaces of (a,a,o) and (a,o,o) are significantly suppressed. ('a' and 'o' in (a,a,o)
indicate acoustic and optical phonons in the three phonon process.)
Q
-PbS
--- AIS
05aA
45s,
CL
fcM
CL
0
AISb
r-PbS
0.4Cu
M
E2 Q.
0
0.3 -
-C/
-C a
GaP
GaSb
(DG
0.C
In
Sn~..N/e&
PbTe/
m~
0.0
2:
0.2
0
0.4
0.6
0
0
~
0.8
1.0
Z
K
X
F
L
aaa aao aoo oo6 total
mass ratio
Figure 3-16 Phase space volume for three-phonon scattering. (a) Phase space volumes for three
phonon scattering of IV-VI and III-V, showing a large scattering phase space for PbSe and PbS. The solid
line is for a guide to the eyes. (b) Comparison of the phonon dispersion of PbS and AlSb, showing
significantly dispersed optical phonons of PbS. (c) Contribution of each scattering process to total
scattering phase space volume. The scattering phase space and phonon dispersion data are normalized by
the inverse of the largest optical phonon frequency of each material for comparison.
The three-phonon scattering pathway in IV-VI materials is much different from that of
III-V materials since (ao,o) channel significantly contribute to the total phase space volume in
IV-VI materials. The difference in the scattering pathway of Il1-V and IV-VI materials can be
74
easily observed in Fig. 3-16a; the phase space volume of IV-VI materials increases slightly as the
mass ratio decreases, an opposite trend to the case of 111-V materials. The reason for this opposite
trend is that (ao,o) channel is a large contributor to the total phase space volume in IV-VI
materials, and this scattering channel is affected by the overlap between acoustic and optical
bands. The significant (a,o,o) channel in IV-VI materials is due to the soft TO modes spanning a
large band width, and lead to the large phase space volume of PbS and PbSe despite of their
large mass contrast between Pb and S (or Se). As a result, PbS and PbSe have much larger phase
space volumes than other Ill-V materials such as AlSb and InP, which have similar mass ratios
as PbSe and PbS. For PbS, the phase space volume of (a,a,a) scattering is similar to the AlSb
case, but (a,o,o) and (a,a,o) scattering channels have markedly larger phase space volumes. The
large phase space volume of (a,o,o) process can be explained with the wide spectrum of optical
modes. The bandwidth of the optical phonons is comparable to the band width of the acoustic
phonons. As a result, most acoustic phonons from very low frequencies to high frequencies can
participate in the (a,o,o) process, widening (a,o,o) channel in IV-VI materials. In particular, the
(a,o,o) processes in PbS contribute more than half of the total phase space from Fig. 3-16c. The
wide (a,a,o) process channel is due to the reduced phonon band gap by soft TO mode. With the
reduced phonon band gap, most acoustic phonons, regardless of frequency, can be scattered by a
TO mode contributing to the (a,a,o) process, while only high frequency acoustic phonons can
participate in the (a,ao) process when the gap is large. The large (ao,o) and (a,ao) scattering
phase space of PbSe and PbS suggests that acoustic phonons are effectively scattered by optical
phonons, exhibiting low thermal conductivity values despite of the small mass ratio as shown in
Fig. 3-1.
3.5.Conclusion
We have presented the effects of resonant bonding on the lattice dynamics characteristics
and thermal conductivity. We revealed that materials with resonant bonding (lead chalcogenides,
SnTe, Bi 2Te 3, Bi and Sb) commonly have long-range interactions along the <100> direction in
75
rocksalt structure. This long-range interaction is significant in
LV-VI materials due to the strong
resonant bonding, and results in the near ferroelectric instability in these materials. However, the
long-range interaction is less significant with increasing distortion of the crystal structures as in
Bi. The near ferroelectric behavior caused by the resonant bonding reduces the phonon thermal
conductivity through two mechanisms: strong anharmonic scattering and a large scattering phase
space volume, both due to softened optical phonons, resulting in the lower thermal conductivity
of IV-VI materials compared to III-V compounds. Therefore, the low thermal conductivity of
these materials is traced back to their crystal structure and electronic occupation (the resonant
nature of the bonding). Our findings have significant implications
for designing better
thermoelectric and phase change materials. The fundamental understanding of lattice dynamics
from chemical bonding points to the potential to search for good thermoelectric materials
through the resonant bonding picture. Also, we showed a deep connection among ferroelectric,
thermoelectric and phase change materials. These insights help researchers to explore better
thermoelectric and phase-change materials.
76
4. Experimental Characterization of Electron
Filtering Effect in Nanocomposite Bi2 Te2 .7 Seo.3
So far, we have discussed phonon transport in bulk three-dimensional thermoelectric
materials. In this chapter, we study electron transport in thermoelectric materials with twodimensional discontinuities, such as grain boundaries. We experimentally characterize the
electron transport across grain boundaries by measuring the various transport coefficients of
electrons. The electron transport across the grain boundaries in a Bi 2Te 2.7Seo. 3 nanocomposite
sample is characterized by the method of four coefficients. The analysis of measured transport
coefficients show that the grain boundaries preferentially scatter the electrons with low energy,
leading to the electron filtering effect which can significantly increase the Seebeck coefficient.
4.1. Background
The introduction of nanostructures in thermoelectric materials has led to significant
improvements of the thermoelectric figure-of-merit over the past two decades [6, 7]. These
improvements are primarily due to the reduced thermal conductivity. The nanostructures provide
many discontinuities in the lattice, such as grain boundaries that strongly scatter acoustic
phonons and thereby reduce thermal conductivity.
The grain boundaries also can alter electron transport and possibly provide a way to
increase the thermoelectric power factor. At the grain boundaries, there are many crystal defects
such as dangling bonds, correspondingly causing surface states as shown in Fig. 4-1. If the
surface state energy level is lower than the Fermi level, electrons are trapped in these surface
77
states and a space charge region occurs. These trapped charges bend the conduction band,
forming an electrical potential barrier.
e------------
states
~surface
EC
- - - - - - -
EF
EV
an n-type
Figure 4-1. A schematic picture of a potential barrier at a grain boundary in
semiconductor. Ec, EF, and Ey represent a conduction band edge, Fermi level, and a valence band edge,
respectively.
This potential barrier can increase the Seebeck coefficient by scattering low energy
4-1. This
electrons more strongly than high energy electrons as schematically shown in Fig.
effect is called the electron filtering effect [94]. This preferential scattering of electrons with
respect to the electron energy can increase the Seebeck coefficient because the physical meaning
carried
of the Seebeck coefficient is an average of the entropy (or energy divided temperature)
the
by electrons, as will be discussed in the following section. The more strongly scattered are
electrons with low energy compared to the electrons with high energy, the larger is the average
of the entropy that electrons carry, finally leading to the increase of the Seebeck coefficient.
However, it should be noted that the potential barrier can also decrease the electrical
an
conductivity by increasing the overall electron scattering rates. Therefore, in order to increase
overall thermoelectric power factor, the potential barrier should be carefully engineered such that
the increase of the Seebeck coefficient is large enough to compensate for the decrease of the
electron
electrical conductivity. Developing such a potential barrier requires characterizing the
78
transport in detail, including the energy dependence of electron transport. In this chapter, we
provide an empirical estimation of several important electron transport parameters, such as the
electron mobility, density-of-state effective mass, Fermi level, and the energy dependence of the
electron scattering rates, in a Bi 2Te 2 7 Seo. nanocomposite sample. For this characterization, we
use the method of four coefficients, which is briefly explained in the following section.
4.2.The Method of Four Coefficients
The method of four coefficients was developed to roughly estimate those transport
parameters that cannot be directly measured [95]. This method was used for thermoelectric
materials [16, 96] and also for other semiconducting materials [97, 98]. In this method, we first
measure the four transport coefficients (electrical conductivity, Seebeck coefficient, Hall
coefficient, and Nernst coefficient). The four transport parameters (electron mobility, density-ofstates effective mass, Fermi level, and the energy dependence of scattering rates) are estimated
by fitting the measured four coefficients.
We briefly introduce the assumptions and models that are used for fitting the four
transport parameters. We assume that transport of electrons is semi-classical, which means that
electrons behave like particles with an effective mass. In many semiconductors with a wide band
gap, a carrier pocket can be described with a single value of the effective mass because the
electronic band structure is almost parabolic:
E(k) =
h21k1 2
2
2m*
(4.1)
where E(k) is a relative energy level of an electron with wavevector, k, from the band edge. In
Eq. (4.1), we assumed an isotropic band structure, and the effective mass is represented by m*.
79
However, most of the good thermoelectric materials, in particular Bi2Te3..xSex that we study in
this chapter, have a very narrow band gap of around 0.2 eV. Because of the narrow band gap, the
electron and hole pockets in these materials cannot be described by a simple parabolic band
structure. In order to describe the non-parabolicity of those carrier pockets, we use the Kane's
non-parabolic model [99]:
c~k)
=
=
E(k) = E(k)(1 + aeE(k))
h 21k1 2
(42
2m*
(4.2)
where ae is an inverse of the band gap.
Under the assumption of semi-classical transport, we use the Boltzmann transport
equation to describe the transport of an electron:
Vks
(
LV
Nk)kN,
(VT
aN s
dk
y Vt kscatt
=
(4.3)
where Nks is the distribution function of the electron with wavevector, k, and band number, s. It
is noteworthy that the Boltzmann transport equation for electrons (Eq. (4.3)) has one additional
term, compared to the Peierls-Boltzmann transport equation for phonons (Eq. (2.10)). The
additional term, (dk/dt) - VkNks, describes a driving force from an external electric or magnetic
field. The scattering term of the Boltzmann transport equation can be simplified by the relaxation
time approximation:
v
- VT ( T
+-
dt
VNks k --
90
(4.4)
Tks
44
where N' is the Fermi-Dirac distribution, and
Tr
is a relaxation time. The relaxation time can
be approximated with a simple power law with respect to the electron energy,
rk
= TOZr
(4.5)
where TO is a numerical constant representing the overall strength of electron scattering, and z is
the electron energy in a dimensionless form, defined as E(k)/kBT. The scattering exponent, r,
in Eq. (4.5) represents the energy dependence of the electron scattering rate. If the scattering
exponent is positive, the electrons with low energy are more strongly scattered than the electrons
with high energy. From the Boltzmann transport equation with a simple scattering model in Eq.
(4.5) and using the Kane's non-parabolic band model in Eq. (4.2), the mathematical expressions
for the four transport coefficients (electrical conductivity, Seebeck coefficient, Hall coefficient,
and Nermst coefficient) can be derived. The details of derivation can be found in the literature
[100, 101].
Below, we present the physical meaning of and the mathematical expressions for the four
transport coefficients. The electrical conductivity, Ue, is expressed as
e
= neepe
where ne, e, and Me denote the carrier density, unit charge, and electron mobility,
respectively.
(4.6)
The Seebeck effect occurs when a temperature gradient exists in electrically conducting
materials, as shown in Fig. 4-2. In this figure, the hot side electrons have a relatively large
kinetic energy compared to the cold side electrons, resulting in the accumulation of charge at the
cold side. Here we assume that the material is n-type. This charge accumulation by the diffusion
process is balanced with the electrostatic field resulting from the charge accumulation. We can
then define the Seebeck coefficient, S, as the resulting electrostatic field per unit temperature
gradient:
81
S = E
(4.7)
VaT
where Ea is electric field along the direction a. In some thermoelectric materials with highly
anisotropic electron transport features, the Seebeck coefficient tensor has large values for its offdiagonal components. However, in nanocomposite materials, many nanograins are randomly
oriented and thus the off-diagonal terms are canceled out. Applying the definition of the Seebeck
coefficient into the Boltzmann transport equation gives the expression for the Seebeck
coefficient:
k
S = k (
/'1r
e 312,1
EF
-2 EF
(4.8)
kBT
where EF denotes Fermi level and I!7,k is the Fermi-Dirac integral for a non-parabolic band,
defined as
N
I
~k
f =z
fZ
dz
z'(z+aez2)n
(4.9)
(1 + 2a,. Z)k
The term, IJg+T1//2+1, in Eq. (4.8) clearly shows that the Seebeck coefficient means an average
of entropy that electrons carry.
82
I
(
Thermal Gradient
Electrochemical Potential Gradient
Figure 4-2. A schematic picture of the Seebeck effect.
The Hall coefficient describes the charge accumulation along the transverse direction
when charge carriers flow under an external magnetic field. In Fig. 4-3, we schematically show
the Hall effect. The charge flow is deflected by the Lorentz force, and charge carriers are
accumulated at one side. The charge accumulation driven by the Lorentz force is balanced with
the electrostatic force resulting from the charge accumulation. The Hall coefficient, H, is defined
as this electrostatic field per unit magnetic field E,/B and the charge current density jp:
_Ea
H = ---
where
(4.10)
j# is charge current density along the # direction and B is an external magnetic field. It
should be noted that the Hall coefficient is defined under an isothermal condition; this isothermal
condition avoids any thermoelectric effect in the Hall coefficient. The Boltzmann transport
equation can give an expression for the Hall coefficient as follows [100].
H =
3/2,2 3/2,0
nee
83
Ir/2
(4.11)
It is noteworthy that the expression for the Hall coefficient in Eq. (4.11) is slightly different from
a well-known expression,
1
(4.12)
ne e
The well-known expression in Eq. (4.12) is based on the Drude model of electron transport in
which the electron's relaxation time is assumed to be a constant and the relaxation time does not
depend on electron energy. The Fermi-Integral term in Eq. (4.11), which is a difference of Eq.
(4.11) from Eq. (4.12), includes the effect of the energy dependent scattering rate.
electron
electron
Figure 4-3. A schematic picture of the Hail effect
The Nernst effect is similar to the Hall effect described above. In the Nernst effect,
however, the charge flow is driven by a thermal gradient, not by an electrostatic force. Figure 4-4
schematically describes the Nernst effect. First, when the electron scattering rate does not depend
on the electron energy (r = 0), the hot side electrons and the cold side electrons have the same
electron mean free path. Since the deflection amount of charge flow is roughly proportional to
the square of electron mean free path, both hot and cold electrons have the same amount of
84
deflection in their trajectory, resulting in no charge accumulation along the transverse direction.
However, when the electrons with large energy (or from the hot side) have a longer relaxation
time (r > 0), the hot side electrons experience larger deflection in their trajectory, leading to the
charge accumulation along the transverse direction. In the opposite case of r < 0, the electric
field along the transverse direction is opposite the case of r > 0, as shown in Fig. 4-4. Therefore,
roughly speaking, the transverse electric field direction in the Nernst effect indicates whether the
scattering exponent is positive or negative.
The Nernst coefficient, Ne, is defined as the electric field per unit temperature gradient
and magnetic field:
Ne =
a
VflTB
(4.13)
As in the definition of the Hall coefficient, the Nernst coefficient also assumes isothermal
condition along the transverse direction to exclude any thermoelectric effect. Applying the
Boltzmann transport theory to the definition of Nernst coefficient gives an expression:
11+2r
N = H kB
3/2,Z
e I-
85
2
1l+r
3/2L
'3/2,1)
(4.14)
when r = 0
when r > 0
0B
when r < 0
0B
Figure 4-4. Schematic pictures of Nernst effect depending on the energy dependence of electron
scattering rates. Note that there is no transverse electric field when r = 0; the hot electrons are
preferentially deflected upward when r > 0 and the cold electrons tend to go downward when r <0.
4.3. Experimental Setup
We use the method of four coefficients
to characterize electron transport in a
nanocomposite thermoelectric material. A nanocomposite sample of n-type Bi 2Te 2 .7 Seo. 3 was
prepared by Dr. Weishu Liu in Prof. Zhifeng Ren's group at Boston College (now at the
University of Houston). For the sample preparation, the bulk material is ball-milled into
nanoparticles and then the nanoparticles are hot-pressed into a pellet-type bulk sample [7]. The
pellet is diced with a diamond saw to prepare a rectangular shape sample with the size of Imm x
86
1mm x 6mm. The large aspect ratio of the sample was intended for the accurate measurements of
the Hall and the Nernst coefficients. When the aspect ratio is less than 4, the Hall and the Nernst
coefficients can be underestimated due to the edge effect [102].
Several wires are attached to the sample for the measurements as shown in Fig. 4-5a.
Two sets of type-T thermocouple wires are attached on one side of the sample using silver epoxy
paste. On the other side, two platinum wires are attached using a spark-welding method at the
same height as the thermocouple wires. On the top and the bottom surfaces of the sample, two
copper wires are attached using silver epoxy paste. All these wires are fine gauge wires with a
diameter of less than 25 gm. These fine gauge wires have a relatively large thermal resistance
and provide nearly thermal adiabatic condition to the sample. In addition, a miniature electric
heater was attached on the top side using silver epoxy paste, and the sample-heater assembly is
attached on a ceramic plate using silver epoxy paste. This ceramic plate serves as a heat sink.
The prepared sample assembly is then mounted on the cold finger of a cryostat using thermally
conductive epoxy paste to ensure good thermal contact between the ceramic plate and the cold
finger (Fig. 4-6). The cryostat used for this experiment was an ST-300 cryostat by Janis and is
equipped with two thermal radiation shields for thermal insulation. The cryostat including the
sample assembly is placed between two electromagnets. A turbo vacuum pump (Agilent TPS81V) is used to provide a vacuum condition in the cryostat. The temperature of the cold finger in
the cryostat is controlled from 77 K to 400 K with liquid nitrogen and a pre-installed electric
heater.
Using these wires and the miniature heater that we installed on the sample, we can
measure the four transport coefficients. For the measurement of electrical conductivity, we use a
four-point probe method. We apply an electric current using the copper wires and we measure
the voltage difference between the two platinum wires. The applied current is AC to avoid any
error from thermoelectric effects. The distance between the two platinum wires is precisely
measured by using an optical microscope with a reticle. For the measurement of the Seebeck
coefficient, thermal gradient is applied by the miniature heater, and the temperature difference
between the two thermocouple wires is measured. The Seebeck voltage is also measured using
the copper wires of the type-T thermocouples. For the measurement of the Hall coefficient, we
apply an electric current using the copper wires and we measure a voltage difference between
87
For the
one of the platinum wires and one copper wire of the type-T thermocouple wires.
heater.
measurement of the Nernst coefficient, we apply a thermal gradient using the miniature
the Hall
Then, we measure a voltage difference along the transverse direction as we do for
coefficient.
b
a
Current
Magnetkc
Raddation
Shield
Sample
Thermocouple
meter
Voltage Probe
Magn et
TE
Isothermr
Magnet
Cryostat
Radiation Shield
at
Cold finger
Figure 4-5. A sample with various probe wires and the configuration of the measurement setup.
88
Figure 4-6. A prepared sample assembly on the cold finger, showing the heater location and the
ceramic plate.
4.4. Results and Discussions
In Fig. 4-7, we present the measurement data of the four transport coefficients. The
electrical resistivity values (Fig. 4-7a), an inverse of electrical conductivity, increases with
temperature due to the stronger scattering of electrons by phonons as the density of phonons
increases. The Seebeck coefficients also increase with temperature almost linearly up to ~ 200 K,
but start to saturate above 300 K and have a peak around 350 K. The saturation of the Seebeck
coefficients is due to the bipolar effect. As the temperature increases further, minority carriers,
holes in this case, become more populated. Since the transport of holes contribute to the Seebeck
coefficient with an opposite sign to the electron contribution, the overall Seebeck coefficient
decreases. The expressions for the four coefficients in Eqs. (4.6) to (4.14) do not include this
bipolar effect, and thus our analysis of the experimental data is limited to the range below 300 K.
89
b
a
2 6x10
4
A A
.1
0x10
OX10,
IF2
e-0
01
5X10
4
'v
.2 6x10'
100
200
300
100
400
Ton parature (K)
d
1O
C
300
200
Tomperature (K)
400
300
400
20x0
00
--2
OxIO
2 0x10
0%
-4
0x10
-6
0x10
8
A OI
100
200
200
100
TemperatUre (K)
200
Tem perature (K)
Figure 4-7. Measurement data of the four transport coefficients. (a) electrical resistivity, (b) Seebeck
coefficient, (c) Hall coefficient, and (d) Nemst coefficient
We fit the four transport parameters to the measured four transport coefficients using the
expressions in Eqs. (4.6), (4.8), (4.11), (4.14). In Fig. 4-8, we present values for Fermi level with
respect to temperature. The Fermi level in the figure is measured relative to the conduction band
.
edge. Figure 4-8 shows that Fermi level is located above the conduction band edge, confirming
3
19
that our sample is degenerately doped. The corresponding carrier density is around 2x10 cm~
We also plot the Fermi level of a similar material, Bi2Te2 .85 Seo.1 5, but with a slightly smaller
doping concentration, Ix1019 cm-3, from the literature [103]. Since our sample has a larger
carrier concentration, the Fermi level of our sample is located deeper into the conduction band
than the Fermi level from the literature [103]. Another observation is that Fermi level decreases
as temperature increases. This is a common behavior of most semiconducting materials. As the
90
temperature increases, the minority carriers, holes in this case, are thermally activated, and the
Fermi level moves towards the valence band.
)
Nanocomposite Bi 2Te 2 7Se0 3 (n=2x1O cm
0.020
0
0
Single Crystal Bi 2Te2.85 e0.1s (n=x10 9 cm 3 )
(by Kaibe et al.)
-0.02
50
-
100
-
-
150
0
0
I
200
250
.
0.00
-
%ft.0
300
Temperature (K)
Figure 4-8. Fermi level from the method of four coefficients. The black points are from Ref. [103]
for
comparison.
Shown in Fig. 4-9 is the density-of-states effective mass from the method of four
coefficients. The estimated density-of-states effective mass is 0.6mo to 0.8mO
depending on
temperature, where mo is the physical mass of a free electron (Mo-9. Ix 10-31 kg).
The estimated
effective mass is much larger than 0.27mo from other literature values [104]. This
discrepancy
can be explained by the second heavy band that was previously predicted for
Bi2Te 3 [105, 106].
In Bi 2 Te3 , the second band with a heavy effective mass exists slightly above the first band
with a
light effective mass. The energy difference between the edges of the first and second
bands is
estimated to be only 10 to 30 meV, as schematically shown in the inset of Fig.
4-9 [105]. This
second heavy band effect was not included in the literature reporting the small
density-of-states
effective mass because the samples used in the literature are lightly doped [104].
However, in
91
our case, the sample is highly doped and the second heavy band can be activated. As a result, the
estimated density-of-states effective mass from the transport coefficients can be much larger than
that of the first light conduction band.
1.0
I
I
nanocomposite Bi.Te Se
0.8 I4D
i
i
0.6
0
E(k)
EI
0.4
-/
IO.O1-O.03eV
0.2
m'=027m,
0.01
)
5
I
I
I
100
150
200
250
300
Temperature (K)
Figure 4-9. Density-of-states effective mass from the method of four coefficients. The blue line is for
eye-guide. The inset schematically shows the first light carrer pocket and the second heavy carrier pocket.
3
The density-of-states effective mass is in unit of mo, physical mass of a free electron (m6 =9. IIx 10~ kg).
We plot electron mobility data in Fig. 4-10. The mobility decreases as the temperature
increases due to the increasing scattering by phonons. We also plot electron mobility data for a
relatively lightly doped polycrystalline Bi 2 Te 2 .7 SeO.3 sample from the literature [107]. Our
mobility data is much smaller than the literature data. The much smaller electron mobility in our
case is probably because 1) our overall effective mass is larger due to the second heavy band, 2)
more dopants and/or grain boundaries strongly scatter electrons.
92
1
)
Polycrystal Bi 2Te2.7 Se0.3 (n=4x10' cm3
.1
E
0
0.1 --
Nanocomposite Bi2Te2.7 Se0. 3 (n=2x10
0.01
50
100
150
I
200
cm)
250
300
Temperature (K)
Figure 4-10. Electron mobility from the method of four coefficients. The black line is from Ref. [107]
for comparison.
The energy dependence of the electron scattering rate is plotted in Fig. 4-11. In this figure,
the y-axis shows the scattering exponent, r, in Eq. (4.5). The scattering exponent in bulk
materials, where the scattering by phonons dominates over other scattering mechanisms, is
theoretically estimated to be -0.5 [100].
This theoretical estimation is confirmed by the
experimental data for single crystalline and polycrystalline samples in which the scattering by
phonons is dominant [103, 107]. However, our Bi 2 Te 2 .7 Seo 3 nanocomposite sample exhibits
much larger scattering exponents than those of bulk materials. The scattering exponent of the
nanocomposite sample is positive, indicating that the electrons with high energy are less
scattered than the electrons with low energy. This positive scattering exponent might lead to a
higher Seebeck coefficient through the electron filtering effect. The large scattering exponent in
the nanocomposite sample is probably due to the scattering by grain boundaries.
93
0.5-
4)
I
I
I
I
I
Nanocomposite Bi 2Te2 7Se 03
Ce
o 0.0
C.
X
Bulk Polycrystal Bi2Te
Se
(by Kutasov)
-0.5
---1.0
(by Kaibe)
- .
-I
50
100
Scattering
by Phonon
Bi 2Te2.8 S eO1 s Single Crystal
0-~~~
U)
-
---
0
0
200
150
I
250
-
I
300
Temperature (K)
Figure 4-11. Scattering exponent representing the energy dependence of the electron scattering
rates from the method of four coefficients. The black line and the circles are from Refs. [ 103, 107] for
comparison. The brown
line represents the case where the scattering by phonons is predominant over
other scattering mechanisms.
In conclusion, we use the method of four coefficients to estimate the four important
parameters regarding electron transport, such as the Fermi level, the density-of-states effective
mass, electron mobility, and the energy dependence of electron scattering rates, in a Bi2 Te 2.7 Seo3
nanocomposite sample. These four transport parameters provide the details of electron transport,
particularly the energy dependence of electron scattering rate. Our nanocomposite sample
exhibits much larger scattering exponents than other bulk samples from the literature, probably
due to the scattering by many grain boundaries in the nanocomposite sample.
94
5. Hydrodynamic Phonon Transport in Suspended
Graphene
In many materials like thermoelectric materials we have discussed in the previous
chapters, scattering of phonons usually cause resistance of phonon transport. This resistive nature
of phonon scattering is reflected in Fourier's law which describes diffusive heat flow at a given
temperature gradient. In this chapter, we discuss hydrodynamic phonon transport where phonon
scattering does not directly cause thermal resistance and thus the Fourier's law is not valid. We
show using the first principles calculations that hydrodynamic phonon transport can be
significant in a two-dimensional material, particularly in suspended graphene. The first
principles calculations demonstrate the hydrodynamic phonon transport in suspended graphene
through drift motion of phonons, phonon Poiscuille flow, and second sound. The significant
hydrodynamic phonon transport in suspended graphene is associated with graphene's twodimensional features as well as its high Debye temperature. The hydrodynamic features of
phonon transport that are not possible within the diffusive Fourier's law provide a new degreeof-freedom in manipulating heat flow.
5.1. Background
The transport of phonons is usually diffusive and describable by Fourier's law of heat
conduction.
Regimes where
Fourier's
law breaks down,
such as ballistic[108]
and
hydrodynamic[109] phonon transport, were discovered in bulk materials more than 50 years ago,
but these phenomena were observed only at extremely low temperatures[l 10-112]. Recent
studies of low-dimensional materials, however, have highlighted the practical importance of
95
ballistic phonon transport in applications such as thermoelectric materials[5] and electronic
devices[ 113, 114]. In this chapter, we discuss how hydrodynamic phonon transport, as well as
ballistic phonon transport, can be significant in a two-dimensional material, particularly in
graphene.
The term hydrodynamic phonon transport arose from its similarity with macroscopic
transport phenomena in fluids. In fluid flow, mass transport is mainly due to the macroscopic
motion of molecules with a drift velocity. Likewise, phonons in the hydrodynamic regime
exhibit macroscopic drift motion. In this sense, hydrodynamic phonon transport is different from
the more well-known diffusive or ballistic phonon transport. During diffusive transport, heat is
transferred through multiple scattering events among phonons without macroscopic drift motion.
During ballistic transport, it is assumed that there is no internal scattering. Hydrodynamic
transport, on the other hand, includes many phonon scattering events. The drift motion of
phonons in the hydrodynamic regime causes two interesting hydrodynamic transport phenomena
that cannot occur in either diffusive or ballistic regimes: phonon Poiseuille flow (Fig. 5-1 a) and
second sound (Fig. 5-ic), which are analogous to Poiseuille flow and ordinary sound in a fluid,
respectively, which will be discussed later.
96
ab
h drod namic
diffusive
N
N
N
N
C
d
hydrodynamic
diffusive
Figure 5-1. Different macroscopic transport phenomena in the hydrodynamic and diffusive regimes.
(a-b) The steady state heat flux profiles in hydrodynamic and diffusive regimes, respectively, under a
temperature gradient. (c-d) The propagation of a heat pulse in the hydrodynamic and diffusive regimes,
respectively. The width and length of the sample are assumed to be much larger than the phonon mean
free path.
Despite the interesting features of hydrodynamic phonon transport, the temperature range
where it was observed was too low and narrow to consider for practical applications. For
example, the reported temperature range for phonon Poiseuille flow is 0.5 to 1.0 K [112] and for
second sound the range is 10 to 20 K [115]. The extremely stringent temperature conditions for
hydrodynamic phonon transport are due to U-scattering, which destroys crystal momentum. In
contrast, scatterings between molecules conserve total momentum. As such, for hydrodynamic
transport to occur, U-scattering should be negligibly weak compared to the other three-phonon
scattering processes which conserves crystal momentum, N-scattering. One way to suppress Uscattering is to consider low temperatures, but at too low temperature transport becomes ballistic
without internal scattering, leaving only a very narrow temperature range for hydrodynamic
transport. In addition to U-scattering, the scattering of phonons by impurities such as isotopes
97
does not conserve crystal momentum, and isotope enrichment imposes another difficulty for
hydrodynamic
phonon
transport
(hereafter
R-scattering
denotes
Umklapp
and
isotope
scatterings).
In this chapter, we show that suspended graphene (hereafter graphene), unlike threedimensional materials, is remarkably well-suited for hydrodynamic phonon transport. Using first
principles calculations, we show drift motion of phonons, phonon Poiseuille flow, and second
sound in graphene at significantly higher and wider temperature ranges compared to those seen
in three-dimensional materials. Then, we discuss how the significant hydrodynamic phonon
transport in graphene stems from its two-dimensional features.
5.2. Drift Motion of Phonons
The most prominent feature of hydrodynamic transport is a drift motion of particles. The
molecules in fluid flow move along seemingly random directions, but all those molecules in a
small finite volume have a same average velocity regardless of their amount of momentum and
moving direction. This average velocity is called a drift velocity and represents an actual flow
velocity.
The macroscopic drift motion can be found in the distribution function. For example, for
molecules with a drift velocity, u, the displaced Maxwell distribution is considered an
equilibrium one.
(r
Nd =
(3/ - )2
1
exp
k27mkBT
-
27rmkB T
(5.1)
where m, T, and v are the mass, temperature, and velocity, respectively, of a molecule. Similarly,
if there is no R-scattering, the displaced Bose-Einstein distribution is usually assumed as the
equilibrium distribution for phonons with a drift velocity u [116].
98
q
(5.2)
BE
1
exp h(o-
u))
1
where h, w, and q represent the Planck constant, phonon frequency, and phonon wavevector,
respectively. The drift velocity is represented by the displacement, u, in the phonon distribution.
In the displaced distribution, the displacement is a constant for all phonon modes regardless of
polarization and wavevector, describing the macroscopic drift motion of phonons with the same
velocity. Assuming a small drift velocity, this displaced distribution can be linearized by the
Taylor's expansion to
N.' = N&E +h
NBE(NE + 1)q - u
kaT
(5.3)
where NB0 E is the equilibrium Bose-Einstein distribution.
Based on the assumption of the displaced phonon distribution in the absence of Rscattering, past work derived macroscopic governing equations that describe hydrodynamic
phonon transport [117]. However, it has remained elusive to our knowledge whether the absence
of R-scattering necessarily leads to the displaced distribution. Moreover, the validity of the
displaced distribution in real materials, where R-scattering cannot be completely avoided, has not
been explicitly confirmed. In this work, we demonstrate from first principles calculations that a
displaced distribution of phonons can occur in graphene even in the presence of weak Rscattering, which validates the assumption of the displaced distribution used in the past studies
[117, 118] and correspondingly shows hydrodynamic phonon transport in graphene.
5.2.1. Details of First Principles Calculations
The calculation for graphene is similar to the details already discussed in Chapter 2. The
second- and third-order force constants of graphene were calculated from density functional
perturbation theory [46, 47] using the Quantum-Espresso
package [70]. The pseudopotential
contains 2s2p2 as valence states with the Perdew-Zunger exchange-correlation functional [71].
99
The calculated force constants of graphene were validated by comparing the mode Grnneisen
parameters as in previous studies [119, 120]. In Fig. 5-2, we show that the mode GrUneisen
parameters from second- and third-order force constants are almost the same as those calculated
by the finite difference of phonon frequency as the crystal volume is changed by 1%. The force
constants of diamond were adopted from the literature [121].
4
finite difference of frequency
0
second- and third-order force constants
E
0
S
-2
O.
-
22D
C- 4
)
-6
-~8
-10
.
K
M
F
Figure 5-2. The mode GrOneisen parameters of graphene. The circles are calculated from the finite
difference of phonon frequencies with different crystal volumes by 1% and the lines are calculated using
both second- and third-order force constants.
The three-phonon scattering rates were calculated using perturbation theory as presented
in Chapter 2. The strong renormalization effect for long-wavelength ZA phonons in graphene
may affect the phase space of three-phonon scattering in graphene. In the quasi-harmonic
approximation, the ZA phonon dispersion of graphene is exactly quadratic in the limit of long
wavelength. However, the strong renormalization effect slightly stiffens the phonon dispersion
and makes the phonon dispersion similar to linear rather than quadratic in the very long
wavelength limit. The slightly changed phonon dispersion may affect the phase space of threephonon scattering. This renormalization effect is included by using phonon stiffening parameters
100
from Ref. [122]. For the isotope scattering calculation, the
13 C
isotope was treated as a point
defect [123].
For solving the Peierls-Boltzmann transport equation, we used the iterative method
explained in Chapter 2 without the commonly used relaxation time approximation [54]. Use of
the relaxation time approximation should be avoided in order to capture the significant role of Nscattering in graphene. For calculating scattering rates and solving the Peierls-Boltzmann
equation, the first Brillouin zone was sampled with 70x70 and 30x30x30 meshes for graphene
and diamond, respectively.
5.2.2. Displaced Phonon Distribution
The first principles calculation results in Fig. 5-3 clearly show the drift motion of
phonons in graphene (0.1 % concentration of isotope
13C)
at 100 K under a static temperature
gradient. The temperature gradient is required to maintain the drift motion since R-scattering
cannot be completely ignored in real materials. In Fig. 5-3, we plot the normalized deviation of
the
distribution
from
the
stationary
equilibrium
distribution,
defined
as
(N - NaE)/{NBE(NBE + 1)}, to examine the existence of a drift velocity. It is clearly seen in Fig.
5-3b that the normalized deviation is linear in wavevector q., along the temperature gradient
direction with the same slope for all three acoustic modes over a wide range of wavevectors.
This indicates that the distribution is indeed displaced, giving the macroscopic motion of
phonons with the same velocity as in Eq. (5.3). The nonlinear behavior in the large wavevector
region of Fig. 5-3b is significantly exaggerated because the deviation is normalized by
NBOE(NJBF + 1), which is negligibly small in this high phonon frequency range. Moreover, these
phonon modes in the range where the normalized deviation is nonlinear to q, contribute
negligibly to actual thermal transport, as presented in Fig. 5-4.
101
b a
6.0x10'
TA
normalized
deviation
of distribution
IO
LA
M,
L
0
0.0
p4
-X10,x
E
A
XS
Ad&0x1
1.5
0!5
140
-1
0050!0
-
(A-)
q,
VT
reciprocal space of a graphene
Figure 5-3. The displaced distribution of phonons at 100 K in the
normalized deviation of the
sheet. The 3C isotope concentration is 0.1%. (a) A contour plot of the
represents the first Brillouin
distribution of flexural acoustic (ZA) phonons in graphene. The hexagon
normalized deviation of the
zone and a temperature gradient is applied along the x-direction. (b) The
at %,=0(M-IF-M). The linear
distribution of the three acoustic branches in graphene along the x-direction
to Eq. (5.3). The same slope for all
dependence on q-, indicates drift motion of acoustic modes, according
drift velocity, regardless of
three acoustic branches means that acoustic phonons have the same
polarization and wavevector.
a
mode thermaI conductivity (Wm-K)
P
ZA
.
CIO10
7.5.10'~~
TA
E
un:
LA-
8.Ox1w -
0
4**
00xx1
0
S
4.0x10
-1.5
-1.0
-0.5
0.0
0.5
1.0
15
q, (A-,)
100 K. (a) A
Figure 5-4. The phonon mode thermal conductivity of graphene with 0.1 % C at
mode thermal
contour plot of the mode thermal conductivity of ZA phonon modes in graphene. (b) The
conductivity of the three acoustic modes in graphene along the x-direction at q,=O.
102
This strong correlation of the distribution among phonon modes in graphene is
remarkably different from the usual cases without strong hydrodynamic transport features. In Fig.
5-5, we show the normalized deviation of the distribution from the stationary equilibrium
distribution, (N - NBOE)/{NBE(NBE + 1)), of pure Bi along the trigonal direction. Unlike the
case of graphene shown in Fig. 5-3, the phonon distribution in Fig. 5-5 does not show any
correlation among phonon modes; i) the phonon distributions for the TA and LA branches are
much different from one another and ii) the phonon distributions do not clearly show any linear
dependence on the wavevector, q., within each branch.
C
0
2.OxlO*"
x1T
--o- LA
C
40
'4)
0
0
.
.....
............
...................
C
S-1.Ox10-
N
-2.Ox1l
r-0.8
-0.4
0.0
0.4
0.8
Figure 5-5. The normalized deviation of the distribution of the acoustic branches in pure bismuth at
300 K along the trigonal-direction (T-F-T). A temperature gradient is applied in the same direction.
Only one TA branch is included in the plot since the two TA branches are degenerate along the line, T-FT. The inset shows the first Brillouin zone of bismuth with the high symmetry points.
The macroscopic drift motion of phonons can be explained by strong N-scattering
compared to R-scattering shown in Fig. 5-6. The phonon distribution under a temperature
gradient is slightly displaced, meaning that phonons gain excess momentum from the
temperature gradient. Then, the excess momentum is affected by N- and R-scatterings in very
103
Through N-scatterings, the excess momentum is exchanged among
phonons such that all phonon modes exhibit the same drift velocity and approach the displaced
Bose-Einstein distribution in Eq. (5.2). However, R-scattering destroys the excess momentum
and induces phonon modes to relax to the stationary equilibrium Bose-Einstein distribution. In
different ways [124].
graphene, N-scattering is much stronger than R-scattering by at least two orders of magnitude
over a wide phonon spectrum, as shown in Fig. 5-6. Therefore, the excess momentum gained
from a temperature gradient is redistributed by strong N-scattering without any considerable
reduction by R-scattering such that most phonon modes have the same drift velocity, regardless
of polarization and wavevector.
1012
10
C])
100
N-scattering
10,1
6
Uo
ZA
TA
LA
R-scattering
<9
CO
M 101
OM
o
0n
@o
10 4
0
10
5
Phonon frequency (THz)
%
Figure 5-6. Comparison of N-scattering and R-scattering rates in graphene with a 0.1
concentration of isotope 3 C at 100 K. The Matthiessen's rule is used to combine U-scattering and
isotope scattering rates into the R-scattering rate.
In addition to the macroscopic collective motion of phonons, it is also important to find
the specific length and time scales for this hydrodynamic transport. If length and time scales
approach infinity, the transport is not hydrodynamic unless R-scattering is completely avoided.
Due to the existence of R-scattering, as we previously stated, a temperature gradient is required
to maintain the drift motion of phonons in Fig. 5-3. In an infinitely large graphene sheet under a
static temperature gradient, the drift velocity of phonons exhibits a uniform distribution across
104
the sample and its magnitude scales as the magnitude of the temperature gradient, both of which
can be explained by the Fourier's law. However, roughly speaking, if the sample size is smaller
than the phonon mean free path of R-scattering or the temperature gradient changes with time
faster than the R-scattering rate, the transport cannot be described by the diffusive law. In such
length and time scales, the transport having macroscopic drift motion shown in Fig. 5-3 can be
hydrodynamic. These considerations regarding length and time scales for hydrodynamic
transport lead to the discussion on phonon Poiseuille flow and second sound in the following
sections.
The displaced distribution we present here is the first explicit confirmation of the
assumption used in past theoretical studies regarding hydrodynamic phonon transport [117, 118,
125, 126]. It also provides a theoretical basis that we will exploit to examine phonon Poiscuille
flow and second sound in the following sections (Chapter 5.3-5.4), further highlighting the
significant hydrodynamic phonon transport in graphene under certain circumstances.
5.3.Phonon Poiseuille Flow
Phonon Poiseuille flow refers to steady-state hydrodynamic phonon transport under a
temperature gradient in a sample. The temperature gradient plays a similar role as pressure
gradient for the molecular Poiseuille flow. Here, it is assumed that the sample is long enough for
the heat flux to be fully developed and to be invariant along the flow direction. Due to their
differing sources of thermal resistance, the phonon Poiscuille flow is distinctly different from the
more well-known diffusive phonon transport. In diffusive phonon transport, the thermal
resistance is mostly due to R-scattering, which can occur anywhere inside a sample. Therefore,
the heat flux in diffusive phonon transport is uniform, as shown in Fig. 5-lb. On the other hand,
the thermal resistance in phonon Poiseuille flow is due to diffuse boundary scattering combined
with many N-scatterings, analogous to viscous effects in fluid flow. The drift velocity is small
near the boundary because of the diffuse boundary scattering, leading to the formation of a drift
velocity gradient along the direction perpendicular to heat flow (Fig. 5-1a). The boundary
scattering can be assumed to be diffuse rather than specular even at a low temperature, such as
105
100 K [127]. The excess momentum of phonons from the temperature gradient is then
transferred to the boundary through the drift velocity gradient and many N-scattering events, and
finally destroyed by diffuse boundary scattering. Therefore, the thermal conductivity largely
depends on the rate of momentum transfer to the boundary, a quantity that is determined by the
N-scattering rates and a sample width, just as resistance in fluid flow depends on viscosity and a
pipe diameter. The significance of the extrinsic momentum loss mechanism in the hydrodynamic
regime, diffuse boundary scattering, implies that a material's thermal conductivity largely
depends on the sample shape and geometry in contrast to the diffusive case. This is also much
different from the ballistic case since the sample size in the hydrodynamic regime is assumed to
be much larger than phonon mean free path.
5.3.1. Criteria for Phonon Poiscuille Flow
The momentum loss mechanism in phonon Poiseuille flow imposes constraints on sample
width in order for phonon Poiseuille flow to occur [126]. If the width of a sample is too large, the
excess momentum is more likely to be destroyed by R-scattering before being transferred to the
boundary. In this case, the transport is close to the diffusive regime rather than to the
hydrodynamic regime. On the other hand, if the sample width is less than the phonon mean free
path, the transport is ballistic. These considerations determine the upper and the lower bounds,
respectively, of the sample width for phonon Poiseuille flow, as formally described in the
following paragraphs.
If phonons exhibit the displaced distribution as shown in Fig. 5-3, the transport of all the
phonon modes can be described by a single parameter, the displacement. Exploiting this feature,
the macroscopic momentum balance equation can be derived from the Peierls-Boltzmann
equation by taking crystal momentum as a moment [126, 128]. Assuming a two-dimensional
material, the momentum balance equation is
-(11V 2-rN12)V 2 a1(x) + (1ITR
Il)a1 (x) = -(01V -VI1)ao(x)
106
(5.4)
where v is a group velocity and an isotropic phonon dispersion is assumed. The above equation
contains inner products of the eigenstates of the N-scattering operator, 10) and 11). The
eigenstates, 10) and |1), represent the deviation of phonon distribution due to the temperature
variation in real space and to a drift velocity, respectively [1251. Both eigenstates are expressed
as [118, 125]
( X
10) = px (2sinh
2) ~
Ila) = (Oaqa/kBT) (2sinh
(5.5)
)
(5.6)
where x is the dimensionless phonon frequency, hw/kBT. The Vp and 0 represent normalization
factors for each state, and a denotes a direction. The ao(x) and a1 (x) in Eq. (5.4) are the weights
of the two states in the actual distribution at position x in real space. The physical meanings of
ao(x) and a1 (x) are temperature and drift velocity, respectively. The detailed derivation and
discussion can be found in literature [118, 125, 126, 128]. Eq. (5.4) was originally derived for an
isotropic and linear dispersion relation [118, 126], and was then extended to general cases [128].
Equation (5.4) looks similar to the Stokes equation, which is the macroscopic momentum
balance equation for molecules when there is no change in the drift velocity along the flow
direction, and thus the inertia term in the Navier-Stokes equation is removed. The right hand side
of Eq. (5.4) shows the momentum gain by the temperature gradient, analogous to the pressure
gradient term in the Stokes equation. The first term on the left-hand side means the momentum
transfer by N-scattering, analogous to the viscous term in the Stokes equation. The second term
on the left hand side reflects the momentum loss by R-scattering and does not have any
counterpart in the Stokes equation. Therefore, for phonon Poiseuille flow to occur, the viscous
effect, (1IV 2 rNI1)V2 , should be larger than the R-scattering effect, (11rR-'|1). In order to
compare the strength of the first and the second terms on the left hand side, we estimate the V 2
operator as (L/2)- 2 , where L is a sample width. Then, the required condition for the viscous
effect being larger than the R-scattering effect is
107
-2) > OR-1)(5.7)
1V2TN
Adding the condition for non-ballistic transport to the above gives,
L
(1vYTN
(11VTN
< (TR
1
1/2
i))(5.8)
where vy is a group velocity along the sample width direction.
The above criteria in Eq. (5.8) can be qualitatively understood with a random walk
picture of phonon transport [126, 129]. A phonon mode experiencing many N-scatterings can be
described with a random walk picture in Fig. 5-7. We assume that the phonon mean free path, AN,
is much smaller than the sample width, L, to avoid ballistic transport. Using the random walk
picture, the required time for the phonon mode to encounter the boundary can be estimated to
(.
)
T
N, where TN is the relaxation time of N-scattering. If the rate of R-scattering, TR-
smaller than the boundary scattering rate,
()
TN'1,
is
the momentum loss by the boundary
scattering is larger than that by R-scattering, indicating phonon Poiseuille flow. This condition
together with the condition for non-ballistic transport leads to the criteria for phonon Poiseuille
flow,
AN
2 im
t
N
N
s
which is similar to Eq. (5.8) obtained from the formal transport theory.
108
(5.9)
A
~L/2
Figure 5-7. A schematic picture describing the random walk of phonons
A window of sample widths for phonon Poiseuille flow, calculated from first
principles,
as a function of temperature is shown in Fig. 5-8. This figure shows that graphene has a much
wider window of sample widths for phonon Poiseuille flow than diamond. The possible range of
sample widths in diamond with an extremely enriched isotope content (0.01%
1C)
is too narrow
for phonon Poiseuille flow to occur even at the lowest temperature we examined (50 K). With a
moderately enriched isotope condition (0.1% '3C), there is no possible sample width for which
phonon Poiseuille flow would occur. The results for diamond confirm the significant difficulty in
observing hydrodynamic phonon transport in three-dimensional materials
[112]. In graphene,
however, there is a large window of sample widths at temperatures below 100 K. When the
isotope content is increased from 0.01 % to 0.1 %, the window becomes narrower, but is still
wide enough at temperatures below 100 K.
109
VV"
10-2
0 Lower bound
--
0
diamond
10-3
-
-
0 Upper bound (0.01%1
3
C)
0 Upper bound (0.1% C)
<> Upper bound (1.1%"C)
0
104
graphene
0
4-J
10-5
E
100
50
150
200 250 300
Temperature (K)
Figure 5-8. The wide window of sample widths for phonon Poiseuille flow in graphene as compared
to diamond.
Graphene's exceptionally wide range of temperatures and sample widths for phonon
Poiseuille flow is due to the strong N-scattering shown in Fig. 5-9. We compare scattering rates
of graphene and diamond in Fig. 5-9 to highlight the strong N-scattering in graphene. We chose
diamond for the comparison since diamond features weak U-scattering compared to N-scattering
owing to its large Debye temperature [35, 130]. The N-scattering rates in graphene are around
1010 s1 , whereas in diamond they are 108 s-' at 100 K. Owing to the strong N-scattering in
graphene, R-scattering remains comparatively small even when the isotope content matches the
naturally occurring case, 1.1%
13 C
(Fig. 5-9b). In diamond, however, R-scattering rates are
comparable to N-scattering rates at the same isotope content (Fig. 5-9d). The underlying reasons
for the strong N-scattering in graphene will be further discussed later in this chapter.
110
a
b
1012
10'0
1010
N-scatterin
N-scattering
10
00x co
4-,
C000t
0
graphene (1C 0.1%)
1lo
0
5
o0
ZA
e0
TA
o
LA
100
Phonon frequency (THz)
d
1
#
ZA
.*o
(3 Cl.1%)
eo
TA
LA
Phonon frequency (THz)
10
in
N-scattering
S
,
N-scattering ,ee
graphene
100
10
- ~.1t
1
Rscatterin
R-scattering
0
106
<p **
108
108
106
106
r
C
M.
C
U)
104
0
R-scattering
10
eo
0.
diamond (OC 0.1%)
102
0
5
R-scattering
10 4
TA1
*0
TA2
10
102
10
Phonon frequency (THz)
5
TAI
.0oLA TA2
1.1%)
diamond(1C
diamond
0 LA
10
Phonon frequency (THz)
Figure 5-9. Comparison between N- and R-scattering rates in graphene and diamond at 100 K,
showing extremely strong N-scattering in graphene. The condition of isotope content is specified in
the plots. The isotope content of 1.1% 13 C in (b,d) represents the naturally occurring case. The figure (a)
is duplicated from Fig. 5-6 for comparison.
5.3.2. Characteristics of Phonon Poiseuille Flow
Phonon Poiseuille flow and molecular Poiseuille flow are very similar in terms of driving
forces and damping mechanisms;
in both cases, transport is driven by a gradient of
thermodynamic forces (temperature gradient for phonons and pressure gradient for molecules)
and the transport is damped by momentum transfer to the boundary and then diffuse scattering at
the boundary. The total mass transfer rate of the molecular Poiseuille flow in the three111
of the tube.
dimensional tube scales as the pressure gradient and as the fourth power of the radius
phonon Poiseuille
The previous work[126] already showed that the total heat transport rate of
Poiseuille
flow in a three-dimensional cylindrical sample follows the same trend as the molecular
flow and
flow in a circular tube. Here, we discuss the similarity between molecular Poiseuille
phonon Poiseuille flow for the two-dimensional geometry case.
per
For the molecular Poiseuille flow in a two-dimensional duct, the mass flow rate (Qm)
unit depth (d) is given by
Qm
d
L3
i_
dP
d
(5.10)
pressure gradient
where L and it are the width of the duct and the viscosity, respectively. The
in Fig. 5along the flow direction (x) is represented by dP/dx. The duct geometry is illustrated
10.
ZZ
d
zx
Flow,
Q
L
V
ZZ
x
duct are represented as L
Figure 5-10 Geometry of two-dimensional duct. The width and depth of the
and d, respectively.
transfer in a
For directly comparing the mass transfer in a molecular system to the heat
system, the
phonon system, we introduce the mass conductivity (Km) because for the phonon
112
thermal conductivity, rather than heat transfer rate, is typically used for describing the resistance
to transport. The mass conductivity (KCm) for the molecular system is defined in the same way as
the thermal conductivity (K) is defined for the phonon system.
QM
Km =
(5.11)
According to Eq. (5.10), the mass conductivity (Km) scales as the second power of the duct width,
L.
Like the molecular Poiseuille flow, the thermal conductivity in two-dimensional phonon
Poiseuille flow scales as the second power of the sample width. The relation between thermal
conductivity and sample width can be derived from the momentum balance equation, Eq (5.4).
-(11v
2
TNI1)V'al(y)+(11rR'I1)aCy)
=2-(OjvV1)a
0
(5.12)
where y is perpendicular to heat flow direction as shown in Fig. 5-10. The physical meanings of
a0 and a1 (y) are the local temperature and displacement in the phonon distribution, respectively.
The first and second terms on the left hand side of Eq. (5.12) show the momentum transfer by Nscattering and the momentum loss by R-scattering, respectively. The right hand side of Eq. (5.12)
shows the momentum gain from the temperature gradient. The above simple differential equation
can be solved by assuming a no slip boundary condition at the boundaries, y=O and y=L. The
solution is
113
exp(L/A) -1
exp
exp(L/A) - exp(- L/A)
where 1/A
-
2
y
(5.13)
+
y
exp(- L/A) - 1
eXp(L/A) - exp(- L/A) exp
=( Iv - V|1)ao
-ITR~11)
a,(y) =
+V
.______
The term in the first parenthesis of Eq. (5.13) represents the
driving force due to the temperature gradient, Vao, and momentum loss due to R-scattering,
(11R-1 1), both together determining the overall magnitude of the heat flux. The second
parenthesis in Eq. (5.13) represents the shape of the heat flux profile. It is noteworthy that L/A
shows the relative strength between R-scattering and the viscous effect by N-scattering.
Therefore, the value of L/A affects the heat flux profile shape as plotted in Fig. 5-11 a. When
L/A has a small value such as 0.1, the transport becomes close to the ideal hydrodynamic
transport and the heat flux profile shows parabolic shape along the y-direction. In contrast, as
L/A increases to 20, the actual transport becomes close to diffusive transport and the heat flux
profile is almost uniform along the y-direction. In the limit as L/A goes to infinity, a,(y) shows
a uniform heat flux profile, a feature of diffusive transport.
Using the solution (Eq. (5.13)) of the momentum balance equation, the thermal
conductivity is
K
~f
1L
2
4exp(-L/A)
al(y)dy ~-1
+
LJ
1a-YL/A
(L /A)(1 + exp(- L/A))
Then, the exponent, a, in the simple power law relation,
_d
a
d(
(logKx)
)
d(log(L/A))
114
(5.14)
~La, can be calculated by
(5.15)
The calculated a is plotted in Fig. 5-1 lb as a function of LIA. In the figure, as L/A becomes
vanishingly small which implies significant hydrodynamic features, the exponent value, a,
approaches to 2, which is the value for the ideal phonon Poiseuille flow without R-scattering and
molecular Poiseuille flow. When L/A is around 1, the exponent value, a, is still much larger than
1, giving the superlinear dependent behavior of the thermal conductivity with respect to sample
width. However, as L/A increases further, a becomes almost zero, indicating that there is no
significant effect from diffuse boundary scattering and the actual transport is very close to the
diffusive regime.
115
a
Boundary
L/..N=20
L/A=O.1
Boundary
Shape of heat flux profile
b
diffusive
hydrodynamic
I-
2
0
1
0
A
10 3
102
101
10
10'
102
101
LIiA
Figure 5-11 Effects of sample width on the heat flux profile and thermal conductivity (a) The shape
of the heat flux profiles when transport is close to the hydrodynamic limit (LIA-A.l) and close to the
diffusive limit (LIA=20) (b) Dependence of the thermal conductivity on sample width, L. The vertical axis
represents the exponent value (a) in the simple power law relation, K~L".
116
5.3.3. Possible Experiments for Observing Phonon Poiseuille Flow
For the experimental
confirmation of the phonon Poiseuille flow, the thermal
conductivity can be measured by varying the sample width. Here we briefly discuss several
transport regimes in various sample widths. When the sample width is much larger than the mean
free path of R-scattering, transport is diffusive and the thermal conductivity does not vary with
sample width. As the sample width decreases such that it is smaller than the mean free path of Rscattering but larger than the mean free path of N-scattering, the phonon transport can be
described by Poiseuille flow. It is well known that the mass flow rate of molecular Poiseuille
flow in a two-dimensional duct scales as the third power of the duct width. Similarly, in phonon
Poiseuille flow, the heat flow rate and thermal conductivity scale as the third power and the
second power of sample width, respectively, as discussed in the previous section. The thermal
conductivity that is superlinearly proportional to the sample width makes phonon Poiseuille flow
distinguished from the diffusive transport. As the sample width is further decreased and
comparable to the mean free path of N-scattering, the transport regime is in between the
hydrodynamic and ballistic limits. In this case, the phonon system is similar to a rarefied gas in a
molecule system and it may be possible to observe a phonon Knudsen minimum [131]. The
phonon Knudsen minimum was observed in liquid helium where phonons carry most of heat at
extremely low temperature [132, 133]. If the sample width is further decreased and much smaller
than the phonon mean free path for N-scattering, the transport is ballistic. The thermal
conductivity in the ballistic regime that is described by the Casimir limit cannot be superlinearly
proportional to the sample width. The thermal conductivity should always be sublinearly or
linearly proportional to the sample width. Therefore, the superlinear dependence of the thermal
conductivity on sample width can be used for confirming the presence of phonon Poiseuille flow.
However, there are several possible challenges in carrying out actual experiments. For
example, one may need several graphene samples that have different sample widths but have the
same quality in terms of isotope content or other defect density. The possible variations in the
quality between several samples can make it untrustworthy to deduce any characteristic trend of
the thermal conductivity as a function of sample width. In addition, the superlinear dependence
of the thermal conductivity on sample width may not provide a decisive evidence for
117
hydrodynamic transport. The superlinear dependence was also observed in the ballistic transport
regime in silicon nanowires with a roughened surface [134].
Another indication of the presence of phonon Poiseuille flow, which has been used for
identification in the past work [112, 135, 136], is found in how the thermal conductivity changes
with temperature. An increase in the thermal conductivity with an exponent in the temperature
that is larger than that for the ballistic case is regarded as a direct evidence of phonon Poiscuille
flow[126, 129]. In the ballistic regime, the thermal conductivity,
K,
can be expressed with a
simple formula, K-cvL, where c is the specific heat, v is the group velocity, and L is the
characteristic size of the sample that is limiting the phonon mean free path. Since the sample size,
L , is not much changed with temperature, an increase of the thermal conductivity with
temperature should be associated only with specific heat and group velocity, both together
determining the thermal conductance in the ballistic limit. In the hydrodynamic regime, however,
the effective sample size for the boundary scattering increases with temperature. This is because
the N-scattering rate increases with temperature, leading to a longer travel distance of phonons to
the boundary as discussed in the previous section (Chapter 5.3.1). The effective sample size for
boundary scattering as well as the ballistic thermal conductance, cv, increase with temperature.
Therefore, the thermal conductivity in the hydrodynamic regime should increase more rapidly
than the ballistic thermal conductance increases with temperature. Considering that the thermal
conductance of graphene in the ballistic limit increases as T .68 (Ref. [137]), an observation of the
thermal conductivity increasing with an exponent in temperature much larger than 1.68 can
indicate phonon Poiseuille flow.
In recent years, there have been many successful measurements of the thermal
conductivity of graphene, but more advances would be required to observe phonon Poiseuille
flow. For thermal conductivity measurements using Raman spectroscopy, the temperature range
was limited to temperatures above room temperature because of large uncertainties near room
temperature [138-141]. Alternatively, a micro-fabricated heater-sensor-assembly was used [142144], but the samples were not isotopically enriched and were too small to observe phonon
Poiseuille flow.
118
5.4. Second Sound
5.4.1. Criteria for Second Sound
Second sound refers to the propagation of a temperature wave (or a phonon density wave)
provoked by a heat pulse, analogous to ordinary sound in a fluid, which is the propagation of a
pressure wave. The propagation of a heat pulse in the hydrodynamic regime is much different
from the propagation of a heat pulse in the diffusive regime as shown in Fig. 5-1. The pulse in
the hydrodynamic regime is transmitted by many N-scatterings [111, 115, 145, 146], whereas the
pulse in the diffusive regime is largely damped by R-scattering and cannot propagate. The
transmission of a heat pulse can also be observed in ballistic transport, but the sample size is
limited to below phonon mean free path. Therefore, the phenomenon of second sound can
provide a unique way to transmit a heat pulse without leaving a temperature trace behind the
wave front in a sample larger than phonon mean free path. It is important to note that second
sound is different from acoustic sound in a solid because the former is a phonon density wave
which is a collective motion of phonons in a wide spectrum maintained by many N-scattering
processes, whereas the latter is just ballistic transport of extremely long-wavelength phonons.
The required conditions on temperature and sample size for second sound are determined
from the relative strength between N-scattering and momentum-destroying scatterings [126]. The
frequency of second sound or the inverse of a pulse duration time (D) should be larger than the
combined rate of U-scattering, isotope scattering, and boundary scattering (TRB-
1
, hereafter RB-
scattering refers to U-scattering, isotope scattering, and boundary scattering combined), but
smaller than the N-scattering rate
(TN-1)-
TRB
<0<TN~1
(5.16)
The former condition minimizes the damping due to RB-scatterings. In particular,
boundary scattering is not desirable for second sound while it provides an important feature of
phonon Poiseuille flow. The latter condition in Eq. (5.16) allows enough time to form a welldefined phonon density pulse. Otherwise, the phonon excitation would be randomized by N119
scattering and would not form a pulse, analogous to the situation where an ordinary sound in a
fluid is damped by a strong viscous effect when the sound frequency is high and comparable to
the rate of scattering between molecules [147].
In Eq. (5.16), the N- and RB-scattering rates calculated from first principles were
averaged with the displaced state, 11) (Ref. [126, 128]):
TN
TRBI =
(lITu
= O
+ Tisotope
Tboundary
(5.17)
N41)1
Tboundary
(1)
(5.18)
(5.19)
In the above expressions, we made a small modification from the original expression given by
Ref. [126]. In the second sound criteria given in Ref. [126], the effect of diffuse boundary
scattering is taken into account by a geometrical factor that describes momentum loss by diffuse
boundary scattering. However, the geometrical factor is defined for the fully developed phonon
flow which cannot be usually assumed for the case where a temporal change in transport occurs
on a fast time scale as in second sound. Therefore, instead of using the geometrical factor, we
simply add boundary scattering rates, Tboundary-', to the Umklapp and isotope scattering rates.
The boundary scattering rates are taken to be IvI/L, where v and L are a group velocity and a
sample size, respectively. Unlike the phonon Poiseuille flow, we do not assume any specific
shape of a graphene sample for second sound, and L represents the characteristic size of the
arbitrarily shaped sample. The simple empirical formula for the boundary scattering rates largely
overestimates the significance of boundary scattering in the hydrodynamic regime, giving a
conservative estimation of the second sound frequency range. This is because the effective
sample size for boundary scattering in the hydrodynamic regime is much larger than the actual
sample size due to the many N-scattering processes[126, 129]. Unlike the ballistic regime,
phonons in hydrodynamic regime experience many N-scattering processes and the travel
120
distance of phonons until they encounter a boundary is much longer than the actual sample width,
as described by the random walk picture in Fig. 5-7.
In Figs. 5-12(a) and 5-12(b), we show a wide range of second sound frequencies in
graphene below 100 K. The frequency range of second sound in graphene becomes narrow upon
the inclusion of isotope and boundary scatterings, but the frequency range is still considerable
when the sample is larger than 100 pm and the isotope content is less than 0.1%. However, the
frequency range of second sound in diamond does not exist for the given conditions, since Eq.
(5.16) cannot be satisfied. Shown in Figs. 5-12(c) and 5-12(d) are contour plots of the second
sound frequency range for various sample sizes, isotope contents, and temperature conditions. In
the same examined range of sample sizes, isotope concentration, and temperature for Figs. 512(c) and 5-12(d), we found that second sound is not possible at all in diamond. Similar to
phonon Poiseuille flow, the main difference between graphene and diamond is attributable to the
extremely strong N-scattering compared to R-scattering in graphene.
121
b
a
101
Ila
Sample size: 1000im
lsotope:0 01% "C
10
10'
raphe
dgraphene
C
03 10'
10'
0
YV
Ln
0
10
-
-
Upper bound
0
Lower bound (1000pm)
Lower bound (100pn)
10b
-
10e
0
50
100
150
200 250 300
50
150
100
200 250 300
Temperature (K)
Temperature (K)
d
C
o Lower bound ("C 001%
o Lower bound V'C 0.1%)
Lower bound VIC 1.1%)
1000
1000
bandwidth of
second sound
frequency
2S dB
100
E-,100
20
is
E
E
10
s
10
0.01
10o
W. c
0.01
0.1
"C
'"Ccontent (%)
content(%
Figure 5-12 The possible frequency ranges of second sound in graphene and diamond. (a) The
content of isotope "C is fixed at 0.01 %. (b) The sample size is fixed at 1000 pm. (c-d) Contour plots of
second sound frequency range in graphene with respect to sample size and isotope content for 50 and 100
are
where fl,,, and 4
K, respectively. The second sound frequency range is defined as f,,/fo,
a
on
plotted
the upper and lower bounds of second sound frequency, respectively. The frequency range is
log scale in the contour plots. Second sound in diamond is not possible in the given range of temperature,
sample size, and isotope content.
5.4.2. Possible Experiments for Observing Second Sound
The second sound has been experimentally confirmed by measuring the transient
temperature response after applying a heat pulse [111, 115, 145, 146, 148]. One can apply a heat
122
pulse at one side of a sample and measure the temperature change with respect to time, dT/dt, at
the other side of the sample. If a clear peak in dT/dt is observed, it can be attributed to either
ballistic or hydrodynamic transport. The second sound peak can be distinguished from a ballistic
pulse using the fact that the propagation of second sound is slower than the propagation of
acoustic sound or ballistic phonon transport. For three-dimensional materials, it was theoretically
estimated that the speed of second sound, vH, is v 1 /V3 where v, is the speed of acoustic sound in
the Debye model [109, 149, 1501. In past experiments, a clear peak in dT/dt was observed with
a delay time that could be explained well with the theoretical prediction of the speed of second
sound, vl~ vi/Nf-.
The second sound in graphene can be measured in a similar way as in the case of threedimensional materials. However, the speed of second sound cannot be estimated as vI/d
because the Debye model is not valid for graphene due to the quadratic ZA branch. Instead of
assuming a Debye model, we calculated the speed of second sound using the phonon dispersion
determined from first principles. Here, we derive the wave equation for second sound from the
Peierls-Boltzmann equation for the case of arbitrary phonon dispersion to estimate the speed of
second sound. We assume that R-scattering is negligibly weak and thus crystal momentum is
approximately conserved.
We start from the Peierls-Boltzmann transport equation for phonon transport:
aN
&t
aN
--
_
ax
N
at+)V
(ODc
(5.20)
where the right hand side represents the change in the phonon distribution due to scattering.
Taking energy and crystal momentum along the flow direction as a moment leads to the
macroscopic equations for energy and momentum balance, respectively.
123
a(IfqxNdq)
f
+
=0
(5.21)
qxvNdq =0
(5.22)
v_,Ndq
f
foNd q +
where s denotes the phonon polarization. The right hand side of Eq. (5.21) is zero because
energy is always conserved upon scattering. The right hand side of Eq. (5.22) is also zero
because here we assume R-scattering is negligibly weak and thus crystal momentum is also
conserved. The actual phonon distribution ( N ) under very weak R-scattering can be
approximated as the displaced distribution function (N
displaced distribution (NgdE)
d)
as we confirmed in Fig. 5-3. The
can be linearized because the displacement in the phonon
distribution from the equilibrium value is very small:
+
NN ~z: dE N~N +kBT NBE(NBOE +1)qxu.
(5.23)
In addition, we can neglect higher order terms associated with the small displacement (ux)
in Eqs. (5.21) and (5.22). The resulting equations are
dqY
T2+
f
x
d
0
N E(N
+ kN
x ~f
124
=0
(5.24)
dqy- =O0
(5.25)
+ 1)qxdq
E0
d
Taking the time derivative of Eq. (5.24) and the spatial derivative of Eq. (5.25), and then
subtracting these two equations results in the hyperbolic wave equation for second sound.
a2T
t=
2
2
2T
X2
(5.26)
where vi represents the speed of second sound and can be expressed as
s f qxvx
V
1
=o
aT dq) (Z f CVX NE(NE + 1) qxdq)
aB
sOT
(Es)
dq) E
f
(5.27)
qxNBO(NBE + 1)qxdq)
The above expression can be simplified using the notation,
I0) and 11), which are defined in Eq.
(5.5) and (5.6):
V= (0|)( 11)2)/
2
(5.28)
Applying the Debye model for three-dimensional materials into Eq. (5.28) gives
VI = v 1/V
(5.29)
as previously derived [109, 149, 150]. For graphene, we calculate the speed of second sound
using Eq. (5.28) and the phonon dispersion of the ZA branch from first principles. We also
calculated the speed of second sound by including all three acoustic branches for the inner
products in Eq. (5.28), but the speed of second sound is almost the same as that obtained by
including only ZA branch.
The calculated speed of second sound in graphene is plotted in Fig. 5-13. In the figure,
the speed of second sound ranges from 2000 to 3000 m s-1 and increases with temperature. The
125
dependence on temperature arises because the group velocity of the ZA phonon modes increases
with frequency. At low temperature where the phonons with the highest probability are low
frequency phonons, the average group velocity is small, resulting in slower propagation of
second sound than that of the higher temperature.
3500
-o
C
.
I
I
100
150
3000-
0
2500
-
0
tA
-0
2000-
15001
50
Temperature (K)
Figure 5-13 The speed of second sound in graphene with respect to temperature
It would be interesting to see how the speed of second sound compares to the speed of
ballistic transport in graphene. In Fig. 5-14, we compare the delay time of the ballistic transport
pulse and second sound in the heat pulse experiment mentioned above. For the delay time of a
ballistic transport pulse, we suppose that an experiment for observing second sound is carried out
that is similar to the past experiments that were done for three-dimensional materials [111, 115,
146, 148]. Suppose that we have a rectangular shape graphene sheet with a point heat source at
the left edge and a point thermal sensor at the right edge, as illustrated in the inset of Fig. 5-14.
At time t--O, a heat pulse is generated at the point source with a delta function profile with
respect to time. Assuming that all phonon modes are transported ballistically, then the
temperature change at the sensor can be expressed using Landauer's formalism [151]:
126
dT
dt
f(t)~AT
f
hov,8(vy)
og
'S
T
t-
W
dq
(5.30)
X,
where W is a distance between the source and the sensor, while AT is the temperature difference
between the source and the sensor at t-O. The delta function of vy is included because phonon
modes propagating with an oblique angle to the x-direction cannot contribute to the ballistic
thermal transport between the point source and the point sensor. This constraint prevents any
additional broadening of the ballistic heat pulse from the finite size of the source and the sensor.
In the actual cases where the source and the sensor have a finite size, the temperature signal
should be broader than that given by Eq. (5.30).
Using Eq. (5.30) and the phonon dispersion of ZA modes obtained from first principles
calculations, we plot the temperature signal for ballistic transport in Fig. 5-14. In the figure, there
is a significant broadening in the ballistic transport pulse because the group velocity of the ZA
phonon modes largely depends on frequency. In addition, the ballistic heat pulse becomes faster
at higher temperature because higher frequency phonons with larger group velocities are excited
at higher temperature. We also plot the delay time of second sound based on the calculated speed
of second sound in Fig. 5-14. The delay time of second sound is around three times longer than
that of the peak of the ballistic heat pulse. The difference in delay time by a factor of three,
together with the estimated speed of second sound, then can be used to separate the second sound
signal from the ballistic heat pulse.
127
W~
Y
E
-
ballistic
(100 K)
f-7sample
ballistic
(50 K)
x
second sound
(100 K)
U
second sound
-
-
(50 K)
0.0
4.0x10
2.0x104
6.0x10
8.Ox10
Time delay (s)
second sound
Figure 5-14 Comparison between the delay time by ballistic transport and
source and the
the
between
distance
per
propagation in the heat pulse experiment. The delay time is
and a point
sensor, W, in [m]. The inset illustrates the configuration of the sample with a point heat source
heat sensor for observing second sound.
5.5. Origin of the Hydrodynamic Phonon Transport in Graphene
The large contrast between graphene and diamond regarding the occurrence of
hydrodynamic transport indicates that there are more reasons for the significant hydrodynamic
that the
transport in graphene, in addition to its large Debye temperature. Here, we show
extremely large anharmonicity and density-of-states of the long-wavelength ZA phonons, both
originating from their two-dimensional
characteristics, are responsible for the significant
hydrodynamic transport in graphene.
The strong three-phonon scattering in graphene reflects a large phonon mode
and
anharmonicity. The mode Grdneisen parameters give a measure of this anharmonicity [152],
extremely
their magnitudes become large as the phonon wavelength increases (Fig. 5-2). The
128
large magnitude of the Gruneisen parameters of the ZA modes near the zone center is a
characteristic of two-dimensional materials as explained by elasticity theory [122]. The elasticity
theory predicts the divergence of the mode Gruneisen parameter, as yq~ -1/|q1
2
where yq is a
mode Grineisen parameter at wavevector, q. The actual divergence, however, is prevented by
the strong phonon renormalization effect or any in-plane strain that stiffens the ZA phonon
dispersion very near the zone center [122, 153, 154]. In general, at large wavelengths, the threephonon scattering processes are dominated by N-scattering, since U-processes by definition
require large-wavelength phonons.
In particular, due to the high graphene Debye temperature,
phonons in graphene are predominantly populated in the large-wavelength region, further
making N-scattering much stronger than U-scattering. This strong N-scattering is consistent with
the behavior of the Gruneisen parameters for the ZA phonon modes in Fig. 5-2 and this behavior
is also confirmed in Fig. 5-9, and also has been previously reported [23, 1201.
The large gap between N- and R-scattering rates in graphene shown in Fig. 5-9 is further
highlighted when considering the frequency range that mostly contributes to the thermal
transport. The transport regime, such as hydrodynamic or diffusive, is determined by comparing
the average scattering rates of the N- and R-processes as seen in second sound. It is noteworthy
that the frequency ranges largely contributing the average scattering rates are different for
graphene and diamond, because of their strongly differing phonon density-of-states. The densityof-states is a constant for the quadratic dispersion in two-dimensional materials, while it
increases with the square of frequency for the linear dispersion in three-dimensional materials.
Therefore, the density-of-states of low frequency phonons in the former case is much larger than
that of the latter case, implying that the role of low frequency phonons in graphene is very
significant relative to the situation in diamond.
In Fig. 5-15, we present the cumulative weighting factor for averaging the scattering rates
as a function of phonon frequency. In the macroscopic transport equation like Eq. (5.4), the
scattering rate is averaged using the displaced state, 11):
(1Ir-1
1) =2
f tm -1x (2sinh X
dx
(5.31)
0
xf (2sinh z)
129
dx
Similarly, we define the cumulative weighting factor for averaging scattering rate as
X
X
-2
(1I1)X = f~'xf (2sinhf2 dx
0' xf (2sinhx)
where the exponent
2
(5.32)
dx
is one in Eqs. (5.31) and (5.32) for quadratic dispersion in two-
dimensional materials and is four for linear dispersion in three-dimensional materials. The values
of
can be derived from the definition of the state, 11), in Eqs. (5.5) and (5.6) by assuming an
isotropic phonon dispersion. In fact, for the case of perfectly quadratic dispersion in twodimensional materials ( =1), the low frequency contribution to the averaged scattering rate
diverges as the frequency approaches zero. Such a singular behavior is prevented by phonon
stiffening from the renormalization effect. The spectral contribution in Fig. 5-15 includes the
renormalization effect by using phonon stiffening parameters given in Ref. [122].
The cumulative weighting factor shows that the frequency ranges that are important for
the averaged scattering rate are very different for diamond and graphene. From Fig. 5-15, we see
that low-frequency phonons are most important for the quadratic dispersion in two-dimensional
materials, whereas mid-frequency phonons give a larger contribution for the linear dispersion in
three-dimensional materials. The significant role of low frequency phonons in graphene results
in a robust hydrodynamic phonon transport since low frequency phonons in graphene exhibit a
larger gap between N- and R-scattering rates as shown in Fig. 5-9.
130
I
I
0 1.0
U)
..
C
(>
0.8
0.6
4-J
'4-
CU
E
U
0.4
0.2
quadratic in 2D
linear in 3D
nn
0
<
5
10
Dimensionless phonon energy (hw/kB 7
Figure 5-15 Cumulative weighting factors for averaging scattering rates for quadratic dispersion in
two-dimensional materials and for linear dispersion in three-dimensional materials.
5.6.Conclusion
In conclusion, we predict hydrodynamic phonon transport in suspended graphene that is
clearly distinguishable from the usual diffusive or ballistic phonon transport. The significant
hydrodynamic transport in graphene is attributed to two-dimensional features such as extremely
large scattering rates for momentum conserving N-processes and a large density-of-states of
long-wavelength ZA phonons. The significant hydrodynamic phonon transport in graphene
provides a new perspective beyond the diffusive and ballistic transport pictures on how to
understand thermal transport in two-dimensional materials. In this work, we have focused on the
sub-room temperature range, where hydrodynamic transport dominates over
diffusive transport,
but the hydrodynamic transport is also important for room temperature cases. The phonon
transport in graphene at room temperature cannot be solely described by the diffusion limit
due
to strong N-scattering [23], and the hydrodynamic transport presented here indicates another
limit one may need to consider to fully understand phonon transport in graphene at room
temperature. In addition, the significant hydrodynamic features imply practical importance. For
131
example, considering the importance of boundary scattering shown in phonon Poiscuille flow,
thermal rectification would be achievable in a tapered graphene sheet. The fast thermal transport
without damping featured in second sound also shows a potential usage of graphene for thermal
interconnects or thermal signal transmitters.
132
6. Summary and Future Directions
6.1. Summary
In this thesis, we have made contributions towards better understanding of transport of
phonons and electrons in thermoelectric materials and graphene. Our study covers phonon
transport in three-dimensional bulk materials, electron transport across or near two-dimensional
discontinuities, and phonon transport in an atomically thin two-dimensional material.
Combined, Chapter 2 and Chapter 3 provided an in-depth understanding of phonon
transport in many good thermoelectric materials. In Chapter 2, we began by showing that a
strong long-range interaction exists in Bi and Sb along a specific crystallographic direction. The
inclusion of the long-range interaction is necessary to accurately predict phonon thermal
conductivity values for Bi, Sb, and Bi-Sb alloys. Using the set of force constants including the
long-range interaction from first principles calculations, we could accurately calculate phonon
thermal conductivity values for Bi, Sb, and Bi-Sb alloys. By comparing the calculated phonon
thermal conductivity to the total thermal conductivity that is experimentally measured, we
discussed the relative contributions from phonons and electrons to the thermal transport in those
materials. We also provided phonon mean free path distributions, which can be used to develop
nanostructures that can significantly reduce thermal conductivity.
Chapter 3 was concerned with establishing a relation between low thermal conductivity
and chemical bonding. We showed that the long-range interaction observed in Bi and Sb is also
observed in many good thermoelectric materials, such as group IV-VI and V2 -V1 3 materials. The
long-range interaction commonly observed in those seemingly different materials was explained
with resonant bonding. The easily polarizable p-electrons in resonant bonding cause the longrange interaction. We established a connection between resonant bonding and low thermal
133
conductivity of the rocksalt IV-VI materials, where the long-range interaction is most significant
among the materials we examined. The significant long-range interaction causes the softening of
the transverse optical phonon modes. The soft transverse optical phonon modes finally lead to a
low thermal conductivity through their large anharmonicity and large phase space for threephonon scattering, both of which contribute to the strong three-phonon scattering.
Chapter 4 characterized electron transport in a nanocomposite thermoelectric sample. In
particular, we examined the effect of two-dimensional discontinuities, such as grain boundaries,
on the electron transport. We examined the electron transport across many grain boundaries in
Bi 2 Te 2 .7 Se.
3
nanocomposite materials. We measured the four transport coefficients (electrical
conductivity, Seebeck coefficient, Hall coefficient, and Nernst coefficient), then we roughly
estimated the energy dependence of electron scattering rates by fitting the measured four
transport coefficients. The estimated energy dependence of electron scattering rates indicate that
many grain boundaries in the nanocomposite sample preferentially scatter the electrons with low
energy rather than the electrons with high energy, thereby contributing to the large Seebeck
coefficient through the electron filtering effect.
Chapter 5 disclosed a new regime of phonon transport in an atomically thin material,
graphene. The phonon transport in suspended graphene had been previously studied in the
ballistic and diffusive phonon transport regimes, but the previous study could not provide a
complete explanation for the extremely high thermal conductivity of graphene. We showed that
hydrodynamic phonon transport, in which the intrinsic thermal resistance is very small compared
to the diffusive transport case, is the reason behind this extremely high thermal conductivity. The
hydrodynamic phonon transport in graphene is possible due to the fact that most of phonon
scattering in graphene conserves crystal momentum unlike the phonon scattering in most threedimensional materials. The strong momentum-conserving scattering gives rise to several features
of hydrodynamic phonon transport, such as a drift motion of phonons, phonon Poiseuille flow,
and second sound. We associated the hydrodynamic phonon transport in graphene with
graphene's two dimensional features, such as large anharmonicity and large phonon-density-ofstates of long wavelength flexural acoustic phonon modes.
134
6.2. Future Directions
Our study in Chapters 2 and 3 discovered a close relation between good thermoelectric
materials and displacive-type ferroelectricity. The displacive ferroelectric behavior, soft optical
phonon modes, can strongly scatter acoustic phonons, leading to very low thermal conductivity.
In this regard, it is worthwhile to study several ferroelectric materials for thermoelectric
applications. However, there are several possible challenges. One possible challenge is that most
ferroelectric materials are electrical insulators with a wide band gap. Finding ferroelectric
materials with a narrow band gap would be important. Another challenge is to check whether the
ferroclectricity is preferred for achieving a high thermoelectric power factor. Most ferroelectric
materials have extremely large dielectric constants because the large dielectric constant is closely
related to their ferroelectric behavior. The large dielectric constant can lead to a high electron
mobility by strongly screening the impurity potential, and thus can be advantageous for
achieving a high thermoelectric power factor. On the other hand, the soft transverse optical
modes have a large amplitude for atomic vibrations and can strongly scatter electrons, possibly
reducing electron mobility. In-depth study of these two opposite effects using first principles will
help to develop a better understanding of electron transport in potential thermoelectric materials
with ferroelectric behavior.
The experimental work presented in Chapter 4 can be further pursued to increase the
thermoelectric power factor. As we discussed in Chapter 4, the electron filtering effect by grain
boundaries can increase the Seebeck coefficient, but has an adverse effect on the electrical
conductivity. In order to increase the thermoelectric power factor, the potential barrier at a grain
boundary needs to be carefully engineered so that the Seebeck coefficient is largely increased,
while the electrical conductivity remains high. One possible way to engineer the grain boundary
would be by adding some elements with low solubility to the thermoelectric materials. The
elements with low solubility will be dispersed homogeneously when the material is processed at
high temperature. Then, once the material is cooled down, the elements with low solubility will
be segregated at grain boundaries and would affect the shape and height of the potential barriers.
The hydrodynamic phonon transport presented in Chapter 5 needs further research to be
experimentally confirmed. The hydrodynamic phonon transport can be experimentally confirmed
135
by observing second sound or phonon Poiseuille flow as we discussed in Chapter 5.3.3 and
Chapter 5.4.2. For the experimental observation of the hydrodynamic phonon transport, it is
essential to have a graphene sheet with large area (> 1 pm) and minimized defects, such as grain
boundaries and surface contaminations. The recent success in exfoliating a large area graphene
sheet would be one possible way to achieve this [155].
Finally, the investigation of phonon transport under phase instability condition can bring
innovations in engineering phonon transport. An important lesson we learned from this thesis
work is that phase instability can lead to extraordinary phonon transport. Chapter 3 showed that
the low thermal conductivity of the resonant bonding materials is due to a ferroelectric instability
and Chapter 5 showed that the high thermal conductivity of graphene is due to the intrinsic
structural instability of two-dimensional materials, represented by the diverging anharmonicity in
the long wavelength limit. An interesting future direction would be in-situ control of phonon
transport in materials with phase instability. Several materials, such as ferroelectric materials and
liquid crystals, have phase instability that can largely affect thermal transport.
The phase
instability of those materials can be controlled by applying an electric field, and it would be
possible to invent a solid-state thermal switch that can be operated by applying an electric field.
This thermal switch will have a broad impact on thermal management of electronic devices and
energy conversion devices that require variable heat transfer coefficients depending on their
modes of operation.
136
Bibliography
[1] L. E. Bell, "Cooling, heating, generating power, and recovering waste heat with thermoelectric
systems", Science 321 (2008).
[2] D. M. Rowe, "Recent advances in silicon-germanium alloy technology and an assessment of the
problems of building the modules for a radioisotope thermo-electric generator", Journal of Power Sources
19 (1987).
[3] D. Kraemer, B. Poudel, H.-P. Feng, J. C. Caylor et al., "High-performance flat-panel solar
thermoelectric generators with high thermal concentration", Nature Mater. 10 (2011).
[4] C. B. Vining, "An inconvenient truth about thermoelectrics", Nature Mater. 8 (2009).
[5] M. Dresselhaus, G. Chen, M. Tang, R. Yang et al., "New directions for low-dimensional
thermoelectric materials", Adv. Mater. 19 (2007).
[6] G. Joshi, H. Lee, Y. Lan, X. Wang et al., "Enhanced Thermoelectric Figure-of-Merit in
Nanostructured p-type Silicon Germanium Bulk Alloys", Nano Letters 8 (2008).
[7] B. Poudel, Q. Hao, Y. Ma, Y. Lan et al., "High-thermoelectric performance of nanostructured
bismuth antimony telluride bulk alloys", Science 320 (2008).
[8] D. A. Broido, M. Malorny, G. Birner, N. Mingo, and D. A. Stewart, "Intrinsic lattice thermal
conductivity of semiconductors from first principles", Appl. Phys. Lett. 91 (2007).
[9] K. Esfarjani, G. Chen, and H. T. Stokes, "Heat transport in silicon from first-principles calculations",
Phys. Rev. B 84 (2011).
[10] W. Liu, X. Tan, K. Yin, H. Liu et al., "Convergence of Conduction Bands as a Means of Enhancing
Thermoelectric Performance of n-Type Mg 2Si 1 ,Snx Solid Solutions", Phys. Rev. Lett. 108 (2012).
[11] H. Liu, X. Shi, F. Xu, L. Zhang et al., "Copper ion liquid-like thermoelectrics", Nature Mater. 11
(2012).
[12] J. Carrete, W. Li, N. Mingo, S. Wang, and S. Curtarolo, "Finding Unprecedentedly Low-ThermalConductivity Half-Heusler Semiconductors via High-Throughput Materials Modeling", Phys. Rev. X 4
(2014).
[13] S. Curtarolo, G. L. W. Hart, M. B. Nardelli, N. Mingo, S. Sanvito, and 0. Levy, "The highthroughput highway to computational materials design", Nature Mater. 12 (2013).
[14] G. A. Slack, "The thermal conductivity of nonmetallic crystals", Solid State Phys. 34 (1979).
[15] D. P. Spitzer, "Lattice thermal conductivity of semiconductors: A chemical bond approach", J. Phys.
Chem. Solids 31 (1970).
[16] J. Heremans, C. Thrush, and D. Morelli, "Thermopower enhancement in lead telluride
nanostructures", Phys. Rev. B 70 (2004).
[ 17] C. Prasad, L. Jiang, D. Singh, M. Agostinelli et al., in Reliability Physics Symposium (IRPS), 2013
IEEE Internationat2013),pp. 5D. 1.1.
[18] E. Pop, S. Sinha, and K. E. Goodson, "Heat Generation and Transport in Nanometer-Scale
Transistors", Proceedings of the IEEE 94 (2006).
[19] I. Chowdhury, R. Prasher, K. Lofgreen, G. Chrysler et al., "On-chip cooling by superlattice-based
thin-film thermoelectrics", Nature Nanotech. 4 (2009).
137
[20] Y.-M. Lin, C. Dimitrakopoulos, K. A. Jenkins, D. B. Farmer, H.-Y. Chiu, A. Grill, and P. Avouris,
100-GHz Transistors from Wafer-Scale Epitaxial Graphene", Science 327 (2010).
[21] P. G. Klemens, and D. F. Pedraza, "Thermal conductivity of graphite in the basal plane", Carbon 32
(1994).
[22] P. G. Klemens, "Theory of Thermal Conduction in Thin Ceramic Films", Int. J. Thermophys. 22
(2001).
[23] L. Lindsay, D. A. Broido, and N. Mingo, "Flexural phonons and thermal transport in graphene",
Phys. Rev. B 82 (2010).
[24] J. H. Seol, I. Jo, A. L. Moore, L. Lindsay et al., "Two-dimensional phonon transport in supported
graphene", Science 328 (2010).
[25] G. S. Nolas, J. Sharp, and H. J. Goldsmid, Thermoelectrics: Basic Principlesand New Materials
Developments (Springer, New York, 2001), Springer Series in Material Science.
[26] W. M. Yim, and A. Amith, "Bi-Sb alloys for magneto-thermoelectric and thermomagnetic cooling",
Solid-State Electron. 15 (1972).
[27] K. Behnia, M.-A. Mdasson, and Y. Kopelevich, "Nernst Effect in Semimetals: The Effective Mass
and the Figure of Merit", Phys. Rev. Lett. 98 (2007).
[28] J. P. Heremans, C. M. Thrush, D. T. Morelli, and M.-C. Wu, "Thermoelectric Power of Bismuth
Nanocomposites", Phys. Rev. Lett. 88 (2002).
[29] Y.-M. Lin, X. Sun, and M. S. Dressethaus, "Theoretical investigation of thermoelectric transport
properties of cylindrical Bi nanowires", Phys. Rev. B 62 (2000).
[30] C. Uher, and H. J. Goldsmid, "Separation of the electronic and lattice thermal conductivities in
bismuth crystals", Phys. Stat. Sol. (b) 65 (1974).
[31] V. Kagan, and N. Red'ko, "Phonon thermal conductivity of bismuth alloys", Sov. Phys. JETP 73
(1991).
[32] C. F. Gallo, B. S. Chandrasekhar, and P. H. Sutter, "Transport properties of bismuth single crystals",
J. Appl. Phys. 34 (1963).
[33] G. S. Nolas, and H. J. Goldsmid, in Thermal conductivity: Theory, Properties, andApplications,
edited by T. M. Tritt (Kluwer Academic Publishers, New York, USA, 2004).
[34] A. Ward, and D. A. Broido, "Intrinsic phonon relaxation times from first-principles studies of the
thermal conductivities of Si and Ge", Phys. Rev. B 81 (2010).
[35] A. Ward, D. A. Broido, D. A. Stewart, and G. Deinzer, "Ab initio theory of the lattice thermal
conductivity in diamond", Phys. Rev. B 80 (2009).
[36] T. Luo, J. Garg, J. Shiomi, K. Esfarjani, and G. Chen, "Gallium arsenide thermal conductivity and
optical phonon relaxation times from first-principles calculations", Europhysics Letters 101 (2013).
[37] L. Lindsay, D. A. Broido, and T. L. Reinecke, "Ab initio thermal transport in compound
semiconductors", Phys. Rev. B 87 (2013).
[38] J. Garg, N. Bonini, B. Kozinsky, and N. Marzari, "Role of Disorder and Anharmonicity in the
Thermal Conductivity of Silicon-Germanium Alloys: A First-Principles Study", Phys. Rev. Lett. 106
(2011).
[39] L. E. Diaz-Sainchez, A. H. Romero, and X. Gonze, "Phonon band structure and interatomic force
constants for bismuth: Crucial role of spin-orbit interaction", Phys. Rev. B 76 (2007).
[40] E. D. Murray, S. Fahy, D. Prendergast, T. Ogitsu, D. M. Fritz, and D. A. Reis, "Phonon dispersion
relations and softening in photoexcited bismuth from first principles", Phys. Rev. B 75 (2007).
[41] K. Esfarjani, and H. T. Stokes, "Method to extract anharmonic force constants from first principles
calculations", Phys. Rev. B 77 (2008).
[42] X. Gonze, B. Amadon, P. M. Anglade, J. M. Beuken et al., "ABINIT: First-principles approach to
material and nanosystem properties", Comput. Phys. Commun. 180 (2009).
138
[43] C. Hartwigsen, S. Goedecker, and J. Hutter, "Relativistic separable dual-space Gaussian
pseudopotentials from H to Rn", Phys. Rev. B 58 (1998).
[44] L. E. Diaz-Sinchez, A. H. Romero, M. Cardona, R. K. Kremer, and X. Gonze, "Effect of the SpinOrbit Interaction on the Thermodynamic Properties of Crystals: Specific Heat of Bismuth", Phys. Rev.
Lett. 99 (2007).
[45] Z. Tian, K. Esfarijani, and G. Chen, "Enhancing phonon transmission across a Si/Ge interface by
atomic roughness: First-principles study with the Green's function method", Phys. Rev. B 86 (2012).
[46] X. Gonze, and C. Lee, "Dynamical matrices, Born effective charges, dielectric permittivity tensors,
and interatomic force constants from density-functional perturbation theory", Phys. Rev. B 55 (1997).
[47] X. Gonze, "First-principles responses of solids to atomic displacements and homogeneous electric
fields: Implementation of a conjugate-gradient algorithm", Phys. Rev. B 55 (1997).
[48] J. L. Yarnell, J. L. Warren, R. G. Wenzel, and S. H. Koenig, "Phonon Dispersion Curves in
Bismuth", IBM J. Res. Dev. 8 (1964).
[49] R. I. Sharp, and E. Warming, "The lattice dynamics of antimony", J. Phys. F: Met. Phys. 1 (1971).
[50] G. P. Srivastava, The physics ofphonons (CRC Press, 1990).
[51] B. Abeles, "Lattice Thermal Conductivity of Disordered Semiconductor Alloys at High
Temperatures", Phys. Rev. 131 (1963).
[52] P. Cucka, and C. S. Barrett, "The crystal structure of Bi and of solid solutions of Pb, Sn, Sb and Te
in Bi", Acta Crystallogr. 15 (1962).
[53] P. Lambin, and J. Vigneron, "Computation of crystal Green's functions in the complex-energy plane
with the use of the analytical tetrahedron method", Phys. Rev. B 29 (1984).
[54] M. Omini, and A. Sparavigna, "Beyond the isotropic-model approximation in the theory of thermal
conductivity", Phys. Rev. B 53 (1996).
[55] K. Biswas, J. He, I. D. Blum, C.-I. Wu et aL., "High-performance bulk thermoelectrics with all-scale
hierarchical architectures", Nature 489 (2012).
[56] J.-P. Issi, "Low Temperature Transport Properties of the Group V Semimetals", Aust. J. Phys. 32
(1979).
[57] C. Dames, and G. Chen, in Handbook of Thermoelectrics: Macro to Nano, edited by D. M. Rowe
(CRC Press, Boca Raton, FL, 2006).
[58] A. S. Henry, and G. Chen, "Spectral phonon transport properties of silicon based on molecular
dynamics simulations and lattice dynamics", J. Comput. Theor. Nanos. 5 (2008).
[59] Y. Pei, X. Shi, A. LaLonde, H. Wang, L. Chen, and G. J. Snyder, "Convergence of electronic bands
for high performance bulk thermoelectrics", Nature 473 (2011).
[60] N. Yamada, E. Ohno, K. Nishiuchi, N. Akahira, and M. Takao, "Rapid-phase transitions of GeTeSb 2Te3 pseudobinary amorphous thin films for an optical disk memory", J. Appl. Phys. 69 (1991).
[61] T. Siegrist, P. Merkelbach, and M. Wuttig, "Phase Change Materials: Challenges on the Path to a
Universal Storage Device", Annu. Rev. Cond. Mat. Phys. 3 (2012).
[62] D. Damon, "Thermal Conductivity of SnTe between 1000 and 5000 K", J. Appl. Phys. 37 (1966).
[63] G. A. Slack, "Nonmetallic crystals with high thermal conductivity", J. Phys. Chem. Solids 34 (1973).
[64] G. Lucovsky, and R. M. White, "Effects of Resonance Bonding on the Properties of Crystalline and
Amorphous Semiconductors", Phys. Rev. B 8 (1973).
[65] E. A. Albanesi, C. M. I. Okoye, C. 0. Rodriguez, E. L. Peltzer y Blanca, and A. G. Petukhov,
"Electronic structure, structural properties, and dielectric functions of IV-VI semiconductors: PbSe and
PbTe", Phys. Rev. B 61 (2000).
[66] D. Lencer, M. Salinga, B. Grabowski, T. Hickel, J. Neugebauer, and M. Wuttig, "A map for phasechange materials", Nature Mater. 7 (2008).
139
,
[67] K. Shportko, S. Kremers, M. Woda, D. Lencer, J. Robertson, and M. Wuttig, "Resonant bonding in
crystalline phase-change materials", Nature Mater. 7 (2008).
[68] S. K. Mishra, and et al., "Electronic structure and thermoelectric properties of bismuth telluride and
bismuth selenide", J. Phys.: Condens. Matter 9 (1997).
[69] M. H. Cohen, L. M. Falicov, and S. Golin, "Crystal Chemistry and Band Structures of the Group V
Semimetals and the IV-VI Semiconductors", IBM J. Res. Dev. 8 (1964).
[70] P. Giannozzi, S. Baroni, N. Bonini, M. Calandra et al., "QUANTUM ESPRESSO: a modular and
open-source software project for quantum simulations of materials", J. Phys.: Condens. Matter 21 (2009).
[71] J. P. Perdew, and A. Zunger, "Self-interaction correction to density-functional approximations for
many-electron systems", Phys. Rev. B 23 (1981).
[72] Y. Zhang, X. Ke, C. Chen, J. Yang, and P. R. C. Kent, "Thermodynamic properties of PbTe, PbSe,
and PbS: First-principles study", Phys. Rev. B 80 (2009).
[73] W. Kullmann, G. Eichhorn, H. Rauh, R. Geick, G. Eckold, and U. Steigenberger, "Lattice Dynamics
and Phonon Dispersion in the Narrow Gap Semiconductor Bi 2Te3 with Sandwich Structure", Phys. Stat.
Sol. (b) 162 (1990).
[74] J. R. Lowney, and S. D. Senturia, "Optical dielectric constant of Pb1..,SnTe in the narrow-gap
region", J. Appl. Phys. 47 (1976).
[75] W. Richter, and C. R. Becker, "A Raman and far-infrared investigation of phonons in the
rhombohedral V2-V1 3 compounds Bi 2Te], Bi 2Se 3 , Sb2Te 3 and Bi 2(Tei.Sex) 3 (0 < x < 1), (Bi1 ySby) 2 Te 3 (0
< y < 1)", Phys. Stat. Sol. (b) 84 (1977).
[76] D. K. Biegelsen, "Frequency dependence of the photoelastic coefficients of silicon", Phys. Rev. B
12 (1975).
[77] W. Cochran, R. Cowley, G. Dolling, and M. Elcombe, "The crystal dynamics of lead telluride",
Proc. R. Soc. Lon. Ser. A 293 (1966).
[78] M. M. Elcombe, "The crystal dynamics of lead sulphide", Proc. R. Soc. Lon. Ser. A 300 (1967).
[79] P. D. DeCicco, and F. A. Johnson, "The Quantum Theory of Lattice Dynamics. V", Proc. R. Soc.
Lon. Ser. A 310 (1969).
[80] R. M. Pick, M. H. Cohen, and R. M. Martin, "Microscopic Theory of Force Constants in the
Adiabatic Approximation", Phys. Rev. B 1 (1970).
[81] P. B. Littlewood, "The crystal structure of IV-VI compounds. I. Classification and description", J.
Phys. C: Solid State 13 (1980).
[82] M. D. Nielsen, V. Ozolins, and J. P. Heremans, "Lone pair electrons minimize lattice thermal
conductivity", Energy Environ. Sci. 6 (2013).
[83] R. E. Peierls, Quantum Theory of Solids (Oxford University Press, 1955).
[84] P. Ghosez, E. Cockayne, U. V. Waghmare, and K. M. Rabe, "Lattice dynamics of BaTiO 3, PbTiO 3
and PbZrO 3:A comparative first-principles study", Phys. Rev. B 60 (1999).
[85] 0. Delaire, J. Ma, K. Marty, A. F. May et al., "Giant anharmonic phonon scattering in PbTe",
Nature Mater. 10 (2011).
[86] J. An, A. Subedi, and D. J. Singh, "Ab initio phonon dispersions for PbTe", Solid State Commun.
148 (2008).
[87] Y. Zhang, X. Ke, P. R. C. Kent, J. Yang, and C. Chen, "Anomalous Lattice Dynamics near the
Ferroelectric Instability in PbTe", Phys. Rev. Lett. 107 (2011).
[88] Z. Tian, J. Garg, K. Esfarijani, T. Shiga, J. Shiomi, and G. Chen, "Phonon conduction in PbSe, PbTe,
and PbTe.-Sex from first-principles calculations", Phys. Rev. B 85 (2012).
[89] T. Shiga, J. Shiomi, J. Ma, 0. Delaire et al., "Microscopic mechanism of low thermal conductivity
in lead telluride", Phys. Rev. B 85 (2012).
[90] 1. U. 1. Ravich, B. A. Efimova, and 1. A. Smirnov, Semiconducting lead chalcogenides (Plenum
Publishing Corporation, 1970), Vol. 5.
140
[91] A. V. Inyushkin, A. N. Taldenkov, A. Y. Yakubovsky, A. V. Markov, L. Moreno-Garsia, and B. N.
Sharonov, "Thermal conductivity of isotopically enriched 7 'GaAs crystal", Semicond. Sci. Tech. 18
(2003).
[92] L. Lindsay, and D. Broido, "Three-phonon phase space and lattice thermal conductivity in
semiconductors", J. Phys.: Condens. Matter 20 (2008).
[93] J. M. Ziman, Electrons and Phonons: the Theory of TransportPhenomena in Solids (Oxford
University Press, UK, 1960).
[94] D. Vashaee, and A. Shakouri, "Improved thermoelectric power factor in metal-based superlattices",
Phys. Rev. Lett. 92 (2004).
[95] I. Chernik, V. Kaidanov, M. Vinogradova, and N. Kolomoets, "Investigation of the valence band of
lead telluride using transport phenomena", SOV PHYS SEMICONDUCTORS 2 (1968).
[96] V. Jovovic, S. J. Thiagarajan, J. West, J. P. Heremans et al., "Transport and magnetic properties of
dilute rare-earth--PbSe alloys", J. Appl. Phys. 102 (2007).
[97] D. Young, T. Coutts, V. Kaydanov, A. Gilmore, and W. Mulligan, "Direct measurement of densityof-states effective mass and scattering parameter in transparent conducting oxides using second-order
transport phenomena", J. Vac. Sci. Technol., A (2000).
[98] D. L. Young, J. F. Geisz, and T. J. Coutts, "Nitrogen-induced decrease of the electron effective
mass in GaAs[sub 1 - x]N[sub x] thin films measured by thermomagnetic transport phenomena", Appl.
Phys. Lett. 82 (2003).
[99] E. 0. Kane, "Band structure of indium antimonide", J. Phys. Chem. Solids 1 (1957).
[100] A. C. Smith, J. F. Janak, and R. B. Adler, Electronic conduction in solids (McGraw-Hill New
York, 1967).
[101] B. M. Askerov, Electron transportphenomena in semiconductors (World Scientific, 1994), Vol.
394.
[102] E. H. Putley, The Hall Effect and Related Phenomena (Butterworths, 1960).
[103] H. Kaibe, Y. Tanaka, M. Sakata, and I. Nishida, "Anisotropic galvanomagnetic and thermoelectric
properties of n-Type Bi 2Te3 single-crystal with the composition of a useful thermoelectric cooling
material", J. Phys. Chem. Solids 50 (1989).
[104] H. Kohler, "Non-Parabolic E(k) Relation of the Lowest Conduction Band in Bi2 Te3", Phys. Stat.
Sol. (b) 73 (1976).
[105] G. E. Shoemake, J. A. Rayne, and R. W. Ure, "Specific Heat of n- and p-Type Bi_{2}Te_{3} from
1.4 to 90&deg;K", Phys. Rev. 185 (1969).
[106] B.-L. Huang, and M. Kaviany, "Ab initio and molecular dynamics predictions for electron and
phonon transport in bismuth telluride", Phys. Rev. B 77 (2008).
[107] V. A. Kutasov, and L. N. Lukyanova, "The Conduction Band Parameters and Scattering
Mechanisms in Solid-Solutions Based on Bi 2Te 3", Phys. Stat. Sol. B-Basic Research 154 (1989).
[108] H. B. G. Casimir, "Note on the conduction of heat in crystals", Physica 5 (1938).
[109] J. C. Ward, and J. Wilks, "III. Second sound and the thermo-mechanical effect at very low
temperatures", Philos. Mag. 43 (1952).
[110] W. J. De Haas, and T. Biermasz, "The thermal conductivity of KBr, KCI and Si0 2 at low
temperatures", Physica 4 (1937).
[11] C. C. Ackerman, B. Bertman, H. A. Fairbank, and R. A. Guyer, "Second sound in solid helium",
Phys. Rev. Lett. 16 (1966).
[112] L. Mezhov-Deglin, "Measurement of the thermal conductivity of crystalline He 4", J. Exp. Theor.
Phys. 49 (1965).
[113] D. G. Cahill, W. K. Ford, K. E. Goodson, G. D. Mahan et al., "Nanoscale thermal transport", J.
Appl. Phys. 93 (2003).
141
[114] D. G. Cahill, P. V. Braun, G. Chen, D. R. Clarke et al., "Nanoscale thermal transport. 11. 20032012", Appl. Phys. Rev. 1 (2014).
[115] H. E. Jackson, C. T. Walker, and T. F. McNelly, "Second sound in NaF", Phys. Rev. Lett. 25
(1970).
[116] P. Klemens, "Thermal conductivity of solids at low temperatures", Handbuch der Physik 14 (1956).
[117] J. A. Sussmann, and A. Thellung, "Thermal conductivity of perfect dielectric crystals in the
absence of Umklapp processes", Proc. Phys. Soc. 81 (1963).
[118] R. A. Guyer, and J. A. Krumhansl, "Solution of the linearized phonon Boltzmann equation", Phys.
Rev. 148 (1966).
[119] S. Lee, K. Esfarijani, J. Mendoza, M. S. Dresselhaus, and G. Chen, "Lattice thermal conductivity of
Bi, Sb, and Bi-Sb alloy from first principles", Phys. Rev. B 89 (2014).
[120] N. Bonini, J. Garg, and N. Marzari, "Acoustic phonon lifetimes and thermal transport in freestanding and strained graphene", Nano Lett. 12 (2012).
[121] D. A. Broido, L. Lindsay, and A. Ward, "Thermal conductivity of diamond under extreme pressure:
A first-principles study", Phys. Rev. B 86 (2012).
[122] R. Roldin, A. Fasolino, K. V. Zakharchenko, and M. 1. Katsnelson, "Suppression of
anharmonicities in crystalline membranes by external strain", Phys. Rev. B 83 (2011).
[123] S.-i. Tamura, "Isotope scattering of dispersive phonons in Ge", Phys. Rev. B 27 (1983).
[124] J. Callaway, "Model for lattice thermal conductivity at low temperatures", Phys. Rev. 113 (1959).
[125] J. A. Krumhansl, "Thermal conductivity of insulating crystals in the presence of normal processes",
Proc. Phys. Soc. 85 (1965).
[126] R. A. Guyer, and J. A. Krumhansl, "Thermal conductivity, second Sound, and phonon
hydrodynamic phenomena in nonmetallic crystals", Phys. Rev. 148 (1966).
[127] A. K. Gupta, T. J. Russin, H. R. Gutierrez, and P. C. Eklund, "Probing Graphene Edges via Raman
Scattering", ACS Nano 3 (2008).
[128] V. L. v. Gurevich, Transportin phonon systems (Elsevier Science Publishers, Amsterdam, 1986),
Modem Problems in Condensed Matter Sciences.
[129] R. Gurzhi, "Thermal conductivity of dielectrics and ferrodielectrics at low temperatures", J. Exp.
Theor. Phys. 46 (1964).
[130] L. Wei, P. K. Kuo, R. L. Thomas, T. R. Anthony, and W. F. Banholzer, "Thermal conductivity of
isotopically modified single crystal diamond", Phys. Rev. Lett. 70 (1993).
[ 131 ] G. Chen, Nanoscale energy transportand conversion: a paralleltreatment of electrons, molecules,
phonons, andphotons (Oxford University Press, USA, 2005).
[132] D. S. Greywall, "Thermal-conductivity measurements in liquid 4 He below 0.7 K", Phys. Rev. B 23
(1981).
[133] R. W. Whitworth, "Experiments on the Flow of Heat in Liquid Helium below 0.7 K", Proc. R. Soc.
Lon. Ser. A 246 (1958).
[134] A. I. Hochbaum, R. Chen, R. D. Delgado, W. Liang et al., "Enhanced thermoelectric performance
of rough silicon nanowires", Nature 451 (2008).
[135] W. C. Thomlinson, "Evidence for anomalous phonon excitations in solid He 3", Phys. Rev. Lett. 23
(1969).
[136] E. M. Hogan, R. A. Guyer, and H. A. Fairbank, "Thermal conductivity of oriented single crystals
of hexagonal close-packed helium 4", Phys. Rev. 185 (1969).
[137] M.-H. Bae, Z. Li, Z. Aksamija, P. N. Martin et al., "Ballistic to diffusive crossover of heat flow in
graphene ribbons", Nature Commun. 4 (2013).
[138] A. A. Balandin, S. Ghosh, W. Bao, 1. Calizo, D. Tewcldebrhan, F. Miao, and C. N. Lau, "Superior
thermal conductivity of single-layer graphene", Nano Lett. 8 (2008).
142
[139] S. Ghosh, 1. Calizo, D. Teweldebrhan, E. P. Pokatilov et al., "Extremely high thermal conductivity
of graphene: Prospects for thermal management applications in nanoelectronic circuits", Appl. Phys. Lett.
92 (2008).
[140] S. Chen, Q. Wu, C. Mishra, J. Kang et al., "Thermal conductivity of isotopically
modified graphene", Nature Mater. 11 (2012).
[141] W. Cai, A. L. Moore, Y. Zhu, X. Li, S. Chen, L. Shi, and R. S. Ruoff, "Thermal transport in
suspended and supported monolayer graphene grown by chemical vapor deposition", Nano Lett. 10
(2010).
[142] M. T. Pettes, I. Jo, Z. Yao, and L. Shi, "Influence of polymeric residue on the thermal conductivity
of suspended bilayer graphene", Nano Lett. 11 (2011).
[143] Z. Wang, R. Xie, C. T. Bui, D. Liu, X. Ni, B. Li, and J. T. L. Thong, "Thermal transport in
suspended and supported few-layer graphene", Nano Lett. 11 (2010).
[144] X. Xu, L. F. C. Pereira, Y. Wang, J. Wu et al., "Length-dependent thermal conductivity in
suspended single-layer graphene", Nature Commun. 5 (2014).
[145] C. C. Ackerman, and W. C. Overton, "Second sound in solid helium-3", Phys. Rev. Lett. 22 (1969).
[146] T. F. McNelly, S. J. Rogers, D. J. Channin, R. J. Rollefson et al., "Heat pulses in NaF: onset of
second sound", Phys. Rev. Lett. 24 (1970).
[147] L. E. Kinsler, A. R. Frey, A. B. Coppens, and J. V. Sanders, Fundamentalsof acoustics (WileyVCH, 1999), 4th edn.
[148] V. Narayanamurti, and R. C. Dynes, "Observation of second sound in bismuth", Phys. Rev. Lett.
28 (1972).
[149] L. Landau, "The theory of superfluidity of helium 11", J. Phys USSR 5 (1941).
[150] J. Ward, and J. Wilks, "The velocity of second sound in liquid helium near the absolute zero",
Philos. Mag. 42 (1951).
[151] Y. Imry, and R. Landauer, "Conductance viewed as transmission", Rev. Mod. Phys. 71 (1999).
[152] G. Leibfried, and G. Schlomann, "Heat conduction in electrically insulating crystals", Nach. Akad.
Wiss. Gottingen. Math. Phys. Klasse. 2a 71 (1954).
[153] P. L. de Andres, F. Guinea, and M. I. Katsnelson, "Bending modes, anharmonic effects, and
thermal expansion coefficient in single-layer and multilayer graphene", Phys. Rev. B 86 (2012).
[154] E. Mariani, and F. von Oppen, "Flexural Phonons in Free-Standing Graphene", Phys. Rev. Lett.
100 (2008).
[155] J. Kim, H. Park, J. B. Hannon, S. W. Bedell, K. Fogel, D. K. Sadana, and C. Dimitrakopoulos,
"Layer-resolved graphene transfer via engineered strain layers", Science 342 (2013).
143