Long-term activity-induced changes in the brain Arvind Govindarajan
advertisement

Long-term activity-induced changes in the brain
A study of translationalregulation and structural plasticity
by
Arvind Govindarajan
B.Sc. Specialist in Biochemistry & Chemistry
Specialist in Computer Science
University of Toronto, 1998
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF
'MASJEiCHUSErS
ISTUE
OF TECHNOLOGY
DOCTOR OF PHILOSOPHY
AT THE
MAY 2 3 2006
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
LIBRARIES
FEBRUARY 2005
-
-
© 2004 Massachusetts Institute of Technology. All rights reserved.
/
Signature of Author:
·-
IV
Department of Biology
November 30, 2004
6'
Certified by:
2
r..})
Susumu Tonegawa
Picower Professor of Biology and Neuroscience
Director, The Picower Center for Learning and Memory
Thesis Supervisor
Accepted by
-V
Stephen P. Bell
Professor of Biology
Chair, Graduate Committee
ARCHIES
I
Long-term activity-induced changes in the brain
A study of translational regulation and structural plasticity
by
Arvind Govindarajan
Submitted to the Department of Biology
on November 30, 2004 in Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in
Biology
Abstract
Long-lasting changes must take place in the brain to store the skills and memories
that have been learned by the organism throughout its history. Long-term memory (LTM),
and its cellular correlate, the late-phase of long-term potentiation (L-LTP), require protein
synthesis. It has generally been assumed that the regulation of transcription underlies LLTM and LTM. Chapter 2 of this thesis demonstrates that transgenic mice with inhibited
ERK MAP Kinase pathway activity in the cortex and CA1 region of the hippocampus show
inhibited LTM and L-LTP. Moreover, the L-LTP phenotype resembles a deficit in translation
and not transcription. Experiments using hippocampal cultured cells demonstrate that
neuronal activity induces translation of a large number of mRNAs in an ERK MAP
Kinase pathway-dependent manner via phosphorylation of the translation factors S6,
elF4E and 4E-BP1. Increased phosphorylation of translation factors was observed along
with increased translation after the induction of L-LTP; lastly, LTM formation occurred
concomitantly with ERK MAP Kinase pathway-dependent phosphorylation of S6 and
elF4E. Chapter 3 extends these findings to demonstrate that other stimuli that cause
protein synthesis-dependent forms of plasticity also upregulate protein synthesis via the
same mechanisms. As some of these stimuli cause L-LTP and others cause L-LTD, it is
proposed that induction of these forms of plasticity, though opposite in terms of synapticweight changes, produce the same proteins, and that activated synapses capture the
appropriate proteins to express the appropriate form of synaptic plasticity.
Cellularly, it is believed that these long-lasting changes involve structural plasticity
in the neurons. However, as it has generally been difficult to show such a correlation
between structural changes and behavioral changes after behavioral training, a correlation
between structural changes and affective behaviors after chronic-stress treatment was
sought. Chapter 4 shows that transgenic mice overexpressing BDNF only in forebrain
pyramidal cells have reduced chronic stress-induced atrophy in the apical dendrites of
CA3 pyramidal cells. This structural change is correlated with improved performance
in the Porsolt forced-swim test. Increased spinogenesis was observed in the amygdala
concomitantly with an increase in anxiety, thus demonstrating a strong correlation between
structural change and behavior.
Thesis Supervisor: Susumu Tonegawa
Title: Director, The Picower Center for Learning & Memory,
Picower Professor of Biology and Neuroscience
2
Acknowledgements
The work in this thesis could not have been done without a lot of help and
assistance. Firstly, I would like to thank Ray Kelleher, and Sumantra "Shona" Chattarji
with whom the vast majority of the work was done. I would also like to thank Shu Huang
who I have had the joy of working with for the last year of my thesis, Lorene Leiter, who
has helped significantly with a collaborative project with Guosong Liu, and Frank Bushard
who I have worked with on various behavioral projects. Candy Carr and Sean Perry
were also of tremendous help with mouse colony management and technical assistance.
Undergraduate students (UROPs) have played a part in the science that is described here,
and an even greater part in various projects that are not described here. These talented
undergraduates include Nadya Mawjee, Mimi Trinh, Colin Galbraith, and Caroline Lee.
Helpful advice was obtained from Hae-yoon Jung, who taught me electrophysiology, Tom
McHugh, Frank Bushard, David Gerber, Lorene Leiter, as well as other members of the
Tonegawa laboratory. I would also like to thank Ray Kelleher, Tom McHugh, and Hongkui
Zeng, who influenced me to join the lab when I was rotating in 1998 - a momentous
decision that I have never regretted. Ray, in particular, was key to my success as he
taught me most of the molecular biological techniques that I needed. I would also like to
thank Guosong Liu, and his lab, who introduced me to cell-culture, and who remain an
influence in my work. In addition, I have had the pleasure of a lot of insightful comments
from Chip Quinn, both in informal meetings, as well as in the classes I TA'd for him. Matt
Wilson and Scott Waddell also were a part of a lot of useful discussions.
I would like to extend my appreciation to my thesis committee - David Housman
for being its chair, Martha Constantine-Paton for providing a physiologist's view, and Mark
Bear whose work is related more directly to mine. I would like to extend a special thankyou to Bruce McEwen who I have had the pleasure of discussing my work with, and who
agreed to come to Boston on the day before thanksgiving to be my external reviewer.
3
Acknowledgements
I would also like to thank Zheng Huang, C. Ramachandran, Philip Tagari, Mike
Gresser, Drew Woolley, Ann Verner, and Karen Henderson who introduced me to scientific
research, and modified my career trajectory from medicine to science.
I also had the pleasure of the company of friends, particularly, John Doll, Emily
Gillett, Philina Lee, Ken Olive, Matt Sullivan, and the rest of my class - "bio98" - for
enjoying the highs, and helping me deal with the lows of graduate school.
This thesis could not have been finished without the support of my family, namely,
my parents, my brother, and particularly, my wife, Annette. Her never-ending support was
essential when experiments were not working and when deadlines were looming.
Lastly, and definitely not leastly, I have to thank Susumu Tonegawa for taking me
into his lab, even though I had no molecular biology or neurobiology experience, and then
trusting me with running projects that I chose in the way that I wanted to run them. He was
generous with resources, provided a great environment in which to work, and helped me
to crystallize my ideas through numerous "brain-storming" sessions.
4
Table of Contents
Title ...........................................................................................
Abstract ...........................................
....... .........................
Acknowledgements
...........................................
I
2
......................3
Table of Contents .............................................
5
Table of Figures .............................................
9
List of Abbreviations .........
.................................... ....... 11
Chapter 1 - Review of long-term memory changes in the
brain
.........................................................................
13
Abstract .............................................................................................
Protein-Synthesis and Long-term Memory ..................................
Vertebrates ....................................................
13
14
14
Invertebrates..................................................................................
16
Extinction and Reconsolidation ....................................................
Protein-Synthesis Dependent Synaptic Plasticity .........................
Vertebrate Long-Term Potentiation ...............................................
Vertebrate Long-Term Depression and Priming ............................
Chemically induced L-LTP and L-LTD ...........................................
17
18
18
19
19
Invertebrates..................................................................................
20
The differential role of transcription and translation .....................
Transcriptional control mechanisms ..............................................
Translational Control Mechanisms ................................................
20
22
24
Translation in Neurons ........................................
25
............
Local Protein Synthesis ........................................
............
Synaptic Tag Hypothesis ........................................
............
RNA Tag Hypothesis ......................................................................
RNA Targeting Mechanisms ....................................................
Dendritic vs. Somatic Protein Synthesis ........................................
StructuralPlasticity........................................
............
25
28
30
30
31
32
Structural Changes after Behavior ................................................
Structural Changes after Synaptic Plasticity ..............................
Structural Changes in Stress related disorders .............................
Structural Plasticity and Affective Disorders ..................................
Conclusion & Specific Aims ........................................
............
Bibliography ......................................................................................
32
33
34
36
37
39
Chapter 2 - Translational Control by MAPK signaling in
long-term synaptic plasticity and memory ........................
53
Abstract.
.............................................................................................53
Introduction ....................................................
54
Protein Synthesis Dependent Plasticity ........................................
Regulation of Translation ........................................
............
MAP Kinase Pathway ....................................................
5
54
55
58
Table of Contents
Materials & Methods: ...................................................
63
Hippocampal Culture ...................................................
63
Plasmid constructions ...................................................
Reporter mRNAs ...................................................
64
65
In situ Hybridization...................................................
Metabolic Labeling.
.................. ..........................................
65
66
Western Blotting ...................................................
66
Synaptoneurosomes .....................................................................
67
Electrophysiology ...................................................
67
Fear conditioning ...................................................
68
Results ...................................................
69
Minimal poly A tail length required for translation of reporter ......... 69
Translation of Reporter is Activity dependent and MEK
dependent ...................................................
69
Different forms of neuronal activity induce translation in a MEK
dependent manner ...................................................
70
MEK Dependent Translation Induction is not dependent on CPE. 72
MEK Dependent Translation is not dependent on
Polyadenylation ......................................................
72
Effects of MEK inhibition are not due to toxicity or transfection
level ......................................................
73
MEK is sufficient to induce translation of reporter ......................... 75
Metabolic Labeling studies show a general upregulation of
translation by BDNF and bicuculline .
............................
76
Phosphorylation of translation initiation factors and ribosomal
proteins accompanies stimulated translation ................................ 77
Translation in synaptoneurosomes and phosphorylation of
translation factors is MEK dependent ......... ......... .................. 79
Spaced Tetani induce translation in a MEK dependent manner .... 80
Fear-conditioning training induces phosphorylation of translation
factors...................................................................
Discussion
......................................................
Bibliography ......................................................
82
83
88
Chapter 3 - Characterization of Neuronal Activity- ............. 93
induced Translation
............................................. 93
Abstract ......................................................
Introduction .......................................................................................
Material and Methods .......................................................................
Hippocampal Culture .....................................................................
93
94
96
96
Metabolic Labeling .....................................................................
96
Western Blotting ............................................................................
97
Results ...............................................................................................
97
Translation in DIV 8 cells grown in serum-free media are
stimulated by BDNF and bicuculline .........
....................................
97
Translational upregulation by BDNF and bicuculline also occurs
6
Table of Contents
in mature cells ........................................
............
99
Dopamine receptor activation and spaced depolarization
upregulate translation in a MAP Kinase dependent manner ...... 100
mGluR activation upregulates translation in a MAP Kinase
dependent manner ....................................................
100
Protein Degradation is not affected by LTP or LTD stimuli .......... 101
Increased translation lasts at least 45 minutes ............................ 103
D iscussion.......................................................................................
103
Bibliography ....................................................
107
Chapter 4 - BDNF prevents depression and chronic
stress-induced hippocampal atrophy, but facilitates
anxiety and amygdalar spinogenesis................................
Abstract .
...........................................................................................
110
110
Introduction
.....................................................
111
Methods ....................................................
113
Experimental Animals ...............................................................
Chronic Immobilization Stress ....................................................
Morphological Analysis .....................................................
Porsolt Swim Test .....................................................
Open Field Test .....................................................
Elevated Plus Maze .....................................................
113
113
114
114
115
115
Biochemical Analysis .........
115
..................................................
Statistical Analysis .....................................................
R esults .............................................................................................
Chronic-Stress causes CA3 dendritic atrophy in mice .................
Chronic-Stress does not cause CA3 dendritic atrophy in
transgenic mice .....................................................
Transgenic mice show decreased immobility in Porsolt forced
swim test.....................................................
Transgenic animals have normal stress hormone levels ..............
Transgenic mice show chronic-stress induced weight loss ..........
Chronic-Stress reduces hippocampal BDNF protein levels but
not mRNA levels ....................................................
Transgenic mice show increased amygdalar spine density.........
Transgenic mice show increased anxiety-like behavior ...............
115
115
115
D iscussion .......................................................................................
Bibliography ....................................................
126
131
Chapter 5 - Conclusion ...........................................
116
118
118
119
120
123
123
136
Role of translation in protein synthesis-dependent synaptic
plasticity and memory ....................................................
137
Structural Plasticity in Affective Behaviors...............................
Conclusion.......................................................................................
Bibliography ....................................................
141
143
146
Credits and Notes............................
.........
7
........... 150
Table of Contents
Chapter 1.......................................
150
Chapter 2 ..........................................................................................
150
Chapter 3.......................................
150
Chapter 4 ..........................................................................................
Chapter 5.......................................
Bibliography....................................................................................
Biography.............................................................................
8
150
151
151
152
Table of Figures
Figure
Figure
Figure
Figure
Figure
Figure
Figure
Figure
1-1:
1-2:
2-1:
2-2:
2-3:
2-4:
2-5:
2-6:
Figure 2-7:
Figure 2-8:
Figure 2-9:
Figure 2-10:
Figure 2-11:
Figure 2-12:
Figure 2-13:
Figure 2-14:
Figure 2-15:
Figure 2-16:
Figure 2-17:
Figure 2-18:
Figure 3-1:
Figure 3-2:
Figure 3-3:
Figure 3-4:
Figure
Figure
Figure
Figure
3-5:
3-6:
3-7:
4-1:
Model for local-protein synthesis ........................................
28
Model for synaptic tagging and capture .............................. 29
Schematic of dnMEK1 mice .
...................................
59
Expression of dnMEK1 transgene in transgenic mice ........ 60
MEK activity in dnMEK1 transgenic mice ........................... 60
Fear conditioning results in dnMEK1 transgenic mice ........ 61
L-LTP phenotype in dnMEK1 transgenic mice .................... 62
Translational regulation underlies L-LTP deficit
in dnM EK1 mice ...............................................................
63
Translational efficiency as a function of poly-A length ........ 70
Activity and MEK inhibitors block translation of reporter ..... 71
Neuronal activity induces translation of reporter
in a MEK dependent manner ..............................................
72
Role of MEK on neuronal activity induced translation of
reporter is not via CPE.
....................................
73
Role of MEK on neuronal activity induced translation of
reporter is not via polyadenylation ......................................
74
Neuronal activity and MEK blockade do not alter
transfection efficiency or toxicity .........................................
74
MEK activity is sufficient to induce translation of reporter .. 75
Metabolic labeling shows that activity induces MEK and
mTOR mediated translation of diverse set of mRNAs ....... 76
Activity induces MEK and mTOR mediated phosph.
of S6, elF4E and 4E-BP1 ..........................................
78
Synaptoneurosomal translation and S6 & elF4E phosph.
is MEK dependent ..........................................
80
L-LTP inducing tetani stimulate translation and ERK, S6
& elF4E phosphorylation in a MEK dependent manner ...... 81
Behavioral training induces ERK, S6 and elF4E phosph.
in a MEK dependent manner .........................................
83
Neuronal activity induces translation in cells grown in
serum-free media .................
.........................................
98
Neuronal activity induces translation in mature cells .......... 99
Dopamine and spaced depolarization but not single
depolarization stimulates translation ................................. 101
DHPG stimulates translation of the same molecules in a
MEK dependent manner via phosph. of S6 & elF4E........ 102
Neuronal activity does not stimulate protein degradation. 103
Stimulated translation lasts at least 45 minutes ................ 104
Model for L-LTP and L-LTD association ............................ 106
BDNF overexpression prevents chronic-stress induced
hippocampal atrophy and improves performance on the
9
Table of Figures
Figure 4-2:
Figure 4-3:
Figure 4-4:
Figure 4-5:
Figure 5-1:
Porsolt Forced Swim Test task........................................
BDNF overexpression does not affect commonly used
endocrine indicators of stress ........................................
Chronic-stress reduces BDNF protein, but not mRNA .....
BDNF overexpression increases BLA spine density,
but does not cause dendritic hypertrophy .........................
BDNF overexpression causes anxiety ..............................
Model for MEK and mTOR dependent translation ............
10
117
121
122
124
125
138
List of Abbreviations
4EBP
elF4E binding protein
5' TOP
5' terminal oligopyrimidine tract
aCSF
artificial cerebrospinal fluid
ACTH
adrenocorticotropic hormone
ACX
acetoxycycloheximide
AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazole
ANI
anisomycin
AP5/APV
2-amino-5-phosphonovaleric acid
BDNF
brain-derived neurotrophic factor
BLA
basolateral amygdala
CaMK
calcium/calmodulin dependent kinase
cAMP
cyclic adenosine-mono-phosphate
propionic acid
CBP
CREB binding protein
CIS
chronic immobilization stress
cMEK
constitutively active MEK
CPE
cytoplasmic polyadenylation element
CPEB
CPE binding protein
CPSF
cleavage and polyadenylation specificity factor
CRE
cAMP responsive element
CREB
CRE binding protein
CRF
corticotropin releasing factor
CS
conditioned stimulus
CYC
cycloheximide
DHPG
(RS)-3,5-dihydroxyphenylglycine
DIV
days in vitro
dnMEK
dominant negative MEK
DNQX
5,6-dinitroquinoxalinedione
eEF
eukaryotic elongation factor
EGFP
enhanced green fluorescent protein
elF
eukaryotic initiation factor
ELISA
enzyme-linked immunosorbent assay
E-LTP
early phase of long-term potentiation
ERK
extracellular-signal regulated kinase
FBS
fetal bovine serum
GR
glucocorticoid receptor
HAT
Histone acetyltransferase
HFS
high-frequency stimulation
HPA
hypothalamic-pituitary-adrenal gland
IEF
isoelectric focusing
ITF
intermediate-term facilitation
11
List of abbreviations
ITM
intermediate-term memory
L-LTP
late phase of long-term potentiation
LTF
long-term facilitation
LTM
long-term memory
LTP
long-term potentiation
MAP2
microtuble-associated protein 2
MAPK
mitogen activated protein kinase
MAPKK
MAPK kinase
MAPKKK
MAPKK kinase
mGluR
metabotropic glutamate receptor
MR
mineralocorticoid receptor
mTOR
mammalian target of rapamycin
NMDA
N-methyl-D-aspartic acid
NT-3/NT3
neurotrophin 3
NT-4/NT4
neurotrophin 4
PABP
poly-A binding protein
PAGE
polyacrylamide gel electrophoresis
PAP
poly-A polymerase
PFA
paraformaldehyde
PPF
paired-pulse facilitation
PP-LFS
paired-pulse low-frequency stimulation
PTSD
posttraumatic stress disorder
SDS
sodium dodecyl sulphate
STF
short-term facilitation
STM
short-term memory
TBS
theta burst stimulation
trk
tropomyosin related kinase
TTX
tetrodotoxin
US
unconditioned stimulus
UTR
untranslated region
YFP
yellow fluorescent protein
ZBP
zipcode binding protein
12
Chapter 1 - Review of long-term memory changes in
the brain
Abstract
Long-term memory can be distinguished from short-term memory by its requirement
for protein synthesis. Converging evidence implicates a short period around the time of
training during which new proteins are produced that are important for the consolidation
of short-term memory into long-term memory. These processes have cellular correlates
- L-LTP is dependent on translation whereas E-LTP is not. Though traditional studies
of LTP have pointed to transcription as a primary control point in these phenomena,
increasing evidence suggests translation as a key regulatory step during the induction of
these processes. Changes in the morphology of neurons may be the consequence of the
protein synthesis required for long-term memory. These changes in morphology may also
underlie the behavioral changes seen after chronic stress and in stress-related disorders
like major depression, PTSD, anxiety disorders and Cushing's syndrome.
13
Chapter 1 - Review of long-term changes in the brain
Protein-Synthesisand Long-term Memory
A role for protein-synthesis in memory was first postulated in 1950, when it was
proposed that crystalline protein lattices could be the substrate underlying memory traces
(Katz and Halstead, 1950). Since then, hundreds of experiments using protein-synthesis
inhibitors have been used to study the role of protein synthesis in learning and memory,
concluding that translation is required for long-term memory, but not for task acquisition or
short-term memory. This process by which short-term memory is converted to long-term
memory in a protein-synthesis dependent manner is termed consolidation. This process
occurs in both vertebrates and invertebrates, as will be discussed below.
Vertebrates
In general, protein-synthesis inhibition during training does not impair acquisition
of the task. When mice are trained to escape a mild foot-shock in a left-right discrimination
task (Barondes and Cohen, 1967; Oliver et al., 1979), or a light-dark discrimination task
(Cohen and Barondes, 1968), they show identical kinetics of learning and performance,
even when protein-synthesis inhibition of 95% is reached using acetocycloheximide
(ACX) (Barondes and Cohen, 1967; Cohen and Barondes, 1968) and cycloheximide
(CYC) (Oliver et al., 1979). Inhibition of protein synthesis with anisomycin (ANI) also does
not impair acquisition of an avoidance response in a left-right discrimination task (Flood
et al., 1975a; Flood et al., 1975b). More recently, contextual as well as cued auditory fearconditioning results have shown that training does not require protein synthesis (Schafe
et al., 1999).
Similarly, protein-synthesis inhibitors have no effect on short-term memory retention
(less than 3 hrs. after training). This has been shown using different tasks including leftright discrimination (Barondes and Cohen, 1966; Barondes and Cohen, 1967; Oliver et
al., 1979), light-dark discrimination (Barondes and Cohen, 1968a; Barondes and Cohen,
1968b), passive-avoidance (Davis et al., 1976; Watts and Mark, 1971a; Watts and Mark,
14
Chapter 1 - Review of long-term changes in the brain
1971b), fear conditioning (Schafe et al., 1999), and appetitive-discrimination training
(Cohen and Barondes, 1968).
However, all protein-synthesis inhibitors affect long-term memory. In all of the tests
in which both short-term memory and long-term memory have been tested, there is a
consistent finding that short-term memory is unaffected, whereas long-term memory is
(Barondes and Cohen, 1966; Barondes and Cohen, 1967; Barondes and Cohen, 1968a;
Barondes and Cohen, 1968b; Cohen and Barondes, 1968; Davis et al., 1976; Oliver et
al., 1979; Schafe et al., 1999; Watts and Mark, 1971a; Watts and Mark, 1971b). This has
led to the model that behavioral training induces synthesis of molecules that are important
for long-term memory, whereas various post-translational mechanisms account for shortterm memory.
Since ACX, CYC and ANI represent two structurally different classes of protein
synthesis inhibitors, it is unlikely that these results can be explained by effects that are
independent of the effects of the inhibitors on protein synthesis. Numerous results have
also been done using the drug puromycin (Flexner et al., 1963; Flexner et al., 1962;
Flexner et al., 1965a; Flexner et al., 1964; Flexner et al., 1965b); those results are not
described here, as it is unclear if some of the effects seen are due to non-specific effects of
the drug on the animal (Davis and Squire, 1984). However, as protein-synthesis inhibition
causes a variety of different effects including sickness, changes in locomotor activity,
changes in cerebral electrical activity, inhibition of adrenal steroidogenesis, and changes
in catecholamine biosynthesis (Davis and Squire, 1984), does the data support the view
that synthesis of "memory" molecules are required for long-term memory?
There are two possible ways in which these non-specific effects could affect longterm memory. The inhibitors could either affect training or the test for retention of the
memory. The most important evidence pointing against an effect of these inhibitors on
training is that neither training nor short-term memory is affected. In addition, proteinsynthesis inhibition immediately after training also prevents formation of long-term memory
15
Chapter I - Review of long-term changes in the brain
(Agranoff et al., 1966; Barondes and Cohen, 1968b; Davis et al., 1981), thus arguing for
a post-training effect of protein-synthesis inhibition on long-term memory formation.
The other possibility is that protein-synthesis inhibitors affect expression of longterm memory. This is unlikely to be true as inhibition of protein synthesis some time after
training (- 1hr.), but well before testing for retention of the memory does not cause any
amnesia (Barondes and Cohen, 1968b; Geller et al., 1969; Neale et al., 1973; Schafe and
LeDoux, 2000; Schafe et al., 1999). This also argues against the effect of protein synthesis
inhibition being solely to reduce constitutive brain protein levels, which would cause a
general reduction in neuronal function. Since the period during which protein synthesis
is required is small (from the time of training to approximately one hour after training), it
is unlikely that a general reduction in protein levels is the cause of the deficit seen in the
animals. Prolonged inhibition of translation well before training (Davis et al., 1980; Flexner
et al., 1963) or well after training, but before testing, have no effect on long-term memory,
even though constitutive protein levels are reduced (Squire and Davis, 1975).
One important point to note is that neither inhibition of translation after training nor
prolonged inhibition of translation prior to training has any effect - in essence, translation
during training is sufficient for long-term memory formation, suggesting that increased
syntheses of one or more molecules during training are necessary and sufficient for longterm memory formation.
Invertebrates
The effect of protein synthesis inhibitors on long-term memory formation is not
restricted to vertebrates; this has been shown best in the sea-slug Aplysia californica
and the fruitfly Drosophila melanogaster. Long-term sensitization of the gill withdrawal
reflex in Aplysia is inhibited when protein-synthesis inhibitors are applied to the ganglia
(Castellucci et al., 1989). In Drosophila, the most common measure of memory uses an
associative odor-conditioning paradigm, where flies are taught to avoid an odor by pairing
that odor with a foot-shock. In this paradigm, short-term memory (10 minutes) is unaffected
16
Chapter 1 - Review of long-term changes in the brain
when the flies are fed cycloheximide, whereas long-term memory is severely though not
entirely inhibited; this incomplete inhibition occurs because flies have a second proteinsynthesis independent consolidation mechanism that is sensitive to anesthesia (Tully et
al., 1990; Tully et al., 1994).
Extinctionand Reconsolidation
In addition to a role for protein-synthesis on consolidation, other behavioral
phenomena also have protein-synthesis dependent phases. When animals that have
been trained to associate a conditioned-stimulus (CS) with an unconditioned-stimulus
(US), are re-exposed for an extended period of time to the CS, they no longer exhibit
the unconditioned-response - a process known as extinction. In rodents, consolidation
of extinction of contextual fear-conditioning (Suzuki et al., 2004) and conditioned tasteaversion (Berman and Dudai, 2001; Berman et al., 2003) have been shown to be proteinsynthesis dependent. This process has also been confirmed in the invertebrate Lymnae
(Sangha et al., 2003b).
Another interesting process that involves protein synthesis is that of reconsolidation.
It was shown that retrieval of a consolidated memory makes it labile (Przybyslawski
and Sara, 1997; Roullet and Sara, 1998), and a second round of protein-synthesis is
required to reconsolidate it. This was shown first in rodent fear-conditioning models,
where treatment with anisomycin at the time of retrieval of a memory results in loss of
the memory (Debiec et al., 2002; Nader et al., 2000; Schafe et al., 2001). Results from
conditioned taste-aversion (Berman and Dudai, 2001; Berman et al., 2003), as well as
from fear-conditioning in fish (Eisenberg et al., 2003), and Lymnae (Sangha et al., 2003a)
have confirmed this conclusion.
Thus translational processes are important for consolidation, consolidation of
extinction, and reconsolidation, leading to the hypothesis that the formation of any type of
long-term memory in the brain requires protein production at the time of training.
17
Chapter I - Review of long-term changes in the brain
Protein-SynthesisDependent Synaptic Plasticity
Vertebrate Long-Term Potentiation
A cellular correlate of memory formation was demonstrated in 1973 by Bliss and
Lomo, who discovered that high-frequency stimulation of the perforant path in vivo elicited
a long-lasting potentiation (LTP) of synaptic strength in the dentate gyrus (Bliss and
Lomo, 1973); interestingly, maintenance of this LTP was shown to be protein-synthesis
dependent (Krug et al., 1984). Similarly, a long-term potentation (LTP) can be elicited at
Schaffer collateral synapses between the CA3 and CA1 regions in hippocampal slices.
Interestingly, this in vitro form of LTP also has multiple phases that have differential
requirements for protein synthesis.
When a single 100 Hz. tetanus is given to the slices, a long-lasting potentiation is
observed that decays to baseline within -2 hrs. This form of potentation has been termed
E-LTP (early-phase of LTP). In contrast, when four spaced trains of 100 Hz. tetani are
given, the potentiation lasts many hours (Frey et al., 1988; Huang and Kandel, 1994);
this form of potentation, called L-LTP (late-phase of LTP), is sensitive to protein-synthesis
inhibition. Whereas translation inhibitors have no effect on E-LTP (Huang and Kandel,
1994), they convert L-LTP to a form that resembles E-LTP (Frey et al., 1988; Huang and
Kandel, 1994).
In a manner analogous to protein-synthesis inhibitors having an effect only around
the time of training during behavioral tasks, the protein-synthesis inhibitors only have an
effect when they are applied during induction of L-LTP.When applied after the tetanization,
no effect is seen (Frey and Morris, 1997). The most parsimonious explanation of these
results is that increased syntheses of one or more molecules during tetanization are
required for the expression of L-LTP, and not that the effect of translation inhibitors is due
simply to lower levels of constitutively present proteins.
In general, it is impossible to guarantee that mechanisms observed in vitro in
hippocampal slices underlie behavior measured in vivo. However, the fact that disruption
18
Chapter 1 - Review of long-term changes in the brain
of pathways that disrupt L-LTP without disrupting E-LTP also disrupt long-term memory
without disrupting short-term memory suggest that the study of pathways in reduced
preparations will yield results likely to be relevant to behavior (Abel et al., 1997; Bach et
al., 1999; Bourtchouladze et al., 1998; Kang et al., 2001).
Vertebrate Long-Term Depression and Priming
Bidirectional modifiability of synapses has been thought to be important for efficient
information storage. A complementary electrophysiological phenomenon to LTP found in
vertebrate synapses is that of long-term depression (LTD). Like LTP, there are multiple
phases of LTD, and maintenance of LTD beyond approximately 1 hour requires protein
synthesis. Inhibitors of protein-synthesis at the time of LTD induction block both NMDA
receptor-dependent LTD (Kauderer and Kandel, 2000; Manahan-Vaughan et al., 2000),
as well as metabotropic glutamate-receptor (mGluR)-dependent LTD (Huber et al., 2000;
Huber et al., 2001).
One other form of synaptic plasticity that is protein-synthesis dependent is priming.
When mGluR receptors are activated prior to an E-LTP inducing tetanus, the E-LTP is
converted to L-LTP. This conversion of E-LTP to L-LTP does not occur when protein
synthesis is blocked (Raymond et al., 2000). Though it is not certain how these different
forms of LTD and priming are relevant for memory, it is interesting to note that protein
synthesis is important for many different enduring forms of synaptic plasticity.
Chemically induced L-LTP and L-LTD
A long-term potentiation can also be elicted in hippocampal slices by the action of
several chemical agents including the neurotrophins BDNF and NT-3, the PKA pathway
activators forskolin and Sp-cAMPs, and dopamine receptor activation (Frey et al., 1993;
Huang and Kandel, 1994; Huang and Kandel, 1995; Kang and Schuman, 1995). These
forms of potentiation differ from electrically induced L-LTP, in that their expression
is completely blocked by translation inhibitors, and they take between 1-2 hrs. to fully
develop. Thus, it is possible that electrically induced L-LTP expression is a summation of
19
Chapter
- Review of long-term changes in the brain
protein-synthesis independent E-LTP type expression, and a protein-synthesis dependent
expression mechanism of the sort that can be induced chemically by neurotrophin, PKA
pathway and dopamine pathway activation.
Invertebrates
Similar electrophysiological phenomena have also been described in invertebrates,
particularly Aplysia. In a manner similar to short-term sensitization of the gill withdrawal
reflex, a relatively short-term increase in synaptic strength lasting minutes can be obtained
at an in vitro sensory neuron-motor neuron synapse by application of a pulse of serotonin.
This increase, termed short-term facilitation (STF), is not blocked by protein-synthesis
inhibition. A longer lasting form of facilitation (LTF) is obtained if 5 spaced applications
of serotonin are presented to the synapse. This LTF is converted to an STF if proteinsynthesis inhibitors are used (Montarolo et al., 1986). Unlike mammals, where the link
between hippocampal electrophysiological results and in vivo behavioral results has
been difficult to make, the mechanism behind STF and LTF are likely to mediate shortterm and long-term sensitization, as the identical physiological changes can be observed
after behavioral training in vivo, and induction of LTF in vivo mimics behavioral training
(Castellucci et al., 1989).
Thus, protein-synthesis dependent synaptic plasticity is an evolutionarily conserved
process that is likely relevant to behavioral phenomena observed in vivo.
The differential role of transcriptionand translation
Most studies examining the role of protein-synthesis dependent memory and
synaptic plasticity have used translation inhibitors as their experimental tool. These
studies do not dissociate between the role of translational regulation and transcriptional
regulation subserving L-LTP and long-term memory. Theoretically, transcription and
translation regulatory mechanisms both have advantages. Transcriptional regulation is
the most efficient assuming that protein-synthesis dependent events occur rarely - by
only transcribing genes as needed, unnecessary production of macromolecules would be
20
Chapter 1 - Review of long-term changes in the brain
reduced. Translational regulation would have the advantage of speed. If macromolecules
are required soon after a stimulus, it would take time for proteins from the cell body to
reach some of the synapses, especially very distal synapses, whereas proteins translated
in dendritic ribosomes (see later section) would be produced near the required site quickly.
In addition, transcriptional responses have the ability to respond to an integrated cell-wide
response, whereas translational responses can response to more localized events, due to
the presence of ribosomes throughout the dendrite. Local translation could also possibly
explain the input-specificity of LTP and LTD, whereas another mechanism would have to
explain the input-specificity with transcription dependent processes.
A few studies have attempted to determine if transcriptional regulation is important
for memory formation, using the transcription inhibitor actinomycin D. In general,
(Agranoff et al., 1967; Appel, 1965; Barondes and Jarvik, 1964; Cohen and Barondes,
1966; Nakajima, 1969; Reinis, 1968; Squire and Barondes, 1970), it was discovered that
inhibition of transcription at the time of training inhibited long-term memory formation, but
not short-term memory formation. Unfortunately, a lot of the studies using actinomycin D
were difficult to interpret because of the sickness it caused during testing; unlike protein
synthesis inhibitors, actinomycin D does not leave cells readily, and thus, transcription
was inhibited continuously from the time of presentation of the drug.
Similar studies have also been performed using L-LTP as an in vitro correlate of
long-term memory. These studies showed that transcription blockade also inhibited LLTP, but not E-LTP (Frey et al., 1996; Nguyen et al., 1994). Interestingly, the inhibition
of L-LTP by transcriptional blockade was always delayed, whereas most studies using
translational blockade showed an immediate blockade of L-LTP (Frey et al., 1988; Otani
et al., 1989; Scharf et al., 2002), though this intriguing kinetic difference has not yet been
discussed in the literature (see also Chapter 2). One caveat has been that the studies
using transcriptional inhibitors and translational inhibitors were not done side-by-side, and
thus, though unlikely, methodological differences may have accounted for the difference.
21
Chapter I - Review of long-term changes in the brain
Interestingly, the case in LTD and priming is clearer, as both mGluR dependent LTD
and priming are not dependent on transcription, as actinomycin D has no effect on either
(Huber et al., 2000; Huber et al., 2001; Raymond et al., 2000). Also, isolated dendrites are
able to support maintenance of LTD, thus arguing against a role for transcription in LTD
(Huber et al., 2000). The case with NMDA dependent LTD is less clear, as the literature
both supports and refutes a role for transcription in the maintenance of NMDA dependent
LTD (Kauderer and Kandel, 2000; Manahan-Vaughan et al., 2000).
In invertebrates, LTF, and long-term sensitization require both transcription and
translation; however, there have been reports of an intermediate-term facilitation (ITF),
and an intermediate-term memory that require translation but not transcription (Sutton
and Carew, 2000; Sutton et al., 2001).
Transcriptional control mechanisms
As transcriptional control mechanisms have generally been better understood,
transcriptional control of learning and memory has been the focus of the learning and
memory field until recently. The first studies to prove a causative role for transcription
focused on the transcription-factor CREB in Aplysia. It had been known that increases
in cAMP and the PKA pathway were involved in LTF and long-term memory in Aplysia.
Furthermore, results from mammalian cell lines had shown a 8 nucleotide sequence,
termed the cAMP response element (CRE) in the promoter-proximal region of the
somatostatin gene that stimulated transcription of the gene in response to elevated cAMP
(Montminy and Bilezikjian, 1987). Theorizing that the CRE was involved in LTF and LTM,
Kandel and colleagues injected oligonucleotides encoding the CRE into Aplysia neurons
in an attempt to outcompete endogenous CRE sites for binding to CREB, thus reducing
transcription of CRE-dependent
genes. They discovered that, in a manner analogous
to translation inhibition, LTF was selectively inhibited, whereas control-oligonucleotide
inhibition had no effect (Dash et al., 1990).
22
Chapter I - Review of long-term changes in the brain
This result was extended into fruit-flies, where transgenic overexpression of
a dominant-negative CREB abrogated long-term odor-association memory (Yin et al.,
1994). Interestingly, there is also a report that overexpression of a CREB activator allows
for long-term memory to be formed even with only one training episode - a type of training
that normally only forms short-term memory (Yin et al., 1995). Experiments in mice were
soon to follow, and yielded similar results. A partial knockout of CREB, which selectively
removes the a and A isoforms of CREB was found to have specific deficits in long-term
memory and L-LTP, consistent with a role for CREB and transcriptional processes, in
general, in persistent forms of synaptic plasticity and memory (Silva et al., 1998).
Though CREB is the best studied example of a transcriptional factor important
for long-term memory, other transcription factors have also been implicated in these
processes. For example, Elk-1 has been shown to be activated by LTD inducing stimuli in
vivo (Thiels et al., 2002), and Notch is required for L-LTP and long-term memory (Costa
et al., 2003; Wang et al., 2004) in mice, as well as long-term memory in Drosophila (Ge
et al., 2004; Presente et al., 2004). In addition, CREB activation induces transcription
of several immediate-early genes (IEGs) including c-fos, C/EPP and zif268, which in
turn are also transcription factors. Thus, plasticity mechanisms likely recruit multiple
transcriptional cascades that produce the proteins required L-LTP and long-term memory.
Interestingly, there are subtle differences in the role of the IEGs in behavior. For example,
the neurotrophin BDNF and the transcription factor zif268 are both CREB dependent IEGs.
Yet, there is evidence that BDNF is involved in consolidation and zif268 in reconsolidation
(Lee et al., 2004). Whether transcription is a unitary process or it is differentially activated
by different neuronal stimuli remains to be seen. In addition, the different roles of the
transcribed genes in behavior have yet to be fully elucidated, though the BDNF / zif268
result mentioned above is a start. Lastly, the proportion of the transcriptome of the neuron
that is upregulated by plasticity inducing events has yet to be seen. Microarray data that
is currently underway in many labs will surely shed light on these questions.
23
Chapter I - Review of long-term changes in the brain
Transcriptional regulation also has been shown via mechanisms including histone
acetylation and deacetylation. Indeed CREB requires an accessory protein, known as
CREB Binding Protein (CBP) with histone acetyl transferase
(HAT) activity. This HAT
activity is activated by LTP in the hippocampus and is essential for long-term memory
(Korzus et al., 2004; Levenson et al., 2004). Thus, a multitude of nuclear events are
activated by plasticity inducing stimuli.
Translational Control Mechanisms
The mechanisms underlying translational control has not been as well studied in
cognitive neurosciences, as it has generally been assumed that translation was essentially
constitutive. However, three different cases of activity induced translation have been
suggested. The first case involves regulated polyadenylation. Certain mRNAs have a cisacting element, called a cytoplasmic polyadenylation element (CPE), which binds a protein
called the CPE binding protein (CPEB). This causes those mRNAs to lie in a translationally
dormant phase with a short poly-A tail. When activated, CPEB becomes phosphorylated,
which derepresses translation of the mRNAs, while concomitantly increasing the poly-A
tail length, which, in turn, increases translational efficiency of the mRNAs. Though, this
mechanism has best been elucidated in Xenopus, there is evidence that exposure to
light of dark-reared rats induces polyadenylation of the CPE-containing mRNA a-CaMKII
in the visual cortex (Wu et al., 1998). In addition, there is evidence that NMDA receptor
activation activates translation of a CPE-containing reporter (Wells et al., 2001).
Another translational mechanism postulated to have neuronal function involves the
fragile-X mental retardation protein (FMRP), which is mutated in fragile-X patients. FMRP
is a translational inhibitor, which acts by binding to specific mRNAs (Laggerbauer et al.,
2001; Li et al., 2001) that either have G-quartets (Darnell et al., 2004) or via adaptor RNAs
like BC1 (Zalfa et al., 2003). Interestingly, mice with null mutations in fmr-1 (the gene that
produces FMRP) have increased mGluR-dependent LTD, but normal LTP (Huber et al.,
2002). How this process is regulated is yet to be elucidated.
24
Chapter 1 - Review of long-term changes in the brain
Whereas translational regulation at the level of initiation is the mode of action
of CPEB and FMRP, translational regulation at the level of elongation has also been
shown. Data from superior collicular synaptoneurosomes has shown that NMDA receptor
activation can cause transient decreases in translation followed by a more persistent
increase in translation. The transient decrease, which is correlated with a decrease in
elongation, has the paradoxical effect of increasing production of certain well-initiated
mRNAs like a-CaMKII (Scheetz et al., 2000).
Translationin Neurons
When discussing translation, two important questions arise: 1) Where in the neuron
does translational regulation take place, and 2) How is one to explain the input-specificity
of L-LTP? These issues will be covered in the next section.
Local Protein Synthesis
It was generally assumed, in spite of evidence that ribosomes could be found
in dendrites (Bodian, 1965), that the soma was the sole center of macromolecular
synthesis in the neuron, and that the dendrites and axons of the neuron depended on this
synthesis for their function. This view was first challenged in the early 1980s by Steward
and colleagues (Steward and Fass, 1983; Steward and Levy, 1982) who showed, using
electron micrographs of dentate gyrus granule neurons, that dendritic polyribosomes
were not randomly distributed, but instead were present near synapses; specifically, at
spine synapses, the polyribosomes were present at the base of the spine, and at nonspine synapses, the polyribosomes were localized to the area immediately beneath the
postsynaptic membrane. The authors termed these synapse-associated polyribosome
complexes (SPRCs), and suggested that the synapse-specific localization of the SPRCs
might be due to neurons synthesizing proteins in a synaptically localized manner (Steward
et al., 1996).
These early observations were extended in subsequent years by various
laboratories who showed that many components of the endoplasmic reticulum (ER),
25
Chapter I - Review of long-term changes in the brain
including ribophorin I and BiP, as well as components of the Golgi complex, such as
TGN38, rabl and CTR433, were at synapses (Gardiol et al., 1999; Lowenstein et al.,
1994; Torre and Steward, 1992; Torre and Steward, 1996); furthermore, it was shown
that the synaptic compartment, isolated as synaptosomes, was capable of synthesizing
proteins, as well as glycosylating membrane proteins (Rao and Steward, 1991; Torre
and Steward, 1992; Torre and Steward, 1996). These results were particularly intriguing,
because they showed that dendrites were capable of synthesizing not just cytosolic
proteins, but membrane molecules as well; indeed, the repertoire of proteins that the
dendrite could synthesize may have been limited only by the mRNAs present.
These studies raised the question of what proteins were translated in dendrites.
In an attempt to answer this question, a number of groups began cataloging the mRNAs
localized to synapses. Initially, the methodology of choice was in situ hybridization
which yielded several dendritically localized mRNAs, including MAP2, and a-CaMKII,
that had known neuronal function; however, more recently, with improvements in mRNA
amplification techniques and microarray technology, the list of dendritically localized
mRNAs, though not yet exhaustive, has grown and contains mRNAs encoding proteins
of many different types including receptor subunits such as the NMDAR1, cytoplasmic
kinases including (a-CaMKII, receptor kinases such as trkB, and cytoskeletal components
such as y-actin (Benson, 1997; Burgin et al., 1990; Tongiorgi et al., 1997); intriguingly, the
mRNA of the transcription factors CREB and zif268 may be localized to dendrites (Crino
et al., 1998), leading to the suggestion that synaptic events may be able to influence
nuclear processes in a manner that bypasses somatic cell signaling.
In general, since most of the studies searching for dendritically localized mRNAs
have not examined spatial segregation of the mRNAs within the dendrite, it has generally
been assumed that most dendritic mRNAs are localized throughout the dendritic
segment with no synapse specificity. However, there is evidence that the trafficking of
mRNAs may be regulated by activity. One piece of evidence was provided by Kosik and
26
Chapter 1 - Review of long-term changes in the brain
colleagues, who used, as a model mRNA, a chimeric mRNA containing the 3' UTR of
a-CaMKII, which is necessary and sufficient for localization of a-CaMKII mRNA to the
dendrites. By tagging the mRNA using the bacteriophage MS2 system, they showed that
depolarization of hippocampal cultured cells increased the anterograde transport of RNA
granules containing the chimeric RNA (Rook et al., 2000). In addition, it is theorized that
the Arc/Arg3.1 mRNA is targeted only to stimulated synapses (Steward et al., 1998). The
mechanism behind this regulated dendritic transport is still being elucidated - to date,
many different proteins including staufen (Kiebler et al., 1999; Kohrmann et al., 1999;
Tang et al., 2001), ZBP1 (Tiruchinapalli et al., 2003), and CPEB (Huang et al., 2003) have
been implicated (see below).
Another important issue raised by the dendritic-synthesis hypothesis was the role
of this potentially synapse-specific protein synthesis in neuronal function. One tantalizing
possibility was that the input-specificity of protein-synthesis dependent plasticity was
mediated by local-synthesis; i.e. the tetanized synapse synthesized locally, and used
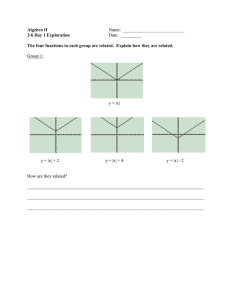
locally, components required by the synapse for potentiation (Fig. 1-1). This point of view
was supported by evidence from Aplysia that showed that when a single synapse of a
bifurcated sensory neuron was stimulated with five pulses of serotonin, protein synthesis
at the stimulated synapse was required for the long-term facilitation (LTF) of the synapse
(Martin et al., 1997). In cultured rat hippocampal cells, using transected dendrites from
cells transfected with a GFP reporter construct, Aakalu et al showed that stimulation of
the dendrites with BDNF led to an increase in GFP levels (Aakalu et al., 2001). Similarly,
in isolated dendrites, it was shown that exponential translation in dendrites could be
stimulated by treatment with DHPG, an mGluR Type I agonist (Job and Eberwine, 2001).
In addition, potentiation in slices by BDNF, a form of protein-synthesis dependent plasticity
similar to L-LTP, could also occur in the absence of the cell body (Kang and Schuman,
1996). Using fluorescently labeled techniques, it was shown that activity can upregulate
translation of GluR1 in dendrites (Ju et al., 2004). These results all have contributed to
27
Chapter 1 - Review of long-term changes in the brain
the hypothesis that mRNAs are diffusely localized throughout the dendritic compartment,
and that local synapse-specific translation may account for the input-specificity of proteinsynthesis dependent forms of LTP and LTF.
One important note is that, though the Aplysia experiments indicate that stimulated
synapse-specific translation can occur, in the mammalian system this has not been
shown - instead stimulated dendritic synthesis has been demonstrated. This is, in part, a
limitation of the mammalian system, and it remains to be seen how localized the spread of
translation in mammalian dendrites is, since the validity of the local-synthesis hypothesis
depends crucially upon increased translation being localized only to the stimulated
synapse.
I f
4. mRNA
1. Stro
Teta
n
2. Stimulation
Translation
Figure 1-1: The model for local protein-synthesis as the substrate for input-specificity. When a
strong tetanus is given to a synapse (1), local-translation (2), and transcription (3) are activated.
mRNAs (4) produced as a result of the transcription travel throughout the neuron, but are only
translated at the activated synapse. Thus structural change is localized (5), leading to input-specific
expression of L-LTP
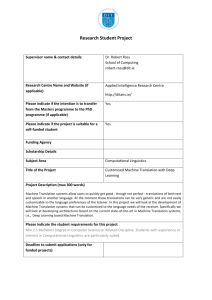
Synaptic Tag Hypothesis
Another prevalent model explaining the input-specificity of protein-synthesis forms
of plasticity
is the synaptic tag hypothesis (Fig 1-2); this theory, states that tetanized
28
Chapter
- Review of long-term changes in the brain
synapses are "tagged", and one consequence of the tag is that the tetanized synapses
"capture" proteins, which in turn causes expression of LTP. Thus, this theory has no
requirement for local protein-synthesis. Evidence for this hypothesis was first obtained by
Frey and Morris (Frey and Morris, 1997), who tetanized one input with an L-LTP inducing
stimulus. Later, they tetanized a separate input in the presence of the protein-synthesis
inhibitor anisomycin. They found that the second input also was potentiated in the same
way as the first input, which is incompatible with the local protein-synthesis hypothesis
which would have predicted that only the first input would be potentiated. This led to the
synaptic-tag hypothesis with the important feature that the tag and capturing process is
protein-synthesis independent. These experiments were later confirmed by Eric Kandel
and colleagues, both in Aplysia as well as in mouse hippocampus (Barco et al., 2002;
Martin et al., 1997).
4. Stimulation of Transcription
I
.'
1'. Weak
Tetanus
Figure 1-2: Synaptic Tag and Capture model as a substrate for input specificity: When a strong
tetanus is applied to an input (1), a tag is created at that input (2). Translation (3) and transcription
(4) are activated, and proteins are produced. Tagged inputs capture the proteins leading to structural change only at that input (5) leading to the expression of L-LTP. If another input is weakly
tetanized (1') within a relatively short temporal and spatial distance from the strongly tetanized
input, only a tag is produced (2'). This input can capture available proteins, leading to structural
change (5'), leading to expression of L-LTP. Note that only the tag, whose production is translation
independent, is localized to activated synapses.
29
Chapter 1 - Review of long-term changes in the brain
Since L-LTP requires protein synthesis, and the second input was tetanized in the
absence of protein-synthesis, these results indicate that local-protein synthesis is not
sufficient for input-specificity, and another translation-independent tag is required for the
input-specific induction of L-LTP. Interestingly, this tag has a life-time of 1-2 hrs., and can
be induced by E-LTP inducing stimuli as well. (Barco et al., 2002; Frey and Morris, 1997;
Martin et al., 1997).
RNA Tag Hypothesis
The RNA-tagging hypothesis theorizes that transcriptional products are
localized to the specific synapses that were tetanized, and that these localized RNAs
mediate input-specificity by synapse-specific interactions with proteins and the translational
machinery. The archetypical localized mRNA is the IEG Arc/Arg3.1; it was shown that
tetanic stimulation of the dentate gyrus caused an increase in Arc transcription, and
furthermore, unlike most dendritic RNAs which are localized throughout the dendrite, the
newly synthesized mRNA was transported from the soma to the specific layer tetanized,
leading to the speculation that Arc is specifically transported to stimulated synapses
(Steward et al., 1998). Since the spatial resolution of those experiments was not high
enough, it remains to be proven that Arc translocation is directed to only those synapses
which are tetanized. Assuming that Arc is translocated to stimulated synapses only, an
interesting question is whether the tag that allows for the specific targeting of Arc is the
same as the "induction" tag responsible for the input specificity of L-LTP.
RNA Targeting Mechanisms
Mechanistically, the natures of the proteins involved in the transport and localization
of some mRNAs to the dendrite are being investigated. mRNA localization is not unique
to the neuronal system - indeed, it has been extensively studied during development
in mammals, Drosophila, and Xenopus. For example, the bicoid, oskar and prospero
mRNAs are localized to specific intracellular compartments; this specific localization
is required for the function of these mRNAs. Much work has gone into identifying the
30
Chapter 1 - Review of long-term changes in the brain
mechanisms leading to this localization. One important molecule that is responsible for
the localization of the bicoid, oskar and prospero mRNAs is staufen (Kiebler et al., 1999;
Roegiers and Jan, 2000). In mammalian neurons, Staufen is localized to dendrites, and
promotes transport of mRNA into the dendrites (Tang et al., 2001). However, the specific
mRNAs whose transport is dependent on Staufen is not known, though the MAP2 mRNA
is one likely target of Staufen.
Another important molecule implicated in mRNA transport is the zipcode binding
protein 1 (ZBP1). Though all the targets of this RNA are not known, it is clear that the 03actin mRNA is one of the mRNAs whose transport into the dendrites depends on ZBP1.
Evidence from Tiruchinapalli et al shows that depolarization causes an increase in ZBP1
colocalization with 13-actin mRNA, and an increase in dendritic targeting of ZBP1, leading
to the model that strong activity causes an increase in f3-actin mRNA in the dendrites in
a ZBP1 dependent manner (Tiruchinapalli et al., 2003). Recent evidence has shown that
CPEB, normally thought to be involved only in translational regulation, also facilitates
transport of CPE containing mRNAs, such as a-CaMKII, into dendrites (Huang et al.,
2003).
Dendritic vs. Somatic Protein Synthesis
It has been shown that dendritic protein-synthesis is sufficient for many
forms of protein-synthesis dependent forms of plasticity in hippocampal slices including
BDNF and cAMP dependent forms of L-LTP. However, unlike the Aplysia system the
question as to its necessity has not been fully demonstrated. One experiment that
provided a partial answer was the creation of mice where the 3' UTR of (a-CaMKII was
replaced by the 3'UTR from bovine growth hormone. The resulting mice, which express
an a-CaMKII transcript that is not dendritically localized, show deficits in L-LTP and longterm memory (LTM). This result is consistent with the hypothesis that dendritic proteinsynthesis is required for L-LTP and LTM (Miller et al., 2002). One caveat is that removal
of the 3' UTR also removes the cytoplasmic-polyadenylation elements (CPEs), which
31
Chapter 1 - Review of long-term changes in the brain
would affect translational regulation of the mRNA, which might affect L-LTP and LTM.
Analysis of animals with more selective disruption of the dendritic localization sequences
(Blichenberg et al., 2001; Mori et al., 2000), and animals with mutations in RNA trafficking
proteins will shed further light on the necessity of dendritic protein synthesis for L-LTP and
LTM.
StructuralPlasticity
Long-term memory necessarily involves changes that are longer lived than any
molecule; in essence, it the result of a change in state brought about by the action of
many molecules that appear to have been produced as a result of transcriptional and
translational stimulation by plasticity-inducing stimuli. One possible "state-change" is a
change in structure, and this has often been postulated as a mechanism behind the longlasting changes that underlie long-term functionality in the brain.
Such an idea was proposed in modern neuroscience by Tanzi, Ramon y Cajal,
and Sherrington, and later expanded upon by Hebb, and Konorski (Lamprecht and
LeDoux, 2004). However, the key evidence that structural changes are brought about
by learning and/or plasticity inducing phenomena, and that this structural change has
functional consequences has only been recently sought. Conceptually, these structural
changes may manifest themselves as a change in the density, size and shape of spines
and presynaptic boutons (i.e. synapses), changes in axonal and dendritic length and
arborization, and changes in the number of cells. Some of these will be considered and
explored in this section.
Structural Changes after Behavior
Structural changes have been observed after different kinds of behavioral training,
including in the visual cortex of rats raised in an "enriched" environment (Greenough et
al., 1985), cerebellum after motor learning (Anderson et al., 1996), hippocampus after
eye-blink conditioning (Geinisman et al., 2001; Leuner et al., 2003), and piriform cortex
after olfactory conditioning (Knafo et al., 2001). However, the requirement for protein
32
Chapter 1 - Review of long-term changes in the brain
synthesis in these changes has not yet been studied. As long-term memory has a critical
requirement for protein synthesis, demonstration of a requirement for protein-synthesis
would bolster the argument that the role of protein-synthesis is to produce proteins that
are required for structural change, and that it is this structural change that is required for
L-LTP and long-term functional changes in the brain.
Structural Changes after Synaptic Plasticity
Morphological changes after changes in synaptic plasticity have been observed
after LTF in Aplysia. When five pulses of serotonin are applied to sensory neuron-motor
neuron synapses, an increase in presynaptic varicosities occurs that depends on the
presence of the postsynaptic cell (Glanzman et al., 1990). Interestingly, the serotonin
mediated increase in synaptic density, and FMRFamide (a chemical that causes longterm depression in Aplysia) induced reduction in synaptic density both require translation
and transcription (Bailey et al., 1992). This has provided a nice link between LTF, protein
synthesis, structural changes and behavioral changes.
In vertebrates, the evidence is not as clear. Initial evidence in favor of the model
came from a live-cell imaging study in slice-culture that showed that one form of LTP
produced an increase in spines that lasted many hours. The difficulty with this study
was that no evidence was provided that the LTP produced was L-LTP, and whether the
formation of the spines, as well as their maintenance was dependent on protein synthesis.
Indeed, the protocol used resembled an E-LTP protocol, and the spines shown resembled
filopodia (Engert and Bonhoeffer, 1999). Asimilar study also showed that an E-LTP inducing
stimulus caused growth of filopodia in slice cultures (Maletic-Savatic et al., 1999). One
possible explanation for this finding is that E-LTP causes the formation of filopodia, but
various protein-synthesis dependent processes are used by L-LTP to convert filopodia
into mature, stable synaptic contacts. One major caveat of these studies is that they
were done using slice cultures, which are obtained from juvenile rats; as it is known that
morphological changes in juvenile and adult rats are very different (Trachtenberg et al.,
33
Chapter I - Review of long-term changes in the brain
2002), it is yet to be seen if the changes seen in these studies will be duplicated in adult
slices.
There have also been reports that claim that LTP causes splitting of dendritic spines
(Toni et al., 1999), though this is still controversial (Fiala et al., 2002). LTP induction in
slices also causes an increase in the size of spines in an acute slice preparation, but
the dependence of the size change on protein synthesis was not explored (Ostroff et
al., 2002). Changes in spine size, and spine-neck size have also been reported (Fifkova
and Anderson, 1981; Fifkova and Van Harreveld, 1977; Van Harreveld and Fifkova,
1975) in the dentate gyrus after perforant-path high-frequency stimulation. Interestingly,
anisomycin treatment at the time of high-frequency stimulation inhibits these changes;
however, as the anisomycin washes out, and protein synthesis resumes, the changes
in spine morphology returned (Fifkova et al., 1982). This is in contrast to L-LTP or LTM,
neither of which reappear after the anisomycin washes out. Thus, there are still some
discrepancies between these structural changes and the facts about the protein-synthesis
dependence of L-LTP and LTM, implying that other protein-synthesis dependent changes
that are necessary for L-LTP and LTM.
Structural Changes in Stress related disorders
Structural changes have also been seen as a result of more chronic perturbations
like chronic-stress. Chronic immobilization-stress, and chronic restraint-stress have been
shown to reduce dendritic length, and arborization in the apical dendrites of the CA3 region
of the rat and tree shrew hippocampi. This chronic-stress induced decreases in dendritic
arborization is reversible over a period of time, if the stressor is removed (Magarinos and
McEwen, 1995; Magarinos et al., 1996; Vyas et al., 2002). Similar reversible changes in
structure are also seen in the CA1 region through the estrous cycle, and stress-induced
changes in the brain are different in male and female rats (Shors et al., 2001; Shors et al.,
2004). Interestingly, there is a marked loss of spines during hibernation of squirrels that
34
Chapter I - Review of long-term changes in the brain
is rapidly reversed within hours of waking up (Popov et al., 1992); whether this process is
related to the estrous cycle and chronic-stress induced changes is not yet known.
Chronic-stress induced morphological changes are likely to be relevant to plasticity
induced structural changes for two key reasons: 1) the molecular signatures of the two
strongly overlap, and 2) chronic-stress affects behavior through its actions on regions of
the brain important for cognitive behavior, such as the hippocampus, amygdala, striatum
and pre-frontal cortex. One key molecular similarity between the two is that NMDA receptor
activity is required for LTP (Harris et al., 1984), some forms of LTD (Dudek and Bear,
1992; Kirkwood et al., 1993), LTP induced structural changes (Toni et al., 1999), and
chronic-stress induced morphological changes (McEwen, 1999). In addition, reducing
serotoninergic transmission prevents chronic-stress induced CA3 atrophy, as does
increasing GABAergic transmission. Both of these systems are also intimately associated
with learning and memory.
One key system that is intimately involved with stress-related phenomena is that
of the neuroendocrine system. Specifically, when an animal is subjected to stress, the
hypothalamus releases corticotropin releasing factor (CRF), which stimulates release
of ACTH from the pituitary gland; this, in turn, activates release of glucocorticoids
(corticosterone in rodents and cortisol in humans) from the adrenal gland. The hippocampus
is enriched in glucocorticoid receptors (GR) and mineralocorticoid receptors (MR); chronic
upregulation of glucocorticoids, as observed under chronic-stress conditions, is required
for the dendritic atrophy seen in chronically-stressed rodents. These receptors are also
required for learning and memory and LTP. Interestingly, the hippocampus, normally
thought of as being important only for cognition, is involved in the activity of hypothalamuspituitary-adrenal axis (HPA axis) as an inhibitor of the axis. The amygdala, a region of key
importance for emotional processes, stimulates the axis (McEwen, 1999).
35
Chapter I - Review of long-term changes in the brain
Structural Plasticity and Affective Disorders
Mood and affect disorders are debilitating diseases that affect a significant
proportion of the population. For example, major depression affects 2-5% of the U.S.
population, while lesser forms may affect as much as 20% (Nestler et al., 2002). Many
of these disorders, including anxiety, depression, and post-traumatic stress disorder
(PTSD) share considerable overlap, both in their symptomatology, as well as in currently
available treatments (Mineka et al., 1998). For example, many commonly administered
antidepressants, such as fiuoexetine, also improve symptoms of anxiety and PTSD
(Strohle and Holsboer, 2003). This, in addition to the fact that many of these disorders
share similar neuroendocrine features (Strohle and Holsboer, 2003), and the fact that
stressful experiences have been linked with several cases of depression and anxiety
(Kessler, 1997), has led to the classification of these disorders as stress-related disorders
(Nestler et al., 2002).
The neural circuits underlying stress-related disorders are still under investigation.
Brain imaging studies have pointed to changes in depressed patients in the function of
several brain areas including the prefrontal cortex, hippocampus, amygdala and striatum
(Drevets, 2001; Liotti and Mayberg, 2001). It has been suggested that the hippocampus
and prefrontal cortex might mediate cognitive aspects of affect and mood disorders,
including memory impairments and feelings of hopelessness and worthlessness. On the
other hand, the amygdala and striatum are postulated to mediate the anhedonia, anxiety
and reduced motivation seen in patients with mood disorders (Nestler et al., 2002).
However, the exact contribution of each of these structures to the behavioral symptoms
in these patients is still debated.
The cellular basis of stress-related disorders is not yet understood. Several pieces
of evidence point to a change in brain structure as playing a key role in these diseases.
Firstly, treatment with antidepressants for many months is required for resolution of
depressive symptoms, pointing to long-term changes such as structural changes as
36
Chapter 1 - Review of long-term changes in the brain
being important (Nestler et al., 2002). Secondly, changes in hippocampal volume have
been found in patients suffering from major depression (Bremner et al., 2000; Sheline et
al., 2003; Sheline et al., 1996), PTSD (Bremner et al., 1995; Bremner et al., 1997), and
Cushing's Syndrome (Starkman et al., 1992; Starkman et al., 1999), which shares some
neuroendocrine similarities to depression and anxiety disorders; in addition, medication
can reverse some of these volume changes (Sheline et al., 2003). Lastly, animal models
of chronic-stress have shown decreases in dendritic length and branching of hippocampal
CA3 cells (McEwen, 1999), which may be the cause of the volume loss phenomenon seen
in human patients as well. Supporting this model is the fact that antidepressants have also
been shown to increase the dendritic length of these cells. Data from the amygdala has
pointed to increased amygdalar volume in depression and anxiety disorders (Anand and
Shekhar, 2003), and animal models of chronic-stress show an increase in the dendritic
length and spine density in spiny pyramidal cells of the basolateral amygdala (Vyas et al.,
2003; Vyas et al., 2002).
Conclusion& Specific Aims
Organisms use memories and skills that have been acquired throughout their
history, in their daily actions. These skills and memories must exist in the brain for an
extended period of time. This requires protein synthesis, and these proteins are thought
to be used in changing the structure of neurons in a long-lasting way, leading to longlasting skills and memories. These structural changes may also take place during certain
disease states leading to altered behaviors.
The working hypothesis is that the changing structures of neurons play a critical part
in the changing behavior of the organism. Furthermore, it is likely that both transcriptional
and translational regulation create altered bidirectional plasticity at synapses, which is
important for optimized coding and cognitive function.
The aim of this thesis is to:
37
Chapter 1 - Review of long-term changes in the brain
1. Characterize activity-induced translational control in neurons and determine if
it is important for long-term synaptic plasticity and memory. The MAP Kinase
pathway was selected for analysis due to its role in translation in other celltypes, and for its described role in learning and memory.
2. Characterize a mouse overexpressing the neurotrophin BDNF to study the
correlation between BDNF, chronic stress-induced structural change, and
affective behaviors. This approach was used to overcome the fact that structural
changes after behavioral training would likely involve only a limited number of
synapses making it difficult to obtain a structural phenotype.
38
Chapter I - Review of long-term changes in the brain
Bibliography
Aakalu, G., Smith, W. B., Nguyen, N., Jiang, C., and Schuman, E. M. (2001). Dynamic
visualization of local protein synthesis in hippocampal neurons. Neuron 30, 489502.
Abel, T., Nguyen, P. V., Barad, M., Deuel, T. A., Kandel, E. R., and Bourtchouladze,
R.
(1997). Genetic demonstration of a role for PKA in the late phase of LTP and in
hippocampus-based long-term memory. Cell 88, 615-626.
Agranoff, B. W., Davis, R. E., and Brink, J. J. (1966). Chemical studies on memory
fixation in goldfish. Brain Res 1, 303-309.
Agranoff, B. W., Davis, R. E., Casola, L., and Lim, R. (1967). Actinomycin
D blocks
formation of memory of shock-avoidance in goldfish. Science 158, 1600-1601.
Anand, A., and Shekhar, A. (2003). Brain imaging studies in mood and anxiety
disorders: special emphasis on the amygdala. Ann N Y Acad Sci 985, 370-388.
Anderson, B. J., Alcantara, A. A., and Greenough, W. T. (1996). Motor-skill learning:
changes in synaptic organization of the rat cerebellar cortex. Neurobiol Learn
Mem 66, 221-229.
Appel, S. H. (1965). Effect of inhibition of RNA synthesis on neural information storage.
Nature 207, 1163-1166.
Bach, M. E., Barad, M., Son, H., Zhuo, M., Lu, Y. F., Shih, R., Mansuy, I., Hawkins,
R. D., and Kandel, E. R. (1999). Age-related defects in spatial memory are
correlated with defects in the late phase of hippocampal long-term potentiation
in vitro and are attenuated by drugs that enhance the cAMP signaling pathway.
Proc Natl Acad Sci U S A 96, 5280-5285.
Bailey, C. H., Montarolo, P., Chen, M., Kandel, E. R., and Schacher, S. (1992). Inhibitors
of protein and RNA synthesis block structural changes that accompany long-term
heterosynaptic plasticity in Aplysia. Neuron 9, 749-758.
Barco, A., Alarcon, J. M., and Kandel, E. R. (2002). Expression of constitutively active
CREB protein facilitates the late phase of long-term potentiation by enhancing
synaptic capture. Cell 108, 689-703.
Barondes, S. H., and Cohen, H. D. (1966). Puromycin effect on successive phases of
memory storage. Science 151, 594-595.
Barondes, S. H., and Cohen, H. D. (1967). Delayed and sustained effect of
acetoxycycloheximide on memory in mice. Proc Natl Acad Sci U S A 58, 157-164.
Barondes, S. H., and Cohen, H. D. (1968a). Arousal and the conversion of "short-term"
to "long-term" memory. Proc Natl Acad Sci U S A 61, 923-929.
39
Chapter I - Review of long-term changes in the brain
Barondes, S. H., and Cohen, H. D. (1968b). Memory impairment after subcutaneous
injection of acetoxycycloheximide. Science 160, 556-557.
Barondes, S. H., and Jarvik, M. E. (1964). The Influence of Actinomycin-D on Brain Rna
Synthesis and on Memory. J Neurochem 11, 187-195.
Benson, D. L. (1997). Dendritic compartmentation of NMDA receptor mRNA in cultured
hippocampal neurons. Neuroreport 8, 823-828.
Berman, D. E., and Dudai, Y. (2001). Memory extinction, learning anew, and learning
the new: dissociations in the molecular machinery of learning in cortex. Science
291, 2417-2419.
Berman, D. E., Hazvi, S., Stehberg, J., Bahar, A., and Dudai, Y. (2003). Conflicting
processes in the extinction of conditioned taste aversion: behavioral and
molecular aspects of latency, apparent stagnation, and spontaneous recovery.
Learn Mem 10, 16-25.
Blichenberg, A., Rehbein, M., Muller, R., Garner, C. C., Richter, D., and Kindler, S.
(2001). Identification of a cis-acting dendritic targeting element in the mRNA
encoding the alpha subunit of Ca2+/calmodulin-dependent protein kinase II. Eur
J Neurosci 13, 1881-1888.
Bliss, T. V., and Lomo, T. (1973). Long-lasting potentiation of synaptic transmission in
the dentate area of the anaesthetized rabbit following stimulation of the perforant
path. J Physiol 232, 331-356.
Bodian, D. (1965). A Suggestive Relationship of Nerve Cell Rna with Specific Synaptic
Sites. Proc Natl Acad Sci U S A 53, 418-425.
Bourtchouladze,
R., Abel, T., Berman, N., Gordon, R., Lapidus, K., and Kandel, E. R.
(1998). Different training procedures recruit either one or two critical periods for
contextual memory consolidation, each of which requires protein synthesis and
PKA. Learn Mem 5, 365-374.
Bremner, J. D., Narayan, M., Anderson,
E. R., Staib, L. H., Miller, H. L., and Charney, D.
S. (2000). Hippocampal volume reduction in major depression. Am J Psychiatry
157, 115-118.
Bremner, J. D., Randall, P., Scott, T. M., Bronen, R. A., Seibyl, J. P., Southwick, S. M.,
Delaney, R. C., McCarthy, G., Charney, D. S., and Innis, R. B. (1995). MRI-
based measurement of hippocampal volume in patients with combat-related
posttraumatic stress disorder. Am J Psychiatry 152, 973-981.
Bremner, J. D., Randall, P., Vermetten,
E., Staib, L., Bronen, R. A., Mazure, C., Capelli,
S., McCarthy, G., Innis, R. B., and Charney, D. S. (1997). Magnetic resonance
imaging-based measurement of hippocampal volume in posttraumatic stress
40
Chapter I - Review of long-term changes in the brain
disorder related to childhood physical and sexual abuse--a preliminary report.
Biol Psychiatry 41, 23-32.
Burgin, K. E., Waxham, M. N., Rickling, S., Westgate, S. A., Mobley, W. C., and Kelly,
P. T. (1990). In situ hybridization histochemistry of Ca2+/calmodulin-dependent
protein kinase in developing rat brain. J Neurosci 10, 1788-1798.
Castellucci, V. F., Blumenfeld, H., Goelet, P., and Kandel, E. R. (1989). Inhibitor of
protein synthesis blocks long-term behavioral sensitization in the isolated gillwithdrawal reflex of Aplysia. J Neurobiol 20, 1-9.
Cohen, H. D., and Barondes, S. H. (1966). Further studies of learning and memory after
intracerebral actinomycin-D. J Neurochem 13, 207-211.
Cohen, H. D., and Barondes, S. H. (1968). Effect of acetoxycycloheximide on learning
and memory of a light-dark discrimination. Nature 218, 271-273.
Costa, R. M., Honjo, T., and Silva, A. J. (2003). Learning and memory deficits in Notch
mutant mice. Curr Biol 13, 1348-1354.
Crino, P., Khodakhah, K., Becker, K., Ginsberg, S., Hemby, S., and Eberwine, J. (1998).
Presence and phosphorylation of transcription factors in developing dendrites.
Proc Natl Acad Sci U S A 95, 2313-2318.
Darnell, J. C., Warren, S. T., and Darnell, R. B. (2004). The fragile X mental retardation
protein, FMRP, recognizes G-quartets. Ment Retard Dev Disabil Res Rev 10, 4952.
Dash, P. K., Hochner, B., and Kandel, E. R. (1990). Injection of the cAMP-responsive
element into the nucleus of Aplysia sensory neurons blocks long-term facilitation.
Nature 345, 718-721.
Davis, H. P., Rosenzweig,
M. R., Bennett, E. L., and Squire, L. R. (1980). Inhibition
of cerebral protein synthesis: dissociation of nonspecific effects and amnesic
effects. Behav Neural Biol 28, 99-104.
Davis, H. P., Rosenzweig,
M. R., Kinkade, P. T., and Bennett, E. L. (1981). Effects of
anisomycin on retention of the passive-avoidance habit as a function of age. Exp
Aging Res 7, 33-44.
Davis, H. P., Spanis, C. W., and Squire, L. R. (1976). Inhibition of cerebral protein
synthesis: performance at different times after passive avoidance training.
Pharmacol Biochem Behav 4, 13-16.
Davis, H. P., and Squire, L. R. (1984). Protein synthesis and memory: a review. Psychol
Bull 96, 518-559.
41
Chapter 1 - Review of long-term changes in the brain
Debiec, J., LeDoux, J. E., and Nader, K. (2002). Cellular and systems reconsolidation in
the hippocampus. Neuron 36, 527-538.
Drevets, W. C. (2001). Neuroimaging and neuropathological studies of depression:
implications for the cognitive-emotional features of mood disorders. Curr Opin
Neurobiol 11, 240-249.
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic
long-term depression
in area CA1
of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc
Natl Acad Sci U S A 89, 4363-4367.
Eisenberg, M., Kobilo, T., Berman, D. E., and Dudai, Y. (2003). Stability of retrieved
memory: inverse correlation with trace dominance. Science 301, 1102-1104.
Engert, F., and Bonhoeffer, T. (1999). Dendritic spine changes associated with
hippocampal long-term synaptic plasticity. Nature 399, 66-70.
Fiala, J. C., Allwardt, B., and Harris, K. M. (2002). Dendritic spines do not split during
hippocampal LTP or maturation. Nat Neurosci 5, 297-298.
Fifkova, E., and Anderson, C. L. (1981). Stimulation-induced changes in dimensions of
stalks of dendritic spines in the dentate molecular layer. Exp Neurol 74, 621-627.
Fifkova, E., Anderson, C. L., Young, S. J., and Van Harreveld, A. (1982). Effect of
anisomycin on stimulation-induced changes in dendritic spines of the dentate
granule cells. J Neurocytol 11, 183-210.
Fifkova, E., and Van Harreveld, A. (1977). Long-lasting morphological changes in
dendritic spines of dentate granular cells following stimulation of the entorhinal
area. J Neurocytol 6, 211-230.
Flexner, J. B., Flexner, L. B., and Stellar, E. (1963). Memory in mice as affected by
intracerebral puromycin. Science 141, 57-59.
Flexner, J. B., Flexner, L. B., Stellar, E., De La Haba, G., and Roberts, R. B. (1962).
Inhibition of protein synthesis in brain and learning and memory following
puromycin. J Neurochem 9, 595-605.
Flexner, L. B., Flexner, J. B., De La Haba, G., and Roberts, R. B. (1965a). Loss of
memory as related to inhibition of cerebral protein synthesis. J Neurochem 12,
535-541.
Flexner, L. B., Flexner, J. B., Roberts, R. B., and Delahaba, G. (1964). Loss of Recent
Memory in Mice as Related to Regional Inhibition of Cerebral Protein Synthesis.
Proc Natl Acad Sci U S A 52, 1165-1169.
42
Chapter I - Review of long-term changes in the brain
Flexner, L. B., Flexner, J. B., and Stellar, E. (1965b). Memory and cerebral protein
synthesis in mice as affected by graded amounts of puromycin. Exp Neurol 13,
264-272.
Flood, J. F., Bennett, E. L., Orme, A. E., and Rosenzweig, M. R. (1975a). Effects of
protein synthesis inhibition on memory for active avoidance training. Physiol
Behav 14, 177-184.
Flood, J. F., Bennett, E. L., Orme, E., and Rosenzweig, M. R. (1975b). Relation of
memory formation to controlled amounts of brain protein synthesis. Physiol
Behav 15, 97-102.
Frey, U., Frey, S., Schollmeier, F., and Krug, M. (1996). Influence of actinomycin
D, a
RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in
vivo and in vitro. J Physiol 490 ( Pt 3), 703-711.
Frey, U., Huang, Y. Y., and Kandel, E. R. (1993). Effects of cAMP simulate a late stage
of LTP in hippocampal CA1 neurons. Science 260, 1661-1664.
Frey, U., Krug, M., Reymann, K. G., and Matthies, H. (1988). Anisomycin, an inhibitor of
protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1
region in vitro. Brain Res 452, 57-65.
Frey, U., and Morris, R. G. (1997). Synaptic tagging and long-term potentiation. Nature
385, 533-536.
Gardiol, A., Racca, C., and Triller, A. (1999). Dendritic and postsynaptic protein synthetic
machinery. J Neurosci 19, 168-179.
Ge, X., Hannan, F., Xie, Z., Feng, C., Tully, T., Zhou, H., and Zhong, Y. (2004). Notch
signaling in Drosophila long-term memory formation. Proc Natl Acad Sci U S A
101, 10172-10176.
Geinisman, Y., Berry, R. W., Disterhoft, J. F., Power, J. M., and Van der Zee, E. A.
(2001). Associative learning elicits the formation of multiple-synapse boutons. J
Neurosci 21, 5568-5573.
Geller, A., Robustelli,
F., Barondes, S. H., Cohen, H. D., and Jarvik, M. E. (1969).
Impaired performance by post-trial injections of cycloheximide in a passive
avoidance task. Psychopharmacologia 14, 371-376.
Glanzman, D. L., Kandel, E. R., and Schacher, S. (1990). Target-dependent structural
changes accompanying long-term synaptic facilitation in Aplysia neurons.
Science 249, 799-802.
43
Chapter 1 - Review of long-term changes in the brain
Greenough, W. T., Hwang, H. M., and Gorman, C. (1985). Evidence for active synapse
formation or altered postsynaptic metabolism in visual cortex of rats reared in
complex environments. Proc Natl Acad Sci U S A 82, 4549-4552.
Harris, E. W., Ganong, A. H., and Cotman, C. W. (1984). Long-term potentiation in the
hippocampus involves activation of N-methyl-D-aspartate receptors. Brain Res
323, 132-137.
Huang, Y. S., Carson, J. H., Barbarese, E., and Richter, J. D. (2003). Facilitation of
dendritic mRNA transport by CPEB. Genes Dev 17, 638-653.
Huang, Y. Y., and Kandel, E. R. (1994). Recruitment of long-lasting and protein kinase
A-dependent long-term potentiation in the CA1 region of hippocampus requires
repeated tetanization. Learn Mem 1, 74-82.
Huang, Y. Y., and Kandel, E. R. (1995). D1/D5 receptor agonists induce a protein
synthesis-dependent late potentiation in the CA1 region of the hippocampus.
Proc Natl Acad Sci U S A 92, 2446-2450.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic
plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U
S A 99, 7746-7750.
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein
synthesis in hippocampal mGluR-dependent long-term depression. Science 288,
1254-1257.
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and
protein synthesis--dependent long-term depression in hippocampal area CA1. J
Neurophysiol 86, 321-325.
Job, C., and Eberwine, J. (2001). Identification of sites for exponential translation in
living dendrites. Proc Natl Acad Sci U S A 98, 13037-13042.
Ju, W., Morishita, W., Tsui, J., Gaietta, G., Deerinck, T. J., Adams, S. R., Garner, C.
C., Tsien, R. Y., Ellisman, M. H., and Malenka, R. C. (2004). Activity-dependent
regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci
7, 244-253.
Kang, H., and Schuman, E. M. (1995). Long-lasting neurotrophin-induced enhancement
of synaptic transmission in the adult hippocampus. Science 267, 1658-1662.
Kang, H., and Schuman, E. M. (1996). A requirement for local protein synthesis in
neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402-1406.
44
Chapter 1 - Review of long-term changes in the brain
Kang, H., Sun, L. D., Atkins, C. M., Soderling, T. R., Wilson, M. A., and Tonegawa, S.
(2001). An important role of neural activity-dependent CaMKIV signaling in the
consolidation of long-term memory. Cell 106, 771-783.
Katz, J. J., and Halstead, W. C. (1950). Protein orgainzation and mental function. Comp
Psychol Monogr 20, 1-38.
Kauderer, B. S., and Kandel, E. R. (2000). Capture of a protein synthesis-dependent
component of long-term depression. Proc Natl Acad Sci U S A 97, 13342-13347.
Kessler, R. C. (1997). The effects of stressful life events on depression. Annu Rev
Psychol 48, 191-214.
Kiebler, M. A., Hemraj, I., Verkade, P., Kohrmann,
M., Fortes, P., Marion, R. M., Ortin,
J., and Dotti, C. G. (1999). The mammalian staufen protein localizes to the
somatodendritic domain of cultured hippocampal neurons: implications for its
involvement in mRNA transport. J Neurosci 19, 288-297.
Kirkwood, A., Dudek, S. M., Gold, J. T., Aizenman,
C. D., and Bear, M. F. (1993).
Common forms of synaptic plasticity in the hippocampus and neocortex in vitro.
Science 260, 1518-1521.
Knafo, S., Grossman, Y., Barkai, E., and Benshalom, G. (2001). Olfactory learning is
associated with increased spine density along apical dendrites of pyramidal
neurons in the rat piriform cortex. Eur J Neurosci 13, 633-638.
Kohrmann,
M., Luo, M., Kaether, C., DesGroseillers,
L., Dotti, C. G., and Kiebler, M. A.
(1999). Microtubule-dependent recruitment of Staufen-green fluorescent protein
into large RNA-containing granules and subsequent dendritic transport in living
hippocampal neurons. Mol Biol Cell 10, 2945-2953.
Korzus, E., Rosenfeld, M. G., and Mayford, M. (2004). CBP histone acetyltransferase
activity is a critical component of memory consolidation. Neuron 42, 961-972.
Krug, M., Lossner, B., and Ott, T. (1984). Anisomycin blocks the late phase of long-term
potentiation in the dentate gyrus of freely moving rats. Brain Res Bull 13, 39-42.
Laggerbauer,
B., Ostareck, D., Keidel, E. M., Ostareck-Lederer,
A., and Fischer, U.
(2001). Evidence that fragile X mental retardation protein is a negative regulator
of translation. Hum Mol Genet 10, 329-338.
Lamprecht, R., and LeDoux, J. (2004). Structural plasticity and memory. Nat Rev
Neurosci 5, 45-54.
Lee, J. L., Everitt, B. J., and Thomas, K. L. (2004). Independent cellular processes for
hippocampal memory consolidation and reconsolidation. Science 304, 839-843.
45
Chapter I - Review of long-term changes in the brain
Leuner, B., Falduto, J., and Shors, T. J. (2003). Associative memory formation increases
the observation of dendritic spines in the hippocampus. J Neurosci 23, 659-665.
Levenson, J. M., O'Riordan,
K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt,
J. D. (2004). Regulation of histone acetylation during memory formation in the
hippocampus. J Biol Chem 279, 40545-40559.
Li, Z., Zhang, Y., Ku, L., Wilkinson, K. D., Warren, S. T., and Feng, Y. (2001). The
fragile X mental retardation protein inhibits translation via interacting with mRNA.
Nucleic Acids Res 29, 2276-2283.
Liotti, M., and Mayberg, H. S. (2001). The role of functional neuroimaging in the
neuropsychology of depression. J Clin Exp Neuropsychol 23, 121-136.
Lowenstein,
P. R., Morrison, E. E., Bain, D., Shering, A. F., Banting, G., Douglas, P.,
and Castro, M. G. (1994). Polarized distribution of the trans-Golgi network
marker TGN38 during the in vitro development of neocortical neurons: effects of
nocodazole and brefeldin A. Eur J Neurosci 6, 1453-1465.
Magarinos, A. M., and McEwen, B. S. (1995). Stress-induced atrophy of apical dendrites
of hippocampal CA3c neurons: comparison of stressors. Neuroscience 69, 83-88.
Magarinos, A. M., McEwen, B. S., Flugge, G., and Fuchs, E. (1996). Chronic
psychosocial stress causes apical dendritic atrophy of hippocampal CA3
pyramidal neurons in subordinate tree shrews. J Neurosci 16, 3534-3540.
Maletic-Savatic, M., Malinow, R., and Svoboda, K. (1999). Rapid dendritic
morphogenesis in CA1 hippocampal dendrites induced by synaptic activity.
Science 283, 1923-1927.
Manahan-Vaughan, D., Kulla, A., and Frey, J. U. (2000). Requirement of translation but
not transcription for the maintenance of long-term depression in the CA1 region
of freely moving rats. J Neurosci 20, 8572-8576.
Martin, K. C., Casadio, A., Zhu, H., Yaping, E., Rose, J. C., Chen, M., Bailey, C. H., and
Kandel, E. R. (1997). Synapse-specific, long-term facilitation of aplysia sensory
to motor synapses: a function for local protein synthesis in memory storage. Cell
91, 927-938.
McEwen, B. S. (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22, 105122.
Miller, S., Yasuda, M., Coats, J. K., Jones, Y., Martone, M. E., and Mayford, M. (2002).
Disruption of dendritic translation of CaMKllalpha impairs stabilization of synaptic
plasticity and memory consolidation. Neuron 36, 507-519.
46
Chapter I - Review of long-term changes in the brain
Mineka, S., Watson, D., and Clark, L. A. (1998). Comorbidity of anxiety and unipolar
mood disorders. Annu Rev Psychol 49, 377-412.
Montarolo,
P. G., Goelet, P., Castellucci, V. F., Morgan, J., Kandel, E. R., and
Schacher, S. (1986). A critical period for macromolecular synthesis in long-term
heterosynaptic facilitation in Aplysia. Science 234, 1249-1254.
Montminy, M. R., and Bilezikjian, L. M. (1987). Binding of a nuclear protein to the cyclicAMP response element of the somatostatin gene. Nature 328, 175-178.
Mori, Y., Imaizumi, K., Katayama, T., Yoneda, T., and Tohyama, M. (2000). Two cisacting elements in the 3' untranslated region of alpha-CaMKII regulate its
dendritic targeting. Nat Neurosci 3, 1079-1084.
Nader, K., Schafe, G. E., and Le Doux, J. E. (2000). Fear memories require protein
synthesis in the amygdala for reconsolidation after retrieval. Nature 406, 722726.
Nakajima, S. (1969). Interference with relearning in the rat after hippocampal injection of
actinomycin D. J Comp Physiol Psychol 67, 457-461.
Neale, J. H., Klinger, P. D., and Agranoff, B. W. (1973). Camptothecin blocks memory of
conditioned avoidance in the goldfish. Science 179, 1243-1246.
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia,
L. M.
(2002). Neurobiology of depression. Neuron 34, 13-25.
Nguyen, P. V., Abel, T., and Kandel, E. R. (1994). Requirement of a critical period of
transcription for induction of a late phase of LTP. Science 265, 1104-1107.
Oliver, G. W., Rose, G. P., Brimblecombe,
R. W., and Livett, B. H. (1979). Analysis of Y-
maze learning in mice using cycloheximide. Gen Pharmacol 10, 489-497.
Ostroff, L. E., Fiala, J. C., Allwardt, B., and Harris, K. M. (2002). Polyribosomes
redistribute from dendritic shafts into spines with enlarged synapses during LTP
in developing rat hippocampal slices. Neuron 35, 535-545.
Otani, S., Marshall, C. J., Tate, W. P., Goddard, G. V., and Abraham, W. C. (1989).
Maintenance of long-term potentiation in rat dentate gyrus requires protein
synthesis but not messenger RNA synthesis immediately post-tetanization.
Neuroscience 28, 519-526.
Popov, V. I., Bocharova, L. S., and Bragin, A. G. (1992). Repeated changes of dendritic
morphology in the hippocampus of ground squirrels in the course of hibernation.
Neuroscience 48, 45-51.
47
Chapter I - Review of long-term changes in the brain
Presente, A., Boyles, R. S., Serway, C. N., de Belle, J. S., and Andres, A. J. (2004).
Notch is required for long-term memory in Drosophila. Proc Natl Acad Sci U S A
101, 1764-1768.
Przybyslawski, J., and Sara, S. J. (1997). Reconsolidation of memory after its
reactivation. Behav Brain Res 84, 241-246.
Rao, A., and Steward, 0. (1991). Evidence that protein constituents of postsynaptic
membrane specializations are locally synthesized: analysis of proteins
synthesized within synaptosomes. J Neurosci 11, 2881-2895.
Raymond, C. R., Thompson, V. L., Tate, W. P., and Abraham, W. C. (2000).
Metabotropic glutamate receptors trigger homosynaptic protein synthesis to
prolong long-term potentiation. J Neurosci 20, 969-976.
Reinis, S. (1968). Block of "memory transfer" by actinomycin D. Nature 220, 177-178.
Roegiers, F., and Jan, Y. N. (2000). Staufen: a common component of mRNA transport
in oocytes and neurons? Trends Cell Biol 10, 220-224.
Rook, M. S., Lu, M., and Kosik, K. S. (2000). CaMKllalpha 3' untranslated regiondirected mRNA translocation in living neurons: visualization by GFP linkage. J
Neurosci 20, 6385-6393.
Roullet, P., and Sara, S. (1998). Consolidation of memory after its reactivation:
involvement of beta noradrenergic receptors in the late phase. Neural Plast 6,
63-68.
Sangha, S., Scheibenstock, A., and Lukowiak, K. (2003a). Reconsolidation of a longterm memory in Lymnaea requires new protein and RNA synthesis and the soma
of right pedal dorsal 1. J Neurosci 23, 8034-8040.
Sangha, S., Scheibenstock, A., Morrow, R., and Lukowiak, K. (2003b). Extinction
requires new RNA and protein synthesis and the soma of the cell right pedal
dorsal 1 in Lymnaea stagnalis. J Neurosci 23, 9842-9851.
Schafe, G. E., and LeDoux, J. E. (2000). Memory consolidation of auditory pavlovian
fear conditioning requires protein synthesis and protein kinase A in the amygdala.
J Neurosci 20, RC96.
Schafe, G. E., Nadel, N. V., Sullivan, G. M., Harris, A., and LeDoux, J. E. (1999).
Memory consolidation for contextual and auditory fear conditioning is dependent
on protein synthesis, PKA, and MAP kinase. Learn Mem 6, 97-110.
Schafe, G. E., Nader, K., Blair, H. T., and LeDoux, J. E. (2001). Memory consolidation
of Pavlovian fear conditioning: a cellular and molecular perspective. Trends
Neurosci 24, 540-546.
48
Chapter I - Review of long-term changes in the brain
Scharf, M. T., Woo, N. H., Lattal, K. M., Young, J. Z., Nguyen, P. V., and Abel, T. (2002).
Protein synthesis is required for the enhancement of long-term potentiation and
long-term memory by spaced training. J Neurophysiol 87, 2770-2777.
Scheetz, A. J., Nairn, A. C., and Constantine-Paton, M. (2000). NMDA receptormediated control of protein synthesis at developing synapses. Nat Neurosci 3,
211-216.
Sheline, Y. I., Gado, M. H., and Kraemer, H. C. (2003). Untreated depression and
hippocampal volume loss. Am J Psychiatry 160, 1516-1518.
Sheline, Y. I., Wang, P. W., Gado, M. H., Csernansky, J. G., and Vannier, M. W. (1996).
Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A
93, 3908-3913.
Shors, T. J., Chua, C., and Falduto, J. (2001). Sex differences and opposite effects
of stress on dendritic spine density in the male versus female hippocampus. J
Neurosci 21, 6292-6297.
Shors, T. J., Falduto, J., and Leuner, B. (2004). The opposite effects of stress on
dendritic spines in male vs. female rats are NMDA receptor-dependent. Eur J
Neurosci 19, 145-150.
Silva, A. J., Kogan, J. H., Frankland, P. W., and Kida, S. (1998). CREB and memory.
Annu Rev Neurosci 21, 127-148.
Squire, L. R., and Barondes, S. H. (1970). Actinomycin-D: effects on memory at different
times after training. Nature 225, 649-650.
Squire, L. R., and Davis, H. P. (1975). Cerebral protein synthesis inhibition and
discrimination training: effect of extent and duration of inhibition. Behav Biol 13,
49-57.
Starkman, M. N., Gebarski, S. S., Berent, S., and Schteingart,
D. E. (1992).
Hippocampal formation volume, memory dysfunction, and cortisol levels in
patients with Cushing's syndrome. Biol Psychiatry 32, 756-765.
Starkman, M. N., Giordani, B., Gebarski, S. S., Berent, S., Schork, M. A., and
Schteingart, D. E. (1999). Decrease in cortisol reverses human hippocampal
atrophy following treatment of Cushing's disease. Biol Psychiatry 46, 1595-1602.
Steward, O., Falk, P. M., and Torre, E. R. (1996). Ultrastructural basis for gene
expression at the synapse: synapse-associated polyribosome complexes. J
Neurocytol 25, 717-734.
49
Chapter 1 - Review of long-term changes in the brain
Steward, O., and Fass, B. (1983). Polyribosomes associated with dendritic spines in
the denervated dentate gyrus: evidence for local regulation of protein synthesis
during reinnervation. Prog Brain Res 58, 131-136.
Steward, O., and Levy, W. B. (1982). Preferential localization of polyribosomes under
the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci 2,
284-291.
Steward, O., Wallace, C. S., Lyford, G. L., and Worley, P. F. (1998). Synaptic activation
causes the mRNA for the IEG Arc to localize selectively near activated
postsynaptic sites on dendrites. Neuron 21, 741-751.
Strohle, A., and Holsboer, F. (2003). Stress responsive neurohormones in depression
and anxiety. Pharmacopsychiatry 36 Suppl 3, S207-214.
Sutton, M. A., and Carew, T. J. (2000). Parallel molecular pathways mediate expression
of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in
Aplysia. Neuron 26, 219-231.
Sutton, M. A., Masters, S. E., Bagnall, M. W., and Carew, T. J. (2001). Molecular
mechanisms underlying a unique intermediate phase of memory in aplysia.
Neuron 31, 143-154.
Suzuki, A., Josselyn, S. A., Frankland, P. W., Masushige, S., Silva, A. J., and Kida,
S. (2004). Memory reconsolidation and extinction have distinct temporal and
biochemical signatures. J Neurosci 24, 4787-4795.
Tang, S. J., Meulemans, D., Vazquez, L., Colaco, N., and Schuman, E. (2001). A role for
a rat homolog of staufen in the transport of RNA to neuronal dendrites. Neuron
32, 463-475.
Thiels, E., Kanterewicz,
B. I., Norman, E. D., Trzaskos, J. M., and Klann, E. (2002).
Long-term depression in the adult hippocampus in vivo involves activation of
extracellular signal-regulated kinase and phosphorylation of Elk-1. J Neurosci 22,
2054-2062.
Tiruchinapalli,
D. M., Oleynikov, Y., Kelic, S., Shenoy, S. M., Hartley, A., Stanton, P.
K., Singer, R. H., and Bassell, G. J. (2003). Activity-dependent trafficking and
dynamic localization of zipcode binding protein 1 and beta-actin mRNA in
dendrites and spines of hippocampal neurons. J Neurosci 23, 3251-3261.
Tongiorgi, E., Righi, M., and Cattaneo, A. (1997). Activity-dependent dendritic targeting
of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci 17, 9492-9505.
Toni, N., Buchs, P. A., Nikonenko,
I., Bron, C. R., and Muller, D. (1999). LTP promotes
formation of multiple spine synapses between a single axon terminal and a
dendrite. Nature 402, 421-425.
50
Chapter 1 - Review of long-term changes in the brain
Torre, E. R., and Steward, 0. (1992). Demonstration of local protein synthesis within
dendrites using a new cell culture system that permits the isolation of living axons
and dendrites from their cell bodies. J Neurosci 12, 762-772.
Torre, E. R., and Steward, 0. (1996). Protein synthesis within dendrites: glycosylation
of newly synthesized proteins in dendrites of hippocampal neurons in culture. J
Neurosci 16, 5967-5978.
Trachtenberg,
J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., and
Svoboda, K. (2002). Long-term in vivo imaging of experience-dependent synaptic
plasticity in adult cortex. Nature 420, 788-794.
Tully, T., Boynton, S., Brandes, C., Dura, J. M., Mihalek, R., Preat, T., and Villella, A.
(1990). Genetic dissection of memory formation in Drosophila melanogaster.
Cold Spring Harb Symp Quant Biol 55, 203-211.
Tully, T., Preat, T., Boynton, S. C., and Del Vecchio, M. (1994). Genetic dissection of
consolidated memory in Drosophila. Cell 79, 35-47.
Van Harreveld, A., and Fifkova, E. (1975). Swelling of dendritic spines in the fascia
dentata after stimulation of the perforant fibers as a mechanism of post-tetanic
potentiation. Exp Neurol 49, 736-749.
Vyas, A., Bernal, S., and Chattarji, S. (2003). Effects of chronic stress on dendritic
arborization in the central and extended amygdala. Brain Res 965, 290-294.
Vyas, A., Mitra, R., Shankaranarayana Rao, B. S., and Chattarji, S. (2002). Chronic
stress induces contrasting patterns of dendritic remodeling in hippocampal and
amygdaloid neurons. J Neurosci 22, 6810-6818.
Wang, Y., Chan, S. L., Miele, L., Yao, P. J., Mackes, J., Ingram, D. K., Mattson, M.
P., and Furukawa, K. (2004). Involvement of Notch signaling in hippocampal
synaptic plasticity. Proc Natl Acad Sci U S A 101, 9458-9462.
Watts, M. E., and Mark, R. F. (1971a). Drug inhibition of memory formation in chickens.
II. Short-term memory. Proc R Soc Lond B Biol Sci 178, 455-464.
Watts, M. E., and Mark, R. F. (1971b). Separate actions of ouabain and cycloheximide
on memory. Brain Res 25, 420-423.
Wells, D. G., Dong, X., Quinlan, E. M., Huang, Y. S., Bear, M. F., Richter, J. D., and
Fallon, J. R. (2001). A role for the cytoplasmic polyadenylation element in NMDA
receptor-regulated mRNA translation in neurons. J Neurosci 21, 9541-9548.
Wu, L., Wells, D., Tay, J., Mendis, D., Abbott, M. A., Barnitt, A., Quinlan, E., Heynen,
A., Fallon, J. R., and Richter, J. D. (1998). CPEB-mediated cytoplasmic
51
Chapter 1 - Review of long-term changes in the brain
polyadenylation and the regulation of experience-dependent translation of alphaCaMKII mRNA at synapses. Neuron 21, 1129-1139.
Yin, J. C., Del Vecchio, M., Zhou, H., and Tully, T. (1995). CREB as a memory
modulator: induced expression of a dCREB2 activator isoform enhances longterm memory in Drosophila. Cell 81, 107-115.
Yin, J. C., Wallach, J. S., Del Vecchio, M., Wilder, E. L., Zhou, H., Quinn, W. G., and
Tully, T. (1994). Induction of a dominant negative CREB transgene specifically
blocks long-term memory in Drosophila. Cell 79, 49-58.
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., Oostra, B., and
Bagni, C. (2003). The fragile X syndrome protein FMRP associates with BC1
RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317327.
52
Chapter 2 - Translational control by MAPK signaling in
long-term synaptic plasticity and memory
Abstract
Long-term memory can be distinguished from short-term memory by its requirement
for protein synthesis. Traditionally, it has been assumed that transcription is the key
regulatory point behind L-LTP and long-term memory, and that translation is constitutive.
Using a transgenic mouse with impaired MAP Kinase signaling in the hippocampus, we
show that translational regulation is essential to L-LTP. Furthermore, neuronal activity
and L-LTP inducing tetani stimulate the MAP Kinase pathway, causing it to upregulate
translation of a large number of mRNAs via phosphorylation of the translation initiation
factors elF4E and 4EBP1, and the ribosomal protein S6. The phosphorylation of elF4E
and S6 is also upregulated after behavioral training in a MEK dependent manner. Thus,
upregulation of a large number of mRNAs via general translational stimulation likely
underlies the expression of L-LTP and LTM.
53
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Introduction
Protein Synthesis Dependent Plasticity
Memory in both invertebrates and vertebrates can be classified temporally into a
short-term component and a long-term component. These components can be separated
on the basis of their requirement for RNA and protein synthesis; short-term memory (STM)
is insensitive to inhibitors of translation and transcription, whereas inhibition of translation
and transcription block long-term memory (LTM) in both invertebrates and vertebrates.
The cellular correlate of memory, long-term potentiation (LTP) can also be classified in
this manner; late-LTP (L-LTP) requires both transcription and translation, whereas earlyLTP (E-LTP) does not (Kandel, 2001; McGaugh, 2000).
Considerable effort over the last decade has been expended in elucidating
the molecular mechanisms behind the requirement for macromolecular synthesis.
However, much of the research has concentrated on the transcriptional requirement
for L-LTP and LTM. Multiple immediately-early genes whose transcription is activated
by synaptic plasticity, including c-fos, zif268, C/EPP, and Arc (Silva, 2003; Steward and
Worley, 2001) have been found, and various second messenger pathways, such as
the calcium/calmodulin-dependent protein kinase (CaMK), protein kinase A (PKA), and
mitogen-activated protein kinase (MAPK) pathways acting via transcription factors such
as cAMP-Responsive-Element Binding Protein (CREB) and Elk-1 (Silva, 2003; Steward
and Worley, 2001) have been implicated in this transcriptional activation.
In contrast, there is less evidence linking specific pathways to translational
regulation, in spite of some evidence that translational regulation is an important
mechanism behind synaptic plasticity. For example, although it is not commented upon
by the authors, work by Frey et al (Frey et al., 1996; Frey et al., 1988), and Nguyen et al
(Huang et al., 1996; Nguyen et al., 1994) suggests a dissociation between the requirement
for transcription and the requirement for translation in L-LTP - inhibition of translation
produces an immediate deficit in L-LTP wherease inhibition of transcription produces a
54
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
delayed inhibition of L-LTP implicating translational upregulation of pre-existing mRNAs as
an important mechanism underlying L-LTP. In addition, some forms of plasticity including
mGluR-dependent long-term depresstion (LTD) (Huber et al., 2000; Huber et al., 2001),
and priming of LTP (Raymond et al., 2000) are susceptible to inhibition by translation
inhibitors, but not transcriptional inhibitors. However, neither the second messenger
pathways, nor the substrate of the translational regulation have been identified in any of
these cases. In addition, the translational requirements behind non-mGluR dependent
LTD have not been determined.
Regulation of Translation
In mitotic cells, where the study of translational regulation is more advanced,
multiple loci of regulation have been identified. Translational initiation, a key regulatory
point in translational control (Gingras et al., 1999), starts with the binding of elF4E to the
mRNA cap; this recruits the elF4G complex, which along with elF4A forms the initiation
complex elF4F. Next, elF2 binds to the complex, followed by the 40S ribosomal subunit
which begins scanning for the initiation codon. Once the initiation codon has been
identified, the 60S ribosomal subunit attaches to the 40S subunit-mRNA complex, starting
the elongation phase of translation (Gray and Wickens, 1998).
The limiting factor in translation, elF4E (Raught and Gingras, 1999), is regulated
in multiple ways. Firstly, a family of proteins called 4E binding proteins (4EBPs) bind
elF4E, and prevent its association with elF4G, thus inhibiting translation; phosphorylation
of 4EBP releases elF4E, allowing translational initiation to begin. The mTOR pathway is
the main pathway implicated in the phosphorylation of 4EBP; however, the ERK MAPK,
P13K, and PKB/Akt pathways are also likely to play a role (Gingras et al., 1999).
In addition, phosphorylation of elF4E is correlated with increased translation, though
the exact mechanism behind this phenomenon has still not been clarified. Phosphorylation
of elF4E occurs mainly via Mnkl, which in turn is activated by the ERK MAPK pathway
(Gingras et al., 1999).
55
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
The length of the mRNA polyA tail can also affect the efficiency of translation. A
protein called poly-A Binding Protein (PABP), binds the mRNA poly-A tail, and the number
of PABP molecules per mRNA molecule is proportional to the length of the poly-Atail. PABP,
in turn, can help recruit elF4G to the cap, which in turn increases the rate of translation.
Regulation of polyadenylation requires cis-acting elements on the mRNA, unlike the
previously mentioned regulatory mechanisms which operate on all mRNAs. mRNAs
regulated via polyadenylation contain a uridine rich sequence, called the Cytoplasmic
Polyadenylation Element (CPE) in the 3'-untranslated region (UTR) of the mRNA within
100 bases upstream of the canonical AAUAAA cleavage and polyadenylation sequence.
These mRNAs contain very short poly-A tails, and are thus inefficiently translated. In
addition, the CPE is bound by a protein called the CPE binding protein (CPEB), which, in
turn, is bound by a 4E binding protein called Maskin; this interaction with elF4E prevents
translation. Unlike conventional 4EBPs, Maskin is not known to be phosphorylated; instead,
activation of translation of CPE containing RNAs requires phosphorylation of CPEB, which
dissociates maskin from elF4E, thus activating translation. In addition, phosphorylated
CPEB recruits, via cytoplasmic and polyadenylation specificity factor (CPSF), poly-A
polymerase (PAP), which extends the poly-A tail, which increases translational efficiency
of the mRNA. This mechanism has been primarily studied in development - specifically
in frog and mouse oocyte maturation, in response to progesterone, and during Drosophila
development. CPEB is phosphorylated on multiple sites, and only one site has been
characterized; this site requires phosphorylation by the Aurora kinase. However, the
CPEB sequence contains sites for phosphorylation by multiple kinases including the ERK
substrate Rsk-2, and CPE and polyadenylation mediated translational upregulation of the
cyclin-B1 mRNA in frog oocytes has been shown to be dependent on the oocyte specific
MAP kinase kinase kinase Mos (Gray and Wickens, 1998).
The Fragile-X Mental Retardation Protein (FMRP), the gene whose dysfunction
is implicated in fragile-X mental retardation, is also a translational inhibitor that acts by
56
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
binding specific mRNAs (Laggerbauer et al., 2001; Li et al., 2001). The nature of the
mRNAs bound by FMRP has not been fully elucidated, as FMRP appears to bind mRNAs
with G-quartets by itself (Darnell et al., 2004), and other mRNAs via accessory noncoding RNAs like BC1 (Zalfa et al., 2003). Fmr-1 knockout mice show increased mGluR
dependent LTD, with no changes in LTP (Huber et al., 2002). The regulation of FMRP is
not yet understood.
Translational regulation can also take place at the level of elongation. One of
the primary loci of regulation is elongation factor 2 (eEF2). Phosphorylation of this site
decreases the elongation rate, thus reducing protein synthesis - however, this mechanism
can be used, via initiation competition, to increase the synthesis rate of specific proteins,
while overall protein synthesis decreases (Scheetz et al., 2000; Walden et al., 1981).
The total translational capacity of the cell is also a target of regulation. Many of the
components required for translation, including many ribosomal components and elF4E
are themselves subject to translational regulation; mRNAs encoding these components
contain a polypyrimidine tract in their 5'-untranslated region, and are called 5'-TOP RNAs).
Phosphorylation of the ribosomal protein S6 increases translation of 5'-TOP RNAs,
which leads to increased translation via an increased number of translational machinery
components. Phosphorylation of S6 was thought to occur exclusively via the mTOR
pathway by S6 kinase (Proud, 2002); however, recently, a second S6 kinase (S6K2),
which is activated by MAP Kinase phosphorylation, has been found (Wang et al., 2001;
Wang and Proud, 2002).
With regards to neuronal plasticity, only regulations of phosphorylation of eEF2,
and increased polyadenylation have been reported. Scheetz et al (Scheetz et al., 2000)
report that activation of the NMDA receptor in superior-collicular synaptoneurosomes
initially inhibits total translation, while stimulating translation of a-CaMKII; the authors
implicate phosphorylation of eEF2 for both effects. Wu et al (Wu et al., 1998) reported
that exposure to light of previously dark-reared rats activates polyadenylation of the CPE
57
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
containing a-CaMKII and total levels of (a-CaMKII; in addition, it has been shown that
certain kinds of stimulation transiently activate translation of a CPE containing reporter in
cultured hippocampal cells; however, no mechanism behind this action was demonstrated
(Wells et al., 2001).
In terms of signaling pathways, the MAP Kinase and Akt-mTOR pathways have
been implicated in translational regulation (Gingras et al., 1999; Gray and Wickens, 1998).
Our experiments concentrated on the MAP Kinase pathway.
MAP Kinase Pathway
There are three MAP Kinase pathways, namely the ERK, JNK, and p38 MAPK
pathways. All of the pathways consist of a MAP Kinase Kinase Kinase (MAPKKK) which
is the entry point into the pathway. The MAPKKK phosphorylates a MAP Kinase Kinase
(MAPKK), which in turn phosphorylates a MAP Kinase (MAPK), which in turn phosphorylates
various effector molecules and regulatory molecules (Johnson and Lapadat, 2002). The
ERK pathway, where the MAPKK is MEK and the MAPKKK is Raf, is a highly conserved
pathway linking growth factor receptors to various effector mechanisms including
transcription and translation. In neurons, the ERK pathway is activated by many diverse
stimuli including neurotrophins and calcium influx, both of which have been implicated in
synaptic plasticity (Sweatt, 2001).
A number of studies have implicated a role for the ERK pathway in synaptic
plasticity. For example, LTP has been shown to activate ERK phosphorylation (English
and Sweatt, 1996); in addition, treatment of hippocampal slices with the MEK inhibitor
PD98059 attenuates both E-LTP and L-LTP (Atkins et al., 1998; English and Sweatt,
1997). In addition, both contextual and cued fear conditioning induces phosphorylation of
ERK, and inhibition of MEK with SL327 blocks both contextual and cued fear conditioning
(Atkins et al., 1998). The main effector of the ERK pathway postulated to affect neuronal
excitability and E-LTP is the Kv4.2 potassium channel (Adams et al., 2000; Selcher et al.,
2003; Yuan et al., 2002), whereas the transcription factor CREB is the primary molecule
58
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
associated with L-LTP (Sweatt, 2001). However, no published studies have implicated the
ERK pathway in activity induced translational regulation.
A role for ERK pathway mediated translational regulation during L-LTP was observed
in the lab during analysis of a transgenic animal expressing dominant-negative MEK1
(dnMEK1) in the CA1 region of the hippocampus and the cortex. To restrict the expression
of dnMEK1, a Cre-loxP approach was used with one mouse expressing dnMEK1 under
the dual control of the chicken -actin promoter and a floxed 'stop' cassette (Fig. 2-1). This
approach allows for the expression of dnMEK1 only in those cells where Cre recombinase
is also expressed. By mating the floxed-dnMEK1 mice to mice expressing Cre only in
the forebrain, mice expressing dnMEK1 only in the cortex and the CA1 region of the
hippocampus were obtained, as demonstrated by the in situ hybridization profile of the
transgene (Fig. 2-2). The double transgenic mice were henceforth designated 'dnMEK1'
mice whereas the floxed 'stop' cassette mice were designated 'control' in all subsequent
experiments.
1 0x P
ch. 3-actin
In
.oxP
STO
dnMEK
c CaMKII-Cre
ch. -actin
In
dnMEK1
H
Figure 2-1: Schematic of dnMEK1 mice. In floxed transgenic mice (top), as well as non-forebrain
pyramidal neurons of floxed dnMEK1 / a-CaMKII Cre mice (double transgenic), the "stop" cassette
prevents expression of dnMEK1. In forebrain pyramidal neurons of double transgenic mice, Cre
expression removes the "stop" cassette by recombination allowing for dnMEK1 expression
59
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Single Transgenic
Double Transgenic
"dnMEKl" mice
Figure 2-2: In situ hybridization images of floxed dnMEK1 mice (single transgenic; left panel), and
floxed dnMEK1 mice / a-CaMKII Cre mice (double transgenic; right panel) show expression of the
transgene in the cortex and CA1 region of the hippocampus. Note the lack of expression in the
hippocampal CA3 subfield. The probe is specific to the transgene
In the dnMEK1
mice, MEK activity as measured
by ERK1 and ERK2
phosphorylation was severely, though not entirely inhibited - basal ERK1 and ERK2
phosphorylation in slices prepared from transgenic and control animals were identical;
however, depolarization-induced phosphorylations of ERK1 and ERK2 were inhibited in
the dnMEK1 animals as compared to the control animals (Fig. 2-3). The partial inhibition
is due, in part, to the expression of the transgene only in the CA1, and, in part, due to an
incomplete inhibition of MEK activity by dnMEK1.
control
{
-
12
I dnMEK1
dnMEK1
+
+
I I
.-.
control
8
KCI
*
I
__
pERK
4
ERK
0
unstimulated
stimulated
Figure 2-3: MEK activity, as measured by phosphorylation of the MEK substrates ERK1/2, in
hippocampal slices derived from control and dnMEK1 animals: Left panel shows sample Western
blots for basal and stimulated phospho-ERK and total ERK. Right panel shows quantification of
Western blot results. Basal MEK activity is unaffected in the dnMEK1 animals; however, MEK activity stimulated by 4 spaced depolarizations of the slices with 90mM KCI was inhibited in slices
derived from the dnMEK1 mutant animals as compared to slices from control animals.
60
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Behavioral analysis using contextual fear-conditioning showed a deficit in long-term
memory as measured by a decreased level of freezing in the mutant mice compared to
the control mice 24 hrs. after training (Fig. 2-4). However, short-term memory was not
affected, since the level of freezing in the control and mutant mice were indistinguishable
1 hr. after training (Fig. 2-4). Since short-term memory is not protein-synthesis dependent,
whereas long-term memory is (Schafe et al., 1999), this implicates a deficit in transcription
and/or translational regulation as the primary basis for the long term memory phenotype
in the mutant animals. Furthermore, as cued fear-conditioning showed no deficit in
the mutant mice as compared to control mice, the locus of the deficit is likely to be the
hippocampus.
75
control
*dnMEK1
75
50
=
50
1 25
.
25
.
hr.
24 hr
I hr.
24 hr
control
* dnMB(1
pre-CS
CS
Figure 2-4: Fear-conditioning using contextual fear-conditioning (left panel), and cued fearconditioning (right panel): Contextual fear-conditioning data show a deficit at 24 hrs., but not 1 hr.
after training in dnMEK1 mice implying a hippocampal long-term memory deficit (left panel). Cued
fear-conditioning (right panel) shows no deficit upon presentation of the tone (CS), confirming that
the deficit is likely to be hippocampal in origin.
Electrophysiology confirmed that the transgenic animals had a specific deficit
in hippocampal protein-synthesis dependent plasticity, as L-LTP induced by four tetani
was inhibited, whereas E-LTP induced by two tetani was not (Fig 2-5). Importantly,
the time course of potentiation in the transgenic mice was identical to the potentiation
induced in control mice treated with the translation inhibitor anisomycin, and furthermore,
anisomycin did not further reduce the level of potentiation in the transgenic mice (Fig 2-
61
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
5), suggesting that the L-LTP deficit seen in the transgenic mice was due to a deficit in
protein-synthesis.
A
B
E
300
300
3250
r
20 ms
*drEK1
I
I
contrl
controlnot;anied
dnllEK1 nonodtanizd
150
[ 150
J00
Cm
q r q~ ~~ 4 10
100
.-.
xi
-15
0
15
30
Time(min)
45
60
0
C
50
100
Time(rin)
150
200
D
300 ,,.
i 250-
300=
! 250
* control+ anisomycin
[o dnMEK1
n dnMEK1
* dnMEKl+anisomycin
[on
-o L
m
u 200m
,. 150co
n 150-
.
0.
L 100lI
I
0
50
1
I
100
150
I
I
200
0
Time (min.)
I
I
I
I
50
100
150
200
Time (min.)
Figure 2-5: L-LTP phenotype in dnMEK1 mice resembles effect of anisomycin on L-LTP in control
mice: (A) E-LTP (two 100 Hz. 1 sec. tetani spaced 15 sec. apart) is identical in slices from control
and dnMEK1 mice. (B) L-LTP (four 100 Hz. 1 sec. tetani spaced 5 min. apart) in dnMEK1 mice
decays to baseline within 2.5 hrs. (C) L-LTP in dnMEK1 mice is indistinguishable from L-LTP in
anisomycin treated control slices. (D) Anisomycin treatment does not alter L-LTP in dnMEK1
mice.
To differentiate between the possible roles for the ERK pathway in translation vs.
transcription, we compared the kinetics of L-LTP in the mutant mice with the kinetics
of L-LTP in actinomycin D (transcription inhibitor) treated slices from control mice. As
shown in Fig. 2-6, the time course of L-LTP in slices from dnMEK1 mice did not match the
delayed decrease in L-LTP seen in control slices treated with the transcription inhibitor,
actinomycin D. This is inconsistent with the model that the deficit in L-LTP in the mutant
mice is due to reduced transcriptional regulation in the mutant mice, and instead points
62
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
to a role for the ERK pathway in translational regulation, which in turn, is important for
synaptic plasticity as measured by L-LTP.
--JUU-
-
*"
~*r
E
·
·
_ ._
control
control+ anisomycin
_
' 250-
*
m
--n
JUU---
* dnMEK1
250-
200200
o
" 150a.
0.
o15
0I 150
AL
c
womb
_
r
W
I
0
I
I
50
100
Time (min.)
I4nn
Iv-
111_
I
I
I
I
150
200
0
50
I
100
Time(min.)
I
I
150
200
Figure 2-6: L-LTP in slices from dnMEK1 mice resembles L-LTP in anisomycin treated control
slices, but not actinomycin D treated control slices. Left Panel: Anisomycin (red) causes an early
decremental reduction in L-LTP, whereas actinomycin D (black) causes a delayed inhibition of
L-LTP as compared to vehicle treated (blue) slices from control mice. Right Panel: Slices from
dnMEK1 mice (blue) show an early reduction in L-LTP that is not further inhibited by either anisomycin (red) or actinomycin D (black).
Thus, we chose to analyze whether the ERK pathway was affecting translation,
and to determine the mechanism by which it regulated protein synthesis. Two possibilities
were envisioned. As one of the few translation regulatory points implicated in neuronal
plasticity is polyadenylation of CPE containing mRNAs (Wells et al., 2001; Wu et al.,
1998), and, as mentioned earlier, polyadenylation of the CPE containing mRNA cyclin B1
is regulated by the MAPKKK Mos during Xenopus oocyte maturation (Gray and Wickens,
1998), we sought to determine if the MAP Kinase pathway controlled translation via the
CPEB mechanism. We also examined the role of the MAP Kinase pathway on general
translational regulation via phosphorylation of elF4E, 4EBP1, and S6.
Materials & Methods:
Hippocampal Culture
High-density hippocampal pyramidal cell (CA1-CA3) cultures were prepared from
P1-P2 rat pups as previously described (Liu and Tsien, 1995). Briefly, neurons were plated
on Matrigel (Becton Dickinson) substrate coated glass coverslips in MEM supplemented
with glucose, sodium bicarbonate, glutamine, insulin, penicillin/streptomycin and 10%
63
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
FBS. Conditioned medium was diluted 1:1 with serum-free media containing B27
supplement on DIV (days in vitro) 2 and DIV 6, and transfections were performed on
DIV 8. RNA constructs (0.8 pg / coverslip) were transfected using a liposomal method
(TransMessenger reagent, Qiagen) as per manufacturer's instructions. Neurons were
pre-treated with pharmacological inhibitors (1 pM tetrodotoxin, 100 pM AP5, 10 pM DNQX,
20 pM U0126, 20 pM U0124, 40 pM rapamycin all from Tocris) for 10-12 hours prior
to transfection, unless otherwise indicated. Stimulations (100 ng/mL BDNF for 4 hours;
40pM bicuculline for 8 min., 90 mM KCI for 3 min. 4 times spaced by 10 min.) were
applied immediately following transfection. Coverslips were fixed in 4% PFA/PBS 4 hours
following transfection and mounted for analysis. EGFP reporter expression was quantified
by determination of the total number of EGFP-positive neurons for each transfection. All
experiments were normalized using in-experiment controls (number of EGFP positive
cells in U0126 treated culture) to minimize differences in cell density and viability across
experiments.
Plasmid constructions
EGFP reporter vectors were derived from pEGFP-N1 by deletion of the Apal-AfIll
fragment encoding EGFP and the SV40 polyadenylation signal, followed by insertion of a
160-bp PCR fragment encoding the distal sequences of the a-CaMKII 3' UTR (including
both CPEs and the hexamersequence; (Wu et al., 1998)) into the Smal site, and subsequent
insertion of an EGFP PCR fragment into the Ndel-Xhol sites. The distal sequences from
the aCaMKII 3' UTR were amplified by PCR from a rat brain Marathon cDNA library
(Clontech), and CPE mutations were subsequently introduced, using previously described
primers (Wu et al., 1998). The hexamer mutation AAGAAA was similarly introduced by
PCR. pFC-MEK1 (Stratagene) contains a constitutively active form (AN32-51, S218E,
S222E) of human MEK1 in a derivative of pCMV-Script.
64
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Reporter mRNAs
Capped reporter mRNAs were synthesized in vitro using the T7 Message Machine
kit according to the manufacturer's instructions (Ambion). Templates were generated by
Pfu (Stratagene, according to manufacturer's instructions) mediated PCR amplification of
EGFP-aCaMKII 3' UTR fragments from pCMV-EGFP-CKUTR using primers that encoded
an upstream T7 promoter and downstream oligo(dT) stretches of specific lengths. The
resulting mRNAs comprised (from 5' to 3') a 5' cap, a short synthetic 5' UTR, the EGFP
open reading frame, the distal 160-bp aCaMKII 3' UTR segment (including dual CPEs
and hexamer), and poly(A) tails of specific lengths (0, 20, 40, 60 adenosines). Reporter
mRNA generated by enzymatic tailing (Ambion) contained -150 poly(A) residues.
In situ Hybridization
Fluorescent in situ hybridization of cultured neurons was performed with a
digoxigenin-labeled cRNA probe using immunodetection. The probe was prepared by in
vitro transcription of a PCR-generated template containing a T7 promoter linked to the
EGFP coding region. Four hours following transfection/stimulation, the cells were fixed in
4% paraformaldehyde in phosphate-buffered saline (PFA/PBS). After washing with PBS
and 0.1M TEA, the cells were incubated in 0.25% acetic anhydride / TEA for 10 min.
before being washed in PBS, and blocked in hybridization buffer at 60°C for 1 hr. The cells
were hybridized with 20ng/mL DIG labeled probe in hybridization buffer at 60°C overnight.
After washing with 5X SSC at 60°C for 30 min., and briefly with PBS, the cells were
incubated with DNAse I for 30 min at 37'C. The cells were then washed twice in 2x SSC /
50% formamide for 30 min., washed briefly in TNE buffer, and incubated with 12.5pg/mL
RNase A in TNE buffer for 30 min. at 37'C. After a brief wash in TNE, two 30 min. washes
in 2x SSC at 60°C, two 30 min. washes in 0,2x SSC at 60'C, and a brief wash in PBS,
endogenous peroxidase activity was destroyed by incubating the cells in 1% H2 0 2/PBS
for 10 min. After a brief wash in 0.05% Tween-20 / PBS, the cells were blocked in TNB
buffer, and incubated with anti-DIG HRP conjugated antibody (1:2000) in TNB buffer at
65
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
4°C overnight. The tyramide amplication reaction was then performed according to the
manufacturer's recommendations (Roche, NEN). Fluorescence intensities were quantified
with ImageJ (NIH). Relative mRNA levels were quantified as the mean fluorescence
intensity of randomly selected neurons, and transfected cell number as the mean cell
count from randomly selected low power fields.
Metabolic Labeling
Hippocampal primary cells were used for labeling on DIV8. The cells were treated
with U0126 12 hrs. prior to the labeling experiment. 1 hr. before labeling, the cells were
transferred to a sulphur-free D-MEM media (Specialty Media), and 15. min. before labeling,
40 pM actinomycin D and 200 pg/mL chloramphenicol were added to inhibit transcription
and mitochondrial translation respectively. The stimulation of the cells with BDNF (1
00Ong/
mL) or bicuculline (40pM, 8min) was started at the same time as the addition of the 35Smethionine/3 5 S-cystine (100pCi/mL; Perkin Elmer). 8 min. after the onset of stimulation,
the cells were harvested in ice-cold RIPA buffer, and 30pg of protein were electrophoresed
under denaturing conditions using a 10% SDS-PAGE gel (Biorad). The gels were then
blotted onto nitrocellulose (Hybond, Amersham), the blots washed in TBS, dried and
exposed to X-ray film (Kodak). The resulting autoradiogram was then scanned and the
individual lanes analyzed using ImageJ (NIH). Each gel contained one lane that was
unstimulated and untreated, and all lanes were normalized to the unstimulated untreated
lane. Ponceau-S staining of the blot was used to confirm equal protein loading levels.
Western Blotting
The experiments were done in a similar manner to the 3 5 S-methionine radiolabeling
experiments, with the following differences: Prior to stimulation, the cells were not starved
in sulphur-free media, and no 35 S-methionine was used. After the proteins were transferred
to the nitrocellulose membranes, they were blocked using 1% I-Block (NEN)/0.1 %Tween
20/TBS for 1 hr.; this was followed by incubation in the primary phospho-specific antibody
(phospho-ERK 1:1000, phospho-S6 Ser235/236
66
1:1000, phospho-elF4E Ser209 1:300,
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
phospho-4EBP1 Ser65 1:300, all from Cell Signaling) in blocking buffer overnight at 40 C.
After washing three times in 0.1%TTBS, the blots were incubated for 2 hr. in blocking
buffer containing 1:1000 HRP conjugated anti-rabbit antibody (Cell Signaling). This was
followed by washing 3 times in 0.1%TTBS, and the blots developed using LumiGlo (Cell
Signaling) and Kodak Biomax film according to manufacturer's recommendations. The
blots were stripped using 2% SDS, 100pM 2-mercaptoethanol, 67.5mM Tris-HCI pH 6.8
at 70C for 30 min. followed by extensive washing in 0.1% TTBS. They were reprobed for
total protein (ERK 1:1000, S6 1:1000, elF4E 1:300, 4EBP1 1:300) using the same methods
as described above. The film was scanned and individual protein bands quantified using
ImageJ. Ponceau-S staining prior to blocking confirmed that relative total protein levels
were similar to relative total ERK levels. Since total levels of elF4E and S6 are themselves
translationally regulated, all normalization was done using total ERK2 in the same lane.
The values were renormalized to the data from the unstimulated untreated cells.
Synaptoneurosomes
Cultured hippocampal cells with or without U0126 treatment were used on DIV8.
The cells were scraped into synaptoneurosome buffer (Tyrode buffer with 200pg/mL
chloramphenicol, RNasin and Complete protease inhibitors). The synaptoneurosomes
were prepared by passing the cells through a 10,pm filter (Millipore). 35
g of
synaptoneurosomes were used at 1mg/mL, and labeling was performed for 15 min. with
100OpCi/mL35 S-methionine. The synaptoneurosomes were lysed in SDS loading buffer,
and treated in a similar manner as the cultured cells.
Electrophysiology
Control and mutant mice were deeply anaesthetized, and sacrificed by decapitation.
The hippocampi were isolated in cold oxygenated artificial cerebrospinal fluid (aCSF),
and 400 pm slices were cut using a chopper. The slices were left in an interface
chamber for 4 hrs., after which they were placed in a submersion chamber containing
oxygenated aCSF flowing at a rate of 1mL/min. The recording electrode was placed in
67
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
the CA1 region, while the stimulating electrode was placed in the stratum radiatum at the
CA1/CA3 border. Stimulation at 0.033 Hz at a stimulation intensity that produced 35%
of the maximal response was used to collect a baseline. Once a stable baseline was
established, four 100 Hz. tetani spaced 5 min. apart was used to induce L-LTP. For the
35 S-methionine
incorporation assay, immediately after the final tetanus, the slices were
moved into a submersion chamber containing oxygenated aCSF spiked with
mCi/mL
35Smethionine. After 30 min., the slices were frozen on dry ice, and the CA1 and CA3
regions microdissected. For the western blotting assay, the slices were frozen 5 min.
after the last tetanus, and the CA1 and CA3 regions microdissected. The tissue was then
sonicated in ice-cold RIPA buffer, 30pg of protein was electophoresed on a SDS-PAGE
gel, and blotted onto nitrocellulose. For the 35Smethionine incorporation assay, the blots
were imaged using a Phosphorlmager (Fuji), whereas for the western blotting assay,
the blots were processed as described earlier. The lanes were quantified using ImageJ
(NIH). One tetanized slice and one non-tetanized slice were used in parallel in the same
electrophysiology apparatus to standardize effects such as perfusion conditions, and
recovery time after slice cutting before L-LTP was induced; normalization was done by
calculating the ratio of the tetanized slice to the non-tetanized slice for each experiment.
Thus, the translation level in the non-tetanized slices is set at 1.
Fear conditioning
The training sessions for contextual fear conditioning consisted of a three-minute
exploration period followed by three CS-US pairings separated by one minute each (footshock intensity 0.75 mA, duration 0.5 sec; tone 75 db white noise, 30-sec. duration.). 30
min. after training, the mice were sacrificed, the hippocampi isolated and solubilized in
ice-cold RIPA buffer. 30pg of protein were electrophoresed, blotted onto nitrocellulose
and processed for Western blotting using methods mentioned above.
68
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Results
Minimal poly A tail length required for translation of reporter
To determine if the CPE regulated translation is regulated by the ERK pathway,
an approach using transfection of a reporter construct into primary hippocampal cultured
cells was used. Unlike conventional transfection using DNA constructs, RNA transfection
was used for most of the experiments to obviate concerns about effects on transfection,
mRNA transport and export. Specifically, we engineered in vitro transcribed capped
RNAs containing a short 5' UTR, the coding region of EGFP, and the distal portion of
the a-CaMKII 3' UTR, which contains the UTR's two CPEs, and the canonical AAUAAA
hexamer.
It has been reported in some systems including the mouse oocyte system (Stutz et
al., 1998) that mRNAs do not require polyadenylation for translation; however, increasing
the length of the polyadenosine tail increases the efficiency of translation. We sought to
determine if there was a minimal poly-A tail length for translation in neurons, and thus,
we transfected cultured hippocampal cells with reporter mRNAs containing tails of 0, 20,
40, 60 and -150 adenosines. As Figure 2-7 shows, an mRNA with a tail length of 20 was
competent for translation. However, an mRNA without a poly-A tail was not translated. As
a tail of 20 adenosines was sufficient for translation, all subsequent experiments were
performed using a reporter mRNA with a poly-A tail length of 20. It is to be noted that
there was an increase in the level of translation, as measured by the number of GFP
expressing cells, as the length of the poly A tail is increased, confirming the correlation
between poly-A tail length and translational efficiency.
Translation of Reporter is Activity dependent and MEK dependent
To determine if the translation of the reporter was activity dependent, primary
hippocampal cells were treated for 12 hrs. before transfection either with the sodium
channel blocker tetrodotoxin (TTX) to block action potentials, or with a combination of
DNQX and AP5 to block AMPAR and NMDAR mediated glutamatergic transmission
69
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Figure 2-7: Representative fields from hippocampal cells transfected with capped in vitro
transcribed EGFP-CPE reporter mRNA containing poly-A tail of lengths 0, 20, 60 and approximately 150
respectively. In both cases, there was a significant reduction in the number of EGFP
expressing cells (Fig. 2-8). To determine the role of the ERK MAPK signaling pathway, the
hippocampal cells were pretreated with the MEK inhibitor U0126; this resulted in a further
reduction in the number of EGFP expressing cells. Treatment of the cells with U0126 and
TTX, or with U0126, DNQX and AP5 resulted in no further decrease in the expression
of GFP, indicating that the expression of EGFP induced by spontaneous activity is likely
mediated via the ERK MAPK pathway (Fig. 2-8). The effects of U0126 were likely caused
by specific inhibition of MEK, as the structurally similar, but inactive, conformer of U0126,
U0124, had no effect on the level of GFP expression. It is to be noted that the spontaneous
activity induced translation was also repressed by rapamycin (1.4 +/- 0.05, p < 0.05),
which is an inhibitor of the mTOR pathway, a pathway that is known to also be involved
in translational regulation
(Gingras et al., 1999), and required for L-LTP (Tang et al.,
2002).
Different forms of neuronal activity induce translation in a MEK dependent
manner
Todetermine if differentstimulations of hippocampal neuronswould inducetranslation
of the reporter, the primary hippocampal neurons were stimulated after transfection with
different stimuli. The cells were pretreated for 12 hrs. prior to transfection with U0126 to
reduce the background translation rate. The three stimuli used were a) 1 OOng/mLBDNF, b)
70
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
*
C 4.5
_
0
· 4.0
_
.I
lk,.
*
3.5
r
XL 3.0
W 2.5
T
LL 2.0
(O 1.5
()
> 1.0
0.5
TTX
-
_+
-_
+
DNQX+AP5
U0126
- -+
-
- -
+
-_
-
+
+
+
Figure 2-8: Spontaneous activity dependent translation of reporter RNA is activity and MEK dependent. Both TTX and DNQX/AP5 causes a significant decrease in EGFP expression. U0126 causes
an even greater inhibition in EGFP expression. Further addition of TTX or DNQX/AP5 does not
increase this inhibition
8 min. treatment with 40pM bicuculline, and c) four 3 min. depolarizations with 90mM KCI
spaced 10 min. apart. These three stimuli were chosen because they produce molecular
changes, namely sustained phosphorylation of CREB, similar to those produced by LLTP (Gaiddon et al., 1996; Hardingham et al., 2002; Wu et al., 2001); in addition, BDNF
has been shown to elicit a long-lasting potentiation in slices (Kang and Schuman, 1995).
As shown in Fig. 2-9, all three stimulations induced increased translation of the reporter.
Furthermore, the increased translation of the reporter was blocked by application of the
MEK inhibitor U0126. This evidence supports a role for the ERK MAP Kinase pathway in
activity induced translation.
71
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
.
_
.
!
.
0_
_I
.
.
age
.
I
I1
I
I
A,
_
nnt
Activity
Figure 2-9: Three different forms of neuronal activity all induce translation of the EGFP-CPE
reporter in a MEK dependent manner. The three stimulations are BDNF (100 ng/mL); 8 min. of
40M bicuculline and four 3 min. depolarizations with 90 mM KCI spaced 10 min. apart. Left panel:
Representative images from stimulated cells. Right panel: Quantification of relative EGFP expression. Inhibition of translation in the presence of the MEK inhibitor U0126 implicates the ERK MAP
kinase pathway as being necessary for neuronal activity dependent translation. All three non-drug
treated stimulations are significantly different relative to the non-drug treated unstimulated case.
MEK Dependent Translation Induction is not dependent on CPE
We next sought to test the hypothesis that the action of the ERK pathway on
reporter translation was mediated via the cytoplasmic polyadenylation element (CPE).
Thus, we performed the same experiment again, except that a synthetic mRNA reporter
with both CPEs mutated was used. As shown in Fig. 2-10, all three stimulations induced
translation of the reporter compared to the unstimulated case. Furthermore, this increased
translation was blocked by U0126, suggesting that the action of the ERK pathway on
translation is largely independent of the CPE and polyadenylation. It is to be noted that
the magnitude of stimulation of the mutated CPE containing reporter was lower than that
of the non-mutated CPE containing reporter, suggesting a minor role for the CPE in MEK
dependent translation.
MEK Dependent Translation is not dependent on Polyadenylation
To verify that the effect of the ERK pathway on reporter translation did not act via
effects on polyadenylation, we used a reporter in which the canonical hexamer (AAUAAA)
required for polyadenylation was mutated. This reporter showed the same MEK sensitivity
as both the wild-type reporter and the reporter containing the mutated CPE (Fig. 2-11). In
72
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
addition, the translation of a reporter in which both CPEs and the hexamer were mutated
was also reduced with treatment of U0126 (Fig. 2-11).
0- ·-
·--
1--
35 *UUZLO
o0 3.
c 2.5
X 2'
w.
LL 1.5
O
( 1.0M0.5I
Spont.
Unstim. BDNF Bicuc.
KCI
Activity
11
Figure 2-10: Three different forms of neuronal activity all induce translation of the EGFP-mutCPE
reporter (mutated CPEs) in a MEK dependent manner. The three stimulations are BDNF (100
ng/mL); 8 min. of 40pM bicuculline and four 3 min. depolarizations with 90mM KCI spaced 10 min.
apart. Left Panel: Representative images from stimulated cells. Right Panel: Quantification of
relative EGFP expression. Inhibition of translation in the presence of the MEK inhibitor U0126 implicates the ERK MAP kinase pathway as being necessary for neuronal activity dependent translation
in a CPE independent manner. All three stimulations in the absence of U0126 are significantly
different relative to the unstimulated sample.
Effects of MEK inhibition are not due to toxicity or transfection level
To verify that the effects of U0126 on GFP levels were not mediated by effects on
transfection efficiency or cell toxicity, we performed in situ hybridization on transfected cells
using an anti-GFP oligonucleotide as the probe. As shown in Fig. 2-12, pharmacologic
treatment of the cells with U0126 had no effect on either the number of cells transfected
or the levels of mRNA within the transfected cells. Furthermore, U0126 had no effect on
the morphology of cells as shown in Fig. 2-12. This, along with the fact that U0126 treated
cells were still competent for reporter translation after washout of the U0126 (Figs. 2-9,
2-10), suggests that the effect of U0126 was on translation itself, and not on cell survival.
It is also to be noted that the intracellular distribution of reporter mRNA was the same in
both U0126 untreated and treated cells - the mRNA was visible in the cell soma and the
dendrites.
73
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
,,
C
-
c.
-
U0126
O
3.0
2.0
L 2.5
TT
7-
Lf
X
a.
LL 1.5
1.5
*
Ol
U
T
a)
2 2.5
x
3.0
'i
C)
LL
,,O.3D · U0126
C
|
*k
An
T
%0 -
> 1.u
1.0-
I
_
I
0.5
0.5n-
-'1
n
I
Spont.
Activity
Spont.
Activity
Figure 2-11: Spontaneous activity induced translation of the reporter with mutated hexamer
sequences is dependent on MEK activity. The left panel shows data using a reporter with mutated
hexamer, whereas the right panel shows data using a reporter with both mutated hexamer and
mutated CPEs. Both panels confirm that MEK's role in activity dependent translation is not on the
regulation of polyadenylation.
14
|2
180
I
4A
r_
:3
1.
12
dl
IOU
T
C140
10
X 120
I :!
100
-U
-J
a,
80
60
I> 4
i 40
20
Z< 2
U
0
IE 0
1
Activity
KCI
spor. UOlLb
Activity
Figure 2-12: In situ hybridization with a probe against the reporter mRNA shows that relative
mRNA levels within cells (middle panel), and the number of transfected cells (right panel) does not
depend on the treatment. The left panel shows representative cells. Thus, the role of MEK in activity induced translation of the reporter is via translation of the reporter, and not via effects on mRNA
stability or transfection efficiency.
74
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
MEK is sufficient to induce translation of reporter
We have demonstrated that the ERK pathway is necessary for activity dependent
translation. To see if the ERK pathway was sufficient for increased translation of the
reporter, we used a DNA transfection strategy. The reason for the use of a DNA reporter
is the relatively short lived nature of RNA transfections compared to DNA transfections,
and the lower level of transfection. Thus, we cotransfected hippocampal cells with the
EGFP reporter under the control of a CMV promoter with a second plasmid encoding
constitutively active MEK (cMEK), also under the control of a CMV promoter. To allow
for the buildup of cMEK protein, without a feedforward effect of cMEK on transcription of
cMEK or the reporter genes, the cells were treated with U0126 for 12 hrs. from the time
of transfection to block the activity of the cMEK. 12 hrs. after transfection, the cells were
washed to remove the U01 26, and simultaneously, the transcription inhibitor actinomycin D
was added to prevent effects of the cMEK on transcription of the reporter or the cMEK
gene. The control experiment consisted of a cotransfection of the DNA reporter with the
parent plasmid of the cMEK. The same experiment was also done with cotransfection
of the reporter and control plasmids, but with a 1OOng/mL BDNF stimulation. As Fig. 213 shows, BDNF and cMEK both induced a significant increase in the translation of the
reporter, thus demonstrating that the ERK pathway is sufficient for induction of translation
of the reporter in neurons.
20
kA
16
*
cL 12
CL_
Lu 8
ra
Z'
4
=,
0'
Unstimulated
BDNF
cMEK1
Figure 2-13: Co-transfection with constitutively active MEK stimulates translation of the reporter to
the same extent as stimulation with BDNF (100 ng/mL). The experiment used DNA transfection
with U0126 and actinomycin D (described in the text) to preclude effects of transcription on the
assay.
75
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Metabolic Labeling studies show a general upregulation of translation by
BDNF and bicuculline
Our results pointed to a MAP Kinase dependent increase in translation induced
by BDNF, bicuculline, and spaced depolarization. However, the regulated stimulation of
translation was not dependent on the CPEs. As no other known cis-acting elements were
present in the reporter RNA, we hypothesized that a general MAP Kinase dependent
increase in translation was induced by the stimulations. We sought to test this hypothesis
by using metabolic labeling with a short 10 minute pulse of
35 S-methionine/35 S-cystine.
To differentiate between changes in translation affecting a large number of mRNAs, and
large changes affecting a few mRNAs, we ran the protein products on a gel, and obtained
an autoradiogram to label the newly translated proteins.
2.5 o-
A
+
-+
- ---i
t _ ,
I
·
A..
:
!:-4
,~ ~
+
-
-
-
-
+
-
- -
+
+
+
+
.
U0126
Rapamycin
BDNF
Bicuculline
*U0126
. .
2.0
J, 4
Rapamycin
c1.o-
IT
I
I
t~ 1.0e
.Ii0.5dij
oI
L.
L
Spontaneous
BDNFBicuculline
Activity
Figure 2-14: Metabolic labeling with 35S-Methionine and 35S-Cystine shows that the levels of all
resolvable proteins are upregulated by treatment with BDNF and bicuculline. Furthermore, this
translation as well as translation by spontaneous activity is blocked by inhibition of MEK by U0126.
Rapamycin also blocks spontaneous activity induced translation. Left panel shows a sample gel,
indicating that all resolvable protein species are equally affected by the treatments. Right panel
shows a quantification of the 35S incorporation data in the lanes of the blot. For each case, the
entire lane was used for the quantification.
As shown in Figure 2-14, metabolic labeling results confirmed the hypothesis that
the ERK pathway controls activity induced translation of not just a few dominant proteins,
but all resolved proteins across the entire molecular weight spectrum of proteins resolved
by the gel. Spontaneous activity, BDNF and bicuculline all stimulated the translation of
proteins; this stimulated translation was blocked by the MEK inhibitor U0126. These
76
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
results also validate the translation reporter assay and the conclusion from the reporter
assay that the ERK signaling pathway is involved in translation of the vast majority of
mRNAs. The mTOR pathway inhibitor, rapamycin also inhibited spontaneous activity
induced translation of all resolved proteins, which is consistent with the role of the mTOR
pathway in control of the translation machinery. Thus, we concluded that the primary
role of the ERK pathway in activity induced translation is via its general effect on capdependent translation, which would affect all mRNAs.
Phosphorylation of translation initiation factors and ribosomal proteins
accompanies stimulated translation
We next sought to determine the molecular mechanism that underlies activity
dependent MEK dependent translation. Though translational regulation has not been
extensively studied, there is, as described earlier, prior evidence from mitotic cells for
the role of the ERK pathway on translational initiation via effects on elF4E (Gingras et
al., 1999), 4EBP-1 (Gingras et al., 1999), and to a lesser degree S6 (Wang et al., 2001;
Wang and Proud, 2002). Thus, Western blotting using phospho-specific antibodies was
used to determine if phosphorylation of key residues on the three proteins was activated
by activity in a MEK dependent manner.
As hypothesized, stimulation of the hippocampal cells by BDNF and bicuculline
led to phosphorylation of ERK; in addition, phosphorylation of the initiation factor, elF4E,
its negative regulator, 4EBP1, and the ribosomal component S6 were all increased by
stimulation (Figure 2-15). Furthermore, all of the stimulated phosphorylation events were
inhibited by the MEK inhibitor U0126, confirming the central role of the ERK signaling
pathway in activity induced cap dependent translation. It is to be noted that the role
of ERK in spontaneous activity induced phosphorylation of S6 was not significant,
whereas its role in the BDNF and bicuculline stimulated phosphorylation of S6 was. The
effects of ERK can be compared with the results of rapamycin on spontaneous activity
induced phosphorylation of the various translation proteins. Rapamycin also blocked the
77
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
B
A
a)
CD
0
rCL
a
i.
W:
Spontaneous
Activity
BDNF
Bicuculline
Spontaneous
Activity
BDNF
Bicuculline
D
C
'a)2.0[· -U0126
LUw
uLL
1.5
0
_
.
't b.u
cu
*~~~~~~
Rapamycin
a,(P
-a~~~~~~~~a
5.0
~~~~~~
T
c
~~~~~W
T
4.0
Io
) 1.0
-o
.
Cl)
0
~~~~a-
> 0.5
"U
a,
3.0
=~~~~
2.0
*
= 1.0
co
n
-_,'
Spontaneous
Activity
t
BDNF
Bicucuine
*
n
"
Spontaneous BDNF
Activity
Bicuculline
Figure 2-15: Western blotting shows an increase in phosphorylation of A) ERK, B) S6, C) elF4E
and D) 4EBP1 in response to stimulation by BDNF and bicuculline as compared to spontaneous
activity. This increase is MEK dependent, as it is blocked by U0126. Rapamycin stimulates ERK
phosphorylation, but inhibits S6, elF4E and 4EBP1 phosphorylation.
phosphorylationof the three translation machineryproteins,though the relative magnitude
of the U0126 mediated inhibition and rapamycin mediated inhibition are not the same. For
example, rapamycin was a much more potent inhibitor of spontaneous activity induced S6
phosphorylation, whereas U0126 was a better inhibitor of spontaneous activity induced
elF4E
and 4EBP1 phosphorylation. Interestingly, rapamycin stimulated phosphorylation
of ERK. We hypothesize that this may be due to a feedback loop which detects the lower
translation rate in the cells. The role of rapamycin on elF4E phosphorylation may be
78
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
indirect, as elF4E phosphorylation may be blocked by the binding of elF4E to 4EBP1, as
would be the case when the cells are treated with rapamycin.
We have shown thatstimulation ofthe hippocampal neurons leads to phosphorylation
of 4EBP1. As described earlier, this would lead to an increase in cap-dependent translation
by releasing elF4E to bind to the mRNA cap (Gingras et al., 1999). Furthermore, stimulation
also led to phosphorylation of elF4E itself, which has also been correlated with increased
translation (Gingras et al., 1999), though the mechanism by which this occurs has not
been determined. The phosphorylation of S6 would lead to an increase in the translation
of 5'-TOP containing mRNAs including elF4E and various ribosomal components such as
S6, which in turn would lead to an increase in the translational capacity of the cell (Proud,
2002).
Translation in synaptoneurosomes and phosphorylation of translation
factors is MEK dependent
The exact locus of the stimulated translation was not determined in the previous
experiments, as the entire contents of the cell were analyzed. Under conditions of
synaptic plasticity, and in vivo behavior, dendrites might be the key location of translation.
To determine if translation in the synaptic compartment was dependent on the ERK
signaling pathway via phosphorylation of elF4E, 4EBP1 and S6, a synaptoneurosome
preparation was used to isolate the synaptic compartment, and metabolic labeling and
Western blotting using phospho-specific antibodies against translation factors was used
As shown in Figure 2-16, the synaptoneurosomes from cells treated with
U0126 had lower levels of translation as compared to untreated cells. In addition, the
synaptoneurosomes from treated cells also had lower levels of phosphorylated ERK,
S6, and elF4E. These results demonstrate that MEK dependent translation occurs in
synaptoneurosomes, and suggest that the role of the ERK signaling pathway in the
synaptic compartment is via S6 and elF4E. 4EBP1 results were not obtainable due to
technical reasons.
79
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Our results show that neuronal activity in cultured cells induces translation via
the ERK signaling pathway mediated phosphorylation of S6, elF4E and 4EBP1. Since
LTM and L-LTP both require translation and both are dependent on the ERK pathway,
we propose that LTM and L-LTP activate the ERK signaling pathway which, in turn,
phosphorylates elF4E via Mnkl (Gingras et al., 1999), S6 via S6K2 (Wang et al., 2001;
Wang and Proud, 2002) and 4EBP1 directlyby ERK (Gingras et al., 1999), which in turn
leads to increased translation of a wide variety of mRNAs producing proteins required for
structural plasticity and the expression of L-LTP and LTM.
A
B
C
1.2-
0
i
,0.6
*
*0.8
i
0.2
>01.0.
(0)
c
w0.8
0.6
a0.4
m0.4
O0.4
*
0
- U0126
0.8
0.6
c0.2
U0126 -
E
1.2-
>
.0
0.
D
1.2-
0.20
- U0126
- U0126
- U0126
Figure 2-16: Metabolic Labeling and Western blotting show that spontaneous activity induced
translation is MEK dependent in the synaptodendritic compartment. (A) Autoradiogram from synaptoneurosomes prepared from untreated and U0126 treated cultured hippocampal cells, and (B)
quantification of autoradiogram data show reduced levels of all resolved proteins after inhibition of
MEK. (C-E) Western blotting shows that spontaneous activity induced phosphorylation of ERK, S6
and elF4E is MEK dependent.
Spaced Tetani induce translation in a MEK dependent manner
One prediction of this hypothesis is that inducing L-LTP and LTM would lead to
a MEK dependent increase in translation of all mRNAs and a MEK dependent increase
in phosphorylation of S6, elF4E and 4EBP1. To determine if L-LTP induces increased
translation of all mRNAs in a MEK dependent manner, and to determine if L-LTP
induces phosphorylation of elF4E, S6 and 4EBP-1, we applied L-LTP inducing tetani to
hippocampal slices from control mice (floxed dnMEK1 mice), and dnMEK1 mutant mice
(floxed dnMEK1 / a-CaMKII Cre).
80
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
The hypothesis was that L-LTP in the CA1 would induce increased translation in the
CA1 region of the control mice. In addition, due to antidromic effects of the stimulation, the
CA3 region is also expected to undergo L-LTP at the commissural / associational synapses
(though L-LTP at this synapse has never been reported, a single tetanus induces NMDA
dependent E-LTP in a manner analogous to the Schaffer collateral synapse (Zalutsky
and Nicoll, 1990; Zalutsky and Nicoll, 1991); thus, it is plausible that L-LTP might occur
at the commissural / associational synapse in an equally analogous manner). In mutant
mice, the hypothesis predicts that there would be no L-LTP induced translation in the CA1
(where the dnMEK1 transgene is expressed), but that L-LTP induced translation would
occur in the CA3.
A
3.0
B
o control
C
_ 3.0, o control
*dnMEK1
>
-°2.5-
Z
A 4.0co
9
a3.01
Y2.
Er
Y 2.0-
2.0-
(
*
V)a1.5
o
._ 1.5-
D
5.0o o control
0
2.0-
· 1.0-
1.0-
0.5
0.5CAl
CA31
nn I
nn'- CA1
CA3
CA3
-'-
CA1
CA3
Figure 2-17: Metabolic labeling and Western blotting show that L-LTP inducing tetani stimulate
cap-dependent translation in a MEK-dependent manner. (A) Quantification of metabolic labeling
(35S-Met and 35S-Cys) shows a tetanus induced increase in labeled protein in slices from the CA1
of control slices but not slices from dnMEK1 mice. In the CA3 region, there is a robust increase in
radioactive incorporation in slices from both control and dnMEK1 mice. Each datum from the
tetanized slices is normalized to the corresponding untetanized slice; thus, the radioactive incorporations into untetanized slices are defined as 1, and are not shown for clarity. (B-D) Western blotting
shows a tetanus induced increase in phosphorylation of ERK, S6, and elF4E in slices from the CA1
of control animals, but not mutant animals. In contrast, a significant level of increased phosphorylation is seen in CA3 slices from both control and mutant animals. The normalization is performed in
the same way as (A).
As shown in Figure 2-17A, induction of L-LTP stimulates translation in the CA1
region of control mice, whereas no increase in translation occurred in the mutant animals.
In contrast, L-LTP stimulated translation in the CA3 region of both control and mutant
animals; this serves as an internal control and precludes any general effects, such as
81
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
slice viability, perfusion conditions, etc. on the lack of translational increase seen in the
mutant animals. In addition, the fact that the CA3 showed an increase in translation after
induction of L-LTP provides evidence that protein-synthesis dependent plasticity might
also occur in the CA3 region.
The Western blotting results also support the translation hypothesis. As shown in
Figure 2-17B-D, L-LTP induced phosphorylation of ERK, S6 and elF4E in the CA1 region
of the control mice, but not in the CA1 region of the mutant mice. However, increased
phosphorylation was seen in the CA3 region of both control mice and mutant mice. We
were unable to obtain a signal for 4EBP-1; however, the data on phosphorylation of S6
and elF4E is consistent with the model that protein synthesis dependent plasticity inducing
stimuli increase translation of all mRNAs in a MEK dependent manner via phosphorylation
of S6, elF4E, and probably 4EBP-1.
Fear-conditioning training induces phosphorylation of translation factors
We sought evidence that the same mechanisms that were demonstrated in
cultured cells and in hippocampal slices were also active in vivo. Thus, control and mutant
mice were subjected to fear conditioning, and hippocampal tissue from the animals were
subjected to Western analysis for phosphorylation of elF4E and S6, along with tissue
from untrained animals. Since the previous data points to the phosphorylation status
of S6 and elF4E being a good measure of translation rate in the neurons, and because
measurement of translation rates in vivo after behavioral training using metabolic labeling
was not feasible, the phosphorylation state of S6 and elF4E was measured after fear
conditioning to assess whether behavioral training could increase the translation rate.
As shown in Figure 2-18, there was no difference in phosphorylation of ERK, S6
and elF4E between control and dnMEK1 untrained mice. However, training induced an
increase in phosphorylation of all three proteins in control mice, whereas no increase was
seen in the dnMEK1 mice. This data confirms the data from cultured cells and slices, and
82
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
further supports the model that protein synthesis dependent plasticity requires translation
of all mRNAs via MEK dependent phosphorylation of S6, elF4E and 4EBP1.
A
B
C
2.0 -
control
U dnMEK1
0>
C1.5 Cn
o
_
1 I
-
T
T
II
0)
LI
Q)
n,
-
v ·
untrained trained
' '
_ ,
,
' ' '
,
,
_.
,
untrained trained
' '
_, _ _
' ' '
_ _ J
__
_ _~~~~~~~~~~~~~~~~~~~~~
untrained trained
Figure 2-18: Western blotting shows that phosphorylation of ERK, S6 and elF4E respectively are
upregulated by fear-conditioning training in the hippocampi of control, but not in dnMEK1 mice.
Note that basal phosphorylation levels are the same in control and dnMEK1 hippocampi.
Discussion
Our results indicate a role for the ERK MAP Kinase pathway in the stimulation
of cap-dependent translation affecting the majority of mRNAs. 3 different experiments
using the cell culture system support this view: 1) The translation of the reporter RNA
is upregulated by activity in a MEK dependent manner irrespective of the presence or
absence of any cis-acting elements on the synthetic mRNA. Indeed, after mutation of
the two CPE sequences and the hexamer sequence, no known cis-acting elements were
present in the in vitro transcribed reporter, and thus, the mRNA likely represents a "generic"
mRNA. Since all of the reporters are translated in a MEK dependent manner, it is likely that
the ERK pathway controls translation of all mRNAs. 2) Metabolic labeling studies show
that all resolvable proteins are upregulated by stimulation by BDNF and bicuculline in a
MEK dependent manner. Though it is possible that some mRNAs have other regulatory
mechanisms superimposed upon MEKdependenttranslational regulation, itseems unlikely
83
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
that all neuronal mRNAs are subject to such gene-specific mechanisms. For example, in a
large-scale analysis of the transcriptome, the CPE element, which is the only gene specific
element described to date in mature neurons, was found in only 804 genes out of 33,409
'transcription units' (Okazaki et al., 2002). Thus, the role of MEK in translation is likely
to be via generalized regulation of cap-dependent translation. 3) Mechanistically, BDNF
and bicuculline stimulate phosphorylation of S6, elF4E and 4EBP1. As mentioned earlier,
elF4E and 4EBP1 phosphorylation increase initiation efficiency at the cap, and thus, all
mRNAs subject to cap dependent translation will see a translational efficiency increase
owing to these phosphorylation events (Gingras et al., 1999). The phosphorylation of S6
leads to an increase in translation of certain RNAs bearing 5'-TOP sequences; many of
these proteins are components of the ribosomal machinery, and other translation factors
including elF4E itself. This would lead to an increase in the translational capacity of the
cell itself, which would affect translation of all mRNAs (Proud, 2002).
These results were extended by showing that both the levels of all resolvable
proteins, as well as the phosphorylation of S6 and elF4E was upregulated by L-LTP
inducing tetani in both the CA1 and CA3 regions; in the dnMEK1 mutant mice, which
express the dnMEK1 transgene only in the CA1, this increased translation and
phosphorylations were inhibited in the CA1, but not in the CA3 region. These results
support the case that synaptic plasticity induces a general upregulation of cap-dependent
translation. In addition, after fear conditioning, the phosphorylation levels of S6 and elF4E
were increased in trained control mice but not in trained dnMEK1 mice, indicating that the
same mechanisms that operate in the cell culture and slice systems also operate in vivo
during behavioral training.
The locus for translational upregulation is still unresolved. Our results, using
synaptoneurosomes, show that the upregulation can occur in the dendritic compartments.
However, many, if not all, of the same translation factors present in the dendrites are
also present in the soma (Tang et al., 2002); in addition, since CREB in the nucleus can
84
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
be activated in a MEK dependent manner upon induction of L-LTP (Hardingham et al.,
2002; Wu et al., 2001), it is clear that ERK pathway signaling can occur in the nucleus in
response to a synaptic event. Thus, since the ERK pathway controls both transcription
(Hardingham et al., 2002; Wu et al., 2001) and translation (our results), it is quite likely that
translation upregulation in the soma occurs as a result of L-LTP, and thus, we hypothesize
that both somatic and dendritic translation is necessary for L-LTP. In addition, as the
translational capacity of the dendrite is smaller than the soma (O. Steward, personal
communication), it would appear that somatic translation may be required to provide all
the proteins required for the expression of L-LTP.
It has generally been believed that synaptic plasticity increases levels of certain key
proteins necessary for plasticity using gene specific mechanisms at both the transcriptional
and translational regulatory stages. Our results point to an increase in general capdependent translation as a mechanism behind plasticity, though we cannot conclude that
all of the proteins that are translated via that mechanism contribute to synaptic plasticy.
In contrast to our findings regarding translation, transcription appears to be more genespecific, depending on the specific activation of certain transcription factors by plasticity
inducing phenomena. However, the level of specificity has not been determined, since
most studies have concentrated on an examination of only a few transcription factors
such as CREB and C/EP3P.It remains possible that, via the activation of a large number of
transcription factors, a significant number of neuronal mRNAs are upregulated by activity,
and that the specificity of transcription may be less than currently perceived.
Our results point to an increase in generalized translation upon induction of L-LTP.
However, we have not looked at L-LTD, the mGluR dependent form of which is similar to
L-LTP in that it requires MAP Kinase activity and translation (Gallagher et al., 2004; Huber
et al., 2000; Huber et al., 2001). Further data has to be obtained to determine if the set
of proteins upregulated during L-LTP is different than the set of proteins upregulated by
L-LTD.
85
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
What is the purpose of generalized translational stimulation? A few possibilities
present themselves: 1) The upregulation of the vast majority of proteins upon induction
of L-LTP is simply because a large number of proteins are required for these forms
of structural plasticity, and it may be more efficient, from a regulatory point of view, to
upregulate translation of most mRNAs (with gene-specific mechanisms controlling the
other mRNAs) instead of having a very elaborate regulatory mechanism for each of
the required proteins. In such a case, the relative concentration of the various mRNAs,
and their localization may be mechanisms that allow for specificity, and the keeping of
stoichiometric levels of the various neuronal proteins. 2) An evolutionary artifact may be
to blame for the effect and 3) Some computational function may be a consequence of the
generalized translational increase seen after L-LTP (see Chapter 3). Further experiments
examining L-LTD, the spatial and temporal extent of translational upregulation, and the
mechanisms controlling input-specificity will shed further light on the role of translation in
long-term synaptic plasticity and long-term memory.
It appears that the expression of plasticity (at the least LTP) can be divided into three
different phases - one that does not depend on either transcripton or translation (E-LTP),
one that depends on translation but not transcription (the early phase of L-LTP), and one
that depends on transcription and translation (the late phase of L-LTP). Interestingly, the
same phenomena has also been described in Aplysia, where STF (short-term facilitation)
requires neither, ITF (intermediate-term facilitation) that requires only translation and LTF
(long-term facilitation) which requires both (Sutton and Carew, 2000; Sutton et al., 2001).
In Aplysia, corresponding forms of memory also exist, where STM (short-term memory)
requires neither transcription nor translation, ITM (intermediate-term memory) requires
only translation, and LTM (long-term memory) requires both (Sutton et al., 2001). No such
process has been described in vertebrates to date; further investigation may yet unveil
such a process in vertebrates as well.
86
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Our results, along with some results from the literature (Frey et al., 1996; Frey et al.,
1988; Nguyen et al., 1994; Scharf et al., 2002), support a model where transcription at the
time of induction is required for the maintenance of L-LTP at later points. What is the role
of this transcription? Two possibilities exist: 1) the mRNAs upregulated by transcriptional
activation simply replace the mRNAs that are already present, but decay due to mRNA
turnover processes, or 2) at least some of the mRNAs produced by transcriptional
activation represent new mRNAs, that provide function not present in naive neurons.
Evidence for the latter hypothesis comes from studies of the IEG Arc. Arc expression
is low in the hippocampal CA1 region, and exposure to novelty drastically upregulates
expression of Arc mRNA (Guzowski et al., 1999).
In conclusion, the results presented here point to a key role for MAP Kinase
dependent stimulation of generalized translation in the expression of LTM and L-LTP.
Thus, translation and transcription seem to have differing roles in persistent forms of
synaptic plasticity, and elucidating the exact consequences of these different processes
would go far in determining the mechanism behind long-term memory.
87
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Bibliography
Adams, J. P., Anderson, A. E., Varga, A. W., Dineley, K. T., Cook, R. G., Pfaffinger, P. J.,
and Sweatt, J. D. (2000). The A-type potassium channel Kv4.2 is a substrate for
the mitogen-activated protein kinase ERK. J Neurochem 75, 2277-2287.
Atkins, C. M., Selcher, J. C., Petraitis, J. J., Trzaskos, J. M., and Sweatt, J. D. (1998).
The MAPK cascade is required for mammalian associative learning. Nat
Neurosci 1, 602-609.
Darnell, J. C., Warren, S. T., and Darnell, R. B. (2004). The fragile X mental retardation
protein, FMRP, recognizes G-quartets. Ment Retard Dev Disabil Res Rev 10, 4952.
English, J. D., and Sweatt, J. D. (1996). Activation of p42 mitogen-activated protein
kinase in hippocampal long term potentiation. J Biol Chem 271, 24329-24332.
English, J. D., and Sweatt, J. D. (1997). A requirement for the mitogen-activated protein
kinase cascade in hippocampal long term potentiation. J Biol Chem 272, 1910319106.
Frey, U., Frey, S., Schollmeier, F., and Krug, M. (1996). Influence of actinomycin
D, a
RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in
vivo and in vitro. J Physiol 490 ( Pt 3), 703-711.
Frey, U., Krug, M., Reymann, K. G., and Matthies, H. (1988). Anisomycin, an inhibitor of
protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1
region in vitro. Brain Res 452, 57-65.
Gaiddon, C., Loeffler, J. P., and Larmet, Y. (1996). Brain-derived neurotrophic factor
stimulates AP-1 and cyclic AMP-responsive element dependent transcriptional
activity in central nervous system neurons. J Neurochem 66, 2279-2286.
Gallagher, S. M., Daly, C. A., Bear, M. F., and Huber, K. M. (2004). Extracellular signal-
regulated protein kinase activation is required for metabotropic glutamate
receptor-dependent long-term depression in hippocampal area CA1. J Neurosci
24, 4859-4864.
Gingras, A. C., Raught, B., and Sonenberg, N. (1999). elF4 initiation factors: effectors of
mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem
68, 913-963.
Gray, N. K., and Wickens, M. (1998). Control of translation initiation in animals. Annu
Rev Cell Dev Biol 14, 399-458.
Guzowski, J. F., McNaughton,
B. L., Barnes, C. A., and Worley, P. F. (1999).
Environment-specific expression of the immediate-early gene Arc in hippocampal
neuronal ensembles. Nat Neurosci 2, 1120-1124.
88
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs
oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways.
Nat Neurosci 5, 405-414.
Huang, Y. Y., Nguyen, P. V., Abel, T., and Kandel, E. R. (1996). Long-lasting forms of
synaptic potentiation in the mammalian hippocampus. Learn Mem 3, 74-85.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic
plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U
S A 99, 7746-7750.
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein
synthesis in hippocampal mGluR-dependent long-term depression. Science 288,
1254-1257.
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and
protein synthesis--dependent long-term depression in hippocampal area CA1. J
Neurophysiol 86, 321-325.
Johnson, G. L., and Lapadat, R. (2002). Mitogen-activated protein kinase pathways
mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911-1912.
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between
genes and synapses. Science 294, 1030-1038.
Kang, H., and Schuman, E. M. (1995). Long-lasting neurotrophin-induced enhancement
of synaptic transmission in the adult hippocampus. Science 267, 1658-1662.
Laggerbauer, B., Ostareck, D., Keidel, E. M., Ostareck-Lederer, A., and Fischer, U.
(2001). Evidence that fragile X mental retardation protein is a negative regulator
of translation. Hum Mol Genet 10, 329-338.
Li, Z., Zhang, Y., Ku, L., Wilkinson, K. D., Warren, S. T., and Feng, Y. (2001). The
fragile X mental retardation protein inhibits translation via interacting with mRNA.
Nucleic Acids Res 29, 2276-2283.
Liu, G., and Tsien, R. W. (1995). Properties of synaptic transmission at single
hippocampal synaptic boutons. Nature 375, 404-408.
McGaugh, J. L. (2000). Memory--a century of consolidation. Science 287, 248-251.
Nguyen, P. V., Abel, T., and Kandel, E. R. (1994). Requirement of a critical period of
transcription for induction of a late phase of LTP. Science 265, 1104-1107.
Okazaki, Y., Furuno, M., Kasukawa, T., Adachi, J., Bono, H., Kondo, S., Nikaido,
I., Osato, N., Saito, R., Suzuki, H., et al. (2002). Analysis of the mouse
89
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature
420, 563-573.
Proud, C. G. (2002). Regulation of mammalian translation factors by nutrients. Eur J
Biochem 269, 5338-5349.
Raught, B., and Gingras, A. C. (1999). elF4E activity is regulated at multiple levels. Int J
Biochem Cell Biol 31, 43-57.
Raymond, C. R., Thompson, V. L., Tate, W. P., and Abraham, W. C. (2000).
Metabotropic glutamate receptors trigger homosynaptic protein synthesis to
prolong long-term potentiation. J Neurosci 20, 969-976.
Schafe, G. E., Nadel, N. V., Sullivan, G. M., Harris, A., and LeDoux, J. E. (1999).
Memory consolidation for contextual and auditory fear conditioning is dependent
on protein synthesis, PKA, and MAP kinase. Learn Mem 6, 97-110.
Scharf, M. T., Woo, N. H., Lattal, K. M., Young, J. Z., Nguyen, P. V., and Abel, T. (2002).
Protein synthesis is required for the enhancement of long-term potentiation and
long-term memory by spaced training. J Neurophysiol 87, 2770-2777.
Scheetz, A. J., Nairn, A. C., and Constantine-Paton, M. (2000). NMDA receptormediated control of protein synthesis at developing synapses. Nat Neurosci 3,
211-216.
Selcher, J. C., Weeber, E. J., Christian, J., Nekrasova, T., Landreth, G. E., and Sweatt,
J. D. (2003). A role for ERK MAP kinase in physiologic temporal integration in
hippocampal area CA1. Learn Mem 10, 26-39.
Silva, A. J. (2003). Molecular and cellular cognitive studies of the role of synaptic
plasticity in memory. J Neurobiol 54, 224-237.
Steward, O., and Worley, P. F. (2001). A cellular mechanism for targeting newly
synthesized mRNAs to synaptic sites on dendrites. Proc Natl Acad Sci U S A 98,
7062-7068.
Stutz, A., Conne, B., Huarte, J., Gubler, P., Volkel, V., Flandin, P., and Vassalli, J. D.
(1998). Masking, unmasking, and regulated polyadenylation cooperate in the
translational control of a dormant mRNA in mouse oocytes. Genes Dev 12, 25352548.
Sutton, M. A., and Carew, T. J. (2000). Parallel molecular pathways mediate expression
of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in
Aplysia. Neuron 26, 219-231.
90
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Sutton, M. A., Masters, S. E., Bagnall, M. W., and Carew, T. J. (2001). Molecular
mechanisms underlying a unique intermediate phase of memory in aplysia.
Neuron 31, 143-154.
Sweatt, J. D. (2001). The neuronal MAP kinase cascade: a biochemical signal
integration system subserving synaptic plasticity and memory. J Neurochem 76,
1-10.
Tang, S. J., Reis, G., Kang, H., Gingras, A. C., Sonenberg,
N., and Schuman, E.
M. (2002). A rapamycin-sensitive signaling pathway contributes to long-term
synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A 99, 467-472.
Walden, W. E., Godefroy-Colburn, T., and Thach, R. E. (1981). The role of mRNA
competition in regulating translation. . Demonstration of competition in vivo. J
Biol Chem 256, 11739-11746.
Wang, L., Gout, I., and Proud, C. G. (2001). Cross-talk between the ERK and p70
S6 kinase (S6K) signaling pathways. MEK-dependent activation of S6K2 in
cardiomyocytes. J Biol Chem 276, 32670-32677.
Wang, L., and Proud, C. G. (2002). Ras/Erk signaling is essential for activation
of protein synthesis by Gq protein-coupled receptor agonists in adult
cardiomyocytes. Circ Res 91, 821-829.
Wells, D. G., Dong, X., Quinlan, E. M., Huang, Y. S., Bear, M. F., Richter, J. D., and
Fallon, J. R. (2001). A role for the cytoplasmic polyadenylation element in NMDA
receptor-regulated mRNA translation in neurons. J Neurosci 21, 9541-9548.
Wu, G. Y., Deisseroth,
K., and Tsien, R. W. (2001). Activity-dependent
CREB
phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and
a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad
Sci U S A 98, 2808-2813.
Wu, L., Wells, D., Tay, J., Mendis, D., Abbott, M. A., Barnitt, A., Quinlan, E., Heynen,
A., Fallon, J. R., and Richter, J. D. (1998). CPEB-mediated cytoplasmic
polyadenylation and the regulation of experience-dependent translation of alphaCaMKII mRNA at synapses. Neuron 21, 1129-1139.
Yuan, L. L., Adams, J. P., Swank, M., Sweatt, J. D., and Johnston, D. (2002). Protein
kinase modulation of dendritic K+ channels in hippocampus involves a mitogenactivated protein kinase pathway. J Neurosci 22, 4860-4868.
Zalfa, F., Giorgi, M., Primerano,
B., Moro, A., Di Penta, A., Reis, S., Oostra,
B., and
Bagni, C. (2003). The fragile X syndrome protein FMRP associates with BC1
RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317327.
91
Chapter 2: Role of the MAP Kinase pathway in activity-induced neuronal translation, L-LTP and LTM
Zalutsky, R. A., and Nicoll, R. A. (1990). Comparison of two forms of long-term
potentiation in single hippocampal neurons. Science 248, 1619-1624.
Zalutsky, R. A., and Nicoll, R. A. (1991). Comparison of two forms of long-term
potentiation in single hippocampus neurons. Correction. Science 251, 856.
92
Chapter 3 - Characterization of neuronal activityinduced translation
Abstract
Previous experiments (Chapter 2) have implicated activity-induced translation as a
key regulatory point for the expression of L-LTP and long-term memory. This process was
further characterized by examining whether translation occurs in mature cells as well as
in immature cells. In addition, various stimuli that activate this translation were identified.
Interestingly, L-LTP and L-LTD inducing stimuli upregulate translation of a diverse set of
mRNAs via the same mechanism - namely phosphorylation of ERK, translation initiation
factors and ribosomal proteins. In contrast to protein synthesis, protein degradation is
not affected by the stimuli tested. In addition, translation upregulation lasts at least 45
minutes.
93
Chapter 3 - Characterization of neuronal activity-induced translation
Introduction
Previously, neuronal activity-induced translation was characterized in a hippocampal
culture system with a radioactive metabolic-labeling assay, and a Western blotting assay,
using phospho-specific antibodies, for phosphorylation of translation initiation factors. The
results from those experiments (Chapter 2) showed that BDNF and bicuculline induced
translation of a large number of mRNAs via general translational upregulation.
A few issues were left unresolved by those experiments. The first was technical:
translation levels under spontaneous-activity conditions were very high, and preincubation
for -8hrs in U0126 was required to reduce the translation rates to a level where stimulation
of the translation was possible. Since the growth factors in serum are known to activate
the MAP Kinase pathway (Buchwalter et al., 2004), and the cells were grown in a serumcontaining media, the role of serum in MAP Kinase dependent translation has to be
determined.
The dependence of translational activation on culture age was also unknown. It
has been previously shown that plasticity in vivo is very different in young animals as
compared to adult animals. For example, spine motility is vastly higher in juvenile mice
(Trachtenberg et al., 2002); LTD that is obtained at the Schaffer collateral synapses in
young animals has different electrical and molecular characteristics as compared to
LTD at older synapses (Kemp and Bashir, 2001). Such age dependence has also been
seen in culture. For example, synaptic transmission in young cultures (prior to 10 DIV)
is more inefficient (Renger et al., 2001), and CREB phosphorylation after stimulation by
depolarization more persistent (Sala et al., 2000). Thus, as a neuronal network, be it in
culture, slice or in vivo, ages, drastically different effects to various stimuli can be seen.
Thus it is critical to determine if mature cultures also undergo MEK-dependent activityinduced translation.
Previous experiments used bicuculline and BDNF as stimulations, both of which
provide correlates of L-LTP inducing tetani (Aakalu et al., 2001; Hardingham et al., 2002).
94
Chapter 3 - Characterization of neuronal activity-induced translation
In addition to BDNF, protein-synthesis dependent potentiation, resembling L-LTP, has
also been shown with dopamine (Huang and Kandel, 1995), and activation of the PKA
pathway (Frey et al., 1993). Though the electrophysiological consequences of L-LTP,
dopamine and activation of the PKA pathway are similar, the biochemical mechanism
underlying these forms of potentiation may be much more different. It is thus of interest to
determine the overlap between the translational regulatory mechanisms underlying these
forms of plasticity.
In addition, mGluR dependent LTD also requires translation and the MAP Kinase
pathway (Gallagher et al., 2004; Huber et al., 2000). This last example is particularly
interesting, since LTD and LTP have opposing effects on synaptic weights but appear
to use overlapping pathways. Thus, it is entirely possible that the translational control
mechanisms are also similar, since the MAP Kinase pathway is necessary for L-LTP
induced translation (Chapter 2).
Whereas translation is a key regulatory control step during the induction of L-LTP,
the opposite process of protein degradation may also be important for synaptic plasticity
(Hegde, 2004). Some evidence suggests that chronic changes in activity can alter protein
degradation rates via the ubiquitin-proteosome system (Ehlers, 2003). Though it is unlikely
that the MAP Kinase dependent upregulation of metabolically labeled protein that we
have previously observed were due to decreases in degradation rates, a measurement of
the protein degradation rate after acute stimulation would shed light on the importance of
this alternate mechanism of increasing protein levels.
To solve these questions, various parameters affecting translation were tested in
the cell-culture system using metabolic labeling as a marker for translation rates. Firstly,
a serum-free system was used to determine if the background translation previously
observed was dependent on serum, and to create an assay system that did not require
exogenous agents to attain a low background translation rate. Secondly, cells at different
ages were tested to determine if translational stimulation also occurred in mature cells,
95
Chapter 3 - Characterization of neuronal activity-induced translation
which would better resemble adult cells. Thirdly, different chemical agents were used to
examine if they activated translation, and fourthly, protein degradation after stimulation
was tested. Lastly, a preliminary time-course of translation was determined.
Material and Methods
Hippocampal Culture
High-density hippocampal pyramidal cell (CAl-CA3) cultures were prepared from
P1-P2 rat pups as previously described (Liu and Tsien, 1995). Briefly, neurons were plated
on Matrigel (Becton Dickinson) substrate coated glass coverslips in MEM supplemented
with glucose, sodium bicarbonate, glutamine, insulin, penicillin/streptomycin and 10%
FBS. The serum-containing media was replaced with MEM media supplemented with
glucose, and B27 on DIV 2. The media was diluted 1:1 on DIV 5.
Metabolic Labeling
Hippocampal primary cells were used for labeling on DIV8 or DIV16. 15 minutes
before labeling, the cells were transferred to a sulphur-free D-MEM media (Specialty
Media) containing 40 pM actinomycin D, 200 pg/mL chloramphenicol, and either vehicle
(DMSO) or U0126 to a concentration of 25pM to inhibit transcription and mitochondrial
translation respectively. The stimulation of the cells with BDNF (100ng/mL), bicuculline
(40pM), dopamine (100pM), or DHPG (50pM) was started at the same time as the addition
of the
3 5 S-methionine / 35 S-cystine
(100pCi/mL). 5 min. after the onset of stimulation, the
cells were harvested in Laemmili sample buffer, and electrophoresed under denaturing
conditions using a 15% SDS-PAGE gel (Biorad). The gels were then blotted onto
nitrocellulose (Hybond, Amersham), the blots washed in TBS, dried and exposed to Xray film (Kodak). The resulting autoradiogram was then scanned and the individual lanes
analyzed using ImageJ (NIH). Each gel contained one lane that was unstimulated and
untreated, and all lanes were normalized to the unstimulated untreated lane. Ponceau-S
staining of the blot was used to confirm equal protein loading levels.
96
Chapter 3 - Characterization of neuronal activity-induced translation
Western Blotting
After the autoradiograms from the blots were obtained, they were blocked using 1 %
I-Block (NEN)/0.1 %Tween 20/TBS for 1 hr.; this was followed by incubation in the primary
phospho-specific antibody (phospho-ERK 1:1000, phospho-S6 Ser235/236
1:1000,
phospho-elF4E Ser209 1:300 all from Cell Signaling) in blocking buffer overnight at 40C.
After washing three times in 0.1%TTBS, the blots were incubated for 2 hr. in blocking
buffer containing 1:1000 HRP conjugated anti-rabbit antibody (Cell Signaling). This was
followed by washing 3 times in 0.1%TTBS, and the blots developed using LumiGlo (Cell
Signaling) and Kodak Biomax film according to manufacturer's recommendations. The
blots were stripped using 2% SDS, 100pM 2-mercaptoethanol, 67.5mM Tris-HCI pH 6.8
at 70C for 30 min. followed by extensive washing in 0.1% TTBS. They were reprobed
for total protein (ERK 1:1000, S6 1:1000, elF4E 1:300) using the same methods as
described above. The film was scanned and individual protein bands quantified using
ImageJ. Ponceau-S staining confirmed that relative total protein levels were similar to
relative total ERK levels. Since total levels of elF4E and S6 are themselves translationally
regulated, all normalization was done using total ERK2 in the same lane. The values were
renormalized to the data from the unstimulated untreated cells.
Results
Translation in DIV 8 cells grown in serum-free media are stimulated by
BDNF and bicuculline
To test the hypothesis that the high background translation in previous experiments
was due to serum-stimulated MAP Kinase levels, cells were grown in serum-free media
from DIV 2. Growing the cells in serum-free media from DIV 0, as well as growing the
cells in serum-free media after DIV 4 caused massive cell death (data not shown). We
used metabolic labeling to determine if these cells' translation could be stimulated by
BDNF and bicuculline. As Fig. 3-1A shows, BDNF and bicuculline significantly increased
the incorporation of radioactive methionine and cystine into the proteome of the cell;
97
Chapter 3 - Characterization of neuronal activity-induced translation
furthermore, this increase was blocked by U0126, indicating that the translation was
MAP Kinase dependent. Thus, under low-serum conditions, an increase in translation is
possible, in support of the view that the serum present in earlier experiments resulted in
high background translation, which could not be further stimulated. Interestingly, though the
background translation was not blocked by acute treatment with U0126, chronic blockade
of the MEK with U0126 did reduce the translation rate (Relative
35
S
incorporation: 0.27
+/- 0.01; p < 0.05). This data also indicates that the previous data (Chapter 2) was not an
artifact of growing the cells in serum-containing media.
The phosphorylation status of the translation initiation factors was also tested
after stimulation by BDNF and bicuculline by Western blotting using phospho-specific
B
A
_
2.0g.
v.._
lvv iA4s
. _v
_
_
° 1.
.)
T
I
I
T
I n1 9A
T
1.51I
JI
.>
71
nv-u
.n.
BDNF
Spont.Activity
C
___
....
g ZUU1
g1.00
^^uz~~~~~~~~
e
~T
I
_.
~~111r"
in
2.25.
T
~~c
-17
.
'
D
II
"
I
I
-
-i
...
-.
BICUCUIIIlne
OUNI
Stimulation
U0126
T
,4)
*
U.
*
0.
if
.
0.25
0.00
Spont.Activity
-. 1
15
~~~~l -
..
spont.
Activity
Bicuculline
Stimulation
rr
*
._
CX
to
X 0.5-
_ 0.75
· 0.50
,r-"
E
±1
Spont.Activity
BDNF
Bicuculline
Stimulation
BDNF
Bicuculline
Stimulation
Figure 3-1: Neuronal activity induced upregulation of translation. (A) Metabolic labeling of DIV8 cells grown
without serum shows that BDNF, bicuculline and DHPG cause stimulation of translation in a MEK dependent
manner. (B-D) Western blotting shows that BDNF, bicuculline and DHPG stimulate phosphorylation of ERK,
S6 and elF4E in a MEK dependent manner. In all cases, labeling and Western signal is significantly higher
after stimulation as compared to Spont. Activity (p < 0.05), but no marks are shown for clarity.(* p < 0.05)
98
Chapter 3 - Characterization of neuronal activity-induced translation
antibodies. The phosphorylation of S6 and elF4E were upregulated by both BDNF and
bicuculline in a MAP Kinase dependent manner. In addition, ERK phosphorylation was
also upregulated by BDNF and bicuculline confirming previous experiments done in
serum-containing
media (Fig. 3-1 B-D).
Translational upregulation by BDNF and bicuculline also occurs in mature
cells
It has been previously shown that synapses in culture initially form in an immature
fashion with a high proportion of silent synapses (Renger et al., 2001); furthermore,
the synapses form on dendritic shafts (Sala et al., 2001). Since we hypothesized that
translational upregulation is important for L-LTP in adult rodent hippocampal slices, and
long-term memory formation in adult mice (Chapter 2), we wanted to test the effects
of BDNF and bicuculline on mature spine-containing neurons to verify that translational
regulation could also occur in cell-culture correlates of adult neurons. We chose DIV 16
neurons because they contain mature spines and synapses (data not shown). BDNF
and bicuculline both significantly upregulated translation in hippocampal cultured cells
1.5
I
T
h.
L
·
I
I
.
U0126
*
1.0-
C)
> 0.5
n
V-V
I
1
Spont. Activity
BDNF
Bicuculline
Stimulation
Figure 3-2: Metabolic labeling of DIV16 cells grown without serum shows that BDNF, bicuculline and DHPG
cause stimulation of translation in a MEK dependent manner. In all cases, labeling is significantly higher after
stimulation (p < 0.05), but no asterisks are shown for clarity.
99
Chapter 3 - Characterization of neuronal activity-induced translation
in a MAP Kinase dependent manner (Fig. 3-2). Further experiments will determine if the
mechanisms uncovered first in DIV 8 cells are the same in DIV 16 cells
Dopamine receptor activation and spaced depolarization upregulate
translation in a MAP Kinase dependent manner
As discussed previously, upregulation of the PKA pathway by forskolin or dopamine
cause a protein-synthesis dependent potentiation (Frey et al., 1993; Huang and Kandel,
1995), similar to BDNF (Kang and Schuman, 1995). In addition, spaced depolarization
with KCI has been shown to activate the MAP Kinase pathway and CREB phosphorylation
in a manner that resembles L-LTP (Wu et al., 2001a; Wu et al., 2001b). Thus, we tested
dopamine and spaced depolarization (4x 3 min. depolarizations with 90mM KCI, separated
by 10 min.) to determine if translation is also similarly upregulated. As a control, a single
depolarization stimulus (3 min. depolarization with 90mM KCI) was used, as a single
depolarization does not activate CREB in the same manner as L-LTP (Wu et al., 2001a;
Wu et al., 2001 b). Remarkably, translation was significantly upregulated by both dopamine
and spaced depolarization, but not single depolarization, in a MAP Kinase dependent
manner (Fig. 3-3). Thus, MAP Kinase dependent translation appears to be stimulated by
a wide range of agents that are known to cause protein-synthesis dependent translation.
mGluR activation upregulates translation in a MAP Kinase dependent
manner
All of the previously tested agents (BDNF, bicuculline, dopamine, and spaced
depolarization) are known to cause protein-synthesis dependent potentiation resembling
L-LTP. How about L-LTD inducing stimuli? When Schaffer collateral synapses are
stimulated with a paired-pulse low-frequency stimulation protocol (PP-LFS; 900 1Hz
paired-pulses with a 50 ms. inter-stimulus interval), a long-lasting L-LTD that depends on
mGluR5 and translation, but not transcription is induced (Huber et al., 2000; Huber et al.,
2001; Kemp and Bashir, 1999). DHPG, an mGluR Type I agonist, mimics and occludes
this form of L-LTD; conversely, the PP-LFS stimulation occludes the effects of DHPG
100
Chapter 3 - Characterization of neuronal activity-induced translation
A,
2.0-
1J U0126
X
T
2.0-
oC1.
-F
1.5co
u,
>
1.0-
nl 0.5n nv.v
I
Spont.
Activity
I
T
*
I
Dopamine
Spaced
Depol.
Single
Depol.
Stimulation
Figure 3-3: Metabolic labeling of DIV8 cells grown without serum shows that dopamine and spaced depolarization (4x 3 min. depolarizations with 90mM KCI, spaced 10 min. apart) significantly increase translation
rates in a MEK dependent manner. Single depol. (3 min. depolarization with 90mM KCI) has no effect on
translation.
(Huber et al., 2001). Since this mGluR dependent form of LTD is also blocked by the MAP
kinase inhibitor U0126 (Gallagher et al., 2004), we sought to determine if the mechanisms
underlying the BDNF and bicuculline induced translation were also stimulated by DHPG.
As shown in Fig. 3-4A, DHPG also upregulates translation in a MAP Kinase
dependent manner. Remarkably, the level of translation induced, as well as the proteins
upregulated by DHPG is the same as that of BDNF and bicuculline. In addition, we
confirmed previous reports that ERK, elF4E and S6 phosphorylation are upregulated by
the DHPG treatment in a MAP Kinase dependent manner (Fig 3-4C-E; Banko and Klann,
personal communication).
Protein Degradation is not affected by LTP or LTD stimuli
It is a priori possible that changes in protein degradation would accompany
changes in protein translation. Thus, we measured protein degradation rates using pulsechase techniques. Briefly, proteins were labeled with a pulse of radioactive methionine.
101
Chapter 3 - Characterization of neuronal activity-induced translation
A
B
80.
I0
+
+
+
-+
u,
C
co
0
(u
.5
+
+
U0126
Bicuculline
DHPG
DHPG
Activity
Stimulation
C
D
I1U0126
!
2.25i
1.5
2.00
T
X M
0,
U0126
1
*
1.
u,
0
*
*
cL
1.
I
'
.
.>
0.
y 0.
O.
ixi
m
m
Spont.
DHPG
0.
0.
'I
Bicuculline
BDNF
Spont.
Activity
DHPG
BDNF
I
Bicuculline
Activity
Stimulation
Stimulation
E
I
2.0.
U0126
1.5'
1.5.
-
U-
0. 1
I
*
*
41
0
(U
.5 0
0
±1
Spont.
DHPG
BDNF
Bicuculline
Activity
Stimulation
Figure 3-4: Metabolic labeling of DIV8 cells grown without serum shows that DHPG, BDNF and Bicuculline
significantly increase translation rates in a MEK dependent manner. A) Representative autoradiograms show
that all resolvable bands whose translation is upregulated by DHPG are also upregulated by BDNF. B) Quantification of autoradiogram data shows that the relative stimulation of cells by BDNF, DHPG and bicuculline are
similar. C-E) Western analysis shows that DHPG upregulates phosphorylation of C) ERK, D) S6 and E) elF4E
in a MEK dependent manner. Note that in B-E), the BDNF, and Bicuculline data are the same as that shown
in Figure 4-1. *, p < 0.05. In all cases, 35S incorporation and Western signal were significantly higher after
DHPG, BDNF and bicuculline stimulation, but no marks are shown for clarity.
102
Chapter 3 - Characterization of neuronal activity-induced translation
After a large excess of non-radioactive methionine was added to the culture media, the
cells were stimulated with BDNF, bicuculline and DHPG, as previously discussed. None
of the stimulations altered the distribution or level of proteins labeled by the radioactive
methionine/cystine, indicating that, over the short time-span of the experiment, protein
degradation rates were unchanged across stimulations (Fig. 3-5).
1.50-
vehicle
rU0126
o 1.25-
T
u) 1.00eo
'o 0.75N
_r,
= 0.50
E
0.25
+
-
+
+
+
+
+
+
+
0.00
UU12b
- BDNF
- Bicuculline
- - +- DHPG
+
Spont.
Act.
BDNF BicucullineDHPG
Stimulation
Figure 3-5: Pulse-chase experiments show that neither stimulation of cultured cells by BDNF, Bicuculline and
DHPG nor blockade of stimulation by the MEK inhibitor U0126 have any effect on stability of proteins. Left
Panel: Representative autoradiogram. Right Panel: Quantification of data from n=4 autoradiograms
Increased translation lasts at least 45 minutes
We next sought to determine the time-course of increased translation. Thus,
we pulsed the cells with radioactive methionine at different times after stimulation. We
discovered that the increased translation lasted at least 45 minutes. Unfortunately, our
experiment did not determine when the translational upregulation ended (Fig. 3-6), and
future experiments will have to be done with pulses of
35 S-methionine/35 S-cystine
at even
later time points after stimulation.
Discussion
Neuronal activity induced translation was further characterized in these set of
experiments. First, a new culture system was designed that allowed for the analysis of
stimulated translation without having to lower background translation with the MEK inhibitor
103
Chapter 3 - Characterization of neuronal activity-induced translation
-%
e ,
-i- Spont.
Activity
Ift
-0- Bicuculline
d.
o 1.5:
-
I
Mf1.04
,_
m
0.5-
0.0
I
01
10
I
20
I
I
30
40
I
i--- 50
Time (min)
Figure 3-6: Stimulation with bicuculline (t=-5 to t=O) causes an increase in translation that lasts at least 45
minutes. 35S incorporation for all points is normalized to incorporation at t=O under spontaneous activity
conditions.
U0126. This system, which lowers background MAP Kinase activity by eliminating serum
from the media, allowed for the demonstration that the results from previous experiments
were not artifacts of serum in the culture media.
We have also demonstrated that the bicuculline and BDNF mediated stimulation
of translation in hippocampal culture is seen in DIV 16 cultures. This data suggests that
increases in MAP Kinase dependent neuronal translation also occurs in mature neurons,
supporting the model that neuronal activity induced translational increases underlie the
L-LTP seen in adult hippocampal slices and long-term memory observed in adult mice.
In addition, chemicals that induced protein-synthesis dependent plasticity, including
dopamine (Huang and Kandel, 1995) and spaced depolarization (Wu et al., 2001a; Wu
et al., 2001b) also activated translation in a MAP Kinase dependent manner. These
results will need to be extended into slices in order to confirm that the dopamine induced
translational increase is also seen in a more physiological situation.
Interestingly, an L-LTD stimulus, DHPG, also induced translation in a MAP
Kinase dependent manner. Furthermore, DHPG also activated ERK, S6 and elF4E
phosphorylation in a MEK dependent manner; also, the set of proteins whose translation
104
Chapter 3 - Characterization of neuronal activity-induced translation
was upregulated was indistinguishable between bicuculline and DHPG as visualized by
the labeled bands on the SDS-PAGE gel. One caveat is that the relatively low resolution
offered by 1D gel electrophoresis may account for the lack of any difference observable
between the conditions. 2D gel electrophoresis would provide further resolving power that
would shed light on the differences between the stimuli. However, given the combination
of the 1D gel, and the elF4E & S6 phosphorylation results, it is quite likely that a diverse,
largely overlapping set of mRNAs' translation would be upregulated both by the L-LTP
and L-LTD inducing stimuli.
Why would L-LTP and L-LTD, which result in opposing changes to synaptic
weights, induce synthesis of a similar set of proteins? One possible explanation is that
L-LTP and L-LTD both require an overlapping set of proteins, and this mechanism is the
evolutionarily easiest way of reaching that goal. Another possible explanation is that it is
simply an evolutionary artifact. A third possibility is that so-far undetermined gene-specific
mechanisms produce the proteins that differentiate between L-LTP and L-LTD, and that
the general translation mechanisms are used to produce only those proteins that are
required for the expression of both processes.
There is an alternate explanation. It is possible that L-LTP and L-LTD inducing
stimuli upregulate translation of the same proteins - namely the entire set of proteins
required for the expression of both processes. Assuming that LTD also uses the synaptic
tag and capture method for input-specificity, the tag induced by LTP (both E-LTP and LLTP) inducing stimuli and the tag induced by LTD (both E-LTD and L-LTD) inducing stimuli
must be different. This would result in an interesting associativity between LTP and LTD
processes (Figure 3-7). This model would predict that, if, at one input, an L-LTD inducing
stimulus is presented, and at another input, an E-LTP inducing stimulus is presented, the
LTP tag at the second synapse would allow for the capture of proteins required for LTP
from the pool of proteins upregulated by the first synapse. Thus, the second synapse
would express L-LTP!
105
Chapter 3 - Characterization of neuronal activity-induced translation
4. Stimulation of Transcription
LP & LTD
13-t.;-
A
1. Strong
a
(I
le
ulrsrarBI·
2.LTP1
P Proteins
5. Struc
Chan!
1'. Weak
LFS
Figure 3-7: Proposed interaction between L-LTP and L-LTD: When a strong tetanus is presented
to an input (1), a LTP tag (2), translation (3) and transcription (4) are activated. The produced
proteins, some of which are required for LTP (LTP proteins), some of which are required for LTD
(LTD proteins), and some of which are required for both (LTP & LTD proteins) diffuse within some
distance from the site of translation, and are thus available to many synapses. The activated
synapse captures proteins, leading to structural change (5), leading to the expression of L-LTP If
a second synapse, within a certain spatial and temporal distance, is given a weak low-frequency
stimulation (1'), an LTD tag (2') is produced, but no translation or transcription is activated. However, this synapse captures LTD proteins from the pool of proteins produced by the first input, leading to structural change (5') and the expression of L-LTD. Note that the converse can also occur,
and that the process is temporally bidirectional - i.e. the weak stimulus can occur before or after
the strong stimulus and tagging and capturing will still occur.
This process, termed either cross-tagging or cross-capturing, would allow for useful
computational phenomena. Several computational models propose that bidirectional
plasticity is important for optimizing neuronal function (Bear, 1996). Since synaptic
capture allows for the conversion of short-term processes (E-LTP or E-LTD) to long-term
processes (L-LTP or L-LTD), it would be optimal for neuronal function if the direction of
synaptic weight change remains unchanged by the capture process. This requires the
presence of proteins required for the expression of both L-LTP and L-LTD, which would
be the result of generalized translational upregulation.
Recent evidence has shown that induction of L-LTD allows for E-LTP to be
converted to L-LTP,and vice versa (Sajikumar and Frey, 2003; Sajikumar and Frey, 2004a;
106
Chapter 3 - Characterization of neuronal activity-induced translation
Sajikumar and Frey, 2004b). This also suggests that any gene-specific mechanisms must
either provide proteins not essential for L-LTP or L-LTD, or be activated by both L-LTP and
L-LTD inducing stimuli. It will be interesting to see into which category CPEB and FMRP
mediated translation fall.
Acute stimulation of neurons via inhibition of GABAAreceptors (bicuculline), activation
of trkB receptors (BDNF), multiple spaced depolarizations, but not single depolarizations,
stimulation of the dopamine pathway (dopamine) and the mGluR type I pathway (DHPG)
all stimulate translation. Unfortunately, the time-course of the translational upregulation
remains unknown. Our results indicate that translation levels remain high for at least 45
minutes. Additional experiments will delineate the exact time-course of the translational
upregulation.
Another issue that remains unknown is the dependence of BDNF, dopamine and
DHPG mediated translation on glutamatergic activity. It is possible that coincidence of
modulatory signals and glutamatergic activity is essential for long-term processes to occur
- indeed, it has been postulated that the MAP Kinase pathway is the coincidence detector
for these different signals (Sweatt, 2001). Also, what is the role of the mTOR pathway in
these functions? As neuronal activity induced translation may be important for certain
computational features of neuronal function, an accurate determination of the stimuli that
can activate translation, and the time course of translation will be important in elucidating
how long-term changes in neurons take place.
107
Chapter 3 - Characterization of neuronal activity-induced translation
Bibliography
Aakalu, G., Smith, W. B., Nguyen, N., Jiang, C., and Schuman, E. M. (2001). Dynamic
visualization of local protein synthesis in hippocampal neurons. Neuron 30, 489502.
Bear, M. F. (1996). A synaptic basis for memory storage in the cerebral cortex. Proc Natl
Acad Sci U S A 93, 13453-13459.
Buchwalter, G., Gross, C., and Wasylyk, B. (2004). Ets ternary complex transcription
factors. Gene 324, 1-14.
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via
the ubiquitin-proteasome system. Nat Neurosci 6, 231-242.
Frey, U., Huang, Y. Y., and Kandel, E. R. (1993). Effects of cAMP simulate a late stage
of LTP in hippocampal CA1 neurons. Science 260, 1661-1664.
Gallagher, S. M., Daly, C. A., Bear, M. F., and Huber, K. M. (2004). Extracellular signal-
regulated protein kinase activation is required for metabotropic glutamate
receptor-dependent long-term depression in hippocampal area CA1. J Neurosci
24, 4859-4864.
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs
oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways.
Nat Neurosci 5, 405-414.
Hegde, A. N. (2004). Ubiquitin-proteasome-mediated local protein degradation and
synaptic plasticity. Prog Neurobiol 73, 311-357.
Huang, Y. Y., and Kandel, E. R. (1995). D1/D5 receptor agonists induce a protein
synthesis-dependent late potentiation in the CA1 region of the hippocampus.
Proc Natl Acad Sci U S A 92, 2446-2450.
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein
synthesis in hippocampal mGluR-dependent long-term depression. Science 288,
1254-1257.
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and
protein synthesis--dependent long-term depression in hippocampal area CA1. J
Neurophysiol 86, 321-325.
Kang, H., and Schuman, E. M. (1995). Long-lasting neurotrophin-induced enhancement
of synaptic transmission in the adult hippocampus. Science 267, 1658-1662.
Kemp, N., and Bashir, Z. I. (1999). Induction of LTD in the adult hippocampus by the
synaptic activation of AMPA/kainate and metabotropic glutamate receptors.
Neuropharmacology 38, 495-504.
108
Chapter 3 - Characterization of neuronal activity-induced translation
Kemp, N., and Bashir, Z. . (2001). Long-term depression: a cascade of induction and
expression mechanisms. Prog Neurobiol 65, 339-365.
Liu, G., and Tsien, R. W. (1995). Properties of synaptic transmission at single
hippocampal synaptic boutons. Nature 375, 404-408.
Renger, J. J., Egles, C., and Liu, G. (2001). A developmental switch in neurotransmitter
flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron
29, 469-484.
Sajikumar, S., and Frey, J. U. (2003). Anisomycin inhibits the late maintenance of longterm depression in rat hippocampal slices in vitro. Neurosci Lett 338, 147-150.
Sajikumar, S., and Frey, J. U. (2004a). Late-associativity, synaptic tagging, and the role
of dopamine during LTP and LTD. Neurobiol Learn Mem 82, 12-25.
Sajikumar, S., and Frey, J. U. (2004b). Resetting of 'synaptic tags' is time- and activitydependent in rat hippocampal CA1in vitro. Neuroscience 129, 503-507.
Sala, C., Piech, V., Wilson, N. R., Passafaro,
M., Liu, G., and Sheng, M. (2001).
Regulation of dendritic spine morphology and synaptic function by Shank and
Homer. Neuron 31, 115-130.
Sala, C., Rudolph-Correia, S., and Sheng, M. (2000). Developmentally regulated NMDA
receptor-dependent dephosphorylation of cAMP response element-binding
protein (CREB) in hippocampal neurons. J Neurosci 20, 3529-3536.
Sweatt, J. D. (2001). The neuronal MAP kinase cascade: a biochemical signal
integration system subserving synaptic plasticity and memory. J Neurochem 76,
1-10.
Trachtenberg,
J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., and
Svoboda, K. (2002). Long-term in vivo imaging of experience-dependent synaptic
plasticity in adult cortex. Nature 420, 788-794.
Wu, G. Y., Deisseroth,
K., and Tsien, R. W. (2001a). Activity-dependent
CREB
phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and
a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad
Sci U S A 98, 2808-2813.
Wu, G. Y., Deisseroth, K., and Tsien, R. W. (2001b). Spaced stimuli stabilize MAPK
pathway activation and its effects on dendritic morphology. Nat Neurosci 4, 151158.
109
Chapter 4 - BDNF prevents depression and chronic
stress-induced hippocampal atrophy, but facilitates
anxiety and amygdalar spinogenesis
Abstract
The neurotrophic hypothesis states that a reduction in neurotrophin signaling
underlies the behavioral symptoms of depression, and that an increase in this signaling
would improve those symptoms. Transgenic overexpression of the neurotrophin BDNF
inhibited the chronic stress-induced atrophy of hippocampal CA3 pyramidal cells, and
improved performance in the Porsolt forced swim test, thus confirming the hypothesis.
However, the transgenic mice showed an increase in spine density of pyramidal cells
in the basolateral amygdala, and an increase in anxiety. Thus, we show a strong link
between BDNF, structural changes in the brain and chronic stress-induced behavior.
110
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
Introduction
Prolonged stress is associated with various cognitive and affective behavioral
abnormalities. The hippocampus, which plays a key role in the formation of declarative
memories, has been a primary focus of research on the cognitive effects of chronic stress
(McEwen and Sapolsky, 1995). A reduction in the length and complexity of dendritic arbors
of CA3 pyramidal neurons in the hippocampus is a well characterized morphological
feature of various rodent models of repeated stress (McEwen, 1999); this stress-induced
dendritic remodeling, in turn, has been hypothesized to underlie the hippocampal volume
loss seen in patients suffering from major depression, Cushing's syndrome and posttraumatic stress disorder (PTSD) (Bremner et al., 2000; Bremner et al., 1995; Bremner
et al., 1997; Sapolsky, 2002; Sheline et al., 2003; Sheline et al., 1996; Starkman et al.,
1992; Starkman et al., 1999).
Neurotrophins, particularly brain-derived neurotrophic factor (BDNF), have provided
a point of convergence for studies investigating the molecular mechanisms underlying
stress and depression because structural change is a key marker for both. BDNF, a ligand
that functions by binding the receptor tyrosine kinase trkB, has been implicated in neuronal
survival, dendritic and axonal morphogenesis, and spine development. It has also been
shown that chronic antidepressant treatment increases the levels of BDNF mRNA in the
rat hippocampus (Dias et al., 2003; Nibuya et al., 1995). Further, acute administration of
BDNF into the dentate gyrus improves the performance of rats in the learned helplessness
paradigm (Shirayama et al., 2002), a well-characterized animal model of depression
which is effective at predicting the efficacy of antidepressant treatments (Porsolt, 2000).
In addition, some, though not all, reports have shown that chronic stress reduces the
levels of BDNF mRNA in rats (Kuroda and McEwen, 1998; Smith et al., 1995a; Smith et
al., 1995b; Ueyama et al., 1997). Furthermore, a mutation in the BDNF gene is correlated
with bipolar disorder (Neves-Pereira et al., 2002).
111
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
These finding have led to the "neurotrophic hypothesis" which states that symptoms
associated with depression, both pathological and behavioral, are a result of decreased
neurotrophic support, and, conversely, that increasing neurotrophic support would lead
to the resolution of these symptoms. Moreover, since BDNF has been shown to regulate
dendritic growth (McAllister et al., 1996; McAllister et al., 1995), it would provide a possible
link between depression and structural changes in the brain (Nestler et al., 2002). Despite
the appeal of this hypothesis, a direct link between all three components of the neurotrophic
hypothesis - BDNF, chronic stress-induced neuronal atrophy in the hippocampus, and
depression - has not been made, partly because mice lacking BDNF die shortly after
birth (Ernfors et al., 1994). Further, previous reports on the antidepressant effect of BDNF
were based on acute administration in normal animals, whereas the clinical efficacy of
antidepressants requires chronic treatment. The neurotrophic hypothesis also implies
that BDNF would help reverse cellular correlates of depressive symptoms, such as those
caused by severe stress, whereas earlier studies have used normal animals, rather than
animals that have undergone stress-induced hippocampal atrophy.
While hippocampal plasticity has, over the years, provided a useful framework
for studying the cognitive effects of stress at multiple levels of neural organization, the
affective aspects of stress-related disorders may involve the amygdala, which plays a
pivotal role in processing aversive or stressful experiences. Recent evidence points to
contrasting effects of chronic stress on the hippocampus and amygdala. For example,
the same antidepressant treatment, that blocks hippocampal CA3 atrophy, fails to prevent
enhanced anxiety and fear after repeated restraint-stress (Conrad et al., 1999). On the
other hand, acquisition of conditioned fear has been shown to require BDNF signaling
(Rattiner et al., 2004). Moreover, the same chronic immobilization-stress that causes
hippocampal atrophy, can elicit dendritic growth in the basolateral amygdala, as well
as enhanced anxiety-like behavior (Vyas et al., 2003; Vyas et al., 2002). Finally, recent
reports have identified an important role for several signaling pathways in fear memory
112
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
formation (Lamprecht and LeDoux, 2004; Rattiner et al., 2004) and development of anxiety
(Matys et al., 2004; Pawlak et al., 2003), and many of these have also been implicated in
structural plasticity.
Hence, we sought to test the neurotrophic hypothesis, in the context of
structural plasticity in the hippocampus and amygdala, using genetically modified mice
overexpressing BDNF in excitatory neurons of the forebrain (Huang et al., 1999). Here,
we provide evidence that, consistent with the neurotrophic hypothesis, overexpression of
BDNF in pyramidal cells results in the prevention of chronic stress-induced hippocampal
CA3 dendritic atrophy and depressive symptoms without affecting neuroendocrine
responses to chronic stress. Interestingly, we also discovered that the mutant mice have
increased spine density in the basolateral amygdala, as well as increased anxiety. Thus,
structural changes in the hippocampus and amygdala, caused by genetic manipulation
of the same molecule, BDNF, give rise to contrasting effects on depressive and anxiety
symptoms, which are two key behavioral correlates of stress-related disorders.
Methods
Experimental animals
Male BDNF overexpressing C57/BL6 mice and theircontrol littermates, as previously
described (Huang et al., 1999), were used at 8-10 weeks of age for all experiments. The
mice were housed in groups of 3-5 with ad libitum access to food and water. The light/
dark cycle ran for 12 hrs (lights on at 7:00 AM), and all experiments were done between
8:30 AM and 1:30 PM. All procedures were performed in accordance with NIH guidelines
and after approval by the MIT Committee for Animal Care.
Chronic immobilization-stress
Mice were randomly assigned to unstressed and stressed groups. Chronic
immobilization-stress (CIS) was performed on the stressed group by immobilizing the
animals in rodent immobilization bags without access to food or water. Unstressed animals
were deprived of food and water while the stressed animals were undergoing CIS. Weight
113
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
gain was monitored by determining the weight of the animals immediately prior to the
beginning of the experiment on day 1, day 6, and day 11 (one day after the end of the
stress induction). Blood for hormone level quantification was obtained on day 11 and
ACTH and corticosterone measurements were performed by Analytics (Bethesda, MD).
Morphological Analysis
After completion of stress protocols, all groups of mice were killed under deep
anesthesia. The brain was removed quickly, and fixed in Golgi-Cox fixative for 6 weeks.
Blocks of tissue containing the hippocampus and amygdala were dissected and processed
for Golgi staining technique as described earlier (Shankaranarayana Rao et al., 2001).
All analysis was done in a blind fashion with the code broken only after quantification
was complete. Only Golgi-impregnated neurons that satisfied the following criteria were
analysed: (1) presence of untruncated dendrites, (2) consistent and dark impregnation
along the entire extent of all of the dendrites, and (3) relative isolation from neighboring
impregnated neurons to avoid interfering with analysis. 10 pyramidal neurons from
the CA3 region, and 10 pyramidal cells from the basolateral complex of the amygdala
(BLA) were selected from each animal for analysis on the basis of morphological criteria
described in the literature (McDonald, 1982). Using the center of the soma as reference
point, dendritic length and branch points were measured as a function of radial distance
from the soma by adding up all values in each successive concentric segment (Sholl's
analysis; segment diameter: 50 pm for CA3 pyramidal neurons, 20 pIm for BLA neurons)
(Shankaranarayana Rao et al., 2001).
Porsolt Swim Test
The mice were tested on two consecutive days by forcing them to swim in a beaker
of water (diameter: 13 cm., temperature 23-25°C). The animals were marked as being
immobile if they were performing no more than the minimal movement required to remain
afloat for at least 1 second. The data were binned into 1 minute intervals.
114
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
Open Field Test
The mice were tested for 10 minutes using the Accuscan system (Dayton, OH).
Locomotor activity, as well as time spent in the center of the apparatus versus the margin,
were measured automatically by the apparatus using infrared beams.
Elevated Plus Maze
The apparatus consisted of two open arms (30 cm. x 5 cm.), and two closed arms
(30 cm. x 5 cm., with a wall height of 10 cm.) elevated 60 cm. in the air. Images were
taken and digitized every second, and the data binned by minute. Numbers of entries as
well as stay time were measured by custom macros written in ImageJ (NIH, Bethesda,
MD).
Biochemical Analysis
For in situ hybridization analysis, the brains were removed on day 11 and processed
as described previously (Kelleher et al, 2004). For ELISA analysis of BDNF levels, the
hippocampus was dissected from mice on day 11, lysed and processed for BDNF ELISA
according to manufacturer's instructions (Promega, Madison, WI).
StatisticalAnalysis
For hippocampal morphometric analysis, hormone analysis, weight measurements,
in situ hybridization and ELISA results, two-way ANOVAwas used with post hoc tests using
a 95% confidence interval. For the Porsolt Forced Swim Test, amygdalar morphometric
analysis, open-field test, and elevated plus maze, a Mann-Whitney test was used to
evaluate significance.
Results
Chronic stress causes CA3 dendritic atrophy in mice
It has been proposed that the hippocampal volume loss seen in patients suffering
from various stress-related disorders such as PTSD, major depression and Cushing's
syndrome could be manifested by CA3 pyrimadal cell dendritic atrophy (McEwen, 1999).
Furthermore, it has been shown that different forms of chronic stress, including chronic
115
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
restraint-stress, chronic immobilization-stress, dominance-hierarchy stress, and intruder
stress cause atrophy of the dendrites of CA3 pyramidal cells in various rodents (McEwen,
1999). However, no published study exists for mice; thus, we sought to extend the results
from other rodents using chronic immobilization-stress (CIS; 2 hrs. immobilization / day
for 10 days) in mice. We measured the atrophy of CA3 pyramidal cells by subjecting the
brains from unstressed and stressed control animals to Golgi-Cox histology (Pasternak
and Woolsey, 1975) one day after the end of the chronic stress induction period.
As shown in Figure 4-1A, CIS induced a shrinking of the apical dendritic tree of CA3
pyramidal cells in control animals. Quantification of this data showed a 12.5% decrease
in total dendritic length (control unstressed: 3620 pm +/- 51 pm, n = 80; control stressed:
3170 pm +/- 59 pm, n = 60; p <
10-7),
as well as a 29% decrease in the number of branch
points (control unstressed: 16.3 +/- 0.3, n = 80; control stressed: 11.5 +/- 0.3; n = 60; p <
10-18).
In contrast, when the basal dendrites were examined, there was no change in either
the dendritic length (control unstressed: 2451 pm +/- 50 pm, n = 80; control stressed:
2350 pm +/- 63 pm; n = 60; p > 0.2) or number of branch points (control unstressed: 14.1
+/- 0.3; control stressed: 13.6 +/- 0.4; n = 60; p > 0.2). This selective atrophy of apical
dendrites is consistent with data from other rodents (McEwen, 1999).
In an effort to better understand the nature of the atrophy, we performed Sholl's
analysis to determine where along the apical dendrite the atrophy was taking place. As
shown in Figure 4-1 B and 4-1 C, and in accordance with data from rats, the dendritic length
and branch point number differences between the unstressed control and stressed control
animals lay only in the regions more than 50pm away from the soma, corresponding to
the stratum radiatum.
Chronic stress does not cause CA3 dendritic atrophy in transgenic mice
We compared the dendritic morphology data obtained from control mice with
data obtained from littermate BDNF overexpressing transgenic mice. In agreement with
the neurotrophic hypothesis, we found no differences between the morphology of CA3
116
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
A
-a- Unstressed
B
-0- Stressed
*
_ 700-
*
**
*
5600
r
C
*
400'
*
2003
control
control
transgenic
unstressed stressed unstressed
transgenic
stressed
5S 100oo
I1
I
.
o
C
.
.
.
.
-
.
D
*
S
-0- Stressed
*r
C
I
*r
I
Stressed
--
9 400= 300Mr200n 100
*
z
e
0
.
.
C 500-
*
E
--
-
Unstressed
900E 800o
- 700-
-4- Unstressed
-
.
50 100 150 200 250 300 350 400 450
Distance from soma (lm)
,~)II
50
.
Distance from soma (m)
E
.
.
.
.
i
-
-
s
i
i
50 100 150 200 250 300 350 400 450
Distance from soma ( m)
0
F
300.
i
!
S
Control
Transgenic
250-
C
*
*
E
0
, 2000 150-
E
z
E
1 M.
*.
0
50 100 150 200 250 300 350 400 450
Distance from soma ( m)
II.
Day2
Day
Day
Figure 4-1: Forebrain BDNF overexpression prevents chronic stress-induced dendritic atrophy,
and reduces immobility in the Porsolt forced-swim test. A) Representative camera lucida tracings
of CA3 pyramidal neurons from control and BDNF overexpressing transgenic mice with and without
stress. B, C) Sholl's analysis of apical dendrites from control animal neurons shows chronic stressinduced reduction in B) dendritic length and C) number of branches. Note that there is no atrophy
in the proximal 50pm of the dendrite. D, E) Sholl's analysis of apical dendrites from transgenic
animal neurons shows no chronic stress-induced atrophy. *, p < 0.05. F) Forebrain BDNF overexpression improves performance on the Porsolt forced-swim test. On both day 1 and day 2, the
transgenic animals show less immobility as compared to control littermates. *, p< 0.05 between
control and transgenic animals. Control, but not transgenic animals exhibit a statistically significant
increase in immobility from day 1 to day 2.
117
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
pyramidal cells in unstressed and stressed animals (Fig. 4-1A, D, E) either in dendritic
length (transgenic unstressed: 4387 pm +/- 83 pm, n = 80; transgenic stressed: 4342 pm
+/- 62 pm; n = 100; p > 0.6) or number of branch points (transgenic unstressed: 19.1 +/0.5, n = 80; transgenic stressed: 18.1 +/- 0.4, n = 100; p > 0.05). Sholl's analysis confirmed
that there was no change in dendritic length or number of branch points anywhere along
the length of the dendrite (Fig. 4-1 D, E).
Transgenic mice show decreased immobility in Porsolt forced swim test
Since we had evidence that BDNF overexpression prevented chronic stressinduced hippocampal damage, we wanted to determine if BDNF overexpression would
also improve a behavioral symptom of depression. The Porsolt forced swim test has
been commonly used as a behavioral correlate of helplessness in rodent models (Porsolt,
2000). In this test, exposure to a water pool with no exit results in increased immobility
during re-exposure in the same water pool 24 hrs. later. This increase in immobility, which
is thought to represent a state of learned helplessness, can be blocked by antidepressant
action.
As shown in Figure 4-1 F,the transgenic mice showed decreased immobility both on
Day 1 and Day 2 of the test. There was no difference between the control and transgenic
mice in locomotor activity (Fig. 4-7A) indicating that the decreased immobility shown by
the mice was not due to increased hyperactivity. Thus, the decreased immobility shown
by the mice is consistent with an antidepressant property of BDNF, and is consistent with
the neurotrophic hypothesis that increasing neurotrophin function can prevent symptoms
associated with depression, both at the cellular and behavioral levels.
Transgenic animals have normal stress-hormone levels
We next decided to examine the mechanism by which BDNF prevents chronic
stress induced hippocampal damage and improves performance on the Porsolt forced
swim test. Specifically, two alternate hypotheses were tested - 1) Because of the potent
role of the HPA axis and glucocorticoids on dendritic morphology and behavior, and
118
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
because the hippocampus is a major negative regulator of the HPA axis, it is possible that
the role of BDNF is to increase the hippocampal inhibition of stress-hormone release,
which in turn would prevent chronic stress induced dendritic atrophy and helplessness
behavior; 2) the role of BDNF is downstream of glucocorticoid secretion and that BDNF
plays a more direct neuroprotective role in preventing the chronic stress induced dendritic
atrophy.
We differentiated between the two hypotheses by measuring the blood plasma
levels of the stress-hormones, ACTH and corticosterone, one day after the end of the
chronic stress period. As shown in Figure 4-2A, there was no significant difference in ACTH
concentration between unstressed control and transgenic animals (control: 326 pg/mL
+/- 82 pg/mL, n = 6; transgenic:
251 pg/mL +/- 61 pg/mL, n = 7; p > 0.05). CIS increased
the levels of ACTH in both control and transgenic animals (p < 0.02), but there was no
significant genotype effect (control: 520 pg/mL +/- 95 pg/mL, n = 6; transgenic: 386 pg/mL
+/- 58 pg/mL, n = 6; p > 0.05). Though corticosterone was slightly, but significantly, lower
in unstressed transgenic animals as compared to unstressed control animals (control:
146 ng/mL +/- 46 ng/mL, n = 6; transgenic:
61 ng / mL +/- 16 ng / mL, n = 7; p < 0.05),
there was no difference between stressed control and transgenic mice (control: 470 ng/
mL +/- 42 ng/mL, n = 6; transgenic: 479 ng/mL 84 ng/mL, n = 6; p > 0.05; Fig. 4-2B). Thus
BDNF overexpression in forebrain excitatory cells has no effect on stress-hormone levels
in animals that have undergone chronic stress.
Transgenic mice show chronic stress-induced weight loss
The stress-hormone data indicate that both the control and transgenic animals
have a similar perception of the stress that they have been subject to, as measured
by their neuroendocrine response. Loss of body weight is another key measure of the
efficacy of a stress-protocol. Hence, we examined the weight loss that the animals
undergo when subjected to 10 days of chronic immobilization-stress. As shown in Figure
4-2C, D, there was almost no change over time in the body weight of unstressed animals.
119
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
However, there was a significant decrease (p < 0.05) in the body weight of both control
and transgenic animals undergoing stress that was evident by day 6 of the stress period
(Fig. 4-2C). This decrease continued and there was a larger loss in body weight (p < 0.05)
when the animals' weight was measured on day 11, one day after the end of the stress
protocol (Fig. 4-2D). However, there was no statistically significant difference between
control and transgenic stressed animals on day 6 (control: 1.1% +/- 0.4% weight loss, n
= 6; transgenic:
1.6% +/- 0.3% weight loss, n = 6; p > 0.05). On day 11, the transgenic
animals showed a statistically insignificant trend towards greater weight loss than the
control animals (control: 1.6% +/- 0.5%, n = 6; transgenic: -3.1% +/- 0.8%, n = 6; p > 0.05).
This shows that the transgenic animals suffered the same degree of body weight loss as
a result of the stress protocol, and, along with the neuroendrocrine data, demonstrate that
the decreased depressive symptoms seen in the transgenic animals were not due to a
decreased perception of stress as compared to control animals.
Chronic stress reduces hippocampal BDNF protein levels but not mRNA
levels
We have shown that increasing BDNF levels in the forebrain could prevent chronic
stress-induced hippocampal damage, and improved performance on the Porsolt forced
swim test. However, there is still controversy over whether the effects of chronic stress are
mediated via decreases in BDNF levels, as different groups have showed contradictory
results (Kuroda and McEwen, 1998; Smith et al., 1995a; Smith et al., 1995b). To determine
if BDNF protein levels are affected by chronic stress, ELISA analysis of lysates from the
hippocampus was performed in stressed and unstressed control and transgenic animals.
In line with expectations, the transgenic animals had significantly (p < 0.0001) higher
BDNF levels than the control animals. Chronic immobilization-stress significantly reduced
(p < 0.05) the levels of BDNF protein in the hippocampus of control animals by 26% (Fig. 43A). The hippocampi from transgenic animals that underwent the stress protocol showed
a similar loss of BDNF protein as compared to their unstressed transgenic counterparts
120
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
A
*
I
E 500-
*
0.
-
300-
Te
Unstressed
Stressed
600-
4500-
[ Stressed
*
B
_ Unstressed
.
S 400-
r%-
300-
IT
0 200-
I
100-
100-
0-
t
0-
o-
Contrrol
Transgenic
Control
1 Unstressed
= Stressed
C
A
v.;
Transgenic
Genotype
Genotype
Unstressed
D
= Stressed
4
0
0.0
o
.S
~0
= -0.5
._
0
. -1.0
.
.3
.5
-1.5
_0
0
(I)
t
-IV
Control
Transgenic
Transgenic
Control
Genotype
Genotype
Figure 4-2: Forebrain BDNF overexpression does not affect commonly used endocrine indicators
of stress level in response to chronic immobilization-stress. A, B) Forebrain BDNF overexpression
does not affect chronic stress-induced increases in blood plasma concentrations of A) ACTH and
B) corticosterone. A) In unstressed animals, there is no significant difference (p > 0.05) between
control and transgenic animals in ACTH concentration. Chronically stressed animals have a statistically significant (p < 0.05) increase in ACTH concentration as compared to unstressed animals,
but again there is no significant genotype difference (p > 0.05). B) In unstressed animals, there is
a significant (p < 0.05) difference between control and transgenic animals. Chronically stressed
animals have a statistically significant (p < 0.05) increase in corticosterone concentration, but there
is no significant genotype effect (p > 0.05) in the stressed animals. C, D) Forebrain BDNF overexpression does not affect chronic stress-induced weight loss. Unstressed control and transgenic
animals show no change in weight, whereas stressed control and transgenic animals weight loss
at both C) 6 and D) 11 days after the onset of chronic stress. There is no genotype effect in either
unstressed or stressed animals. (*, p < 0.05 between unstressed and stressed animals; $, p < 0.05
in comparison between unstressed control and unstressed transgenic animals)
121
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
(48%, p < 0.05). However, the levels of BDNF in the transgenic stressed animals were
still significantly higher than the levels of BDNF in the control unstressed animals (p <
0.01), which would explain why there were no chronic stress-induced effects visible in the
hippocampus of transgenic animals.
The decrease in BDNF protein levels after chronic stress in control animals could be
a result of a corresponding decrease in BDNF mRNA levels, or changes in translation and
protein degradation rates. To examine the possibility of a chronic stress induced change
in the levels of BDNF mRNA in the hippocampus, we performed in situ hybridization
on hippocampal sections. As BDNF can be released both postsynaptically as well as
presynaptically (Lu, 2003), changes in BDNF mRNA levels in the dentate gyrus, CA3, and
the CA1 could all affect the CA3 region. However, we were unable to detect any changes
in BDNF mRNA levels between stressed and unstressed animals (Fig. 4-3B) anywhere in
the hippocampus. As expected, the transgenic animals had an increased level of BDNF
_
Unstressed
,U-
IStressed
---
::
I
+..
1
7
.sed
,d
300-
*
E
_
ac
200.
I0
r
m
O100-
n.
.Sw
*
v·
Control
Transgenic
CA1
Genotype
CA3
DG
CTX
Thai.
Brain Region
Figure 4-3: Chronic immobilization stress reduces BDNF protein, but not BDNF mRNA levels. A)
ELISA shows a statistically significant decrease in BDNF levels after chronic immobilization-stress
in control and transgenic animals. As expected, the level of BDNF protein in transgenic animals is
higher compared to control animals. Note that the BDNF protein level in stressed transgenic
animals is higher than BDNF levels in unstressed control animals. B) Quantification of the in situ
hybridization data shows no statistically significant difference in mRNA levels between unstressed
and stressed cases in either control or transgenic animals. As a positive control, transgenic animals
show a statistically significant increase in mRNA levels in hippocampal regions and cortex, which
are regions expressing the transgene. In contrast, no such difference is seen in the thalamus,
where the transgene is not expressed. Abbreviations: DG - dentate gyrus; CTX - cortex; Thai thalamus. (*, p < 0.05 between control and transgenic animals; X, p < 0.05 in comparison between
control and transgenic animals)
122
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
mRNA. Thus, chronic stress mediates its effect on BDNF levels in a post-transcriptional
manner.
Transgenic mice show increased amygdalar spine density
As the amygdala has also been associated with stress-related disorders, we
examined the effect of BDNF overexpression on amygdalar function. Since BDNF
promotes dendritic and spine growth (McAllister et al., 1996; McAllister et al., 1995), we
hypothesized that there would be increased dendritic length and spine density in the
BDNF overexpressing transgenic animals. Thus, we performed Golgi-Cox analysis on
spiny pyramidal neurons of the basolateral amygdala (BLA). There was a 20% increase in
the spine density between control and transgenic animals on apical dendrites (control: 6.6
spines/1 0 pm +/- 0.1 spines/1 0 pm, n = 24; transgenic:
pm, n = 30; p <
10-7).
7.9 spines/1 0 pm +/- 0.1 spines/1 0
Sholl's analysis (Fig. 4-4B) showed that there was a significant
increase in spine density along the entire length of the apical dendrite. There was also a
17% increase in basal dendrite spine density (control: 6.0 spines/10 pm +/- 0. 2 spines/10
pm, n = 24; transgenic: 7.0 spines / 10 pm +/- 0.3 spines / 10 pm, n = 30; p < 0.01).
Once again, Sholl's analysis showed an increase in spine density in neurons derived
from transgenic animals along the entire length of the dendrite (Fig. 4-4C), suggestive of
increased connectivity and increased amygdalar activity in the transgenic animals.
In contrast, there was no difference in the length and number of branches in
basolateral amygdala spiny pyramidal neurons; neurons derived from control and
transgenic animals have the same total dendritic length (control: 2502 pm +/- 128 pm, n =
25; transgenic: 2345 pm +/- 100 pm, n = 20; p > 0.3) and branch number (control: 14.1 +/0.5, n = 25; transgenic: 13.4 +/- 0.5, n = 20; p > 0.3) along the entire length of the apical
dendrite (Fig. 4-4D-F).
Transgenic mice show increased anxiety-like behavior
Chronic stress is known to cause increased anxiety, and this anxiety is postulated to
be mediated by the amygdala. Previous reports have pointed to an increase in amygdalar
123
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
A
B
*
i*
10-
* *
*
*
8'
E
6'
g* 4-
._
Il
NI
I
-
Transgenic
C
*
10-
*
E
C
'..
-
-
-
I
I
I
-
-
\.,.
I
*
I
I
-
* * *
i;
6-
CL
c
-
I
-
D
*
8-
I
10 20 30 40 50 60 70 80 90
Distance from soma (lm)
0
Control
Control
Transgenic
---
2-
4-
x ...
a!,
..
2-
;nic
-I
.
0
.
.
.
.
.
.
.
10 20 30 40 50 60 70 80
Distance from soma (m)
/,.
/
/
..
.
90
E
Control
Transgenic
F
I
E
:L
0
30
--
Control
---
Transgenic
C
.0
c
9 2C
'I-
.2
C
a
E
zZ
i
0
50
100 150
200 250
Distance from soma (m)
0
300
25
50 75 100 125 150 175 200
Distance from soma ( gm)
Figure 4-4: Forebrain BDNF overexpression increases amygdalar spine density, without affecting
dendritic arbors in the basolateral amygdala (BLA). A) Representative photomicrographs of apical
and basal dendrites from BLA spiny pyramidal neurons. B, C) Transgenic mice show an increase
in spine density along the entire length of the B) apical and C) basal dendrite. D) Representative
camera lucida tracings of BLA spiny pyramidal cells. E, F) Sholl's analysis of E) dendritic length and
F) branch number, shows no statistically significant differences between control and transgenic
dendrites. (* p < 0.05 between control and transgenic animals.)
124
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
spine density and branch number that correlates with the increase in anxiety behavior seen
after chronic stress in rats (Vyas et al., 2003; Vyas et al., 2002). Thus, we hypothesized
that the increased anxiety seen in chronically stressed animals was a function of the
increased spine density and dendritic hypertrophy visible in the BLA of those animals. We
tested for anxiety in the BDNF overexpressing mice, hypothesizing that we would see an
increase in anxiety even without the induction of chronic stress. The open-field test and
the elevated plus-maze test were performed to test for anxiety.
A
·
45.
40'
100-
35.
80-
30'
0
E
.a 25
1
Control
I
Transgenic
*
60-
0
2 20.
40-
*
15
10'
20-
5.
l_
i
0'
I
Control
=I
0-
Ceenter
Transgenic
B
20'
60-
40-
T
'15
*
0-
ri
Openfield region
Genotype
E 50-
I
Margin
LU
E
w
0 30-
l
10.
1 1
0
E 20-
o
0
10-
5'
0 .
0Control
Control
Transgenic
I
Transgenic
Genotype
Genotype
Figure 4-5: Forebrain BDNF overexpressing transgenic mice show increased anxiety-like behavior. A) Transgenic mice show an increase in time spent in the margin of the open-field, as compared
to control animals. There is no difference in total movement indicating that BDNF overexpression
has no effect on locomotor activity. B) Elevated plus-maze data shows that transgenic mice spend
significantly less time in the open arm as compared to control animals (left panel). There is no
change in activity levels as measured by number of closed-arm entries (right panel). (* p < 0.05
between control and transgenic animals.)
125
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
As shown in Figure 4-5A, BDNF overexpressing transgenic mice spent significantly
more time in the sides (control: 70% +/- 2%, n=170, N=17 mice; transgenic: 85% +/2%, n=110, N=11 mice; p < 0.01) of the open-field, and less time (control: 30% +/- 2%,
n=170, N=17 mice; transgenic: 15% +/- 2%, n=110, N=11 mice; p < 0.01) in the center
compared to control littermates. Importantly, total activity was the same in both groups of
animals (control: 23.3% +/- 0.7%, n = 170, N=17 mice; transgenic: 24% +/- 1%, n = 110,
N=10 mice; p > 0.05), ensuring that changes in total activity were not responsible for the
difference between the animals.
These results were confirmed by the open-field test where, as shown in Figure 45B, the BDNF overexpressing transgenics spent less time in the open arm as compared
to the controls (controls: 43% +/- 7%, N=10 mice; transgenic: 24% +/- 4%, N=10 mice; p <
0.05). There were no differences in number of closed arm entries (controls: 12 +/- 1, N=10
mice; transgenics: 13 +/- 2, N=10 mice; p > 0.05) indicating that differences in total activity
were not responsible for the differences between the animals. Thus, results using the
open-field and the elevated plus maze behavioral paradigms both demonstrate increased
anxiety in the BDNF overexpressing mice as compared to the control animals.
Discussion
The neurotrophic hypothesis, states that a reduction in neurotrophic pathway
signaling underlies the behavioral state that is characteristic of mood disorders, such
as depression, and that conversely, an increase in neurotrophin pathway signaling can
reverse these symptoms. We sought to test the neurotrophic hypothesis by using a
mouse that had chronically elevated levels of BDNF. This approach improved upon earlier
experiments using injected BDNF because a) BDNF has been shown to be released by
neurons in an activity dependent manner (Lu, 2003); thus, BDNF upregulation in these
mice better replicates increased BDNF function in a physiologically relevant manner;
b) though BDNF has been suggested as a candidate antidepressant, the experiments
supporting that view have come from acute upregulation of BDNF (Shirayama et al., 2002)
126
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
which does not match the requirement for chronic administration of antidepressants in the
resolution of depressive symptoms (Nestler et al., 2002); c) when BDNF was injected into
the brain, there will have been a gradient of BDNF from the site of injection; this would not
occur in the genetically modified mice; and d) since BDNF in the transgenic mice was only
expressed by excitatory neurons, the hypothesis that BDNF overexpression by excitatory
neurons is sufficient for the resolution of depressive symptoms was demonstrated.
It has been reported that patients suffering from PTSD, Cushing's syndrome and
major depression have smaller hippocampal volumes and hippocampal function (Anand
and Shekhar, 2003; Bremner et al., 2000; Bremner et al., 1995; Bremner et al., 1997;
Sheline et al., 2003; Sheline et al., 1996; Starkman et al., 1992; Starkman et al., 1999).
It has been postulated that this volume decrease may be due to smaller dendritic arbors
in the CA3 region (McEwen, 1999). Thus, we examined the effects of chronic stress on
the CA3 of mice, and demonstrated that chronic immobilization-stress caused atrophy
of the apical hippocampal CA3 neurons, thus extending prior work done in other rodents
(McEwen, 1999); interestingly, we found that the atrophy was most pronounced in the
stratum radiatum. This chronic stress induced apical dendritic atrophy is abolished in the
transgenic mice in which the BDNF pathway is chronically upregulated. In addition, when
these mice were tested using the Porsolt forced swim test paradigm (Porsolt, 2000),
they showed decreased immobility. These results support the neurotrophic hypothesis
- namely that BDNF can act as an antidepressant. Importantly, this is the first correlation
of chronic BDNF upregulation, prevention of dendritic atrophy and improvement in the
resolution of depressive behavior, thus strengthening the link between structural changes
in the hippocampus and depression.
Our work also showed a role for BDN F in the control of amygdalar spine development;
specifically, we found an increase in spine density in the spiny cells of the basolateral
amygdala. The mice also showed an increase in anxiety-like behaviors, which supports
the notion that increased amygdalar activity would increase anxiety. This idea is also
127
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
supported by data that shows that chronic stress causes an increase in the dendritic arbor
size and spine density of cells in the BLA, while concomitantly increasing anxiety (Vyas
et al., 2002). Since chronic stress causes a decrease in hippocampal dendritic arbor in
control animals, it is unlikely that the increased anxiety that we see in the transgenic
animals is due to their increased dendritic arbor in the hippocampus. It has been noted
that patients suffering from anxiety disorders such as PTSD, manias and panic attacks
have increased amygdalar function, and one report shows an increase in amygdalar
volume in some of these patients (Anand and Shekhar, 2003). It will be interesting to see
if these patients show an increase in spine density in the amygdala, in a manner similar
to the rodent studies.
From a mechanistic point of view, we found that the hypertrophy seen in BDNF
overexpressing transgenic mice did not lead to a greater inhibition of the HPA axis under
stress conditions. However, there was a trend towards lower levels of both ACTH and
corticosterone in the unstressed case. Though conventional theories predict that higher
hippocampal output would result in lower stress-hormone levels, there are reports of
hippocampal lesions having no effect on those levels (Tuvnes et al., 2003). In addition,
the amygdala is a very potent activator of the HPA axis (Duman et al., 1997), and thus,
we hypothesize that under low-stress conditions, when amygdala output is low, there
might be a weak inhibition of the HPA axis by the hippocampus, but that under high-stress
conditions, strong amygdalar output would overcome the weaker hippocampal inhibition
resulting in no difference between the control and transgenic animals in stress-hormone
levels.
The similarity of stress-hormone levels in both control and transgenic animals
indicates that the role of BDNF in ameliorating the depressive symptoms is downstream
of the stress-hormones, and furthermore, that the effect of increases in stress-hormones
can be reversed without affecting the stress-hormones themselves. This is further
supported by the data that weight loss, which is governed by the hypothalamus, occured
128
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
to a similar extent in both the control and transgenic animals. This specificity of BDNF
function, where the effect of overexpression only manifests itself in those regions where
the overexpression occurs, may be important therapeutically, as stress-hormones are
required for normal cognitive function (Oitzl et al., 1997), and thus, therapies that act
downstream of the stress-hormones may allow for a resolution of depressive symptoms
without negatively affecting cognition.
We also found that BDNF mRNA levels were not downregulated anywhere in the
hippocampus by chronic stress, which is in agreement with some (Kuroda and McEwen,
1998), but not all reports (Smith et al., 1995a; Ueyama et al., 1997). Various factors
such as different stress conditions, and interspecies differences may account for this
difference; however, BDNF protein levels were lower after chronic immobilization-stress,
and thus, our report is consistent with a role for BDNF for the manifestation of chronic
stress induced dendritic atrophy.
The mechanism by which BDNF controls dendritic and spine morphology is still
under active investigation. It has been proposed that the Rho GTPases, which have also
been shown to affect dendritic arbor development an spinogenesis, might be activated by
neurotrophins such as BDNF (Huang and Reichardt, 2003). Analysis of mice with these
GTPases mutated in vivo would shed further light on whether these pathways have a
bearing on chronic stress induced pathology and stress-related behaviors. In addition,
other molecules that affect the cytoskeleton, such as the p21-activated kinase PAK1,
have been implicated in cognitive function (Hayashi et al., 2004), and it is probable that
PAK1 mutant animals would show changes in stress-related pathologies and behaviors.
Mice with conditional deletion of BDNF have also been tested for affective
behavioral effects by other researchers. When BDNF is deleted in all neurons, the most
obvious phenotype was an increase in hyperactivity, anxiety and obesity, all of which
are correlated with changes in the hypothalamus (Rios et al., 2001). This increase in
hyperactivity was also seen in mice with the BDNF receptor, trkB, deleted in the forebrain
129
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
(Zorner et al., 2003), which prevented the researchers from analyzing effects on anxiety and
depression. Our mice, which overexpress BDNF in the forebrain, do not have the confound
of hyperactivity, allowing us to analyze the effects of forebrain BDNF overexpression on
these stress-related disorders. Mice overexpressing trkB, which would be hypothesized
to have the same phenotype as mice overexpressing BDNF, have been produced and
show a decrease in anxiety (Koponen et al., 2004). As trkB is a receptor for both BDNF
and NT4, the decreased anxiety may be due to an increase in NT4 signaling in the trkB
transgenics which could explain the differences between the BDNF overexpressing
transgenics and the trkB overexpressing transgenics. In addition, BDNF binds both to
full-length trkB and truncated trkB (T1), and thus, overexpression of BDNF would result
in increased signaling via both trkB and T1, whereas overexpression of full-length trkB
would result in increased signaling via the trkB pathway only (Saarelainen et al., 2000).
Strain differences between the two different mice, as well as different expression levels
and patterns may also play a role in these differences.
Our work used transgenic animals in which BDNF overexpression was restricted
to pyramidal neurons throughout the forebrain. As such, we cannot exclude effects of
BDNF on other forebrain structures such as the prefrontal cortex that could mediate some
of the behavioral effects that we observe. An extension of the work would be to make
mice that are even more restricted in their overexpression of BDNF, specifically to the
CA3, amygdala or prefrontal cortex to confirm the role of a specific region on depression,
anxiety and other stress-related disorders. In addition, further research on the molecular
mechanism behind the differential effects of chronic stress on hippocampus and amygdala
would allow for the development of therapies that selectively ameliorate the symptoms of
one affective disorder without affecting other behaviors.
Our data provide a compelling case for the role of BDNF in stress-related disorders,
and provide evidence for a strong correlation between structural changes in the brain
and behavior. This will ensure that adequate therapies can be found that can ameliorate
130
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
the symptoms of diseases like depression without worsening other symptoms such as
anxiety.
131
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
Bibliography
Anand, A., and Shekhar, A. (2003). Brain imaging studies in mood and anxiety
disorders: special emphasis on the amygdala. Ann N Y Acad Sci 985, 370-388.
Bremner, J. D., Narayan, M., Anderson,
E. R., Staib, L. H., Miller, H. L., and Charney, D.
S. (2000). Hippocampal volume reduction in major depression. Am J Psychiatry
157, 115-118.
Bremner, J. D., Randall, P., Scott, T. M., Bronen, R. A., Seibyl, J. P., Southwick, S. M.,
Delaney, R. C., McCarthy, G., Charney, D. S., and Innis, R. B. (1995). MRI-
based measurement of hippocampal volume in patients with combat-related
posttraumatic stress disorder. Am J Psychiatry 152, 973-981.
Bremner, J. D., Randall, P., Vermetten,
E., Staib, L., Bronen, R. A., Mazure, C., Capelli,
S., McCarthy, G., Innis, R. B., and Charney, D. S. (1997). Magnetic resonance
imaging-based measurement of hippocampal volume in posttraumatic stress
disorder related to childhood physical and sexual abuse--a preliminary report.
Biol Psychiatry 41, 23-32.
Conrad, C. D., LeDoux, J. E., Magarinos, A. M., and McEwen, B. S. (1999). Repeated
restraint stress facilitates fear conditioning independently of causing hippocampal
CA3 dendritic atrophy. Behav Neurosci 113, 902-913.
Dias, B. G., Banerjee, S. B., Duman, R. S., and Vaidya, V. A. (2003). Differential
regulation of brain derived neurotrophic factor transcripts by antidepressant
treatments in the adult rat brain. Neuropharmacology 45, 553-563.
Duman, R. S., Heninger, G. R., and Nestler, E. J. (1997). A molecular and cellular theory
of depression. Arch Gen Psychiatry 54, 597-606.
Ernfors, P., Lee, K. F., and Jaenisch, R. (1994). Mice lacking brain-derived neurotrophic
factor develop with sensory deficits. Nature 368, 147-150.
Hayashi, M. L., Choi, S. Y., Rao, B. S., Jung, H. Y., Lee, H. K., Zhang, D., Chattarji, S.,
Kirkwood, A., and Tonegawa, S. (2004). Altered cortical synaptic morphology
and impaired memory consolidation in forebrain- specific dominant-negative PAK
transgenic mice. Neuron 42, 773-787.
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal
transduction. Annu Rev Biochem 72, 609-642.
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., Maffei,
L., and Tonegawa, S. (1999). BDNF regulates the maturation of inhibition and the
critical period of plasticity in mouse visual cortex. Cell 98, 739-755.
Koponen, E., Voikar, V., Riekki, R., Saarelainen, T., Rauramaa, T., Rauvala, H., Taira,
T., and Castren, E. (2004). Transgenic mice overexpressing the full-length
132
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
neurotrophin receptor trkB exhibit increased activation of the trkB-PLCgamma
pathway, reduced anxiety, and facilitated learning. Mol Cell Neurosci 26, 166181.
Kuroda, Y., and McEwen, B. S. (1998). Effect of chronic restraint stress and tianeptine
on growth factors, growth-associated protein-43 and microtubule-associated
protein 2 mRNA expression in the rat hippocampus. Brain Res Mol Brain Res 59,
35-39.
Lamprecht, R., and LeDoux, J. (2004). Structural plasticity and memory. Nat Rev
Neurosci 5, 45-54.
Lu, B. (2003). BDNF and activity-dependent synaptic modulation. Learn Mem 10, 86-98.
Matys, T., Pawlak, R., Matys, E., Pavlides, C., McEwen, B. S., and Strickland, S. (2004).
Tissue plasminogen activator promotes the effects of corticotropin-releasing
factor on the amygdala and anxiety-like behavior. Proc Natl Acad Sci U S A.
McAllister, A. K., Katz, L. C., and Lo, D. C. (1996). Neurotrophin regulation of cortical
dendritic growth requires activity. Neuron 17, 1057-1064.
McAllister, A. K., Lo, D. C., and Katz, L. C. (1995). Neurotrophins regulate dendritic
growth in developing visual cortex. Neuron 15, 791-803.
McDonald, A. J. (1982). Neurons of the lateral and basolateral amygdaloid nuclei: a
Golgi study in the rat. J Comp Neurol 212, 293-312.
McEwen, B. S. (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22, 105122.
McEwen, B. S., and Sapolsky, R. M. (1995). Stress and cognitive function. Curr Opin
Neurobiol 5, 205-216.
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M.
(2002). Neurobiology of depression. Neuron 34, 13-25.
Neves-Pereira, M., Mundo, E., Muglia, P., King, N., Macciardi, F., and Kennedy, J.
L. (2002). The brain-derived neurotrophic factor gene confers susceptibility to
bipolar disorder: evidence from a family-based association study. Am J Hum
Genet 71, 651-655.
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB
mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug
treatments. J Neurosci 15, 7539-7547.
133
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
Oitzl, M. S., de Kloet, E. R., Joels, M., Schmid, W., and Cole, T. J. (1997). Spatial
learning deficits in mice with a targeted glucocorticoid receptor gene disruption.
Eur J Neurosci 9, 2284-2296.
Pasternak, J. F., and Woolsey, T. A. (1975). On the "selectivity" of the Golgi-Cox method.
J Comp Neurol 160, 307-312.
Pawlak, R., Magarinos, A. M., Melchor, J., McEwen, B., and Strickland, S. (2003).
Tissue plasminogen activator in the amygdala is critical for stress-induced
anxiety-like behavior. Nat Neurosci 6, 168-174.
Porsolt, R. D. (2000). Animal models of depression: utility for transgenic research. Rev
Neurosci 11, 53-58.
Rattiner, L. M., Davis, M., French, C. T., and Ressler, K. J. (2004). Brain-derived
neurotrophic factor and tyrosine kinase receptor B involvement in amygdaladependent fear conditioning. J Neurosci 24, 4796-4806.
Rios, M., Fan, G., Fekete, C., Kelly, J., Bates, B., Kuehn, R., Lechan, R. M., and
Jaenisch, R. (2001). Conditional deletion of brain-derived neurotrophic factor in
the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15, 17481757.
Saarelainen, T., Pussinen, R., Koponen, E., Alhonen, L., Wong, G., Sirvio, J., and
Castren, E. (2000). Transgenic mice overexpressing truncated trkB neurotrophin
receptors in neurons have impaired long-term spatial memory but normal
hippocampal LTP. Synapse 38, 102-104.
Sapolsky, R. M. (2002). Chickens, eggs and hippocampal atrophy. Nat Neurosci 5, 11111113.
Shankaranarayana
Rao, B. S., Govindaiah,
Laxmi, T. R., Meti, B. L., and Raju, T. R.
(2001). Subicular lesions cause dendritic atrophy in CA1 and CA3 pyramidal
neurons of the rat hippocampus. Neuroscience 102, 319-327.
Sheline, Y. I., Gado, M. H., and Kraemer, H. C. (2003). Untreated depression and
hippocampal volume loss. Am J Psychiatry 160, 1516-1518.
Sheline, Y. I., Wang, P. W., Gado, M. H., Csernansky, J. G., and Vannier, M. W. (1996).
Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A
93, 3908-3913.
Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S., and Duman, R. S. (2002).
Brain-derived neurotrophic factor produces antidepressant effects in behavioral
models of depression. J Neurosci 22, 3251-3261.
134
Chapter 4 - Role of BDNF in hippocampal and amygdalar structural plasticity, depression and anxiety
Smith, M. A., Makino, S., Kvetnansky, R., and Post, R. M. (1995a). Effects of stress on
neurotrophic factor expression in the rat brain. Ann N Y Acad Sci 771, 234-239.
Smith, M. A., Makino, S., Kvetnansky, R., and Post, R. M. (1995b). Stress and
glucocorticoids affect the expression of brain-derived neurotrophic factor and
neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15, 1768-1777.
Starkman, M. N., Gebarski, S. S., Berent, S., and Schteingart,
D. E. (1992).
Hippocampal formation volume, memory dysfunction, and cortisol levels in
patients with Cushing's syndrome. Biol Psychiatry 32, 756-765.
Starkman, M. N., Giordani, B., Gebarski, S. S., Berent, S., Schork, M. A., and
Schteingart, D. E. (1999). Decrease in cortisol reverses human hippocampal
atrophy following treatment of Cushing's disease. Biol Psychiatry 46, 1595-1602.
Tuvnes, F. A., Steffenach, H. A., Murison, R., Moser, M. B., and Moser, E. . (2003).
Selective hippocampal lesions do not increase adrenocortical activity. J Neurosci
23, 4345-4354.
Ueyama, T., Kawai, Y., Nemoto, K., Sekimoto, M., Tone, S., and Senba, E. (1997).
Immobilization stress reduced the expression of neurotrophins and their
receptors in the rat brain. Neurosci Res 28, 103-110.
Vyas, A., Bernal, S., and Chattarji, S. (2003). Effects of chronic stress on dendritic
arborization in the central and extended amygdala. Brain Res 965, 290-294.
Vyas, A., Mitra, R., Shankaranarayana Rao, B. S., and Chattarji, S. (2002). Chronic
stress induces contrasting patterns of dendritic remodeling in hippocampal and
amygdaloid neurons. J Neurosci 22, 6810-6818.
Zorner, B., Wolfer, D. P., Brandis, D., Kretz, O., Zacher, C., Madani, R., Grunwald, I.,
Lipp, H. P., Klein, R., Henn, F. A., and Gass, P. (2003). Forebrain-specific trkBreceptor knockout mice: behaviorally more hyperactive than "depressive". Biol
Psychiatry 54, 972-982.
135
Chapter 5 - Conclusion
136
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Role of translationin protein synthesis-dependentsynapticplasticity
and memory
Long-term memory and the late-phase of long-term potentiation (L-LTP) are both
characterized by their dependence on protein synthesis (Davis and Squire, 1984; Frey et
al., 1988). Though previously not discussed, prior experiments suggested that the effects
of transcriptional and translational blockades on L-LTP were dissociable (Frey et al.,
1996; Frey et al., 1988; Nguyen et al., 1994; Scharf et al., 2002). In Chapter 2, it is shown
that the early phase of L-LTP is dependent only on translation, whereas the late phase of
L-LTP is dependent on both transcription and translation. In addition, mGluR dependent
L-LTD and priming of LTP both depend on translation and not transcription (Huber et al.,
2000; Huber et al., 2001; Kauderer and Kandel, 2000; Manahan-Vaughan et al., 2000;
Raymond et al., 2000).
We studied the role of translation in L-LTP and long-term memory, using mice with
a targeted expression of a dominant-negative MEK1 transgene; the mutant mice which
express the transgene in the cortex and hippocampal area CA1, but not CA3 have a
long-term memory deficit in contextual fear-conditioning, while short-term memory and
cued fear-conditioning are normal. Interestingly, the electrophysiological deficit in the
dnMEK1 mutant mice resembles the deficit seen in control slices treated with anisomycin
implicating a role for the MAP Kinase pathway in translational regulation. Cell culture
experiments determined that the role of the MAP Kinase pathway on neuronal activity
induced translation is not dependent on cytoplasmic polyadenylation of CPE containing
mRNAs; on the other hand, the MAP Kinase pathway affects a large number of mRNAs
possibly via phosphorylation of the translation initiation factor elF4E, its inhibitor 4EBP1,
and the ribosomal protein S6 (Chapter 2). As the level of elF4E, which is essential
for all capped translation, is the rate-limiting step in translation in most cells (Gingras
et al., 1999), an increase in elF4E levels mediated by 4EBP1 phosphorylation would
increase translation of all mRNAs. Similarly, phosphorylation of elF4E is also correlated
137
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
with increased translation of all mRNAs (Gingras et al., 1999). S6 phosphorylation is
associated with the translation of a certain class of mRNAs with polypyrimidine tracts in
their 5' UTRs (known as 5' TOP mRNAs). These mRNAs generally code for ribosomal
proteins, including S6 and elF4E. Thus, S6 phosphorylation results in an increase in the
total translational capacity of the cell which affects the translation of all mRNAs (Proud,
2002). These MAP Kinase dependent mechanisms are also found after L-LTP inducing
tetani in hippocampal slices, and after behavioral training (Chapter 2). Thus, we propose
a model where plasticity inducing events activate the MAP Kinase pathway, leading to
increased translation and production of proteins, leading, in turn, to the expression of LLTP and the expression of long-term memory (Figure 5-1).
A UTR
ERK
Cap
oO.PABIOFABP
O
3'
UT
5' UTR
mTOR
PABPAB
AUAM
i TAAAAAAAAAA'UTR
AAUAAAAAAAAAADM AAAAAAAAAA)'A
Figure 5-1: Proposed mechanism behind role of MAP Kinase (and mTOR) pathway in translation:
Neuronal activity, L-LTP and L-LTD inducing stimuli, and long-term memory inducing stimuli
activate the ERK MAP Kinase and mTOR pathways, which cause phosphorylation of 4E-BP1; this
allows elF4E to bind the cap, thus activating translation. Phosphorylation of elF4E is also correlated with increased translation. Phosphorylation of the ribosomal protein S6 causes an increase
in the translational capacity of the cell, by increasing the levels of translation proteins including S6
and elF4E itself. These cause an increase in translation of a large number of mRNAs, leading to
an increase in a diverse set of proteins, a subset of which are required for L-LTP, and a subset that
is required for L-LTD.
Our results show that type I mGluR activation of cultured hippocampal cells by
DHPG also activates translation of a diverse set of proteins via the phosphorylation of
S6 and elF4E (Chapter 4); similar data has been confirmed by the results of Banko and
Klann (personal communication) in acute hippocampal slices. These experiments suggest
that the translational apparatus recruited by L-LTP and L-LTD inducing stimuli are the
same - indeed, the set of proteins synthesized by the LTP stimuli BDNF and bicuculline
appears identical to the set of proteins synthesized by the LTD stimulus DHPG given the
resolving power of 1D SDS-PAGE analysis. These data point to a model where L-LTP
138
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
and L-LTD inducing stimuli activate translation of a very similar set of proteins by a similar
mechanism, namely the phosphorylation of translation initiation factors and ribosomal
proteins.
These data have led us to propose that L-LTP and L-LTD inducing stimuli both
stimulate translation of the entire repertoire of proteins required for the expression of both
L-LTP and L-LTD. According to this model, LTP and LTD stimuli create distinct "synaptic
tags" at the activated synapse that dictate whether that synapse "captures" LTP or LTD
proteins, ultimately deciding the form of plasticity expressed at that synapse (Chapter 3;
Figure 3-7). This model allows for strong input of one input to allow for the conversion of
another input from a short-lasting form of plasticity (E-LTP/E-LTD) to a long-lasting form
(L-LTP/L-LTD), without affecting the direction of plasticity. This preservation of the direction
of synaptic-weight changes would allow for the maintenance of the pattern of synaptic
weight changes within a dendrite, changing only the persistence of those changes. We
propose that these changes would manifest themselves behaviorally by allowing for a
short-term memory to be converted to a long-term memory, if another long-term memory
event occurs in close temporal proximity to the short-term memory.
Based on our model, the associative features of protein synthesis- dependent
plasticity will depend on certain parameters. Since synaptic tagging and capturing requires
the presence of both proteins and a tag, the lifetime of the tag, and the time course of
upregulated translation will be important. In addition, the availability of proteins will be
dependent on the distance between the locus of translation, and the tagged synapse.
Thus, the spatial extent of translation and the distance over which translated proteins are
available will also have to be determined in order to fully understand the nature of this
form of hetero-association..
The key question raised by our model regards the nature of the tag. A number
of different processes may contribute to the tag, and thus, the tag need not be a single
molecule; however, based on experiments by various researchers, the tag has to satisfy
139
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
a number of properties: 1) The tag has to be induced in a protein-synthesis independent
manner, 2) the tag has a finite life-time of 1-2 hrs., 3) the tag has to be induced both by ELTP and by L-LTP, 4) the tag has to be induced in an input-specific manner, 5) the tag has
to interact with the proteins required for L-LTP and facilitate capture, and that 6) different
tags are created by L-LTP and L-LTD inducing stimuli (Frey and Morris, 1997; Frey and
Morris, 1998; Sajikumar and Frey, 2004).
There are many different possibilities that could satisfy these criteria; the most
obvious of the candidates are the activity of kinases and phosphatases. For example, it
is known that various kinases such as a-CaMKII are activated during LTP, and various
phosphatases such as calcineurin (CaN) are activated during LTD. The phosphorylation
state of some key molecules may be the key tag determinant. Another possibility could
be a change in cytoskeletal dynamics. There is evidence that cytoskeletal changes occur
during LTP and LTD; these changes, along with changes in the various motors that interact
with the cytoskeleton could form the mechanism behind the tag and capture process.
Changes in membrane receptor number, molecular architecture of the synapse, localized
protein degradation or changes in structure of some molecule, among other possibilities,
could also form the basis for the tag (Martin and Kosik, 2002).
There is recent evidence that the CPEB isoform in Aplysia, ApCPEB, is capable of
changing state into a prion-like state. Since prions are extremely state, and ApCPEB is
induced by increasing translation of the ApCPEB mRNA locally, it has been proposed that
activation of a synapse causes an increase, via translational upregulation, of ApCPEB,
which converts to a prion state and thus tags the synapse; Since ApCPEB causes an
increase in translation of CPE containing mRNAs, the tagged synapse would remain in a
facilitated state (Si et al., 2003a; Si et al., 2003b).
One issue with discussing synaptic tagging is the ambiguity of the term. The original
synaptic tag, as described first by Frey and Morris (Frey and Morris, 1997) is important
for induction of L-LTP and L-LTD, and is independent of translation (Barco et al., 2002;
140
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Frey and Morris, 1997; Martin et al., 1997; Sajikumar and Frey, 2004). The ApCPEB tag
appears to affect maintenance of LTF, is dependent on the translation of ApCPEB itself,
and effects its function via upregulation of translation. Thus, it is a "maintenance" tag,
and different from the originally described "induction" tag. One curious issue is that if
ApCPEB is a maintenance tag, continued upregulation of translation is important for the
maintenance of LTF. However, at extended periods, inhibition of translation has no effect
on LTF, a fact that is at odds with the ApCPEB prion hypothesis (Casadio et al., 1999).
Structural Plasticity in Affective Behaviors
Protein synthesis is the key molecular signature of long-term changes in the brain;
however, the cellular changes that are the consequence of this synthesis have not been
deduced. It has been hypothesized that structural changes, which presumably would
depend on protein synthesis, are the substrate of long-term behavioral changes. However,
conventional behavioral training has not provided much evidence for such changes
(Lamprecht and LeDoux, 2004), whereas extreme environmental perturbations, such as
chronic stress, do produce robust changes in neuronal structure. Particularly, dendritic
atrophy in the CA3 region of the hippocampus is a particularly salient change that occurs
after chronic-stress (McEwen, 1999), and thus, we chose chronic stress-induced structural
changes as a model to explore the behavioral consequences of structural plasticity.
It has been hypothesized that hippocampal dendritic atrophy may be a cellular
substrate of the cognitive dysfunction seen in depression (McEwen, 1999). The
neurotrophin BDNF, a ligand that functions by binding the receptor tyrosine kinase TrkB,
has been implicated in neuronal survival, dendritic and axonal morphogenesis, as well
as dendritic spine development, and synaptic plasticity (McAllister et al., 1999; Schuman,
1999). It has also been shown that chronic antidepressant treatment increases the levels
of BDNF mRNA in the rat hippocampus (Dias et al., 2003; Nibuya et al., 1995). Further,
acute administration of BDNF into the dentate gyrus improves the performance of rats
in the forced swim test (Shirayama et al., 2002), a well-characterized animal model of
141
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
depression which is effective at predicting the efficacy of antidepressant treatments
(Porsolt, 2000). In addition, some, though not all, reports have shown that chronic stress
reduces the levels of BDNF mRNA in rats (Kuroda and McEwen, 1998; Smith et al.,
1995a; Smith et al., 1995b; Ueyama et al., 1997). Furthermore, a mutation in the BDNF
gene is correlated with bipolar disorder (Neves-Pereira et al., 2002). These results have
led to the neurotrophic hypothesis, which states that a deficiency in neurotrophic support
underlies the pathology and behavioral abnormalities behind stress-related disorders
such as depression, and that increasing neurotrophic signaling would resolve both the
cellular and behavioral symptoms.
Thus, we used BDNF overexpression transgenic mice to test this hypothesis. We
show that the transgenic animals, in contrast to control animals, do not exhibit chronic
stress-induced CA3 dendritic atrophy; furthermore, these mice have improved performance
on the Porsolt forced swim test. These data support the neurotrophic hypothesis, and
support the possible use of activators of the BDNF signaling pathway as candidate
antidepressants (Chapter 4).
Interestingly, BDN F overexpression did not change chronic-stress induced increases
in stress hormones, or chronic-stress induced weight loss. As the transgene is expressed
only in forebrain pyramidal cells, it is not expected to affect the hypothalamus, a region
partially responsible for the neuroendocrine response and feeding-related behaviors, and
any observed changes would have been due to effects on hypothalamic function by the
brain. Our data thus shows that the prevention of hippocampal atrophy did not result in
increased inhibition of the HPA axis, as was suggested in the literature (McEwen, 1999),
and thus, the transgenic mice have the same level of stress as the control animals.
BDNF is known to promote dendritic growth and spinogenesis (Huang and
Reichardt, 2003; Lu, 2003), and chronic-stress promotes basolateral amygdalar (BLA)
dendritic hypertrophy and spinogenesis concomitantly with increasing anxiety (Vyas
et al., 2003; Vyas et al., 2002). Since, our transgenic mice overexpress BDNF in the
142
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
BLA (Huang et al., 1999), we hypothesized that the transgenic mice would have BLA
hypertrophy, spinogenesis and anxiety. Though, we do not find evidence for hypertrophy
in BLA pyramidal neurons, increased spinogenesis is observed. In addition, both the openfield and elevated plus-maze tests showed increased anxiety in the transgenic animals
(Chapter 4).
BDNF in the amygdala has been implicated in the acquisition of fear-conditioning
(Rattiner et al., 2004); interestingly the data shows a transient increase in the level of
BDNF shortly after fear-conditioning. We propose that such transient increases in BDNF
cause structural changes leading to changes in synaptic weights, specifically at those
synapses activated at the time of the training; this, in turn, leads to cue-specific behavioral
changes. When fear-conditioning pathways are upregulated chronically, either via chronicstress, or genetically via chronic upregulation of molecules like BDNF, structural changes
occur throughout the dendrite, leading to a cue-non-specific fear response, manifested
as anxiety.
A few questions remain unresolved. Firstly, do chronic-stress induced changes
depend on translation and transcripton? In addition, the correlation between structural
changes and behavioral output will need to be strengthened by the further manipulation of
other molecules that also affect dendritic morphology. Changes in the structure of neurons
might be expected to profoundly change the electrophysiological characteristics of neurons
as well as the network in they are present. A decrease in LTP has been described, primarily
in the CA1 region of the hippocampus, though changes in vivo have been described at
the mossy fiber synapses as well (Pavlides et al., 2002; Pavlides et al., 1996; Pavlides
et al., 1993). However, dendritic changes in the hippocampus are observed specifically
in the CA3 region (McEwen, 1999). Thus, electrophysiogical consequences would be
expected at associational / commissural synapses, and this has yet to be tested.
143
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Conclusion
Organisms use their brain to integrate environmental and internal inputs occurring
throughout their history in order to plan and execute the appropriate actions. Thus, longlasting changes must take place in the brain to store the skills and memories that have
been learned by the organism.
In contrast to short-term memory, long-term memory requires protein synthesis.
The traditional viewpoint has been that transcription is the key regulatory step behind
protein synthesis-dependent forms of synaptic plasticity and long-term memory, whereas
translation has been thought of as being essentially constitutive. In Chapter 2 of this
thesis, it was demonstrated that translation is a key regulatory point in the early part
of the late-phase of long-term potentiation (L-LTP), and likely, long-term memory. Mice
expressing a dominant-negative MEK1 in the cortex and CA1, thus having reduced ERK
MAP Kinase pathway activity in those regions, showed inhibited long-term memory,
and L-LTP; interestingly, the L-LTP deficiency resembled the phenotype obtained by a
translational blockade of L-LTP. In Chapters 2 & 3, it was shown that neuronal translation
activated by BDNF, bicuculline, spaced depolarization, dopamine, and DHPG all depended
on MAP Kinase pathway activity. Furthermore, this translation caused an increase in the
translation of the vast majority of mRNAS concomitant with the phosphorylation of the
translation factors S6, elF4E and 4E-BP1. Hippocampal slice experiments confirmed that
the ERK MAP Kinase dependent phosphorylation of translation factors was correlated
with increased translation and the expression of L-LTP; in addition, ERK MAP Kinase
dependent phosphorylation of translation factors also occurred in response to behavioral
training (Chapter 2). It was proposed that all protein synthesis-inducing stimuli, including
L-LTP and L-LTD inducing stimuli, activate translation of the same set of proteins via
generalized translational upregulation, and that these proteins can be captured by all
activated synapses, leading to cross-tagging & cross-capturing, and cross-facilitation of
the persistent forms of LTP and LTD.
144
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
It is generally believed that the long-lasting changes that occur in the brain,
presumably via protein synthesis, involve structural changes in the activated neurons.
However, it has generally been difficult to show such a correlation since traditional
behavioral training is expected to produce changes in a limited number of neurons, and
affecting an even smaller number of synapses. Thus, in Chapter 4, a correlation between
chronic stress-induced disorders and affective behaviors was sought, and demonstrated.
Specifically, it was shown that transgenic mice overexpressing BDNF only in forebrain
pyramidal cells had reduced chronic stress-induced atrophy in the apical dendrites of CA3
pyramidal cells. Correlated with this structural change was an inhibition of immobility in the
transgenic animals in the Porsolt forced swim test, a reliable predictor of antidepressant
action. Interestingly, the transgenic animals did not show an inhibition of the HPA axis,
or of chronic stress-induced weight loss, suggesting that the transgenic animals had the
same "level" of stress as the control animals. The transgenic animals had higher levels
of BDNF in the amygdala, correlated with increased spinogenesis in the pyramidal cells
of the basolateral amygdala. Interestingly, the animals showed a higher level of anxiety,
a form of cue-non-specific fear. Thus, it was demonstrated that structural changes in
different parts of the brain were tightly correlated with affective behavioral changes.
While further experiments will be needed to further characterize these long-term
changes, this thesis provides compelling evidence that translation plays a key role in
providing the proteins necessary for bidirectional synaptic plasticity that underlies longterm memory. These proteins likely produce structural changes in activated neurons,
leading to the appropriate behaviors, or when hyperactivated chronically, to affective
disorders such as depression and anxiety. More generally, this thesis demonstrates how
the study of neuroscience from molecular perturbations through an understanding of
cellular processes, and relevant physiology, can shed light on the computational processes
underlying neuronal function, ultimately leading a better understanding of the behavior of
an organism.
145
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Bibliography
Barco, A., Alarcon, J. M., and Kandel, E. R. (2002). Expression of constitutively active
CREB protein facilitates the late phase of long-term potentiation by enhancing
synaptic capture. Cell 108, 689-703.
Casadio, A., Martin, K. C., Giustetto, M., Zhu, H., Chen, M., Bartsch, D., Bailey, C. H.,
and Kandel, E. R. (1999). A transient, neuron-wide form of CREB-mediated longterm facilitation can be stabilized at specific synapses by local protein synthesis.
Cell 99, 221-237.
Davis, H. P., and Squire, L. R. (1984). Protein synthesis and memory: a review. Psychol
Bull 96, 518-559.
Dias, B. G., Banerjee, S. B., Duman, R. S., and Vaidya, V. A. (2003). Differential
regulation of brain derived neurotrophic factor transcripts by antidepressant
treatments in the adult rat brain. Neuropharmacology 45, 553-563.
Frey, U., Frey, S., Schollmeier,
F., and Krug, M. (1996). Influence of actinomycin
D, a
RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in
vivo and in vitro. J Physiol 490 ( Pt 3), 703-711.
Frey, U., Krug, M., Reymann, K. G., and Matthies, H. (1988). Anisomycin,
an inhibitor of
protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1
region in vitro. Brain Res 452, 57-65.
Frey, U., and Morris, R. G. (1997). Synaptic tagging and long-term potentiation. Nature
385, 533-536.
Frey, U., and Morris, R. G. (1998). Synaptic tagging: implications for late maintenance of
hippocampal long-term potentiation. Trends Neurosci 21, 181-188.
Gingras, A. C., Raught, B., and Sonenberg, N. (1999). elF4 initiation factors: effectors of
mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem
68, 913-963.
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal
transduction. Annu Rev Biochem 72, 609-642.
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., Maffei,
L., and Tonegawa, S. (1999). BDNF regulates the maturation of inhibition and the
critical period of plasticity in mouse visual cortex. Cell 98, 739-755.
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein
synthesis in hippocampal mGluR-dependent long-term depression. Science 288,
1254-1257.
146
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and
protein synthesis--dependent long-term depression in hippocampal area CA1. J
Neurophysiol 86, 321-325.
Kauderer, B. S., and Kandel, E. R. (2000). Capture of a protein synthesis-dependent
component of long-term depression. Proc Natl Acad Sci U S A 97, 13342-13347.
Kuroda, Y., and McEwen, B. S. (1998). Effect of chronic restraint stress and tianeptine
on growth factors, growth-associated protein-43 and microtubule-associated
protein 2 mRNA expression in the rat hippocampus. Brain Res Mol Brain Res 59,
35-39.
Lamprecht, R., and LeDoux, J. (2004). Structural plasticity and memory. Nat Rev
Neurosci 5, 45-54.
Lu, B. (2003). BDNF and activity-dependent synaptic modulation. Learn Mem 10, 86-98.
Manahan-Vaughan, D., Kulla, A., and Frey, J. U. (2000). Requirement of translation but
not transcription for the maintenance of long-term depression in the CA1 region
of freely moving rats. J Neurosci 20, 8572-8576.
Martin, K. C., Casadio, A., Zhu, H., Yaping, E., Rose, J. C., Chen, M., Bailey, C. H., and
Kandel, E. R. (1997). Synapse-specific, long-term facilitation of aplysia sensory
to motor synapses: a function for local protein synthesis in memory storage. Cell
91, 927-938.
Martin, K. C., and Kosik, K. S. (2002). Synaptic tagging -- who's it? Nat Rev Neurosci 3,
813-820.
McAllister, A. K., Katz, L. C., and Lo, D. C. (1999). Neurotrophins and synaptic plasticity.
Annu Rev Neurosci 22, 295-318.
McEwen, B. S. (1999). Stress and hippocampal plasticity. Annu Rev Neurosci 22, 105122.
Neves-Pereira, M., Mundo, E., Muglia, P., King, N., Macciardi, F., and Kennedy, J.
L. (2002). The brain-derived neurotrophic factor gene confers susceptibility to
bipolar disorder: evidence from a family-based association study. Am J Hum
Genet 71, 651-655.
Nguyen, P. V., Abel, T., and Kandel, E. R. (1994). Requirement of a critical period of
transcription for induction of a late phase of LTP. Science 265, 1104-1107.
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB
mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug
treatments. J Neurosci 15, 7539-7547.
147
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Pavlides, C., Nivon, L. G., and McEwen, B. S. (2002). Effects of chronic stress on
hippocampal long-term potentiation. Hippocampus 12, 245-257.
Pavlides, C., Ogawa, S., Kimura, A., and McEwen, B. S. (1996). Role of adrenal steroid
mineralocorticoid and glucocorticoid receptors in long-term potentiation in the
CA1 field of hippocampal slices. Brain Res 738, 229-235.
Pavlides, C., Watanabe, Y., and McEwen, B. S. (1993). Effects of glucocorticoids on
hippocampal long-term potentiation. Hippocampus 3, 183-192.
Porsolt, R. D. (2000). Animal models of depression: utility for transgenic research. Rev
Neurosci 11, 53-58.
Proud, C. G. (2002). Regulation of mammalian translation factors by nutrients. Eur J
Biochem 269, 5338-5349.
Rattiner, L. M., Davis, M., French, C. T., and Ressler, K. J. (2004). Brain-derived
neurotrophic factor and tyrosine kinase receptor B involvement in amygdaladependent fear conditioning. J Neurosci 24, 4796-4806.
Raymond, C. R., Thompson, V. L., Tate, W. P., and Abraham, W. C. (2000).
Metabotropic glutamate receptors trigger homosynaptic protein synthesis to
prolong long-term potentiation. J Neurosci 20, 969-976.
Sajikumar, S., and Frey, J. U. (2004). Late-associativity, synaptic tagging, and the role of
dopamine during LTP and LTD. Neurobiol Learn Mem 82, 12-25.
Scharf, M. T., Woo, N. H., Lattal, K. M., Young, J. Z., Nguyen, P. V., and Abel, T. (2002).
Protein synthesis is required for the enhancement of long-term potentiation and
long-term memory by spaced training. J Neurophysiol 87, 2770-2777.
Schuman, E. M. (1999). Neurotrophin regulation of synaptic transmission. Curr Opin
Neurobiol 9, 105-109.
Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S., and Duman, R. S. (2002).
Brain-derived neurotrophic factor produces antidepressant effects in behavioral
models of depression. J Neurosci 22, 3251-3261.
Si, K., Giustetto, M., Etkin, A., Hsu, R., Janisiewicz, A. M., Miniaci, M. C., Kim, J. H.,
Zhu, H., and Kandel, E. R. (2003a). A neuronal isoform of CPEB regulates local
protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia.
Cell 115, 893-904.
Si, K., Lindquist, S., and Kandel, E. R. (2003b). A neuronal isoform of the aplysia CPEB
has prion-like properties. Cell 115, 879-891.
148
Chapter 5 - Outstanding issues in understanding of long-term changes in the brain
Smith, M. A., Makino, S., Kvetnansky, R., and Post, R. M. (1995a). Effects of stress on
neurotrophic factor expression in the rat brain. Ann N Y Acad Sci 771, 234-239.
Smith, M. A., Makino, S., Kvetnansky, R., and Post, R. M. (1995b). Stress and
glucocorticoids affect the expression of brain-derived neurotrophic factor and
neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15, 1768-1777.
Ueyama, T., Kawai, Y., Nemoto, K., Sekimoto, M., Tone, S., and Senba, E. (1997).
Immobilization stress reduced the expression of neurotrophins and their
receptors in the rat brain. Neurosci Res 28, 103-110.
Vyas, A., Bernal, S., and Chattarji, S. (2003). Effects of chronic stress on dendritic
arborization in the central and extended amygdala. Brain Res 965, 290-294.
Vyas, A., Mitra, R., Shankaranarayana Rao, B. S., and Chattarji, S. (2002). Chronic
stress induces contrasting patterns of dendritic remodeling in hippocampal and
amygdaloid neurons. J Neurosci 22, 6810-6818.
149
Credits and Notes
Chapter
1
Some of the information in Chapter 1 has been published in the review (Kelleher et
al., 2004b), including Figure 1-1. Figure 1-2 is modified from figures in the review.
Chapter 2
Most of the information in Chapter 2 has been published in (Kelleher et al.,
2004a); however the chapter was written independently of the text. The dnMEK1 mice
were engineered by R. J. Kelleher, and the electrophysiology contributed by H. Jung and
H. Kang. The behavioral data were performed by R. J. Kelleher. Thus, these data are
given as part of the introduction. The data for figures 2-16 and 2-18 were part of a joint
experiment by R. J. Kelleher and myself. All other data from Fig. 2-7 - 2-18 were obtained
by me, with input from R. J. Kelleher. Figure 2-6 was obtained by H. Jung with input from
R. J. Kelleher and myself.
Chapter 3
I was assisted in obtaining the results in Chapter 3 by Shu Huang. Valuable advice
was provided by R. J. Kelleher. The model shown in Figure 3-7 has been published in
(Kelleher et al., 2004b).
Chapter 4
The information in Chapter 4 has been submitted for publication in slightly
modified form (Govindarajan et al, submitted). Golgi-Cox staining was performed by B.
S. Shankaranarayana Rao, and Deepti Nair from the lab of Dr. Sumantra Chattarji at
the NCBS in Bangalore, India. I also had the assistance of two UROPs, Nadya Mawjee
and Mimi Trinh, who aided in the production of Figure 4-3. Two other UROPs, Caroline
Lee and Colin Galbraith assisted me on occasion. Francis Bushard and Lorene Leiter
provided technical advice on the behavioral apparati, and Chanel Lovett aided with the in
situ hybridization.
150
Chapter 5
Parts of this conclusion have been published, in modified form, including Figure
5-1 (Kelleher et al., 2004a; Kelleher et al., 2004b).
Bibliography
Kelleher, R. J., 3rd, Govindarajan,
A., Jung, H. Y., Kang, H., and Tonegawa, S. (2004a).
Translational control by MAPK signaling in long-term synaptic plasticity and
memory. Cell 116, 467-479.
Kelleher, R. J., 3rd, Govindarajan, A., and Tonegawa, S. (2004b). Translational
regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44, 5973.
151
Arvind Govindarajan
Tonegawa Lab, Massachusetts Institute of Technology
Address: 77 Massachusetts Ave. Room E18-313, Cambridge, MA 02139, USA
Phone: +1 617 258 9410
Email: guv@mit.edu
EDUCATION
1998-
Massachusetts Institute of Technology, Cambridge, MA, USA
Doctoral Candidate in Biology
Topic: "Long-term activity-induced changes in the brain"
1993-1998
University of Toronto at Scarborough, Toronto, ON, Canada
B.A. Honors in Physical Sciences, Graduation with High Distinction
Specialist in Biochemistry & Chemistry, Specialist in Computer Science
SELECTED AWARDS
* 1999-2004
Howard Hughes Medical Institute Predoctoral Fellowship
* 2004
D)ean's Educational and Student Advising Award
* 1998
(GovernorGeneral's Silver Medal, Graduation Prize in Physical Sciences
* 1998
Society of Chemical Industry Student of Merit Award
* 1993-1997
Canada Science & Technology Scholarship
* 1997
SHL Systemhouse Award in Computer Sciences
* 1993-1996
University of Toronto Scholar
* 1994-1998
University of Toronto Honor Roll
(Other award information available upon request)
PUBLICATIONS
* Govindarajan, A., et al "BDNF prevents depression and chronic-stress induced hippocampal
atrophy, but facilitates anxiety and amygdalar spinogenesis", submitted.
* Kelleher, R.J., Govindarajan, A., and Tonegawa S. "Translational Regulatory Mechanisms in
Persistent Forms of Synaptic Plasticity", Neuron 44(1): 59-73
* Kelleher, R.J., Govindarajan, A., Jung, H., Kang, H., and Tonegawa, S. "Translational Control
by MAPK Signaling in Long-Term Synaptic Plasticity and Memory", Cell 116(3): 467-479,
2004
152
* Laliberte, F., Hian,Y., Govindarajan, A. et al, "Conformational difference between PDE4
apoenzyme and holoenzyme", Biochemistry 39(21): 6449-6458, 2000
SELECTED LIST OF POSTERS PRESENTED
* 2003 Society for Neuroscience Annual Meeting (USA): "BDNF overexpression protects the
hippocampus from chronic stress induced damage"
* 2003 RIKEN BSI Annual Retreat (Japan): "Role of ERK MAP Kinase in Neuronal
Translation, L-LTP and Memory"
* 2003 Cold Spring Harbor Laboratory Learning & Memory Meeting (USA): "Role of ERK
MAP Kinase in Neuronal Translation, L-LTP and Memory"
* 2003 RIKEN-MIT Symposium (USA): "BDNF overexpression protects the hippocampus
from chronic stress induced damage"
* 2003 National Center for Biological Sciences Networks & Behavior Symposium (India):
"BDNF overexpression protects the hippocampus from chronic stress induced damage"
* 2001 Picower Center for Learning & Memory Annual Retreat (USA): "Role of ERK MAP
Kinase in Neuronal Translation"
SELECTED LIST OF LECTURES GIVEN
* 2004 Society for Neuroscience LTP Slide Session: " Characterization of Neuronal Activity
Induced Translation"
* 2004 MIT Center for Cancer Research Annual Retreat: "Characterization of Neuronal
Activity Induced Translation"
* 2003 Dept. of Brain & Cognitive Sciences Brain Lunch: "Role of ERK MAP Kinase in
Neuronal Translation, L-LTP and Memory"
* 2003 MIT Center for Cancer Research Annual Retreat: "Role of ERK MAP Kinase in
Neuronal Translation and L-LTP"
* 2003 Picower Center for Learning & Memory Plastic Lunch: "Role of ERK MAP Kinase
in Neuronal Translation, L-LTP and Memory"
* 2002 Dept. of Brain & Cognitive Sciences Brain Lunch: "Role of ERK MAP Kinase in
Neuronal Translation"
* 2002 MIT Center for Cancer Research Annual Retreat: "Role of ERK MAP Kinase in
Neuronal Translation"
153
CONFERENCES ATTENDED
* 2001-2004 Society for Neuroscience Annual Meeting (USA)
* 2001, 2003 Cold Spring Harbor Laboratory Learning & Memory Meeting (USA)
* 2003 National Center for Biological Sciences Networks & Behavior Meeting (India)
* 2004 Banbury Meeting on MAP Kinase (USA)
* 2004 RIKEN BSI Learning and Memory Course (Japan)
TEACHING EXPERIENCE
* 2002-2004 Teaching Assistant - "Neural Plasticity" (MIT)
* 2001 Teaching Assistant - "Graduate Biochemistry" (MIT)
* 2000 Teaching Assistant - "Cellular Neurobiology" (MIT)
* 1997 Teaching Assistant - "General Chemistry" (Lab Section, University of Toronto)
PREVIOUS WORK EXPERIENCE
* Undergraduate Research, Dept. of Chemistry, U. of Toronto, Toronto, ON, 1997 - 1998
Dr. G. A. Woolley Lab: Use of unnatural amino acids to create photo-controlled enzymes.
* Merck-Frosst Center for Therapeutic Research, Montreal, PQ, 1994-1996, 1998 summers
Tyrosine phosphatase biochemistry & cell biology; apoptosis biochemistry; proteomics
* Lead Programmer, Merck & Co, Rahway, NJ, Sep. 1994-Apr. 1995
Prototype for large-scale data analysis and storage software
154