Energy-resolved positron annihilation rates for molecules 兲
advertisement

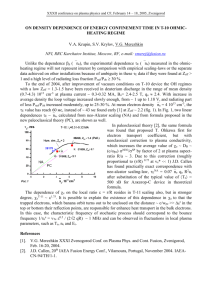
PHYSICAL REVIEW A 74, 012706 共2006兲 Energy-resolved positron annihilation rates for molecules L. D. Barnes, J. A. Young, and C. M. Surko Department of Physics, University of California, San Diego, California 92093-0319, USA 共Received 17 March 2006; published 10 July 2006兲 The development of high resolution positron beams has enabled measurements of annihilation rates for molecules as a function of incident positron energy. Vibrational Feshbach resonances in these spectra provide evidence for the existence of positron-molecule bound states. In this paper we present further studies of this phenomenon. Evidence is presented for positronically excited bound states 共i.e., in addition to the ground state兲 in C12H26 and C14H30. Measurements of the annihilation spectra of the halomethanes, CH3F, CH3Cl, and CH3Br, exhibit strong resonances that vary significantly with the substituted halogen. Annihilation spectra for linear alkanes and ring molecules are compared. Annihilation spectra and infrared absorption spectra are compared for a number of molecules. Finally, annihilation rate measurements are presented for a variety of molecules at energies 艌0.5 eV 共i.e., above the vibrational resonances兲. These provide a measure of the annihilation rates in the absence of vibrational resonances. DOI: 10.1103/PhysRevA.74.012706 PACS number共s兲: 34.85.⫹x, 34.50.⫺s, 78.70.Bj, 71.60.⫹z I. INTRODUCTION As antimatter has become more widely available in the laboratory, it is increasingly important to understand in detail the interactions between antiparticles and ordinary matter. Obtaining such an understanding presents significant challenges to modern atomic and molecular physics. The presence of the light electron and positron, absent the exchange interaction, can lead to strong electron-positron correlations difficult to model using the quantum chemical techniques so successful in conventional electron systems. In particular, the positronium channel, whether open or closed, is essential for many processes of interest but difficult to incorporate into theoretical calculations. Early measurements of positron-on-molecule annihilation using thermal positrons revealed a number of unusual phenomena. One of the most important observations was that annihilation rates for some molecules exceed the free electron annihilation rate by many orders of magnitude 关1–3兴. The dimensionless parameter Zeff is a quantitative measure of this enhancement. This quantity is defined by normalizing the measured annihilation rate by the rate of annihilation of positrons in a free electron gas of the same number density 关4兴, namely: Zeff = ⌫ . r20cnm 共1兲 In this expression, ⌫ is the annihilation rate, r0 is the classical electron radius, c is the speed of light, and nm is the molecular number density. The parameter Zeff has been crudely interpreted as the effective number of electrons per molecule which contribute to annihilation, assuming that the positron and electron motions are not correlated. However, for molecular species such as hydrocarbons, Zeff is found to be so much larger than Z, the number of electrons in the molecule, that interpretation of Zeff as an effective number of electrons clearly fails. In fact, in the alkanes 共CnH2n+2兲, this rate grows exponentially with the number of electrons 关5,6兴. As we discuss below, this is due to highly correlated states in 1050-2947/2006/74共1兲/012706共10兲 which the positrons form long-lived resonances with target molecules. The development of the Penning-Malmberg buffer-gas trap has led to a new class of experiments by transforming the standard warm, continuously moderated positron beam into a colder 共25 meV FWHM兲, pulsed positron beam 关8兴. The unusual annihilation effects observed when thermal positrons interact with hydrocarbons can now be resolved as a function of positron energy. Experiments of this type 关6,7兴 revealed remarkable new features, such as those illustrated in Fig. 1. This figure shows the normalized annihilation rate, Zeff, as a function of positron energy for propane and hexane. In propane, for example, the annihilation is enhanced near the energies of vibrational modes. The most prominent peak is associated with the C-H stretch vibrational mode at 370 meV, but similar effects are observed throughout the region of energies of the other vibrational modes. In larger alkane molecules, such as hexane, the C-H peak is significantly larger and much further shifted below the C-H stretch mode energy. These observations are qualitatively consistent with a model involving vibrational Feshbach resonances 共VFR兲, proposed by a number of authors 关3,9兴, and developed in detail by Gribakin 关10兴. The VFR enhancement of annihilation rates requires the existence of a bound state of the positron and the target molecule. At specific incident positron energies, the ground state molecule plus an unbound positron is nearly degenerate with the vibrationally excited molecule with a bound positron, namely Ev=0 + ⑀k = Ev=1 − ⑀b , 共2兲 where Ev=0 and Ev=1 are the ground and vibrationally excited energies of the molecule, ⑀k is the kinetic energy of the positron, and ⑀b is the binding energy of the positron to the molecule. In this resonant state, the overlap between the positron and electron wave functions is greatly enhanced, which in turn greatly enhances the probability of annihilation. As shown in Fig. 1, this effect is manifested in the measured annihilation spectra by large resonant enhancements associated with the molecular vibrational modes. Due to the 012706-1 ©2006 The American Physical Society PHYSICAL REVIEW A 74, 012706 共2006兲 BARNES, YOUNG, AND SURKO FIG. 1. The energy-resolved Zeff spectra 共쎲兲 for 共a兲 propane 共C3H8兲; and 共b兲 hexane 共C6H14兲, both scaled by a factor of 10−3 关6,7兴. Also shown in 共a兲 is the vibrational mode spectrum for propane 共---兲, arbitrarily scaled in amplitude and broadened by a Lorentzian line shape with a width of 10 meV. The vertical dashed line indicates the position of the C-H stretch mode in these molecules. binding of the positron and molecule, these resonant features occur at an energy lower than that of the vibrational mode by the positron-molecule binding energy, ⑀b, which ranges from a few tens of meV for smaller alkane molecules 共e. g., propane and butane兲 to more than 200 meV for large molecules 共e. g., C12H26兲. In the alkanes, the binding increases linearly with molecular size, while the annihilation rate, Zeff, grows exponentially with molecular size, reaching ⬃107 for C12H26 关6兴. The mechanism for exponential growth of Zeff is still open to debate. However, it is believed that in the case of alkane molecules with more than two carbon atoms, vibrational energy redistribution is responsible 关3,10,11兴. According to this model, the positron is captured into a “doorway state,” usually by exciting a fundamental vibrational mode of the molecule. This vibrational energy is then rapidly redistributed into one of the many degenerate combination modes, greatly enhancing the number of available resonant states and hence the overall annihilation rate. If the total density of vibrational states is included, the estimated growth of Zeff is too large 关11兴. One way to moderate this is to include fewer modes or to allow for inelastic detrapping. In the latter case, a component vibration with energy greater than positron binding deexcites, ejecting the positron before annihilation can occur. In this paper, the results of studies of the energy-resolved annihilation spectra for a range of molecules are presented with the goal of gaining further insight into this phenomenon of enhanced annihilation rates in molecules. A primary focus of the present study is understanding the effect of structural variations beyond those observed in previous studies of alkane molecules. It will be shown that small changes frequently affect not only the local mode strengths and positions but also global features such as binding energies and the overall magnitudes of Zeff. Data are presented for the largest alkane molecules studied to date, C12H26 and C14H30, that provide evidence for a second positron bound state 共i.e., a positronically excited bound state兲. Since large molecules are expected to be more difficult to treat theoretically, the results of additional studies of small molecules are presented, namely data for the halogenated methanes CH3F, CH3Cl, and CH3Br. These molecules exhibit large resonant enhancements with positions and strengths that depend on the particular halogen. To test the dependence of the observed VFR on molecular conformation, the spectra of three-carbon and six-carbon alkanes are compared with the analogous ring-shaped molecules 共including benzene in the six-carbon case兲. For further insight into the positron capture process, annihilation rate spectra are compared to infrared absorption spectra. The infrared data provide a quantitative measure of the long-range dipole coupling to vibrational modes. Continuing this theme, measurements of the infrared absorption spectra for hexane, 1-fluorohexane, nonane and 1-fluorononane were made and compared with previously published Zeff data. While the addition of a fluorine atom to these molecules produces a large effect on the annihilation peak associated with the C-H vibrational mode, very little change is observed in the corresponding infrared spectra. Finally, the annihilation spectra were measured for a number of molecules in the region of energies above that of the vibrational modes but below the positronium formation threshold. These spectra, which are approximately constant in this region of energies, provide a measure of the direct annihilation rate, absent vibrational resonance effects. The paper ends with a brief set of conclusions and suggestions for future work on problems of interest. II. EXPERIMENT The experimental arrangement used to measure Zeff is described in detail elsewhere 关6,7兴. Positrons from a radioactive 22 Na source are slowed by a solid neon reflection moderator and magnetically guided into a three-stage PenningMalmberg buffer-gas trap. A 1.5 kG magnetic field confines the positrons in the radial direction. Positrons are trapped in a potential well in the axial direction using an N2 buffer gas. The trapped positrons cool to the temperature of the electrodes in the third stage of the trap by collisions with N2 and CF4 molecules. They are then forced out of the trap by raising the electrostatic potential in the third trapping stage and are magnetically guided through a gas cell filled with a low pressure 共e.g., ⬃10−4 Torr兲 of the gas to be studied. As the positrons pass through this gas cell, annihilation events are recorded by detecting the resulting 511 keV annihilation radiation. The annihilation rate is calculated from the number of annihilation events, the number of positrons transiting the gas cell, the length of the cell, the pressure of the test gas, 012706-2 PHYSICAL REVIEW A 74, 012706 共2006兲 ENERGY-RESOLVED POSITRON ANNIHILATION RATES¼ and the energy of the positrons in the gas cell. It should be noted that in all plots of Zeff, there is an estimated overall uncertainty of ⬃10%, due largely to uncertainties in the testgas pressure. Some of the infrared 共ir兲 absorption spectra presented in this paper were taken using a Fourier-transform infrared spectrometer. The samples were diluted to 0.2 molar concentration in a carbon disulfide solvent. Since the exact path length in the sample cell was not known, the absolute molar absorptivities could not be determined. However, since the same cell geometry was used for all molecules studied in this way, the relative magnitudes are reliable. The background due to the solvent was measured and subtracted to obtain the reported ir spectra. In addition to these spectra, where indicated, ir spectra for other species were taken from the NIST chemistry webbook database 关12兴. These NIST data have arbitrary absorptivity scales that are independent of each other. Finally, for other molecules, computations of oscillator strengths for dipole-allowed infrared transitions were performed using the GAMESS quantum computational package 关13兴 using a 6-31G共d兲 basis set both for optimizing the geometry and computing the Hessian matrix. FIG. 2. The energy-resolved Zeff spectrum for 共䊊兲 dodecane 共C12H26兲; and 共쎲兲 tetradecane 共C14H30兲, with the former scaled by a factor of 10−6 and the latter scaled arbitrarily. Arrows indicate the locations of the second peaks interpreted here as evidence for positronically excited bound states. See text for details. B. Tetradecane III. EVIDENCE FOR A SECOND POSITRON BOUND STATE A. Dodecane It is plausible that, as the attraction between the positron and a target molecule increases with increasing molecular size, the resulting attractive potential well might become deep enough to support a second bound state. In this case, new resonances might be expected to appear in larger alkanes just below the energies of the molecular vibrational modes. These resonances would correspond to the first 共positronically兲 excited states of the vibrationally excited positron-molecule complex. As the size of the molecule is increased, these resonances would be expected to move to lower energy 共i.e., corresponding to increased binding兲. Pursuing this hypothesis, we examined the Zeff spectrum for dodecane 共C12H26兲, shown in Fig. 2. This spectrum exhibits a large peak associated with the C-H stretch mode at an incident positron energy ⬃150 meV, downshifted by about 220 meV from the C-H stretch energy and consistent with the trend for other alkanes studied 关6兴. It also has a second peak at 360 meV, much smaller in amplitude than the peak associated with the positronic ground state. As there are no fundamental modes above the C-H stretch mode, this would appear to be the excited state postulated above. To rule out possible interactions with multiple target particles, the annihilation rate Zeff in this energy range was measured for several test-gas pressures. These measurements confirmed that the peak is a two-body effect 共involving a positron and a single molecule兲. While it cannot be entirely ruled out, there are reasons to believe that this second peak is not simply a combination mode. In particular, no other combination modes have been observed in alkanes to date, and the energy of the peak does not correspond to a simple, twomode combination. Intrigued by the dodecane result, we measured the spectrum for tetradecane 共C14H30兲. It is also shown in Fig. 2. In this case, the tetradecane pressure is uncertain due to the extremely low pressure required for this high Zeff material. Since an absolute normalization of Zeff cannot be determined without knowledge of this pressure, the spectrum in Fig. 2 has been scaled arbitrarily for comparison with the dodecane spectrum. In the tetradecane spectrum, both the higher and lower energy resonances are clearly visible 共i.e., corresponding to the positron ground state and first excited state兲. As shown in Fig. 3, the binding energy corresponding to the primary C-H stretch peak in tetradecane is consistent with the trend reported for the other alkanes 关6兴, namely a linear relationship between binding and molecular size. The higher lying, excited-state peak also shifts a similar amount, appearing to maintain a fixed energy above the first peak. These observations led Gribakin and Lee to study this effect in the alkanes using a zero-range potential model 关14兴. FIG. 3. Binding energy as a function of the number of carbons in alkane molecules 共쎲兲. The binding energies of two positronically excited bound states 共䊊兲 are also shown. 012706-3 PHYSICAL REVIEW A 74, 012706 共2006兲 BARNES, YOUNG, AND SURKO In particular, they extended a model used previously to describe krypton dimers 关15兴 and represented the CH2 and CH3 groups in alkanes by zero-range potentials with identical scattering lengths. The binding energy could then be determined from the total scattering length of the composite molecule. Using this approach, they calculated positron binding energies as a function of the number of carbons, with the monomer scattering length chosen such that propane would be very weakly bound. Their predicted binding energies agree within a factor of 2 with those observed experimentally. They also found that a second bound state solution first appears in the 10 carbon alkane, which is slightly smaller than the onset number of carbons observed in experiment. Essentially, increasing the number of carbon potentials increases the binding of all positronic states until the next excited state solution is drawn below the continuum to become a true bound state. Considering the simplifications made in the model, this degree of agreement is encouraging. The resonance associated with the positron-molecule ground state is much larger than the excited state resonance, even though both are mediated by the same C-H vibrational mode. This effect occurs naturally in the VFR model if the positron kinetic energy is quickly redistributed among the vibrational modes of the molecule and detrapping is allowed 关10兴. The deeper the binding, the less likely it is that a vibration 共or a nonlinearly coupled combination of vibrations兲 will have sufficient energy to eject the positron. Hence Zeff is enhanced. In addition, deeper binding results in a larger positron density in the vicinity of the molecule, which can also enhance Zeff. IV. ANNIHILATION ON HALOGENATED METHANES While VFR effects are very large and well documented in large molecules, the size of these molecules virtually precludes any first-principles theoretical description in the foreseeable future. Hence, we were motivated to study smaller molecules that exhibit resonances in Zeff 共i.e., molecules with considerably fewer vibrational modes兲. The halogenated methanes fit this description well and provide an opportunity to examine further the local and global factors that affect Zeff spectra. By substituting various halogen atoms for a hydrogen, one can change the physical properties of the molecule and alter its mode frequencies. Like deuterium substitution, this can help in the identification of particular modes responsible for the VFR. The Zeff spectra of CH4, CH3F, CH3Cl, and CH3Br are shown in Fig. 4. The methane molecule, CH4, is relatively featureless, likely due to its high degree of symmetry, which results in weak infrared activity and no dipole moment. A featureless spectrum is also observed for CF4, probably for similar reasons 关6兴. This featureless spectrum is not, however, shared by the rest of the halomethanes studied here. It should be noted that they all have dipole moments exceeding the critical value, Dcr = 1.625 D, required for dipole-bound states 关16,17兴. When methane’s symmetry is broken by the substitution of a fluorine atom to form CH3F, a peak is observed at FIG. 4. Energy-resolved Zeff spectra for 共a兲 CH4 共䉭兲 and CH3F 共쎲兲; 共b兲 CH3Cl 共쎲兲; and 共c兲 CH3Br 共쎲兲. The lines 共—兲 represent the respective experimental ir absorption spectra from the NIST data base 共normalized arbitrarily and collected under different conditions兲. The bottom panel in each plot indicates the experimentally determined energies of infrared- and Raman-active vibrational modes 关12兴. Note that the ir modes and spectra in 共a兲 correspond to CH3F, whose Raman modes are unavailable. ⬃150 meV and a second, much weaker, peak at ⬃395 meV. The first is likely due to either the C-H bend mode or the C-F stretch and bend modes, which are too close to resolve. The second is about 20 meV above the C-H stretch, which is the 012706-4 PHYSICAL REVIEW A 74, 012706 共2006兲 ENERGY-RESOLVED POSITRON ANNIHILATION RATES¼ highest energy fundamental mode. This is an exceptional result in that, to date, no other molecule has exhibited such a positive energy shift. This peak position would seem incompatible with VFR theory, which requires a positive binding energy for the existence of a resonance. This inconsistency could, in principle, be resolved if the energy of the molecular vibrations were sufficiently increased by the presence of a positron. As the size of the halogen is increased, a number of new trends can be seen. The C-H stretch peak shifts systematically to lower energies: 395 meV for CH3F, 352 meV for CH3Cl and 323 meV for CH3Br. This indicates increased binding. In addition, both the high- and low-energy peaks grow dramatically. The peak around 150 meV grows nearly an order of magnitude from CH3F to CH3Cl and becomes significantly wider. In contrast with results for the alkanes, this study of the halogenated methanes provides insight into the importance of other parameters besides the number or type of vibrational modes in determining the magnitude and the energy spectrum of Zeff. In the case of the halogenated methanes, their large molecular dipole moments and the halogen atom polarizabilities are likely relevant and potentially important parameters. It is hoped that these data could be used to benchmark theoretical calculations of the Zeff spectra for relatively small molecules. The Zeff spectrum of CH3Cl shows three distinct peaks. The high energy peak is due to one or more of the C-H stretch modes. The remaining two low energy peaks, which nearly overlap, are likely due to the C-Cl modes and C-H bend modes, respectively. The low energy modes spread apart between CH3F and CH3Cl which may explain why the two peaks can be resolved in CH3Cl and not CH3F. The spectrum of CH3Br is more difficult to evaluate as its lowest energy peak occurs near the lower end of our measurement range, at ⬃50 meV. With this in mind, the second low energy peak still appears to be missing if a uniform shift due to binding is assumed. The ir absorption spectra in Fig. 4 do not appear to be strong indicators of the Zeff peak heights. Nevertheless, each peak can still be associated with a particular ir-active mode. The data shown in Fig. 4 motivated a calculation of Zeff for CH3Cl and CH3Br assuming the positrons are captured by infrared-active modes 关18兴. It indicates that all modes contribute nearly equally to Zeff and estimates binding energies of 8.5 and 12 meV, respectively. V. EFFECT OF STRUCTURAL CHANGES—RINGS VS CHAINS A. Three carbon chains and rings The molecules propane 共C3H8兲 and cyclopropane 共C3H6兲 provide a comparison of a relatively small linear alkane molecule with the analogous ring molecule. The cyclopropane ring is formed by eliminating the terminal methyl groups on the alkane chain, so that the end carbons can be joined. These two molecules have similar C-C and C-H stretch and bend modes, albeit cyclopropane has fewer of them. Their Zeff annihilation and infrared spectra are shown in Fig. 5. These data potentially provide insight into the importance of FIG. 5. The energy-resolved Zeff spectra 共쎲兲 for 共a兲 propane 共C3H8兲; and 共b兲 cyclopropane 共C3H6兲, both scaled by a factor of 10−3. The lines 共—兲 represent the experimental ir absorption spectra, arbitrarily normalized. The lower panels indicate the energies of ir and Raman active vibrational modes 关12兴. terminal CH3 groups and carbon backbone motion. The C-H stretch annihilation peaks in propane and cyclopropane occur at similar locations, as do their vibrational mode energies. The small downshifts that are observed indicate that these molecules have similar, small positron binding energies. As discussed in the next section, the similar binding energies of propane and cyclopropane are consistent with results for hexane and cyclohexane. The C-H stretch annihilation peak in cyclopropane is only about one-third the height of the analogous peak in propane. Forming a ring from propane involves reducing the number of C-H stretch modes by 25%. Considering the measurements and models for alkanes in which the Zeff peak height grows exponentially with the number of C-H stretch modes, one might expect the peak height to decrease faster than the number of modes. Alternatively, this difference between propane and cyclopropane could indicate the importance of the C-H modes associated with terminal methyl groups in determining the C-H peak height. An unusual feature in the cyclopropane spectrum, as compared to propane, is the peak at ⬃100 meV which is the same magnitude in Zeff as the broad plateau at ⬃100 meV in 012706-5 PHYSICAL REVIEW A 74, 012706 共2006兲 BARNES, YOUNG, AND SURKO propane. Rather than exhibiting a decrease in magnitude with decreasing mode density, this feature, presumably due to the C-H bend modes is much narrower in cyclohexane, more closely approximating the ir spectrum of this molecule 共i.e., as compared to propane兲. As discussed below, this behavior is qualitatively different than that observed in cyclohexane, where the amplitude of the entire spectrum is proportionately lower than that in hexane at all vibrational energies. Perhaps propane is at a critical size where the mode density is low and individual modes become more important. Thus, resonances in the spectrum might be switched off as certain modes disappear, as is the case when the ring molecule is formed. The infrared spectra of propane and cyclopropane are also shown in Fig. 5. The ir and Zeff spectra of cyclopropane are relatively similar. However this is not true for propane. The ir spectrum of propane has a C-H peak that dominates the rest of the spectrum by more than an order of magnitude in amplitude, with virtually no ir activity below 180 meV. In contrast, the Zeff spectrum has more evenly weighted peaks throughout the energy range of the vibrational modes. This is another example where there is seemingly little connection between the ir magnitudes and Zeff magnitudes. B. Six carbon chains and rings Continuing the comparison of chain and ring hydrocarbons, the annihilation spectra of hexane 共C6H14兲, cyclohexane 共C6H12兲, and benzene 共C6H6兲 are shown in Fig. 6. Cyclohexane, like cyclopropane is formed by connecting the terminal carbons of the alkane. It has nearly the same number of C-H stretch modes as hexane 共12, compared with 14 for hexane兲 and, as can be seen in Fig. 7, the modes are predicted to be similarly infrared active. Note that these ir spectra are computer generated 共rather than measurement based兲 in order to ensure a common normalization. Benzene has the same number of carbons as cyclohexane, but fewer hydrogen atoms by a factor of 2, resulting in double bonds shared among the carbon atoms. This does not significantly affect the energy of its C-H stretch modes but does reduce their number. This is reflected in a significantly weaker ir absorption. In comparison with hexane, both benzene and cyclohexane have significantly smaller Zeff than the linear molecule. For cyclohexane, this effect cannot be attributed to a decrease in mode number or ir strength. On the other hand, it is an additional indication that the terminal methyl 共CH3兲 groups may be important in the VFR enhancement of Zeff. Another important issue is the location of the C-H stretch peaks in the Zeff spectra. While the large peaks for hexane and cyclohexane are at approximately the same energy, the largest peak in benzene is significantly lower in energy. If these are all C-H stretch peaks, in the context of the vibrational Feshbach resonance model, this would indicate that positrons bind more strongly to benzene than to the other two molecules. In this interpretation, the binding energy for benzene is ⬃130 meV as compared to ⬃80 meV for hexane and cyclohexane. While this would appear to break the trend of stronger binding leading to larger Zeff, the combination of fewer C-H FIG. 6. The energy-resolved Zeff spectra 共쎲兲 for 共a兲 cyclohexane 共C6H12兲 and 共b兲 benzene 共C6H6兲, both scaled by 10−4. In both figures, the open circles 共䊊兲 are the data for hexane 共C6H14兲, scaled by 0.5 for ease of comparison. For reference, the thermal Zeff’s of hexane, cyclohexane, and benzene are 120 000; 20 000; and 15 000, respectively 关19兴. FIG. 7. The infrared absorption spectrum for hexane 共—兲, cyclohexane 共--兲 and benzene 共-·-兲 as computed using the GAMESS quantum computational package 关13兴 using a 6-31Gd basis set. 012706-6 PHYSICAL REVIEW A 74, 012706 共2006兲 ENERGY-RESOLVED POSITRON ANNIHILATION RATES¼ FIG. 8. Energy-resolved Zeff spectra for 共a兲 1-fluorohexane 共C6H13F兲 共䊊兲 compared with hexane 共C6H14兲 共쎲兲 scaled by 10−4; and 共b兲 1-fluorononane 共C9H19F兲 共䊊兲, compared with nonane 共C9H20兲 共쎲兲 scaled by 10−6. In 共a兲, the data for 1-fluorohexane are also plotted multiplied by a factor of 10 共䊐兲. The arrows on the ordinate indicate the value of Zeff for the fully hydrogenated species in a 300 K thermal distribution of positrons. The thermal Zeff of 1-fluorohexane is 269 000, which is off the scale of the plot. The thermal Zeff of 1-fluorononane has not been measured. modes and the lack of terminal methyl groups in benzene may suppress any enhancements in Zeff gained from deeper binding. However, one must bear in mind that benzene is not a small change from cyclohexane. The shape of the ir and Zeff spectra in benzene is quite different, and so other factors may be relevant. Nevertheless, alternative explanations for the location of the main Zeff peak in the benzene spectrum are even less compelling. In particular, it is difficult to associate the largest Zeff peak in benzene with any mode other than the C-H stretch and still get positive binding. The second strongest mode in the ir spectrum, for example, is a ring stretch at ⬃200 meV, which is about 30 meV below the main peak in the Zeff spectrum. VI. COMPARISON OF Zeff AND INFRARED SPECTRA FOR FLUOROALKANES Substitution of a fluorine atom for a hydrogen in alkane molecules has been shown to produce very large effects on annihilation rates and annihilation spectra. Figure 8 shows Zeff spectra for 1-fluorohexane 共C6H13F兲 and 1-fluorononane 共C9H19F兲 关6兴. These molecules differ from the alkanes, hexane 共C6H14兲 and nonane 共C9H20兲, by the substitution of a FIG. 9. 共a兲 Infrared 共ir兲 absorption for hexane 共—兲 and 1-fluorohexane 共---兲 over the range of the C-H stretch vibrational energies; 共b兲 ir absorption for nonane 共—兲 and 1-fluorononane 共---兲. The units of absorbtivity are arbitrary, but are the same for all four molecules. See text for details. fluorine atom for one of the hydrogen atoms in either of the two methyl 共CH3兲 groups at an end of the molecule. The effect of this substitution on Zeff is dramatic. In both of these fluorinated molecules, there is a large reduction in the magnitude of the peak associated with the C-H stretch vibrational mode. In 1-fluorohexane, the amplitude of the peak is reduced by more than a factor of 20, while in 1-fluorononane the peak is reduced by ⬃2.5. In both cases, the position of C-H stretch peak is not significantly different from that in the nonfluorinated 共fully hydrogenated兲 species. In the context of the VFR model, this means the positron-molecule binding energy is unchanged by fluorination. As discussed above, a potentially important mechanism for producing vibrational excitation and the observed resonances is long-range dipole coupling, which can be measured using infrared absorption. The infrared spectra for the molecules studied in Fig. 8 were measured to investigate the degree to which the observed reductions in Zeff with fluorination can be attributed to changes in these dipole oscillator strengths. Figure 9 shows the infrared absorption spectra for 1-fluorohexane, hexane, 1-fluorononane and nonane. The in- 012706-7 PHYSICAL REVIEW A 74, 012706 共2006兲 BARNES, YOUNG, AND SURKO frared absorption spectra in the region of the C-H stretch modes are largely unchanged with single-fluorine substitution. The slight reduction in coupling magnitude for the fluorinated molecules can be attributed to the reduction by one in the number of C-H stretch modes. The conclusion is that factors other than changes in dipole coupling 共as reflected in changes in the ir spectra兲 are required to explain the large changes in magnitude of the C-H stretch peaks shown in Fig. 9. These fluorination results reinforce the idea that seemingly small changes in molecular structure 共e.g., those that barely affect the vibrational mode energies and ir absorption spectra兲 can produce very large changes in the Zeff spectra. The other major effect of single fluorination substitution in large hydrocarbon molecules is an increase in Zeff at lower energies 共i.e., 艋100 meV兲. This is immediately apparent in Fig. 8共b兲, where the Zeff of 1-fluorononane rises more rapidly than the Zeff of nonane as the positron energy is decreased. This trend is not apparent in the spectrum of 1-fluorohexane shown in Fig. 8共a兲. However, the Zeff value for 1-fluorohexane for a thermal distribution of positrons is 269 000, which is significantly larger than in any of the structure observed at higher energies 关19兴. More importantly, it is larger than the thermal Zeff of hexane. The physical origin of this effect is unclear. If this large Zeff value is to be attributed to low-lying Feshbach resonances, it remains to be explained why the resonances at low energies are enhanced while the resonances at higher energies are suppressed. Regarding these large values of Zeff at small positron energies, one may be tempted by the proposition that Zeff could be enhanced at low energies by the presence of a low-lying positron-molecule resonance or bound state 关10,20兴. This model appears to have merit in the case of heavier noble gas atoms and small molecules. However, in large molecules, the magnitude of the observed enhancements are much larger than can be explained by this mechanism. VII. NONRESONANT Zeff IN MOLECULES It is of interest to consider the annihilation rates of molecules in the substantial range of energies between that of the vibrational modes 共i.e., where vibrational Feshbach resonances are observed兲 and the thresholds for positronium formation. Our measurements of a number of molecules in this range of energies show that Zeff depends only weakly on positron energy. This is in agreement with the energy dependence seen in previous theoretical calculations and models of Zeff in noble gases and polyatomic molecules 关10,21–25兴. In this range of energies, the interaction of the positron with the molecule is relatively independent of the details of the molecular vibrational structure. An example of our data for butane is shown in Fig. 10. Although a more detailed study of Zeff would be useful, we present here only a single, average value of Zeff in the plateau region of each target. These values, which we refer to 共d兲 as Zeff , are summarized in Table I. In each case, the average annihilation rate was calculated over the range of energies, ⌬⑀, indicated in the table. The errors given in the table represent the standard deviations of the measurements made in this energy range. Typically, these errors are large since other FIG. 10. Energy-resolved Zeff spectrum for butane 共C4H10兲, scaled by 10−4, illustrating the relatively small values of Zeff above 共d兲 the energy range of the molecular vibrations. The value for Zeff for butane in Table I is the average of all data at energies greater than 0.7 eV and less than 3.0 eV. The inset is a close up of Zeff in this energy range. scattering processes require that test-gas pressure be kept low to avoid multiple scattering. This in turn reduces the achievable signal to noise. As shown in Table I and Fig. 11, large molecules tend to 共d兲 have large values of Zeff . These molecules typically also th , which are have large values of Zeff for thermal positrons, Zeff 共d兲 also given in Table I. The results in Table I indicate that Zeff increases faster than linearly with molecular size, at least for the alkanes. With regard to the historical association of Zeff with the total number of target electrons, Z, we note the 共d兲 relatively poor correlation of Zeff with Z. This is not surprising since inner shell electrons contribute little to the annihilation for positrons with energies less than a few electron volts 关26兴. It is important to note that unlike Zeff measured at lower 共d兲 energies, where VFR phenomena are important, Zeff may be more amenable to calculation 共e.g., using simplified models in which the nuclei are fixed兲. Fixed-nuclei calculations are available for at least three of the molecules listed in Table I: 共d兲 Zeff values for CH4, CF4, and NH3 were predicted to be 28, 90, and 18, respectively, as compared with measured values 共d兲 of 21, 11, and 47 关22兴. It is hoped that these data for Zeff will stimulate further theoretical calculations of Zeff. VIII. CONCLUDING REMARKS At this point, there is considerable evidence that positrons bind to a wide variety of molecules via vibrational Feshbach resonances, particularly in alkanes containing more than two carbon atoms. However, the factors that determine the magnitude of Zeff for each vibrational mode and the magnitudes of the positron binding energies are still largely unknown. To some degree, this is due to a lack of energy resolution to identify specific modes. But it is also due to a paucity of energy-resolved experimental Zeff spectra beyond the alkanes and a few other targets. This paper attempts to address the latter issue. In the case of the largest alkanes studied thus far, dodecane and tetradecane, a resonant feature was observed above 012706-8 PHYSICAL REVIEW A 74, 012706 共2006兲 ENERGY-RESOLVED POSITRON ANNIHILATION RATES¼ 共d兲 TABLE I. Average “direct” annihilation rates, Zeff , for positrons in the indicated energy range, ⌬⑀, below the threshold for positronium th formation. Also shown are the total number of target electrons, Z, and where available, the values of Zeff for thermal positrons, Zeff . Target Z 共d兲 Zeff ⌬⑀ 共eV兲 共th兲 Zeff 共d兲 Zeff /Z Argon Xenon Methane 共CH4兲 CF4 CH3F CHF3 Ethane Propane Butane 共C4H10兲 d-butane 共C4D10兲 Pentane 共C5H12兲 Ammonia 18 54 10 13.4± 0.6 54± 6 21± 3 0.7–2.6 0.6–3.6 0.6–3.6 33.8 401 142 0.74 1.0 2.1 42 18 34 18 26 34 11± 2 22± 2 28± 5 32± 4 55± 43 118± 37 0.7–1.1 0.6–2.0 0.5–3.6 1.3–2.3 1.8–2.8 0.7–2.8 54.4 1390 247 660 3500 11300 0.26 1.2 0.82 1.8 2.1 3.5 34 132± 56 0.8–2.8 42 246± 56 1.0–2.0 37800 5.9 10 47± 8 0.5–2.3 1600 4.7 the principal C-H stretch resonance. We interpret this as evidence of a second, positronically excited bound state mediated by excitation of the C-H stretch vibrational mode. The effect of small structural alterations on the Zeff spectra was investigated for several molecules. Methane exhibits both local and global changes in Zeff spectra as a result of halogen substitution. Data presented for three halomethanes, CH3F, CH3Cl, CH3Br reveal striking changes as the size of the halogen is increased. The binding energy increases nearly linearly with electron shell number, and the resonant peak heights increase nonlinearly by over an order of magnitude. In contrast with similar trends in the alkane series, trends in the halomethanes cannot be attributed to changes in the number of vibrational modes. Of particular interest are the data for CH3Cl, where the vibrational modes are sufficiently well separated that resonances of three distinct mode symmetries th FIG. 11. Zeff as a function of Zeff d 共쎲兲 for the molecules listed in Table I. The open circles 共䊊兲, where shown, represent the values at the C-H stretch peak for the given molecule. 3.9 can be observed. It is hoped that these halomethane Zeff spectra can be used to motivate and benchmark theoretical calculations, since they are likely more amenable to firstprinciples theoretical calculations as compared with spectra for larger molecules. A second study of changes in Zeff spectra with changes in molecular structure compared linear alkane molecules with ring hydrocarbons containing the same number of carbons. In the case of propane and cyclopropane, ring creation actually eliminates some low energy resonances rather than reducing their magnitude. This is perhaps an indication of an underlying selection rule. It is interesting to note that these resonant modes appear to “switch on” with a Zeff of a few thousand. Similarly, the halogenated methanes, which have relatively few modes, have Zeff values that do not exceed a few thousand. Perhaps this indicates that there is a maximum Zeff which can be produced in a relatively small molecule by a single mode. Further study of “borderline” small molecules similar to cyclopropane may help to test this idea. Studies of the six-carbon molecules, hexane, cyclohexane and benzene, provided further insight. The Zeff value of cyclohexane is half that of hexane at all energies in spite of the fact that both molecules have similar numbers of vibrational modes. The Zeff value of benzene is smaller still. However, in addition to having no terminal methyl groups, benzene has fewer vibrational modes. The benzene Zeff spectrum is quite unusual in that the main annihilation peak is downshifted from the C-H stretch mode by 130 meV as compared with 80 meV for hexane and cyclohexane. Assuming this peak is due to the C-H stretch mode, this indicates increased binding in benzene. In the alkanes, increased binding energy is strongly correlated with larger Zeff values. The fact that benzene has a smaller Zeff than both cyclohexane and hexane appears to contradict this trend. Study of a molecule such as 1-3-5 hexatriene, another six-carbon planar molecule with C-C bonding similar to ben- 012706-9 PHYSICAL REVIEW A 74, 012706 共2006兲 BARNES, YOUNG, AND SURKO zene but without the ring shape, may shed light on this behavior. A systematic comparison was made between the infrared absorption spectra and the Zeff spectra of a number of molecules. Infrared absorption provides a quantitative measure of electric dipole coupling to the vibrational modes and, presumably, VFR. While one cannot rule out the possibility that Zeff peaks are associated with ir-active modes, there is little evidence that dipole coupling alone is responsible for the observed VFR. In the alkanes, the replacement of a single hydrogen with a fluorine on a terminal methyl group decreases Zeff dramatically. Such substitutions in hexane and nonane did not result in commensurate reductions in ir absorption. This lack of correlation between the ir and Zeff magnitudes was also seen in hexane, cyclohexane, propane, and cyclopropane. It is clear that ir coupling strengths alone are not consistent predictors of Zeff spectra—other factors are clearly important. The final topic addressed in this paper is the magnitude and spectrum of annihilation in molecules above the energy range of the vibrational modes, but below the threshold for positronium formation. Studies of a number of molecules indicate that Zeff is approximately constant in this range of energies. The values of Zeff are much smaller than those observed in the VFR region. It would be interesting to compare these “nonresonant” values of Zeff with the predictions of fixed-nuclei calculations, rather than with values measured at lower positron energies where VFR are typically important and often dominant. In this paper we have presented studies of positron annihilation spectra, positron-induced VFR, and positron binding to molecules. We believe that these phenomena merit further theoretical and experimental exploration. In the case of large molecules, while the experimental signatures of positron VFR are fairly clear, consistent quantitative agreement between theoretical predictions and experimental measurements has yet to be achieved. In the case of small molecules, many open questions remain, including in many cases, the underlying mechanism or mechanisms responsible for observed large values of Zeff. 关1兴 D. A. L. Paul and L. Saint-Pierre, Phys. Rev. Lett. 11, 493 共1963兲. 关2兴 G. R. Heyland, M. Charlton, T. C. Griffith, and G. L. Wright, Can. J. Phys. 60, 503 共1982兲. 关3兴 C. M. Surko, A. Passner, M. Leventhal, and F. J. Wysocki, Phys. Rev. Lett. 61, 1831 共1988兲. 关4兴 P. A. Fraser, Adv. At. Mol. Phys. 4, 63 共1968兲. 关5兴 K. Iwata, R. G. Greaves, T. J. Murphy, M. D. Tinkle, and C. M. Surko, Phys. Rev. A 51, 473 共1995兲. 关6兴 L. D. Barnes, S. J. Gilbert, and C. M. Surko, Phys. Rev. A 67, 032706 共2003兲. 关7兴 S. J. Gilbert, L. D. Barnes, J. P. Sullivan, and C. M. Surko, Phys. Rev. Lett. 88, 043201 共2002兲. 关8兴 S. J. Gilbert, C. Kurz, R. G. Greaves, and C. M. Surko, Appl. Phys. Lett. 70, 1944 共1997兲. 关9兴 P. M. Smith and D. A. L. Paul, Can. J. Phys. 48, 2984 共1970兲. 关10兴 G. F. Gribakin, Phys. Rev. A 61, 022720 共2000兲. 关11兴 G. F. Gribakin and P. M. W. Gill, Nucl. Instrum. Methods Phys. Res. B 221, 30 共2004兲. 关12兴 NIST Chemistry WebBook 共2005兲, URL http:// webbook.nist.gov/chemistry/ 关13兴 M. W. Schmidt et al., J. Comput. Chem. 14, 1347 共1993兲. 关14兴 G. F. Gribakin and C. M. R. Lee, Nucl. Instrum. Methods Phys. Res. B 247, 31 共2006兲. 关15兴 G. F. Gribakin, Nucl. Instrum. Methods Phys. Res. B 192, 26 共2002兲. 关16兴 M. Tachikawa, I. Shimamura, R. J. Buenker, and M. Kimura, Nucl. Instrum. Methods Phys. Res. B 192, 40 共2002兲. 关17兴 R. J. Buenker, H. P. Liebermann, V. Melnikov, M. Tachikawa, L. Pichland, and M. Kimura, J. Phys. Chem. A 109, 5956 共2005兲. 关18兴 G. F. Gribakin and C. M. R. Lee 共private communication兲. 关19兴 K. Iwata, Ph.D. thesis, University of California, San Diego 共unpublished兲. 关20兴 V. I. Goldanskii and Y. S. Sayasov, Phys. Lett. 13, 300 共1964兲. 关21兴 R. P. McEachran, A. G. Ryman, and A. D. Stauffer, J. Phys. B 12, 1031 共1979兲. 关22兴 F. A. Gianturco, T. Mukherjee, and A. Occhigrossi, Phys. Rev. A 64, 032715 共2001兲. 关23兴 J. Mitroy and I. A. Ivanov, Phys. Rev. A 65, 042705 共2002兲. 关24兴 J. Ludlow, Ph.D. thesis, Queen’s Univerisity, Belfast, U.K. 共unpublished兲. 关25兴 M. T. do N. Varella, C. R. C. de Carvalho, and M. A. P. Lima, Nucl. Instrum. Methods Phys. Res. B 192, 225 共2002兲. 关26兴 K. Iwata, R. G. Greaves, and C. M. Surko, Phys. Rev. A 55, 3586 共1997兲. ACKNOWLEDGMENTS The authors thank John Crowell and Charles Perrin for use of their infrared equipment, Peter Langhoff and Jeff Mills for sharing their GAMESS expertise, Gleb Gribakin and Joan Marler for helpful conversations, and Gene Jerzewski for technical assistance. This work is supported by National Science Foundation Grant No. PHY 02-44653. 012706-10