Crescentic IgA Nephropathy
advertisement

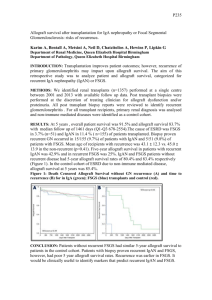
Nephrology Grand Rounds March 16th, 2010 Aditya Mattoo Outline Background Epidemiology Clinical Presentation Prognosis Pathogenesis Histology Treatment Recurrence in Transplant Background First described by Berger et al in 1968. Characterized by predominant IgA deposition in the glomerular mesangium. Most common form of primary glomerulonephritis around the world. Berger et al. J Urol Nephrol, 74:p694, 1968. Demographics Clinical onset in second and third decades of life. 80% of patients are between the ages of 16-35 years at the time of diagnosis. Male predominance of 2:1 in Japan to as high as 6:1 in northern Europe and US. Asians and Caucasians more prone to develop IgAN than people of African descent. Demographics There appears to be a familial clustering of IgAN which shows strong family predisposition in about 10% of cases. In the U.S., regions in Kentucky, Alabama and other parts of the Southeast exhibit a higher incidence of IgAN. In other parts of the world, familial clustering of IgAN seems to be more common in Southern France and Italy. Many genetic studies are underway, trying to establish common susceptibility genes in familial IgA. IgAN Nationwide Epidemiology IgAN prevalence as a percentage of primary GN: In Japan, 50% of new cases of GN are IgAN (causing 40% of all ESRD). 30% of new GN cases in Western Europe and Australia. 10% in general US population (exception Native Americans from New Mexico with prevalence of rate of 38%) Crescentic IgAN (CIgAN) is seen in approximately 7% of patients with IgAN. However, a study conducted by Shouno et al reported that by increasing the number of serial sections examined for any single biopsy specimen from the standard 20 to 100 sections, the finding of a segmental necrotizing lesion increases to 30%. Donadio et al. NEJM, 347:p738, 2002. Shouno et al. Acta Pathol Jpn, 43:p723, 1993. Clinical Presentation IgAN is highly variable, both clinically and pathologically. Clinical features range from asymptomatic hematuria to RPGN. Classic flare includes painless hematuria concurrent with the onset of viral illness (e.g. pharyngitis, gastroenteritis, etc.) Clinical Presentation Approximately 40-50% of patients present with one or recurrent episodes of gross hematuria. Another 30-40% have microscopic hematuria and usually mild proteinuria incidentally detected on a routine examination. Gross hematuria will eventually occur in 2025% of these patients. Clinical Presentation Of the patients with gross hematuria secondary to IgAN, up to 40% will develop transient renal failure. Less than 10% present with either nephrotic syndrome or RPGN (characterized by edema, hypertension, and renal dysfunction). Clinical Presentation: Crescentic vs Non-crescentic Crescentic IgAN Non–crescentic IgAN 35 229 Sex male/female 26/9 147/82 NS Mean age years 33 ± 12.5 32 ± 13 NS Serum creatinine mg/dL 1.3 ± 0.5 1.2 ± 0.4 NS Proteinuria g/day 2.3 ± 2.1 1.1 ± 1.2 < 0.003 20% 31.2% NS 37% 41% NS Number of patients History of recurrent macroscopic P value Hematuria % of patients Arterial hypertension % of patients Ferarrio et al. 3rd Congress of Nephrology, 2003. Prognosis Between 5-30% of patients with mild proteinuria, hematuria or mild renal dysfunction undergo spontaneous remission of abnormal laboratory findings. A Chinese study of 72 consecutive patients with IgAN performed diagnostic biopsies on patients with hematuria, but with no or minimal proteinuria (defined as less than 0.4 g/day). After a seven year follow up period, protein excretion >1g/d, HTN, and serum Cr ≥1.4 mg/d developed in 33%, 26%, and 7%, respectively. Hotta et al. AJKD, 39:p493, 2002. Szeto et al. Am J Med. 110:434, 2001. Prognosis Approximately 25-30% of patients will reach ESRD at 10 years. Clinical risk factors associated with progressive disease are: HTN > 1g/d proteinuria Male gender Persistent microscopic hematuria Histologic risk factors include cellular crescents and endocapillary proliferation. Donadio et al. NEJM, 347:p738, 2002. Prognosis – Crescentic IgAN Some correlation between crescents and clinical risk factors exists (in one case series all patients who had at least 10% cellular crescents had hypertension and > 1g proteinuria). Furthermore, prospective studies have shown that 40% of patients with as little at 10% cellular crescents will progress to ESRD within 3 years. Tumlin et al. Seminars in Nephrol 24:p256, 2006. Pathogenesis IgA is an antibody that plays a critical role in mucosal immunity. IgA has two subclasses (IgA1 and IgA2) and can exist in a dimeric form called secretory IgA. It exists in two isotypes, IgA1 (90%) and IgA2 (10%): IgA1 is found in serum and made by bone marrow B cells. IgA2 is made by B cells located in the mucosa and is the major immunoglobulin found in mucosal secretions. IgA2 provides a key first line of defense against invasion by inhaled and ingested pathogens at the vulnerable mucosal surfaces. IgA1 provides a second line of defense in the serum, mediating elimination of pathogens that have breached the mucosal surface Pathogenesis Panel A – Normal IgA1 Molecule Panel B - Structure of carbohydrates O-linked to serine (Ser) or threonine (Thr) residues on IgA1. The IgA1 heavy chain contains a hinge region (a 19-residue sequence between CH1 and CH2, which consisted entirely of serine, threonine and proline). Glycosylation is restricted to the hinge region of IgA1. N-acetyl galactosamine (GalNAc) is Olinked to Ser or Thr residues. GalNAc is linked to Gal through the action of the enzyme β1,3-galactosyl transferase. Sialic acid is linked to Gal through an α2,3 link and to GalNac through an α2,6 link. Donadio et al. NEJM, 347:p738, 2002. Pathogenesis – Mesangial Deposition Although the pathogenesis of IgAN is not completely clear, it is well accepted that aberrant glycosylation pattern of IgA is involved. This is supported by the fact that in IgAN, mesangial deposits of IgA contain high concentrations of abnormally undergalactosylated IgA1. Furthermore it has been demonstrated that enzymatic removal of complex oligosaccharides from the hinge region of IgA1 antibodies from normal individuals significantly enhanced IgA deposition in the mesangium. Sano et al. NDT, 17:p50, 2002. Pathogenesis – Mesangial Deposition Leukocyte β1,3-galactosyl transferase activity is decreased in patients with IgAN which may be responsible for deficient galactosylation of IgA1. Abnormally glycosylated IgA has a higher tendency to self-aggregate and form complexes with IgG antibodies directed at epitopes in the hinge region of IgA1. Novak et al. KI 62:p465, 2006. Allen et al. NDT. 12:p701, 1997. Pathogenesis – Decreased Clearance Leukocyte Fc-receptor for IgA (CD89) is downregulated, furthermore the receptor binding site is in the CH2 domain close the hinge (possibly affected by deficient galactosylation). Altered IgA1 clearance from circulation, particularly via the hepatic asialoglycoprotein receptor (ASGPR) whose chief ligand is the terminal galactose of IgA1 (the principle site of IgA catabolism). Pathogenesis - Summary Floege et al. JASN, 11:p2395, 2000. Pathogenesis – Inflammatory Response IgA elicits a phenotypic transformation in mesangial cells in vitro, with mesangial cell proliferation and secretion of extracellular matrix component. IgA appears to stimulate the production of a variety of proinflammatory and profibrotic molecules, such as interleukin-6. Increased renal expression of TGF-beta which correlates with severity of tubulointersitial damage in IgAN. Barratt et al. Seminars in Nephrol 24:p197, 2004. Taniguchi et al. Scand J of Urol Nephrol. 33:p243, 1999. Pathogenesis – Inflammatory Response Studies have suggested that mesangial IgA probably activates C3, leading to the generation of C5b-9 (MAC), which then promotes the production of inflammatory mediators and matrix proteins by mesangial cells. Systemically, low-grade complement activation through the alternative pathway can be seen in patient’s with IgAN as well. Zwriner at al. KI, 51:p1257, 1997. Diagnosis The suspicion of IgAN is generally based upon the clinical history and laboratory data. The diagnosis can be confirmed ONLY by kidney biopsy demonstrating IgA deposition. Given the generally benign course of patients with IgAN who have isolated hematuria, biopsies are usually performed only if there are signs suggestive of more severe or progressive disease (HTN, proteinuria, elevated Cr, etc.) Diagnosis A skin biopsy, looking for IgA deposition in the dermal capillaries, has not proven to be sufficiently predictive in IgAN. Plasma polymeric IgA1 levels are elevated in 30-50% of cases, but this suggestive finding is not sufficiently specific to establish the diagnosis. Circulating IgA-rheumatoid factors and IgAimmune complexes have been tested as diagnostic markers but are not specific nor can they be reliably correlated with disease activity. Susuki at al J Clin Invest. 119:p1668, 2009. Diagnosis Increased serum levels of Gal-deficient IgA1, present in IgAN, may suggest the diagnosis. However, this assay has not been validated by testing non-IgAN patients with GN who present similarly to IgAN. Gal-deficient IgA1-specific IgG may be prove to be a clinically useful diagnostic marker as serum levels of IgG specific for Gal-def IgA1 are elevated in patients with IgAN. Susuki at al J Clin Invest. 119:p1668, 2009. Diagnosis (A) Gal-deficient IgA1 incubated with IgG from healthy controls, non-IgAN disease controls and IgAN patients. The rIgG from an IgAN patient served as a positive control. Serum IgG from IgAN patients bound more to Gal-deficient IgA1 compared with the IgG from disease controls or healthy controls. (B) The intensity of signal in each well was measured by densitometry as compared to rIgG Serum IgG from IgAN patients has significantly higher reactivity to Gal-def IgA1 compared with that from healthy (P < 0.0001) and disease controls (P < 0.0001). Serum IgG from 54 of the 60 patients with IgAN showed values greater than the 90th percentile of the values for healthy controls. (C) ROC for serum IgG binding to Galdeficient IgA1. The area under the curve is 0.9644. These data indicate a sensitivity of 88.3% and a specificity of 95.0% (D)/(E) The intensity of IgG binding to Galdeficient IgA1 correlated with urine Pr/Cr ratio as well as with urinary IgA-IgG immune complexes. Susuki at al J Clin Invest. 119:p1668, 2009 Histology The major finding on light microscopy is mesangial proliferation and matrix expansion (arrows) that can be focal, but more often seen diffusely. Histology Light microscopy of a glomerulus from a patient with IgAN showing increased mesangial matrix and cellularity. Histology A. B. C. D. E. F. H&E demonstrating mesangial hypercellularity and matrix expansion. H&E with mesangial cell hypercellularity and focal area of endocapillary proliferation (bold arrow). H&E demonstrating diffuse endocapillary proliferation and mesangial hypercellularity. H&E -silver demonstrating cellular crescent with partial collapse of glomerular tuft. H&E demonstrating diffuse endocapillary proliferation and fibrinoid necrosis. Silver stain demonstrating crescent and focal glomerular tuft adhesion to Bowman’s capsule. Tumlin et al. CJASN. 2:p1054, 2004. Histology Segmental crescents are relatively common, although they may be missed by sampling error if only a few glomeruli are obtained. (as mentioned earlier as many of 30% of IgAN on biopsy may have crescents). Although there is usually little or no glomerulosclerosis on initial biopsy, patients may eventually develop glomerulosclerosis, by which time they have clinically advanced disease (i.e. decreased GFR and increased proteinuria). Histology - Immunoflourescence IF demonstrates prominent, globular deposits of IgA (often accompanied by C3 and IgG) in the mesangium and, to a lesser degree, along the glomerular capillary wall. Histology - IF IF demonstrating large, globular mesangial IgA deposits. Note that the capillary walls are not outlined, since the deposits are primarily limited to the mesangium. Histology – Electron Microscopy EM typically reveals electron-dense deposits that are primarily limited to the mesangium, but may also occur in the subendothelial and subepithelial spaces. The number and size of these deposits generally correlates well with the severity of changes seen on light microscopy. Histology - EM Low power electron micrograph in IgAN. The primary finding is electron dense deposits that are limited to the mesangial regions (D). The glomerular basement membrane (GBM) is normal and there are no glomerular capillary wall deposits. Histology - EM Higher power EM with significant expansion of mesangial matrix and presence of large mesangial dense deposits (arrow). Histology – Oxford Classification A consensus on the pathologic classification of IgA nephropathy has been developed by the International IgAN Network with the Renal Pathology Society: Mesangial hypercellularity: 0 = < 4 mesangial cells are present per mesangial area 1 = 4-5 mesangial cells are present per mesangial area 2 = 6-7 mesangial cells are present per mesangial area 3 = >8 mesangial cells are present per mesangial area. Scores for all glomeruli are averaged and the resulting assigned hypercellularity score is either M0 if the mean score is less than 0.5 or M1 if the mean score is greater than 0.5. Segmental glomerulosclerosis: S1 = any part of the glomerular tuft is involved in sclerosis S0 if no segmental glomerulosclerosis is present. Endocapillary hypercellularity: E1 = if hypercellularity is present within the capillary lumina resulting in narrowing. E0 if no hypercellularity is present within lumina. Tubular atrophy/interstitial fibrosis — The percentage of the cortical area involved by tubular atrophy or interstitial fibrosis. A score of T0, T1 or T2 is given if the percentage of involved cortical area is 0-25; 26-50 or >50 percent, respectively. Histology – Haas Classification Based on the histological features of 244 cases of IgAN over a 14 year period at one institution: Class I (39 cases): minimal or no mesangial hypercellularity, without glomerulosclerosis Class II (18 cases): FSGS without active cellular proliferation Class Ill (110 cases): focal proliferative GN Class IV (42 cases): diffuse proliferative GN Class V (35 cases): any biopsy showing > 40% globally sclerotic glomeruli and/or > 40% estimated cortical tubular atrophy or loss. Haas et al. AJKD, 29:p829, 1997. Histology – Haas Classification Haas et al. AJKD, 29:p829, 1997. Histology – Haas Classification Haas showed a statistically significant correlation between histologic subclass and renal survival, with an order I, II (greatest survival) > Ill > IV, V. Histology – Haas Classification with Crescents Haas also reported the probability of renal survival when crescents were present in Haas subclass III and IV. Treatment Patients with isolated hematuria, no or minimal proteinuria, and a normal GFR are typically not treated (and often not biopsied), unless they have evidence of progressive disease such as increasing proteinuria, blood pressure, and/or serum creatinine. Treatment – ACE/ARB Patients with persistent proteinuria (500-1000 mg/day), mildly reduced GFR that is not declining rapidly, and only mild to moderate histologic findings on renal biopsy are traditionally managed with ACE/ARB. In one trial, 44 patients with proteinuria (≥0.5 g/day, mean 1.9 g/day) and a Cr ≤1.5 mg/dL at baseline were randomly assigned to either enalapril or antihypertensive agents other than ACE inhibitors or ARBs. At follow-up of about six years, renal survival, defined as <50% increase in the Cr, was significantly more likely in the enalapril group (92% versus 55%). A significant decrease in proteinuria was only observed in the enalapril group (2 g/day at baseline to 0.9 g/day). Blood pressure control was similar in the two groups. Praga et al. JASN, 14:p1578, 2003. Treatment - Immunosuppressives Patients with more severe or rapidly progressive disease (e.g. nephrotic range proteinuria or proteinuria persisting despite ACE/ARB therapy, rising serum creatinine, and/or renal biopsy with more severe histologic findings) may benefit from immunosuppressive therapy in addition to ACE/ARB slow disease progression. Treatment – Glucocorticoids Glucocorticoid therapy is recommended in patients with clinical and histologic evidence of active inflammation (eg, hematuria and/or proliferative or necrotizing glomerular changes). The potential benefit of glucocorticoid therapy alone in IgAN has been examined in uncontrolled studies, retrospective observations, and a few relatively small, randomized controlled trials. The applicability of these trials to current practice is unclear, since most trials predated widespread use of ACE/ARBs. Treatment - Glucocorticoids A prospective trial from Italy included 86 adults with proteinuria (1 to 3.5 g/day) and at most mild renal insufficiency (median serum creatinine 1 mg/dL). The patients were randomly assigned to supportive therapy alone, or glucocorticoids (1.0 gram of IV methylprednisolone for 3 consecutive days at the beginning of months 1, 3, and 5, combined with 0.5 mg/kg of oral prednisone given on alternate days for 6 months). At five and ten years, the glucocorticoid treated patients had a markedly lower incidence of the primary end point, which was a doubling of Cr (2% vs. 21% at five years and 2% vs. 30% at 10 years). The effect of ACE/ARB was not assessed. Pozzi et al. Lancet, 353:p883, 1999. Rx – Combined Immunosuppressive Therapy Combined immunosuppressive therapy is recommended in patients with more severe active disease as defined by a more rapidly progressive clinical course and/or histologic evidence of severe active inflammation (eg, crescent formation). Several trials have suggested a possible benefit from combined immunosuppressive therapy in these patients, however, most did not include a comparison group treated with prednisone alone. Similarly, the studies were primarily performed prior to the widespread use of aggressive ACE/ARB therapy. Rx – Combined Immunosuppressive Therapy In a randomized control trial of 38 patients with rapidly progressive disease (without crescents) combined treatment with prednisone and oral cyclophosphamide for 3 months, followed by azathioprine for two years or more, resulted in better preservation of renal function. At 2, 3, 4, and 5 years renal function was preserved in 82%, 82%, 72% and 72% of treatment patients, respectively, when compared with 68%, 47%, 26%, and 6% in controls who received placebo. A lower degree of proteinuria was also observed in treatment group compared to controls. Ballardie et al. JASN, 13:p142 2002. Treatment – Crescentic IgAN Uncontrolled reports in patients with IgAN causing crescentic RPGN suggest possible benefit from regimens similar to those used in other forms of crescentic GN: IV pulse methylprednisolone followed by oral prednisone, IV or PO cyclophosphamide, and/or plasmapheresis. Treatment – CIgAN – Roccatello et al One report evaluated the efficacy of combination therapy (steroids, oral cyclophosphamide and plasmapheresis) in six patients with crescentic glomerulonephritis due to IgAN (entry required >40% crescents). After two months of therapy, there was substantial clinical improvement characterized by reductions in Cr and proteinuria. However, repeat renal biopsy at 2 months showed persistence of crescents in all patients and 50% of patients had progressive disease after therapy was discontinued. Roccatello et al. NDT, 10:p2054, 1995. Treatment – CIgAN – Tumlin et al A more prolonged course of aggressive immunosuppressive therapy was evaluated in 12 patients with CIgAN who had a mean serum Cr of 2.7 mg/dL and proteinuria of 4 g/day at baseline. The treatment regimen consisted of the following: Pulse methlyprednisolone (15 mg/kg/d for 3 days) PO prednisone ○ 1 mg/kg/d for 60 days, then slow taper with all patients on 10 mg/d at the time of repeat biopsy Monthly IV cyclophosphamide (0.5 g/m2) for six months. Tumlin et al. NDT, 18:p1321, 2003. Treatment – CIgAN – Tumlin et al After the six month course, there was significant improvement in the serum Cr concentration (from 2.7 to 1.5 mg/dL) and in proteinuria (from 4 to 1.4 g/day). Repeat biopsy at six months revealed the absence of cellular crescents and endocapillary proliferation in all patients. Throughout a three-year follow-up, all patients continued prednisone (0.15 mg/kg per day), and the blood pressure was controlled to a goal of <130/70 mmHg with ACE inhibitors and other agents as needed. Compared with 12 untreated historic controls (matched for age, gender, baseline serum Cr and histologic severity), the incidence of ESRD at three years was significantly lower in the treated group (1 of 12 = 8% versus 5 of 12 = 42%). Tumlin et al. NDT, 18:p1321, 2003. Transplant IgAN Recurrence In 1975, only 7 years after his initial description of the entity of IgAN, Berger et al reported the first case of recurrent IgA in a renal allograft. The recurrence of IgA in transplants among patients with IgAN in their native kidneys occurred in 40-60% of cases when protocol biopsies were performed. In one study of 240 recipients, after a mean follow up of 5 years, 13% of exhibited recurrence related graft dysfunction with 5% losing the graft secondary to recurrent IgAN. Wang et al. AJKD, 38:p588, 2001. CIgAN in Transplants – Kowalewska et al A study reviewed 2959 renal biopsies over a period of 14 years and found 33 cases of glomerulonephritis with crescents (1.1%). Of these 33 cases, 8 had the diagnosis of IgAN (0.2% of total). 6 of the 8 cases were the result of recurrent IgAN, and 2 cases were presumptive de novo IgAN. 6 patients had 10-30% crescents in the glomeruli, the 2 remaining cases about 7%. Despite intensified therapy, 4 patients developed renal failure and returned to hemodialysis within 1 year. Kowalewska et al. AJKD, 45:p167, 2005. CIgAN in Transplants – Tang et al Another retrospective study reviewed 1742 allograft biopsies over a period of 9 years at a Chinese University hospital and found 18 cases with crescent formation, of which 10 patients (0.5% of total) were diagnosed with recurrent or de novo IgAN. 9 cases progressed to ESRD and returned to dialysis after 6 to 36 months. Tang et al. Renal Failure. 30:p611, 2008. CIgAN in Transplants – Mousson et al Over a 15 year period, 42 patients with biopsy proven IgAN received kidney transplants, they were followed for a mean 9 year period and had sequential allograft biopsies. In their native kidneys, 5 patients (12%) had more then 20% crescents, and only 2 (5%) had more than 50% of the glomeruli involved. 52.4% of recipients showed recurrent IgA deposits in their grafts. The 2 patients with diffuse crescentic IgAN in their native kidneys, experienced acute graft dysfunction at 15 and 47 months post transplant. No crescentic proliferation was observed during follow up in any other case. The authors suggest that only diffuse crescentic IgAN in the native kidneys was associated with occurrence of crescents in the kidney transplants. Mousson et al. Transplantation Proceedings, 39:p2595, 2007. The End