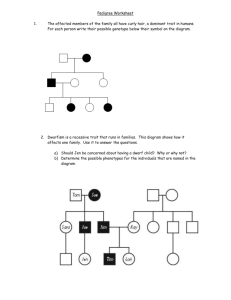
Stature in adolescent twins
advertisement

Genetics of complex human disease traits. Daniel T . OХConnor, M.D. Department of Medicine. Nopm-252. First year curriculum in human genetics. Wed Apr 1, 2009. CMME-2047. PURPOSE. In the ne xt two hours, we plan to cover the role of he redity and genes in very c ommon, n on-Mendelian traits that are frequent ly s een by primary c are physicians. We will illustrate how w e establish the role of heredity on any trait, and then methods to position the particular genes that influence such a trait. WHAT IS A ТCOMPLEX TRAITУ? Trait = phenotype. Disease causation/etiology/origin: The old conund rum o f: ТNature (heredity) versus nurture (environment)У. How to solve this riddle: Family/pedigree or twin studies (see below). Frequency. Most (>95%) of the disease encount ered in i nternal medicine, family medicine, p ediatrics, neurology, or psychiatry is c omplex, and its origin is not well understood. Not clearly c ompletely hereditary (Mendelian) or environmental. Read: Hy pertension, coronary artery disease, arrhythmia, stroke, aneurysm , asthma, COPD, diabetes, obesity, schizophrenia, bipolar disorder . . . Multifactorial: Genes, environment, g ene-by-environment interactions. Non-Mendelian. Mendelian: Gene Trait (1:1; high penetrance ~100%). Non-Mendelian: Only partial penetrance. Some people with the gene do not get the t rait. Some people who do not ha ve the g ene still get the t rait. Bimodality: Hallmark of a major gene effect on a quantitative trait. Bimodality: Hallmark of a major gene effect on a quantitative trait. Polygenic Traits 1 Gene 2 Genes 3 Genes 4 Genes 3 Genotypes 3 Phenotypes 9 Genotypes 5 Phenotypes 27 Genotypes 7 Phenotypes 81 Genotypes 9 Phenotypes 3 3 2 2 1 1 0 0 7 6 5 4 3 2 1 0 20 15 10 5 0 Complex Trait Model Linkage Marker Gene1 Linkage disequilibrium Linkage Association Mode of inheritance Gene2 Disease Phenotype Individual environment Common environment Gene3 Polygenic background APPROACHES TO COMPLEX TRAITS. (Genetic) epidemiology. Demographics (age, sex, ethnicity, geography, fam hx). ТRiskУ (susceptibility) factors (see above). Relative risk (RR): Given a risk factor, what is the increase in trait prevalence? Estimator of RR: Odds ratio (OR). Given a risk factor, OR = (have trait/do not have trait). OR + /- confidence interval (+/-95% CI) v ersus reference (no risk) =1. No risk: RR or OR = 1. OR + /- CI >>1 Risk (susceptibility) factor. OR + /- CI <<1 Protective factor. Test by 2 (2x2 contingency table). CHGA genetic variation: Risk factor for hypertensive ESRD in blacks Estimator of RR: Odds ratio (OR). Given a risk factor, OR = ( have trait/do not have trait). J Am Soc Nephrol. 2008 Mar;19(3):600-14. Chromogranin A polymorphisms are associated with hypertensive renal disease. Salem RM, Cadman PE, Chen Y, Rao F, Wen G, Hamilton BA, Rana BK, Smith DW, Stridsberg M, Ward HJ, Mahata M, Mahata SK, Bowden DW, Hicks PJ, Freedman BI, Schork NJ, O'Connor DT. ТIntermediateУ (risk) traits (phenotypes). Int ermediate in time and mechanism. Bridging genotype and ultimate, late disease trait. Ideally: Greater h 2, earlier penetrance. If biochemical assays: ТBiomarkersУ. Gene “Intermediate” in time and causality Cardiorenal disease trait (later life) Mechanism Twins (fixed at conception) Twins: window into heritability (h2) of any phenotype Monozygotic (MZ, identical) twins: Billy and Benny . . . VP = VG + VE h2 = VG/VP = 2(RMZ - RDZ) Source: Guinness Book of World Records. Total mole count for MZ and DZ twins DZ twins - 199 pairs, r = 0.60 400 400 300 300 Twin 1 Twin 1 MZ twins - 153 pairs, r = 0.94 200 200 100 100 0 0 0 100 200 300 Twin 2 400 0 100 200 300 Twin 2 400 2 Heritability (h ) of traits in human twin pairs 100 87 +/-2 91 +/-1 Physiological Biochemical 71 +/-4 60 70 +/-4 67 +/-4 59 +/-6 2 40 47 +/-7 50 +/-6 43 +/-7 Trait Plasma epi Plasma norepi Baroreflex down Cardiac output DBP SBP Weight Height 0 Baroreflex up 33 +/-9 20 SVR Heritability (h ), mean +/- SEM 80 Physical Family studies (twin pairs, pedigrees). Fam hx as a risk factor. In 1st degree relatives: Parents, siblings. Heritability (h2): Fraction of trait variance accounted for by genetic variance. VP = VG + VE h2 = VG/VP Estimate h2 from twin pair (or pedigree) studies: Type of twin pair Allele sharing across the genome. MZ = monozygotic = identical. 100% DZ = dizygotic = fraternal ~50% (on average), like any sib pair Quick-and-dirty algorithm: h2 = VG/VP = 2(RMZ – RDZ) Family history as a risk factor for complex traits. Family history West J Med. 1984 Dec;141(6):799-806. Understanding genetic and environmental risk factors in susceptible persons. Williams RR. (Genetic) ТlinkageУ: Co-segregation of marker and trait. Versus independent segregation: Different chromosome, or far apart on s ame chromosome. Thus, marker and trait loci are within ~50 cM of ТgeneticУdistance. cM: Just count the meiotic recombinants v ersus non-recombinants cM = (R ecombinant meioses / Total m eioses)*(100) Lower cM means clos er (marker and trait loci) Calibration. cM (genetic/meiotic distance) vs Mb (physical distance): 1 cM = ~1 Mb (actually ~ 0.5-2.0). Markers to span the genome for linkage: 3000 cM/50 cM = 60 fully i nformative (heterozygosity) markers in theo ry In practice: ~400-800 highly i nformative markers (multiallelic microsatelli tes) ~2000-10,000 less informative markers (biallelic SNPs) Linkage = Meiotic co-segregation A3A4 A1A2 A1A3 A1A2 A1A4 A2A4 A3A4 A2A3 A3A2 Marker allele A1 cosegregates with dominant disease Linkage Markers… Thomas Hunt Morgan – discoverer of linkage Idiosyncratic features of genetic linkage (= meiotic co-segregation). Units. Metric = meiotic recombination (~50 meioses/generation). Units of genetic distance = recombination during meiosis (cM). cM = (recombinant meioses/total meioses)*100 E.g.: [8/(8+86)]*100 =(8/94)*100 = 8.5 cM 1 cM ~ 1 Mb Range ~0.5-2.0 Varies by species, sex, chromosomal region (meiotic “hot spots”) Significance. “LOD” scores. LOD = Log10 of the odds ratio for linkage Odds ratio: Co-segregation (marker and trait) Not Significant: LOD >3.0 (i.e., odds ratio > 1000/1) Why 3.0? ~50 “linkage groups” (meiotic breaks/generation), target =0.05. 1/50*1/20=1/1000. Genetic linkage: Meiotic recombination distance in cM cM = (recombinant meioses/total meioses)*100 [8/(8+86)]*100 =(8/94)*100 = 8.5 cM 1 cM ~ 1 Mb Range ~0.5-2.0 Varies by species, sex, chromosomal region (meiotic “hot spots”) Mahata SK, Kozak CA, Szpirer J, Szpirer C, Modi WS, Gerdes HH, Huttner WB, O'Connor DT. Dispersion of chromogranin/secretogranin secretory protein family loci in mammalian genomes. Genomics. 1996 Apr 1;33(1):135-9. Mouse SBP crosses: “Genome scan” linkage. Wright FA, O'Connor DT, Roberts E, Kutey G, Berry CC, Yoneda LU, Timberlake D, Schlager G. Genome scan for blood pressure loci in mice. Hypertension. 1999 Oct;34(4 Pt 1):625-30. LOD score: Log10 of the O R for linkage (marker, trait). Log10 (non-recombinant s / total m eioses). ТSignificantУ LOD > 3 (i.e., OR > 1000). Why? ~50 linkage groups (meiotic breaks/generation), target =0.05. 1/50*1/20=1/1000. Quantitative trait, non-parametric: Regression. Plot: Y = (Trait difference)2 within each sib pair of DZ twin pair. X = Alleles shared IB D (identical by descent), 0 12. Linkage is about loci, not particular alleles! Terminology: ТQTLУ(Quantitative Trait Locus). LOD >3 peak for that t rait. Problems: Need families based on probands (but r elatives may not be available). Ideal for Mendelian disorders, but limited success for c omplex traits. Meiosis yi elds v ery large chromosomal s haring blocks by first-degree relatives, but this can give rise to low s patial r esolution of involve d genes (very broad LOD peaks). Genetic linkage: What the data (marker, trait) look like. J Clin Invest. 1996 May 1;97(9):2111-8. Quantitative trait locus mapping of human blood pressure to a genetic region at or near the lipoprotein lipase gene locus on chromosome 8p22. Wu DA, Bu X, Warden CH, Shen DD, Jeng CY, Sheu WH, Fuh MM, Katsuya T, Dzau VJ, Reaven GM, Lusis AJ, Rotter JI, Chen YD. Genetic linkage: What the data (marker, trait) look like. J Clin Invest. 1996 May 1;97(9):2111-8. Quantitative trait locus mapping of human blood pressure to a genetic region at or near the lipoprotein lipase gene locus on chromosome 8p22. Wu DA, Bu X, Warden CH, Shen DD, Jeng CY, Sheu WH, Fuh MM, Katsuya T, Dzau VJ, Reaven GM, Lusis AJ, Rotter JI, Chen YD. (Allelic) association: Marker Trait. ТCandidateУ gene: Specific prior hypothesis for one gene (e.g., hemoglobin thalassemia). First, systematic polym orphism discovery (by r esequencing). Define haplotype/LD (linkage disequilibrium) blocks. Blocks ~3-50 kbp in unrelated individuals. Origin is ancestral meiotic recombination. Vary in l ength by ethnicity. Assay S NPs (or SNP haplotypes) in phenot yped individuals. Haplotypes: Inf er from individual diploid genotypes by probability. Dichotomous trait: Disease cases v ersus c ontrols. Analyz e by 2 on 3x2 contingency table: 3 (diploid genotype classes; e.g., A/A, A/G, G/G ) x 2 (case/control) contingency t ables. Continuous trait: Analyz e by ANOVA, with gen otype (diploid genotype classes; e.g., A/A, A/G, G/G ) as i ndependent variable. Association derives effects of particular alleles, not just loci. Advantage: Unrelated individuals (do not need families). Problems: Population stratification (artifactual association as a result of allele frequency differences across populations). The catecholamine biosynthetic pathway. T Flatmark. Regulation of catecholamine biosynthesis. Acta Physiol Scand 168:1-17, 2000. Tyrosine hydroxylase promoter haplotype 2: Pleiotropy. Coordinate effects on both catecholamine excretion and stress blood pressure response in twins 18 2 Norepinephrine h =49.6+/-6.7%, p=0.0001* Haplotype 2 on norepinephrine: p=0.0125* , 4.06% variation explained Change in DBP during cold stress, mmHg 2 16 DBP h =32+/-8%, p=0.0003* Haplotype 2 on DBP: p=0.0004* , 3.73% variation explained Pleiotropy: bivariate likelihood ratio test 2 =14.2, p=0.0002* Haplotype 2 n=2 copies (n=32 individuals) 14 12 Haplotype 2 n=1 copy (n=164 individuals) 10 8 Haplotype 2 n=0 copies (n=131 individuals) 6 2.4 10 4 2.6 10 4 2.8 10 4 3 104 3.2 10 4 3.4 10 4 3.6 10 4 Norpinephrine excretion, ng/gm Figure 8: TH haplotypes in vivo Tyrosine hydroxylase regulatory polymorphism: Interaction of genotype and sex to affect DBP 100 95 90 2-way ANOVA: (Covariates: age, BMI) Overall F=12.4, p<0.001* Genotype F=1.30, p=0.273 Sex F=31.6, p<0.001* Genotype * Sex F=3.14, p=0.044* C-824T explains 3.4% of DBP variance Alleles: C=61%, T=39% Male DBP Female DBP Males alone: Genotype F=3.12, p=0.045* Females alone: Genotype F=0.015, p=0.985 2 DBP, mmHg (mean +/- SEM) HWE: =0.54, p=0.46 85 80 75 70 85.3 +/-2.6 (n=75) 82.8 +/-1.3 (n=285) 78.3 +/-1.5 (n=234) 74.9 +/-1.5 (n=233) 74.9 +/-1.2 (n=329) 73.2 +/-2.1 (n=110) 65 60 C/C C/T T/T Tyrosine hydroxylase (TH) promoter C-824T diploid genotype Disease trait Mechanism Gene Biochemical trait Physiological trait Time Hypertension: “Intermediate” phenotypes and candidate genes. Tyrosine hydroxylase C-824T Catecholamines Baroreceptor function Stress blood pressure Hypertension Figure 7: Intermediate phenotypes GWAS (Genome Wide Association Study). Hypothesis: Common disease/Common Variant. Markers to span the g enome for association: ~3.3 Gbp (1 Gbp = 109 bp) genome / 500K SNPs = ~ 6000 bp (~6 kbp). The only va riants spaced this closely (i.e., the common) are SNPs. Spacing based on H apMap <www.hapmap.org> and LD (li nkage disequilibrium) blocks. HapMap: 270 people world-wide typed at ~4 million SNPs across the genome. Within an LD block, SNPs are highly c orrelated (r2 ~0.6-1.0). Try to ТtagУeach LD block across the genome. Advantage: Unrelated individuals (do not need families). Problems: Population stratification (artifactual association as a result of allele frequency differences across populations). Statistical c hallenges to GWAS: Many LD blocks tested, modified target p=5x10-7. Solution: Replication in an independent sample, for joint ( multiplicative, ) probability. Haplotype: Ordered array of alleles along a single chromosome. Biallelic SNPs (single nucleotide polymorphisms). Typically “transitions”: Purine Purine (G A) Pyrimidine Pyrimidine (C T) Chromosome T C A G C T G A Paternal Maternal pter qter 5’ 3’ “Linkage disequilibrium” (LD): Local, marker-on-marker locus Equilibrium = randomness (no correlation, r2=0) Disequilibrium = non-random (correlated, r2>0) Marker-on-trait locus: Mapping tool 0.0 0.9 Paternal Maternal 0.5 0.5 0.9 0.9 0.0 0.9 0.9 r2 0.9 T A C G T A C G C G T C C G T A 5’ 3’ Ancestral (shared) Meiotic recombination Biallelic SNPs Linkage disequilibrium (LD). Marker trait Marker marker In population genetics, linkage disequilibrium is the non-random association of alleles at two or more loci. Linkage disequilibrium describes a situation in which some combinations of alleles or genetic markers occur more or less frequently in a population than would be expected from a random formation of haplotypes from alleles based on their frequencies. Non-random associations between polymorphisms at different loci are measured by the degree of linkage disequilibrium (LD). The level of linkage disequilibrium is influenced by a number of factors including the rate of meiotic recombination (crossovers) and the rate of mutation. HapMap: View variation patterns Triangle plot shows LD values using r2 or D’/LOD scores in one or more HapMap population The International HapMap Project (Identification of SNPs that ‘tag’ haplotypes within blocks) Daly, M.J., Rioux, J.D., Schaffner, S.F., Hudson, T.J. and Lander, E.S. (2001). High-resolution haplotype structure in the human genome. Nature Genet. 29: 229-232. Linkage disequilibrium (LD) “blocks” on human chromosome 14q32 100 kbp displayed, from <www.HapMap.org> 3 shortrange (~30 kbp) LD blocks No long-range (~100 kbp) LD Gene-by-Environment (GxE) interaction probed by MZ twin intra-pair trait differences: HDL-cholesterol effect of T-cadherin (CDH13, novel adiponectin receptor) genetic variation revealed by dense, genome-wide profiling in 1662 MZ pairs p=8.5x10-8 QuickTime™ and a TIFF (LZW) decompressor are needed to see this picture. 100 kbp Region of the genome around around SNP rs9941339 in CDH13 (T-cadherin = novel adiponectin receptor) on 16q24 associated with intra MZ pair differences in HDL cholesterol (GWAS in n=1662 MZ pairs). Black points represent SNPs genotyped in the study and gray points represent SNPs whose genotypes were imputed. In middle panel, red line shows the fine-scale recombination rate (centimorgans per Mb) estimated from Phase II HapMap and the black line shows the cumulative genetic distance (in cM). Association p=8.5x10^-8. Common disease / Rare variant hypothesis. Or, accumulation of excess rare variants: Non-synonymous (amino acid replacement) cSNPs. Techno logy: Extensive re-sequencing in large numbers of cases vs controls. Typically resequence a ТpathwayУin population tr ait extreme individuals (boost statistical power). Analys es: Summed 2, cases versus controls. C omputational ass essment of amino acid chang e functionality (SIF T, PolyPhen). Ultimately functional studies. Accumulation of deleterious rare amino acid substitution variants at extremes of human body mass index (BMI) Ahituv N, Kavaslar N, Schackwitz W, Ustaszewska A, Martin J, Hebert S, Doelle H, Ersoy B, Kryukov G, Schmidt S, Yosef N, Ruppin E, Sharan R, Vaisse C, Sunyaev S, Dent R, Cohen J, McPherson R, Pennacchio LA. Medical sequencing at the extremes of human body mass. Am J Hum Genet. 2007 Apr;80(4):779-91. ТDrillin g down to the ТQTNУ (Quantitative Trait Nucleotide)У. Problem: Even after successful allelic association, the lower limit of resolution is the LD block (~3-50 kbp). So where, within that LD block, is the causative variant? Solution: Studies of the putative responsible variant in a sys tem outside of the hum an organism: in vitro (test-tube enzy mology), in cella (transfection into cultured cells) , or in viv o (transgenic mice). Drilling down to the “QTN” (“Quantitative Trait Nucleotide”) • Haplotype “block” is the lower limit of resolution of marker-on-trait mapping. • Switch to studies of associated variants: – In cella. E.g., transfected/expressed variants. – In vitro. E.g., kinetic properties of variants. – In vivo: transgenic mice (BAC haplotype variant expression on knockout background). Positional candidate genetic loci. “Positional candidate” locus Wong C, Mahapatra NR, Chitbangonsyn S, Mahboubi P, Mahata M, Mahata SK, O'Connor DT. The angiotensin II receptor (Agtr1a): functional regulatory polymorphisms in a locus genetically linked to blood pressure variation in the mouse. Physiol Genomics. 2003 Jun 24;14(1):83-93. “Positional candidate” locus Wong C, Mahapatra NR, Chitbangonsyn S, Mahboubi P, Mahata M, Mahata SK, O'Connor DT. The angiotensin II receptor (Agtr1a): functional regulatory polymorphisms in a locus genetically linked to blood pressure variation in the mouse. Physiol Genomics. 2003 Jun 24;14(1):83-93. “Positional candidate” locus Wong C, Mahapatra NR, Chitbangonsyn S, Mahboubi P, Mahata M, Mahata SK, O'Connor DT. The angiotensin II receptor (Agtr1a): functional regulatory polymorphisms in a locus genetically linked to blood pressure variation in the mouse. Physiol Genomics. 2003 Jun 24;14(1):83-93. Promoter variant characterization Transcription Transcription Promoter/reporter plasmid (pGL3-Basic) Promoter/reporter plasmid (pGL3-Basic) Transfection Luciferase transcription Luciferase translation Nucleus Cytosol Chromaffin cell Cell lysis Firefly luciferase enzymatic activity assay “Positional candidate” locus Wong C, Mahapatra NR, Chitbangonsyn S, Mahboubi P, Mahata M, Mahata SK, O'Connor DT. The angiotensin II receptor (Agtr1a): functional regulatory polymorphisms in a locus genetically linked to blood pressure variation in the mouse. Physiol Genomics. 2003 Jun 24;14(1):83-93. ТRiskУ allele (gene) versus ТmodifierУ allele (gene). Risk allele: Allele that increases risk/susceptibility for disease, in longitudinal s tudies. Example: CFTR (Cl - channel) F508 Cystic fibrosis. Modifier allele: Allele that influences course of the d isease, once that disease has occurred. Example: KCNMB1 Glu65Lys Rapid progression of renal dysfunction. Genomics in Dz risk: Two kinds of Gene-by-Environment interactions. Gene: Susceptibility Gene: Modifier Stable Dz Exposure Switch Rapid decline Switch Not Initiation (case/control study) Time (decades) Drug Outcome (longitudinal study) KCNMB1 Glu65Lys effect on rate of GFR decline in hypertensive nephrosclerosis (NIDDK AASK) 1 Overall: p<0.001 KCNMB1: p=0.030 Covariate: Pro_cr p<0.001; Mb_GFR p<0.001; BP goal p=0.196; Drug p=0.065 Permutation test: p=0.017 Alleles: E=95%, K=5%. HWE: p=0.552 0 -1 -1.88+/-0.08 (704) -2.46+/-0.25 (72) -2 -3 E/E1 2 E/K+K/K KCNMB1 E65K genotype Figure 3A KCNMB1 Glu65Lys predicts long-term loss of renal function in hypertensive nephrosclerosis (NIDDK AASK) Cumulative survival Log rank p=0.0190 Event: ESRD requiring dialysis or doubling of serum creatinine E/E E/K+K/K E/K+K/K censored E/K+K/K E/E E/E censored Months after enrollment Figure 3B KCNMB1 Glu65Lys: Hypothesis for effect on GFR VOCC K+ Ca2+ K+ Ca2+ VOCC BK BK Wild-type Variant b 1 subunit (65Lys) b 1 subunit (Glu65) 1 subunit K+ K+ Contracted mesangial or smooth muscle cell Higher flux Lower flux 1 subunit Relaxed mesangial or smooth muscle cell : Ca2+ ions : Inhibition Figure 4 WHAT HAVE WE LEARNED? Ґ ТComplexУ trait definition. Ґ Risk (s usceptibility) factors. Ґ ТIntermediateУ(risk) traits (phenotypes). Ґ Family/twin s tud ies and he ritability. Ґ Genetic li nkage: Co-segregation of marker and tr ait. Ґ Allelic association: Marker trait. Ґ ТCandidateУgene. Ґ GWAS (Genome Wide Association Stud y). Hypothesis: Common disease/Common Variant. Ґ Common d isease / Rare variant hypothesis. Ґ ТDrilli ng down to t he ТQTNУ (Quant itative Trait Nucleotide)У. Ґ ТRiskУ allele (gene) versus ТmodifierУallele (gene). SAMPLE QUESTIONS. The idea has been to develop a feeling for concepts, rather than details. The responses I w ould make are indicated by Т*Уor ТУ. Which of these phenot yped datasets w ould allow y ou to establish heritability (h2) of a trait? Pick all t hat a re correct. Twin pairs * Nuclear families (parents, siblings, children) * Extended pedigrees (i ncluding second degree relatives) * Random sample of the US urban population taken from Chicago, Illi nois. Probands from a case/control s tudy of asthma. Which of the f ollowing data on fa milies and disease would allow estimation of heritability? Pick one. Street and city address wi th zip code. Trait measurements in MZ and DZ twin pairs. * Match the gene-finding method wi th the type of information discovered. Method Inf ormation Linkage Loci Association Alleles What biological process gives rise to haplotype (LD) blocks? (Pick one). Meiotic recombination * X-irradiation Cosmic rays Site-directed mutagenesis Which of the f ollowing epidemiologic parameters can he lp to estimate whether heredity in fluences a trait? Pick one. Familial relative risk, estimated by odds ratio * Family i ncome Federal and state income tax r eturns College tuition payments If you wished to scroll through the e ntire genome searching fo r an allelic ass ociation to a complex trait in cases ve rsus c ontrols, how many SNP genotypes w ould you need to type, in order to capture the correlated LD blocks across the genome? Pick one. 5K 50K 500K * 5 million