disorders of the systemic blood pressure. hypertension and
advertisement

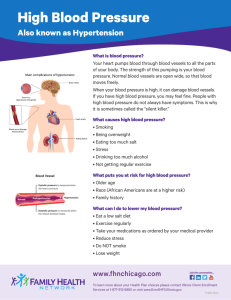
DISORDERS OF THE SYSTEMIC BLOOD PRESSURE. HYPERTENSION AND HYPOTENSION CONTROL OF MEAN ARTERIAL BLOOD PRESSURE Blood pressure is proportional to cardiac output and peripheral vascular resistance. (Ohm’s law). BP = CO×PR where, for the cardiovascular system, BP is the mean arterial blood pressure, CO is the cardiac output (which equals HR × SV). HR is the heart rate in beats per minute and SV is the stroke volume in milliliters of blood pumped per beat PR is the total peripheral resistance. Cardiac output is highly dependent on blood volume, itself greatly influenced by the whole body sodium homeostasis. Peripheral vascular resistance is determined mainly at the level of the arterioles and is affected by neural and hormonal factors. The variable regulated by the body, and usually measured clinically, is the systemic arterial blood pressure (BP). Blood pressure control depends on sensors that continually measure blood pressure and send the information to the brain. The brain integrates all incoming information and responds by sending efferent (outgoing) stimulation to the heart and vasculature through the autonomic nerves. Various hormones and locally released chemical mediators add to the control of blood pressure. Sensors Blood pressure is continually monitored by sensors called baroreceptors (pressure receptors). There are baroreceptors in the carotid artery (in the neck) and in the aortic arch where the aorta leaves the heart; these sensors are called the carotid and aortic baroreceptors, respectively. There are baroreceptors located in the arterioles supplying the kidney nephrons. Receptors in both atria and in the pulmonary artery also respond to changes in pressure. Because the atrial and pulmonary artery receptors are in low-pressure areas of the vasculature, they are called low-pressure receptors. All baroreceptors act as stretch receptors that respond to changes in blood pressure. Their stretch increases with increased blood pressure. This stretch increase causes afferent neurons receiving information from the receptors to increase their rate of firing. These neurons travel to the brain and innervate its cardiovascular center. A decrease in blood pressure decreases the stretch of the baroreceptors, which reduces the firing of the afferent nerves innervating the cardiovascular center. Integrating Center for the Control of Blood Pressure The cardiovascular center in the brain is part of the reticular formation and is located in the lower medulla and pons. The signals concerning blood pressure are integrated here. If a change in blood pressure has occurred, the cardiovascular center activates the autonomic nervous system, leading to changes in sympathetic and parasympathetic stimulation to the heart and sympathetic stimulation to the entire vascular system. Resistance of the vasculature is altered and blood flow and blood pressure are affected. Efferent Neural Innervation of the Vascular System Sympathetic nerves stimulate heart rate and contractility by binding to ОІ1 receptors in the heart. Parasympathetic nerves decrease heart rate by binding to cholinergic receptors. In addition, sympathetic nerves influence blood pressure by exerting control over virtually the entire peripheral vascular system (except the capillaries) through innervation of the tunica media (the smooth muscle). At most blood vessels, sympathetic nerves release norepinephrine, which binds to specific receptors on the smooth muscle cells, called alpha receptors. Stimulation of the alpha receptors causes the smooth muscle to contract, constricting the vessel, which increases PR and therefore increases blood pressure. Blood vessels supplying skeletal muscle have a different type of receptor, called beta2 (ОІ2) receptors, which, when stimulated by norepinephrine, cause the vessels to relax. It appears that this sympathetic vasodilatory response plays a significant role only in the anticipatory response to exercise, perhaps serving to prime the skeletal muscle with oxygen and nutrient support before exercise onset. Skeletal muscle blood vessels also possess receptors for acetylcholine. These receptors are called muscarinic receptors and do not appear to be innervated by parasympathetic neurons. However, they respond to acetylcholine released by certain sympathetic cholinergic neurons. These neurons also supply the vascular smooth muscle in skeletal muscle and cause relaxation of the vessels, thus increasing blood flow through these vessels. Hormonal Control of the Vascular System There are several hormones that control the resistance of the vascular system. These hormones are released directly in response to changes in blood pressure, in response to neural stimulation, or both. Norepinephrine (noradrenalane) and Epinephrine (adrenaline) Norepinephrine and epinephrine are released from the adrenal medulla in response to activation of the sympathetic nervous system. Both substances act like norepinephrine released from nerve terminals and bind to alpha receptors to cause vasoconstriction, or to ОІ2 receptors to cause vasodilation of arterioles supplying skeletal muscles. Norepinephrine and epinephrine from the adrenal medulla also bind to ОІ1 receptors and increase heart rate. Aldosterone Aldosterone circulates to the kidney and causes cells of the distal tubule to increase sodium reabsorption. Under many circumstances, reabsorption of water follows that of sodium, leading to an increase in plasma volume. An increase in plasma volume increases stroke volume, and hence cardiac output. It also causes increased blood pressure. Antidiuretic Hormone Antidiuretic hormone (ADH), also called vasopressin, is released from the posterior pituitary in response to increased plasma osmolality (decreased water concentration) or decreased blood pressure. ADH is a potent vasoconstrictor with the potential to increase blood pressure by increasing the resistance to blood flow. Under most circumstances, except perhaps during severe hemorrhage, levels of circulating ADH are too low to affect the arterioles. However, ADH controls the reabsorption of water across the collecting ducts of the kidney back into the bloodstream. This effect influences blood pressure by increasing plasma volume and therefore cardiac output. Without ADH, water does not follow sodium reabsorption in the kidney and severe dehydration may occur. Reabsorption of water in response to ADH reduces the stimuli (increased plasma osmolality and decreased blood pressure) for ADH release. Atrial Natriuretic Peptide Atrial natriuretic peptide (ANP) is a hormone released from cells of the right atrium in response to an increase in blood volume. ANP acts on the kidney to increase the excretion of sodium ion (natriuresis). Because water will follow sodium in the urine, ANP serves to decrease blood volume and blood pressure. Renin-Angiotensin System Changes in blood pressure are sensed by the renal baroreceptors. If blood pressure is high, release of the hormone renin is decreased. If blood pressure decreases, renin release increases. Renin release is also stimulated by sympathetic nerves to the kidney. Renin controls the production of another hormone, angiotensin II. Renin circulates in the blood and acts as an enzyme to convert the protein angiotensinogen to angiotensin I. Angiotensin I is a amino acid protein, which is immediately split by angiotensinconverting enzyme (ACE) into the amino acid peptide, angiotensin II. ACE is the same enzyme that breaks down (and inactivates) the vasodilator hormone bradykinin. Blocking the action of ACE blocks the production of angiotensin II and the breakdown of bradykinin. Angiotensin II is a powerful vasoconstrictor that primarily causes constriction of the small arterioles. This causes an increase in resistance to blood flow and an increase in blood pressure. The increase in blood pressure then acts in a negative feedback manner to reduce the stimulus for further renin release. Angiotensin II also circulates to the adrenal gland and causes cells of the adrenal cortex to synthesize another hormone, aldosterone. Renin-angiotensin-aldosterone feedback cycle It should be emphasized that the stimuli causing the release of renin decreased blood pressure and decreased plasma sodium concentration are reversed by the actions of angiotensin II and aldosterone. This is an excellent example of a negative feedback cycle. Autoregulation of Blood Flow In general, blood pressure is controlled through neural and hormonal influences. However, individual tissues have mechanisms that regulate their own blood flow. This is accomplished by local vasodilation or vasoconstriction of meta-arterioles and precapillary sphincters. This local control of blood flow is called autoregulation, and is the means by which some organs can maintain constant blood flow over a wide range of blood pressure, from approximately 70 to 180 mmHg. Theories of Autoregulation. Several theories are proposed to explain local control of blood flow. The most widely accepted is that chemical mediators are released by metabolizing cells that bind to metaarterioles or precapillary sphincters, causing them to open or shut to blood flow. Chemical mediators that control local blood flow include adenosine (a metabolite of ATP), carbon dioxide, histamine, lactic acid, potassium ions, and hydrogen ions. All of these substances except histamine are byproducts of metabolism; as cell metabolism increases, so do their concentrations. This serves to match increased metabolic activity with increased blood flow. Mast cells present throughout the interstitial space release histamine in response to immune stimulation or local injury. Adenosine appears to particularly regulate local blood flow in the heart. An alternative theory concerning local control suggests that the meta-arterioles and capillary sphincters sense an oxygen or nutrient deficit that causes them to relax, thereby increasing blood flow to the surrounding cells. Other Chemical Mediators Influencing Blood Flow Various other chemicals are released by the blood vessels or by mediators of inflammation or healing, which affect blood flow to an area. Nitric Oxide Endothelial cells of the small arteries and arterioles respond to the binding of various vasoactive substances such as acetylcholine with the production of the vasodilator nitric oxide (previously called endothelial-derived relaxing factor). Nitric oxide diffuses through endothelial cells to underlying smooth muscle cells, causing endothelial-dependent relaxation of the smooth muscle. Nitric oxide release also occurs with increased blood flow through a vessel, allowing local dilation of the microvasculature to be matched by dilation of the small arteries and arterioles. Drugs that boost nitric oxide synthesis or prevent its breakdown are used to improve blood flow in a variety of clinical situations. Endothelin Endothelial cells also release endothelin, amino acid peptide that acts as a potent constrictor of vascular smooth muscle. Endothelin release is stimulated by angiotensin II, ADH, thrombin, cytokines, reactive oxygen species, and shearing forces acting on the vascular endothelium. Its release is inhibited by prostacyclin and nitric oxide. Deleterious effects of overproduction of endothelin on vascular smooth muscle include extended vasoconstriction, vascular hypertrophy, cell proliferation, and fibrosis. In addition, by binding to receptors on endothelial cells that are different from the ones on vascular smooth muscle, endothelin increases capillary permeability and contributes to inflammation. Endothelin has a long half-life (length of presence in the circulation or interstitial fluid), so even small changes in its production or clearance have significant impact on the vascular system. Overproduction of endothelin is implicated in numerous pathologies, including pulmonary hypertension and heart failure. Drugs to block endothelin receptors are being tested in a variety of clinical situations. Serotonin Serotonin (5-hydroxytryptamine) is primarily released by platelets drawn to an area of injury or inflammation. Effects of serotonin may be vasodilatory or vasoconstricting, depending on the site of release. Serotonin's ability to vasoconstrict and decrease blood flow appears to be one mechanism whereby platelets control or reduce bleeding. Bradykinin Bradykinin, like all members of the kinin family, is a small polypeptide that acts as a potent vasodilator of arterioles and as a mediator to increase capillary permeability. Bradykinin is produced in the plasma or interstitial fluid by enzymatic splitting of a serum globulin in response to vascular or tissue injury or inflammation. The half-life of bradykinin is short. Normally, it is broken down rapidly by circulating ACE or another enzyme, carboxypeptidase. The effects of bradykinin are increased local blood flow, increased capillary permeability, and decreased vascular resistance. Blockage of angiotensin-converting enzyme by various pharmaceutical agents prolongs the half-life of bradykinin and its effects. Prostaglandins There are many different types of prostaglandins. Some cause dilation of the vascular system and some cause constriction. Prostaglandins are derived from the metabolism of arachidonic acid by cyclooxygenase (COX) enzymes one (COX1) and two (COX2). Arachidonic acid is present in all cell membranes and is released with tissue injury. Prostaglandins work to control local blood flow. They may circulate to affect distant cells. One main group of prostaglandins, those of the E series, cause local vasodilation and increased blood flow, which makes them important mediators of inflammation. Prostaglandins of the I series, especially PGI2, called prostacyclin, also are vasodilatory. PGI2 inhibits platelet aggregation and blood clotting. Thromboxane A2 is an important prostaglandin that causes vasoconstriction and blood clotting. Summary of Blood Pressure Control With a decrease in blood pressure, baroreceptor information is transmitted to the cardiovascular center in the brain. This causes the stimulation of sympathetic output to the heart and vascular system, increasing heart rate and PR. Parasympathetic output is decreased, also increasing heart rate. Renin release increases, causing increased angiotensin II, which directly increases PR and aldosterone synthesis. Increased aldosterone increases sodium reabsorption and, in the presence of ADH, water reabsorption. Increased plasma volume, stroke volume, and cardiac output result. Capillary pressure decreases directly with blood pressure and indirectly from sympathetic constriction of the arteriole feeding the capillary. This serves to decrease filtration of fluid out of the capillary. All of these responses serve to increase blood pressure toward normal, by increasing heart rate, stroke volume, and PR. In contrast, if blood pressure increases, baroreceptor responses cause a decrease in sympathetic stimulation to the heart and vascular smooth muscle, and heart rate and PR decrease. Increased parasympathetic stimulation to the heart contributes to the decrease in heart rate. There is a decrease in renin and ADH release, reducing TPR and plasma volume. ANP release increases. All these responses serve to decrease blood pressure toward normal. The integrated function of these systems ensures adequate perfusion of all tissues, despite regional differences in demand. Arterial hypertension occurs when the relationship between cardiac output and total peripheral resistance is altered. Hypertension may result from a disturbance in one of the regulatory mechanisms listing earlier HYPERTENSIONS This disorder consists of protracted, abnormally high arterial blood pressure defined as systolic pressure exceeding 140 mm Hg and diastolic pressure exceeding 90 mm Hg. Hypertension is abnormally high blood pressure measured on at least three different occasions from a person who has been at rest at least 5 minutes. Normal blood pressure varies with age, thus any diagnosis of hypertension must be age specific. The 7th Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure has published revised guidelines on optimal and hypertensive values of systolic and diastolic pressures. In general, optimal pressures are considered less than 120 mm Hg systolic and 80 mm Hg diastolic, while pressures considered hypertensive are higher than 140 mm Hg systolic, and higher than 90 mm Hg diastolic. A state of prehypertension includes blood pressures between 120 and 139 mm Hg systolic and 80 and 89 mm Hg diastolic. For those with especially significant cardiovascular risk factors, including a strong family history of myocardial infarct or stroke, or a personal history of diabetes, even these prehypertensive values are too high. Types of Hypertension Hypertension is often classified as either primary or secondary, based on whether a cause can be identified. Most cases of hypertension have no known cause and are called primary or essential hypertension. When a clear cause of hypertension can be identified, it is called secondary hypertension. Arterial hypertensions may be also classified using various characteristics: 1) Cardiac output: Arterial hypertension with normal cardiac output is termed eukinetic, with high cardiac output - hyperkinetic, and with low - hypokinetic. 2) Peripheral resistence: There are arterial hypertensions with normal or high absolute peripheral resistence. 3) Blood volume: Arterial hypertension may be associated with normal (volume-independent) or high (volume-dependent) blood volume. 4) Increase in arterial pressure may be limited to the diastolic pressure, so-called isolated diastolic hypertension or systolic pressure, so-called isolated systolic hypertension. Often, arterial hypertension is combined, systolic-diastolic. 5) Renin activity: Arterial hypertension may be accompanied by high renin activity in the peripheral blood (high-renin hypertension), or by low renin activity (low-renin hypertension). 6) Clinical course: Arterial hypertension may progress slowly over decades with minimal complications (benign form), or rapidly, with renal failure and severe retinal pathology (malignant form). 7) Pathogenesis: arterial hypertension may be primary or essential, and secondary or symptomatic. Essential, primary, or idiopathic hypertension Essential, primary, or idiopathic hypertension is systemic hypertension of unknown cause. More than 95% of all cases of hypertension are in this category. Secondary hypertension is systemic hypertension of known cause. Fewer than 5% of all cases of systemic hypertension are in the latter category. Risk factors for primary hypertension include: family history, advancing age, obesity, tobacco use, high intake of sodium, high intake of saturated fat, excessive alcohol consumption, sedentary lifestyle, stress, excess rennin, mineral deficiencies (calcium, potassium, and magnesium), diabetes mellitus. The theories helping to explain the development of hypertension The hypertension development as respons to excess salt consumption, which result in a prolonged increase in plasma volume. Epidemiological, migration, and genetic studies in humans and animals provide compelling evidence of a relationship between high salt intake and elevated blood pressure. From an evolutionary perspective, humans are adapted to ingest and excrete less than 1 gram of salt per day, which is at least ten times less than the average salt consumption in industrialized countries. Besides excess dietary intake of salt, abnormally increased renin or aldosterone levels or decreased blood flow to the kidneys also may alter renal handling of salt and water. A prolonged increase in plasma volume may occur as a result of renal mishandling of salt and water. According to Guyton’s hypothesis a defect in renal sodium homeostasis implies the existence of genetic factors that predispose to decreased level of Na+ excretion at normal arterial pressure when the affected individuals consume standard diet with high salt content. Accumulation of Na+ leads to fluid retention, and, thus, increases the circulating volume. In the face of increasing cardiac output, peripheral vasoconstriction occurs, as an autoregulation response, to prevent the overperfusion of tissues. Autoregulation leads to increased peripheral resistance, and along with it an elevation of blood pressure. The hypertension development as respons to increased peripheral vascular resistance (PR) The possible causes of hypertension mentioned may result from increased sympathetic nervous system activity. This may result from a prolonged stress response, which is known to involve sympathetic activation, or from a genetic excess of receptors for norepinephrine in the heart or vascular smooth muscle. Chronically increased PR may occur with an overresponsiveness of the arterioles to normal sympathetic or hormonal stimulation, which would cause a narrowing of the vessels. With increased PR, the heart has to pump more forcefully, and therefore exert more pressure, to drive the blood through the narrow vessels. This is referred to as an increase in the afterload of the heart, and is usually associated with an increased diastolic pressure reading. With a prolonged increase in afterload, the left ventricle may begin to hypertrophy (increase in size). As a result, the ventricle's own oxygen demands increase further, causing it to pump even more forcefully to meet those demands. Disorder of sodium-calcium pumping. There is evidence that African Americans, who generally report more frequent and more severe hypertension, demonstrate an alteration in sodium-calcium pumping such that calcium accumulates in the smooth muscle cells, increasing muscle contraction and resistance The modern theorie of pathogenesis Essential hypertension is multifactorial disorder exhibiting polygenic inheritance. Defects may be present in one or more factors that increase cardiac output or rise peripheral resistance. Environmental factors, especially increased salt intake, may potentiate the effects of genetic factors. The resultant increases in cardiac output and peripheral resistance contribute to hypertension. Cardiac output is increased by conditions that increase heart rate or stroke volume, or both. Peripheral resistance is increased by factors that increase blood viscosity or reduce the lumen size of vessels, especially the arterioles. Exhibit two congenital forms of hypertension, a neural form due to sympathetic hyperactivity and a renal form due to defective excretion of sodium Often hypertension may result from a disturbance in one of the following intrinsic mechanisms. Sympathetic nervous hyperactivity Renal retention of excess sodium The Renin-angiotensin system Defects in cell transport or binding Endothelial cell dysfunction Hyperinsulinemia/insulin resistance Sympathetic nervous hyperactivity Young hypertensives tend to have increased levels of circulating catecholamines, augmented sympathetic nerve traffic in muscles, faster heart rate, and increased vascular reactivity to norepinephrine. These could raise blood pressure by causing arteriolar and venous constriction, by increasing cardiac output, or by altering the normal renal pressure-volume relationship. Renal retention of excess sodium To induce hypertension, excess sodium must be retained by the kidneys. Such retention could arise in a number of ways: 1. A decrease in filtration surface by a congenital or acquired deficiency in nephron number or function, this disorders result in accumulation of Na+ and lead to fluid retention, and, thus, increases the circulating volume. 2. A resetting of the normal pressure-natriuresis relationship, including disorders of the reninangiotensin system. In normal, a rise in pressure invokes an immediate increase in renal sodium excretion, shrinking of fluid volume and returning the pressure to normal. The Renin-angiotensin system. The renin-angiotensin system has the central roles of sodium metabolism There many genetic and acquired disorders that alter blood pressure by altering activity of this system Activation of the system increases blood volume by conserving sodium and water. Defects in cell transport or binding The smooth muscle cells from hypertensive patients have a defective Na+-K+-Cl- cotransport system in the plasma membrane. This system is coupled with the Na+Ca2+ exchange mechanism. As a result, two processes are deranged: the removal of Ca2+ from cytosol and pumping out of Na+ to the extracellular space. This defect leads to an abnormal accumulation of calcium in the vascular smooth muscle cells, resulting in enhanced vascular responsiveness to any vasoconstrictory stimuli. Endothelial cell dysfunction Some hypertensive patients have a reduced vasodilatory response to various stimuli of NO release. In addition, hypertensives display a decreased basal release of NO by the endothelial cells. Hyperinsulinemia/insulin resistance Insulin exerts vasodilatory effect, mediated through increased synthesis of nitric oxide. The latter normally counteracts the multiple pressor effects of insulin. Insulin resistence promotes pressor effects of insulin, a trophic action on vascular hypertrophy and increased renal sodium reabsorption. Pathogeneses In this condition stress factors such as emotional and psychological conflicts can trigger hypertension. This stimulates vascular baroreceptors, decreasing the sensitivity of the cerebral cortex and pain sensitivity, and result in hypertension. Arterial hypertension is therefore a “learnable” strategy for coping with stress. Fluid volume and/or cardiac output are increasing. A number of other factors can cause both functional contraction and structural remodeling and hypertrophy of the vessel wall. Multiple vasoactive substances act as growth factors for vascular hypertrophy. Hypertrophy of the vessel increase peripheral vascular resistance Essential hypertension usually begins insidiously as a benign disease, slowly progressing to a malignant state. Malignant hypertension follows the accelerated course and is characterized by extreme blood pressure elevations. If untreated, even mild cases can cause major complications and death. Prolonged hypertension increases the workload of the heart as resistance to left ventricular ejection increases. To increase contractile force, the left ventricle hypertrophies. Clinical Manifestations In the 1997 the Sixth Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure designate stage of hypertension according to the patient's blood pressure, risk factors, and target organ damage. RISK GROUP The patient with the risk factors, which were listened above Blood pressure is high then norma - (130–139/85–89) Stage 1- (140–159/90–99) Stages 2 and 3 - (>160/>100) Most clinical manifestations occur after years of hypertension, and include: Waking headache, sometimes with nausea and vomiting, caused by increased intracranial blood pressure. Blurred vision caused by hypertensive damage to the retina. Unsteadiness in the gait caused by central nervous system damage. Nocturia caused by increased renal blood flow and glomerular filtration. Dependent edema and swelling caused by increased capillary pressure. Hypertensive crisis. Hypertensive crisis is a severe rise in arterial blood pressure caused by a disturbance in one or more of the regulating mechanisms. If untreated, hypertensive crisis may result in renal, cardiac, or cerebral complications and, possibly, death. Complication Stroke may result from a high-pressure hemorrhage in the brain or from an embolus broken off a noncerebral vessel exposed to high pressure. Strokes may occur with long-standing hypertension if arteries supplying the brain become hypertrophied and thickened, thereby reducing blood flow to areas of the brain that depend on them. The cerebral arteries that are atherosclerotic may become weak, increasing the likelihood of an aneurysm. A myocardial infarct (MI) may occur if the atherosclerotic coronary arteries cannot supply adequate oxygen to the myocardium or if a thrombus develops that blocks flow through a vessel. With chronic hypertension and the development of ventricular hypertrophy, the oxygen demands of the myocardium may not be met, and cardiac ischemia leading to an infarct may occur. Likewise, ventricular hypertrophy may cause changes in the timing of electrical conductance through the ventricle, leading to dysrhythmia, cardiac hypoxia, and an increased risk of clot formation. Renal failure may occur with progressive high-pressure damage to the renal capillaries, the glomeruli. With glomerular injury, blood flow to the functional units of the kidney, the nephrons, is impaired, and these can become hypoxic and die. With damage to the glomerular membranes, proteins will be lost in the urine, decreasing the plasma colloid osmotic pressure and contributing to edema, which is often seen with long-standing hypertension. Encephalopathy (brain damage) may occur, especially with malignant (swiftly progressing, dangerous) hypertension. The dramatically high pressure seen in this condition causes increased cerebral capillary pressure and drives fluid into the interstitial space throughout the central nervous system. Surrounding neurons collapse, and coma and death may result. Secondary Hypertension The pathophysiology of secondary hypertension is related to the underlying disease. Renal arterial hypertension The most common cause of secondary hypertension is chronic renal disease. The causes of renal arterial hypertention Acute glomerulonephritis Chronic renal disease Polycystic disease Renal artery stenosis Renal artery fibromuscular dysplasia Renal vasculitis Renin-producing tumors Hypertension produced by renal disease is the result of either 1 alteration in renal secretion of vasoactive materials (Renovascular) 2 derangement in the renal handling of sodium and fluids leading to volume expansion (Parenchymal) An example of secondary hypertension resulting from alteration in renal secretion of vasoactive materials is renal vascular hypertension, which develops as a result of renal artery stenosis. This condition may be congenital or a result of atherosclerosis. Renal artery stenosis reduces blood flow to the kidney, leading to activation of renal baroreceptors, stimulation of renin release, and production of angiotensin II. Angiotensin II increases blood pressure directly by increasing PR, and indirectly by increasing aldosterone synthesis and sodium reabsorption. If repair of the stenosis is possible or the affected kidney is removed, blood pressure returns to normal. Deficiency in nephron number result in accumulation of Na+ and lead to fluid retention, and, thus, increases the circulating volume. Besides, often patchy areas of one or both kidneys are diseased and become ischemic because of local vascular constrictions, whereas other areas of the kidneys may be normal. The patchy ischemic kidney tissue secretes renin, and it acting through the formation of angiotensin II. Other vasoconstrictors (e.g., endothelin) and loss of vasodilators (nitric oxide) may also contribute to vasoconstriction. Aldosterone levels are also elevated, and salt retention undoubtedly contributes to the elevation of blood pressure. Neurogenic hypertension The causes Psychogenic (various stressful factors) Increased intracranial pressure, at organic lesion of a brain as result of a trauma, encephalitis, tumours, ischemic and hemorrhagic stroke. 1. Arterial hypertension may develop as a result of damage to the brain regions involved in the regulation of blood pressure. 2. Arterial hypertension may develop when the flow of impulses from baroreceptors of the aortic arc and sinocarotid zone is reduced. This may occur in patients with atherosclerotic lesion of aorta which may lose its normal The cases may lead to a massive rise in blood pressure via central nervous stimulation of the sympathetic nervous system. An increase in sympathetic activity of central nervous system origin can cause an increase in cardiac output. Increase of cardiac output (hyperdynamic hypertension) leads to an increased venous return and thus an increased stroke volume (Frank–Starling mechanism). Cardiovascular The causes Coarctation of aorta Diseases associate with increased intravascular volume (At some patients with cardiac failure) Diseases associate with increased cardiac output (e.g. At disorders of a rhythm)- see hyperdynamic hypertension Coarctation of aorta That is, when а constrictor is placed on the aorta above the renal arteries, the blood pressure in both kidneys at first falls, renin is secreted, angiotensin is formed, and acute hypertension осcurs in the upper body because of the vasoconstrictor effects of the angiotensin on the arterioles throughout the body. Within а few days, salt and water retention also occurs so that the arterial pressure in the lower body at the level of the kidneys now rises approximately to normal, but high pressure persists in the upper body. In coarctation of the aorta, the arterial pressure in the lower body is usually almost normal, whereas the pressure in the upper body is far higher than normal. Endocrine hypertension Adrenocortical hyperfunction (Cushing syndrome, primary aldosteronism) Exogenous hormones (glucocorticoids, estrogen [including pregnancy-induced and oral contraceptives]) Pheochromocytoma Acromegaly Hyperthyroidism (thyrotoxicosis) Increased secretion of vasopressin Pregnancy-induced Adrenocortical hyperfunction Hypertension is a feature of a variety of adrenal cortical abnormalities. In patients with primary aldosteronism, aldosterone levels are high. Aldosterone causes increasing in sodium reabsorption by the kidney tubules, thus increasing the total body extracellular fluid sodium. This increased sodium then causes water retention and increasing the extracellular fluid volume. Increased intravascular volume, altered sodium concentrations in vessel walls, or very high aldosterone levels cause vasoconstriction and increased resistance. Thus, arterial pressure increases Pituitary-dependent Cushing's syndrome results from ACTH excess. The increased secretion of corticotropin in the anterior pituitary leads to hyperplasia of the adrenals and hypersecretion of glucocorticoids. At chronic hypercortisolism (Cushing's syndrome, caused by excess of glucocorticoids (i.e. cortisol), production of glucocorticoids by the adrenals increase too. Increased levels of glucocorticoids raise blood pressure by increasing renal sodium retention, angiotensin II levels, and vascular response to norepinephrine Pheochromocytoma In patients with pheochromocytoma, increased secretion of epinephrine and norepinephrine by a tumor causes excessive stimulation of adrenergic receptors, which results in peripheral vasoconstriction and cardiac stimulation. Hyperthyroid hypertension Hyperthyroidism, associated with increased blood levels of thyroxine and triiodothyronine or the increased sensititivity of the peripheral receptors of thyroid hormones, is accompanied by arterial hypertension. The mechanism of arterial hypertension includes cardiotonic action of thyroid hormones with sinus tachycardia and wide pulse pressure Vasopressin Vasopressin, also called antidiuretic hormone, plays little role as vasoconstrictor, but has an important function to increase greatly water reabsorption into the blood from the renal tubules . Arterial hypertension may be caused by an increased secretion of vasopressin. The ensuing retention of water leads to elevation of blood volume. Arterial hypertension associated with changes in the blood Arterial hypertension develops in patients with chronic myeloproliferative disorders, such as polycythemia vera. In this disease the increased red cell mass causes an increase in total blood volume with ensuing increase in stroke volume and sustained elevation of blood pressure. Hypertension in Pregnancy Hypertension in a pregnant woman carries risk to both the mother and fetus. Four categories of hypertension in pregnancy have been identified by the National Institutes of Health Working Group on High Blood Pressure in Pregnancy: gestational hypertension, chronic hypertension, preeclampsiaeclampsia, and preeclampsia superimposed on chronic hypertension. Gestational hypertension is a type of secondary hypertension because, by definition, the elevation in blood pressure (140 mmHg systolic; 90 mmHg diastolic) occurs after 20 weeks' gestation in a previously non-hypertensive woman, and reverses within 12 weeks postpartum. Gestational hypertension appears to result in part from a combination of increased cardiac output and increased TPR. If hypertension persists beyond 12 weeks postpartum, or was present before 20 weeks' gestation, it is categorized as chronic hypertension. With preeclampsia, high blood pressure is accompanied by proteinuria (urinary excretion of at least 0.3 g protein in 24 hours). Preeclampsia typically develops after 20 weeks' gestation and is associated with decreased placental blood flow and the release of chemical mediators that cause dysfunction of vascular endothelial cells throughout the body. It is a very serious disorder, as is preeclampsia superimposed on chronic hypertension ARTERIAL HYPOTENSION Arterial hypotension is defined as a decrease in blood pressure below 60 mm Hg diastolic and 90 mm Hg systolic. Hypotension may be classified as essential and symptomatic. Pathogenesis of the essential hypotension is poorly understood. The contributing factors may be decreased sensitivity of the peripheral alfa-adrenoreceptors of the vessel walls; the inherited low level of renin production by the juxtaglomerular cells, or the decreased conversion of dopamine to norepinephrine (e.g. lack of dopamine beta-hydroxylase). Symptomatic arterial hypotension can be caused by various factors, including the impaired central regulation of the vascular tone, abnormal operating of the baroreflex, chronic cardiac failure, adrenal insufficiency, or decreased renin production in the kidneys.