AA metabolismI
advertisement

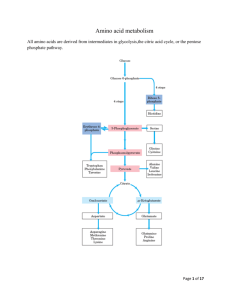
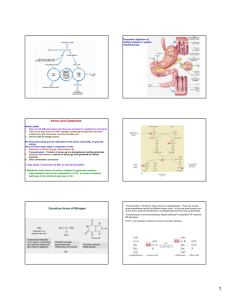
Amino acid metabolism I proteins proteolysis (digestion) proteosynthesis specific synthesis (nonessencial AA) amino acids pool common metabolic pathways transamination deamination decarboxylation keto acids NH3 biogenic amines ketone bodies glukose TCA CO2, H2O, ATP UC urea H2N-C-NH2 O specific pathways ↓ hormones neurotransmiters koenzymes porphyrins purines pyrimidines creatine physiologically important nitrogenous compounds Protein digestion - proteolytic enzymes (proteinases/proteases) ↓Phe ↓ ↓ Tyr ↓ endopeptidasses stomach Trypsin Chymotrypsin Elastase Carboxypeptidases Lys ↓, Arg ↓ Phe ↓, Tyr ↓, Trp ↓, Met ↓, Leu ↓ Ala ↓, Gly ↓,Ser ↓ A: ↓, Ala ↓ Ile, ↓ Leu, ↓Val B: ↓ Lys, ↓ Arg brush border membrane enzymes absorption - enterocyte (active transport) portal blood liver exopeptidases small intestine Specificity and activation of pancreatic proteases Trypsin = activator of all digestive proteolytic enzymes in the small intestine Proteinases (proteases) 1. Synthesis and secretion: proenzymes (zymogenes) 2. Activation: cleavage of several peptide bonds or/and removal of a short peptide → openning of an active site pepsinogen HCl pepsin + 41 AA trypsinogen enteropeptidase trypsin + 6 AA trypsinogen 3. Inhibition: protein inhibitors → interaction with proteinases active sites substrate (polypeptide) trypsin Proteinases inhibitors • pancreatic trypsin-inhibitor: small protein → very tight binding at the active site of trypsinu ⇒ inactive complex (half life: several months) ↓ protection of intestinal walls and cells against proteolytic cleavage • α1-antitrypsin (α1-antiproteinase, antielestase): plasma protein – irreversible binding at the active site of trypsin and elastase → protection of tissues against digestion by elastase (trypsin) genetic mutants ↑ smokers slow secretion from liver low blood level ↓ destruction of alveolar walls (degradation of connective structures by elastase) ↓ emphysema Protein turnover Dietary proteins 100 g/day equivalent amount of nitrogenous metabolites (mainly urea) excreted Body proteins 400 g/day constant degradation and resynthesis AA – mostly metabolised in liver; Leu, Ile, Val pass without transformation – utilized in muscle and brain glutamine, valine, alanine, glycine - most abundant AA in the blood circulation biological half-life of proteins: <2h < 2- 200 h > 200 h several months, years digestive enzymes, ornithine dekarboxylase, HMG-CoA-reductase most proteins hemoglobin, acetylcholine receptor collagen, structural proteins Intracellular protein degradation 1. Lysosomal proteases (cathepsins, collagenase, dipeptidases..): most extracellular proteins, long-lived proteins 2. Proteasomes: abnormal proteins, short-lived proteins Ubiquitination cytosolic protein ubiquitin – kiss of death - binding with a protein predestines it for degradation in proteasome = oligomer – supramolecular structure, several subunits = proteinases ubiquitin-COOH + H2N-Lys-protein mark for degradation Signals for degradation: 1.oxidation: Lys, Trp, His, Cys 2. PEST-sequence: Pro, Glu, Ser, Thr 3. NH3-end of AA: Arg, Lys, Asp, Phe Catabolism of amino acids – common reactions Pyridoxal phosphate – unique role in amino acid metabolism - cofactor of enzymes catalyzing different kinds of AA transformations in the active site held by noncovalent interactions or linked covalently to ε-amino group of lysine a derivative of vitamin B6 3 2 pyridoxine pyridoxal phosphate (PLP) 1 Schiff base AA - PLP essential intermediate of AA metabolism 1. Decarboxylation common reactions of amino acid metabolism 2. Transamination 3. Transformation of a carbon chain → specific for each amino acid 1. Decarboxylation biogenic amines; enzymes - decarboxylases H R-C-COOH NH2 PLP R-CH2-NH2 + CO2 neuromediators histidine → histamine: mediator serine → ethanolamine → phospholipids cysteine → cysteamine → CoA-SH Trp → 5-OH-Trp → serotonine glutamic acid→ GABA Polyamines – spermidine, spermine – initial substrates ornithine and methionine stabilization of RNA, DNA, membranes - regulation of cell growth and proliferation, regeneration of tissues 2 ornithine decarboxylase putrescine 2. Transamination → keto acids; enzymes – aminotransferases H R1-C-COOH NH2 ! reaction reversible – involved both in catabolism and biosynthesis of aminoacids H PLP + R2-C-COOH R1-C-COOH + R2-C-COOH NH2 O O Example: alanine aminotransferase - - alanine 2-oxoglutarate pyruvate glutamate = α-ketoglutarate exclusive acceptor of amino group in transaminase reaction in AA catabolism AK -NH2 of any AA incorporated into molecule of glutamate 1 1 1 Mechanism of transamination 1 2 2 2 2 ALT, AST – important aminotransferases/transaminases ALT – alanine aminotransferase: alanine + 2-oxoglutarate PLP pyruvate + glutamate AST – aspartate aminotransferase: aspartate + 2-oxoglutarate PLP oxaloacetate + glutamate Localization: mainly liver, muscle, kidney ALT – cytosol, AST – cytosol and mitochondria ! Metabolic significance: ALT, AST – catabolism andi biosynthesis of alanine/aspartate, ALT – important for utilization of muscle protein AA for gluconeogenesis (glucose-alanine cycle) AST – replenishment of oxalacetate for CC (anaplerotic reaction), transport of oxalacetate across mitochondrial membrane (gluconeogenesis), important for the function of malate dehydrogenase shuttle and urea syntesis (formation of aspartate as a donor of one nitrogen atom of the urea) Diagnostic value: ALT, AST – markers of liver injury (hepatocellular necrosis), AST – blood level increases at myocardial infarction (not used as a marker in the present IM diagnostics) Deamination: removal of -NH2 as NH3 1. Direct deamination → serine, threonine H H2O HO serine (threonine) dehydratase 2 CH2-C-COOH OH NH2 CH2= C-COOH NH2 OH H NH3 2. Oxidative deamination – other amino acids L-amino acid oxidases, cofactor FMN - liver, kidney, low activity – little value for AA metabolism D-amino acid oxidases, cofactor FAD - ?deamination of D-amino acids of bacterial and plant origin + CH2=C-COOH OH CH3-C-COOH O Oxidative deamination of glutamate - kee reaction for removal of nitrogen from amino acids amino acid +α-ketoglutarate → keto acid + glutamate -a product of transamination oxidative deamination NH3 + α-ketoglutarate ! Glutamate dehydrogenase (GDH) (NAD+/ NADP+) (high amount – liver mitochondria – used in diagnostics of liver diseases) NAD+ HOOC-CH2-CH2-C-COOH glutamate NADH+H+ NH2 H2O HOOC-CH2-CH2-C-COOH O NH HOOC-CH2-CH2- C-COOH + NH3 O H2 α−ketoglutarate Ammonia - NH3 • sources: 1. Deamination of amino acids (glutamate) – all tissues 2. Hydrolysis of glutamine - kidney, small intestine, liver 3. Bacterial degradation of proteins and urea (urease) in the intestine 4. Catabolism of purines, pyrimidines, catecholamines - liver, brain • detoxification: 1. Urea synthesis - !liver! (exclusively) → kidney → urine 2. Synthesis of glutamine – extrahepatic tissues 3.Formation of NH4+ - kidney → urine NH3 + HOOC-CH-CH2-CH2-COOH glutamate NH2 muscle, brain NH4+ kidney H+ glutamine synthetase urine HOOC-CH-CH2-CH2-C NH2 glutamine NH2 NH3 + glutamate kidney, liver ad 2, 3 O glutaminase glutamine Ammonia metabolism - overview Urea synthesis = Ureagenesis: major pathway of NH3 detoxification Localization: liver ⇒ mitochondria, cytosol of periportal hepatocytes O H2N-C-NH2 precursors: aspartate NH3 oxidative deamination of glutamate (GDH) transamination of oxaloacetate (AST) CO2 TCA cycle glutamate NAD+ glutamate NADH+H+ GDH NH3 + α-ketoglutarate AST aspartate oxaloacetate α-ketoglutarate Energy needs of ureagenesis: 4 ATP Proteins ↓ ↓ amino acids aminotransferase α-ketoglutarate α-keto acid NAD+ glutamate glutamate degydrogenase aspartate aminotransferase α-ketoglutarate NADH+H+ CO2 NH3 oxaloacetate α-ketoglutarate aspartate citrulline carbamoyl phosphate ornithine urea urea cycle arginine argininosuccinate fumarate Urea cycle – formation of urea - NH3 enters intu the cycle after formation of carbamoyl phosphate - carbamoyl phosphate formation from NH3, CO2, ATP catalyzes carbamoyl phosphate synthetase I – enzyme located in hepatocyte mitochondria only ! -carbamoyl phosphate is also precursor of pyrimidine nucleotides – formation in cytosol – catalyzed by carbamoyl phosphate synthetase II, source of its NH2-group is glutamine (! not NH3) Cooperation of urea cycle and citric acid cycle - utilization of fumarate, a side product of ureagenesis oxaloacetate Regulation of urea synthesis 1. A substrate delivery - low-protein diet - decreased AA intake decreased NH3 formation low activity of urea cycle, decreased urea level in urine - high-protein diet, fasting (degradation of muscle proteins) - increased AA delivery into liver increased activity of urea cycle 2. N-acetylglutamate – allosteric activator of carbamoyl phosphate synthetase I (essential for the enzyme activity) formation from glutamate – increased at higher AA delivery = at protein degradation - explanation of increased ureagenesis in fasting Genetic defects of urea cycle enzymes hyperammonemia – partial deficit can be treated by low-protein diet and suplementation of food with arginine (precursor of ornithine) - significantly decreased activities of ureagenetic enzymes incompatible with life Hyperammonemia 1. liver cirrhosis (alcoholic), hepatitis, obstruction of bile duct ⇒ NH3 from GIT enters via portal blood → directly into systemic circulation 2. genetic defects of enzymes of ureagenesis→ inhibition of urea synthesis → NH3 cannot be detoxified ⇒ vomiting, lethargy, coma , serious brain damage, death Ammonia toxicity (namely for CNS) – suggested mechanisms Brain: NH3 + α−ketoglutarate GDH glutamate NADPH+H+ NADP+ decreased mitochondrial level → inhibition of citric acid cycle (substrate depletion) → decreased ATP synthesis NH3 + glutamate glutamine x depletion of glutamate (neurotransmitter) (excess of NH3 inhibits glutaminase) alteration of nerve impuls transmission Fate of ammonia in tissues brain + most tissues (also muscle) Liver amino acid amino acid transamination transamination glutamate glutamate GDH GDH NH3 NH3 urea cycle glutamine synthetase urea glutamine Kidney glutamine glutaminase urea glutamate + NH3 urine Glucose-alanine cycle - connection of muscle glycolysis and liver gluconeogenesis – analogy to Cori cycle - kee role of ALT - important in starvation - gluconeogenesis from AA - dominant - uses also alanine released during breakdown of contractile proteins