dN/dS
advertisement

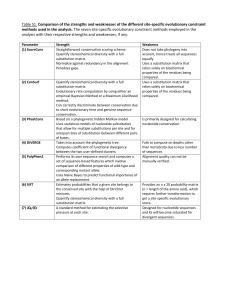
Advanced in Phylogenetic Analysis Group 4 53A36 Group 3 Group 5 ** ** ** ** Group 6 ** 53A26 ** Group 1 Group 2 53A37 .2 Hsin-Fu Liu, Ph.D. Department of Medical Research, Mackay Memorial Hospital, Taipei, Taiwan E-mail: hsinfu@ms1.mmh.org.tw Application of Phylogenetic Analysis • • • • • • • • Evolutionary studies Classifications Phylogeography Phylogenomics Molecular Epidemiology Phylodynamics Forensic Sciences, Criminal Investigation etc.……. Some of the applications need to be very precise! Some key points for performing a phylogenetic analysis Select an informative region to analyze coding or non-coding region length of the fragment phylogenetic signal Make an optimal sequence alignment the bias in selecting the taxa sequence homology insertions or deletions Use different methods to construct the trees Neighbour-Joining Maximum Parsimony Maximum Likelihood Bayesian Statistical test for phylogenetic trees bootstrapping (1000 replicates) http://evolution.genetics.washington.edu/phylip/software.html This site lists more than 392 phylogeny packages and 54 free servers. Perhaps the best-known programs are PAUP* (David Swofford and colleagues) and PHYLIP (Joe Felsenstein). Distance matrix methods 1 TCAAGTCAGGTTCGA 2 TCCAGTTAGACTCGA 3 TTCAATCAGGCCCGA Convert dissimilarity to evolutionary distance by correcting for multiple events per site according to a certain model of evolution, e.g. Jukes and Cantor. 2 3 1 0.266 0.333 2 3 1 0.33 0.44 2 3 dissimilarity 0.333 2 0.44 3 evolutionary distance A first idea about the relationship among different taxa can be viewed by their evolutionary distance. The simplest way to estimate the evolutionary distance is to count the total number of nucleotide differences between them and divide by the total number of available sites. An evolutionary distance obtained in such a way is called uncorrected distance or Hamming distance. A more realistic estimation can be achieved by applying a particular model of evolution that is making assumptions on how nucleotides change during the evolutionary process. The simplest approach to measuring distances between sequences is to align pairs of sequences, and then to count the number of differences. The degree of divergence is called the Hamming distance. For an alignment of length N with n sites at which there are differences, the degree of divergence D is: D=n/N But observed differences do not equal genetic distance! Genetic distance involves mutations that are not observed directly. Uncorrected distance vs. evolutionary distance Jukes and Cantor (1969) proposed a corrective formula: D = (- 3 ) ln (1 – 4 p) 3 4 Consider an alignment where 3/60 aligned residues differ. The normalized Hamming distance is 3/60 = 0.05. The Jukes-Cantor correction is 4 D = (- 3 ) ln (1 – 0.05) = 0.052 3 4 When 30/60 aligned residues differ, the Jukes-Cantor correction is more substantial: D = (- 3 ) ln (1 – 4 0.5) = 0.82 4 3 Ancestral Sequence Present Sequences 1 A C T G A A C G T A A C G C A C T G G A G G A A T C G C 2 A A T G A A A G A A T C G C Although only 3 nucleotide differences can be observed, 13 mutations have occurred during evolution. During Evolution Sequence 1 A C T G T A C A C G G T A A T A C C G C G Sequence 2 A A C T G A A A C G A T A A T C G T C C Single substitution Sequential substitutions Coincidental substitutions Parallel substitutions Convergent substitutions Back substitutions Correcting for unobserved mutations Expected difference Sequence difference Correction Observed difference Time/Evolutionary distance How to correct the difference? Apply a substitution model that tries to estimate the correct number of substitutions. Nucleotide substitution models Jukes and Cantor’s A C T pyrimidines equal substitution rate Jukes-Cantor (JC or JC69) Kimura 2-Parameter (K2P or K80) A purines G Kimura’s 2 parameter C G T unequal substitution rate transition transversion one rate of substitution equal base frequencies two types of substitution equal base frequencies Felsenstein 1981 (F81) one rate of substitution unequal base frequencies Hasegawa-Kishino-Yano, or Felsenstein (HKY85 or F84) two types of substitution unequal base frequencies Tamura and Nei (TN93) three types of substitution unequal base frequencies General Time Reversible (GTR) six types of substitution unequal base frequencies Among-site rate variation (Γ distribution) α<1 is L-shaped: most of the sites are considered to have a low substitution rate, or they are virtually invariable, while few of them evolve at very high rate. α>1 is bell-shaped: most of the sites have intermediate rates, while few of them evolve at very low or very high rate. All the models described in the previous section assume each site in the aligned nucleotide sequences mutates exactly at the same rate. However, different selective pressure at different positions of the genome can be responsible for some sites to evolve slower or faster than others. In coding regions, mutations in first and second position generally lead to amino acid replacement and they occur normally at slower rate due to purifying selection. On the other hand, non-coding regions tend to change faster, because mutations can occur with less constraints than in coding ones. In a classical approach among-site rate heterogeneity can be approximated by a statistical function called g-distribution, which represents the nucleotide substitution rate as a function of the proportion of sites having a particular rate. The use of g-distribution can account for two different levels of rate variation in real data. The inclusion of g-distributed nucleotide substitution rates in the models described above is a more realistic description of the evolutionary process and can improve the estimation of the evolutionary distance. Genetic Distances: Example Genetic distance between SIVcpz and HIV-1 (p24 gene) • observed # changes / # sites = 0.406 • Jukes-Cantor = 0.586 • Kimura 2 parameter = 0.602 • Hasegawa-Kishino-Yano = 0.611 • General reversible = 0.620 • General reversible + gamma = 1.017 Likelihood Ratio Test (LRT) The performances of the different substitution models can be tested using the likelihood ratio test •For particular questions (e.g. molecular clock analysis) it is important to have the correct branch length and thus it is worth to put effort in investigating the most appropriate evolutionary model. •Whether or not the different lineages in a phylogenetic trees are evolving at constant rate it is possible to evaluate employing the LRT. •Analyses of real data suggest that while different models of nucleotide substitutions often have led to drastic changes in the likelihood of a tree or in the distance matrix of the taxa, they have only minor effect on tree topology. •The LRT rejects a simpler model in favor of a more complex one when the double of the difference between the two log likelihood, calculated employing the two different models, is significant in a 2 test. The degrees of freedom for the 2 test is given by the difference between the number of free parameters in the more complex model and the number of constraints in the simpler one. Model selection Akaike information criterion The Akaike information criterion is a measure of the relative goodness of fit of a statistical model. It was developed by Hirotsugu Akaike, under the name of "an information criterion" (AIC) first published in 1974. It is grounded in the concept of information entropy, in effect offering a relative measure of the information lost when a given model is used to describe reality. It can be said to describe the tradeoff between bias and variance in model construction, or loosely speaking between accuracy and complexity of the model. Where k is the number of parameters in the statistical model, and L is the maximized value of the likelihood function for the estimated model. AICc AICc is AIC with a correction for finite sample sizes. AICc was first proposed by Hurvich & Tsai in 1989. When all the models in the candidate set have the same k, then AICc and AIC will give identical (relative) valuations. In that situation, then, AIC can always be used. Model selection Bayesian information criterion Bayesian information criterion (BIC) is a criterion for model selection among a finite set of models. It is based, in part, on the likelihood function, and it is closely related to AIC. When fitting models, it is possible to increase the likelihood by adding parameters, but doing so may result in over-fitting. The BIC resolves this problem by introducing a penalty term for the number of parameters in the model. The penalty term is larger in BIC than in AIC. The BIC was developed by Gideon E. Schwarz. In fact, Akaike was so impressed with Schwarz's Bayesian formalism that he developed his own Bayesian formalism, now often referred to as the ABIC for "a Bayesian Information Criterion" or more casually "Akaike's Bayesian Information Criterion". Parameter in the model More parameters the model uses, more powerful it does? Bias: distance between the average estimate and truth Variance: spread of the estimates around the truth Statistical test for phylogenetic trees The tree topology obtained by any of the tree-making methods should be viewed as only an estimate of the phylogenetic relation among the taxa. Therefore, it is essential to know how much confidence can be associated with the branch length or the appearance of a set of taxa as a monophyletic group in a given tree. Bootstrap analysis Bootstrap analysis is the most often used method for statistical evaluation of phylogenies. It is used to test the effects of sampling error on tree inference and the stability of tree nodes by calculating how often a particular cluster in a tree appears when nucleotide sites are re-sampled with replacements many times. In general: Bootstrap value > 95% : often close to 100% confidence in that branch Bootstrap value > 75% : often close to 95% confidence in that branch Branch can be considered as a monophyletic group. Note: If the original data set is biased for some reasons, a clade may be regarded as statistically significant even if it is a wrong one. Conversely, a clade may be a correct one even if its bootstrap value is less than 75%. This is because the original bias cannot be corrected by the re-sampling process. Possible consequence of insufficient bootstrapping samples Random number seed Alignment not modified Modified alignment Bootstrapping times Bootstrapping times 100 times 1000 times 100 times 1000 times 5 62% 69.4% 67% 70.2% 15 68% 70.1% 77% 72.7% 23 71% 67.8% 85% 74.0% Neighbor-joining trees Random number seed Alignment not modified Modified alignment Bootstrapping times Bootstrapping times 100 times 1000 times 100 times 1000 times 5 37.6% 35.1% 85% 80% 15 35.9% 37.2% 84% 80.5% 23 31.8% 33.7% 81% 80.5% Maximum parsimony trees Likelihood-mapping analysis (quartet puzzling method) Likelihood-mapping analyses can be used as a complementary approach to solve the controversial phylogenies. •The method is based on an analysis of the maximum likelihoods for the three fully resolved tree topologies that can be computed for four sequences. •The three likelihoods are represented as points inside an equilateral triangle. •The triangle is partitioned into different regions. •The centre of the triangle represents a star-like evolution whereas the three corners represent wellresolved phylogeny and the three intermediate regions between the corners reflect the difficulty in distinguishing between two of the three trees. •For more than four sequences, the different strains can be grouped into four different subsets and all possible quartets generated by drawing one sequence from each subset can be evaluated. •The more points distribute in a certain region of a particular corner, the bigger the support for the tree topology joining the four subsets represented by that corner. •If most points locate in the center of the triangle, the four subsets are independent and related by a star-like tree. L1 A C B D L3 L2 C A D L2 B L1 B A C L3 D Quartet puzzling method The three corners of the triangle represent the three possible unrooted tree topologies for four taxa. L1, L2, and L3 represent the three likelihoods of the three trees, respectively. Each length of the perpendicular from point P to the triangle side is equal to the likelihood of the tree represented by the opposite corner. Quartet Puzzling Support Values The quartet puzzling support values can be interpreted in a similar way as bootstrap values (though they should not confused with them). Branches showing a quartet puzzling reliability > 90% can be considered strongly supported. Branches with lower reliability (> 70%) can in principle be also trusted but in this case it is advisable to check how well the respective internal branch does in comparison to other branches in the tree (i.e. check relative reliability). If you are interested in a branch with a low confidence it is also important to check the alternative groupings that are not included in the quartet puzzling tree. Analysis of the phylogenetic signal The quartet puzzling approach can be used also to visualize the phylogenetic content of a particular data set of n aligned sequences. For n sequences (n!/4!) possible quartets, exist. When n≥11, a random sample of 10000 quartets is sufficient to obtain a comprehensive picture of the kind of phylogenetic signal present. The more the dots in the center of the triangle, the more the phylogenetic noise, reflecting star-like evolution, in the data set. 1 ALLELE FREQUENCY fixed mutation polymorphis m maintained lost mutation 0 TIME • • • • • Positive selective pressure: mutant is more fit Negative selective pressure: mutant is less fit Balancing selection: heterozygote is more fit Most synonymous mutations can be considered neutral Non-synonymous mutations are always subject to selective pressure dN/dS •Are the substitutions we observe neutral or beneficial? •First, classify mutations as synonymous or non-synonymous •Synonymous substitutions are likely to be neutral •Non-synonymous substitutions may be neutral, beneficial or deleterious •Beneficial mutations spread faster than neutral ones •dN/dS=ratio of non-synonymous & synonymous substitution rates. •dN/dS = 1 if all non-synonymous substitutions are neutral •dN/dS < 1 if some non-synonymous substitutions are deleterious •dN/dS = 0 if all non-synonymous substitutions are deleterious •dN/dS > 1 only if some non-synonymous substitutions are beneficial Synonymous substitutions are not always neutral •Overlapping genes and reading frames Very common in viruses (e.g. HTLVs) •Secondary RNA or DNA structure e.g. stem-loops •Codons for the same amino acid differ in fitness May result from different translational efficiencies of tRNAs •Untranslated or regulatory regions Detecting recombination in viral sequences Consistent genome-wide HIV-1 M-group clustering Phylogenetic analysis results in similar general clustering on HIV strains irrespective of which region of the genome is used for the analysis. Jumping of recombinant isolates Recombinant isolate KAL153. Similarity and bootscanning analysis followed by phylogenetic analysis (K2P+NJ) of the identified segments. Networks for analysis of recombination Split decomposition method Evolutionary relationships between taxa are most often represented as phylogenetic trees and this is justified by the assumption that evolution is a branching or tree-like process. However, a set of real data often contains a number of different and sometimes conflicting signals and thus does not always clearly support a unique tree. The split decomposition method is a transformation-based approach. Essentially, evolutionary data is transformed or, more precisely, "canonically decomposed", into a sum of "weakly compatible splits" and then represented by a so-called splits graph. For ideal data, this is a tree, whereas less ideal data will give rise to a tree-like network that can be interpreted as possible evidence for different and conflicting phylogenies. Neighbor-Net Neighbor-Net, a distance based method for constructing phylogenetic networks that is based on the Neighbor-Joining (NJ) algorithm of Saitou and Nei. Neighbor-Net provides a snapshot of the data that can guide more detailed analysis. Unlike split decomposition, Neighbor-Net scales well and can quickly produce detailed and informative networks for several hundred taxa. Multilocus sequence typing (MSLT) to classify several hundreds Salmonella isolates. J Clin Microbiol 2002; 40: 1627-35. Comparison of K80-based NJ tree and split-decomposition phylogenetic analysis Split decomposition of the Human Bocavirus in Taiwan References Graur D and Li W-H. 2000. Fundamentals of Molecular Evolution, 2nd edition, Sinauer Associates, Sunderland, MA. Hillis D, Moritz C, and Mable BK (eds). 1996. Molecular Systematics, 2nd edition, Sinauer Associates, Sunderland, MA. Masatoshi Nei and Sudhir Kumar. 2000. Molecular Evolution and Phylogenetics, Oxford University Press, New York, NY. Philippe Lemey, Marco Salemi and Anne-Mieke Vandamme (eds). 2009. The Phylogenetic Handbook : A Practical Approach to phylogenetic analysis and hypothesis testing, Second Edition, Cambridge University Press. Pevsner J. 2009. Bioinformatics and Functional Genomics, 2nd edition, Wiley-Blackwell. International Bioinformatics Workshop on Virus Evolution and Molecular Epidemiology, 1995-2009, Rega Institute for Medical Research, Katholieke Universiteit Leuven, Leuven, Belgium. Workshop on Molecular Evolution, 1998-2008, Marine Biological Laboratory, Woods Hole, Massachusetts, USA. Liu Hsin-Fu. 1996. Genomic diversity and molecular phylogeny of human and simian T-cell lymphotropic viruses. Thesis submitted for the degree of “Doctor in Medical Sciences”, Faulty of Medicine, Katholieke Universiteit Leuven, Leuven, Belgium. Salemi Macro. 1999. Molecular investigation of the origin and genetic stability of the human T-cell lymphotropic viruses. Thesis submitted for the degree of “Doctor in Sciences”, Faulty of Science, Katholieke Universiteit Leuven, Leuven, Belgium. Note: This teaching material was prepared for the workshop only, not for formal publication, therefore did not specifically address to the references. They were all taken from the list above.