Theoretical Mechanisms Associated With Increases in HDL Levels
advertisement

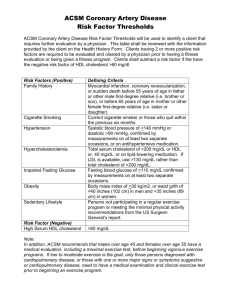
John Steele Taylor November 14, 2007 Pharmacology 201 Lifestyle Factors and Pharmacological Agents that Manage Hyperlipidemia Through Activation of Peroxisome Proliferator Activated Receptors Part 1: Introduction and Overview This paper concerns a specific class of drugs used in the treatment of hypertriglyceridemia and hypercholesterolemia that operate through activating a class of transcriptional activators known as peroxisome proliferator activated receptors (PPARs). PPARs are transcriptional activators distributed throughout a diversity of tissues that operate through orchestrated mechanisms primarily to regulate carbohydrate and lipid metabolism. Therefore, these transcriptional activators play a central role in treatment of atherosclerotic diseases and general metabolic syndromes that are highly prevalent in western populations. Clinical and epidemiological studies have thoroughly established a definite correlation between atherosclerotic related events and certain serum lipid profiles. These serum lipid profiles represent independent risk factors; however, they often accompany insulin resistance and other features of metabolic syndrome. Due to the complexity of factors and pathways involved in lipid metabolism and the pathogenesis of atherosclerosis, an introductory section is included to summarize these mechanisms for the lay-reader. Additionally, mechanisms through which diet and exercise regiments can favorably influence serum lipid profile and prevent atherosclerosis will be discussed at considerable length, both for the purpose of emphasizing their role as a first line of treatment and for elucidating some of the mechanisms that are targeted by the drugs discussed later in this paper. Once this has been accomplished, a second standalone section is devoted entirely to the discussion of PPAR activating drugs, including 1 proposed mechanisms of action, pharmacokinetic considerations, and adverse effects/toxicities. Lipoprotein Metabolism: Normal and Dysfunctional The transport of cholesterol and triglycerides through the bloodstream is complicated by the fact that these are hydrophobic molecules that need to be moved through a watery environment. Transport is therefore achieved through packaging lipids into lipoproteins, which have the general characteristic of having a protein hydrophilic outer layer and a lipid core (hence the name lipo-protein). Three important outer proteins, or apoproteins, will be considered in this paper and include apoprotein-A1 (apoA1), apoprotein-B-100 (apo-B-100), and apoprotein C-3 (apo-C3). These apoproteins are discretely associated with certain lipoproteins and are major determinants in the characteristics and function of these lipoproteins. Regulation of apoprotein expression, degradation, and exchange is an important drug target due to their influence on mechanisms involved in the development of arterial diseases (1). Two pathways of lipoprotein metabolism exist: an exogenous pathway where dietary triglycerides and cholesterol are packaged into chylomicrons in the intestinal mucosaand distributed throughout the circulation; and an endogenous pathway where triglycerides and cholesterol are packaged in to lipoproteins in the liver for distribution through the bloodstream. The endogenous pathway is our primary concern, especially due to the fact that a major fate of chylomicrons in post-prandial (post-meal) metabolism is hepatic conversion into very-low-density-lipoprotein (VLDL), which are the primary endogenous triglyceride transporter, (2). 2 Circulating VLDLs are hydrolyzed by lipoprotein lipase (LPL), an enzyme fixed on the luminar surface of vascular endothelium, into free fatty acids (FFAs) for uptake into muscle and adipose tissue. VLDL is converted to intermediate-density-lipoprotein (IDL) as it releases FFAs. IDL has two fates: degradation in the liver and conversion to low-density lipoprotein through donating apolipoprotein C-II (Apo-C2) to high density lipoproteins (HDL). In this manner, increasing LPL activity causes a decrease in VLDL and an associated increase in HDL. This pathway is a major drug target. (2). Low density lipoprotein (LDL) is responsible for the transfer of cholesterol to peripheral tissues for utilization in numerous functions including membrane synthesis and steroid hormone production. Under normal conditions most of the circulating LDL is cleared by the liver for synthesis of bile acids, which are important for emulsifying fats in the digestive tract, and represent the only avenue for removal of cholesterol from the body. Apoprotein-B (apo-B) is a major component of LDL, and cellular uptake of LDL occurs through recognition of apo-B at the receptor binding sites (2). Dysregulation of LDL function occurs when LDL suffers oxidative damage, which alters its composition to the extent that it is poorly recognized by hepatocytes. Conversely, uptake of oxidized LDL is greatly increased in scavenger cells in peripheral tissues, most notably in macrophages that are situated in the vascular endothelium (2). For this reason LDL is referred to as ‘bad-cholesterol’, and increased levels are a major risk factor for cardiovascular disease. (3) However it is probably more appropriate to refer to oxidized LDL as the bad cholesterol. Mechanisms involved in oxidation of LDL, and the pathogenesis that ensues from macrophage uptake, are discussed below. Without 3 further mention, this should highlight the role of antioxidants in foods as cardiovascular protective agents. HDL is synthesized in the liver and is primarily composed of apoprotein-a-1 (apoa1) (2). Synthesis of apo-a1 is largely regulated by PPARα, and thus exogenous activators of PPARα can directly contribute to increases in apo-a1 levels and consequently increased HDL levels (4). HDL is responsible for reverse transport of cholesterol from the periphery to the liver for elimination. This process of reverse transport is primarily mediated by apo-A1 stimulation of lecithin-cholesterol-acetyltransferase (LCAT), which esterifies free cholesterol in the periphery for removal by HDL. It also follows that apo-A1 stimulates ATP-binding cassette transporter A-1 (ABCA1), which is a transporter protein that enables this exchange of lipids from foam cells to HDL(2). Because of this mechanism, HDL is referred to as ‘good cholesterol’ and decreased levels represent a risk factor for cardiovascular disease. Increasing HDL levels is an important drug target and often occurs through degradation of VLDL, as mentioned above. Atherosclerosis Pathogenesis Atherosclerosis essentially involves the inappropriate deposition of soft lipidbased masses, or atheromas, into the walls of the arterial lumen. These lipids are primarily derived from circulating LDL. Atheromas start as fatty streaks and then progress to plaques, which are characterized by accumulation of foam cells (macrophages that are filled with lipids), proliferation of smooth muscle cells, and increased deposition of collagen and other debris that progressively narrow and ultimately occlude the artery (1). Consequences of this narrowing include impaired capacity to respond to vasodilators 4 such as nitric oxide, diminished nutrient supply to associated tissues; and the preparation of an environment for circulating blood clots to lodge thus causing acute ischemic events. Finally, destabilized plaque can rupture, which initiates a clotting cascade that can suddenly and completely occlude the artery (3). Chronic mild vascular inflammation is an independent risk factor for cardiovascular events. Cytokines (immune signaling chemicals) activate proinflammatory genes and vascular cell adhesion molecules (VCAMs), which allow neutrophils to adhere to vascular walls and exert their inflammatory activity thus furthering the local atherogenesis (5). Cytokine release from macrophages may be induced by transcriptional activators released by oxidized LDL (2), however in the obese state adipose tissue also releases proinflammatory compounds (7). Mechanisms for reducing inflammation via diet, exercise, and drug therapy will also be explored in later sections and is a major feature of cardioprotective therapy mediated through non-lipid pathways. Atherosclerosis, particularly in the coronary and cerebral arteries, is a silent killer because no pain or prior symptoms need be necessary for a lethal event to occur as a result. Indeed, over 50% of sudden death from cardiovascular disease occurs in individuals with no previously recognized symptoms, and an artery can be over 70% obstructed before symptoms (such as angina) develop. Alarmingly, although most cardiovascular events are suffered in adulthood, atherosclerosis starts in childhood and progresses throughout life. Autopsies performed on children and adolescents reveal formation of fatty streak as early as 3 years of age, and adolescents with arterial obstruction so significant that they could have suffered myocardial infarction (3). Based 5 on this information the importance of regular preventative measures achieved through exercise and diet cannot be overemphasized. Effects of Exercise Regular physical activity can profoundly reduce cardiovascular risk through altering the dynamics of lipoprotein metabolism. This is best explained by summarizing the mechanisms through which type two diabetes and metabolic syndrome (a complex characterized by insulin resistance, obesity, hypertension, and dyslipidemia) can profoundly increase cardiovascular risks, because these conditions are associated with a sedentary lifestyle. Not all people with cardiovascular disease have diabetes, however, it is estimated by the American Heart Association that around 50 million people in the United States have metabolic syndrome (6), while 65 million people in the US suffer from some sort of cardiovascular disease (3). The discussion that follows will highlight this close correlation. Type 2 diabetes and metabolic syndrome are both characterized by insulin resistance. Insulin is essential for the cellular uptake of dietary fat, protein, and glucose following a meal (during the post-prandial state) (3). This is important not only for the effective storage and utilization of nutrients for tissue synthesis and cellular energy, but for the often overlooked protection against the pathogenic consequences of pronounced and prolonged increases in blood glucose levels becomes toxic to tissues. The interaction of glucose with tissues and circulating compounds leads to the formation of advanced glycated end-product (AGE) complexes, which disrupt normal tissue function in a broad variety of ways. For our purposes, the glycation of circulating LDL renders it far more susceptible to oxidative damage, thus increasing its deposition into vascular endothelium. 6 Additionally, gylcation of HDL may inhibit its ability for reverse transport of cholesterol (2). AGE complex formation is greatly accelerated in a hyperglycemic environment, which is a hallmark of insulin resistance. Under normal responsiveness to insulin, glucose uptake is dramatically increased in skeletal muscle and hepatic tissue, which stabilizes blood glucose levels following a post-prandial spike. This occurs through the translocation of GLUT-4 receptors to the surface of these cells, which facilitate the diffusion of glucose into the cytoplasm. Regular exercise favorably enhances this mechanism, resulting in increased sensitivity to insulin, increased number and translocation of GLUT-4, and increased glycogen synthase activity (3). Therefore regular exercise profoundly reduces post-prandial hyperglycemia and consequently reduces excessive glycation of LDL and HDL. On the other side of the equation, a sedentary lifestyle results in an insulin resistant state, in which post-prandial blood glucose levels are slow to stabilize and remain high post-absorptively, and thus LDL and HDL are excessively glycated. Fat metabolism is heavily dictated by insulin activity as well as through adaptations to exercise. LPL activity is regulated by insulin in order to rapidly clear postprandial triglycerides: insulin stimulates LPL to liberate free fatty acids (FFA’s) from VLDL for uptake into adipose tissue for storage. As fat reserves increase, adipose tissue releases factors that promote insulin resistance (7). In the insulin resistant state, circulating triglycerides increase and glycogen reserves are diminished due to insufficient uptake by in skeletal muscle and hepatic tissue. As a consequence highly metabolically active tissues in the central nervous system and cardiac muscle must rely 7 more on FFA’s and ketone bodies as a fuel (3). FFA flux to the liver signals the liver to increase output of apo-B (and thus increased LDL formation). This combined with reduced LPL activity leads to significant increases in VLDL and LDL (2). These trends and vicious cycles are reversible through regular exercise. As already mentioned, skeletal muscle increases its sensitivity to insulin thus improving post-prandial glucose clearance. Trained muscle also increases its LPL activity thus reducing VLDL levels. A major training response is improved utilization of FFA’s as an energy source at higher levels of intensity. Recent studies suggest that this effect is likely mediated via PPARδ activation in skeletal muscle. This results in increased LPL activity and improved mitochondrial populations, which is accompanied by enhanced oxidation of fatty acids as mediated by upregulation of carnitine palmitoyl transferase activity (CPT1) – a protein responsible for the transport of fatty acids across mitochondrial membranes. Because these mechanisms ultimately lead to increases HDL levels, PPARδ agonist drugs mimic favorable exercise-induced alterations in lipid metabolism (8). FFA utilization increases in proportion to exercise intensity as intensity approaches 50% of Vo2 max, and then FFA utilization declines as higher intensities are achieved (7). Thus exercise at this moderate intensity level is desirable. However higher intensity resistance training-induced increases in muscle mass also confer benefits in metabolic syndrome via separate mechanisms not discussed. Reduction of fat stores in adipose tissue through negative energy balance achieved through regular exercise leads to decreased output of proinflammatory compounds such as resistin and interleukin-6. The level of secretion stands in direct relationship to fat reserves such that decreased reserves also decrease proinflammatory 8 output (7). This is crucially important because, as discussed above, inflammation is a prominent risk factor for cardiovascular events. Effects of Diet The ultimate goal of lifestyle and pharmacological interventions is to increase hepatic clearance of cholesterol from the general circulation. However, once this has been accomplished, cholesterol must still be eliminated, which is problematic because cholesterol cannot be degraded in the body. Elimination occurs through hepatic conversion of cholesterol to bile acids, which are delivered to the gallbladder and ultimately secreted into the small intestine to assist in the emulsification of fats. However, most bile acids are reabsorbed at the ileum of the small intestine and returned to the liver – a process known as enterohepatic cycling. In fact, bile acids may be recycled up to 18 times before they are finally eliminated in the feces(1). Bile-acid binding resins are a major class of lipid-lowering drugs that work well in combination with other interventions because they bind with bile-acids in the small intestine and increase their elimination in the feces. Consequently the liver must upregulate LDL receptors in order to pull more cholesterol from the circulation in order to maintain bile-acid levels (9). Clearance is enabled through upregulation of cholesterol 7 alpha-hydroxylase, which is the rate limiting enzyme in bile-acid synthesis, and this mechanism is a prominent indirect effect of these drugs (10). A similar mechanism has been demonstrated for dietary fiber, such that dietary fiber (particularly soluble fibers – lignans, gums, pectins, etc.) can prevent the reabsorption of bile-acids and therefore also result in increased LDL clearance and cholesterol 7 alpha-hydroxylase activity. Additionally, the binding of nutrients such as 9 starches and sugars in the small intestine by dietary fibers slows their absorption thus mitigating post-prandial spikes in blood glucose levels (11). Reductions in dietary cholesterol and saturated fat intake are generally recognized preventative dietary strategies for reducing the risk of heart disease. However, supplementation with certain omega-3 fatty acids may confer much more specific benefits. Most clinical evidence suggests that these effects are specifically derived from two omega-3 fatty acids found in fish oils – eicosapentanoic acid (EPA) and docosahexanoic acid (DHA) - and not necessarily from vegetable sources of omega-3’s (although more research is necessary regarding potential benefits of vegetable omega3’s). The primary mechanism of action involves gradual displacement of arachidonic acid from phospholipid membranes by DHA and EPA. As a consequence, eicosanoids become increasingly derived from these fatty acids instead of from arachidonic acid. A 3series of prostanoids and thromboxanes are derived from EPA and DHA as opposed to the 2-series derived from arachidonic acid, and as a general trend these 3-series derivatives tend to have a far less pronounced vasoconstricting and platelet-aggregating effect of the 2-series derived from arachidonic acid because they are much less active at receptor sites (12). Inflammatory pathways are also altered as EPA/DHA yield a 5-series of leukotrienes that competitively inhibit AA-derived 4-series leukotrienes at their binding sites, thus producing an anti-inflammatory and anti-allergenic effect. EPA and DHA may also operate as ligands for PPARα, thus promoting increased levels of apo-A1, decreased levels of apo-B, and increasing LPL activity. Activation of PPARα may also inhibit smooth muscle proliferation, which is a feature of the pathogenesis atherosclerotic 10 plaques. Adverse effects of omega-3 fat supplementation include increased bleeding due to an anti-aggregating effect and the potential for increased oxidation of LDL as they are polyunsaturated fats and thus highly prone to oxidative damage (12). Part 2: PPAR Active Drugs Peroxisome proliferator activated receptors are transcriptional activators distributed throughout a diversity of tissues that operate together to produce orchestrated effects that regulate lipid and energy metabolism. Three major subunits have been identified: alpha, gamma, and delta – the former two have been exploited by marketed pharmaceuticals, while drugs that operate on PPARδ are still in the advanced phases of development. PPARγ coactivator-1-alpha (PGC1-A) is also of interest as it is activated by nitric oxide and mediates exercise-induced proliferation of mitochondria in skeletal muscle(13). PPARα and Fibric Acid Derivatives The class of lipid lowering drugs known as fibric acid derivatives (FAD’s), or fibrates, are activating ligands for PPARα. Activation results in increased expression of LPL in multiple tissue and consequently decreases circulating VLDL. Accompanying this mechanism is a moderate increase in circulating HDL, both through stabilization of HDL by VLDL hydrolysis, and increased hepatic expression of apo-A1 which improves reverse transport of cholesterol. Improved LPL activity with FAD’s may occur via reduction in the levels of apoliprotein C-III (apo-CIII), which is a component of VLDL’s and has been demonstrated to inhibit the activity of LPL (2). Similar to the statin drugs, FAD’s have non-lipid benefits for prevention of cardiovascular disease. PPARα expression in vascular endothelial cells regulates 11 inflammatory processes through complex mechanisms that suppress proinflammatory pathways, vasoconstriction, and platelet aggregation (5). Gemfibrozil has two approved clinical uses in the US: 1) the treatment of patients with high triglyceride levels who are at increased risk for developing pancreatitis and 2) patients with high serum triglycerides and low HDL levels. FAD’s should not be the first line of defense for patients with hypercholesterolemia but low serum triglycerides. FAD’s are an excellent combination drug, either with statin drugs or bile-acid binding resins (11) PPARgamma and Thiazolidinediones The class of lipid lowering and anti-diabetic drugs known as thiazolidinediones or glitazones are activators of PPARγ. In adipose and muscle tissue, this effect appears to increase the output of LPL and thus improves insulin sensitivity and post-prandial clearance of glucose and triglycerides. GLUT-4 transporter activity in adipocytes is also enhanced. Additionally, PPARγ activation may lead to favorable redistribution of fat stores from visceral adipose tissue to subcutaneous adipose tissue (14). Similar to FAD’s and statins, glitazones also provide non-lipid benefits through suppression of proinflammatory signaling pathways, mediated through their activity at PPARγ binding sites in vascular endothelial cells and macrophages. Specifically PPARγ expression in macrophages and endothelial cells increases the efflux of cholesterol for reverse transport via HDL (4). Major side effects primarily concern increased plasma volume and consequently an increased risk for hospitalization due to congestive heart failure (14). 12 Currently in the developmental phases is a drug referred to as propionic acid derivative 8 (compound 8). Compound 8 has both PPARα and PPARγ activity, and is thus a promising drug for reducing atherosclerosis and dyslipidemia in patients with type 2 diabetes mellitus (4). PPARδ and GW501516 In contrast to PPARα and PPARγ activators, PPARδ activating drugs are still in the developmental stages. GlaxoSmithKline is developing a drug currently referred to as GW501516, which is in phase II of development. GW501516 shows promise for treating hyperlipidemia through a variety of mechanisms. Dramatic increases in HDL levels have been observed in human and primate studies with GW501516 (4, 8). This effect is primarily achieved through increased hydrolysis of VLDL that ultimately occurs through upregulation in oxidative activity of skeletal muscle mitochondria such that FFA utilization as a fuel is enhanced. Additional lipid associated benefits include increased expression of apo-1A and ATP-binding cassette transporter A-1 (ABCA1). ABCA1 is a protein that enables the exchange of lipids from foam cells to HDL. Thus HDL levels and efficacy are increased by GW501516 (8). Activation of PPARδ may also provide important cardioprotection with specific regard to reperfusion-induced oxidative stress. As blood flow is returned to ischemic tissue, oxidative stress and inflammation can occur to the extent that they are more damaging than the actual ischemia. PPARδ mediated protection occurs through upregulation in antioxidant mechanisms and inhibition of cardiac myocyte apoptosis triggered by oxidative stress (15). Additionally, GW501516 may inhibit cardiac fibrosis 13 through the inhibition of cardiac fibroblast collagen synthesis and fibroblast proliferation. Thus, this drug has a potential role in the treatment and prevention of congestive heart failure (16). Possible adverse effect for GW501516 involves its role as an angiogenic agent. Excessive angiogenesis is associated with increased inflammatory activity and tumor growth. In contrast to PPARα and PPARγ, which appear to be anti-angionenic through their impact on eicosanoid activity, PPARδ stimulates vascular endothelial growth factor (VEGF), which suggests that patients at risk for angiogenesis be increasingly monitered if taking PPARδ’s such as GW501516. However, GW501516 may also represent a novel and inadvertent pharmacological agent for promoting wound healing and treating ischemic heart disease precisely due to its angiogenic properties (17). Conclusions Exciting new lines of therapy are emerging for the treatment of dyslipidemia, and appear to operate through similar mechanisms and receptor activity. It is likely that as advances are made in exercise physiology to understand the role of these receptors in favorable adaptations to physical activity, increasingly precise and effective pharmacological agents can be developed. It has also been demonstrated that combination of exercise and diet produces a broader range of benefit that are generally more potent and without side-effect. This justifies the generally accepted protocol of diet and exercise as the first line of treatment in hyperlipidemic individuals, and pharmacological intervention only if lipid levels fail to respond to these lifestyle changes. 14 References 1. McKee T and McKee JR. (2003) Biochemistry: The Molecular Basis of Life. 3rd edition. Boston: McGraw Hill 2. Levy DM and GaltonD. (2006) Diabetes, Lipids, and Atherosclerosis. In DeGroot LJ and Jameson JL Endocrinology 5th edition. Volume 3. (pp. 2587-607) Philadelphia: Elsevier Saunders. 3. McArdle WD, Katch FI, and Katch VL. (2007) Exercise Physiology: Energy, Nutrition, and Human Performance. 6th Edition. Philadelphia: Lippincott Williams & Wilkins. 4. Frishman WH, Choi AY, and Guh A. (2003) Innovative Medial Approaches for the Treatment of Hyperlipidemia. In Frishman WH, Sonnenblick EH, and Sica DA. Cardiovascular Pharmacotherapeutics. 2nd edition. (pp. 841-53) New York: McGraw Hill. 5. Cuzzocrea S, Mazzon E, Di Paola R, Peli A, Bonato A, Britti D, Genovese T, Muià C, Crisafulli C and Caputi AP. The role of the peroxisome proliferator-activated receptor(PPAR- ) in the regulation of acute inflammation. Journal of Leukocyte Biology. 2006;79:999-1010.) Internet: http://www.jleukbio.org/cgi/content/full/79/5/999 (accessed 14 November 2007) 6. The American Heart Association. (2007) Metabolic Syndrome. Internet: http://www.americanheart.org/presenter.jhtml?identifier=4756 (accessed 14 November 2007) 7. Houston M. (2006) Biochemistry Primer For Exercise Science. 3rd Edition. Blacksburg: Human Kinetics. 8. Sprecher DL, Massien C, Pearce G, Billin AN, Perlstein I, Willson TM, Hassall DG, Ancellin N, Patterson SD,Lobe DC, Johnson TG. (2006) Triglyceride:High-Density Lipoprotein Cholesterol Effects inHealthy Subjects Administered a Peroxisome Proliferator Activated Receptor _ Agonist. Arterioscler Thromb Vasc Biol. February 2007. 359-65 Internet: http://atvb.ahajournals.org/cgi/reprint/27/2/359 9. Malloy MJ & Kane JP. (2007) Agents Used In Hyperlipidemia. In Katzung BG. Basic & Clinical Pharmacology, 10th Edition McGraw Hill. Internet: http://www.accessmedicine.com/content.aspx?aID=2507047 (Accessed 15 November 2007) 10. Trautwein EA, Kunath-Rau A and Erbersdobler HF. Increased Fecal Bile Acid Excretion and Changes in the Circulating Bile Acid Pool Are Involved in the Hypocholesterolemic and Gallstone-Preventive Actions of Psyllium in Hamsters. Journal of Nutrition. 1999;129:896-902. Internet: 15 http://jn.nutrition.org/cgi/content/full/129/4/896?maxtoshow=&HITS=10&hits=10&RES ULTFORMAT=&searchid=1&FIRSTINDEX=0&minscore=5000&resourcetype=HWCI T (Accessed 15 November 2007) 11. Schocter NS, Zemetbaum P, and Frishman WH. (2003) Lipid Lowering Drugs In Frishman WH, Sonnenblick EH, and Sica DA. Cardiovascular Pharmacotherapeutics. 2nd edition. (pp. 317-53)) New York: McGraw Hill. 12. Kruger NA, Frishman WH, and Hussain J. (2003) Fish Oils, the B-Vitamins, and Folic Acid as Cardiovascular Protective Agents. In Frishman WH, Sonnenblick EH, and Sica DA. Cardiovascular Pharmacotherapeutics. 2nd edition. (pp. 381-406) New York: McGraw Hill. 13. Brown GC. NO Says Yes To Mitochondria. Science 7 February 2003: Vol. 299. no. 5608, pp. 838 - 839 Internet: http://www.sciencemag.org/cgi/content/full/299/5608/838 (Accessed 15 November 2007) 14. Martha S. Nolte MS & Karam JH. (2007) Pancreatic Hormones & Antidiabetic Drugs. In Katzung BG. Basic & Clinical Pharmacology, 10th Edition McGraw Hill. Internet: http://www.accessmedicine.com/content.aspx?aID=2507047 (Accessed 15 November 2007) 15. Pesant M, Sueur S, Dutartre P, Tallandier M, Grimaldi PA, Rochette L, Connat JL. (2006) Peroxisome proliferator-activated receptor delta (PPARdelta) activation protects H9c2 cardiomyoblasts from oxidative stress-induced apoptosis. Cardiovasc Res. 2006 Feb 1;69(2):440-9. Internet: http://www.ncbi.nlm.nih.gov/sites/entrez?Db=pubmed&Cmd=ShowDetailView&TermT oSearch=16337160&ordinalpos=1&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_Res ultsPanel.Pubmed_RVAbstractPlus (Accessed 15 November 2007) 16. Teunissen BEJ, Smeets PJH, Willemsen PHM, Windt LJD, der Vusse GJVand Bilsen MV.(2007) Activation of PPARδ inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovascular Research Volume 75, Issue 3, 1 August 2007, Pages 519-529 Internet: http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T14-4NMSRX92&_user=1563816&_coverDate=08%2F01%2F2007&_rdoc=1&_fmt=&_orig=search&_ sort=d&view=c&_acct=C000053744&_version=1&_urlVersion=0&_userid=1563816& md5=a1b66c39e658e799380d3fe353113357 (Accessed 15 November 2007) 17. Laura Piqueras, Andrew R. Reynolds, Kairbaan M. Hodivala-Dilke, Ara´ntzazu Alfranca, Juan M. Redondo, Toshihisa Hatae, Tadashi Tanabe, Timothy D. Warner, David BishopBailey. (2006) Activation of PPARβ/δ Induces Endothelial Cell Proliferation and Angiogenesis. Arterioscler Thromb Vasc Biol. January 2007 pp.63-9. Internet: http://atvb.ahajournals.org/cgi/reprint/27/1/63 (Accessed 15 November 2007) 16