Supplementary Information (doc 68K)
advertisement
")
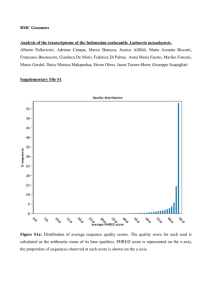
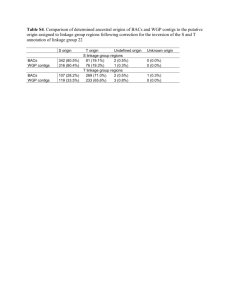
STRAIN-LEVEL GENOMIC VARIATION IN NATURAL POPULATIONS OF LEBETIMONAS FROM AN ERUPTING DEEP-SEA VOLCANO Julie L Meyer and Julie A Huber Josephine Bay Paul Center, Marine Biological Laboratory, Woods Hole, MA, USA Supplementary Material: Growth of Lebetimonas under different conditions The enrichment media consisted of anaerobic saltwater with H2 as an electron donor, elemental sulfur as an electron acceptor, ammonia as a nitrogen source, CO2 as a carbon source, trace elements, vitamins, and 0.1g/L yeast extract. All six isolated strains grew with or without the low level of yeast extract used in the enrichment media, but did not grow without vitamins. Lebetimonas strain JS085 was transferred to media without yeast extract prior to testing the following alternative electron donor/acceptor pairs: H2/NO3, formate/S0, formate/NO3, thiosulfate/NO3, and thiosulfate/O2. Growth of Lebetimonas strain JS085 was evaluated at pH levels of 5, 5.5, 6, 6.5, 7, 7.5, 8, and 8.5 and at temperatures from 25°C to 70°C, at 5°C intervals. Lebetimonas strain JS085 could not use thiosulfate as an electron donor nor use nitrate or oxygen as an electron acceptor under the conditions tested. Growth in strain JS085 occurred at pH 6 to 8 and at temperatures from 40°C to 60°C. Cultures grown with the original enrichment media at 55°C became turbid within 24 hours, with a doubling time of 3 hours in strain JS085 under these conditions. Cultures without yeast extract grew much slower, taking up to a week to become turbid. Cells were motile, short, curved rods, about 1 µm in length. Genome assembly Prior to assembly, Illumina sequences were quality filtered using adaptive window trimming and a quality threshold of 30 using the script Trim.pl (http://wiki.bioinformatics.ucdavis.edu/index.php/Trim.pl). All reads were screened for adaptor, barcorde, primer, and transposan sequences and trimmed as needed using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html). De novo genome assembly was performed with several assembly programs. Sequences generated through the 454 platform were first assembled with Roche’s GS De Novo Assembler v 2.6 (“Newbler”) 2 using default parameters. De novo assemblies of 454 reads were also performed using mira 3 with the default settings for normal quality de novo genome assembly. De novo assembly of subsets of Illumina reads was performed with velvet 4, using an estimated coverage of 1000x, kmer size of 21, and a coverage cutoff of 5). Large contigs from Newbler , mira, and velvet were consolidated using Geneious Pro v 5.6.6 (Biomatters, Ltd, http://www.geneious.com) and aligned with progressiveMauve 5 to visualize the relationship of large contigs from different assemblies and to identify gaps to close. Primers were designed at the ends of contigs using either Geneious Pro or CLC Genomics Workbench v 5.1 (CLCbio, http://www.clcbio.com) to amplify gaps between contigs. Positive PCR amplification products linking contigs were cleaned using a Min-Elute PCR Purification kit (Qiagen) and Sanger sequenced. A nearly complete draft genome from strain JS085 served as a reference genome for the remaining five strains. Both Illumina and 454 reads were mapped to the reference genome with CLC Genomics Workbench. Unmapped reads were then assembled de novo to ensure that novel genomic content in the mapped strains was not overlooked. De novo assembly of 454 and/or Illumina reads for each strain was also performed in CLC Genomics Workbench and compared to the mapped assemblies using progressiveMauve. Four of the strains were sequenced using both 454 and Illumina and two strains were sequenced only with Illumina. The sequencing coverage depth of quality-filtered reads ranged from 22X to 50X for 454 and up to 3618X for Illumina (Table 2 in main text). Lebetimonas strain JS085 had the highest coverage of 454 reads and was assembled into 33 large contigs with Newbler and 1747 contigs with mira. The 20 largest contigs from each of these assemblies were consolidated using de novo assembly in Geneious to 10 contigs. An additional round of assembly in Geneious with the 10 consolidated contigs and velvet contigs greater than 10 Kbp further consolidated the draft genome to 6 contigs. Primers were designed for all possible combinations between the 6 contigs. One gap was closed using Sanger-sequenced positive pcr products. Finally, all 454 and Illumina reads for strain JS085 were mapped to the draft genome consisting of 5 contigs and the resulting consensus was used as the final draft genome. The five remaining genomes were assembled by mapping 454 and Illumina reads to the JS085 reference genome in CLC Genomics Workbench. Hybrid de novo assemblies in CLC Genomics Workbench of each strain did not extend contigs or close gaps between the 5 contigs of the draft genomes. Assemblies of unmapped reads produced only short contigs with no significant similarities using nucleotide BLAST 6. 1. Mehta, M. P., Butterfield, D. A. & Baross, J. A. Phylogenetic Diversity of Nitrogenase ( nifH ) Genes in Deep-Sea and Hydrothermal Vent Environments of the Juan de Fuca Ridge. Applied and Environmental Microbiology 69, 960–970 (2003). 2. Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–80 (2005). 3. Chevreux, B., Wetter, T. & Suhai, S. Sequence assembly with MIRA3 The Definitive Guide. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB) 45–56 (1999). 4. Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18, 821–9 (2008). 5. Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS one 5, e11147 (2010). 6. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410 (1990). Table S1. Proportion of shared genes with >60% identity in Lebetimonas strains. JH292 JH292 JH369 JS032 JS085 JS138 JS170 96.0% 98.8% 98.8% 99.5% 99.7% JH369 92.8% 99.0% 99.8% 98.9% 100.7% JS032 90.2% 93.2% 96.0% 96.2% 96.9% JS085 90.5% 94.0% 96.4% 96.4% 97.8% JS138 89.1% 91.5% 94.1% 94.1% JS170 89.8% 93.3% 95.7% 96.2% 95.6% 94.9% Table S2. Average nucleotide identity (ANI) of shared genes in Lebetimonas strains. JH292 JH292 JH369 JS032 JS085 JS138 JS170 95.7% 95.8% 95.8% 96.0% 95.8% JH369 96.1% 98.5% 99.2% 99.3% 99.3% JS032 98.8% 98.4% 98.9% 98.3% 98.9% JS085 96.3% 99.3% 99.0% 99.0% 99.9% JS138 96.5% 99.2% 98.4% 98.9% JS170 96.4% 99.3% 99.0% 99.9% 99.0% 98.9% Figure S1. Photos of representative diffuse flow vents at NW Rota-1 Seamount. A) Arrowhead. B and C) Marker 109. D) Sulfur Crust. Figure S2. Dotplot created with nucmer comparing the nucleotide sequences of the two least similar genomes, Lebetimonas strains JS085 and JH292, showing the synteny of the draft genomes.