jws-prot.21160.lgd
advertisement

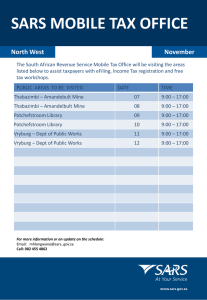
Insight into the Activity of SARS Main Protease: Molecular Dynamics Study of Dimeric and Monomeric Form of Enzyme Kewen Zheng1 2*, Guozheng Ma1, Jinming Zhou2, Min Zen1, Wenna Zhao1, Yongjun Jiang1, Qingsen Yu1 1 Key Laboratory for Molecular Design and Nutrition Engineering of Ningbo City, Ningbo Institute of Technology, Zhejiang University, Ningbo 315100, China 2 Department of Computer Chemistry and Cheminformatics, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China Author for correspondence. E-mail:zhengkw@126.com. Phone: 86-21-54925266. Fax: 86-21-54925264. The rmsd of the backbone heavy atoms during MD simulations of protomer A and protomer B relative to the crystal structure are shown in Fig. 1. It can be seen that the fluctuation of rmsd values is small after 2.5 ns, meaning that protomer A and protomer B have become stable in solution. Fig.2 compares the calculated B-factors of protomer A with the protomer B. It is obvious from the plot in Fig.2 that the B-factor of the segment 1-170 of protomer A is higher than that of protomer B, meaning that the segment 1-170 of protomer A shows greater motions than protomer B, especially for the key residues mentioned in article. 3.5 4.0 (b) 3.5 2.5 3.0 R.M.S.D. ( Å) 3.0 2.0 1.5 1.0 0.5 2.5 2.0 1.5 0 1000 2000 3000 4000 1.0 5000 0 1000 2000 3000 4000 5000 Simulation times(ps) Simulation times(ps) Fig 1. Root-mean-square deviations (rmsd) of the heavy atoms of the backone of (a) protomer A and (b) protomer B relative to the SARS Mpro crystal structure during simulation. 200 B-factor( Å2) R.M.S.D. ( Å) (a) 150 100 50 0 50 100 150 200 250 300 Residue number Fig 2. Fluctuation of SARS Mpro Ca atom computed from dynamics dimulations (blue line corresponding to protomer A and red line corresponding to protomer B). Dynamics Behavior of Catalytic Site In the MD simulation of protomer B complexes, the distance between NE2 of His41 and SG of Cyx145 averages 4.51 Å (Fig. 3b), close to the averaged distance in protomer A, 4.75 Å. These longer 10 Distance NE2(41)…SG(145)( Å) Distance NE2(41)…SG(145)( Å) distances both deviate a lot from the experimental observations of the active enzyme made by X-ray (a) 8 6 4 2 0 0 1000 2000 3000 4000 5000 10 (b) 8 6 4 2 0 0 1000 Simulation times(ps) 2000 3000 4000 5000 Simulation times(ps) Fig 3. Time-dependent variation of distance between NE2 of His41 and SG of Cys145 in (a) SARS Mpro protomer A complex and (b) protomer B complex. crystallography and might be unfavorable to substrate hydrolysis by SARS main proteinase by a general-base mechanism. This might decrease the capability of substrate hydrolysis by SARS main proteinase. Conformational Characteristics of His163 and Glu166 From the analysis in this article, we have known that in order to interact with the glutamine at substrate P1 site, the imidazole ring of His163 has to maintain the conformational stability. This is achieved by two interactions: i) stacking onto the phenyl ring of Phe140; and ii) accepting a hydrogen bond from the Tyr161 hydroxyl group which has no other hydrogen-bonding partner. (b) (a) His163 His163 Tyr161 Tyr161 Phe140 Phe140 Fig 4. The structures of the residues around His163. Comparison of the residues around His163 in the MD averaged structure of (a) protomer A and (b) protomer B complexed with substrate analog. In the crystal structure of protomer B, in contrast to protomer A, Phe140 no longer stacks against His163, but undergoes a dramatic conformation variation, with the phenyl ring moving by as much as ~14 Å. During the MD simulation of protomer B complex, Phe140 remains in the conformation and cannot stack against His163 (Fig. 4b). Despite the efficient stack with His163 in the crystal structure of protomer A, the active form of SARS Mpro, during the MD simulation of protomer A complex, the phenyl ring of Phe140 has a ~40°increment in the dihedral angle CD1-CG-CB-CA and rotates away from the imidazole ring of His163, resulting in the destruction of stacking against His163 (Fig. 4a). This is in general similar with protomer B and the conformational difference is probably due to that the time scale of both simulations is not long enough. The distances between OH of Tyr161 and ND1 of His163 during MD simulations of protomer A and protomer B are shown in Fig. 5. It can be seen that the distance in protomer B is near that in protomer A, with averaged distances of 4.01 Å and 4.21 Å, Distance OH(161)-ND1(163) (Å) Distance OH(161)-ND1(163) (Å) both cannot form the hydrogen bond with His163. 10 (a) 8 6 4 2 0 0 1000 2000 3000 4000 10 (b) 8 6 4 2 0 5000 0 1000 Simulation times(ps) 2000 3000 4000 5000 Simulation times(ps) Fig 5. Separation between OH(Tyr161) and ND1(His163) for the simulation time from 0 to 5000 ps of (a) SARS Mpro protomer A complex and (b) protomer B complex. From above analysis, we have known that the interactions required for maintaining the conformational stability of His163 in averaged MD structures of both protomer A and protomer B are all destroyed and the conformation of His163 becomes unstable, which should be one possible reason that makes SARS main proteinase inactive. There also exist similar conformational variations of Glu166 between protomer A and protomer B. In the crystal structure of protomer B, in contrast to protomer A, Glu-166 no longer forms a salt bridge with His172 but reorientates to interact instead with His163 [1]. During the MD simulation of protomer B, Glu166 keeps the reorient conformation, which can be monitored by the distance between Glu166 and His172. For the majority of the simulation time (Fig. 6b), the distance concentrates mainly at the value around 6.87Å, which is considered as one of the properties of inactive SARS M pro [1]. Fig. 6a is a plot of the distance between Glu166 and His172 in protomer A, although Glu166 in protomer A doesn’t turn in the initial crystal structure, it does so in MD simulation till it is similar to protomer B and makes Distance OH(161)-ND1(163) (Å) Distance OH(161)-ND1(163) (Å) SARS main proteinase inactive. 12 (a) 10 8 6 4 2 0 1000 2000 3000 Simulation times(ps) 4000 5000 12 (b) 10 8 6 4 2 0 1000 2000 3000 4000 5000 Simulation times(ps) Fig 6. Separation between OE2(Glu166) and NE2(His172) for the simulation time from 0 to 5000 ps of (a) SARS Mpro protomer A complex and (b) protomer B complex. Conformational Variations of Substrate-Binding Pocket In the crystal structure of protomer B, the whole 138–143 loop undergoes a dramatic rearrangement and has consequences for the collapse of “oxyanion hole”, comprising Gly-143 and Cys-145. Gly143 moves by ~3 Å toward the active site, relative to its counterpart in protomer A. During the MD simulation of protomer B, the “oxyanion hole” remains collapsed and leaves no space to accommodate a tetrahedral reaction intermediate (Fig. 7b). Fig. 7a is the averaged structure of protomer A, although “oxyanion hole” in protomer A doesn’t collapse in the initial crystal structure, it does so in MD simulation till it is similar to protomer B, which might make the SARS main proteinase inactive. (a) (b) Oxyanion hole Oxyanion hole Fig 7. Schematic presentation of the MD averaged structures of substrate-analog binding site of the SARS Mpro (a) protomer A complex and (b) protomer B complex. From the above analyses, we have known that despite the differences between the crystal structures of protomer A and Protomer B, the similar ultimate conformations of the residues key to the activity of SARS Mpro have been got through the molecular dynamics simulation. The reasons that make monomer inactive might be the same as that for inactive protomer B in the crystal structure. References 1. Yang HT, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z.The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. PNAS 2003; 100:13190-95.