The reference genome annotation project
advertisement

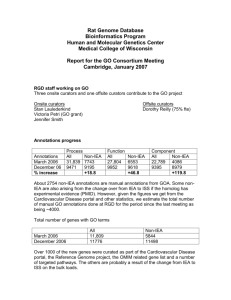
The GO Reference Genome Annotation Project The Reference Genome Group of the Gene Ontology Consortium° *Corresponding author: Pascale Gaudet Northwestern University, Chicago, Illinois, USA pgaudet@northwestern.edu Page 1 Abstract The goal of the Reference Genome project is to provide a set of high quality gene annotations from a number of biologically diverse model organisms selected because of their considerable bodies of experimental literature and expert bio-curation teams. Towards this end we are engaged in a focused effort to significantly improve the depth and breadth of Gene Ontology (GO) annotations for twelve genomes through coordinated annotation. Concurrent annotation by multiple curators is improving annotation consistency across genome databases, and is providing important improvements to GO's logical structure and biological content. We expect this effort to increase the confidence of transitive annotation, that is, annotation by inference of newly sequenced genomes, since a comprehensive body of experimentally based annotations will be available for these twelve well-studied organisms. Our purpose in this paper is to introduce this project and discuss preliminary results. Background and Motivation The functional analysis of gene products (both proteins and RNAs) is a major endeavor that requires a judicious mix of experimental and computational tools. The Gene Ontology (GO) Consortium is a collaborative effort committed to providing structured vocabularies for the annotation of the functional role of gene products in a controlled, systematic way and in a species-neutral manner (Ashburner et al. 2000, Gene Ontology Consortium 2008). Experimentally determined functions from the biomedical literature are manually curated by database curators using GO terms to create descriptive annotations of the gene products. This annotation task is carried out by curators, a word whose root comes from the Latin cure: to look after and preserve. A curator in the sense used here is a Ph.D. trained professional life scientist whose task is to preserve published, and in some cases unpublished, biological data by abstracting and integrating them into a database. Future biological research is dependent on the availability of well-structured representations of biological data with detailed, accurate descriptions (Bourne 2006, Hower et al. 2008) provided by the curators of the data repositories. The annotations created by the curators provide a solid, dependable substrate for downstream computational analyses to automatically infer the functions of gene products that are as yet uncharacterized experimentally. GO is an essential semantic resource for curators and one of the most widely used tools for functional annotation. Each uniquely defined term in the hierarchically structured GO can be used across organisms and research areas, thus supporting powerful computational and comparative analyses of high-throughput genomic datasets. Highquality manual annotation by experts is an absolute prerequisite for seeding this system and, other than the major model organism database (MOD) projects, very few research communities have the resources or expertise to perform this labor-intensive task. Therefore, the functional annotation of other genomes typically relies on automated methods that provide the transitive inheritance of annotations from related genes for which reliable annotations are available. To address the important and much-discussed issue of errors arising from transitive annotation (see, for example, Smith 1996; Smith 1997, Wheeler and Boguski 1998; Iliopoulos et al. 2003; Artamonova et al. 2005), the GO Consortium was determined to provide the larger research community with a comprehensive set of complete annotations for key reference organisms. The nine organisms selected to provide this gold-standard reference set have the following characteristics: each represents a major clade from the phylogenetic spectrum; there exists a significant body of scientific literature on the organism; a reasonably sized community of researchers study the organism; and the organism is an important experimental model for the study of human disease, or for economically important activities such as agriculture. Thus the GO Consortium is committed to providing complete annotation of the human genome as well as those of eight important model organism genomes, the GO ‘Reference Genomes’: Arabidopsis thaliana, Caenorhabditis elegans, Danio rerio, Dictyostelium discoideum, Drosophila melanogaster, Escherichia coli, Mus musculus, and Saccharomyces cerevisiae. In addition, the MOD curators for Gallus gallus, Rattus norvegicus, and Schizosaccharomyces pombe have joined the Reference Genome initiative and are fully participating in these efforts. Each model organism has its own advantages for studying different aspects of gene function, ranging from basic metabolic reactions to cellular processes, development, physiology, behavior, and Page 2 disease. All of these organisms are supported by database resources with GO curators who have the expertise to annotate gene products in these genomes according to rigorous standards set by the groups participating in the Reference Genome project (see below). We expect these reference annotations to have two important applications: they will provide improved annotations for researchers who use GO for analysis of both large and small scale datasets, and they will facilitate the annotation of new genomes where extensive experimental data on gene function is often unavailable. Methods The aim of the Reference Genome project is to annotate the gene products of the twelve participating genomes as completely as possible. There are two complementary aspects to achieving annotation completeness: “depth” and “breadth”. For maximal depth, annotations should be as precise and specific as possible; in other words, all experimentally determined information (primarily from the biomedical literature) about the gene products from each of these organisms should be curated. For maximal breadth, the annotations should cover every gene product in the genome, which means we should ensure that computational inferences are included in the annotation sets. In practice, because the inferred annotations are dependent upon the experimentally derived annotations, curation is carried out in two passes: first, literature -based annotation of experimental data followed by curator-approved computational inference to maximize both depth and breadth of annotation across all of Reference Genomes. Annotation collaboration Although the development of the GO has been a collaborative effort since its inception, each participating group initially worked independently in assigning GO annotations, with only limited coordination between the curators of the different databases. Thus, prior to this project, specific protocols for annotation varied between the database groups. Much of the challenge in this project arises from bringing twelve disparate annotation groups together to jointly determine common protocols for annotation and quality assurance. Agreement is critical if the Reference Genome resource is to satisfy our goal of providing semantically consistent functional annotations; therefore, the curators for the different organisms must curate in a consistent and reliable manner, otherwise the resulting annotations will not be systematically comparable and downstream analysis based upon these data will be skewed. Overall, the elements involved in managing and tracking this project are familiar to each of the database resource projects involved. Curators are responsible for capturing information from two sources: the literature, and the output of computational analyses. In the first case, a curator reads a research article and captures several key pieces of information: the organism being studied; the gene product to be annotated; the type of experiment performed; the GO term(s) that best describes the gene product function/process/location that has been determined; and an identifier (typically a PubMed ID) for the citation. Curators similarly review the output of computational analyses and approve or reject these data, once again capturing the organism; the gene product; the type of computational analysis, the GO term(s) describing what can be inferred from that computational analysis; an identifier referencing the computational algorithm; and a reference to the original entity, for example protein domain or related gene product, upon which the inference was based. Variation in annotation occurs because curators differ in their decisions as to what is appropriate to annotate and in choosing which GO terms to employ (Camon et al. 2005; Dolan et al. 2005). Following extensive discussions among the curation groups, two key decisions were made. First, the groups would simultaneously curate a number of homologous genes, to provide an opportunity for checking the consistency of the annotations provided by different groups. This strategy, coincidently, also improves the ontology, since several curators working simultaneously with particular nodes of the GO structure can collaboratively identify any omissions, ambiguities or logical inconsistencies in the GO and work towards their resolution with the ontology developers. Second, the homologous sets of genes would be prioritized based on a number of criteria, to be described below. Page 3 Annotation approach The organisms represented in the GO Reference Genome project span over 500 million years of evolutionary divergence. The premise that underpins comparative genomics is that homologous genes descended from a common ancestor, will have related functions. This is not, of course, to deny that genes will diverge in function over time, but it is a sound null hypothesis. For our purposes this means that a critical first step is to establish a standard approach to determining sets of homologous genes. Ideally, the evolutionary history of each gene in all organisms would be analyzed and stored in a single resource that could be used as the definitive reference for gene family relationships and homologous gene sets. However, generating this resource is a non-trivial problem, both theoretically and practically. At present no single such resource providing a fully satisfactory solution exists (Dolinski and Botstein 2007, Alexeyenko et al. 2006). One central confounding problem has been the lack of a ‘gold standard’ protein set: a shared resource used by all databases and homology prediction tools for analyzing the proteomes of the organisms used in this project. Because the different homology prediction tools do not use common, shared protein sets as inputs their results cannot be compared. Moreover, the protein sets that are being annotated by the GO Consortium members may, and often do, differ from those used by the different homology prediction programs. By and large it is the MODs that have the most authoritative protein datasets for the species they annotate. While at present the total number of gene products is imprecisely known (largely because the full extent of post-translational modifications and alternative splicing remain uncertain) there are reasonable estimates available from the MODs for the numbers of genes encoding protein products in each genome, ranging from 4,389 in E. coli (data from EcoCyc Version 12.1, http://ecocyc.org) to 27,029 in Arabidopsis (Rhee et al. 2008). The GO Consortium is now providing lists of protein sequence accessions for each organism to those who compute homology sets. This initiative first began in collaboration with the InParanoid group, and are now used as input to InParanoid, OrthoMCL, the P-POD database (Heinicke et al. 2007), and the PANTHER database (Mi et al. 2007). Having agreed to use standardized protein sequence datasets as inputs, we next considered the existing algorithmic approaches to the determination of homology that would best meet our objectives. We came to the conclusion that accurately resolving the relationships between genes in highly-duplicated gene families requires an accurate, though computationally expensive, model based on phylogenetic trees. Phylogenetic tree-based approaches to orthology prediction have both theoretical and practical advantages for the Reference Genome project. Theoretical, because they are based on an explicit evolutionary model and can be computationally evaluated, practical because they are amenable to attractive graphical output that facilitates the rapid identification of homology sets by multiple curators. To this end we are collaborating with the PANTHER database (http://www.pantherdb.org/) to produce a curated set of gene trees derived from the standardized protein-coding gene sets for each of the twelve organisms. The curators select a number of homologous sets to annotate in each round of concurrent annotation. Each member of the set is first annotated to the highest level of detail possible based on available experimental results. Then, related proteins are annotated based on inference from the functions of the characterized members of the family, in order to provide a predicted function for each protein whenever possible. Those inferences are made in very conservative manner, as we are very aware that even orthologous genes may have evolved new physiological functions layered on top of more ancient functions. For example in single cell eukaryotes, proteins of the syntaxin family are required for vesicle transport, and that function is conserved up the evolutionary tree. Yet in the higher eukaryotes these proteins are required for the novel function of neurotransmitter release. Large gene families and lineage-specific duplications also pose a problem, in that some members of a set may have diverged and assumed distinct functions from those of their ancestors. It is for these reasons that curators are conservative in drawing inferences concerning gene function from sequence similarity. Annotation priorities While the goal of the Reference Genome project is to cover all gene families, and subsequently maintain these annotations as a part of normal curation, this work will take time. Even by initially concentrating solely on protein Page 4 gene products1 this still presents a large and formidable target annotation list, especially given the limited resources of the contributing database groups. The magnitude of the curation task is somewhat offset by the fact that for many of these proteins there is little or no experimental data available. For example, only ~30% of mouse and human genes (out of ~25,000 total) have experimentally determined GO annotations while the proportion of S. cerevisiae genes (out of ~6000 total) that are experimentally annotated is over 70%. Nevertheless, it is clear that coordination of the Reference Genome project demands a coherent prioritization of targets for curation. Accordingly, Reference Genome curators are selecting targets using the following principles: 1. 2. 3. 4. Genes known to be implicated in human disease, and their orthologs in other taxa, e.g. the gene MSH6 (a DNA mismatch repair protein) which is known to be involved in a hereditary form of colorectal cancer in humans. Genes whose products are involved in known biochemical and signaling pathways, e.g. the gene PYGB (a phosphorylase) that participates in glycogen degradation. Genes whose products are very highly conserved during evolution, e.g. GLYA (a serine hydroxymethyltransferase) is a human gene that is conserved in E. coli. Genes identified from recently published literature as having an important or new scientific impact, e.g. POU5F1 (POU class 5 homeobox 1 gene) that is important for stem cell function. Annotation Consistency An important aspect of the Reference Genome project is to provide consistently generated annotations across the different participating groups. This requires that GO terms are applied with the same intended meaning; that evidence codes are used uniformly; and that ‘inferred from sequence similarity’ (ISS) annotations are based on the same methods. For the Reference Genome project curators review existing annotations as well as add new annotations based on more recent information. We have implemented appropriate procedures, employing a mix of automated and manual methods, to ensure the highest possible quality of annotations for the Reference Genome project, whether based on experimental data found in the literature or inferred from these experimental annotations. The review includes the following: (1) A periodic assessment of consistency using a peer review system in which a curator evaluates the experimentally determined annotations provided by other curators for a gene family; (2) A computational verification that all associations “inferred based on sequence similarity” (ISS) explicitly reference a gene in which experimental evidence is available; (3) The replacement of older annotations that are not based on direct experimental evidence or reliable inferences with annotations that adhere to these new standards. Literature curation consistency The quality control process for consistency and completeness of annotations begins when curators indicate that the targeted gene(s) in their organism has been comprehensively annotated based on the information available in the biomedical literature. The time and effort required for literature-based curation is a function of the amount of information available. If there is no literature, then the genes are immediately considered completely annotated. For genes with little literature, the curator reviews all available papers, but for genes for which hundreds of papers are available this is impractical. In these cases, curators assess the comprehensiveness of curation based upon recent reviews, and curate key primary publications accordingly. The curation consistency review process also identifies problems with the interpretation of particular GO terms. These terms are then flagged within the GO with a comment that a curator must be careful when using these terms. For example, "cell growth" is sometimes misused because it is confused with “cell proliferation” which contributes to the growth of organs. Certain concepts, such as "development", "differentiation" and "morphogenesis" are used with various, overlapping meanings in the literature. In GO they are specifically Although some GO curation teams are recording isoform details (when available) these protein annotations are not generally presented at this level, however we expect to incorporate and reflect isoform level annotations for all twelve organisms in the future. 1 Page 5 defined, and we verify that all annotations use terms as defined by the GO. The review of curation also identified several GO terms that proved to be ambiguously defined, or that were not logically consistent within the GO. These terms have been made obsolete by the GO and replaced by more appropriate terms. Examples of such terms include "electron transport" (replaced by two terms: "electron transport chain" and "oxidation reduction"), and "secretory pathway" (replaced by two terms: "exocytosis" and "vesicle-mediated transport"). At the beginning of the GO Consortium project, a number of annotations were made, usually from textbooks or review articles, which were solely supported by author statements and often represented what could be thought of as common knowledge. For example, ‘DNA polymerase [mouse Poln] (UniProt:Q7TQ07)’ performs 'DNA replication (GO:0006260)' in the 'nucleus (GO:0005634)'. Although this information is valuable and in most cases accurate, we soon realized that often such annotations could either not be traced to an original experiment, or the original experiment was actually carried out on a homologous gene product from another organism. Therefore, the Reference Genome project has made it standard practice to avoid making annotations based solely on author (generic) statements that lack direct experimental verification for the specific gene being annotated, and we are systematically replacing such annotations with new annotations supported by experimental or sequence similarity evidence as they arise. Consistent transitive curation of proteins in a homolog set To ensure maximal breadth of genome annotation, we transfer GO annotations from experimentally studied proteins to homologous proteins for which no experimental data are available. These inferences take place after all the experimental data have been captured. When all curators have completed their literature-based annotations for a given target gene set, a second iteration of annotation is done, but now based on inference. This was initially an idiosyncratic process unique to each database group. However, the use of tree-based methods to examine homologous groups allows for a more rigorous and systematic transference of this information. This is not an automatic process, rather a curator reviews each inferred annotation with care since the function of a gene can diverge during evolution, particularly after gene duplication events that may free one of the duplicated copies from selection constraints and allow the evolution of new functionality. One of the gold standards of the Reference Genome project is that inferential annotations are made only in cases where there is experimental data from the organism from which the annotation is inferred. For example, orthologs of the mismatch repair MSH2 are present in all eukaryotes. However, the role in mismatch repair (GO:0000710) has only been experimentally demonstrated in a subset of species: e.g. human, mouse, Arabidopsis, the yeasts, and C. elegans. Therefore, inferential annotations in other organisms must refer to a corresponding ortholog from one these species in which experimental work has been done (this need not necessarily be a species from the Reference Genome project). Hence, after making inferences, queries are run to verify that this is the case. Progress As of October 2008, we have annotated nearly 400 homology sets, comprising approximately 2500 genes. In the upcoming months, we plan to scale up the effort by having all the proteins from the twelve organisms organized into the PANTHER family database and annotation tool. The annotations may be viewed using AmiGO, the GOC browser (http://amigo.geneontology.org/, Carbon et al. 2008). In the upcoming release of AmiGO (end of 2008) a number of new displays will be available that are specifically designed for public browsing of data from the Reference Genome project. The additions include a table listing all gene families that have complete Reference Genome annotations. For each entry in the table there will be a link to a “Comparison Graph” as shown in (Figure 1) to highlight the common functions for each gene family as well as those particular to certain organisms or groups of organisms. In addition to the graphical representation of the annotation data, individual pages for each gene family will also be available which list all annotations in that family as shown in (Figure 2). Page 6 Improvements to annotations Gene products selected for concurrent annotation in the course of the Reference Genome project have improved the breadth and depth of annotation coverage of gene products as shown in Figure 3. Those genes have a higher percentage of annotations derived from published experimental research. Moreover, the annotation information for those genes is significantly higher relative to the annotation information prior to this effort. The number of genes annotated by inference through homology has also increased, further contributing to increased breadth and depth of genome coverage in the annotations. In all cases, the percentage of genes annotated only with noncurator reviewed computed annotation is reduced, demonstrating that this effort does contribute to improving the breadth and quality of annotation for each genome. Improvements to GO The collaborative annotation of a group of similar gene products has also proven to be useful for the development of the GO itself. For example, as a direct consequence of the Reference Genome project, 223 ontology change requests were made. These changes were incorporated in the ontologies making up slightly more than 10% of the total ontology change requests during this period. Examples of requested new terms include regulation of NAD(P)H oxidase activity, DNA 5'-adenosine monophosphate hydrolase activity, neurofilament bundle assembly, and quinolinate metabolic process. We have also enhanced the ontology by adding synonyms, improving definitions, and correcting inconsistencies. Discussion The aim of the Reference Genome project is to provide a source of reliable GO annotations for twelve key genomes based upon rigorous standards. This endeavor faces many difficult challenges, such as: the determination and provision of reference protein sets for each genome; the establishment of gene families for curation; the application of consistent best practices for annotation; and the development of methodologies for evaluating progress towards our goal. Although this is a laborious effort, steady progress is being made in developing this resource for the research community. This initiative has propelled the GOC into the provision of standardized protein sets for these genomes, which we expect to be of broad utility beyond the Reference Genome project. By engaging curators from across the MODs in joint discussions we are observing improvements in curational consistency and refinement of the GOC best practices guidelines (see http://geneontology.org/GO.annotation.conventions.shtml). The genes that have been targeted by the Reference Genome project have significantly improved annotation specificity as compared to their previous annotation, and the number of genes annotated by inference through homology has also increased. This increased breadth and depth of genome coverage in the annotations is one of the major goals of the project. An additional beneficial side effect has been the improvements to the GO itself, which will consequently improve the accuracy of inferences based on these annotations. Genomes that are fully and reliably functionally annotated empower scientific research in the community as they are essential for use in automated inferential annotation of other genomes, and this motivates the Reference Genome project's work. We encourage users to communicate with the GO Consortium (send e-mail to gohelp@genome.stanford.edu) with questions or suggestions for improvements to better achieve this aim. Data availability Access to all GOC software and data is free and without constraints of any kind. An overview of the project as well as links to all resources described below can be found at http://www.geneontology.org/GO.refgenome.shtml. Annotations made by the databases participating in the Reference Genome project are available from the GOC website in gene_association file format (http://www.geneontology.org/GO.current.annotations.shtml). The protein sequence datasets are available for the community as a standardized resource from http://www.geneontology.org/gp2protein/. The exact queries used to gather statistics for the annotation improvement reports can be found at: http://www.geneontology.org/GO.database.schema-with-views.shtml. Page 7 A. B. C. Figure 1. [ UPDATE] The Comparison Graph shows all annotations, both experimental (evidence codes: IDA, IMP, IGI, IPI, IEP) as well as those inferred from sequence similarity to an experimentally characterized gene (ISS) and by curators (IC). Colored wedges are used to indicate where in the GO graph there are direct annotations to specific terms for the different species. The species key (panel B) is presented to the user in the upper left corner of the browser window and allows the user to select which species to show. Panel A presents the zoomed out window for an overview of the complete annotation picture. In this particular screen capture “[e] experimental” has been selected, which is presented as a bulls-eye placed over those terms with experimentally based annotations. Panel C presents the zoomed in view with the detailed annotation on each Page 8 term. At either level of zoom mousing over term provides a pop-up window with these same annotation details. Figure 2.[ UPDATE] The Categories List highlights common or related annotations. Page 9 Figure 3. UPDATE AND PROVIDE LEGEND XXX Acknowledgements The Reference Genome effort is overseen by Pascale Gaudet2dictyBase and includes these representatives from the curational staff: Tanya BerardiniTAIR, Emily DimmerGOA, , Stacia R. EngelSGD, Petra Fey2, David P. HillMGI, Doug HoweZFIN, Jim HuE.coliDB, Rachael HuntleyGOA, Varsha K. KhodiyarUCL, Ranjana KishoreWormBase, Donghui Li3, Ruth C. LoveringGOA, Fiona McCarthyAgBase, Li NiMGI ,Victoria PetriRGD, Susan TweedieFlyBase, Kimberly Van AukenWormBase, and Valerie WoodGeneDB—as well as the following computational staff representatives: Siddhartha Basu3dictyBase, Seth CarbonBBOP, Mary DolanMGI, and Christopher J. MungallBBOP—those establishing the protein dictyBaseNorthwestern University, Chicago, IL, USA; TAIRCarnegie Institution, Department of Plant Biology, Stanford, CA, USA; EBI, Hinxton, UK; MGIThe Jackson Laboratory, Bar Harbor, ME, USA; SGDDepartment of Genetics, Stanford University, Stanford, CA, USA; ZFINUniversity of Oregon, Eugene, OR, USA; E.coliDBTexas A&M University, College Station, TX, USA; UCLDept of Medicine, University College London, London, UK; WormBaseCalifornia Institute of Technology, Pasadena, CA, USA; AgBaseMississippi State University, Starkville, MS, USA; RGDMedical College of Wisconsin, Milwaukee, WI, USA; FlyBaseDepartment of Genetics, University of Cambridge, Cambridge, UK; GeneDBWellcome Trust Sanger Institute, Hinxton, UK; BBOPLawrence Berkeley National Laboratory, Berkeley, CA, USA; PPODPrinceton University, Princeton, NJ, USA; PANTHERSRI, Palo Alto, CA, USA, GOEOEBI, Hinxton, UK. GOAUniProt, Page 10 families to be annotated: Kara DolinskiPPOD, and Paul ThomasPANTHER—and, of course, the four PIs of the GO Consortium: Michael AshburnerFlyBase, Judith A. BlakeMGI, J. Michael CherrySGD, and Suzanna LewisBBOP. The authors particularly wish to thank and recognize the invaluable contributions of their curator colleagues in the GO Consortium whose work ensures that the objectives of the Reference Genome project are fully realized: Rama BalakrishnanSGD, Gail BinkleySGD, Karen R. ChristieSGD, Maria C. CostanzoSGD, Jennifer DeeganGOEO, Alexander D. DiehlMGI, Qing DongSGD, Harold DrabkinMGI, Dianna G. FiskSGD, Midori HarrisGOEO, Jodi E. HirschmanSGD, Benjamin C. HitzSGD, Eurie L. HongSGD, Amelia IrelandGOEO, Cynthia J. KriegerSGD, Jane LomaxGOEO, Stuart R. MiyasatoSGD, Robert S. NashSGD, Julie ParkSGD, Debby SiegeleE.coliDB, Dmitry SitnikovMGI, Marek S. SkrzypekSGD, Shuai WengSGD, Edith D. WongSGD, and Kathy K. ZhuSGD. We also thank these Principal Investigators for their enthusiastic support of this effort that is taking place within their research groups: Rolf ApweilerGOA, Carol BultMGI, Rex ChisholmdictyBase, Janan EppigMGI, Howard JacobRGD, Julian ParkhillGeneDB, Seung RheeTAIR, Martin RingwaldMGI, Paul SternbergWormBase, and Monte WesterfieldZFIN. The Gene Ontology Consortium is supported by a grant, NIH-NHGRI P41 grant HG002273. Curation at the model organism databases is supported as follows: FlyBase, Medical Research Council grant G0500293; dictyBase: National Institutes of Health (NIH) grants GM64426 and HG00022; MGI, NIH-NHGRI P41 grant HG000330 and NIH grant HD XXXXX; WormBase: US NIH- NHGRI P41 grant HG02223; Human Cardiovascular GO team: British Medical Research Council; British Heart Foundation grant SP/07/007/23671; ZFIN, NIH-NCRR P41 grant HG002659-06; GOA, core EMBL funding; E. coli XXX; SGD XXX; chicken XXX; RGD XXX; Pombe XXX; TAIR XXX; and chicken XXX. We are very grateful to the following for stimulating discussions: Richard Durbin and Erik Sonnhammer. References Alexeyenko A, Linberg J, Perz-Bercoff A and Sonnhammer ELL, 2006. Overview and comparison of ortholog databases. Drug Discovery Today: Technologies. 3: 137-143. Artamonova, G. Frishman, M. S. Gelfand and D. Frishman. 2005. Mining sequence annotation databanks for association patterns. Bioinformatics 21 Suppl. 3: iii49-iii57. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene Ontology: Tool for the unification of biology. Nature Genetics 25: 25-29. Berglund AC, Sjölund E, Ostlund G and Sonnhammer EL. 2008. InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 36: D263-D266. Bourne PE, McEntyre J (2006) Biocurators: Contributors to the World of Science. PLoS Comput Biol 2(10): e142. Camon EB, Barrell DG, Dimmer EC, Lee F, Magrane M, Maslen J, Binns D, and Apweiler R, 2005. An evaluation of GO annotation retrieval for BioCreAtIvE and GOA. BMC Bioinformatics 6: S17. Dolan, M.E., Ni, L., Camon, E., and Blake, J.A. 2005. A procedure for assessing GO annotation consistency. Bioinformatics Suppl 1:i136-i143. Dolinski K and Botstein D, 2007. Orthology and functional conservation in eukaryotes. A. Review Genetics 41: 465507. Fitch WM, 1970. Distinguishing homologous from analogous proteins. Systematic Biol. 19: 99-113. Heinicke S, Livstone MS, Lu C, Oughtred R, Kang F, Angiuoli SV, White O, Botstein D, Dolinski K. 2007. The Princeton Protein Orthology Database (P-POD): a comparative genomics analysis tool for biologists. PloS ONE 2: e766. Howe, Costanzo, Fey, Gojobori, Hannick, Hide, Hill, Kania, Schaeffer, St Pierre, Twigger, White & Rhee. 2008 Big data: The future of biocuration. Nature 455: 47. Page 11 Iliopoulos I, Tsoka S, Andrade MA, Enright AJ, Carroll M, Poullet P, Promponas V, Liakopoulos T, Palaios G, Pasquier C, Hamodrakas S, Tamames J, Yagnik AT, Tramontano A, Devos D, Blaschke C, Valencia A, Brett D, Martin D, Leroy C, Rigoutsos I, Sander C, Ouzounis CA. 2003. Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 19: 717-726. Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13: 178-189. Mi H, Guo N, Kejariwal A, Thomas PD. PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 2007 Jan;35:D247-52. Nature Publishing Group (2007) The database revolution. Nature 445, 229-230. Pena-Castillo L, Tasan M, Myers CL, Lee H, Joshi T, Zhang C, Guan Y, Leone M, Pagnani A, Kim WK, Krumpelman C, Tian W, Obozinski G, Qi Y, Mostafavi S, Lin GN, Berriz GF, Gibbons FD, Lanckriet G, Qiu J, Grant C, Barutcuoglu Z, Hill DP, Warde-Farley D, Grouios C, Ray D, Blake JA, Deng M, Jordan MI, Noble WS, Morris Q, Klein-Seetharaman J, Bar-Joseph Z, Chen T, Sun F, Troyanskaya OG, Marcotte EM, Xu D, Hughes TR, Roth FP. A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol 2008; 9:S2 http://genomebiology.com/2008/9/S1/S2 Penkett CJ, Morris JA, Wood V and Bahler J. 2006. YOGY: A web-based, integrated database to retrieve protein orthologs and associated Gene Ontology terms. Nucleic Acids Res. 34: W330-W334. Rhee SY, Wood V, Dolinski K, Draghici S. 2008. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 9: 509-515. Ruan J, Li H, Chen Z, Coghlan A, Coin LJ, Guo Y, Hériché JK, Hu Y, Kristiansen K, Li R, Liu T, Moses A, Qin J, Vang S, Vilella AJ, Ureta-Vidal A, Bolund L, Wang J, Durbin R. TreeFam: 2008 Update. Nucleic Acids Res. 2008 Jan;36(Database issue):D735-40. Smith, RF. 1996. Perspectives: sequence database searching in the era of large-scale genomic sequencing. Genome Res. 6: 653-660. Smith, TF and Zhang, X. 1997. The challenges of genome sequence annotation or “The devil is in the details”. Nat. Biotechnology 15:1222-1223 Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003 4: 41. The Gene Ontology Consortium. 2008. The Gene Ontology Project in 2008. Nucleic Acids Res. 36: D440-4. Wheelan, SJ and Boguski MS. 1998. Late-Night Thoughts on the Sequence Annotation Problem. Genome Res. 8: 168-169. Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. 2008. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 36: D13-D21. Page 12