Advanced Characterization methods lectures
advertisement

Advanced Characterization methods
1. Administration (syllabus, grading , etc.)
Syllabus:
Overview of course
Error analysis
Spectroscopy
Underlying physics – characterization of energy transitions in materials
Optical (absorption and emission, Raman, time dependent)
XPS,
NMR, EPR
Electrochemical
Scattering/diffraction
Fundamental idea – structure determination through scatter of radiation or
particles
Optical
X-ray, neutron, electron
Fractionation
Separation of complex mixture according to specific quantity, followed by
detection
Mass spec
Chromatography
Imaging
Optical (transmission, fluorescence, confocal, TP)
Electron microscopy (SEM, TEM, STM)
AFM & variations
Direct
Direct measurement of quantity
Electrical
Magnetic
Thermal
Date
10/20
10/21
10/22
10/24
10/27
10/28
10/29
10/31
11/03
11/04
11/05
Topic
Intro, error analysis
Lab – fluorescence
spectroscopy
Optical spectroscopy
Optical spectroscopy
Surface spectroscopy (XPS)
Lab – XPS
NMR
EPR, EC, etc.
Intro to
scattering/diffraction, XRD
Lab – EC detection
XRD, E-diffraction, neutron
Assignment
Lab notebook
Choose presentation topic
1st homework
1
11/07
11/10
11/11
11/12
11/14
11/17
11/18
11/19
11/21
11/24
11/25
11/26
11/28
12/01
12/02
12/03
12/05
Intro to imaging, optical
Electron microscopy
Lab-DLS
Scanning probe microscopy
Fractionation – MS
Mid-term exam
Lab – SEM
Chromatography
Chromatography, cont.
Direct measurements –
electrical
Lab – AFM
Magnetic, thermal
Thanksgiving holiday
Elemental analysis
No lab – oral presentations
On-line vs. lab-based
1st lab due
2nd homework
3rd homework
2nd lab due
Grading
Labs – 6 labs, notebooks should be kept, two lab reports, will not know
beforehand which ones. One report technical, the other less technical. 20%
Three homeworks 15%
One mid term exam 15%
One topic for presentation – three types of presentation (15-30 second, 2-3
minute, 10-15 minute) 15 %
Final 20%
Integrative experience 15%
2. Overview of course
More of a survey course – not in much depth about specific techniques, but will
cover a lot of ground
However, will still explore underlying physics of general classes of technique
(spectroscopy, scattering) as well as specific examples in some detail
Techniques will be described in terms of:
What exactly is being measured?
What is the underlying physics of the method?
How is the raw data analyzed, what is the end result (after analysis)?
What are the assumptions being made in both the measurement and
analysis?
What sample preparation is needed?
What are the (dis)advantages compared to other techniques?
What are the limitations?
2
Characterization methods may be for determination of:
Composition
Structure
Process
Specific properties (thermal, electrical, etc.)
Can subdivide the course accordingly, but from the underlying physics, this
doesn’t make mush sense (and often hard to separate, many techniques cover
more than one)
The course will be divided according to some underlying principle of the method,
in particular, we’ll cover the following
1. Spectroscopy – dispersion, energy resonant processes, e.g. optical absorption &
fluorescence, NMR, ESR, XPS, SIMS, Auger, DSC, rheology.
2. Scattering and diffraction – interaction of radiation with material, not energy
dispersion, but relating output to some structure or composition, X-ray, light
scattering, electron, neutron diffraction.
3. Fractionation – Subdivide the sample according to some physical property, e.g.,
mass spec, GC, GPC, HPLC, electrophoresis.
4. Imaging – Create visual representation of some property – optical microscopy,
TEM, SEM ,scanning probe.
5. Specific properties & other – electrical, mechanical, BET.
6. Process – measurement of something that changes with some reaction of
system, generally more empirical.
For the specific properties, the analysis is straightforward. For example, in a
measurement of electrical conductivity the current response to an applied voltage is
measured. The assumptions and analysis may need to consider the relation between the
actual response we measure (needle deflection) to the underlying quantity.
For others, we are using some raw data to infer other characteristics, which is likely to
require both more analysis as well as assumptions about the underlying physics. For
example, X-ray diffraction itself might be interesting but the main purpose is to reveal
underlying crystal structure.
3. Spectroscopy (spectrometry) – material response vs. input energy
Materials have some energy response determined by composition and structure – the
location and strength of these will be unique to the particular sample. Spectroscopy deals
with looking at this dependence by applying some energy to a system and looking at the
response as a function of this energy.
A. Optical – input is in the form of EM radiation, change in the energy is a change
in the photon energy, i.e., the frequency or wavelength (not the irradiance).
3
Absorption
UV-VIS
IR, FTIR
Emission
Fluorescence
Interaction
Raman
Variations
Nonlinear
Time dependent
Combinations
Background
Interactions of EM radiation with matter
Photons can be absorbed and emitted, but also interact non-resonantly (scatter –
all optical interaction with matter can be described in terms of scattering).
Semiclassically, the optical electric field of the radiation interacts with the charges
in the material. For now, we consider the interaction with the electrons, although
this can easily be generalized. Interaction with the electrons is the main one for
optical properties in the ultraviolet and visible. Macroscopically and to first order,
the complex polarization vector is the key quantity. Usually, the electric dipole
approximation is invoked, which says that the important interaction between
material and radiation is simply the interaction of a dipole with the field. The term
in the Hamiltonian is
H int er E
This is often much weaker than other effects and can be calculated with
perturbation theory. The term that comes out of this is the matrix element of r,
which is proportional to the transition dipole moment
nm e n r n
Note that this is not the (permanent) dipole moment of the ground state – this is a
dipole moment of the transition itself. On the macro scale, this is the difference
between a permanent and induced polarization.
How is this connected to absorption?
We can wave our hands and use the following argument. The applied (optical)
electric field induces a dipole. The energy of the dipole will be proportional to the
square of the polarization and so the square of the dipole moment. So energy
remove from the beam (& given to the dipole) is proportional to the square of the
dipole moment as defined above. The irradiance of the incident beam is the
energy per cross sectional area per time, so the absorption (change in this energy
per unit cross section) will also be proportional to the square of the dipole
moment.
The propagation of EM radiation through material can be characterized by the
complex refractive index. The real part is the “usual” refractive index that relates
4
to the speed of light in the material. The complex part is related to the absorption.
The infinitesimal change in irradiance is
dI Idx
This equation tells us that the relative change will be proportional to a constant
() and the infinitesimal distance the beam travels. Solving it gives
I I 0 e L
(Beer’s law)
Where L is the distance traveled through the medium.
Often we want an absorption per unit concentration of the absorbing species.
Also, logs to base 10 are more practical for applications. The molar extinction
coefficient is defined as
C ln( 10)
where C is the concentration, usually molar (short aside on molar concentrations
if needed). The corresponding equation for the irradiance is
I I 0 10 CL
{Note – be careful about the use of and , since they are defined by different
people different ways. Chemists usually use to be absorption coefficient divided
by the concentration, and occasionally is used with the natural log instead of the
base 10 log.}
The exponent is the Absorbance, or Optical Density of the sample, and is a
unitless quantity.
As we’ll see later, most transitions are not narrow lines, but rather broad peaks. A
typical absorption peak in the visible looks like (OHP)
in other words, the absorption coefficient is some function of frequency or
wavelength. An integrated absorption coefficient can be defined as
A ( )d
where is the frequency. A unitless quantity called the oscillator strength can be
defined as
4m c
f 2 e 0 A
e CN A
where me is the electron mass, c is the speed of light, 0 is the free space
permittivity, L is the path length and e is the electron charge. Finally, one can
show that the relation to the dipole moment defined earlier is
8 2 me
2
nm
f nm
2
3he
for a transition between states n and m.
The transition dipole moment leads to selection rules for optical transitions. If the
symmetry of the initial and final states are the same, the transition dipole moment
will be zero, and so there will be no interaction with the field. This results in a socalled forbidden transition – in more intuitive terms, transitions are forbidden
when charge distribution of ground and excited states does not change symmetry
5
A forbidden transition will have orders of magnitude weaker absorption than an
allowed one.
Why weak and not completely gone?
- have neglected other parts of wave function (nuclear) – remove symmetry – e.g.
spin-orbit coupling allowing triplet state excitation
- have used the dipole approximation – higher order terms may allow transition,
but are weaker
In semiconductor and insulating crystals, the picture is similar. If the bottom of
the conduction band and the top of the valence band are at different values of k,
optical absorption or emission can only occur with the absorption or emission of a
phonon, in a so-called indirect transition. This can be much weaker, in particular
for emission.
Einstein A, B coefficients (detailed derivation will come next semester)
The rate of stimulated emission will be proportional to the input irradiance. The
coefficient of proportionality is called the Einstein B-coefficient. The rate of
spontaneous emission is constant for an excited state and is referred to the
Einstein A-coefficient. These two are not independent, one can use energy
conservation to find that
A 8h( / c) 3
B
Fate of excited states (OHP)
1. Luminescence
2. Inter- and Intra-molecular transfer
3. Quenching
4. Ionization
5. Isomerization
6. Dissociation
7. Direct reaction or charge transfer
We’ll talk more about these when we come to fluorescence emission
spectroscopy.
Absorption spectroscopy
UV-VIS-NIR – UV radiation technically runs from about 10 nm (~100 eV) to
about 390 nm (~3.2 eV), but most absorption spectroscopy is done in the lower
end of energy, in the wavelength range 180 to 390 nm. Visible is defined as
between 390 and 780 nm. Infrared is separated into three regions: NIR (780 nm to
3.0 microns) MIR (3.6-6.0 microns) FIR (6 to 15 microns). Sometimes an
additional region called the extreme IR is defined (15 to 1 mm) but recently this
has received more attention as the so-called THz region.
UV-VIS are usually grouped together because they involve transitions in the outer
shell electrons. Not only are these important for identifying materials
6
(composition) but are much more sensitive to surrounding changes such as
bonding (structure). Infrared spectroscopy usually involves transitions between
vibrational states, with energies corresponding to wavelength ranges from 800 nm
to several microns.
UV-VIS – broad, often featureless, so not useful for identification of composition.
More likely to be used for process control or as basic property – where
something absorbs. Visual aspect of application, e.g., paper, colorants.
Photometric titration.
Time scales (excited state lifetimes) – 10-11 to 10-8 sec
Causes of nonzero peak widths:
1. Intrinsic (lifetime, natural) broadening (uncertainty principle – in excited state
for finite time, so the specification of it cannot be arbitrarily precise).
2. Vibrational and rotational sublevels
3. Doppler broadening – Doppler shifts due to motion of scatterers, direction and
speed are distributions, so shifts have cumulative effect of broadening
4. Interactions with surroundings
Pressure (collisional) broadening – collisions with other scatterers induce
shifts in frequency
Interactions with solvent – similar to pressure
Most of this discussion has been relevant to atomic and molecular spectra. Similar
effects are also present in bulk materials such as semiconductors. In this case, it is
often difficult to measure transmission spectra, so reflection spectra can be taken
instead. These will probe the surface of the sample.
In ATR = Attenuated Total Reflection spectroscopy, the sample is in contact with
a prism. Light is incident on the sample through the prism, where it is reflected at
the prism/sample interface. At this interface, the light penetrates into the sample,
even for total reflection. At absorbing wavelengths, this removes energy from the
beam, which is measured in the output (reflected) beam spectrum.
Practical application
Spectrometers (spectrophotometers) – dispersive (moving, diode array), FT
Broadband sources, usually cw
Deuterium – UV
Tungsten – Vis
Xe – both
heater (blackbody) – IR
Detectors
PMTs
Photodiodes (arrays)
For UV-VIS, two calibrations are usually required – dark and reference. The dark
removes the baseline signal from ambient light, detector noise or electronic offset, and
7
the reference determines the I0 term in Beer’s law. This includes the spectral input of the
source, any absorption by the cuvette and solvent, and the spectral response of the
detector.
FT spectrometers
Instead of dispersive (either many detectors or moving grating, prism) all light from a
broadband source is used. In absorption spectrometers, this is sent through an
interferometer (usually Michelson). Changing the length of one of the interferometers
arms modulates the output and creates a Fourier transform of the input light.
(see handout)
In an FTIR, this is used to modulate the input broadband IR source. The output in the FT
of the source, with the displacement of the arm being the FT variable. This is sent
through the material, and the output is FT’ed to give the transmitted light, and then the
spectrum is determined.
IR absorption
The energy of photons in the IR region corresponds mostly to transitions between
vibrational and rotational states in materials. The optical absorption bands for these
transitions are much narrower than typical ones in UV-VIS spectroscopy, and so are
much more useful for identification of the constituents of a sample.
The basic physics underlying these bands is easily understood at a simple level using the
mass and spring model. The response of a system of masses attached by springs will
depend on the excitation (initial conditions), the masses and the spring constants. In this
case, the masses are the atoms and the springs the bonds, so the IR spectrum will
uniquely determine the constituents and bonds between them, i.e., the molecules in the
sample. Since calculation from first principles is difficult/time consuming, most analysis
is empirical – bands of known compounds or functional groups have been measured and
categorized in the literature.
(OHPs– energy levels, typical IR spectra)
Solid samples: powder samples are usually pressed in KBr pellets – these give very IR
transparent samples with little scattering.
Liquids: Cells are usually very narrow, since IR absorption coefficients are generally
high. Sodium chloride is a typical material for the cell windows, since it has good
transparency in the IR.
Most current IR absorption spectrometers are of the FT type.
(Handout)
There are many advantages to FT-type spectrometers, the main one being that all the light
transmitted through the sample is measured by the detector.
8
Raman Spectroscopy
IR spectra have similar selection rules as for optical spectroscopy – this means that many
bands can not be measured with usual IR absorption. Raman spectroscopy (scattering)
offers a way around this - since the interactions involve more than one photon, the
selection rules are different than in standard absorption spectroscopy. Raman
spectroscopy also has the useful advantage of much fewer problems due to water than IR
spectroscopy (water gives a large broad signal conventional IR spectroscopy, which can
hide other lines of interest).
The Raman effect is a parametric effect in which a photon is “absorbed” and re-emitted,
with the energy difference being taken up by or released from a vibrational state of the
material. When the final state has higher energy than the initial, the results are the socalled Stokes lines, when the final state has lower energy, they are the anti-Stokes lines.
The Stokes lines arise from the final state being a higher vibrational state of the ground
state manifold, the anti-Stokes line occurs when the initial state is one of these and the
final state a lower energy vibrational level or the ground state. Since these rely on a finite
population in these vibrational levels, they are generally much weaker and also
temperature dependent.
X-ray absorption. The absorption of higher energy photons, in the X-ray region, usually
causes transitions involving inner shell electrons, which are less affected by environment.
This can give elemental information, rather than molecular. These electrons are generally
ejected from the atom. (OHP of typical spectrum).
Appearance of X-ray absorption – now kinetic energy of electrons released plays a role –
the edges are sharp with tails as more KE is given to the electrons.
The absorption can be measured in the usual way, or by monitoring the resulting
fluorescence.
The measurement of the emitted electrons constitutes XPS – more on this later.
Photoluminescence
There are a number of possible fates of the excited state atom or molecule in terms of
how it loses its energy, usually decaying back to the ground state. We can broadly divide
these into two groups of transitions: radiative and non-radiative, depending on whether or
not a photon is emitted in the process. (OHP)
The emission of photons in the UV-VIS range is referred to generally as luminescence.
When the excitation is by a photon, the process is called photoluminescence (general
term) or fluorescence/phosphorescence, depending on the state the emission comes from
(and the time scale of the decay). Other processes can excite the material which then
emits photons, for example chemical processes (chemiluminescence) biological
(bioluminescence), electron injection (electroluminescence), etc.
9
The spin multiplicity of a state, defined as 2S+1, can yield information on the probability
of a radiative transition. The usual stable (lowest energy) state is one where the electron
spins are paired, S=0 and the multiplicity 2S+1=1 – this is termed the singlet state. When
two electrons are unpaired, S=1, 2S+1=3 and the state is called a triplet state. The
ordering of the state in energy is often added as a subscript, i.e., ground state S0, 1st
excited singlet state S1, etc. Triplet states start at T1 being the lowest energy triplet state.
The T1 state is lower in energy that S1.
Molecules will maintain spin multiplicity during transitions, meaning that transitions
between singlet and triplet states are technically forbidden. However, since spin is usually
not a pure quantity in a real system, transitions do occur but with low probability. This
means that molecules in an excited S1 state may decay into a lower energy T1 state rather
than the S0 ground state. This process is called intersystem crossing (ISC). Since this state
and the ground state do not have the same spin multiplicity, the probability of decay is
also small and the state consequently has a long lifetime (ms to seconds, minutes, hours).
This radiative decay, when it does occur, is called phosphorescence, and is responsible
for “glow in the dark” objects.
The emission of photons is nearly always from the lowest excited state (Kasha’s rule). In
other words, the excited state will decay to the lowest singlet state before radiating. It
also will generally emit from the lowest vibrational sublevel of the excited state by
nonradiative processes, then emit the photon and decay to the ground state (manifold).
The hand-waving argument supporting this is that vibrational relaxation is generally
much faster (~femtoseconds) than electronic (~nanoseconds), so that it can happen before
the electron has a chance to decay radiatively.
What does this have to do with spectroscopy? In fluorescence emission spectroscopy, the
radiative emission due to a S1 to S0 transaction is measured as a function of the
wavelength (energy) of the emitted photon. The final state will be a vibrational sublevel
of the ground state, and vibrational structure is often seen in fluorescence emission
spectra. However, there is also a finite probability that the emission will come from a
triplet state – the spectral information will be the same, since all transitions come form
one initial state and the final state manifold is the same, but of course the entire spectrum
is shifted in this case.
Since the fluorescence emission comes from the bottom of the excited state manifold to
the various vibrational sublevels, while the absorption is from the bottom of the g.s.
manifold to the sublevels of the excited state, the transitions are not the same. However,
in most cases the vibrational spectrum is nearly the same for both states. Even then,
though, there is a shift of energy between the peak locations for absorption and emission,
known as the Stokes shift. This is most easily explained in a diagram (OHP).
Time dependence of emission
In contrast to absorption, which occurs on the fs time scale, the decay of the excited by
emission of photons is usually much longer. The decay time will be dependent on a
number of factors, including of course the molecule and transition, but also on the
surroundings. Time-resolved fluorescence spectroscopy is a useful technique for
10
determining these. For nanosecond decay times, this can be measured directly in the time
domain with PMTs (response times ~nanoseconds) fast photodetectors (ps response
times). Often the electronics to process the detector response in the limiting factor in the
minimum time that can be measured. For faster decay times, the measurements can be
done using nonlinear optical (NLO) techniques. Measurements are also routinely done in
the frequency domain by modulating the input light and measuring both the in and out of
phase components of the output.
Nonlinear Optical (NLO) techniques
Up to now we considered only the linear response of a material to light, i.e. the first order
polarization. Higher order terms exists, and can be used to characterize materials in a
number of ways, as well as integral parts of other techniques.
Without going into too much detail, the polarization of a material in response to an
optical electric field can be expanded in a power series in the field E. The linear term is
the one we’ve been using so far. The next two terms, proportional to the square or cube
of the field, are the source terms in second-order and third-order nonlinear optics,
respectively. The effects due to them are progressively weak, but with various
experimental techniques they can be measured easily.
We’ll mainly discuss second order effects, and then only briefly. In this case, there is a
symmetry of the output that can be exploited to gain information about molecular
orientation in a material. A polarization in response to the square of the electric field will
only be non-zero for a material not having inversion symmetry. In a second order
process, because of the quadratic field dependence, the polarization vector will be the
same for the field in one direction as for the exact opposite field. Now if we inverted the
material, and it had inversion symmetry, the response should be the same, but this can’t
be since the polarization vector should be in the opposite direction. Therefore, we need
this non-centrosymmetric property for second order NLO.
This can be useful in spectroscopy. Observation of a second order effect in an ensemble
of molecules will imply that there is a molecular ordering giving a non-centrosymmetric
material on the length scale of the wavelength or above.
Another useful application is for time dependent fluorescence (or other optical)
properties. Second order processes allow one to combine two beams to produce a third at
a different wavelength. Normally, photons do not interact, so two beams passing through
a material will not affect each other. In a second order material, the polarization is
proportional to the square of the applied field. If this comes from two different sources,
there will be cross terms, giving a resulting polarization depending on both beams. By
measuring the effect of this polarization (radiation),
Third order nonlinear spectroscopy
Pump-probe – in this method, excited states are studied by looking at the time
dependence of the absorption after the material has been excited. First a very short
monochromatic pulse excites the material, then usual absorption spectroscopy is done,
11
but also with a short, broadband pulse at a fixed delay from the first pulse. This gives a
view of both the absorption from the excited state as well as the depletion of the ground
state (decrease in the usual absorption).
Electron spectroscopy
Instead of just letting the electrons fly off, they can be used to determine the
energy levels of the molecule they came from. We’ll concentrate on two of these types of
spectroscopy: X-ray Photoelectron Spectroscopy (XPS) and Auger electron spectroscopy.
Both are mainly used for surface characterization due to the large absorption or scattering
in most materials of the exciting beam (X-rays or electrons) and the emitted electrons.
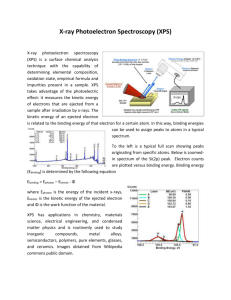
XPS
In XPS the input energy is from X-rays in the keV region. This ejects electrons from the
inner shells of the material being characterized, which are measured spectroscopically
(energy). Standard electron spectrometers use a magnetostatic field to bend the electron
beam through a semicircle to the detector; the field strength dictates the energy of the
electrons needed to stay on this path to reach the detector. (OHP)
Source: For XPS, typically K lines of Magnesium (=9.8900 Å or E=1.254 keV) or
Aluminum ( = 8.3393Å or E=1.487 keV) (OHP)
A related technique, Ultraviolet Photoelectron Spectroscopy (UPS) uses the same
principle, but with the exciting photons in the UV range (10’s of eV).
XPS (and UPS) can give information about composition, but also structure and oxidation
state. The reaction is written
A+hA+*+ewhere A is an atom or molecule and A+* is the electronically excited ion. The electron
emitted will have a kinetic energy
Ek=h-Eb-
where Eb is the binding energy of the electron and is the work function of the material
(energy needed for charge to escape surface). The work function can be measured in
other ways, and the binding energy is the energy of the atomic or molecular electron
orbitals relative to zero at infinite distance from the nucleus. The energy spectrum of the
emitted electrons, minus the input photon energy and work function, therefore, gives the
electron energy spectrum. The energy of the emitted electrons is in the range 250 to 1500
eV, with binding energies in the 0 t o1250 eV range.
The approximate sampling depth for XPS is d=3sin, with values typically 1 to 10 nm.
The spatial (cross sectional) resolution down to 10 m, so that surface imaging is
possible at the microscopic level.
Applications of XPS
Low resolution XPS is often used for elemental analysis at surfaces. This is usually a
qualitative analysis in the sense that the elements are identified only, but the relative
12
amounts present are not. This limitation stems from the need to calibrate the detectors in
terms of the peak heights or peak areas as a function of energy, which is difficult to do
accurately.
Chemical shifts: High resolution XPS can be used to investigate molecular environments,
because slight shifts in peak locations give information about the chemical environment.
This is due to the weak effects of the outer shell electrons on the energy levels of the
inner shell electrons. The outer shell electrons are affected by bonding and oxidation
state. This can also yield structural information, depending on the electron withdrawing
(or donating) abilities of the other atoms bonding to a particular atom. (OHP)
Limitations
Due to the short penetration depth, XPS is mainly limited to being a surface technique.
Chemical structure near a surface may not also represent the bulk, for example many
metals oxidize, so XPS spectrum are of the oxide and not the metal itself. This can be
very useful, though, in characterizing these surface phenomena.
The difficulty of calibrating peak heights also makes this technique difficult for
quantitative analysis, as described earlier.
There also can be interference from Auger electrons (see next) although this is easily
accounted for.
Finally, some materials are sensitive to high energy irradiation, so there is a danger of
either damaging the sample, or worse, changing its chemical composition during
measurement without knowing it.
Auger electron spectroscopy (AES)
AES is a related technique based on a similar, two step excitation process. In it, an
electron in an atom or molecule is ejected by an incident electron beam or X-ray. There is
now a vacant inner orbital state. This excited ion can decay by either an electron dropping
down from a higher level with the emission of a photon (usually X-ray – this is X-ray
fluorescence) or by the Auger process. In this process, an upper level electron also drops
down, but the energy it releases is given to a second electron, which is ejected from the
ion. The first process can be written as
A+ e-i (or h)A+*+e-i‘+e-A
Where e-i is the incident electron before (unprimed) or after (primed) the interaction and
e-A is the “first” Auger electron that is ejected from the inner orbitals.
The second process can be either X-ray fluorescence
A+*A++ h
or the Auger process
A+*A+++e-A
where the ion is now doubly ionized and the emitted Auger electron is the one that is
spectroscopically analyzed. The advantage with using this process over XPS is that the
kinetic energy of the Auger electron is now independent of the original excitation. The
kinetic energy will simply be the difference between the energy of relaxation (Eb- Eb‘
where the prime is the initial and unprimed the final energy level) and the energy needed
to remove the second electron, Eb‘. Note that these are the binding energies of the
13
electrons, i.e., we are going from zero at infinity to larger values as we go closer to the
nucleus (need more energy to remove). The kinetic energy will then be
Ek=(Eb-Eb’) - Eb‘ = Eb-2Eb’
In Auger spectroscopy, the independence of the process on the source energy means that
broadband sources can be used. The excitation generally produces both Auger and XPS
electrons – they can be differentiated by this dependence on the excitation source. In
Auger spectroscopy with broadband sources, the XPS electrons will just be a broad
background, but in XPS, the Auger electrons can be accounted for only by using a
different source.
NMR spectroscopy
Nuclear magnetic resonance is an extremely useful technique for materials
characterization and imaging. The principle is based on the absorption of radio frequency
radiation by nuclei in a material. Normally these nuclei would not absorb, but the
application of a strong static magnetic field causes a splitting of nuclear energy levels
due to nuclear spin. This allows one to vary both the energy levels by changing the static
field, as well as scanning the radio frequency to determine the level splitting. The
splitting will depend on both the field strength and the nucleus itself, but also on the
surrounding electrons, which shield the nucleus from the applied magnetic field.
Theory
A nucleus has a magnetic dipole moment m proportional to its angular momentum, p:
m =p. The constant of proportionality, , is called the gyromagnetic (or magnetogyric)
ratio. This ratio has a unique value for every nucleus and is the final quantity used for the
analysis in NMR spectroscopy. The angular momentum vector is proportional to the
nuclear spin I: p=I, giving m =I. The interaction of an external field B with this
moment gives rise to an interaction energy
U=-m ·B = -I·B
The direction of B specifies a direction (B=B0k), so the spins (angular momenta) along
this (z-) axis will be quantized with quantum numbers: mz=I, I-1, ... –I, where I is the
magnitude of the vector I. The resulting energy term is
U= -mzB0
In a nucleus with I=½, the two possible values of mz are ±½, giving a splitting of the
energy level E=-B0
which corresponds to EM absorption at a frequency =E/h=-B0/2. This is the basic
equation for NMR. It says that the frequency of absorption of EM radiation by a nucleus
in the presence of an applied magnetostatic field will be proportional to the field strength
and the magnetogyric ratio.
For example, for a proton, = 2.675104gauss-1s-1, giving an absorption frequency in Hz
of 4.258103 B0 (gauss) – typical absorption frequencies are in the radio range. By
applying either a constant field and scanning the radio frequency or a constant frequency
and changing the applied field (usual method), the magnetogyric ratio can be found and
the material identified.
Since the energy level difference between the two split levels depends on the applied
field, it can be (usually is) small compared to kT at room temperature (kT~40meV). To
14
determine the dynamics of a nuclear spin system under a field plus EM radiation, we
need to use statistical mechanics/thermodynamics.
In thermal equilibrium, the proportion of spins in the two states can be calculated using
Boltzmann’s equation as
N2
B
e E / kT e B / kT 1
N1
kT
The total magnetization will then be
M=(N2-N1) = Ntanh(B/kT)
When the magnetization is no longer in thermal equilibrium, we assume that it
approaches the equilibrium value exponentially, with a rate proportional to the deviation
from the equilibrium
dM z M 0 M z
dt
T1
T1 is called the longitudinal relaxation time. If a sample in thermal equilibrium is placed
in a field, the magnetization will realign to a new equilibrium value. Integrating the
equation above gives:
M z (t ) M 0 (1 e t / T1 )
Semiclassically, the torque on the moment m will be m × B giving the equation of
motion for the moment as
dp/dt = dI/dt = m × B
which leads to
dm/dt = m × B
If we rewrite this in terms of the macroscopic magnetization vector, M=mi where the
sum is over all spins in the material, we can write
dM/dt = M × B
where we’ve assumed only one isotope is important, so the magnetogyric ratios are the
same for all species.
Now we also need to include the relaxation term. The result for the z-component of
magnetization is
M Mz
dM z
(M B) z 0
dt
T1
Likewise the transverse components of the magnetization will decay to their equilibrium
value of zero, if they were not at zero when the field was applied
dM x
M
(M B) x x
dt
T2
dM y
My
(M B) y
dt
T2
where T2 is called the transverse relaxation time. These three equations together describe
the motion of the magnetization vector. It will precess about the applied field, depending
on its direction when the field was applied, but also relax to an equilibrium alignment
with the field. Since Mx and My are perpendicular to the field, no energy flows in or out
15
of the system, so the relaxation will be different than for Mz (hence different relaxation
time). T2 can be thought of as the time required for the spins to lose their phase.
{There is a similar description of the resonant absorption and decay of optical radiation
by a two level system. In it, the T1 relaxation time is called the population decay time
and T2 the phase decay time}
These three equations are called the Bloch equations – from them, we can determine the
response of a system to an applied field in terms of line width and intensity.
The longitudinal relaxation is often called the spin-lattice relaxation, after the main
mechanism for it: the interaction of the spins with the lattice. The transverse relaxation is
called spin-spin relaxation.
Typical values for T1 are on the order of seconds, while T2 values are generally smaller.
As described earlier, the positions of the resonances in the spectra are unique to each
species. The electrons will affect these resonances, so information on the bonds can also
be extracted from the spectra. These so-called chemical shifts are central to the use of
NMR spectroscopy in materials analysis. They can give information not only on bonds
themselves, but also their orientations.
Application
Standard NMR is preformed in the time-domain by application of short duration RF
pulses, followed by measurement of the relaxation vs. time of the “Free Induction Decay”
(FID). By Fourier transforming this data, a spectrum is obtained. Two main types of
NMR are proton (H1) and carbon, referring to the specific nuclear moment being
measured. Since the nuclear spin of carbon-12 is zero, the line for carbon-13 (~1% of
naturally occurring carbon) is used. For quantitative analysis of the material present, the
chemical shifts are the quantity of interest, so these shifts are measured for the proton or
carbon lines.
Since the splitting is B-field dependent, and calibrating the field exactly is difficult, a
reference is usually needed. This is usually accomplished by adding a well characterized
material to the sample - Tetramethylsilane, (CH3)4Si, usually referred to as TMS, is the
standard for this. Solvents used for samples must also have no strong lines to interfere
with the sample signal. This is often achieved by using deuterated solvents for proton
NMR.
General point about lines in spectroscopy – if two states are separated by an energy
corresponding to frequency and if the decay time of the states is on the order of , the
lines will no longer be capable of being resolved when ~1 (1/2). One way to think
about this is in terms of the uncertainty principle – the energy of states will be only
defined to within the inverse lifetime, so if the states are short-lived, the uncertainty in
energy means the spectral lines are correspondingly broad and overlap. In this case, only
a single broad feature is measured instead of separate lines. Another way to think about
this is that over some characteristic time of the measurement, the molecule will rapidly
change between states, so the spectrum will be some average of the two states.
Due to the close spacing of the spectral lines in NMR, this consideration is particularly
important for these measurements.
16
Limitations
NMR is limited to nuclei that have nonzero magnetic moments. It is also less sensitive
compared to other methods for analysis.
2D NMR
Multiple RF pulses can also be used to reorient dipoles that are losing phase by
precession. This allows one to independently determine both T1 and T2 relaxation times.
This can help resolving lines that cannot be isolated in standard 1-D NMR spectroscopy.
ESR
Electron Spin Resonance spectroscopy (also know as EPR – electron paramagnetic
resonance) is similar to NMR but uses the interaction of an applied field with magnetic
moments of unpaired electrons to split energy levels. This is useful for materials such as
radicals, metal containing complexes and semiconductors.
In contrast to NMR, this technique measures effects associated with the unpaired
electrons in the valence (outer) electron bands of a material, and so it is sensitive to
environmental effects. In particular, the interaction of this electron with its own nucleus
leads to hyperfine structure in the resonance spectrum, which can yield information about
the electron density in relation to the nucleus.
Applications
ESR is extremely sensitive, and since it detects materials with unpaired spins, it can be
used to detect the presence of impurities such as metal ions in a polymer or
semiconductor.
Mössbauer spectroscopy
Absorption spectroscopy extended to extremely high energies (-rays, 1019 Hz). Samples
are embedded in a crystal lattice. The lines measured here are extremely sharp, and give
similar information to that obtained with NMR. The sharpness of the lines is due to 1)
long lifetimes for nuclear excited states, so small lifetime broadening, 2) momentum of
photon is small since recoil is taken up by lattice – this means the Doppler shift will be
small.
Elemental analysis
One final, non-spectroscopic method for analyzing composition of materials is elemental
analysis. This is simply using a stoichiometric chemical reaction to produce a product
that can be more easily quantified. Some of these final quantification steps are simple, for
example gravimetric analysis. In this method, the reaction causes the precipitation of a
known product, which is weighed to calculate the total amount. Other techniques include
all of those we’ve talked about or will talk about for identifying composition.
Since the reaction must be chosen to react with a particular species, this method is most
often used when the presence of a particular species is known or suspected, and the
analysis is simply to determine the amount.
17
4. Scattering and diffraction techniques
As we’ve seen, spectroscopic techniques yield information on energy levels in materials.
This has most application for determination of composition or for assessing changes in
materials as a function of some processing. While structural information can also be
found, it usually involves some assumed models of the relation of the energy levels to the
structure.
A more direct characterization of the structure can be obtained from scattering methods.
(General use of the term scattering).
Scattering, general
In principle, the propagation of radiation (or other wave-like things) in material can
always be discussed purely in terms of scattering. At a molecular or atomic level, an atom
sees an incident EM field, polarizes (interacts nonresonantly with the field) and this
oscillating dipole radiates in all directions. The sum of the incident field and all the
radiating fields is the EM radiation field in the material. For example, the refractive index
of a material (speed of light) can be determined from this type of calculation.
Starting out with this simple idea, we assume the dipole moment of the scatterer as
(t)=0cos(t); where (t) and 0 may be complex.
The resulting radiation field will be
02 k 02 sin cos( kr t )
E
40
r
which gives an irradiance
I
04 4 sin 2
32 2 c 3 0 r 2
Note the inverse square law and the dependence of the irradiance on the fourth power of
the frequency.
This scattering is elastic – there is no change in energy (frequency) between the incident
and scattered photon.
Now consider a collection of scatterers. The phases from all of these will add up to the
resulting field at any point in space. If the scatterers are randomly placed, the scattering at
directions other than forward will be random. If their distribution is tenuous enough, the
radiation from individual scatterers will arrive at some point away from the forward
direction completely incoherent with the radiation from the other scatterers. In this case,
we can simply add the irradiances, ignoring phase (interference) effects to determine the
18
total scattered irradiance. For small particles or molecules, this is called Rayleigh
scattering.
Since the dipole moment of a particle (a collection of molecules) will go as the volume of
the scatterer, the irradiance will have a sixth power dependence on the particle size. This
allows one to use static particle scattering for determining particle size in dilute
dispersions.
Note that in the forward direction, the scattering from separate scatterers will always be
in phase.
Now we’ll start discussing diffraction/interference effects, which use the wave nature of
EM radiation (or particles) scattered from a material to understand its structure.
The starting point involves a consideration of a monochromatic plane EM wave.
E E0 e i ( k r t )
Consider a simple atomic plane in a material. X-rays have wavelengths on the order of
sub 1Å, which is the same order of magnitude as the atomic spacing. First consider what
happens to the individual atoms. Each experiences an applied EM field, has a
polarizability, and so a dipole (& higher order) is induced. This radiates out in all
directions. If the radiation is coherent with the input (probably non resonant) there is a
fixed phase relation between the input and output waves. Now consider the next plane
down. An atom here sees the same EM field, plus some phase difference due to the path
length difference. We assume that the phase relation between input and output is the
same. Therefore the rays scattered from the two will be in phase if the optical path length
is an integral multiple of the wavelength. This leads to the Bragg condition
2d sin n
This is the basis of X-ray (neutron, electron) diffraction.
[review of FT]
Reciprocal lattice
Crystal structures are generally specified according to the real-space intercepts of the
crystal planes. For example, a plane that intercepts the Cartesian (x,y,z) axes at ha0, kb0,
lc0, where a0, b0 and c0 are the unit cell dimensions, is referred to as the hkl plane. As a
results, planes in real space correspond to points in this reciprocal space, with the set of
these points making up the reciprocal lattice.
The axis vectors of the reciprocal lattice are constructed from the primitive vectors of the
crystal lattice:
An arbitrary vector in reciprocal space may be written
19
G
X-ray diffraction is a map of the reciprocal space.
Under invariant crystal transformations, the Fourier series for the electron density should
be invariant. It will be for wave vectors of the form of G above, e.g., the Fourier series of
the electron density can be expanded as
For X-ray scattering from two points within the sample, the phase factor will be
Now for the scattering over the entire material, the electron density times this phase
factor must be integrated over the sample. This gives
The result of the integration will be effectively zero when the reciprocal lattice vector is
not equal to the scattering vector. In this case, the amplitude will equal the sample
volume times the electron density coefficient for vector G (V*ng).
Using the fact that the magnitudes of the wave vectors are equal for elastic scattering, we
can derive the equation
This is in essence a generalized version of the Bragg condition.
We can subdivide the effects resulting in the X-ray scattering into three groups: scattering
by the electrons themselves, scattering from the atoms and scattering by the unit cell.
1) electron
2) Atom
The scattering by the nucleus is much smaller than that from the electrons due to the 1/m
factor in the equation above. Therefore, the atomic scattering will be the sum of the
scattering by the electrons with the appropriate relative phases determined by the
electronic positions within the atom. This calculation gives the atomic scattering factor f,
equal to the amplitude of the wave scattered by the atom divided by the amplitude
scattered by a single electron. For forward scattering (=0) where the contribution from
all electrons is in phase, the atomic scattering factor is equal to Z, the total number of
electrons.
3) Unit cell
20
Likewise, the scattering from the unit cell will equal the sum of the scattering from
individual atoms multiplied by the appropriate form factors. For a unit cell with N atoms
in it, this factor is
N
fe
i 1
2 i ( hui kvi lwi )
i
where ui, vi and wi are the positions of the ith atoms.
Multiple planes in crystal – rotate the crystal – at each angle such that the Bragg
condition is fulfilled, there will be strong scattering.
This allows a full determination of the crystal structure.
There are several ways to actually perform this measurement. In Laue diffraction, a
stationary bulk crystal is irradiated with polychromatic X-rays. The broad band of the
incident radiation insures that the Bragg condition will be fulfilled at some angles. The
angular distribution of the diffracted waves is measured and using the Bragg equation, the
spacing of the molecular planes is calculated. This method has the advantage of allowing
the determination of the planes with respect to the crystal habit.
This method has a simple setup of source and detector and is easy to perform, but
requires a bulk crystal. This does lead to an additional application, though. By varying
the input beam size and observing the scattering intensity, quantitative information about
the crystal grain size can be found.
A second method is the Debye-Scherrer powder method. This uses monochromatic Xrays incident on a powder sample – now the random orientation of the powder insures
that the Bragg condition will be met. The scattering intensity as a function of the
incidence angle is again measured.
In general, the planar spacing alone does not give the crystal structure directly. A number
of pacing values must be determined and the ratios considered in order to form a model
of the structure. Often the results are compared to known structures.
Sources: same as XPS
(Brehmstrahlung, atomic fluorescence lines, synchrotron)
Structure factor:
Real crystals are never perfect. The following OHP shows what one might expect from
various materials. The x-axis is the magnitude of the so-called scattering vector
q=2ksin.
21
Small angle scattering
We
the Bragg diffraction condition can be written as
saw
k G
where G is a reciprocal vector of the lattice. diffraction at grazing incidence implies
incident and scattered wave vectors k that are almost the same. Therefore k will be
small and the observed diffraction will correspond to small reciprocal lattice vectors, or
large spatial features in the sample.
Two examples of small angle diffraction techniques are Small angle X-ray scattering
(SAXS) and small-angle neutron scattering (SANS). Both of these probe features on the
order of 10 to 1000 Å, compared to <10 Å for usual X-ray or neutron scattering. These
techniques are especially suited to characterizing small particles or voids within
materials.
Neutron diffraction
Neutrons have a number of advantages as diffracting particles, compared to X-ray
photons or electrons. The fact that they have no charge means they interact with the
nucleus only. This also implies that they have a high penetration depth, as so can be used
for bulk studies, as compared to electrons, which are absorbed or scattered with a short
distance into a material.
Many of the disadvantages stem from the difficulty of producing and measuring them.
Some type of nuclear reaction is needed for the production, and the resulting neutrons are
polychromatic, and usually must be diffracted by germanium or lead crystals to yield
monochromatic beams. Detection is usually with a gas-type scintillation counters.
Neutron diffraction can be considered in terms of the deBroglie wavelength of the
particles. This wavelength is related to the particle momentum by
=h/p
where h is Planck’s constant and p is the particle momentum. For non-relativistic
particles, p is just mv=(2mE)½ where E is the kinetic energy. This gives the wavelength
in terms of the energy as
=h(2mE) -½
this leads to another advantage of neutron diffraction – the large mass of the neutron
means that small distances can be probes (small ) with relatively low energies. Neutron
diffraction energies generally lie in the meV to eV range, with wavelengths 10-13 to 10-4
cm.
Imaging
Optical imaging is the transfer from the object to image plane by an optical system. We’ll
first consider this in the simple geometric optics limit, i.e. with ray tracing/lens equation.
The object (so) and image distance (si) for a thin lens are related by the equation
1 1 1
so si
f
22
where f is the focal length of the lens. The sign convention for si and so is that the bema
propagates from left to right, so is positive for an object located to the left of the lens, si is
positive for an image to the right of the lens, and the focal length is positive for a
focusing lens.
From this equation, the image location can be determined. The size of the image is also
determined (in the thin lens approximation) by the same variables. The transverse
magnification is the size increase in the plane perpendicular to the optical axis.
y
s
f
MT i i
yo
so
xo
where y denotes the transverse size of the object or image, and xo is the distance of the
object from the focus.
It is also possible to show that the longitudinal size of the image (along the optical axis)
will not have the same magnification. The longitudinal magnification is
dx
M L i M T2
dxo
More complex optical systems can be analyzed by successive application of these ideas,
i.e., by propagating the image through each optical element.
One other important consideration for optical systems is the light gathering power.
Apertures are critical parts to optical systems, since they restrict the total amount of light
getting through the system. Even for no intentional aperture, the boundaries of the lenses
are apertures that must be considered. The effective aperture for light entering an optical
system is called the entrance pupil – it is the image of the aperture stop seen looking into
the system. When there are more than one stops, it is the smallest of these images.
Essentially, this is what you would see as the boundary when you looked into the system.
The area of the light entering the system will be proportional to the square of this
entrance pupil. The area of the image (for a fixed object distance) will be proportional to
the square of the focal length of the system. The total flux density of light at the image,
therefore, will be proportional to (D/f)2, where D is the radius of the entrance pupil. The
ratio D/f is called the relative aperture, and its inverse f/D is called the f-number,
sometimes written f/#. The relative aperture is measure of the light gathering power at the
image plane, and the f number is referred to as the speed of a lens – the smaller it is, the
faster an image can be formed for a given detector or film sensitivity.
In general, for optical systems one wants to specify the f/# of a lens and match
components with the same f/#’s, otherwise light is being “wasted” in passing through the
system.
Compound microscope
The basic setup for a compound microscope is shown in the OHP. Objective forms
inverted, magnified image, eyepiece further magnifies.
Definition of numerical aperture in terms of maximum angle that light can still enter the
sample at.
NA=nisin
High NA objectives – oil immersion (oil provides index match to objective)
23
Aberrations
5 primary plus chromatic (other notes)
Resolution
Even with perfect objects, there is a fundamental limit to the resolution an optical system
is capable of. A beam of light passing through an aperture (i.e. any beam not infinite in
cross section) will diffract – this is essentially interference of the bema with itself. A
point imaged through a lens will not give a point image, rather a diffraction pattern (Airy
disk)
The Rayleigh diffraction criterion states that two objects may still be resolved if the
maximum of the diffraction pattern of one coincides with the first minimum of the other.
Quantitatively, the criterion is
l=1.22f/D
where l is the minimum spacing of two points such that they can be resolved. We see
immediately that the minimum size that can be resolved is proportional to the f/# and the
wavelength. For higher power, we need shorter wavelength and smaller (faster) lens
systems. The disadvantage to smaller f/# is that the focal length will be very small (hard
to work with) and the aperture must be large (also difficult).
Types of microscopy
Transmission
Bright field – reflection microscopy with direct backscatter
Contrast
Dark field – reflection microscopy, grazing incidence of illumination
Fluorescence emission
Confocal
etc.
Detection
Eye
CCD camera
High sensitivity photodetector (PMT, APD) for confocal
Specific property characterization
Electrical
(More from Prof. Currie)
The transport of current through a material in response to an applied field is a critical
phenomenon for many applications, especially in the electronics industry, but also in
other areas such as electrochemical synthesis and electro optic displays.
The starting place for this is Ohm’s law for an ideal resistor, where the current response
to an applied potential is linear
V=IR=I/
where we can characterize the response using R, the resistance of the material, or , the
conductance.
24
This equation is only an approximation, valid for specific materials and under limited
range of currents and voltages. In most real life cases, one needs to characterize the
response by measuring the current as a function of applied voltage. This can also be
“non-Markovian” – it may depend on the history of the applied potential as well as the
potential at a given instant. The measurement is straightforward – a potential is applied
and the resulting current is measured. One must be careful with the instrumentation to
make sure to account for resistances other than that of the sample.
With solid samples, care must also be taken with respect to the contacts to the material. If
the sample is not a metal, there can be a barrier to the injection of charge into the
material, depending on the relative work functions of the probe and material. This leads
to definitions of the internal (or intrinsic) and external (or extrinsic) conductivity, where
the intrinsic is the actual material conductivity at a molecular scale, and extrinsic is that
measured in a particular device implementation.
In liquid samples, the conductivity measurement can be critical for understanding
electrochemical reactions. These are reactions in an electrochemical cell – one that relies
on the injection of charges through electrodes placed in the reaction mixture.
(Solar cell application)
A history dependence of the current on applied voltage can be characterized by the
response to an applied harmonic voltage as a function of frequency (time-frequency). The
measurement of this is a technique called impedance spectroscopy (see links). The
current in response to this voltage will have both an amplitude and a phase shift, which
contains details about the time dependence. One application this is in the study of charge
transport mechanisms in solids. One can tell from the spectral response whether the
resistance is from scattering of electrons (phonons) or because the charges must “hop”
between sites.
Magnetic
For many applications, one would like to measure the magnetization in response to an
applied field. In particular, for recording applications, this is crucial.
The basic magnetostatic equations are
B=M+0H
where B is the magnetic induction, or flux density vector, M is the magnetization
(magnetic dipole per unit volume) and H is the magnetic field vector and 0 is the
vacuum permeability. The linear response of a material to an applied field can be written
M=H
where is the magnetic susceptibility. Rearranging the equations we can also write
B=H; = (+0) is the magnetic permeability.
Many different types of magnetization are possible in response to an applied field,
depending on the origin and functionality of the response (diamagnetism, para-, antiferro, ferri-, ferro- , etc.)
As in electrical conductivity measurements, a rigorous characterization requires
consideration of the time (or frequency) dependence, as well as the response over a wide
range of applied magnetic fields. In addition, there also may be a difference between the
intrinsic and extrinsic magnetization, especially because of the demagnetizing field. The
25
induced magnetization will set up a reverse field inside the sample, which will depend on
the sample geometry as well as the magnetization. This must be properly accounted for to
determine the intrinsic magnetization of the sample.
Many materials will also display history dependent properties, which can be quantified in
hysteresis curves of the magnetization or B-field as a function of the applied magnetic
field. (OHP)
Optical scattering
Optical scattering can be used to characterize both composition and structure.
Composition is generally found through spectral measurements of the scattered radiation
– we’ve already covered that, so we’ll concentrate more on the structure. Two general
areas we’ll discuss are scattering from surfaces and ordered structures, which is related to
the diffraction techniques we’ve talked about, and scattering from particles. Furthermore,
we’ll discuss several types of scattering from particles, depending on what is measured.
Surfaces
As an EM wave, light will scatter/diffract from a surface
We spoke earlier about dipole scattering in general.
Fractionation techniques
- spearation of species comprsing a complex mixture by a specific propoerty
Mass spectroscopy
In mass spec, molecules are ionized and often fragmented, then accelerated by an electric
field. An ion of a given charge will receive a set amount of (kinetic) energy for a fixed
electric field. The resulting velocity will then depend on the mass of the molecule or
fragment. Measurement of this velocity, which will yield the mass/charge ratio, is done
by separating the ions with a combination of electric and magnetic fields. Mass spec
instruments differ in the methods used to ionize the molecules and the way in which the
resulting velocities are measured.
There are two versions of mass spec methods that basically differ in the sample
preparation. In the first type, the molecules in the sample are simply ionized, as described
above and below. In order to get more information about the chemical moeties
comprising a molecule, the molecules may also be borken into parts, and the m/z ratio of
these measured. The so-called tandem mass spec methods are based on applying this
second method after the m/z ratios of the molecular species have been determined.
26
Generally, low pressures are needed for mass spec, since the ions must travel unscattered
through the chambers for the mass analysis. Typically pressures on the order of 10-5 torr
and lower are used. Care must be taken to ensure proper vacuum seals when introducing
the sample into the ionization chamber, in particular for combination techniques in which
the sample is the output of some other fractionation technique.
After ionization, the molecules and fragments are accelerated by an electric field, giving
them a kinetic energy
KE qV zeV 1 mv 2
2
2 zeV
v
m
Here the accelerating potential is on the order of kV.
A number of methods exist for the actual detection and analysis of the mass. Common
detectors include electron multipliers and Faraday cups. The analysis (separation) of mass
also is done with a number of different methods, including time of flight, quadrupole and
magnetic sector spectrometers.
For all of these, the resolution of the spectrometer is defined as
R m / m
where m is the mass difference between adjacent peaks that can be resolved with the
spectrometer and m is the mass of the first of these peaks. The higher the resolution, the
better.
Ionization methods:
Gas-phase
Electron impact (EI) – molecules are first thermally vaporized, then ionized by energetic
electrons (usually 70 eV). This often causes fragmentation, resulting in multiple peaks in
the mass spectrum.
Chemical ionization (CI) A chemical reaction produces the ions. The reactant (commonly
methane) is itself usually an ion produced by electron bombardment. Some fragmentation
may also occur, but much less than in EI.
Field ionization (FI) – ionization by large E-field
Desorption – direct formation of gaseous ions from solid or liquid
MALDI (matrix assisted laser desorption ionization) – useful for smaller molecular
weight materials. The sample is dispersed in a polymer binder. Pulsed laser radiation is
abostrbed by the matrix, and the (thermal) energy is transferred to the sample. This
ionizes it and may also break it into molecular fragments, which are then analyzed in
standard m/z analyzers (often TOF).
SIMS (secondary ion mass spectroscopy) – bombardment of surface with positive ions –
causes secondary ionization. Used primarily as surface analysis technique.
PDMS (plasma desorption mass spec) – desorption of molecules from a thin metal (Al)
target by fission fragments of 252Cf, little fragmentation
FAB (fast atom bombardment) – bombardment of sample by high energy Xe or Ar ions.
27
ES (electrospray) – Spray of droplets, heating causes solvent loss, individual ions. Good
for large biomolecules
Mass analyzers
Magnetic sector
These use a permanent (static) or electromagnet to cause the ions to travel in a circular
path (usually 90 or 180 degrees) through a slit to the detector. The particular ions that
travel in this circular path will depend on the magnetic field strength and the velocity of
the ions. The Lorentz force on the ions is given by
mv 2
F Bzev
r
Bzer
2 zeV
v
m
m
2 2
m B r e
z
2V
We see that the particular ions (values of m/z) that reach the detector can be varied by
changing B, r or V.
Quadrupole
These are more common commercially due to being more rugged and compact. These use
differences in trajectories of the ions in an electric quadrupole to sort the various m/z
values. DC and AC fields are applied between the poles; only ions of a fixed m/z value
will have stable trajectories through the poles.
Time of flight
These use a timing mechanisms to determine the time for ions to reach the detector. They
are used with ionization sources that allow this, for example PDMS, where one of the
fission fragments starts the timing and the other ionizes the sample. Other methods use
pulses ionization to start the timing.
Chromatography
In chromatographic methods, complex mixtures are spearated according to time. A
sample undergoes some process in which the total time that individual species take to
complete the process differs.
Most commonly, the sample is transported in a mobile phase through an immiscible
stationary phase. The sample partitions itself between the two phases, and its transport
through the instrument will depend on this partition and the amount of retention in the
stationary phase.
Elution – continuous washing speices through column by addition of fresh solvent. The
sample moves down the column in the mobile phase only, the average rate will depend on
the fraction of time it spends in the that phase. Separation into zones.
The partioning is a thermodynamic equilibrium between two phases, and can be
descriebd by a simple rate reation equations, with rate constant K=cs/cm. The numerator
28
cs is the molar concentration of solute in the statiinoary phase and cm in the mobile
phase. This rate is generally constant. One measures the retention times of the solute in
the column; to calculate the molecular weights or other quantities, one needs to
understand the underlying physics of the particular retention process, or perform
sufficient calibrations with known substances.
Direct measurement techniques:
These are methods that depend on specific properties – the property itself is measureed
directly or indirecly. Some examples follow.
Measurement of magnetization
1. Force acting on sample
magnetic balance – deflection of sample in field
2. Magnetic field produced by sample
vibrating sample magnetometer – sample in field, also pickup coils. Sample vibrated &
current induced in coils used to determine magnetization.
3. Current produce by magnetization – material in coil, changing applied field or
removing from coils induces current in coil.
Measurement of magnetic field
1. Pickup coils (for changes in field)
2. Hall probe – Lorentz force on electrons
3. Squid – measures magnetic flux through superconducting loop.
Thermal measurements
Calorimetry
DSC
29