Supplementary Information (doc 1553K)
advertisement
")
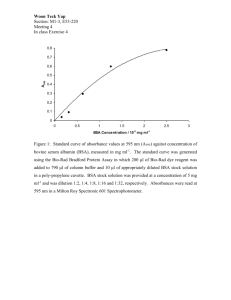
[Supplementary Information] A novel approach for the parallel synthesis of glycopeptides by combining solid-phase peptide synthesis and dendrimer-supported enzymatic modifications Takahiko Matsushita,1 Seiji Handa,1 Kentaro Naruchi,2 Fayna Garcia-Martin,1 Hiroshi Hinou,1,2 and Shin-Ichiro Nishimura*1,2 1 Graduate School of Life Science, Hokkaido University, N22, W11, Kita-ku, Sapporo 001-0021, Japan. 2 Medicinal Chemistry Pharmaceuticals, Co. Ltd., N22, W12. Kita-ku, Sapporo 001-0021, Japan Corresponding author: Shin-Ichiro Nishimura, E-mail: shin@sci.hokudai.ac.jp; Fax: +81-11-706-9042; Tel: +81-11-706-9043 S1 Materials and Methods All solid-phase reactions for glycopeptide synthesis were performed manually in a polypropylene tube equipped with a filter (LibraTube; Hipep Laboratories). Coupling reactions of Fmoc- amino acid derivatives and Fmoc removal reactions were conducted under microwave irradiation (0-120 w) at 50°C, and acetyl capping reactions after coupling were carried out by conventional manner at ambient temperature. Microwave-assisted solid-phase reactions were performed manually using a single mode microwave reactor (IDX Corp., GreenMotif I). The microwave reactor was a customized reaction cavity for installation of LibraTube and introduced an additional function of mechanically shaking for the reaction cavity in our laboratories. Bis-Boc-aminooxyacetyl-G7 PAMAM dendrimer (1). To a solution of 5 wt% G7 PAMAM dendrimer (ethylenediamine core) in methanol (400 l, 0.058 mol, 29.7 mol of amino group) were added bis-Boc-aminooxyacetic acid N-hydroxysuccinimide ester (115 mg, 297 mol) and N-methylmorpholine (3.3 l, 29.7 mol) and stirred at ambient temperature for 24 h. The crude product was purified by size-exclusion chromatography (Sephadex LH-20, GE Healthcare Bio-Science) eluted with MeOH to give 1 (8.5 mg, 57%). 600 MHz 1H-NMR spectrum of 1 (methanol-d4, 300 K) is shown in Figure S4. Aminooxy-functionalized G7 PAMAM dendrimer (2). 4M HCl (1.88 ml) was added to dendrimer 1 (8.5 mg, 16.9 mol peripheral amino group) and stirred at ambient temperature for 12h. After quenching with 4M NaOH (1.8 ml) and 1M NaOH (50 l) to adjust at pH 7.0, water (7.53 ml) was added to prepare a stock solution of 2 (theoretically 1.5 mM aminooxy group). The 600 MHz 1H-NMR spectrum of 2 (D2O, 300 K) is shown in Figure S5. Solid-phase synthesis of molecular shuttle (model glycopeptide) (3). The solid-phase synthesis of molecular shuttle carrying model glycopeptide 3 was carried out using TentaGel S RAM resin (Leading: 0.26 mmol/g, 100 mg, 26 mol). To the pre-swollen resin in DMF for 30 min at ambient temperature was added 20% S2 piperidine in DMF (3 ml) and shaken under microwave irradiation (0-120W) at 50°C for 6 min. After washing with DMF (5×3 ml), Fmoc-amino acid (130 μmol, 5.0 equiv) activated with HBTU (130 μmol, 5 equiv), HOBt (130 μmol, 5 equiv), and DIPEA (260 μmol, 10 equiv) in DMF (1.5 mL) was coupled to the resin under microwave irradiation (0-120W) at 50°C for 12 min. For incorporation of Fmoc-glycosylated amino acid to the resin, Fmoc-Ser(Ac3GlcNAc)-OH (75 μmol, 1.5 equiv) was used in the presence of HBTU (75 μmol, 1.5 equiv), HOBt (75 μmol, 1.5 equiv), and DIPEA (150 μmol, 3 equiv) in DMF (1.5 mL) under microwave irradiation (0-120W) at 50 °C for 20 min. After filtration and washing with DMF (5×3 mL), unreacted amino group on the resin were capped with acetyl group by treatment of acetic anhydride (4.75%, v/v), DIPEA (2.25%, v/v), and HOBt (13 mM) in DMF (4 mL). After shaking under microwave irradiation (0-120W) at 50 °C for 3 min, the resin was filtered and washed with DMF (5×3 mL). Fmoc-removal, coupling, and capping procedures were carried out repeatedly. At the final step of glycopeptide elongation on the resin, extra Fmoc- Glu(OtBu)-OH, Fmoc-Phe-OH, and 5-oxohexanoic acid were assembled under the same coupling condition of Fmoc amino acids. Upon completion of the synthesis, the glycopeptidyl-resin was treated with 90% aqueous TFA (3 ml) at ambient temperature for 1 h, and the resin was filtered off. The resin was washed with 90% aqueous TFA (2×1 ml) and the combined filtrate was concentrated by a flow of nitrogen gas. The crude glycopeptide was precipitated using cold tert-butylmethylether. After collecting the precipitate by centrifugation (3,000×g, 15 min, 4°C) and removal of supernatant, the dried precipitate was dissolved in 50% acetonitrile aqueous (4 ml) and lyophilized. Next, to a solution of the lyophilized material in methanol (2 ml) was added 1 M NaOH in methanol (1:1, v/v) to adjust pH to 12.5 and kept at room temperature for 40 min. After quenching with acetic acid, solvent was removed under reduced pressure. The crude glycopeptide was purified by preparative RP-HPLC (tR = 25.4 min) to give 3 (14.2 mg, 8.8 mol) in 34% overall yield. The RP-HPLC purification condition was described below: column, Inertsil ODS-3 (250×20 mmI.D.); eluent A, water containing 0.1% TFA; eluent B, acetonitrile containing 0.1% TFA; Eluent (A/B = 98/2) was employed and kept over 10 min, then the ratio of eluent B was increased lineally from 2% to 10% over 40 min with a flow rate of 5.0 mL/min. Preparative RP-HPLC and MALDI-TOFMS of 3 were shown in Figure S1. MALDI-TOFMS: C70H111N18O25 [M+H]+ calcd (m/z) 1603.797, found (m/z) 1603.565. S3 (A) concentration of eluent B (%) 3 time (min) 1603.565 Intens . [a .u.] (B) 5000 3 [M+H]+ 4000 1641.636 3000 2000 2356.480 2233.138 2029.813 2119.387 1895.467 1700.783 1546.504 1441.912 1271.483 1117.343 955.237 839.015 777.501 684.592 597.299 538.014 396.184 346.589 1000 0 500 750 1000 1250 1500 1750 2000 2250 2500 m /z Figure S1. (A) Preparative RP-HPLC of crude glycopeptide 3. (B) MALDI-TOFMS of the fraction corresponding to the peak at 25.48 min shown in (A). S4 BLase-catalyzed enzymatic cleavage for molecular shuttle (model glycopeptides) 3 (Scheme S1). To the stock solution of 3 in water (4 mM, 35 l) were added 500 mM ammonium acetate buffer, pH 6.8 (14 l), 17.4 gml-1 BLase (1.6 l) and water (89 l). After standing at room temperature, 10 l of the reaction mixtures after 0, 1, 2, 3, 6 and 12 h were analyzed by RP-HPLC (Figure S2). The analytical RP-HPLC condition was described below: column, Inertsil ODS-3 (φ4.6x250 mm); eluent A, 25 mM ammonium acetate buffer, pH 5.0; eluent B, 10% eluent A in acetonitrile. Eluent (A/B = 95/5) was employed and the ratio of eluent B was increased lineally from 5% to 40% over 40 min and from 40% to 90% over 2 min, and kept 90% over 2min; then the ratio of eluent B was decreased lineally from 90% to 5% over 1 min and kept 5% over 14 min with a flow rate of 1.0 mL/min. S5 Scheme S1. BLase-catalyzed enzymatic cleavage for molecular shuttle (model glycopeptides) 3. 3 5 26 time (min) Figure S2. RP-HPLC stack plot of reaction mixtures after 0, 1, 2, 3, 6 and 12 h of BLase-catalyzed enzymatic cleavage for 3. The peaks at 17 min, 24 min, and 36 min are correspondeding to 5, 26, and 3. S6 Figure S3. SEC-HPLC stack plots of the reaction mixtures after 0, 1, 2, 3, 6, 12, 24 and 48 h of conjugation reaction between dendrimer 2 and molecular shuttle 3 under different pH condition (pH 3.5, 4.0, 4.5, 5.0 and 5.5). The SEC-HPLC condition is as indicated bellow; column: YMC-Pack Diol-200 column, 500×8.0 mm I.D., flow rate: 0.7 ml/min, eluent: 50 mM sodium phosphate buffer, 0.3 M NaCl (pH 7.0). S7 10 9 3 8 buffer 7 1.0 eq 0.8 eq 0.6 eq 0.4 eq 0.2 eq 6 time (min) Figure S4. SEC-HPLC stack plot of conjugation reaction after 48 h for preparing glycopeptide-dendrimer conjugates 6, 7, 8, 9 and 10 using 0.2, 0.4, 0.6, 0.8 and 1.0 equivalent molecular shuttle 3 to 1.0 equivalent aminooxy group on dendrimer 2, respectively. The analytical SEC-HPLC condition is as indicated bellow: column; YMC-Pack Diol-200 column, 500×8.0 mmI.D., flow rate; 0.7 ml/min, eluent; 50 mM sodium phosphate buffer, 0.3 M NaCl (pH 7.0). S8 Figure S5. 600 MHz 1H-NMR spectrum of 1 in methanol-d4 at 300 K. Figure S6. 600 MHz 1H-NMR spectrum of 2 in D2O at 300 K. S9 S10