Haem Revision Notes
advertisement

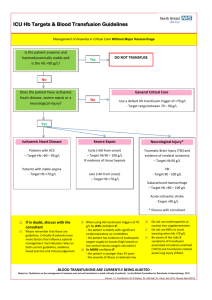
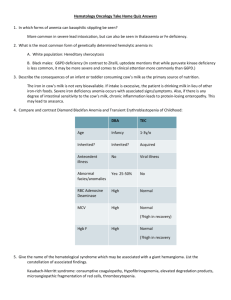
Haematology Notes Neutrophilia Primary Neoplasia e.g. CML Eosinophilia Neoplasia e.g. HL, T-cell NHL Monocytosis Neoplasia e.g. CML Basophilia Lymphocytosis Neoplasia e.g. CML Neoplasia e.g. CML/ lymphoma Coagulation Cascade Secondary Infection - Bacterial infection Inflammation - autoimmune Infection - Parasitic infection Inflammation - allergic disease Infection - usually chronic (e.g. TB, CMV) Infection - pox virus Infection - EBV, CMV Others Steroids DKA Idiopathic hyperoesinophilic syndrome Haemostasis Vasoconstriction Formation of loose platelet plug (primary) Stabilisation (secondary) Fibrinolysis Key Diagnosic Tests Common Pathway Intrinsic Pathway Extrinsic Pathway Factors X, V, platelets, prothrombin/thrombin, fibrinogen/fibrin, VIII, IX, XI, XII TF, VII Investigations PT APTT PT Haemorrhagic Disorders Site of bleeding Petechiae Ecchymoses Hemarthrosis/muscle Bleeding after surgery Platelet problem Skin, mucous membranes Yes Small, superficial No Immediate Platelet Disorders Normal count: 150-400 < 50 - petechiae, ecchymoses < 20 - mucocutaneous bleeds (GI bleeding, haematuria) Coagulation problem Soft tissues, joints, muscles No Large, deep Yes Delayed (days) Clotting Disorders Disease Haemophilia A (VIII deficiency) Haemophilia B (IX deficiency) vWD Inheritance X-linked Recessive X-linked Type 1 - AD + partial deficiency Type 2 - AD + loss of multimers altered quality Type 3 - AR + complete deficiency Liver disease Vitamin K deficiency DIC Presentation < 1%: haemarthroses + deep muscle haematoma 1-5%: severe bleeding after injury > 5%: mild bleeding after injury. Late dx Same features as haemophilia A ↓Platelet adhesion ↓Stabilising factor for VIII:C Presents as a platelet problem Investigations Tx: Factor concentrate, Desmopressin (mild disease) Desmopressin ineffective Bleeding time prolonged PT & APTT – normal Tx: Desmopressin, VIII + vWF concentrates Malnutrition, drugs or Malabsorption causes ↓ Vit K, ↓ factor production + functional abnormalities ↓ II, VII, IX and X Sepsis, Malignancy, Severe liver disease, Haemolytic transfusion reactions, Obstetric causes and Trauma causes Widespread intravascular coagulation (Consumption of platelets + Consumption of clotting factors and fibrin generation), Secondary fibrinolysis Leads to bleeding and clotting ↑ PT + APTT Prolonged PT and APTT Prolonged TT ↑FDPs/D-dimer Thrombocytopenia Tx: Support + treat underling cause Thrombotic Disorders Virchow’s Triad Hypercoagulability Stasis Vessel wall Inherited Thrombophilia Factor V Leiden Prothrombin G20210A Protein C deficiency Protein S deficiency Anti-thrombin deficiency Features Normal pro-coagulant, abnormal anti-coagulation Elevated prothrombin Autosomal dominant – early thrombosis (<40y) Tx – protein C/protein S concentrate Inherited (AD) or acquired (nephrotic syndrome) Inherited present early Acquired Thrombophilia Immobility/stasis Surgical (tissue trauma -- pro-coagulant) Malignancy (? pro-coagulant molecule) Anti-phospholipid syndrome Hyper-oestrogen state Anti-coagulant therapy Heparin Immediate effect Potentiates anti-thrombin III 2 types: o Unfractionated heparin o LMWH (1 daily s.c.) Aim for APPT 1.5-2.5 Features Hospital in-patients, elderly with loss of mobility Orthopaedics and gynaecology Young women with: Recurrent DVT, miscarriages and stroke Pregnancy/COCP/HRT Increased coagulation factors and reduced anticoagulation factors Warfarin Long term therapy Vitamin K antagonist Initial procoagulant effect Monitored with PT/INR Anaemia Hb < reference range for age, sex and gender of an individual. o Men < 13.5 g/dl o Women < 11.5 g/dl Reduced red cell count (RCC) Reduced packed cell volume (PCV) Classical Signs Conjunctival pallor Koilonychia Glossitis Angular stomatitis Post-cricoid webs (Plummer-Vinson Syndrome) High-flow murmur May be classified according to: Size o Microcytic (IDA, thalassaemia) o Normocytic (Acute blood loss, chronic disease, leukoerythroblastic) o Macrocytic (Vitamin B12 deficiency, folate deficiency, alcoholism, hypothyroidism, liver disease) Pathophysiology o Reduced production (Haematinic deficiency, bone marrow infiltration, chronic disease) o Reduced life-Span (Haemolysis) o Pooling (Splenomegaly) Reduced Production Pathology ↓Fe Causes Reduced intake Malabsorption Blood loss ↑Demand ↓Vit B12 ↓ intake Impaired absorption Pernicious Anaemia (AI, ↓IF) ↓Folate ↓ intake Malabsorption ↑Demand Drugs (methotexate, ETOH, anticonvulsants) Destruction/competition with normal bone marrow Can be cellular or hypocellular Bone Marrow Infiltration Myelodysplasia Anaeamia of Chronic Disease Mild malignancy of early myeloid progenitor cells Elderly Chronic infection Chronic inflammation Malignancy Neoplasia Increased Destruction Extravascular o Common o Occurs in spleen (macrophages) Clinical Features Variable No anaemia Chronic haemolytic anaemia Folate deficiency Key haematological features Raised unconjugated bilirubin ↓ haptoglobin ↓ LDH Features ↓ MCV, ↓ ferritin, ↑ TIBC Hypochromia, poikilocytosis, anisocytosis Tx: Ferrous Sulphate, Paraenteral Fe Macrocytosis (megaloblast) Hypersegmented neutrophils Thrombocyopaenia Peripheral neuropathy Tx: Hydroxycobalamin ↓ RBC folate Tx: Folate Cellular Myeloproliferation, myelofibrosis, malignant spread Hypocellular Idiopathic (50%), Cytotoxic drugs + radiation, Infections, Immune Reduction in cell line function BM: increased cellularity + abnormal myeloid precursors ↓Hb, ↔/↓MCV, ↔/↑Ferritin Tx: Erythropoietin Intravascular o Rare o Occurs in blood Acute episodes Crises Aplastic anaemia ↑ urinary urobilinogen Reticulocytes Inherited: Spherocytosis Inheritance AD Features Variable Elliptocytosis AD Mild Course G6PD X-linked Enzyme needed to produce NADPH Used with glutathione to protect from oxidative damage Sickle cell Thalassaemia Point Mutation (β globin chain) α - Gene deletions β- Point mutations Very common Africa/Americans/Mediterraneans Chronic haemolytic anaemia Acute haemolytic episodes - Drugs - Fava beans - Infection Shortened red cell survival • Chronic haemolysis • Splenomegaly • Gallstones • Folate defiency • Vulnerable to aplastic anaemia Vaso-occlusive disease • Impaired passage of cells in microcirculation • Obstruction of small vessel + infarction • Consequences • Crises • Organ dysfunction β: Major - homozygous (Cooley’s anaemia), present in first year, require blood transfusion Minor/Intermedia – heterozygous, asymptomatic, mild haemolytic picture α: Severity deficiency 4 - incompatible with life (hydrops fetalis) 3 - moderate anaemia + splenomegaly 1or2 - asymptomatic with mild anaemia Haematological Malignancies Leukaemia: malignant cells in peripheral blood/bone marrow Lymphoma: solid tumour (malignant cells in lymph nodes) Myeloma: malignant cells in bone marrow and production of paraprotein Diagnostic tests DAT neg, osmotic fragility Tx: Splenectomy + folate Blood film Tx: Nil Heinz Bodies Bite Cells DAT test – Negative Beutler Fluorescent Spot test Dx: Hb electrophoresis + sick solubility tests Tx: Acute sickle crisis prevention, Vaccination, penicillin prophylaxis + folic acid, hydroxycarbamide Leukaemias Subtypes ALL B cell T cell Burkitt’s leukaemia AML M0-M7/WHO classification CLL B cell (95%) T cell (5%) Features Children De novo Presentation: Bone marrow failure, Lymphadenopathy, hepatosplenomegaly, Testicular infiltration Adults De novo or secondary Presentation: Bone marrow failure, Bleeding tendency, GUM/Skin/CNS infiltration Elderly Indolent course with good prognosis Usually presents on FBC Myeloproliferative Disorders Disease Polycythaemia rubra vera Cell line RBCs Essential thrombocythaemia Platelets Chronic myeloid leukaemia Granulocytes Myelofibrosis Megakaryocyte proliferation Investigations Full blood count: Anaemia, thrombocytopenia + leucopenia, Leucocytosis Blood film: Blasts Bone marrow biopsy: Hypercellular with blasts Immunophenotyping: B cell vs. T cell Cytogenetics: Prognosis Auer Rods FBC: ↑lymphocytes Film: smear cells Marrow: lymphocytic infiltration Features Elderly Non-specific - Tiredness, depression, vertigo, visual disturbance, Hypertension, angina, intermittent claudication Specific – Erythomyalgia, Gout + peptic ulceration, Plethoric Ix:↑ Hb & PCV, Erythroid hyperplasia Tx: (keep PCV < 0.45), Venesection, Hydroxyurea +/radioactive 32P Benign natural history Features: Thrombosis (increased number), Bleeding (reduced quality) Ix: Platelets > 1000 + hypercellular marrow Tx - hydroxyurea 40 -60y Slowly progressive course Non-specific presentation (Generally unwell with splenomegaly+ hepatomegaly) Ix: ↑WBC, Blood film: Granulocytes, Philadelphia chromosome Tx: Imatinib, stem cell transplant Megakaryocyte proliferation leading to Fibrosis + Ineffective Erythropoiesis Features: BM failure + splenomegaly BM very helpful: • Dry tap • Fibrotic trephine biopsy • Lack of Philadelphia chromosome Myelodysplastic Syndromes Disease Lymphoma Features Hodgkin’s - Reed-Sternberg Cells Non-Hodgkin’s – B-cell or T-cell/NK cell Non-specific - Fatigue, loss of appetite, Lymphadenopathy Specific - Fever/night sweats, Itching, ETOH-induced pain, Splenomegaly Multiple Myeloma Bone resorption • Vertebral collapse • Spinal cord compression • Hypercalcaemia + associations Paraprotein • Infections • Pneumonia + pyelonephritis • Hyperviscosity • Visual loss • Renal failure Monoclonal Gammopathy of Undetermined Significance Investigations FBC: nil specific BM: Normal Hodgkin Radiotherapy + chemotherapy Non-Hodgkin Watch and wait (follicular) Radiotherapy Chemotherapy (e.g. CHOP) Monoclonal antibodies (rituximab) Stem cell transplants FBC: BM infiltration, ↑calcium BM: abnormal plasma cell infiltration Serum/urine electrophoresis Tx: Treat complications, Suppress the disease < 65 – chemo then stem cell transplant > 65 - chemo roglobulin Often occurs in elderly Paraprotein present but no other symptoms/signs or laboratory findings to suggest myeloma Natural history is not well defined Porphyria Group of disorders caused by deficiencies in haem synthesis enzymes The major problem is the build up of toxic haem precursors Type Neurovisceral Cutaneous Severity Severe Mild Features Psychosis, seizures ,abdominal pain, polyneuropathy Photo-sensitive vesicular rash Acute Intermittent Porphyria Archetypal neurovisceral porhyria Autosomal dominant inheritance Precipitated by stress Port urine Tx - analgesia and carbohydrates Transfusion Complications Type Febrile non-haemolytic reaction M.O.A - white cell antibodies vs. donor blood Bacterial infection Transfusion related acute lung injury (rare) M.O.A. - HLA Ab in donor vs. HLA antigen on patient WBC Post-transfusion purpura M.O.A - Patient ab. vs. donor platelet antigen Delayed haemolytic transfusion reaction M.O.A. - Red cell ab. not detected Iron overload Viral Infections Graft vs. host disease (rare) M.O.A - lymphocytes from donor vs. receipient Features Fever only No features of haemolysis Treatment Slow down the transfusion Sudden onset fever Very unwell Sudden onset SOB & hypoxia No features of haemolysis/other anaphylactic features Stop transfusion Broad spectrum antibiotics Stop blood Ventilate ~ 1 week after transfusion Brusing as platelets very low IVIG Delayed Features of haemolysis (may cause renal failure) Transfusions for many years Damage to heart, liver and gonads Many possible Hepatitis e.g. C/B, HIV, CMV, HTLV Must be already immunosuppressed Skin rashes, organ failure, marrow failure Treat renal failure Repeat CXM Avoid transfusion, Chelating agent e.g. desferioxamine Screen blood Treat infection Irradiate blood