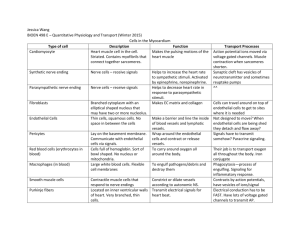
Open Access version via Utrecht University Repository
advertisement

Regulation of endothelial cell-cell junctions and vascular permeability by Rho GEFs and GAPs Master thesis Karin Prummel (Student number: 3257975) Master program: Cancer Genomics and Developmental Biology, Utrecht University Supervisor: Stephan Huveneers, PhD Examiner UU: Fried Zwartkruis, PhD Date: 11/03/2013 1 Abstract The endothelial monolayer covers the luminal side of blood and lymphatic vessels and functions as a physical barrier. The cell-cell junctions between the endothelial cells are important to regulate and maintain the barrier function. Adherens and tight junctions, the main adhesion molecules found in endothelial cells, are regulated by Rho GTPases RhoA, Rac1 and Cdc42. All three Rho GTPases can either induce endothelial permeability as protect the barrier, depending on their spatiotemporal activation and upstream regulators. Rho GTPases are regulated by several guanine exchange factors (GEFs) and GTPase activating proteins (GAPs), which switch the GTPases between the active GTP-bound state and the inactive GDP-bound state respectively. In turn, GEFs and GEFs are under control of inflammatory cytokines and vascular growth factors. In this review is outlined which GEFs and GAPs are associated with the regulation of cellular junctions. For some GEFs/GAPs is already established that they control the vascular permeability in vivo. A specific attention is given to GEFs/GAPs involved in the regulation of junctions in other cell types, but known to be expressed in endothelium. It is important to unravel how GEFs/GAPs are activated and what their subcellular localization is, because this will give more insight in the complex regulation of Rho GTPases. 2 Summary for layman All organs and tissues are provided by oxygen and nutrients, and get rid of waste via a dense network of blood vessels and lymphatic vessels. The blood facing side of these vessels is covered by a layer of single cells, called endothelial cells. This layer of cells forms a barrier between the lumen of the vessel and the surrounding tissues. The endothelial cells in the barrier are connected to each other via protein complexes on the plasma membrane, called cell-cell junctions. Adherens junctions, containing VE-cadherin, and tight junctions are the main endothelial junctions contributing to cell-cell contacts. These protein complexes are intracellulary connected to the skeleton of the cell: the actin filaments. Less cell-cell junctions between endothelial cells will increase the permeability of the barrier. Control of these junctions is important for several biological processes, such as the formation of new blood vessels and the transport of immune cells over the blood vessel wall after an infection. Dysfunction in cell-cell junction control can cause for example leakage of the vessels, fluid buildup (edema) in tissues and a declined response to inflammation. Several permeability inducing or barrier protecting factors circulate in the blood or are secreted by neighboring cells and can regulate Rho GTPases. Rho GTPases are enzymes known to regulate cell-cell junctions. The main Rho GTPases in this review are RhoA, Rac1 and Cdc42. Their activity is regulated by guanine exchange factors (GEFs) and GTPase activating proteins (GAPs): Rho GTPases switch between an inactive and active state. In this review, an overview is made of the Rho GEFs and GAPs that might be involved in the control of cell-cell junctions and consequently can regulate the permeability of the endothelial monolayer. Firstly, we elucidated how six different inflammatory mediators and growth factors can activate or inactive the three Rho GTPases and what is the eventual effect on the endothelial barrier. It becomes clear that each GTPase can either protect or disrupt the barrier function. This indicates that their activity is tightly regulation and will depend on the level of activation, the timing of activating and its location in the cell. For example, RhoA can be located at different sites in the cell: around VE-cadherin, but also near actin filaments attached to the cell-cell junctions. GEFs and GAPs play a role in this complex (in)activation of Rho GTPases. Secondly, we looked into the GEFs and GAPs known to regulate cell-cell contacts in all types of cells. For some Rho GEFs and GAPs is already proven via in experiments in mice that they can control the endothelial barrier function. However, the majority of the GEFs/GAPs is studied in cultured endothelial, epithelial or cancer cells. Many GEFs/GAPs known to regulate cell-cell junctions in culture are studied in epithelial cells. Epithelial cells can also form barriers in for example the intestine. Almost all GEFs/GAPs found to regulate cell junctions in epithelial cells are also expressed in endothelial cells, however, not for all these GEFs/GAPs is yet established whether they cause (the same) effects in endothelial monolayers. Of note, tight junctions can be more organized in epithelial cells than in endothelial cells, so it might be possible that a GEF or GAP can have a different effect on the tight junctions in endothelial than in epithelial cells. To give more understanding of the signaling networks controlling the activity of Rho GTPases in endothelial cells, more experiments should be performed in mice. Moreover, the biological relevance must be taken in account as well. For most GEFs/GAPs is described which inflammatory mediators can regulate them in culture, however, can the same factors regulate them also in a living animal? These kinds of experiments can lead to the discovery of interesting therapeutic targets to for example reduce vascular leakage in patients with atherosclerosis. 3 Introduction The vascular barrier: regulation of endothelial cell-cell junctions The endothelium is a monolayer of cells covering the luminal side of blood and lymphatic vessels and acts as a physical barrier between the intravascular fluid compartment and the surrounding tissues. The ability of monolayer cells to form barriers between tissues is also occurring in epithelial tissues, for example in play a crucial role in the formation and maintenance of the barrier function. These cell-cell adhesions are not simply static adhesion structures; in contrast, dynamic regulation of cell-cell junctions is required during inflammation and the formation of new blood vessels (angiogenesis). The regulation of these junctions is also involved in immune surveillance when immune cells migrate from the blood stream across the endothelial layer towards tissues and vice versa (Vestweber et al., 2009). Upon certain stimuli, leukocytes, macromolecules and fluids in the blood can enter the tissue through intercellular spaces in the endothelium layer, caused by destabilized cell-cell junctions. The migration of leukocytes over the endothelial layer is referred to as paracellular transendothelial migration (Bazzoni, 2006; Vestweber et al., 2009). Without the ability to destabilize endothelial cell-cell junctions such leukocyte transmigration events are strongly inhibited (Schulte et al., 2011) and will prevent sprouting during angiogenesis (Abraham et al., 2009). When endothelial permeability is induced inappropriately, for example during chronic infections or autoimmune disease, pathophysiological effects may occur, including prolonged vascular leakage, acute lung injury, tissue edema, and atherosclerosis (Wojciak-Stothard and Ridley, 2002). Molecular adhesion complexes at cell-cell junctions Adherens junctions (AJs), nectin-based junctions and tight junctions (TJs) are the main cell-cell adhesion complexes involved in the formation of cell-cell adhesions and in the maintenance of barrier function (Meng and Takeichi, 2009). All junction types are intracellulary linked to the actin cytoskeleton and may function as signaling scaffolds, locally regulating signal transduction cascades in control of cytoskeletal remodeling, cell growth and differentiation. The major function of AJs is to physically connect neighboring cells and to maintain these contacts. Loss of cell-cell junctions will loosen the cell-cell contacts, resulting in disrupted tissue organization. Therefore, they are crucial for cell-cell adhesion (Meng and Takeichi, 2009). TJs tightly link the membranes of neighboring cells, forming a barrier that even prevents small molecules and fluid to pass. They consist of transmembrane and intracellular proteins: occludins, claudins and small junctional adhesion molecules (JAMs). Occludin and claudin bind to zona occludens proteins (ZO), which can interact with actin filaments (Bazzoni and Dejana, 2004; Shin et al., 2006). In epithelial cells, TJs divide the membrane in an apical (lumen facing) and basolateral region. In contrast, in endothelial cells TJs do not divide the plasma membrane in such distinct regions in vitro and are more evenly distributed over the plasma membrane than in epithelial cells (Bazzoni and Dejana, 2004). Interestingly, in the vasculature is found that the density of TJs differs between the various segments of the vascular network, what may relate to the variability in permeability of these segments (Bazzoni and Dejana, 2004). Variance occurs due to the molecular composition of TJs and cellular expression levels of occludins and claudins. The membrane of post-capillary venules contains many receptors for permeability factors and less TJs. This is also the location for leukocyte transmigration. In veins, the organization of the 4 TJs less organized as well. Additionally, in the blood-brain-barrier (BBB), the endothelial cells contain many and complex TJs (Bazzoni and Dejana, 2004). In addition, vascular endothelial (VE-) cadherin is the most important component of AJs in endothelium (Vestweber et al., 2009), contributing to adhesion between endothelial cells, whereas epithelial cells mainly form epithelial (E-) cadherin-based adherens junctions. VEcadherin knockout studies have shown that VE-cadherin is a crucial protein for the formation of endothelial cell-cell adhesions (Carmeliet et al., 1999; Gory-Fauré et al., 1999). Also neuronal (N) and placental (P-) cadherin can be found in endothelial cells (Bazzoni and Dejana, 2004). Homophilic interactions between the extracellular domains of these cadherins mediate adhesion between cells. The cytoplasmatic domain of cadherins associate with catenin proteins, linking the AJs to the actin cytoskeleton and consequently stabilizing the cadherins at cell-cell contacts. The proteins p120-catenin and β-catenin bind directly to cadherin’s cytoplasmatic domain. In turn, β-catenin binds to α-catenin, which in turn associated to actin filaments via actin binding proteins. Consequently, α- and β-catenin link cadherin complexes to the actin cytoskeleton (Vestweber, 2008; Meng and Takeichi, 2009; Gottardi and Gumbiner, 2001; Yamada et al., 2005; Nelson, 2008). Control of endothelial cell-cell adhesion complexes by permeability regulators Several inflammatory mediators and growth factors, circulating in the blood or locally secreted by cells, tightly regulate the balance between the opening and closure of endothelial cell-cell junctions to facilitate paracellular migration or avoid aberrant leakage of the vessels respectively (Vestweber et al., 2009). Thrombin, histamine, tumor necrosis factor α (TNFα) and vascular endothelial growth factor (VEGF) are well known factors that increase the permeability of the endothelium by destabilizing endothelial cell-cell junctions. On the other hand, sphingosine 1-phosphate (S1P) and angiopoietin 1 (Ang-1) enhance the endothelial barrier function through stabilization of cell-cell junctions (Vestweber et al., 2009; Gavard et al., 2008; Spindler et al., 2010). Thus, several growth factors and cytokines can control the endothelial barrier function. Next, we will focus on how they can regulate the vascular permeability. Vascular growth factors and inflammatory cytokines can increase the permeability of blood vessels in several ways. First, for example VEGF may induce phosphorylation of components of the VE-cadherin complex and stimulate recruitment of β-arrestin towards VE-cadherin, enhancing endocytosis of the VE-cadherin-complex (Bazzoni and Dejana, 2004; Gavard and Gutkind, 2006). Increased internalization of the VE-cadherin complex destabilizes AJs and consequently results in loss of cell-cell contacts. Second, permeability regulators like thrombin, VEGF and TNFα induce increased contraction of the actin/myosin II complex (actomyosin) (Bryan et al., 2010; Mckenzie, 2007). Contraction of actomyosin produces physical tension on adhesion structures like AJs to which the actin bundles are attached to (De Rooij et al., 2005). These actomyosin dependent physical forces destabilize endothelial AJs by pulling them apart (Huveneers et al., 2012) (Figure 1). Elevated tension on AJs, induced by growth factors or cytokines, contributes to permeability of the endothelial monolayer (Fischer et al., 2010; Mckenzie, 2007; Bryan et al., 2010). Adhesion of cells to the extracellular matrix influences the cytoskeletal tension as well: thrombin induces larger intercellular gaps in endothelial monolayers grown on a stiff substrate compared to cells grown on less rigid cellular microenvironments in vitro (Krishnan et al., 2011; de Rooij et al., 5 2005). In drosophila, it is recognized that the generated force on AJs, induced by actomyosin contraction, is crucial to direct epithelial morphogenesis (Rauzi et al., 2010). On the other hand, increased tension on AJs can also induce stabilization and growth of junctions in endothelial monolayers not exposed to growth factors and hormones (Liu et al., 2010), corresponding with the notion that the formation of E-cadherin based junctions depends on myosin II activity (Yamada and Nelson, 2007). Thus, inflammatory mediators can disrupt the endothelial barrier via stimulation of endocytosis of adhesion complexes and via induction of actomyosindependent tension on adhesion structures. Figure 1. Regulation of adherens junctions by inflammatory mediators. Cadherin binds homophylically to the extracellular domain of cadherin on neighboring cells (left). Permeability inducing factors such as thrombin and VEGF induce contraction of the actin/myosin II complex (actomyosin). Contraction of actomyosin creates strong actin bundles (stress fibers), associating to αcatenin via actin binding proteins (right). Pulling by these stress fibers provides tension on the adherens junctions, leading to destabilization of the junctions. This induces weakening of cell-cell contacts, consequently resulting in more permeability of the endothelial monolayer. 6 Aim of the thesis The regulation of vascular permeability is important for angiogenesis and inflammatory responses. Adherens and tight junctions are the cell-cell adhesion structures underlying the endothelial barrier function, and they are, in turn, controlled by the actin cytoskeleton. Inflammatory cytokines and vascular growth factors control the actin-connected cell-cell junctions through several signaling routes, and in this thesis I will address the role of activating Rho GTPases. Rho GTPases are molecular switches that can control the actin cytoskeleton and cell-cell junctions. The activity of Rho GTPases is controlled by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs), activating and inactivating Rho GTPases respectively. I will focus on which GEFs and GAPs are known or proposed to regulate Rho GTPases in control of endothelial cell-cell junctions. This thesis gives an overview of the Rho GTPase signaling networks that lie in between the by endothelial growth factors/cytokines induced signaling and cell-cell junction regulation. After briefly introducing the Rho GTPases and their regulators GEFs and GAPs, I will first summarize the effects of six key inflammatory mediators and growth factors on the endothelial barrier function. Also, I will explain how these effects relate to the activity of three main Rho GTPases Rac1, Cdc42 and RhoA. Second, I will explain in detail which GEFs and GAPs are associated with regulation of cellular junctions and I will point out the upstream regulators of GEFs/GAPs if known. Finally, I will define which GEFs/GAPs are shown to control endothelial junctions and are likely to play a role in inflammation or angiogenesis by regulating endothelial permeability. 7 Rho GTPases The actin cytoskeleton and the organization of AJs and TJs can be tightly regulated by Ras homologous GTPases (Rho GTPases). Rho GTPases, consisting of 20 family members, transduce signals from receptors on the plasma membrane to specific intracellular effector proteins (Wennerberg et al., 2005). Besides regulation of junctions and the cytoskeleton, they are also implicated in regulating the cell-cycle and gene transcription. Rho GTPases can stimulate a variety of cell processes, such as adhesion, migration, vesicle transport, cellular remodeling, phagocytosis and cancer (proliferation) (Heasman and Ridley, 2008; Rossman et al., 2005; Sahai and Marshall, 2002a). Rho GTPases are often activated via G-protein coupled receptors (GPCRs), which can activate heterotrimeric G-protein complexes, made up of an α-subunit and tightly associated β/γ-subunits. Many classes of Gα-subunit are known; Gα12/13 and Gα11/q are the main subunits involved in direct or indirect activation of Rho GTPases respectively (Rossman et al., 2005; Oldham and Hamm, 2008). The best-studied members of the Rho family in regard to actin cytoskeletal organization are RhoA, Rac1 and Cdc42. Although all three Rho GTPases are intracellular proteins, their specific location in the cell and especially their spatiotemporal activation pattern differ. All can localize at the plasma membrane once activated (Ridley, 2006). In general, activated RhoA is involved in actomyosin contractions and the formation of stress fibers, whereas activated Cdc42 and Rac1 are involved in the formation of lamellipodial protrusions by producing branched actin at the leading edge of cells. Cdc42 is also the main activator of filopodia formation, which are actin-rich protrusions of the plasma membrane beyond lamellipodia (Ridley, 2006). Figure 2. Rho GTPases signaling pathways with their downstream effectors. In these potential pathways, Cdc42, Rac1 and RhoA (highlighted) influence the actin cytoskeleton and the organization of cell-cell junctions (TJs and AJs) in non-muscle cells. The dotted arrows indicate indirect activation. Activated Rho GTPases interact with multiple downstream effectors involved in contractility and polymerization of the actin cytoskeleton. Cdc42 and Rac1 stimulate branching of actin filaments. RhoA stimulates the formation and polarization of non-branching actin filaments. Downstream of active RhoA and upon activation of myosin II, actin filaments slide over each other, leading to contraction of the actin cytoskeleton. Important downstream effectors of RhoA are Rho8 associated kinase (ROCK) and Diaphanous-related formins (Dia). ROCK can activate the Ca2+dependent myosin light chain kinase (MLCK) and can also directly phosphorylate the myosin light chain (MLC) (Amano et al., 2010; Citi et al., 2011). It also phosphorylates the MLC phosphatase (MLCP), inhibiting MLCP’s phosphatase activity (Figure 1) (Amano et al., 2010; Citi et al., 2011). Dia1 stimulates the formation and polarization of non-branching actin filaments (Ridley, 2006; Citi et al., 2011; Lammers et al., 2008). To sum up, RhoA activates Dia1, resulting in actin polymerization, and activates ROCK, leading to actomyosin contractions. An overview of key effectors of Rho GTPases involved in actin remodeling is displayed in Figure 2. Regulation of Rho GTPases by GEFs and GAPs Rho GTPases cycle, like other small GTPases, between the inactive GDP-bound state and the active GTP-bound state. These switches are controlled by several proteins. Guanine nucleotide exchange factors (GEFs) stimulate the replacement of the nucleotide GDP for GTP in inactive GTPase. GTPase-activating proteins (GAPs) accelerate the GTPase’s intrinsic GTP hydrolysis reaction to become in the GDP-bound conformation. Lastly, guanine nucleotide dissociation inhibitors (GDIs) bind inactive GTPases to keep them in their inactive state in the cytoplasm (Figure 2) (Bos et al., 2007). Figure 3. Regulation of Rho GTPases. GDI sequesters the inactive Rho GTPase (GDP-bound). When released from the Rho GDI, Rho GEFs can induce the replacement of GDP for GTP. The GTPbound Rho GTPase is active and enhances activation of several target and effector proteins. Rho GAPs mediate hydrolysis of GTP into GDP, inducing the Rho GTPase to become in the inactive state. The Rho GEF family, also called Dbl-family, contains approximately 69 members (Rossman et al., 2005). GEFs consist of several domains, including a Dbl homology (DH) domain and a pleckstrin homology (PH) domain. The PH domain is for most Rho GEFs necessary to localize at and attach to the plasma membrane (Stam et al., 1997), however, some GEFs have other domains that influence their cellular localization (Rossman et al., 2005). GTPases bind the nucleotides GTP and GDP between two loops, called switch 1 and 2. GEFs interacts via their DH domain with these switches and modify their conformation, resulting in release of GDP (Bos et al., 2007; Rossman et al., 2005). Since small GTPases have a similar affinity for GDP and GTP, and the cellular GTP concentration is 10x higher than the GDP concentration, there is an increased likelihood that the GTPase binds GTP over GDP after modification by GEFs. Moreover, GEFs can be regulated by for example posttranslational modifications. These regulations can result in activation and/or translocation of the GEF towards a specific GTPase (Bos et al., 2007). 9 Additionally, GAPs are needed to enhance the slow intrinsic GTP hydrolysis of Rho GTPases. Most Rho GAPs are ubiquitously expressed (Su et al., 2004) and stabilize the catalytic glutamine of the GTPase by lowering the transition state energy. Moreover, they stabilize the switch regions (Bos et al., 2007). GAPs can be regulated via similar mechanisms like GEFs (Bos et al., 2007). In this review, we will focus on the GEFs and GAPs that may be involved in cell-cell junction regulation and vascular permeability induced by inflammatory cytokines and growth factors. Rho GTPases and cell-cell contacts As mentioned before, small Rho GTPases can regulate cell junctions. RhoA and Rac1 play a role in the formation of cell-cell junctions: Rac1 can initiate the formation of AJs and RhoA is needed for completion of the cell-cell contact (Yamada and Nelson, 2007). In epithelial cells, Rac1 can be activated near E-cadherin-based adhesions, which is required for the association of the actin cytoskeleton and the cadherin complex, supporting cell-cell adhesion (Braga et al., 1997; Takaishi et al., 1997; Jou and Nelson, 1998). RhoA is involved in both the formation of AJs and TJs (Takaishi et al., 1997; Braga et al., 1997). In more detail is shown that the activities of RhoA, Rac1 and their effectors are restricted to specific regions of (growing) cell-cell contacts. First, Rac1 is found active near the tips of lamellipodia in expanding cell-cell contacts (Yamada and Nelson, 2007). Thereafter, RhoA and its effector ROCK become activated at the distal edges of growing cell-cell contacts, what is in accordance with the high levels of activated myosin II found at the edges. This drives the accumulation of E-cadherin-based adhesions (Yamada and Nelson, 2007; Shewan et al., 2005). The role of RhoA in cell-cell junction regulation is complex. Whether RhoA stabilizes or destabilizes AJs will depend on its spatiotemporal regulation and the level of activity (Van Nieuw Amerongen et al., 2007; Yamada and Nelson, 2007). Enhanced ROCK and myosin II activity can induce contractile forces, pulling neighboring cells apart. On the other hand, basal ROCK and myosin II activity supports integrity of AJs. Moreover, via Dia1, RhoA promotes localization of αcatenin towards cadherin and clustering of AJs, resulting is more stable cadherin-based junctions (Sahai and Marshall, 2002b; van Nieuw Amerongen et al., 2007). Furthermore, Rac1 and Cdc42 play a role in the stabilization of AJs. They inhibit IQGAP, a scaffolding protein that in its active form dissociates α-catenin from β-catenin. This disables the actin cytoskeleton to bind to cadherin and will weaken the AJs. So, inhibition of IQGAP by Rac1/Cdc42 results in strong cadherin-mediated cell junctions (Figure 1) (Kuroda, 1998; Kaibuchi et al., 1999). Moreover, Cdc42 is also implicated in the transport of E-cadherin from the Golgi to plasma membrane in epithelial cells to support the formation of cell-cell adhesions (Wang et al., 2005). 10 Effect of inflammatory mediators and growth factors on Rho GTPases and endothelial permeability Growth factors and inflammatory mediators regulate endothelial cell-cell junctions. As mentioned in the Introduction, thrombin, histamine, TNFα and VEGF are known factors that increase the permeability of the endothelium. In contrast, S1P and Ang1 stabilize the cell-cell junctions and restore the endothelial barrier. This chapter will focus on the known effects of these factors on endothelial or, where relevant on, epithelial permeability. Moreover, I will explain which Rho GTPases function downstream of the different factors. It will become clear that each Rho GTPase has the ability to either induce or inhibit endothelial permeability, depending on their spatiotemporal activation. Thrombin Thrombin is serine protease, functioning as a blood coagulation factor (converting fibrogen into fibrin) and as a remodeler of endothelial junctions. This inflammatory mediator increases endothelial permeability. Thrombin can cleave the extracellular N-terminus of proteaseactivated receptor 1 (PAR-1) on endothelial cells, consequently activating this receptor (McLaughlin et al., 2005). PAR-1 is a GPCR and activates heterotrimeric G-proteins. Thrombin induces fast and slower responses in in vitro experiments. Subunits of these G-proteins, Gα11/q, Gα12/13 and Gαi, can rapidly activate RhoA (Figure 4). During this phase of a thrombin response, Rac1 is inhibited after PAR-1 activation. By contrast, Cdc42 and Rac1 are activated at later phases of the response (1 hour past thrombin treatments), because they are needed for recovery of the thrombin-mediated endothelial barrier destabilization. Quickly after thrombin stimulation, several signaling mechanisms are initiating RhoA activation and induce disruption of endothelial cell-cell junctions. RhoA remains active for roughly 30 minutes (Van Nieuw Amerongen, 2002; Mehta et al., 2003). One of the signaling routes induced by thrombin activates Gα11/q, inducing phospholipase C to convert phospholipids into inositol triphosphate (IP3) and diacyl glycerol (DAG) (Figure 4). IP3 and DAG can induce endothelial permeability through RhoA activation dependent and independent routes. IP3 increases cytosolic Ca2+-concentration, activating calmodulin, which in turn activates MLCK. The latter phosphorylates MLC, thereby enhancing actomyosin contraction. This destabilizes the cell-cell contacts and consequently induces endothelial permeability (Satpathy et al., 2004). DAG also elevates the cytosolic Ca2+ levels. Moreover, DAG activates Protein Kinase Cγ (PKCγ), promoting MLC phosphorylation via two ways. First, PKCγ can inactivate MLCP via phosphorylation (Satpathy et Figure 4. Thrombin-mediated regulation of RhoA, Rac1 and Cdc42. The G-protein pathways are in more detailed discussed in the text. The signaling downstream of PKCα (green arrow) is depicted in Figure 5. 11 al., 2004). Second, PKCγ activates RhoA through inhibition of RhoGDI and stimulation of a RhoA GEF (Mehta et al., 2001; Holinstat et al., 2003). A second signaling route directly downstream of thrombin induced PAR1 activation occurs via Gα12/13, activating RhoA via a GEF in a Ca2+-independent matter (Birukova et al., 2004). The third route is via Gαi, a subunit that inhibits the production of cAMP through inhibition of adenylate cyclase (Hung et al., 1992) (Figure 4). Consequently, Protein Kinase A (PKA) is not activated, resulting in inhibition of RhoGDI. RhoA is released and free to be activated (Qiao et al., 2008). Thus, thrombin can activate RhoA via all three signaling routes, resulting in increased phosphorylation of MLC and enhances actomyosin contraction. The increased tension on cell-cell adhesions, including cell-cell junctions, is the major cause of endothelial permeability induction by thrombin. In conjunction with the rapid RhoA activation, Rac1 is inactivated by thrombin for approximately 1 hour, which is thought to weaken the cell-cell junctions. It is established that Rac1 is inactivated after thrombin stimulation because Gαi reduces of cAMP levels in endothelial cells (Baumer et al., 2008, 2009; Hung et al., 1992). Moreover, it is proposed that effectors of RhoA can activate two Rac1 GAPs, leading to Rac1 inhibition as well (Sanz-Moreno et al., 2008; Ohta et al., 2006). However, this mechanism to inactive Rac1 is only observed in melanoma cells and not yet confirmed to take place in endothelial cells. After approximately 1 hour, Rac1 and Cdc42 are (re)activate and recover the endothelial barrier, an important repairing function, which in vivo is important to prevent e.g. severe leakage of the blood vessel. Thrombin induces sphingosine-1-phosphate (S1P) activation via activation of the sphingosine kinase (SPHK), consequently activating Rac1 (Tauseef et al., 2008). See the paragraph below concerning the role of S1P for a detailed description of this signaling route affecting cell-cell junctions. Also elevated cAMP levels increase Rac1 activity (Baumer et al., 2009, 2008). Moreover, Cdc42 is activated 1 hour after thrombin treatments, preceding junction formation. This is required for the barrier function of endothelial cells, including the as observed in mice lung endothelium. Subsequently, association of the actin cytoskeleton with the VEcadherin complex is thought to stabilize the junction at this stage (Kouklis et al., 2004). Furthermore, the disassembly event of AJs itself might activate Cdc42 as well, since subconfluent endothelial monolayers have an increased Cdc42 activity compared to confluent monolayers (Kouklis et al., 2004). These observations indicate that Cdc42 and Rac1 recover the endothelial barrier function after thrombin by stabilizing cell-cell junctions. In conclusion, activation of PAR-1 by thrombin activates three known pathways, initiated by Gα11/q, Gα12/13 and Gαi that induces endothelial permeability which may occur with or without RhoA activation. Effectors of RhoA subsequently induce actomyosin contraction. After an hour, the inhibited Rac1 and Cdc42 become activated and inhibit RhoA. This enhances cell-cell junction stability and repairs the endothelial barrier. 12 S1P Sphingosine is a member of a class of plasma membrane lipids and can be phosphorylated by sphingosine kinases (SPHK), leading to the generation of S1P, a signaling molecule (Strub et al., 2010). S1P can function as an intracellular lipid second messenger or can be secreted to subsequently function as an extracellular ligand for the GPCR S1P1 till -5 in an autocrine or paracrine manner (Hla, 2003; Alvarez et al., 2007; Strub et al., 2010). S1P can be secreted by for example platelets (Mehta et al., 2005). As observed in endothelial and rat pancreatic islet cells, SPHK can be activated downstream of thrombin, TNFα and histamine (Figure 5) (Tauseef et al., 2008; Mastrandrea et al., 2005; Huwiler et al., 2006). This results in activation of Rac1 and inhibition of RhoA, consequently leading to mitigation of the monolayer permeability (Mehta et al., 2005; Schaphorst et al., 2003). Knocking down SPHK in endothelial cells induces a constant RhoA activation and lowers Rac1 activity (Tauseef et al., 2008), inducing permeability of endothelial monolayers. Additionally, it was shown in vivo that S1P stimulates activation of endothelial Rac1. Administration of S1P to lungs of rats indeed stabilized their endothelial cell-cell adhesions in lung microvessels (Adamson et al., 2010). Figure 5. S1P-mediated regulation of RhoA and Rac1. S1P can be formed after thrombin and TNFα stimulation, but can also be secreted by platelets. The G-protein subunit Gαi induces RhoA inhibition via two pathways: one via Ca2+ and RhoGDI, the other via Rac1 and p190RhoGAP activation, which are more detailed described in the text. The dashed arrow indicates indirect activation. The S1P1 receptor can activate the Gαi subunit via the PI3-kinase (PI3K)-Akt pathway, subsequently activating Rac1 (Figure 5) (Adamson et al., 2010). In human umbilical vein endothelial cells (HUVECs), S1P induces translocation of Rac1 and two Rac1 GEFs towards cellcell contacts, resulting in AJ assembly (Lee et al., 1999). Moreover, the G-protein subunit increase the cytosolic levels of second messengers such as calcium and cAMP (Hla, 2003). This elevated calcium level contributes to Rac1 activation as well. It has been shown that calcium induces RhoGDI to release from Rac1, enabling GEFs to activate Rac1 (Mehta et al., 2005; Price et al., 2003). Rac1 in its turn induces the formation of lamellipodia at the cell-cell contacts, initiating accumulation of VE-cadherin and thereby promoting cell-cell adhesion. Moreover, S1P can promote the formation of TJs via Rac1. Active Rac1 mediates the distribution of ZO-1 towards the cell-cell junctions in endothelial cells (Lee et al., 2006). Furthermore, RhoA might be suppressed via RhoGDI and via Rac1-mediated p190RhoGAP activation (Figure 5) (Papaharalambus et al., 2005). In addition, several studies showed that elevated levels of intracellular S1P can decrease the endothelial permeability as well. However, the signaling routes downstream of intracellular S1P are not yet elucidated (Strub et al., 2010). To sum up, S1P formation can be induced by inflammatory mediators such as thrombin and TNFα. S1P can activate in an autocrine/paracrine manner the S1P receptors. This active Rac1 and inactivate RhoA, provoking stabilization of AJs and TJs. This supports the endothelial barrier 13 and prevents vascular leakage. Also increased intracellular concentrations of S1P seem to protect the barrier function. Histamine Histamine is a vasoactive mediator and is known to rapidly induce vascular permeability after a trauma or allergy. This mediator exerts its function via four different histamine receptors (HRH1-4), which are all GPCRs. The HRH1 is particularly highly expressed on endothelial cells (Clough et al., 1998). Activation of the receptor leads to RhoA activation, inducing increased actomyosin contractions and subsequently disrupting junctions (Figure 6). HRH1 activates the G-protein subunit Gα11/q, mediating activation of a RhoA specific GEF in endothelial cells (Pfreimer et al., 2012). More details about the specific RhoA activation pathway remain unclear and are not yet established in endothelium. Through Gα11/q signaling, the cytosolic calcium levels elevate and active MLCK (Figure 6), resulting in phosphorylation of MLC and consequently causing instable cell-cell junctions through actomyosin contractions of the connected actin filaments (Wei et al., 2011; Wojciak-Stothard and Ridley, 2002; Guo et al., 2007; Wójciak-stothard et al., 2001). Furthermore, histamine induces significant loss of TJs and AJs between endothelial cells (Wójciak-stothard et al., 2001). Figure 6. Histamineinduced activation of RhoA. Via the Gα11/q pathway, a RhoA GEF and elevated Ca2+ levels contribute to increased RhoA activation. However, these histamine effects on endothelial cell-cell junctions are transient. Histamine reduces the endothelial barrier function for only 3-5 min, whereas e.g. thrombin induces a prolonged effect of more than 40 min in HUVECs. There are several differences between the responses of endothelial cells to histamine and thrombin. For example, less MLCs are phosphorylated in histamine stimulated HUVECs compared to thrombin stimulated cells (Moy et al., 1996). Moreover, thrombin induces bigger gaps between the individual endothelial cells in vitro, whereas histamine induces smaller intercellular gaps (Wójciak-stothard et al., 2001). Summarizing, histamine induces activation of RhoA, resulting in disruption of AJs and TJs. This contributes to endothelial permeability. VEGF The permeability factor VEGF is strongly involved in angiogenesis during embryogenesis and adult life. Endothelial cells express two VEGF receptors: VEGFR1 and VEGFR2. In adult endothelial cells, VEGFR2 is the main receptor that recognizes and binds VEGF, whereas both receptors are involved during embryogenesis. VEGFR2 is a tyrosine kinase receptor that dimerizes after binding of VEGF, resulting in autophosphorylation of its intracellular domain. Accordingly, several adaptor proteins bind. VEGFR2 mediates the activation of the MAPK pathway and PKC via phospholipase Cγ (PLCγ). PLCγ also increases the cytosolic Ca2+ concentration. Moreover, VEGFR2 activates the PI3K pathway (Olsson et al., 2006). Intriguingly, VEGF is able to induce activation of RhoA, Rac1 and Cdc42, although their timing of activation is different. 14 Activation of VEGFR2 by VEGF is followed by a fast Rac1 activation for approximately 15 min in vitro (Figure 7A) (Beckers et al., 2010; Garrett et al., 2007). The rapid Rac1 activation is followed by a more sustained Rac1 activation at 30 min (Garrett et al., 2007). This biphasic Rac1 activation by VEGF corresponds with the biphasic reaction of human pulmonary artery endothelial cells after stimulation with VEGF in vitro. First, the endothelial barrier function briefly increases (for 10 min), followed by a sustained decrease, indicating an increased endothelial permeability (Figure 7B) (Mirzapoiazova et al., 2006). A B Figure 7. Effect of VEGF addition on Rho GTPases in endothelial cells and on the resistance of the endothelial monolayer. A) The activation of RhoA, Rac1 and Cdc42 after VEGF stimulation is plotted against the time after VEGF addition (in minutes). The response of Rac1 is biphasic. The figure is adapted from Beckers et al. (Beckers et al., 2010). B) The transendothelial electrical resistance (TER) is measured after stimulating a monolayer of human pulmonary artery endothelial cells with VEGF (at time point 0 min). The cells were stimulated with two different concentrations of VEGF or a vehicle. The cells showed a biphasic response after treatment with 100ng/ml VEGF. The resistance across the monolayer first increased, followed by a decreased in resistance. The figure is adapted from Mirzapoiazova et al. (Mirzapoiazova et al., 2006). Several studies showed that VEGF-induced Rac1 activates PAK1. Dominant-negative Rac1 and RNA interferences approaches for Rac1 reduce the endothelial permeability (Eriksson, 2003; Gavard and Gutkind, 2006; Garrett et al., 2007). On the other hand, a constitutive active form of Rac1 can disrupt VE-cadherin based cell junctions (Gavard and Gutkind, 2006). Gavard and Gutkind showed that the serine/threonine kinase PAK1 phosphorylates VE-cadherin at Ser665, inducing β-arrestin recruitment towards AJs and eventually inducing internalization of the total VE-cadherin complex, destabilizing cell-cell junctions (Figure 8) (Gavard and Gutkind, 2006). This function of Rac1 is in contrast with its function established downstream of e.g. thrombin, where it enhances cell-cell junction stability. Of note, a recent study looking into the role of phosphorylation of VE-cadherin in vivo could not corroborate the data that Ser665 phosphorylation is necessary for VE-cadherin internalization upon VEGF stimulation (Orsenigo et al., 2012). This study showed that VE-cadherin can be (tyrosine) phosphorylated in venous endothelial cells even in the absence of permeability inducing factors. Possibly, the phosphorylation of VE-cadherin makes cell-cell junctions more dynamic and sensitive for inflammatory mediators (Orsenigo et al., 2012). Furthermore, VEGF-induced Rac1 activation stimulates the production of Reactive Oxygen Species (ROS) via NADPH oxidase. The generation of ROS may have distinct effects on cell-cell 15 junctions; either destabilizing or stabilizing. First, ROS can induce phosphorylation of VEcadherin and β-catenin in microvascular endothelial cells, leading to a reduced affinity of βcatenin for VE-cadherin and consequently in decreased AJ integrity (Monaghan-Benson and Burridge, 2009). Secondly, ROS production results in translocation of the RhoA GAP p190RhoGAP towards the plasma membrane and AJs. P190RhoGAP interacts with p120-catenin and has the ability to block RhoA activity at the AJs (Wildenberg et al., 2006; Nimnual et al., 2003) (Figure 8). This inhibits internalization of N-cadherin and disrupts AJs in fibroblasts. However, thus far a link between ROS-p190RhoGAP was only found in fibroblasts, although the p190RhoGAP is expressed in endothelial cells as well. It still needs to be clarified how ROS-production regulates RhoA’s spatiotemporal activation. In addition, VEGF activates the RhoA/ROCK pathway (Bryan et al., 2010), suggesting that this pathway is involved in VEGF-mediated permeability (Bryan et al., 2010). Moreover, Cdc42 is activated after binding of VEGF to VEGFR2 in endothelial cells, which could play a, as yet unclear, role in the regulation of cell-cell junctions by VEGF as well (Kusuhara et al., 2012; Lamalice et al., 2004). Although thrombin and VEGF are both vascular permeability factors, their downstream signaling pathways are very different. The levels of activated RhoA are low at 1 min after Figure 8. VEGF-mediated regulation of Rac1 and RhoA. VEGFR, a RTK, mediates activation of Rac1, resulting in phosphorylation of cell junction proteins. RhoA is inactivated after VEGF stimulation. The pathways are in more detail described in the text. Dotted arrows indicate indirect regulation. stimulation VEGF compared to the major increase in activated RhoA after thrombin stimulation (Van Nieuw Amerongen, 2002). This is probably caused by the lack of Gα12/13 activation in VEGF stimulated endothelial cells. This G-protein subunit is in thrombin stimulated cells a crucial mediator in RhoA activation. Where thrombin primarily initiates actomyosin contraction, the by VEGF-mediated RhoA activation seems more involved in stimulation of cell migration which might be relevant for the induction of sprouts during VEGF driven angiogenesis (Van Nieuw Amerongen, 2002). TNFα Tumor necrosis factor α (TNFα) is an inflammatory cytokine produced by monocytes and macrophages. Within 30 min., increased stress fibers are formed, followed by sustained formation of interendothelial gaps for many hours in vitro (Wójciak-Stothard et al., 1998). TNFα can bind to its two plasma membrane receptors TNFR1 and TNFR2, activating the PI3K/Akt and MAPK pathway, and several phospholipids (Wójciak-Stothard et al., 1998) (Figure 9). As previously mentioned, TNFα can also increase the activity of SPHK, supporting increase in intracellular S1P levels in pancreatic cells (Mastrandrea et al., 2005). It has been proposed that RhoA, Rac1 and Cdc42 function downstream of TNFα in HUVECs and are responsible for the induced changes of the actin cytoskeleton and destabilization of cell-cell junctions (WójciakStothard et al., 1998). However, it should be noted here that several follow-up studies could not substantiate the finding that RhoA is essential for TNFα-induced permeability (Mckenzie, 2007; 16 Schlegel and Waschke, 2009). By contrast, Rac1 seems to be a major player; although also for this GTPase conflicting data are published (Cain et al., 2010; Papaharalambus et al., 2005; Schlegel and Waschke, 2009). First, it has been shown that TNFα induces RhoA activation, resulting in activated ROCK and Dia1. However, the level of phosphorylated MLC does not rise above basal levels when TNFα stimulated cells are compared to unstimulated cells, suggesting that the RhoA/ROCK signaling towards MLC is not involved in the endothelial response to TNFα (Mckenzie, 2007; Schlegel and Waschke, 2009). It is a possibility that the improved Dia1 activation contributes to stress fiber formation. On the other hand, it is possible that the RhoA/ROCK pathway indeed plays a role in the TNFα reaction, but that the effects are local and no significant changes in activation can be detected in total lysate of cells. Within 30 min after TNFα, no significant increase in endothelial permeability can be observed, whereas thrombin already induces strong permeability at this time point. In contrast, TNFα induces endothelial permeability in the long run (8-24 hr). After sustained TNFα stimulation, stress fibers are accumulated, occludin and JAM-A are removed from the TJs, and ZO-1 is not located properly at the TJs. This results in weakened TJs and in increased permeability of the HUVEC monolayer (Mckenzie, 2007). It is not clear if and how RhoA is involved in this long term effect. Several studies show, however, that TNFα induces activation of Rac1 in endothelial and epithelial cells underlying its effect on permeability (Cain et al., 2010; Papaharalambus et al., 2005; Wójciak-Stothard et al., 1998). In HUVECs, this is initiated via activation of PI3K pathway (Figure 9). It is suggested that the p110α subunit stimulates recruitment of a Rac1 GEF towards VE-cadherin. As a result, Rac1 is activated at cell-cell contacts (Cain et al., 2010). However, conflicting data are published by Schlegel and Waschke. They used human dermal microvascular endothelial cells and found that Rac1 was inactivated after TNFα stimulation as a consequence of decreased cAMP levels. This inhibition of Rac1 disrupted the junctions and increased the endothelial permeability (Schlegel and Waschke, 2009). Figure 9. TNFαinduced activation of RhoA and Rac1. Via PI3K and Akt, Rac1 is activated. In conclusion, TNFα induces prolonged permeability compared to other inflammatory mediators. Findings concerning the role of RhoA and Rac1 in the TNFα-induced response on endothelial cell-cell junctions are contradictory. It is not yet established whether RhoA’s role is crucial. Furthermore, a majority of the studies on the role of Rac1 show that Rac1 activation induces endothelial and epithelial permeability, although again, opposite effects are reported. Ang-1 Angiopoietin-1 (Ang-1) is a growth factor maintaining the vascular integrity. It is established that Ang-1 attenuates the by inflammatory mediators induced vascular permeability (Van der Heijden et al., 2011; Gavard et al., 2008). Ang-1 binds to the RTK Tie2 on endothelial cells, activating MAPK (ERK) and the PI3K/Akt pathway, consequently leading to sustainability of the junctions and prevents actomyosin contractions, thereby stabilizing the endothelial barrier function (Figure 10) (Fukuhara et al., 2008; Saharinen et al., 2008). In confluent HUVECs, Tie2 is 17 located at AJs and binds to Tie2 on neighboring cells. This localization towards cell junctions is induced by Ang-1 (Saharinen et al., 2008). Ang-1 signals via Rho GTPases to enhance the endothelial barrier function: Rac1 is activated after approximately 15 minutes and RhoA is inactivated (David et al., 2012). PI3K has several downstream effects, such as activating Akt, but also GEFs, subsequently targeting and activating Rac1 at cell-cell junctions. It is suggested that IQGAP is required for efficient and sustainable activation of Rac1 in endothelial cells (David et al., 2012). Subsequently, Rac1 can mediate activation of p190RhoGAP (a finding which was confirmed in vivo), inhibiting RhoA. P190RhoGAP activation is essential for enhancing the barrier function (Mammoto et al., 2007). However, it remains remarkable that TNFα/PI3K-mediated Rac1 activation induces breakdown of the cell-cell contacts, whereas S1P or Ang-1/PI3K-mediated Rac1 activation supports restoration of cell adhesion. The effect of Rac1 activation on cell-cell junctions probably depends on its specific location in the cell, and may depend on which cytokine activated Rac1. Figure 10. Ang-1-mediated regulation of Rac1 and RhoA. Near the plasma membrane, Ang-1 stimulates activation of RhoA, whereas signaling via active Rac1 inhibits RhoA in the cytosol. The pathways are more detailed explained in the text. The dashed lines indicate indirect activation. Surprisingly, Ang-1 can also stimulate RhoA close to where VEGFRs localizes (Gavard et al., 2008). RhoA in turn activates Dia. Active Dia will bind Src kinase and prevents Src from binding to the VEGFR, leading to reduced phosphorylation of VE-cadherin by Src (Figure 10). Consequently, this mitigates the disruption of cell-cell contacts induced by VEGF, thus protecting the endothelial barrier (Gavard et al., 2008). To conclude, Ang-1 can restore the endothelial barrier via initiating dephosphorylation of VEcadherin and via PI3K-mediated activation of Rac1. In contrast, Ang-1 can also enhance RhoA activity near VEGFRs, reducing phosphorylation of VE-cadherin as well, which may stabilize cellcell junctions too. 18 Rho GEFs and GAPs and cell-cell junction regulation As described in the previous chapter, each Rho GTPase can either initiate protection of the endothelial barrier or can promote vascular permeability, which will depend on the level, timing, and cellular location of their activation. Possibly GEFs and GAPs, functioning upstream of RhoGTPases and activate and inhibit GTPases respectively, play an important role in the spatiotemporal control of Rho GTPases. In this chapter we will focus on the GEFs and GAPs known to control cell-cell junctions in all cell types in order to comprehend the regulation of endothelial cell-cell junctions downstream of vascular cytokines. GEFs and GAPs controlling Rac1 signaling involved in junction regulation. The proposed roles of Rac1 in regulating AJs are conflicting. On one hand, Rac1 induces via its effector PAK1 or via ROS formation phosphorylation of VE-cadherin, leading to VE-cadherin internalization and loss of AJs. On the other hand, Rac1 is involved in barrier restoration after Ang-1 stimulation and via the S1P pathway. Rac1 induces junction stabilization through dislocation of IQGAP from cell junctions and activation p190RhoGAP, inhibiting RhoA. In both situations, similar Rac1 GEFs are suggested to be involved as I will discuss here. One of the GEFs that activates Rac1 in endothelial and epithelial cells is Tiam1 (Habets et al., 1994; Sander et al., 1998). Tiam1 is recruited to the plasma membrane and associates with VEcadherin (Lampugnani et al., 2002) depending on the p110α subunit of PI3K downstream of e.g. TNFα (Lee et al., 1999). Knockdown of this subunit results in decreased codistribution of the GEF with VE-cadherin in HUVECs, suggesting that PI3K regulates Rac1 at AJs via recruitment of Tiam1 (Cain et al., 2010). This leads to increase endothelial permeability (Cain et al., 2010). In contrast, Tiam1 can be activated through increased cAMP concentrations, correlating with increased barrier function Elevated cAMP levels, induced by prostaglandins, lead to activation of PKA1, phosphorylating and activating Tiam1 in HUVECs (Kobayashi et al., 2013). Using lung endothelial cells, similar results are reported: Tiam1 is involved in the recovery of cell-cell junctions after thrombin exposure (Birukova et al., 2012). In support for a junction protection function for Tiam1, in epithelial cells was found that Tiam1 is needed to maintain E-cadherin mediated cell-cell contacts (Sander et al., 1998; Malliri et al., 2004). In addition, Tiam1 is required for TJ formation in epidermal keratinocytes. Keratinocytes isolated from Tiam1 knockout mice display disturbed Rac1 activation and failure in TJ maturation (Mertens et al., 2005). Thus, Tiam1 can either induce destabilization or stabilization of cell-cell junctions, but is reported at least to be involved in junction regulation downstream of vascular cytokines. The Rac-GEF Vav2 is responsive to VEGF (Garrett et al., 2007). Vav2-mediated activation of Rac1 leads to PAK1 activation, resulting in phosphorylation of VE-cadherin and subsequently in internalization of VE-cadherin in mouse vascular endothelial cells (Gavard and Gutkind, 2006). In human mammary epithelial cells is shown that Vav2 co-localizes with E-cadherin and that activation of the Vav2/Rac1 pathway results in AJ disruption after stimulation with the growth factor EGF (Duan et al., 2011). Overexpression of constitutively active Vav2 elevates Rac1 activity and remodels the actin cytoskeleton near cell-cell junctions. Vav2 knockdown experiments significantly reduced the Rac1 activity and cells have less junctional F-actin, resulting in destabilized junctions (Duan et al., 2011). Besides Rac1 activation, Vav2 may activate Cdc42 as well (Liu et al., 2000). This signaling route is suggested to be involved in cell migration, although, a role in cell-cell adhesion is not yet established. However, similar as 19 described for Tiam1, another study established that Vav2 is involved in the formation of junctions and recovery of the barrier function after stimulation with thrombin (Birukova et al., 2012). So, Vav2 can be involved in barrier function protection or destabilization. A third GEF for Rac1 that is shown to be involved in junctional control is P-Rex1. P-Rex1 is detected in vascular lung endothelial cells and can be activated via the TNFα/PI3K pathway, resulting in Rac1 activation. P-Rex1 stimulates Rac1-mediated ROS production, inducing VEcadherin phosphorylation. Knockdown of P-Rex1 significantly reduces the level of Rac-GTP in TNFα stimulated cells. The in vivo effects are studied as well in P-Rex1 knockout mice. The knockout mice have reduced lung endothelial permeability and less lung edema after intratracheal exposure to TNFα, compared to wild type mice. Moreover, P-Rex1-/- mice had less transendothelial migration of leukocytes than wild type mice, suggesting that endothelial cellcell junctions had stabilized (Naikawadi et al., 2012). This clearly shows that TNFα-based activation of the PI3K/P-Rex1/Rac1 pathway destabilizes endothelial cell-cell junctions. P-Rex2b is also expressed in endothelial cells and is GEF for Rac1, however, a role for P-Rex2b in AJ regulation is not yet reported (Li et al., 2005). To sum up, at least three GEFs for Rac1 are expressed in endothelial cells. P-Rex1 functions downstream of PI3K and disrupts junctions via Rac1 signaling. Both Vav2 and Tiam1 play a role in junction disassembly as well. However, contradictory results are published. Tiam1 can also become activate by cAMP, leading to enhancement of cell-cell contacts. Moreover, Tiam1 enhances TJs in epidermal keratinocytes. Intriguingly, the described regulation mechanisms for these GEFs also do not explain how Ang-1 and S1P induced junction stabilization via Rac1. Probably, it depends on the spatiotemporal control of the GEFs and their upstream activators, but convincing data that deals with this dilemma is not published. Cdc42 GEF and GAPS regulating cell-cell junctions Cdc42 becomes active after thrombin stimulation and is shown to be essential in the restoration of AJs, (Broman et al., 2007; Ramchandran et al., 2008). Mice expressing constitutively active Cdc42 are more resistant to the induction of endothelial permeability by LPS (Ramchandran et al., 2008). Moreover, Cdc42 is shown to assemble TJs, however, a role in destabilizing TJs is reported as well (Wells et al., 2006; Otani et al., 2006). Tuba is a Cdc42-specific GEF concentrated at the apical zone of epithelial cells, where it interacts with the TJ-protein ZO-1 and contributes to TJ-stability (Otani et al., 2006). TJs and AJs are disordered after silencing Tuba. However, the recruitment of ZO-1 to TJs on the plasma membrane is not affected by Tuba depletion , in contrast, the organization of E-cadherin on the membrane is strongly dependent on Tuba (Otani et al., 2006). Tuba is the first reported Cdc42 regulator that directly interacts with junctional proteins. It is not yet established whether Tuba is expressed in endothelial cells. Furthermore, the GAP Rich1 is found to regulate Cdc42. Rich1 localizes at TJs and AJs and regulates the activity of Cdc42 at junctions in MDCK cells (Wells et al., 2006). A GAP-deficient mutant of Rich1 induces improper localization of TJ proteins at the membrane, consequently leading to instable TJs and increasing the permeability of the monolayer. However, Rich1 is not necessary for the initiation of TJ formation. Moreover, silencing Rich1 by shRNA does not disturb AJ organization. Additionally, the scaffold angiomotin (Amot) binds Rich1 and is recruited to TJs. Overexpression of Amot leads to disruption of TJs, (Wells et al., 2006). Since Rich1 can also 20 accelerates the intrinsic GTP hydrolysis of Rac1, Rich1 might be involved in Rac1 inactivation at junctions as well (Richnau and Aspenström, 2001). Furthermore, in mice and zebrafish Amot depletion leads to inactive Rac1 and vascular defects (Aase et al., 2007). Thus, Rich1 activation, consequently inhibiting Cdc42 and Rac1, results in stable TJs and in decreased permeability of cellular monolayers. It is reported that Rich1 is expressed in mouse embryonic microvascular endothelial cells too (Aase et al., 2007) A second Cdc42 GAP involved in cell-cell junction regulation is PX-RICS. In contrast to Rich1, this GAP possibly regulates the activity of Cdc42 at the Golgi and not at the plasma membrane (Nakamura et al., 2008). It is suggested that in HeLA cells, PX-RICS is involved in the transport of the N-cadherin/β-catenin complex from the Golgi towards the plasma membrane (Nakamura et al., 2008). This indicates that PX-RICS’s induced inactivation of Cdc42 supports repair of AJs. Similar involvement of Cdc42 in the transport of E-cadherin from Golgi to membrane was reported using MDCK cells (Wang et al., 2005).Thus far, PX-RICS expression is not reported to be expressed in endothelial cells. In conclusion, the Cdc42 GEF Tuba and the GAPs Rich1 and PX-RICS can all stabilize cell-cell junctions. This is surprising, since the GEF activates Cdc42 and the GAPs inactivate Cdc42. This again supports the notion that local regulation by GEFs or GAPs may differ at distinct junction types and cell types. Tuba and Rich1 are involved in the regulation of TJs at the membrane, whereas PX-RICS regulates the secretory pathway of N-cadherin towards the plasma membrane near the Golgi. It will be interesting to study whether PX-RICS is also involved in the transport of E- and VE-cadherin, although it is not yet established if PX-RICS is expressed in endothelial cells. RhoA GEFs and GAPs controlling cell-cell junctions and the barrier function The Rho GTPase RhoA acts downstream of several permeability inducing and protecting factors (see previous chapter). The effectors of RhoA can either induce stabilization or disruption of cellular junctions. Likely, the effect on the endothelial permeability will depend on the spatiotemporal RhoA activation. RhoA specific GEFs and GAPs play an important role in this tight regulation of RhoA. Some GEFs induce RhoA-mediated endothelial permeability, whereas other GEFs stabilize junctions and have the ability to protect the barrier function. Also two GAPs are known to regulate RhoA. The first RhoA GEF we describe is GEF-H1, activated via the MAPK pathway downstream of TNFα and required for TJ dissociation. Knockdown of GEF-H1 prevented activation of RhoA by TNFα in renal epithelial cells (Kakiashvili et al., 2008). It is established that GEF-H1 is associated with the TJ component ZO-1 in epithelial cells (Benais-Pont et al., 2003; Samarin et al., 2007) and endothelial cells (Mckenzie, 2007). Overexpression of GEF-H1 does not induce changes in the cytoskeleton and disruption of TJs in MDCKs, however, the intercellular permeability of the epithelial monolayer is elevated (Benais-Pont et al., 2003). Another study found that GEF-H1 is involved in the disassembly of the apical junctions in calcium depleted epithelial cells. RhoA and ROCK increase the contractility of the actin cytoskeleton, providing force that disrupts junctions (Samarin et al., 2007). The RhoA GEF P115RhoGEF is activated via the G-protein subunit Gα12/13 downstream of thrombin’s receptor PAR-1 (Birukova et al., 2004; Holinstat et al., 2003). In addition, p115RhoGEF is phosphorylated within 1 min after thrombin through activated PKCα, an intermediate downstream of TNFα and Gα11/q (Peng et al., 2011; Holinstat et al., 2003). 21 Knockdown of PKCα results in reduced phosphorylation of p115RhoGEF. As established in lung endothelial cells, p115RhoGEF and RhoA both translocate towards the plasma membrane after stimulation with thrombin. Subsequently, RhoA actives ROCK and induces actomyosin contraction and phosphorylation of TJ proteins (Birukova et al., 2004; Yamamoto et al., 2008). However, it has been shown that prostate cancer cells overexpressing PAR-1 still react on thrombin after knockdown of p115RhoGEF (Wang et al., 2004), suggesting that also other GEFs are able to activate RhoA. Moreover, in this cell type, depleting p115RhoGEF in mouse brain microvascular endothelial cells inhibits RhoA activation and less intercellular gaps are formed by thrombin (Peng et al., 2011). Another study in mouse brain endothelial cells showed that knockdown of p115RhoGEF partly inhibits activation of RhoA, reducing the formation of stress fibers and degradation of TJ proteins (Xiaolu et al., 2011). These data show that p115RhoGEFmediated RhoA activation is involved in breakdown of TJs in endothelial cells, contributing to increased permeability. RhoA activation through the above described GEFs disrupts junctions in endothelial and epithelial cells. By contrast, Terry et al. suggest that the opposite occurs when RhoA is activated via p114RhoGEF in epithelial cells. P114RhoGEF is found to be recruited towards TJs via cingulin, a scaffold interacting with ZOs, JAM-A and actin (Bazzoni and Dejana, 2004; Shin et al., 2006). Deletion of this GEF is associated with disassembly of TJs and activation of non-junctional RhoA, resulting in MLC phosphorylation (Terry et al., 2011). However, another group found in a different epithelial cell line that knockdown of P114RhoGEF did not result in disorganization of the TJs, suggesting that the regulation of RhoA may vary between different cell types (Itoh et al., 2012). Interestingly, p114RhoGEF is expressed in lung microvascular endothelial cells (Niu et al., 2003). Thus, p114RhoGEF induces spatially restricted RhoA activation, supporting TJ formation in some types of epithelial cells. Additionally, the RhoA GEF PDZ-RhoGEF is shown to stabilize TJs. PDZ-RhoGEF binds directly to ZO-1 and is expressed in both endothelium and epithelium (Basile et al., 2007; Itoh et al., 2012), however, its function is only thoroughly studied in epithelial cells. PDZ-RhoGEF becomes activated through Gα12/13, downstream of several GPCRs. Knockdown of PDZ-RhoGEF does not result in diminished levels of ZO-1 at TJs. Although, depletion of PDZ-RhoGEF does affect the cell-cell junction formation, resulting in attenuated barrier function. Moreover, MLC phosphorylation is increased in the absence of PDZ-RhoGEF at apical TJs, leading to contraction of the actomyosin cytoskeleton near cell-cell junctions. In epithelial cells, this actomyosin contraction induces clustering of TJs and consequently polarizes the plasma membrane in an apical and basolateral side (Itoh et al., 2012). Thus, this GEF also spatially stimulates RhoA to regulate TJ formation in epithelium. Since the function of TJs in endothelium is not the same as in epithelium, PDZ-RhoGEF might have other effects on the endothelial barrier, for which further experimentation is necessary. Another RhoA GEF supporting junctional integrity via junction-associated actomyosin is Ect2 (Ratheesh et al., 2012). During cell division the centralspindlin complex attracts this GEF towards AJs at the cleavage furrow, where it activates RhoA. Centralspindlin is also involved in inhibition of the p190RhoGAP function, thereby enhancing RhoA activity. As a result, active RhoA initiates myosin II activation, leading to actomyosin contraction and stabilization of apical AJs (Ratheesh et al., 2012). Interestingly, both PDZ-RhoGEF and Ect2 seem to underlie the integrity of the zonula adherens in epithelial cells via RhoA mediated ROCK/myosin II activation. Still, it is poorly understood how if these GEFs are regulated by inflammatory mediators. Both 22 GEFs are known to be expressed by endothelial cells, but no functions have been reported (Basile et al., 2007; Nacak et al., 2007). LARG, related to PDZ-RhoGEF and P115RhoGEF, can also be activated by Gα12/13, downstream of thrombin and histamine signaling (Wang et al., 2004; Pfreimer et al., 2012). In podocytes, epithelial cells in the glomerulus in the kidney, is shown that LARG can play a role in cell-cell adhesion. It is suggested that a scaffolding protein recruits LARG appropriately at the AJs, consequently activation RhoA at the junctions. RhoA on its turn can link actin to cadherin (Kim et al., 2012). LARG expression is also detected in HUVECs (Geneprof, 2013a). Furthermore, the GEF Synectin-binding RhoA exchange factor (Syx) is expressed in several cell types, including endothelial cells and associates with AJ and TJ remodeling. Syx can be recruited towards ZO-1 in HUVECs and MDCK cells (Garnaas et al., 2009; Ngok et al., 2012). The localization of Syx can be regulated by VEGF and Ang-1 and is crucial for the regulation of junction stability. VEGF promotes removal of Syx from TJs, preventing RhoA to become active near TJs, whereas Ang-1 preserves Syx at the junctions and accordingly stabilizes the intercellular interactions via RhoA effectors (Ngok et al., 2012). It is indicated that Dia1 functions downstream of Syx, since impaired ZO-1 localization at the plasma membrane in Syx depleted cells can be restored through expression of constitutively active Dia1 (Ngok et al., 2012). After Syx depletion, increased VE-cadherin internalized occurs, probably initiated by increased Src-mediated VE-cadherin phosphorylation. Moreover, by controlling expression of Syx in zebrafish, it was shown that this GEF is required for the sprouting at the intersomatic vessels, showing that Syx is involved in angiogenesis which depends on endothelial cell-cell junction remodeling. (Garnaas et al., 2009). Moreover, Syx KO mice showed malformed or absent intercellular endothelial cell-cell junctions and vascular leakage compared to wild type mice. To sum up, Syx is shown to induce RhoA-mediated activation of Dia and inhibition of Src near the AJs and TJs, mediating reannealing of junctions, which potentially explains the crucial role of Syx in angiogenesis. Interestingly, not only Rac1 is suggested to initiate cadherin internalization. Also RhoA can play a role the endocytosis of AJs, as established in drosophila (Levayer et al., 2011). The RhoA GEF RhoGEF2 can initiate disruption of cell-cell adhesion during epithelial morphogenesis in drosophila embryos. RhoGEF2 has three mammalian homologs: p115-RhoGEF, PDZ-RhoGEF and LARG (Brumby et al., 2011). It localizes in epithelial cells near the plasma membrane, where it activates Rho1 (drosophila ortholog of RhoA). It has been proposed that Rho1 activates both Dia and ROCK near AJs, which in cooperation initiate the endocytosis of E-cadherin at the plasma membrane. Dia and active myosin II are necessary for appropriate clathrin localization at the junctions, since E-cadherin is internalized via clathrin-coated-endocytosis. Moreover, drosophila embryos carrying a dominant negative RhoGEF2 have lower levels of clathrin around the cellcell junctions (Levayer et al., 2011). Reduction of RhoGEF2 activity also results in limited Ecadherin turnover during tracheal tube morphogenesis in drosophila (Warrington et al., 2013). RhoGEF2 has been shown to be involved in the elongation of the epithelial tissue, a process that requires remodeling of cell junctions (Warrington et al., 2013). Thus, it is established that RhoGEF2 induces internalization of E-cadherin during early drosophila development, eventually contributing to less cell-cell junctions. In addition, also some GAPs are known to be involved in the RhoA-mediated cell-cell junction regulation. One of them is GRAF2, a GAP colocalizing with α-catenin at AJs in several types of 23 epithelial cells. Without cadherin expression, GRAF2 is detected at the Golgi (Sousa et al., 2005). Like RhoGEF2, this GAP is involved in E-cadherin internalization. The function of GRAF2 is studied in the context of bacteria internalization via the cadherin-mediated endocytosis. Overexpression of GRAF2 results in elevated α-catenin levels and disrupted actin fibers, whereas knockdown via siRNA leads to impaired α-catenin recruitment towards AJs and weak junctions. GRAF2 is suggested to coordinate AJ formation in epithelial cells (Sousa et al., 2005) and it is also found to be expressed in HUVECs (Geneprof, 2013b). P190RhoGAP is activated after stimulation of endothelial cells with inflammatory mediators such as thrombin. Activated Rac1 induces to production of ROS, resulting in several responses including translocation of p190RhoGAP towards AJs, where it interacts with p120-catenin (Nimnual et al., 2003; Wildenberg et al., 2006). Also Ang-1 signals via Rac1 and p190RhoGAP to inactivate RhoA. Knockdown of p190RhoGAP in endothelial cells eliminates the ability of Ang-1 to mitigate lung vascular permeability induced by inflammatory cytokines. However, depletion of p190RhoGAP does not alter the permeability (in vivo and in vitro) in non-stimulated cells (Mammoto et al., 2007). To sum up, many GEFs and GAPs are suggested to regulate RhoA activity in epithelial and/or endothelial cells. It is clear that the activation of RhoA by GEFs and GAPs is spatially restricted. GEF-H1 and p115-RhoGEF induce the formation of stress fibers, providing tension on cell-cell junctions. . The GEFs p114-RhoGEF and PDZ-RhoGEF are located near TJs in epithelial cells. In this subcellular location, they activate RhoA, resulting in the stabilization of the TJs and so mediating cell-cell adhesion. It has to be taken in account that there is a difference between endothelial and epithelial cells. Both PDZ-RhoGEF and Ect2 enhance junctional stability via induced actomyosin contractions near TJs. This results in clustering of the TJs and strengthening of the cell-cell contacts. Since endothelial TJs are not organized similarly as in polarized epithelial cells, it might be possible that these GEFs have different effects on endothelial cell-cell junctions. Another important GEF stabilizing both AJs and TJs in endothelial cells is Syx, which crucial function is also established in the vasculature of zebrafish and mice. Furthermore, in drosophila embryos the p115-RhoGEF, PDZ-RhoGEF and LARG homologue RhoGEF2 induces internalization of E-cadherin, which may contribute to regulation of cell-cell junctions in mammalian cells. Lastly, two GAPs are associated with RhoA regulation and support cell-cell contacts. GRAF2 inhibits RhoA near the plasma membrane and blocks internalization of Ecadherin. p190RhoGAP inhibits RhoA and prevents disassembly of junctions. P190RhoGAP lies downstream of Rac1 signaling and once activated will restore cell-cell adhesion in endothelial monolayers. 24 Discussion Rho GEFs and GAPs involved in remodeling endothelial cell junctions, supporting endothelial permeability Rho GTPases are proteins that regulate the actin cytoskeleton and remodel cell junctions, such as AJs and TJs. I focused on the role of Rho GTPases RhoA, Rac1 and Cdc42 in regulating the endothelial permeability. Opening of the junctions between the endothelial cells in the vessel wall allows leukocytes and molecules to pass the barrier and enter the surrounding tissue, regulated by several inflammatory mediators. Rho GTPases play an important role downstream of these factors. They can initiate the phosphorylation of AJ and TJ components, contraction of the junctional-associated actomyosin cytoskeleton or regulate endocytosis of AJs. It becomes clear that the activation/inactivation of RhoGTPases is complex: it depends on their cellular localization and the available effectors whether a GTPase induces permeability of protects against permeability. Each Rho GTPase is reported to induce both, depending on the biological context. GEFs and GAPs are necessary in this complex regulation: their activation and subcellular localization is crucial in the activating and inactivating of Rho GTPases respectively, but our knowledge concerning the regulation of GEFs and GAPs is still in its infancy. In this review, I paid special attention to the GEFs and GAPs known to regulate the junctions in all cell types and animals to try to understand if they might be involved in regulation of cell-cell junctions in vascular biology. It is shown that some GEFs/GAPs are involved in the regulation of the endothelial barrier function in vivo. For example, Syx localizes near junctions after exposure to Ang-1, resulting in RhoA/Dia activation. This stabilizes cell junctions and maintains of the barrier function. Furthermore, P-Rex1 is activated via the TNFα/PI3K pathway, activating Rac1. P-Rex1 knockout mice have less transendothelial leukocyte migration in the lungs after exposure to TNFα, indicating that P-Rex1 reduces endothelial barrier function. The two Rac1 GEFs Vav2 and Tiam1 seem to be involved in the regulation of endothelial cell-cell junctions as well. Their functions are widely studied in endothelial cells; however, it is still not clear whether Vav2 and Tiam1 induce disruption or stabilization of junctions. Two RhoA GEFs and one RhoA GAP are established to regulate endothelial junctions in vitro via RhoA activation. GEF-H1 and p115-RhoGEF induce the formation of stress fibers, creating force on junctions. Both effects cause an improved barrier function. Moreover, p115-RhoGEF stimulates breakdown of TJs. p190RhoGAP is necessary for barrier protection, indicating that p190RhoGAP plays an important role in supporting endothelial permeability. Mouse models may confirm whether these regulatory proteins indeed regulate the endothelial barrier function via RhoA activation or inactivation in vivo. Additionally, the GEFs and GAPs that may be be involved in junction regulation are PDZ-RhoGEF, p114RhoGEF, LARG, GRAF1, Rich1 and PX-RICs. They are detected in endothelial cells; however, their function in the regulation of cell-cell junction is only established in epithelial cells. PDZRhoGEF, p114RhoGEF and LARG activate RhoA near junctions and it has been shown that this signaling stabilizes AJs and TJs. Since TJs are more organized in epithelium than in endothelium (Itoh et al., 2012), spatial activation of RhoA by PDZ-RhoGEF and LARG could have different effects on endothelial cell-cell adhesions. Another RhoA regulation protein is the GAP GRAF2, stabilizing cell-cell contacts via inhibition of RhoA. Furthermore, one GEF and two GAPs are 25 suggested to regulate Cdc42; however, they have contrasting effects. The GEF Tuba activates Cdc42, whereas the GAPs Rich1 and PX-RICS inhibit Cdc42 at TJs and AJs. As illustrated in this thesis, all three Rho GTPases can have various functions on the endothelial barrier function, depending on their spatiotemporal activation. For example, local Rac1 activation seems critical, since the same upstream pathway (PI3K) can induce either Rac1/Tiam1-mediated barrier weakening as barrier strengthening. Also for RhoA is shown that different GEFs can control RhoA at different subcellular locations, leading to distinct effects on the cell-cell junctions. RhoA can either regulate junctional actomyosin, non-junctional actomyosin, the phosphorylation of junctional proteins and the internalization of cell junction complexes, making the interpretation of biochemical experiments on MLCK and MLC even more complex. Also the regulation and localization of the upstream Rho GEFs and GAPs is crucial. Not for all GEFs/GAPs is yet established how their activation is induced by vascular cytokines (Table 1). Exploring the upstream regulation of GEFs/GAPs will uncover whether a GEF/GAP might be biological relevant. Can it be activated by inflammatory mediators? To gain more insight in the regulation of the Rho GTPases in endothelial cells, it is necessary to perform experiments that can clarify where and by which GEF/GAP a GTPase is (in)activated. To establish their regulation in space and time, bimolecular fluorescence resonance energy transfer (FRET)-probes that visualize local Rho GTPase activities would be useful. The role of the GEFs/GAPs can be studied by using knockdown approaches, in combination with vascular cytokine stimuli. Of note, cultured in vitro cells can have other characteristics than the same cell type in vivo. In addition, HUVECs are often used for studying the endothelial permeability; however, they may not represent the exact responses to inflammatory mediators of other endothelial cells in the vascular tree. It will be interesting to determine whether the reaction of GEFs/GAPs is different between specific subtypes of endothelial cells (microvascular, arterial, venal) and organs (brain, lung, kidney, etc.). Furthermore, using also an in vivo model can give more understanding of the signaling networks. Knockout mice can be used for investigating the effect of GEFs and GAPs on e.g. vascular leakage. When embryonic lethal, a conditional knockout system can be the solution. With these techniques, new GEFs/GAPs involved in angiogenesis and/or inflammation can be explored. These GEFs/GAPs can be interesting therapeutic targets to reduce vascular leakage in patients with e.g. atherosclerosis or to prevent cancer metastasis via angiogenesis. 26 GEF Rho GTPase Upstream regulator Junction type (AJ/TJ) Tiam1 Rac1 TNFα, S1P, AJ, TJ Vav2 Rac1, Cdc42 VEGF, EGF AJ P-Rex1 Rac1 TNFα AJ RhoGEF2 RhoA ? AJ Ect2 Syx RhoA RhoA P115RhoGEF/ ARHGEF1 RhoA ? VEGF, Ang-1 TNFα, Thrombin GEF-H1/ ARHGEF2/ Lfc RhoA PDZ-RhoGEF/ ARHGEF11 LARG/ ARHGEF12 P114RhoGEF/ ARHGEF18 Tuba RhoA RhoA TNFα Contractility actin cytoskeleton TJ TJ x TJ x TJ x Cell type(s) used for experiments In vivo support Expressed in endothelium Epithelium, HUVECs Lung endothelium, HUVECs, epithelium Lung endothelium Epithelium (epidermis) Epithelium Endothelium mice yes yes mice yes drosophila ? mice, zebrafish ? yes Lung and brain endothelium, HUVECs Epithelium (MDCK and renal), HUVECs Epithelium yes Epithelium yes yes yes ? RhoA Histamine, Thrombin ? TJ Epithelium yes Cdc42 ? TJ epithelium ? GAP Rho GTPase Upstream regulator Junction type (AJ/TJ) GRAF2/ ARHGAP10 P190RhoGAP RhoA ? AJ Epithelium RhoA Thrombin, Ang-1 AJ RICH1/ ARHGAP17 PX-RICS/ p250RhoGAP/ p200RhoGAP/ Grit Cdc42, Rac1 Cdc42 ? TJ ? AJ Epithelium, endothelium, fibroblasts Epithelium (MDCK) HeLa cells Contractility actin cytoskeleton Cell type(s) used for experiments In vivo support Expressed in endothelium yes mice yes yes ? Table 1. Rho GEFs and GAPs regulating cell junctions. The GEFS and GAPs mentioned are suggested to play a role in the regulation of cell-cell junctions. For each GEF/GAP is depicted which Rho GTPase(s) it can activate or inactivate respectively. If known, the upstream regulators are shown. In the third and fourth columns is described which type of junction the GEF/GAP can regulate and whether they induce contractility of the actin cytoskeleton. Next is shown which cell types are used to study the function of the GEF/GAP and whether the function is also studied in an animal model. In the last column is mentioned whether the GEF/GAP is expressed in endothelial cells. 27 Abbreviations AJ Ang-1 DH domain DRFs E-cadherin GAP GDI GEF GPCR GTPase HUVEC JAM LIMK MLC MLCK N-cadherin PH domain PLCγ Rho GTPase ROCK ROS S1P SPHK TER TJ TNFα TRK VE-cadherin VEGF VEGFR ZO Adherens junction Angiopoietin-1 Dbl homology domain Diaphanous related formins Epithelial cadherin GTPase activating protein Guanine nucleotide dissociation inhibitor Guanine nucleotide exchange factor G-protein-coupled receptor Guanine triphosphate phosphohydrolase Human umbilical vein endothelial cell Junctional adhesion molecule LIM kinase Myosin light chain MLC kinase Neuronal cadherin Pleckstrin homology domain Phospholipase Cγ Ras homologous GTPase Rho-associated kinase Reactive oxygen species Sphingosine 1-phosphate Sphingosine kinase Transendothelial/-epithelial electrical resistance Tight junction Tumor necrosis factor α Tyrosine kinase receptor Vascular endothelial cadherin Vascular endothelial growth factor VEGF receptor Zona occludens Acknowledgements I thank Stephan Huveneers for the opportunity to write my thesis under his supervision and for his feedback on my literature study and ideas. 28 References Aase, K., M. Ernkvist, L. Ebarasi, L. Jakobsson, A. Majumdar, C. Yi, O. Birot, Y. Ming, A. Kvanta, D. Edholm, P. Aspenström, J. Kissil, L. Claesson-Welsh, A. Shimono, and L. Holmgren. 2007. Angiomotin regulates endothelial cell migration during embryonic angiogenesis. Genes & development. 21:2055–68. Abraham, S., M. Yeo, M. Montero-Balaguer, H. Paterson, E. Dejana, C.J. Marshall, and G. Mavria. 2009. VECadherin-mediated cell-cell interaction suppresses sprouting via signaling to MLC2 phosphorylation. Current biology : CB. 19:668–74. Adamson, R.H., R.K. Sarai, A. Altangerel, T.L. Thirkill, J.F. Clark, and F.-R.E. Curry. 2010. Sphingosine-1phosphate modulation of basal permeability and acute inflammatory responses in rat venular microvessels. Cardiovascular research. 88:344–51. Alvarez, S.E., S. Milstien, and S. Spiegel. 2007. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends in endocrinology and metabolism: TEM. 18:300–7. Amano, M., M. Nakayama, and K. Kaibuchi. 2010. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken, N.J.). 67:545–54. Basile, J.R., J. Gavard, and J.S. Gutkind. 2007. Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. The Journal of biological chemistry. 282:34888–95. Baumer, Y., D. Drenckhahn, and J. Waschke. 2008. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochemistry and cell biology. 129:765–78. Baumer, Y., V. Spindler, R.C. Werthmann, M. Bünemann, and J. Waschke. 2009. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. Journal of cellular physiology. 220:716–26. Bazzoni, G. 2006. Endothelial tight junctions : permeable barriers of the vessel wall. Thrombosis and haemostasis. 95:36–42. Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiological reviews. 84:869–901. Beckers, C.M.L., V.W.M. van Hinsbergh, and G.P. van Nieuw Amerongen. 2010. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thrombosis and haemostasis. 103:40–55. Benais-Pont, G., A. Punn, C. Flores-Maldonado, J. Eckert, G. Raposo, T.P. Fleming, M. Cereijido, M.S. Balda, and K. Matter. 2003. Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. The Journal of cell biology. 160:729–40. Birukova, A. a, K. Smurova, K.G. Birukov, K. Kaibuchi, J.G.. Garcia, and A.D. Verin. 2004. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvascular Research. 67:64– 77. Birukova, A. a, Y. Tian, O. Dubrovskyi, N. Zebda, N. Sarich, X. Tian, Y. Wang, and K.G. Birukov. 2012. VEcadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. Journal of cellular physiology. 227:3405–16. Bos, J.L., H. Rehmann, and A. Wittinghofer. 2007. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 129:865–77. 29 Braga, V.M., L.M. Machesky, a Hall, and N. a Hotchin. 1997. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. The Journal of cell biology. 137:1421–31. Broman, M.T., D. Mehta, and A.B. Malik. 2007. Cdc42 regulates the restoration of endothelial adherens junctions and permeability. Trends in cardiovascular medicine. 17:151–6. Brumby, A.M., K.R. Goulding, T. Schlosser, S. Loi, R. Galea, P. Khoo, J.E. Bolden, T. Aigaki, P.O. Humbert, and H.E. Richardson. 2011. Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics. 188:105–25. Bryan, B. a, E. Dennstedt, D.C. Mitchell, T.E. Walshe, K. Noma, R. Loureiro, M. Saint-Geniez, J.-P. Campaigniac, J.K. Liao, and P. a D’Amore. 2010. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 24:3186–95. Cain, R.J., B. Vanhaesebroeck, and A.J. Ridley. 2010. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. The Journal of cell biology. 188:863–76. Carmeliet, P., M.G. Lampugnani, L. Moons, F. Breviario, V. Compernolle, F. Bono, G. Balconi, R. Spagnuolo, B. Oosthuyse, M. Dewerchin, a Zanetti, a Angellilo, V. Mattot, D. Nuyens, E. Lutgens, F. Clotman, M.C. de Ruiter, a Gittenberger-de Groot, R. Poelmann, F. Lupu, J.M. Herbert, D. Collen, and E. Dejana. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98:147–57. Citi, S., D. Spadaro, Y. Schneider, J. Stutz, and P. Pulimeno. 2011. Regulation of small GTPases at epithelial cell-cell junctions. Molecular membrane biology. 28:427–44. Clough, G.F., a R. Bennett, and M.K. Church. 1998. Effects of H1 antagonists on the cutaneous vascular response to histamine and bradykinin: a study using scanning laser Doppler imaging. The British journal of dermatology. 138:806–14. David, S., C.C. Ghosh, A. Mukherjee, and S.M. Parikh. 2012. Angiopoietin-1 requires IQGAP1 to activate Rac1 and promote endothelial barrier defense. Arteriosclerosis, thrombosis, and vascular biology. 31:2643– 2652. Duan, L., S.M. Raja, G. Chen, S. Virmani, S.H. Williams, R.J. Clubb, C. Mukhopadhyay, M. a Rainey, G. Ying, M. Dimri, J. Chen, A.L. Reddi, M. Naramura, V. Band, and H. Band. 2011. Negative regulation of EGFR-Vav2 signaling axis by Cbl ubiquitin ligase controls EGF receptor-mediated epithelial cell adherens junction dynamics and cell migration. The Journal of biological chemistry. 286:620–33. Eriksson, a. 2003. Small GTP-Binding Protein Rac Is an Essential Mediator of Vascular Endothelial Growth Factor-Induced Endothelial Fenestrations and Vascular Permeability. Circulation. 107:1532–1538. Fischer, R.S., M. Gardel, X. Ma, R. Adelstein, and M. Clare. 2010. Myosin II mediates local cortical tension to guide endothelial cell branching morphogenesis and migration in 3D. Current biology : CB. 19:260–265. Fukuhara, S., K. Sako, T. Minami, K. Noda, H.Z. Kim, T. Kodama, M. Shibuya, N. Takakura, G.Y. Koh, and N. Mochizuki. 2008. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nature cell biology. 10:513–26. Garnaas, M.K., K.L. Moodie, M. Liu, G. V Samant, K. Li, J.M. Baraban, A. Horowitz, and R. Ramchandran. 2009. Syx, a RhoA guanine exchange factor, is essential for angiogenesis in vivo. Circulation research. 103:710–716. Garrett, T. a, J.D. Van Buul, and K. Burridge. 2007. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Experimental cell research. 313:3285–97. 30 Gavard, J., and J.S. Gutkind. 2006. VEGF controls endothelial-cell permeability by promoting the betaarrestin-dependent endocytosis of VE-cadherin. Nature cell biology. 8:1223–34. Gavard, J., V. Patel, and J.S. Gutkind. 2008. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Developmental cell. 14:25–36. Geneprof. 2013a. ARHGEF12. http://www.geneprof.org/GeneProf/record.jsp?id=20005&ds_id=PUB_HS_ENS59_GRCH37. Geneprof. 2013b. ARHGAP10. http://www.geneprof.org/GeneProf/record.jsp?id=15812&ds_id=PUB_HS_ENS59_GRCH37. Gory-Fauré, S., M.H. Prandini, H. Pointu, V. Roullot, I. Pignot-Paintrand, M. Vernet, and P. Huber. 1999. Role of vascular endothelial-cadherin in vascular morphogenesis. Development (Cambridge, England). 126:2093–102. Gottardi, C.J., and B.M. Gumbiner. 2001. Adhesion signaling: how beta-catenin interacts with its partners. Current biology : CB. 11:R792–4. Guo, Y., C. Ramachandran, M. Satpathy, and S.P. Srinivas. 2007. Histamine-induced myosin light chain phosphorylation breaks down the barrier integrity of cultured corneal epithelial cells. Pharmaceutical research. 24:1824–33. Habets, G.G., E.H. Scholtes, D. Zuydgeest, R. a van der Kammen, J.C. Stam, a Berns, and J.G. Collard. 1994. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 77:537–49. Heasman, S.J., and A.J. Ridley. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nature reviews. Molecular cell biology. 9:690–701. Van der Heijden, M., G.P. van Nieuw Amerongen, J. van Bezu, M. a Paul, a B.J. Groeneveld, and V.W.M. van Hinsbergh. 2011. Opposing effects of the angiopoietins on the thrombin-induced permeability of human pulmonary microvascular endothelial cells. PloS one. 6:e23448. Hla, T. 2003. Signaling and biological actions of sphingosine 1-phosphate. Pharmacological Research. 47:401–407. Holinstat, M., D. Mehta, T. Kozasa, R.D. Minshall, and A.B. Malik. 2003. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. The Journal of biological chemistry. 278:28793–8. Hung, D.T., Y.H. Wong, T.K. Vu, and S.R. Coughlin. 1992. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. The Journal of biological chemistry. 267:20831–4. Huveneers, S., J. Oldenburg, E. Spanjaard, G. van der Krogt, I. Grigoriev, A. Akhmanova, H. Rehmann, and J. de Rooij. 2012. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. The Journal of cell biology. 196:641–52. Huwiler, A., F. Doll, S. Ren, S. Klawitter, A. Greening, I. Romer, S. Bubnova, L. Reinsberg, and J. Pfeilschifter. 2006. Histamine increases sphingosine kinase-1 expression and activity in the human arterial endothelial cell line EAhy 926 by a PKC-α-dependent mechanism. Biochimica et biophysica acta. 357–376. Itoh, M., S. Tsukita, Y. Yamazaki, and H. Sugimoto. 2012. Rho GTP exchange factor ARHGEF11 regulates the integrity of epithelial junctions by connecting ZO-1 and RhoA-myosin II signaling. Proceedings of the National Academy of Sciences of the United States of America. 109:9905–10. 31 Jou, T.S., and W.J. Nelson. 1998. Effects of regulated expression of mutant RhoA and Rac1 small GTPases on the development of epithelial (MDCK) cell polarity. The Journal of cell biology. 142:85–100. Kaibuchi, K., S. Kuroda, M. Fukata, and M. Nakawaga. 1999. Regulation of cadherin-mediated cell – cell adhesion by the Rho family GTPases. Current Opinion in Cell Biology. 5:591–596. Kakiashvili, E., P. Speight, F. Waheed, R. Seth, M. Lodyga, S. Tanimura, M. Kohno, O.D. Rotstein, a. Kapus, and K. Szaszi. 2008. GEF-H1 Mediates Tumor Necrosis Factor- -induced Rho Activation and Myosin Phosphorylation: ROLE IN THE REGULATION OF TUBULAR PARACELLULAR PERMEABILITY. Journal of Biological Chemistry. 284:11454–11466. Kim, J.H., A. Mukherjee, S.M. Madhavan, M. Konieczkowski, and J.R. Sedor. 2012. WT1-interacting protein (Wtip) regulates podocyte phenotype by cell-cell and cell-matrix contact reorganization. American journal of physiology. Renal physiology. 302:F103–15. Kobayashi, K., Y. Tsubosaka, M. Hori, S. Narumiya, H. Ozaki, and T. Murata. 2013. Prostaglandin D2-D Prostanoid Signaling Promotes Endothelial Barrier Function via the cAMP/Protein Kinase A/Tiam1/Rac1 Pathway. Arteriosclerosis, thrombosis, and vascular biology. Kouklis, P., M. Konstantoulaki, S. Vogel, M. Broman, and A.B. Malik. 2004. Cdc42 regulates the restoration of endothelial barrier function. Circulation research. 94:159–66. Krishnan, R., D.D. Klumpers, C.Y. Park, K. Rajendran, X. Trepat, J. van Bezu, V.W.M. van Hinsbergh, C. V Carman, J.D. Brain, J.J. Fredberg, J.P. Butler, and G.P. van Nieuw Amerongen. 2011. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. American journal of physiology. Cell physiology. 300:C146–54. Kuroda, S. 1998. Role of IQGAP1, a Target of the Small GTPases Cdc42 and Rac1, in Regulation of ECadherin- Mediated Cell-Cell Adhesion. Science. 281:832–835. Kusuhara, S., Y. Fukushima, S. Fukuhara, L.M. Jakt, M. Okada, Y. Shimizu, M. Hata, K. Nishida, A. Negi, M. Hirashima, N. Mochizuki, S.-I. Nishikawa, and A. Uemura. 2012. Arhgef15 promotes retinal angiogenesis by mediating VEGF-induced Cdc42 activation and potentiating RhoJ inactivation in endothelial cells. PloS one. 7:e45858. Lamalice, L., F. Houle, G. Jourdan, and J. Huot. 2004. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene. 23:434–45. Lammers, M., S. Meyer, D. Kühlmann, and A. Wittinghofer. 2008. Specificity of interactions between mDia isoforms and Rho proteins. The Journal of biological chemistry. 283:35236–46. Lampugnani, M.G., A. Zanetti, F. Breviario, G. Balconi, F. Orsenigo, M. Corada, M. Betson, V. Braga, and E. Dejana. 2002. VE-Cadherin Regulates Endothelial Actin Activating Rac and Increasing Membrane Association of Tiam. Molecular biology of the cell. 13:1175–1189. Lee, J.-F., Q. Zeng, H. Ozaki, L. Wang, A.R. Hand, T. Hla, E. Wang, and M.-J. Lee. 2006. Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. The Journal of biological chemistry. 281:29190–200. Lee, M., S. Thangada, K.P. Claffey, N. Ancellin, C.H. Liu, M. Kluk, M. Volpi, R.I. Sha, T. Hla, T.E.- Endothelial, and D. Gene-. 1999. Assembly and Morphogenesis Induced by Sphingosine-1-Phosphate. Cell. 99:301–312. Levayer, R., A. Pelissier-Monier, and T. Lecuit. 2011. Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nature cell biology. 13:529– 40. 32 Li, Z., J.-H. Paik, J.-H. Paik, Z. Wang, T. Hla, and D. Wu. 2005. Role of guanine nucleotide exchange factor PRex-2b in sphingosine 1-phosphate-induced Rac1 activation and cell migration in endothelial cells. Prostaglandins & other lipid mediators. 76:95–104. Liu, B.P., N. Carolina, and C. Hill. 2000. Vav2 Activates Rac1 , Cdc42 , and RhoA Downstream from Growth Factor Receptors but Not Beta-1 Integrins. Molecular and cellular biology. 20:7160–7169. Liu, Z., J.L. Tan, D.M. Cohen, M.T. Yang, N.J. Sniadecki, S. Alom, C.M. Nelson, and C.S. Chen. 2010. Mechanical tugging force regulates the size of cell – cell junctions. PloS one. 107:9944–9949. Malliri, A., S. van Es, S. Huveneers, and J.G. Collard. 2004. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. The Journal of biological chemistry. 279:30092–8. Mammoto, T., S.M. Parikh, A. Mammoto, D. Gallagher, B. Chan, G. Mostoslavsky, D.E. Ingber, and V.P. Sukhatme. 2007. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. The Journal of biological chemistry. 282:23910–8. Mastrandrea, L.D., S.M. Sessanna, and S.G. Laychock. 2005. Sphingosine Kinase Activity and SPhingosine-1 Phosphate Production in rat Pancreatic Islets and INS-1 Cells. Diabetes. 54:1429–1436. Mckenzie, J.A.G. 2007. Roles of Rho / ROCK and MLCK in TNF-a-Induced Changes in Endothelial Morphology and Permeability. Journal of cellular physiology. 502935:221–228. McLaughlin, J.N., L. Shen, M. Holinstat, J.D. Brooks, E. Dibenedetto, and H.E. Hamm. 2005. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. The Journal of biological chemistry. 280:25048–59. Mehta, D., G.U. Ahmmed, B.C. Paria, M. Holinstat, T. Voyno-Yasenetskaya, C. Tiruppathi, R.D. Minshall, and A.B. Malik. 2003. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. The Journal of biological chemistry. 278:33492–500. Mehta, D., M. Konstantoulaki, G.U. Ahmmed, and A.B. Malik. 2005. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. The Journal of biological chemistry. 280:17320–8. Mehta, D., a Rahman, and a B. Malik. 2001. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. The Journal of biological chemistry. 276:22614–20. Meng, W., and M. Takeichi. 2009. Adherens junction: molecular architecture and regulation. Cold Spring Harbor perspectives in biology. 1:a002899. Mertens, A.E.E., T.P. Rygiel, C. Olivo, R. van der Kammen, and J.G. Collard. 2005. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. The Journal of cell biology. 170:1029–37. Mirzapoiazova, T., I. Kolosova, P. V Usatyuk, V. Natarajan, and a D. Verin. 2006. Diverse effects of vascular endothelial growth factor on human pulmonary endothelial barrier and migration. American journal of physiology. Lung cellular and molecular physiology. 291:L718–24. Monaghan-Benson, E., and K. Burridge. 2009. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. The Journal of biological chemistry. 284:25602–11. 33 Moy, a B., J. Van Engelenhoven, J. Bodmer, J. Kamath, C. Keese, I. Giaever, S. Shasby, and D.M. Shasby. 1996. Histamine and thrombin modulate endothelial focal adhesion through centripetal and centrifugal forces. The Journal of clinical investigation. 97:1020–7. Nacak, T.G., A. Alajati, K. Leptien, C. Fulda, H. Weber, T. Miki, F.S. Czepluch, J. Waltenberger, T. Wieland, H.G. Augustin, and J. Kroll. 2007. The BTB-Kelch protein KLEIP controls endothelial migration and sprouting angiogenesis. Circulation research. 100:1155–63. Naikawadi, R.P., N. Cheng, S.M. Vogel, F. Qian, D. Wu, A.B. Malik, and R.D. Ye. 2012. A critical role for phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger 1 in endothelial junction disruption and vascular hyperpermeability. Circulation research. 111:1517–27. Nakamura, T., T. Hayashi, Y. Nasu-Nishimura, F. Sakaue, Y. Morishita, T. Okabe, S. Ohwada, K. Matsuura, and T. Akiyama. 2008. PX-RICS mediates ER-to-Golgi transport of the N-cadherin/beta-catenin complex. Genes & development. 22:1244–56. Nelson, W.J. 2008. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 36:149–155. Ngok, S.P., R. Geyer, M. Liu, A. Kourtidis, S. Agrawal, C. Wu, H.R. Seerapu, L.J. Lewis-Tuffin, K.L. Moodie, D. Huveldt, R. Marx, J.M. Baraban, P. Storz, A. Horowitz, and P.Z. Anastasiadis. 2012. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. The Journal of cell biology. 199:1103–15. Van Nieuw Amerongen, G.P. 2002. Involvement of RhoA/Rho Kinase Signaling in VEGF-Induced Endothelial Cell Migration and Angiogenesis In Vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 23:211–217. Van Nieuw Amerongen, G.P., C.M.L. Beckers, I.D. Achekar, S. Zeeman, R.J.P. Musters, and V.W.M. van Hinsbergh. 2007. Involvement of Rho kinase in endothelial barrier maintenance. Arteriosclerosis, thrombosis, and vascular biology. 27:2332–9. Nimnual, A.S., L.J. Taylor, and D. Bar-Sagi. 2003. Redox-dependent downregulation of Rho by Rac. Nature cell biology. 5:236–41. Niu, J., J. Profirovic, H. Pan, R. Vaiskunaite, and T. Voyno-Yasenetskaya. 2003. G Protein betagamma subunits stimulate p114RhoGEF, a guanine nucleotide exchange factor for RhoA and Rac1: regulation of cell shape and reactive oxygen species production. Circulation research. 93:848–56. Ohta, Y., J.H. Hartwig, and T.P. Stossel. 2006. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nature cell biology. 8:803–14. Oldham, W.M., and H.E. Hamm. 2008. Heterotrimeric G protein activation by G-protein-coupled receptors. Nature reviews. Molecular cell biology. 9:60–71. Olsson, A.-K., A. Dimberg, J. Kreuger, and L. Claesson-Welsh. 2006. VEGF receptor signalling - in control of vascular function. Nature reviews. Molecular cell biology. 7:359–71. Orsenigo, F., C. Giampietro, A. Ferrari, M. Corada, A. Galaup, S. Sigismund, G. Ristagno, L. Maddaluno, G. Young Koh, D. Franco, V. Kurtcuoglu, D. Poulikakos, P. Baluk, D. McDonald, M. Grazia Lampugnani, and E. Dejana. 2012. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nature communications. 3:1208. Otani, T., T. Ichii, S. Aono, and M. Takeichi. 2006. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. The Journal of Cell Biology. 175:135–146. 34 Papaharalambus, C., W. Sajjad, A. Syed, C. Zhang, M.O. Bergo, R.W. Alexander, and M. Ahmad. 2005. Tumor necrosis factor alpha stimulation of Rac1 activity. Role of isoprenylcysteine carboxylmethyltransferase. The Journal of biological chemistry. 280:18790–6. Peng, J., F. He, C. Zhang, X. Deng, and F. Yin. 2011. Protein kinase C-α signals P115RhoGEF phosphorylation and RhoA activation in TNF-α-induced mouse brain microvascular endothelial cell barrier dysfunction. Journal of neuroinflammation. 8. Pfreimer, M., P. Vatter, T. Langer, T. Wieland, P. Gierschik, and B. Moepps. 2012. LARG links histamine-H1receptor-activated Gq to Rho-GTPase-dependent signaling pathways. Cellular signalling. 24:652–63. Price, L.S., M. Langeslag, J.P. ten Klooster, P.L. Hordijk, K. Jalink, and J.G. Collard. 2003. Calcium signaling regulates translocation and activation of Rac. The Journal of biological chemistry. 278:39413–21. Qiao, J., O. Holian, B.-S. Lee, F. Huang, J. Zhang, and H. Lum. 2008. Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. American journal of physiology. Cell physiology. 295:C1161–8. Ramchandran, R., D. Mehta, S.M. Vogel, M.K. Mirza, P. Kouklis, and A.B. Malik. 2008. Critical role of Cdc42 in mediating endothelial barrier protection in vivo. American journal of physiology. Lung cellular and molecular physiology. 295:L363–9. Ratheesh, A., G.A. Gomez, R. Priya, S. Verma, E.M. Kovacs, K. Jiang, N.H. Brown, A. Akhmanova, S.J. Stehbens, and A.S. Yap. 2012. Centralspindlin and α-catenin regulate Rho signalling at the epithelial zonula adherens. Nature cell biology. 14:818–830. Rauzi, M., P.-F. Lenne, and T. Lecuit. 2010. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468:1110–4. Richnau, N., and P. Aspenström. 2001. Rich, a rho GTPase-activating protein domain-containing protein involved in signaling by Cdc42 and Rac1. The Journal of biological chemistry. 276:35060–70. Ridley, A.J. 2006. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends in cell biology. 16:522–9. De Rooij, J., A. Kerstens, G. Danuser, M. a Schwartz, and C.M. Waterman-Storer. 2005. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. The Journal of cell biology. 171:153–64. Rossman, K.L., C.J. Der, and J. Sondek. 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nature reviews. Molecular cell biology. 6:167–80. Sahai, E., and C.J. Marshall. 2002a. RHO-GTPases and cancer. Nature reviews. Cancer. 2:133–42. Sahai, E., and C.J. Marshall. 2002b. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nature cell biology. 4:408–15. Saharinen, P., L. Eklund, J. Miettinen, R. Wirkkala, A. Anisimov, M. Winderlich, A. Nottebaum, D. Vestweber, U. Deutsch, G.Y. Koh, B.R. Olsen, and K. Alitalo. 2008. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nature cell biology. 10:527–37. Samarin, S.N., A.I. Ivanov, G. Flatau, C.A. Parkos, and A. Nusrat. 2007. Rho / Rho-associated Kinase-II Signaling Mediates Disassembly of Epithelial Apical Junctions. Molecular and cellular biology. 18:3429– 3439. Sander, E.E., S. van Delft, J.P. ten Klooster, T. Reid, R. a van der Kammen, F. Michiels, and J.G. Collard. 1998. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. The Journal of cell biology. 143:1385–98. 35 Sanz-Moreno, V., G. Gadea, J. Ahn, H. Paterson, P. Marra, S. Pinner, E. Sahai, and C.J. Marshall. 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 135:510–23. Satpathy, M., P. Gallagher, M. Lizotte-Waniewski, and S.P. Srinivas. 2004. Thrombin-induced phosphorylation of the regulatory light chain of myosin II in cultured bovine corneal endothelial cells. Experimental eye research. 79:477–86. Schaphorst, K.L., E. Chiang, K.N. Jacobs, A. Zaiman, V. Natarajan, F. Wigley, and J.G.N. Garcia. 2003. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. American journal of physiology. Lung cellular and molecular physiology. 285:L258–67. Schlegel, N., and J. Waschke. 2009. Impaired cAMP and Rac 1 Signaling Contribute to TNF-α-induced Endothelial Barrier Breakdown in Microvascular Endothelium. Microcirculation. 16:521–533. Schulte, D., V. Küppers, N. Dartsch, A. Broermann, H. Li, A. Zarbock, O. Kamenyeva, F. Kiefer, A. Khandoga, S. Massberg, and D. Vestweber. 2011. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. The EMBO journal. 30:4157–70. Shewan, A.M., M. Maddugoda, A. Kraemer, S.J. Stehbens, S. Verma, E.M. Kovacs, and A.S. Yap. 2005. Myosin 2 Is a Key Rho Kinase Target Necessary for the Local Concentration of E-Cadherin at Cell – Cell Contacts □. Molecular and cellular biology. 16:4531–4542. Shin, K., V.C. Fogg, and B. Margolis. 2006. Tight junctions and cell polarity. Annual review of cell and developmental biology. 22:207–35. Sousa, S., D. Cabanes, C. Archambaud, F. Colland, E. Lemichez, M. Popoff, S. Boisson-Dupuis, E. Gouin, M. Lecuit, P. Legrain, and P. Cossart. 2005. ARHGAP10 is necessary for alpha-catenin recruitment at adherens junctions and for Listeria invasion. Nature cell biology. 7:954–60. Spindler, V., N. Schlegel, and J. Waschke. 2010. Role of GTPases in control of microvascular permeability. Cardiovascular research. 87:243–53. Stam, J.C., E.E. Sander, F. Michiels, F.N. van Leeuwen, H.E. Kain, R. a van der Kammen, and J.G. Collard. 1997. Targeting of Tiam1 to the plasma membrane requires the cooperative function of the N-terminal pleckstrin homology domain and an adjacent protein interaction domain. The Journal of biological chemistry. 272:28447–54. Strub, G.M., M. Maceyka, N.C. Hait, S. Milstien, and S. Spiegel. 2010. Extracellular and intracellular actions of Sphingosine-1-phosphate. Adv Exp Med Bio. 688:141–155. Su, Z., C.N. Hahn, G.J. Goodall, N.M. Reck, A.F. Leske, A. Davy, G. Kremmidiotis, M.A. Vadas, and J.R. Gamble. 2004. A vascular cell-restricted RhoGAP, p73RhoGAP, is a key regulator of angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 101. Takaishi, K., T. Sasaki, H. Kotani, H. Nishioka, and Y. Takai. 1997. Regulation of cell-cell adhesion by rac and rho small G proteins in MDCK cells. The Journal of cell biology. 139:1047–59. Tauseef, M., V. Kini, N. Knezevic, M. Brannan, R. Ramchandaran, H. Fyrst, J. Saba, S.M. Vogel, A.B. Malik, and D. Mehta. 2008. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circulation research. 103:1164– 72. Terry, S.J., C. Zihni, A. Elbediwy, E. Vitiello, I. V Leefa Chong San, M.S. Balda, and K. Matter. 2011. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nature cell biology. 13:159–66. 36 Vestweber, D. 2008. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arteriosclerosis, thrombosis, and vascular biology. 28:223–32. Vestweber, D., M. Winderlich, G. Cagna, and A.F. Nottebaum. 2009. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends in cell biology. 19:8–15. Wang, B., F.G. Wylie, R.D. Teasdale, and J.L. Stow. 2005. Polarized trafficking of E-cadherin is regulated by Rac1 and Cdc42 in Madin-Darby canine kidney cells. American journal of physiology. Cell physiology. 288:C1411–9. Wang, Q., M. Liu, T. Kozasa, J.D. Rothstein, P.C. Sternweis, and R.R. Neubig. 2004. Thrombin and lysophosphatidic acid receptors utilize distinct rhoGEFs in prostate cancer cells. The Journal of biological chemistry. 279:28831–4. Warrington, S.J., H. Strutt, and D. Strutt. 2013. The Frizzled-dependent planar polarity pathway locally promotes E-cadherin turnover via recruitment of RhoGEF2. Development (Cambridge, England). 1054:1045–1054. Wei, X.N., B.C. Han, J.X. Zhang, X.H. Liu, C.Y. Tan, Y.Y. Jiang, B.C. Low, B. Tidor, and Y.Z. Chen. 2011. An integrated mathematical model of thrombin-, histamine-and VEGF-mediated signalling in endothelial permeability. BMC systems biology. 5:112. Wells, C.D., J.P. Fawcett, A. Traweger, Y. Yamanaka, M. Goudreault, K. Elder, S. Kulkarni, G. Gish, C. Virag, C. Lim, K. Colwill, A. Starostine, P. Metalnikov, and T. Pawson. 2006. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 125:535–48. Wennerberg, K., K.L. Rossman, and C.J. Der. 2005. The Ras superfamily at a glance. Journal of cell science. 118:843–6. Wildenberg, G. a, M.R. Dohn, R.H. Carnahan, M. a Davis, N. a Lobdell, J. Settleman, and A.B. Reynolds. 2006. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 127:1027–39. Wojciak-Stothard, B., and A.J. Ridley. 2002. Rho GTPases and the regulation of endothelial permeability. Vascular Pharmacology. 39:187–199. Wójciak-Stothard, B., a Entwistle, R. Garg, and a J. Ridley. 1998. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. Journal of cellular physiology. 176:150–65. Wójciak-stothard, B., S. Potempa, T. Eichholtz, and A.J. Ridley. 2001. Rho and Rac but not Cdc42 regulate endothelial cell permeability. Journal of cell science. 114:1343–1355. Xiaolu, D., P. Jing, H. Fang, Y. Lifen, W. Liwen, Z. Ciliu, and Y. Fei. 2011. Role of p115RhoGEF in lipopolysaccharide-induced mouse brain microvascular endothelial barrier dysfunction. Brain research. 1387:1–7. Yamada, S., and W.J. Nelson. 2007. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. The Journal of cell biology. 178:517–27. Yamada, S., S. Pokutta, F. Drees, W.I. Weis, and W.J. Nelson. 2005. Deconstructing the cadherin-cateninactin complex. Cell. 123:889–901. Yamamoto, M., S.H. Ramirez, S. Sato, T. Kiyota, R.L. Cerny, K. Kaibuchi, Y. Persidsky, and T. Ikezu. 2008. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. The American journal of pathology. 172:521–33. 37