Robinson_JALSResearchPaper
advertisement

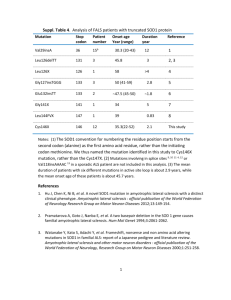
Running head: AMYOTROPHIC LATERAL SCLEROSIS Amyotrophic Lateral Sclerosis Jessica Robinson The University of Montevallo 1 AMYOTROPHIC LATERAL SCLEROSIS 2 History Amyotrophic lateral sclerosis is a disease of the nervous system that causes motor neuron degradation (“What is ALS?”). It is also referred to as Lou Gehrig’s disease after Lou Gehrig, a famous baseball player diagnosed with amyotrophic lateral sclerosis in the 1930s (“Lou Gehrig’s Disease”). That same site also went on to describe that the first publication about amyotrophic lateral sclerosis was written in 1869 by a French doctor named Jean-Martin Charcot; in France the disease is called maladie de Charcot in honor of his findings. Related syndromes and development Wijesekera & Leigh (2009) describe various syndromes that are covered under the amyotrophic lateral sclerosis description: Other syndromes related to this spectrum of disorders include, Progressive bulbar palsy (PBP), Progressive muscular atrophy (PMA), Primary lateral sclerosis (PLS), Flail arm syndrome (Vulpian-Bernhardt syndrome), Flail let syndrome (Pseudopolyneuritic form) and ALS with multi-system involvement (e.g. ALS Dementia). (p. 2) Amyotrophic lateral sclerosis can disrupt motor neurons in the bulbar, cervical, and lumbar regions of the body in both the upper and lower levels of the neuron from the cortex to the anterior horn of the spinal cord (Mitchell & Borasio, 2007) The most affected areas of the body include neurons of the skeletal muscle system, which includes anything controlling voluntary movement. Breathing can also be affected because an individual can consciously hold their breath, so eventually those neurons may degrade (“What is ALS?”). Different symptoms may present initially. Presentation of the cervical and bulbar sections often arises at the same time (Wijesekera & Leigh, 2009). Bulbar onset symptoms may include AMYOTROPHIC LATERAL SCLEROSIS 3 “slurring of speech (dysarthria), difficulty swallowing (dysphagia), or both” (Mitchell & Borasio, 2007, p. 2031). Often sialorrhoea, or uncontrollable drooling, may occur due to the dysarthria (Wijesekera & Leigh, 2009). Cervical onset symptoms involve the upper limbs, such as the arms and shoulders. Upper and lower neurons may both be involved (Mitchell & Borasio, 2007). With Lumbar onset the lower extremities are affected. The anterior horns of the neurons in the lumbar region of the spinal cord degrade (Mitchell & Borasio, 2007). Mitchell & Borasio go on to explain that individuals may have problems walking, holding up their feet and legs, and tripping over objects (2007). Two thirds of individuals who are diagnosed with amyotrophic lateral sclerosis will have a spinal form that presents with symptoms “distally or proximally in the upper limbs and lower limbs” (Wijesekera & Leigh, 2009, p. 3). Mitchell and Borasio also describe the disease as it progresses, explaining that patients may lose any function of facial muscles, making chewing food impossible. The ability to speak will also fade. All limb movement will cease to exist. Dyspnoea, or breathing difficulty, is a main factor in the death of patients due to respiratory failure. Patients may also have heavy mucous secretions, uncontrollable giggling or crying, pain, and depression (2007). The life expectancy after diagnosis is between three and five years, with 50 percent dying within three (Mitchell & Borasio, 2007). Classification There are two different categories of amyotrophic lateral sclerosis: sporadic and familial. The most common is sporadic amyotrophic lateral sclerosis, or SALS. The sporadic form occurs in 90 to 95 percent of cases (“What is ALS?”). This means there is no found genetic cause. Males have been observed in more cases than females, with the ratio being approximately one and a half males to every one female (Wijesekera & Leigh, 2009). The article went on to say that AMYOTROPHIC LATERAL SCLEROSIS 4 the average for the population is approximately 2 per 100,000 people. The average age of incidence is 55-64 years, with only 5 percent happening before the age of 30 (Wijesekera & Leigh, 2009). Unfortunately, there has not been a conclusive answer as to what causes sporadic amyotrophic lateral sclerosis. There has been much research in the possibility of methylaminoalanine being a possible factor. There is an unusually high instance of the disease in Guam, where a primary food is the cyad seed. Cyad seeds are also a primary food of the fruit bat, which “has been reported to bioaccumulate methylaminoalanine” (Mitchell & Borasio, 2007, p. 2032). Unfortunately, there has been no conclusive evidence in the matter. There is ongoing research to correlate environmental factors with the disease. Familial amyotrophic lateral sclerosis, or FALS, occurs in the remaining five to ten percent of cases, which generally have an “autosomal-dominant pattern of inheritance” (Mitchell & Borasio, 2007, p. 2033). This means that offspring have a 50 percent chance of inheriting the disease. Mitchell & Borasio have also found research to link 10 to 20 percent of familial cases with a mutation in the copper/zinc superoxide dismutase (SOD1) gene (2007). AMYOTROPHIC LATERAL SCLEROSIS 5 In the research found by Wijeskera & Leigh (2009), mutations were found in other genes as well which is detailed in the table below: Pathogenesis Though there is not a definitive answer as to what causes amyotrophic lateral sclerosis, it is believed that there are conglomerates of factors that contribute to the pathogenesis of the disease. One concern is the excitotoxicity of nerve cells. Excitotoxicity is the path by which nerve cells are damaged and killed by tremendous stimulation of neurotransmitters. In this case, overstimulation of the glutamate receptors by the neuromodulator glutamate causes an influx of calcium. Too much nitric oxide is then accumulated and neuronal death occurs (Wijesekera & Leigh, 2009). The article goes on to express the notation of higher levels of glutamate in amyotrophic lateral sclerosis. Mitchell & Borasio also noted the possibility of α-amino-hydroxy-5methylisoxasole-4 (AMPA) and kainite being excitotoxins (2007). Another cause that has AMYOTROPHIC LATERAL SCLEROSIS 6 previously been correlated to neurodegeneration is oxidative stress. As previously noted, mutations in the SOD1 gene are linked to familial amyotrophic lateral sclerosis, and with the mutation free radicals are often produced which cause toxicity to the nerve cells and eventually lead to their death (Mitchell & Borasio, 2007). Wijesekera & Leigh found several more possible links to pathogenesis: Impaired mitochondrial activity and axonal transport are possible pathogenic factors by mutations in the SOD1 gene. “Neurofilament proteins together with Peripherin (an intermediate filament protein) are found in the majority of axonal inclusions motor neurons of ALS patients. A toxic isoform of peripherin, (peripherin 61), has been found to be toxic to motor neurons even when expressed at modest levels and is detectable in spinal cords of ALS patients but not controls. (p.7) Other possibilities such as inflammatory dysfunction and deficits in neurotrophic factors & signaling pathways are also being pursued, but no foundational evidence has been found to consistently link them to the disease. Diagnosis In order to diagnose amyotrophic lateral sclerosis, other possible conditions, or mimic syndromes, must first be ruled out. Some conditions that are common in diagnostic errors are cerebral lesions, cervical spondylotic myelopathy, inclusion body myositis (IBM), multifocal motor neuropathy (MFMN), and Kennedy’s disease (Wijesekera & Leigh, 2009). The El Escorial diagnostic criteria were created in the late 1980s to define the different levels of possibility of having the disease. Mitchell and Borasio (2007) have shown it in their research and can be seen below: AMYOTROPHIC LATERAL SCLEROSIS 7 Electrophysiological studies Electrophysiological studies are conducted to rule out other disorders and to “document lower motor dysfunction in clinically involved and uninvolved regions” (Wijesekera & Leigh, 2009, p. 8). There are four types of electrophysiological tests that can be performed. The first is a nerve conduction study, which is mainly to exclude the possibility of other diseases that could be of similar nature. Next, conventional electromyography is used and is beneficial in determining lower motor neuron dysfunction and diagnosis of amyotrophic lateral sclerosis. There must be evidence of dysfunction in at least two of the four regions of the central nervous system: brainstem, cervical, thoracic, or lumbosacral spinal cord (2009) To assess the upper motor neuron function, transcranial magnetic stimulation and central motor conduction studies are performed. Medical personnel are looking for lesions by monitoring “motor amplitude, cortical threshold, central motor conduction time and silent periods” (Wijesekera & Leigh, 2009, p. 8). Indications of amyotrophic lateral sclerosis would AMYOTROPHIC LATERAL SCLEROSIS 8 include increase in central motor conduction time and below average firing rates of motor unit potentials (2009). Finally, quantitative electromyography may be used to measure the number of axons innervating muscles. In doing so, it is also possible to monitor the progression of the disease to see the number of axons depleted over time (2009). Neuroimaging studies Neuroimaging studies such as magnetic resonance imaging are used to detect lesions in the corticospinal tract. Lesions may be indicative of a mimicking disease. However, demyelination of neurons may also be detected, which is a common sign in amyotrophic lateral sclerosis (Wijesekera & Leigh, 2009). Muscle biopsy and neuropathalogical studies Biopsies are not a common or required measure to be taken in diagnosing amyotrophic lateral sclerosis. However, they may be beneficial, especially to determine cause of death (Wijesekera & Leigh, 2009, p. 8). The possibility of replacing lost neurons is being investigated with stem cell research. In the past, there have been ethical issues that correlate with using embryonic stem cells, but now scientists are looking at using induced pluripotent stem cells. This process requires skin cells that are reversed to a pluripotent state and then reprogrammed to become the cells needed. In a study conducted by Dimos, et al. (2008), researchers were able to successfully reprogram skin cells from a human to induced pluripotent stem cells. Two control subjects were initially found to compare against, and two sisters with motor neuron disease were included as the experimental group in the study. Both women in the experimental group were heterozygous for the L144F dominant allele of SOD1 gene. The first subject, identified as A29, had obvious symptoms of disease, which included “difficulty in swallowing, and weakness of the arms and AMYOTROPHIC LATERAL SCLEROSIS 9 legs” (Dimos, et al., 2008, p. 2). The second, identified as A30, was found to have signs of upper motor disease after examination in her lower extremities. iPS research Both individuals A29 and A30 had a skin biopsy, and characteristics of fibroblasts were found. The process which Dimos, et. al. (2008) used to reverse those fibroblasts to a stem cell state is described below: Transgenes encoding KLF4, SOX2, OCT4, and c-MYC were introduced into these fibroblasts by means of vesicular stomatitis virus glycoprotein (VSVg)-pseudotyped Moloney- based retroviruses. About 30,000 fibroblasts were transduced twice over 72 hours, cultured for 4 days in standard fibroblast medium, and then passaged onto a feeder layer of mouse embryonic fibroblasts in an ES cell-supportive medium. (p.2) After a two-week period, colonies were observed with embryonic stem cell morphology. Dimos, et al. (2008), decided to focus primarily on patient A29 because of her definitive case of classic amyotrophic lateral sclerosis. It was verified that the colonies being studied were from A29 through DNA fingerprinting analysis, direct sequencing, and polymerase chain reaction analysis. To verify that reversal of the fibroblasts to iPS cells had occurred and were indeed pluripotent, they were compared to embryonic stem cells. It was concluded that the A29 supposed iPS cells had an “active cell cycle profile, with 35 percent of cells in S or G2/M phases” (Dimos, et al., 2008, p. 2). Also, the karyotype was standard, alkaline phosphatase activity was demonstrated, and antigens correlated with embryonic stem cells were identified. It was found through quantitative reverse transcription that “genes expressed in pluripotent cells were transcribed at levels comparable to human embryonic stem cells in each of the three putative iPS cell lines” (Dimos, et al., 2008, p. 3). It was also discovered that in the AMYOTROPHIC LATERAL SCLEROSIS 10 induced pluripotent stem cells, stem cell marker genes SOX2 and OCT4 were expressed and had not been while the cell was in a fibroblast state. After cells are reversed back to the pluripotent state, it is hoped for that all retroviral transgenes will be silenced and no longer expressed, however two of the transgenes were still being produced: OCT4 and c-MYC. When introduced to a suspension culture, these induced pluripotent stem cells from A29 formed embryonic bodies, which differentiated into the three embryonic germ layers after being cultured for 13 to 16 days (Dimos, et al., 2008, p. 3). This officially verified that the fibroblasts from individual A29 with amyotrophic lateral sclerosis had been successfully reversed to induce pluripotent stem cells. The objective was then to generate these cells into spinal motor neurons and glia. The induced pluripotent stem cells were treated with “an agonist of the sonic hedgehog (SHH) signaling pathway and retinoic acid (RA)” (Dimos, et al., 2008, p. 3). The researchers then placed these treated cells on a laminin-coated surface where neuron-like growths were observed. These neuron-like growths were tested for a globular protein, tubulin, which is expressed exclusively in neurons. Β-Tubulin III was confirmed to be present by researchers, indicating that these were neurons. 651 out of 3262 nuclei examined were found to have the neuromarker HB9. They then found that over 90 percent of the 651 were also identified to have ISLET ½, a transcription factor associated with motor neuron development. Dimos, et al. (2008) also found progenitor markers OLIG2 and PAX6, which indicated the induced pluripotent stem cell-derived “motor neurons arose from progenitors similar to those found in the developing spinal cord” (Dimos, et al., 2008, p. 4). AMYOTROPHIC LATERAL SCLEROSIS 11 Future research There are still obstacles to overcome, however. The oncogenic genes and retroviruses need to be replaced with safer genes, and any defects in the individual’s neurons or glia must be corrected prior to being used in cell therapy (Dimos, et al., 2008, p. 4). This study was also conducted using patients with familial forms of amyotrophic lateral sclerosis, and the majority of people with the disease have the sporadic form. AMYOTROPHIC LATERAL SCLEROSIS 12 References Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., Croft, G. F., Saphier, G., Leibel, R., Goland, R., Wichterle, H., Henderson, C.E., & Eggan, K. Induced pluripotent stem cells generated from patients with als can be differentiated into motor neurons. (2008). Science, 321(5893), 1218-1221. doi: 10.1226/science.1158799 Genetic testing for als. Retrieved from http://www.alsa.org/about-als/genetic-testing-for-als.html Mitchell, J. D., & Borasio, G. D. Amyotrophic lateral sclerosis. (2007). Lancet, 369, 2031-2041. Retrieved from www.thelancet.com What is als?. Retrieved from http://www.alsa.org/about-als/what-is-als.html Wijesekera, L. C., & Leigh, P. N. Amyotrophic lateral sclerosis. (2009). Orphanet journal of rare diseases, 1-22. doi: 10.1186/11750-1172-4-3